Globally recognized as the second major cause of
mortality, stroke also stands as the predominant source of enduring
disability in adults (1). The
repercussions of a stroke extend beyond the immediate physical
manifestations; numerous survivors grapple with significant
impediments to their long-term physical functions. A substantial
proportion of these survivors, as a result, find themselves reliant
on external support, and are predisposed to additional neurological
complications, including depression (2,3).
An ischemic stroke (IS) delineates into two
principal regions: The core ischemic infarct and the surrounding
ischemic penumbra. While neural cells within the infarcted core are
often irreversibly damaged, the penumbral tissue, albeit
compromised, retains a temporal viability. Swift therapeutic
interventions can facilitate cerebral blood flow restoration within
the penumbral zone, forestalling further tissue degeneration and
subsequent necrosis. Contemporary treatments, notably intravenous
thrombolysis deploying recombinant tissue plasminogen activator
(rt-PA) and endovascular strategies such as intra-arterial
thrombolysis and mechanical thrombectomy, have shown efficacy in
preserving functionality within the ischemic penumbra (1). However, the rapid reperfusion
inherent to these treatments, bound by a constrained therapeutic
window, risks potential irreversible neurological impairments.
Cerebral ischemia/reperfusion injury (CI/RI) spans
both the initial ischemic damage and the ensuing ischemia
instigated by reperfusion (4).
Intriguingly, the pathophysiological trajectory of CI/RI exhibits
associations with gut flora and their metabolic byproducts. The
evolution in understanding of the brain-gut dynamics has now
shifted focus from the traditionally recognized gut-brain axis
(GBA) to the more encompassing gut-brain-microbiota axis (GBMA).
This paradigm emphasizes the intricate, bidirectional linkage
tethering the gut and brain. GBMA provides an avenue to elucidate
the symbiotic interactions at the microbiota-gut-brain interface,
facilitated by signaling conduits such as cyclic GMP-AMP synthase
(cGAS)-stimulator of interferon genes (STING) and modulated by
intestinal microbial metabolites. Such insights underscore the
potential for innovative therapeutic strategies targeting CI/RI via
intestinal microbial metabolite modulation.
The methods for selecting articles were as follows:
The main things that determined which studies were included in the
present review were how they related to the brain-gut axis and how
they played a part in neuroinflammation after cerebral
ischemiareperfusion injury. Preference was given to peer-reviewed
research articles and reviews published in reputable journals,
focusing on those that provided original data, comprehensive
reviews, or significant theoretical contributions.
The initial search generated a large number of
articles, from which titles and abstracts were screened for
relevance. A full-text review was then conducted to assess the
depth of the content and the soundness of the methodology. The
selection process was iterative, refining search terms and
inclusion criteria as needed to ensure comprehensive coverage of
the topic.
Each article underwent a rigorous quality assessment
focusing on the significance of the study design, methodology,
sample size, statistical analyses and findings. Special attention
was given to studies that provided novel insights or challenged
existing paradigms.
Articles were excluded if they did not address the
brain-gut axis associated with CI/RI, lacked scientific rigor or
were not peer-reviewed. Duplicate studies and those with outdated
or superseded information were also omitted.
The selection process was a collaborative effort
that included discussions with co-authors and consultation with
other experts in neurology and internal medicine specialists to
ensure a multidisciplinary perspective.
As the review progressed, search and selection
criteria were continually refined based on emerging themes and
findings to ensure comprehensive and up-to-date analyses. The
approach to selecting material for review was designed to provide a
comprehensive and unbiased overview of current understanding of the
effects of the brain-gut axis on neuroinflammation in CI/RI.
Microglia, the resident immune cells of the brain,
constitute between 6-21% of the brain's total glial cell population
(5). Historically, the
understanding of microglia's origins underwent a paradigm shift in
the early 1990s. Contrary to the previously held belief that
microglia solely originated from peripheral macrophages, it was
unveiled that they could also arise from bone marrow progenitor
cells located in the yolk sac. This novel classification of
macrophages was aptly christened 'microglia' by Pío del
Río-Hortega, a terminology that has since gained universal
acceptance (6,7).
Despite their behavioral and functional parallels
with macrophages, which led to their initial identification as such
(8), microglia maintain a
distinct and irreplaceable role within the central nervous system
(CNS). A mere h post-ischemic insult, resident microglia spring
into action. These cells undergo notable morphological
transformations: Retracting their external protrusions, they adopt
an amoeboid form (9).
Additionally, based on temporal progression post-injury, microglia
can be categorized into acute, subacute and chronic phases, each
presenting unique pathological characteristics (10).
Within mere h post-ischemic injury, microglia are
promptly activated by the infiltration of plasma proteins. Once
stimulated, they infiltrate both the infarct core and the
peri-infarct region. This activation is mediated by a suite of
downstream signaling effectors, notably including nuclear factor κB
(NF-κB), hypoxia-inducible factor 1 (HIF-1) and activator of
transcription 3 (STAT3). Morphologically, peri-infarct microglia
exhibit enlarged cell bodies, short branches and amoeboid
structures. Adjacent to the ischemic core, particularly large,
round microglia display heightened activity, attributed to their
low activation thresholds.
Historically, microglia were perceived to polarize
into two distinct phenotypes: M1 and M2, in response to activation.
The M1 phenotype, characterized by its pro-inflammatory and
neurotoxic activities, emerges within the initial 24 h post-injury.
This induction is primarily steered by the Toll-like receptor (TLR)
and interferon-gamma (IFN-gamma) signaling pathways (11). M1 microglia secrete a plethora of
pro-inflammatory factors such as tumour necrosis factor alpha
(TNF-alpha), IL-6, IL-12 and IL-1β, alongside reactive oxidative
agents such as reactive oxygen species (ROS) and inducible nitric
oxide synthase (12,13). These agents, particularly ROS and
TNF-alpha, can disrupt the integrity of the blood-brain barrier
(BBB), inflicting severe endothelial damage and potentially leading
to hemorrhagic transformation (14,15).
Conversely, while the pro-inflammatory M1 phenotype
emerges, certain chemokines, including CCL2 and CXCL4, initiate the
polarization of microglia towards the anti-inflammatory M2
phenotype post the 24-h mark (16,17). Distinct from M1, the M2 phenotype
secretes anti-inflammatory agents such as IL-4, IL-10 and
transforming growth factor beta, aiming to curb inflammation
(18). These cells also release
growth factors and neurotrophic agents including insulin-like
growth factor 1, vascular endothelial growth factor and
brain-derived neurotrophic factors to foster vascular repair and
prevent neuronal cell apoptosis in the ischemic zone (19). Furthermore, the M2 phenotype is
nuanced, with subcategories M2a, M2b and M2c (20). Each subtype is characterized by
its induction mechanism and function (21).
However, the once clear-cut M1/M2 distinction is now
acknowledged as an oversimplification. Contemporary understanding
recognizes the vast functional overlaps and asserts that the
complete array of microglial activation states in vivo
extends beyond just the conventional M1 or M2 categories (22). For instance, post-ischemia, the
presence of M1-like microglia amplifies from day one and peaks
around the second week, whereas M2-like microglia peak around day
five and subsequently decline (23). Notably, older mice exhibit
significantly fewer M2 microglia compared with their younger
counterparts, underscoring the adverse influence of advanced age on
post-stroke neuroinflammation (24-26). The intricate nature of microglial
responses is epitomized by findings where markers of both
inflammatory resistance and promotion co-exist within the same
cell. This complexity is a key factor influencing the challenges in
treating IS injury with whole microglial cells (27).
While research efforts into the acute phase of
post-stroke neuroinflammation have burgeoned over the years,
studies exploring the subacute and chronic phases remain relatively
sparse. During these later phases, damage-associated molecular
patterns activate microglia and infiltrating macrophages. These
activated cells subsequently release inflammatory cytokines such as
TNF-alpha and IL-1beta, exacerbating neuronal cell death in the
ischemic penumbra (28).
Remarkably, even after six months post-injury, blood samples from
stroke patients persistently exhibited elevated levels of TNF-alpha
(29).
Anti-inflammatory M2-type microglia play a pivotal
role during this phase by secreting cytokines, most notably IL-10,
which counteract the inflammatory effects incited by IL-1beta and
TNF-alpha, providing a protective barrier against further ischemic
damage (30). Supporting this, a
study by Ooboshi et al (31) demonstrated that administering an
adenoviral vector encoding the human IL-10 gene into the lateral
ventricles of stroke-affected rats substantially reduced cerebral
infarct sizes and mitigated hippocampal ischemic injuries. The
likely mechanism involves the capacity of IL-10 to dampen the
production of inflammatory cytokines and enhance the activity of
anti-inflammatory cytokine antagonists (31).
Interestingly, microglia can sustain a state of
prolonged activity. While their activity tends to wane after six
months within the infarct core, it often radiates to more distant
regions, following the brain's corticospinal tracts (32). Modern imaging techniques such as
positron emission tomography and magnetic resonance imaging have
unveiled that, for certain patients, neuroinflammation migrates
from the infarct's initial site to remote brain areas, encompassing
the hippocampus, thalamus and basal ganglia, even those situated
contralaterally (33). Echoing
this, Price et al (34)
observed a decline in microglial activity in the infarct core three
to four weeks post-stroke. Conversely, there was a conspicuous
surge of microglia in distant regions of the ipsilateral hemisphere
and even in areas within the contralateral cerebral hemisphere
(34).
Astrocytes, akin to microglia, contribute
significantly to the brain's innate immune response. These
versatile cells govern ionic homeostasis, modulate neurotransmitter
dynamics and contribute to the structural integrity of the BBB,
thus playing pivotal roles in safeguarding both neuronal and
vascular functionalities. In the context of CI/RI, astrocytes have
been linked to the release of Kruppel-like factor 4 (KLF4), which
not only curtails the infarcted area and suppresses oxidative
stress but also fortifies the BBB. Intriguingly, KLF4 amplifies the
levels of Nrf2 and Trx1 mRNA in the oxygen glucose
deprivation/reperfusion (OGD/R) model, resulting in improved BBB
repair, diminished malondialdehyde levels and elevated antioxidant
SOD levels (35).
Astrocytes are swiftly activated post-brain injury
and exhibit a dual-functional paradigm (36). Classically activated A1-like
astrocytes, stimulated by IL1 alpha, TNF alpha, and complement
component 1q (C1q) from microglia, unleash pro-inflammatory
molecules, manifesting neurotoxic implications (37). By contrast, alternatively
activated A2-like astrocytes confer neuroprotection via the
secretion of anti-inflammatory mediators (38). Notably, post-brain I/R,
lipopolysaccharide (LPS)-driven neuroinflammation precipitates the
transformation of astrocytes to the A1 type. Research by Li et
al (39) illuminated that
hydrogen sulfide (H2S) curbs this LPS-induced A1-like astrocyte
metamorphosis in the murine hippocampus, simultaneously promoting
the shift of reactive astrocytes to the A2 type. This
transformation was hypothesized to be modulated by the upregulation
of the BKCa channel (39). In a
complex cascade, A1 astrocytes release LCN2, which subsequently
binds to 24p3R on astrocytes, instigating focal death of astrocytes
via the NLRP3 mechanism. Yet, mitigating this astrocytic focal
death curbs neuronal damage, particularly through attenuating
caspase-1 activation, subsequently diminishing neuronal apoptosis
(40).
Reactive astrocytes, once activated in the wake of
cerebral ischemic injury, orchestrate a neuroglial scar within the
peri-infarct cortex (PIC). This scar acts as a protective barrier,
isolating the injury site from healthy tissues and impeding the
spread of deleterious agents (41). Nevertheless, as cerebral ischemia
advances, this very neuroglial scar evolves into an obstacle,
hampering post-ischemic brain repair. Yuan et al (42) uncovered that
p-Hydroxybenzaldehyde, derived from asparagus, curtailed
astrocytosis in middle cerebral artery occlusion (MCAO) rat PICs.
This led to a reprogramming of astrocytes, transitioning the
neuroglial scar back to neuronal progenitor cells, thus promoting
neural and vascular rejuvenation within the PIC (42). Another noteworthy study revealed
that miR-124, encapsulated within the small extracellular vesicles
(EVs) of M2-type microglia, expedited the reprogramming of
astrocytes. This reprogramming counteracted the impediments posed
by the glial scar in the PIC, catalyzing stroke recuperation
(43).
The BBB is a complex and multifaceted structure,
comprising astrocytes, pericytes, the extracellular matrix and the
endothelial cells that line the brain's microvasculature. Its
pivotal role is to safeguard the CNS. Not only does the BBB
facilitate the entry of vital metabolic substrates such as oxygen
and glucose into the brain, but it also acts as a vigilant
sentinel, preventing the infiltration of pathogens, non-CNS cells,
and other potentially harmful agents. In doing so, the BBB plays an
indispensable role in preserving the delicate homeostasis of the
brain (44).
Astrocyte-derived Matrix metalloproteinase-2/9
(MMP-2/9) have been implicated in disrupting the integrity of BBB.
Specifically, these enzymes catalyze the degradation of tight
junction protein (TJP) ZO-1, thereby compromising BBB permeability
(45). Corroborating this,
research has revealed that MMPs degrade the BBB's TJPs. However,
agents such as BB-1101 have been shown to counteract this
degradation, thereby bolstering BBB integrity (46).
In the context of I/R, the cyclic RNA of FoxO3
(circ-FoxO3) has been identified as a crucial player. It interacts
synergistically with mTOR and E2F1, enhancing autophagic activity
through the inhibition of mTORC1. This cascade of molecular events
ultimately attenuates BBB-associated neuro-pathologies stemming
from damage (47).
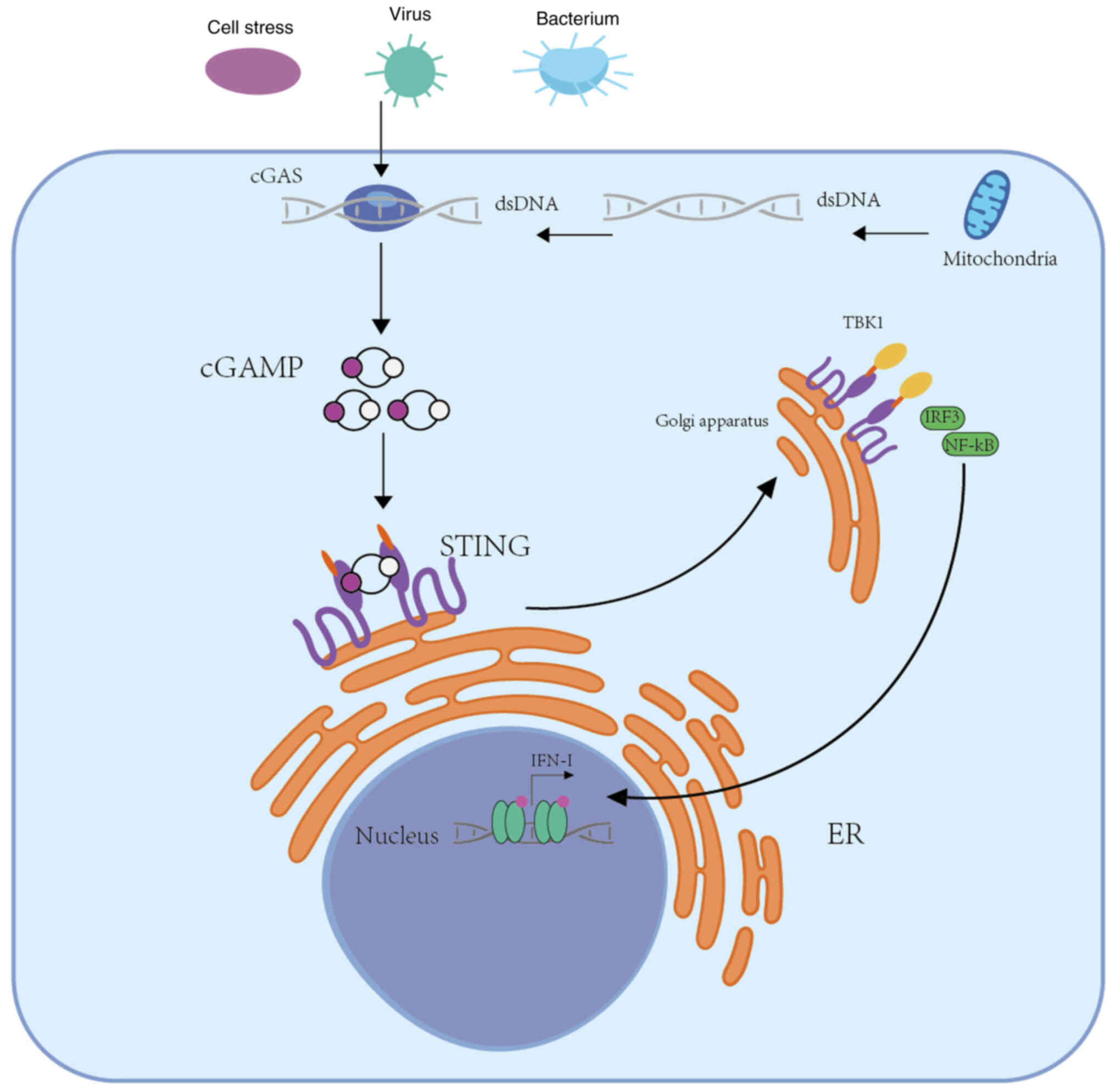
The cGAS-STING pathway is a central molecular
signaling cascade underpinned by two primary proteins: Cyclic
GMP-AMP synthase (cGAS) and the stimulator of interferon genes
(STING). Acting as an intrinsic immune sensor, cGAS detects an
array of cytoplasmic double-stranded DNA (dsDNA) sources,
encompassing viral, bacterial, mitochondrial, micronuclear and
reverse transcription-derived DNA (48). Once bound to dsDNA, cGAS
undergoes an enzymatic transformation, leading to the synthesis of
the secondary messenger, cGAMP, from ATP and GTP. This messenger is
subsequently identified by STING, a homodimeric protein anchored in
the endoplasmic reticulum (ER) membrane. Upon cGAMP binding, STING
undergoes a conformational shift, facilitating its oligomerization.
This modified STING then translocates to the Golgi apparatus
through the ER-Golgi intermediate compartment (ERGIC). Within the
Golgi, a palmitoylation event occurs at STING's cysteine residues
(Cys88 and Cys91), enabling its C-terminus to liaise with
TANK-binding kinase 1 (TBK1). This interaction culminates in the
phosphorylation of the STING protein at Ser366, leading to the
recruitment and subsequent phosphorylation of interferon-regulating
factor 3 (IRF3). As a result, IRF3 dimerizes, undergoes nuclear
translocation, and induces target gene expression (49). Parallelly, STING also stimulates
the nuclear factor kappaB (NF-kB) pathway, further influencing type
1 IFN expression (50).
Experimental data derived from IS models, wherein cGAS was deleted
or STING silenced, revealed reduced cerebral infarct injury and
enhanced neuronal survival (51). By contrast, overactivation of
this cGAS-STING axis has been linked to heightened
neuroinflammation (52)
(Fig. 1).
Modulating the cGAS-STING pathway has demonstrated
therapeutic potential in mitigating neuroinflammation.
Specifically, suppressing this pathway diminishes the inflammatory
response elicited by LPS/IFN-gamma in BV-2 microglia (53). Such modulation has been noted to
ameliorate learning and memory deficits in Alzheimer's disease,
making cognitive impairments less pronounced. In addition,
downregulation of the cGAS-STING pathway not only inhibited the
attenuation of microglia M1 polarization but also reduced the
expression of A1-type astrocytes to achieve an anti-inflammatory
effect (54,55). The inhibitor RU.521, targeting
cGAS, has shown promise in several contexts. It curbs the
expression of NF-kB-associated inflammatory cytokines, thereby
safeguarding neurons from potential damage. Furthermore, RU.521
effectively counteracts microglial pyroptosis, which is typically
incited by the activation of cytosolic gasdermin-D and NLRP3 post
cerebral venous sinus thrombosis (CVST) (52). Notably, RU.521 can also
counteract the mtDNA-driven activation of the cGAS-STING pathway
and modulate autophagy, further restraining the M1 polarization of
microglia (56). Consequently,
RU.521's modulation of the STING pathway offers a therapeutic
avenue for mitigating IS. The evolving understanding and
manipulation of the cGAS-STING signaling in microglia underscore
its potential as a therapeutic target for managing
neuroinflammation following cerebral I/R.
In the context of cerebral I/R, the downstream
STING-TBK1-IRF3 pathway of cGAS is adeptly regulated by histone
deacetylase 3 (HDAC3). The BCl-2-associated X-gene (BAX) is known
to complex with IRF3, promoting immune cell apoptosis-a process
that, if unchecked, can amplify post-stroke neuroinflammation
(57,58). Both IRF3 and NF-kB, being
downstream effectors of STING, can intensify neuroinflammation,
especially evident in the OGD/R model (51). However, pharmacological
intervention using HDAC3 inhibitors, such as Entinostat and
Trichostatin A, has revealed efficacy in curtailing I/R injuries
and the consequent neuroinflammation (59). Post-I/R, HDACs undergo oxidative
activation. Different HDAC subclasses can either bolster or hinder
cell viability and neurogenesis. For instance, inhibiting HDAC1,
HDAC2, HDAC3 and HDAC6 enhances cell survival and dampens
neuroinflammation (60,61). Conversely, augmenting the
activity of HDAC4 and HDAC5 promotes both cell viability and
neurogenesis (62).
Neutrophil extracellular traps (NETs) have emerged
as central players in the vascular dynamics of stroke. They
stimulate platelets, fostering a conducive environment for
thrombosis, impede the repair of damaged vessels, breach the
sanctity of the BBB, and underlie degenerative shifts observed in
the protracted aftermath of stroke (63). Notably, tissue plasminogen
activator (tPA), the primary therapeutic agent for thrombolysis and
recanalization in IS, instigates neutrophils to yield NETs.
Coincidentally, tPA treatment amplifies the cGAS expression,
synchronously boosting STING functionality. This chain of events
precipitates enhanced phosphorylation of TANK-binding kinase 1
(pTBK1) and ignites TBK1-facilitated activation of interferon
regulatory factor 3 (IRF3), culminating in a surge of type I
interferon (IFN-I) and interleukin-6 (IL-6) in ischemic zones. A
study led by Rosell et al (64) underscored the instrumental role
of NETs activation in the cGAS-STING pathway, especially post-tPA
administration (64).
In a notable experiment, neutrophils treated with
A23187 spawned EVs laden with DNA via NETs. These EV-DNAs, upon
internalization by macrophages, activate an alternate cGAS-STING
trajectory that propels NF-kB to synthesize IL-6-a process
distinctly independent of the conventional cGAS-STING-TBK1-IRF3
signaling mechanism (65). These
A23187-induced EVs provide invaluable insights into
neutrophil-driven inflammatory cascades under ischemic conditions,
offering potential therapeutic avenues to address
neuroinflammation, infections, and autoimmunity in stroke
patients.
Currently, tPA thrombolytic therapy is the only
available treatment for reperfusion in areas affected by cerebral
infarction. However, this treatment still carries the risk of
cerebral hemorrhage (66,67).
NETs or cGAS as a treatment could reduce the risk of hemorrhage
post-tPA thrombolysis in IS, offering a new strategy to enhance the
safety of tPA thrombolysis (68). By targeting NETs, it is also
possible to influence cGAS-STING activation, further reducing
M1-type microglia activation induced by STING/IFN-gamma (53). This reduction can alleviate
neurotoxic effects, protect the BBB, and prevent further death of
neurons in the ischemic penumbra (14,37). Additionally, this approach
appears capable of addressing the prolonged elevated levels of
TNF-alpha in the serum of CI/RI patients, which can last up to six
months (29).
The gastrointestinal (GI) tract stands as a
cornerstone of the body's immunological defense, harboring an
extensive congregation of immune cells. Remarkably, over 70% of the
human immune system finds its home within the GI immune system.
Resident within this GI tract is a diverse community of
microorganisms collectively termed the GI microbiota (GM). Advances
in microbiome research have led to the evolution of the classical
GBA concept, ushering in the more comprehensive GBMA. This holistic
perspective encompasses the GI neuroendocrine system, the GI immune
system, the enteric nervous system, and, pivotally, the GM.
Within the GBMA, GM emerges as a pivotal player,
intricately influencing metabolic regulation, nutritional
physiology, anticancer responses, immunomodulation, and crucially,
neurological functions (76,77). Dominating the GI flora are
Firmicutes (~48%) and Bacteroidetes (roughly 51%),
with the residual composition being a mix of Actinobacteria,
Proteobacteria, and other microorganisms (78). Notably, a marked reduction in the
abundance of Proteobacteria and certain Firmicutes
(such as Clostridia and Lachnospiraceae) has been
observed in post-stroke cognitive impairment patients, setting them
apart from non-stroke counterparts (79).
The human gut serves as a sanctuary for an array of
microorganisms pivotal in either bolstering health or predisposing
to disease, additionally playing a central role in immune
homeostasis (47). Deviations in
the GM's composition and function, termed dysbiosis, carry potent
associations with a plethora of health concerns. Internally, within
the GI domain, GM dysregulation correlates with inflammatory bowel
diseases (IBD) such as ulcerative colitis, Crohn's disease,
colorectal cancers and irritable bowel syndrome (80). Externally, beyond the confines of
the gut, an imbalanced GM has links with neurodegenerative ailments
including AD, IS and Parkinson's disease (PD) (81-83). Intriguingly, IBD patients exhibit
a heightened prevalence of PD compared with those without IBD
(84). Beyond merely being
bystanders, GM can actively sway the onset of stroke by influencing
risk factors including hypertension, diabetes, hyperlipidemia and
atherosclerosis (85).
Furthermore, the role of GM extends to modulating stroke outcomes
through its interactions with neural pathways, endocrine-mediated
neurotransmitter pathways, metabolite production and intricate
immune signaling cascades.
The intricate interplay of microorganisms
significantly influences neuroinflammatory responses in the host.
These effects emanate from their ability to metabolize substrates
that interact with receptors both within and beyond the GI
environment, orchestrating changes in affiliated signaling
pathways.
Short-chain fatty acids (SCFAs) epitomize the
pivotal products resulting from the microbial fermentation of
dietary fibers within the GI realm, exerting considerable
protective influences on IS. The triumvirate of principal SCFA
constituents encompasses acetate, propionate and butyrate (86). The synthesis of butyrate in SCFAs
is orchestrated from diverse precursors such as carbohydrates,
organic acids, glutamic acid and lysine, facilitated predominantly
by microbial communities within the gut. Renowned producers of
butyrate include bacterial families Lachnospiraceae
(including Eubacterium rectale, Roseburia inulinivorans and
others) and Ruminococcus (spanning species such as
Faecalibacterium prausnitzii and Coprococcus comes)
(87). Vancomycin treatment is
known to suppress these butyrate-producing bacteria, namely
Lachnospiraceae and Ruminococcus (88). Comparative analyses revealed that
SCFA levels, particularly butyrate, were appreciably diminished in
Sprague-Dawley (SD) rats afflicted by MCAO in contrast to their
healthy counterparts. This decline underscores the potential of
butyrate to ameliorate GM during IS by mitigating harmful bacteria
and bolstering beneficial lactic acid bacterial populations
(89). Intriguingly, an inverse
correlation emerges between SCFA levels and the prognosis of
reperfused stroke, particularly in elderly patients contending with
pronounced stroke events. Empirical studies demonstrated that aged
mice subjected to MCAO exhibited attenuated neuroinflammation and
reduced neurological deficits when administered fecal transplants
from young, SCFA-rich donors (90). Given the presence of SCFA
receptors on microglial cells, SCFAs are poised to modulate
microglial functionality (91).
Sodium butyrate, identified as a histone deacetylase inhibitor, is
known to confer neuroprotective benefits against neonatal
hypoxic-ischemic brain afflictions, primarily via the BDNF-TrkB
signaling cascade (92).
Furthermore, the capability of SCFAs to traverse the BBB
underscores their influence on both central and peripheral immune
cells, which, in turn, modulates post-ischemic cerebral recovery
dynamics (93).
Trimethylamine N-oxide (TMAO) emerges from the
transformation of trimethylamine, a derivative of GM, facilitated
by hepatic flavin monooxygenases. This metabolite has garnered
attention due to its association with adverse outcomes in IS
patients (94,95). The biosynthesis of TMAO primarily
roots from dietary sources, encompassing marine fish, shellfish
(96), mushrooms, specific meat
sources such as liver and kidneys, various fruits, cruciferous
vegetables and chicken (97).
The GI flora, including bacterial phyla such as Firmicutes,
Proteobacteria and Actinobacteria, play a pivotal
role by metabolizing ingested choline, L-carnitine, and betaine
into trimethylamine, which subsequently gets converted to TMAO in
the liver (98). Central to the
hepatic conversion process is the enzyme flavin monooxygenase 3.
Interestingly, certain nutritional supplements, such as L-alpha
glycerol-phosphoryl-choline (alpha-GPC), can metabolize TMAO
(99).
Empirical evidence underscores that serum TMAO
concentrations exhibit a positive correlation with the risk factors
of stroke and the ensuing neurological deficits post-ischemic brain
injuries. Notably, average serum TMAO levels were observed to be
~49% elevated in stroke-afflicted patients compared with their
healthy counterparts (100).
Beyond being a standalone predictor for IS, TMAO has been
identified to correlate positively with atherosclerotic tendencies
and the incidence of atrial fibrillation (101-103). Reinforcing its clinical
significance, TMAO not only enhances traditional cardiovascular
disease risk predictions but also stands out as a more sensitive
and specific independent prognostic marker (104).
Commonly perceived as beneficial, foods including
seafood, mushrooms and animal liver, as well as nutritional
supplements such as alpha-GPC, can be metabolized in the liver into
TMAO (96,97,99). This indicates the importance of
diet in stroke patients, as the conversion of the aforementioned
foods to TMAO can impact the recovery from CI/RI through the GBMA.
However, the specific quantities for consumption still require
further experimental validation. Cytidine diphosphate-choline
(CDP-choline), commonly used in clinical settings, is typically
regarded as a neuroprotective and neural restorative agent
(105). Yet, recent studies
have questioned its efficacy (106,107). There is also evidence that
CDP-choline can be converted to TMAO (108). However, there is no consensus
on the rate of this conversion. Further investigation into the
clinical efficacy of CDP-choline is needed in the future.
Vitamin B12 is pivotal in safeguarding the
intestinal epithelium, especially in inflammatory bowel conditions
such as colitis. Its contributions encompass nurturing beneficial
intestinal microbes, curbing pathogenic strains and thereby
ensuring the stability of the GM (109). Methyl-cobalamin, often in
synergy with adenosyl-cobalamin or paired with hydroxocobalamin and
cyanocobalamin, serves as a supplement to vitamin B12 (VB12)
(110). Deficiencies in both
VB12 and folic acid have been linked to heightened stroke risk
(111). As VB12 is essential
for fatty acid synthesis in the citric acid-pyruvic acid cycle, its
absence can impede the recuperation of myelinated nerve fibers
post-ischemic events (112).
Homocysteine (Hcy) is converted into methionine through the enzyme
methionine synthase. VB12 and folate, key cofactors for this
enzyme, when deficient, escalate Hcy levels. This in turn, as found
by Wang et al (113),
during I/R injury, impedes the PI3K-AKT- and ERK-dependent mTOR
pathways, detrimentally amplifying autophagy and compromising the
viability of neural stem cells in specific brain regions (113). Additionally, VB12 fosters the
growth of gut bacteria including Akkermansia and E.
faecalis, known for promoting SCFA production (114).
LPS, a key component of the outer membrane of
Gram-negative bacteria, is a potent modulator of innate immunity.
Structurally, it is comprised of lipid A, core oligosaccharides,
and extended polysaccharides (O antigens) (115). Recognized by the intricate
TLR4-MD-2 complex, LPS can instigate an inflammatory response
within the GM and associated lymphoid tissues (116). Following an IS, LPS can enter
the brain via the bloodstream, through compromised gut and BBB.
This promotes the transformation of astrocytes into the A1 subtype,
while inhibiting their transformation into the A2 subtype (117). Notably, folic acid can mitigate
LPS synthesis (118), providing
relief from LPS-induced cognitive impairments and modulating
post-injury inflammatory responses (119).
5-Hydroxytryptamine (5-HT), a pivotal monoamine
neurotransmitter, facilitates communication between nerve cells in
both central and peripheral nervous systems. Selective serotonin
reuptake inhibitors (SSRIs) have demonstrated efficacy in treating
post-stroke depression (PSD) (120). The onset of PSD might be
attributed to diminished neurotransmission owing to damage to the
mood-regulating frontal/temporal-basal ganglia-ventral brainstem
loop and interruptions in 5-HTergic neurons and pathways. This
damage, in turn, compromises neurotransmitter synthesis (121). Notably, 5-HT's precursor is the
essential amino acid tryptophan, and a staggering 90% of the body's
5-HT is localized within the GI tract cells (122). Intriguingly, there is a
positive feedback mechanism between GI-produced 5-HT and NLRP3,
with excessive 5-HT amplifying the activation of NLRP3 inflammatory
vesicles (123). NLRP3 is
implicated in triggering inflammatory signaling cascades and
exacerbating neuronal damage (124).
Scutellarin has demonstrated its potential in
reversing neuronal deterioration and cognitive impairments by
acting through the cAMP-PKA-CREB-HDAC3 pathway within the GBMA
(125). LPS is known to
stimulate HDAC3 in the GBMA circulation. By contrast, butyrate
attenuates HDAC3 expression, counteracting LPS-induced neurotoxic
effects. Additionally, melatonin appears to ameliorate cognitive
deficits either by downregulating LPS or enhancing butyrate levels
(126). Fiber-rich diets such
as high-inulin promote butyrate production to inhibit HDAC3
(127). These findings
underscore the pivotal role of HDAC3 in modulating cerebral I/R
injury via the GBMA. The aforementioned content is summarized in
Table I.
During ischemia and reperfusion, the gut's immune
cells play an instrumental role. The intestinal mucosal lamina
propria (LP) is replete with a variety of specialized immune cells,
including regulatory T (T-reg) cells, gammadelta T cells, innate
lymphocytes and helper T 17 cells. Remarkably, this region boasts
the most extensive T cell population in the human body. Within the
gut microbiota, gammadelta T cells stand out, as they transmit
diverse signals that adjust the host's immune response (128). These immune cells, while
influenced by the GM, traverse through peripheral circulation to
exert their influence in the brain. Research has noted an uptick in
the frequency of gammadelta T cells within the meningeal zones of
brains afflicted by ischemic injury (129). The overexpression of IL-17
protein in these cells incites a surge in chemokines in the brain
tissue. This results in significant neutrophil infiltration at the
injury site, thereby compromising the structural cohesiveness of
BBB (130). Furthermore, IL-17
stimulates the production of ROS and elevates MMP-3 and MMP-9
levels, which further erode BBB integrity by weakening its tight
junctions (131,132). Ultimately, IL-17 accentuates
neuronal death, through processes including apoptosis and
autophagy, exacerbating neuroinflammatory damage (133,134).
Following an IS, neutrophils can be detected in the
soft meninges as early as 6 h. Their presence peaks between days 3
to 7 post-stroke and then gradually diminishes, yet they can still
be observed up to 14 days after the ischemic incident (63). In the clinical context,
neutrophils are predominantly detrimental during IS. These cells
are potent catalysts for the abundant secretion of ROS through
myeloperoxidase (MPO), intensifying the deleterious impacts of
cerebral I/R. Notably, inhibiting MPO has been demonstrated to
diminish neuronal death, enhance the prognosis for
neuroinflammation, reduce the infarct size, and decrease the levels
of the pro-apoptotic protein, p53 (139). Additionally, neutrophils can
produce MMP-9, which, when found in elevated concentrations in the
core of ischemic regions, can inflict damage on brain tissue and
possibly precipitate hemorrhagic transformations (140).
The GM produces an array of dsDNA that activates the
STING pathway. The GM has a pivotal role in orchestrating the
synthesis of type I IFN through the cGAS-STING-dependent pathway
(48). Lactobacillus
rhamnosus GG, a prevalent probiotic in the GI system, has the
potential to enhance the cGAS-STING-TBK1-IRF7-IFN-beta signaling
cascade via dendritic cells (DCs), fostering a potent adaptive
immune response (141).
Conversely, Helicobacter pylori infections have been
identified to reduce the host's STING expression by 50%, leading to
the downregulation of IRF3-dependent type I IFN (142).
Moreover, STING plays a role in precipitating
intestinal epithelial cell (IEC) apoptosis. IEC apoptosis is a
central factor contributing to the dysfunction of the intestinal
barrier. This dysfunction results in heightened intestinal
permeability, allowing for the abnormal positioning of intestinal
flora (143). Research has
highlighted that suppressing STING leads to an augmentation in the
TJPs, which bolsters defense against increased intestinal
permeability. However, research by Canesso et al (144) highlighted the complexity of
STING pathway functions. It was found that intact STING signaling
is essential for maintaining intestinal homeostasis and regulating
CD4+Foxp3+ T regulatory cells in the gut
through type I IFN. In STING knockout mice, a reduction in T
regulatory cell function and expression was observed, indicating
the multifaceted role of the STING pathway in intestinal
permeability and homeostasis (144). This cGAS-STING signaling
pathway's multifaceted regulatory actions on intestinal
permeability, mucosal protection and immune modulation have
repercussions on cerebral I/R through the GBMA. Interestingly,
butyrate has been identified to inhibit TBK1 and IRF3
phosphorylation in the downstream segments of the cGAS-STING
pathway, thereby constraining STING-activated type I IFN expression
in DCs (145).
Mitochondria, often termed the 'powerhouses' of
cells, are chiefly tasked with energy synthesis. Paradoxically,
instead of aiding in the restoration of ischemic areas after
reperfusion brought about by the reintroduction of oxygen and
nutrients, further cellular damage is catalyzed. This damage is
propelled by an accumulation of mitochondrial Ca2+, a
surge in open mitochondrial permeability transition pores and a
pronounced release of ROS (146). Mitophagy stands as a sentinel,
governing both the number and functionality of mitochondria. It
achieves this by systematically eliminating dysfunctional or
overabundant mitochondria, leading to enhanced neuronal viability
and improved neurobehavioral outcomes. A deficiency in XBP1 hampers
the efficiency of mitochondrial autophagy. When mitochondria
sustain extensive damage, they release ROS and initiate
mitochondrial (mt)DNA clustering. The ensuing recognition of mtDNA
by macrophage cGAS instigates STING activation, intensifying
cGAS-dependent cellular pyroptosis (147). Notably, the collaboration of
the Parkin (an E3 ubiquitin ligase) and PINK1 (a ubiquitin kinase)
pathways fosters mitochondrial autophagy. This synergy curtails
mtDNA release, suppresses cGAS activation and dampens STING-driven
neuroinflammation (148).
Innovative formulations such as Fucoidan-proanthocyanidin
nanoparticles have showcased potential in kindling mitochondrial
autophagy while simultaneously inhibiting the cGAS/STING expression
(149). Harnessing and
modulating the intricacies of mitophagy post-cerebral I/R has
revealed promising avenues for mitigating neuroinflammation and
catalyzing neurological rejuvenation.
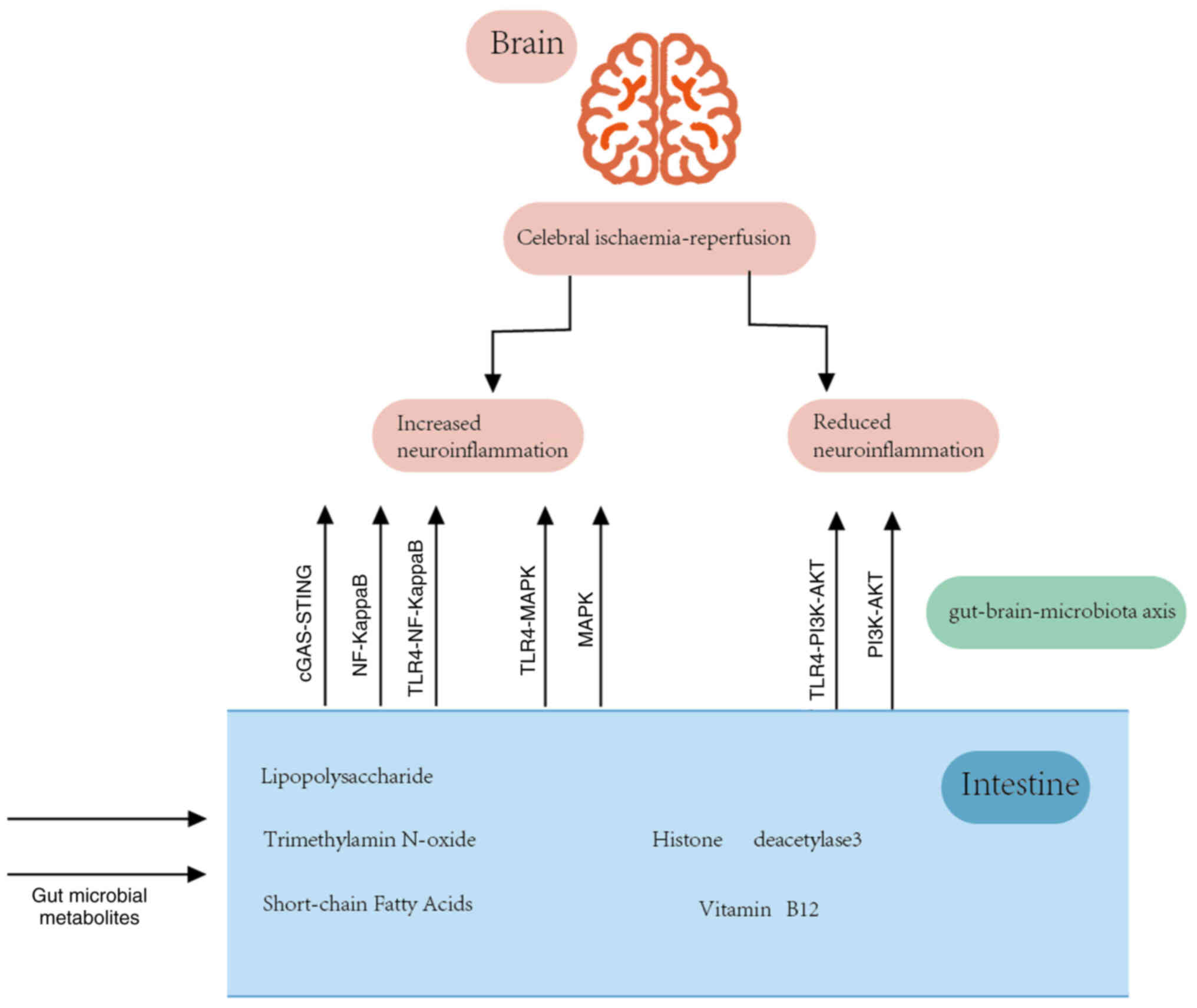
Emerging research underscores the profound interplay
between cerebral I/R and various signaling pathways, influenced by
the GBMA. Several pathways have been identified that amplify the
severity of cerebral infarction and intensify neuroinflammation
following cerebral I/R. These pathways include: The cGAS-STING
signaling pathway (51), the
NF-kB signaling cascade (71),
the MAPK signaling conduit (150), and the integrated
TLR4-NF-kB/PI3K-AKT/MAPK signaling mechanism (151).
Conversely, the PI3K-AKT signaling pathway has been
spotlighted for its neuroprotective role, demonstrating potential
in mitigating neurological injury post-cerebral I/R (152,153). Intriguingly, pathways
downstream of cGAS-STING exhibit the capacity to activate either
NF-kB or IRF3, signifying a multifaceted modulation of the
neuroinflammatory response in cerebral I/R dynamics.
Butyrate effectively inhibits the phosphorylation of
both TBK1 and IRF3, a process that is activated in the presence of
STING. This action in turn impedes the production of IFN-I
(145). Upon stimulation by
LPS, Kupffer cells release mtDNA through the voltage-dependent
anion channel 1, thereby initiating the cGAS-STING cascade.
Notably, within this cascade, IRF3 is revealed to elevate the
expression of NLRP3, instigating inflammatory reactions and
apoptosis. It is noteworthy that by suppressing STING, the
inflammatory and apoptotic effects of LPS are significantly
diminished (154).
However, there remains an ambiguity regarding the
role of IRF3 in the LPS-driven TLR4-NF-kB-NLRP3 signaling pathway.
Further comprehensive studies are essential to elucidate the
interrelation between the STING-IRF3 pathway and the TLR4 signaling
mechanisms.
Fluvoxamine, recognized as an SSRI, exhibits a dual
inhibitory action on both cGAS and STING (155). The modulation of the cGAS-STING
signaling pathway by these specific gut microbial metabolites
within the GBMA framework opens promising avenues for potential
targeted pharmaceutical interventions or nutritional strategies to
address cerebral I/R.
Pathology NF-kB mediates neuroinflammation by
modulating the expression of multiple inflammatory factors such as
IL-6, TNF-α and COX2 (156,157). L-alpha-GPC, a dietary
supplement, undergoes metabolic transformation in vivo,
leading to the formation of TMAO, which in turn activates the NF-kB
and MAPK signaling pathways (99). Notably, the effects of
cholinergic and anti-cholinergic agents on IS might be attributed
to their modulation of TMAO. Furthermore, LPS and homocysteine
(Hcy) has been identified as activators of the NF-kB signaling
cascade (158,159).
The phosphatidylinositol 3-kinase (PI3K)/protein
kinase B (PKB, also known as AKT) signaling pathway has been
identified as a key regulator of apoptosis-associated protein
expression and cell survival in CI/RI, thus playing a significant
neuroprotective role (162,163). Conversely, the
mitogen-activated protein kinase (MAPK) signaling pathway is
implicated in inflammatory processes in CI/RI (164). Activation of the MAPK pathway
triggers a substantial release of inflammatory factors (165). Inhibiting this pathway can
reduce astrocyte apoptosis, attenuate astrocyte-mediated
inflammatory responses, and protect brain neurons from I/R injury
(166). Additionally, blocking
the MAPK pathway may alleviate oxidative stress-induced damage and
reduce microglial activation and the overexpression of inflammatory
factors (167).
Aloe-emodin acts as a potent modulator, attenuating
the LPS-induced TLR4-NF-kB pathway while simultaneously enhancing
the TLR4-PI3K-AKT signaling. This dual activity offers promising
therapeutic potential for mitigating neurological damage and
inflammation associated with IS (168). Several other agents have
displayed similar inhibitory effects on the LPS-triggered
TLR4-NF-kB pathway. Notably, the depletion of Polymerase
δ-interacting protein 2 (169),
reduction in Cofilin (170),
neuroprotective agents (171)
and Escin (172) have all shown
substantial inhibitory effects on this pathway.
The MAPK signaling pathway, particularly its p38
branch, plays a crucial role in the cellular responses post-IS. A
notable influencer of this pathway is Hyperhomocysteinemia (hHcy).
Recognized as a standalone risk factor for stroke, hHcy's
interactions with the MAPK pathway heighten its implications.
Specifically, hHcy's activation of the p38 MAPK pathway exacerbates
neurotoxic effects after an ischemic event (179,180). This correlation underscores the
importance of understanding the mechanistic details of hHcy and the
MAPK pathway. Exploring this further could unveil potential
therapeutic targets that can mitigate the detrimental aftermath of
stroke events.
The PI3K)-AKT signaling pathway stands as a crucial
component in the cellular responses following IS. One noteworthy
mediator of this pathway is SCFA. Research has illuminated SCFA's
potential in mitigating the adverse effects of IS by activating the
PI3K-AKT signaling route, thereby inhibiting neuronal apoptosis,
curbing neuroinflammation and enhancing cognitive function
(181). Additionally,
experiments involving intranasal administration of sodium butyrate
have revealed a significant reduction in infarct volume and overall
improvement in neurological function, outcomes attributed to the
activation of the PI3K-AKT pathway (182). Conversely, hHcy is a noteworthy
antagonist in this context. By suppressing the PI3K-AKT pathway,
hHcy amplifies neurotoxic effects post-ischemic damage (113), highlighting the pathway's
double-edged nature in the realm of IS (Fig. 2). The aforementioned content is
summarized in Table II.
Patients with CI/RI exhibit a more disordered gut
microbiome compared with healthy individuals. Through the complex
bidirectional GBMA communication between the gut and the brain,
exploring the connection between gut microbiota and
neuroinflammation post CI/RI can illuminate promising pathways for
targeted treatment of post-CI/RI neuroinflammation. These gut
microbial communities, via signaling pathways such as cGAS-STING,
produce cytokines that positively or negatively affect microglia,
astrocytes and the BBB in CI/RI patients, thereby influencing the
neuroinflammation associated with CI/RI.
Within the GBMA, SCFAs block the cGAS-STING pathway
while simultaneously upregulating the PI3K-AKT signaling pathway.
This dual action reduces neuroinflammatory damage factors following
CI/RI and enhances protective factors (145,182). Future treatments could focus on
increasing the presence of gut bacterial families including
Lachnospiraceae and Ruminococcus in IS patients.
Additionally, increasing the infusion of VB12 and the intake of
high-inulin, fiber-rich diets can elevate SCFA levels (127). TMAO, an independent predictor
for IS, can upregulate NF-kB and MAPK signaling pathways,
exacerbating neurological damage. In the context of CI/RI's GBMA,
on one hand, serotonin (5-HT) can inhibit the cGAS-STING/NF-kB
pathway, offering neuroprotection (155,160,161), while on the other hand,
excessive 5-HT can enhance NLRP3 inflammasome activation, worsening
brain injury (123). More
importantly, LPS can activate the TLR4-dependent NF-kB/MAPK
signaling pathways, exacerbating neuroinflammatory damage
post-CI/RI (151,167), but can also activate
TLR4-dependent PI3K/AKT signaling pathways, participating in the
protection of the brain from CI/RI-induced damage (162,183).
In summary, these gut microbial metabolites,
produced by various gut microbial communities, suggest that broad
anti-inflammatory strategies are not suitable for treating
neuroinflammatory damage in CI/RI. The development and application
of targeted antibiotics are crucial. A new balance between
different gut microbial communities and their metabolites and the
signaling pathways of cGAS-STING/NF-kB/MAPK/PI3K-AKT is sought.
This balance aims to improve post-CI/RI neuroinflammation. Through
the GBMA, especially via various neural pathways, understanding of
how to reduce detrimental microbial communities and factors
(including LPS, TMAO, hcy, NET and HDAC3) or enhance beneficial
ones (such as SCFA, VB12 and 5-HT) can improve. This approach could
target treatment and even prevent neuroinflammation in patients,
promote early recovery in the ischemic penumbra, and reduce the
disease burden. Future research should validate these findings from
animal models to human diseases, identify the minimum levels needed
for therapeutic efficacy, and ultimately introduce new and
promising strategies for treating neuroinflammation following
CI/RI.
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
YZ and HY reviewed the literature, drafted and
revised the manuscript. SH and YX discussed and revised the
manuscript. YQW provided critical comments. YZ and HY drew figures
and translated the manuscript. YZ and YQW revised and finalized the
manuscript. All authors read and approved the final manuscript.
Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the Yuan Du Scholars,
Clinical Research Center of Affiliated Hospital of Weifang Medical
University (grant no. 2022WYFYLCYJ02) and Weifang Science and
Technology Development Plan Project Medical category (grant no.
2022YX093).
|
1
|
Knap D, Honkowicz M, Kirmes T, Koroński M,
Bukański M, Kysiak M, Kadłubicki B, Dymon I, Sieroń D and Baron J:
Endovascular treatment of acute ischemic stroke-own experience.
Neurol Neurochir Pol. 49:81–89. 2015.
|
|
2
|
Wang HE, Kabeto MM, Gray M, Wadley VG,
Muntner P, Judd SE, Safford MM, Kempker J and Levine DA: Trajectory
of cognitive decline after sepsis. Crit Care Med. 49:1083–1094.
2021.
|
|
3
|
Feigin VL, Roth GA, Naghavi M, Parmar P,
Krishnamurthi R, Chugh S, Mensah GA, Norrving B, Shiue I, Ng M, et
al: Global burden of stroke and risk factors in 188 countries,
during 1990-2013: A systematic analysis for the global burden of
disease study 2013. Lancet Neurol. 15:913–924. 2016.
|
|
4
|
Sun Y, Yang X, Xu L, Jia M, Zhang L, Li P
and Yang P: The role of Nrf2 in relieving cerebral
ischemia-reperfusion injury. Curr Neuropharmacol. 21:1405–1420.
2023.
|
|
5
|
Jayaraj RL, Azimullah S, Beiram R, Jalal
FY and Rosenberg GA: Neuroinflammation: Friend and foe for ischemic
stroke. J Neuroinflammation. 16:1422019.
|
|
6
|
Boullerne AI and Feinstein DL: History of
neuroscience I. Pío del Río-Hortega (1882-1945): The discoverer of
microglia and oligodendroglia. ASN Neuro.
12:17590914209532592020.
|
|
7
|
Del Río-Hortega Bereciartu J: Pío del
Río-Hortega: The revolution of glia. Anat Rec (Hoboken).
303:1232–1241. 2020.
|
|
8
|
Kanazawa M, Ninomiya I, Hatakeyama M,
Takahashi T and Shimohata T: Microglia and monocytes/macrophages
polarization reveal novel therapeutic mechanism against stroke. Int
J Mol Sci. 18:21352017.
|
|
9
|
Ito D, Tanaka K, Suzuki S, Dembo T and
Fukuuchi Y: Enhanced expression of Iba1, ionized calcium-binding
adapter molecule 1, after transient focal cerebral ischemia in rat
brain. Stroke. 32:1208–1215. 2001.
|
|
10
|
Wu P, Zeng F, Li YX, Yu BL, Qiu LH, Qin W,
Li J, Zhou YM and Liang FR: Changes of resting cerebral activities
in subacute ischemic stroke patients. Neural Regen Res. 10:760–765.
2015.
|
|
11
|
Guruswamy R and ElAli A: Complex roles of
microglial cells in ischemic stroke pathobiology: New insights and
future directions. Int J Mol Sci. 18:4962017.
|
|
12
|
Perego C, Fumagalli S and De Simoni MG:
Temporal pattern of expression and colocalization of
microglia/macrophage phenotype markers following brain ischemic
injury in mice. J Neuroinflammation. 8:1742011.
|
|
13
|
West AP, Brodsky IE, Rahner C, Woo DK,
Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS and
Ghosh S: TLR signalling augments macrophage bactericidal activity
through mitochondrial ROS. Nature. 472:476–480. 2011.
|
|
14
|
Chen AQ, Fang Z, Chen XL, Yang S, Zhou YF,
Mao L, Xia YP, Jin HJ, Li YN, You MF, et al: Microglia-derived
TNF-α mediates endothelial necroptosis aggravating blood
brain-barrier disruption after ischemic stroke. Cell Death Dis.
10:4872019.
|
|
15
|
Perez-de-Puig I, Miró-Mur F, Ferrer-Ferrer
M, Gelpi E, Pedragosa J, Justicia C, Urra X, Chamorro A and Planas
AM: Neutrophil recruitment to the brain in mouse and human ischemic
stroke. Acta Neuropathol. 129:239–257. 2015.
|
|
16
|
Roca H, Varsos ZS, Sud S, Craig MJ, Ying C
and Pienta KJ: CCL2 and interleukin-6 promote survival of human
CD11b+ peripheral blood mononuclear cells and induce M2-type
macrophage polarization. J Biol Chem. 284:34342–34354. 2009.
|
|
17
|
Gleissner CA, Shaked I, Little KM and Ley
K: CXC chemokine ligand 4 induces a unique transcriptome in
monocyte-derived macrophages. J Immunol. 184:4810–4818. 2010.
|
|
18
|
Ortega-Gómez A, Perretti M and Soehnlein
O: Resolution of inflammation: An integrated view. EMBO Mol Med.
5:661–674. 2013.
|
|
19
|
Li C, Wu Z, Zhou L, Shao J, Hu X, Xu W,
Ren Y, Zhu X, Ge W, Zhang K, et al: Temporal and spatial cellular
and molecular pathological alterations with single-cell resolution
in the adult spinal cord after injury. Signal Transduct Target
Ther. 7:652022.
|
|
20
|
Ma Y, Wang J, Wang Y and Yang GY: The
biphasic function of microglia in ischemic stroke. Prog Neurobiol.
157:247–272. 2017.
|
|
21
|
Orihuela R, McPherson CA and Harry GJ:
Microglial M1/M2 polarization and metabolic states. Br J Pharmacol.
173:649–665. 2016.
|
|
22
|
Bell-Temin H, Culver-Cochran AE, Chaput D,
Carlson CM, Kuehl M, Burkhardt BR, Bickford PC, Liu B and Stevens
SM Jr: Novel molecular insights into classical and alternative
activation states of microglia as revealed by stable isotope
labeling by amino acids in cell culture (SILAC)-based proteomics.
Mol Cell Proteomics. 14:3173–3184. 2015.
|
|
23
|
Jiang CT, Wu WF, Deng YH and Ge JW:
Modulators of microglia activation and polarization in ischemic
stroke (review). Mol Med Rep. 21:2006–2018. 2020.
|
|
24
|
Suenaga J, Hu X, Pu H, Shi Y, Hassan SH,
Xu M, Leak RK, Stetler RA, Gao Y and Chen J: White matter injury
and microglia/macrophage polarization are strongly linked with
age-related long-term deficits in neurological function after
stroke. Exp Neurol. 272:109–119. 2015.
|
|
25
|
Ritzel RM, Lai YJ, Crapser JD, Patel AR,
Schrecengost A, Grenier JM, Mancini NS, Patrizz A, Jellison ER,
Morales-Scheihing D, et al: Aging alters the immunological response
to ischemic stroke. Acta Neuropathol. 136:89–110. 2018.
|
|
26
|
Badan I, Buchhold B, Hamm A, Gratz M,
Walker LC, Platt D, Kessler Ch and Popa-Wagner A: Accelerated glial
reactivity to stroke in aged rats correlates with reduced
functional recovery. J Cereb Blood Flow Metab. 23:845–854.
2003.
|
|
27
|
Yenari MA: Microglia, the brain's double
agent. J Cereb Blood Flow Metab. 40(1 Suppl): S3–S5. 2020.
|
|
28
|
Kim JW, Park MS, Kim JT, Kang HJ, Bae KY,
Kim SW, Shin MG, Cho KH and Kim JM: The impact of tumor necrosis
factor-α and interleukin-1β levels and polymorphisms on long-term
stroke outcomes. Eur Neurol. 79:38–44. 2018.
|
|
29
|
Pascotini ET, Flores AE, Kegler A, Gabbi
P, Bochi GV, Algarve TD, Prado AL, Duarte MM, da Cruz IB, Moresco
RN, et al: Apoptotic markers and DNA damage are related to late
phase of stroke: Involvement of dyslipidemia and inflammation.
Physiol Behav. 151:369–378. 2015.
|
|
30
|
Wu L, Xiong X, Wu X, Ye Y, Jian Z, Zhi Z
and Gu L: Targeting oxidative stress and inflammation to prevent
ischemia-reperfusion injury. Front Mol Neurosci. 13:282020.
|
|
31
|
Ooboshi H, Ibayashi S, Shichita T, Kumai
Y, Takada J, Ago T, Arakawa S, Sugimori H, Kamouchi M, Kitazono T
and Iida M: Postischemic gene transfer of interleukin-10 protects
against both focal and global brain ischemia. Circulation.
111:913–919. 2005.
|
|
32
|
Yu C, Zhu C, Zhang Y, Chen H, Qin W, Wang
M and Li K: A longitudinal diffusion tensor imaging study on
Wallerian degeneration of corticospinal tract after motor pathway
stroke. Neuroimage. 47:451–458. 2009.
|
|
33
|
Thiel A, Radlinska BA, Paquette C, Sidel
M, Soucy JP, Schirrmacher R and Minuk J: The temporal dynamics of
poststroke neuroinflammation: A longitudinal diffusion tensor
imaging-guided PET study with 11C-PK11195 in acute subcortical
stroke. J Nucl Med. 51:1404–1412. 2010.
|
|
34
|
Price CJ, Wang D, Menon DK, Guadagno JV,
Cleij M, Fryer T, Aigbirhio F, Baron JC and Warburton EA: Intrinsic
activated microglia map to the peri-infarct zone in the subacute
phase of ischemic stroke. Stroke. 37:1749–1753. 2006.
|
|
35
|
Huang T, Yin J, Ren S and Zhang X:
Protective effects of KLF4 on blood-brain barrier and oxidative
stress after cerebral ischemia-reperfusion in rats through the
Nrf2/Trx1 pathway. Cytokine. 169:1562882023.
|
|
36
|
Liddelow SA and Barres BA: Reactive
astrocytes: Production, function, and therapeutic potential.
Immunity. 46:957–967. 2017.
|
|
37
|
Liddelow SA, Guttenplan KA, Clarke LE,
Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS,
Peterson TC, et al: Neurotoxic reactive astrocytes are induced by
activated microglia. Nature. 541:481–487. 2017.
|
|
38
|
John GR, Lee SC and Brosnan CF: Cytokines:
powerful regulators of glial cell activation. Neuroscientist.
9:10–22. 2003.
|
|
39
|
Li X, Yin X, Pang J, Chen Z and Wen J:
Hydrogen sulfide inhibits lipopolysaccharide-based
neuroinflammation-induced astrocyte polarization after cerebral
ischemia/reperfusion injury. Eur J Pharmacol. 949:1757432023.
|
|
40
|
Li J, Xu P, Hong Y, Xie Y, Peng M, Sun R,
Guo H, Zhang X, Zhu W, Wang J and Liu X: Lipocalin-2-mediated
astrocyte pyroptosis promotes neuroinflammatory injury via NLRP3
inflammasome activation in cerebral ischemia/reperfusion injury. J
Neuroinflammation. 20:1482023.
|
|
41
|
Okada S, Hara M, Kobayakawa K, Matsumoto Y
and Nakashima Y: Astrocyte reactivity and astrogliosis after spinal
cord injury. Neurosci Res. 126:39–43. 2018.
|
|
42
|
Yuan Y, Liu L, Du Y, Fan R, Zhang R and
Zhou N: p-hydroxy benzaldehyde revitalizes the microenvironment of
peri-infarct cortex in rats after cerebral ischemia-reperfusion.
Phytomedicine. 105:1543792022.
|
|
43
|
Li Z, Song Y, He T, Wen R, Li Y, Chen T,
Huang S, Wang Y, Tang Y, Shen F, et al: M2 microglial small
extracellular vesicles reduce glial scar formation via the
miR-124/STAT3 pathway after ischemic stroke in mice. Theranostics.
11:1232–1248. 2021.
|
|
44
|
Patel JP and Frey BN: Disruption in the
blood-brain barrier: The missing link between brain and body
inflammation in bipolar disorder? Neural Plast.
2015:7083062015.
|
|
45
|
Zhang S, An Q, Wang T, Gao S and Zhou G:
Corrigendum to 'Autophagy- and MMP-2/9-mediated reduction and
redistribution of ZO-1 contribute to hyperglycemia-increased
blood-brain barrier permeability during early reperfusion in
stroke' [Neuroscience 377 (2018) 126-137]. Neuroscience.
386:3512018.
|
|
46
|
Yang Y, Estrada EY, Thompson JF, Liu W and
Rosenberg GA: Matrix metalloproteinase-mediated disruption of tight
junction proteins in cerebral vessels is reversed by synthetic
matrix metalloproteinase inhibitor in focal ischemia in rat. J
Cereb Blood Flow Metab. 27:697–709. 2007.
|
|
47
|
Clemente JC, Ursell LK, Parfrey LW and
Knight R: The impact of the gut microbiota on human health: An
integrative view. Cell. 148:1258–1270. 2012.
|
|
48
|
Erttmann SF, Swacha P, Aung KM, Brindefalk
B, Jiang H, Härtlova A, Uhlin BE, Wai SN and Gekara NO: The gut
microbiota prime systemic antiviral immunity via the
cGAS-STING-IFN-I axis. Immunity. 55:847–861.e10. 2022.
|
|
49
|
Yu H, Liao K, Hu Y, Lv D, Luo M, Liu Q,
Huang L and Luo S: Role of the cGAS-STING pathway in aging-related
endothelial dysfunction. Aging Dis. 13:1901–1918. 2022.
|
|
50
|
Yu CH, Davidson S, Harapas CR, Hilton JB,
Mlodzianoski MJ, Laohamonthonkul P, Louis C, Low RRJ, Moecking J,
De Nardo D, et al: TDP-43 triggers mitochondrial DNA release via
mPTP to activate cGAS/STING in ALS. Cell. 183:636–649.e18.
2020.
|
|
51
|
Kong L, Li W, Chang E, Wang W, Shen N, Xu
X, Wang X, Zhang Y, Sun W, Hu W, et al: mtDNA-STING axis mediates
microglial polarization via IRF3/NF-κB signaling after ischemic
stroke. Front Immunol. 13:8609772022.
|
|
52
|
Ding R, Li H, Liu Y, Ou W, Zhang X, Chai
H, Huang X, Yang W and Wang Q: Activating cGAS-STING axis
contributes to neuroinflammation in CVST mouse model and induces
inflammasome activation and microglia pyroptosis. J
Neuroinflammation. 19:1372022.
|
|
53
|
Gao D, Hao JP, Li BY, Zheng CC, Miao BB
and Zhang L, Li YL, Li L, Li XJ and Zhang L: Tetrahydroxy stilbene
glycoside ameliorates neuroinflammation for Alzheimer's disease via
cGAS-STING. Eur J Pharmacol. 953:1758092023.
|
|
54
|
Wu W, Zhang X, Wang S, Li T, Hao Q, Li S,
Yao W and Sun R: Pharmacological inhibition of the cGAS-STING
signaling pathway suppresses microglial M1-polarization in the
spinal cord and attenuates neuropathic pain. Neuropharmacology.
217:1092062022.
|
|
55
|
Chen Y, Hu Y, He X, Zang H, Sun R, Zhu C
and Yao W: Activation of mitochondrial DNA-mediated cGAS-STING
pathway contributes to chronic postsurgical pain by inducing type I
interferons and A1 reactive astrocytes in the spinal cord. Int
Immunopharmacol. 127:1113482024.
|
|
56
|
Han C, Qian X, Ren X, Zhang S, Hu L, Li J,
Huang Y, Huang R, Ooi K, Lin H and Xia C: Inhibition of cGAS in
paraventricular nucleus attenuates hypertensive heart injury via
regulating microglial autophagy. Mol Neurobiol. 59:7006–7024.
2022.
|
|
57
|
Paludan SR, Reinert LS and Hornung V:
DNA-stimulated cell death: Implications for host defence,
inflammatory diseases and cancer. Nat Rev Immunol. 19:141–153.
2019.
|
|
58
|
Kim HY, Kim TJ, Kang L, Kim YJ, Kang MK,
Kim J, Ryu JH, Hyeon T, Yoon BW, Ko SB and Kim BS: Mesenchymal stem
cell-derived magnetic extracellular nanovesicles for targeting and
treatment of ischemic stroke. Biomaterials. 243:1199422020.
|
|
59
|
Liao Y, Cheng J, Kong X, Li S, Li X, Zhang
M, Zhang H, Yang T, Dong Y, Li J, et al: HDAC3 inhibition
ameliorates ischemia/reperfusion-induced brain injury by regulating
the microglial cGAS-STING pathway. Theranostics. 10:9644–9662.
2020.
|
|
60
|
Nieto-Estevez V, Changarathil G, Adeyeye
AO, Coppin MO, Kassim RS, Zhu J and Hsieh J: HDAC1 regulates
neuronal differentiation. Front Mol Neurosci. 14:8158082021.
|
|
61
|
Wang M, Zhou C, Yu L, Kong D, Ma W, Lv B,
Wang Y, Wu W, Zhou M and Cui G: Upregulation of MDH1 acetylation by
HDAC6 inhibition protects against oxidative stress-derived neuronal
apoptosis following intracerebral hemorrhage. Cell Mol Life Sci.
79:3562022.
|
|
62
|
Gao Y, Liu Y, Zheng D, Ho C, Wen D, Sun J,
Huang L, Liu Y, Li Q and Zhang Y: HDAC5-mediated Smad7 silencing
through MEF2A is critical for fibroblast activation and
hypertrophic scar formation. Int J Biol Sci. 18:5724–5739.
2022.
|
|
63
|
Kang L, Yu H, Yang X, Zhu Y, Bai X, Wang
R, Cao Y, Xu H, Luo H, Lu L, et al: Neutrophil extracellular traps
released by neutrophils impair revascularization and vascular
remodeling after stroke. Nat Commun. 11:24882020.
|
|
64
|
Rosell A, Ortega-Aznar A, Alvarez-Sabín J,
Fernández-Cadenas I, Ribó M, Molina CA, Lo EH and Montaner J:
Increased brain expression of matrix metalloproteinase-9 after
ischemic and hemorrhagic human stroke. Stroke. 37:1399–1406.
2006.
|
|
65
|
Allen ER, Whitefoot-Keliin KM, Palmatier
EM, Mahon AR and Greenlee-Wacker MC: Extracellular vesicles from
A23187-treated neutrophils cause cGAS-STING-dependent IL-6
production by macrophages. Front Immunol. 13:9494512022.
|
|
66
|
Powers WJ, Rabinstein AA, Ackerson T,
Adeoye OM, Bambakidis NC, Becker K, Biller J, Brown M, Demaerschalk
BM, Hoh B, et al: Guidelines for the early management of patients
with acute ischemic stroke: 2019 Update to the 2018 guidelines for
the early management of acute ischemic stroke: A guideline for
healthcare professionals from the american heart
association/American stroke association. Stroke. 50:e344–e418.
2019.
|
|
67
|
Whiteley WN, Slot KB, Fernandes P,
Sandercock P and Wardlaw J: Risk factors for intracranial
hemorrhage in acute ischemic stroke patients treated with
recombinant tissue plasminogen activator: A systematic review and
meta-analysis of 55 studies. Stroke. 43:2904–2909. 2012.
|
|
68
|
Wang R, Zhu Y, Liu Z, Chang L, Bai X, Kang
L, Cao Y, Yang X, Yu H, Shi MJ, et al: Neutrophil extracellular
traps promote tPA-induced brain hemorrhage via cGAS in mice with
stroke. Blood. 138:91–103. 2021.
|
|
69
|
Yang LY, Luo Q, Lu L, Zhu WW, Sun HT, Wei
R, Lin ZF, Wang XY, Wang CQ, Lu M, et al: Increased neutrophil
extracellular traps promote metastasis potential of hepatocellular
carcinoma via provoking tumorous inflammatory response. J Hematol
Oncol. 13:32020.
|
|
70
|
Liu M, Xu Z, Wang L, Zhang L, Liu Y, Cao
J, Fu Q, Liu Y, Li H, Lou J, et al: Cottonseed oil alleviates
ischemic stroke injury by inhibiting the inflammatory activation of
microglia and astrocyte. J Neuroinflammation. 17:2702020.
|
|
71
|
Luo L, Liu M, Fan Y, Zhang J, Liu L, Li Y,
Zhang Q, Xie H, Jiang C, Wu J, et al: Intermittent theta-burst
stimulation improves motor function by inhibiting neuronal
pyroptosis and regulating microglial polarization via
TLR4/NFκB/NLRP3 signaling pathway in cerebral ischemic mice. J
Neuroinflammation. 19:1412022.
|
|
72
|
Zhu H, Jian Z, Zhong Y, Ye Y, Zhang Y, Hu
X, Pu B, Gu L and Xiong X: Janus kinase inhibition ameliorates
ischemic stroke injury and neuroinflammation through reducing NLRP3
inflammasome activation via JAK2/STAT3 pathway inhibition. Front
Immunol. 12:7149432021.
|
|
73
|
Ning L, Wei W, Wenyang J, Rui X and Qing
G: Cytosolic DNA-STING-NLRP3 axis is involved in murine acute lung
injury induced by lipopolysaccharide. Clin Transl Med.
10:e2282020.
|
|
74
|
Liu X, Lv X, Liu Z, Zhang M and Leng Y:
MircoRNA-29a in astrocyte-derived extracellular vesicles suppresses
brain ischemia reperfusion injury via TP53INP1 and the NF-κB/NLRP3
axis. Cell Mol Neurobiol. 42:1487–1500. 2022.
|
|
75
|
He M, Fan J, Zhou R, Gao G, Li R, Zuo Y,
Li B, Li Y and Sun T: NLRP3/Caspase-1-mediated pyroptosis of
astrocytes induced by antipsychotics is inhibited by a histamine H1
receptor-selective agonist. Front Aging Neurosci.
14:8475612022.
|
|
76
|
Grenham S, Clarke G, Cryan JF and Dinan
TG: Brain-gutmicrobe communication in health and disease. Front
Physiol. 2:942011.
|
|
77
|
Vogt NM, Kerby RL, Dill-McFarland KA,
Harding SJ, Merluzzi AP, Johnson SC, Carlsson CM, Asthana S,
Zetterberg H, Blennow K, et al: Gut microbiome alterations in
Alzheimer's disease. Sci Rep. 7:135372017.
|
|
78
|
Qin J, Li R, Raes J, Arumugam M, Burgdorf
KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al: A
human gut microbial gene catalogue established by metagenomic
sequencing. Nature. 464:59–65. 2010.
|
|
79
|
Ling Y, Gong T, Zhang J, Gu Q, Gao X, Weng
X, Liu J and Sun J: Gut microbiome signatures are biomarkers for
cognitive impairment in patients with ischemic stroke. Front Aging
Neurosci. 12:5115622020.
|
|
80
|
Huang QX, Liang JL, Yang CH, Li K, Niu MT,
Fan JX and Zhang XZ: Stimulation-responsive mucoadhesive probiotics
for inflammatory bowel disease treatment by scavenging reactive
oxygen species and regulating gut microbiota. Biomaterials.
301:1222742023.
|
|
81
|
Zhou Y, Xie L, Schröder J, Schuster IS,
Nakai M, Sun G, Sun YBY, Mariño E, Degli-Esposti MA, Marques FZ, et
al: Dietary fiber and microbiota metabolite receptors enhance
cognition and alleviate disease in the 5xFAD mouse model of
Alzheimer's disease. J Neurosci. 43:6460–6475. 2023.
|
|
82
|
Qiao CM, Zhou Y, Quan W, Ma XY, Zhao LP,
Shi Y, Hong H, Wu J, Niu GY, Chen YN, et al: Fecal microbiota
transplantation from aged mice render recipient mice resistant to
MPTP-induced nigrostriatal degeneration via a
neurogenesis-dependent but inflammation-independent manner.
Neurotherapeutics. 20:1405–1426. 2023.
|
|
83
|
Liu C, Cheng X, Zhong S, Liu Z, Liu F, Lin
X, Zhao Y, Guan M, Xiao T, Jolkkonen J, et al: Long-term
modification of gut microbiota by broad-spectrum antibiotics
improves stroke outcome in rats. Stroke Vasc Neurol. 7:381–389.
2022.
|
|
84
|
Peter I, Dubinsky M, Bressman S, Park A,
Lu C, Chen N and Wang A: Anti-tumor necrosis factor therapy and
incidence of parkinson disease among patients with inflammatory
bowel disease. JAMA Neurol. 75:939–946. 2018.
|
|
85
|
Huang PY, Liu HM, Ko YR, Chang ZY and Lee
TY: Electroacupuncture relieves portal hypertension by improving
vascular angiogenesis and linking gut microbiota in bile duct
ligation rats. Front Microbiol. 14:12071372023.
|
|
86
|
Martin-Gallausiaux C, Marinelli L,
Blottière HM, Larraufie P and Lapaque N: SCFA: Mechanisms and
functional importance in the gut. Proc Nutr Soc. 80:37–49.
2021.
|
|
87
|
Louis P, Hold GL and Flint HJ: The gut
microbiota, bacterial metabolites and colorectal cancer. Nat Rev
Microbiol. 12:661–672. 2014.
|
|
88
|
Awoniyi M, Wang J, Ngo B, Meadows V, Tam
J, Viswanathan A, Lai Y, Montgomery S, Farmer M, Kummen M, et al:
Protective and aggressive bacterial subsets and metabolites modify
hepatobiliary inflammation and fibrosis in a murine model of PSC.
Gut. 72:671–685. 2023.
|
|
89
|
Chen R, Xu Y, Wu P, Zhou H, Lasanajak Y,
Fang Y, Tang L, Ye L, Li X, Cai Z and Zhao J: Transplantation of
fecal microbiota rich in short chain fatty acids and butyric acid
treat cerebral ischemic stroke by regulating gut microbiota.
Pharmacol Res. 148:1044032019.
|
|
90
|
Lee J, d'Aigle J, Atadja L, Quaicoe V,
Honarpisheh P, Ganesh BP, Hassan A, Graf J, Petrosino J, Putluri N,
et al: Gut microbiota-derived short-chain fatty acids promote
poststroke recovery in aged mice. Circ Res. 127:453–465. 2020.
|
|
91
|
Erny D, Hrabě de Angelis AL, Jaitin D,
Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T,
Jakobshagen K, Buch T, et al: Host microbiota constantly control
maturation and function of microglia in the CNS. Nat Neurosci.
18:965–977. 2015.
|
|
92
|
Jaworska J, Zalewska T, Sypecka J and
Ziemka-Nalecz M: Effect of the HDAC inhibitor, sodium butyrate, on
neurogenesis in a rat model of neonatal hypoxia-ischemia: Potential
mechanism of action. Mol Neurobiol. 56:6341–6370. 2019.
|
|
93
|
Sadler R, Cramer JV, Heindl S, Kostidis S,
Betz D, Zuurbier KR, Northoff BH, Heijink M, Goldberg MP, Plautz
EJ, et al: Short-chain fatty acids improve poststroke recovery via
immunological mechanisms. J Neurosci. 40:1162–1173. 2020.
|
|
94
|
Fryc J and Naumnik B: Thrombolome and its
emerging role in chronic kidney diseases. Toxins (Basel).
13:2232021.
|
|
95
|
Wu C, Li C, Zhao W, Xie N, Yan F, Lian Y,
Zhou L, Xu X, Liang Y, Wang L, et al: Elevated trimethylamine
N-oxide related to ischemic brain lesions after carotid artery
stenting. Neurology. 90:e1283–e1290. 2018.
|
|
96
|
Cho CE, Taesuwan S, Malysheva OV, Bender
E, Tulchinsky NF, Yan J, Sutter JL and Caudill MA:
Trimethylamine-N-oxide (TMAO) response to animal source foods
varies among healthy young men and is influenced by their gut
microbiota composition: A randomized controlled trial. Mol Nutr
Food Res. 61:16003242017.
|
|
97
|
Garcia-Perez I, Posma JM, Gibson R,
Chambers ES, Hansen TH, Vestergaard H, Hansen T, Beckmann M,
Pedersen O, Elliott P, et al: Objective assessment of dietary
patterns by use of metabolic phenotyping: A randomised, controlled,
crossover trial. Lancet Diabetes Endocrinol. 5:184–195. 2017.
|
|
98
|
Wang Z, Klipfell E, Bennett BJ, Koeth R,
Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al:
Gut flora metabolism of phosphatidylcholine promotes cardiovascular
disease. Nature. 472:57–63. 2011.
|
|
99
|
Wang Z, Hazen J, Jia X, Org E, Zhao Y,
Osborn LJ, Nimer N, Buffa J, Culley MK, Krajcik D, et al: The
nutritional supplement l-alpha glycerylphosphorylcholine promotes
atherosclerosis. Int J Mol Sci. 22:134772021.
|
|
100
|
Rexidamu M, Li H, Jin H and Huang J: Serum
levels of trimethylamine-N-oxide in patients with ischemic stroke.
Biosci Rep. 39:BSR201905152019.
|
|
101
|
Zheng L, Zheng J, Xie Y, Li Z, Guo X, Sun
G, Sun Z, Xing F and Sun Y: Serum gut microbe-dependent
trimethylamine N-oxide improves the prediction of future
cardiovascular disease in a community-based general population.
Atherosclerosis. 280:126–131. 2019.
|
|
102
|
Zhu W, Gregory JC, Org E, Buffa JA, Gupta
N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, et al: Gut microbial
metabolite TMAO enhances platelet hyperreactivity and thrombosis
risk. Cell. 165:111–124. 2016.
|
|
103
|
Svingen GFT, Zuo H, Ueland PM, Seifert R,
Løland KH, Pedersen ER, Schuster PM, Karlsson T, Tell GS,
Schartum-Hansen H, et al: Increased plasma trimethylamine-N-oxide
is associated with incident atrial fibrillation. Int J Cardiol.
267:100–106. 2018.
|
|
104
|
Qi J, You T, Li J, Pan T, Xiang L, Han Y
and Zhu L: Circulating trimethylamine N-oxide and the risk of
cardiovascular diseases: A systematic review and meta-analysis of
11 prospective cohort studies. J Cell Mol Med. 22:185–194.
2018.
|
|
105
|
Dávalos A and Secades J: Citicoline
preclinical and clinical update 2009-2010. Stroke. 42(1 Suppl):
S36–S39. 2011.
|
|
106
|
Agarwal A, Vishnu VY, Sharma J, Bhatia R,
Garg A, Dwivedi S, Upadhyay A, Goyal V, Singh MB, Gupta A, et al:
Citicoline in acute ischemic stroke: A randomized controlled trial.
PLoS One. 17:e02692242022.
|
|
107
|
Martí-Carvajal AJ, Valli C, Martí-Amarista
CE, Solà I, Martí-Fàbregas J and Bonfill Cosp X: Citicoline for
treating people with acute ischemic stroke. Cochrane Database Syst
Rev. 8:CD0130662020.
|
|
108
|
Synoradzki K and Grieb P: Citicoline: A
superior form of choline? Nutrients. 11:15692019.
|
|
109
|
Lurz E, Horne RG, Määttänen P, Wu RY,
Botts SR, Li B, Rossi L, Johnson-Henry KC, Pierro A, Surette MG and
Sherman PM: Vitamin B12 deficiency alters the gut microbiota in a
murine model of colitis. Front Nutr. 7:832020.
|
|
110
|
Thakkar K and Billa G: Treatment of
vitamin B12 deficiency-methylcobalamine? Cyancobalamine?
Hydroxocobalamin?-clearing the confusion. Eur J Clin Nutr. 69:1–2.
2015.
|
|
111
|
Van Guelpen B, Hultdin J, Johansson I,
Stegmayr B, Hallmans G, Nilsson TK, Weinehall L, Witthöft C,
Palmqvist R and Winkvist A: Folate, vitamin B12, and risk of
ischemic and hemorrhagic stroke: A prospective, nested
case-referent study of plasma concentrations and dietary intake.
Stroke. 36:1426–1431. 2005.
|
|
112
|
Arora K, Sequeira JM, Alarcon JM, Wasek B,
Arning E, Bottiglieri T and Quadros EV: Neuropathology of vitamin
B12 deficiency in the Cd320−/− mouse. FASEB
J. 33:2563–2573. 2019.
|
|
113
|
Wang M, Liang X, Cheng M, Yang L, Liu H,
Wang X, Sai N and Zhang X: Homocysteine enhances neural stem cell
autophagy in in vivo and in vitro model of ischemic stroke. Cell
Death Dis. 10:5612019.
|
|
114
|
Gurwara S, Ajami NJ, Jang A, Hessel FC,
Chen L, Plew S, Wang Z, Graham DY, Hair C, White DL, et al: Dietary
nutrients involved in one-carbon metabolism and colonic
mucosa-associated gut microbiome in individuals with an
endoscopically normal colon. Nutrients. 11:6132019.
|
|
115
|
Zhang Q, Wei J, Liu Z, Huang X, Sun M, Lai
W, Chen Z, Wu J, Chen Y, Guo X and Huang Q: STING signaling sensing
of DRP1-dependent mtDNA release in kupffer cells contributes to
lipopolysaccharide-induced liver injury in mice. Redox Biol.
54:1023672022.
|
|
116
|
Zhao Z, Ning J, Bao XQ, Shang M, Ma J, Li
G and Zhang D: Fecal microbiota transplantation protects
rotenone-induced Parkinson's disease mice via suppressing
inflammation mediated by the lipopolysaccharide-TLR4 signaling
pathway through the microbiota-gut-brain axis. Microbiome.
9:2262021.
|
|
117
|
Ryu KY, Lee HJ, Woo H, Kang RJ, Han KM,
Park H, Lee SM, Lee JY, Jeong YJ, Nam HW, et al: Dasatinib
regulates LPS-induced microglial and astrocytic neuroinflammatory
responses by inhibiting AKT/STAT3 signaling. J Neuroinflammation.
16:1902019.
|
|
118
|
Wang P, Zhang X, Zheng X, Gao J, Shang M,
Xu J and Liang H: Folic acid protects against hyperuricemia in
C57BL/6J mice via ameliorating gut-kidney axis dysfunction. J Agric
Food Chem. 70:15787–15803. 2022.
|
|
119
|
Darbandi ZK, Amirahmadi S, Goudarzi I,
Hosseini M and Rajabian A: Folic acid improved memory and learning
function in a rat model of neuroinflammation induced by
lipopolysaccharide. Inflammopharmacology. Aug 23–2023.Epub ahead of
print.
|
|
120
|
Yi J, Lu J, Yang A and Marsh EB:
In-hospital predictors of post-stroke depression for targeted
initiation of selective serotonin reuptake inhibitors (SSRIs). BMC
Psychiatry. 22:7222022.
|
|
121
|
Liu F, Song M, Huang X, Yi H, Chen H and
Tian F: Symptomatic plaque enhancement is associated with
early-onset post-stroke depression. J Affect Disord. 306:281–287.
2022.
|
|
122
|
Chen Z, Luo J, Li J, Kim G, Stewart A,
Urban JF Jr, Huang Y, Chen S, Wu LG, Chesler A, et al:
Interleukin-33 promotes serotonin release from enterochromaffin
cells for intestinal homeostasis. Immunity. 54:151–163.e6.
2021.
|
|
123
|
Li T, Fu B, Zhang X, Zhou Y, Yang M, Cao
M, Chen Y, Tan Y and Hu R: Overproduction of gastrointestinal 5-HT
promotes colitis-associated colorectal cancer progression via
enhancing NLRP3 inflammasome activation. Cancer Immunol Res.
9:1008–1023. 2021.
|
|
124
|
Franke M, Bieber M, Kraft P, Weber ANR,
Stoll G and Schuhmann MK: The NLRP3 inflammasome drives
inflammation in ischemia/reperfusion injury after transient middle
cerebral artery occlusion in mice. Brain Behav Immun. 92:223–233.
2021.
|
|
125
|
Zhang S, Wei D, Lv S, Wang L, An H, Shao
W, Wang Y, Huang Y, Peng D and Zhang Z: Scutellarin modulates the
microbiota-gut-brain axis and improves cognitive impairment in
APP/PS1 mice. J Alzheimers Dis. 89:955–975. 2022.
|
|
126
|
Wang X, Wang Z, Cao J, Dong Y and Chen Y:
Gut microbiota-derived metabolites mediate the neuroprotective
effect of melatonin in cognitive impairment induced by sleep
deprivation. Microbiome. 11:172023.
|
|
127
|
Zhao C, Bao L, Zhao Y, Wu K, Qiu M, Feng
L, Zhang N, Hu X and Fu Y: A fiber-enriched diet alleviates
Staphylococcus aureus-induced mastitis by activating the
HDAC3-mediated antimicrobial program in macrophages via butyrate
production in mice. PLoS Pathog. 19:e10111082023.
|
|
128
|
Round JL and Mazmanian SK: The gut
microbiota shapes intestinal immune responses during health and
disease. Nat Rev Immunol. 9:313–323. 2009.
|
|
129
|
Gelderblom M, Weymar A, Bernreuther C,
Velden J, Arunachalam P, Steinbach K, Orthey E, Arumugam TV,
Leypoldt F, Simova O, et al: Neutralization of the IL-17 axis
diminishes neutrophil invasion and protects from ischemic stroke.
Blood. 120:3793–3802. 2012.
|
|
130
|
Lim K, Hyun YM, Lambert-Emo K, Capece T,
Bae S, Miller R, Topham DJ and Kim M: Neutrophil trails guide
influenza-specific CD8+ T cells in the airways. Science.
349:aaa43522015.
|
|
131
|
Huppert J, Closhen D, Croxford A, White R,
Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann HJ, Waisman A
and Kuhlmann CR: Cellular mechanisms of IL-17-induced blood-brain
barrier disruption. FASEB J. 24:1023–1034. 2010.
|
|
132
|
Ni P, Dong H, Wang Y, Zhou Q, Xu M, Qian Y
and Sun J: IL-17A contributes to perioperative neurocognitive
disorders through blood-brain barrier disruption in aged mice. J
Neuroinflammation. 15:3322018.
|
|
133
|
Li T, Zhang YM, Han D, Hua R, Guo BN, Hu
SQ, Yan XL and Xu T: Involvement of IL-17 in secondary brain injury
after a traumatic brain injury in rats. Neuromolecular Med.
19:541–554. 2017.
|
|
134
|
Liu T, Han S, Dai Q, Zheng J, Liu C, Li S
and Li J: IL-17A-mediated excessive autophagy aggravated neuronal
ischemic injuries via Src-PP2B-mTOR pathway. Front Immunol.
10:29522019.
|
|
135
|
Benakis C, Brea D, Caballero S, Faraco G,
Moore J, Murphy M, Sita G, Racchumi G, Ling L, Pamer EG, et al:
Commensal microbiota affects ischemic stroke outcome by regulating
intestinal γδ T cells. Nat Med. 22:516–523. 2016.
|
|
136
|
Park KP, Rosell A, Foerch C, Xing C, Kim
WJ, Lee S, Opdenakker G, Furie KL and Lo EH: Plasma and brain
matrix metalloproteinase-9 after acute focal cerebral ischemia in
rats. Stroke. 40:2836–2842. 2009.
|
|
137
|
Li TT, Zhao DM, Wei YT, Li JB, Li XF, Wan
Q, Zhang X, Liu XN, Yang WC and Li WZ: Effect and mechanism of
sodium butyrate on neuronal recovery and prognosis in diabetic
stroke. J Neuroimmune Pharmacol. 18:366–382. 2023.
|
|
138
|
Yuan C, Shi L, Sun Z, Xu F, Wang C, Shan
J, Hitchens TK, Foley LM, Ye Q, Chen J, et al: Regulatory T cell
expansion promotes white matter repair after stroke. Neurobiol Dis.
179:1060632023.
|
|
139
|
Kim HJ, Wei Y, Wojtkiewicz GR, Lee JY,
Moskowitz MA and Chen JW: Reducing myeloperoxidase activity
decreases inflammation and increases cellular protection in
ischemic stroke. J Cereb Blood Flow Metab. 39:1864–1877. 2019.
|
|
140
|
Rosell A, Cuadrado E, Ortega-Aznar A,
Hernández-Guillamon M, Lo EH and Montaner J: MMP-9-positive
neutrophil infiltration is associated to blood-brain barrier
breakdown and basal lamina type IV collagen degradation during
hemorrhagic transformation after human ischemic stroke. Stroke.
39:1121–1126. 2008.
|
|
141
|
Si W, Liang H, Bugno J, Xu Q, Ding X, Yang
K, Fu Y, Weichselbaum RR, Zhao X and Wang L: Lactobacillus
rhamnosus GG induces cGAS/STING-dependent type I interferon and
improves response to immune checkpoint blockade. Gut. 71:521–533.
2022.
|
|
142
|
Dooyema SDR, Noto JM, Wroblewski LE,
Piazuelo MB, Krishna U, Suarez G, Romero-Gallo J, Delgado AG and
Peek RM: Helicobacter pylori actively suppresses innate
immune nucleic acid receptors. Gut Microbes. 14:21051022022.
|
|
143
|
Hu Q, Ren H, Li G, Wang D, Zhou Q, Wu J,
Zheng J, Huang J, Slade DA, Wu X and Ren J: STING-mediated
intestinal barrier dysfunction contributes to lethal sepsis.
EBioMedicine. 41:497–508. 2019.
|
|
144
|
Canesso MCC, Lemos L, Neves TC, Marim FM,
Castro TBR, Veloso ÉS, Queiroz CP, Ahn J, Santiago HC, Martins FS,
et al: The cytosolic sensor STING is required for intestinal
homeostasis and control of inflammation. Mucosal Immunol.
11:820–834. 2018.
|
|
145
|
Yang K, Hou Y, Zhang Y, Liang H, Sharma A,
Zheng W, Wang L, Torres R, Tatebe K, Chmura SJ, et al: Suppression
of local type I interferon by gut microbiota-derived butyrate
impairs antitumor effects of ionizing radiation. J Exp Med.
218:e202019152021.
|
|
146
|
Wang H, Chen S, Zhang Y, Xu H and Sun H:
Electroacupuncture ameliorates neuronal injury by
Pink1/Parkin-mediated mitophagy clearance in cerebral
ischemia-reperfusion. Nitric Oxide. 91:23–34. 2019.
|
|
147
|
Liu Z, Wang M, Wang X, Bu Q, Wang Q, Su W,
Li L, Zhou H and Lu L: XBP1 deficiency promotes hepatocyte
pyroptosis by impairing mitophagy to activate mtDNA-cGAS-STING
signaling in macrophages during acute liver injury. Redox Biol.
52:1023052022.
|
|
148
|
Sliter DA, Martinez J, Hao L, Chen X, Sun
N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, et al: Parkin
and PINK1 mitigate STING-induced inflammation. Nature. 561:258–262.
2018.
|
|
149
|
Gao X, Yin Y, Liu S, Dong K, Wang J and
Guo C: Fucoidan-proanthocyanidins nanoparticles protect against
cisplatin-induced acute kidney injury by activating mitophagy and
inhibiting mtDNA-cGAS/STING signaling pathway. Int J Biol Macromol.
245:1255412023.
|
|
150
|
Tian T, Cao L, He C, Ye Q, Liang R, You W,
Zhang H, Wu J, Ye J, Tannous BA and Gao J: Targeted delivery of
neural progenitor cell-derived extracellular vesicles for
anti-inflammation after cerebral ischemia. Theranostics.
11:6507–6521. 2021.
|
|
151
|
Dong X, Wang L, Song G, Cai X, Wang W,
Chen J and Wang G: Physcion protects rats against cerebral
ischemia-reperfusion injury via inhibition of TLR4/NF-kB signaling
pathway. Drug Des Devel Ther. 15:277–287. 2021.
|
|
152
|
Hou Y, Wang K, Wan W, Cheng Y, Pu X and Ye
X: Resveratrol provides neuroprotection by regulating the
JAK2/STAT3/PI3K/AKT/mTOR pathway after stroke in rats. Genes Dis.
5:245–255. 2018.
|
|
153
|
Fu C, Wu Y, Liu S, Luo C, Lu Y, Liu M,
Wang L, Zhang Y and Liu X: Rehmannioside A improves cognitive
impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2
and SLC7A11/GPX4 signaling pathway after ischemia. J
Ethnopharmacol. 289:1150212022.
|
|
154
|
Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W
and Tang Q: STING-IRF3 contributes to lipopolysaccharide-induced
cardiac dysfunction, inflammation, apoptosis and pyroptosis by
activating NLRP3. Redox Biol. 24:1012152019.
|
|
155
|
Xie X, Wu X, Zhao D, Liu Y, Du Q, Li Y, Xu
Y, Li Y, Qiu Y and Yang Y: Fluvoxamine alleviates bleomycin-induced
lung fibrosis via regulating the cGAS-STING pathway. Pharmacol Res.
187:1065772023.
|
|
156
|
Sun J, Zhou YQ, Xu BY, Li JY, Zhang LQ, Li
DY, Zhang S, Wu JY, Gao SJ, Ye DW and Mei W:
STING/NF-κB/IL-6-mediated inflammation in microglia contributes to
spared nerve injury (SNI)-induced pain initiation. J Neuroimmune
Pharmacol. 17:453–469. 2022.
|
|
157
|
Zhou Q, Zhang Y, Lu L, Zhang H, Zhao C, Pu
Y and Yin L: Copper induces microglia-mediated neuroinflammation
through ROS/NF-κB pathway and mitophagy disorder. Food Chem
Toxicol. 168:1133692022.
|
|
158
|
Osanai T, Fujiwara N, Sasaki S, Metoki N,
Saitoh G, Tomita H, Nishimura T, Shibutani S, Yokoyama H, Konta Y,
et al: Novel pro-atherogenic molecule coupling factor 6 is elevated
in patients with stroke: A possible linkage to homocysteine. Ann
Med. 42:79–86. 2010.
|
|
159
|
Somensi N, Rabelo TK, Guimarães AG,
Quintans-Junior LJ, de Souza Araújo AA, Moreira JCF and Gelain DP:
Carvacrol suppresses LPS-induced pro-inflammatory activation in RAW
264.7 macrophages through ERK1/2 and NF-kB pathway. Int
Immunopharmacol. 75:1057432019.
|
|
160
|
Tian M, Yang M, Li Z, Wang Y, Chen W, Yang
L, Li Y and Yuan H: Fluoxetine suppresses inflammatory reaction in
microglia under OGD/R challenge via modulation of NF-κB signaling.
Biosci Rep. 39:BSR201815842019.
|
|
161
|
Rahimian R, Fakhfouri G, Ejtemaei Mehr S,
Ghia JE, Genazzani AA, Payandemehr B, Dehpour AR, Mousavizadeh K
and Lim D: Tropisetron attenuates amyloid-beta-induced inflammatory
and apoptotic responses in rats. Eur J Clin Invest. 43:1039–1051.
2013.
|
|
162
|
Feng C, Wan H, Zhang Y, Yu L, Shao C, He
Y, Wan H and Jin W: Neuroprotective effect of danhong injection on
cerebral ischemia-reperfusion injury in rats by activation of the
PI3K-Akt pathway. Front Pharmacol. 11:2982020.
|
|
163
|
Li Y, Xiang L, Wang C, Song Y, Miao J and
Miao M: Protection against acute cerebral ischemia/reperfusion
injury by Leonuri Herba Total Alkali via modulation of
BDNF-TrKB-PI3K/Akt signaling pathway in rats. Biomed Pharmacother.
133:1110212021.
|
|
164
|
Pan B, Sun J, Liu Z, Wang L, Huo H, Zhao
Y, Tu P, Xiao W, Zheng J and Li J: Longxuetongluo Capsule protects
against cerebral ischemia/reperfusion injury through endoplasmic
reticulum stress and MAPK-mediated mechanisms. J Adv Res.
33:215–225. 2021.
|
|
165
|
Tsai YT, Huang HC, Kao ST, Chang TT and
Cheng CY: Neuroprotective effects of alpinia oxyphylla Miq against
mitochondria-related apoptosis by the interactions between
upregulated p38 MAPK signaling and downregulated JNK signaling in
the subacute phase of cerebral ischemia-reperfusion in rats. Am J
Chin Med. 50:2057–2083. 2022.
|
|
166
|
Xu D, Kong T, Shao Z, Liu M, Zhang R,
Zhang S, Kong Q, Chen J, Cheng B and Wang C: Orexin-A alleviates
astrocytic apoptosis and inflammation via inhibiting OX1R-mediated
NF-κB and MAPK signaling pathways in cerebral ischemia/reperfusion
injury. Biochim Biophys Acta Mol Basis Dis. 1867:1662302021.
|
|
167
|
Chen B, Cao P, Guo X, Yin M, Li X, Jiang
L, Shao J, Chen X, Jiang C, Tao L, et al: Maraviroc, an inhibitor
of chemokine receptor type 5, alleviates neuroinflammatory response
after cerebral Ischemia/reperfusion injury via regulating
MAPK/NF-κB signaling. Int Immunopharmacol. 108:1087552022.
|
|
168
|
Xian M, Cai J, Zheng K, Liu Q, Liu Y, Lin
H, Liang S and Wang S: Aloe-emodin prevents nerve injury and
neuroinflammation caused by ischemic stroke via the PI3K/AKT/mTOR
and NF-κB pathway. Food Funct. 12:8056–8067. 2021.
|
|
169
|
Kikuchi DS, Campos ACP, Qu H, Forrester
SJ, Pagano RL, Lassègue B, Sadikot RT, Griendling KK and Hernandes
MS: Poldip2 mediates blood-brain barrier disruption in a model of
sepsis-associated encephalopathy. J Neuroinflammation.
16:2412019.
|
|
170
|
Alhadidi Q and Shah ZA: Cofilin mediates
LPS-induced microglial cell activation and associated neurotoxicity
through activation of NF-κB and JAK-STAT pathway. Mol Neurobiol.
55:1676–1691. 2018.
|
|
171
|
Cui Y, Wu J, Jung SC, Kim GO, Kyeong Ko R,
Lee HJ, Yoo ES, Kang HK, Suk K and Eun SY: Neuroprotective effect
of methyl lucidone against microglia-mediated neurotoxicity. Eur J
Pharmacol. 690:4–12. 2012.
|
|
172
|
Li M, Wang S, Zhang C, Chi C, Liu R, Wang
T and Fu F: Escin alleviates stress-induced intestinal dysfunction
to protect brain injury by regulating the gut-brain axis in
ischemic stroke rats. Int Immunopharmacol. 115:1096592023.
|
|
173
|
Yang H, Chen Y, Yu L and Xu Y:
Esculentoside A exerts anti-inflammatory activity in microglial
cells. Int Immunopharmacol. 51:148–157. 2017.
|
|
174
|
Ge JW, Deng SJ, Xue ZW, Liu PY, Yu LJ, Li
JN, Xia SN, Gu Y, Bao XY, Lan Z, et al: Imperatorin inhibits
mitogen-activated protein kinase and nuclear factor kappa-B
signaling pathways and alleviates neuroinflammation in ischemic
stroke. CNS Neurosci Ther. 28:116–125. 2022.
|
|
175
|
Lei B, Mace B, Dawson HN, Warner DS,
Laskowitz DT and James ML: Anti-inflammatory effects of
progesterone in lipopolysaccharide-stimulated BV-2 microglia. PLoS
One. 9:e1039692014.
|
|
176
|
Jung YS, Park JH, Kim H, Kim SY, Hwang JY,
Hong KW, Bae SS, Choi BT, Lee SW and Shin HK: Probucol inhibits
LPS-induced microglia activation and ameliorates brain ischemic
injury in normal and hyperlipidemic mice. Acta Pharmacol Sin.
37:1031–1044. 2016.
|
|
177
|
Lv Y, Qian Y, Ou-Yang A and Fu L:
Hydroxysafflor yellow A attenuates neuron damage by suppressing the
lipopolysaccharide-Induced TLR4 pathway in activated microglial
cells. Cell Mol Neurobiol. 36:1241–1256. 2016.
|
|
178
|
Kim DC, Quang TH, Oh H and Kim YC:
Steppogenin isolated from cudrania tricuspidata shows
antineuroinflammatory effects via NF-κB and MAPK pathways in
LPS-stimulated BV2 and primary rat microglial cells. Molecules.
22:21302017.
|
|
179
|
Zou CG, Zhao YS, Gao SY, Li SD, Cao XZ,
Zhang M and Zhang KQ: Homocysteine promotes proliferation and
activation of microglia. Neurobiol Aging. 31:2069–2079. 2010.
|
|
180
|
Jindal A, Rajagopal S, Winter L, Miller
JW, Jacobsen DW, Brigman J, Allan AM, Paul S and Poddar R:
Hyperhomocysteinemia leads to exacerbation of ischemic brain
damage: Role of GluN2A NMDA receptors. Neurobiol Dis. 127:287–302.
2019.
|
|
181
|
Guo HH, Shen HR, Tang MZ, Sheng N, Ding X,
Lin Y, Zhang JL, Jiang JD, Gao TL, Wang LL and Han YX:
Microbiota-derived short-chain fatty acids mediate the effects of
dengzhan shengmai in ameliorating cerebral ischemia via the
gut-brain axis. J Ethnopharmacol. 306:1161582023.
|
|
182
|
Zhou Z, Xu N, Matei N, McBride DW, Ding Y,
Liang H, Tang J and Zhang JH: Sodium butyrate attenuated neuronal
apoptosis via GPR41/Gβγ/PI3K/Akt pathway after MCAO in rats. J
Cereb Blood Flow Metab. 41:267–281. 2021.
|
|
183
|
Xu S, Wang J, Jiang J, Song J, Zhu W,
Zhang F, Shao M, Xu H, Ma X and Lyu F: TLR4 promotes microglial
pyroptosis via lncRNA-F630028O10Rik by activating PI3K/AKT pathway
after spinal cord injury. Cell Death Dis. 11:6932020.
|