Introduction
Nod-like receptor (NLR) family pyrin domain
containing 3 (NLRP3) inflammasome is mainly expressed in the
cytoplasm of natural immune cells such as macrophages, and it is
the most deeply studied inflammatory corpuscle complex at present.
When NLRP3 is specifically stimulated, it recruits the connexin
apoptosis-associated spot-like protein (ASC) and the effector
protein pro-caspase-1 to start the assembly and activation of the
inflammatory complex. The activated inflammatory body of NLRP3
mediates the self-shearing of pro-caspase-1 and produces caspase-1
with enzyme activity. On the one hand, caspase-1 can cleave
pro-IL-1β and pro-IL-18, and promote the maturation and secretion
of IL-1β and IL-18; on the other hand, caspase-1 can cleave
gasdermin D (GSDMD) and release its N-terminus. The N-terminus of
GSDMD forms a hole in the cell membrane and induces inflammatory
cell death, that is, pyroptosis (1). The NLRP3 inflammasome is activated
not only by a variety of ligands from pathogens and environmental
sources, such as microbial cell wall components, nucleic acids,
Alum, silica, aluminium hydroxide, nanoparticles, crystalline
silicon dioxide, carbon nanotubes and chitosan, but also by
endogenous danger signals, such as lipopolysaccharides (LPS),
adenosine triphosphate (ATP), uric acid crystal, serum amyloid,
prion protein, biglycan, hyaluronan, islet amyloid polypeptide,
hydroxyapatite, haeme, oxidized mitochondrial DNA and membrane
attack complex (2-4). The activation of the NLRP3
inflammasome plays an important role in pathogen clearance and
adaptive immune response induction. However, the abnormal
activation of the NLRP3 inflammasome leads to excessive
inflammatory response, which in turn promotes the occurrence and
development of numerous inflammatory diseases. Therefore, it is
involved in the occurrence and development of many major human
diseases, including Alzheimer's disease (AD), autoimmune diseases
and arteriosclerosis (5,6). Therefore, the NLRP3 inflammasome is
an important target to treat these major human diseases. It is of
great significance to clarify the activation and regulation
mechanism of NLRP3 inflammatory corpuscles and develop therapeutic
drugs for the NLRP3 inflammasome. In this paper, the activation
mechanism of the NLRP3 inflammasome and a series of related
diseases are reviewed in order to provide a theoretical basis and
new ideas for the prevention and treatment of inflammation-related
diseases.
Structural characteristics of the NLRP3
inflammasome
The NLRP3 inflammasome is a high-molecular-weight
intracellular multi-protein complex, which consists of NLRP3,
apoptosis-associated speck-like protein containing card (ASC) and
pro-caspase-1. NLRP3 is a member of the NLRs protein family,
including three domains: An N-terminal pyrin domain (PYD), a
central NBD-containing ATPase domain called NACHT and a C-terminal
Leucine-rich repeat (LRR). ASC is a scaffold protein connecting
NLRP3 and pro-caspase-1, and pro-caspase-1 is an inactive precursor
of caspase-1. Caspase-1 produced by its activation is the effector
protein of the NLRP3 inflammatory body. NLRP3 recruits ASC under
the action of various agonists to form the NLRP3 inflammasome by
combining ASC with pro-caspase-1 (7-9).
Activation mechanism of NLRP3
inflammasome
The NLRP3 inflammasome is closely related to
numerous major human diseases, so it is important to clarify its
activation mechanism. Since the concept of the NLRP3 inflammasome
was first put forward in 2002, Kelley et al (10) discovered the classical,
non-classical and alternative pathways of NLRP3 inflammasome
activation. In this chapter, the activation mechanisms of these
three pathways will be briefly introduced.
Classical activation pathway of the NLRP3
inflammasome
The classical activation pathway of the NLRP3
inflammasome includes priming and activation. The priming process
means that cells are exposed to various stimuli, such as Toll-like
receptors (TLRs) and NLD-like receptors, and then the transcription
factor NF-ĸB is activated, thus upregulating the expression of
pro-IL-1β and NLRP3 (10,11).
The activation process means that cells treated by the priming
process are activated by pathogen-associated molecular patterns
(PAMPs) or damage-associated molecular patterns (DAMPs), and
proteins such as NLRP3, ASC and pro-caspase-1 are assembled into
inflammatory corpuscles, thus inducing cell apoptosis and IL-1β and
IL-18 to activate the NLRP3 inflammasome. It is generally thought
that there are three kinds of molecular and cellular signaling
events induced by NLRP3 agonists (11,12) (Fig. 1).
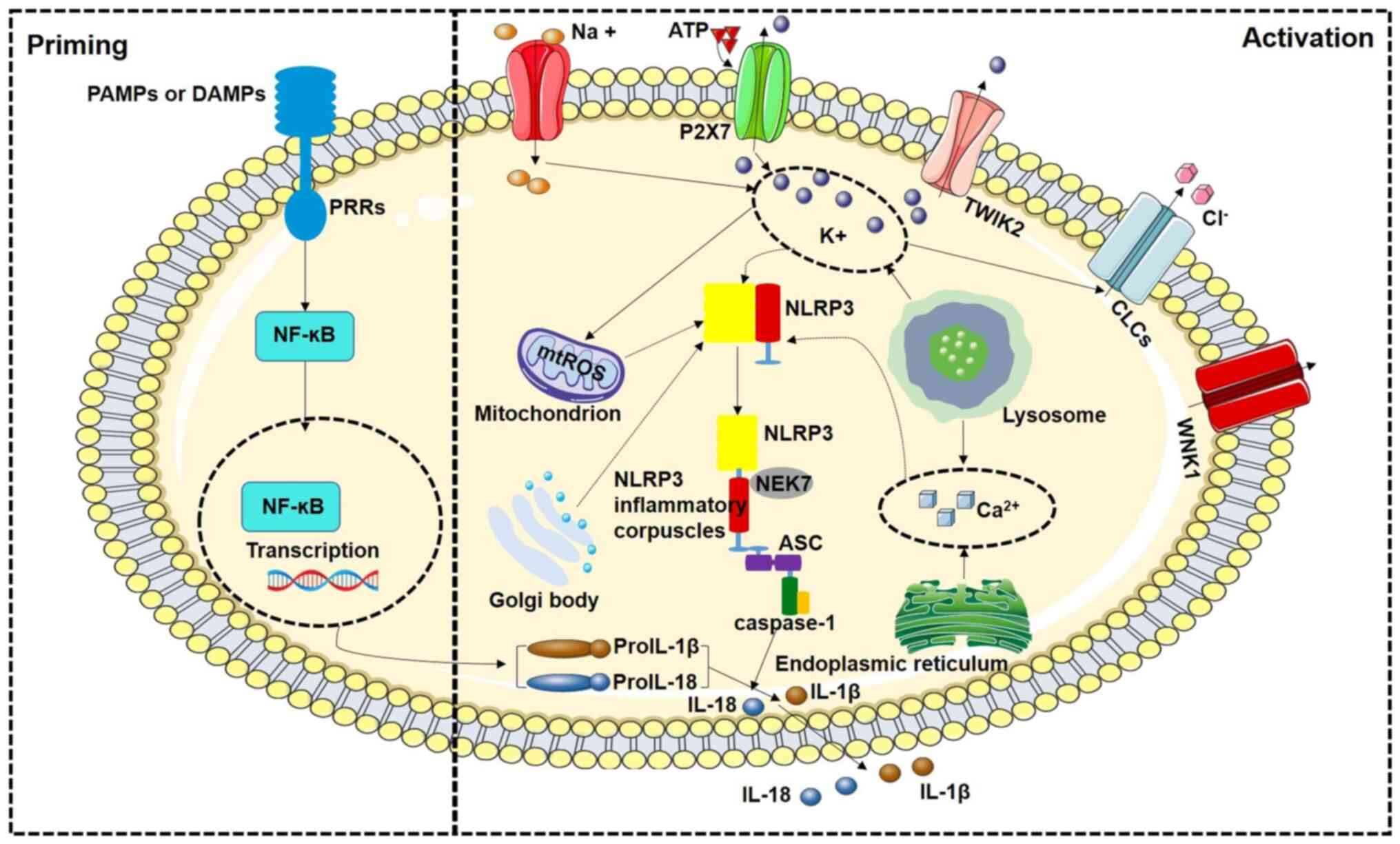 | Figure 1Schematic diagram of classical
pathway regulation mechanism of NLRP3 inflammasome activation. The
activation of NLRP3 inflammasome includes two processes: Priming
and activation. Priming process: NF-κB signal transduction induced
by PAMPs or DAMPs upregulates the transcription levels of NLRP3,
Pro-IL-1β and Pro-IL-18. Activation process: It includes the
composition of NLRP3 inflammasome and the activation of Caspase-1.
After activation of Caspase-1, Caspase-1 transforms Pro-IL-1β and
Pro-IL-18 stored in cells into active IL-1β and IL-18 and releases
them to the outside of cells. PAMPs, pathogen-associated molecular
patterns; DAMPs, damage-associated molecular patterns; NLRP3,
Nod-like receptor family pyrin domain containing 3; mtROS,
mitochondrial reactive oxygen species; NF-κB, nuclear factor κB;
ASC, connexin apoptosis-associated spot-like protein; PRRs, pattern
recognition receptors; TWIK2, the two-pore domain Weak Inwardly
rectifying K+ channel 2; CLC, chloride intracellular
channels; WNK1, recombinant WNK lysine deficient protein kinase 1;
NEK7, NIMA-related kinase 7. |
Ion flow
Intracellular K+ outflow is the first
signaling pathway to activate the NLRP3 inflammasome (13). Previous studies have confirmed
that low-K solution promotes the activation of the NLRP3
inflammasome, while high-K solution can inhibit the activation of
the NLRP3 inflammasome by blocking K+ outflow, and
numerous NLRP3 activators, such as bacteriocin, ATP, granular
molecules and crystalline substances, directly cause intracellular
to extracellular flow of K+, thus activating the NLRP3
inflammasome (13). Further
mechanistic research confirmed that the purinergic receptor (P2X7)
on the cell membrane and the K family member 6/TWIK2 of the
K+ double-pore channel are jointly responsible for
regulating the intracellular K+ outflow, which leads to
the activation of the NLRP3 inflammasome (14). However, previous research shows
that certain small molecules activate the NLRP3 inflammasome
without relying on potassium ion outflow, such as imiquimod, CL097
and peptidoglycan-induced activation of the NLRP3 inflammasome, and
accordingly, K+ outflow is a sufficient condition for
activation of the NLRP3 inflammasome, but it is not a necessary
condition (15,16). It is worth noting that the
outflow of K+ is closely related to the potassium-acid
balance in the body, and acidosis will lead to the release of
K+ from the inside to the outside of the cell, thus
increasing the concentration of K+ in the blood.
Rajamäki et al (17)
confirmed that extracellular acidosis activates NLRP3 inflammatory
corpuscles in macrophages, which leads to enhanced caspase-1
processing and IL-1β secretion. Acute exposure to an acidic
environment did not induce the activation of NLRP3 inflammatory
bodies, which indicates that an acidic pH is not a direct
activation factor (18).
The role of Ca2+ flow in the activation
of inflammatory corpuscles in NLRP3 is still controversial. As a
second messenger, Ca2+ has an important role in cell
signal transduction and the disorder of Ca2+ flow has
disastrous consequences for cells. Ca2+ receptor
activator C promotes the expression of inositol 1,4,5-triphosphate
and induces the release of Ca2+ from the endoplasmic
reticulum, thus activating the NLRP3 inflammasome (19). Early studies have confirmed that
Ca2+ chelating agent BAPTA-AM inhibited the activation
of the NLRP3 inflammasome and the secretion of IL-1β (20-22). Furthermore, Katsnelson et
al (23) also found that
numerous NLRP3 agonists cause changes of intracellular
Ca2+. However, certain studies have also found that the
Ca2+ chelating agent BAPTA-AM is independent of
Ca2+ flow, indicating that Ca2+ flow may not
be necessary for the activation of NLRP3 inflammasome, but it may
play a regulatory role in this process.
One study indicated that a decrease in the
CI− concentration enhanced the activation of the NLRP3
inflammasome and the secretion of IL-1β (24). On the contrary, an increase in
the concentration of extracellular CI− inhibited the
secretion of IL-1β. Certain studies further confirmed that
CI−channel inhibitors inhibit the activation of NLRP3
inflammasome (24,25). Furthermore, chloride
intracellular channels (CLIC), the CI− channel, is
involved in the activation of the NLRP3 inflammasome. As the next
event of mitochondrial reactive oxygen species (ROS), the outflow
of CI− mediated by CLICs induces the activation of NLRP3
inflammasome by promoting the interaction between NIMA-related
kinase 7 (NEK7) and NLRP3 (24).
However, it remains elusive how the outflow of CI−
enhances the interaction between NEK7 and NLRP3. In addition,
another study found that WNK lysine-deficient protein kinase 1
regulates Cl−/cation synergistic transporter and reduces
the outflow of CI− to inhibit the activation of NLRP3
inflammasome, but Cl−/cation co-transporter not only
regulates the outflow of CI−, but also affects the
outflow of K+ (26).
Furthermore, the intracellular Cl− efflux induced by
low-Cl− solution does not activate the NLRP3
inflammasome, and accordingly, Cl− efflux may trigger
the activation of NLRP3 inflammasome by cooperating with other
signaling pathways (27).
Na+ was previously reported to be
involved in the activation of the NLRP3 inflammasome. The results
showed that decreasing the concentration of extracellular
Na+ could inhibit the activation of the NLRP3
inflammasome induced by Nigericin and bacitracin, but
Na+ could not affect the activation of NLRP3
inflammasome induced by ATP and other factors, indicating that
Na+ influx alone is not sufficient to activate the NLRP3
inflammasome, which may be due to the synergistic effect of
Na+ influx and intracellular K+ outflow, and
the activation of the NLRP3 inflammasome by Na+ influx
depends on K+ (13).
Therefore, Na+ influx may indirectly activate the NLRP3
inflammasome by triggering intracellular K+ outflow.
Generation of ROS
The production of ROS is an important signal for the
activation of the NLRP3 inflammasome. Most NLRP3 agonists induce
cells to produce ROS. Under normal physiological conditions, there
is a low level of ROS in the body, which participate in the process
of cell proliferation and apoptosis. However, when the body is in a
pathological state, mitochondrial damage leads to an increase in
ROS production, thus activating the NLRP3 inflammasome. In 2011,
Zhou et al (28)
discovered for the first time that the accumulation of damaged
mitochondria would increase the production of ROS, which in turn
would activate the NLRP3 inflammasome. Subsequently, a large number
of studies confirmed that the increase of ROS induced by various
factors would lead to the activation of NLRP3 inflammasome
(29). In the follow-up study,
Shimada et al (30) found
that certain NLRP3 agonists promote the release of mitochondrial
DNA into the cytoplasm in a way that depends on mitochondrial ROS,
and oxidized mitochondrial DNA was indicated to be the key
substance to activate NLRP3 inflammasome. However, certain scholars
have found that linezolid and bacitracin promote the activation of
the NLRP3 inflammasome in a way independent of mitochondrial ROS,
and even certain serum β-amyloid proteins and viruses can activate
NLRP3 inflammasome independent of ROS (13,29,31).
Lysosome rupture
In 2008, it was found for the first time that
amyloid β protein induced lysosomes to dissolve, which in turn
activated the NLRP3 inflammasome (32). Furthermore, Lima et al
(33) found that a series of
granular substances, such as urate crystals and asbestos, could
damage lysosomes after being swallowed by cells, which led to the
leakage of lysosomes into the cytoplasm, and then activated the
NLRP3 inflammasome. When particulate matter (such as urate
crystals) is swallowed into lysosomes, ion flow and intracellular
osmotic pressure will change, bile will lead to K+
outflow and activation of NLRP3 inflammasome, and tissue protease
inhibitors also inhibit particle-induced activation of the NLRP3
inflammasome (34). Cathepsin B
was originally considered the key lysosomal enzyme, as its
inhibitor inhibits the activation of the NLRP3 inflammasome, and
particulate matter also promotes the release of cathepsin when
activating the NLRP3 inflammasome (32,35). However, in a cell experiment with
cathepsin B deficiency, Dostert et al (36) found the activation level of NLRP3
inflammasome to be equivalent to that of wild-type cells, which is
also contrary to previous research results. Orlowski et al
(37) have also found that,
besides inhibiting cathepsin B, the expression of cathepsin X, L or
S genes has no effect on the activation of the NLRP3 inflammasome.
However, the activation of NLRP3 inflammasome by exogenous
particles is accompanied by K+ outflow and
Ca2+ inflow, which indicates that lysosomal cleavage may
trigger an ion current in the process of activation of the NLRP3
inflammasome, thus activating the NLRP3 inflammasome, so the signal
of K+ and Ca2+ slurry flow is a downstream
event of lysosomal rupture (37).
Non-classical activation pathway of the
NLRP3 inflammasome
In addition to the above classical pathways, more
and more scholars have found that there are other activation
pathways for the NLRP3 inflammasome. The molecular mechanism of the
activation pathway of the non-classical NLRP3 inflammasome was
studied and the researchers found that mouse caspase-11 activates
the NLRP3 inflammasome by directly recognizing LPS released into
the cytoplasm during infection by Gram-negative bacteria. This
process is known as the non-classical activation pathway of the
NLRP3 inflammasome (38,39). In further research, related
researchers found that the secretion of IL-1β was significantly
inhibited in mice with caspase-11 gene knockout infected with
Gram-negative bacilli. Kayagaki et al (39) reasoned that this was because the
cells with caspase-11 gene knockout lacked the step of caspase-11
to cut its substrate protein GSDMD. In general, only after this
step is completed can the substrate protein form a cavity on the
cell membrane. Then it causes K+ outflow, apoptosis and
activation of the NLRP3 inflammasome (the function of human
caspase-4/5 is similar to that of mouse caspase-11) (39). Gram-negative bacteria in the
cytoplasm release LPS directly, thus activating caspase-4/5/11.
Activated caspase-4/11 can cleave GSDMD, resulting in the formation
of cell membrane pores and cell death, but caspase-4/5/11 itself
does not have the function of cleaving pro-IL-1β and pro-IL-18.
Cell membrane pores formed by GSDMD promote the activation of NLRP3
inflammasome by triggering K+ outflow, thus inducing the
activation of caspase-1 and the mature secretion of IL-1β and IL-18
(40-42) (Fig. 2).
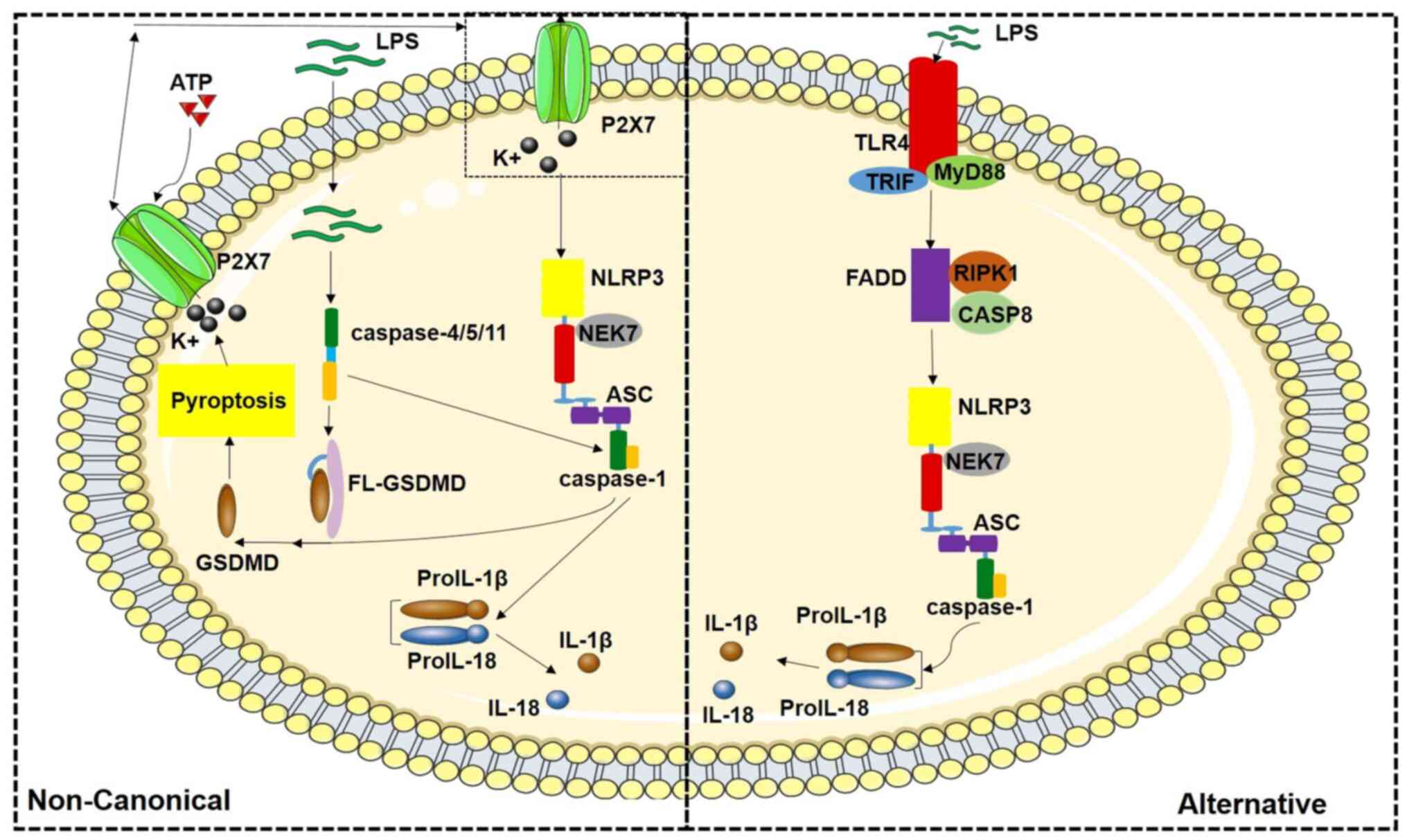 | Figure 2Schematic diagram of the
non-classical pathway of NLRP3 inflammasome activation and
regulation mechanism of the alternative pathway. LPS in cytoplasm
can activate non-classical NLRP3 inflammatory corpuscles mediated
by human caspase-4/5 or mouse caspase-11. LPS stimulation alone can
activate NLRP3 inflammasome of the alternative pathway through TLR4
signaling. NLRP3, Nod-like receptor family pyrin domain containing
3; LPS, lipopolysaccharide; TLR, Toll-like receptor; GSDMD,
gasdermin D; ASC, connexin apoptosis-associated spot-like protein;
CASP, caspase; FL-GSDMD, full-length GSDMD; NEK7, NIMA-related
kinase 7; TRIF, TIR-domain-containing adaptor-inducing
interferon-β; MyD88, myeloid differentiation factor 88; RIPK1,
serine/threonine kinase-1; FADD, Fas-associated protein with death
domain. |
Alternative activation pathways of the
NLRP3 inflammasome
In addition to the classical and non-classical
pathways, there is an alternative activation of the NLRP3
inflammasome. It has been shown that LPS stimulation activates
inflammatory bodies in monocytes through its receptor TLR4
signaling pathway. Most scholars find that LPS stimulation causes
ATP to be released outside the cells, and then combine with
ATP-gated K+ channel P2X7, resulting in K+
outflow, which is the key to the activation of the NLRP3
inflammasome (43). Others have
confirmed that LPS stimulation activates NLRP3 inflammasome through
the TLR4/TIR-domain-containing adaptor-inducing
interferon-β/serine/threonine kinase-1/Fas-associated protein with
death domain/caspase 8 signaling axis, promoting the activation of
caspase-1 and the mature secretion of IL-1β and IL-18 (44). However, it is interesting that
the alternative pathways have obvious cell and species
characteristics, which has only been verified in human and pig
monocytes and mouse dendritic cells (43,45) (Fig. 2).
NLRP3 inflammasome-related diseases
NLRP3 is an important aseptic inflammatory signal
receptor and a key regulator of chronic inflammatory diseases. It
was first discovered in 2001 and then further confirmed,
researchers have found that the autosomal dominant mutation of the
NLRP3 gene leads to a series of inflammatory diseases and the NLRP3
inflammasome has attracted extensive attention in related disease
research fields. The NLRP3 inflammasome is subject to research on
diseases in almost every system, but there is no doubt that the
diseases of the respiratory system, cardiovascular system,
digestive system, bone and joint system and central nervous system
are the most important diseases.
NLRP3 inflammasome drives respiratory
system diseases
Various pathogens in the air get in contact with
pattern recognition receptors co-expressed by airway epithelial
cells, alveolar macrophages or neutrophils in the lung, activating
downstream signal transduction and triggering innate immune
response. Studies have shown that the early immune response of the
lung to harmful stimuli can be mediated by the NLRP3 inflammasome,
which promotes the release of inflammatory factors, thus playing a
protective role, but excessive inflammatory response will aggravate
tissue damage and lead to a series of respiratory diseases. This
part summarizes the research on NLRP3 inflammasome in the
respiratory system (Table
I).
 | Table INLRP3 inflammasome drives respiratory
system diseases. |
Table I
NLRP3 inflammasome drives respiratory
system diseases.
| Disease | Related
mechanism | (Refs.) |
|---|
| COPD | Epithelial cells,
macrophages and dendritic cells in the lung, stimulated by harmful
particles or gases, activate the NLRP3 inflammasome through the
NF-ĸB signaling pathway or P2X7 receptor pathway, thus promoting
the release of inflammatory factors such as IL-1β and IL-18, and
leading to inflammatory reaction. | (46-54) |
| Bronchial
asthma | Airway macrophages,
epithelial cells and dendritic cells are stimulated by allergens to
activate the NLRP3 inflammasome and then promote the release of
IL-1β and IL-18 to induce inflammation. | (55-70) |
| Silicosis | SiO2 can
stimulate the assembly of adaptor factors such as NLRP3 and ASC in
macrophages, promote the secretion of IL-1β under the mediation of
caspase-1 and participate in the occurrence of diseases. | (71-73) |
| Bacterial
infectious pneumonia | NLRP3 inflammasome
mediates the lung injury induced by α-hemolysin, and α-hemolysin
activates the processing and secretion of IL-1β in a way similar to
pore-forming toxin, which further aggravates the progress of
Staphylococcus aureus pneumonia. Streptococcus
pneumoniae hemolysin can also interfere with the plasma
membrane and cause K+ outflow, and then activate the
NLRP3 inflammasome, trigger the secretion of IL-1β and IL-18, and
aggravate the course of Streptococcus pneumoniae
pneumonia. | (74-77)
(78-80) |
NLRP3 inflammasome participates in
chronic obstructive pulmonary disease (COPD)
COPD is an irreversible chronic progressive lung
disease characterized by continuous airflow limitation. The exact
cause of COPD has so far remained elusive. Inhaling harmful
particles or gases is an important cause of COPD and airway
inflammation. These substances act on pulmonary epithelial cells,
macrophages and dendritic cells, accelerating the synthesis and
release of inflammatory factors and tissue-degrading enzymes,
accelerating the development of inflammation and the destruction of
lung structure. Previous studies have confirmed that inhalation of
harmful substances directly activates the lung recognition receptor
TLR, thus activating the downstream NF-ĸB signaling pathway, and
promotes the transcription of NLRP3 and related inflammatory
factors (46). Furthermore,
these harmful substances also cause massive death of lung cells,
lead to the release of a variety of endogenous dangerous molecules
and further promote the activation of the NLRP3 inflammasome. The
decrease of extracellular nucleotidase expression and the increase
of ATP in patients with COPD further activates the NLRP3
inflammasome through the P2X7 receptor and ATP also promotes the
activation of inflammatory cells through the P2X7 receptor, which
aggravates the inflammatory reaction in the lung (47,48). At the same time, oxidative stress
is another important mechanism of activation of the NLRP3
inflammasome. A large number of studies have found that during the
occurrence and aggravation of COPD, the level of ROS in the lung is
significantly increased and the infiltration of immune cells in the
airway further increases the production of oxygen free radicals and
oxidative stress in the lung, which has been confirmed in animal
experiments. IL-1β production and confirmed cell infiltration were
significantly inhibited in NLRP3 gene knockout mice. In addition,
the activity of caspase-1 and the levels of inflammatory factors
such as IL-1β and IL-18 in lung tissue of COPD patients was also
significantly increased, suggesting that the NLRP3 inflammasome was
indeed activated during the occurrence and development of COPD
(49,50).
So far, there is little research on the role of
NLRP3 inflammasome in the progression of COPD. Certain scholars
have found that the specific expression of IL-1β in mouse lung
epithelial cells leads to increased infiltration of macrophages and
neutrophils in the lung, destruction of elastic fibers in the
alveolar septum, fibrosis of the airway wall and dilatation of
small airways at the distal end of the lung (51). In addition, overexpression of
IL-18 increased the level of inflammation in the lungs of mice and
promoted the formation of emphysema and pulmonary hypertension in
mice. However, knocking out IL-1R or IL-18 and intervention with
IL-1β neutralizing antibody significantly reduced lung injury and
inflammatory reaction in mice (52). Similarly, application of
caspase-1-specific inhibitors also reduced infiltration of
inflammatory cells in the lung of mice and reduced the level of
IL-1β (53). Furthermore, it has
been observed that P2X7 receptor-specific inhibitors or knockout of
P2X7 can inhibit the activation of caspase-1 and the release of
IL-1β in a smoking-induced lung injury model in mice, thus
alleviating the symptoms of COPD (54).
NLRP3 inflammasome involved in bronchial
asthma
Bronchial asthma is a kind of airway hyperreactive
chronic inflammation involving numerous cells and its main feature
is extensive and variable reversible expiratory airflow restriction
(55). It has been shown that
asthma pathogens promote macrophages, epithelial cells and
dendritic cells to release ATP, activate the NLRP3 inflammasome and
enhance airway inflammation. In addition, high levels of ATP and
P2X7 receptor were detected in the lungs of asthmatic patients
(56). Inhalation of allergens
is one of the important causes of asthma. Yazdi et al
(57) found that allergens
promote the production of ROS in the airways and ROS activates the
NLRP3 inflammasome, thus increasing the maturity and release of
IL-1β and IL-18. An associated study detected an increase in
caspase-1 activity and IL-1β levels in a mouse asthma model induced
by ovalbumin and alumina, and the IL-1β level was positively
correlated with caspase-1 activity (58). In loss-of-function experiments,
researchers found that neutralizing lung ATP or applying
non-selective purinergic receptor antagonists reduced the
infiltration of inflammatory cells and airway hyperresponsiveness
in asthmatic mice induced by ovalbumin and alumina, and the
application of caspase inhibitors also reduced airway inflammatory
reaction in asthmatic mice (56,59). Overexpression of IL-1R antagonist
by recombinant adenovirus or direct deletion of IL-1 may also
significantly reduce the airway hyperresponsiveness caused by
allergens in mice and alleviate inflammation around the airway
(60,61). In clinical research, Harada et
al (62) found that high
levels of IL-1β can also be detected in the sputum of asthmatic
patients, and it is positively associated with the severity of the
disease.
Type 2 T-helper (Th2) cells are closely related to
bronchial asthma, which increases airway reactivity of sensitizing
eosinophils (63). Knocking out
NLRP3 or caspase-1 inhibits the activation of Th2 cells and reduces
airway eosinophils and factors related to the activation of Th2
cells. Accordingly, knocking out IL-1β or IL-R can also
significantly inhibit the activation of Th2 cells, which proves
that NLRP3 inflammatory corpuscles participate in the occurrence
and development of bronchial asthma (64-66). IL-18 has been proved to promote
Th2 cells to secrete IL-4, IL-5, IL-9 and IL-13. Lung-specific
overexpression of IL-18 increases the infiltration of inflammatory
cells such as T cells, eosinophils and neutrophils in the airway of
asthmatic mice induced by ovalbumin and enhances airway
hyperresponsiveness (67). On
the contrary, knocking out IL-18 significantly alleviates asthma
symptoms induced by allergens such as ovalbumin in mice (68). Of note, the NLRP3 inflammasome
may not be involved in airway hyperresponsiveness caused by
ovalbumin and Allen et al (69) also confirmed that knocking out
NLRP3 did not affect airway allergic response of mice with four
different allergic asthma models. Even Hartwig et al
(70) found that IL-18 has
nothing to do with the occurrence of asthma and can even inhibit
the progress of asthma, which also shows that the role of the NLRP3
inflammasome in asthma requires further study.
Role of NLRP3 inflammasome in
silicosis
Silicosis is one of the most important occupational
diseases in the world, which is caused by crystalline silicon
dioxide (SiO2) (71).
In the whole disease development process of silicosis, NLRP3
inflammasome is involved and occupies a large proportion. Studies
have shown that SiO2 can be used as an activating
molecule to stimulate the assembly of NLRP3 and ASC in macrophages,
promote the secretion of IL-1β under the mediation of caspase-1 and
participate in the occurrence of diseases. At the same time, ROS
produced by cells is also used as the upstream signal of the NLRP3
signaling pathway to activate the NLRP3 inflammasome. In subsequent
animal experiments, Cassel et al (72) found that compared with wild-type
mice, ASC−/− and NLRP3−/− mice had less lung
inflammation and pulmonary fibrosis, which further confirmed that
the NLRP3 inflammasome is involved in the development of silicosis.
Jessop et al (73) also
found that autophagy of macrophages can inhibit the activation of
the NLRP3 inflammasome induced by SiO2 stimulation by
chelating or degrading inflammatory corpuscles and cytokines, thus
delaying the progress of subsequent diseases.
NLRP3 inflammasome is involved in the
course of bacterial infectious pneumonia
Community-acquired and hospital-acquired infectious
pneumonia are mainly caused by Staphylococcus aureus
(74). It has been proved that
α-hemolysin is the key virulence factor to induce lung
inflammation, which has the function of activating the processing
and secretion of IL-1β, and its function of inducing cell necrosis
requires the signal transduction of NLRP3 inflammasome (75). Animal experiments have confirmed
that NLRP3 inflammasome mediates α-hemolysin-induced lung injury.
Compared with wild-type mice, NLRP3−/− mice have no
decreased lung compliance and no obvious inflammatory reaction
after being infected with pneumonia, which proves that NLRP3
inflammasome plays a role in mediating lung injury caused by
pneumonia (76).
Streptococcus pneumoniae hemolysin is the
main virulence factor of Streptococcus pneumoniae, which
causes the formation of cavities on the cell surface and leads to
cell lysis tissue damage (77).
Furthermore, Streptococcus pneumoniae hemolysin can also
interfere with the plasma membrane and cause K+ outflow,
which further activates the NLRP3 inflammasome, triggering the
secretion of IL-1β and IL-18, and leads to a series of downstream
inflammatory reactions. It has been shown that IL-1β is necessary
to resist pneumococcal infection and the production of IL-1β
depends on NLRP3 inflammasome and TLR2; accordingly, the NLRP3
inflammasome is an important mediator of the innate immune response
to pneumococcal infection (78,79). van Lieshout et al
(80) also found that compared
with wild-type mice, the levels of cytokines and chemokines
expressed by NLRP3−/− mice decreased, among which the
level of IL-1β decreased significantly and the animals died within
48 h after being infected with bacteremia, while wild-type mice
only showed mild lung inflammation. It shows that NLRP3 plays a
beneficial role in pneumonia caused by Streptococcus
pneumoniae in the early stage (80). In short, the NLRP3 inflammasome
is widely involved in the occurrence and development of bacterial
infectious pneumonia.
NLRP3 inflammasome and digestive system
diseases
As a part of the innate immune system, the NLRP3
inflammasome can be activated by various metabolic stimulation
signals, among which the main effector molecules, IL-1β and IL-18,
play an important role in the occurrence and development of
digestive system diseases. As the key link of IL-1β- and
IL-18-mediated inflammatory reaction, the NLRP3 inflammasome plays
an essential role in the occurrence of digestive system diseases
(Table II).
| Table IINLRP3 inflammasome and digestive
system diseases. |
Table II
NLRP3 inflammasome and digestive
system diseases.
| Disease | Related
mechanism | (Refs.) |
|---|
| Hp-related stomach
diseases | Hp infection may
lead to the activation of intracellular NLRP3 inflammasome in
gastric epithelial cells, mononuclear macrophages and lymphocytes,
and then produce a large number of active IL-1β and IL-18 to induce
diseases. | (81-89) |
| Liver diseases | Activation of NLRP3
inflammasome in liver tissue can promote the production of active
IL-1β and IL-18, and finally lead to the occurrence and development
of liver fibrosis. | (92-105) |
| Inflammatory
intestinal diseases | Activation of NLRP3
inflammasome in intestinal mucosa or epithelial cells can promote
the production of active IL-1β and IL-18, which leads to intestinal
inflammation and hinder the repair mechanism of intestinal
epithelium. | (107-125) |
| Acute pancreatitis/
acute severe pancreatitis | Activation of NLRP3
inflammasome in acinar cells leads to the massive production of
IL-1β and IL-18, which eventually leads to inflammation and injury
of the pancreas and peripancreatic tissues. | (127-132) |
NLRP3 inflammasome and Helicobacter
pylori (Hp)-related stomach diseases
Hp is an aerobic gram-negative bacterium that causes
gastric mucosal inflammation in the stomach. The infection rate of
Hp was reported to be high and it has been colonized in the gastric
epithelial cells of healthy individuals accounting for >50% of
the world population (81). Hp
infection is closely related to numerous kinds of stomach diseases
and persistent Hp infection is related to complications such as
chronic gastritis, ulcer and even gastric cancer, among which the
relationship between Hp infection and gastric cancer has been
studied the most. However, so far, the initiation of the
pro-inflammatory signaling cascade in gastric epithelial cells
caused by Hp infection has remained elusive and the specific
pathogenesis of gastrointestinal diseases caused by Hp infection
remains to be further studied (82). However, Pachathundikandi et
al (83) have confirmed that
the activation of the NLRP3 inflammasome and the production of a
large number of pro-inflammatory factors, including IL-1β, are the
key factors for the occurrence and development of Hp-related
gastrointestinal diseases (83).
Hp infection induces the production of various cytokines, which
promotes the aggregation and infiltration of a series of immune
cells, such as monocytes, macrophages and lymphocytes, into the
gastric mucosa, thus leading to related gastric diseases. In this
process, cytokines play the role of the initiator and IL-1β is one
of the most critical cytokines (84-86). Caspase-1 is the key enzyme for
the differentiation and maturation of IL-1β, and IL-1β can
transform the precursor of IL-1β into mature IL-1β through its
shearing action. However, the production of caspase-1 depends on
the activation of the NLRP3 inflammasome. In this process,
pro-caspase-1 is cleaved to form caspase-1 with biological
activity, which leads to massive secretion of IL-1β and induces
diseases (11,12). In a groundbreaking study, Kameoka
et al (87) found that Hp
infection leads to increased IL-1β secretion and this process is
achieved through the NLRP3 inflammasome, which initially confirmed
the possibility that Hp infection can induce gastric diseases by
activating NLRP3 inflammasome. A clinical study on gastric cancer
confirmed that Hp induces gastric cancer by activating the NLRP3
inflammasome in the mononuclear phagocyte system and promoting the
release of inflammatory cytokines (87). Of course, this process can also
be achieved by activating NLRP3 inflammasome in gastric mucosal
epithelial cells GES-1 through ROS pathway or Hp toxin-related
protein CagA (88). Of note,
during the study of peptic ulcer disease induced by Hp infection,
Davari et al (89) found
that the decrease of IL-1β protein expression and the increase of
NLRP3 expression promoted Hp to develop digestive tract ulcer.
Therefore, it is of great significance to explore the mechanism of
activation of the NLRP3 inflammasome in Hp-related stomach diseases
to further the understanding of the prevention of Hp-related
stomach diseases and even gastric cancer.
NLRP3 inflammasome and liver
diseases
Liver disease has become a major global health
problem. According to relevant surveys, millions of individuals
worldwide die of liver disease every year. Liver diseases are
mainly divided into nonalcoholic fatty liver disease (NAFLD) and
alcohol-related liver disease (ALD), among which NAFLD is the most
common chronic liver disease. Both NAFLD and ALD can lead to
fibrosis and cirrhosis and increase the risk of hepatocellular
carcinoma if they are not properly controlled (90,91). Wree et al (92) found that the NLRP3 inflammasome,
pro-IL-1β, pro-IL-18 and pro-caspase-1 were significantly increased
in the liver tissue of patients with NAFLD. In addition, NAFLD is
often associated with obesity, type 2 diabetes and hyperlipidemia
(93). Lee et al
(94) found that NLRP3 and IL-1β
are increased in peripheral blood mononuclear cells and serum of
patients with type 2 diabetes. Furthermore, the expression of NLRP3
inflammasome was also increased in the liver tissues of obese
patients and mice (95). In a
further experimental study, it was found that the NLRP3
inflammasome may play different roles in the process of NAFLD
developing into nonalcoholic steatohepatitis (NASH). In the early
NAFLD animal model, increased NLRP3 inflammasome components in the
liver was only observed at the mRNA level. However, after NAFLD
completely developed into NASH, the mRNA and protein levels of
NLRP3 inflammasome (caspase-1 and IL-1β) in the liver tissue and
serum of the animal model increased significantly. In the reversal
experiment of mice with NLRP3, ASC and caspase-1 gene deficiency,
Csak et al (96) further
confirmed that NLRP3 inflammasome did participate in the occurrence
of NAFLD.
Excessive drinking and alcoholism are usually the
direct causes of ALD (97). ALD
and NAFLD have certain similarities in disease characteristics,
both of which develop from liver degeneration to liver fibrosis,
and may eventually develop into liver cancer (98). A clinical study showed that the
levels of IL-1β, IL-18 and caspase-1 in the liver of patients with
ALD were higher than those of healthy controls (99). Furthermore, in animal
experiments, compared with the control mice, the protein levels of
NLRP3 and IL-1β and caspase-1 activity in the liver of alcohol-fed
mice were significantly enhanced, which was consistent with the
clinical research conclusions (100). In addition, in the reverse
experiment of mice with NLRP3, ASC and caspase-1 gene deletion, it
was found that the degree of fat infiltration and damage in the
liver of mice was reduced (100,101). Therefore, NLRP3 inflammasome is
involved in the pathogenesis of ALD.
Hepatocirrhosis is the ultimate development trend of
the above two kinds of liver diseases (102). Boaru et al (103) detected a high level of NLRP3
expression in the liver tissue of a mouse liver fibrosis model. In
addition, in the mouse model with NLRP3 or ASC gene deficiency, the
degree of liver fibrosis was weakened (104). Clinicians also found an
increase of IL-1β, IL-18 and active caspase-1 in ascites
macrophages of patients with hepatocirrhosis (105). Therefore, NLRP3 inflammasome is
involved in the occurrence and development of numerous liver
diseases, and effective intervention is likely to be a new strategy
to treat these diseases.
NLRP3 inflammasome and inflammatory
intestinal diseases
Inflammatory bowel disease (IBD) includes Crohn's
disease and ulcerative colitis, and its main clinical features are
recurrent abdominal pain, diarrhea and even mucus pus and bloody
stool. If IBD cannot be effectively treated, it increases the risk
of rectal cancer. Previous studies have found that the pathogenesis
of IBD mainly includes genetic factors, environmental factors and
immune factors, of which immune factors may be the most critical
pathogenic factors, and inflammatory factors may play an important
role (106). Previous studies
have found high expression levels of IL-1β and IL-18 in the
intestinal mucosa of patients with Crohn's disease and ulcerative
colitis, allowing for the conclusion that the expression level of
IL-1β or IL-18 may be related to the susceptibility to IBD
(107-109). Further research indicated that
NLRP3 was highly expressed in the early stage of IBD, and then the
processing of pro-IL-18 and pro-IL-1β by caspase-1 led to an
increase of pro-inflammatory factors IL-1β and IL-18, which
eventually led to the aggravation of the course of IBD (110,111). By comparing the secretion of
IL-1β in peritoneal macrophages of NLRP3 knockout mice and
non-knockout mice, it was confirmed that NLRP3 inflammasome played
a role in promoting the progression of the disease in the colitis
model induced by dextran sulfate sodium (DSS) (112). Of note, animal studies also
found that NLRP3 gene knockout mice are more susceptible to colitis
induced by DSS than wild-type mice, and mice with ASC or caspase-1
gene deficiency also have the same susceptibility to colitis
(113-115). These gene knockout mice have
more serious intestinal pathological changes and higher mortality
in the acute and chronic stages of DSS-induced colitis. This shows
that the NLRP3 inflammasome may also play a certain role in the
process of intestinal inflammatory reaction, which is characterized
by the increase of stem cell division at the bottom of the crypt to
replace damaged intestinal cells (116). Generally speaking, activation
of the NLRP3 inflammasome leads to a large number of secretion
traps of IL-1β and IL-18, which hinders the intestinal repair
mechanism, increases the permeability of the intestinal epithelium
and eventually leads to the deterioration of the disease.
If IBD develops into chronic inflammation, it
becomes an important cause of IBD-related intestinal tumors (e.g.
colon or rectal cancer) (117).
According to reports, IL-1β is not only a proinflammatory factor,
but also a cancer-promoting factor (118). A high-cholesterol diet
increases the risk of colon cancer. Du et al (118) found that cholesterol crystals
promote inflammation and tumor development in the intestine of a
mouse colon cancer model. Furthermore, cholesterol crystallization
can activate the NLRP3 inflammasome, leading to an increase of
IL-1β secretion to promote the development of colon cancer. In
addition, it was found that knocking out the NLRP3 gene reversed
this process, which also preliminarily confirmed that the NLRP3
inflammasome-related pathway did participate in the course of
IBD-related tumors (118).
However, IL-18, another downstream inflammatory factor of the NLRP3
inflammasome, may have a role in inhibiting the progression of the
disease. Studies have found that IL-18 plays an anti-tumor role in
several experimental tumor models of sarcoma and melanoma (119-121). IL-18 can not only inhibit tumor
angiogenesis, but also promote the repair and regeneration of ulcer
epithelium (122,123). It has been indicated that,
although IL-18 can repair colon epithelial injury by promoting the
proliferation of intestinal cells, it inhibits proliferation in the
chronic stage of colitis, so IL-18 may play an important role in
preventing and inhibiting IBD-related tumors (124,125). In a word, the NLRP3
inflammasome is closely related to IBD and IBD-related tumors, and
further study of its specific mechanism may be helpful in treating
these diseases.
NLRP3 inflammasome and acute pancreatitis
(AP)/acute severe pancreatitis (SAP)
AP is one of the most common acute abdominal
conditions in the clinic and it is also the most common digestive
system disease in patients. As a classic inflammatory disease,
patients usually have the main clinical features of elevated serum
amylase and lipase. >20% of AP is SAP (126). SAP has an acute onset and rapid
development, which easily leads to multiple organ failure, with a
mortality rate of >20% (127). The essence of pancreatic
inflammation is that acinar cells secrete a large number of
inflammatory factors after being damaged to trigger the
inflammatory process. These inflammatory factors also lead to the
recruitment and activation of neutrophils and monocytes, thus
releasing oxidants and cytotoxic substances, further damaging
pancreatic tissue (127).
Various inflammatory factors have been proved to be important
indexes to evaluate the severity of AP and IL-1β and IL-18 are
among them (128). Different
from other inflammatory factors, IL-1β and IL-18 are synthesized as
precursor proteins and need to be cleaved to produce their
bioactive forms, and it has been speculated that NLRP3 inflammasome
may be involved in this process. Fu et al (129) found that the inflammatory
corpuscles of NLRP3 were obviously activated during AP in mice, and
in further experiments, they found that the edema and inflammation
of pancreatic tissue in mice were obviously reduced in the absence
of caspase-1, ASC or NLRP3. It has also been confirmed that the
secretion and maturation of IL-1β in the AP model of mice with
NLRP3 gene deficiency are obviously inhibited, which also impairs
the further development of the AP inflammatory cascade (129). Studies also found that TLR4 can
aggravate the development of AP by mediating the activation of the
NLRP3 inflammasome (130,131). Therefore, NLRP3 inflammasome
may be involved in the occurrence and development of AP.
Similarly, in SAP, inflammatory reaction of the
pancreas and peripancreatic tissues is mainly caused by local
proinflammatory factors such as IL-1β, and the production of mature
IL-1β is completed by the shearing process of caspase-1, and the
biological activity of caspase-1 is obtained on the basis of the
formation of NLRP3 inflammasome. In an animal model of SAP, Ren
et al (132) found that
the expression of various components of NLRP3 inflammasome
increased, which in turn promoted the activation of caspase-1 and
eventually led to the production of proinflammatory factors such as
IL-1β, which caused damage to the pancreas and peripancreatic
tissues and participated in the occurrence and development of SAP
(132).
NLRP3 inflammasome is involved in bone
and joint system diseases
With the continuous progress in the research on the
structure and function of NLRP3 inflammasome, increasing evidence
shows that the NLRP3 inflammasome is a key factor mediating tissue
injury in numerous bone and joint diseases, and its
pro-inflammatory factor release and cell apoptosis effect can
aggravate bone and joint injury. Various studies have confirmed
that NLRP3 has an important role in common diseases such as
osteoarthritis and certain autoimmune-related orthopedic diseases.
The NLRP3/caspase-1/IL-1β/IL-18 axis is a good target for the
treatment of bone and joint diseases (Table III).
| Table IIINLRP3 inflammasome is involved in
bone and joint system diseases. |
Table III
NLRP3 inflammasome is involved in
bone and joint system diseases.
| Disease | Related
mechanism | (Refs.) |
|---|
| Rheumatoid
arthritis | After synovial
tissue is stimulated, the NLRP3 inflammasome can be activated
through NF-ĸB signaling pathway or HIF-1α-related pathway, thus
promoting the production of IL-1β and finally causing the
destruction of synovial tissue of joints. | (134-143) |
| Osteoarthritis | Activation of the
NLRP3 inflammasome in articular chondrocytes or macrophages and
monocytes induces the activation and release of IL-1β and IL-18,
which eventually leads to degenerative diseases of articular
cartilage. | (144-149) |
| Osteoporosis | NLRP3 inflammasome
in osteoclasts can directly regulate osteoclast activity through
proteolysis, and NLRP3 inflammasome may also thicken chondrocyte
maturation and osteoblast activity, thus increasing the risk of
osteoporosis. | (151-157) |
| Gouty
arthritis | Stimulators such as
uric acid crystals activate NLRP3 inflammasome, which leads to an
increase in the expression of pro-inflammatory cytokines IL-1β and
IL-18, and finally leads to joint swelling and pain. | (159-163) |
| Intervertebral disc
degeneration | Mitochondrial
dysfunction, endoplasmic reticulum stress and ROS damage can
activate the NLRP3 inflammasome and finally lead to increased
release of IL-1 and other factors, which in turn leads to disc
degeneration. | (165-167) |
Role of NLRP3 inflammasome in rheumatoid
arthritis (RA)
RA is an autoimmune disease characterized by joint
injury. The initial pathological manifestation is synovitis. With
the development of the disease, synovitis can invade cartilage and
bone, which may lead to irreversible joint malformation and
dysfunction. The pathogenesis of RA is mainly related to rheumatoid
factor and anti-citrulline protein antibody (133).
Studies have shown that the NLRP3 inflammasome may
have a role in the pathogenesis of RA. NLRP3 and ASC were expressed
in myeloid cells, endothelial cells and lymphocytes of patients
with RA, but the expression level of NLRP3 did not increase in
fibroblast-like synovial cells (134,135), and it also increased in
peripheral blood cells of patients with RA (136). Zhang et al (137) also found that the expression of
NLRP3 in the serum and synovium of mice with collagen-induced
arthritis (CIA) was significantly higher than that of the control
group and the expression of NLRP3 in the synovium of the articular
capsule was positively associated with the severity of CIA/disease
imaging score in mice. A further study indicated that the
expression of NLRP3 inflammasome in synovium was obviously
inhibited and its clinical symptoms were alleviated after Guo et
al (138) used
NLRP3-specific inhibitors on CIA model mice. The expression of
caspase-1 in vivo was downregulated and IL-1β in synovium
and serum of joints was significantly decreased.
When cells are stimulated to a certain extent, the
NF-ĸB pathway is activated and they can enter the stage of
activation of the NLRP3 inflammasome. Therefore, targeting the
NF-ĸB pathway may have potential therapeutic effects on
NLRP3-related diseases. A20, TNF-α-inducible protein 3 is a kind of
protein involved in the negative feedback regulation of NF-ĸB
signaling and is also considered a susceptible gene of RA. A20
knockout mice show spontaneous erosive arthritis related to the
increase of NLRP3 inflammasome and the secretion of IL-1β (139). A20-deficient bone
marrow-derived macrophages secreted inflammatory factors and
apoptosis related to the overactivation of NLRP3, which indicated
that A20 could inhibit RA by weakening the activation of NLRP3
inflammasome (140).
The imbalance of microRNA (miR) in synovial cells
and synovial fluid and hypoxia microenvironment in synovium also
have an important role in the occurrence and development of RA
diseases. Li et al (141) found that miR-20a can reduce the
production of ROS introduced by thioredoxin binding protein (TXNIP)
and the activation of NLRP3 inflammasome by targeting the 3′-UTR of
rat TXNIP. In further animal experiments, it was found that the RA
rat model could be improved by receiving miR-223 from exosomes, and
the related mechanism may be that miR-223 can target the 3′-UTR of
NLRP3 and inhibit the expression of NLRP3 (142). The hypoxia environment in the
synovium may also induce the activation of NLRP3. In the rat model,
synovial succinate accumulates and activates NLRP3 inflammasome by
regulating the transcription of hypoxia-inducible factor-1α
(HIF-1α), and the level of HIF-1α is increased in fibroblast-like
synoviocytes of patients with RA (143). Therefore, the NLRP3
inflammasome can be activated by various upstream signals and its
expression tends to increase in patients with RA or animal models,
thus participating in the occurrence and development of RA
diseases. Therefore, targeting NLRP3 shows a good therapeutic
prospect, which effectively inhibits the secretion of downstream
inflammatory factors and apoptosis.
Role of NLRP3 inflammasome in
osteoarthritis (OA)
OA is a degenerative joint disease whose main
symptom is joint pain. The etiology is still unclear, and the
pathological manifestations are mainly deformation and destruction
of articular cartilage fibrosis. Numerous inflammatory factors
induce local synovitis and cartilage matrix degradation and then
promote the progress of knee OA. The expression of NLRP3, ASC and
caspase-1 in synovial tissue of patients with knee OA is increased.
In early in vitro experiments, Han et al (144) found that knocking out NLRP3
could weaken inflammation and apoptosis of synovial cells induced
by LPS and ATP.
Alkaline calcium phosphate and calcium phosphate
crystals usually exist in synovial fluid and tissues of patients
with OA and play an important role in the pathogenesis of OA
(145). Both alkaline calcium
phosphate and calcium phosphate crystals can activate NLRP3
inflammasome, and alkaline calcium phosphate promotes the
production of IL-1β by macrophages and monocytes by activating the
NLRP3 inflammasome (146). It
is well-known that IL-1β and IL-18 in synovial fluid are positively
related to the severity of OA (147). In a mouse OA model experiment,
oral selective NLRP3 inflammasome inhibitor obviously improved the
inflammation of mouse joint tissue and the inflammatory factors in
the synovium tissue of the control mice were obviously reduced,
which indicates that targeting NLRP3 can also improve the progress
of OA diseases (148).
In the chondrocyte OA model stimulated by IL-1β,
Senkyunolide A (SenA) inhibited the pathway of NLRP3 inflammasome
and significantly reduced the expression of related proteins, such
as NLRP3 and ASC. It was found that SenA inhibits the NLRP3
signaling pathway of chondrocytes in an OA model stimulated by
IL-1β and OA mice (148).
Further clinical drug research found that metformin, a first-line
treatment drug for diabetes, has similar effects, which has
attracted much attention for its future prospects in OA treatment.
Lordén et al (149)
performed medial meniscus instability surgery on the knee joint of
experimental mice to build a mouse model of OA and the experimental
group of mice received metformin intervention. After
administration, the researchers found that the expression of NLRP3,
caspase-1, IL-1β and other related factors in mouse articular
cartilage decreased significantly. In addition, at the subchondral
bone level, metformin can inhibit the formation of osteophytes,
increase bone density and reduce trabecular separation. Therefore,
metformin can inhibit the activation of NLRP3 inflammasome and the
focal death of chondrocytes, thereby delaying the progress of OA
(149).
As the most common degenerative disease of joints,
OA is characterized by degenerative changes of articular cartilage.
The characteristics of releasing inflammatory factors and promoting
apoptosis of NLRP3 are closely related to the pathological changes
of OA such as synovitis and chondrocyte apoptosis, and accordingly,
targeting NLRP3 has the potential to delay the progress of OA.
NLRP3 inflammasome involved in
osteoporosis (OP)
OP is a systemic metabolic bone disease
characterized by decreased bone mass and destruction of the bone
microstructure, which leads to increased bone fragility and easy
fracture (150). There are
numerous factors leading to OP. According to certain scholars, the
NLRP3 inflammasome may play an important role in this process. In a
previous study, Alippe et al (151) found that NLRP3 deficiency can
prevent bone loss caused by ovariectomy (OVX) in mice and play an
important role in bone resorption. In vivo studies of NLRP3
gene knockout mice showed that NLRP3 participated in bone
induction; NLRP3 knockout mice are shorter than normal mice and
this growth defect is mainly characterized by growth plate defect
and osteopenia of trabecular bone. The researchers speculated that
this effect is achieved by regulating the maturation of
hypertrophic chondrocytes and the activity of osteoblasts (152). Recently, Jiang et al
(153) also found that NLRP3
inflammasome can cause dysfunction of osteoclasts by reducing
differentiation, and at the same time accelerate the proliferation
and differentiation of osteoclasts, which promotes bone absorption
and destroys bone formation. NLRP3 inflammasome in myeloid cells
indirectly regulates the activity of osteoclasts through systemic
inflammation, while NLRP3 inflammasome in osteoclasts directly
regulates the activity of osteoclasts through proteolysis of
poly(ADP-ribose) polymerase 1, which is a negative regulator of
osteoclast formation. Inhibition of the NLRP3 inflammatory
corpuscle pathway promoted osteoblast differentiation and restored
bone volume in OVX mice (154,155). The involvement of NLRP3
inflammatory corpuscles in the course of OP has also been
preliminarily confirmed in clinical research. Guaraná et al
(156) 's research on
menopausal women with OP showed that NLRP3 inflammatory corpuscles
activate pro-inflammatory cytokines IL-1β and IL-18, and the single
nucleotide variation caused by NLRP3 inflammasome is closely
related to the severity of OP. Therefore, NLRP3 inflammasome not
only accelerates bone resorption, but also inhibits bone formation,
thus increasing the risk of OP (157). Therefore, inhibiting NLRP3
inflammasome is a promising therapeutic strategy to prevent and
control OP.
Relationship between NLRP3 inflammasome
and gouty arthritis (GA)
GA is a disease in which the amount of uric acid in
the body increases due to excessive purine metabolism or
insufficient uric acid excretion, which leads to the
crystallization and accumulation of uric acid in joints, thus
leading to joint pain and swelling (158). As the upstream pathway of IL-1β
and IL-18, NLRP3 inflammasome is firstly activated by uric acid
crystallization and LPS in this process, and the self-activation of
caspase-1 is mediated by the activated NLRP3 inflammasome, which
leads to the increased expression of pro-inflammatory cytokines
IL-1β and IL-18 (159). It is
reported that calcium pyrophosphate dihydrate (CPPD) crystals cause
pseudo-RA attack and participate in the activation of NLRP3
inflammasome. Zhao et al (160) confirmed that the deposition of
monosodium urate (MSU) crystals can lead to inflammatory reaction
of human macrophages, which is the classic pathogenesis of acute
gout. Macrophage phagocytosis of MSU crystals activates NLRP3
inflammasome and then upregulates the expression of IL-1β and
IL-18, thus inducing the occurrence of acute GA (160). In addition, inhibiting the
activation of NLRP3 inflammasome can prevent acute GA, and
therefore, it is thought that the activation of NLRP3 inflammasome
is the core process of gout attack (161,162). Blocking the activation of
NLRP3/caspase-1/IL-1β pathway induced by ATP has a significant
effect on relieving GA inflammation in rats (163). Therefore, inhibiting the
activation of NLRP3 inflammasome and promoting the relief of RA
inflammation represent a new direction in the treatment of GA.
NLRP3 inflammasome induces intervertebral
disc degeneration (IDD)
IDD is an age-related degenerative disease of the
spine, which has a great influence on social economy and human
life. It is also one of the main causes of low back pain, which can
cause serious nerve damage and disability. The main pathological
changes are the decrease of proteoglycan content, height, endplate
sclerosis and osteophyte formation, which eventually leads to a
decrease of the ability of the intervertebral disc to withstand
compression load (164). The
pathogenesis of IDD is complex and the efficacy of current
treatments is not obvious, and accordingly, it is urgent to find
new therapeutic targets. It has been indicated that during the
occurrence and development of IDD, NLRP3 inflammasome may be
activated, thus mediating the production of a variety of
inflammatory cytokines and further promoting the progress of IDD. A
study also showed that this process may depend on NLRP3′s ability
to perceive change in intracellular homeostasis, the activation of
NLRP3 inflammasome, mitochondrial dysfunction related to IDD,
endoplasmic reticulum stress and ROS damage (165). At the same time, this study
also confirmed that the NLRP3 inflammasome participated in the
inflammatory reaction of IDD mice. The researchers found that,
compared with normal rat intervertebral disc tissues, the
expression of NLRP3, caspase-1 and IL-1 in degenerated
intervertebral disc were significantly upregulated and the
expression of caspase-1 and IL-1 was similar (165,166). In further research, Zhao et
al (167) found that the
lactic acid content in intervertebral disc tissue increased during
IDD, thus stimulating nucleus pulposus cells to drive the
activation of NLRP3 inflammatory bodies and increase the release of
IL-1β, which further induced the IDD process, which they thought
was realized through ROS and NF-κB-related pathways. These findings
fully indicate that NLRP3 inflammatory corpuscles may play an
important role as a potential target for IDD treatment in the
future.
NLRP3 inflammasome participates in
cardiovascular diseases
The NLRP3 inflammasome is a protein complex that
can be activated by crystal or granular PAMPs and ischemic hypoxia
risk-related molecular patterns, DAMPs, which promote the secretion
of IL-1β and IL-18. Through these mechanisms, it can promote
diseases such as atherosclerosis and heart failure. Therefore,
NLRP3 inflammasome may have a key role in the physiology and
pathology of cardiovascular diseases and play the role of
pro-inflammatory mediators. In recent years, it has become one of
the focuses of relevant research (Table IV).
| Table IVNLRP3 inflammasome participates in
cardiovascular diseases. |
Table IV
NLRP3 inflammasome participates in
cardiovascular diseases.
| Disease | Related
mechanism | (Refs.) |
|---|
|
Atherosclerosis | The activation of
NLRP3 inflammasome in arterial tissue leads to the increase of
IL-1β expression, which in turn leads to the early inflammatory
reaction of AS. | (169-173) |
| Heart failure | The activation of
NLRP3 inflammasome in myocardial cells can lead to a large amount
of secretion of IL-1β and IL-18, which will lead to cardiac
contraction dysfunction and myocardial cell death, which may
eventually lead to ventricular remodeling. | (174-180) |
| Myocardial
ischemia-reperfusion injury | The outflow of ROS
and K+ mediates the activation of NLRP3 inflammasome in
myocardial fibroblasts or microvascular endothelial cells, which
leads to the increase of IL-1β and IL-18, and finally induces
myocardial apoptosis and myocardial infarction. | (181-183) |
NLRP3 inflammasome and atherosclerosis
(AS)
AS is a chronic inflammatory disease and it is also
one of the most important causes of death from cardiovascular
diseases. The pathogenesis of AS is closely related to inflammatory
reaction and lipid accumulation, but the specific pathogenesis of
AS has not been fully clarified, and its prevention and treatment
are limited (168). In 2010,
researchers proposed for the first time that the NLRP3 inflammasome
may be involved in the occurrence and development of AS. Duewell
et al (169) believed
that oxidized low-density lipoprotein could promote cholesterol
crystallization, and then cholesterol crystallization activated
NLRP3 inflammasome AS and endogenous signaling molecules, which
eventually led to the increase of IL-1β expression, which was also
the reason for the early inflammatory reaction of AS. In subsequent
clinical experiments, Paramel Varghese et al (170) also found that the expression
levels of NLRP3, ASC, IL-1 β and IL-18 mRNA in atherosclerotic
plaques of patients with AS were significantly higher than those in
normal arterial tissues. In the AS model of mice, Usui et al
(171) found that compared to
the control mice, the area of aortic plaque was significantly
reduced after caspase-1 knockout, and the number of macrophages and
vascular smooth muscle cells in the plaque and the level of plasma
inflammatory factors were also reduced (171). At the same time, silencing the
expression of the NLRP3 gene inhibited the progress of AS and the
expression of inflammatory cytokines, reduced the contents of
macrophages and lipids in plaques and promoted the stability of
plaques (172). A previous
clinical study confirmed that the levels of NLRP3 mRNA, IL-1β and
IL-18 in peripheral blood mononuclear cells of patients with
coronary heart disease were higher than those of patients without
coronary heart disease and atorvastatin was observed to reduce the
levels of IL-1β and IL-18 through the NLRP3 inflammasome to treat
AS (173). Therefore, the NLRP3
inflammasome may be a potential target for the prevention and
treatment of AS. Further clarifying the role of the NLRP3
inflammasome in AS and developing inhibitors of NLRP3 inflammasome
are expected to provide new intervention measures for the
prevention and treatment of AS.
Relationship between NLRP3 inflammasome
and heart failure
Heart failure is a group of clinical syndromes
caused by the impairment of ventricular filling and ejection
function caused by structural or functional diseases of the heart.
In the last decade, great progress has been made in drug treatment
of heart failure, but its main pathogenesis has not been fully
explained, so most patients still suffer from symptoms such as
dyspnea and decreased fatigue tolerance. Previous studies have
suggested that the NLRP3 inflammasome has an important role in
regulating chronic inflammation and influencing heart failure
(174).
Researchers have found that NLRP3, pro-caspase-1
and serum IL-1β in heart failure mice are significantly higher than
those in the control group. However, after the NLRP3 gene is
knocked out or NLRP3 inhibitor is given, the myocardial
inflammatory response and myocardial contraction function of heart
failure mice are significantly improved and the heart failure mice
can also gain similar effects after applying IL-1β inhibitor
(175). In further cell
research, Mezzaroma et al (176) found that the related components
ASC and caspase-1 of the NLRP3 inflammasome in myocardial cells of
mice with heart failure were higher than those in the sham-operated
group, which further confirmed that NLRP3 inflammasome could
mediate the death of myocardial cells and lead to ventricular
remodeling, and promote the occurrence of heart failure after
myocardial infarction.
IL-1β, an inflammatory factor produced by the
activation of NLRP3 inflammasome, can cause myocardial reversible
systolic dysfunction, and its mechanism is to induce the activation
of IL-18, which leads to cardiac systolic dysfunction (177,178). Clinicians have translated these
research achievements to clinical treatment. For instance,
anakinra, an antagonist of IL-1R, can alleviate the inflammatory
reaction after myocardial infarction in patients with heart failure
(179,180). IL-1β and IL-18 produced by
activation of NLRP3 inflammasome are the focus of immunotherapy for
heart failure at present. Immunosuppressants, such as anakinra,
have shown good application prospects in clinical research, and
NLRP3 inflammasome and their products will become new targets of
immunotherapy for heart failure in the future.
Relationship between NLRP3 inflammasome
and myocardial ischemia-reperfusion injury
Myocardial ischemia-reperfusion injury is a common
pathophysiological process in ischemic cardiomyopathy,
cardiopulmonary bypass and heart transplantation. Its mechanism
includes free radical damage, Ca2+ overload, energy
metabolism disorder and inflammatory reaction. Kawaguchi et
al (181) have suggested
that NLRP3 inflammasome may also be involved in this process. The
release of IL-1β, the infiltration of inflammatory cells and the
expression of inflammatory cytokines in myocardial tissue of mice
with myocardial injury and ischemia-reperfusion increased, which
was caused by the activation of NLRP3 inflammasome of myocardial
fibroblasts mediated by ROS and K+ outflow (181). After establishing the model of
myocardial ischemia in mice, researchers found that the expression
of NLRP3, IL-1β and IL-18 mRNA in mouse myocardial fibroblasts
increased significantly, while the heart function of NLRP3-related
gene knockout mice improved significantly (181-183). In myocardial microvascular
endothelial cells of mice with myocardial ischemia, the expression
of NLRP3 increased, the activity of caspase-1 increased, and the
production of IL-1β and IL-18 increased (183). Cardiac myocyte apoptosis and
cardiac function were significantly improved after intracardiac
injection of NLRP3 inhibitor (183). Furthermore, Kawaguchi et
al (181) found that the
inflammatory reaction, infarct size and degree of myocardial
fibrosis in mice with myocardial ischemia were significantly
reduced after silencing ASC or caspase-1 gene expression.
A large number of inflammatory mediators are being
released during myocardial ischemia, which mediates the cascade
amplification effect of inflammatory reactions. It has been proved
that NLRP3 inflammasome plays an important role in myocardial
ischemia. However, its specific signaling pathways and mechanisms
require to be further clarified. It has a good research prospect to
prevent and treat myocardial ischemia-reperfusion injury with NLRP3
inflammasome as the target.
Relationship between NLRP3 inflammasome
and central nervous system diseases
In recent years, with the deepening of the research
on NLRP3 inflammasome, its role in nervous system diseases has been
increasingly studied. Although there have been numerous studies on
nervous system diseases and inflammatory factors, research on the
relationship between nervous system diseases and NLRP3 inflammatory
corpuscles is still in its infancy. It is of great significance to
clarify the pathogenesis of central nervous system diseases and
improve the therapeutic effect to find out the specific function
and relationship between them. Therefore, the role of NLRP3
inflammasome in common nervous system diseases was summarized in
the following chapter (Table
V).
| Table VRelationship between NLRP3
inflammasome and central nervous system diseases. |
Table V
Relationship between NLRP3
inflammasome and central nervous system diseases.
| Disease | Related
mechanism | (Refs.) |
|---|
| Traumatic brain
injury | The specific
mechanism remains to be fully elucidated. NLRP3 inflammasome in
neurons, astrocytes and microglia was activated in large quantities
after brain injury, which induced neuroinflammatory reaction and
neuronal death. | (185-188) |
| PD | The specific
mechanism remains to be fully elucidated. The activation of NLRP3
inflammasome in microglia of patients with PD and PD mouse models
may be related to the occurrence and development of PD. | (191,192) |
| Alzheimer's
disease | The activation of
NLRP3 inflammasome in brain tissue may enhance Aβ aggregation and
promote the development of neuroinflammation and Tau lesions. | (195-198) |
| MS | NLRP3 inflammasome
in MS plaque may be activated to promote the production of IL-1β
and caspase-1, eventually leading to a series of neuropathy. | (200) |
| Cerebral
ischemia | After cerebral
ischemia, NLRP3 inflammasome is activated and pro-inflammatory
factors IL-1β and IL-18 are produced to aggravate the
neuroinflammatory response. | (202-206) |
NLRP3 inflammasome participates in
traumatic brain injury (TBI)
TBI is one of the most serious diseases of the
nervous system in the world, which is usually caused by external
forces, such as normal brain function damage or pathological brain
tissue damage. Due to the irreversible loss of functional neurons
and nerve tissue damage, the central nervous system is difficult to
repair and regenerate after trauma, resulting in poor prognosis for
patients with TBI. To date, the mechanism of TBI has not been
thoroughly studied, so although a large number of successful
preclinical studies have been used for TBI treatment, they are
rarely used in transformation (184).
After TBI, neuroinflammation is a key factor that
obviously aggravates brain tissue damage and leads to functional
defects. It has been confirmed that inflammatory corpuscles have an
important significance in regulating the secondary injury of TBI. A
variety of inflammatory corpuscles that damaged brain tissue,
particularly NLRP3, were detected in neurons, astrocytes and
microglia, which can induce neuroinflammatory reaction and neuron
death, aggravate brain tissue damage and was closely related to the
pathogenesis of TBI (185). It
has been shown that the gene and protein levels of the NLRP3
inflammasome are obviously upregulated after moderate TBI (186). It was also found that the level
of NLRP3 in cerebrospinal fluid increased on the first day after
brain injury in infants, decreased the next day and increased again
at 3-4 days. Wallisch et al (187) indicated that the early increase
of NLRP3 may be due to cell necrosis or traumatic dissolution,
while the late increase may be due to infection, cell stress or
activation of other inflammatory bodies. The NLRP3 inflammasome is
activated after brain injury and scholars have found that
inhibiting the activation of the NLRP3 inflammasome reduced
inflammation and improved neurological function in brain injury
model mice. Geng et al (188) observed that a high
concentration of oxygen can inhibit the activation of the NLRP3
inflammasome and the inflammatory response after traumatic brain
injury in mice. It should be noted that hypoxia therapy is a
classic treatment for TBI, which indicates that the NLRP3
inflammasome is involved in the occurrence and development of TBI,
and its inhibitors are likely to be a new important treatment for
TBI.
Role of NLRP3 inflammasome in Parkinson's
disease (PD)
PD is a progressive chronic neurodegenerative
disease with a complex pathogenesis, which is closely related to
numerous pathogenic factors, such as oxidative stress,
neuroinflammation and mitochondrial dysfunction. At present,
levodopa, dopamine receptor agonist and anticholinergic drugs are
the main drugs used to treat PD in the clinic, but there are no
clear treatment methods to improve the pathological progress
(189). Studies have shown that
the activation of microglia and neuroinflammation may be the key
regulatory factors for the loss of dopaminergic neurons in PD. The
activation of microglia in the brain and excessive release of
proinflammatory mediators can lead to the expression of
costimulatory molecules, which leads to neuroinflammation and nerve
dysfunction (190). It has been
found that the activation of NLRP3 inflammasome and the increase of
ASC expression can be observed in microglia of patients with PD and
PD mouse models. In PD mice, daily oral administration of NLRP3
inhibitor had a neuroprotective effect, reducing the loss of
dopamine in the striatum and the deformation of dopaminergic
substance in the substantia nigra (191,192). These studies show that the
NLRP3 inflammasome has an important relationship with the
occurrence and development of PD, but at present, only a small
number of studies on this topic are available. Further study on the
relationship between microglia, NLRP3 inflammasome and PD will lay
a foundation for further elucidating its pathogenesis.
Role of NLRP3 inflammasome in AD
AD is a chronic and progressive neurodegenerative
disease caused by numerous factors. Its main pathological features
include amyloid β protein (Aβ) deposition, Tau protein
hyperphosphorylation and neuronal loss. At present, AD has become a
major disease that seriously endangers human health all over the
world, but with the current treatment methods, it cannot be cured
and these methods have toxic side effects (193,194).
According to Abbott (195), the NLRP3 inflammasome has an
important role in the pathogenesis of AD. It can stimulate the
innate immune response to trigger the inflammatory response,
release the pro-inflammatory factors IL-1β and IL-18, and continue
to activate during the occurrence and development of the disease,
leading to neurological dysfunction and neuronal apoptosis
(195). The NLRP3 inflammasome
can regulate the activation of caspase-1 and increase the
expression of IL-1β and IL-18 in AD brain tissue, which is related
to the occurrence and development of diseases (196). The NLRP3 inflammasome has been
proved to be co-located with amyloid plaques and its level is
significantly increased in the brains of patients with AD.
Activation of the NLRP3 inflammasome can enhance the aggregation of
Aβ by weakening the phagocytosis of Aβ by microglia, and promote
the development of neuroinflammation and Tau lesions. It has been
reported in the literature that the NLRP3 inflammasome was
significantly activated after Aβ was added to cultured astrocytes,
and it was confirmed by experiments that the phosphorylation level
and pathological features of Tau decreased significantly after Aβ
was injected into the brain of ASC- or NLRP3-deficient mice,
indicating that Aβ-induced Tau lesions were dependent on the NLRP3
inflammasome (197). Of note,
certain researchers found that loss of NLRP3 and the subsequent
activation of inflammatory corpuscles did not significantly affect
the distribution of microglia in Aβ-related pathological reactions.
It was indicated that the NLRP3 inflammasome did not play a
significant role in forming microglia activation or driving
Aβ-induced neurodegeneration (198). Therefore, although its role in
the pathogenesis of AD is controversial, targeting the NLRP3
inflammasome is likely to become a potential treatment for AD.
Role of the NLRP3 inflammasome in
multiple sclerosis (MS)
MS is an autoimmune disease of the central nervous
system, which is characterized by inflammatory cell infiltration,
neuronal degeneration, axonal injury and reactive glial
hyperplasia. The causes and mechanisms of the disease have not been
fully clarified, and it is generally thought that it is the result
of multiple factors, such as genetic susceptibility, environment,
viral infection and immunity (199).
Inoue et al (200) found that peripheral immune
cells could enter the nervous system through the damaged
blood-brain barrier during the pathological process of MS. IL-1β
and caspase-1 are present in MS plaques and the expression of these
proteins in peripheral blood mononuclear cells of patients with MS
is also increased. Experimental autoimmune encephalomyelitis (EAE)
is an ideal animal model of MS and numerous studies have confirmed
that the NLRP3 inflammasome has an important role in EAE. In mice
with silenced expression of the NLRP3 gene, the severity of
pathological changes was reduced, the proliferation of astrocytes
and the infiltration of inflammatory cells were reduced, and the
symptoms were improved (200).
The above research indicates that the neuroprotective effect of
inhibiting inflammasome activation of NLRP3 in MS may provide a new
breakthrough for the effective treatment of MS, and exploring drugs
and methods to inhibit inflammasome activation of NLRP3 will
provide new hope for the effective treatment of MS.
Role of NLRP3 inflammasome in cerebral
ischemia
Cerebral ischemia is a destructive cerebrovascular
disease, which is highly associated with neurological dysfunction,
cognitive dysfunction and severe brain injury. The mechanism of its
occurrence and development has been proved to be closely related to
neuroinflammation, which is the main factor of secondary brain
damage after cerebral ischemia (201).
At present, the clinical efficacy of treating
cerebral ischemia is limited and its pathogenesis needs further
discussion. As an important mediator of neuroinflammatory response
after cerebral ischemia, NLRP3 mediates nerve cell injury and
neuroinflammatory response, and linker ASC and effector protein
caspase-1 are important components of the NLRP3 inflammatory
complex. It has been shown that the expression levels of ASC and
caspase-1 increase significantly after cerebral ischemia, which
plays an important role in regulating the maturation and
differentiation of IL-1β and IL-18 (202). Therefore, Wang et al
(203) preliminarily concluded
that the NLRP3 inflammasome produced proinflammatory factors IL-1β
and IL-18 after cerebral ischemia. In animal experiments,
researchers found that NLRP3-selective inhibitor MCC950 blocked the
activation of NLRP3 inflammasome that can relieve cerebral
ischemia-reperfusion injury (203), and previous studies have
confirmed that TLR4 is an important mediator of neuroinflammatory
cascade reaction in the central nervous system, which activates the
NF-ĸB pathway to promote the transcription of NLRP3 components, and
further regulates the release of downstream inflammatory mediators
through inflammatory reaction. Subsequent studies further confirmed
that cerebral ischemia can induce high-level phosphorylation of
TLR4, NLRP3 and NF-ĸB (204,205). These findings indicate that
NLRP3 inflammasome plays an important role in the pathogenesis of
cerebral ischemia, and inhibiting the activation of NLRP3
inflammasome may provide new ideas for the treatment of cerebral
ischemia (206).
Study of the NLRP3 inflammasome in clinical
treatment
Abnormal activation of NLRP3 inflammasome will
drive the occurrence and development of inflammatory diseases in
numerous systems, so the NLRP3 inflammasome provides important
targets for the treatment of these inflammatory diseases, and
inhibiting the activity of the NLRP3 inflammasome is an important
strategy to alleviate these inflammatory diseases. Scientists have
found numerous biological inhibitors that can prevent the
activation of NLRP3, and found that direct NLRP3 protein inhibitors
have obvious advantages and good therapeutic potential compared
with other types of inhibitors. However, there are no clinical
drugs specifically targeting the NLRP3 inflammasome at present.
Although related inhibitors have entered the clinical trial stage,
their safety and effectiveness need to be further verified and
there is still a lot of work to be done before their clinical
application. Certain studies have made great breakthroughs in gene
therapy. MiRNA and gene editing have become important in the study
of this disease. A series of research results have proved that gene
therapy can improve the course of this disease, but these methods
have not yet entered the clinical trial stage. Therefore, it is of
far-reaching significance to find an NLRP3 inflammasome inhibitor
or gene editing method that can effectively treat NLRP3-related
diseases with high efficiency, specificity and safety.
NLRP3 inflammasome inhibitors
Components of the complex signaling cascade during
the activation of the NLRP3 inflammasome are being targeted to
inhibit NLRP3 inflammasome. According to current development
strategies, these inhibitory effects can be mainly divided into
indirect inhibitors, direct inhibitors and protein inhibitors
related to NLRP3 inflammasome (Table VI).
| Table VIClassical inhibitors of the NLRP3
inflammasome. |
Table VI
Classical inhibitors of the NLRP3
inflammasome.
A, Indirect
inhibitors
|
|---|
| Name | Mechanism of
action | (Refs.) |
|---|
| Auranofin | Inhibition of the
differentiation and maturation of NLRP3 and pro-IL-1β by inhibiting
NF-ĸB activation. | (207) |
| FC11A-2 | Inhibition of the
expression of NLRP3, ASC, caspase-1 and pro-IL-1β. | (207,208) |
| β-Hydroxybutyrate
Celastrol Glyburide | Inhibition of
activation of NLRP3 inflammatory corpuscles is achieved by
inhibiting K+ channels. | (207-211) |
|
Epigallocatechin-3-gallate | Inhibition of the
production of ROS in mitochondria, which is the upstream signal
activated by NLRP3 inflammatory corpuscles, and inhibition of the
synthesis of mitochondrial DNA. | (207) |
| Licochalcone A | Inhibition of the
upstream signal mitochondrial ROS production of NLRP3 inflammatory
corpuscle activation. | (207) |
|
| B, Direct
inhibitors |
|
| Name | Mechanism of
action | (Refs.) |
|
| Provitamin A | Direct binding to
the PYD structure of NLRP3 to inhibit the activation of NLRP3
inflammatory bodies. | (212) |
| CY-09 | Inhibition of
ATPase activity of NLRP3. | (213) |
| MNS | Direct binding to
LRR and NACHT domains of NLRP3 and subsequent inhibition of ATPase
activity of NLRP3. | (214) |
| Bay11-7082 | Alkylation
modification of the ATPase active region of NLRP3 can be carried
out and NF-ĸB signaling can be inhibited. | (215,216) |
|
| C, Protein
inhibitors of other components of NLRP3 inflammasome |
|
| Name | Mechanism of
action | (Refs.) |
| ASC inhibitor
CAPE | Direct binding of
ASC and blocking of the interaction between NLRP3 and ASC. | (217) |
| Caspase-1 inhibitor
VX-740 | The activity of
caspase-1 is inhibited by covalent modification of a cysteine
residue at the active site of caspase-1. | (218) |
| IL-1β
inhibitors | | |
| Anakinra | Antagonist of IL-1β
receptor. | (219) |
| Cankinumab | IL-1β-neutralizing
antibody. | (219) |
| Rilonacept | An IL-1 trapping
agent, which connects two molecules of IL-1β receptors with an
immunoglobulin Fc segment. | (219) |
Indirect inhibitors
Indirect inhibitors mainly inhibit the activation
of NLRP3 inflammasome in three ways: Inhibiting the protein
expression of NLRP3 inflammasome, blocking the upstream signaling
pathway of the NLRP3 inflammasome and inhibiting the
post-translational modification of regulatory proteins of NLRP3.
For instance, auranofin inhibits the differentiation and maturation
of NLRP3 and pro-IL-1β by inhibiting NF-ĸB activation, and
inhibitors such as FC11A-2 inhibit the expression of NLRP3, ASC,
caspase-1 and pro-IL-1β. β-Hydroxybutyrate, celastrol and glyburide
inhibit the activation of NLRP3 inflammasome by inhibiting
K+ channels. Epigallocatechin-3-gallate (EGCG) and
Licochalcone A inhibit the production of ROS in the upstream
signaling of NLRP3 inflammasome activation, and EGCG can also
inhibit mitochondrial DNA synthesis (207-211). These indirect inhibitors have
been proven to have significant therapeutic effects in specific
NLRP3 inflammasome-related disease models in previous studies, but
the defects are also obvious: i) The mechanism of most indirect
inhibitors is unclear; ii) the signaling pathways inhibited by
numerous inhibitors lack specificity, and these signaling pathways
may not be unique to NLRP3 inflammasome, which may easily lead to
immunosuppression and even infection; iii) the upstream signaling
pathway network of the NLRP3 inflammasome is complex and it is
difficult to effectively inhibit the activation process of NLRP3
inflammasome only by inhibiting one of the signaling processes; iv)
this type of inhibitor is only in the experimental stage of disease
models and has not been applied in the clinic.
Direct inhibitors
Direct inhibitors of NLRP3 inflammasome have the
advantages of clear mechanisms of action and specific inhibition of
NLRP3 inflammasome activation. For instance, in the study of GA,
Yang et al (212) found
that provitamin A can directly combine with the PYD structure of
NLRP3, inhibit the activation of NLRP3 inflammasome and directly
reduce the expression level of IL-1β secreted by synovial cells
isolated from patients with GA. CY-09 also inhibits the ATPase
activity of NLRP3, which has also been proven to have the effect of
alleviating GA in mice (213).
There are many such direct inhibitors. For instance, 3,4-meth
ylenedioxy-β-nitrostyrene can directly bind to the LRR and NACHT
domains of NLRP3 and then inhibit the activation of the NLRP3
inflammasome by inhibiting the ATPase activity of NLRP3 (214). Bay11-7082 can alkylate the
ATPase active region of NLRP3, inhibit the NF-ĸB signal and further
inhibit the activation of NLRP3 inflammasome (215,216). Although direct inhibitors of
NLRP3 have obvious advantages and show good therapeutic potential,
and many inhibitors directly targeting NLRP3 protein have been
identified, there are still no therapeutic drugs that specifically
target NLRP3 inflammasome in the clinic. Although a few inhibitors
have been promoted to clinical trials of inflammatory diseases
related to NLRP3, their clinical efficacy and safety still need to
be further verified, which is still a long time from their
potential clinical application.
Protein inhibitors of other components of
NLRP3 inflammasome
Protein inhibitors of other components of NLRP3
inflammasome include ASC inhibitors, caspase-1 inhibitors and
IL-1β/IL-18 inhibitors. Caffeine acid phenethyl ester (CAPE) is the
only reported ASC inhibitor at present. CAPE binds directly to ASC
and then inhibits the activation of NLRP3 inflammasome by blocking
the interaction between NLRP3 and ASC (217). VX-740 is a typical inhibitor of
caspase-1, which impairs the activity of caspase-1 by covalently
modifying the cysteine residue at the active site of caspase-1,
thus inhibiting the activation of the NLRP3 inflammasome, and has
shown good anti-inflammatory effects in the first and second
clinical trials for patients with RA. However, both ASC and
caspase-1 are component proteins needed for the activation of other
inflammatory corpuscles, and the lack of specificity may lead to
immunosuppression, which will eventually increase the risk of
infection (218). IL-1β
inhibitor is the only strategy that can be used to treat NLRP3
inflammasome-related diseases. Its three related biological agents,
anakinra, IL-1β-neutralizing antibody cankinumab and soluble
inducible receptor rilonacept, have been approved by the Food and
Drug Administration, and previous studies have confirmed that it
can effectively reduce the inflammatory response of joint, bone and
muscle diseases, hereditary systemic autoinflammatory diseases and
systemic and local inflammatory diseases (219). The treatment with IL-18
antagonist is similar to that with IL-1β antagonist, but it has not
been used in clinical treatment. Although anti-IL-1β therapy has
been proven to be applicable in the clinic, the related side
effects are quite serious: i) IL-1β is not the specific downstream
pathway of NLRP3 inflammatory corpuscles and other cytokines such
as IL-18 may be involved in the occurrence and development of
inflammatory diseases. ii) IL-1β can be produced by other
inflammatory corpuscles or independent of inflammatory corpuscles.
Blocking IL-1β signaling may increase the risk of infection.
Studies have confirmed that certain infections are even fatal. iii)
The curently available biological agents targeting IL-1β have a
weak ability to penetrate the blood-brain barrier, which leads to
the related treatment methods possibly not being suitable for
central nervous system diseases (220). In short, simply blocking
inflammatory cytokine signals may not be an effective or specific
treatment.
Research on targeted gene therapy
Most inhibitors only inhibit the upstream signaling
of the NLRP3 inflammasome, but not NLRP3 itself, which limits the
effectiveness of inhibitors and may lead to unexpected side
effects. Therefore, gene editing is probably an effective measure
for NLRP3 and numerous scientists have performed large amounts of
research on it.
Role of miRNA in the regulation of NLRP3
inflammasome
Studies have proved that miRNAs regulate
inflammatory corpuscles after transcription. They can inhibit or
increase the expression of inflammatory genes by binding to their
mRNA targets. In the literature, it is known that different miRNAs
regulate different inflammatory corpuscles and NLRP3 is one of the
star members (Table VII).
| Table VIIList of miRNAs known to target NLRP3
inflammasome genes and associated diseases. |
Table VII
List of miRNAs known to target NLRP3
inflammasome genes and associated diseases.
| miRNA | Target gene | Disease | (Refs.) |
|---|
| miR-223 | NLRP3 | Inflammatory bowel
diseases, respiratory diseases and hepatocellular carcinoma | (221-223) |
| miR-133a | NLRP3 | Inflammatory
diseases | (224) |
| miR-22 | NLRP3 | Gastric cancer | (225) |
| miR-30e | NLRP3 | PD | (226) |
| miR-7 | NLRP3 | PD | (227) |
| miR-146a-5p | NLRP3 | MS | (228) |
| miR-20b-5p | NLRP3 | MS | (229) |
| miR-495-3p | NLRP3 | Cardiovascular
disease | (230) |
MiR-233-3p is the first human miRNA that has been
proven to regulate NLRP3. Related studies found that NLRP3
contained the conserved binding site of miR-223-3p in its 3′-UTR.
The binding between this conserved region and miR-223-3p leads to
decreased NLRP3 activity and mutation of this region leads to the
complete loss of apoptosis regulation mediated by miR-223 (221-223). In the process of studying the
role of miRNA in NLRP3 inflammasome, Bandyopadhyay et al
(224) overexpressed and
inhibited miR-133a-1 in differentiated THP1 cells and found that
overexpression of miR-133a-1 increased the levels of caspase-1 and
IL-1β. They demonstrated that miR-133a-1 did not change the basic
expression of individual components of the NLRP3 complex but could
regulate IL-1β (224). Other
scholars identified the constitutive expression of miR-22 in
gastric mucosa and found that miR-22 directly targeted NLRP3 at the
transcription level, which weakened the carcinogenic effect of
NLRP3 in vitro and in vivo. As mentioned above, NLRP3
inflammatory corpuscles play an important role in the process of Hp
infection, and Hp infection significantly inhibits the expression
of miR-22, which directly inhibits the biological effect of miR-22,
leading to the activation of NLRP3 inflammatory corpuscles, thus
promoting cell proliferation, aggravating the course of disease and
even leading to the transformation into gastric cancer (225).
Other miRNAs can affect the NLRP3 inflammasome. For
instance, Li et al (226) found that NLRP3 showed a
conserved binding site for miR-30e in its 3′-UTR, indicating that
there is a connection between miR-30e and neuroinflammation
mediated by NLRP3 inflammatory corpuscles in the pathogenesis of
PD. Animal experiments have confirmed that in the induced PD mouse
model, miR-30e can improve neuronal damage by negatively regulating
the expression of NLRP3 and inhibiting the activation of NLRP3
inflammatory bodies (226).
MiR-7 and neuroinflammation mediated by the NLRP3 inflammasome are
also directly related to the pathogenesis of PD, which directly
regulates the expression of α-Synuclein in dopaminergic neurons
through post-transcriptional regulation, which is related to the
pathophysiology of PD (227).
During the research of multiple sclerosis, Boxberger et al
(228) and Tezcan et al
(229) also found that
miR-146a-5p and miR-20b-5p can also target and regulate NLRP3
inflammasome to control the progress of the disease. MiR-495-3p may
regulate the progression of cardiovascular diseases by targeting
NLRP3 (230).
Gene editing targeting NLRP3
inflammasome
As the third-generation genome editing tool,
clustered regularly interspaced short palindromic repeats
(CRISPR)/CRISPR-associated protein 9 (Cas 9) can specifically and
effectively destroy or repair pathogenic genes through a single
guide (g)RNA-directed Cas 9 nuclease. To date, T cells modified by
CRISPR/Cas 9 in vitro have entered the clinical trials stage
for the treatment of metastatic non-small cell lung cancer
(231). Adenovirus-associated
or adeno-associated virus-mediated CRISPR/Cas9 delivery has been
used to treat diseases such as hypercholesterolemia and Duchenne
muscular dystrophy (232,233). Jiang et al (213) used CRISPR/Cas9 to directly
destroy the key molecule NLRP3 at the genomic level and the results
showed that it completely inhibits the activation of NLRP3 while
avoiding the potential risk of inhibiting the off-target pathway of
anti-inflammatory biological agents and inhibitors. In a mouse
model experiment, Xu et al (234) encapsulated mCas9 and gNLRP3 in
cationic lipid-assisted nanoparticle (CLAN) and then delivered Cas9
mRNA (mCas9)/gNLRP3 to macrophages, which prevented septic shock
and peritonitis, and improved diabetes-related inflammation and
insulin resistance by destroying NLRP3 in macrophages. To date,
only a small number of studies on gene editing of NLRP3 have been
published, but scholars have successfully verified that CLAN
carrying mCas9/gNLRP3 can alleviate acute and chronic inflammatory
diseases, which is undoubtedly expected to be a potential treatment
for NLRP3-dependent inflammatory diseases.
Discussion
With the development of research on the structure
and function of the NLRP3 inflammasome, more and more evidence
shows that the NLRP3 inflammasome is a key molecule mediating
tissue damage in numerous systemic diseases, and their
pro-inflammatory factor release and cell death effects can
aggravate the corresponding tissue damage. As mentioned above,
NLRP3 plays an important role in common diseases of the respiratory
system, cardiovascular system, digestive system, bone joint system
and central nervous system. The NLRP3/caspase-1/IL-1β/IL-18 axis is
a good target for treating these diseases.
Small molecular inhibitors against NLRP3
inflammasome have shown certain therapeutic prospects. Among the
known indirect inhibitors, direct inhibitors and other protein
inhibitors of NLRP3 inflammasome (ASC, caspase-1, IL-1β/IL-18),
only IL-1β inhibitors can be used in clinical treatment and others
are still in the experimental stage. In addition to specific small
molecules, the effective components of classical drugs and plant
extracts have also been verified to inhibit the activation of NLRP3
nonspecifically. Targeting NLRP3 through exosomes and miRNA or
certain gene editing methods may also provide more ideas for
related research.
However, research on NLRP3 inflammasome in the
field of diseases is still in its infancy and the in-depth
mechanism of activation and assembly of NLRP3 inflammasome and its
mediation of cell death requires further clarification. The defects
of various inhibitors, such as low specificity, likeliness to
produce immunosuppression and possible infection, need further
improvement. Therefore, the role of the NLRP3 inflammasome in the
occurrence and development of numerous systemic inflammatory
diseases has been confirmed. However, there is still a long way to
go for its clinical treatment and transformation. Furthermore,
although gene editing or non-coding RNA have successfully confirmed
the feasibility of the new targeted therapy of NLRP3 in basic
research, they are only of great significance for the diagnosis of
diseases in clinical activities from the perspective of epigenetics
and are still rare in clinical application. How to improve
patients' symptoms and delay the progression of the disease as much
as possible while ensuring biological safety requires further
experimental models and clinical research to confirm. Further
elucidation of the mechanisms of the NLRP3 inflammasome will lay
the foundation for fully understanding the occurrence and
development of related diseases and proposing specific targeted
therapies with NLRP3 inflammasome inhibitors and NLRP3 gene
intervention as the core.
It is worth noting that natural small molecular
compounds of traditional Chinese medicine, such as polyphenols,
flavonoids and alkaloids, have attracted the wide attention of
scholars because of their rich sources, complex and diverse
structures, high biocompatibility, few adverse reactions and
incomparable biochemical valence diversity of synthetic compounds.
Although the specific mechanism of action remains to be clarified,
it has been preliminarily confirmed that dihydromyricetin (235) and amygdalin (236) can inhibit the NF-κB signaling
pathway in the start-up stage; oxyphyllin (237) and piperine (238) can block the interaction between
NEK7 and NLRP3; tanshinone I (239) can block the interaction between
ASC and NLRP3; chrysotin (240)
and silybin (241) can inhibit
the activation of caspase-1. Related research is still in the early
stage and more evidence is needed in the future to clarify the
specific molecular mechanisms, as well as to evaluate the
bioavailability, effectiveness and safety of the compounds, so as
to determine whether they are suitable for treating related
diseases and beneficial to improve clinical applications.
There have been numerous studies and summaries on
the NLRP3 inflammasome and various systemic diseases in the past,
but the relevant results are scattered and there is no systematic
summary. There is still no comprehensive summary and analysis of
the NLRP3 inflammasome and the whole clinical disease research. The
role of NLRP3 in various systemic diseases is becoming increasingly
significant. How to prepare related inhibitors or targeting methods
with strong efficacy, long action time and high specificity has
undoubtedly become the focus of current clinical research. In this
paper, the diseases of various systems and the current treatment
methods were comprehensively analyzed, so as to attract the
attention of clinical workers in this field and provide a reference
for follow-up researchers.
Availability of data and materials
Not applicable.
Authors' contributions
JL and NX designed the study; HW wrote the
manuscript; WS and YL prepared the figures; and LM edited the
manuscript. All authors have read and approved the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The study was supported by grants from the National Natural
Science Foundation of China (grant nos. 81671563 and 82172486),
Jiangsu Provincial Commission of Health and Family Planning, the
'Six One' Project of Jiangsu Province (grant no. LGY2016018) and
the Jiangsu Provincial Personnel Department 'the Great of Six
Talented Man Peak' Project (grant no. WSW-04).
References
|
1
|
Li Y, Huang H, Liu B, Zhang Y, Pan X, Yu
XY, Shen Z and Song YH: Inflammasomes as therapeutic targets in
human diseases. Signal Transduct Target Ther. 6:2472021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Duncan JA, Bergstralh DT, Wang Y,
Willingham SB, Ye Z, Zimmermann AG and Ting JP: Cryopyrin/NALP3
binds ATP/dATP, is an ATPase, and requires ATP binding to mediate
inflammatory signaling. Proc Natl Acad Sci USA. 104:8041–8046.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jo EK, Kim JK, Shin DM and Sasakawa C:
Molecular mechanisms regulating NLRP3 inflammasome activation. Cell
Mol Immunol. 13:148–159. 2016. View Article : Google Scholar :
|
|
4
|
Swanson KV, Deng M and Ting JP: The NLRP3
inflammasome: Molecular activation and regulation to therapeutics.
Nat Rev Immunol. 19:477–489. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ozaki E, Campbell M and Doyle SL:
Targeting the NLRP3 inflammasome in chronic inflammatory diseases:
Current perspectives. J Inflamm Res. 8:15–27. 2015.PubMed/NCBI
|
|
6
|
Leemans JC, Cassel SL and Sutterwala FS:
Sensing damage by the NLRP3 inflammasome. Immunol Rev. 243:152–162.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schroder K, Zhou R and Tschopp J: The
NLRP3 inflammasome: A sensor for metabolic danger? Science.
327:296–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Inoue M and Shinohara ML: NLRP3
Inflammasome and MS/EAE. Autoimmune Dis. 2013:8591452013.PubMed/NCBI
|
|
9
|
Zhen Y and Zhang H: NLRP3 inflammasome and
inflammatory bowel disease. Front Immunol. 10:2762019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kelley N, Jeltema D, Duan Y and He Y: The
NLRP3 inflammasome: An overview of mechanisms of activation and
regulation. Int J Mol Sci. 20:33282019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu J and Núñez G: The NLRP3 inflammasome:
Activation and regulation. Trends Biochem Sci. 48:331–344. 2023.
View Article : Google Scholar
|
|
12
|
Paik S, Kim JK, Silwal P, Sasakawa C and
Jo EK: An update on the regulatory mechanisms of NLRP3 inflammasome
activation. Cell Mol Immunol. 18:1141–1160. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Muñoz-Planillo R, Kuffa P, Martínez-Colón
G, Smith BL, Rajendiran TM and Núñez G: K+ efflux is the
common trigger of NLRP3 inflammasome activation by bacterial toxins
and particulate matter. Immunity. 38:1142–1153. 2013. View Article : Google Scholar
|
|
14
|
Xu Z, Chen ZM, Wu X, Zhang L, Cao Y and
Zhou P: Distinct molecular mechanisms underlying potassium efflux
for NLRP3 inflammasome activation. Front Immunol. 11:6094412020.
View Article : Google Scholar
|
|
15
|
Groß CJ, Mishra R, Schneider KS, Médard G,
Wettmarshausen J, Dittlein DC, Shi H, Gorka O, Koenig PA, Fromm S,
et al: K(+) efflux-independent NLRP3 inflammasome activation by
small molecules targeting mitochondria. Immunity. 45:761–773. 2016.
View Article : Google Scholar
|
|
16
|
Sanman LE, Qian Y, Eisele NA, Ng TM, van
der Linden WA, Monack DM, Weerapana E and Bogyo M: Disruption of
glycolytic flux is a signal for inflammasome signaling and
pyroptotic cell death. Elife. 5:e136632016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rajamäki K, Nordström T, Nurmi K, Åkerman
KE, Kovanen PT, Öörni K and Eklund KK: Extracellular acidosis is a
novel danger signal alerting innate immunity via the NLRP3
inflammasome. J Biol Chem. 288:13410–13419. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chae BJ, Lee KS, Hwang I and Yu JW:
Extracellular acidification augments NLRP3-mediated inflammasome
signaling in macrophages. Immune Netw. 23:e232023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee GS, Subramanian N, Kim AI,
Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner
DL and Chae JJ: The calcium-sensing receptor regulates the NLRP3
inflammasome through Ca2+ and cAMP. Nature. 492:123–127. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brough D, Le Feuvre RA, Wheeler RD,
Solovyova N, Hilfiker S, Rothwell NJ and Verkhratsky A: Ca2+ stores
and Ca2+ entry differentially contribute to the release of IL-1
beta and IL-1 alpha from murine macrophages. J Immunol.
170:3029–3036. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Feldmeyer L, Keller M, Niklaus G, Hohl D,
Werner S and Beer HD: The inflammasome mediates UVB-induced
activation and secretion of interleukin-1beta by keratinocytes.
Curr Biol. 17:1140–1145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chu J, Thomas LM, Watkins SC, Franchi L,
Núñez G and Salter RD: Cholesterol-dependent cytolysins induce
rapid release of mature IL-1beta from murine macrophages in a NLRP3
inflammasome and cathepsin B-dependent manner. J Leukoc Biol.
86:1227–1238. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Katsnelson MA, Rucker LG, Russo HM and
Dubyak GR: K+ efflux agonists induce NLRP3 inflammasome activation
independently of Ca2+ signaling. J Immunol. 194:3937–3952. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang T, Lang X, Xu C, Wang X, Gong T, Yang
Y, Cui J, Bai L, Wang J, Jiang W and Zhou R: CLICs-dependent
chloride efflux is an essential and proximal upstream event for
NLRP3 inflammasome activation. Nat Commun. 8:2022017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Verhoef PA, Kertesy SB, Lundberg K,
Kahlenberg JM and Dubyak GR: Inhibitory effects of chloride on the
activation of caspase-1, IL-1beta secretion, and cytolysis by the
P2X7 receptor. J Immunol. 175:7623–7634. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mayes-Hopfinger L, Enache A, Xie J, Huang
CL, Köchl R, Tybulewicz VLJ, Fernandes-Alnemri T and Alnemri ES:
Chloride sensing by WNK1 regulates NLRP3 inflammasome activation
and pyroptosis. Nat Commun. 12:45462021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Green JP, Yu S, Martín-Sánchez F, Pelegrin
P, Lopez-Castejon G, Lawrence CB and Brough D: Chloride regulates
dynamic NLRP3-dependent ASC oligomerization and inflammasome
priming. Proc Natl Acad Sci USA. 115:E9371–E9380. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar
|
|
29
|
Dominic A, Le NT and Takahashi M: Loop
between NLRP3 inflammasome and reactive oxygen species. Antioxid
Redox Signal. 36:784–796. 2022. View Article : Google Scholar
|
|
30
|
Shimada K, Crother TR, Karlin J, Dagvadorj
J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et
al: Oxidized mitochondrial DNA activates the NLRP3 inflammasome
during apoptosis. Immunity. 36:401–414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Iyer SS, He Q, Janczy JR, Elliott EI,
Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, et
al: Mitochondrial cardiolipin is required for Nlrp3 inflammasome
activation. Immunity. 39:311–323. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Halle A, Hornung V, Petzold GC, Stewart
CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ and
Golenbock DT: The NALP3 inflammasome is involved in the innate
immune response to amyloid-beta. Nat Immunol. 9:857–865. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lima H Jr, Jacobson LS, Goldberg MF,
Chandran K, Diaz-Griffero F, Lisanti MP and Brojatsch J: Role of
lysosome rupture in controlling Nlrp3 signaling and necrotic cell
death. Cell Cycle. 12:1868–1878. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schorn C, Frey B, Lauber K, Janko C,
Strysio M, Keppeler H, Gaipl US, Voll RE, Springer E, Munoz LE, et
al: Sodium overload and water influx activate the NALP3
inflammasome. J Biol Chem. 286:35–41. 2011. View Article : Google Scholar :
|
|
35
|
Hornung V, Bauernfeind F, Halle A, Samstad
EO, Kono H, Rock KL, Fitzgerald KA and Latz E: Silica crystals and
aluminum salts activate the NALP3 inflammasome through phagosomal
destabilization. Nat Immunol. 9:847–856. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dostert C, Guarda G, Romero JF, Menu P,
Gross O, Tardivel A, Suva ML, Stehle JC, Kopf M, Stamenkovic I, et
al: Malarial hemozoin is a Nalp3 inflammasome activating danger
signal. PLoS One. 4:e65102009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Orlowski GM, Colbert JD, Sharma S, Bogyo
M, Robertson SA and Rock KL: Multiple cathepsins promote Pro-IL-1β
synthesis and NLRP3-mediated IL-1β activation. J Immunol.
195:1685–1697. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kayagaki N, Stowe IB, Lee BL, O'Rourke K,
Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT,
et al: Caspase-11 cleaves gasdermin D for non-canonical
inflammasome signalling. Nature. 526:666–671. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kayagaki N, Warming S, Lamkanfi M, Vande
Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al:
Non-canonical inflammasome activation targets caspase-11. Nature.
479:117–121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li
P, Hu L and Shao F: Inflammatory caspases are innate immune
receptors for intracellular LPS. Nature. 514:187–192. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ding J, Wang K, Liu W, She Y, Sun Q, Shi
J, Sun H, Wang DC and Shao F: Pore-forming activity and structural
autoinhibition of the gasdermin family. Nature. 535:111–116. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ramirez MLG, Poreba M, Snipas SJ, Groborz
K, Drag M and Salvesen GS: Extensive peptide and natural protein
substrate screens reveal that mouse caspase-11 has much narrower
substrate specificity than caspase-1. J Biol Chem. 293:7058–7067.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gaidt MM, Ebert TS, Chauhan D, Schmidt T,
Schmid-Burgk JL, Rapino F, Robertson AA, Cooper MA, Graf T and
Hornung V: Human monocytes engage an alternative inflammasome
pathway. Immunity. 44:833–846. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang L and Hauenstein AV: The NLRP3
inflammasome: Mechanism of action, role in disease and therapies.
Mol Aspects Med. 76:1008892020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
He Y, Franchi L and Núñez G: TLR agonists
stimulate Nlrp3-dependent IL-1β production independently of the
purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol.
190:334–339. 2013. View Article : Google Scholar
|
|
46
|
Wang C, Zhou J, Wang J, Li S, Fukunaga A,
Yodoi J and Tian H: Progress in the mechanism and targeted drug
therapy for COPD. Signal Transduct Target Ther. 5:2482020.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Birrell MA and Eltom S: The role of the
NLRP3 inflammasome in the pathogenesis of airway disease. Pharmacol
Ther. 130:364–370. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cicko S, Lucattelli M, Müller T,
Lommatzsch M, De Cunto G, Cardini S, Sundas W, Grimm M, Zeiser R,
Dürk T, et al: Purinergic receptor inhibition prevents the
development of smoke-induced lung injury and emphysema. J Immunol.
185:688–697. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
De Nardo D, De Nardo CM and Latz E: New
insights into mechanisms controlling the NLRP3 inflammasome and its
role in lung disease. Am J Pathol. 184:42–54. 2014. View Article : Google Scholar :
|
|
50
|
Hosseinian N, Cho Y, Lockey RF and
Kolliputi N: The role of the NLRP3 inflammasome in pulmonary
diseases. Ther Adv Respir Dis. 9:188–197. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pauwels NS, Bracke KR, Dupont LL, Van
Pottelberge GR, Provoost S, Vanden Berghe T, Vandenabeele P,
Lambrecht BN, Joos GF and Brusselle GG: Role of IL-1α and the
Nlrp3/caspase-1/IL-1β axis in cigarette smoke-induced pulmonary
inflammation and COPD. Eur Respir J. 38:1019–1028. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hoshino T, Kato S, Oka N, Imaoka H,
Kinoshita T, Takei S, Kitasato Y, Kawayama T, Imaizumi T, Yamada K,
et al: Pulmonary inflammation and emphysema: Role of the cytokines
IL-18 and IL-13. Am J Respir Crit Care Med. 176:49–62. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Churg A, Zhou S, Wang X, Wang R and Wright
JL: The role of interleukin-1beta in murine cigarette smoke-induced
emphysema and small airway remodeling. Am J Respir Cell Mol Biol.
40:482–490. 2009. View Article : Google Scholar
|
|
54
|
Eltom S, Stevenson CS, Rastrick J, Dale N,
Raemdonck K, Wong S, Catley MC, Belvisi MG and Birrell MA: P2X7
receptor and caspase 1 activation are central to airway
inflammation observed after exposure to tobacco smoke. PLoS One.
6:e240972011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xu S, Panettieri RA Jr and Jude J:
Metabolomics in asthma: A platform for discovery. Mol Aspects Med.
85:1009902022. View Article : Google Scholar :
|
|
56
|
Müller T, Vieira RP, Grimm M, Dürk T,
Cicko S, Zeiser R, Jakob T, Martin SF, Blumenthal B, Sorichter S,
et al: A potential role for P2X7R in allergic airway inflammation
in mice and humans. Am J Respir Cell Mol Biol. 44:456–464. 2011.
View Article : Google Scholar
|
|
57
|
Yazdi AS, Guarda G, Riteau N, Drexler SK,
Tardivel A, Couillin I and Tschopp J: Nanoparticles activate the
NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause
pulmonary inflammation through release of IL-1α and IL-1β. Proc
Natl Acad Sci USA. 107:19449–19454. 2010. View Article : Google Scholar
|
|
58
|
Ather JL, Ckless K, Martin R, Foley KL,
Suratt BT, Boyson JE, Fitzgerald KA, Flavell RA, Eisenbarth SC and
Poynter ME: Serum amyloid A activates the NLRP3 inflammasome and
promotes Th17 allergic asthma in mice. J Immunol. 187:64–73. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Idzko M, Hammad H, van Nimwegen M, Kool M,
Willart MA, Muskens F, Hoogsteden HC, Luttmann W, Ferrari D, Di
Virgilio F, et al: Extracellular ATP triggers and maintains
asthmatic airway inflammation by activating dendritic cells. Nat
Med. 13:913–919. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nakae S, Komiyama Y, Yokoyama H, Nambu A,
Umeda M, Iwase M, Homma I, Sudo K, Horai R, Asano M and Iwakura Y:
IL-1 is required for allergen-specific Th2 cell activation and the
development of airway hypersensitivity response. Int Immunol.
15:483–490. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang CC, Fu CL, Yang YH, Lo YC, Wang LC,
Chuang YH, Chang DM and Chiang BL: Adenovirus expressing
interleukin-1 receptor antagonist alleviates allergic airway
inflammation in a murine model of asthma. Gene Ther. 13:1414–1421.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Harada M, Obara K, Hirota T, Yoshimoto T,
Hitomi Y, Sakashita M, Doi S, Miyatake A, Fujita K, Enomoto T, et
al: A functional polymorphism in IL-18 is associated with severity
of bronchial asthma. Am J Respir Crit Care Med. 180:1048–1055.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Luo W, Hu J, Xu W and Dong J: Distinct
spatial and temporal roles for Th1, Th2, and Th17 cells in asthma.
Front Immunol. 13:9740662022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Besnard AG, Guillou N, Tschopp J, Erard F,
Couillin I, Iwakura Y, Quesniaux V, Ryffel B and Togbe D: NLRP3
inflammasome is required in murine asthma in the absence of
aluminum adjuvant. Allergy. 66:1047–1057. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Eisenbarth SC, Colegio OR, O'Connor W,
Sutterwala FS and Flavell RA: Crucial role for the Nalp3
inflammasome in the immunostimulatory properties of aluminium
adjuvants. Nature. 453:1122–1126. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Demento SL, Eisenbarth SC, Foellmer HG,
Platt C, Caplan MJ, Mark Saltzman W, Mellman I, Ledizet M, Fikrig
E, Flavell RA and Fahmy TM: Inflammasome-activating nanoparticles
as modular systems for optimizing vaccine efficacy. Vaccine.
27:3013–3021. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sawada M, Kawayama T, Imaoka H, Sakazaki
Y, Oda H, Takenaka S, Kaku Y, Azuma K, Tajiri M, Edakuni N, et al:
IL-18 induces airway hyperresponsiveness and pulmonary inflammation
via CD4+ T cell and IL-13. PLoS One. 8:e546232013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yamagata S, Tomita K, Sato R, Niwa A,
Higashino H and Tohda Y: Interleukin-18-deficient mice exhibit
diminished chronic inflammation and airway remodelling in
ovalbumin-induced asthma model. Clin Exp Immunol. 154:295–304.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Allen IC, Jania CM, Wilson JE, Tekeppe EM,
Hua X, Brickey WJ, Kwan M, Koller BH, Tilley SL and Ting JP:
Analysis of NLRP3 in the development of allergic airway disease in
mice. J Immunol. 188:2884–2893. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Hartwig C, Tschernig T, Mazzega M, Braun A
and Neumann D: Endogenous IL-18 in experimentally induced asthma
affects cytokine serum levels but is irrelevant for clinical
symptoms. Cytokine. 42:298–305. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Leung CC, Yu IT and Chen W: Silicosis.
Lancet. 379:2008–2018. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Cassel SL, Eisenbarth SC, Iyer SS, Sadler
JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA and
Sutterwala FS: The Nalp3 inflammasome is essential for the
development of silicosis. Proc Natl Acad Sci USA. 105:9035–9040.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Jessop F, Hamilton RF, Rhoderick JF, Shaw
PK and Holian A: Autophagy deficiency in macrophages enhances NLRP3
inflammasome activity and chronic lung disease following silica
exposure. Toxicol Appl Pharmacol. 309:101–110. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tong SY, Davis JS, Eichenberger E, Holland
TL and Fowler VG Jr: Staphylococcus aureus infections:
Epidemiology, pathophysiology, clinical manifestations, and
management. Clin Microbiol Rev. 28:603–661. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Craven RR, Gao X, Allen IC, Gris D, Bubeck
Wardenburg J, McElvania-Tekippe E, Ting JP and Duncan JA:
Staphylococcus aureus alpha-hemolysin activates the NLRP3
inflammasome in human and mouse monocytic cells. PLoS One.
4:e74462009. View Article : Google Scholar
|
|
76
|
Kebaier C, Chamberland RR, Allen IC, Gao
X, Broglie PM, Hall JD, Jania C, Doerschuk CM, Tilley SL and Duncan
JA: Staphylococcus aureus α-hemolysin mediates virulence in a
murine model of severe pneumonia through activation of the NLRP3
inflammasome. J Infect Dis. 205:807–817. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bewley MA, Naughton M, Preston J, Mitchell
A, Holmes A, Marriott HM, Read RC, Mitchell TJ, Whyte MK and
Dockrell DH: Pneumolysin activates macrophage lysosomal membrane
permeabilization and executes apoptosis by distinct mechanisms
without membrane pore formation. mBio. 5:e01710–14. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Witzenrath M, Pache F, Lorenz D, Koppe U,
Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, et
al: The NLRP3 inflammasome is differentially activated by
pneumolysin variants and contributes to host defense in
pneumococcal pneumonia. J Immunol. 187:434–440. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
McNeela EA, Burke A, Neill DR, Baxter C,
Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, McLoughlin RM,
Mori A, et al: Pneumolysin activates the NLRP3 inflammasome and
promotes proinflammatory cytokines independently of TLR4. PLoS
Pathog. 6:e10011912010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
van Lieshout MH, Scicluna BP, Florquin S
and van der Poll T: NLRP3 and ASC differentially affect the lung
transcriptome during pneumococcal pneumonia. Am J Respir Cell Mol
Biol. 50:699–712. 2014. View Article : Google Scholar
|
|
81
|
Gravina AG, Zagari RM, De Musis C, Romano
L, Loguercio C and Romano M: Helicobacter pylori and extragastric
diseases: A review. World J Gastroenterol. 24:3204–3221. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Venerito M, Vasapolli R, Rokkas T,
Delchier JC and Malfertheiner P: Helicobacter pylori, gastric
cancer and other gastrointestinal malignancies. Helicobacter.
22(Suppl 1)John Wiley and Sons; 2017, View Article : Google Scholar
|
|
83
|
Pachathundikandi SK, Blaser N, Bruns H and
Backert S: Helicobacter pylori avoids the critical activation of
NLRP3 inflammasome-mediated production of oncogenic mature IL-1β in
human immune cells. Cancers (Basel). 12:8032020. View Article : Google Scholar
|
|
84
|
Li X, Liu S, Luo J, Liu A, Tang S, Liu S,
Yu M and Zhang Y: Helicobacter pylori induces IL-1β and IL-18
production in human monocytic cell line through activation of NLRP3
inflammasome via ROS signaling pathway. Pathog Dis. 73:ftu0242015.
View Article : Google Scholar
|
|
85
|
Shigematsu Y, Niwa T, Rehnberg E, Toyoda
T, Yoshida S, Mori A, Wakabayashi M, Iwakura Y, Ichinose M, Kim YJ
and Ushijima T: Interleukin-1β induced by Helicobacter pylori
infection enhances mouse gastric carcinogenesis. Cancer Lett.
340:141–147. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Serizawa T, Hirata Y, Hayakawa Y, Suzuki
N, Sakitani K, Hikiba Y, Ihara S, Kinoshita H, Nakagawa H, Tateishi
K and Koike K: Gastric metaplasia induced by helicobacter pylori is
associated with enhanced SOX9 expression via interleukin-1
signaling. Infect Immun. 84:562–572. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kameoka S, Kameyama T, Hayashi T, Sato S,
Ohnishi N, Hayashi T, Murata-Kamiya N, Higashi H, Hatakeyama M and
Takaoka A: Helicobacter pylori induces IL-1β protein through the
inflammasome activation in differentiated macrophagic cells. Biomed
Res. 37:21–27. 2016. View Article : Google Scholar
|
|
88
|
Yu Q, Shi H, Ding Z, Wang Z, Yao H and Lin
R: The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome
activation in Helicobacter pylori-associated gastritis by
regulating ROS and autophagy. Cell Commun Signal. 21:12023.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Davari F, Shokri-Shirvani J, Sepidarkish M
and Nouri HR: Elevated expression of the AIM2 gene in response to
Helicobacter pylori along with the decrease of NLRC4 inflammasome
is associated with peptic ulcer development. APMIS. 131:339–350.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Asrani SK, Devarbhavi H, Eaton J and
Kamath PS: Burden of liver diseases in the world. J Hepatol.
70:151–171. 2019. View Article : Google Scholar
|
|
91
|
Sharma A and Nagalli S: Chronic Liver
Disease. StatPearls [Internet] Treasure Island, FL: 2023
|
|
92
|
Wree A, McGeough MD, Peña CA, Schlattjan
M, Li H, Inzaugarat ME, Messer K, Canbay A, Hoffman HM and
Feldstein AE: NLRP3 inflammasome activation is required for
fibrosis development in NAFLD. J Mol Med (Berl). 92:1069–1082.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Arab JP, Arrese M and Trauner M: Recent
insights into the pathogenesis of nonalcoholic fatty liver disease.
Annu Rev Pathol. 13:321–350. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ and
Jo EK: Upregulated NLRP3 inflammasome activation in patients with
type 2 diabetes. Diabetes. 62:194–204. 2013. View Article : Google Scholar
|
|
95
|
Vandanmagsar B, Youm YH, Ravussin A,
Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM and Dixit
VD: The NLRP3 inflammasome instigates obesity-induced inflammation
and insulin resistance. Nat Med. 17:179–188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Csak T, Ganz M, Pespisa J, Kodys K,
Dolganiuc A and Szabo G: Fatty acid and endotoxin activate
inflammasomes in mouse hepatocytes that release danger signals to
stimulate immune cells. Hepatology. 54:133–144. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Mellinger JL: Epidemiology of alcohol use
and alcoholic liver disease. Clin Liver Dis (Hoboken). 13:136–139.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Ohashi K, Pimienta M and Seki E: Alcoholic
liver disease: A current molecular and clinical perspective. Liver
Res. 2:161–172. 2018. View Article : Google Scholar
|
|
99
|
Voican CS, Njiké-Nakseu M, Boujedidi H,
Barri-Ova N, Bouchet-Delbos L, Agostini H, Maitre S, Prévot S,
Cassard-Doulcier AM, Naveau S and Perlemuter G: Alcohol withdrawal
alleviates adipose tissue inflammation in patients with alcoholic
liver disease. Liver Int. 35:967–978. 2015. View Article : Google Scholar
|
|
100
|
Petrasek J, Bala S, Csak T, Lippai D,
Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA and Szabo G:
IL-1 receptor antagonist ameliorates inflammasome-dependent
alcoholic steatohepatitis in mice. J Clin Invest. 122:3476–3489.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Dixon LJ, Berk M, Thapaliya S, Papouchado
BG and Feldstein AE: Caspase-1-mediated regulation of fibrogenesis
in diet-induced steatohepatitis. Lab Invest. 92:713–723. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Boaru SG, Borkham-Kamphorst E, Tihaa L,
Haas U and Weiskirchen R: Expression analysis of inflammasomes in
experimental models of inflammatory and fibrotic liver disease. J
Inflamm (Lond). 9:492012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Alyaseer AAA, de Lima MHS and Braga TT:
The role of NLRP3 inflammasome activation in the epithelial to
mesenchymal transition process during the fibrosis. Front Immunol.
11:8832020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Lozano-Ruiz B, Bachiller V,
García-Martínez I, Zapater P, Gómez-Hurtado I, Moratalla A, Giménez
P, Bellot P, Francés R, Such J and González-Navajas JM: Absent in
melanoma 2 triggers a heightened inflammasome response in ascitic
fluid macrophages of patients with cirrhosis. J Hepatol. 62:64–71.
2015. View Article : Google Scholar
|
|
106
|
Liu X, Zhou W, Zhang X, Lu P, Du Q, Tao L,
Ding Y, Wang Y and Hu R: Dimethyl fumarate ameliorates dextran
sulfate sodium-induced murine experimental colitis by activating
Nrf2 and suppressing NLRP3 inflammasome activation. Biochem
Pharmacol. 112:37–49. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Sivakumar PV, Westrich GM, Kanaly S, Garka
K, Born TL, Derry JM and Viney JL: Interleukin 18 is a primary
mediator of the inflammation associated with dextran sulphate
sodium induced colitis: Blocking interleukin 18 attenuates
intestinal damage. Gut. 50:812–820. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Siegmund B, Fantuzzi G, Rieder F,
Gamboni-Robertson F, Lehr HA, Hartmann G, Dinarello CA, Endres S
and Eigler A: Neutralization of interleukin-18 reduces severity in
murine colitis and intestinal IFN-gamma and TNF-alpha production.
Am J Physiol Regul Integr Comp Physiol. 281:R1264–R1273. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Siegmund B: Interleukin-1beta converting
enzyme (caspase-1) in intestinal inflammation. Biochem Pharmacol.
64:1–8. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Zhao S, Gong Z, Zhou J, Tian C, Gao Y, Xu
C, Chen Y, Cai W and Wu J: Deoxycholic acid triggers NLRP3
inflammasome activation and aggravates DSS-induced colitis in mice.
Front Immunol. 7:5362016. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Zherebiatiev A and Kamyshnyi A: Expression
levels of proinflammatory cytokines and NLRP3 inflammasome in an
experimental model of oxazolone-induced colitis. Iran J Allergy
Asthma Immunol. 15:39–45. 2016.PubMed/NCBI
|
|
112
|
Bauer C, Duewell P, Lehr HA, Endres S and
Schnurr M: Protective and aggravating effects of Nlrp3 inflammasome
activation in IBD models: Influence of genetic and environmental
factors. Dig Dis. 30(Suppl 1): S82–S90. 2012. View Article : Google Scholar
|
|
113
|
Allen IC, TeKippe EM, Woodford RM, Uronis
JM, Holl EK, Rogers AB, Herfarth HH, Jobin C and Ting JP: The NLRP3
inflammasome functions as a negative regulator of tumorigenesis
during colitis-associated cancer. J Exp Med. 207:1045–1056. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zaki MH, Boyd KL, Vogel P, Kastan MB,
Lamkanfi M and Kanneganti TD: The NLRP3 inflammasome protects
against loss of epithelial integrity and mortality during
experimental colitis. Immunity. 32:379–391. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Dupaul-Chicoine J, Yeretssian G, Doiron K,
Bergstrom KS, McIntire CR, LeBlanc PM, Meunier C, Turbide C, Gros
P, Beauchemin N, et al: Control of intestinal homeostasis, colitis,
and colitis-associated colorectal cancer by the inflammatory
caspases. Immunity. 32:367–378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Radtke F and Clevers H: Self-renewal and
cancer of the gut: two sides of a coin. Science. 307:1904–1909.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Itzkowitz SH and Yio X: Inflammation and
cancer IV. Colorectal cancer in inflammatory bowel disease: The
role of inflammation. Am J Physiol Gastrointest Liver Physiol.
287:G7–G17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Du Q, Wang Q, Fan H, Wang J, Liu X, Wang
H, Wang Y and Hu R: Dietary cholesterol promotes AOM-induced
colorectal cancer through activating the NLRP3 inflammasome.
Biochem Pharmacol. 105:42–54. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Micallef MJ, Tanimoto T, Kohno K, Ikeda M
and Kurimoto M: Interleukin 18 induces the sequential activation of
natural killer cells and cytotoxic T lymphocytes to protect
syngeneic mice from transplantation with Meth A sarcoma. Cancer
Res. 57:4557–4563. 1997.PubMed/NCBI
|
|
120
|
Micallef MJ, Yoshida K, Kawai S, Hanaya T,
Kohno K, Arai S, Tanimoto T, Torigoe K, Fujii M, Ikeda M and
Kurimoto M: In vivo antitumor effects of murine
interferon-gamma-inducing factor/interleukin-18 in mice bearing
syngeneic Meth A sarcoma malignant ascites. Cancer Immunol
Immunother. 43:361–367. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Osaki T, Péron JM, Cai Q, Okamura H,
Robbins PD, Kurimoto M, Lotze MT and Tahara H: IFN-gamma-inducing
factor/IL-18 administration mediates IFN-gamma- and
IL-12-independent antitumor effects. J Immunol. 160:1742–1749.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Coughlin CM, Salhany KE, Wysocka M, Aruga
E, Kurzawa H, Chang AE, Hunter CA, Fox JC, Trinchieri G and Lee WM:
Interleukin-12 and interleukin-18 synergistically induce murine
tumor regression which involves inhibition of angiogenesis. J Clin
Invest. 101:1441–1452. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Hegardt P, Widegren B, Li L, Sjögren B,
Kjellman C, Sur I and Sjögren HO: Nitric oxide synthase inhibitor
and IL-18 enhance the anti-tumor immune response of rats carrying
an intrahepatic colon carcinoma. Cancer Immunol Immunother.
50:491–501. 2001. View Article : Google Scholar
|
|
124
|
Zaki MH, Vogel P, Body-Malapel M, Lamkanfi
M and Kanneganti TD: IL-18 production downstream of the Nlrp3
inflammasome confers protection against colorectal tumor formation.
J Immunol. 185:4912–4920. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Reuter BK and Pizarro TT: Commentary: the
role of the IL-18 system and other members of the IL-1R/TLR
superfamily in innate mucosal immunity and the pathogenesis of
inflammatory bowel disease: Friend or foe? Eur J Immunol.
34:2347–2355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Swaroop VS, Chari ST and Clain JE: Severe
acute pancreatitis. JAMA. 291:2865–2868. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Yang ZW, Meng XX and Xu P: Central role of
neutrophil in the pathogenesis of severe acute pancreatitis. J Cell
Mol Med. 19:2513–2520. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Janiak A, Leśniowski B, Jasińska A,
Pietruczuk M and Małecka-Panas E: Interleukin 18 as an early marker
or prognostic factor in acute pancreatitis. Prz Gastroenterol.
10:203–207. 2015.
|
|
129
|
Fu Q, Zhai Z, Wang Y, Xu L, Jia P, Xia P,
Liu C, Zhang X, Qin T and Zhang H: NLRP3 deficiency alleviates
severe acute pancreatitis and pancreatitis-associated lung injury
in a mouse model. Biomed Res Int. 2018:12949512018. View Article : Google Scholar
|
|
130
|
Hoque R, Farooq A, Ghani A, Gorelick F and
Mehal WZ: Lactate reduces liver and pancreatic injury in Toll-like
receptor- and inflammasome-mediated inflammation via GPR81-mediated
suppression of innate immunity. Gastroenterology. 146:1763–1774.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Wu BU, Hwang JQ, Gardner TH, Repas K,
Delee R, Yu S, Smith B, Banks PA and Conwell DL: Lactated Ringer's
solution reduces systemic inflammation compared with saline in
patients with acute pancreatitis. Clin Gastroenterol Hepatol.
9:710–717.e1. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Ren JD, Ma J, Hou J, Xiao WJ, Jin WH, Wu J
and Fan KH: Hydrogen-rich saline inhibits NLRP3 inflammasome
activation and attenuates experimental acute pancreatitis in mice.
Mediators Inflamm. 2014:9308942014. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Smolen JS, Aletaha D, Barton A, Burmester
GR, Emery P, Firestein GS, Kavanaugh A, McInnes IB, Solomon DH,
Strand V and Yamamoto K: Rheumatoid arthritis. Nat Rev Dis Primers.
4:180012018. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Kolly L, Busso N, Palmer G, Talabot-Ayer
D, Chobaz V and So A: Expression and function of the NALP3
inflammasome in rheumatoid synovium. Immunology. 129:178–185. 2010.
View Article : Google Scholar :
|
|
135
|
Rosengren S, Hoffman HM, Bugbee W and
Boyle DL: Expression and regulation of cryopyrin and related
proteins in rheumatoid arthritis synovium. Ann Rheum Dis.
64:708–714. 2005. View Article : Google Scholar
|
|
136
|
Choulaki C, Papadaki G, Repa A, Kampouraki
E, Kambas K, Ritis K, Bertsias G, Boumpas DT and Sidiropoulos P:
Enhanced activity of NLRP3 inflammasome in peripheral blood cells
of patients with active rheumatoid arthritis. Arthritis Res Ther.
17:2572015. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Zhang Y, Zheng Y and Li H: NLRP3
Inflammasome plays an important role in the pathogenesis of
collagen-induced arthritis. Mediators Inflamm. 2016:96562702016.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Guo C, Fu R, Wang S, Huang Y, Li X, Zhou
M, Zhao J and Yang N: NLRP3 inflammasome activation contributes to
the pathogenesis of rheumatoid arthritis. Clin Exp Immunol.
194:231–243. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Matmati M, Jacques P, Maelfait J,
Verheugen E, Kool M, Sze M, Geboes L, Louagie E, Mc Guire C,
Vereecke L, et al: A20 (TNFAIP3) deficiency in myeloid cells
triggers erosive polyarthritis resembling rheumatoid arthritis. Nat
Genet. 43:908–912. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
140
|
Vande Walle L, Van Opdenbosch N, Jacques
P, Fossoul A, Verheugen E, Vogel P, Beyaert R, Elewaut D,
Kanneganti TD, van Loo G and Lamkanfi M: Negative regulation of the
NLRP3 inflammasome by A20 protects against arthritis. Nature.
512:69–73. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Li XF, Shen WW, Sun YY, Li WX, Sun ZH, Liu
YH, Zhang L, Huang C, Meng XM and Li J: MicroRNA-20a negatively
regulates expression of NLRP3 inflammasome by targeting TXNIP in
adjuvant-induced arthritis fibroblast-like synoviocytes. Joint Bone
Spine. 83:695–700. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Huang Y, Lu D, Ma W, Liu J, Ning Q, Tang F
and Li L: miR-223 in exosomes from bone marrow mesenchymal stem
cells ameliorates rheumatoid arthritis via downregulation of NLRP3
expression in macrophages. Mol Immunol. 143:68–76. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Li Y, Zheng JY, Liu JQ, Yang J, Liu Y,
Wang C, Ma XN, Liu BL, Xin GZ and Liu LF: Succinate/NLRP3
inflammasome induces synovial fibroblast activation: Therapeutical
effects of clematichinenoside AR on arthritis. Front Immunol.
7:5322016. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Han X, Lin D, Huang W, Li D, Li N and Xie
X: Mechanism of NLRP3 inflammasome intervention for synovitis in
knee osteoarthritis: A review of TCM intervention. Front Genet.
14:11591672023. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Derfus BA, Kurian JB, Butler JJ, Daft LJ,
Carrera GF, Ryan LM and Rosenthal AK: The high prevalence of
pathologic calcium crystals in pre-operative knees. J Rheumatol.
29:570–574. 2002.PubMed/NCBI
|
|
146
|
Pazár B, Ea HK, Narayan S, Kolly L,
Bagnoud N, Chobaz V, Roger T, Lioté F, So A and Busso N: Basic
calcium phosphate crystals induce monocyte/macrophage IL-1β
secretion through the NLRP3 inflammasome in vitro. J Immunol.
186:2495–2502. 2011. View Article : Google Scholar
|
|
147
|
An S, Hu H, Li Y and Hu Y: Pyroptosis
plays a role in osteoarthritis. Aging Dis. 11:1146–1157. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Shao M, Lv D, Zhou K, Sun H and Wang Z:
Senkyunolide A inhibits the progression of osteoarthritis by
inhibiting the NLRP3 signalling pathway. Pharm Biol. 60:535–542.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Lordén G, Sanjuán-García I, de Pablo N,
Meana C, Alvarez-Miguel I, Pérez-García MT, Pelegrín P, Balsinde J
and Balboa MA: Lipin-2 regulates NLRP3 inflammasome by affecting
P2X7 receptor activation. J Exp Med. 214:511–528. 2017. View Article : Google Scholar :
|
|
150
|
Compston JE, McClung MR and Leslie WD:
Osteoporosis. Lancet. 393:364–376. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Alippe Y, Wang C, Ricci B, Xiao J, Qu C,
Zou W, Novack DV, Abu-Amer Y, Civitelli R and Mbalaviele G: Bone
matrix components activate the NLRP3 inflammasome and promote
osteoclast differentiation. Sci Rep. 7:66302017. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Detzen L, Cheat B, Besbes A, Hassan B,
Marchi V, Baroukh B, Lesieur J, Sadoine J, Torrens C, Rochefort G,
et al: NLRP3 is involved in long bone edification and the
maturation of osteogenic cells. J Cell Physiol. 236:4455–4469.
2021. View Article : Google Scholar
|
|
153
|
Jiang N, An J, Yang K, Liu J, Guan C, Ma C
and Tang X: NLRP3 Inflammasome: A new target for prevention and
control of osteoporosis? Front Endocrinol (Lausanne).
12:7525462021. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Rocha FRG, Delitto AE, de Souza JAC,
González-Maldonado LA, Wallet SM and Rossa Junior C: Relevance of
caspase-1 and Nlrp3 inflammasome on inflammatory bone resorption in
a murine model of periodontitis. Sci Rep. 10:78232020. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Wang C, Xiao J, Nowak K, Gunasekera K,
Alippe Y, Speckman S, Yang T, Kress D, Abu-Amer Y, Hottiger MO and
Mbalaviele G: PARP1 hinders histone H2B occupancy at the NFATc1
promoter to restrain osteoclast differentiation. J Bone Miner Res.
35:776–788. 2020. View Article : Google Scholar
|
|
156
|
Guaraná WL, Lima CAD, Barbosa AD, Crovella
S and Sandrin-Garcia P: Can polymorphisms in NLRP3 inflammasome
complex be associated with postmenopausal osteoporosis severity?
Genes (Basel). 13:22712022. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Xu L, Zhang L, Wang Z, Li C, Li S, Li L,
Fan Q and Zheng L: Melatonin suppresses estrogen deficiency-induced
osteoporosis and promotes osteoblastogenesis by inactivating the
NLRP3 inflammasome. Calcif Tissue Int. 103:400–410. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Dehlin M, Jacobsson L and Roddy E: Global
epidemiology of gout: Prevalence, incidence, treatment patterns and
risk factors. Nat Rev Rheumatol. 16:380–390. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Mangan MSJ, Olhava EJ, Roush WR, Seidel
HM, Glick GD and Latz E: Targeting the NLRP3 inflammasome in
inflammatory diseases. Nat Rev Drug Discov. 17:6882018. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Zhao Q, Xia N, Xu J, Wang Y, Feng L, Su D
and Cheng Z: Pro-Inflammatory of PRDM1/SIRT2/NLRP3 axis in
monosodium urate-induced acute gouty arthritis. J Innate Immun.
15:614–628. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Renaudin F, Orliaguet L, Castelli F,
Fenaille F, Prignon A, Alzaid F, Combes C, Delvaux A, Adimy Y,
Cohen-Solal M, et al: Gout and pseudo-gout-related crystals promote
GLUT1-mediated glycolysis that governs NLRP3 and interleukin-1β
activation on macrophages. Ann Rheum Dis. 79:1506–1514. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Lee HE, Yang G, Park YB, Kang HC, Cho YY,
Lee HS and Lee JY: Epigallocatechin-3-gallate prevents acute gout
by suppressing NLRP3 inflammasome activation and mitochondrial DNA
synthesis. Molecules. 24:21382019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Li X, Liu Y, Luo C and Tao J: Z1456467176
alleviates gouty arthritis by allosterically modulating P2X7R to
inhibit NLRP3 inflammasome activation. Front Pharmacol.
13:9799392022. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Xin J, Wang Y, Zheng Z, Wang S, Na S and
Zhang S: Treatment of intervertebral disc degeneration. Orthop
Surg. 14:1271–1280. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Chen F, Jiang G, Liu H, Li Z, Pei Y, Wang
H, Pan H, Cui H, Long J, Wang J and Zheng Z: Melatonin alleviates
intervertebral disc degeneration by disrupting the
IL-1β/NF-κB-NLRP3 inflammasome positive feedback loop. Bone Res.
8:102020. View Article : Google Scholar
|
|
166
|
Chen S, Wu X, Lai Y, Chen D, Bai X, Liu S,
Wu Y, Chen M, Lai Y, Cao H, et al: Kindlin-2 inhibits Nlrp3
inflammasome activation in nucleus pulposus to maintain homeostasis
of the intervertebral disc. Bone Res. 10:52022. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Zhao K, An R, Xiang Q, Li G, Wang K, Song
Y, Liao Z, Li S, Hua W, Feng X, et al: Acid-sensing ion channels
regulate nucleus pulposus cell inflammation and pyroptosis via the
NLRP3 inflammasome in intervertebral disc degeneration. Cell
Prolif. 54:e129412021. View Article : Google Scholar
|
|
168
|
Wojtasińska A, Frąk W, Lisińska W, Sapeda
N, Młynarska E, Rysz J and Franczyk B: Novel insights into the
molecular mechanisms of atherosclerosis. Int J Mol Sci.
24:134342023. View Article : Google Scholar
|
|
169
|
Duewell P, Kono H, Rayner KJ, Sirois CM,
Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr
M, et al: NLRP3 inflammasomes are required for atherogenesis and
activated by cholesterol crystals. Nature. 464:1357–1361. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Paramel Varghese G, Folkersen L,
Strawbridge RJ, Halvorsen B, Yndestad A, Ranheim T, Krohg-Sørensen
K, Skjelland M, Espevik T, Aukrust P, et al: NLRP3 inflammasome
expression and activation in human atherosclerosis. J Am Heart
Assoc. 5:e0030312016. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Usui F, Shirasuna K, Kimura H, Tatsumi K,
Kawashima A, Karasawa T, Hida S, Sagara J, Taniguchi S and
Takahashi M: Critical role of caspase-1 in vascular inflammation
and development of atherosclerosis in Western diet-fed
apolipoprotein E-deficient mice. Biochem Biophys Res Commun.
425:162–168. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Zheng F, Xing S, Gong Z, Mu W and Xing Q:
Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques
in apolipoprotein E-deficient mice. Mediators Inflamm.
2014:5072082014. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Satoh M, Tabuchi T, Itoh T and Nakamura M:
NLRP3 inflammasome activation in coronary artery disease: Results
from prospective and randomized study of treatment with
atorvastatin or rosuvastatin. Clin Sci (Lond). 126:233–241. 2014.
View Article : Google Scholar
|
|
174
|
Butts B, Gary RA, Dunbar SB and Butler J:
The importance of NLRP3 inflammasome in heart failure. J Card Fail.
21:586–593. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Bracey NA, Beck PL, Muruve DA, Hirota SA,
Guo J, Jabagi H, Wright JR Jr, Macdonald JA, Lees-Miller JP, Roach
D, et al: The Nlrp3 inflammasome promotes myocardial dysfunction in
structural cardiomyopathy through interleukin-1β. Exp Physiol.
98:462–472. 2013. View Article : Google Scholar
|
|
176
|
Mezzaroma E, Toldo S, Farkas D, Seropian
IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF and
Abbate A: The inflammasome promotes adverse cardiac remodeling
following acute myocardial infarction in the mouse. Proc Natl Acad
Sci USA. 108:19725–19730. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Van Tassell BW, Seropian IM, Toldo S,
Mezzaroma E and Abbate A: Interleukin-1β induces a reversible
cardiomyopathy in the mouse. Inflamm Res. 62:637–640. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Toldo S, Mezzaroma E, O'Brien L, Marchetti
C, Seropian IM, Voelkel NF, Van Tassell BW, Dinarello CA and Abbate
A: Interleukin-18 mediates interleukin-1-induced cardiac
dysfunction. Am J Physiol Heart Circ Physiol. 306:H1025–H1031.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Abbate A, Kontos MC, Grizzard JD,
Biondi-Zoccai GG, Van Tassell BW, Robati R, Roach LM, Arena RA,
Roberts CS, Varma A, et al: Interleukin-1 blockade with anakinra to
prevent adverse cardiac remodeling after acute myocardial
infarction (Virginia Commonwealth University Anakinra Remodeling
Trial (VCU-ART) Pilot study). Am J Cardiol. 105:1371–1377.e1. 2010.
View Article : Google Scholar
|
|
180
|
Abbate A, Van Tassell BW, Biondi-Zoccai G,
Kontos MC, Grizzard JD, Spillman DW, Oddi C, Roberts CS, Melchior
RD, Mueller GH, et al: Effects of interleukin-1 blockade with
anakinra on adverse cardiac remodeling and heart failure after
acute myocardial infarction (from the Virginia Commonwealth
University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study).
Am J Cardiol. 111:1394–1400. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Kawaguchi M, Takahashi M, Hata T, Kashima
Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J,
et al: Inflammasome activation of cardiac fibroblasts is essential
for myocardial ischemia/reperfusion injury. Circulation.
123:594–604. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Sandanger Ø, Ranheim T, Vinge LE, Bliksøen
M, Alfsnes K, Finsen AV, Dahl CP, Askevold ET, Florholmen G,
Christensen G, et al: The NLRP3 inflammasome is up-regulated in
cardiac fibroblasts and mediates myocardial ischaemia-reperfusion
injury. Cardiovasc Res. 99:164–174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao
C, Xin C, Zhu D, Li Y, Yan W, et al: TXNIP mediates NLRP3
inflammasome activation in cardiac microvascular endothelial cells
as a novel mechanism in myocardial ischemia/reperfusion injury.
Basic Res Cardiol. 109:4152014. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Capizzi A, Woo J and Verduzco-Gutierrez M:
Traumatic brain injury: An overview of epidemiology,
pathophysiology, and medical management. Med Clin North Am.
104:213–238. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Liu W, Chen Y, Meng J, Wu M, Bi F, Chang
C, Li H and Zhang L: Ablation of caspase-1 protects against
TBI-induced pyroptosis in vitro and in vivo. J Neuroinflammation.
15:482018. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Xu X, Yin D, Ren H, Gao W, Li F, Sun D, Wu
Y, Zhou S, Lyu L, Yang M, et al: Selective NLRP3 inflammasome
inhibitor reduces neuroinflammation and improves long-term
neurological outcomes in a murine model of traumatic brain injury.
Neurobiol Dis. 117:15–27. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Wallisch JS, Simon DW, Bayır H, Bell MJ,
Kochanek PM and Clark RSB: Cerebrospinal fluid NLRP3 is increased
after severe traumatic brain injury in infants and children.
Neurocrit Care. 27:44–50. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Geng F, Ma Y, Xing T, Zhuang X, Zhu J and
Yao L: Effects of hyperbaric oxygen therapy on inflammasome
signaling after traumatic brain injury. Neuroimmunomodulation.
23:122–129. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Marino BLB, de Souza LR, Sousa KPA,
Ferreira JV, Padilha EC, da Silva CHTP, Taft CA and Hage-Melim LIS:
Parkinson's disease: A review from pathophysiology to treatment.
Mini Rev Med Chem. 20:754–767. 2020. View Article : Google Scholar
|
|
190
|
Marogianni C, Sokratous M, Dardiotis E,
Hadjigeorgiou GM, Bogdanos D and Xiromerisiou G: Neurodegeneration
and Inflammation-an interesting interplay in Parkinson's disease.
Int J Mol Sci. 21:84212020. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Mao Z, Liu C, Ji S, Yang Q, Ye H, Han H
and Xue Z: The NLRP3 inflammasome is involved in the pathogenesis
of Parkinson's disease in rats. Neurochem Res. 42:1104–1115. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Qiao C, Zhang Q, Jiang Q, Zhang T, Chen M,
Fan Y, Ding J, Lu M and Hu G: Inhibition of the hepatic Nlrp3
protects dopaminergic neurons via attenuating systemic inflammation
in a MPTP/p mouse model of Parkinson's disease. J
Neuroinflammation. 15:1932018. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Villain N and Dubois B: Alzheimer's
disease including focal presentations. Semin Neurol. 39:213–226.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Cummings JL, Tong G and Ballard C:
Treatment combinations for Alzheimer's disease: Current and future
pharmacotherapy options. J Alzheimers Dis. 67:779–794. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Abbott A: Is 'friendly fire' in the brain
provoking Alzheimer's disease? Nature. 556:426–428. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Zhang Y, Dong Z and Song W: NLRP3
inflammasome as a novel therapeutic target for Alzheimer's disease.
Signal Transduct Target Ther. 5:372020. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Ising C, Venegas C, Zhang S, Scheiblich H,
Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM,
Tejera D, et al: NLRP3 inflammasome activation drives tau
pathology. Nature. 575:669–673. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Srinivasan S, Kancheva D, De Ren S, Saito
T, Jans M, Boone F, Vandendriessche C, Paesmans I, Maurin H,
Vandenbroucke RE, et al: Inflammasome signaling is dispensable for
ß-amyloid-induced neuropathology in preclinical models of
Alzheimer's disease. Front Immunol. 15:13234092024. View Article : Google Scholar
|
|
199
|
Dobson R and Giovannoni G: Multiple
sclerosis-a review. Eur J Neurol. 26:27–40. 2019. View Article : Google Scholar
|
|
200
|
Inoue M, Williams KL, Gunn MD and
Shinohara ML: NLRP3 inflammasome induces chemotactic immune cell
migration to the CNS in experimental autoimmune encephalomyelitis.
Proc Natl Acad Sci USA. 109:10480–10485. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Zhao Y, Zhang X, Chen X and Wei Y:
Neuronal injuries in cerebral infarction and ischemic stroke: From
mechanisms to treatment (Review). Int J Mol Med. 49:152022.
View Article : Google Scholar :
|
|
202
|
Zhao J, Piao X, Wu Y, Liang S, Han F,
Liang Q, Shao S and Zhao D: Cepharanthine attenuates cerebral
ischemia/reperfusion injury by reducing NLRP3 inflammasome-induced
inflammation and oxidative stress via inhibiting 12/15-LOX
signaling. Biomed Pharmacother. 127:1101512020. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Wang H, Zhong D, Chen H, Jin J, Liu Q and
Li G: NLRP3 inflammasome activates interleukin-23/interleukin-17
axis during ischaemia-reperfusion injury in cerebral ischaemia in
mice. Life Sci. 227:101–113. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Silveira LS, Antunes Bde M, Minari AL, Dos
Santos RV, Neto JC and Lira FS: Macrophage polarization:
Implications on metabolic diseases and the role of exercise. Crit
Rev Eukaryot Gene Expr. 26:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Luo L, Liu M, Fan Y, Zhang J, Liu L, Li Y,
Zhang Q, Xie H, Jiang C, Wu J, et al: Intermittent theta-burst
stimulation improves motor function by inhibiting neuronal
pyroptosis and regulating microglial polarization via
TLR4/NFκB/NLRP3 signaling pathway in cerebral ischemic mice. J
Neuroinflammation. 19:1412022. View Article : Google Scholar
|
|
206
|
Ye X, Shen T, Hu J, Zhang L, Zhang Y, Bao
L, Cui C, Jin G, Zan K, Zhang Z, et al: Purinergic 2X7
receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic
stroke in the mouse. Exp Neurol. 292:46–55. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Seok JK, Kang HC, Cho YY, Lee HS and Lee
JY: Therapeutic regulation of the NLRP3 inflammasome in chronic
inflammatory diseases. Arch Pharm Res. 44:16–35. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Liu W, Guo W, Wu J, Luo Q, Tao F, Gu Y,
Shen Y, Li J, Tan R, Xu Q and Sun Y: A novel benzo(d)imidazole
derivate prevents the development of dextran sulfate sodium-induced
murine experimental colitis via inhibition of NLRP3 inflammasome.
Biochem Pharmacol. 85:1504–1512. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Youm YH, Nguyen KY, Grant RW, Goldberg EL,
Bodogai M, Kim D, D'Agostino D, Planavsky N, Lupfer C, Kanneganti
TD, et al: The ketone metabolite β-hydroxybutyrate blocks NLRP3
inflammasome-mediated inflammatory disease. Nat Med. 21:263–269.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Yan CY, Ouyang SH, Wang X, Wu YP, Sun WY,
Duan WJ, Liang L, Luo X, Kurihara H, Li YF and He RR: Celastrol
ameliorates propionibacterium acnes/LPS-induced liver damage and
MSU-induced gouty arthritis via inhibiting K63 deubiquitination of
NLRP3. Phytomedicine. 80:1533982021. View Article : Google Scholar
|
|
211
|
Li M, Liu H, Shao H, Zhang P, Gao M, Huang
L, Shang P, Zhang Q, Wang W and Feng F: Glyburide attenuates B(a)p
and LPS-induced inflammation-related lung tumorigenesis in mice.
Environ Toxicol. 36:1713–1722. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Yang G, Lee HE, Moon SJ, Ko KM, Koh JH,
Seok JK, Min JK, Heo TH, Kang HC, Cho YY, et al: Direct binding to
NLRP3 pyrin domain as a novel strategy to prevent NLRP3-driven
inflammation and gouty arthritis. Arthritis Rheumatol.
72:1192–1202. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Jiang H, He H, Chen Y, Huang W, Cheng J,
Ye J, Wang A, Tao J, Wang C, Liu Q, et al: Identification of a
selective and direct NLRP3 inhibitor to treat inflammatory
disorders. J Exp Med. 214:3219–3238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
He Y, Varadarajan S, Muñoz-Planillo R,
Burberry A, Nakamura Y and Núñez G:
3,4-methylenedioxy-β-nitrostyrene inhibits NLRP3 inflammasome
activation by blocking assembly of the inflammasome. J Biol Chem.
289:1142–1150. 2014. View Article : Google Scholar
|
|
215
|
Juliana C, Fernandes-Alnemri T, Wu J,
Datta P, Solorzano L, Yu JW, Meng R, Quong AA, Latz E, Scott CP and
Alnemri ES: Anti-inflammatory compounds parthenolide and Bay
11-7082 are direct inhibitors of the inflammasome. J Biol Chem.
285:9792–9802. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Kerr ID, Lee JH, Farady CJ, Marion R,
Rickert M, Sajid M, Pandey KC, Caffrey CR, Legac J, Hansell E, et
al: Vinyl sulfones as antiparasitic agents and a structural basis
for drug design. J Biol Chem. 284:25697–25703. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Lee HE, Yang G, Kim ND, Jeong S, Jung Y,
Choi JY, Park HH and Lee JY: Targeting ASC in NLRP3 inflammasome by
caffeic acid phenethyl ester: A novel strategy to treat acute gout.
Sci Rep. 6:386222016. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Linton SD: Caspase inhibitors: a
pharmaceutical industry perspective. Curr Top Med Chem.
5:1697–1717. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Dinarello CA, Simon A and van der Meer JW:
Treating inflammation by blocking interleukin-1 in a broad spectrum
of diseases. Nat Rev Drug Discov. 11:633–652. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Ridker PM, Everett BM, Thuren T, MacFadyen
JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker
SD, et al: Antiinflammatory therapy with canakinumab for
atherosclerotic disease. N Engl J Med. 377:1119–1131. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Neudecker V, Haneklaus M, Jensen O,
Khailova L, Masterson JC, Tye H, Tye H, Biette K, Jedlicka P,
Brodsky KS, et al: Myeloid-derived miR-223 regulates intestinal
inflammation via repression of the NLRP3 inflammasome. J Exp Med.
214:1737–1752. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Feng Z, Qi S, Zhang Y, Qi Z, Yan L, Zhou
J, He F, Li Q, Yang Y, Chen Q, et al: Ly6G+ neutrophil-derived
miR-223 inhibits the NLRP3 inflammasome in mitochondrial
DAMP-induced acute lung injury. Cell Death Dis. 8:e31702017.
View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Li X, Zhang Y, Zhang H, Liu X, Gong T, Li
M, Sun L, Ji G, Shi Y, Han Z, et al: miRNA-223 promotes gastric
cancer invasion and metastasis by targeting tumor suppressor
EPB41L3. Mol Cancer Res. 9:824–833. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Bandyopadhyay S, Lane T, Venugopal R,
Parthasarathy PT, Cho Y, Galam L, Lockey R and Kolliputi N:
MicroRNA-133a-1 regulates inflammasome activation through
uncoupling protein-2. Biochem Biophys Res Commun. 439:407–412.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Li S, Liang X, Ma L, Shen L, Li T, Zheng
L, Sun A, Shang W, Chen C, Zhao W and Jia J: MiR-22 sustains NLRP3
expression and attenuates H. pylori-induced gastric carcinogenesis.
Oncogene. 37:884–896. 2018. View Article : Google Scholar
|
|
226
|
Li D, Yang H, Ma J, Luo S, Chen S and Gu
Q: MicroRNA-30e regulates neuroinflammation in MPTP model of
Parkinson's disease by targeting Nlrp3. Hum Cell. 31:106–115. 2018.
View Article : Google Scholar
|
|
227
|
Zhou Y, Lu M, Du RH, Qiao C, Jiang CY,
Zhang KZ, Ding JH and Hu G: MicroRNA-7 targets Nod-like receptor
protein 3 inflammasome to modulate neuroinflammation in the
pathogenesis of Parkinson's disease. Mol Neurodegener. 11:282016.
View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Boxberger N, Hecker M and Zettl UK:
Dysregulation of inflammasome priming and activation by MicroRNAs
in human immune-mediated diseases. J Immunol. 202:2177–2187. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Tezcan G, Martynova EV, Gilazieva ZE,
McIntyre A, Rizvanov AA and Khaiboullina SF: MicroRNA
post-transcriptional regulation of the NLRP3 inflammasome in
immunopathologies. Front Pharmacol. 10:4512019. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Zhou T, Xiang DK, Li SN, Yang LH, Gao LF
and Feng C: MicroRNA-495 ameliorates cardiac microvascular
endothelial cell injury and inflammatory reaction by suppressing
the NLRP3 inflammasome signaling pathway. Cell Physiol Biochem.
49:798–815. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Cyranoski D: CRISPR gene-editing tested in
a person for the first time. Nature. 539:4792016. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Ran FA, Cong L, Yan WX, Scott DA,
Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et
al: In vivo genome editing using Staphylococcus aureus Cas9.
Nature. 520:186–191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Bengtsson NE, Hall JK, Odom GL, Phelps MP,
Andrus CR, Hawkins RD, Hauschka SD, Chamberlain JR and Chamberlain
JS: Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates
pathophysiology in a mouse model for Duchenne muscular dystrophy.
Nat Commun. 8:144542017. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Xu C, Lu Z, Luo Y, Liu Y, Cao Z, Shen S,
Li H, Liu J, Chen K, Chen Z, et al: Targeting of NLRP3 inflammasome
with gene editing for the amelioration of inflammatory diseases.
Nat Commun. 9:40922018. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Wang YC, Liu QX, Zheng Q, Liu T, Xu XE,
Liu XH, Gao W, Bai XJ and Li ZF: Dihydromyricetin alleviates
sepsis-induced acute lung injury through inhibiting NLRP3
inflammasome-dependent pyroptosis in mice model. Inflammation.
42:1301–1310. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Tang F, Fan K, Wang K and Bian C:
Amygdalin attenuates acute liver injury induced by D-galactosamine
and lipopolysaccharide by regulating the NLRP3, NF-κB and Nrf2/NQO1
signalling pathways. Biomed Pharmacother. 111:527–536. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Zhao Q, Bi Y, Guo J, Liu YX, Zhong J, Pan
LR, Tan Y and Yu XJ: Pristimerin protects against inflammation and
metabolic disorder in mice through inhibition of NLRP3 inflammasome
activation. Acta Pharmacol Sin. 42:975–986. 2021. View Article : Google Scholar :
|
|
238
|
Shi J, Xia Y, Wang H, Yi Z, Zhang R and
Zhang X: Piperlongumine Is an NLRP3 Inhibitor With
Anti-inflammatory Activity. Front Pharmacol. 12:8183262021.
View Article : Google Scholar
|
|
239
|
Zhao J, Liu H, Hong Z, Luo W, Mu W, Hou X,
Xu G, Fang Z, Ren L, Liu T, et al: Tanshinone I specifically
suppresses NLRP3 inflammasome activation by disrupting the
association of NLRP3 and ASC. Mol Med. 29:842023. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Liao T, Ding L, Wu P, Zhang L, Li X, Xu B,
Zhang H, Ma Z, Xiao Y and Wang P: Chrysin attenuates the NLRP3
inflammasome cascade to reduce synovitis and pain in KOA rats. Drug
Des Devel Ther. 14:3015–3027. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Liu B and Yu J: Anti-NLRP3 inflammasome
natural compounds: An update. Biomedicines. 9:1362021. View Article : Google Scholar : PubMed/NCBI
|














