1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is a
clinicopathological syndrome, which is characterized by excessive
fat deposition in hepatic cells (1,2),
affecting over 1.7 billion individuals globally and representing a
significant public health concern (3). Lifestyle changes have led to NAFLD
becoming the predominant chronic liver disease in China (4). The advanced stages of NAFLD,
particularly non-alcoholic steatohepatitis (NASH), are associated
with escalating mortality rates (4,5).
Consequently, the condition has garnered increasing global research
attention, emphasizing the necessity of identifying preventative
and therapeutic targets.
The pathogenesis of NAFLD is multifaceted and not
entirely elucidated, encompassing metabolic, environmental, genetic
and microbial factors (6-9).
The prevailing conceptual framework posits NAFLD as a 'multiple
hits' phenomenon (10). Central
to NAFLD's etiology is elevated circulating free fatty acids (FFAs)
and insulin resistance, culminating in excessive triglyceride (TG)
accumulation in hepatocytes. FFAs are also implicated in
lipotoxicity, oxidative stress and inflammation, contributing to
progressive hepatic injury (10,11). Furthermore, recent studies
indicated that a confluence of mechanisms, particularly in
genetically susceptible individuals, plays a crucial role in
NAFLD's onset and progression (12,13). Consequently, devising effective
preventive and therapeutic approaches for NAFLD represents a
pressing scientific challenge.
The pathophysiology of NAFLD is complex, marked by
significant individual heterogeneity. Currently, no efficacious
pharmacological treatments exist. Current therapeutic approaches,
including weight management, intestinal glucagon-like peptide-1
agonists, sodium-glucose cotransporter inhibitors, peroxisome
proliferator-activated receptor (PPAR) agonists, farnesoid
derivative X receptor-bile acid axis modulators, hormonal
therapies, lipid synthesis inhibitors, antioxidants, targeted
apoptosis-targeting agents, anti-inflammatory drugs, microbiome
modulators and anti-fibrotic treatment and combination therapies
(14-17), have demonstrated limited clinical
efficacy and are often accompanied by adverse effects, such as
elevated serum transaminases, edema, gastrointestinal discomfort
and increased liver burden (18,19). Given the complexity and
interconnectedness of NAFLD's pathogenic factors, identifying
effective therapeutic targets and pathways remains a formidable
task.
Phospholipids (PLs) are the principal components of
biofilms, and their biophysical membrane properties are largely
determined by the composition of their fatty acyl chains, which
significantly influences biological processes. It has been
established that lyso-phosphatidyl-choline acyltransferase (LPCAT)
plays a crucial role in catalyzing the integration of fatty acyl
chains into the sn-2 site of phosphatidyl-choline (PC), profoundly
impacting pathophysiology (20,21). Increasing evidence suggests that
alterations in LPCAT activity are implicated in various diseases,
including NAFLD, hepatitis C, atherosclerosis and cancers (22-24). Specifically, LPCAT3 is key in
maintaining lipid homeostasis by regulating hepatic lipogenesis,
intestinal lipid absorption and lipoprotein secretion (21). The liver X receptor (LXR) is
instrumental in regulating metabolism of cholesterol, FAs and PLs,
thereby playing a significant role in lipid homeostasis (25). LXR provides a mechanism to
protect biofilms from lipid stress by altering PL composition in
response to cellular sterol level fluctuations. Concurrently, LXR
activation enhances LPCAT3 expression of LPCAT3 on cell membranes
and increases the abundance of polyunsaturated PLs. This activation
strategy can ameliorate saturated FFA-induced effects in
vitro or endoplasmic reticulum stress (ERS) in vivo,
arising from lipid accumulation in hepatic cells (24,26). Pharmacological regulation
focusing on LXR, LPCAT and membrane PL may offer new avenues in
NAFLD treatment. The present review mainly discussed the potential
role of the LXR-LPCAT3 signaling pathway in NAFLD, aiming to
identify novel targets and pathways that could facilitate the
development of effective therapeutics.
2. Main metabolic pathways of PLs
Characteristics of PLs
PLs are composed of two hydrophobic fatty acyl
chains and one hydrophilic head group. Their bilayer membrane
structure plays a critical role in compartmentalizing intracellular
contents and facilitating the formation of subcellular organelles,
essential for various cellular processes (27,28). In signal transduction, PLs serve
as matrices for bioactive molecules such as eicosanoids,
lyso-phosphatidyl-choline (LPC), lysophosphatidic acid and
diacylglycerol (DAG) (29,30). Glycerophospholipids, including
PC, phosphatidylethanolamine and phosphatidylserine, constitute the
main structure of mammalian cell membranes. PC, particularly
abundant in mammalian cell membranes and organelles, accounts for
~40-50% of total PLs (27). In
the liver, PLs primarily derive from DAG and
phosphatidylethanolamine (Fig.
1).
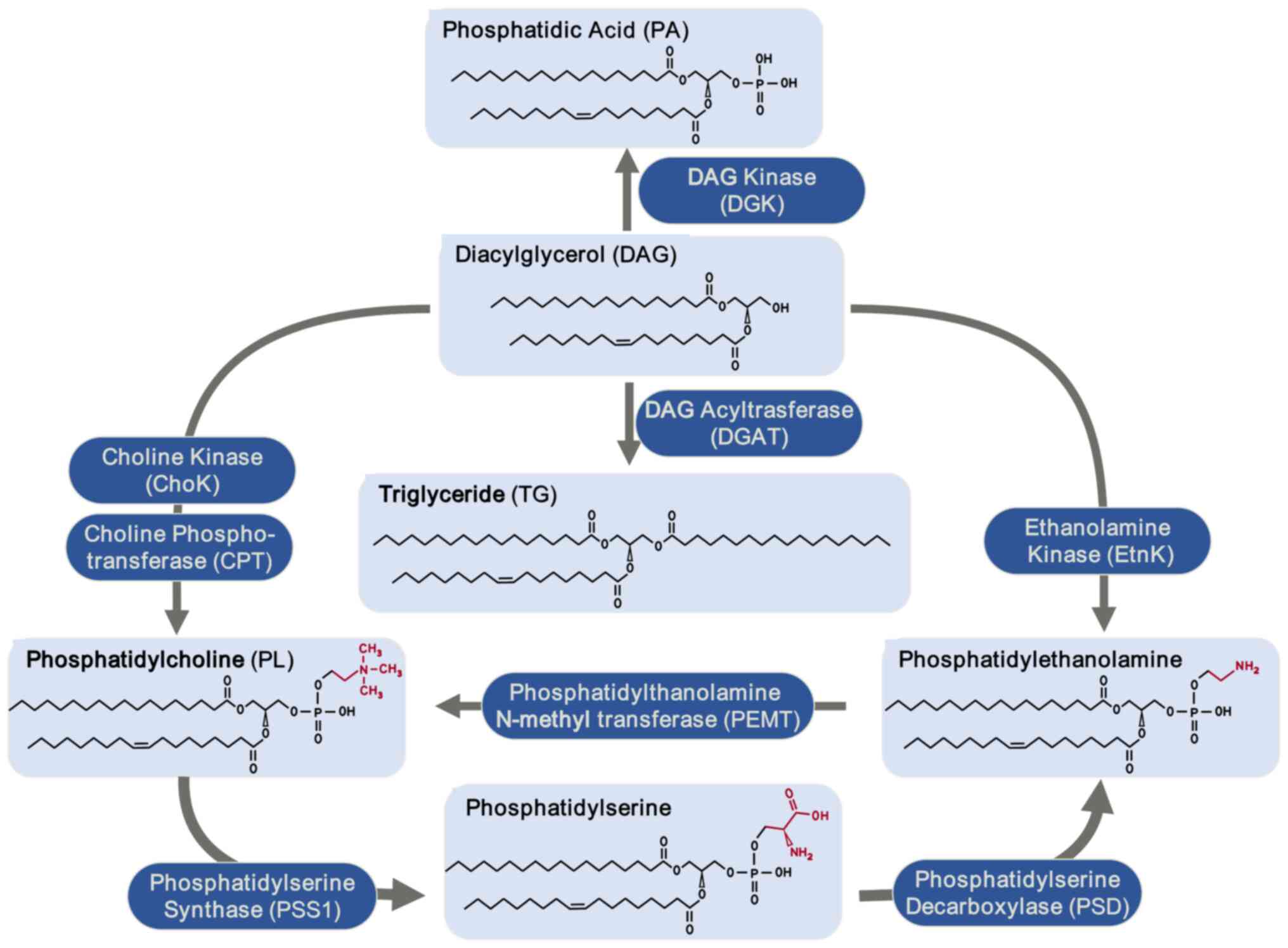 | Figure 1Hepatic phospholipid biosynthesis
pathways. DAG is converted into PA by DGK. TG formation is
catalyzed by DGAT. Phosphatidylethanolamine is synthesized under
the action of EtnK and phospholipids are produced through the
activity of ChoK and CTP: phosphocholine cytidylyltransferase
(CPT). PL forms phosphatidylserine under the catalysis of PSS1,
which in turn is converted into phosphatidylethanolamine by PSD.
Additionally, phosphatidylethanolamine can be converted back into
PL via PEMT. DAG, diacylglycerol; PA, phosphatidic acid; DGK, DAG
kinase; TG, triglyceride; DGAK, DAG acyltrasferase; EtnK,
ethanolamine kinase; ChoK, choline kinase; CPT, choline
phosphotransferase; PL, phosphatidylcholine; PSS1,
phosphatidylserine synthase 1; PSD, phosphatidylserine
decarboxylase; PEMT, phosphatidylethanolamine N-methyl
transferase. |
Metabolic processing of PC
Biosynthesis of PC (Kennedy
pathway)
The Kennedy pathway, the primary route for PC
synthesis, operates predominantly via the CDP choline pathway
(31). This pathway involves
three enzymatic reactions: Choline kinase catalyzes the conversion
of choline to phosphocholine; CTP:phosphocholine
cytidylyltransferase (CCT) then catalyzes the formation of cytidine
diphosphate (CDP) choline, which subsequently forms PC under the
catalysis of 1,2-DAG choline-phosphotransferase (32) (Fig. 2, left).
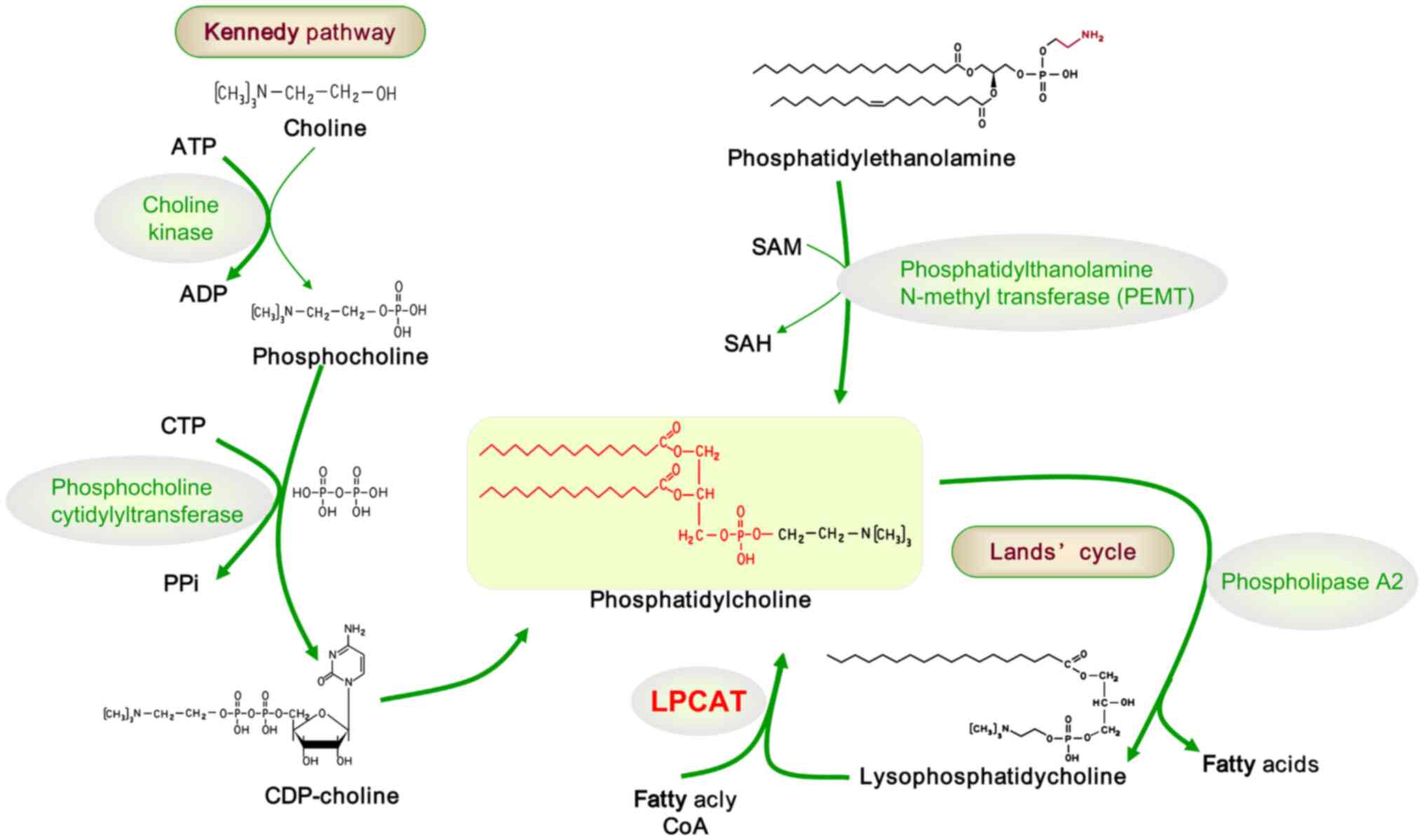 | Figure 2Pathways of phosphatidylcholine
metabolism: Kennedy pathway and Lands' cycle. In the Kennedy
pathway, choline kinase catalyzes the conversion of choline into
PC. PC is then converted to CDP-choline by phosphocholine
cytidylyltransferase, and subsequently, CDP-choline forms PC under
the action of 1,2-diacylglycerol choline-phosphotransferase.
Phosphatidylethanolamine synthesizes PC through PEMT. In
phospholipids, saturated and monounsaturated fatty acids typically
esterify at the sn-1 position, while polyunsaturated fatty acids
are esterified at the sn-2 position. The asymmetric distribution of
fatty acids at sn-1 and sn-2 is established through a
deacylation-reacylation process known as the Lands' cycle. The
deacylation step, catalyzed by phospholipase A2, removes saturated
or monounsaturated fatty acids from the sn-2 position of PCs. The
reacylation step, facilitated by LPCAT, incorporates
polyunsaturated fatty acids at the sn-2 position of PC. PC,
phosphocholine; PEMT, phosphatidylethanolamine N-methyl
transferase; LPCAT, lyso-phosphatidyl-choline acyltransferase; ATP,
adenosine-triphosphate; ADP, adenosine diphosphate; CTP, cytidine
triphosphate; PPi, pyrophosphoric acid; CDP, cytidine diphosphate;
SAM, S-adenosyl methionine; SAH, S-Adenosyl-L-homocysteine; CoA,
coenzyme A. |
Remodeling of PC (Lands' cycle)
Beyond the Kennedy pathway, the liver synthesizes PC
through an alternative route, largely via the methylation of
phosphatidylethanolamine by phosphatidylethanolamine
N-methyltransferase (PEMT), contributing to ~30% of hepatic PC
synthesis (33). Fatty acyl
chains in PLs exhibit not only diversity in structure but also
asymmetric distribution on each side of the molecule. In mammalian
cells, the sn-1 position typically hosts saturated and
monounsaturated FAs, while the sn-2 position is reserved for
polyunsaturated FAs. This asymmetry arises from the
deacylation-reacylation process known as Lands' cycle (33). Calcium-independent A2
[phospholipase A2 (iPLA2] mediates the deacylation step, removing
saturated or monosaturated FAs from the sn-2 position of PCs. The
subsequent reacylation step, characterized by the addition of
polyunsaturated FAs at the sn-2 position, is regulated by LPCAT
(24) (Fig. 2, right).
3. The integral role of LXRs in PL
metabolism
LXRs are categorized into two subtypes: LXR α and
LXR β. LXR α exhibits high expression levels in the liver,
particularly in Kupffer cells, and is also present in the adrenal
gland, small intestine, adipose tissue, lungs and kidneys.
Conversely, LXR β is expressed ubiquitously throughout the body
(34-37). LXRs feature a highly conserved
structure, including a hydrophobic ligand-binding domain, a DNA
binding domain, a ligand-independent N-terminal domain and a
ligand-dependent C-terminal domain. LXRs form a heterodimer with
the retinoic acid receptor (RXR), binding to specific nucleotide
sequences in the LXR response element to regulate the transcription
of the target genes (38). In
the nucleus, LXRs remain inactive and bound to DNA, forming
complexes with corepressors such as the retinoic acid silencing
regulator, thyroid hormone receptor, and nuclear receptor
co-repressor proteins (38-40). These complexes can inhibit the
transcriptional activity of LXR target genes in the absence of
ligands. Ligand binding to the LXR/RXR heterodimer induces a
conformational change in LXR. This change results in corepressor
detachment and coactivator recruitment, thereby activating LXR and
inducing expression of target genes (41-44).
Function of LXRs in lipid metabolism
The pivotal role of LXRs in lipid metabolism
regulation is multifaceted: LXRs, as ligand-activated nuclear
receptors, are central to lipid homeostasis (45); LXRs govern cellular and systemic
cholesterol homeostasis through the regulation of cholesterol
assimilation, cellular uptake and excretion, reverse transport and
biosynthesis in various tissues and cells (39,46); LXRs enhance adipogenesis by
regulating sterol response element-binding protein-1c (SREBP-1c)
and its downstream genes in the hepatic tissues (47,48); LXRs also regulate the expression
of membrane PL components through the induction of LPCAT3 (49) (Fig. 3).
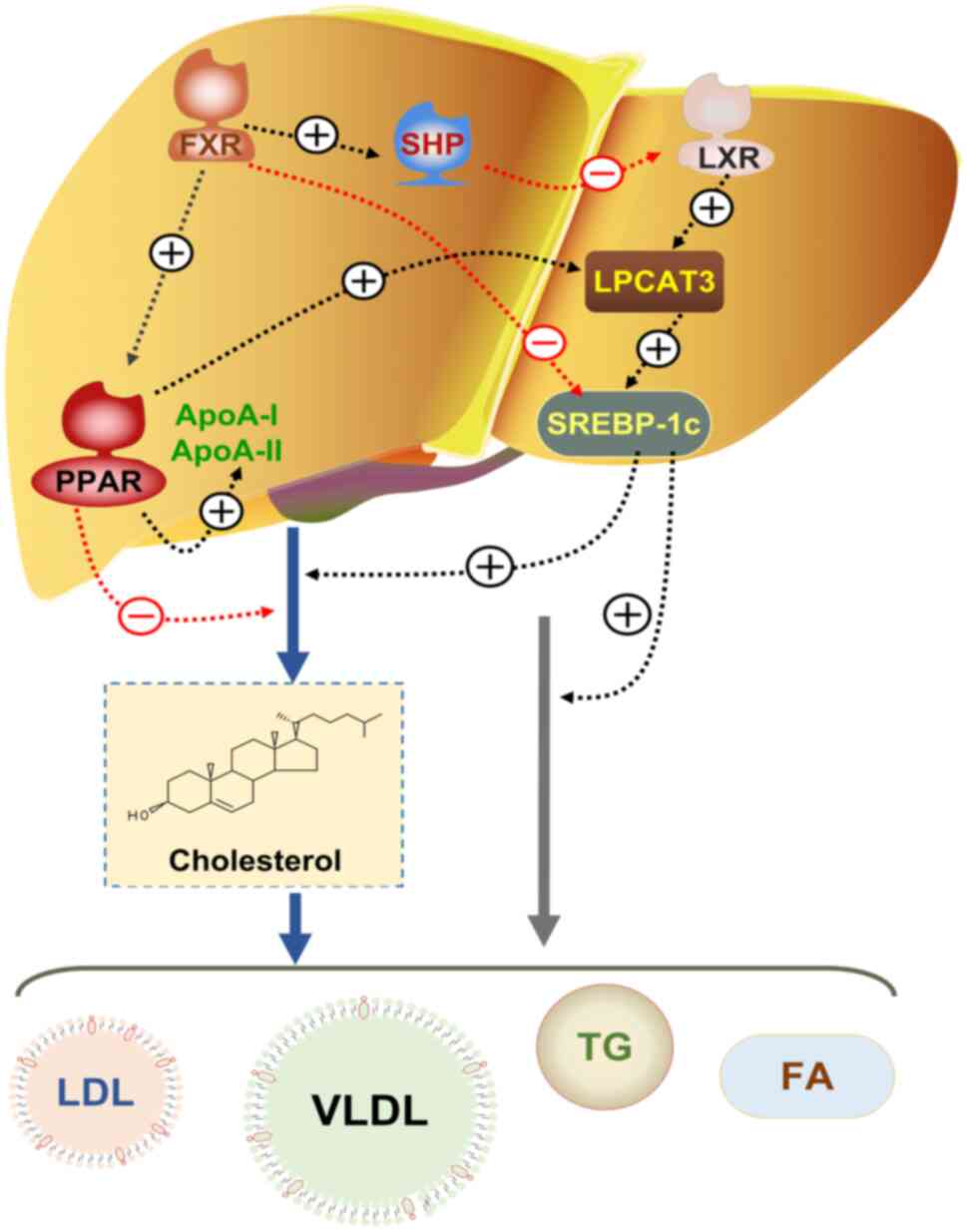 | Figure 3LPCAT3-dependent hepatic lipid
metabolism pathway. In the liver, the FXR regulates cholesterol
metabolism by upregulating the expression of PPAR. PPAR directly
influences LPCAT3, thereby promoting the expression of SREBP-1c to
regulate lipid metabolism. FXR also enhances the expression of SHP,
leading to the downregulation of LXR. LXR, an upstream regulator of
LPCAT3, upregulates LPCAT3 expression, which subsequently elevates
SREBP-1c expression and promotes lipid metabolism. Additionally,
FXR directly inhibits SREBP-1c expression, further influencing
lipid metabolism. LPCAT3, lyso-phosphatidyl-choline acyltransferase
3; FXR, farnesoid X receptor; PPAR, peroxisome
proliferation-activated receptor; SREBP-1c, sterol regulatory
element-binding protein-1c; SHP, small heterodimer partner; LXR,
liver X receptor alpha; FA, fatty acid; LDL, low density
lipoprotein; TG, triglyceride; VLDL, very LDL. |
LXR-dependent PL remodeling in the
liver
Activation of LXRs facilitates very low-density
lipoprotein (VLDL) secretion in mouse livers by regulating SREBF-1c
and its downstream lipogenesis-related genes, and by enhancing the
expression of phospholipid transfer proteins (PLTP). PLTP
contributes to VLDL maturation by incorporating PLs into nascent
VLDL, expanding lipoprotein particles (50). Given that PLs constitute a major
component of lipoprotein particles, their efficiency influences
lipoprotein production (51).
Studies have demonstrated that LPCAT3 and PL remodeling
significantly affect lipoprotein production, underscoring the
importance of PL quantity and composition in lipoprotein secretion
(52-54). Rong et al (26) demonstrated that LXRs regulate the
membrane PL composition by activating LPCAT3, an enzyme that
catalyzes the incorporation of polyunsaturated FAs at sn-2 position
of lyso-phospholipids.
The induction of LPCAT3 expression in hepatic
tissues is a critical aspect of LXR agonists' pharmacological
action. The mice with liver-specific LPCAT3 gene knockout
(LPCAT3-LKO) exhibited reduced VLDL secretion compared with
controls following LXR agonist treatment, indicating that LXRs'
ability to enhance hepatic VLDL production is contingent upon
LPCAT3 activity (52). LPCAT3
mediates the integration of polyunsaturated FAs into PLs, which is
essential for the processing and lipogenesis of SREBP-1c. As liver
lipogenesis largely depends on interaction of LXRs with SREBP-1c,
LPCAT3 activity also plays a role in hepatic lipogenesis (55). Notably, obese mice display
elevated levels of polyunsaturated PLs in the endoplasmic
reticulum, a consequence of increased LPCAT3 activity. Reducing
expression of LPCAT3 in obese mice can attenuate the activation of
the SREBP-1c pathway, thereby decelerating fat production and
accumulation and delaying the progression of fatty liver disease
(56).
4. Regulation of polyunsaturated fatty acid
PLs by LPCAT3
The LPCAT family
LPCATs are categorized based on amino acid sequence
variations into two distinct groups. LPCAT1 and LPCAT2,
characterized by four conserved domain localization sequences known
as LPA acyltransferase motifs 1-4 and an ER localization, are part
of the acyl-glycerol phosphate acyltransferase family (57). LPCAT3 and LPCAT4, classified
under the membrane-bound O-acyltransferase (MBOAT) family as MBOAT5
and MBOAT2 respectively, lack the LPA acyltransferase moiety but
contain the MBOAT moiety (57).
Each LPCAT exhibits specific acyl-CoA substrate
preferences: LPCAT1 favors 16:0-acyl-CoA for palmitoyl PC
synthesis, whereas LPCAT2 utilizes acetyl-CoA or 20:4-acyl-CoA.
LPCAT3 and LPCAT4 preferentially use polyunsaturated fat-acyl-CoA
substrates, such as 18:2-acyl-CoA or 20:4-acyl-CoA and
18:1-acyl-CoA, respectively (57-60). Consequently, tissue-selective
remodeling of membrane PC species may be optimized by leveraging
the distinct substrate preferences of specific LPCAT isoforms in
various tissues.
Impact of LPCAT3 on lipid metabolism via
PLs composition regulation
LPCAT3, a direct transcriptional target of
lipid-activated nuclear receptors such as LXR, PPAR-α and PPAR-γ,
plays a crucial role in lipid homeostasis (26,61) (Fig. 3). Highly expressed in liver,
intestinal and adipose tissues, LPCAT3 accounts for >90% of LPC
acyltransferase activity, making it the most abundant LPCAT in the
liver tissue (53). PC, the
primary PL component of all plasma lipoproteins, and its FA chain
synthesis are key regulators of lipoprotein secretion and lipid
metabolism in hepatic and intestinal tissues (62). The diversity and saturation of
fatty acyl groups in membrane PLs, attributable to the differences
in double bond and single bonds, determine the biophysical
properties of the cell membranes, including its fluidity, curvature
and subdomain structure. These properties, in turn, influence
biological processes involving membranes, such as signaling and
protein transport (63). The
important role of LPCATs in lipid metabolism and homeostasis has
been corroborated through genetic model studies, and the regulation
of a variety of PC species by LPCATs in different cells and
tissues, thereby influencing membrane PL composition and affecting
biological functions (20-22).
Role of LPCAT3 in regulating VLDL
secretion through membrane polyunsaturated PLs composition
Studies have indicated that inducing acute
LPCAT3-LKO in mice via adenovirus elevates plasma TG levels while
concurrently reducing liver TG content (26,64). This phenomenon may be attributed
to an increase in LPC, while enhances the expression of microsomal
TG transfer protein (MTTP) and apolipoprotein B (ApoB), thereby
facilitating the assembly and secretion of VLDL (64). By contrast, mice with acute
overexpression of human LPCAT3 exhibited reduced VLDL secretion and
liver TG levels, potentially due to low LPC levels no longer
inhibiting FA β-oxidation in hepatic cells (65). These mice also presented elevated
plasma levels of high-density lipoprotein (HDL) levels, enriched
with protective ApoE. However, contradictory studies suggested that
LPCAT3-deficient mice display decreased plasma TG levels, increased
VLDL fat deposition and decreased secretion, leading to liver
steatosis. This outcome might relate to the role of LPCAT3 activity
and polyunsaturated PLs in influencing membrane surface fluidity
and curvature, thus diminishing TG mobilization to VLDL (52,53).
The long-term effects of LPCAT3 activation or
overexpression remain unclear. Permanent LPCAT3 deletion in mouse
hepatocytes resulted in a metabolic phenotype distinct from the
acute knockout. Unlike acute LPCAT3 knockout, chronic LPCAT3
knockout mice did not exhibit significantly higher LPC
accumulation, possibly due to the redirection of LPC into the
biosynthesis of both saturated and monounsaturated PCs (52,53).
A previous study revealed that hepatic deletion of
genes involved in de novo PC synthesis in the liver (such as
PEMT and CT-α) impairs VLDL secretion, as evidenced by reduced
plasma ApoB protein levels (66). However, LPCAT3-deficient mice
maintained stable plasma ApoB levels, suggesting preserved ApoB
secretion functionality (52).
Decreased plasma VLDL particles and TG-rich ApoB particles in the
Golgi apparatus of LPCAT3-deficient mice underscore LPCAT3's
influence on VLDL assembly: TG is assembled, low-fat ApoB particles
are incorporated, and mature VLDL is produced. Mechanistically,
these phenotypes are linked to diminished ER membrane mobility and
altered curvature, stemming from the loss of linoleic acid and
arachidonic acid PLs. The presence of these polyunsaturated PLs
enhance membrane fluidity and dynamics (67,68). Proteomic studies have elucidated
the composition of VLDL transport vesicles containing LPCAT3,
indicating that LPCAT3 and the nascent VLDL particles originate
from the ER and transit to the Golgi apparatus (52). Further investigations reveal that
LPCAT3-mediated accumulation of polyunsaturated PLs in membrane,
particularly at high local concentrations, facilitates efficient TG
transfer (52,53).
Therefore, LPCAT3's modulation of linoleic acid and
arachidonic acid PL composition in membranes plays a pivotal role
in creating an environment conducive to lipid transport and
substantial fat deposition during VLDL assembly.
Influence of LPCAT3 on SREBP-1c
production via polyunsaturated PLs levels in the ER membrane
LXR activation significantly stimulates hepatic
lipogenesis, primarily through the upregulation of genes such as
SREBP-1c, fatty acid synthase, and stearoyl-CoA desaturase 1
(SCD-1) (25,48) (Fig. 3). Certain studies have reported
that LXR activation promotes the post-translational processing of
SREBP-1c by inducing LPCAT3 expression (50,55). The integration of polyunsaturated
FAs into PLs in the ER membrane, mediated by LPCAT3, enhances
SREBP-1c expression, thereby promoting adipogenesis. On the
contrary, hepatocytes deficient in LPCAT3 exhibit reduced
polyunsaturated PL levels in the ER, diminished nuclear SREBP-1c
levels, and a muted adipogenic response to LXR agonist treatment
(47,55). The specific role of ER membrane
PL components in regulating the SREBP-1c pathway remains to be
fully elucidated. Notably, the impact of PL metabolism on the
SREBP1 and SREBP2 pathways varies by the tissue specificity. In the
liver, LPCAT3 does not selectively influence SREBP-1c due to the
absence of corresponding target genes, while in the intestine,
SREBP2 is significantly affected due to the presence of
corresponding target genes (20,47,50,63). These findings suggested that
membrane PL remodeling differentially modulates SREBP maturation in
response to cellular lipids. The involvement of LPCAT3 and membrane
lipid acyl chain composition in SREBP-1c processing is considered
to hinge on the SREBP cleavage-activating protein, potentially
influencing the transport of SREBP-1c from the ER to the Golgi
matrix. The total cellular PL level also affects SREBP-1 activity
(55). Inhibiting enzymes in the
PL de novo biosynthesis pathway reduces total cellular PL
levels, activating the SREBP1 process and leading to the
mislocalization of sphingosine 1-phosphate and sphingosine
2-phosphate, thereby disrupting CopII-dependent ER-to-Golgi
transport (69).
Under both physiological (such as feeding) and
pathological (such as obesity) conditions, LPCAT3 is known to
enhance SREBP-1c activity and lipogenesis (69). Mass spectrometry analysis
revealed a selective increase in polyunsaturated PLs in the ER of
both wild-type and obese mice during feeding, a change dependent on
LPCAT3 activity. Inhibition of LPCAT3 activity in obese mice using
adenoviral LPCAT3shRNA has been demonstrated to reduce SREBP-1c
processing, slow down fat production and improve fatty liver
symptoms (69). These findings
suggested that LPCAT3 could be a potential target for NAFLD
treatment.
Impact of LPCAT3 on lipogenesis through
polyunsaturated PL levels
LPCAT3 is the most highly expressed LPCAT in adipose
tissue (21). Eto et al
(70) demonstrated that LPCAT3
may play a role in adipogenesis, with its expression and activity
markedly increasing during adipocyte differentiation, as observed
in vitro in studies using C3H10T1/2 cells. These mesenchymal
stem cells have the capacity to differentiate into adipocyte-like
cells. Correlating with gene expression changes, both PC and PE, as
well as arachidonic PLs, were found to increase in adipocytes.
Arachidonic acid in PLs, a substrate for eicosanoid biosynthesis,
is considered as an endogenous ligand for PPAR-γ. It is posited
that the enhancement of the endogenous lipid ligand PPAR-γ may
result from the incorporation of arachidonic acid into PLs, a
process mediated by LPCAT3 (70). Further studies indicated that
LPCAT3 knockout in 3T3-L1 preadipocytes impairs lipogenesis and
differentiation (71). Limiting
LPCAT3 reduces levels of polyunsaturated PLs, such as linoleic acid
and arachidonic acid PLs, and reduces the expression of
adipogenesis-related genes, including SREBPs, PPAR-γ and C/EBPs.
This mechanism involves regulation of the Wnt/β-catenin signaling
pathway (71). Although these
cell studies confirm the role of LPCAT3 in impaired adipogenesis,
the specific effects of LPCAT3 on adipose tissue and systemic
metabolism in vivo remain to be fully elucidated.
Role of LPCAT3 in intestinal lipid
absorption by regulating PC levels
PC in the intestinal lumen has long been recognized
for its essential role in lipid absorption. LPCAT3 is the
predominant LPCAT in the intestinal tract, accounting for 80-90% of
the total LPC acyltransferase activity (53,54). Previous studies have identified
the significance of LPCAT3 in the lipid metabolism of the small
intestine (54,72). Mice with systemic LPCAT3 knockout
can be born normally but typically succumb to hypoglycemia and
mortality shortly after birth, around postnatal day 2 (P2)
(53). Similarly, mice with
intestinal-specific LPCAT3 knockout, induced by Vilin-Cre, also
experience hypoglycemia and increased mortality during lactation,
highlighting the vital role of intestinal LPCAT3 l in neonatal
survival (72). LPCAT3 activity
is crucial in mediating intestinal fat absorption (54,72). Mice with intestinal LPCAT3
deficiency (LPCAT3Vil-Cre) display reduced serum TG and
TC levels, indicative of impaired uptake of FAs by intestinal cells
and the secretion of smaller chylomicron particles. The absence of
LPCAT3 predominantly affects intestinal lipid metabolism, rather
than hepatic VLDL production. A deficiency in LPCAT3 leads to
decreased levels of ApoB in chylomicrons and its accumulation in
the intestine (73), suggesting
that not only the lipid loading of chylomicrons is compromised, but
their secretion is also impaired. The loss of intestinal LPCAT3
results in altered binding of linoleic acid and arachidonic acids
to membrane PLs. This change, coupled with a significant increase
in saturated and monounsaturated PCs, leads to reduced membrane
fluidity, thereby impairing passive FA transport across the apical
membrane of intestinal epithelial cells (73).
Li et al (54) demonstrated the impact of LPCAT3
deficiency on FA transport in the intestine, noting a decreased
expression of FA transporters, including FA transporter (FAT/CD36)
and FA transporter protein 4 (FATP4), in intestinal cells of LPCAT3
knockout mice. This reduction in transporter expression may
contribute to impaired FA uptake. However, another study indicated
that the loss of CD36 or FATP4 in the intestinal tract of mice does
not appear to significantly alter FA intake (74). The potential connection between
altered membrane fluidity and kinetics due to LPCAT3 deficiency and
the transport and secretion of chylomicrons warrants further
investigation.
In addition to impaired TG absorption,
LPCAT3Vil-Cre mice also exhibit reduced serum
cholesterol levels (54,72,75), potentially resulting from
diminished production of intestinal chylomicrons and HDL.
Niemann-Pick C1-like 1, a crucial protein in intestinal cholesterol
absorption, is significantly reduced in LPCAT3-deficient intestinal
tissues (76). The decrease in
HDL levels in LPCAT3Vil-Cre mice has been attributed to
lower ATP-binding cassette transporter A1 (ABCA1) expression and
ApoA-I secretion (77). Notably,
~30% of total plasma HDL is generated via intestinal ABCA1
activity, as evidenced by mouse experiments (78). However, the extent to which
LPCAT3 deficiency affects the expression of cholesterol
transport-related genes and alters membrane PL components remains
to be fully elucidated.
5. The role of lipo-apoptosis related to the
LXR-LPCAT3-ERS signaling pathway in NAFLD
Connection between PL metabolism and
lipo-apoptosis in NAFLD development
NAFLD has emerged as a significant global health
issue. A proportion of NAFLD patients progress to NASH, marked by
steatosis, hepatocyte death and inflammation. A positive
correlation exists between serum FFAs, hepatocyte death and liver
inflammation (79). Saturated
FFAs can induce hepatocyte apoptosis, a process termed
lipo-apoptosis, which is linked to the severity of NAFLD (80). Numerous studies have revealed
that saturated fatty acids (SFAs) are more toxic than their
unsaturated counterparts, leading to a progressive lipotoxic
cascade (Fig. 4). Malhi et
al (81) confirmed that SFAs
induce JNK-dependent hepatocyte lipo-apoptosis by activating the
pro-apoptotic proteins Bim and Bax.
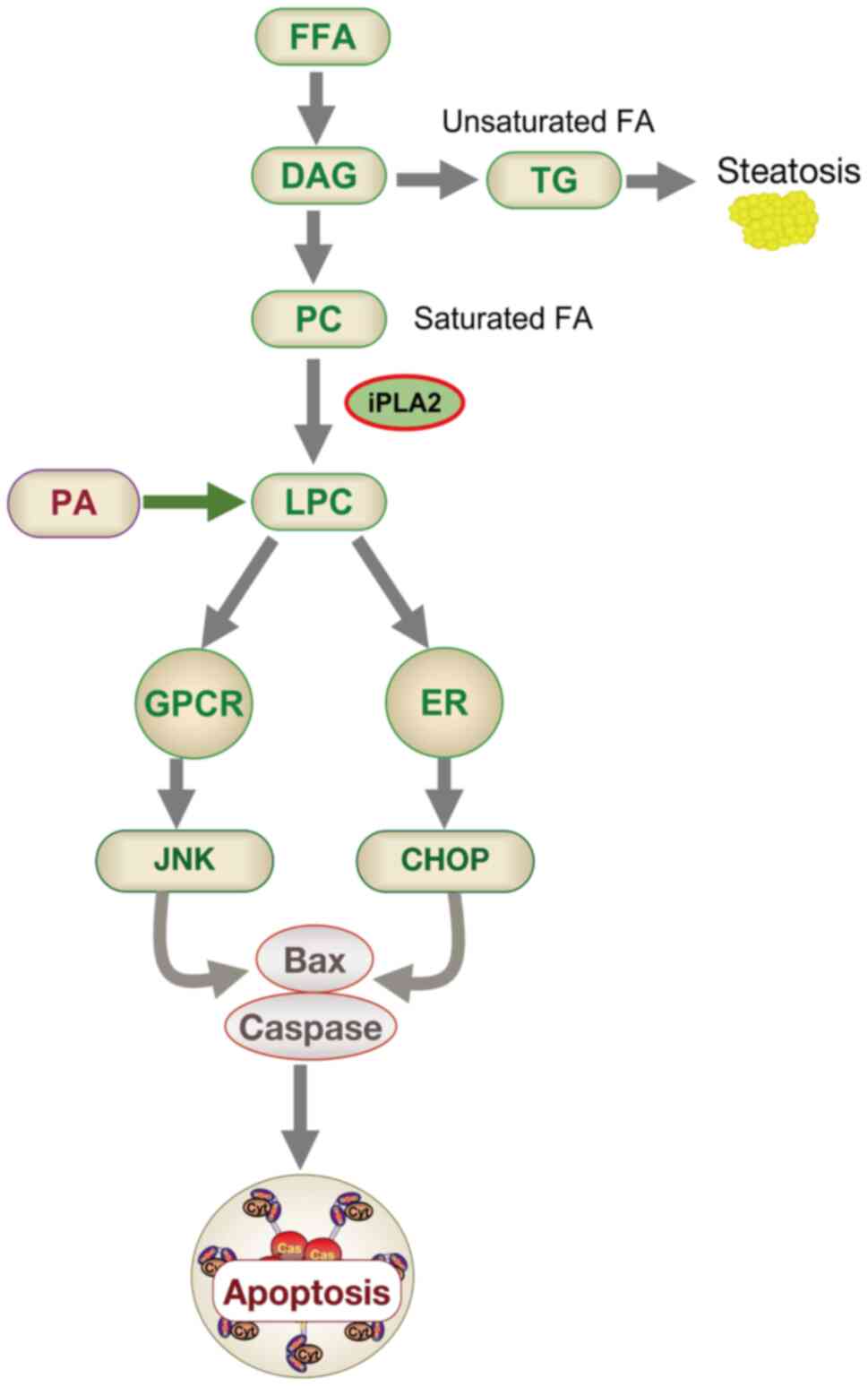 | Figure 4Schematic diagram of apoptosis model
induced by fatty acid metabolism. DAG is produced when FFA entering
the glycerol backbone can be converted into LPC or TG. Steatosis or
hepatitis may occur when the TG pathway or the LPC pathway,
respectively, is dominant. LPC can also be derived from PA. LPC may
activate the JNK pathway by activating GPCR, and may also activate
the ER stress pathway, thereby inducing cell apoptosis. DAG,
diacylglycerol; FFA, free fatty acid; LPC,
lyso-phosphatidyl-choline; TG, triglyceride; PA, phosphatidic acid;
JNK, c-Jun N-terminal kinase; GPCR, G protein-coupled receptor; ER,
endoplasmic reticulum; CHOP, C/EBP homologous protein; PC,
phosphatidylcholine. |
Palmitic acid (PA) is an SFA. Gu et al
(82) found that PA induced
HepG2 cytotoxicity and apoptosis in a dose-dependent manner, and
induced ER stress, which was manifested by increased
phosphorylation of eIF2α and upregulation of IRE1α and CHOP. After
PA treatment, BIP expression levels were slightly downregulated.
Overexpression of Bip attenuated PA-induced ERS and resulted in a
significant reduction in PA-mediated apoptosis, indicating that ERS
is necessary for the lipotoxic effect of hepatocytes (82). Similarly, Guo et al
(83) demonstrated that PA
modulates intracellular signaling in mouse 3T3-L1 and rat primary
preadipocytes, induces ERS, and leads to their apoptosis. It
induces multiple ERS responses, including increased CHOP and GRP78
protein levels, XBP-1 mRNA splicing, and changes in eIF2
phosphorylation. It also increases the phosphorylation of JNK and
ERK1/2 (83). In addition, oleic
acid counteracts PA-induced hepatocyte death by converting PA to
triglycerides and decreasing LPC levels, indicating that FFAs
contribute to steatosis or lipid metabolism depending on the
balance of saturated/unsaturated Fas (84).
Normally, FFAs undergo metabolism into PLs, in
addition to being oxidized in mitochondria, esterified into
triglycerides, and incorporated into lipoprotein complexes
(85). For instance, saturated
FFAs can combine with DAG, subsequently forming PC. LPC is a major
plasma PL derived from PC through PLA2 activity (86). Under physiological conditions,
LPCAT convert LPC back to PC, maintaining a dynamic equilibrium
known as the Lands' cycle. However, previous studies have indicated
that elevated hepatic LPC levels in NAFLD patients correlate with
disease severity (87). In two
small biopsy studies, liver LPC concentrations in NASH patients
were higher compared with healthy controls (88,89). Intravenous LPC administration in
ICR mice significantly increased in vivo AST/ALT levels,
lobular hepatitis and apoptosis, albeit without steatosis (89), suggesting LPC's crucial role in
hepatocyte lipo-apoptosis. Kakisaka et al (84) demonstrated that in vivo
LPC administration induces hepatitis in mice, observing that PA
converts to LPC, triggering hepatocyte apoptosis through
G-protein-coupled receptor activation, mitochondrial events and JNK
activation (Fig. 4). Han et
al (89,90) identified LPC as an active
metabolite of palmitate, noting its activation of ERS and JNK
signaling, which promotes hepatocyte lipo-apoptosis. Exogenous LPC
exposure mimics lipotoxic phenotypes observed in SFA overexposure,
characterized by ERS markers, caspase activation and apoptosis
(84,89). Exploring the role of LPC in
PA-derived SFA-induced lipotoxicity, Kakisaka et al
(84) used Huh-7 cells and
isolated mouse and human primary hepatocytes. Their findings
indicated that substituting LPC for PA leads to caspase-dependent
cell death, c-JUN phosphorylation, JNK activation, increased PUMA
expression and ERS induction, as evidenced by eIF2 activation
(84). It was deduced that
saturated FFA itself may not be necessary to induce hepatocyte
cytotoxicity pathways as long as its downstream lipo-phospholipid
metabolite LPC is present. While investigating LPC in SFA-induced
lipotoxicity is a promising research direction, further studies are
needed to clarify the direct relationship between these two
mechanisms.
The breakdown and transformation of PLs have long
been recognized as having a significant impact on the occurrence of
NAFLD (91,92). Lipidomic analysis of liver
tissues has revealed that levels of polyunsaturated PCs, including
linoleic acid and arachidonic acid PCs, are significantly reduced
in patients with non-alcoholic fatty liver and NASH compared with
control groups (93). Hall et
al (94) observed altered PL
partitioning characteristics in both NASH animal models and human
patients. Given that PC is the main component of PLs, alterations
in the FA composition of structural PLs have been shown to protect
hepatocytes from PA-induced ERS and associated lipotoxicity
(95). In the context of NAFLD,
the specific mechanism between PL metabolites, such as PC, PE, PS
and lipid apoptosis needs further study.
Lipo-apoptosis related to LXR-LPCAT3-ERS
pathway in NAFLD
It has been previously suggested that elevated
levels of FFAs, especially SFAs, are key contributors to
lipotoxicity, both in experimental models and in patients with
NAFLD. In NASH patients, serum SFA levels are notably higher than
in those with simple steatosis (96). SFAs are absorbed by hepatocytes
and can directly induce hepatocellular toxicity, leading to
lipo-apoptosis (97). Increasing
evidence indicates that ERS is an upstream signal of SFA-induced
cellular dysfunction and apoptosis. SFAs can increase the
saturation of cell membrane phospholipids, thereby initiating
unfolded protein response (UPR) and causing ERS (98-100). The JNK signaling pathway
responds to prolonged ERS and activates downstream apoptotic
pathways (99). SFAs have
demonstrated the ability to potentially induce ERS in multiple cell
types in vitro, including hepatocytes (98), pancreatic B cells (101), adipocytes (83) and CHO cells (102). Therefore, previous studies have
identified ERS as a key factor in the pathogenesis of NAFLD.
The ER is crucial for protein processing,
maturation, lipid synthesis and redox stability maintenance
(103). Disruption of ER
homeostasis triggers leads to ERS and activates the UPR. Chronic
UPR activation can lead to inflammation and contribute to metabolic
diseases, including obesity, type 2 diabetes, liver disease and
atherosclerosis (102).
Elevated FFA levels can induce ERS and UPR, potentially linked to
alterations in ER membrane composition (102,104) (Fig. 5). Following FFA exposure, ER
stress is initiated via UPR, primarily mediated by three
transmembrane protein pathways (105-107). Protein kinase RNA-like ER
kinase (PERK) pathway: PERK activation and subsequent C/EBP
homologous protein (CHOP) induction lead to apoptosis.
Inositol-requiring enzyme-1α (IRE-1α) pathway: Activated IRE-1α
interacts with tumor necrosis factor receptor-associated factor 2
and signal-regulating kinase 1, impacting JNK activation, cell
apoptosis, NF-κB activation and downstream inflammatory factors
expression. Activating transcription factor-6 (ATF-6) pathway:
N-terminal phosphorylation of ATF-6 activates NF-κB and triggers
inflammation (107,108). ERS plays a significant role in
NAFLD progression. These pathways induce the expression of
inflammatory cytokines (TNF-α and IL-1β), mediate inflammatory
response, promote cell apoptosis, and exacerbate liver injury and
fibrosis (109) (Fig. 5). Importantly, activation of JNK
has been identified as a central mediator of FFA-induced hepatocyte
apoptosis.
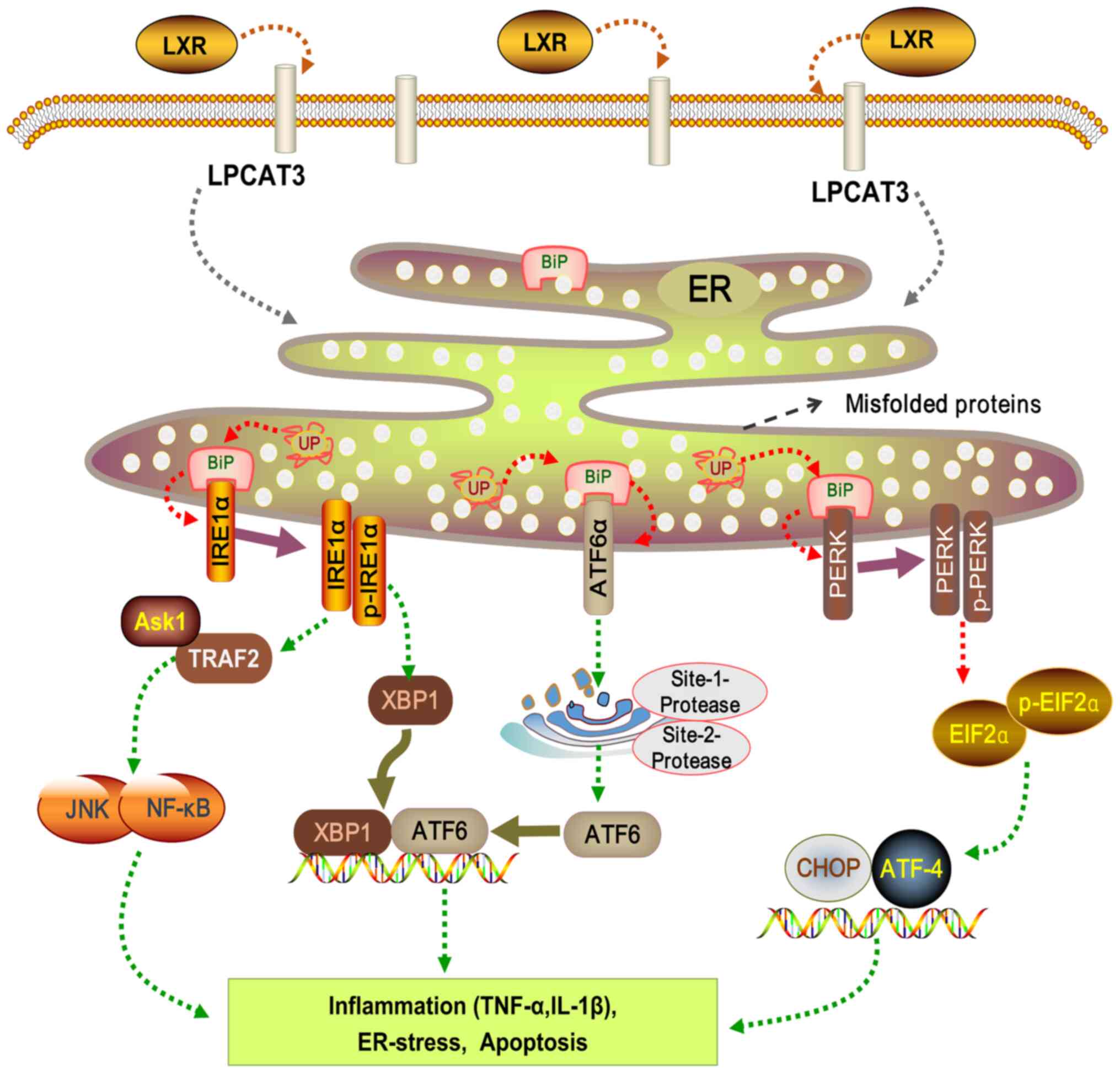 | Figure 5LXR-LPCAT3-ERS signaling pathway. LXR
affects LPCAT3 on the cell membrane, thus regulating signaling
molecules associated with the ER. ER stability is maintained by
three UPR pathways: PERK, ATF6 and IRE1α. Phosphorylated of PERK
activates downstream EIF2α phosphorylation, regulating the
expression of downstream ATF4 and CHOP expression. ATF6 directly
influences XBP1 expression. IRE1α modulates the expression of
downstream molecules such as ASK1, JNK and NF-κB. Under
pathological conditions, aberrant activation of these pathways can
alter inflammatory factor expression, leading to ER stress and cell
apoptosis. LXR, liver X receptor alpha; LPCAT3,
lyso-phosphatidyl-choline acyltransferase 3; ERS, ER stress; ER,
endoplasmic reticulum; UP, unfolded protein; UPR, UP response;
PERK, protein kinase-like ER kinase; ATF, activating transcription
factor; IRE1α, inositol requiring enzyme-1α; EIF2α, eukaryotic
initiation factor 2 alpha; CHOP, C/EBP homologous protein; XBP1,
X-box binding protein 1; ASK1, apoptosis signal-regulating kinase
1; JNK, c-Jun N-terminal kinase; NK-κB, nuclear factor kappa B;
TRAF2, TNF receptor associated factor 2. |
In vitro studies across various cell types
have revealed that excessive exposure to SFAs enhances
pro-inflammatory cytokine expression, disrupts insulin signaling,
and initiates apoptosis, marked by ER damage and oxidative stress
(110-113). Conversely, monounsaturated
fatty acids predominantly lead to steatosis and triglyceride
formation, without inducing apoptosis (111,114). A particular study focused on
the impact of increased PL saturation in cell membranes (115). This research examined the
functions of SCD-1, involved in desaturating SFAs for lipid
biosynthesis, and LPCAT3, known for its preferential integration of
polyunsaturated FAs into PCs. Notably, SCD-1 and LPCAT3 knockout
cells exhibited marked PL saturation and UPR activation even in the
absence of SFA supplementation. This was evidenced by increased
X-box binding protein splicing and PERK phosphorylation, effects
that were intensified with the combined knockout of LPCAT3/SCD-1
and additional palmitate supplementation (115). These findings underscore the
importance of incorporating unsaturated FAs into PLs for
maintaining ER membrane functionality, highlighting that SFA
overexposure can disrupt this balance. Further research on the
mechanism and role of SFA in controlling the composition of cell
membrane phospholipids is of great significance for establishing a
complete lipotoxic cell death mechanism.
Studies have identified that LXR activation can
increase the expression of cell membrane LPCAT3 and the abundance
of polyunsaturated PLs, thereby improving ERS caused by saturated
FFA in vivo and in vitro or liver lipid accumulation
in vivo (26,45). Rong et al (26) discovered that LPCAT3 plays a
pivotal role in maintaining ER homeostasis by regulating membrane
PLs composition. This maintenance is evident both in vitro,
where LPCAT3 inhibits ERS induced by FFAs, and in vivo, as
demonstrated by reduced liver lipid accumulation in ob/ob mice.
These effects are manifestations of LPCAT3's mediation of LXR
activation and its function as an LXR target. Enhanced LPCAT3
expression, prompted by LXR activation, fosters an increase in
polyunsaturated FAs that integrate with PLs, thereby reducing ER
membrane saturation. By contrast, acute liver LPCAT3 knockout in
mice, facilitated via adenovirus, exacerbates ERS (26). Furthermore, impaired LPCAT3
activity can elevate liver inflammation and modulate c-Src activity
by altering membrane microdomain components (116). The activity of LPCAT3
influences arachidonic acid levels, which are instrumental in the
production of prostaglandin E2, a lipid inflammatory mediator
contributing to inflammation (117). A recent study highlighted that
inhibiting the SCAP/SREBP pathway exacerbates liver damage and
carcinogenesis in NASH mice (118). The underlying mechanism
involves not only the inhibition of FA synthesis but also a
disruption in FA incorporation into PC due to downregulation of
LPCAT3. This alteration in FA composition leads to ERS and
hepatocellular damage. It was also found that the activity of LXR,
a key transcription factor that regulates LPACT3 expression, was
downregulated in hepatocytes of PTEN/SCAP double-knockout mice, and
LXR agonists restored the expression of LPCAT3 in hepatocytes of
PTEN/SCAP double-knockout mice. Therefore, LXR-mediated LPCAT3
expression was suppressed in the livers of PTEN/SCAP
double-knockout mice, which may be partly responsible for the
reduced number of PCs containing PUFAs (118).
Jiang et al (119) investigated the impact of LPCAT3
on serum lipid levels through systemic knockdown, as well as
intestinal and liver-specific knockouts in mice. It was revealed
that these alterations were primarily associated with a decrease in
polyunsaturated PC on the plasma membrane. By contrast, Feng et
al (71) posited that
suppression of LPCAT3 attenuates lipid production in 3T3-L1 cells
via activation of the Wnt/β-catenin signaling pathway, a finding
echoed by Rong et al (52). LPCAT3 is also implicated in the
biosynthesis of inflammatory lipid mediators in humans (117). Cell and animal studies have
demonstrated that LPCAT3 expression is related to the mitigation of
ERS and inflammation in response to saturated Fas (26). LPCAT3 knockdown leads to the
accumulation of LPC in the liver, which enhances the production of
VLDL through upregulated expression of MTTP (64). A previous genome-wide association
study highlighted a significant link between genetic variations at
the LPCAT3 locus and cellular FAs composition, underscoring the
essential role of LPCAT3 in human PL component regulation (120).
Rong et al (52) have highlighted the critical role
of LPCAT3-mediated arachidonic acid accumulation in PC for the
production of TG-rich lipoproteins. Their prior research indicated
that an acute decrease in LPCAT3 in the liver of obese mice
exacerbated lipid-induced ERS (26). Interestingly, it was also
observed that LPCAT3 gene deletion in the liver did not affect the
expression of ERS markers (121). These findings suggested a
potential compensatory response in membrane components to mitigate
the induction of ERS when LPCAT3 is chronically deleted. A study
from Xiang et al (122)
demonstrated that the LXR α-LPCAT3 pathway can regulate ER stress
to ameliorate liver damage in NASH.
In a previous study, LPCAT3 expression was reduced
in the liver in a mouse model of HDF-induced NASH (123). To confirm the protective role
of LPCAT3 in lipotoxicity, the LPCAT3-overexpressing Huh7 cells
(LPCAT3-OE) were established. In LPCAT3-OE cells, PA-induced
expression of CHOP, a transcription factor that serves as a marker
of ERS, was significantly reduced. Furthermore, the phosphorylation
of eIF2α and PA-induced cell death were significantly reduced in
LPCAT3-OE cells compared with WT cells. Finally, PA-induced LPC
increase in LPCAT3-OE cells was significantly attenuated compared
with WT cells. In-depth mechanistic research demonstrated that
PA-induced hepatocyte death under LPCAT3 depletion is performed by
a caspase-independent mechanism and is mediated by LPC (123). These results suggested that
LPCAT3 may be a therapeutic target for NASH by reducing hepatocyte
death.
LXR functions as an upstream regulator of LPCAT3.
LPCAT3 itself is pivotal in PL metabolism, governing the
physiological conversion between LPC and PC. Under pathological
conditions, the role of LPCAT3 extends beyond influencing the
composition of the endoplasmic reticulum membrane to impacting FA
metabolism. This impact is a crucial factor in ERS. Therefore, the
stable modulation of ERS by the LXR-LPCAT3 pathway is vital in
maintaining normal adipocyte metabolism in the liver. Among the
existing studies, research on the regulation of endoplasmic
reticulum stress by LXR-LPCAT3 is available, but there is no clear
experimental study to fully elucidate the role of LXR-LPCAT3-ERS
pathway in the development of NAFLD. It is anticipated that more
experiments will be conducted in the future to clarify this
mechanism and provide new targets for the treatment of NAFLD.
6. Controversy and prospects
The significance of the LXR-LPCAT3 signaling
pathway in NAFLD has not been extensively studied, and the existing
research on LPCAT3 presents some controversial findings.
While some researchers posit that LPCAT3 mitigates
ERS and inflammation, others argue that inhibiting LPCAT3
expression exacerbates these conditions. Rong et al
(26) suggested that LXR
activation leads to increased LPCAT3 expression, which in turn
reduces the saturation of ER membrane. On the contrary, acute
liver-knockout of LPCAT3 is associated with aggravated ERS, and
inhibiting LPCAT3 activity appears to enhance liver inflammation.
Adding another dimension to this discourse, Wang et al
(124) observed that
disruptions in LPCAT3-dependent sphingomyelin and cholesterol
metabolism in Apcmin mice promote tumor formation,
suggesting broader implications of LPCAT3 beyond NAFLD.
Some researchers proposed that LPCAT3 facilitates
adipogenesis, and thus suppressing LPCAT3 expression could reduce
adipogenesis. Rong et al (52) highlighted that LPCAT3-mediated
accumulation of arachidonic acid in PC is essential for the
production of TG-rich lipoproteins. Li et al (54) suggested that inhibiting
intestinal LPCAT3 might be an effective strategy for hyperlipidemia
treatment. In obese mice, the increase in LPCAT3 expression leads
to a selective rise in polyunsaturated PLs in ER; consequently,
inhibiting LPCAT3 activity can reduce the activation of SREBP1c
pathway, slow down fat production, and delay the progression of
fatty liver (56). LXR
activation is known to promote the post-translational processing of
SREBP-1c by inducing LPCAT3 expression, where LPCAT3's integration
of polyunsaturated FAs into PLs aids in SREBP1c processing and
lipid formation. Conversely, LPCAT3 deficiency in hepatocytes leads
to reduced levels of polyunsaturated PLs in the ER, lowered nuclear
SREBP-1c levels, and a decreased adipogenic response to LXR agonist
treatment (55). Studies using
LPCAT3shRNA adenovirus in obese mice have demonstrated a reduction
in SREBP-1c processing, a slow-down in fat production, and an
improvement in fatty liver condition (69). Additionally, LPCAT3 knockout in
3T3-L1 preadipocytes impairs fat generation and differentiation
(71). LPCAT3 inhibition
decreases levels of polyunsaturated PLs, such as linoleic acid and
arachidonic acid PLs, and reduces the expression of
adipogenesis-related genes expression including SREBPs, PPAR-γ and
C/EBPs, with the mechanism being related to the regulation of the
Wnt/β-catenin pathway (71).
Feng et al (71) revealed
that LPCAT3 deficiency attenuates lipid production in 3T3-L1 cells
by activating the Wnt/β-catenin signaling pathway, a finding
corroborated by Rong et al (52). Furthermore, LPCAT3 is considered
to be directly involved in the biosynthesis of inflammatory lipid
mediators in humans (117).
LXR can modulate adipogenesis-related factors
through the regulation of LPCAT3. LPCAT3 itself influences FA
metabolism and the saturation level of the ER membrane. Research
has demonstrated that saturated FAs can induce hepatocyte
lipo-apoptosis through JNK-dependent activation of pro-apoptotic
proteins Bim and Bax (81). ERS
is recognized as a critical factor in FFA-induced apoptosis.
However, the specific role of the LXR-LPCAT3-ERS pathway in fat
apoptosis within the context of NAFLD remains incompletely
understood, necessitating further experimental research to
elucidate the underlying mechanisms.
At present, the international and domestic research
on LPCAT3 has not converged to a unified conclusion, highlighting
the need for more studies to clarify its critical role. Questions
such as whether receptors other than LXR, PPAR-α and PPAR-γ
regulate LPCAT3 expression, and whether LPCAT3 is involved in other
physiological and pathological processes beyond ER stress and
inflammation, remain open for investigation. These areas warrant
in-depth exploration. Therefore, a detailed examination of the
LXR-LPCAT3 signaling pathway's role in NAFLD is essential. Such
research could provide a foundational understanding of NAFLD
pathogenesis and hold significant theoretical and practical value
in advancing the treatment of NAFLD.
Availability of data and materials
Not applicable.
Authors' contributions
JW contributed to acquisition, analysis,
interpretation and drafted the manuscript. LL, YF, YZ and JL
contributed to acquisition and analysis. YL contributed to
conception and design and critically revised the manuscript. All
authors read and approved the final manuscript. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
ABCA1
|
ATP-binding cassette transporter
A1
|
|
ApoB
|
apolipoprotein B
|
|
ATF-6
|
activating transcription factor-6
|
|
CTP
|
cytidine triphosphate
|
|
CCT
|
CTP: phosphocholine
cytidylyltransferase
|
|
ERS
|
endoplasmic reticulum stress
|
|
FATP4
|
fatty acid transporter protein 4
|
|
FA
|
fatty acid
|
|
FFA
|
free fatty acid
|
|
HDL
|
high density lipoprotein
|
|
IRE-1α
|
inositol requiring enzyme-1α
|
|
LPC
|
lyso-phosphatidyl-choline
|
|
LPCATs
|
lyso-phosphatidyl-choline
acyltransferases
|
|
LXR
|
liver X receptor
|
|
MBOAT
|
membrane-bound O-acyltransferase
|
|
MTTP
|
microsomal TG transfer protein
|
|
NAFLD
|
non-alcoholic fatty liver disease
|
|
NASH
|
non-alcoholic steatohepatitis
|
|
PC
|
phosphatidylcholine
|
|
PLs
|
phospholipids
|
|
PLTP
|
phospholipid transfer protein
|
|
PERK
|
protein kinase-like ER kinase
|
|
PEMT
|
phosphatidylethanolamine
N-methyltransferase
|
|
PPAR
|
peroxisome proliferation-activated
receptor
|
|
RXR
|
retinoic acid receptor
|
|
SCD-1
|
stearoyl-CoA desaturase 1
|
|
SFA
|
saturated fatty acids
|
|
SREBP-1c
|
sterol response element-binding
protein 1a
|
|
TG
|
triglyceride
|
|
UPR
|
unfolded protein response
|
|
VLDL
|
very low density lipoprotein
|
Acknowledgments
Not applicable.
Funding
The present study was supported by Shanghai Natural Science
Foundation (grant no. 22ZR1459400) and Shanghai Science and
Technology Innovation Project (grant no. 22S21901100).
References
|
1
|
Sanyal AJ: Past, present and future
perspectives in nonalcoholic fatty liver disease. Nat Rev
Gastroenterol Hepatol. 16:377–386. 2019.
|
|
2
|
Leow WQ, Chan AW, Mendoza PGL, Lo R, Yap K
and Kim H: Non-alcoholic fatty liver disease: The pathologist's
perspective. Clin Mol Hepatol. 29(Suppl): S302–S318. 2023.
|
|
3
|
Fan JG, Wei L and Zhuang H; National
Workshop on Fatty Liver and Alcoholic Liver Disease, Chinese
Society of Hepatology, Chinese Medical Association; Fatty Liver
Disease Expert Committee, Chinese Medical Doctor Association:
Guidelines of prevention and treatment for nonalcoholic fatty liver
disease (2018, China). J Dig Dis. 20:163–173. 2019.
|
|
4
|
Zhou J, Zhou F, Wang W, Zhang XJ, Ji YX,
Zhang P, She ZG, Zhu L, Cai J and Li H: Epidemiological Features of
NAFLD From 1999 to 2018 in China. Hepatology. 71:1851–1864.
2020.
|
|
5
|
Younossi Z, Anstee QM, Marietti M, Hardy
T, Henry L, Eslam M, George J and Bugianesi E: Global burden of
NAFLD and NASH: Trends, predictions, risk factors and prevention.
Nat Rev Gastroenterol Hepatol. 15:11–20. 2018.
|
|
6
|
Raza S, Rajak S, Upadhyay A, Tewari A and
Anthony Sinha R: Current treatment paradigms and emerging therapies
for NAFLD/NASH. Front Biosci (Landmark Ed). 26:206–237. 2021.
|
|
7
|
Meroni M, Longo M, Rustichelli A and
Dongiovanni P: Nutrition and Genetics in NAFLD: The Perfect
Binomium. Int J Mol Sci. 21:29862020.
|
|
8
|
Papatheodoridi M and Cholongitas E:
Diagnosis of non-alcoholic fatty liver disease (NAFLD): Current
concepts. Curr Pharm Des. 24:4574–4586. 2018.
|
|
9
|
Safari Z and Gérard P: The links between
the gut microbiome and non-alcoholic fatty liver disease (NAFLD).
Cell Mol Life Sci. 76:1541–1558. 2019.
|
|
10
|
Buzzetti E, Pinzani M and Tsochatzis EA:
The multiple-hit pathogenesis of non-alcoholic fatty liver disease
(NAFLD). Metabolism. 65:1038–1048. 2016.
|
|
11
|
Polyzos SA, Kountouras J, Zavos C and
Deretzi G: Nonalcoholic fatty liver disease: Multimodal treatment
options for a pathogenetically multiple-hit disease. J Clin
Gastroenterol. 46:272–284. 2012.
|
|
12
|
Karkucinska-Wieckowska A, Simoes ICM,
Kalinowski P, Lebiedzinska-Arciszewska M, Zieniewicz K, Milkiewicz
P, Górska-Ponikowska M, Pinton P, Malik AN, Krawczyk M, et al:
Mitochondria, oxidative stress and nonalcoholic fatty liver
disease: A complex relationship. Eur J Clin Invest.
52:e136222022.
|
|
13
|
Tilg H, Adolph TE, Dudek M and Knolle P:
Non-alcoholic fatty liver disease: The interplay between
metabolism, microbes and immunity. Nat Metab. 3:1596–1607.
2021.
|
|
14
|
Albillos A, de Gottardi A and Rescigno M:
The gut-liver axis in liver disease: Pathophysiological basis for
therapy. J Hepatol. 72:558–577. 2020.
|
|
15
|
Filipovic B, Lukic S, Mijac D,
Marjanovic-Haljilji M, Vojnovic M, Bogdanovic J, Glisic T,
Filipovic N, Al Kiswani J, Djokovic A, et al: The new therapeutic
approaches in the treatment of non-alcoholic fatty liver disease.
Int J Mol Sci. 22:132192021.
|
|
16
|
Singh S, Osna NA and Kharbanda KK:
Treatment options for alcoholic and non-alcoholic fatty liver
disease: A review. World J Gastroenterol. 23:6549–6570. 2017.
|
|
17
|
Townsend SA and Newsome PN: Review
article: New treatments in non-alcoholic fatty liver disease.
Aliment Pharmacol Ther. 46:494–507. 2017.
|
|
18
|
Albhaisi SAM and Sanyal AJ: New drugs for
NASH. Liver Int. 41(Suppl 1): S112–S118. 2021.
|
|
19
|
Gofton C and George J: Updates in fatty
liver disease: Pathophysiology, diagnosis and management. Aust J
Gen Pract. 50:702–707. 2021.
|
|
20
|
Klińska S, Jasieniecka-Gazarkiewicz K and
Banaś A: Acyl-CoA: lysophosphatidylcholine acyltransferases
(LPCATs) of Camelina sativa seeds: Biochemical properties and
function. Planta. 250:1655–1670. 2019.
|
|
21
|
Zhang Q, Yao D, Rao B, Jian L, Chen Y, Hu
K, Xia Y, Li S, Shen Y, Qin A, et al: The structural basis for the
phospholipid remodeling by lysophosphatidylcholine acyltransferase
3. Nat Commun. 12:68692021.
|
|
22
|
Law SH, Chan ML, Marathe GK, Parveen F,
Chen CH and Ke LY: An updated review of lysophosphatidylcholine
metabolism in human diseases. Int J Mol Sci. 20:11492019.
|
|
23
|
Shao G, Qian Y, Lu L, Liu Y, Wu T, Ji G
and Xu H: Research progress in the role and mechanism of LPCAT3 in
metabolic related diseases and cancer. J Cancer. 13:2430–2439.
2022.
|
|
24
|
Wang B and Tontonoz P: Phospholipid
remodeling in physiology and disease. Annu Rev Physiol. 81:165–188.
2019.
|
|
25
|
Hong C and Tontonoz P: Liver X receptors
in lipid metabolism: Opportunities for drug discovery. Nat Rev Drug
Discov. 13:433–444. 2014.
|
|
26
|
Rong X, Albert CJ, Hong C, Duerr MA,
Chamberlain BT, Tarling EJ, Ito A, Gao J, Wang B, Edwards PA, et
al: LXRs regulate ER stress and inflammation through dynamic
modulation of membrane phospholipid composition. Cell Metab.
18:685–697. 2013.
|
|
27
|
Morita SY and Ikeda Y: Regulation of
membrane phospholipid biosynthesis in mammalian cells. Biochem
Pharmacol. 206:1152962022.
|
|
28
|
Vance JE: Phospholipid synthesis and
transport in mammalian cells. Traffic. 16:1–18. 2015.
|
|
29
|
Patton-Vogt J and de Kroon AIPM:
Phospholipid turnover and acyl chain remodeling in the yeast ER.
Biochim Biophys Acta Mol Cell Biol Lipids. 1865:1584622020.
|
|
30
|
Vance DE: Phospholipid methylation in
mammals: From biochemistry to physiological function. Biochim
Biophys Acta. 1838:1477–1487. 2014.
|
|
31
|
Kennedy EP and Weiss SB: The function of
cytidine coenzymes in the biosynthesis of phospholipides. J Biol
Chem. 222:193–214. 1956.
|
|
32
|
Kennedy EP: Biosynthesis of
phospholipides. Fed Proc. 16:847–853. 1957.
|
|
33
|
Lands WE: Stories about acyl chains.
Biochim Biophys Acta. 1483:1–14. 2000.
|
|
34
|
Dahlman I, Nilsson M, Jiao H, Hoffstedt J,
Lindgren CM, Humphreys K, Kere J, Gustafsson JA, Arner P and
Dahlman-Wright K: Liver X receptor gene polymorphisms and adipose
tissue expression levels in obesity. Pharmacogenet Genomics.
16:881–889. 2006.
|
|
35
|
Han M, Liang L, Liu LR, Yue J, Zhao YL and
Xiao HP: Liver X receptor gene polymorphisms in tuberculosis:
Effect on susceptibility. PLoS One. 9:e959542014.
|
|
36
|
Yu M, Geiger B, Deeb N and Rothschild MF:
Liver X receptor alpha and beta genes have the potential role on
loin lean and fat content in pigs. J Anim Breed Genet. 123:81–88.
2006.
|
|
37
|
Yonezawa S, Abe M, Kawasaki Y, Natori Y
and Sugiyama A: Each liver X receptor (LXR) type has a different
purpose in different situations. Biochem Biophys Res Commun.
508:92–96. 2019.
|
|
38
|
Ducheix S, Lobaccaro JM, Martin PG and
Guillou H: Liver X Receptor: An oxysterol sensor and a major player
in the control of lipogenesis. Chem Phys Lipids. 164:500–514.
2011.
|
|
39
|
Bilotta MT, Petillo S, Santoni A and
Cippitelli M: Liver X Receptors: Regulators of cholesterol
metabolism, inflammation, autoimmunity, and cancer. Front Immunol.
11:5843032020.
|
|
40
|
Cave MC, Clair HB, Hardesty JE, Falkner
KC, Feng W, Clark BJ, Sidey J, Shi H, Aqel BA, McClain CJ and
Prough RA: Nuclear receptors and nonalcoholic fatty liver disease.
Biochim Biophys Acta. 1859:1083–1099. 2016.
|
|
41
|
Wang B and Tontonoz P: Liver X receptors
in lipid signalling and membrane homeostasis. Nat Rev Endocrinol.
14:452–463. 2018.
|
|
42
|
Goodwin BJ, Zuercher WJ and Collins JL:
Recent advances in liver X receptor biology and chemistry. Curr Top
Med Chem. 8:781–791. 2008.
|
|
43
|
Russo-Savage L and Schulman IG: Liver X
receptors and liver physiology. Biochim Biophys Acta Mol Basis Dis.
1867:1661212021.
|
|
44
|
Hamilton JP, Koganti L, Muchenditsi A,
Pendyala VS, Huso D, Hankin J, Murphy RC, Huster D, Merle U,
Mangels C, et al: Activation of liver X receptor/retinoid X
receptor pathway ameliorates liver disease in Atp7B(-/-) (Wilson
disease) mice. Hepatology. 63:1828–1841. 2016.
|
|
45
|
Schulman IG: Liver X receptors link lipid
metabolism and inflammation. FEBS Lett. 591:2978–2991. 2017.
|
|
46
|
Endo-Umeda K and Makishima M: Liver X
receptors regulate cholesterol metabolism and immunity in hepatic
nonparenchymal cells. Int J Mol Sci. 20:50452019.
|
|
47
|
Higuchi N, Kato M, Shundo Y, Tajiri H,
Tanaka M, Yamashita N, Kohjima M, Kotoh K, Nakamuta M, Takayanagi R
and Enjoji M: Liver X receptor in cooperation with SREBP-1c is a
major lipid synthesis regulator in nonalcoholic fatty liver
disease. Hepatol Res. 38:1122–1129. 2008.
|
|
48
|
Chen G, Liang G, Ou J, Goldstein JL and
Brown MS: Central role for liver X receptor in insulin-mediated
activation of Srebp-1c transcription and stimulation of fatty acid
synthesis in liver. Proc Natl Acad Sci USA. 101:11245–11250.
2004.
|
|
49
|
Lu Y, Shao M, Xiang H, Wang J, Ji G and Wu
T: Qinggan huoxue recipe alleviates alcoholic liver injury by
suppressing endoplasmic reticulum stress through LXR-LPCAT3. Front
Pharmacol. 13:8241852022.
|
|
50
|
Okazaki H, Goldstein JL, Brown MS and
Liang G: LXR-SREBP-1c-phospholipid transfer protein axis controls
very low density lipoprotein (VLDL) particle size. J Biol Chem.
285:6801–6810. 2010.
|
|
51
|
Vance DE: Role of phosphatidylcholine
biosynthesis in the regulation of lipoprotein homeostasis. Curr
Opin Lipidol. 19:229–234. 2008.
|
|
52
|
Rong X, Wang B, Dunham MM, Hedde PN, Wong
JS, Gratton E, Young SG, Ford DA and Tontonoz P: Lpcat3-dependent
production of arachidonoyl phospholipids is a key determinant of
triglyceride secretion. Elife. 4:e065572015.
|
|
53
|
Hashidate-Yoshida T, Harayama T, Hishikawa
D, Morimoto R, Hamano F, Tokuoka SM, Eto M, Tamura-Nakano M,
Yanobu-Takanashi R, Mukumoto Y, et al: Fatty acid remodeling by
LPCAT3 enriches arachidonate in phospholipid membranes and
regulates triglyceride transport. Elife. 4:e063282015.
|
|
54
|
Li Z, Jiang H, Ding T, Lou C, Bui HH, Kuo
MS and Jiang XC: Deficiency in lysophosphatidylcholine
acyltransferase 3 reduces plasma levels of lipids by reducing lipid
absorption in mice. Gastroenterology. 149:1519–1529. 2015.
|
|
55
|
Rong X, Wang B, Palladino EN, de Aguiar
Vallim TQ, Ford DA and Tontonoz P: ER phospholipid composition
modulates lipogenesis during feeding and in obesity. J Clin Invest.
127:3640–3651. 2017.
|
|
56
|
Anderson CD, Upadhya G, Conzen KD, Jia J,
Brunt EM, Tiriveedhi V, Xie Y, Ramachandran S, Mohanakumar T,
Davidson NO and Chapman WC: Endoplasmic reticulum stress is a
mediator of posttransplant injury in severely steatotic liver
allografts. Liver Transpl. 17:189–200. 2011.
|
|
57
|
Hishikawa D, Shindou H, Kobayashi S,
Nakanishi H, Taguchi R and Shimizu T: Discovery of a
lysophospholipid acyltransferase family essential for membrane
asymmetry and diversity. Proc Natl Acad Sci USA. 105:2830–2835.
2008.
|
|
58
|
Valentine WJ, Yanagida K, Kawana H, Kono
N, Noda NN, Aoki J and Shindou H: Update and nomenclature proposal
for mammalian lysophospholipid acyltransferases, which create
membrane phospholipid diversity. J Biol Chem. 298:1014702022.
|
|
59
|
Shindou H, Hishikawa D, Harayama T, Eto M
and Shimizu T: Generation of membrane diversity by lysophospholipid
acyltransferases. J Biochem. 154:21–28. 2013.
|
|
60
|
Matsuda S, Inoue T, Lee HC, Kono N, Tanaka
F, Gengyo-Ando K, Mitani S and Arai H: Member of the membrane-bound
O-acyltransferase (MBOAT) family encodes a lysophospholipid
acyltransferase with broad substrate specificity. Genes Cells.
13:879–888. 2008.
|
|
61
|
Gross B, Pawlak M, Lefebvre P and Staels
B: PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat Rev
Endocrinol. 13:36–49. 2017.
|
|
62
|
Cole LK, Vance JE and Vance DE:
Phosphatidylcholine biosynthesis and lipoprotein metabolism.
Biochim Biophys Acta. 1821:754–761. 2012.
|
|
63
|
Harayama T and Riezman H: Understanding
the diversity of membrane lipid composition. Nat Rev Mol Cell Biol.
19:281–296. 2018.
|
|
64
|
Li Z, Ding T, Pan X, Li Y, Li R, Sanders
PE, Kuo MS, Hussain MM, Cao G and Jiang XC: Lysophosphatidylcholine
acyltransferase 3 knockdown-mediated liver lysophosphatidylcholine
accumulation promotes very low density lipoprotein production by
enhancing microsomal triglyceride transfer protein expression. J
Biol Chem. 287:20122–20131. 2012.
|
|
65
|
Cash JG and Hui DY: Liver-specific
overexpression of LPCAT3 reduces postprandial hyperglycemia and
improves lipoprotein metabolic profile in mice. Nutr Diabetes.
6:e2062016.
|
|
66
|
Jacobs RL, Lingrell S, Zhao Y, Francis GA
and Vance DE: Hepatic CTP:phosphocholine cytidylyltransferase-alpha
is a critical predictor of plasma high density lipoprotein and very
low density lipoprotein. J Biol Chem. 283:2147–2155. 2008.
|
|
67
|
Balla T, Sengupta N and Kim YJ: Lipid
synthesis and transport are coupled to regulate membrane lipid
dynamics in the endoplasmic reticulum. Biochim Biophys Acta Mol
Cell Biol Lipids. 1865:1584612020.
|
|
68
|
Lev S: Nonvesicular lipid transfer from
the endoplasmic reticulum. Cold Spring Harb Perspect Biol.
4:a0133002012.
|
|
69
|
Walker AK, Jacobs RL, Watts JL, Rottiers
V, Jiang K, Finnegan DM, Shioda T, Hansen M, Yang F, Niebergall LJ,
et al: A conserved SREBP-1/phosphatidylcholine feedback circuit
regulates lipogenesis in metazoans. Cell. 147:840–852. 2011.
|
|
70
|
Eto M, Shindou H, Koeberle A, Harayama T,
Yanagida K and Shimizu T: Lysophosphatidylcholine acyltransferase 3
is the key enzyme for incorporating arachidonic acid into
glycerophospholipids during adipocyte differentiation. Int J Mol
Sci. 13:16267–16280. 2012.
|
|
71
|
Feng C, Lou B, Dong J, Li Z, Chen Y, Li Y,
Zhang X, Jiang XC and Ding T: Lysophosphatidylcholine
acyltransferase 3 deficiency impairs 3T3L1 cell adipogenesis
through activating Wnt/β-catenin pathway. Biochim Biophys Acta Mol
Cell Biol Lipids. 1863:834–843. 2018.
|
|
72
|
Wang B, Rong X, Duerr MA, Hermanson DJ,
Hedde PN, Wong JS, Vallim TQ, Cravatt BF, Gratton E, Ford DA and
Tontonoz P: Intestinal phospholipid remodeling is required for
dietary-lipid uptake and survival on a high-fat diet. Cell Metab.
23:492–504. 2016.
|
|
73
|
Repa JJ, Liang G, Ou J, Bashmakov Y,
Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL and
Mangelsdorf DJ: Regulation of mouse sterol regulatory
element-binding protein-1c gene (SREBP-1c) by oxysterol receptors,
LXRalpha and LXRbeta. Genes Dev. 14:2819–2830. 2000.
|
|
74
|
Shim J, Moulson CL, Newberry EP, Lin MH,
Xie Y, Kennedy SM, Miner JH and Davidson NO: Fatty acid transport
protein 4 is dispensable for intestinal lipid absorption in mice. J
Lipid Res. 50:491–500. 2009.
|
|
75
|
Kabir I, Li Z, Bui HH, Kuo MS, Gao G and
Jiang XC: Small intestine but not liver lysophosphatidylcholine
acyltransferase 3 (Lpcat3) deficiency has a dominant effect on
plasma lipid metabolism. J Biol Chem. 291:7651–7660. 2016.
|
|
76
|
Altmann SW, Davis HR Jr, Zhu LJ, Yao X,
Hoos LM, Tetzloff G, Iyer SP, Maguire M, Golovko A, Zeng M, et al:
Niemann-Pick C1 Like 1 protein is critical for intestinal
cholesterol absorption. Science. 303:1201–1204. 2004.
|
|
77
|
Kalhan SC, Guo L, Edmison J, Dasarathy S,
McCullough AJ, Hanson RW and Milburn M: Plasma metabolomic profile
in nonalcoholic fatty liver disease. Metabolism. 60:404–413.
2011.
|
|
78
|
Brunham LR, Kruit JK, Iqbal J, Fievet C,
Timmins JM, Pape TD, Coburn BA, Bissada N, Staels B, Groen AK, et
al: Intestinal ABCA1 directly contributes to HDL biogenesis in
vivo. J Clin Invest. 116:1052–1062. 2006.
|
|
79
|
Feldstein AE, Canbay A, Angulo P, Taniai
M, Burgart LJ, Lindor KD and Gores GJ: Hepatocyte apoptosis and fas
expression are prominent features of human nonalcoholic
steatohepatitis. Gastroenterology. 125:437–443. 2003.
|
|
80
|
Akazawa Y and Nakao K: Lipotoxicity
pathways intersect in hepatocytes: Endoplasmic reticulum stress,
c-Jun N-terminal kinase-1, and death receptors. Hepatol Res.
46:977–984. 2016.
|
|
81
|
Malhi H, Bronk SF, Werneburg NW and Gores
GJ: Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis.
J Biol Chem. 281:12093–12101. 2006.
|
|
82
|
Gu X, Li K, Laybutt DR, He ML, Zhao HL,
Chan JC and Xu G: Bip overexpression, but not CHOP inhibition,
attenuates fatty-acid-induced endoplasmic reticulum stress and
apoptosis in HepG2 liver cells. Life Sci. 87:724–732. 2010.
|
|
83
|
Guo W, Wong S, Xie W, Lei T and Luo Z:
Palmitate modulates intracellular signaling, induces endoplasmic
reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat
primary preadipocytes. Am J Physiol Endocrinol Metab.
293:E576–E586. 2007.
|
|
84
|
Kakisaka K, Cazanave SC, Fingas CD,
Guicciardi ME, Bronk SF, Werneburg NW, Mott JL and Gores GJ:
Mechanisms of lysophosphatidylcholine-induced hepatocyte
lipoapoptosis. Am J Physiol Gastrointest Liver Physiol.
302:G77–G84. 2012.
|
|
85
|
Cohen JC, Horton JD and Hobbs HH: Human
fatty liver disease: Old questions and new insights. Science.
332:1519–1523. 2011.
|
|
86
|
Fu S, Yang L, Li P, Hofmann O, Dicker L,
Hide W, Lin X, Watkins SM, Ivanov AR and Hotamisligil GS: Aberrant
lipid metabolism disrupts calcium homeostasis causing liver
endoplasmic reticulum stress in obesity. Nature. 473:528–531.
2011.
|
|
87
|
Wu X, Zhang Y, Qiu J, Xu Y, Zhang J, Huang
J, Bai J, Huang Z, Qiu X and Xu W: Lipidomics analysis indicates
disturbed hepatocellular lipid metabolism in reynoutria
multiflora-induced idiosyncratic liver injury. Front Pharmacol.
11:5691442020.
|
|
88
|
Puri P, Baillie RA, Wiest MM, Mirshahi F,
Choudhury J, Cheung O, Sargeant C, Contos MJ and Sanyal AJ: A
lipidomic analysis of nonalcoholic fatty liver disease. Hepatology.
46:1081–1090. 2007.
|
|
89
|
Han MS, Park SY, Shinzawa K, Kim S, Chung
KW, Lee JH, Kwon CH, Lee KW, Lee JH, Park CK, et al:
Lysophosphatidylcholine as a death effector in the lipoapoptosis of
hepatocytes. J Lipid Res. 49:84–97. 2008.
|
|
90
|
Han MS, Lim YM, Quan W, Kim JR, Chung KW,
Kang M, Kim S, Park SY, Han JS, Park SY, et al:
Lysophosphatidylcholine as an effector of fatty acid-induced
insulin resistance. J Lipid Res. 52:1234–1246. 2011.
|
|
91
|
Masoodi M, Gastaldelli A, Hyötyläinen T,
Arretxe E, Alonso C, Gaggini M, Brosnan J, Anstee QM, Millet O,
Ortiz P, et al: Metabolomics and lipidomics in NAFLD: biomarkers
and non-invasive diagnostic tests. Nat Rev Gastroenterol Hepatol.
18:835–856. 2021.
|
|
92
|
Sun X, Seidman JS, Zhao P, Troutman TD,
Spann NJ, Que X, Zhou F, Liao Z, Pasillas M, Yang X, et al:
Neutralization of oxidized phospholipids ameliorates non-alcoholic
steatohepatitis. Cell Metab. 31:189–206.e8. 2020.
|
|
93
|
Wattacheril J, Seeley EH, Angel P, Chen H,
Bowen BP, Lanciault C, Caprioli RM, Abumrad N and Flynn CR:
Differential intrahepatic phospholipid zonation in simple steatosis
and nonalcoholic steatohepatitis. PLoS One. 8:e571652013.
|
|
94
|
Hall Z, Bond NJ, Ashmore T, Sanders F,
Ament Z, Wang X, Murray AJ, Bellafante E, Virtue S, Vidal-Puig A,
et al: Lipid zonation and phospholipid remodeling in nonalcoholic
fatty liver disease. Hepatology. 65:1165–1180. 2017.
|
|
95
|
Leamy AK, Egnatchik RA, Shiota M, Ivanova
PT, Myers DS, Brown HA and Young JD: Enhanced synthesis of
saturated phospholipids is associated with ER stress and
lipotoxicity in palmitate treated hepatic cells. J Lipid Res.
55:1478–1488. 2014.
|
|
96
|
Sanyal AJ, Campbell-Sargent C, Mirshahi F,
Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML and Clore
JN: Nonalcoholic steatohepatitis: association of insulin resistance
and mitochondrial abnormalities. Gastroenterology. 120:1183–1192.
2001.
|
|
97
|
Hirsova P, Ibrabim SH, Gores GJ and Malhi
H: Lipotoxic lethal and sublethal stress signaling in hepatocytes:
Relevance to NASH pathogenesis. J Lipid Res. 57:1758–1770.
2016.
|
|
98
|
Wei Y, Wang D, Topczewski F and
Pagliassotti MJ: Saturated fatty acids induce endoplasmic reticulum
stress and apoptosis independently of ceramide in liver cells. Am J
Physiol Endocrinol Metab. 291:E275–E281. 2006.
|
|
99
|
Leamy AK, Egnatchik RA and Young JD:
Molecular mechanisms and the role of saturated fatty acids in the
progression of non-alcoholic fatty liver disease. Prog Lipid Res.
52:165–174. 2013.
|
|
100
|
Wang D, Wei Y and Pagliassotti MJ:
Saturated fatty acids promote endoplasmic reticulum stress and
liver injury in rats with hepatic steatosis. Endocrinology.
147:943–951. 2006.
|
|
101
|
Diakogiannaki E, Welters HJ and Morgan NG:
Differential regulation of the endoplasmic reticulum stress
response in pancreatic beta-cells exposed to long-chain saturated
and monounsaturated fatty acids. J Endocrinol. 197:553–563.
2008.
|
|
102
|
Borradaile NM, Han X, Harp JD, Gale SE,
Ory DS and Schaffer JE: Disruption of endoplasmic reticulum
structure and integrity in lipotoxic cell death. J Lipid Res.
47:2726–2737. 2006.
|
|
103
|
Cao SS and Kaufman RJ: Targeting
endoplasmic reticulum stress in metabolic disease. Expert Opin Ther
Targets. 17:437–448. 2013.
|
|
104
|
Senft D and Ronai ZA: UPR, autophagy, and
mitochondria crosstalk underlies the ER stress response. Trends
Biochem Sci. 40:141–148. 2015.
|
|
105
|
Hetz C: The unfolded protein response:
controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012.
|
|
106
|
Hetz C, Zhang K and Kaufman RJ:
Mechanisms, regulation and functions of the unfolded protein
response. Nat Rev Mol Cell Biol. 21:421–438. 2020.
|
|
107
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011.
|
|
108
|
Wang H, Karnati S and Madhusudhan T:
Regulation of the homeostatic unfolded protein response in diabetic
nephropathy. Pharmaceuticals (Basel). 15:4012022.
|
|
109
|
Lebeaupin C, Vallée D, Hazari Y, Hetz C,
Chevet E and Bailly-Maitre B: Endoplasmic reticulum stress
signalling and the pathogenesis of non-alcoholic fatty liver
disease. J Hepatol. 69:927–947. 2018.
|
|
110
|
Hardy S, El-Assaad W, Przybytkowski E,
Joly E, Prentki M and Langelier Y: Saturated fatty acid-induced
apoptosis in MDA-MB-231 breast cancer cells. A role for
cardiolipin. J Biol Chem. 278:31861–31870. 2003.
|
|
111
|
Okere IC, Chandler MP, McElfresh TA,
Rennison JH, Sharov V, Sabbah HN, Tserng KY, Hoit BD, Ernsberger P,
Young ME and Stanley WC: Differential effects of saturated and
unsaturated fatty acid diets on cardiomyocyte apoptosis, adipose
distribution, and serum leptin. Am J Physiol Heart Circ Physiol.
291:H38–H44. 2006.
|
|
112
|
Wang J, Hu R, Yin C and Xiao Y: Tanshinone
IIA reduces palmitate-induced apoptosis via inhibition of
endoplasmic reticulum stress in HepG2 liver cells. Fundam Clin
Pharmacol. 34:249–262. 2020.
|
|
113
|
Malhi H and Gores GJ: Molecular mechanisms
of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver
Dis. 28:360–369. 2008.
|
|
114
|
Listenberger LL, Han X, Lewis SE, Cases S,
Farese RV Jr, Ory DS and Schaffer JE: Triglyceride accumulation
protects against fatty acid-induced lipotoxicity. Proc Natl Acad
Sci USA. 100:3077–3082. 2003.
|
|
115
|
Ariyama H, Kono N, Matsuda S, Inoue T and
Arai H: Decrease in membrane phospholipid unsaturation induces
unfolded protein response. J Biol Chem. 285:22027–22035. 2010.
|
|
116
|
Holzer RG, Park EJ, Li N, Tran H, Chen M,
Choi C, Solinas G and Karin M: Saturated fatty acids induce c-Src
clustering within membrane subdomains, leading to JNK activation.
Cell. 147:173–184. 2011.
|
|
117
|
Ishibashi M, Varin A, Filomenko R, Lopez
T, Athias A, Gambert P, Blache D, Thomas C, Gautier T, Lagrost L
and Masson D: Liver x receptor regulates arachidonic acid
distribution and eicosanoid release in human macrophages: a key
role for lysophosphatidylcholine acyltransferase 3. Arterioscler
Thromb Vasc Biol. 33:1171–1179. 2013.
|
|
118
|
Kawamura S, Matsushita Y, Kurosaki S,
Tange M, Fujiwara N, Hayata Y, Hayakawa Y, Suzuki N, Hata M, Tsuboi
M, et al: Inhibiting SCAP/SREBP exacerbates liver injury and
carcinogenesis in murine nonalcoholic steatohepatitis. J Clin
Invest. 132:e1518952022.
|
|
119
|
Jiang H, Li Z, Huan C and Jiang XC:
Macrophage lysophosphatidylcholine acyltransferase 3
deficiency-mediated inflammation is not sufficient to induce
atherosclerosis in a mouse model. Front Cardiovasc Med.
5:1922019.
|
|
120
|
Tintle NL, Pottala JV, Lacey S,
Ramachandran V, Westra J, Rogers A, Clark J, Olthoff B, Larson M,
Harris W and Shearer GC: A genome-wide association study of
saturated, mono- and polyunsaturated red blood cell fatty acids in
the Framingham Heart Offspring Study. Prostaglandins Leukot Essent
Fatty Acids. 94:65–72. 2015.
|
|
121
|
Hishikawa D, Hashidate T, Shimizu T and
Shindou H: Diversity and function of membrane glycerophospholipids
generated by the remodeling pathway in mammalian cells. J Lipid
Res. 55:799–807. 2014.
|
|
122
|
Xiang H, Shao M, Lu Y, Wang J, Wu T and Ji
G: Kaempferol alleviates steatosis and inflammation during early
non-alcoholic steatohepatitis associated with liver X Receptor
α-Lysophosphatidylcholine acyltransferase 3 signaling pathway.
Front Pharmacol. 12:6907362021.
|
|
123
|
Kakisaka K, Suzuki Y, Fujiwara Y, Suzuki
A, Kanazawa J and Takikawa Y: Caspase-independent hepatocyte death:
A result of the decrease of lysophosphatidylcholine acyltransferase
3 in non-alcoholic steatohepatitis. J Gastroenterol Hepatol.
34:1256–1262. 2019.
|
|
124
|
Wang B, Rong X, Palladino END, Wang J,
Fogelman AM, Martín MG, Alrefai WA, Ford DA and Tontonoz P:
Phospholipid Remodeling and Cholesterol Availability Regulate
Intestinal Stemness and Tumorigenesis. Cell Stem Cell.
22:206–220.e4. 2018.
|














