Introduction
Chronic lung inflammation induced by inhalable
particles is a major cause of chronic obstructive pulmonary disease
(COPD), asthma, pulmonary fibrosis and occupational pulmonary
diseases (1-3). Silicosis is an irreversible and
incurable lung disease caused by inhalation of crystalline silica;
even when patients are no longer exposed to silica particles,
fibrosis continues to progress (4). Mounting evidence suggests that
silica-induced persistent damage to the distal lung is a major
cause of epithelial remodeling and fibrosis in silicosis (5) and epithelial remodeling is the
final outcome caused by the functional abnormalities of epithelial
cells, fibroblasts and immune cells (6,7).
Histologically, inhaled silica particles are deposited in the
terminal ends of distal bronchioles, contributing to mucous
plugging (8), epithelial
dysfunction (9,10) and peribronchiolar fibrosis and
bronchiolar obstruction (11,12). Moreover, repetitive or recurrent
injury to the distal lung results in dysregulated repair and
regeneration, further promoting the development of pulmonary
fibrosis (13,14).
The distal lung contains terminal bronchioles and
alveoli that facilitate gas exchange and can be compromised by
disorders including interstitial lung disease and coronavirus
disease 2019 (15). The
bronchoalveolar duct junction (BADJ) is a unique region at the
terminal ends of distal bronchioles at which the lung is separated
into the proximal conducting airways and peripheral gas exchange
region (16). At the BADJ, an
abrupt transition occurs in cell type and morphology (airway to
alveolar epithelial cells) (16), which determines the unique
features and key roles of the area. Moreover, studies have
demonstrated the importance of the BADJ as the niche for
bronchioalveolar stem cells (BASCs) co-expressing markers of club
cells and type II alveolar epithelial cells (17,18); this population exhibits a robust
capacity for repair and regeneration when treated with naphthalene
or bleomycin in vivo (19). Therefore, the distal lung is an
appropriate site for study of reparative and regenerative responses
in fibrotic lung injury.
The NLRP3 inflammasome is a large cytosolic protein
complex comprising NLRP3, apoptosis-associated speck-like protein
containing a CARD domain (ASC), and pro-Caspase-1 (20). Once activated, the assembled
complex activates pro-Caspase-1; cleaved Caspase-1 in turn produces
the biologically active forms of pro-interleukin-1β (IL-1β) and
IL-18, as well as pyroptotic cell death (21,22). To date, the NLRP3 inflammasome
has been demonstrated to contribute to the progression of several
types of inflammatory respiratory disease, including lung fibrosis
(23,24). Additionally, silica is a
well-recognized activator of the NLRP3 inflamma-some (25,26), which promotes inflammatory damage
leading to progressive fibrosis in silicosis. MCC950 is a highly
potent specific NLRP3 inhibitor with good pharmacokinetic and
pharmacodynamic properties that can block NLRP3-mediated ASC
oligomerization and inflammasome assembly (27,28). Additionally, inflammasome
activation appears to regulate the balance between tissue repair
and inflammation following inhalation of crystalline silica
(29). However, it remains
unclear whether silica-induced NLRP3 inflammasome activation
mediates epithelial remodeling and dysregulated regeneration in the
distal lung. In the present study, a silica-induced mouse lung
fibrosis model was used to examine the effects and mechanisms of
the NLRP3 inflammasome on regulating the balance between pulmonary
inflammation, epithelial remodeling and dysregulated regeneration
in the distal lung at three time points.
Materials and methods
Animals
All experimental procedures involving mice were
approved by the Institutional Animal Care and Use Committee of
Nanjing Medical University (Nanjing, China; approval no.
NJMU/IACUC-2012034) and complied with the guidelines published by
the National Institutes of Health Guide for the Care and Use of
Laboratory Animals (NIH publication No. 85-23, revised 1996)
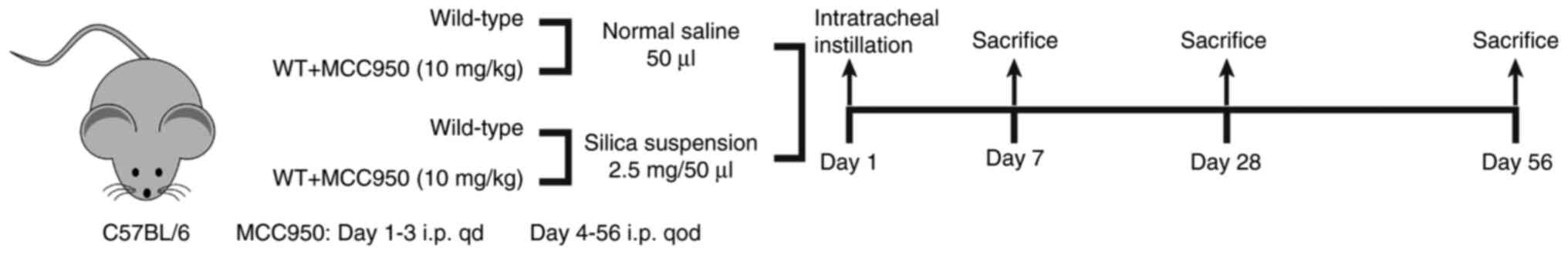
(30). A total of 72 male
C57BL/6 mice (22-25 g; age, 8 weeks) provided by Nanjing Medical
University Experimental Animal Center (Nanjing, China) were given
free access to food and water at 22°C), controlled illumination (12
h light/dark cycles) and suitable humidity (40-60%).
Experimental design
After 1 week of acclimation to the environment,
wild-type C57BL/6 mice were randomly assigned to four groups
(normal saline, normal saline + MCC950, silica and silica + MCC950;
n=18/group) and received a single tracheal instillation of 50
μl sterile saline or 2.5 mg silica crystals (cat. no.
CAS14808-60-7; purity 99%; particle diameter 0.5-10.0 μm;
Sigma-Aldrich; Merck KGaA) in the same volume of sterile saline.
MCC950 (10 mg/kg; Selleck Chemicals) was administered
intraperitoneally every day for the first 3 days and every other
day for the next 4, 25 or 53 days. Sterile saline was
intraperitoneally injected into mice as the negative control. Six
mice from each group were anesthetized with an intraperitoneal
injection of sodium pentobarbital (50 mg/kg) and sacrificed at 7,
28 and 56 days post-instillation (Fig. 1).
Assessment of pulmonary function
Prior to sacrifice of the mice, the Buxco FinePointe
RC system (Data Sciences International) was used to analyze
ventilatory parameters, including static lung compliance, dynamic
lung compliance and airway resistance of mice. Respiratory
frequency was set as 100 breaths/min and tidal volume was set as
0.2 ml with a positive end-expiratory pressure of 2 cm
H2O. The mean values (n=6) were recorded during 3-min
period following ventilation.
Bronchoalveolar lavage fluid (BALF)
collection and cell counting
BALF was collected after the mice were euthanized
with an overdose of sodium pentobarbital (150 mg/kg). BALF was
obtained by infusing 0.5 ml cold sterile saline three consecutive
times with the assistance of tracheal cannulation, followed by
centrifugation at 200 × g and 4°C for 10 min. The supernatant was
separated and stored at −80°C for subsequent analysis, and the cell
pellets were resuspended in 1 ml saline for differential cell
counting using a BTX-1800 hematology analyzer (Zibo Hengtuo
Analytical Instrument Co., Ltd.).
Analysis of IL-1β, IL-18, TNF-α and IL-10
in BALF
The levels of IL-1β (cat. no. KE10003, Proteintech),
IL-18 (CSB-E04609m, CUSABIO), TNF-α (KE10002, Proteintech) and
IL-10 (KE10008, Proteintech) in BALF were measured using
enzyme-linked immunosorbent assay kits according to the
manufacturer's instructions.
Histological, immunohistochemical and
immunofluorescent analyses
Right lung tissue was infused with 4% (w/v)
paraformaldehyde (Sigma-Aldrich; Merck KGaA) using a blunted
30-gauge needle through the trachea and fixed in 4%
paraformaldehyde at 4°C overnight.
For paraffin sectioning, the lungs were dehydrated
using an ethanol gradient, embedded in paraffin and sectioned (5
μm). Hematoxylin and eosin (H&E) and Masson's trichrome
staining were performed following standard protocols (31,32) and examined by an Olympus VS200
slide scanner (Olympus Corporation) to assess mean inflammation or
fibrosis. Lung inflammation was graded as previously described
(33): none (0), no alveolitis;
mild (1+), affected area <20%; moderate (2+), affected area
20-50% and severe (3+), affected area >50%. Lung fibrosis was
graded and quantified by the modified scale (34): alveolar septa: no fibrotic burden
at the most flimsy small fibers in some alveolar walls, lung
structure: normal lung (0); alveolar septa: isolated gentle
fibrotic changes, lung structure: alveoli partly enlarged and
rarefied, but no fibrotic masses present (1); alveolar septa: clearly fibrotic
changes with knot-like formation but not connected to each other,
lung structure: alveoli partly enlarged and rarefied but no
fibrotic masses (2); alveolar
septa: contiguous fibrotic walls predominantly in whole microscopic
field, lung structure: alveoli partly enlarged and rarefied, but no
fibrotic masses (3); alveolar
septa: variable, lung structure: single fibrotic masses (4); alveolar septa: variable, lung
structure: confluent fibrotic masses, and lung structure severely
damaged but still preserved (5);
alveolar septa: variable, mostly not existent, lung structure:
large contiguous fibrotic masses, and lung architecture mostly not
preserved (6); alveolar septa:
non-existent, lung structure: alveoli nearly obliterated with
fibrous masses but still up to five air bubbles (7); alveolar septa: non-existent, lung
structure: microscopic field with complete obliteration with
fibrotic masses (8).
Immunostaining was performed using standard procedures. Briefly,
paraffin sections were dewaxed using xylene and rehydrated using
gradient alcohol, and antigen repair was performed by microwave
heating (95°C, 20 min). The sections were blocked with QuickBlock™
Blocking Buffer (Beyotime Institute of Biotechnology) for 30 min at
room temperature and incubated at 4°C overnight with anti-NLRP3
(1:50, cat. no. NBP2-12446, Novus Biologicals, LLC), anti-Caspase-1
(1:100; cat. no. ab138483, Abcam), anti-IL-1β (1:200, ab205924,
Abcam), anti-Ki67 (1:200, AF0198, Affinity Biosciences),
anti-surfactant protein C (SPC) (1:200, DF6647, Affinity
Biosciences), anti-club cell 10 kDa protein (CC10) (1:200,
sc-365992, Santa Cruz Biotechnology, Inc.), anti-gasdermin D
(GSDMD) (1:400, AF4012, Affinity Biosciences), anti-nerve growth
factor receptor (NGFR) (1:200, ab271290, Abcam) and anti-Vimentin
(1:1,000, ab8978, Abcam) antibodies, then incubated with
corresponding secondary antibodies for 1 h at room temperature:
goat anti-rabbit IgG (H+L) (1:500, 111-035-003, Jackson
ImmunoResearch Laboratories, Inc.), donkey anti-mouse Alexa Fluor™
488 (1:1,000, A-21202, Invitrogen; Thermo Fisher Scientific, Inc.),
donkey anti-rabbit Alexa Fluor™ 555 (1:1,000, A-31572, Invitrogen;
Thermo Fisher Scientific, Inc.) or goat anti-rat Alexa Fluor™ 647
(1:1,000, ab150159, Abcam). Images were captured using an Olympus
VS200 slide scanner and Leica Thunder DMi8 Imager (Leica GmbH;
magnification, ×40).
For cryosectioning (15 μm), fixed tissues
were dehydrated in 20 and 30% sucrose solution before embedding in
the optimal cutting temperature compound (Sakura Finetek).
Immunofluorescence staining was performed following standard
protocols. The sections were blocked with QuickBlock™ Blocking
Buffer (Beyotime Institute of Biotechnology) for 30 min at room
temperature and incubated with antibodies against GSDMD (1:400,
AF4012, Affinity Biosciences), SOX9 (1:400, ab185966, Abcam), SOX2
(1:200, 14-9811-82, Invitrogen; Thermo Fisher Scientific, Inc.),
mucin 5 subtype AC (MUC5AC) (1:100, abs126767, Absin (Shanghai)
Biotechnology Co., Ltd.), MUC5B (1:200, ab77995, Abcam), E-Cadherin
(1:200, 20874-1-AP, Proteintech Group, Inc.) and Vimentin (1:1,000,
ab8978, Abcam) at 4°C overnight, and then incubated with secondary
antibodies for 1 h at room temperature: donkey anti-mouse Alexa
Fluor™ 488 (1:1,000, A-21202, Invitrogen; Thermo Fisher Scientific,
Inc.), donkey anti-rabbit Alexa Fluor™ 555 (1:1,000, A-31572,
Invitrogen; Thermo Fisher Scientific, Inc.) or goat anti-rat Alexa
Fluor™ 647 (1:1,000; cat. no. ab150159, Abcam). Nuclei were
counter-stained with DAPI (Beyotime Institute of Biotechnology) for
10 min at room temperature. All samples were covered with ProLong™
Gold antifade reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
Fluorescence images were captured using a Leica Thunder DMi8 Imager
and Stellaris STED confocal microscope (Leica GmbH; magnification,
×40).
Measurement of hydroxyproline (HYP)
content in peripheral lung tissue
The concentration of HYP was measured using a HYP
assay kit (cat. no. A030-2-1, Nanjing Jiancheng Bioengineering
Institute) according to the manufacturer's protocol. Peripheral
lung tissue (60-90 mg) collected from the left lobe was hydrolyzed,
and the results were calculated as μg HYP per g wet lung
weight.
Western blotting
Peripheral lung tissue from the left lobe was
homogenized in cold RIPA buffer (Thermo Fisher Scientific, Inc.)
supplemented with protease and phosphatase inhibitors. Protein
concentrations were measured using a bicinchoninic acid protein
concentration assay kit according to the instructions (Beyotime
Institute of Biotechnology). Equal amounts of protein (40
μg/lane) were subjected to electrophoresis on 8-12% SDS-PAGE
and electroblotted PVDF membranes (MilliporeSigma). The membranes
were blocked in 5% defatted milk (Beyotime Institute of
Biotechnology) for 1 h at room temperature and then incubated at
4°C overnight with the following primary antibodies: Anti-NLRP3
(1:500, NBP2-12446, Novus Biologicals, LLC), anti-pro-Caspase-1
(1:1,000, 24232, Cell Signaling Technology, Inc.), anti-ASC
(1:1,000, 67824, Cell Signaling Technology, Inc.), anti-Caspase-1
p20 (1:1,000, 22915-1-AP, Proteintech Group, Inc.), anti-GSDMD
(1:1,000, AF4012, Affinity Biosciences), anti-SOX9 (1:1,000, 82630,
Cell Signaling Technology, Inc.), anti-SOX2 (1:1,000, ab97959,
Abcam), anti-E-Cadherin (1:1,000, 14472, Cell Signaling Technology,
Inc.), anti-Vimentin (1:1,000, ab8978, Abcam), anti-Sonic hedgehog
(Shh) (1:500, TA500040, OriGene Technologies, Inc.),
anti-Smoothened (Smo) (1:1,000, ab236465, Abcam),
anti-glioma-associated oncogene homolog-1 (Gli1) (1:2,000,
NB600-600, Novus Biologicals, LLC), anti-Wnt10a (1:1,000, ab106522,
Abcam), anti-phospho-glycogen synthase kinase-3β (p-GSK-3β)
(1:1,000, 9336, Cell Signaling Technology, Inc.), anti-GSK-3β
(1:1,000, 9315, Cell Signaling Technology, Inc.), anti-β-catenin
(1:1,000, 8480, Cell Signaling Technology, Inc.), anti-β-actin
(1:2,000, 20536-1-AP, Proteintech Group, Inc.) and anti-GAPDH
(1:2,000, 10494-1-AP, Proteintech Group, Inc.). After incubation
with horseradish peroxidase-conjugated secondary antibodies
(1:8,000, 111-035-003/115-035-003, Jackson ImmunoResearch
Laboratories, Inc.) for 1 h at room temperature, the membranes were
incubated with WesternBright Quantum (Advansta, Inc.) and protein
expression was quantified using a ChemiDoc™ XRS+ system with Image
Lab 4.0 software (Bio-Rad Laboratories, Inc.).
Statistical analysis
Data were analyzed using SPSS 18.0 software (SPSS,
Inc.) and are presented as the mean ± standard error of the mean of
3-6 independent experimental repeats. Statistical analysis was
performed using one-way ANOVA followed by Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Pharmacological inhibition of NLRP3
inflammasome improves pulmonary function in silica-treated
mice
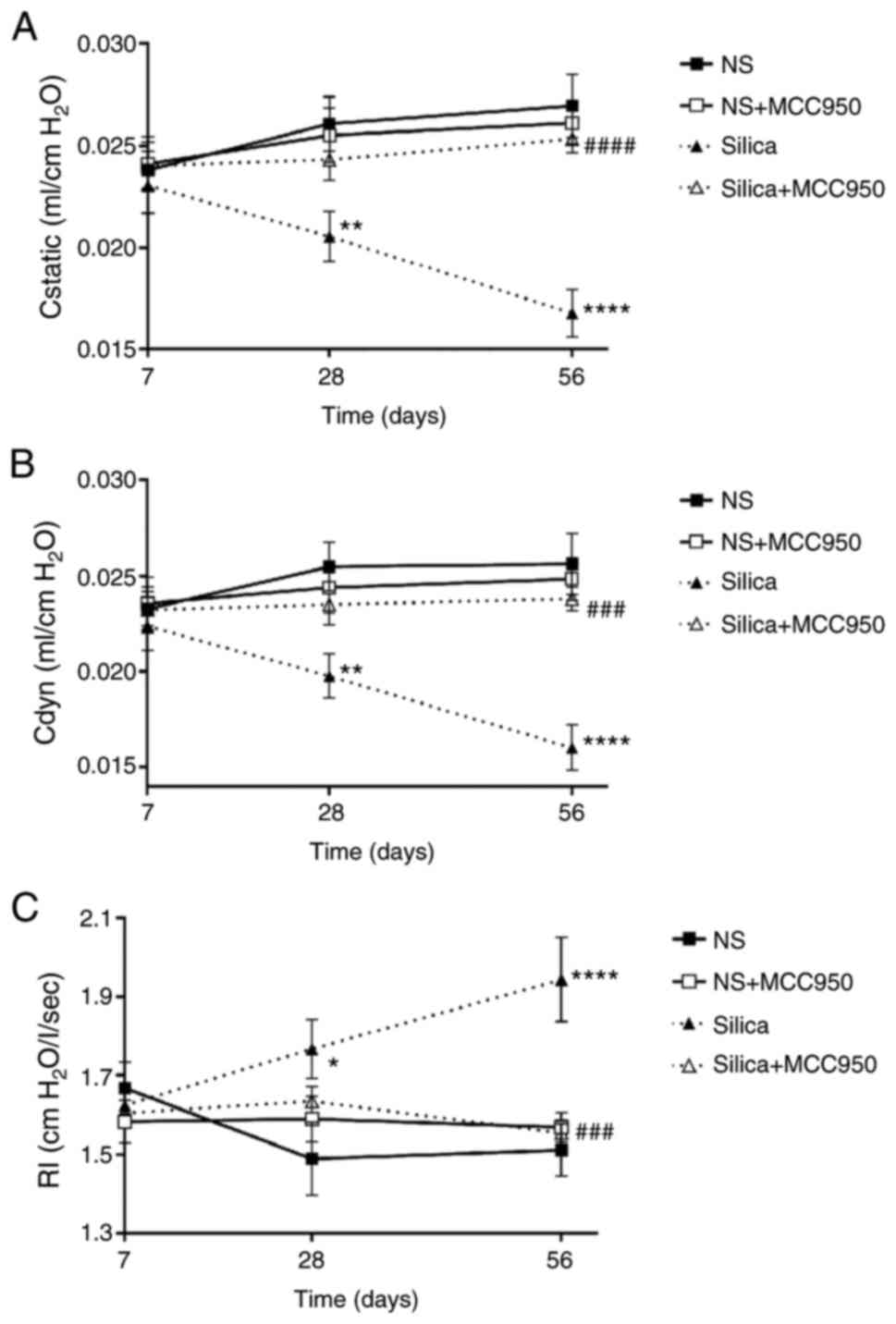
To evaluate the effects of NLRP3 inflammasome on
lung tissue remodeling in silica-exposed mice, pulmonary function
was assessed by measuring lung compliance and resistance (Fig. 2A-C). Although there was no
significant difference in static compliance, dynamic compliance or
airway resistance on day 7, the silica-treated group had worse
pulmonary function than the other groups on days 28 and 56.
MCC950-alone exerted little effect on pulmonary function, while it
rescued the significant decrease in lung compliance and increase in
airway resistance induced by silica at day 56. These results
demonstrated that inhibiting NLRP3 inflammasome activation improved
pulmonary function in silica-treated mice.
Inhibition of NLRP3 inflammasome
alleviates distal lung remodeling by inhibiting the fibrotic
response in silica-treated mice
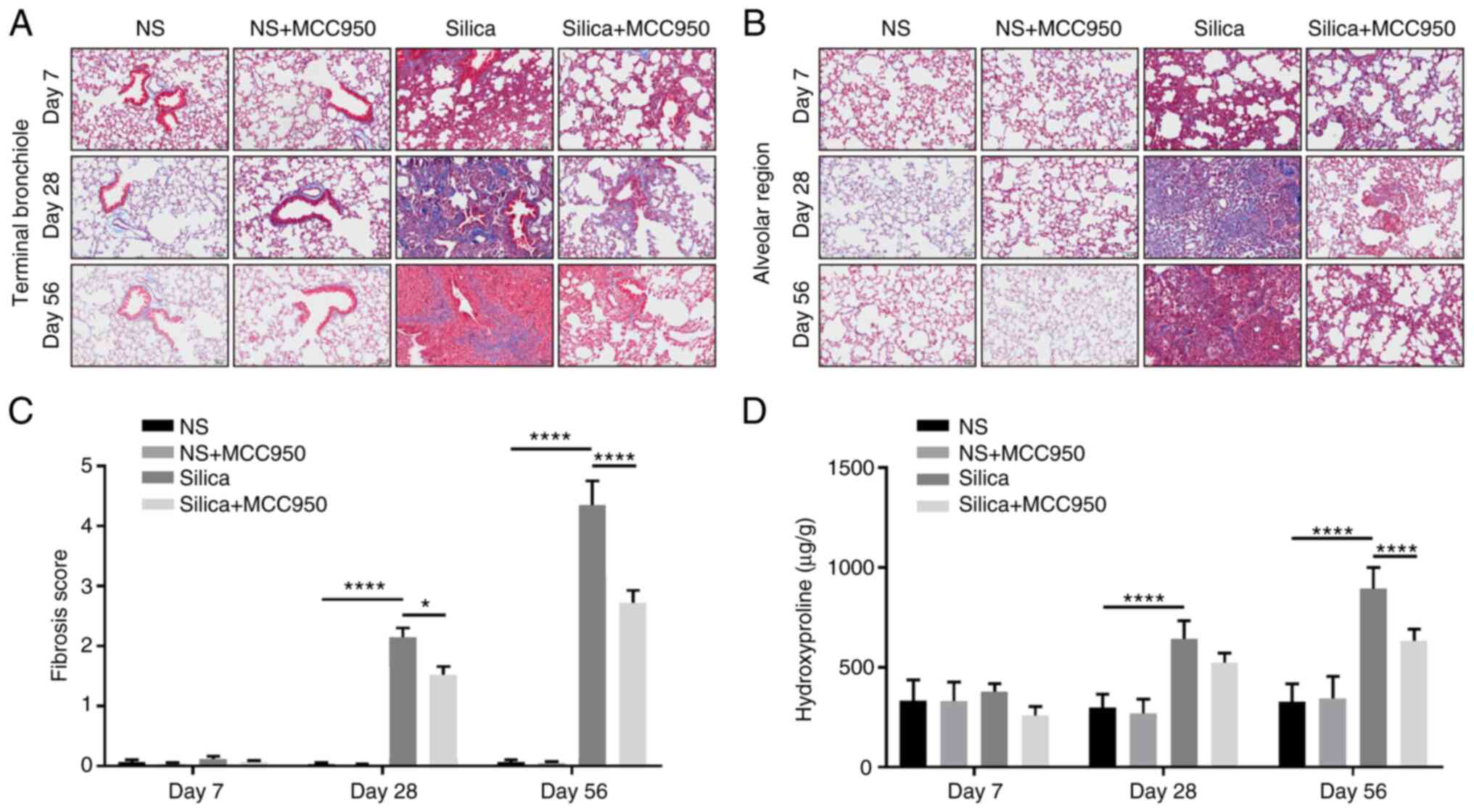
The role of the NLRP3 inflammasome in the
silica-induced fibrotic response in distal lung was further
evaluated. Masson's trichrome staining indicated that silica
induced excess collagen hyperplasia in the terminal bronchiole
(Fig. 3A) and alveolar region
(Fig. 3B). In addition,
intratra-cheal instillation of silica suspension triggered a
significant pulmonary fibrotic response (increases in positive
Masson's trichrome staining and fibrosis score) in the distal lung
starting on day 28 (Fig. 3A-C).
Collagen deposition measured by hydroxyproline assay revealed that
although MCC950-alone had no significant effect, it alleviated the
lung fibrotic response in silica-treated mice, especially during
chronic fibrosis (day 56; Fig.
3D).
Inhibition of NLRP3 inflammasome
ameliorates distal lung inflammatory response in silica-treated
mice
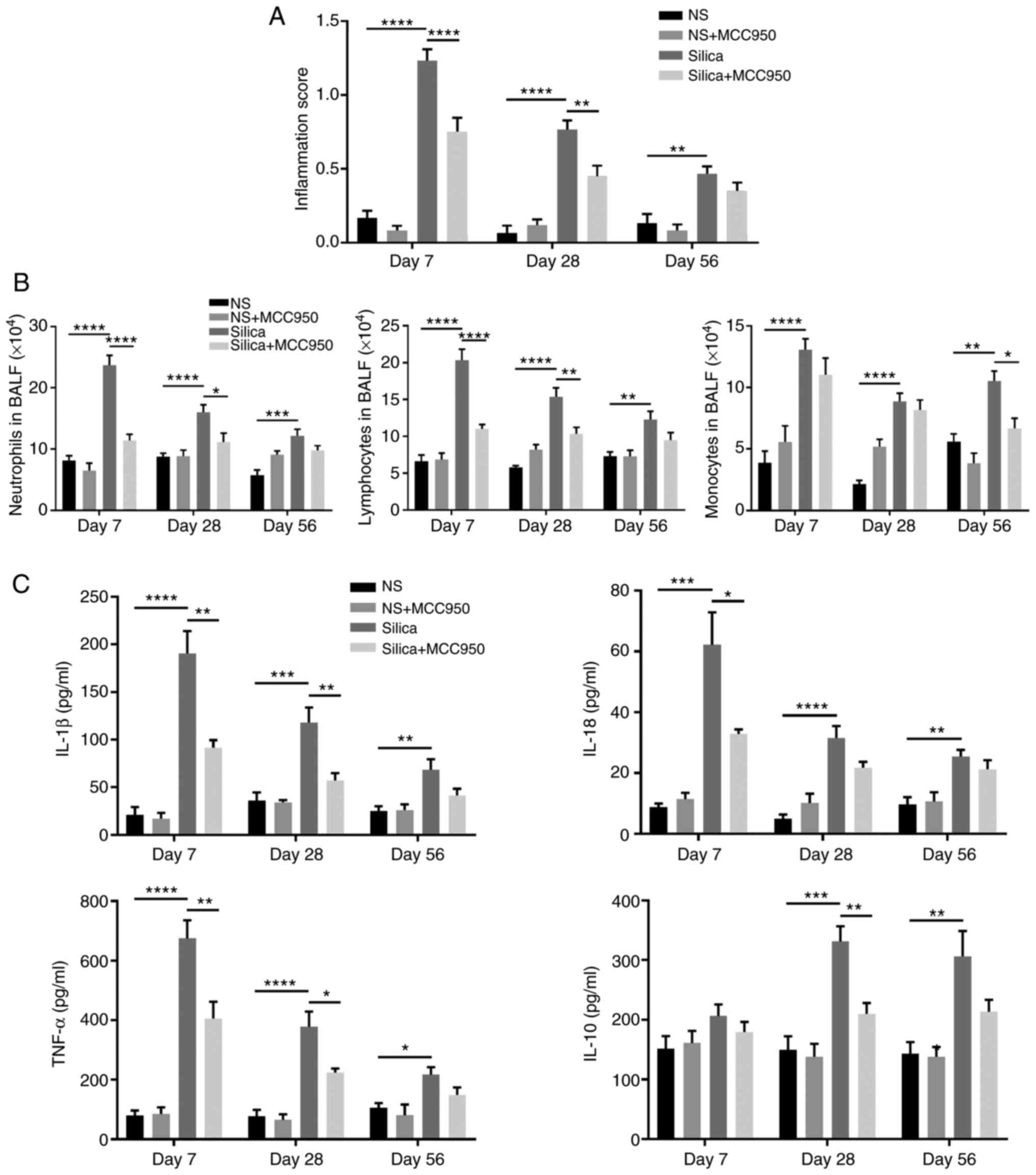
Effects of inhibition of the NLRP3 inflammasome on
the silica-induced inflammatory response in the distal lung were
investigated. H&E staining confirmed that intratracheal
instillation of silica suspension induced diffuse infiltration of
inflammatory cells in the terminal bronchiole (Fig. S1A) and alveolar region (Fig. S1B), which was consistent with
increases in inflammation score on days 7, 28 and 56 (Fig. 4A). Moreover, inflammatory cell
count, including neutrophils, lymphocytes and monocytes in BALF
(Fig. 4B) and the concentrations
of pro-inflammatory cytokines (IL-1β, IL-18 and TNF-α) and
anti-inflammatory factor (IL-10) in BALF (Fig. 4C) revealed that MCC950
ameliorated lung inflammatory responses in silica-treated mice,
especially during acute inflammation (day 7).
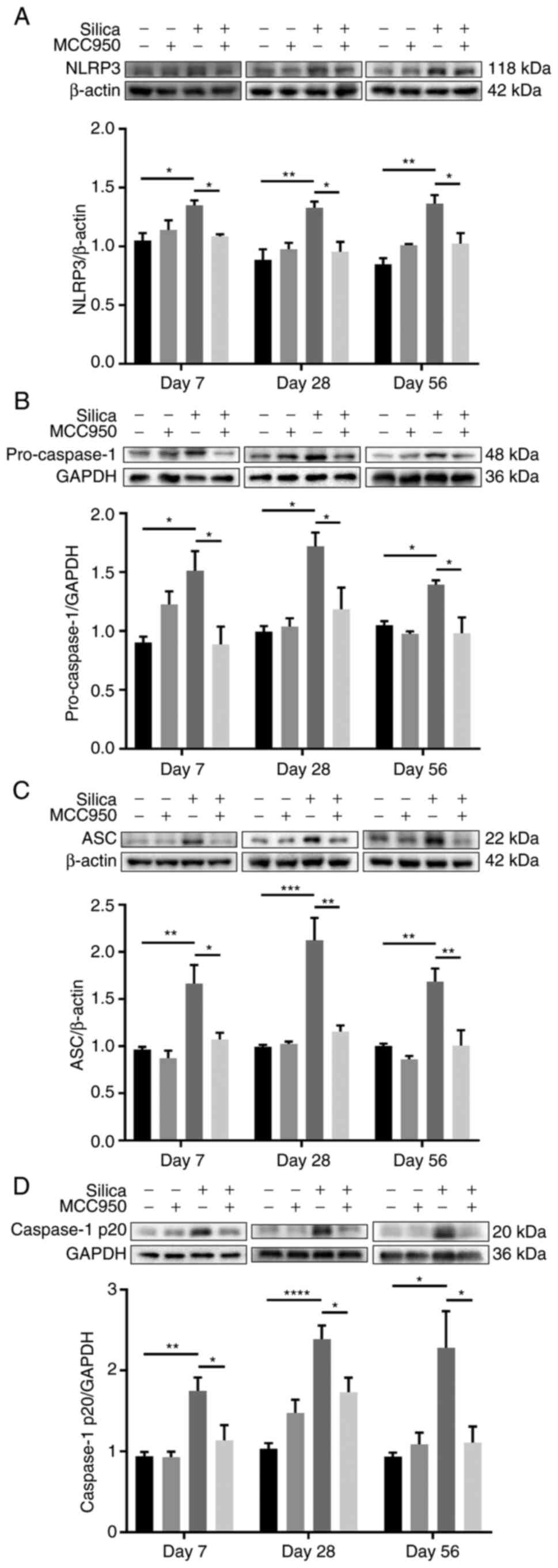
Instillation of a silica suspension leads
to sustained NLRP3 inflammasome activation and pyroptosis in the
distal lung
As silica is a well-recognized activator of the
NLRP3 inflammasome (35,36), NLRP3 inflammasome activation was
investigated. Both western blot (Fig. 5A-D) and immunohistochemical
(Figs. S2A-C and S3A-C)
analysis of components and products of the NLRP3 inflammasome
(including NLRP3, pro-Caspase-1, ASC, Caspase-1 p20 and IL-1β)
confirmed that single intratracheal instillation of silica
suspension resulted in sustained activation of the NLRP3
inflammasome, which was significantly suppressed by pharmacological
inhibition of the NLRP3 inflammasome using MCC950. In addition,
representative immunohistochemical staining of the terminal
bronchiole (Fig. S2A-C) and
alveolar region (Fig. S3A-C)
indicated sustained NLRP3 inflammasome activation in the
development of silica-induced progressive epithelial remodeling of
the distal lung.
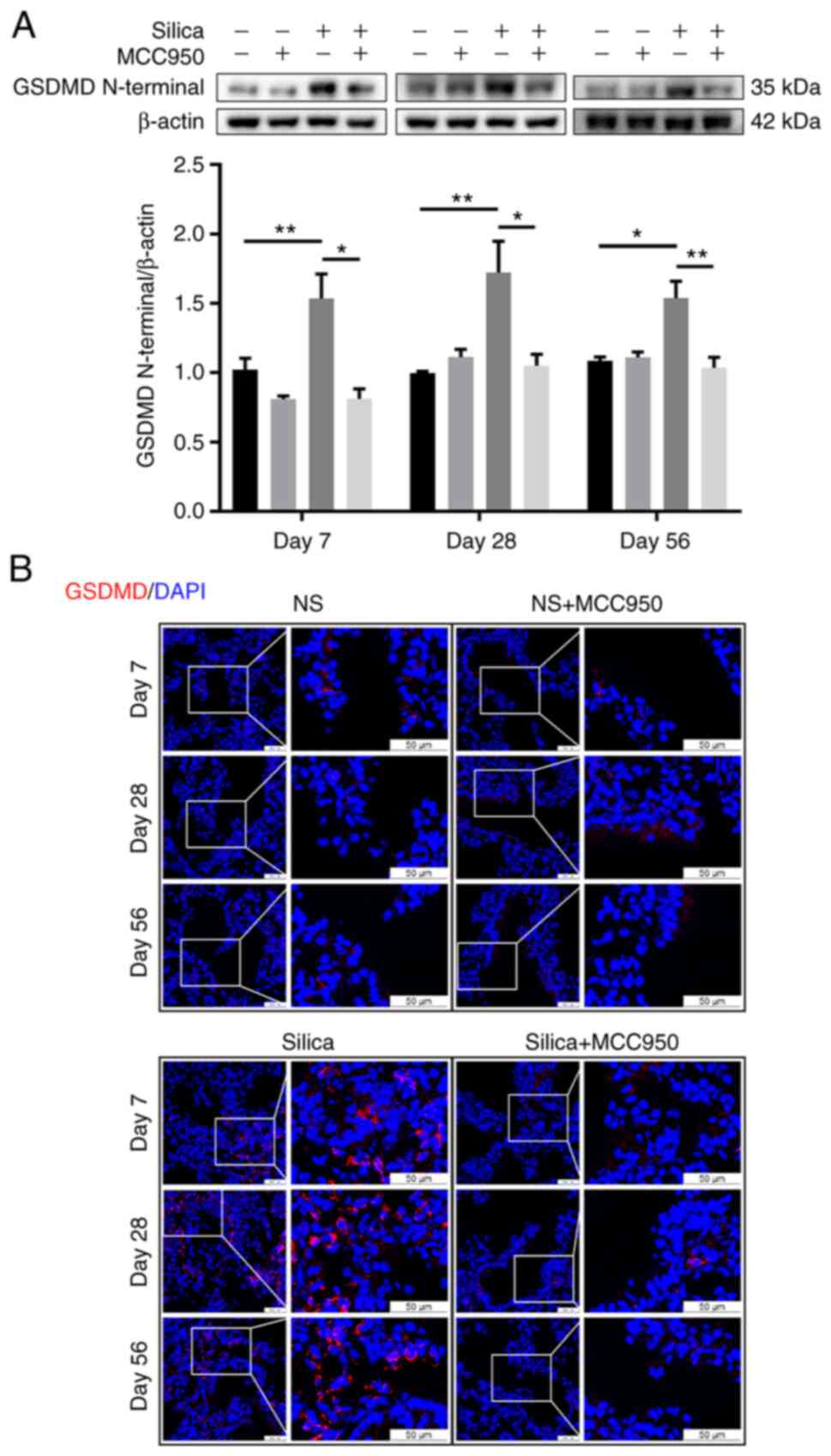
NLRP3-dependent pyroptosis is an autolytic
programmed cell death characterized by membrane rupture and release
of proinflammatory intracellular contents (36). Once activated, NLRP3 inflammasome
downstream of Caspase-1 cleaves cytoplasmic GSDMD to release an
active N-terminal domain to induce pyroptotic cell death (37). Western blotting revealed that,
compared with the control, single silica instillation persistently
upregulated expression of GSDMD N-terminal, which was reversed by
the NLRP3 inflammasome inhibitor MCC950 (Fig. 6A). Similarly, immunofluorescence
analysis showed that silica caused more membrane-distributed
GSDMD+ cells (evidence of pyroptosis activation)
(38) in the terminal
bronchiole, which was alleviated by MCC950 (Fig. 6B). Furthermore, pyroptotic cells
were mainly epithelial cells, including club cells
(CC10+) and ectopic basal cells (NGFR+)
(Fig. S4).
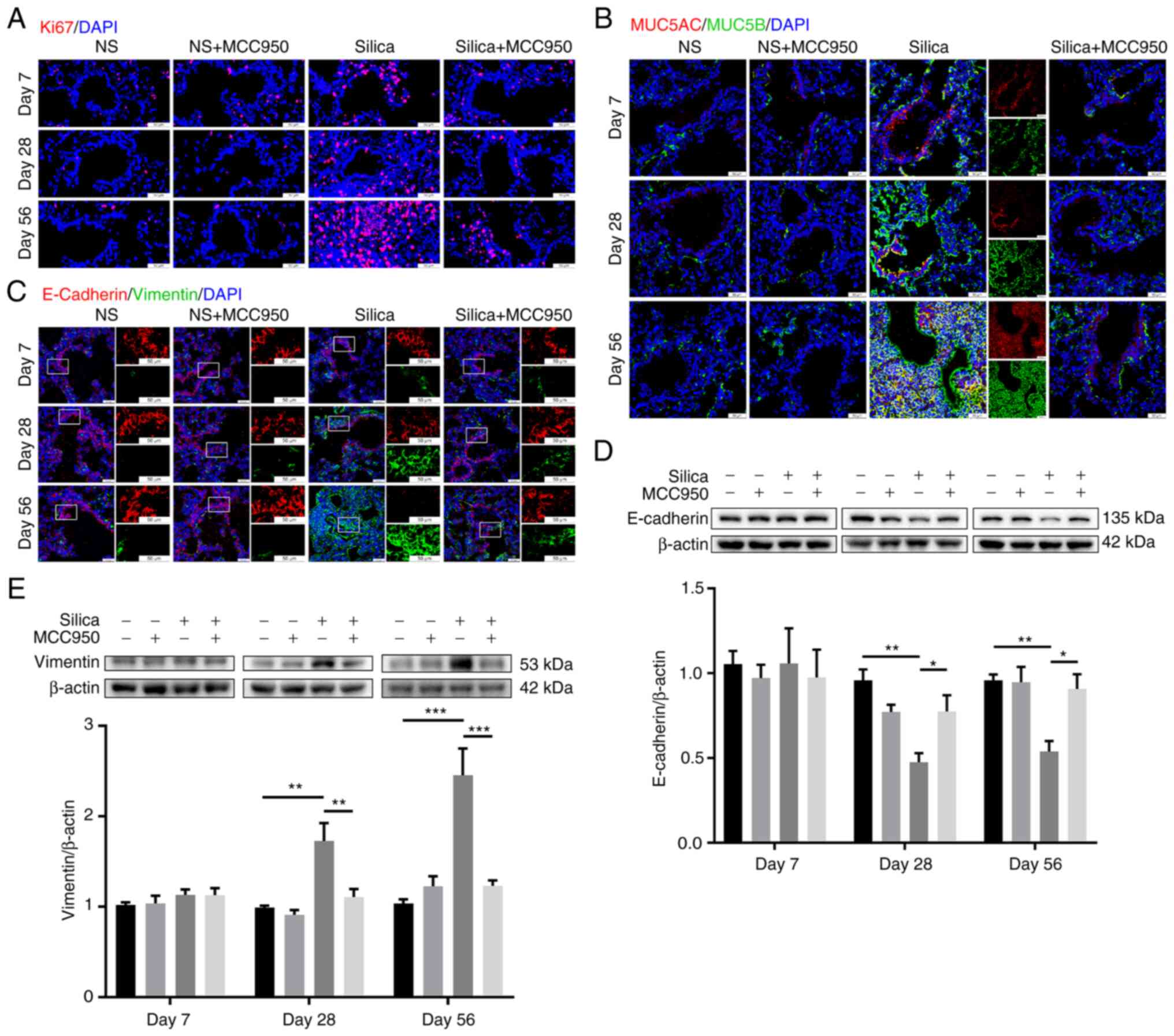
Silica-induced sustained NLRP3
inflammasome activation enhances cell proliferation, mucus
production and epithelial-mesenchymal transition (EMT) in the
distal lung
The effects of silica-induced NLRP3 inflammasome
activation on cell proliferation, mucus production and EMT in the
distal lung were further investigated. Immunofluorescence revealed
that single administration of silica suspension led to
progressively increased cell proliferation and mucus (MUC5AC/MUC5B)
production in the terminal bronchiole, which were significantly
suppressed by NLRP3 inflammasome inhibitor MCC950 (Fig. 7A and B). Moreover, proliferative
cells were fibroblasts (Vimentin+; key effector cells in
the pathogenesis of pulmonary fibrosis (39); Fig. S5). Additionally, silica exposure
promoted EMT of cells in the distal lung during the development of
pulmonary fibrosis, with decreased E-Cadherin (epithelial marker)
and increased Vimentin (mesenchymal marker) (40), especially in the chronic fibrotic
phase (day 56; Fig. 7C). Western
blotting demonstrated consistent results with the
immunofluorescence assays (Fig. 7D
and E). MCC950-alone did not affect the expression of
EMT-associated markers in control mice, but reversed the
silica-induced downregulation of E-Cadherin and upregulation of
Vimentin on days 28 and 56 (Fig. 7D
and E).
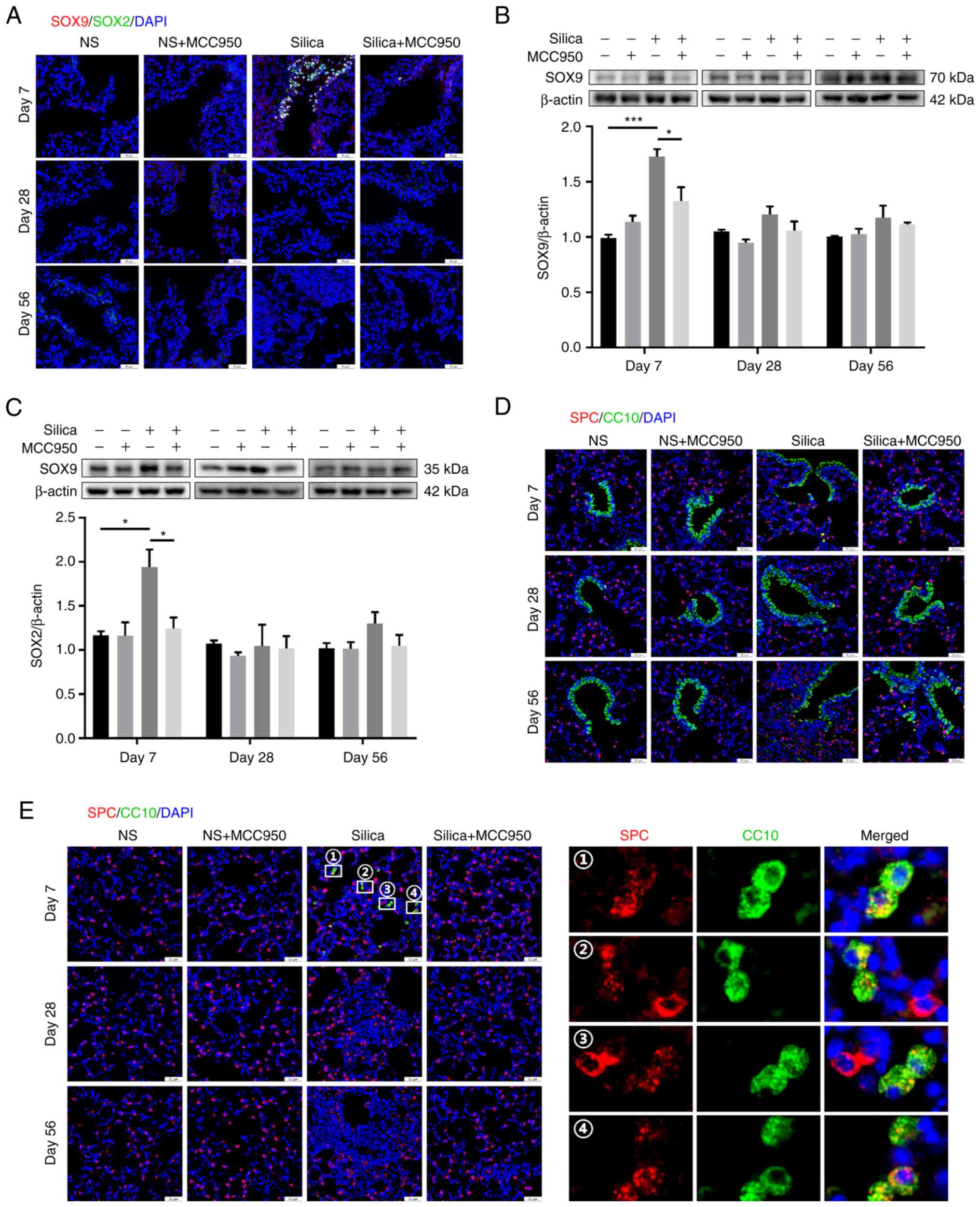
Silica-induced NLRP3 inflammasome
activation causes abnormal repair and regeneration in the distal
lung
Based on the findings that inhibition of the NLRP3
inflammasome alleviated epithelial remodeling in the distal lung,
the reparative and regenerative behaviors of cells in this region
in response to silica challenge were investigated. SOX9 and SOX2
are important factors related to lung repair and regeneration
(41,42), and
SOX9+SOX2+ progenitor cells play a major role
in embryonic lung branching morphogenesis by specifying
proximal-distal fate (43). In
terminal bronchiole, SOX9/SOX2 double-positive cells were not
detected in the control or MCC950-only group but were abundant in
the silica-treated group on day 7 (Fig. 8A). However, MCC950 significantly
suppressed the expression of these double-positive cells.
Consistently, western blotting showed that silica instillation
markedly upregulated levels of SOX9 and SOX2 on day 7, which were
decreased by MCC950 (Fig. 8B and
C). Non-significant alterations in SOX9 and SOX2 were observed
between the four groups on days 28 and 56. Dual fluorescence
staining assay using club cell-specific antibody CC10 and type II
alveolar epithelial cell biomarker SPC to identify BASCs. Although
few co-stained cells were detected at the BADJ (Fig. 8D), these BASCs were distributed
in the alveolar region on day 7 (Fig. 8E). These ectopic BASCs were not
observed following MCC950 treatment, leaving only SPC-positive type
II alveolar epithelial cells (Fig.
8E). Taken together, these data demonstrated that the NLRP3
inflammasome mediated silica-induced dysregulated repair and
regeneration in the distal lung on day 7 after initial exposure,
which were restored by MCC950 treatment.
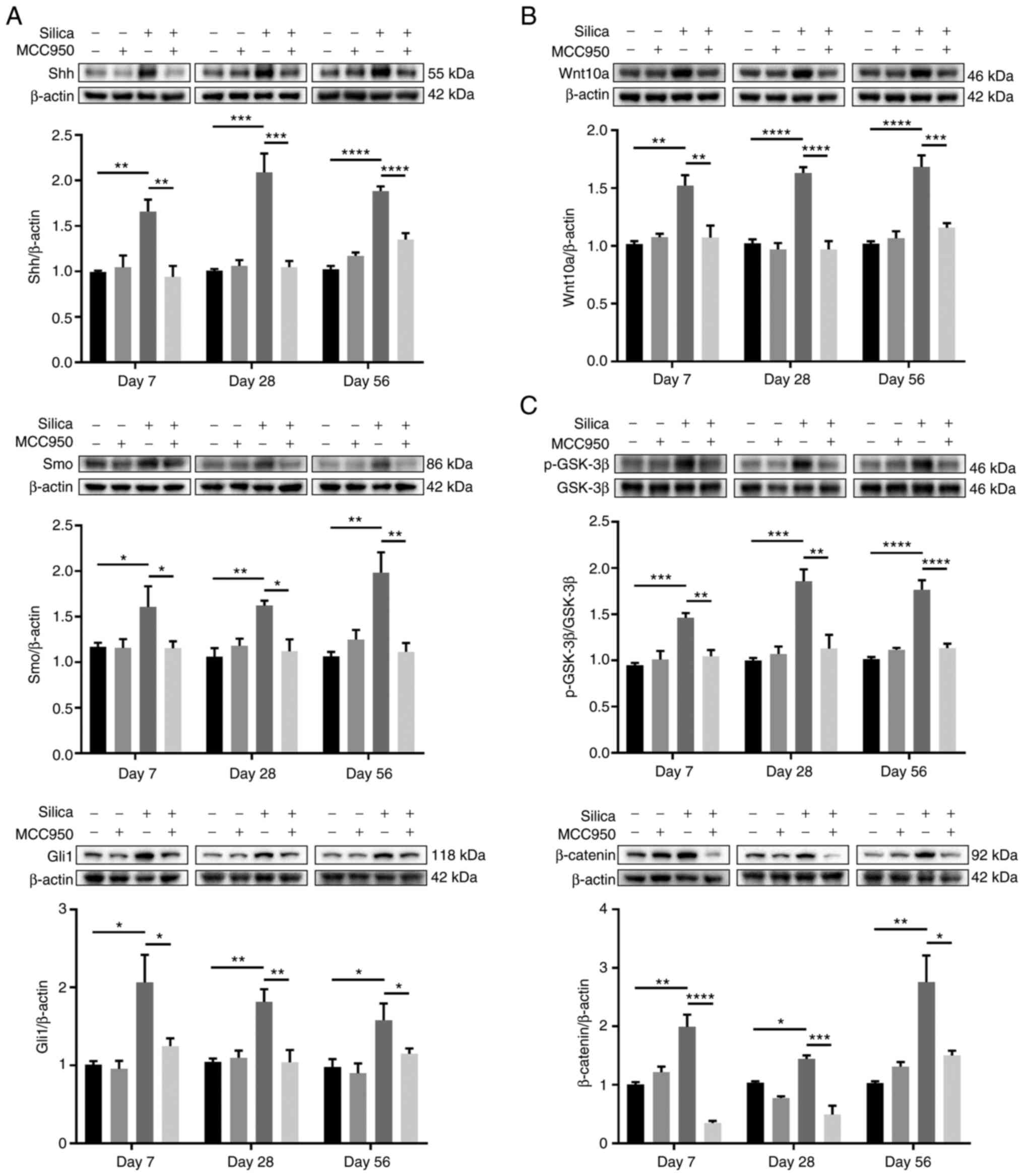
Shh/Gli and Wnt/β-catenin pathways are
involved in NLRP3 inflammasome-mediated epithelial remodeling and
dysregulated regeneration in the distal lung
Previous studies have demonstrated that aberrant
activation of the hedgehog signaling pathway, which serves a
crucial role in lung homeostasis and tissue injury repair, is
linked to pulmonary fibrosis (44,45). Compared with the control, silica
administration significantly increased the expression of Shh, a
major ligand of the hedgehog pathway (46), along with its downstream
transmembrane protein Smo and responsive transcription factor Gli1
at all time points (Fig. 9A).
However, following treatment with MCC950, the upregulation was
effectively rescued compared with that in mice treated only with
silica (Fig. 9A). Wnt pathway is
key for lung homeostasis and tissue repair after injury (47,48). Wnt10a, a member of the Wnt family
and key factor in pulmonary fibrosis (49), was markedly upregulated in the
peripheral lung of silica-treated mice (Fig. 9B). Consistently, silica induced
increases in the levels of Wnt downstream signaling proteins,
including p-GSK-3β and β-catenin, both of which were significantly
downregulated by NLRP3 inflammasome inhibitor MCC950 (Fig. 9C).
Discussion
Long-term inhalation and retention of crystalline
silica particles leads to silicosis, an irreversible occupational
pulmonary disease characterized by pulmonary fibrosis and silicon
nodule formation (50).
Currently, available management strategies are focused on control
of associated symptoms (including chest tightness and dyspnea) and
complications (including respiratory failure and lung cancer);
there is no effective treatment for silicosis. Silica is a strong
activator of the NLRP3 inflammasome (35,36) and enhances the inflammatory
microenvironment by release of IL-1β and IL-18, as well as
pyroptosis with cell swelling and rupture, which further promote
NLRP3 activation and tissue damage (37). Moreover, recent preclinical
findings in mouse models of pulmonary fibrosis confirmed a key role
for the NLRP3 inflammasome in silica-driven chronic inflammation
and irreversible fibrosis (28,51). Once inhaled, respirable silica
particles permeate the lung to reach distal bronchioles and alveoli
and are difficult to remove or degrade due to their physicochemical
properties (crystals; small particles), initiating a cycle of
persistent inflammation and repetitive injury. Silica-induced NLRP3
inflammasome activation leads to excessive release of inflammatory
cytokines such as IL-1β, enhancing the inflammatory
microenvironment surrounding deposited silica crystals and inducing
inflammatory responses, including neutrophil infiltration and
increased production of cytokines such as TNF-α (28,35). Moreover, when stimulated by these
inflammatory cytokines, epithelial cells lose canonical features
and acquire a mesenchymal phenotype, known as EMT, leading to
excessive deposition of extracellular matrix (52). Therefore, the NLRP3 inflammasome
represents a promising therapeutic target for silica-associated
lung injuries and diseases.
Although sufficient evidence indicates that the
NLRP3 inflammasome serves an essential role in silica-induced lung
inflammation and fibrosis (28,29,51,53), its effects on distal lung
remodeling, repair and regeneration in different phases are poorly
understood. The lung exhibits low levels of cell regeneration
during normal homeostasis but displays a notable capacity for
repair and regeneration following injury (54,55). However, dysfunctional or
dysregulated epithelial repair in the distal lung contributes to
tissue remodeling and fibrosis in chronic lung disease, such as
COPD and idiopathic pulmonary fibrosis (IPF) (14,56,57). In particular, the distal lung is
susceptible to silica-induced injury due to deposition in the
terminal regions of the lung (5). In the initial inflammatory
microenvironment, resident stem cells in the distal lung are
recruited to repair damaged tissue. Once damage is eliminated, the
acute inflammatory reaction subsides, allowing the restoration of
tissue structure and functional recovery (58,59). Here, by using a long-term mouse
model of silicosis, the role of the NLRP3 inflammasome in
triggering pulmonary inflammation and fibrosis and its contribution
to epithelial remodeling and dysregulated regeneration in the
distal lung during different periods was investigated. Days 7, 28
and 56 were defined as the early, middle and late phases of
silicosis, respectively. The early phase is dominated by the
inflammatory response, while the final phase is dominated by the
fibrotic response; chronic inflammation initiates fibrosis
(60). In addition, increased
mucin production occurs in response to persistent silica
stimulation in fibrotic development, which presents a challenge for
the function of the local stem/progenitor cells. Here, there was an
increase in SOX9 and SOX2 in the terminal bronchiole only on day 7,
as well as an ectopic distribution of BASCs in the alveolar region,
which may be reparative responses to rebuild functional respiratory
units in the early stage of silica-induced pulmonary fibrosis
(61,62).
Under physiological conditions, pulmonary defense
and function are dependent on normal mucus production and clearance
(63). However, increased mucus
production in the distal lung is observed in response to constant
silica stimulation, which is one of the primary causes of airway
blockage and increased resistance (64). Previous studies have reported
that excessive mucus accumulation in the distal lung leads to
recurrent injury/inflammation/repair cycles with defective
mucociliary clearance and mucosal host defense (65-67). Here, silica-induced sustained
NLRP3 inflammasome activation resulted in MUC5AC or MUC5B
overproduction as a repair response to distal lung injuries during
silica-induced chronic pulmonary inflammation and fibrosis. MUC5AC
and MUC5B are the primary glycoprotein components of airway mucus
that are involved in local defense of the airway and lung
homeo-stasis (68) but their
overexpression is a feature of inflammatory airway diseases and is
associated with adverse pulmonary outcomes (69). Specifically, MUC5AC is a secreted
gel-forming mucin produced by superficial airway goblet cells;
excessive MUC5AC production serves a detrimental role in lung
inflammation and injury (69,70), as well as airway diseases such as
COPD and cystic fibrosis (CF) (71). By contrast, MUC5B is
predominantly expressed in submucosal glands and is key for
homeostatic defense (71).
However, excessive MUC5B aggregation impairs mucosal host defense
and results in excessive lung injury from inhaled substances
(70). For example, accumulation
of MUC5B initiates the muco-obstructive process in CF, leads to
development of idiopathic interstitial pneumonia and causes
mucociliary dysfunction and enhanced lung fibrosis in mouse models
(70,72,73). The present study showed that
targeted suppression of mucin hypersecretion by the NLRP3
inflammasome during the development of pulmonary fibrosis improved
epithelial remodeling and pulmonary fibrosis, which may be
implicated in the functional conservation of local stem/progenitor
cells in the distal lung upon silica challenge. This is consistent
with our previous observations in a mouse lung stem/progenitor
cell-derived organotypic model (40).
In mouse lung development, proximal-distal
patterning is defined by two key transcription factors, SOX2 and
SOX9, which are exclusively localized in the proximal and distal
epithelium, respectively (74,75). The specific distribution promotes
proper branching morphogenesis, including proximal air-conducting
airways and distal gas-exchanging alveoli. Prior to this, when
maximal branching occurs, a progenitor cell population
co-expressing SOX2 and SOX9 is present in the distal tips of the
branching epithelium, which is lost as branching proceeds (74). Furthermore, although the
population exhibits an enhanced proliferative potential in
developing lungs, such potential is rare in adult distal lungs
(74,76). Here, after silica exposure for 7
days, the SOX9/SOX2 double-positive population reappeared in the
terminal bronchiole, with a concurrent increase in levels of SOX9
and SOX2 in the peripheral lungs. Additionally, SOX9/SOX2
double-positive cells in the distal lung were lost on days 28 and
56 and no significant differences in SOX9 and SOX2 levels were
observed. Our previous study demonstrated that the proportion of
SOX9+SOX2+ cells is increased in
silica-treated air-liquid interface cultures, which contributes to
hyperproliferation and abnormal differentiation of the lung
stem/progenitor cell-derived airway epithelium (40). Furthermore, BADJ is a novel
regenerative microenvironment that has a population of rare stem
cells called BASCs that can self-renew over multiple passages and
contribute to maintenance of both bronchiolar and alveolar lineages
(17,18,77). Similar to the aberrant
spatiotemporal expression of SOX2 and SOX9, the silica-induced
NLRP3 inflammasome disrupted distribution of BASCs, which were
ectopically expressed in the alveolar region on day 7 after silica
exposure. By contrast, BASCs were almost undetectable in the
alveolar areas of silica-exposed mice on days 28 and 56 and were
rare at the BADJ in all groups at all time points. The regenerative
potential of stem/progenitor cells relies on correct spatial
localization and temporal expression (78-80) and these specific cell populations
appearing only in the early inflammatory phase represent a
hyperproliferative state to repair the distal lung epithelium in
response to silica stimulation. However, sustained silica
stimulation led to persistent activation of the NLRP3 inflammasome
and caused continuous inflammatory responses, ultimately destroying
inflammatory homeostasis and leading to depletion of stem cells
that promoted epithelial remodeling and pulmonary fibrosis.
However, these abnormalities on day 7 were effectively ameliorated
by NLRP3 inflammasome inhibition, indicating that NLRP3
inflammasome activation may be a central event in dysregulated
regeneration in the distal lung during the early inflammatory phase
of silica-induced epithelial remodeling. Although the role of the
NLRP3 inflammasome in spatiotemporal regulation of these cell
populations is unclear, the initial inflammatory microenvironment
induced by the activated NLRP3 inflammasome may alter
development-associated signals to modulate repair and regeneration
of the distal lung.
Increasing evidence indicates that aberrant
activation of lung developmental signals, including the Shh/Gli and
Wnt/β-catenin pathways, is associated with fibrotic lung disease
(49,75,81). It has been suggested that Shh/Gli
signaling is significant not only in embryonic lung development and
branching morphogenesis but also in repair and regeneration
following injury to adult lungs (82,83). Although the Shh pathway is
maintained at low levels after birth, it is reactivated in the lung
epithelium in response to acute injury, signaling nearby cells and
promoting stem cell proliferation and tissue repair to reestablish
homeostasis and structural integrity (84). Once this is achieved, Shh levels
return to normal. However, in the development of chronic lung
inflammatory disease, higher expression of Shh is maintained, which
signals inflammatory cell populations and supports sustained
inflammatory responses, leading to tissue remodeling and
regeneration failure (83,85,86). Similarly, Wnt/β-catenin signaling
serves critical roles in the pathogenesis of chronic lung diseases,
including lung fibrosis, and in repair and regeneration of the lung
(47,87). Following acute lung injury,
active canonical Wnt signaling is key for the proliferation and
differentiation of lung epithelial stem/progenitor cells (47,81). However, in the fibrotic
environment, chronic Wnt/β-catenin signaling activity induces
senescence in lung epithelial cells, contributing to the
dysfunction and reduction of progenitor cells, as well as impaired
lung repair (88). Moreover, in
IPF, continuous injury of lung epithelium promotes prolonged and
chronic Wnt/β-catenin activity and further stimulates tissue
remodeling and destruction (13,88). Furthermore, Wnt10a is an upstream
activator of β-catenin in the canonical Wnt/β-catenin signaling
pathway and its overexpression in lung-resident mesenchymal stem
cells is associated with Shh/Gli activation (49), indicating signaling crosstalk
between the Shh/Gli and Wnt/β-catenin pathways. In the present
silica-induced mouse lung fibrosis model, key signaling molecules
of the Shh and Wnt pathways were significantly upregulated at all
time points and were effectively suppressed following treatment
with MCC950. These results suggested that in addition to mediating
inflammation and fibrosis, NLRP3 inflammasome activation also
participated in the regulation of development- and
regeneration-associated pathways. Although increased activity of
these signals serves a functional role in guiding repair during
acute inflammation and damage, their continuous upregulation
signals to inflammatory cells promote epithelial remodeling and
dysregulated regeneration. As the restoration or destruction of
tissue structure is dependent on the duration of signal
transduction, precise temporal regulation of NLRP3 inflammasome
assembly may serve a protective role in lung repair and
regeneration following injuries.
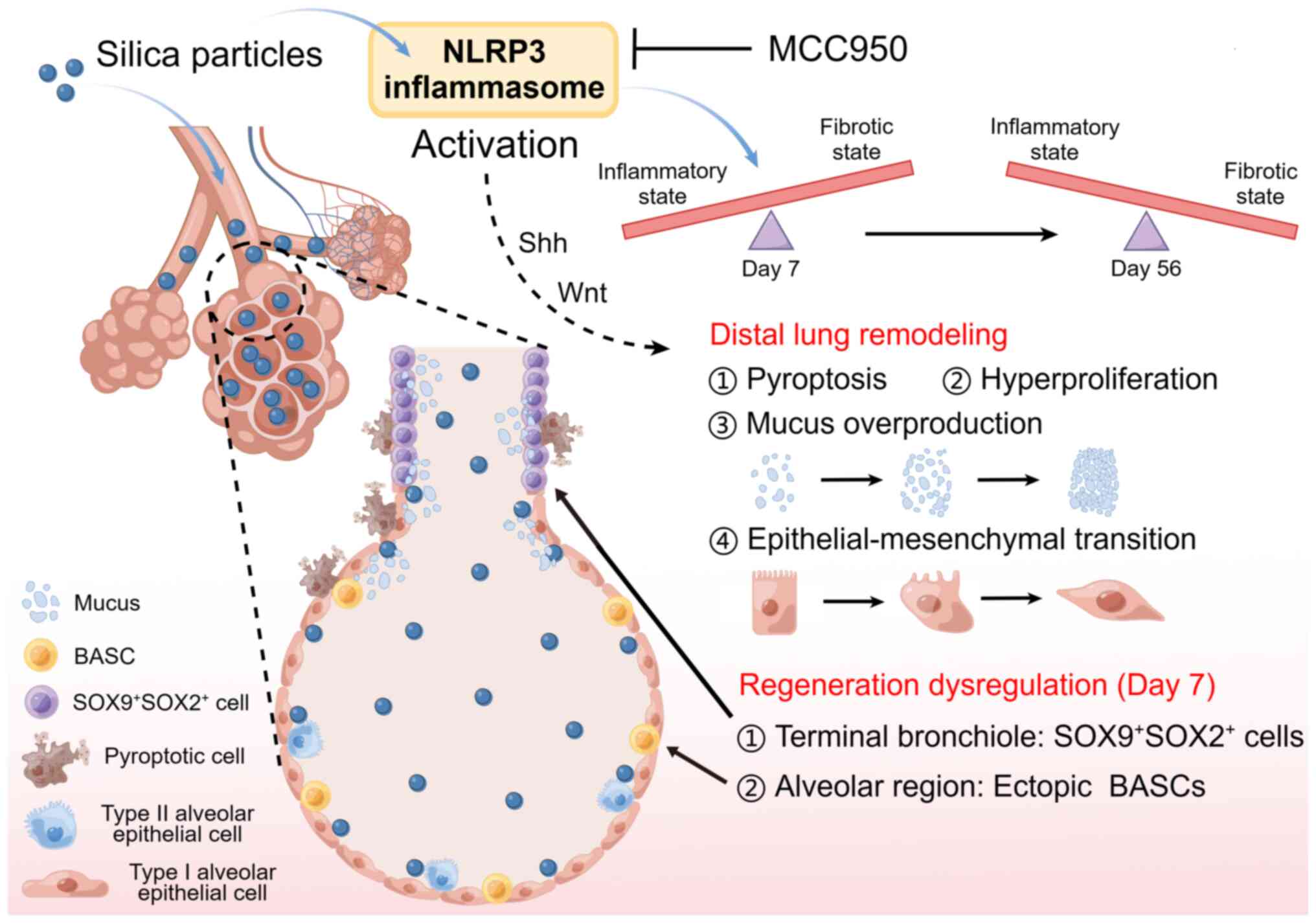
Taken together, the present data demonstrated that
the NLRP3 inflammasome served a crucial role in silica-induced
epithelial remodeling and dysregulated regeneration in a
time-dependent manner in the distal lung of mice (Fig. 10). NLRP3 inflammasome activation
in the early inflammatory phase promotes the migration and
recruitment of stem/progenitor cells to promote tissue repair and
functional recovery but its sustained activation results in
persistent inflammatory reactions, causing depletion of
stem/progenitor cells, subsequent regeneration failure and
epithelial remodeling (40).
Therefore, detection of the early stage of inhalable
particle-related lung disease and precise control of NLRP3
inflammasome activation to enhance normal epithelial repair and
regeneration are of clinical importance to avoid irreversible
damage.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HZ, QZ and HK designed the study. HZ, CL and JF
performed the experiments and analyzed data. HZ wrote the
manuscript. WH, NL and MY collected and interpreted data and
provided technological assistance. HW, WX and HK analyzed and
interpreted data. HZ and HK revised the manuscript and confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
All experimental procedures involving mice were
approved by the Institutional Animal Care and Use Committee of
Nanjing Medical University (approval no. NJMU/IACUC-2012034).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by National Key Research &
Development Program of China (grant no. 2022YFF0710800), National
Natural Science Foundation of China (grant no. 81870054) and the
Key Project of National Science & Technology for Infectious
Diseases of China (grant no. 2018ZX10722301).
References
|
1
|
Christenson SA, Smith BM, Bafadhel M and
Putcha N: Chronic obstructive pulmonary disease. Lancet.
399:2227–2242. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aghapour M, Ubags ND, Bruder D, Hiemstra
PS, Sidhaye V, Rezaee F and Heijink IH: Role of air pollutants in
airway epithelial barrier dysfunction in asthma and COPD. Eur
Respir Rev. 31:2101122022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takada T, Moriyama H and Suzuki E:
Elemental analysis of occupational and environmental lung diseases
by electron probe microanalyzer with wavelength dispersive
spectrometer. Respir Investig. 52:5–13. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leung CC, Yu IT and Chen W: Silicosis.
Lancet. 379:2008–2018. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma J, Cai Q, Yang D, Yang J, Xue J, Yu M,
Liu Y, Ma F, Li F and Liu X: A positive feed forward loop between
Wnt/β-Catenin and NOX4 promotes silicon dioxide-induced
epithelial-mesenchymal transition of lung epithelial cells. Oxid
Med Cell Longev. 2020:34041682020. View Article : Google Scholar
|
|
6
|
Koudstaal T, Funke-Chambour M, Kreuter M,
Molyneaux PL and Wijsenbeek MS: Pulmonary fibrosis: From
pathogenesis to clinical decision-making. Trends Mol Med.
29:1076–1087. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aggarwal K, Arora S and Nagpal K:
Pulmonary Fibrosis: Unveiling the pathogenesis, exploring
therapeutic targets, and advancements in drug delivery strategies.
AAPS PharmSciTech. 24:1522023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu Q, Fu G, Lin H, Zhao Q, Liu Y, Zhou Y,
Shi Y, Zhang L, Wang Z, Zhang Z, et al: Influence of silica
particles on mucociliary structure and MUC5B expression in airways
of C57BL/6 mice. Exp Lung Res. 46:217–225. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li S, Li Y, Zhang Y, Li S, Zhang M, Jin F,
Wei Z, Yang Y, Gao X, Mao N, et al:
N-Acetyl-Seryl-Asparyl-Lysyl-Proline regulates lung renin
angiotensin system to inhibit epithelial-mesenchymal transition in
silicotic mice. Toxicol Appl Pharmacol. 408:1152552020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang J, Wu S, Hu W, Yang D, Ma J, Cai Q,
Xue J, Chen J, Li F, Zeng J and Liu X: Bmi1 signaling maintains the
plasticity of airway epithelial progenitors in response to
persistent silica exposures. Toxicology. 470:1531522022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Churg A and Wright JL: Bronchiolitis
caused by occupational and ambient atmospheric particles. Semin
Respir Crit Care Med. 24:577–584. 2003. View Article : Google Scholar
|
|
12
|
Ferreira TP, de Arantes AC, do Nascimento
CV, Olsen PC, Trentin PG, Rocco PR, Hogaboam CM, Puri RK, Martins
MA and Silva PM: IL-13 immunotoxin accelerates resolution of lung
pathological changes triggered by silica particles in mice. J
Immunol. 191:5220–5229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Crosby LM and Waters CM: Epithelial repair
mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol.
298:L715–L731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ptasinski VA, Stegmayr J, Belvisi MG,
Wagner DE and Murray LA: Targeting alveolar repair in idiopathic
pulmonary fibrosis. Am J Respir Cell Mol Biol. 65:347–365. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Salahudeen AA, Choi SS, Rustagi A, Zhu J,
van Unen V, de la O SM, Flynn RA, Margalef-Català M, Santos AJM, Ju
J, et al: Progenitor identification and SARS-CoV-2 infection in
human distal lung organoids. Nature. 588:670–675. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alanis DM, Chang DR, Akiyama H, Krasnow MA
and Chen J: Two nested developmental waves demarcate a compartment
boundary in the mouse lung. Nature Commun. 5:39232014. View Article : Google Scholar
|
|
17
|
Kawakita N, Toba H, Miyoshi K, Sakamoto S,
Matsumoto D, Takashima M, Aoyama M, Inoue S, Morimoto M, Nishino T,
et al: Bronchioalveolar stem cells derived from mouse-induced
pluripotent stem cells promote airway epithelium regeneration. Stem
Cell Res Ther. 11:4302020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jones-Freeman B and Starkey MR:
Bronchioalveolar stem cells in lung repair, regeneration and
disease. J Pathol. 252:219–226. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Eisenhauer P, Earle B, Loi R, Sueblinvong
V, Goodwin M, Allen GB, Lundblad L, Mazan MR, Hoffman AM and Weiss
DJ: Endogenous distal airway progenitor cells, lung mechanics, and
disproportionate lobar growth following long-term postpneumonectomy
in mice. Stem Cells. 31:1330–1339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Caseley EA, Lara-Reyna S, Poulter JA,
Topping J, Carter C, Nadat F, Spickett GP, Savic S and McDermott
MF: An atypical autoinflammatory disease due to an LRR domain NLRP3
mutation enhancing binding to NEK7. J Clin Immunol. 42:158–170.
2022. View Article : Google Scholar
|
|
21
|
Swanson KV, Deng M and Ting JP: The NLRP3
inflammasome: Molecular activation and regulation to therapeutics.
Nat Rev Immunol. 19:477–489. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Burdette BE, Esparza AN, Zhu H and Wang S:
Gasdermin D in pyroptosis. Acta Pharm Sin B. 11:2768–2782. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liang Q, Cai W, Zhao Y, Xu H, Tang H, Chen
D, Qian F and Sun L: Lycorine ameliorates bleomycin-induced
pulmonary fibrosis via inhibiting NLRP3 inflammasome activation and
pyroptosis. Pharmacol Res. 158:1048842020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zheng R, Tao L, Jian H, Chang Y, Cheng Y,
Feng Y and Zhang H: NLRP3 inflammasome activation and lung fibrosis
caused by airborne fine particulate matter. Ecotoxicol Environ Saf.
163:612–619. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lamkanfi M and Dixit VM: Inflammasomes and
their roles in health and disease. Annu Rev Cell Dev Biol.
28:137–161. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song M, Wang J, Sun Y, Pang J, Li X, Liu
Y, Zhou Y, Yang P, Fan T, Liu Y, et al: Inhibition of gasdermin
D-dependent pyroptosis attenuates the progression of silica-induced
pulmonary inflammation and fibrosis. Acta Pharm Sin B.
12:1213–1224. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tapia-Abellán A, Angosto-Bazarra D,
Martínez-Banaclocha H, de Torre-Minguela C, Cerón-Carrasco JP,
Pérez-Sánchez H, Arostegui JI and Pelegrin P: MCC950 closes the
active conformation of NLRP3 to an inactive state. Nat Chem Biol.
15:560–564. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lam M, Mansell A and Tate MD: Another One
Fights the Dust: Targeting the NLRP3 inflammasome for the treatment
of silicosis. Am J Respir Cell Mol Biol. 66:601–611. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sayan M and Mossman BT: The NLRP3
inflammasome in pathogenic particle and fibre-associated lung
inflammation and diseases. Part Fibre Toxicol. 13:512016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Clark JD, Gebhart GF, Gonder JC, Keeling
ME and Kohn DF: Special Report: The 1996 Guide for the Care and Use
of Laboratory Animals. ILAR J. 38:41–48. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zou Z, Hu X, Luo T, Ming Z, Chen X, Xia L,
Luo W, Li J, Xu N, Chen L, et al: Naturally-occurring spinosyn A
and its derivatives function as argininosuccinate synthase
activator and tumor inhibitor. Nat Commun. 12:22632021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao S, Zhou L, Wang Q, Cao JH, Chen Y,
Wang W, Zhu BD, Wei ZH, Li R, Li CY, et al: Elevated branched-chain
amino acid promotes atherosclerosis progression by enhancing
mitochondrial-to-nuclear H2O 2-disulfide HMGB1 in
macrophages. Redox Biol. 62:1026962023. View Article : Google Scholar
|
|
33
|
Szapiel SV, Elson NA, Fulmer JD,
Hunninghake GW and Crystal RG: Bleomycin-induced interstitial
pulmonary disease in the nude, athymic mouse. Am Rev Respir Dis.
120:893–899. 1979.PubMed/NCBI
|
|
34
|
Hübner RH, Gitter W, El Mokhtari NE,
Mathiak M, Both M, Bolte H, Freitag-Wolf S and Bewig B:
Standardized quantification of pulmonary fibrosis in histological
samples. Biotechniques. 44:507–511. 514–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Campden RI, Warren AL, Greene CJ,
Chiriboga JA, Arnold CR, Aggarwal D, McKenna N, Sandall CF,
MacDonald JA and Yates RM: Extracellular cathepsin Z signals
through the α5 integrin and augments NLRP3 inflammasome
activation. J Biol Chem. 298:1014592022. View Article : Google Scholar
|
|
36
|
Yin H, Fang L, Wang L, Xia Y, Tian J, Ma
L, Zhang J, Li N, Li W, Yao S and Zhang L: Acute silica exposure
triggers pulmonary inflammation through macrophage pyroptosis: An
experimental simulation. Front Immunol. 13:8744592022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shi J, Gao W and Shao F: Pyroptosis:
Gasdermin-Mediated programmed necrotic cell death. Trends Biochem
Sci. 42:245–254. 2017. View Article : Google Scholar
|
|
38
|
Zhou YP, Mei MJ, Wang XZ, Huang SN, Chen
L, Zhang M, Li XY, Qin HB, Dong X, Cheng S, et al: A congenital CMV
infection model for follow-up studies of neurodevelopmental
disorders, neuroimaging abnormalities, and treatment. JCI Insight.
7:e1525512022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu Y, Zhang X, Wang J, Yang F, Luo W,
Huang J, Chen M, Wang S, Li C, Zhang W and Chao J: ZC3H4 regulates
infiltrating monocytes, attenuating pulmonary fibrosis through
IL-10. Respir Res. 23:2042022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou H, Zhang Q, Huang W, Zhou S, Wang Y,
Zeng X, Wang H, Xie W and Kong H: NLRP3 inflammasome mediates
silica-induced lung epithelial injury and aberrant regeneration in
lung Stem/Progenitor Cell-Derived organotypic models. Int J Biol
Sci. 19:1875–1893. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen S, Li K, Zhong X, Wang G, Wang X,
Cheng M, Chen J, Chen Z, Chen J, Zhang C, et al: Sox9-expressing
cells promote regeneration after radiation-induced lung injury via
the PI3K/AKT pathway. Stem Cell Res Ther. 12:3812021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Eenjes E, Tibboel D, Wijnen RMH, Schnater
JM and Rottier RJ: SOX2 and SOX21 in lung epithelial
differentiation and repair. Int J Mol Sci. 23:130642022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Belgacemi R, Danopoulos S, Deutsch G,
Glass I, Dormoy V, Bellusci S and Al Alam D: Hedgehog signaling
pathway orchestrates human lung branching morphogenesis. Int J Mol
Sci. 23:52652022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang J, Liu H, Song C, Zhang J, Wang Y,
Lv C and Song X: Astilbin ameliorates pulmonary fibrosis via
blockade of Hedgehog signaling pathway. Pulm Pharmacol Ther.
50:19–27. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang C, Cassandras M and Peng T: The role
of hedgehog signaling in adult lung regeneration and maintenance. J
Dev Biol. 7:142019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li L, Bao J, Wang H, Lei JH, Peng C, Zeng
J, Hao W, Zhang X, Xu X, Yu C, et al: Upregulation of amplified in
breast cancer 1 contributes to pancreatic ductal adenocarcinoma
progression and vulnerability to blockage of hedgehog activation.
Theranostics. 11:1672–1689. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Raslan AA and Yoon JK: WNT Signaling in
lung repair and regeneration. Mol Cells. 43:774–783.
2020.PubMed/NCBI
|
|
48
|
Skronska-Wasek W, Mutze K, Baarsma HA,
Bracke KR, Alsafadi HN, Lehmann M, Costa R, Stornaiuolo M,
Novellino E, Brusselle GG, et al: Reduced frizzled receptor 4
expression prevents WNT/β-Catenin-driven alveolar lung repair in
chronic obstructive pulmonary disease. Am J Respir Crit Care Med.
196:172–185. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cao H, Chen X, Hou J, Wang C, Xiang Z,
Shen Y and Han X: The Shh/Gli signaling cascade regulates
myofibroblastic activation of lung-resident mesenchymal stem cells
via the modulation of Wnt10a expression during pulmonary
fibrogenesis. Lab Invest. 100:363–377. 2020. View Article : Google Scholar
|
|
50
|
Jiang R, Han L, Gao Q and Chao J: ZC3H4
mediates silica-induced EndoMT via ER stress and autophagy. Environ
Toxicol Pharmacol. 84:1036052021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Song MY, Wang JX, Sun YL, Han ZF, Zhou YT,
Liu Y, Fan TH, Li ZG, Qi XM, Luo Y, et al: Tetrandrine alleviates
silicosis by inhibiting canonical and non-canonical NLRP3
inflamma-some activation in lung macrophages. Acta Pharmacol Sin.
43:1274–1284. 2022. View Article : Google Scholar
|
|
52
|
Pang X, Shao L, Nie X, Yan H, Li C, Yeo
AJ, Lavin MF, Xia Q, Shao H, Yu G, et al: Emodin attenuates
silica-induced lung injury by inhibition of inflammation, apoptosis
and epithelial-mesen-chymal transition. Int Immunopharmacol.
91:1072772021. View Article : Google Scholar
|
|
53
|
Song Z, Wang L, Cao Y, Liu Z, Zhang M,
Zhang Z, Jiang S, Fan R, Hao T, Yang R, et al: Isoandrographolide
inhibits NLRP3 inflammasome activation and attenuates silicosis in
mice. Int Immunopharmacol. 105:1085392022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wujak L, Schnieder J, Schaefer L and
Wygrecka M: LRP1: A chameleon receptor of lung inflammation and
repair. Matrix Biol. 68-69:366–381. 2018. View Article : Google Scholar
|
|
55
|
Beers MF and Morrisey EE: The three R's of
lung health and disease: Repair, remodeling, and regeneration. J
Clin Invest. 121:2065–2073. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Spella M, Lilis I and Stathopoulos GT:
Shared epithelial pathways to lung repair and disease. Eur Respir
Rev. 26:1700482017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Brandsma CA, de Vries M, Costa R, Woldhuis
RR, Königshoff M and Timens W: Lung ageing and COPD: Is there a
role for ageing in abnormal tissue repair? Eur Respir Rev.
26:1700732017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nakao M, Kim K, Nagase K, Grainger DW,
Kanazawa H and Okano T: Phenotypic traits of mesenchymal stem cell
sheets fabricated by temperature-responsive cell culture plate:
Structural characteristics of MSC sheets. Stem Cell Res Ther.
10:3532019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hegdekar N, Sarkar C, Bustos S, Ritzel RM,
Hanscom M, Ravishankar P, Philkana D, Wu J, Loane DJ and Lipinski
MM: Inhibition of autophagy in microglia and macrophages
exacerbates innate immune responses and worsens brain injury
outcomes. Autophagy. 19:2026–2044. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhao Y, Hao C, Bao L, Wang D, Li Y, Qu Y,
Ding M, Zhao A and Yao W: Silica particles disorganize the
polarization of pulmonary macrophages in mice. Ecotoxicol Environ
Saf. 193:1103642020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xu T, Yan W, Wu Q, Xu Q, Yuan J, Li Y, Li
P, Pan H and Ni C: MiR-326 inhibits inflammation and promotes
autophagy in Silica-Induced pulmonary fibrosis through targeting
TNFSF14 and PTBP1. Chem Res Toxicol. 32:2192–2203. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Grazioli S, Gil S, An D, Kajikawa O,
Farnand AW, Hanson JF, Birkland T, Chen P, Duffield J, Schnapp LM,
et al: CYR61 (CCN1) overexpression induces lung injury in mice. Am
J Physiol Lung Cell Mol Physiol. 308:L759–L765. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Whitsett JA: Airway epithelial
differentiation and mucociliary clearance. Ann Am Thorac Soc.
15(Suppl 3): S143–S148. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kim SH, Pei QM, Jiang P, Liu J, Sun RF,
Qian XJ and Liu JB: Upregulation of MUC5AC by VEGF in human primary
bronchial epithelial cells: Implications for asthma. Respir Res.
20:2822019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chen G, Ribeiro CMP, Sun L, Okuda K, Kato
T, Gilmore RC, Martino MB, Dang H, Abzhanova A, Lin JM, et al:
XBP1S Regulates MUC5B in a promoter Variant-Dependent pathway in
idiopathic pulmonary fibrosis airway epithelia. Am J Respir Crit
Care Med. 200:220–234. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kim E, Mathai SK, Stancil IT, Ma X,
Hernandez-Gutierrez A, Becerra JN, Marrero-Torres E, Hennessy CE,
Hatakka K, Wartchow EP, et al: Aberrant multiciliogenesis in
idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol.
67:188–200. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Dobrinskikh E, Estrella AM, Hennessy CE,
Hara N, Schwarz MI, Kurche JS, Yang IV and Schwartz DA: Genes,
other than Muc5b, play a role in bleomycin-induced lung fibrosis.
Am J Physiol Lung Cell Mol Physiol. 321:L440–Ll450. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ma J, Rubin BK and Voynow JA: Mucins,
mucus, and goblet cells. Chest. 154:169–176. 2018. View Article : Google Scholar
|
|
69
|
Singanayagam A, Footitt J, Marczynski M,
Radicioni G, Cross MT, Finney LJ, Trujillo-Torralbo MB, Calderazzo
M, Zhu J, Aniscenko J, et al: Airway mucins promote
immunopa-thology in virus-exacerbated chronic obstructive pulmonary
disease. J Clin Invest. 132:e1209012022. View Article : Google Scholar
|
|
70
|
Koeppen M, McNamee EN, Brodsky KS, Aherne
CM, Faigle M, Downey GP, Colgan SP, Evans CM, Schwartz DA and
Eltzschig HK: Detrimental role of the airway mucin Muc5ac during
ventilator-induced lung injury. Mucosal Immunol. 6:762–775. 2013.
View Article : Google Scholar
|
|
71
|
Evans CM, Raclawska DS, Ttofali F, Liptzin
DR, Fletcher AA, Harper DN, McGing MA, McElwee MM, Williams OW,
Sanchez E, et al: The polymeric mucin Muc5ac is required for
allergic airway hyperreactivity. Nature Commun. 6:62812015.
View Article : Google Scholar
|
|
72
|
Keith JD, Henderson AG, Fernandez-Petty
CM, Davis JM, Oden AM and Birket SE: Muc5b contributes to mucus
abnormality in rat models of cystic fibrosis. Front Physiol.
13:8841662022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hancock LA, Hennessy CE, Solomon GM,
Dobrinskikh E, Estrella A, Hara N, Hill DB, Kissner WJ, Markovetz
MR, Grove Villalon DE, et al: Muc5b overexpression causes
mucociliary dysfunction and enhances lung fibrosis in mice. Nature
Commun. 9:53632018. View Article : Google Scholar
|
|
74
|
Danopoulos S, Alonso I, Thornton ME,
Grubbs BH, Bellusci S, Warburton D and Al Alam D: Human lung
branching morphogenesis is orchestrated by the spatiotemporal
distribution of ACTA2, SOX2, and SOX9. Am J Physiol Lung Cell Mol
Physiol. 314:L144–Ll149. 2018. View Article : Google Scholar
|
|
75
|
Gajjala PR, Kasam RK, Soundararajan D,
Sinner D, Huang SK, Jegga AG and Madala SK: Dysregulated
overexpression of Sox9 induces fibroblast activation in pulmonary
fibrosis. JCI Insight. 6:e1525032021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Danopoulos S, Thornton ME, Grubbs BH, Frey
MR, Warburton D, Bellusci S and Al Alam D: Discordant roles for FGF
ligands in lung branching morphogenesis between human and mouse. J
Pathol. 247:254–265. 2019. View Article : Google Scholar :
|
|
77
|
Basil MC and Morrisey EE: BASC-ing in the
glow: Bronchioalveolar stem cells get their place in the lung. EMBO
J. 38:e1023442019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Aros CJ, Vijayaraj P, Pantoja CJ, Bisht B,
Meneses LK, Sandlin JM, Tse JA, Chen MW, Purkayastha A, Shia DW, et
al: Distinct spatiotemporally dynamic Wnt-secreting niches regulate
proximal airway regeneration and aging. Cell Stem Cell.
27:413–429.e4. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Sivakumar A and Frank DB: Paradigms that
define lung epithelial progenitor cell fate in development and
regeneration. Curr Stem Cell Rep. 5:133–144. 2019. View Article : Google Scholar
|
|
80
|
Rock J and Königshoff M: Endogenous lung
regeneration: Potential and limitations. Am J Respir Crit Care Med.
186:1213–1219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chanda D, Otoupalova E, Smith SR,
Volckaert T, De Langhe SP and Thannickal VJ: Developmental pathways
in the pathogenesis of lung fibrosis. Mol Aspects Med. 65:56–69.
2019. View Article : Google Scholar :
|
|
82
|
Deng M, Li J, Gan Y, Chen Y and Chen P:
Changes in the number of CD31-CD45-Sca-1+ cells and Shh signaling
pathway involvement in the lungs of mice with emphysema and
relevant effects of acute adenovirus infection. Int J Chron
Obstruct Pulmon Dis. 12:861–872. 2017. View Article : Google Scholar :
|
|
83
|
Lau CI, Yánez DC, Papaioannou E, Ross S
and Crompton T: Sonic Hedgehog signalling in the regulation of
barrier tissue homeostasis and inflammation. FEBS J. 289:8050–8061.
2022. View Article : Google Scholar
|
|
84
|
Peng T, Frank DB, Kadzik RS, Morley MP,
Rathi KS, Wang T, Zhou S, Cheng L, Lu MM and Morrisey EE: Hedgehog
actively maintains adult lung quiescence and regulates repair and
regeneration. Nature. 526:578–582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Beachy PA, Karhadkar SS and Berman DM:
Tissue repair and stem cell renewal in carcinogenesis. Nature.
432:324–331. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Chen X, Jin Y, Hou X, Liu F and Wang Y:
Sonic hedgehog signaling: Evidence for its protective role in
endotoxin induced acute lung injury in mouse model. PLoS One.
10:e01408862015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hu HH, Cao G, Wu XQ, Vaziri ND and Zhao
YY: Wnt signaling pathway in aging-related tissue fibrosis and
therapies. Ageing Res Rev. 60:1010632020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lehmann M, Hu Q, Hu Y, Hafner K, Costa R,
van den Berg A and Königshoff M: Chronic WNT/β-catenin signaling
induces cellular senescence in lung epithelial cells. Cellular
Signal. 70:1095882020. View Article : Google Scholar
|