Introduction
Fine particulate matter (PM2.5) is a type of small
particle with a diameter of <2.5 μm. PM2.5 is either a
solid or liquid particle with the ability to be suspended in air.
The results of epidemiological investigations have demonstrated
that exposure to PM2.5 is inextricably associated with the
development of multiple respiratory diseases, including asthma and
chronic obstructive pulmonary disease (COPD), which are exacerbated
by airway inflammation and a decline in pulmonary function
(1-3). However, the mechanisms by which
PM2.5 causes airway inflammation and lung injury are not yet fully
understood.
The results of a previous study demonstrated that
PM2.5 typically contains a large number of ions, polycyclic
aromatic hydrocarbons or lipopolysaccharides, which can induce free
radical production, causing reactive oxygen species (ROS) and
hydroxyl radical production (4).
The overproduction of hydroxyl radicals or ROS may lead to lipid
peroxidation of the cell membrane and an elevation in the
intracellular calcium concentration, thus disrupting the balance of
intracellular calcium homeostasis. In addition, the increase in the
intracellular calcium concentration may further enhance the
manufacturing of hydroxyl radicals or ROS (5). Notably, the PM2.5-induced
accumulation of ROS may play a crucial role in the release of
inflammatory mediators and cytokines, such as interleukin (IL)-6
and tumor necrosis factor (TNF)-α, and may induce mucus secretion
in the lungs (6). PM2.5-induced
lung injury may also cause the hypersecretion of various types of
mucins, such as mucin 5AC (MUC5AC). The results of previous studies
have revealed that MUC5AC levels are increased in the lumen of the
endoplasmic reticulum (ER) when airway cells are stimulated by
mucus-promoting substances, such as ROS (7,8).
Excess mucin and ROS production leads to the accumulation of
misfolded proteins in the ER, leading to ER stress (9). Thus, it was hypothesized that ER
stress may play a notable role in PM2.5-induced airway inflammation
and mucin hypersecretion. Moreover, nuclear factor (NF)-κB plays a
key role in ER stress-mediated inflammation (10). There are three major signaling
pathways which are associated with ER stress in eukaryotes; namely,
inositol-requiring kinase (IRE)1α, protein kinase R-like ER kinase
(PERK) and the activating transcription factor (ATF)6 pathways
(11). Keestra-Gounder et
al (12) demonstrated that
the nucleotide-binding oligomerization domain (NOD)-like receptor
family, including NOD1, activated NF-κB. This activation initiated
the gene expression of cytokines and the release of inflammatory
factors, which may reveal the association between ER stress and
inflammation (12). However, the
specific roles of the aforementioned pathways in regulating
PM2.5-induced airway inflammation, mucin hypersecretion and ROS
overproduction remain to be fully elucidated. Thus, further
investigations into the effects of PM2.5 on ER stress and
subsequent signaling cascades are required.
The results of a previous study by the authors
demonstrated that ER stress may promote airway mucin secretion
under the influence of neutrophil elastase (13). Thus, the present study aimed to
investigate the effects of ER stress on the management of
PM2.5-induced pro-inflammatory factor production, namely ROS and
MUC5AC. The present study also aimed to determine the potential
pathways involved in PM2.5-induced airway damage. The findings of
the present study may provide a basis for the development of novel
therapeutic strategies to relieve airway inflammation and mucin
hypersecretion.
Materials and methods
Cells, reagents and antibodies
The immortalized human bronchial epithelial cell
line, HBE135-E6E7, was purchased from Procell Life Science &
Technology Co., Ltd. The Cell Counting Kit (CCK)-8 (cat. no.
E-CK-A362), human MUC5AC enzyme-linked immunosorbent assay (ELISA)
kit (cat. no. E-EL-H2279c), Cy3-labeled goat anti-rabbit IgG (cat.
no. BA1032) and FITC-labeled goat anti-mouse IgG (cat. no. BA1101)
were obtained from Wuhan Boster Biological Technology Co., Ltd.
Human IL-6 (cat. no. MM-0049H2) and TNF-α (cat. no. MM-0122H2)
ELISA kits were obtained from MMbio. The Annexin V-FITC/PI double
staining cell apoptosis detection kit (cat. no. KGA108) was
purchased from Nanjing KeyGen Biotech Co., Ltd. Mouse anti-human
β-actin monoclonal antibody (cat. no. 66009-1-Ig) was purchased
from ProteinTech Group, Inc. Goat anti-rabbit IgG-HRP (cat. no.
BA1054) and goat anti-mouse IgG-HRP (cat. no. BA1051) antibodies
were purchased from Wuhan Boster Biological Technology, Ltd. Rabbit
anti-human glucose-regulated protein 78 (GRP78) polyclonal antibody
(cat. no. ab21685), and rabbit anti-human PERK monoclonal antibody
(cat. no. ab229912) were obtained from Abcam. Rabbit anti-human
phosphorylated (p-)PERK (cat. no. Bs-3330R) and MUC5AC (cat. no.
Bs-7166R) polyclonal antibodies were purchased from BIOSS. Mouse
anti-human CCAAT-enhancer binding protein homologous protein (CHOP)
monoclonal antibody (cat. no. 2895) was purchased from Cell
Signaling Technology, Inc. Mouse anti-human NOD1 monoclonal
antibody (cat. no. sc-398696) was purchased from Santa Cruz
Biotechnology, Inc. Rabbit anti-human IRE1α (cat. no. DF7709),
p-IRE1α (cat. no. AF7150) and ATF6 (cat. no. DF6009) polyclonal
antibodies were obtained from Affinity Biosciences. The NF-κB
activation-nuclear translocation assay kit (cat. no. SN368) and ROS
assay kit (cat. no. C1022) were purchased from the Beyotime
Institute of Biotechnology. The ER stress inhibitor,
4-phenylbutyric acid (4-PBA; cat. no. HY-A0281), the ATF6
inhibitor, Ceapin-A7 (cat. no. HY-108434), the PERK inhibitor,
ISRIB (cat. no. HY-12495), and the IRE1α inhibitor, 4μ8C
(cat. no. HY-19707), were purchased from MedChemExpress. The NOD1
agonist, C12-iE-DAP (cat. no. tlrl-c12dap), was obtained from
InvivoGen. Small interfering RNA (siRNA) targeting IRE1α and NOD1,
and negative control (NC) siRNA were obtained from GenePharma. The
IRE1α siRNA sequence was 5′-GCA GAU AGU CUC UGC CCA UTT-3′ (sense),
5′-AUG GGC AGA GAC UAU CUG CTT-3′ (antisense), the NOD1 siRNA
sequence was 5′-CCU UCU UUA CAG CCU UCU UTT-3′ (sense), 5′-AAG AAG
GCU GUA AAG AAG GTT-3′ (antisense), and the negative control siRNA
sequence was 5′-UUC UCC GAA CGU GUC ACG UTT-3′ (sense), 5′-ACG UGA
CAC GUU CGG AGA ATT-3′ (antisense).
Cell viability assay
The optimal concentration and duration of PM2.5
exposure were determined using a CCK-8 assay, as previously
described (14). Briefly,
1x104/ml cells in a 200-μl cell suspension were
cultured in a 96-well plate. The cells were subsequently exposed to
10, 50, 100, 200 or 400 μg/ml PM2.5 and cultured at 37°C in
a 5% CO2 environment. Following exposure to PM2.5 for
12, 24 and 48 h, 10 μl CCK-8 were added to each well, and
the cells were incubated at 37°C for a further 2 h. The optical
density (OD) was detected using a microplate reader (Multiskan FC;
Thermo Fisher Scientific, Inc.) at a wavelength of 450 nm.
Cell transfection
Cells were cultured in 24-well plates at a density
of 1.5x105/ml with 0.45 ml serum-free RPMI-1640 in each
well. To reach a final volume of 50 μl, 5 μl siRNA
transfection reagent (Lipofectamine™ 2000; cat. no. 52887;
Invitrogen; Thermo Fisher Scientific, Inc.) were mixed with 45
μl serum-free medium. In total, 1 μg aliquots of
IRE1α siRNA, NOD1 siRNA or NC siRNA were diluted with 50 μl
serum-free medium. Subsequently, the transfection reagent and
diluted siRNA were mixed and incubated at room temperature for an
additional 20 min. The transfection combination was added to each
well drop-by-drop, and vortexed for 10 sec prior to incubation for
16 h at room temperature. The supernatant was removed and the cells
were washed three times with phosphate-buffered saline (PBS). The
cells were subsequently incubated with fresh RPMI-1640 medium
containing 10% fetal bovine serum for 24 h prior to exposure to 100
μg/ml PM2.5.
Cell culture and grouping
The HBE135-E6E7 cells were cultured in RPMI-1640
medium at 37°C in a humidity incubator with 5% CO2. The
cells were grouped as follows: i) The control group, in which the
cells were cultured under normal conditions (37°C in a humidity
incubator with 5% CO2) with no treatment; ii) the PM2.5
group, in which the cells were exposed to 100 μg/ml PM2.5;
iii) the PM2.5 + 4-PBA group, in which the cells treated with 5
mmol/l 4-PBA for 2 h prior to PM2.5 exposure, as previously
described (15); iv) the PM2.5 +
ISRIB group, in which the cells were treated with 200 nmol/l ISRIB
for 2 h prior to PM2.5 exposure, as previously described (16); v) the PM2.5 + Ceapin-A7 group, in
which the cells were treated with 20 μmol/l Ceapin-A7 for 2
h prior to PM2.5 exposure, as previously described (17); vi) the PM2.5 + 4μ8C group,
in which the cells were treated with 10 μmol/l 4μ8C
for 2 h prior to PM2.5 exposure, as previously described (18); vii) the PM2.5 + NC siRNA group, in
which the cells were transfected with NC siRNA and subsequently
exposed to PM2.5; viii) the PM2.5 + IRE1α siRNA group, in which the
cells were transfected with IRE1α siRNA and subsequently exposed to
PM2.5; ix) the PM2.5 + NOD1 siRNA group, in which the cells were
transfected with NOD1 siRNA and subsequently exposed to PM2.5; and
x) the PM2.5 + C12-iE-DAP group, in which cells were treated with
10 μg/ml C12-iE-DAP, as previously described (19), and subsequently exposed to PM2.5.
Prior to subsequent experiments, all the cells were incubated at
37°C for an additional 24 h following grouping.
Western blot analysis
Total protein was extracted from the cells using
RIPA buffer reagent supplemented with PMSF. Total protein was
quantified using a BCA assay and proteins were separated on a 5-10%
SDS-PAGE gel; the amount of protein sampled for each sample was 20
μl. The same amount of protein for each sample was
subsequently transferred onto PVDF membranes and blocked with PBS
containing 0.05% Tween-20 and 5% skimmed milk for 1 h. The
membranes were incubated for 2 h at room temperature with primary
antibodies against GRP78, CHOP, p-PERK, PERK, ATF6, NOD1, p-IRE1α,
IRE1α (dilution for all, 1:500) and β-actin (dilution, 1:1,000).
Following washing three times, the membranes were incubated with an
HRP-conjugated secondary antibody (dilution, 1:5,000) for 2 h at
room temperature. After washing three times, protein bands were
visualized using an enhanced chemiluminescence (ECL) reagent
(Applygen Technologies Inc.). Protein expression was quantified
using ImageJ software (version 1.8.0; National Institutes of
Health) with β-actin as the loading control. The experiment was
performed in triplicate, as previously prescribed (20).
Flow cytometric analysis
The apoptosis of the HBE135-E6E7 cells was examined
using flow cytometry. The cells were inoculated in six-well plates
at a density of 3x105 cells per well. The cells were
digested with 0.25% trypsin without EDTA, and following the
termination of digestion, the treated cells were collected and
centrifuged at 800 x g for 5 min at room temperature. The cells
were subsequently washed three times with PBS, resuspended in 500
μl binding buffer, and incubated with AV-FITC (5 μl)
and PI (5 μl) at room temperature for 15 min in the dark.
Apoptotic cells were evaluated using a flow cytometer (cat. no.
cytoFLEX; Beckman Coulter, Inc.). The experiment was repeated three
times, as previously described (21).
ELISA
Cell supernatants were collected following treatment
as mentioned above. Various dilutions of cell lysates (1:0, 1:1,
1:2 and 1:5) were treated with PBS, and the expression levels of
IL-6, TNF-α and MUC5AC in the cell supernatants were assessed using
the corresponding kits, following the manufacturer's protocols. The
OD was evaluated using a microplate reader (Multiskan FC; Thermo
Fisher Scientific, Inc.) at a wavelength of 450 nm, and the results
are presented as percentages of the baseline controls, as
previously described (22).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the HBE135-E6E7 cells
in each group using TRIzol® reagent (Thermo Fisher
Scientific, Inc.), and the RNA samples were incubated at -20°C for
5 min. The mRNA expression levels of IL-6, TNF-α and MUC5AC were
measured using a two-step RT-PCR kit following the manufacturer's
protocols (cat. no. R223-01; Nanjing Vazyme Biotech Co., Ltd.). The
primer sequences used for PCR (cat. no. Q111-02; Nanjing Vazyme
Biotech Co., Ltd.) are displayed in Table I, which were designed using Primer
Premier 5.0 software. The following thermocycling conditions were
used for qPCR: Initial denaturation at 95°C for 10 min; 40 cycles
of denaturation at 95°C for 35 sec, annealing at 56°C for 55 sec
and extension at 72°C for 3 min. The relative mRNA levels were
quantified using the 2−ΔΔCq method (23). All samples were repeated in
triplicate.
 | Table ISequences of primers used in the
present study. |
Table I
Sequences of primers used in the
present study.
| Gene | Primer | Sequence
(5′-3′) | Accession no. |
|---|
| Homo β-actin | Forward |
CCCTGGAGAAGAGCTACGAG | NM_001101.5 |
| Reverse |
CGTACAGGTCTTTGCGGATG | |
| Homo IL-6 | Forward |
TTCGGTCCAGTTGCCTTCTCCC | NM_000600.5 |
| Reverse |
CCAGTGCCTCTTTGCTGCTTTC | |
| Homo TNF-α | Forward |
CCCATGTTGTAGCAAACCCTC | NM_000594.4 |
| Reverse |
AGAGGACCTGGGAGTAGATGA | |
| Homo MUC5AC | Forward |
CTACAATGGACAGCGCTTCC | NM_001304359.2 |
| Reverse |
AGAAGGAGAAGGTGGTTGGG | |
Immunofluorescence analysis
Following treatment as mentioned above, the cells in
each group were washed three times with PBS and subsequently fixed
with 4% paraformaldehyde at room temperature for 30 min. The cells
were washed three times and permeabilized for 15 min with 0.1%
Triton X100 (cat. no. ST795; Beyotime Institute of Biotechnology).
Subsequently, the cells were blocked with goat serum at room
temperature for 45 min, and incubated with antibodies against
MUC5AC (dilution, 1:100), NOD1 (dilution, 1:100) or IRE1α(dilution,
1:100) overnight at 4°C. Following three 5-min washes with PBS, the
cells were incubated with secondary antibodies labeled with FITC
(dilution, 1:100) or Cy3 (dilution, 1:100) for 1 h, and DAPI (cat.
no. C1002; Beyotime Institute of Biotechnology) at room temperature
for 5 min. The cells were analyzed using a fluorescence microscope
(DS-Fi3; Nikon Corporation) to determine the expression of target
proteins.
Assessment of ROS generation
DCFH-DA (cat. no. S0033; Beyotime Institute of
Biotechnology) was diluted in serum-free medium at 1:1,000 to reach
a final concentration of 10 μmol/l. Subsequently, the cell
culture medium was removed and 10 μmol/l DCFH-DA were added
to the cells for incubation at 37°C for 20 min. The cells were then
washed three times with serum-free medium to remove the DCFH-DA
that was not absorbed by the cells. Images were generated using a
fluorescence microscope (DS-Fi3; Nikon Corporation), and ImageJ
software (version 1.8.0; National Institutes of Health) was used to
assess the fluorescence intensity.
NF-κB activation-nuclear translocation
assay
Following treatment as mentioned above, the cells
were fixed and washed three times with PBS. Subsequently, the cells
were blocked with BSA and were then incubated with p65 primary
antibody (cat. no. SN368; Beyotime Institute of Biotechnology)
(dilution, 1:100) overnight at 4°C. Following three 5-min washes
with PBS, the cells were incubated with the secondary antibodies
labeled with Cy3 for 1 h. DAPI (dilution, 1:200) at room
temperature for 5 min was used to stain the nucleus. The cell
supernatants were removed and mounting medium (Jiangsu CITOTEST
Scientific Co., Ltd.) was added. Images were generated using a
fluorescence microscope (DS-Fi3; Nikon Corporation), and ImageJ
software (version 1.8.0; National Institutes of Health) was used to
assess the fluorescence intensity, indicative of the levels of
NF-κB activation-nuclear translocation.
Statistical analysis
All data are presented as the mean ± standard
deviation. At least three cell cultures were used in each
experiment and repeated in duplicate or triplicate. Multiple groups
were compared using one-way ANOVA followed by the Tukey's post hoc
test. All data were analyzed using SPSS (version 26.0; IBM Corp.)
and R software (version 4.1.2; AT&T Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
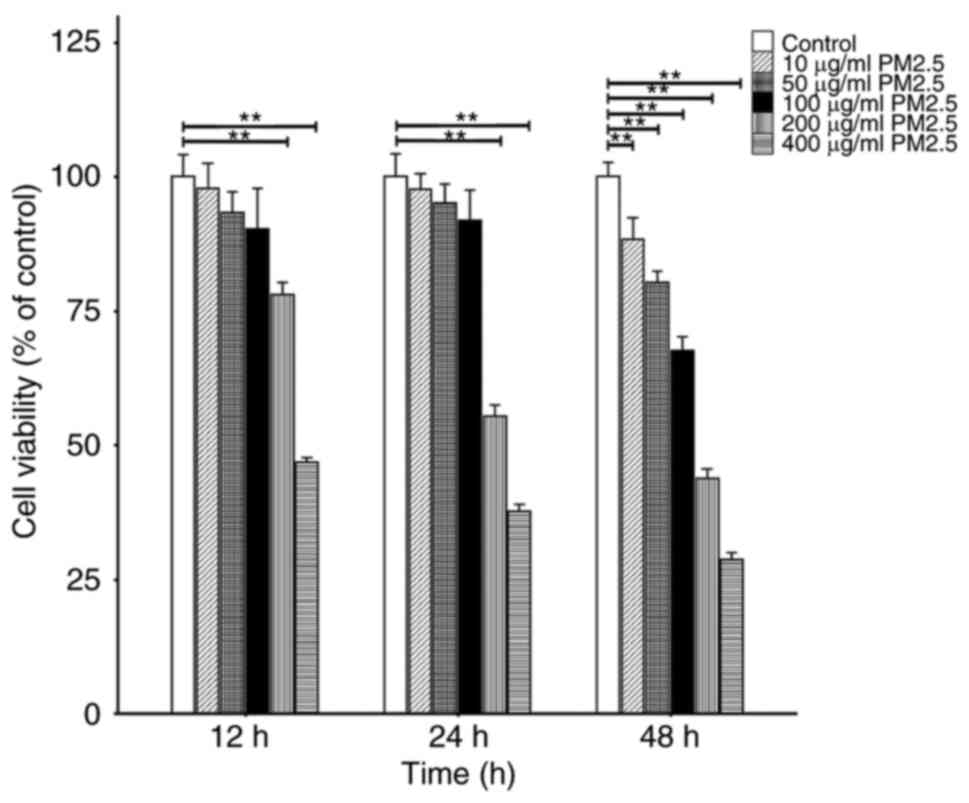
Cell viability following exposure to
PM2.5
To determine the appropriate concentration and
duration for exposure to PM2.5, the cells were exposed to various
concentrations (10, 50, 100, 200 and 400 μg/ml) of PM2.5 for
specific periods of time (12, 24 and 48 h). Cell viability was
determined using a CCK-8 assay. The results demonstrated that the
levels of cell viability were comparable from 12 to 24 h between
the control group and PM2.5-exposed groups, following exposure to
10-100 μg/ml PM2.5. However, at 12 to 48 h, the levels of
cell viability in the groups exposed to 200 and 400 μg/ml
PM2.5 decreased compared with the control group, as well as in the
10-100 μg/ml groups at 48 h. Thus, results of the present
study suggested that when used for up to 24 h and at 100
μg/ml, PM2.5 did not cause toxicity to the HBE135-E6E7 cells
(Fig. 1).
PM2.5 increases ER stress-related protein
expression, which may be reversed by 4-PBA
To determine whether exposure to PM2.5 activated the
ER stress pathway, the expression levels of ER stress-related
proteins were detected following exposure to PM2.5. In addition,
western blot analysis was carried out to evaluate the protein
expression levels of GRP78, CHOP, p-IRE1α, NOD1, ATF6 and p-PERK in
the HBE135- E6E7 cells. As illustrated in Fig. 2, exposure to 100 μg/ml
PM2.5 for 24 h resulted in increased protein expression levels of
GRP78, CHOP, p-IRE1α, NOD1, ATF6 and p-PERK, compared with the
control groups. Moreover, the expression levels of these proteins
were significantly decreased following pre-treatment with 4-PBA, an
ER stress inhibitor, compared with the groups exposed to PM2.5
alone (Fig. 2).
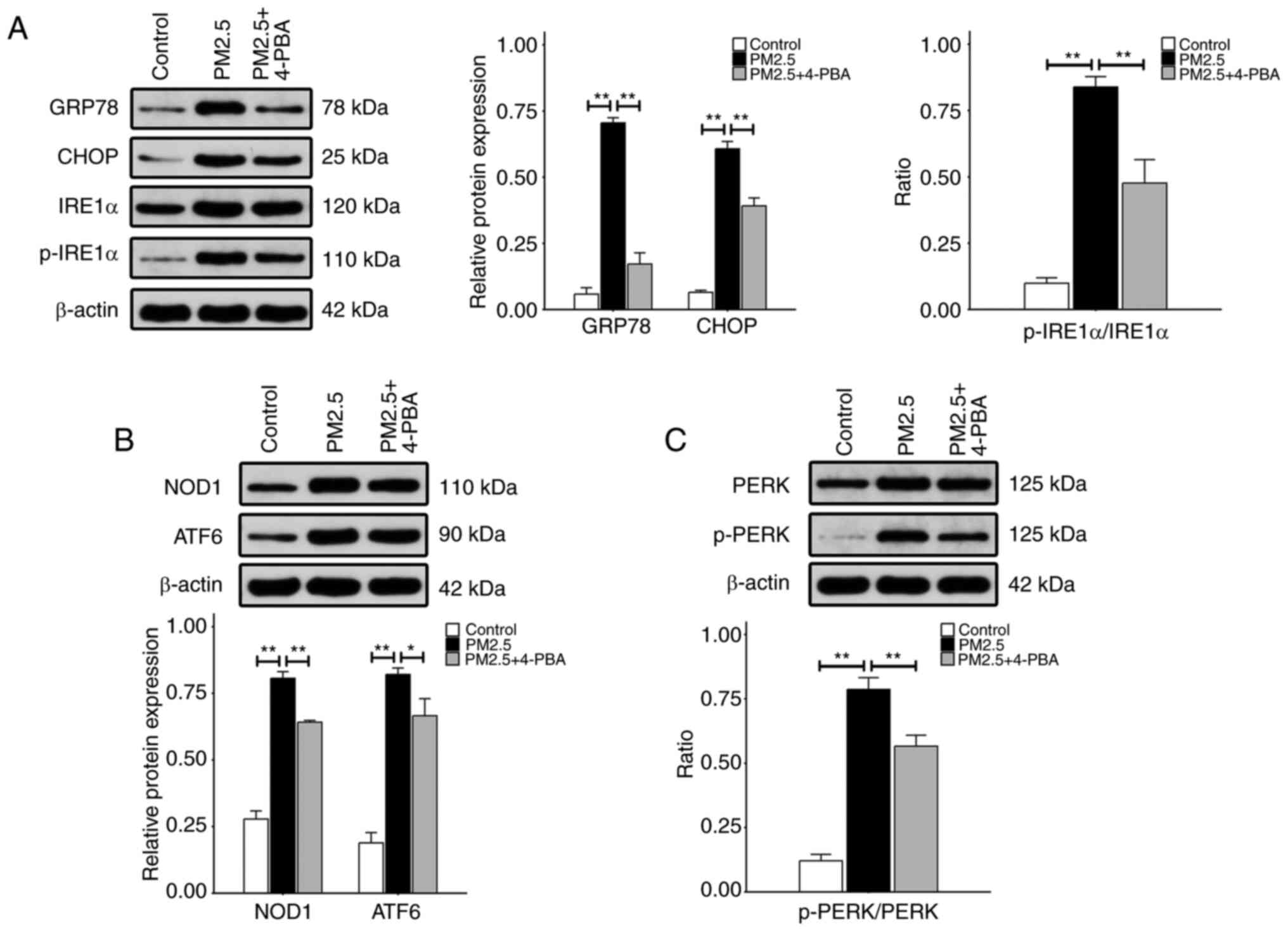 | Figure 2Expression of endoplasmic reticulum
stress-related proteins in HBE135-E6E7 cells. Cells were exposed to
100 μg/ml PM2.5, or treated with 5 mmol/l 4-PBA prior to
PM2.5 exposure. The levels of the aforementioned proteins were
measured using western blot analysis. (A) The relative protein
expression levels of GRP78 and CHOP are depicted as the ratio of
each to β-actin. The relative p-IRE1α protein expression levels are
presented as the ratio of p-IRE1α to IRE1α; β-actin blots were used
as the loading control. (B) The same method was used to indicate
the relative expression levels of NOD1 and ATF6 proteins. (C)
Relative p-PERK protein expression levels. Data are presented as
the mean ± SD (n=3 repeats per group). *P<0.05 and
**P<0.01. PM2.5, fine particulate matter; 4-PBA,
4-phenylbutyric acid; GRP78, glucose-regulated protein 78; CHOP,
CCAAT-enhancer binding protein homologous protein; IRE1α,
inositol-requiring kinase 1α; NOD1, nucleotide-binding
oligomerization domain 1; ATF6, activating transcription factor 6;
PERK, protein kinase R-like endoplasmic reticulum kinase; p-,
phosphorylated. |
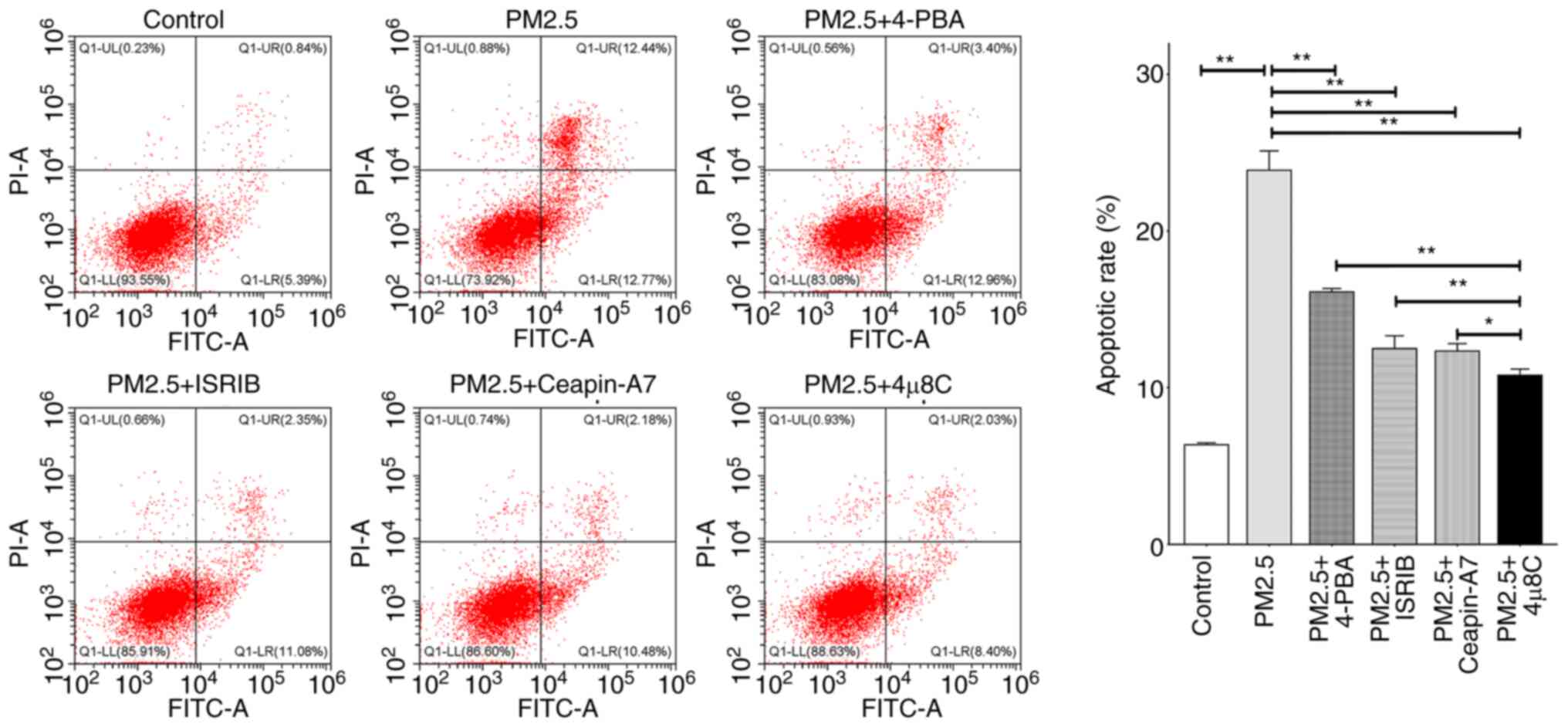
The IRE1α inhibitor, 4μ8C, exerts
suppressive effects on the PM2.5-induced apoptosis of airway
epithelial cells
The results of a previous study demonstrated that ER
stress was involved in the apoptosis of various cells (24); however, the effects of the
inhibition of ER stress-related pathways on the PM2.5-induced
apoptosis of airway epithelial cells remain to be fully elucidated.
Thus, herein, the HBE135-E6E7 cells were exposed to 100
μg/ml PM2.5 alone, or treated with 5 mmol/l 4-PBA (an ER
stress inhibitor), 200 nmol/l ISRIB (a PERK inhibitor), 20
μmol/l Ceapin-A7 (an ATF6 inhibitor) or 10 μmol/l
4μ8C (an IRE1α inhibitor) prior to exposure to PM2.5. The
results of flow cytometric analysis demonstrated that exposure to
PM2.5 significantly increased cell apoptosis compared with the
control group. However, the effects of PM2.5 exposure were
attenuated by the aforementioned ER stress-related inhibitors,
compared with PM2.5 exposure alone. Notably, 4μ8C exerted
the most potent inhibitory effects on PM2.5-induced apoptosis
compared with the other three inhibitors, suggesting that the IRE1α
signaling pathway may play a key role in PM2.5-induced airway
injury (Fig. 3).
PM2.5 promotes the production of airway
inflammatory cytokines and MUC5AC, which may be mitigated by ER
stress-related inhibitors, such as 4μ8C
To determine the role of ER stress in PM2.5-induced
airway inflammation and mucus hypersecretion, the HBE135-E6E7 cells
were treated with matched concentrations of PM2.5, 4-PBA, ISRIB,
Ceapin-A7 and 4μ8C. The results of ELISA and RT-qPCR assays
demonstrated that PM2.5 significantly promoted the secretion and
expression of IL-6, TNF-α and MUC5AC compared with the control
groups. However, following pre-treatment with ER stress-related
inhibitors, the PM2.5-induced hypersecretion and overexpression of
IL-6, TNF-α and MUC5AC were significantly decreased in each group
(Fig. 4A-F). The inhibitory
effects of 4μ8C pre-treatment on IL-6 secretion were more
pronounced than those following 4-PBA or ISRIB pre-treatment
(Fig. 4A); however, the
inhibitory effects of 4μ8C pre-treatment on IL-6 mRNA
expression were only more prominent than those of 4-PBA
pre-treatment (Fig. 4D). Compared
with the other three inhibitors, 4μ8C inhibited the
secretion of TNF-α and MUC5AC at the highest levels (Fig. 4B and C). Moreover, 4μ8C was
only superior to 4-PBA in inhibiting the TNF-α mRNA expression
levels (Fig. 4E), and was only
superior to ISRIB in inhibiting the MUC5AC mRNA expression levels
(Fig. 4F). The cell
immunofluorescence images of MUC5AC revealed that PM2.5 promoted
the intracellular expression of MUC5AC, which was markedly reversed
by the four inhibitors mentioned above (Fig. 4G). Collectively, the results of
the present study demonstrated that 4μ8C, an IRE1α
inhibitor, may exhibit the highest potential in inhibiting
PM2.5-induced airway inflammation and mucin hypersecretion.
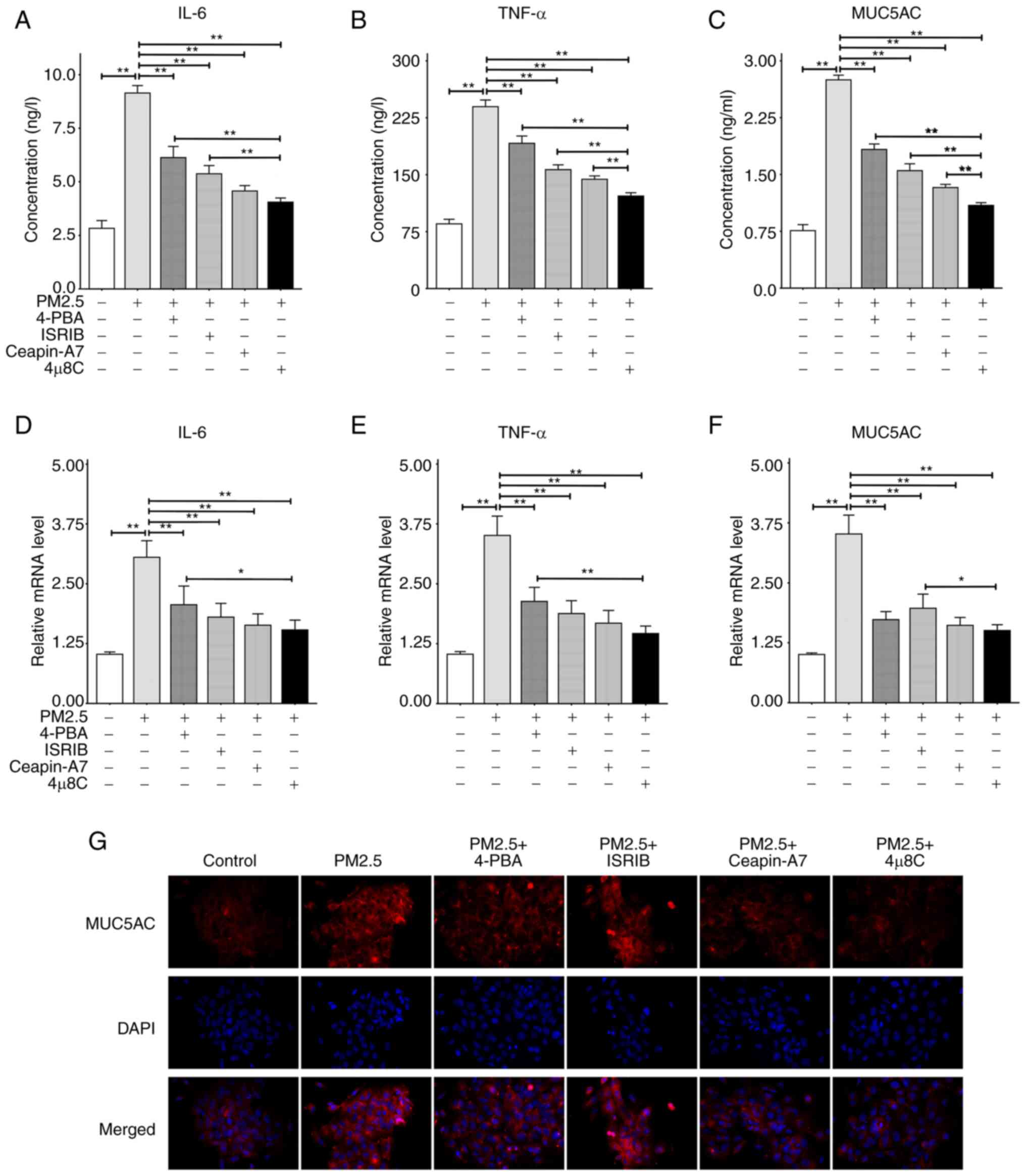 | Figure 4Secretion and mRNA expression levels
of IL-6, TNF-α and MUC5AC. ELISA was used to detect the levels of
inflammatory factors and mucin secretion. RT-qPCR assay was
utilized to measure the mRNA expression level of the target protein
relative to β-actin. Cells were pre-treated with 4-PBA (5 mmol/l),
ISRIB (200 nmol/l), Ceapin-A7 (20 μmol/l) and 4μ8C
(10 μmol/l) for 2 h, and then incubated with 100
μg/ml PM2.5 for 24 h. (A) ELISA results of IL-6. (B) ELISA
results of TNF-α. (C) ELISA results of MUC5AC. (D) RT-qPCR results
of IL-6. (E) RT-qPCR results of TNF-α. (F) RT-qPCR results of
MUC5AC. (G) Intracellular expression of MUC5AC detected using
immunofluorescence at x400 magnification. All data are presented as
the mean ± SD of each group (ELISA and RT-qPCR with n=3 repeatsper
group, and immunofluorescence with n=3 different field images per
group). *P<0.05 and **P<0.01. PM2.5,
fine particulate matter; IL-6, interleukin-6; TNF-α, tumor necrosis
factor-α; MUC5AC, mucin 5AC; 4-PBA, 4-phenylbutyric acid; ISRIB,
integrated stress response inhibitor; RT-qPCR, reverse
transcription-quantitative PCR. |
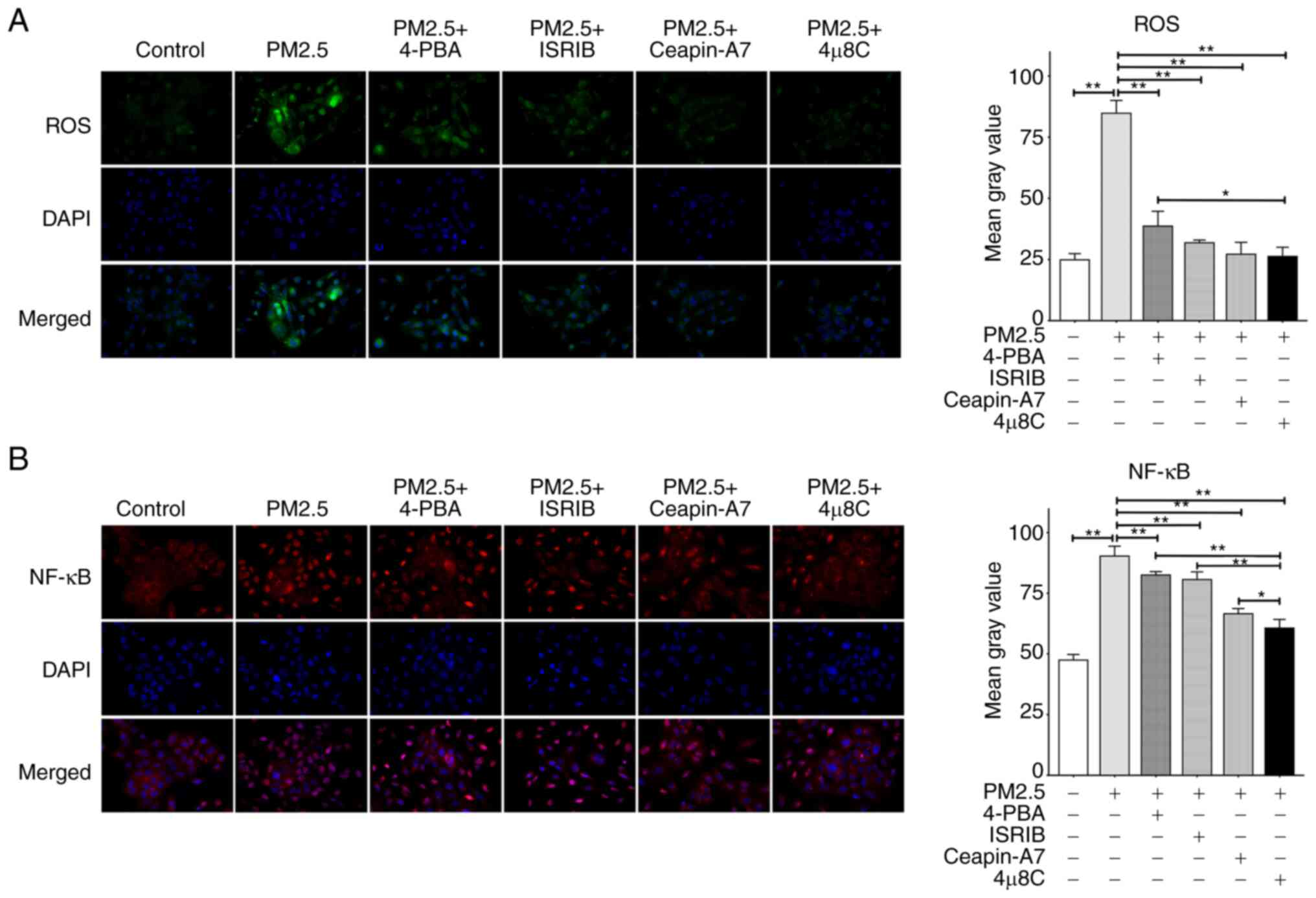
Inhibition of ER stress may alleviate the
increase of ROS and NF-κB induced by PM2.5
ROS affects several critical aspects of ER stress
(25); however, the role of ER
stress in the PM2.5-induced overproduction of ROS remains to be
fully elucidated. In the present study, the results of DCFH-DA
staining demonstrated that ROS production was markedly increased
following exposure to PM2.5, and this effect was mitigated
following treatment with ER stress inhibitors (Fig. 5A). Notably, the inhibitory effects
of 4μ8C on ROS were more pronounced than those of 4-PBA
(Fig. 5A); however, there was no
significant difference in the inhibitory effects between
4μ8C and ISRIB or Ceapin-A7. In addition, the effects of
three ER stress-related pathways (IRE1α, ATF6 and PERK) on NF-κB
activation-nuclear translocation were investigated in the present
study. As shown in Fig. 5B, PM2.5
exposure significantly enhanced NF-κB activation-nuclear
translocation, and this was suppressed by the four ER
stress-related inhibitors. Notably, 4μ8C, an IRE1α
inhibitor, inhibited NF-κB activation-nuclear translocation at a
higher level than ISRIB (a PERK inhibitor), Ceapin-A7 (an ATF6
inhibitor) and 4-PBA (an ER stress inhibitor). These results
demonstrated that IRE1α may play a key role in the regulation of
NF-κB activation.
IRE1α regulates NF-κB via NOD1 in
PM2.5-induced ER stress
The results of the present study demonstrated that
IRE1α exhibited the highest potential in the PM2.5-induced
regulation of ER stress and NF-κB; however, the potential signaling
cascades of IRE1α remain unclear. NOD1, a newly discovered ER
stress-related protein, may interact with IRE1α. Firstly, the
inhibition efficiency of IRE1α siRNA, NOD1 siRNA, or NC siRNA was
evaluated using RT-qPCR and it was found that compared with control
groups, no change in IRE1α or NOD1 mRNA levels was observed when
the HBE135-E6E7 cells were transfected with NC siRNA alone;
however, there was a significant decrease in the mRNA levels of
IRE1α or NOD1 following transfection with IRE1α siRNA or NOD1 siRNA
alone, and the inhibitory efficiencies reached ~70% in both cases
(Fig. 6A and B). The cells were
then transfected with IRE1α siRNA, NOD1 siRNA or NC siRNA prior to
exposure to 100 μg/ml PM2.5. As shown in Fig. 6C, the results of western blot
analysis demonstrated that exposure to PM2.5 promoted the
expression of p-IRE1α, and this was not affected by prior
transfection with NC siRNA. In addition, p-IRE1α was significantly
inhibited following transfection with IRE1α siRNA. Notably, results
of the western blot analysis were comparable with the
immunofluorescence intensity of IREα in each group (Fig. 6D). These results indicated that
IRE1α siRNA inhibited the expression and phosphorylation of IRE1α.
As shown in Fig. 6E, PM2.5
exposure significantly increased the protein expression levels of
NOD1, compared with the control group. There was no significant
difference in NOD1 expression between the PM2.5 + NC siRNA and
PM2.5 groups. However, pre-treatment with IRE1α siRNA markedly
reduced the PM2.5-induced increase in NOD1 expression, indicating
that IRE1α modulates NOD1 in PM2.5-induced ER stress. In addition,
NOD1 expression was markedly decreased in the PM2.5 + NOD1 siRNA
group compared with the PM2.5 group, suggesting that NOD1 siRNA
inhibited the expression of NOD1. Moreover, following pre-treatment
with C12-iE-DAP, a NOD1 agonist, there was a significant increase
in NOD1 expression compared with the PM2.5 group. Similar results
were obtained using immunofluorescence analysis (Fig. 6F). In addition, the effects of
pre-treatment with IRE1α siRNA or NOD1 siRNA on the PM2.5-induced
NF-κB activation-nuclear translocation were then investigated. As
shown in Fig. 6G, there was a
notable decrease in the activation-nuclear translocation of NF-κB
in the PM2.5 + IRE1α siRNA or PM2.5 + NOD1 siRNA groups, compared
with the PM2.5 + NC siRNA group. However, pre-treatment with
C12-iE-DAP further increased the NF-κB activation-nuclear
translocation induced by PM2.5. These results suggested that in
PM2.5-induced ER stress, IRE1α may promote the NF-κB
activation-nuclear translocation by increasing the expression of
NOD1.
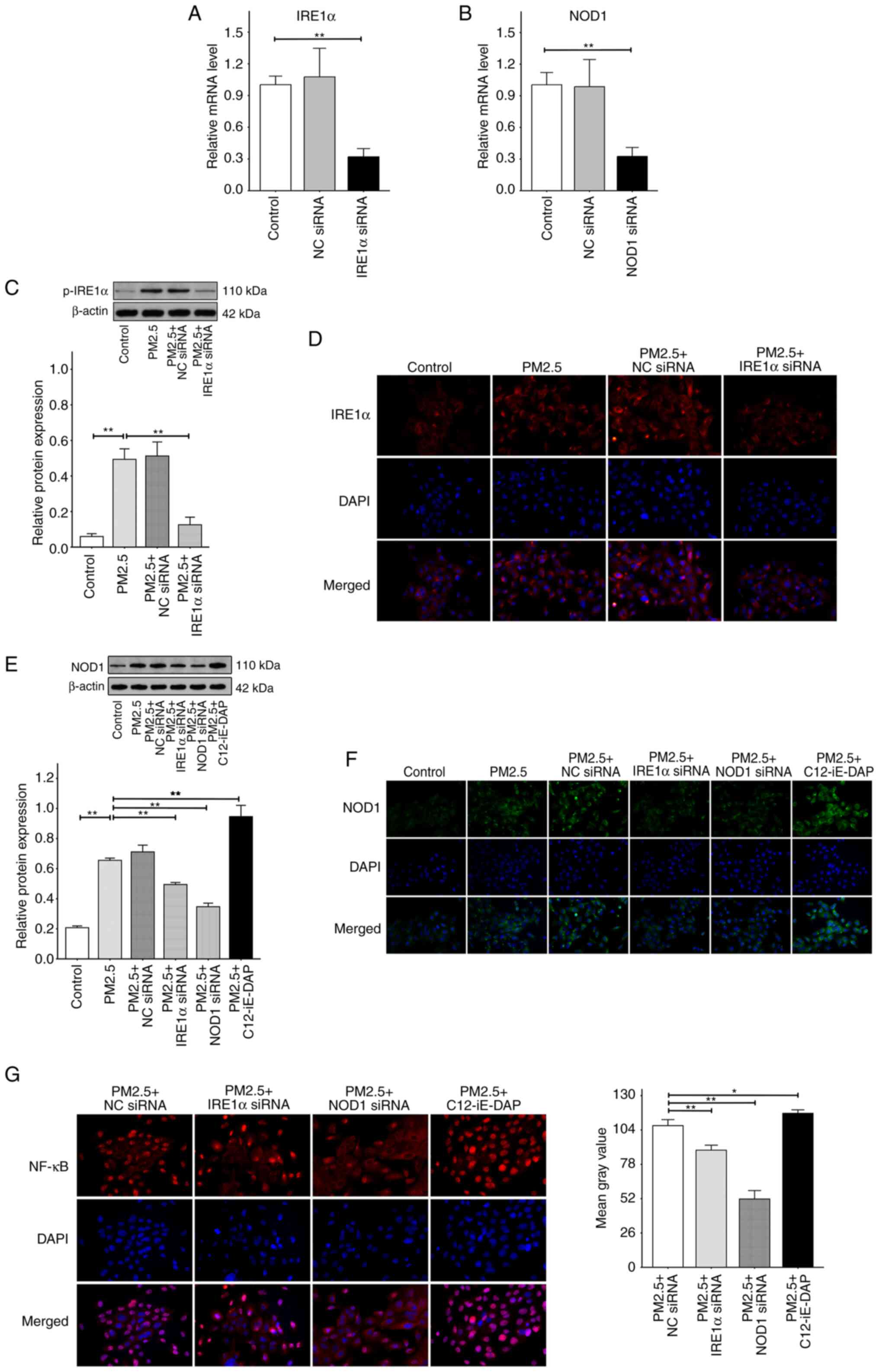 | Figure 6The levels of p-IRE1α, IRE1α and NOD1
expression, and NF-κB activation-nuclear translocation. The cells
were transfected with negative control (NC) siRNA, IRE1α siRNA or
NOD1 siRNA respectively, or were pretreated with 10 μg/ml
C12-iE-DAP (a NOD1 agonist) for 2 h prior to exposure to 100
μg/ml PM2.5 for 24 h. (A) The mRNA expression levels of
IRE1α relative to β-actin measured by RT-qPCR. (B) The mRNA levels
of NOD1 relative to β-actin. (C) Relative protein expression levels
of p-IRE1α, detected by western blot, were depicted as the ratio of
each group to β-actin. (D) The expression levels of IRE1α, assayed
by the immunofluorescence labeled by CY3, were detected using a
fluorescence microscope at x400 magnification. (E) Relative protein
expression levels of NOD1, detected using western blot analysis.
(F) The expression levels of NOD1, assayed using immunofluorescence
labeled with FITC, were detected using a fluorescence microscope at
x400 magnification. (G) The intracellular NF-κB activation-nuclear
translocation, assayed using a specialized kit, were also detected
using a fluorescence microscope at x400 magnification, and the mean
gray values were computed using ImageJ software. Data are presented
as the mean ± SD (RT-qPCR and western blot with n=3 repeats per
group, and immunofluorescence with n=3 different field images per
group). *P<0.05 and **P<0.01. RT-qPCR,
reverse transcription-quantitative PCR; PM2.5, fine particulate
matter; IRE1α, inositol-requiring kinase 1α; NOD1,
nucleotide-binding oligomerization domain 1. |
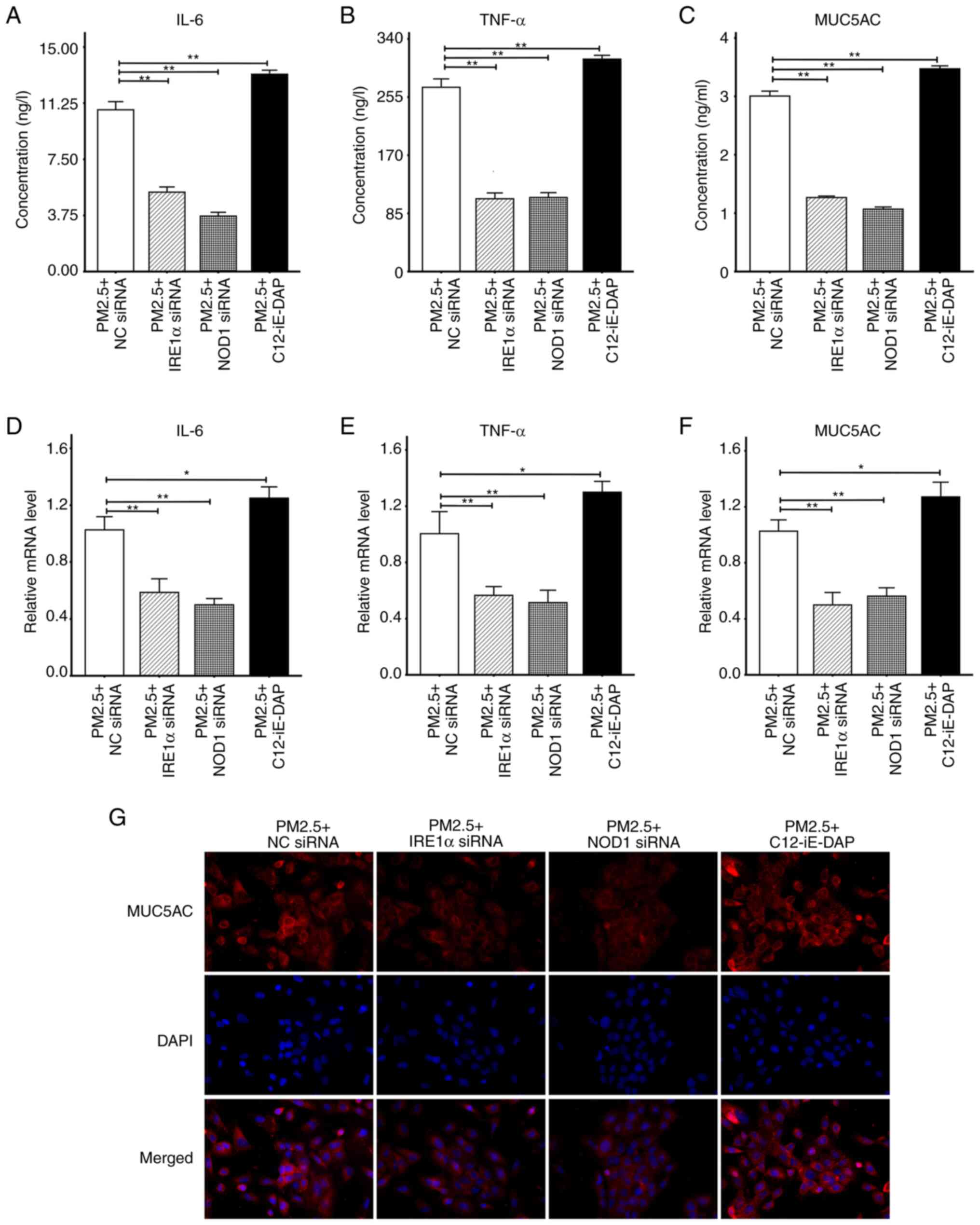
PM2.5 promotes airway inflammation and
mucin production through the IRE1α/NOD1/NF-κB pathway
The results of the present study demonstrated that
the expression levels of IL-6, TNF-α, MUC5AC, IRE1α and NOD1 were
increased following PM2.5 exposure, and IRE1α regulated the
activation-nuclear translocation of NF-κB via NOD1. The results of
previous studies have demonstrated that NF-κB plays a critical role
in regulating airway inflammation and mucus hypersecretion
(26,27). Therefore, herein, the effects of
the IRE1α/NOD1/NF-κB pathway on the expression of inflammatory
cytokines and mucin were investigated following exposure to PM2.5.
As illustrated in Fig. 7A-F,
there was a notable decrease in the secretion and mRNA expression
of IL-6, TNF-α and MUC5AC compared with the PM2.5 + NC siRNA group,
regardless of IRE1α siRNA or NOD1 siRNA pre-treatment. However,
following pre-treatment with C12-iE-DAP, the secretion and mRNA
expression of IL-6, TNF-α and MUC5AC were significantly increased.
The results of the immunofluorescence analysis demonstrated that
MUC5AC was markedly attenuated in the PM2.5 + IRE1α siRNA and PM2.5
+ NOD1 siRNA groups, compared with the PM2.5 + NC siRNA group. In
addition, the fluorescence intensity was markedly increased in the
PM2.5 + C12-iE-DAP group (Fig.
7G), and these results were comparable with those obtained
using ELISA and RT-qPCR (Fig.
7A-F). These results suggested that PM2.5 may promote the
expression of IREα, NOD1 and NF-κB activation-nuclear
translocation, thus, promoting airway inflammation and mucin
production.
Discussion
The findings of the present study demonstrated that
PM2.5 activated ER stress-related proteins, including CHOP, GRP78,
ATF6, PERK, p-PERK, IRE1α, p-IRE1α and NOD1. These proteins
enhanced the apoptosis of airway epithelial cells, resulting in an
increase in the levels of ROS, IL-6, TNF-α and MUC5AC. These
increases were significantly alleviated following pre-treatment
with ER stress-related inhibitors, such as 4-PBA, ISRIB, Ceapin-A7
and 4μ8C. Moreover, the results of the present study
demonstrated that transfection with IRE1α siRNA reduced NOD1,
NF-κB, IL-6, TNF-α and MUC5AC expression, and transfection with
NOD1 siRNA also decreased NF-κB, IL-6, TNF-α and MUC5AC expression.
C12-iE-DAP treatment increased the levels of NOD1, NF-κB, IL-6,
TNF-α and MUC5AC, suggesting that the IREα-induced promotion of
NOD1 increased NF-κB, which may play a role in the overproduction
of inflammatory cytokines and mucin induced by PM2.5.
The results of a previous study demonstrated that
various particles induce tissue necrosis and cell apoptosis,
followed by the release of intracellular contents. This may include
the release of non-zyme lactate dehydrogenase, which is associated
with the release of free radicals (28). Subsequently, the release of
intracellular contents may lead to the recruitment of inflammatory
cells, such as macrophages, monocytes and natural killer cells.
These cells may secrete TNF-α, which is associated with
inflammation in various processes (29). PM2.5 is often generated via the
burning of coal and diesel engine exhaust emissions. As the
aerodynamic diameter of PM2.5 is <2.5 μm (30), a higher proportion of the
particles are deposited in the lungs where they permeate deep
alveoli, thus, affecting the respiratory system and potentially
entering the bloodstream (31).
PM2.5 also promotes airway oxidative stress, inflammation and mucin
hypersecretion, which may cause long-term airway damage. At
present, research is focused on determining the effects of PM2.5 on
respiratory diseases, such as asthma and COPD. However, the
effectiveness of current treatment options for PM2.5 remain
limited, and further investigations are required to determine the
mechanisms responsible for PM2.5-induced airway damage.
Previous studies have demonstrated that the ER
stress response is a fundamental characteristic in numerous
inflammatory diseases, and this may activate the unfolded protein
response (UPR) (32,33). UPR performs an adaptive role in
protein quality assurance to promote optimal protein synthesis,
secretion and folding in ER to reverse ER stress (34). In addition, ER stress is involved
in various airway inflammatory diseases, including lung infections,
pulmonary fibrosis, asthma and COPD (33,35). However, the specific role of ER
stress in PM2.5-induced airway injury is not yet fully understood.
Molagoda et al (36)
reported that the inhibition of ER stress protected HaCaT human
keratinocytes from PM2.5-induced oxidative stress and apoptosis
(36), and similar results were
observed in the present study using HBE135-E6E7 airway epithelial
cells. Kim et al (37)
revealed that the inhibition of ER stress alleviated the
lipopolysaccharide-induced production of inflammatory cytokines in
healthy human bronchial epithelial cells (37), which was also consistent with the
results of the present study. The results of previous studies have
also confirmed that ER stress may be involved in the hypersecretion
of airway mucin (38,39). Notably, the role of ER stress in
PM2.5-induced mucin hypersecretion was demonstrated in the present
study. In summary, ER stress may play a key role in PM2.5-induced
airway injury; however, further investigations into the specific
underlying mechanisms are required.
As ER transmembrane receptors, ATF6, PERK and IRE1
are considered the three major signaling pathways in maintaining ER
homeostasis, and function differently in the pathogeneses of
various airway diseases (40-42). On the one hand, p-PERK promotes
the phosphorylation of eukaryotic translation initiation factor 2α
(eIF2α), inhibiting the transcription of numerous proteins. On the
other hand, IRE1 and ATF6 may localize in the nucleus and enhance
the transcription of UPR chaperone proteins, such as GRP78, to
enhance folding capacity and decimate misfolded proteins for
relieving ER stress (43). As
previously demonstrated, in a PM2.5-exposed macrophage cell line,
UPR was mediated by the PERK-eIF2α axis (44). However, in human airway epithelial
cells, IRE1 and ATF6 play a major role in mucin hypersecretion
(7,13). These results suggest that the
importance of the three aforementioned ER stress-related signaling
pathways may vary under different conditions. Notably, IRE1 exists
in two isoforms; namely, IRE1α and IRE1β. IRE1α may be expressed in
numerous cell types; however, IRE1β is restricted to epithelial
cells, mainly in the gastrointestinal tract (45). The present study demonstrated that
4μ8C, only inhibiting one of the pathways in the process of
ER stress, had a more potent inhibitory effect than 4-PBA
inhibiting ER stress as a whole. The reason for this result may be
that 4μ8C inhibits only one pathway of ER stress, which is
more targeted and specific compared to 4-PBA. The inhibition of
three pathways by 4-PBA may result in a weak inhibitory effect on
each pathway. The IRE1α pathway may be more crucial in promoting
PM2.5-induced ER stress. The results of a previous study confirmed
that IRE1α promoted NF-κB by increasing X-box-binding protein 1,
which plays a key role in the inflammatory response of airway
epithelial cells (46). The
results of the present study demonstrated that ATF6, PERK and IRE1α
exert specific regulatory effects on PM2.5-induced apoptosis and
the overproduction of ROS, IL-6, TNF-α and MUC5AC; however, IRE1α
exhibited a greater regulatory potential. Moreover, in homeostatic
states, GRP78 is attached to the specific domains of ATF6, PERK and
IRE1α to prevent them from activation. Under conditions of stress,
GRP78 is redirected to unfolded proteins of the ER, activating the
three transducers (47). In
addition, CHOP controls the intricate interaction between the
adaptive and apoptotic branches of the UPR (48). Therefore, both GRP78 and CHOP
exhibit potential as ER stress markers. Collectively, the results
of previous studies and those of the present study indicated that
GRP78 and CHOP may be elevated in the airway under the influence of
various stimuli, including ovalbumin, lipopolysaccharide, house
dust mites or PM2.5 (49,50).
Among the multitudinous airway cells, epithelial
cells serve as a physical barrier between the body and the external
environment, and activate the inflammatory response to external
stimuli (51). Epithelial cells
express numerous pattern recognition receptors, including Toll-like
receptors (TLRs), NOD1 and NOD2, to identify different external
triggers, producing chemokines and cytokines (52). A previous study demonstrated that
IRE1α activated TLR2, TLR4 and NF-κB for involvement in ER stress
caused by bacteria and mycobacteria (53). Notably, the results of a previous
study demonstrated that NOD1 and NOD2, which play roles in
detecting bacterial peptidoglycan, may also transduce ER stress
signals to trigger inflammation (54). Keestra-Gounder et al
(12) demonstrated that IRE1α
activation promoted NOD1 and NOD2 to mediate the inflammatory
branch of the UPR. The present study further demonstrated that
PM2.5 activated IREα, promoted NOD1 and enhanced NF-κB to
overproduce airway inflammation and mucin. However, further studies
are required to focus on further verifying the effects of ER stress
on ROS, inflammatory factors and mucin production, and determine
the regulatory role of the IRE1α/NOD1/NF-κB pathway in animal
models of PM2.5-induced lung injury. Moreover, further
investigations into the mutual promotion mechanism between IRE1α
and NOD1 in ER stress are warranted.
In conclusion, the results of the present study
suggested that ER stress was involved in PM2.5-induced apoptosis,
and the overproduction of ROS, inflammatory cytokines and mucin in
airway epithelial cells. Furthermore, the IRE1α/NOD1/NF-κB pathway
may play a critical role in airway inflammation and mucin
hypersecretion caused by PM2.5. Blocking the IRE1α/NOD1/NF-κB
pathway may exhibit potential in the treatment of airway
damage.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LH and CX obtained and evaluated the data, and wrote
the final manuscript. XT, SY and LW carried out the cell
experiments. QL and XZ designed and conceptualized the study. XZ
carefully reviewed this manuscript and managed the progress of the
experiments. LH, CX, QL and XZ confirm the authenticity of all the
raw data. All authors have read and agreed with the final version
of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Hainan Province Clinical
Medical Center, the Hainan Provincial Natural Science Foundation of
China (grant nos. ZDYF2020223, ZDKJ2021036, 820CXTD448 and
GHYF2022011) and the National Natural Science Foundation of China
(grant nos. 82260001, 82160012 and 81860001).
References
|
1
|
Gleason JA, Bielory L and Fagliano JA:
Associations between ozone, PM2.5, and four pollen types on
emergency department pediatric asthma events during the warm season
in New Jersey: A case-crossover study. Environ Res. 132:421–429.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Montoya-Estrada A, Torres-Ramos YD,
Flores-Pliego A, Ramirez-Venegas A, Ceballos-Reyes GM,
Guzman-Grenfell AM and Hicks JJ: Urban PM2.5 activates GAPDH and
induces RBC damage in COPD patients. Front Biosci (Schol Ed).
5:638–649. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davel AP, Lemos M, Pastro LM, Pedro SC, de
André PA, Hebeda C, Farsky SH, Saldiva PH and Rossoni LV:
Endothelial dysfunction in the pulmonary artery induced by
concentrated fine particulate matter exposure is associated with
local but not systemic inflammation. Toxicology. 295:39–46. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miller MR, Shaw CA and Langrish JP: From
particles to patients: Oxidative stress and the cardiovascular
effects of air pollution. Future Cardiol. 8:577–602. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brown DM, Donaldson K, Borm PJ, Schins RP,
Dehnhardt M, Gilmour P, Jimenez LA and Stone V: Calcium and
ROS-mediated activation of transcription factors and TNF-alpha
cytokine gene expression in macrophages exposed to ultrafine
particles. Am J Physiol Lung Cell Mol Physiol. 286:L344–L353. 2004.
View Article : Google Scholar
|
|
6
|
Liu K, Hua S and Song L: PM2.5 exposure
and asthma development: The key role of oxidative stress. Oxid Med
Cell Longev. 2022:36188062022.PubMed/NCBI
|
|
7
|
Kim MH, Bae CH, Choi YS, Na HG, Song SY
and Kim YD: Endoplasmic reticulum stress induces MUC5AC and MUC5B
expression in human nasal airway epithelial cells. Clin Exp
Otorhinolaryngol. 12:181–189. 2019. View Article : Google Scholar :
|
|
8
|
Park SH, Gong JH, Choi YJ, Kang MK, Kim YH
and Kang YH: Kaempferol inhibits endoplasmic reticulum
stress-associated mucus hypersecretion in airway epithelial cells
and ovalbumin-sensitized mice. PLos One. 10:e1435262015. View Article : Google Scholar
|
|
9
|
Martino MB, Jones L, Brighton B, Ehre C,
Abdulah L, Davis CW, Ron D, O'Neal WK and Ribeiro CMP: The ER
stress transducer IRE1β is required for airway epithelial mucin
production. Mucosal Immunol. 6:639–654. 2013. View Article : Google Scholar
|
|
10
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Bio. 13:89–102. 2012. View
Article : Google Scholar
|
|
12
|
Keestra-Gounder AM, Byndloss MX, Seyffert
N, Young BM, Chávez-Arroyo A, Tsai AY, Cevallos SA, Winter MG, Pham
OH, Tiffany CR, et al: NOD1 and NOD2 signalling links ER stress
with inflammation. Nature. 532:394–397. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu X, Li Q, Li L, Zeng M, Zhou X and Cheng
Z: Endoplasmic reticulum stress/XBP1 promotes airway mucin
secretion under the influence of neutrophil elastase. Int J Mol
Med. 47:812021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dai Y, Wang Y, Lu S, Deng X, Niu X, Guo Z,
Qian R, Zhou M and Peng X: Autophagy attenuates particulate matter
2.5-induced damage in HaCaT cells. Ann Transl Med. 9:9782021.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Iordache C and Duszyk M: Sodium
4-phenylbutyrate upregulates ENaC and sodium absorption in T84
cells. Exp Cell Res. 313:305–311. 2007. View Article : Google Scholar
|
|
16
|
Yan C, Zhang L, Lu B, Lyu D, Chen H, Song
F, Wang X, Chen Z, Fu Q and Yao K: Trans, trans-2,4-decadienal
(tt-DDE), a composition of cooking oil fumes, induces oxidative
stress and endoplasmic reticulum stress in human corneal epithelial
cells. Toxicol In Vitro. 68:1049332020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kubra K, Akhter MS, Saini Y, Kousoulas KG
and Barabutis N: Activating transcription factor 6 protects against
endothelial barrier dysfunction. Cell Signal. 99:1104322022.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pinto AR, Américo MF, Terenzi H and
Silveira DB: Inhibiting IRE-1 RNase signaling decreases HIV-1
Tat-induced inflammatory M1 state in microglial cells. Biochim
Biophys Acta Gen Subj. 1866:1302192022. View Article : Google Scholar
|
|
19
|
Porcherie A, Cunha P, Trotereau A, Roussel
P, Gilbert FB, Rainard P and Germon P: Repertoire of Escherichia
coli agonists sensed by innate immunity receptors of the bovine
udder and mammary epithelial cells. Vet Res. 43:142012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Scherbakov AM, Vorontsova SK, Khamidullina
AI, Mrdjanovic J, Andreeva OE, Bogdanov FB, Salnikova DI, Jurisic
V, Zavarzin IV and Shirinian VZ: Novel pentacyclic derivatives and
benzylidenes of the progesterone series cause anti-estrogenic and
antiproliferative effects and induce apoptosis in breast cancer
cells. Invest New Drug. 41:142–152. 2023. View Article : Google Scholar
|
|
21
|
Jurisic V, Srdic-Rajic T, Konjevic G,
Bogdanovic G and Colic M: TNF-α induced apoptosis is accompanied
with rapid CD30 and slower CD45 shedding from K-562 cells. J Membr
Biol. 239:115–122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jurisic V: Multiomic analysis of cytokines
in immuno-oncology. Expert Rev Proteomic. 17:663–674. 2020.
View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Hetz C, Zhang K and Kaufman RJ:
Mechanisms, regulation and functions of the unfolded protein
response. Nat Rev Mol Cell Bio. 21:421–438. 2020. View Article : Google Scholar
|
|
25
|
Cui X, Zhang Y, Lu Y and Xiang M: ROS and
endoplasmic reticulum stress in pulmonary disease. Front Pharmacol.
13:8792042022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Z, Yao N, Fu X, Wei L, Ding M, Pang
Y, Liu D, Ren Y and Guo M: Butylphthalide ameliorates airway
inflammation and mucus hypersecretion via NF-κB in a murine asthma
model. Int Immunopharmacol. 76:1058732019. View Article : Google Scholar
|
|
27
|
Ma B, Athari SS, Nasab EM and Zhao L:
PI3K/AKT/mTOR and TLR4/MyD88/NF-κB signaling inhibitors attenuate
pathological mechanisms of allergic asthma. Inflammation.
44:1895–1907. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jurisic V, Radenkovic S and Konjevic G:
The actual role of LDH as tumor marker, biochemical and clinical
aspects. Adv Exp Med Biol. 867:115–124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jurisic V, Terzic T, Colic S and Jurisic
M: The concentration of TNF-alpha correlate with number of
inflammatory cells and degree of vascularization in radicular
cysts. Oral Dis. 14:600–605. 2008. View Article : Google Scholar
|
|
30
|
Shi Y, Ji Y, Sun H, Hui F, Hu J, Wu Y,
Fang J, Lin H, Wang J, Duan H and Lanza M: Nanoscale
characterization of PM2.5 airborne pollutants reveals high
adhesiveness and aggregation capability of soot particles. Sci Rep.
5:112322015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Marshall J: PM 2.5. Proc Natl Acad Sci
USA. 110:87562013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cao SS, Luo KL and Shi L: Endoplasmic
reticulum stress interacts with inflammation in human diseases. J
Cell Physiol. 231:288–294. 2016. View Article : Google Scholar
|
|
33
|
Hasnain SZ, Lourie R, Das I, Chen AC and
McGuckin MA: The interplay between endoplasmic reticulum stress and
inflammation. Immunol Cell Biol. 90:260–270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wei J, Rahman S, Ayaub EA, Dickhout JG and
Ask K: Protein misfolding and endoplasmic reticulum stress in
chronic lung disease. Chest. 143:1098–1105. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Molagoda IMN, Kavinda MHD, Choi YH, Lee H,
Kang CH, Lee MH, Lee CM and Kim GY: Fisetin protects HaCaT human
keratinocytes from fine particulate matter (PM2.5)-induced
oxidative stress and apoptosis by inhibiting the endoplasmic
reticulum stress response. Antioxidants (Basel). 10:14922021.
View Article : Google Scholar
|
|
37
|
Kim SR, Kim HJ, Kim DI, Lee KB, Park HJ,
Jeong JS, Cho SH and Lee YC: Blockade of interplay between IL-17A
and endoplasmic reticulum stress attenuates LPS-induced lung
injury. Theranostics. 5:1343–1362. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schroeder BW, Verhaeghe C, Park S,
Nguyenvu LT, Huang X, Zhen G and Erle DJ: AGR2 is induced in asthma
and promotes allergen-induced mucin overproduction. Am J Respir
Cell Mol Biol. 47:178–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang X, Yang X, Li Y, Wang X, Zhang Y, Dai
X, Niu B, Wu J, Yuan X, Xiong A, et al: Lyn kinase represses mucus
hypersecretion by regulating IL-13-induced endoplasmic reticulum
stress in asthma. EBioMedicine. 15:137–149. 2017. View Article : Google Scholar :
|
|
40
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View Article : Google Scholar
|
|
41
|
Mijošek V, Lasitschka F, Warth A, Zabeck
H, Dalpke AH and Weitnauer M: Endoplasmic reticulum stress is a
danger signal promoting innate inflammatory responses in bronchial
epithelial cells. J Innate Immun. 8:464–478. 2016. View Article : Google Scholar
|
|
42
|
Delmotte P and Sieck GC: Interaction
between endoplasmic/sarcoplasmic reticulum stress (ER/SR stress),
mitochondrial signaling and Ca(2+) regulation in airway smooth
muscle (ASM). Can J Physiol Pharmacol. 93:97–110. 2015. View Article : Google Scholar
|
|
43
|
Almanza A, Carlesso A, Chintha C,
Creedican S, Doultsinos D, Leuzzi B, Luís A, McCarthy N,
Montibeller L, More S, et al: Endoplasmic reticulum stress
signalling - from basic mechanisms to clinical applications. FEBS
J. 286:241–278. 2019. View Article : Google Scholar
|
|
44
|
Laing S, Wang G, Briazova T, Zhang C, Wang
A, Zheng Z, Gow A, Chen AF, Rajagopalan S, Chen LC, et al: Airborne
particulate matter selectively activates endoplasmic reticulum
stress response in the lung and liver tissues. Am J Physiol Cell
Ph. 299:C736–C749. 2010. View Article : Google Scholar
|
|
45
|
Urano F, Bertolotti A and Ron D: IRE1 and
efferent signaling from the endoplasmic reticulum. J Cell Sci.
21:3697–3702. 2000. View Article : Google Scholar
|
|
46
|
Duan Q, Zhou Y and Yang D: Endoplasmic
reticulum stress in airway hyperresponsiveness. Biomed
Pharmacother. 149:1129042022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Duvigneau JC, Luís A, Gorman AM, Samali A,
Kaltenecker D, Moriggl R and Kozlov AV: Crosstalk between
inflammatory mediators and endoplasmic reticulum stress in liver
diseases. Cytokine. 124:1545772019. View Article : Google Scholar
|
|
48
|
Lei Y, Wang S, Ren B, Wang J, Chen J, Lu
J, Zhan S, Fu Y, Huang L and Tan J: CHOP favors endoplasmic
reticulum stress-induced apoptosis in hepatocellular carcinoma
cells via inhibition of autophagy. PLoS One. 12:e1836802017.
View Article : Google Scholar
|
|
49
|
Kropski JA and Blackwell TS: Endoplasmic
reticulum stress in the pathogenesis of fibrotic disease. J Clin
Invest. 128:64–73. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Siddesha JM, Nakada EM, Mihavics BR,
Hoffman SM, Rattu GK, Chamberlain N, Cahoon JM, Lahue KG, Daphtary
N, Aliyeva M, et al: Effect of a chemical chaperone,
tauroursodeoxycholic acid, on HDM-induced allergic airway disease.
Am J Physiol Lung Cell Mol Physiol. 310:L1243–L1259. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Aghapour M, Ubags ND, Bruder D, Hiemstra
PS, Sidhaye V, Rezaee F and Heijink IH: Role of air pollutants in
airway epithelial barrier dysfunction in asthma and COPD. Eur
Respir Rev. 31:2101122022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gon Y and Hashimoto S: Role of airway
epithelial barrier dysfunction in pathogenesis of asthma. Allergol
Int. 67:12–17. 2018. View Article : Google Scholar
|
|
53
|
Bradley KL, Stokes CA, Marciniak SJ,
Parker LC and Condliffe AM: Role of unfolded proteins in lung
disease. Thorax. 76:92–99. 2021. View Article : Google Scholar
|
|
54
|
Byndloss MX, Keestra-Gounder AM, Bäumler
AJ and Tsolis RM: NOD1 and NOD2: New functions linking endoplasmic
reticulum stress and inflammation. DNA Cell Biol. 35:311–313. 2016.
View Article : Google Scholar : PubMed/NCBI
|