Introduction
Chronic low back and leg pain originating from
intervertebral disc degeneration (IDD) affects daily life, and is
the main cause of adult disability, thus resulting in a substantial
economic burden to society and families (1). Due to the increase in the elderly
population and an increase in sedentary lifestyles, the number of
patients with IDD-related diseases has been continuously increasing
(2). The intervertebral disc is
the largest avascular tissue in the human body, and its nutritional
supply is received mainly via diffusion through the cartilage
endplate (CEP) and annulus fibrosus (AF). The CEP is the main route
for this nutrition supply, with 75% of normal intervertebral disc
nutrition originating from infiltration in this region (3). Studies have shown that aging and
degenerative changes in the CEP can significantly affect the
biomechanics and nutritional supply status of the intervertebral
disc, and are considered to be the initiating factors leading to
IDD (1,4). Chondrocytes are the only cell type
in the CEP, which synthesize and secrete extracellular matrix to
maintain the structure and function of the vertebral CEP. Normal
adult chondrocytes are terminally differentiated cells and cannot
adapt to microenvironmental changes through self-renewal (5). Concomitantly, the CEP lacks nerve
and vascular distribution, which disables its ability to react to
pathological conditions. Therefore, excessive mechanical stress,
inflammation and metabolic abnormalities may lead to oxidative
stress damage or even death of chondrocytes, which in turn can lead
to degeneration and calcification of the CEP (6).
The transcription factor nuclear factor erythroid
2-related factor 2 (NFE2L2/Nrf2) was initially identified as a
central controller of cellular redox homeostasis (7). Nrf2 is normally continuously
ubiquitinated and degraded when bound to Kelch-like ECH-associated
protein 1 (Keap1) in the cytoplasm. However, following induction of
oxidative or electrophilic stress in the cell, the spatial
conformation of Keap1 is altered, enabling the release of Nrf2 from
Keap1 and its translocation to the nucleus. This in turn activates
target genes, including those involved in the cell antioxidant
response and regulation of iron homeostasis, lipid metabolism and
mitochondrial function regulation (8). Controlled activation of Nrf2 in
normal cells serves important roles in redox balance. By contrast,
the inhibition or insufficient activation of Nrf2 is associated
with the onset of oxidative stress. Oxidative stress has been
demonstrated to be the leading cause of IDD. The protective effect
of various phytochemicals extracted from plants, such as
astaxanthin or icariin, against the development of IDD is
attributed to the activation of Nrf2 signaling (9,10). However, to the best of our
knowledge, only one study has assessed the roles of Nrf2 in CEP
degeneration (11).
Iron is the largest trace element in the human body,
and it participates in a wide range of physiological functions and
biochemical reactions. However, excessive iron can lead to the
production of a large number of reactive oxygen species (ROS) by
participating in electron transfer of the mitochondrial respiratory
chain, which can lead to the destruction of mitochondrial structure
and the disruption of mitochondrial function, as well as the
induction of oxidative stress, lipid peroxidation and DNA damage;
these events eventually lead to iron-dependent programmed
ferroptosis (12). Previous
studies have shown that the accumulation of iron in tissues and
cells is a common feature in a series of degenerative diseases,
including IDD (13,14). Recent studies have shown that
iron overload and ferroptosis can lead to oxidative stress injury
and the death of chondrocytes, which are important factors
contributing to articular cartilage degeneration (15,16) and IDD (17,18). As a result, the pursuit of
targeting ferroptosis has emerged as a potential therapeutic
strategy for these diseases. However, the regulatory mechanism of
iron metabolism in CEP chondrocytes under pathological conditions
requires further investigation.
In the present study, the regulatory effects of Nrf2
and its role in CEP chondrocyte degeneration were explored with
regard to cellular iron homeostasis and ferroptosis. The present
study aimed to offer insights into the involvement of the Nrf2
pathway and iron homeostasis in the pathogenesis of CEP
degeneration. The ultimate goal was to assess the potential of
developing effective therapeutic strategies for inhibiting the
progression of IDD.
Materials and methods
Primary CEP chondrocyte isolation and
culture
A total of 20 C57BL/6J male mice (age, 5 days) were
anesthetized by intraperitoneal injection of 2% pentobarbital
sodium (35 mg/kg body weight) and sacrificed via cervical
dislocation. All animals were purchased from the Experimental
Animal Center of Shandong First Medical University and maintained
in ventilated filter-top cages under standard laboratory conditions
at a constant temperature of 25°C and 40% humidity under a 12-h
light/dark cycle, and were given free access to conventional rodent
chow with water. All animal protocols were approved by the
Institutional Animal Care Committee of the Shandong Provincial
Hospital Affiliated to Shandong First Medical University (approval
no. 2022-816; Jinan, China). The tissues were minced into small
pieces using a dissecting microscope, and were then incubated with
0.25% trypsin (Gibco; Thermo Fisher Scientific, Inc.) for 30 min
and with 0.25% type 2 collagenase (MilliporeSigma) for 4-6 h at
37°C. Chondrocytes were harvested following centrifugation at 250 ×
g for 10 min at room temperature and were resuspended in DMEM/F12
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 1%
streptomycin sulfate and 1% penicillin. The cells were cultured in
an incubator maintained with 5% CO2 at 37°C. The culture
medium was changed every other day.
siRNA transfection
siRNAs targeting Nrf2 and a negative control siRNA
were synthesized by Guangzhou RiboBio Co., Ltd. The siRNA sequences
were as follows: Nrf2#1, sense 5′-CCA CCG CCA GGA CTA CAG T-3′;
Nrf2#2, sense 5′-GAT GGA CTT GGA GTT GCC A-3′; Nrf2#3, sense 5′-CAG
GAC TAC AGT CCC AGC A-3′; scrambled siRNA, sense 5′-TTC TGC GAA CGA
GTG ACG T-3′ and antisense 5′-ACC TGA CGC GTA CGG AGA A-3′. When
cells reached 60-70% confluence, they were transfected with 50 nM
siRNA using Lipofectamine® 3000 (cat. no. L3000015;
Invitrogen; Thermo Fisher Scientific, Inc.) for 5 h at 37°C. The
negative control group was transfected with scrambled siRNA.
Subsequently, the medium was replaced with normal growth medium
comprised of DMEM/F12 and 10% FBS. A total of 1 day
post-transfection, chondrocytes were used for further study.
Western blot analysis
CEP chondrocytes were treated with 0, 0.1, 1, 5 and
10 ng/ml of TNF-α (R&D Systems, Inc.) with or without Nrf2
small interfering RNA (siRNA) or 100 μM desferoxamine (DFO;
cat. no. D9533; MilliporeSigma) for 24 h at 37°C. Then, cells were
lysed with RIPA buffer (cat. no. CW2333; CoWin Biosciences)
supplemented with a protease inhibitor cocktail for 15 min on ice;
subsequently, total protein was collected following centrifugation
at 2,500 × g at 4°C for 20 min. The concentration of total protein
was measured using the bicinchoninic acid assay protein assay kit
(Beyotime Institute of Biotechnology). A total of 25 μg
total protein was added and subsequently subjected to separation by
SDS-PAGE on 10% gels, followed by the transfer of the separated
proteins onto polyvinylidene difluoride membranes (MilliporeSigma).
After blocking with 5% BSA (Wuhan Boster Biological Technology,
Ltd.) for 30 min at room temperature, the membranes were incubated
with primary antibodies against Nrf2 (cat. no. 16396-1-AP; 1:1,000;
Proteintech Group, Inc.), heme oxygenase (HO)-1 (cat. no. BM4010;
1:1,000; Wuhan Boster Biological Technology, Ltd.), nuclear
receptor coactivator 4 (NCOA4; cat. no. ab86707; 1:1,000; Abcam),
LC3B (cat. no. 4108; 1:1,000; Cell Signaling Technology, Inc.),
dynamin-related protein 1 (Drp1; cat. no. 12957-1-AP; 1:1,000;
Proteintech Group, Inc.), mitochondrial fission factor (MFF;
1:1,000; cat. no. 84580; Cell Signaling Technology, Inc.), ferritin
heavy chain 1 (FTH1; cat. no. ab75973; 1:1,000; Abcam), type II
collagen (COL2; cat. no. 28459-1-AP; 1:1,000; Proteintech Group,
Inc.), SOX9 (cat. no. A00177-2; 1:500; Wuhan Boster Biological
Technology, Ltd), Parkin (cat. no. 14060-1-AP; 1:1,000; Proteintech
Group, Inc.), matrix metalloproteinase (MMP)3 (cat. no. 17873-1-AP;
1:1,000; Proteintech Group, Inc.), MMP13 (cat. no. 18165-1-AP;
1:1,000; Proteintech Group, Inc.), solute carrier family 7 member
11 (SLC7A11; cat. no. 26864-1-AP; 1:1,000; Proteintech Group,
Inc.), COL10 (cat. no. BA 2023; 1:500; Wuhan Boster Biological
Technology, Ltd.), mitochondrial fission 1 (FIS1; cat. no.
10956-1-AP; 1:1,000; Proteintech Group, Inc.), RUNX2 (cat. no.
PB0171; 1:500; Wuhan Boster Biological Technology, Ltd.),
glutathione (GSH) peroxidase 4 (GPX4; cat. no. 67763-1-Ig; 1:1,000;
Proteintech Group, Inc.) and GAPDH (cat. no. 10494-1-AP; 1:2,000;
Proteintech Group, Inc.). Following an overnight incubation at 4°C,
the membranes underwent three washes with TBS-0.1% Tween 20. The
membranes were subsequently incubated with the corresponding
horseradish peroxidase-conjugated anti-rabbit or anti-mouse
secondary antibodies (1:2,000; cat. nos. BA1061 and BA1062; Wuhan
Boster Biological Technology, Ltd.) for 1 h at room temperature.
The signal intensity of the membranes was visualized with enhanced
chemiluminescence reagent (Wuhan Boster Biological Technology,
Ltd.) and images were captured using a Bio-Radscanner (Bio-Rad
Laboratories, Inc.).
Immunofluorescence staining
CEP chondrocytes were isolated and seeded onto a
12-well plate. The cells were subsequently subjected to 5 ng/ml
TNF-α treatment with or without Nrf2 siRNA when reaching 80%
confluence. Following fixation at room temperature for 20 min with
4% paraformaldehyde and permeabilization with 0.1% Triton X-100
(cat. no. T8787; Sigma-Aldrich; Merck KGaA), the cells were blocked
with 5% BSA (Wuhan Boster Biological Technology, Ltd.) for 1 h at
room temperature and incubated with primary antibodies against COL2
(cat. no. 28459-1-AP, 1:500; Proteintech Group, Inc.), GPX4 (cat.
no. 67763-1-lg; 1:200; Proteintech Group, Inc.) and NCOA4 (cat. no.
ab86707; 1:500; Abcam) at 4°C overnight. Subsequently, the cells
were incubated with Cy3-conjugated goat anti-rabbit secondary
antibody (cat. no. A0516; Beyotime Institute of Biotechnology;
1:500) in the dark for 1 h at 37°C. The cells were then subjected
to a washing step with PBS and labeled with DAPI (cat. no. AR1177;
Wuhan Boster Biological Technology, Ltd.) for 5 min with a
fluorescence microscope (Olympus Corporation).
To examine the colocalization of mitochondria and
Parkin, as well as Drp1, cells were incubated with diluted
Mito-Tracker Red CMXRos solution (cat. no. C1049B; Beyotime
Institute of Biotechnology) in the dark at 37°C for 30 min. After
MitoTracker incubation, cells were subjected to fixation and
permeabilization as aforementioned, then the cells were incubated
with Parkin (cat. no. 14060-1-AP; 1:200; Proteintech Group, Inc.)
and Drp1 (cat. no. 12957-1-AP; 1:200; Proteintech Group, Inc.)
antibodies at 4°C overnight, and were then incubated with
fluorescein isothiocyanate-conjugated goat anti-rabbit secondary
antibodies (cat. no. A0562; Beyotime Institute of Biotechnology;
1:500) in the dark at 37°C for 1.5 h. After washing with PBS and
labeling with DAPI, fluorescence microscopy (Axio Observer 3; Carl
Zeiss AG) was employed to capture the images and detect the
differences in the fluorescence expression of the corresponding
proteins.
Assessment of intracellular ROS and
mitochondrial membrane potential
The primary cause of oxidative stress in IDD is the
excessive accumulation of ROS. To assess intracellular ROS
production, a Reactive Oxygen Species Assay Kit (cat. no. S0033;
Beyotime Institute of Biotechnology) was used, according to the
manufacturer's instructions. The CEP cells were subjected to three
washes with serum-free medium. Subsequently,
dichloro-dihydro-fluorescein diacetate was diluted to 10 μM
in serum-free medium and added to the cells for 30 min at 37°C in
the dark. After washing the cells with serum-free medium, they were
examined under a fluorescence microscope (Axio Observer 3; Carl
Zeiss AG).
Mitochondrial membrane potential was evaluated using
a mitochondrial membrane potential kit (cat. no. C2006; Beyotime
Institute of Biotechnology). Briefly, after incubation with the
JC-1 staining working solution for 20 min at 37°C, CEP chondrocytes
were rinsed with ice-cold JC-1 washing buffer three times.
Multimeric JC-1 with red fluorescence transitions to monomeric JC-1
with green fluorescence, thus indicating the loss of mitochondrial
membrane potential. The changes in mitochondrial membrane potential
were captured using an inverted fluorescence microscope (Axio
Observer A1; Carl Zeiss).
Alizarin red staining
Briefly, chondrocytes derived from the CEP were
seeded in 24-well plates at a density of 1×105
cells/well. The osteogenic differentiation culture medium (Cyagen
Biosciences, Inc.) was added to the cells when cell confluence
reached 80%, and the cells were incubated for 3 weeks at 37°C.
After thorough rinsing with PBS and fixation with 4%
paraformaldehyde at room temperature for 20 min, the cells were
stained with alizarin red solution (Cyagen Biosciences, Inc.) at
room temperature for 30 min. Semi-quantification of the mineralized
nodules was achieved using spectrophotometric analysis.
Specifically, following dissolution in 10% (wt/vol) cetylpyridinium
chloride (Sigma-Aldrich; Merck KGaA) for 1 h at room temperature,
semi-quantification was performed by measuring its optical density
at 570 nm.
Ferrous ion (Fe2+)
detection
Chondrocytes were seeded in a 24-well plate at a
concentration of 1×105 cells/ml. After 1 day, serum-free
medium containing 5 ng/ml TNF-α was added to the cells and they
were cultured for 24 h. Subsequently, medium was replaced with
serum-free medium containing 0.1 mmol/l ammonium ferric citrate
(MilliporeSigma) and cells were cultured for 2 h. After three
washes with Hank's Balanced Salt Solution (HBSS; Wuhan Boster
Biological Technology, Ltd.), the cells were stained with 1
μM FerroOrange (Dojindo Laboratories, Inc.) in HBSS for 40
min at 37°C. Subsequently, the cells underwent three additional
washes with HBSS and were then subjected to imaging using a
fluorescence microscope (Axio Observer 3; Carl Zeiss AG).
Mitochondrial-specific fluorescence
staining
Mito-Tracker Green (Beyotime Institute of
Biotechnology) was employed to assess the morphological changes of
mitochondria. Following treatment of the cells with 5 ng/ml
TNF-αfor 24 h at 37°C, they were rinsed twice with FBS-free
DMEM/F12, and then exposed to FBS-free DMEM/F12 and incubated with
diluted Mito-Tracker Green solution (1:1,000) for 30 min at 37°C in
the absence of light. Subsequently, the images were captured using
a fluorescence microscope (Axio Observer 3; Carl Zeiss AG).
Immunohistochemical (IHC) analysis
To examine the expression pattern of Nrf2 in aging
mice, 20 3-week-old C57/BL6 male mice (weight, 10 g) were randomly
divided into the following groups: 1, 3, 6 and 16 months
(n=5/group), and were maintained in ventilated filter-top cages
under standard laboratory conditions at a constant temperature of
25°C and 40% humidity under a 12-h light/dark cycle, and were given
free access to conventional rodent chow with water. C57/BL6 mice
were purchased from the Experimental Animal Center of Shandong
Provincial Hospital Affiliated to Shandong First Medical
University. At the age of 1, 3, 6 and 16 months, mice were
anesthetized by an intraperitoneal injection of 2% pentobarbital
sodium (35 mg/kg body weight) and were sacrificed via cervical
dislocation. Subsequently, L4/5 intervertebral disc samples were
collected and fixed in 4% paraformaldehyde at 4°C for at least 24
h. To perform IHC analysis, the following steps were executed: The
samples underwent deparaffinization using xylene, followed by
blocking with 5% BSA-containing PBS solution for 30 min at 37°C.
After antigen retrieval in citrate buffer at 100°C for 30 min, the
sections were incubated with primary antibodies against Nrf2 (cat.
no. 16396-1-AP; 1:200; Proteintech Group, Inc.) overnight at 4°C.
The samples were then incubated with biotinylated donkey
anti-rabbit secondary antibodies (cat. no. BA1002; Wuhan Boster
Biological Technology, Ltd.) at room temperature for 30 min.
Finally, the sections were incubated for 10 min with
3,3′-diaminobenzidine at room temperature and counterstained with
hematoxylin at room temperature for 5 min. Images were captured
under a microscope (EVOS FL Auto system; Thermo Fisher Scientific,
Inc.) and Image Pro Plus software (version 6.0; Media Cybernetics)
was used to analyze the images of IHC staining. At least five
randomly selected cartilage fields were used to accurately count
immune-positive cells, and the percentage of immune-positive cells
relative to the overall number of cells was calculated. The animals
were treated and used in accordance with all applicable
international, national and institutional guidelines.
Statistical analysis
Data are presented as the mean ± SD. All analyses
were performed with GraphPad Prism software (version 9.0;
Dotmatics). Comparisons between multiple groups were analyzed using
one-way ANOVA followed by Tukey's test. For data that were
expressed as a relative fold change and for comparisons between the
control group and each of the treatment groups, a one-way ANOVA
with Dunnett's test was performed. P<0.05 was considered to
indicate a statistically significant difference.
Results
IDD is characterized by altered
expression of Nrf2 and mitochondrial dysfunction
Aging is the leading cause of IDD (19). As shown in Fig. 1A and B, degenerated CEP with bony
tissues that contained bone marrow and mineralized bone were
observed in mice that were >3 months old. Furthermore, IHC
analysis indicated that Nrf2 expression was higher in the
degenerated CEP when compared with the CEP in 1-month old mice and
the highest expression of Nrf2 was observed in 3-month-old mice.
The expression of Nrf2 in the CEP was decreased after 3 months and
no significant difference was noted between the CEPs derived from
1-month-old and 16-month-old mice. To further determine the
expression pattern of Nrf2 during CEP degeneration, TNF-α was used
to mimic endplate osteochondritis; the protein expression levels of
Nrf2 and those of its downstream protein heme oxygenase (HO)-1 were
increased following treatment with TNF-α, and were decreased when
the TNF-α concentration reached >1 ng/ml, which was similar to
the results obtained from the in vivo experiments (Fig. 1C and D). These results indicated
that a low dose of TNF-α could activate a Nrf2-mediated antioxidant
effect. To mimic the pathological condition in cartilage
osteochondritis, a higher concentration of TNF-α (5 ng/ml) was used
in the present study.
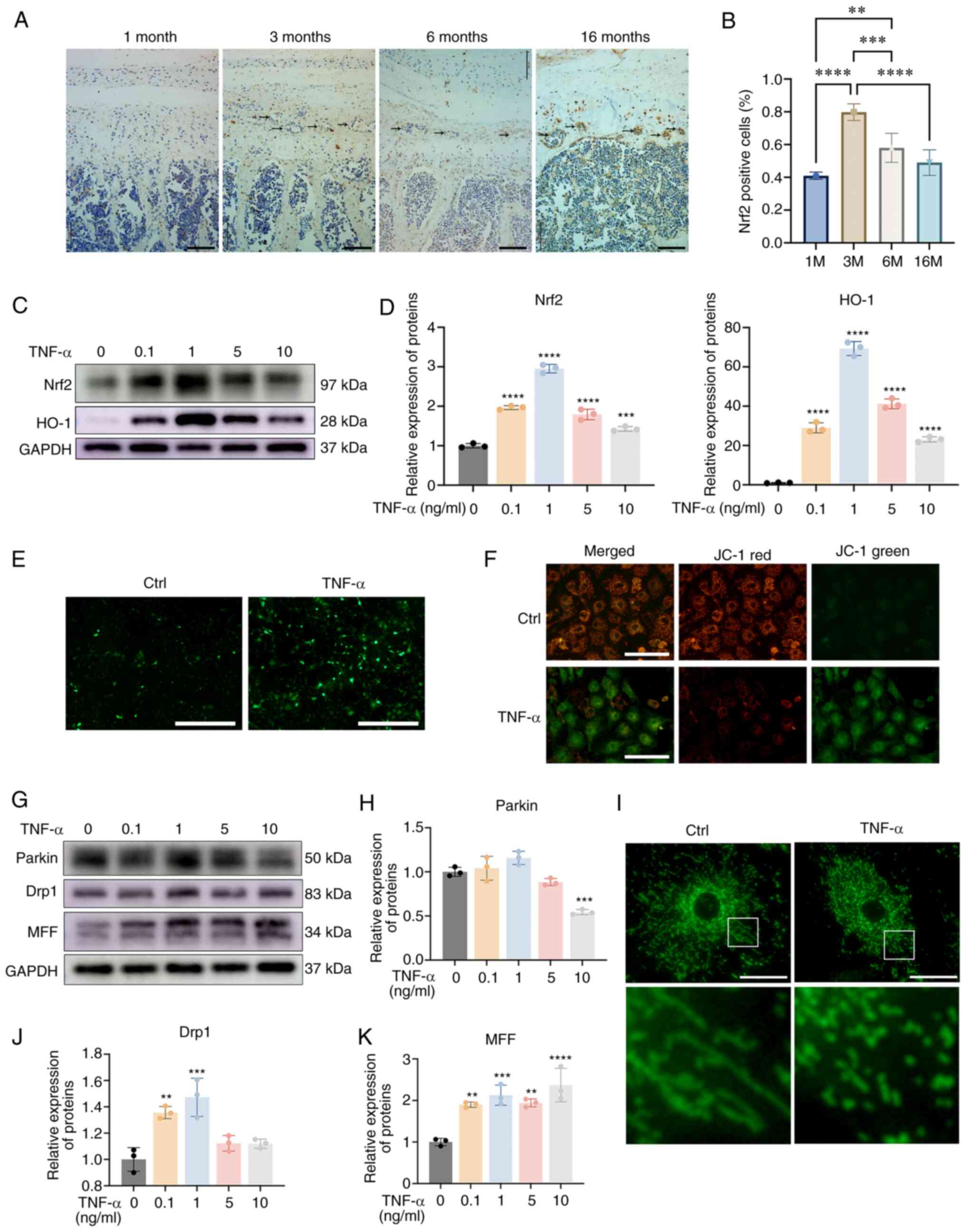 | Figure 1IDD is characterized by altered
expression of Nrf2 and mitochondrial dysfunction. (A)
Immunohistochemistry for Nrf2 in the CEP from mice aged 1, 3, 6 and
16 months. Black arrows indicate bony tissues. Scale bar, 100
μm; magnification, ×200. (B) The ratio of Nrf2-positive
cells was determined under a microscope using five sections from
seven mice. (C) CEP chondrocytes were treated with increasing
concentrations of TNF-α for 24 h, and western blotting was
conducted to examine the protein expression levels of Nrf2, HO-1
and GAPDH. (D) The band density was semi-quantified and normalized
to the control. (E) CEP chondrocytes were treated with 5 ng/ml
TNF-α for 24 h, and representative fluorescence photomicrographs of
intracellular ROS in chondrocytes are shown. Scale bar, 400
μm. (F) Representative fluorescence photomicrographs of
mitochondrial membrane potential following incubation with JC-1.
Red fluorescence was emitted by JC-1 aggregates in healthy
mitochondria with polarized inner mitochondrial membranes, whereas
green fluorescence was emitted by cytosolic JC-1 monomers,
indicating mitochondrial membrane potential collapse. Scale bar,
200 μm. (G) CEP chondrocytes were treated with increasing
concentrations of TNF-α for 24 h, and western blotting was
conducted to examine the protein expression levels of Parkin, Drp1
and MFF. The band density of (H) Parkin was semi-quantified and
normalized to the control. (I) Representative fluorescence images
of mitochondria. CEP chondrocytes were treated with 5 ng/ml TNF-α
for 24 h and the morphology of the mitochondria was visualized
using Mito-Tracker Green staining. Scale bar, 25 μm. The
band density of (J) Drp1 and (K) MFF was semi-quantified and
normalized to the control. Data are presented as the mean ± SD from
three independent experiments. **P<0.01,
***P<0.001, ****P<0.0001 vs. 0 ng/ml
TNF-α group or as indicated. CEP, cartilage endplate; Ctrl,
control; Drp1, dynamin-related protein 1; HO-1, heme oxygenase-1;
MFF, mitochondrial fission factor; Nrf2, nuclear factor erythroid
2-related factor 2. |
Nrf2 serves essential roles in redox homeostasis and
mitochondrial function (20). To
determine the function of Nrf2 in CEP chondrocytes under endplate
osteochondritis conditions, the production of ROS and the
development of mitochondrial dysfunction were examined. As shown in
Fig. 1E, TNF-α treatment
markedly promoted ROS production. Subsequently, JC-1 staining was
utilized to investigate the collapse of the mitochondrial membrane
potential. As shown in Fig. 1F,
the ratio of green JC-1 monomers to red JC-1 aggregates was
increased in the TNF-α treatment group, indicating the collapse of
mitochondrial membrane potential. Mitochondria in healthy
chondrocytes displayed a normal shape. As shown in Fig. 1G-K, TNF-α treatment markedly
promoted mitochondrial destruction with increased expression levels
of the mitochondrial fission proteins, MFF and Drp1. Also the
mitophagy protein Parkin was slightly increased in response to a
low dose of TNF-α (<5 ng/ml) and was significantly decreased
when TNF-α reached 10 ng/ml. In addition, the immunofluorescence
analysis of the mitochondria indicated the destruction of
mitochondria with the presence of short and granulated
mitochondria. These results indicated a negative association
between Nrf2 and CEP degeneration. During the late stage of IDD
development, Nrf2 expression was downregulated alongside excess ROS
production and mitochondrial dysfunction.
TNF-α impairs iron homeostasis and
promotes ferroptosis
Ferroptosis plays important roles in the development
of IDD (21). In the present
study, the labile iron pool (LIP) and the expression levels of
ferroptosis markers were examined in CEP chondrocytes following
TNF-α treatment. As shown in Fig. 2A
and B, TNF-α significantly increased the immunofluorescence
intensity of the Fe2+ probe, indicating that it promoted
the Fe2+ levels of the LIP. Subsequently, the expression
levels of the ferroptosis markers, GPX4, SLC7A11 and FTH1 were
examined. Western blot analysis indicated that the expression
levels of GPX4, a key factor of ferroptosis, were significantly
reduced by TNF-α; similarly, the expression levels of FTH and
SLC7A11 were reduced (Fig. 2C-D,
F-G). The decreased GPX4, SLC7A11 and FTH1 expression indicated
increased CEP chondrocyte ferroptosis. In addition,
immunofluorescence analysis of GPX4 indicated a similar trend; the
red fluorescence activity of GPX4 was decreased following TNF-α
treatment (Fig. 2E). These
results indicated that TNF-α could increase the Fe2+
levels of the LIP and promote ferroptosis.
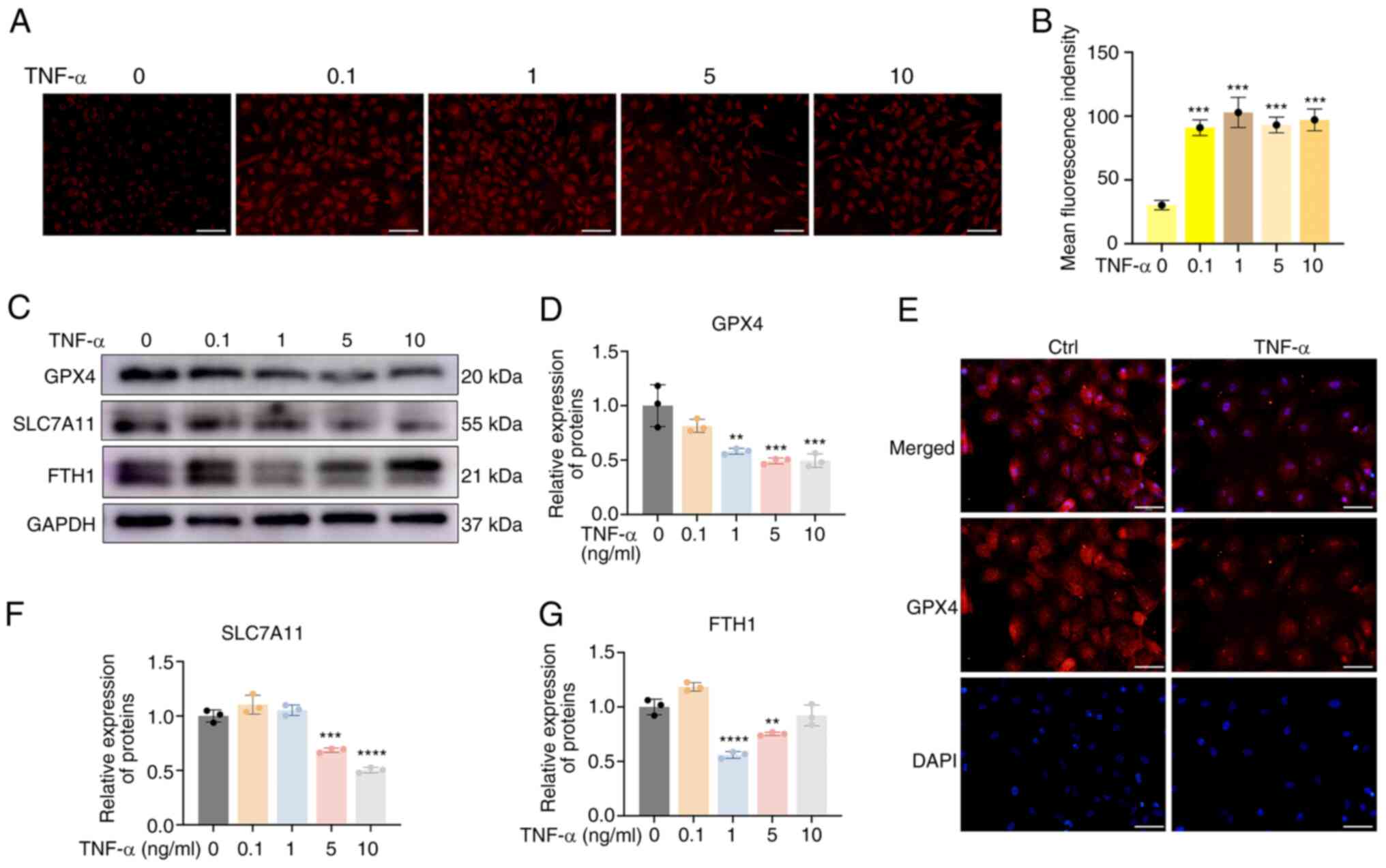 | Figure 2TNF-α impairs iron homeostasis and
promotes ferroptosis. (A) CEP chondrocytes were treated with
increasing concentrations of TNF-α with 100 μM ammonium
ferric citrate for 20 min. Representative staining of
Fe2+ is shown. Scale bar, 100 μm. (B) Statistical
analysis of fluorescence intensity (Fe2+) is shown. (C)
CEP chondrocytes were treated with increasing concentrations of
TNF-α for 24 h, and western blotting was conducted to examine the
protein expression levels of GPX4, SLC7A11 and FTH1. (D) Band
density of GPX4 was semi-quantified and normalized to the control.
(E) CEP chondrocytes were treated with 5 ng/ml TNF-α for 24 h, and
representative fluorescence photomicrographs of GPX4 in CEP
chondrocytes are shown. Scale bar, 50 μm. The band density
of (F) SLC7A11 and (G) FTH1 was semi-quantified and normalized to
the control. Data are presented as the mean ± SD from three
independent experiments. **P<0.01,
***P<0.001, ****P<0.0001 vs. 0 ng/ml
TNF-α. CEP, cartilage endplate; Ctrl, control; Fe2+,
ferrous ion; FTH, ferritin heavy chain; GPX4, glutathione
peroxidase 4; SLC7A11, solute carrier family 7 member 11. |
Nrf2 inhibition sensitizes CEP
chondrocytes to ferroptosis by promoting NCOA4-mediated
ferritinophagy
NCOA4-mediated ferritinophagy has recently been
demonstrated to serve important roles in regulating intracellular
Fe2+ balance, and emerging evidence has reported that
NCOA4 can regulate ferroptosis (22). The present study investigated
whether NCOA4 was involved in CEP chondrocyte ferroptosis and
whether NCOA4 was regulated by Nrf2. As shown in Fig. 3A, the protein expression levels
of NCOA4 were upregulated in CEP chondrocytes following TNF-α
treatment. Subsequently, Nrf2 siRNA was synthesized and transfected
into CEP chondrocytes; the transfection efficiency was evaluated by
western blot analysis and Nrf2#1 was used in subsequent
experiments(Fig. 3D). The levels
of Fe2+ and ferroptosis markers were then examined. As
shown in Fig. 3B and E,
inhibition of Nrf2 expression promoted the expression levels of
NCOA4 and the amount of cellular Fe2+ when compared with
the TNF-α group. Similar results of immunofluorescence analysis of
NCOA4 were obtained, indicating that inhibition of Nrf2 further
promoted CEP chondrocyte NCOA4 expression (Fig. 3E). JC-1 staining results
indicated that inhibition of Nrf2 expression impaired the
mitochondrial membrane potential, as demonstrated by the increased
fluorescence levels of the JC-1 green probe and the decreased
fluorescence levels of the JC-1 red probe (Fig. 3C). Compared with the
TNF-α-treated group, inhibition of Nrf2 expression significantly
enhanced the ferroptosis process, as determined by the reduced
expression of ferroptosis markers, including GPX4, SLC7A11 and FTH
(Fig. 3F and G). These results
indicated that Nrf2 could inhibit the NOCA4-mediated ferritinophagy
process and maintain cellular Fe2+ balance. Inhibition
of Nrf2 expression enhanced NCOA4-mediated ferritinophagy and
increased the levels of active Fe2+ of the LIP and
sensitized CEP chondrocytes to ferroptosis.
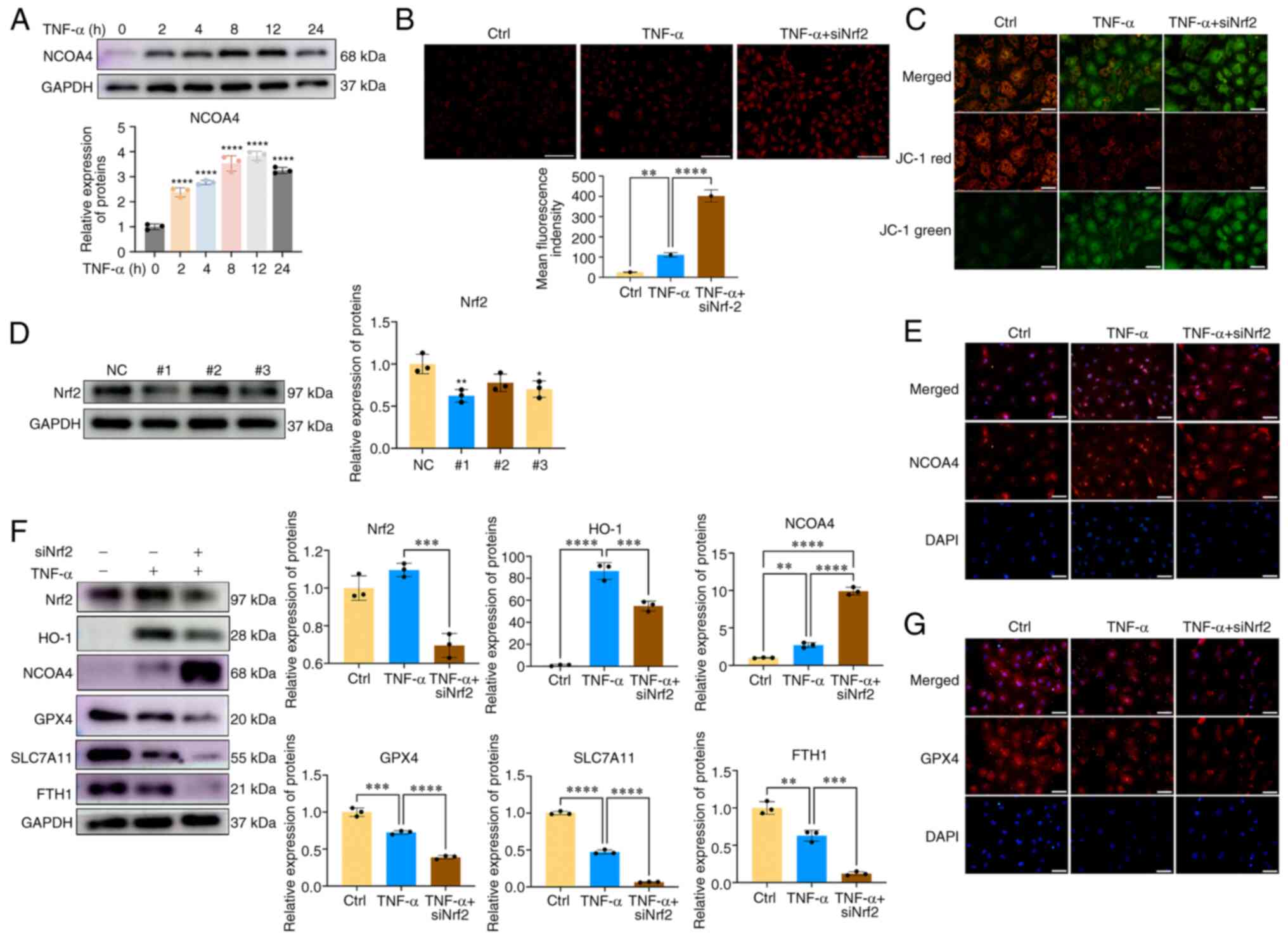 | Figure 3Nrf2 inhibition sensitizes CEP
chondrocytes to ferroptosis via promoting NCOA4-mediated
ferritinophagy. (A) CEP chondrocytes were treated with increasing
concentrations of TNF-α for 24 h, and western blotting was
conducted to examine the protein expression levels of NCOA4 and
GAPDH. The band density was semi-quantified and normalized to the
control. (B) CEP chondrocytes were treated with TNF-α with or
without Nrf2 siRNA transfection. Representative staining for
ferrous ions and statistical analysis of fluorescence intensity
(ferrous ions) are shown. Scale bar, 200 μm. (C)
Representative fluorescence photomicrographs of mitochondrial
membrane potential after incubating with JC-1 in the indicated
group are shown. Scale bar, 50 μm. (D) Transfection
efficiency was evaluated by detecting Nrf2 expression using western
blotting. (E) CEP chondrocytes were treated with TNF-α with or
without Nrf2 siRNA transfection. Representative fluorescence
microscopy photomicrographs of NCOA4 in CEP chondrocytes are shown.
Scale bar, 50 μm. (F) CEP chondrocytes were transfected with
Nrf2 siRNA and were then treated with 5 ng/ml TNF-α for 24 h.
Western blotting was conducted to examine the protein expression
levels of Nrf2, HO-1, NCOA4, GPX4, SLC7A11, FTH and GAPDH. The band
density was semi-quantified and normalized to the control. (G) CEP
chondrocytes were treated with TNF-α with or without Nrf2 siRNA
transfection. Representative fluorescence microscopy
photomicrographs of GPX4 in CEP chondrocytes are shown. Scale bar,
50 μm. Data are presented as the mean ± SD from three
independent experiments. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. CEP, cartilage endplate; Ctrl, control;
FTH, ferritin heavy chain; GPX4, glutathione peroxidase 4; HO-1,
heme oxygenase-1; NC, negative control; NCOA4, nuclear receptor
coactivator 4; Nrf2, nuclear factor erythroid 2-related factor 2;
siRNA, small interfering RNA; SLC7A11, solute carrier family 7
member 11. |
Nrf2 inhibition aggravates CEP
chondrocyte degeneration and calcification
To determine the role of Nrf2 in CEP degeneration
and calcification, CEP chondrocytes were isolated and transfected
with Nrf2 siRNA, and the role of the Nrf2/HO-1 axis in
TNF-α-induced CEP degeneration and calcification was investigated.
As shown in Fig. 4A and B,
transfection of cells with Nrf2 siRNA further promoted
TNF-α-induced CEP degeneration and matrix degradation, as indicated
by increased MMP3, MMP13 and COL10 expression, and decreased SOX9
and COL2 expression when compared with the TNF-α group. Similar
results were obtained by immunofluorescence analysis of COL2,
indicating that Nrf2 siRNA further inhibited the red fluorescence
staining of COL2 protein expression (Fig. 4C). Oxidative stress and iron
overload can promote CEP chondrocyte osteogenic differentiation and
calcification (23). As shown in
Fig. 4D and E, inhibition of
Nrf2 further promoted the formation of calcium nodules. These
results demonstrated the essential role of Nrf2 in ameliorating CEP
degeneration, and indicated that inhibition of Nrf2 expression
could aggravate CEP chondrocyte degeneration and calcification.
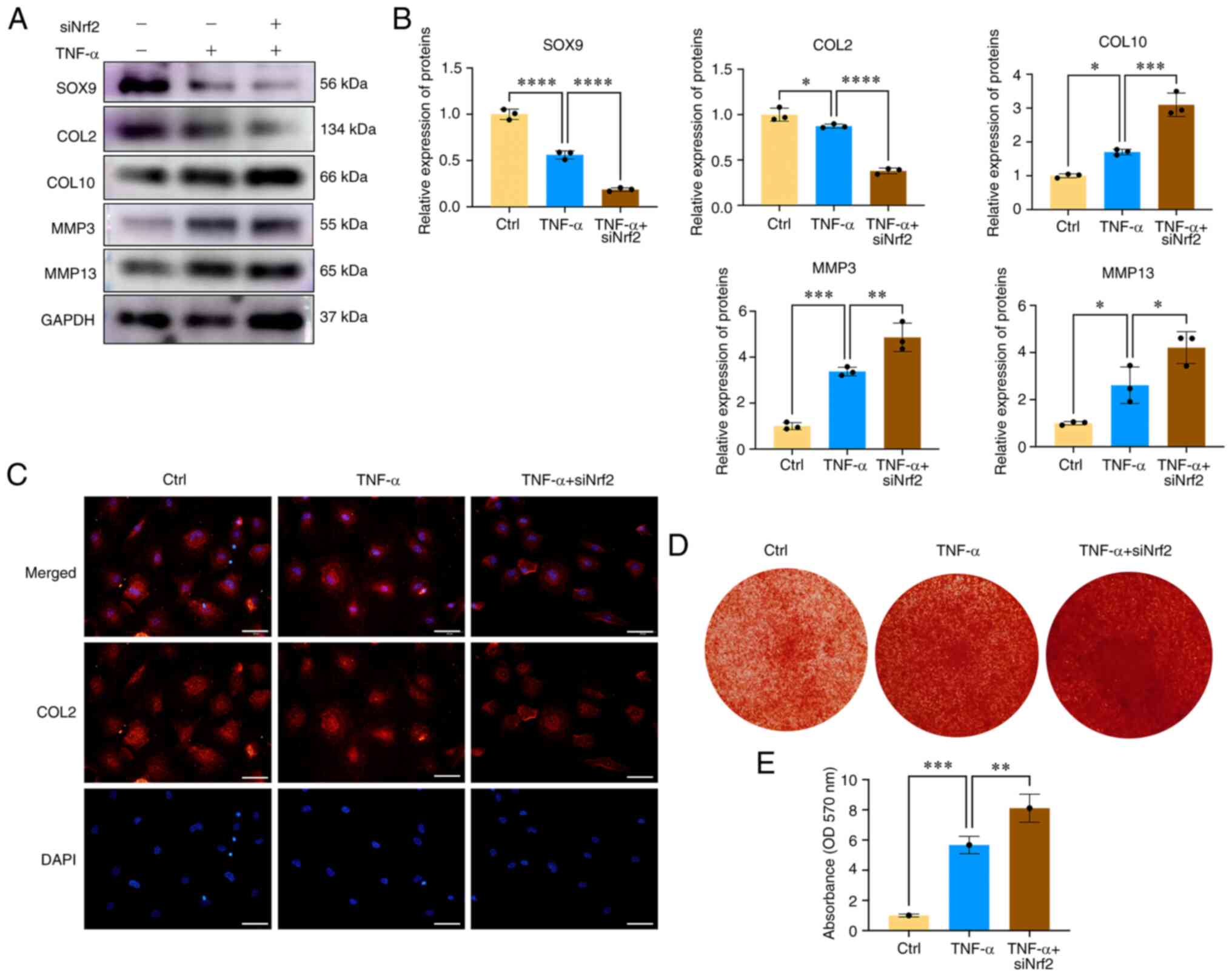 | Figure 4Nrf2 inhibition aggravates CEP
chondrocyte degeneration and calcification. (A) CEP chondrocytes
were transfected with Nrf2 siRNA and then treated with 5 ng/ml
TNF-α for 24 h. Western blotting was conducted to examine the
protein expression levels of SOX9, COL2, COL10, MMP3, MMP13 and
GAPDH. (B) The band density was semi-quantified and normalized to
the control. (C) CEP chondrocytes were treated with TNF-α with or
without Nrf2 siRNA transfection. Representative fluorescence
photomicrographs of COL2 in CEP chondrocytes are shown. Scale bar,
50 μm. (D) Alizarin Red staining for calcium deposition in
CEP chondrocytes. (E) Semi-quantitative analysis of the mineralized
nodules in CEP chondrocytes is shown. Data are presented as the
mean ± SD from three independent experiments.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. CEP,
cartilage endplate; COL, collagen; Ctrl, control; MMP, matrix
metalloproteinase; Nrf2, nuclear factor erythroid 2-related factor
2; siRNA, small interfering RNA. |
Inhibition of Nrf2 results in
mitochondrial dysfunction and decreased mitophagy
Mitochondrial dysfunction and the subsequent
overproduction of ROS serve important roles in the development of
IDD (24). It has been reported
that Parkin activation can eliminate impaired mitochondria and
ameliorate the progression of IDD (25). Next, CEP chondrocytes were
transfected with Nrf2 siRNA before TNF-α co-treatment; scrambled
siRNA was used as negative control. The expression levels of Parkin
and changes in mitochondrial morphology were then investigated. As
shown in Fig. 5A and C, the
expression levels of Parkin in the 5 ng/ml TNF-α + siNC group were
significantly increased when compared with those in the control
group, while following inhibition of Nrf2 expression, the
expression levels of Parkin and LC3B were inhibited, indicating
that inhibition of Nrf2 decreased the mitophagy process. Double
staining of Parkin and mitochondria indicated that Nrf2 siRNA
reduced aggregation of Parkin in the mitochondria of
TNF-α-stimulated CEP chondrocytes (Fig. 5B). In addition, the expression
levels of the mitochondrial fission proteins, including MFF, Drp1
and FIS1 were further increased by Nrf2 siRNA, which suggested the
destruction of the mitochondrial morphology (Fig. 5A and C). As shown in Fig. 5D, immunofluorescence results
indicated that Nrf2 siRNA further led to the destruction of
mitochondrial morphology, with an increased number of short and
granulated mitochondria. Moreover, elevated colocalization of Drp1
and mitochondria was observed in the Nrf2 siRNA treatment group.
These results indicated that Nrf2 inhibition decreased CEP
chondrocyte mitophagy and promoted mitochondrial dysfunction.
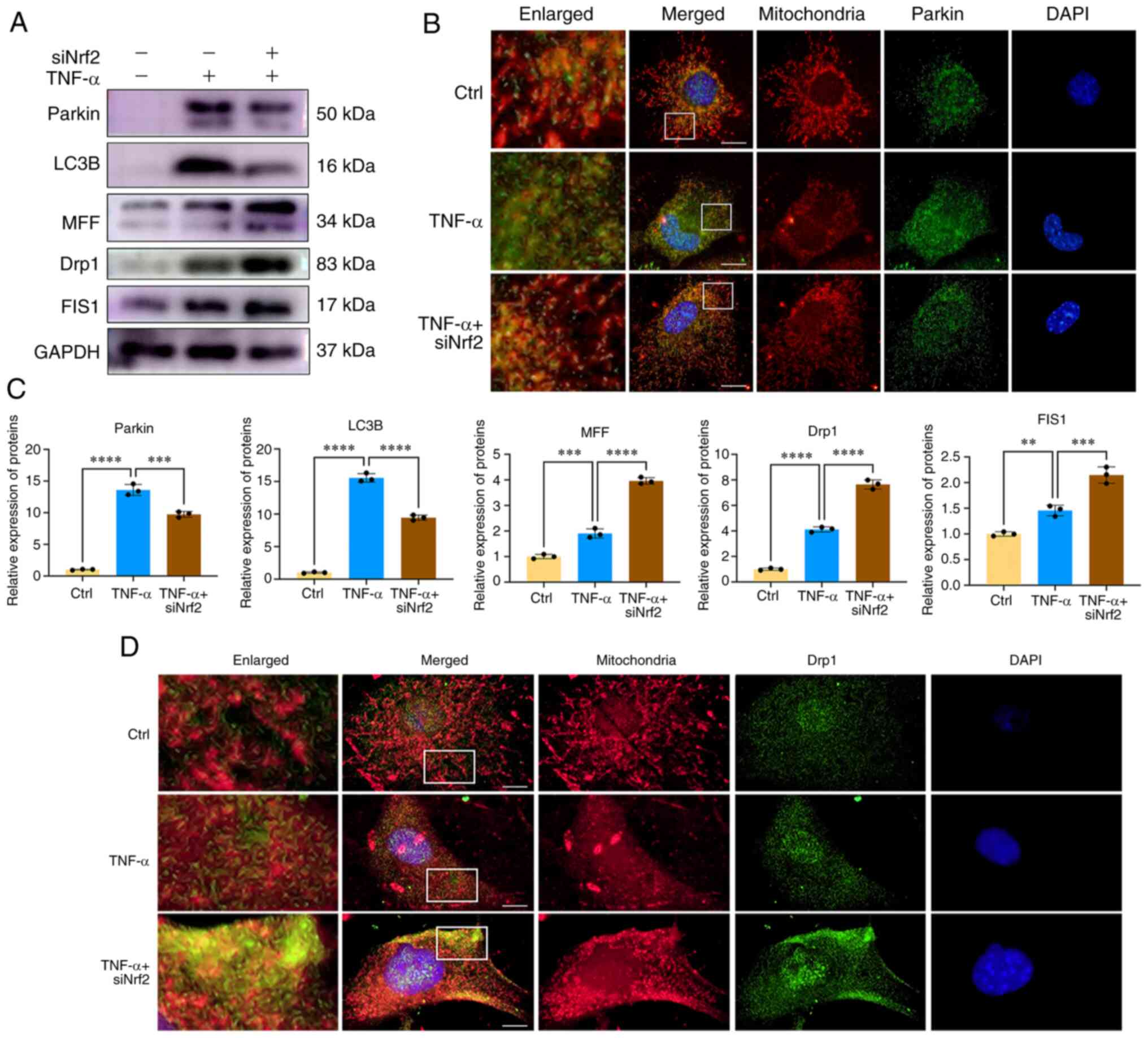 | Figure 5Nrf2 inhibition results in
mitochondrial dysfunction and decreased mitophagy. (A) CEP
chondrocytes were transfected with Nrf2 siRNA and then treated with
5 ng/ml TNF-α for 24 h. Western blotting was conducted to examine
the protein expression levels of Parkin, LC3B, MFF, Drp1, MFF, FIS1
and GAPDH. (B) Immunofluorescence staining was conducted to examine
the expression and localization of Parkin (green), mitochondria
(red). Scale bar, 10 μm. (C) The band density was
semi-quantified and normalized to the control. (D)
Immunofluorescence staining was conducted to examine the expression
and localization of Drp1 (green) and mitochondria (red). Scale bar,
10 μm. Data are presented as the mean ± SD from three
independent experiments. **P<0.01,
***P<0.001, ****P<0.0001. CEP,
cartilage endplate; Ctrl, control; Drp1, dynamin-related protein 1;
FIS1, mitochondrial fission 1; MFF, mitochondrial fission factor;
Nrf2, nuclear factor erythroid 2-related factor 2; siRNA, small
interfering RNA. |
Iron chelation with desferoxamine (DFO)
ameliorates CEP chondrocyte degeneration and calcification
Subsequently, the role of iron ions in CEP
chondrocyte degeneration was assessed via DFO co-treatment. DFO can
chelate iron ions and decrease the levels of cellular iron ions. As
shown in Fig. 6A, TNF-α promoted
CEP chondrocyte degeneration, which was accompanied by decreased
expression levels of the chondrogenesis proteins SOX9 and COL2, and
increased expression levels of cartilage matrix-degrading enzymes,
including MMP3 and MMP13. However, co-treatment of cells with DFO
and TNF-α reversed the detrimental effect of TNF-α, and was
accompanied by increased expression levels of SOX9, and decreased
levels of MMP3 and MMP13. Immunofluorescence of COL2 obtained
similar results, in that DFO promoted COL2 protein expression
(Fig. 6B). The degeneration and
calcification of the CEP can significantly diminish the oxygen and
nutrient supply to intervertebral discs, and is an important
contributing factor in IDD. To explore the potential inhibitory
effects of DFO on CEP calcification, and the expression of
chondrocyte hypertrophic and osteogenic markers, including RUNX2
and COL10, western blot analysis was conducted. As shown in
Fig. 6C, DFO exhibited a
significant inhibitory effect on the expression levels of
TNF-α-induced hypertrophic and osteogenic markers (COL10 and
RUNX2). Furthermore, CEP chondrocytes were treated with DFO to
determine its effects on chondrocyte osteogenic differentiation and
mineralization. Fig. 6D displays
the results of alizarin red staining, which was used to assess
calcium nodule formation. TNF-α treatment markedly promoted the
formation of mineralized deposits in CEP chondrocytes, an effect
that could be reversed following co-treatment of the cells with DFO
and TNF-α. As shown in Fig. 6E,
western blot analysis revealed that DFO treatment restored the
diminished protein expression levels of GPX4 and SLC7A11 in CEP
chondrocytes, indicating that DFO effectively inhibited
TNF-α-induced CEP chondrocyte ferroptosis. These findings suggested
that an excess of iron ions may contribute to CEP chondrocyte
degeneration and calcification, and that reducing cellular iron
ions represents an effective approach to inhibit CEP chondrocyte
degeneration and calcification.
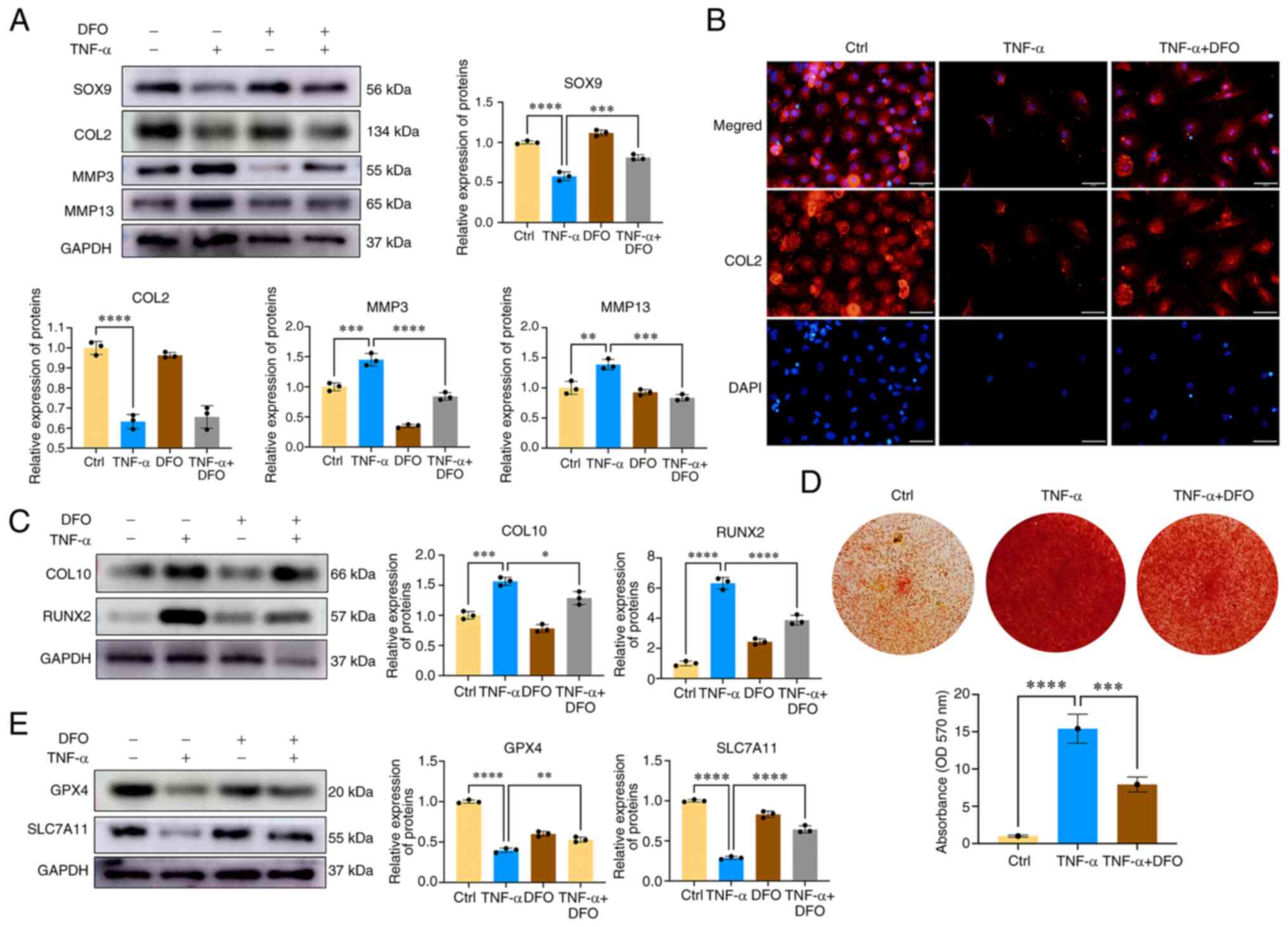 | Figure 6Iron chelation with DFO ameliorates
CEP chondrocyte degeneration and calcification. (A) CEP
chondrocytes were treated with TNF-α with or without 100 μM
DFO for 24 h. Western blotting was conducted to examine the protein
expression levels of SOX9, COL2, MMP3, MMP13 and GAPDH. The band
density was semi-quantified and normalized to the control. (B) CEP
chondrocytes were treated with TNF-α with or without 100 μM
DFO. Representative fluorescence photomicrographs of COL2 in CEP
chondrocytes are shown. Scale bar, 20 μm. (C) CEP
chondrocytes were treated with TNF-α with or without 100 μM
DFO for 24 h. Western blotting was conducted to examine the protein
expression levels of COL10, RUNX2 and GAPDH. The band density was
semi-quantified and normalized to the control. (D) Alizarin Red
staining for calcium deposition in CEP chondrocytes.
Semi-quantitative analysis of the mineralized nodule in CEP
chondrocytes is shown. (E) CEP chondrocytes were treated with TNF-α
with or without 100 μM DFO for 24 h. Western blotting was
conducted to examine the protein expression levels of GPX4, SLC7A11
and GAPDH. The band density was semi-quantified and normalized to
the control. Data are presented as the mean ± SD from three
independent experiments. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. CEP, cartilage endplate; COL, collagen;
Ctrl, control; DFO, desferoxamine; GPX4, glutathione peroxidase 4;
MMP, matrix metalloproteinase; SLC7A11, solute carrier family 7
member 11. |
Discussion
The degeneration and calcification of the CEP can
significantly hinder the nutrient supply to the intervertebral
disc, making it a recognized and substantial contributor to the
initiation and development of IDD (26). Although clinical evidence has
established a close association between endplate osteochondritis
and IDD, previous investigations have predominantly concentrated on
the degeneration of the nucleus pulposus (NP) and AF; however, the
mechanisms underlying CEP degeneration have not yet been fully
elucidated (27). The strategy
of targeting CEP degeneration aims to enhance nutrient and oxygen
supply to the intervertebral disc, and is gaining increasing
attention. It has previously been reported that Nrf2 has a
protective role in inhibiting IDD development (28). In addition, ferroptosis is a
newly identified cell death process that has been shown to serve an
important role in NP degeneration (17). In view of the close relationship
between ferroptosis and cellular oxidation levels, the roles of
Nrf2 signaling were examined with regard to the regulation of CEP
chondrocyte ferroptosis and its contribution to the development of
IDD in the present study. The results showed that Nrf2 serves
important roles in ameliorating CEP degeneration. In addition, Nrf2
can maintain CEP chondrocyte redox homeostasis and mitochondrial
function, decrease the levels of Fe2+ of the LIP and
inhibit ferroptosis by inhibiting NCOA4-mediated
ferritinophagy.
In the present study, CEP chondrocytes were treated
with TNF-α to mimic endplate osteochondritis. Notably, aging is the
leading cause of IDD (19). The
results of the present study demonstrated that Nrf2 expression in
the CEP was increased at the early stage of the IDD process, and
was decreased following 3 months of IDD development. No significant
differences in Nrf2 expression levels were observed in the CEP
between 16-month-old mice and 1-month-old mice. Similar results
were observed in the in vitro experiments of the present
study. Along with the decrease in Nrf2 expression, decreased Parkin
protein expression and increased mitochondrial dysfunction were
observed following treatment of the cells with a high concentration
of TNF-α treatment. To elucidate the role of Nrf2 in CEP
degeneration, CEP chondrocytes were isolated and transfected with
Nrf2 siRNA. The data indicated that inhibition of Nrf2 expression
promoted CEP degeneration and calcification. Furthermore,
inhibition of Nrf2 promoted CEP chondrocyte ferroptosis. The
results were consistent with those of the previous study indicating
that pro-inflammatory cytokines, such as IL-1β and TNF-α, or
oxidative stress could promote Nrf2 expression and activate the
antioxidant system (12,13). The results of the present study
further indicated that Nrf2 expression in the CEP was decreased in
the late stages of IDD, and inadequate activation of the Nrf2
signaling pathway aggravated CEP degeneration via promoting
mitochondrial dysfunction and oxidative stress.
Epidemiological evidence has suggested that due to
the lack of effective ways for the body to excrete iron,
middle-aged and elderly individuals are generally in an iron
overload state (29). Previous
research has found that when estrogen levels in postmenopausal
women decrease by 90% compared with normal levels, serum iron can
rapidly increase by 2-3 fold. In addition, the average serum iron
level in middle-aged and elderly men is 121 ng/ml, which is
considered to be 3-4 fold higher than that noted in adolescents
(30,31). Ferroptosis is closely regulated
by the lipid repair system, including GSH and GPX4. Two main
induction modes of cell ferroptosis have been proposed as follows:
One is the classical ferroptosis mode caused by decrease of the
GPX4 lipid repair system, and the other is the LIP caused by the
increase of cellular Fe2+ (32). Increased levels of
Fe2+ in the LIP can increase sensitivity to ferroptosis
(33). The present results
indicated that TNF-α increased the concentration of Fe2+
in CEP chondrocytes and promoted CEP chondrocyte ferroptosis.
Ferroptosis depends on the imbalance of iron
metabolism and the deposition of iron ions in cells. Cellular iron
overload can affect the sensitivity of cells to ferroptosis by
increasing the concentration levels of iron ions in cells and the
levels of lipid peroxidation (34). Numerous diseases and pathological
conditions in the human body are attributed to iron overload and
ferroptosis, such as chronic renal failure, Parkinson's disease,
Alzheimer's disease, osteoporosis and arteriosclerosis (35). In the present study, DFO was used
to chelate the cellular iron ions and decrease the Fe2+
of the LIP. The data indicated that DFO could inhibit TNF-α-induced
CEP degeneration and ferroptosis, and demonstrated that targeting
iron metabolism and ferroptosis is a promising treatment strategy
for IDD.
Cellular iron homeostasis is achieved by precise
regulation of iron metabolism-related proteins involved in iron
uptake (transferrin receptor protein 1, divalent metal transporter
1), storage (ferritin) and output [ferroportin (FPN)] (30). Previous studies have indicated
that Nrf2 can regulate the iron efflux protein FPN and promote
cellular iron output (36). In
the present study, it was revealed that, in addition to controlling
cellular iron output, Nrf2 could directly regulate the LIP by
controlling ferritin synthesis. NCOA4 serves as a specific cargo
receptor responsible for facilitating the autophagic degradation of
ferritin within lysosomes. This recycling mechanism is commonly
referred to as ferritinophagy (37). It has been demonstrated that
smoke exposure can promote the expression of NCOA4 in bronchial
epithelial cells, which could activate autophagic degradation of
ferritin and increase the intracellular LIP (38). The present study demonstrated
that Nrf2 could inhibit NCOA4-mediated ferritinophagy, thus
decreasing the levels of active Fe2+ of the LIP. By
contrast, Nrf2 inhibition promoted NOCA4 expression and increased
the induction of ferritinophagy, which could increase the levels of
cellular free Fe2+ and enhance cell susceptibility to
ferroptotic cell death.
Parkin-mediated mitophagy serves important roles in
NP degeneration and has been demonstrated to be a potential
therapeutic target for IDD (25). However, the role of Parkin in CEP
degeneration and calcification has been rarely reported (30). The present study demonstrated
that the protein expression levels of Parkin were increased in
response to a low concentration of TNF-α treatment and were
decreased along with the knockdown of Nrf2. Obvious Parkin
expression was observed in the control group and peak levels of
Parkin expression were observed in response to 1 ng/ml TNF-α; the
protein expression levels of Parkin began to decrease when the
concentration of TNF-α reached 5 ng/ml. It was hypothesized that
this may be because incubation with 5 ng/ml TNF-α for 24 h mimics
the pathological condition of OA and begins to cause damage to CEP
chondrocytes. A link between the Nrf2 signaling pathway and Parkin
protein has previously been demonstrated (29). To investigate the regulation of
the Parkin protein by Nrf2, Nrf2 siRNA was synthesized and
transfected into CEP chondrocytes. Notably, the expression levels
of Parkin were low in the group transfected with scrambled siRNA
but were significantly increased by 5 ng/ml TNF-α; it was
hypothesized that this may be attributed to the effect of the
scrambled siRNA and Lipofectamine on cell viability. The data
indicated that the Parkin-mediated mitophagic process was inhibited
and mitochondrial morphology destruction was increased with the
elevated expression of mitochondrial fission proteins, Drp1, MFF
and FIS1. Increased short and granulated mitochondria were observed
following inhibition of Nrf2 expression, indicating the destruction
and dysfunction of mitochondrial morphology.
In conclusion, the findings of the present study
demonstrated the essential role of Nrf2 in controlling CEP
degeneration and revealed that Nrf2 could regulate iron homeostasis
via NCOA4-mediated ferritinophagy. Inhibition of Nrf2 expression
could promote NCOA4-mediated ferritinophagy, thus enhancing the
free LIP, and promoting CEP chondrocyte oxidative stress and
ferroptotic cell death. Activation of Nrf2 and targeting iron
metabolism may be promising strategies for the treatment of
IDD.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZJ, CS and ZKM contributed to the research
conception and design. ZKM, HL, XMF, JHL, QZ and YDS performed the
experiments. XZJ, CS, YDS, TD and XDG performed data analysis. ZKM
and XZJ drafted the manuscript. XZJ, YDS and CS confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
All animal protocols were approved by the
Institutional Animal Care Committee of the Shandong Provincial
Hospital Affiliated to Shandong First Medical University (approval
no. 2022-816).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
CEP
|
cartilage endplate
|
|
NP
|
nucleus pulposus
|
|
AF
|
annulus fibrosus
|
|
LIP
|
labile iron pool
|
|
IDD
|
intervertebral disc degeneration
|
|
ROS
|
reactive oxygen species
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor 2
|
|
GPX4
|
glutathione peroxidase 4
|
|
NCOA4
|
nuclear receptor coactivator 4
|
|
GSH
|
glutathione
|
|
FTH
|
ferritin heavy chain
|
|
FPN
|
ferroportin
|
|
DFO
|
desferoxamine
|
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82002325), the Natural Science
Foundation of Shandong Province (grant nos. ZR2020QH075 and
ZR2022LZY001), the Shandong Province Traditional Chinese Medicine
Science and Technology Project (grant no. M-2022133) and the
Shandong Medical and Health Science and Technology Development Plan
Project (grant no. 202004071188).
References
|
1
|
Kos N, Gradisnik L and Velnar T: A brief
review of the degenerative intervertebral disc disease. Med Arch.
73:421–424. 2019. View Article : Google Scholar
|
|
2
|
Dowdell J, Erwin M, Choma T, Vaccaro A,
Iatridis J and Cho SK: Intervertebral disk degeneration and repair.
Neurosurgery. 80(3S): S46–S54. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhong R, Wei F, Wang L, Cui S, Chen N, Liu
S and Zou X: The effects of intervertebral disc degeneration
combined with osteoporosis on vascularization and microarchitecture
of the endplate in rhesus monkeys. Eur Spine J. 25:2705–2715. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li FC, Zhang N, Chen WS and Chen QX:
Endplate degeneration may be the origination of the vacuum
phenomenon in intervertebral discs. Med Hypotheses. 75:169–171.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ashinsky BG, Bonnevie ED, Mandalapu SA,
Pickup S, Wang C, Han L, Mauck RL, Smith HE and Gullbrand SE:
Intervertebral disc degeneration is associated with aberrant
endplate remodeling and reduced small molecule transport. J Bone
Miner Res. 35:1572–1581. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ding Y, Jiang J, Zhou J, Wu X, Huang Z,
Chen J and Zhu Q: The effects of osteoporosis and disc degeneration
on vertebral cartilage endplate lesions in rats. Eur Spine J.
23:1848–1855. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang X, Yu Y, Lei H, Cai Y, Shen J, Zhu
P, He Q and Zhao M: The Nrf-2/HO-1 Signaling Axis: A ray of hope in
cardiovascular diseases. Cardiol Res Pract. 2020:56957232020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He F, Ru X and Wen T: NRF2, a
transcription factor for stress response and beyond. Int J Mol Sci.
21:47772020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang G, Liu X, Jing X, Wang J, Wang H,
Chen F, Wang W, Shao Y and Cui X: Astaxanthin suppresses oxidative
stress and calcification in vertebral cartilage endplate via
activating Nrf-2/HO-1 signaling pathway. Int Immunopharmacol.
119:1101592023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shao Y, Sun L, Yang G, Wang W, Liu X, Du
T, Chen F, Jing X and Cui X: Icariin protects vertebral endplate
chondrocytes against apoptosis and degeneration via activating
Nrf-2/HO-1 pathway. Front Pharmacol. 13:9375022022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xiang Q, Zhao Y, Lin J, Jiang S and Li W:
The Nrf2 antioxidant defense system in intervertebral disc
degeneration: Molecular insights. Exp Mol Med. 54:1067–1075. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Galaris D, Barbouti A and Pantopoulos K:
Iron homeostasis and oxidative stress: An intimate relationship.
Biochim Biophys Acta Mol Cell Res. 1866:1185352019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeidan RS, Han SM, Leeuwenburgh C and Xiao
R: Iron homeostasis and organismal aging. Ageing Res Rev.
72:1015102021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jing X, Lin J, Du T, Jiang Z, Li T, Wang
G, Liu X, Cui X and Sun K: Iron Overload is associated with
accelerated progression of osteoarthritis: The role of DMT1
mediated iron homeostasis. Front Cell Dev Biol. 8:5945092020.
View Article : Google Scholar
|
|
16
|
Jing X, Du T, Li T, Yang X, Wang G, Liu X,
Jiang Z and Cui X: The detrimental effect of iron on OA
chondrocytes: Importance of pro-inflammatory cytokines induced iron
influx and oxidative stress. J Cell Mol Med. 25:5671–5680. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang RZ, Xu WN, Zheng HL, Zheng XF, Li B,
Jiang LS and Jiang SD: Involvement of oxidative stress-induced
annulus fibrosus cell and nucleus pulposus cell ferroptosis in
intervertebral disc degeneration pathogenesis. J Cell Physiol.
236:2725–2739. 2021. View Article : Google Scholar
|
|
18
|
Yao X, Sun K, Yu S, Luo J, Guo J, Lin J,
Wang G, Guo Z, Ye Y and Guo F: Chondrocyte ferroptosis contribute
to the progression of osteoarthritis. J Orthop Translat. 27:33–43.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang F, Cai F, Shi R, Wang XH and Wu XT:
Aging and age related stresses: A senescence mechanism of
intervertebral disc degeneration. Osteoarthritis Cartilage.
24:398–408. 2016. View Article : Google Scholar
|
|
20
|
Tang Z, Hu B, Zang F, Wang J, Zhang X and
Chen H: Nrf2 drives oxidative stress-induced autophagy in nucleus
pulposus cells via a Keap1/Nrf2/p62 feedback loop to protect
intervertebral disc from degeneration. Cell Death Dis. 10:5102019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Y, Han S, Kong M, Tu Q, Zhang L and
Ma X: Single-cell RNA-seq analysis identifies unique chondrocyte
subsets and reveals involvement of ferroptosis in human
intervertebral disc degeneration. Osteoarthritis Cartilage.
29:1324–1334. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Santana-Codina N, Gikandi A and Mancias
JD: The Role of NCOA4-Mediated ferritinophagy in ferroptosis. Adv
Exp Med Biol. 1301:41–57. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang W, Jing X, Du T, Ren J, Liu X, Chen
F, Shao Y, Sun S, Yang G and Cui X: Iron overload promotes
intervertebral disc degeneration via inducing oxidative stress and
ferroptosis in endplate chondrocytes. Free Radic Biol Med.
190:234–246. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song Y, Lu S, Geng W, Feng X, Luo R, Li G
and Yang C: Mitochondrial quality control in intervertebral disc
degeneration. Exp Mol Med. 53:1124–1133. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Z, Xu T, Chen J, Shao Z, Wang K, Yan
Y, Wu C, Lin J, Wang H, Gao W, et al: Parkin-mediated mitophagy as
a potential therapeutic target for intervertebral disc
degeneration. Cell Death Dis. 9:9802018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ariga K, Miyamoto S, Nakase T, Okuda S,
Meng W, Yonenobu K and Yoshikawa H: The relationship between
apoptosis of endplate chondrocytes and aging and degeneration of
the intervertebral disc. Spine (Phila Pa 1976). 26:2414–2420. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vergroesen PP, Kingma I, Emanuel KS,
Hoogendoorn RJ, Welting TJ, van Royen BJ, van Dieën JH and Smit TH:
Mechanics and biology in intervertebral disc degeneration: A
vicious circle. Osteoarthritis Cartilage. 23:1057–1070. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kang L, Liu S, Li J, Tian Y, Xue Y and Liu
X: Parkin and Nrf2 prevent oxidative stress-induced apoptosis in
intervertebral endplate chondrocytes via inducing mitophagy and
anti-oxidant defenses. Life Sci. 243:1172442020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gozzelino R and Arosio P: Iron homeostasis
in health and disease. Int J Mol Sci. 17:1302016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Anderson CP, Shen M, Eisenstein RS and
Leibold EA: Mammalian iron metabolism and its control by iron
regulatory proteins. Biochim Biophys Acta. 1823:1468–1483. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Masaldan S, Clatworthy SAS, Gamell C,
Meggyesy PM, Rigopoulos AT, Haupt S, Haupt Y, Denoyer D, Adlard PA,
Bush AI and Cater MA: Iron accumulation in senescent cells is
coupled with impaired ferritinophagy and inhibition of ferroptosis.
Redox Biol. 14:100–115. 2018. View Article : Google Scholar
|
|
32
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao
N, Sun B and Wang G: Ferroptosis: Past, present and future. Cell
Death Dis. 11:882020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Siddique A and Kowdley KV: Review article:
The iron overload syndromes. Aliment Pharmacol Ther. 35:876–893.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Han K, Jin X, Guo X, Cao G, Tian S, Song
Y, Zuo Y, Yu P, Gao G and Chang YZ: Nrf2 knockout altered brain
iron deposition and mitigated age-related motor dysfunction in
aging mice. Free Radic Biol Med. 162:592–602. 2021. View Article : Google Scholar
|
|
37
|
Santana-Codina N and Mancias JD: The role
of NCOA4-Mediated ferritinophagy in health and disease.
Pharmaceuticals (Basel). 11:1142018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zi Y, Wang X, Zi Y, Yu H, Lan Y, Fan Y,
Ren C, Liao K and Chen H: Cigarette smoke induces the ROS
accumulation and iNOS activation through deactivation of
Nrf-2/SIRT3 axis to mediate the human bronchial epithelium
ferroptosis. Free Radic Biol Med. 200:73–86. 2023. View Article : Google Scholar : PubMed/NCBI
|














