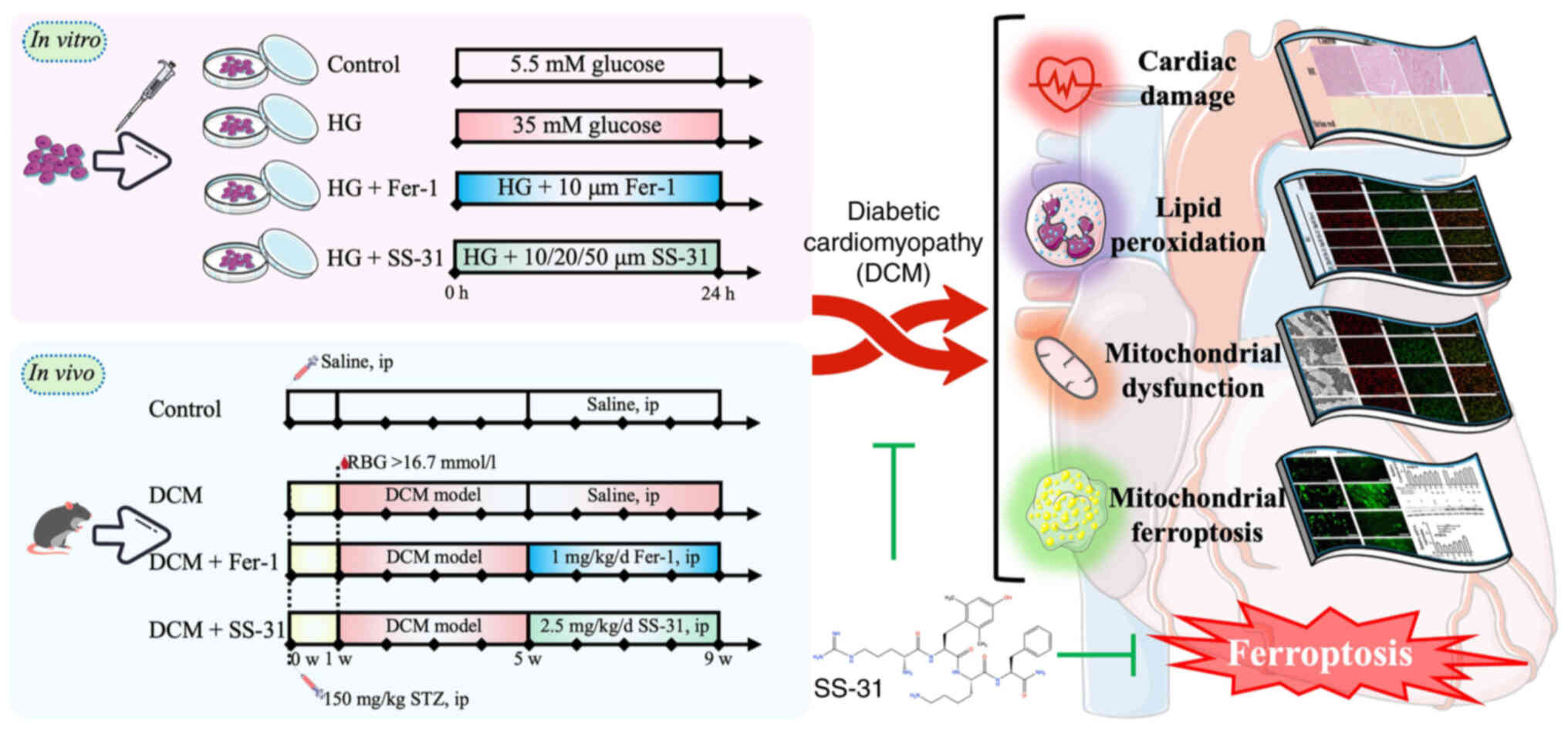
Introduction
Individuals with diabetes are at an increased risk
of developing cardiovascular disease, a risk that is 2-3 times
higher than that of the general population (1). Furthermore, 19-26% of diabetes
patients develop heart failure (HF), as blood glucose levels are
closely associated with the risk of HF (2). Diabetic cardiomyopathy (DCM) is the
prevailing cardiovascular complication of diabetes and has emerged
as a significant cause of death among diabetic patients (3). At present, DCM is defined as a
cardiac disorder based on diabetes, without any other cardiac
disease, and ultimately leads to HF (4).
The molecular mechanisms underlying DCM are
multi-faceted and the incorporation of ferroptosis has propelled
research to a new stage (5). The
concept of ferroptosis was initially proposed by Dixon et al
(6) and was characterized as an
iron-dependent regulated cell death caused by lethal lipid
peroxidation. The lipophilic radical-trapping antioxidant,
ferrostatin-1 (Fer-1), is an inhibitor of ferroptosis (7) and glutathione peroxidase 4 (GPX4)
is an endogenous lipid peroxide (LPO) scavenger that serves a
pivotal role in the defense against ferroptosis (8). Our previous research revealed the
involvement of ferroptosis in DCM, which was concomitant with the
suppression of GPX4 and dysfunction of mitochondria (9). Due to the distinctive
characteristics of myocardial cells, the abundance of mitochondria,
mitochondrial dysfunction (10)
and mitochondrial oxidative stress (11) are particularly important in the
pathogenesis of DCM. The presence of shrunken mitochondria,
increased mitochondrial membrane density and the disappearance of
mitochondrial cristae in cells undergoing ferroptosis indicates the
involvement of the mitochondria in ferroptosis and suggests the
mitochondria as the core site (12). It is noteworthy that GPX4 exists
as isoforms in the cytosol (cytoGPX4), mitochondria (mitoGPX4) and
nucleus (nuclGPX4), exerting its anti-ferroptosis efficacy
(13). Tadokoro et al
(14) confirmed that
mitoGPX4-mediated mitochondria-dependent ferroptosis has a pivotal
role in the progression of doxorubicin-induced cardiomyopathy
(DIC). However, the investigation of mitochondria-dependent
ferroptosis in DCM remains limited.
SS-31, also known as MTP-131, elamipretide and
Bendavia, is a tetrapeptide and a novel mitochondria-targeting
antioxidant (15). Current
research indicates that SS-31 exhibits exceptional biocompatibility
and safety (16) and has
demonstrated notable therapeutic potential for different types of
cardiomyopathy, such as hypertensive cardiomyopathy (17), dilated cardiomyopathy (18) and DIC (19). Nevertheless, to the best of the
authors' knowledge, the effect of SS-31 on DCM remains unexplored.
In addition to the direct effect on mitochondria, SS-31 also
mitigates oxidative stress and ferroptosis by regulating the
related signaling pathways. The relationship between SS-31 and
ferroptosis has also been validated in neurodegenerative diseases
(20,21), and this evidence points SS-31
towards GPX4, which is a key regulatory factor in ferroptosis.
Therefore, the present study investigated the
association between mitochondria-dependent ferroptosis and the
pathogenesis of DCM and explored a potential therapeutic strategy
for DCM by alleviating mitochondria-dependent ferroptosis using
SS-31.
Materials and methods
Cells and treatment
H9C2 rat cardiomyocyte cells were obtained from the
Chinese National Infrastructure of Cell Line Resource (cat. no.
1101RAT-PUMC000219). The H9C2 cells were cultured in DMEM
containing 5.5 mmol/l glucose (cat. no. 10567022; Gibco; Thermo
Fisher Scientific, Inc.), supplemented with 10% fetal bovine serum
(cat. no. 10100147C; Gibco; Thermo Fisher Scientific, Inc.), 100
U/ml penicillin and 100 μg/ml streptomycin (cat. no.
B540732; Sangon Biotech Co., Ltd.). The cell incubator (370; Thermo
Fisher Scientific, Inc.) was set at 37°C with a 5% CO2
environment. An in vitro DCM model was established by
treating H9C2 cells with 35 mmol/l glucose for 24 h, according to
the protocol reported by previous studies (9,10).
As shown in Fig.
1, the H9C2 cells were divided into four groups: i) Control
group, cultured in 5.5 mmol/l glucose for 24 h; ii) high glucose
(HG) group, cultured in 35 mmol/l glucose for 24 h; iii) HG + Fer-1
group, cultured in 35 mmol/l glucose and 10 μM Fer-1 for 24
h; and iv) HG + SS-31 group, cultured in 35 mmol/l glucose and 10,
20 or 50 μM SS-31 for 24 h.
Cell viability assay
The cell viability was assessed by Cell Counting
Kit-8 (CCK-8; cat no. E606335; Sangon Biotech Co., Ltd.). The H9C2
cells were seeded into 96-well plates at a density of
2×103 cells/well and allowed to adhere. Following a 6-h
serum starvation period for cell cycle synchronization, the cells
were treated as aforementioned. After 24 h of treatment, the 100
μl medium in each well was replaced with 10% CCK-8 reagent
and the cells were incubated for another 2 h. The absorbance at 450
nm was then measured using a microplate reader (Multiskan GO;
Thermo Fisher Scientific, Inc.). The cell viability was determined
as follows:
(Aexperiment-Ablank)/(Acontrol-Ablank)
x100%.
Animal model
A total of 40 male C57BL/6J mice (aged 6-8 weeks,
weighing 18-20 g) were purchased from Shanghai Slack Laboratory
Animal Co., Ltd. [production license no. SCXK (Shanghai)
2017-0005]. The mice were housed in the Zhejiang Chinese Medical
University Laboratory Animal Research Center (Hangzhou, China)
under a specific pathogen-free environment (room temperature,
22±2°C; humidity, 55±5%; 12-h light/dark cycle), with unrestricted
access to food and water. All experimental procedures were approved
by the Animal Ethical and Welfare Committee of ZCMU (approval no.
IACUC-20220307-10).
Prior to the experiment, the mice were acclimated
for 1 week. As shown in Fig. 1,
the diabetes model was established by overnight fasting followed by
a single intraperitoneal injection of 150 mg/kg streptozotocin
(cat. no. V900890; Sigma-Aldrich; Merck KGaA). Tail snip was used
to obtain tail-tip blood, and blood-glucose was detected by a
glucose meter (Accu-Chek® Performa test strips and an
Accu-Chek® Performa Combo; both Roche Diabetes Care
GmbH) 3, 5 and 7 days after injection. A random blood-glucose value
of ≥16.7 mmol/l was classified as having diabetes, which was
maintained for 4 weeks to induce DCM (9). Subsequently, the mice were divided
into 4 groups (n=8): i) Control group, healthy mice were
intraperitoneally injected with saline for 4 weeks; ii) DCM group,
DCM model mice were intraperitoneally injected with saline for 4
weeks; iii) DCM + Fer-1 group, DCM model mice were
intraperitoneally injected with 1 mg/kg/day Fer-1 (cat. no.
HY-100579; MedChemExpress) for 4 weeks; and iv): DCM + SS-31 group,
DCM model mice were intraperitoneally injected with 2.5 mg/kg/day
SS-31 (cat. no. B27916; Shanghai Yuanye Biotechnology, Co., Ltd.)
for 4 weeks.
The humane end points, which involve weight loss,
loss of appetite, weakness (inability to eat or drink) and
cardiovascular system, were established for the present study.
However, none of the mice in the present study reached these
endpoints.
Echocardiograms
The mice were anesthetized with 1% isoflurane in
100% oxygen and placed on a heating pad to keep warm (22). Then, transthoracic
echocardiography was performed using an ultrasound system
(VisualSonics Vevo 2100; FUJIFILM VisualSonics, Inc.) with a 30 MHz
probe (MX400; FUJIFILM VisualSonics, Inc.). The left ventricle
internal diameter in diastole (LVIDd), left ventricle internal
diameter in systole (LVIDs), interventricular septum in diastole
(IVSd), interventricular septum in systole (IVSs), left ventricular
posterior wall in diastole (LVPWd) and left ventricular posterior
wall in systole (LVPWs) were measured in M-mode. Subsequently, the
left ventricular end-diastolic volume (LVEDV), left ventricular
end-systolic volume (LVESV), fractional shortening (FS) and
ejection fraction (EF) were calculated using the Teichholtz formula
as follows (22): LVEDV=[7/(2.4
+ LVIDd)] x LVIDd3; LVESV=[7/(2.4 + LVIDs)] x
LVIDs3; FS=[(LVIDd - LVIDs)/LVIDd] x100%; and EF=[(LVEDV
- LVESV)/LVEDV] x100%.
Collection of blood and tissue
samples
At the end of the experiment, the cardiac puncture
was performed before sacrifice. In brief, after being anesthetized
by sodium pentobarbital (50 mg/kg), a needle was inserted at the
top of the sternum to collected 200 μl blood slowly. After
collecting blood, a lethal dose of sodium pentobarbital (150 mg/kg)
was immediately injected, followed by dissection and collection of
cardiac tissue. The serum was then separated by centrifugation
(1,500 x g, 10 min, 4°C) and stored at -80°C. A small piece of the
left ventricle tissues was fixed in 4% paraformaldehyde (cat. no.
E672002; Sangon Biotech Co., Ltd.) at 4°C overnight, or 2.5%
glutaraldehyde (cat. no. A17876; Alfa Aesar; Thermo Fisher
Scientific, Inc.) at 4°C overnight, for histopathological analysis
and transmission electron microscopy (TEM). The remaining samples
were rapidly frozen by liquid nitrogen and stored at -80°C.
Biochemical analysis
The serum samples were examined using an automated
biochemical analyzer (3100; Hitachi High-Technologies Corporation)
for the quantification of blood glucose, lactate dehydrogenase
(LDH) and creatine kinase isoenzymes (CK-MB) levels.
Histopathology
The left ventricular tissues fixed in 4%
paraformaldehyde (4°C overnight) were subjected to
histopathological examination using H&E (hematoxylin for 10
min, eosin for 2 min, room temperature) and Sirius red (30 min,
room temperature) staining to evaluate the pathological changes.
Images were captured under a fluorescence upright microscope
(Axioscope A1; Zeiss GmbH).
TEM
The left ventricular tissues fixed with 2.5%
glutaraldehyde (4°C overnight) underwent post-fixation (1%
OsO4+1.5%K3[Fe(CN)3] for 1 h at
4°C, TCH for 60 min, 1% OsO4 post-fixation for 1 h at
4°C, 2% uranyl acetate solution for overnight at 4°C), dehydration
(50-100% gradient ethanol), resin penetration and embedding
(acetone: EMBed 812=1:1 for 1 h, acetone: EMBed 812=1:3 for
overnight, two changes of pure EMBed 812 for 8 h), polymerization
(60°C for 48 h) and sectioning (60-80 nm, then onto the copper
screen). The myofilament and mitochondria of the myocardial cells
were imaged by a transmission electron microscope (H-7500; Hitachi
High-Technologies Corporation).
Mitochondria isolation
The mitochondria were isolated using a Tissue
Mitochondria Isolation kit (cat. no. C3606; Beyotime Institute of
Biotechnology) and a Mitochondria Isolation kit for Cultured Cells
(cat. no. 89874; Thermo Fisher Scientific, Inc.). Subsequently,
RIPA lysis buffer (cat. no. C500005; Sangon Biotech Co., Ltd.)
containing protease inhibitor (cat. no. C600380; Sangon Biotech
Co., Ltd.) was used to lyse the mitochondria and extract the
mitochondrial proteins.
Redox status determination
In total, 2×105 H9C2 cells/well were
seeded into 6-well plates. The malondialdehyde (MDA),
4-Hydroxynonenal (4-HNE), ferrous iron and glutathione (GSH)
content of the cell and tissue samples were determined using a
Malondialdehyde Content Assay kit (cat. no. D799762; Sangon Biotech
Co., Ltd.), 4-Hydroxynonenal ELISA kit (cat. no. D751041; Sangon
Biotech Co., Ltd.), Ferrous Iron Colorimetric Assay kit (cat. no.
E-BC-K773-M; Wuhan Elabscience Biotechnology Co., Ltd.) and GSH
Content Assay kit (cat. no. D799614; Sangon Biotech Co., Ltd.),
respectively. The protein concentrations were quantified using a
BCA Protein Assay kit (cat. no. C503051; Sangon Biotech Co., Ltd.).
The results were detected using a microplate reader and expressed
in terms of protein concentration.
Total LPO assay
The total LPO assay was conducted using BODIPY
581/591 C11 (cat. no. D3861; Thermo Fisher Scientific, Inc.), which
shifts fluorescence properties from red to green signals upon
oxidation. Following the protocol described by Martinez et
al (23), H9C2 cells were
incubated with 2.5 μM BODIPY 581/591 C11 at 37°C for 30 min.
After washing with PBS, fluorescence images were captured using a
fluorescence inverted microscope (Axio Observer D1; Zeiss GmbH) in
the FITC and Rhodamine channel. ImageJ software (version 1.53t;
National Institutes of Health) was used for fluorescence intensity
analysis.
Adenosine triphosphate (ATP)
detection
The ATP content and protein concentration in the
cell and tissue samples were measured using the ATP Assay kit (cat.
no. S0026; Beyotime Institute of Biotechnology) and the BCA Protein
Assay kit, respectively. Data were obtained using a Luminometer
(GloMax 20/20; Promega Corporation) or a microplate reader, with
the final results expressed in terms of protein concentration.
Mitochondrial membrane potential (MMP)
assay
The MMP assay was conducted using JC-1 (cat. no.
C2006; Beyotime Institute of Biotechnology). In a higher MMP, JC-1
aggregates in the matrix of the mitochondria, forming J-aggregates
and emitting red fluorescence; conversely, once MMP is decreased,
JC-1 remains as a monomer and does not aggregate in the matrix of
the mitochondria and instead emits green fluorescence. Briefly,
H9C2 cells were incubated with 10 μg/ml JC-1 at 37°C for 30
min. After washing with buffer, fluorescence images were captured
using a fluorescence inverted microscope (Axio Observer D1; Zeiss
GmbH) in the FITC and Rhodamine channel. ImageJ software (version
1.53t; National Institutes of Health) was used for fluorescence
intensity analysis.
Mitochondrial LPO (mitoLPO) assay
The fluorescent dye, MitoPeDPP (cat. no. M466;
Dojindo Laboratories, Inc.), localizes to the inner mitochondrial
membrane and emits strong fluorescence upon oxidation. Briefly,
H9C2 cells were incubated with 0.5 μM MitoPeDPP at 37°C for
15 min. After washing with PBS, fluorescence images were captured
using a fluorescence inverted microscope (Axio Observer D1; Zeiss
GmbH) in the FITC channel. ImageJ software (version 1.53t; National
Institutes of Health) was used for fluorescence intensity
analysis.
Mitochondrial ferrous iron assay
The fluorescent dye, Mito-FerroGreen (cat. no. M489;
Dojindo Laboratories, Inc.), can detect ferrous ions specifically
in the mitochondria, with the fluorescence intensity exhibiting a
positive correlation with the concentration of ferrous ions in the
mitochondria. Briefly, H9C2 cells were incubated with 5 μM
Mito-FerroGreen at 37°C for 30 min. After washing with serum-free
medium, fluorescence images were captured using a fluorescence
inverted microscope in the FITC channel. ImageJ software (version
1.53t; National Institutes of Health) was used for fluorescence
intensity analysis.
Reverse transcription-quantitative (RT-q)
PCR
Total RNA from cells on 6-well plate or 50 mg tissue
was extracted by RNAiso Plus reagent (cat. no. 9109; Takara Bio,
Inc.) and quantified using a micro-spectrophotometer (Quickdrop;
Molecular Devices, LLC). Subsequently, cDNA was synthesized using
PrimeScript RT Master Mix (cat. no. RR036; Takara Bio, Inc.). The
qPCR assay was next performed using TB Green® Premix Ex
Taq II (cat. no. RR820; Takara Bio, Inc.) and a Real-Time PCR
system (ABI 7500; Applied Biosystems; Thermo Fisher Scientific,
Inc.) with the following thermocycling conditions: 95°C for 30 sec,
1 cycle; 95°C for 5 sec and 60°C for 34 sec, 40 cycles. All
experiments were performed according to the manufacturers'
instructions and included at least three biological replicates.
β-actin served as the reference gene and the relative expression
levels were calculated using the 2−ΔΔCq method (24). The qPCR primer sequences are
listed in Table I. The primers
for totalGPX4 were designed to target all isoforms of GPX4, while
the primers for mitoGPX4 were specifically designed to recognize
the unique sequence of mitoGPX4.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Species | Gene | Forward sequence
(5′-3′) | Reverse sequence
(5′-3′) |
|---|
| Mouse | totalGPX4 |
CGCGATGATTGGCGCT |
CACACGAAACCCCTGTACTTATCC |
| mitoGPX4 |
GATGAGCTGGGGCCGTCTG |
CGTTGGTGACGATGCACAC |
| β-actin |
GTGCTATGTTGCTCTAGACTTCG |
ATGCCACAGGATTCCATACC |
| Rat | totalGPX4 |
AATTCGCAGCCAAGGACATC |
GGCCAGGATTCGTAAACCAC |
| mitoGPX4 |
GCCGTCTGAGCCGCTTATTG |
GTCGGTTTTGCCTCATTGCG |
| β-actin |
GTCCACCCGCGAGTACAAC |
TATCGTCATCCATGGCGAACTGG |
Western blotting
Western blotting analysis was performed using total
and mitochondrial protein. The total proteins were extracted using
RIPA lysis buffer containing protease inhibitor, and a tissue
grinder (Scientz-48; Ningbo Scientz Biotechnology, Co., Ltd.) for
the tissue samples. After the protein concentration was quantified
by a BCA protein assay kit, 30 μg protein for each sample
was separated using a 10% SDS-PAGE gel, then transferred to a PVDF
membrane (cat. no. F619534; Sangon Biotech Co., Ltd.). After
blocking with 5% Block Buffer (cat. no. A600669; Sangon Biotech
Co., Ltd.) for 1 h at room temperature, the membrane was incubated
with anti-GPX4 (1:1,000; cat. no. ab125066; Abcam), anti-β-actin
(1:1,000; cat. no. ab8227; Abcam) and anti-voltage dependent anion
channel 1 (1:1,000; cat. no. ab15895; Abcam) primary antibodies at
4°C overnight, then with a goat anti-rabbit (1:2,000; cat. no.
ab6721; Abcam) secondary antibody (HRP) at room temperature for 1
h. Finally, the bands was visualized by ECL luminescence reagent
(cat. no. C500044; Sangon Biotech Co., Ltd.) and a chemiluminescent
imaging system (5200multi; Tanon Science and Technology Co., Ltd.).
ImageJ software (version 1.53t; National Institutes of Health) was
used for grayscale analysis.
Statistical analysis
The results were presented as the mean ± standard
deviation. Statistical analysis was performed using SPSS software
(version 19.0; IBM Corp.). Statistical comparisons were conducted
by one-way ANOVA follow by the Bonferroni post hoc test. P<0.05
was considered to indicate a statistically significant
difference.
Results
SS-31 restores mitochondrial dysfunction
induced by high glucose in H9C2 cells
First, in vitro experiments were conducted to
reveal the restorative effect of SS-31 on high glucose-induced
mitochondrial dysfunction in H9C2 cells. Briefly, 35 mM glucose was
employed to induce high glucose damage, then the cells were treated
with 10, 20 or 50 μM SS-31. Meanwhile, a positive control
group was treated with 10 μM Fer-1, a ferroptosis inhibitor.
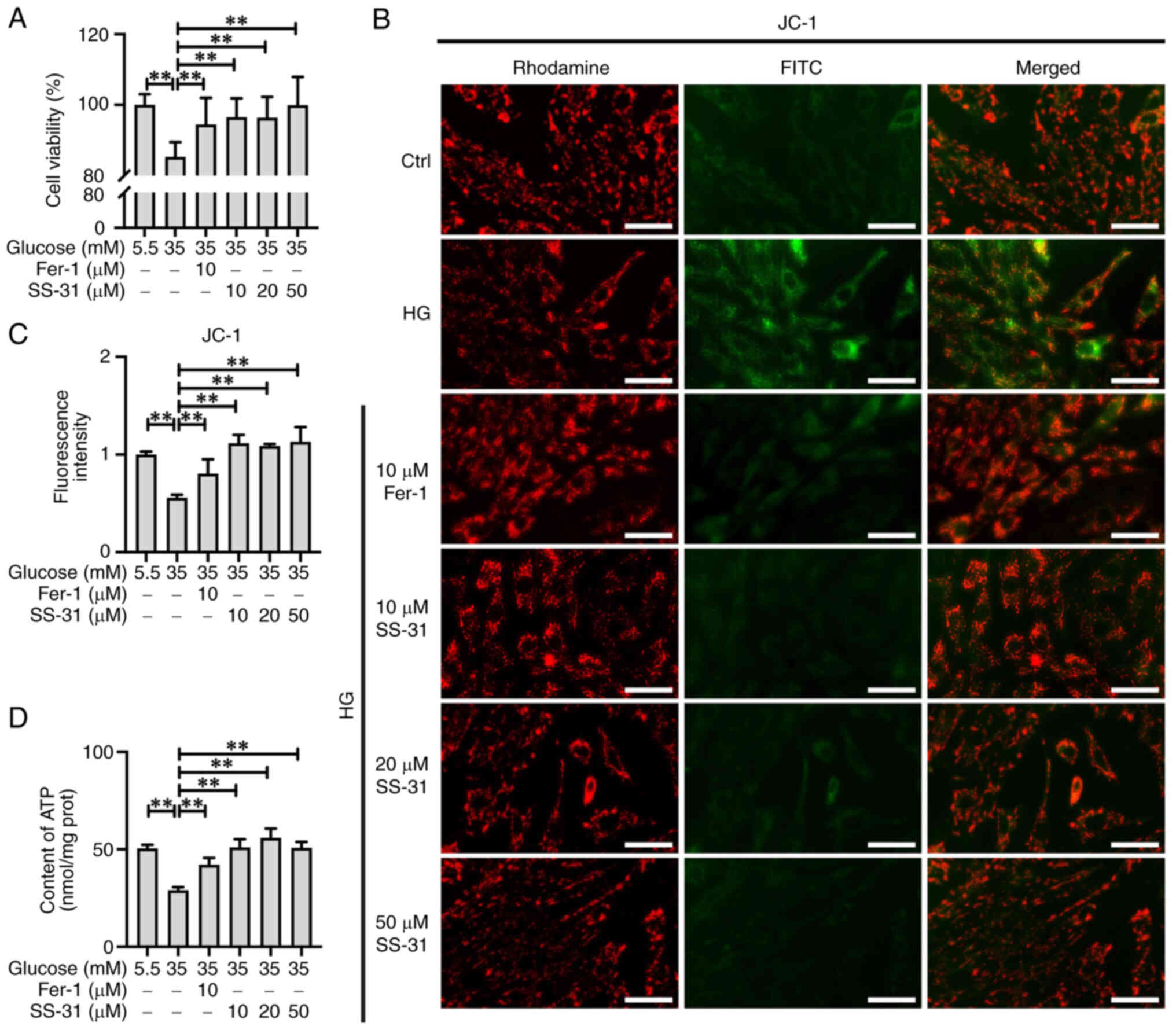
As shown in Fig. 2A, the cell
viability assay results indicated that 35 mM glucose significantly
suppressed H9C2 cell viability (P<0.01), while treatment with
SS-31 or Fer-1 effectively restored this impaired cell viability
(P<0.01). The MMP was assessed by employing JC-1 fluorescence
staining (Fig. 2B and C). It was
observed that high glucose resulted in a decrease in MMP in H9C2
cells (P<0.01), while both Fer-1 and SS-31 enhanced the MMP in
high glucose-treated H9C2 cells (P<0.01) with a stronger
restorative effect observed in the HG + SS-31 group. Furthermore,
the results of the ATP content analysis demonstrated that the HG
group exhibited a significant decrease in ATP (P<0.01), which
was effectively restored by Fer-1 (P<0.01) or SS-31 (P<0.01),
with SS-31 exhibiting a superior efficacy in restoring ATP content
(Fig. 2D). These results
suggested that SS-31 could effectively restore the mitochondrial
dysfunction induced by high glucose in H9C2 cells.
SS-31 alleviates LPO damage induced by
high glucose in H9C2 cells
In the present study, MDA and 4-HNE were detected as
reliable markers of LPO (Fig. 3A and
B). Compared with the control group, the HG group exhibited a
significant elevation in MDA (P<0.01) and 4-HNE (P<0.01)
levels. Furthermore, the treatments with Fer-1 and SS-31 both
significantly suppressed the production of MDA and 4-HNE
(P<0.01). The visualization of totalLPO and mitochondrial
(mito)LPO in H9C2 cells was achieved using BODIPY 581/591 C11 and
mitoPEDPP, respectively (Fig. 3C and
D). The results demonstrated a significant increase in both
totalLPO (P<0.01) and mitoLPO (P<0.01) in high
glucose-treated H9C2 cells, with the mitoLPO exhibiting higher an
accumulation compared with totalLPO. Furthermore, the treatments
with Fer-1 and SS-31 showed effective reductions in the
accumulation of totalLPO (P<0.01) and mitoLPO (P<0.01).
Notably, SS-31 had a stronger capacity to counteract the increase
in mitoLPO compared with Fer-1. These results suggested that both
Fer-1 and SS-31 were effective in alleviating LPO accumulation,
with SS-31 exhibiting an improved efficacy in alleviating
mitoLPO.
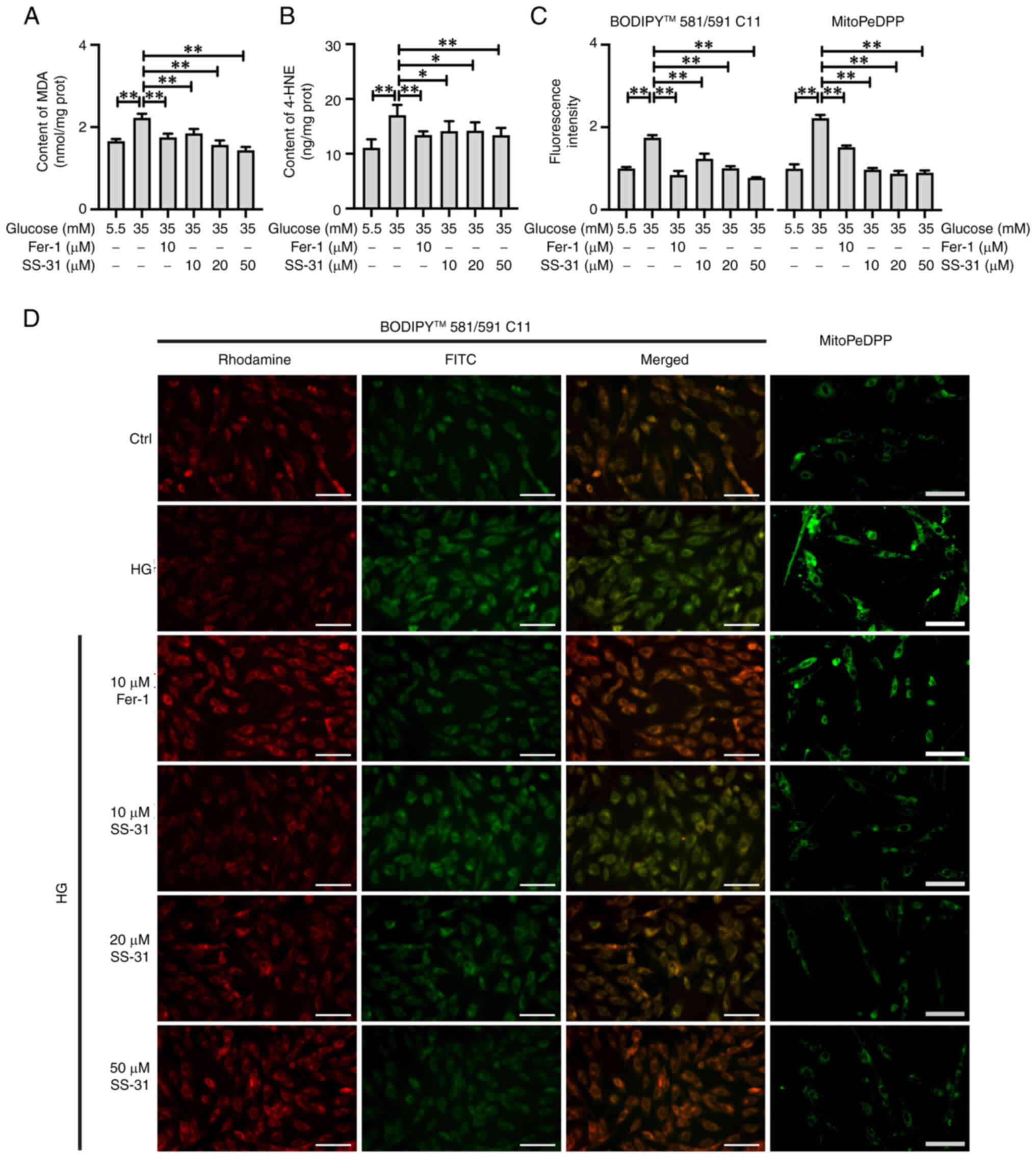 | Figure 3SS-31 mitigates the LPO damage
induced by HG in H9C2 cells. (A) MDA and (B) 4-HNE content of
treated cells. (C) Fluorescence intensity and (D) representative
images of total LPO and mitochondrial LPO detected by BODIPY
581/591 C11 and MitoPEDPP, respectively. The Rhodamine channel
represents the unoxidized form, the FITC channel represents the
oxidized form. Scale bar, 100 μm. *P<0.05,
**P<0.01. LPO, lipid peroxide; HG, high glucose; MDA,
malondialdehyde; HNE, 4-Hydroxynonenal; Ctrl, control; Fer-1,
ferrostatin-1. |
SS-31 promotes mitoGPX4 to alleviate
mitochondria-dependent ferroptosis
Accumulation of labile iron is a pivotal contributor
to ferroptosis; therefore, Mito-FerroGreen was employed for the
visualization and quantification of mitochondrial ferrous ions
(Fig. 4A and B) and the Ferrous
Iron Colorimetric Assay kit was employed for the quantification of
the total ferrous ions (Fig. 4C)
in H9C2 cells. The results showed a significant accumulation of
total (P<0.01) and mitochondrial (P<0.01) ferrous ions in
H9C2 cells treated with high glucose. Although both Fer-1
(P<0.01) and SS-31 (P<0.05) exhibited the capacity to
alleviate the accumulation of total and mitochondrial ferrous ions,
Fer-1 exerted a more notable effect.
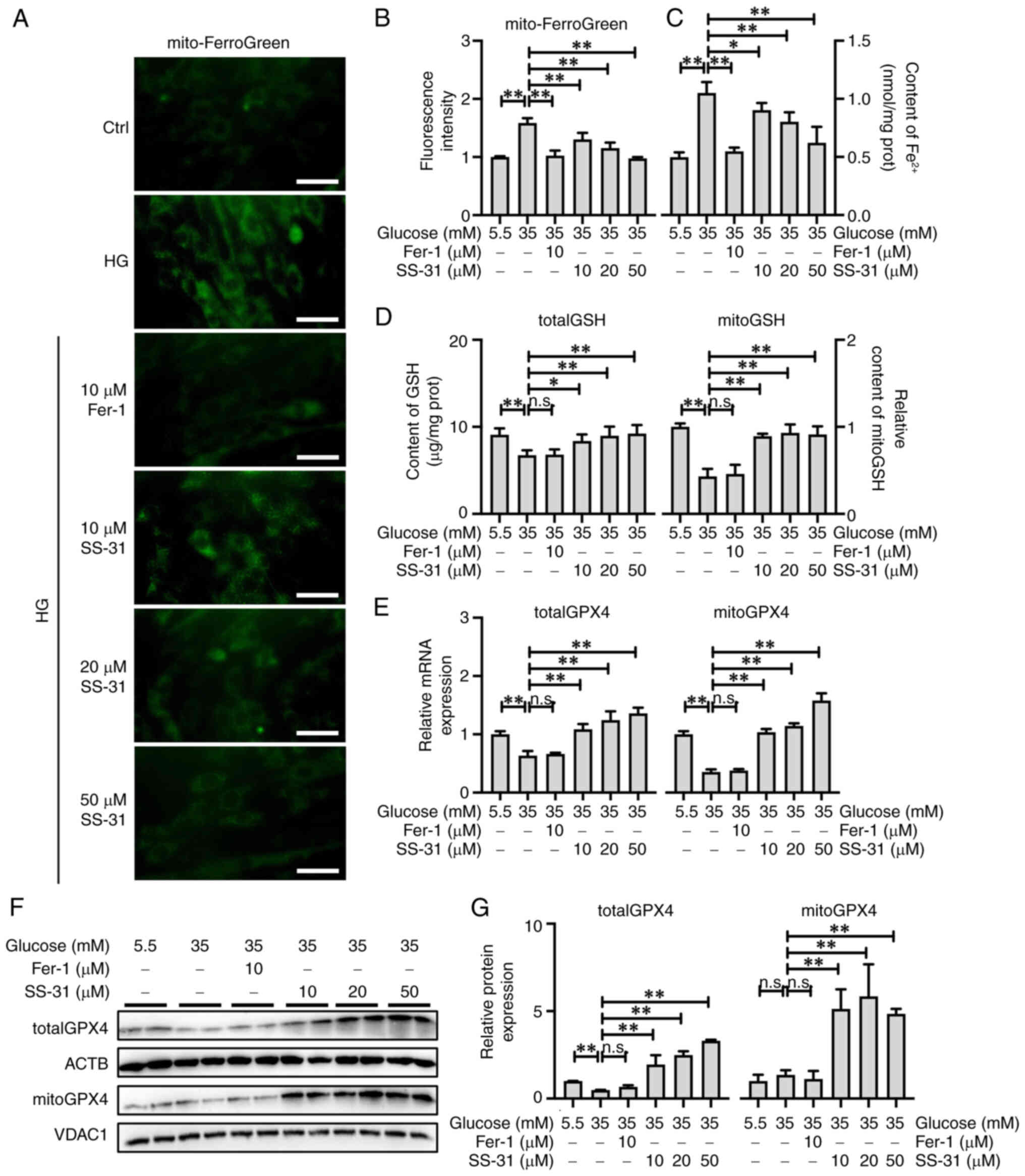 | Figure 4SS-31 promotes mitoGPX4 to alleviate
mitochondria-dependent ferroptosis. (A) Representative images and
(B) fluorescence intensity of mito ferrous ions detect by
Mito-FerroGreen, Scale bar, 50 μm. (C) Total ferrous ions
and (D) totalGSH and mitoGSH content. (E) Relative mRNA expression
of totalGPX4 and mitoGPX4. (F) Western blotting and (G)
semi-quantification of totalGPX4 and mitoGPX4 protein expression.
*P<0.05, **P<0.01; n.s., no
significance; mito, mitochondrial; GPX4, glutathione peroxidase 4;
GSH, glutathione; ACTB, β-actin; Ctrl, control; Fer-1,
ferrostatin-1; HG, high glucose; VDAC1, voltage dependent anion
channel 1. |
GSH serves as the crucial substrate for GPX4 to
effectively exert its anti-LPO function. Similar to GPX4, GSH is
also distributed in the cytosol and mitochondria. As shown in
Fig. 4D, totalGSH (P<0.01)
and mitoGSH (P<0.01) were significantly decreased in high
glucose-treated H9C2 cells, and Fer-1 failed to restore the
suppression in totalGSH (P>0.05) and mitoGSH (P<0.05) levels.
By contrast, SS-31 not only reinstated the totalGSH levels
(P<0.05) but also restored the mitoGSH levels (P<0.01).
Detection of GPX4 expression in high glucose-treated H9C2 cells
demonstrated that totalGPX4 (P<0.01) and mitoGPX4
(P<0.01) mRNA expression was significantly reduced (Fig. 4E); however, only the protein
expression of totalGPX4 was significantly inhibited (P<0.01;
Fig. 4F and G). Additionally,
treatment with Fer-1 did not affect the expression of totalGPX4
(P>0.05) and mitoGPX4 (P>0.05), whereas SS-31 significantly
upregulated both the mRNA and protein expression of totalGPX4
(P<0.01) and mitoGPX4 (P<0.01), with a higher increase in
mitoGPX4 than totalGPX4 protein expression (Fig. 4E-G). Therefore, it was
hypothesized that, although Fer-1 and SS-31 exhibited efficacy in
alleviating ferroptosis induced by high glucose in H9C2 cells,
their underlying mechanisms were different. Specifically, SS-31
demonstrated the capacity to activate the GSH/GPX4 pathway, while
more notably activating the mitoGSH/mitoGPX4 pathway in
mitochondria.
SS-31 alleviates myocardial injury in DCM
mice
To further investigate the therapeutic effects of
SS-31 on DCM in vivo, a DCM mouse model was established and
administered intraperitoneal injections of 2.5 mg/kg/day SS-31 or 1
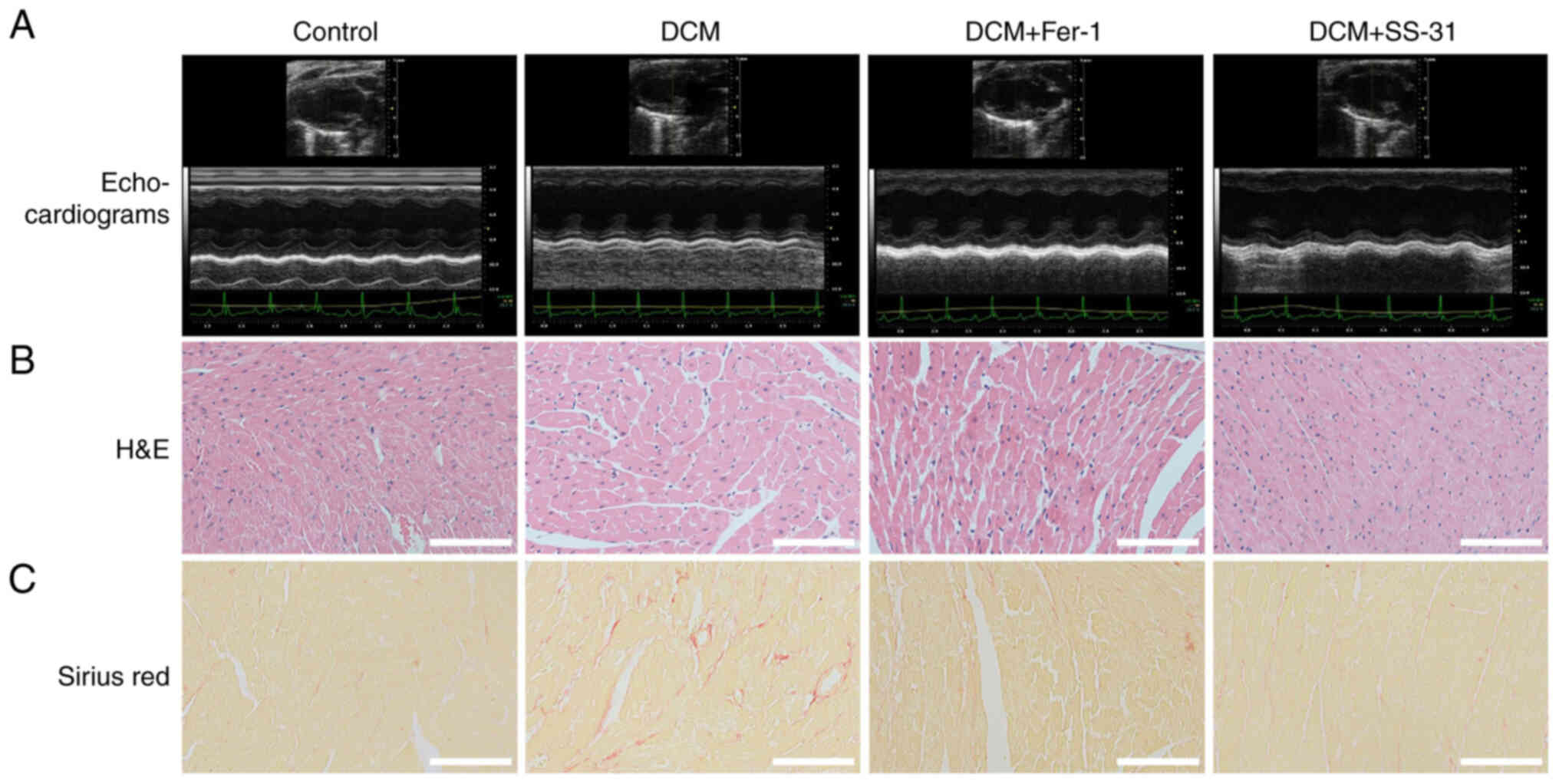
mg/kg/day Fer-1 for 4 weeks. As shown in Table II and Fig. 5A, at the end of the experiment,
the DCM mice exhibited significant diabetic symptoms with
hyperglycemia (P<0.01) and a low body weight (P<0.01).
However, treatment with Fer-1 or SS-31 did not ameliorate
hyperglycemia in the DCM mice (P>0.05), but SS-31 did improve
body weight (P<0.05). In terms of the cardiac parameters, there
was a significant decrease in cardiac weight (P<0.01), FS
(P<0.01) and EF (P<0.01) and a significant increase in LDH
(P<0.01) and CK-MB (P<0.01) in the DCM mice, indicating
cardiac damage. Conversely, treatment with Fer-1 or SS-31
effectively restored the cardiac weight (P<0.01), FS (DCM +
Fer-1, P<0.05; DCM + SS-31, P<0.01) and EF (DCM + Fer-1,
P<0.05; DCM + SS-31, P<0.01) and reduced LDH (P<0.01) and
CK-MB (P<0.01). Notably, the DCM + SS31 group displayed lower
CK-MB levels compared with the DCM + Fer-1 group, highlighting the
improved cardioprotective effect of SS-31 over Fer-1 on DCM mice.
The detailed echocardiographic results are available in Table SI.
| Table IIBasic characteristics of the mice in
each group at the end of the experiment. |
Table II
Basic characteristics of the mice in
each group at the end of the experiment.
| Characteristic | Control | DCM | DCM+Fer-1 | DCM+SS-31 |
|---|
| Body weight, g | 36.7±2.3 | 21.0±5.4b | 25.3±5.8b | 27.7±1.2b,c |
| Cardiac weight,
g | 0.17±0.02 | 0.12±0.02b | 0.15±0.01a,d | 0.16±0.01d |
| GLU (mM) | 7.3±0.6 | 39.7±3.2b | 36.9±2.9b | 37.0±2.4b |
| LDH (U/l) | 467±70 | 1053±104b | 701±105b,d | 655±92b,d |
| CK-MB (U/l) | 343±52 | 652±50b | 505±61b,d | 330±61d,e |
| FS (%) | 32.30±2.77 | 15.31±2.12b | 21.27±2.29b,c | 25.88±3.80a,d |
| EF (%) | 61.65±4.34 | 32.70±4.17b | 43.79±3.94b,c | 51.83±5.58a,d |
The histopathological staining of cardiac tissues is
shown in Fig. 5B and C. The
H&E staining showed that the myocardial fibers were intact and
aligned in the control group, but disorganized and even fractured
in the DCM group. In the DCM + SS-31 group, the disorder of the
myocardial fibers was improved. Sirius red staining revealed
collagen fiber deposition in the myocardial tissues of the DCM
group, whereas treatment with Fer-1 or SS-31 alleviated this
pathology. These results suggested that both Fer-1 and SS-31
exhibited cardioprotective effects in DCM mice.
SS-31 alleviates mitochondria-dependent
ferroptosis in DCM mice
The ultrastructure of the cardiac tissue from DCM
mice was visualized by TEM (Fig.
6A). There were significant alterations in the ultrastructure
of the cardiac tissue in DCM mice, including sarcomere disruption,
mitochondrial swelling, disarray and outer membrane rupture, as
well as disappearance or fragmentation of the mitochondrial
cristae. Treatment with SS-31 effectively corrected the
mitochondrial damage; it restored swelling, preserved the integrity
of the inner and outer membranes and enhanced the abundance of
cristae. Conversely, Fer-1 intervention exhibited a relatively
modest effect on the ultrastructure of mitochondria. The results of
the ATP content examination (Fig.
6B) demonstrated that the DCM cardiac tissue exhibited a
significant decrease in ATP content (P<0.01). However, treatment
with Fer-1 (P<0.05) or SS-31 (P<0.01) restored the ATP
content, with SS-31 exhibiting a more significant efficacy. These
results suggested that SS-31 exhibited an improved efficacy
compared with Fer-1 in alleviating the mitochondrial dysfunction in
the cardiac tissues of DCM mice.
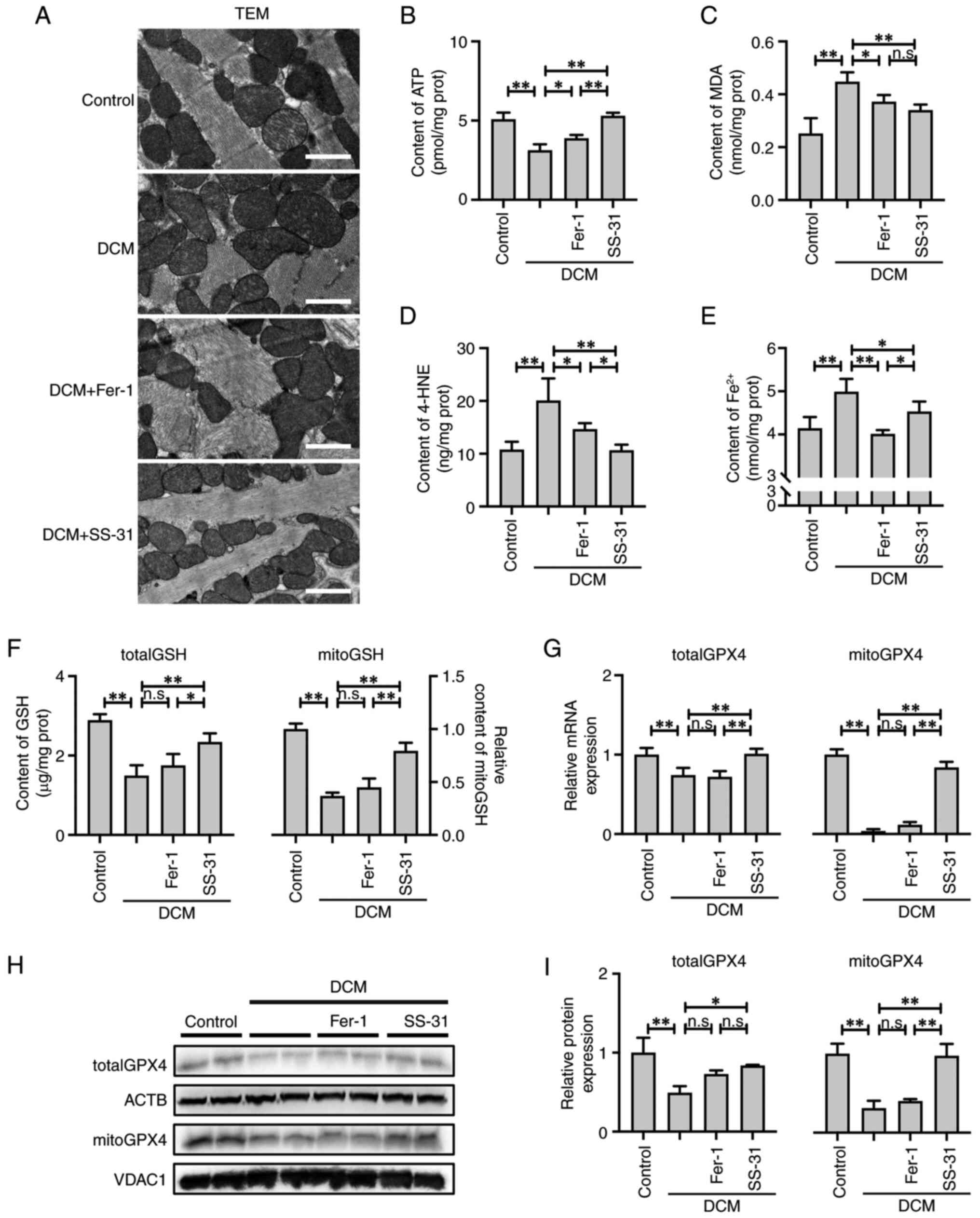 | Figure 6SS-31 alleviates
mitochondria-dependent ferroptosis in DCM mice by activating the
mitoGSH/mitoGPX4 pathway. (A) TEM of the ultrastructure of
myocardial tissues; scale bar, 1 μm. (B) ATP, (C) MDA, (D)
4-HNE, (E) total ferrous ion and (F) totalGSH and mitoGSH content.
(G) Relative mRNA expression of totalGPX4 and mitoGPX4. (H) Western
blotting and (I) semi-quantification of totalGPX4 and mitoGPX4
protein expression. *P<0.05, **P<0.01;
n.s., no significance. DCM, diabetic cardiomyopathy; mito,
mitochondrial; GSH, glutathione; GPX4, glutathione peroxidase 4;
TEM, transmission electron microscopy; ATP, adenosine triphosphate;
MDA, malondialdehyde; 4-HNE, 4-Hydroxynonenal; Fer-1,
ferrostatin-1; ACTB, β-actin; VDAC1, voltage dependent anion
channel 1. |
In the cardiac tissues of DCM mice, there was a
significant increase in MDA (P<0.01) and 4-HNE (P<0.01)
levels and an accumulation in total ferrous ions (P<0.01),
indicating severe LPO damage (Fig.
6C-E). However, treatment with Fer-1 or SS-31 effectively
suppressed the generation of MDA and 4-HNE as well as the
accumulation of total ferrous ions, with SS-31 demonstrating a
stronger inhibition of 4-HNE production (P<0.05) and Fer-1
demonstrating a stronger alleviation of ferrous ion accumulation
(P<0.05).
Similar to the in vitro experiments, the
GSH/GPX4 and mitoGSH/mitoGPX4 pathways in the cardiac tissues of
DCM mice were also analyzed. In the DCM group, both the totalGSH
(P<0.01) and mitoGSH (P<0.01) levels were significantly
reduced, which were only effectively restored by SS-31 treatment
(P<0.01; Fig. 6F). Consistent
with the aforementioned in vitro results, both the totalGPX4
and mitoGPX4 mRNA (Fig. 6G) and
protein (Fig. 6H and I)
expression levels were significantly suppressed in the cardiac
tissues of DCM mice (P<0.01), and Fer-1 failed to reverse this
inhibition (P>0.05); however, SS-31 did counteract this
inhibition (P<0.01). Furthermore, it is noteworthy that the
decrease in mitoGPX4 relative to totalGPX4 was comparatively more
pronounced in the DCM mice during the in vivo experiments,
indicating that mitochondria-dependent ferroptosis may play a more
critical role in the pathogenesis of DCM. The primary mechanism of
SS-31 in alleviating ferroptosis was through activation of the
mitoGSH/mitoGPX4 pathway.
Discussion
SS-31, a mitochondria-targeting antioxidant peptide,
exhibits favorable water solubility and cell permeability, with a
receptor and transporter-independent cellular uptake mechanism
(25). SS-31 selectively binds
to cardiolipin in the inner mitochondrial membrane via both
electrostatic and hydrophobic interactions. This selective binding
demonstrates robust affinity towards the mitochondria while
preventing cardiolipin oxidation, thereby stabilizing the
cytochrome c and respiratory chain complex (26,27). Consequently, SS-31 can directly
alleviate mitochondrial oxidative stress as well as protect
mitochondrial function and ATP synthesis (15). Previously, research on
mitochondria-dependent ferroptosis presented a novel avenue and
trajectory for the prevention and treatment of cardiomyopathy
(14). Mitochondria, as highly
dynamic double-membrane organelles, function as the metabolic
center for carbohydrates, lipids and proteins and have a pivotal
role in energy metabolism, signal transduction and cell death
regulation. Under physiological conditions, mitochondria account
for 30-40% of the volume of cardiomyocytes, and almost all
(>95%) of the ATP generated in the heart is derived from
mitochondrial oxidative phosphorylation. Diabetes, as the
underlying cause of DCM, leads to cellular metabolic dysfunction,
such as iron metabolism and mitochondrial dysfunction. The
subsequent excessive labile iron triggers LPO damage through the
Fenton reaction, while impaired mitochondrial function disrupts
redox homeostasis (28).
Therefore, the heart is susceptible to mitochondria-dependent
ferroptosis. Previously, we established the involvement of
ferroptosis in the pathogenesis of DCM (9). In the present study, it was
hypothesized that mitochondria-dependent ferroptosis was a crucial
determinant in the pathogenesis of DCM and proposed the
administration of SS-31 as a targeted therapeutic approach for
DCM.
In the present study, DCM mouse and high
glucose-treated H9C2 cell models were established to investigate
the potential mechanisms of SS-31 treatment. Meanwhile, Fer-1 (a
ferroptosis inhibitor) was employed as a positive control, which
alleviates ferroptosis by eliminating the labile iron pool
(8). The serum cardiac injury
biomarkers, echocardiograms and histopathological results confirmed
the establishment of the in vivo model, and assessment of
the H9C2 cell viability indicated the establishment of the in
vitro model, consistent with our previous report (9). In the in vivo experiments,
both Fer-1 and SS-31 did not improve hyperglycemia in the DCM mice,
while SS-31 exhibited stronger cardioprotective effects than Fer-1,
indicating that the cardioprotective effect of SS-31 was not
related to glycemic control. The mitochondrial dysfunction of the
DCM group was verified by the decrease in ATP content, the
attenuation of the MMP and the disruption in the mitochondrial
ultrastructure. Treatment with SS-31 exhibited a stronger
restorative effect on ATP and the MMP compared with Fer-1, while
effectively restoring the mitochondrial ultrastructure.
Additionally, the DCM group exhibited severe LPO damage, with more
significant mitochondrial LPO damage than total LPO damage and
treatment with SS-31 effectively alleviated the mitochondrial LPO
damage. Considering the contribution of the labile iron pool to
ferroptosis, intracellular and mitochondrial iron ions were also
assessed in the in vitro experiment. In the model group, the
ferrous ions accumulated both intracellularly and in the
mitochondria; however, SS-31 treatment mitigated the accumulation
of ferrous ions, albeit with a weaker efficacy compared with Fer-1.
These findings suggested that the involvement of
mitochondria-dependent ferroptosis was critical in DCM pathogenesis
and provided evidence for SS-31 alleviating mitochondria-dependent
ferroptosis.
In addition to the direct effects on the
mitochondria, recent research has reported that SS-31 can also
activate the GSH/GPX pathway to alleviate ferroptosis; however, the
research is still restricted and limited to neurological injury
disorders. First, Zhang et al (20) demonstrated that SS-31 effectively
alleviates hippocampal ferroptosis and ameliorates cognitive
dysfunction in sevoflurane-induced neonatal mice. Second, Liu et
al (21) discovered that
SS-31 activates the GSH/GPX4 pathway to suppress hippocampal
ferroptosis in epileptic rats. It is worth noting that before the
concept of ferroptosis was proposed, Dai et al (17) demonstrated that SS-31 improves
hypertensive cardiomyopathy by inhibiting NADPH oxidase 4 (NOX4).
Subsequently, NOX4 was shown to be a facilitator of ferroptosis
(29). The GSH/GPX4 pathway has
a central role in limiting LPO and ferroptosis; intracellular GSH
is synthesized from cystine and serves as the substrate for GPX4
(30). GPX4 eliminates the
cellular toxicity of LPO by oxidizing GSH to glutathione disulfide
(8). GPX4 is a highly
conserved gene that encodes three distinct GPX4 proteins, which are
respectively localized within the mitochondria, cytosol and
nucleus. Previous research has demonstrated the presence of the
GSH/GPX4 pathway not only in the cytosol but also within
mitochondria (mitoGSH/mitoGPX4), where it inhibits
mitochondria-dependent ferroptosis (14). In the present study, total and
mitoGSH were quantified and the expression levels of total and
mitoGPX4 were assessed. In the in vivo DCM group, the
depletion of mitoGSH was more significant than the totalGSH
depletion. Although no significant decrease in mitoGPX4 protein
expression was observed in the HG group during the in vitro
experiment, a reduction in mRNA was detected. It was hypothesized
that this phenomenon may be attributed to the metabolic rate of
mRNA being faster than proteins; therefore, the GPX4 protein
expressed before the 24-h high glucose exposure was possibly
incompletely degraded, resulting in a discordance between mRNA and
protein expression. In addition, the in vivo experimental
results demonstrated a synchronicity between mRNA and protein
expression. These findings of the present study further supported
the hypothesis that the inactivation of mitoGSH/mitoGPX4, leading
to mitochondria-dependent ferroptosis, has a pivotal role in the
pathogenesis of DCM. Furthermore, only SS-31 exhibited the capacity
to activate the mitoGSH/mitoGPX4 pathway.
The complete GPX4 genomic DNA contains 8
exons, with exons 3-8 present in all three isoforms of GPX4
protein. Alternative splicing does not participate in the
production of these three GPX4 isoforms, which are instead
determined by different transcription start sites. Exon 1 of
GPX4 genomic DNA contains two translational start codons,
the first for mitoGPX4 and the second for cytoGPX4; exon 2 contains
the third translational start codon for nuclGPX4. Each
translational start codon has its own distinct transcription start
site (31). This may explain why
the expression of mitoGPX4 was unsynchronized with totalGPX4.
The present study still has certain limitations.
Since the difference between mitoGPX4 and cytoGPX4 only exists in
the first exon, which governs mitochondrial localization, precise
and effective RNA interference targeting mitoGPX4 becomes
unattainable. There is also currently an absence of a precise
inhibitor for mitoGPX4; therefore, further comprehensive
investigations were impeded in the present study. The employment of
Fer-1 in the present study had no effect on the mitoGSH/mitoGPX4
pathway, despite alleviating LPO damage. This observation implied
that activation of the mitoGSH/mitoGPX4 pathway by SS-31 was not
due to the alleviation of LPO damage.
In conclusion, the present study demonstrated that
mitochondria-dependent ferroptosis serves as a pathogenic mechanism
underlying DCM and can be alleviated by SS-31. The specific
mechanism involved activation of the mitoGSH/mitoGPX4 pathway and
mitigating the accumulation of ferrous ions in mitochondria.
Considering the safety profile and multi-organ protective effects
of SS-31, it is a promising drug for the prevention and treatment
of DCM.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ZY and YS conducted conceptualization. HS and LX
performed experiments. LX conducted data analysis and
visualization. HH and FZ confirm the authenticity of all the raw
data. XF and SP, ZY and YS provided medication guidance and ethical
oversight. FZ and JZ provided methodological support. LX wrote
original draft. HH, JZ, ZY and YS reviewed and edited the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Experiments were carried out according to the
Guideline for the Care and Use of Laboratory Animals published by
the National Institute of Health, USA. All experimental procedures
were approved by the Animal Ethical and Welfare Committee of ZCMU
(approval no. IACUC-20220307-10).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests
Acknowledgements
The authors thank Professor Yan Tai (Electron
Microscope Platform, Medical Research Center of Zhejiang Chinese
Medical University, Hangzhou, China) for providing the technical
support of TEM; Professor Lizong Zhang and Professor Xiaoping Xu
(Experimental Animal Center of Zhejiang Chinese Medical University,
Hangzhou, China) for providing the technical support of animal
experiments.
Funding
The present study was funded by Key Research Projects of the
Affiliated Hospital of Zhejiang Chinese Medical University, grant
no. 2022FSYYZZ22; Science and Technology Innovation Special Project
of Jiaxing Science and Technology Bureau, grant nos. 2020AY30003
and 2024AY30006; Medicine and Health Science and Technology Plan
Projects of Zhejiang Province, grant nos. 2020PY029 and 2023KY1227;
Zhejiang Provincial Natural Science Foundation of China, grant no.
LTGC23H150001; Jiaxing Key Laboratory of Diabetic Angiopathy
Research.
References
|
1
|
Bhagani H, Nasser SA, Dakroub A, El-Yazbi
AF, Eid AA, Kobeissy F, Pintus G and Eid AH: The mitochondria: A
target of polyphenols in the treatment of diabetic cardiomyopathy.
Int J Mol Sci. 21:49622020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jia G, Hill MA and Sowers JR: Diabetic
cardiomyopathy: An update of mechanisms contributing to this
clinical entity. Circ Res. 122:624–638. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dillmann WH: Diabetic cardiomyopathy. Circ
Res. 124:1160–1162. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao X, Liu S, Wang X, Chen Y, Pang P,
Yang Q, Lin J, Deng S, Wu S, Fan and Wang B: Diabetic
cardiomyopathy: Clinical phenotype and practice. Front Endocrinol
(Lausanne). 13:10322682022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–72. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miotto G, Rossetto M, Di Paolo ML, Orian
L, Venerando R, Roveri A, Vučković AM, Bosello Travain V, Zaccarin
M, Zennaro L, et al: Insight into the mechanism of ferroptosis
inhibition by ferrostatin-1. Redox Biol. 28:1013282020. View Article : Google Scholar
|
|
8
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156(1-2): 317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Du S, Shi H, Xiong L, Wang P and Shi Y:
Canagliflozin mitigates ferroptosis and improves myocardial
oxidative stress in mice with diabetic cardiomyopathy. Front
Endocrinol (Lausanne). 13:10116692022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Anderson EJ, Rodriguez E, Anderson CA,
Thayne K, Chitwood WR and Kypson AP: Increased propensity for cell
death in diabetic human heart is mediated by
mitochondrial-dependent pathways. Am J Physiol Heart Circ Physiol.
300:H118–H124. 2011. View Article : Google Scholar :
|
|
11
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–70. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gao M, Yi J, Zhu J, Minikes AM, Monian P,
Thompson CB and Jiang X: Role of mitochondria in ferroptosis. Mol
Cell. 73:354–363.e3. 2019. View Article : Google Scholar :
|
|
13
|
Imai H, Matsuoka M, Kumagai T, Sakamoto T
and Koumura T: Lipid peroxidation-dependent cell death regulated by
GPx4 and ferroptosis. Curr Top Microbiol Immunol. 403:143–170.
2017.PubMed/NCBI
|
|
14
|
Tadokoro T, Ikeda M, Ide T, Deguchi H,
Ikeda S, Okabe K, Ishikita A, Matsushima S, Koumura T, Yamada KI,
et al: Mitochondria-dependent ferroptosis plays a pivotal role in
doxorubicin cardiotoxicity. JCI Insigh. 5:e1327472020. View Article : Google Scholar
|
|
15
|
Schiller PW, Nguyen TM, Berezowska I,
Dupuis S, Weltrowska G, Chung NN and Lemieux C: Synthesis and in
vitro opioid activity profiles of DALDA analogues. Eur J Med Chem.
35:895–901. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Du X, Zeng Q, Luo Y, He L, Zhao Y, Li N,
Han C, Zhang G and Liu W: Application research of novel peptide
mitochondrial-targeted antioxidant SS-31 in mitigating
mitochondrial dysfunction. Mitochondrion. 75:1018462024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dai DF, Chen T, Szeto H, Nieves-Cintrón M,
Kutyavin V, Santana LF and Rabinovitch PS: Mitochondrial targeted
antioxidant peptide ameliorates hypertensive cardiomyopathy. J Am
Coll Cardiol. 58:73–82. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Machiraju P, Wang X, Sabouny R, Huang J,
Zhao T, Iqbal F, King M, Prasher D, Lodha A, Jimenez-Tellez N, et
al: SS-31 peptide reverses the mitochondrial fragmentation present
in fibroblasts from patients with DCMA, a mitochondrial
cardiomyopathy. Front Cardiovasc Med. 6:1672019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang L, Feng M, Wang X, Zhang H, Ding J,
Cheng Z and Qian L: Peptide Szeto-Schiller 31 ameliorates
doxorubicin-induced cardiotoxicity by inhibiting the activation of
the p38 MAPK signaling pathway. Int J Mol Med. 47:632021.
View Article : Google Scholar :
|
|
20
|
Zhang P, Chen Y, Zhang S and Chen G:
Mitochondria-related ferroptosis drives cognitive deficits in
neonatal mice following sevoflurane administration. Front Med
(Lausanne). 9:8870622022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu X, Wang FY, Chi S, Liu T, Yang HL,
Zhong RJ, Li XY and Gao J: Mitochondria-targeting peptide SS-31
attenuates ferroptosis via inhibition of the p38 MAPK signaling
pathway in the hippocampus of epileptic rats. Brain Res.
1836:1488822024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zacchigna S, Paldino A, Falcão-Pires I,
Daskalopoulos EP, Dal Ferro M, Vodret S, Lesizza P, Cannatà A,
Miranda-Silva D, Lourenço AP, et al: Towards standardization of
echocardiography for the evaluation of left ventricular function in
adult rodents: A position paper of the ESC Working Group on
Myocardial Function. Cardiovasc Res. 117:43–59. 2021. View Article : Google Scholar
|
|
23
|
Martinez AM, Kim A and Yang WS: Detection
of ferroptosis by BODIPY™ 581/591 C11. Methods Mol Biol.
2108:125–130. 2020. View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–8. 2001.
View Article : Google Scholar
|
|
25
|
Zhao K, Luo G, Zhao GM, Schiller PW and
Szeto HH: Transcellular transport of a highly polar 3+ net charge
opioid tetrapeptide. J Pharmacol Exp Ther. 304:425–32. 2003.
View Article : Google Scholar
|
|
26
|
Zhao K, Zhao GM, Wu D, Soong Y, Birk AV,
Schiller PW and Szeto HH: Cell-permeable peptide antioxidants
targeted to inner mitochondrial membrane inhibit mitochondrial
swelling, oxidative cell death, and reperfusion injury. J Biol
Chem. 279:34682–34690. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Birk AV, Liu S, Soong Y, Mills W, Singh P,
Warren JD, Seshan SV, Pardee JD and Szeto HH: The
mitochondrial-targeted compound SS-31 re-energizes ischemic
mitochondria by interacting with cardiolipin. J Am Soc Nephrol.
24:1250–1261. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park MW, Cha HW, Kim J, Kim JH, Yang H,
Yoon S, Boonpraman N, Yi SS, Yoo ID and Moon JS: NOX4 promotes
ferroptosis of astrocytes by oxidative stress-induced lipid
peroxidation via the impairment of mitochondrial metabolism in
Alzheimer's diseases. Redox Biol. 41:1019472021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Conrad M and Sato H: The oxidative
stress-inducible cystine/glutamate antiporter, system x (c) (-):
Cystine supplier and beyond. Amino Acids. 42:231–246. 2012.
View Article : Google Scholar
|
|
31
|
Imai H, Saito M, Kirai N, Hasegawa J,
Konishi K, Hattori H, Nishimura M, Naito S and Nakagawa Y:
Identification of the positive regulatory and distinct core regions
of promoters, and transcriptional regulation in three types of
mouse phospholipid hydroperoxide glutathione peroxidase. J Biochem.
140:573–590. 2006. View Article : Google Scholar : PubMed/NCBI
|