Introduction
Parkinson's disease (PD) is a prevalent degenerative
disease of the central nervous system (CNS), significantly
affecting the quality of life through both motor and non-motor
symptoms (1). The primary
pathological features of PD include the progressive loss of
dopaminergic neurons in the substantia nigra (SN) pars compacta and
the formation of Lewy bodies, composed of α-synuclein (α-syn)
(2). It has been reported that
factors, such as α-syn aggregation, oxidative stress, ferroptosis,
mitochondrial dysfunction, neuroinflammation and gut dysbiosis are
involved in the degeneration and death of dopaminergic neurons in
PD (3). Although levodopa is
commonly used as a first-line therapy to alleviate motor symptoms,
it cannot halt the progression of PD (4). Therefore, developing interventions
to slow or stop the progression of PD remains a top priority for
both patients and researchers (5).
Ceftriaxone (CEF), a third-generation cephalosporin
antibiotic, has shown neuroprotective effects in recent studies on
PD. In a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
(MPTP)-induced PD animal model, CEF was found to reverse behavioral
deficit and promote neurogenesis (6,7).
It could also mitigate nigral oxidative damage and enhance
neurogenesis (8), reduce
glutamate-mediated neuro-inflammation and restore brain-derived
neurotrophic factor (BDNF) levels (9). At the molecular level, CEF could
bind to α-syn to inhibit its polymerization in vitro
(10). These findings suggested
that CEF could be applied in the treatment of PD. However, the
particular neuroprotective mechanisms of CEF remain unclear.
Emerging evidence has indicated that ferroptosis is a key molecular
mechanism in PD (11). Since
cystine/glutamate transport is closely associated with ferroptosis
and CEF can regulate glutamate transport, it was hypothesized that
CEF could affect PD via modulating ferroptosis.
The current study aimed to investigate the
ferroptosis-related neuroprotective mechanism of CEF and
neuroinflammation in PD. Therefore, both in vivo and in
vitro PD injury models were established to verify whether CEF
could alleviate glial cell activation and neuronal damage in PD via
inhibiting ferroptosis.
Materials and methods
Cell culture and drugs
BV2 (cat. no. SCSP-5208; Cell Bank of Type Culture
Collection of the Chinese Academy of Sciences) and C8-D1A [cat. no.
CRL-2541; American Type Culture Collection (ATCC)] cells were
cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.), while
SH-SY5Y cells (cat. no. CRL-2266; ATCC) in DMEM/F12, both
supplemented with 10% heat-inactivated fetal bovine serum (both
from Gibco; Thermo Fisher Scientific, Inc.) and
penicillin/streptomycin solution (100 μg/ml) at 37°C in a
humidified incubator with 5% CO2. CEF (Roche
Diagnostics) and MPP+ (MedChemExpress) were dissolved in
saline and DMSO, respectively. Liposaccharide (LPS;
MilliporeSigma), MPTP (MedChemExpress) and TNFα (Beyotime Institute
of Biotechnology) were dissolved in saline. Cells were seeded into
12-well plates starting at a concentration of 2×105
cells/ml and incubated overnight. Cells in the experimental group
were pre-treated with 300 μM CEF for 4 h with a confluency
of ~75% at the time of treatment. For inflammatory activation, BV2
and C8-D1A cells were challenged with 100 ng/ml LPS and 10 ng/ml
TNFα, respectively, for 24 h. To establish the
MPP+-induced SH-SY5Y cell model, cells were treated with
1 μM MPP+ for 24 h.
Antibodies
Immunoblot analysis was performed using the
following primary antibodies: Anti-tyrosine hydroxylase (TH;
1:2,000; cat. no. 25859-1-AP), anti-solute carrier family 7 member
11 (SLC7A11; 1:1,000; cat. no. 26864-1-AP), anti-glial fibrillary
acidic protein (GFAP; 1:10,000; cat. no. 60190-1-Ig; all from
Proteintech Group, Inc.), anti-cyclooxygenase-2 (COX-2, 1:1,000;
cat. no. ab179800), anti-inducible nitric oxide synthase (iNOS;
1:1,000; cat. no. ab178945), anti-glutathione peroxidase 4 (GPX4;
1:2,000; cat. no. ab125066,), anti-allograft inflammatory factor 1
(IBA1; 1:1,000; cat. no. ab178846; all from Abcam), anti-GAPDH
(1:1,000; cat. no. 5174S), anti-p65 (1:1,000; cat. no. 8242S) and
anti-phosphorylated (p)-p65 (1:1,000; cat. no. 3033S; all from Cell
Signaling Technology, Inc.). Horseradish peroxidase
(HRP)-conjugated goat anti-mouse (1:10,000; cat. no. 31430) or
anti-rabbit antibodies (1:10,000; cat. no. 31460; Thermo Fisher
Scientific, Inc.) served as secondary antibodies. The proteins were
visualized using an ECL detection kit (Thermo Fisher Scientific,
Inc.).
Animal study
C57BL/6 mice (male, n=72, weight, 25-30 g) were
purchased from Shanghai Slack Laboratory Animal Co., Ltd. Each
group was comprised six mice. All mice were maintained under
controlled temperature (22±1°C) and humidity (50±5%) conditions in
a 12/12-h light/dark cycle with free access to food and water. The
PD mouse model was established by intraperitoneal injection of 5
mg/ml MPTP (dissolved in saline) for 7 days (25 mg/kg per day for
the first three days followed by 30 mg/kg per day for the last 4
days). No mice succumbed after MPTP injection in the present
experiment. After the 7-days period of continuous injections of
MPTP, a behavioral test was conducted to confirm the establishment
of a stable pathological state in the mice. To assess the
successful establishment of the PD model, mice were subjected to a
series of behavioral tests, including the rotarod and climbing rod
tests. Only mice with significant motor deficits were selected for
the subsequent experiments. A total of 12 mice were selected for
the study, and the remaining mice were euthanized according to the
protocol approved by the Institutional Animal Care and Use
Committee. The neuroinflammatory in vivo model was
established following stereotaxic injection of 1 mg/ml
lipopolysaccharide (LPS; volume, 2 μl) into the ventricles.
Pentobarbital sodium was administered at a dose of 50 mg/kg to
anesthetize the mice prior to the injection. Mice in the CEF and
blank control groups were intraperitoneally injected for 7 days
with 200 mg/kg CEF (once a day) and equal volume of saline,
respectively. After the experiment, mice were anesthetized by
pentobarbital sodium and then euthanized by cervical dislocation.
Mouse brain slices were collected for western blot and
immunohistochemistry assays. All experimental operations in animals
were performed simultaneously according to the Institutional
Guidelines for Animal Use and Care and all procedures were approved
by the Animal Committee of Suzhou Institute of Biomedical
Engineering and Technology, Chinese Academy of Sciences (approval
no. 2024-A50; Suzhou, China). For the MPTP-treated C57BL/6 mouse
model of PD, humane endpoints were defined as follows: Severe
weight loss (a reduction of 20% or more from baseline weight),
severe neurological impairment (including inability to move,
tremors, or non-response to being placed on their back), severe
pain or distress (indicated by labored breathing, extreme
dehydration, or abnormal posture), lack of appetite and water
consumption (evidenced by signs of dehydration or malnutrition),
severe tremors or convulsions that impede overall well-being, and
unhealed injuries or wounds resulting in infection or extreme pain.
If any mice exhibited these symptoms, humane euthanasia would be
conducted to prevent prolonged suffering. In the present study, all
mice were euthanized before the conclusion of the experiment;
however, none of the mice reached these humane endpoints.
Behavioral tests
Rotarod and climbing rod tests are commonly used to
assess motor coordination and bradykinesia in animal models of
neurological disorders (12,13). The behavioral instruments were
purchased from SANS Biological Technology (https://www.sansbio.com). Rotarod test: Mice were
trained on the rotarod until they could remain on it for over 2 min
at a speed of 4 revolutions per min (r/min) during the training. On
the day of testing, the speed of the rotarod was gradually
increased from 4 to 40 r/min over 5 min. Each mouse was monitored
freely walking on the rotarod, and the elapsed time until falling
was recorded. This experiment was conducted three times per mouse.
Pole test: A wooden pole with a rough surface, measuring 50 cm in
height and 1 cm in diameter, was inserted into the home cage.
During the training session, every mouse was positioned on top of
the pole and allowed to descend to the home cage from the peak
three times. On the day of testing, each mouse was placed
head-first on top of the pole, and the duration taken for it to
descend back into the cage was timed. The experimentation process
was conducted thrice for every mouse.
Western blot analysis
Total proteins were extracted from tissues or cells
using a RIPA lysis solution supplemented with protease inhibitors
(both from Beyotime Institute of Biotechnology). Protein
quantification was performed using a BCA protein quantification kit
(Beyotime Institute of Biotechnology). Subsequently, ~30 μg
protein extracts were separated by 8-12% SDS-PAGE and were then
transferred onto a PVDF membrane (MilliporeSigma). Following
transfer, the membranes were subjected to blocking with 5% (w/v)
skim milk powder diluted in Tris-buffered saline containing 0.1%
Tween-20 (TBST) under ambient conditions (25±2°C) for 1 h. The
membranes were first incubated with primary antibodies at 4°C
overnight in a refrigerator and then with the corresponding
secondary antibody at room temperature for 2 h. The blots were then
incubated with an ECL reagent and the expression levels of the
target proteins were detected using a chemiluminescence imaging
analysis system (Clinx Science Instruments Co., Ltd.). GAPDH was
used as a reference gene. Quantification was performed with ImageJ
software (version 1.54k; National Institutes of Health).
Immunofluorescence staining
The brains of mice were injected with 4%
paraformaldehyde (PFA), fixed in 4% PFA at 4°C for three days and
were then incubated with 30% sucrose at 4°C for three days.
Subsequently, the 20-μm thick brain sections were collected
and incubated with 5% fetal bovine serum (cat. no. Gibco-10270-106;
Thermo Fisher Scientific, Inc.) to block non-specific protein
binding. The tissue sections were then incubated at 4°C overnight
with the primary antibodies. After washing the tissue sections with
PBS on a decolourisation shaker, they were incubated with the
corresponding secondary antibodies Alexa Fluor 488 (1;500; cat. no.
A-21202) and Alexa Fluor 594 (1:500; cat. no. A-21207) from
Invitrogen (Thermo Fisher Scientific, Inc.) at room temperature for
1 h. Subsequently, the sections were examined using an inverted
fluorescence microscope (IX71; Olympus Corporation). For cell
counts and quantitative statistics for fluorescence intensity,
ImageJ software was used.
Cell viability assay
The Cell Counting Kit-8 (CCK-8) assays were
performed according to the manufacturer's instructions. Briefly,
cells were seeded in a 96-well plate at a concentration of
1×105 cells/well and allowed to adhere overnight.
Following treatment with increasing concentrations of the indicated
drugs for 24 h, each well was supplemented with 10 μl CCK-8
solution (cat. no. C0039; Beyotime Institute of Biotechnology). The
plate was incubated at 37°C for 2 h and the absorbance in each well
was measured at a wavelength of 450 nm using a microplate reader
(CMaxPlus from Molecular Devices, LLC).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). cDNA was
reversely transcribed from total RNA using PrimeScript RT Master
Mix (Takara Bio, Inc.) following the manufacturer's instructions.
qPCR was performed using a LightCycler 480 system (Roche
Diagnostics) with TB Green® Premix Ex Taq™ II (Takara
Bio, Inc.). The thermal cycling protocol comprised an initial
denaturation step at 95°C for 30 sec, followed by 40 cycles of
denaturation at 95°C for 5 sec and combined annealing/extension at
60°C for 10 sec. RT-qPCR analysis was performed using the
2−ΔΔCq method (14).
RT-qPCR primers were designed as follows: mouse iNOS forward,
5′-TCC CAGCCTGCCCCTTCAAT-3′ and reverse,
5′-CGGATCTCTCTCCTCCTGGG-3′; COX-2 forward,
5′-CAGGCTGAACTTCGAAACAG-3′ and reverse,
5′-CTCACGAGGCCACTGATACCTA-3′; and β-actin forward,
5′-GACCTGACTGACTACCTC-3′ and reverse, 5′-GACAGCGAGGCCAGGATG-3′.
Conditioned media (CM)
BV2 cells were pretreated with or without CEF (300
μM) for 4 h and then exposed to LPS (100 ng/ml) for 24 h.
C8-D1A cells were pretreated with or without CEF (300 μM)
for 4 h and then exposed to TNFα (10 ng/ml) for 24 h. CM was used
to culture SH-SY5Y cells.
Data analysis
Prism version 7.0 (GraphPad Software; Dotmatics) was
used for data analysis. Statistical analysis was conducted by
one-way analysis of variance (ANOVA) followed by Tukey's post hoc
test for multiple comparisons, as indicated in the figure legends.
The data are presented as the mean ± standard deviation (SD).
P<0.05 was considered to indicate a statistically significant
difference.
Results
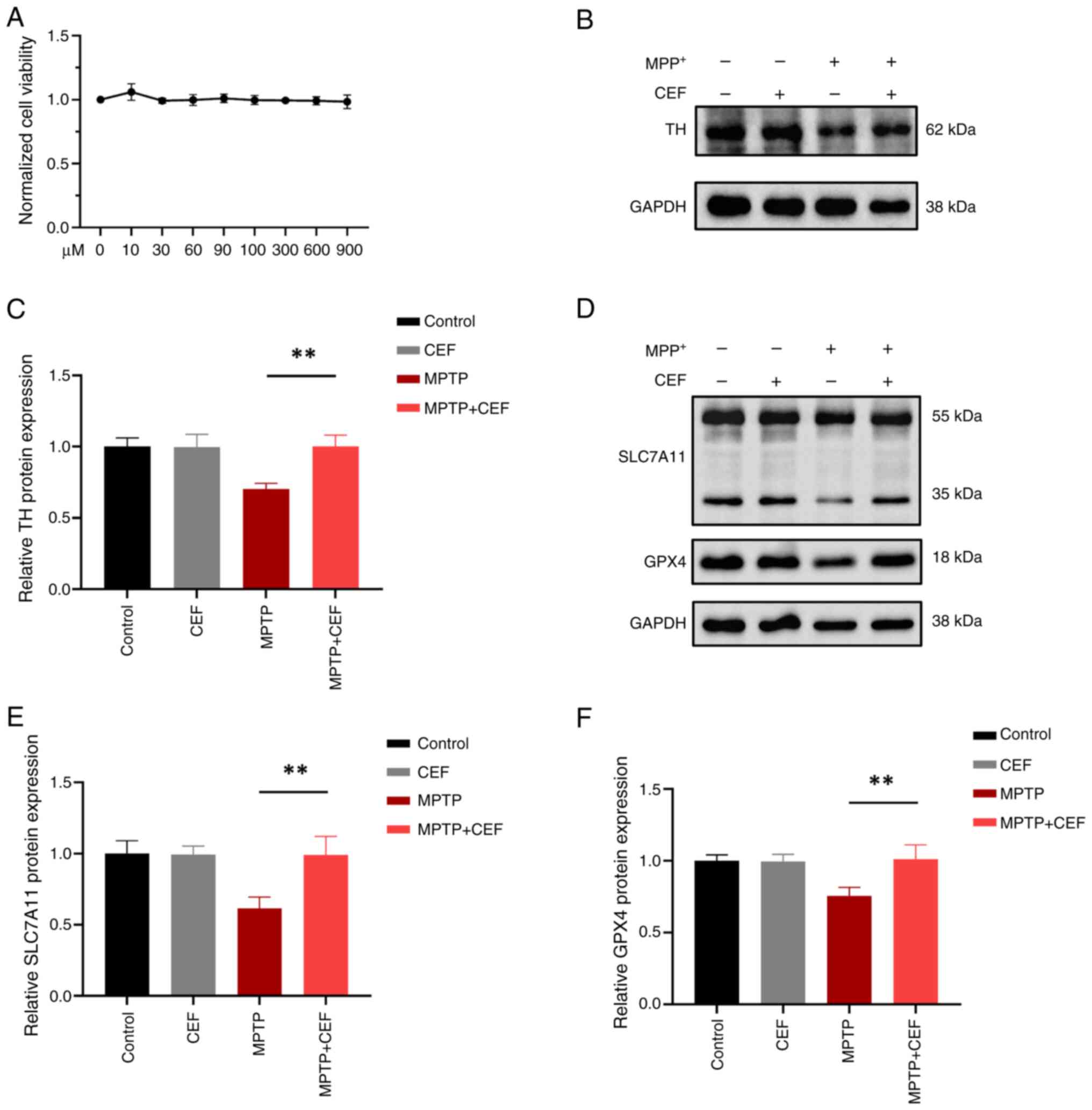
CEF displays a direct protective effect
on neurons via reversing the expression of TH in a MPTP-induced
mouse model
TH is a rate-limiting enzyme, which is involved in
the synthesis of dopamine, while it is widely acknowledged as a
marker of dopaminergic neurons, which transport dopamine from the
SN to the caudal putamen (CPu) via the nigro-striatum pathway
(15). Dopamine is synthesized
in dopaminergic neurons in the SN and then transmitted through
synapses to different brain regions such as CPu, which receives a
large amount of dopamine input from SN (16). Therefore, it can be observed that
both the SN and CPu are capable of detecting the presence of TH. To
evaluate the effect of CEF on PD neuropathology, western blot and
immunofluorescence analyses were carried out to detect the
expression levels of TH in both the SN and CPu in a MPTP mice
model. C57BL/6 mice were administered CEF with intraperitoneal
injections after intraperitoneally administering MPTP (Fig. 1A). Behavioral tests demonstrated
that the MPTP-treated mice had a significant decrease in latency
time on the rotarod and increase in pole test time, indicating a
decline in their motor coordination abilities (Fig. 1B and C). Western blot analysis
revealed that CEF could significantly reverse the MPTP-induced TH
downregulation in the SN and CPu (Fig. 1D-G). Additionally,
immunofluorescence analysis revealed that CEF could substantially
alleviate the loss of MPTP-induced TH immunoreactive neurons in the
SN and CPu (Fig. 1H-K). These
findings suggested that treatment with CEF could promote neuronal
protection in a PD mouse model.
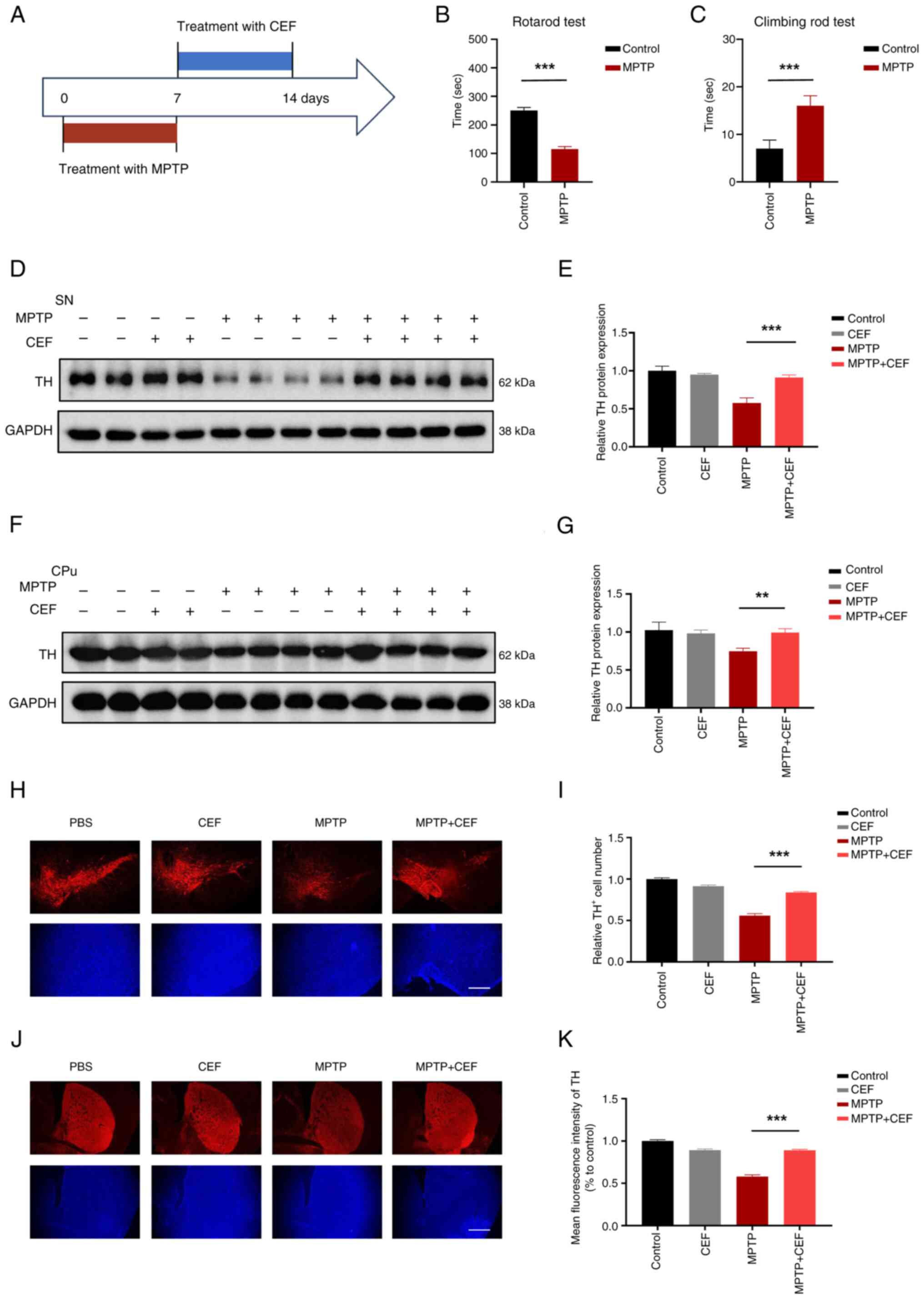 | Figure 1CEF reverses the expression of TH in
a MPTP-induced mouse model. (A) Experimental design of CEF
treatment in attenuating Parkinson's disease in a mouse model. Mice
were intraperitoneally injected with MPTP once a day for 7 days (25
mg/kg for the first 3 days, 30 mg/kg for the last 4 days), followed
by intraperitoneal injection of 200 mg/kg CEF once a day for 7
days. (B) Latencies to fall from the accelerated rotating beams in
the rotarod tests. (C) Climbing time on climbing rod tests. (D) The
protein expresion levels of TH were detected in the SN of brain
tissue sections using western blot analysis. (E) The protein
expression levels of TH in (D) were quantified following
normalization to those of GAPDH. (F) The protein expression levels
of TH were also detected in the CPu region of the brain by western
blot analysis. (G) The protein expression levels of TH in (F) were
quantified after normalization to the expression levels of GAPDH.
(H-K) The brain tissue sections were cut into slices and stained
with an antibody against TH. The corresponding fluorescence images
and statistical analysis of TH expression in (H and I) the SN and
(J and K) CPu are presented. Data are expressed as the mean ± SD.
**P<0.01 and ***P<0.001 (n=3). Scale
bar, 100 μm. Coronal slices. CEF, ceftriaxone; TH, tyrosine
hydroxylase; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine;
SN, substantia nigra; CPu, caudal putamen. |
CEF alleviates MPP+-induced
SH-SY5Y cell injury via resisting ferroptosis
The cystine/glutamate antiporter SLC7A11 (xCT)/GPX4
system is considered as the mainstay to inhibit ferroptosis, since
glutathione (GSH) is the principal antioxidant in mammalian cells
(17). To explore the
neuroprotective mechanism in neurons, the effect of the
SLC7A11/GPX4 axis in the ferroptosis pathway was investigated.
Therefore, the effect of different concentrations of CEF on SH-SY5Y
cell viability was assessed by CCK-8 assay. The results identified
that CEF at a concentration of 900 μM had no toxic effect on
the viability of SH-SY5Y cells (Fig.
2A). In addition, western blot analysis demonstrated that CEF
significantly reversed the reduced expression levels of TH in
MPP+-induced SH-SY5Y cells (Fig. 2B and C). Additionally, CEF
substantially alleviated the MPP+-induced SLC7A11 and
GPX4 downregulation in SH-SY5Y cells (Fig. 2D-F). These findings suggested
that CEF could suppress ferroptosis via regulating the SLC7A11/GPX4
axis in neurons.
CEF attenuates the activation of
microglia and astrocytes in LPS- and MPTP-induced mouse models
Activation of glial cells is accompanied by
morphological changes; proinflammatory microglia and astrocytes
generally exhibit larger volumes and coarser morphologies, while
anti-inflammatory microglia and astrocytes typically feature
smaller volumes and more prominent morphologies (18,19). To investigate whether
neuroinflammation could be involved in the neuroprotective effects
of CEF, tissue-section immunofluorescence staining was utilized to
determine cellular morphological characteristics. By utilizing a
coupling of markers (namely, the microglial activation-associated
marker IBA1 and the astrocyte activation-associated marker GFAP)
and morphological traits, it was observed that microglia and
astrocytes exhibit larger volumes and coarser morphologies in the
SN in both models. However, treatment with CEF considerably
abrogated this effect (Fig.
3A-C). The same effects were also observed in the CPu of the
brain (Fig. 3D-F). The
aforementioned results indicated that CEF could attenuate
neuroinflammation via inhibiting the activation of microglia and
astrocytes in vivo.
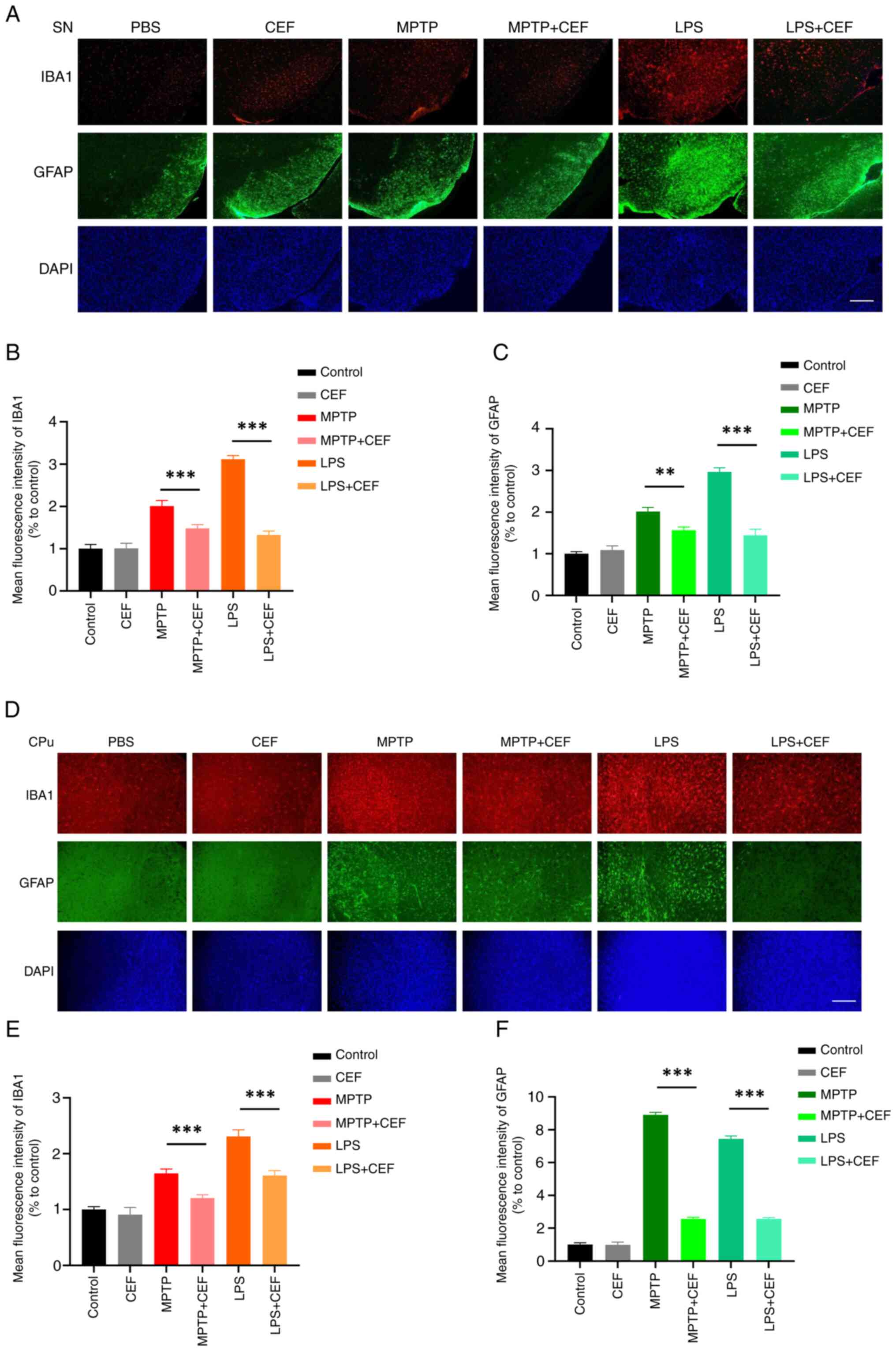 | Figure 3CEF inhibits the activation of glial
cells in LPS- and MPTP-treated mice. (A-C) To establish a
LPS-induced mouse model, mice were stereotaxically injected with 2
μl LPS (1 mg/ml) into the ventricles, followed by
intraperitoneal injection of CEF (200 mg/kg) once a day for 7 days.
For the establishment of the MPTP-induced mouse model, mice were
intraperitoneally injected with 25 mg/kg MPTP per day for the first
3 days and 30 mg/kg per day for the last 4 days. Subsequently, mice
were intraperitoneally injected with 200 mg/kg CEF once a day for 7
days. The brain tissues were cut into slices and stained with
antibodies against IBA1 and GFAP. Fluorescence images and
statistical analysis of the IBA1 and GFAP protein expression levels
in the substantia nigra are shown. (D-F) Fluorescence images and
statistical analysis of IBA1 and GFAP expression in the caudal
putamen are presented. Data are expressed as the mean ± SD.
**P<0.01 and ***P<0.001 (n=3). Scale
bar, 100 μm. Coronal slices. CEF, ceftriaxone; LPS,
lipopolysacharide; MPTP,
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; IBA1, allograft
inflammatory factor 1; GFAP, glial fibrillary acidic protein. |
CEF displays an indirect protective
effect on neurons via inhibiting the activation of microglia and
astrocytes in the LPS-induced mouse model
Given that CEF inhibited the activation of microglia
and astrocytes in both the LPS- and MPTP-induced mouse models, the
current study also aimed to explore whether CEF could protect
neurons in the LPS-induced mouse model. A pro-inflammatory state
can cause changes in an increase in the expression of GFAP in
astrocytes and IBA-1 in microglia (20). Western blotting was utilized to
determine the expression of GFAP and IBA-1. The western blot
analysis results revealed a significant induction in IBA1 and GFAP
expression accompanied by a considerable reduction in TH expression
in LPS-induced mice. However, treatment with CEF reversed the
aforementioned changes (Fig.
4A-D). Furthermore, immunofluorescence analysis showed that CEF
considerably attenuated the loss of TH immunoreactive neurons in
mice treated with LPS (Fig.
4E-H). Overall, these data suggested that CEF could supress
neuroinflammation and inhibit neuroinflammation-mediated
neurotoxicity in vivo.
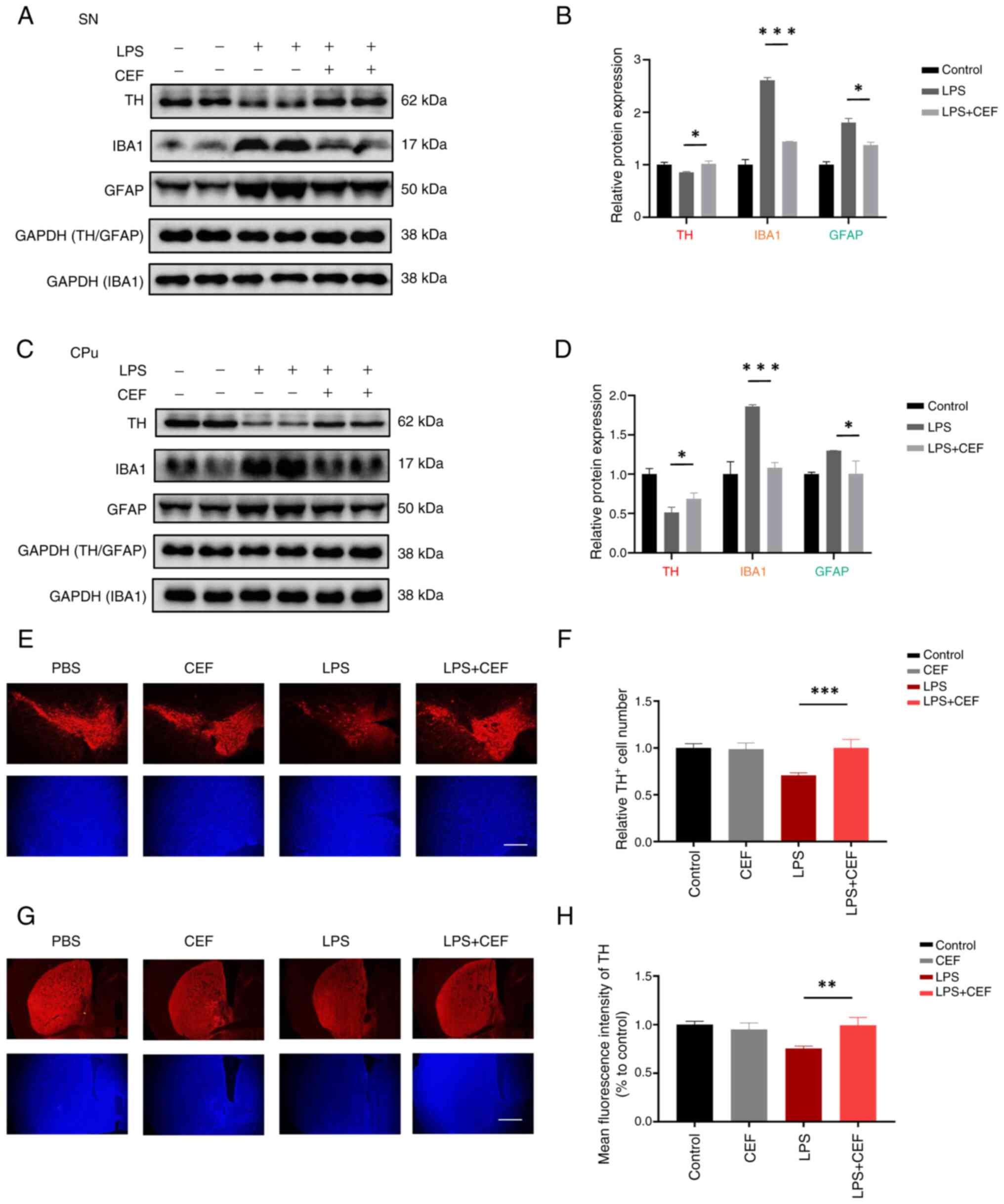 | Figure 4CEF reverses TH expression in
LPS-induced mice. (A-D) Mice were stereotaxically injected with 2
μl LPS (1 mg/ml) into the ventricles, followed by
intraperitoneal injection of 200 mg/kg CEF once a day for 7 days.
The protein expression levels of TH, IBA1 and GFAP were detected in
the SN and CPu of the brain by western blot analysis. (E-H) The
brain tissues were cut into slices and stained with an antibody
against TH. Fluorescence images and statistical analysis of TH
expression in the SN and CPu are shown. Data are expressed as the
mean ± SD. *P<0.05, **P<0.01 and
***P<0.001 (n=3). Scale bar, 100 μm. Coronal
slices. CEF, ceftriaxone; TH, tyrosine hydroxylase; LPS,
lipopolysacharide; IBA1, allograft inflammatory factor 1; GFAP,
glial fibrillary acidic protein; SN, substantia nigra; CPu, caudal
putamen. |
CEF inhibits neurotoxicity via
attenuating microglial activation
The aforementioned in vivo studies
demonstrated that CEF could inhibit glial cell activation and
alleviate neuronal injury. Therefore, subsequently, in vitro
studies were preformed to investigate this mechanism. CCK-8 assays
showed that CEF at a concentration of 900 μM had no toxic
effect on BV2 cell viability (Fig.
5A). In addition, the mRNA and protein expression levels of
iNOS and COX-2 were significantly increased in LPS-treated BV2
cells. However, cell pre-treatment with CEF considerably reversed
this effect (Fig. 5B-D), thus
suggesting that CEF could alleviate microglial activation. Nuclear
factor-κB (NF-κB) is a key transcription factor involved in
regulating the expression of inflammatory genes, while the
increased phosphorylation of NF-κB/p65 is a primary trigger for
inflammatory activation (21).
In the present study, the results demonstrated that p-NF-κB/p65 was
upregulated, while its expression levels were decreased following
cell pre-treatment with CEF (Fig. 5E
and F). The aforementioned finding indicated that CEF could
alleviate microglial activation via regulating the NF-κB pathway.
Emerging evidence has suggested that the activation of inflammation
promotes ferroptosis (22,23). Therefore, whether the ferroptosis
mechanism could be associated with the CEF-mediated inhibition of
microglial activation was also explored. The western blot analysis
results demonstrated that CEF could significantly reverse the loss
of SLC7A11 and GPX4 protein expression levels in LPS-treated BV2
cells (Fig. 5G and H). These
findings suggested that CEF could suppress ferroptosis via
regulating the SLC7A11/GPX4 axis in activated microglia.
Subsequently, whether microglial activation could affect neuronal
survival was determined. Therefore, the conditioned medium (CM)
from LPS-treated BV2 cells induced cellular injury in SH-SY5Y
cells, a neuroblastoma cell line. Additionally, the CM from
LPS-treated BV2 cells pre-treated with CEF reduced cellular
toxicity in SH-SY5Y cells (Fig. 5I
and J). Collectively, these findings suggested that CEF could
inhibit microglial activation in vitro and protect neurons
from microglial activation-induced neuronal damage.
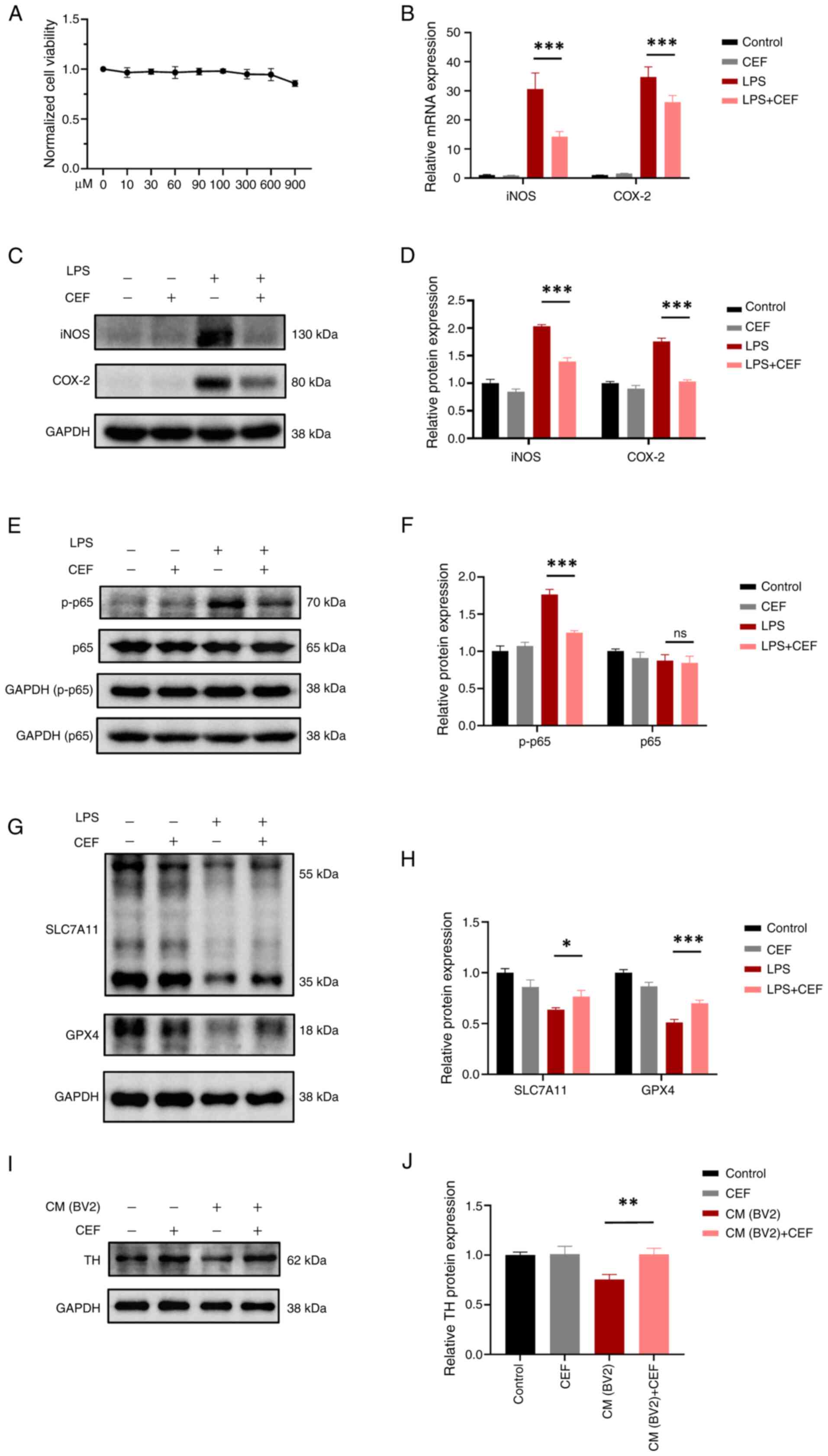 | Figure 5CEF reduces neuronal injury via
inhibiting the activation and ferroptosis of microglia. (A) A Cell
Counting Kit-8 assay was carried out to examine the effect of CEF
on cell viability. BV2 cells were treated with different
concentrations of CEF (0-900 μM) for 24 h. (B) BV2 cells
were pre-treated with 300 μM CEF for 4 h and then with 100
ng/ml LPS for an additional 6 h. The mRNA expression levels of iNOS
and COX-2 were measured by reverse transcription-quantitative PCR.
(C) BV2 cells were pre-treated with CEF for 4 h and then with 100
ng/ml LPS for an additional 24 h. Subsequently, the protein
expression levels of iNOS and COX-2 were detected by western blot
analysis. (D) The relative expression levels of proteins in (C) are
shown. (E) BV2 cells were pre-treated with CEF for 4 h and 100
ng/ml LPS for 15 min. Then the protein expression levels of p65 and
p-p65 were detected by western blot analysis. (F) The relative
expression levels in (E) are shown. (G) BV2 cells were pre-treated
with CEF for 4 h. After treatment with 100 ng/ml LPS for 24 h, the
protein expression levels of SLC7A11 and GPX4 were measured by
western blot assay. Due to the proximity in molecular weight
between SLC7A11/GPX4 and GAPDH, simultaneous detection on the same
membrane was not feasible, but all experiments utilized the same
biological samples and were conducted from the same batch on the
same day. (H) The relative expression levels of proteins in (G) are
presented. (I) SH-SY5Y were cultured in culture medium from BV2
cells for 24 h and the protein expression levels of TH were
detected via western blot analysis. (J) The relative expression
levels of proteins in (I) are shown. Data are expressed as the mean
± SD. *P<0.05, **P<0.01 and
***P<0.001 (n=3). CEF, ceftriaxone; LPS,
lipopolysacharide; iNOS, inducible nitric oxide synthase; COX-2,
cyclooxygenase-2; p-, phosphorylated; SLC7A11, solute carrier
family 7 member 11; GPX4, glutathione peroxidase 4; tyrosine
hydroxylase. |
CEF inhibits neurotoxicity via
alleviating astrocyte activation
To further investigate the role of CEF in astrocyte
activation, further in vitro studies were carried out in
TNFα-treated C8-D1A cells. The CCK-8 assay results revealed that
C8-D1A cell treatment with 900 μM CEF had no toxic effect on
these cells (Fig. 6A). In
TNFα-treated C8-D1A cells, CEF significantly reduced the mRNA and
protein expression levels of iNOS and COX-2, thus suggesting that
CEF could inhibit astrocyte activation (Fig. 6B-D). Additionally, CEF could
alleviate the enhanced expression levels of p-NF-κB/p65 in
TNFα-treated C8-D1A cells, thus indicating that CEF could inhibit
astrocyte activation via regulating the NF-κB pathway (Fig. 6E and F). Furthermore, the western
blot results revealed that CEF could significantly restore the
reduced protein expression levels of SLC7A11 and GPX4 in
TNFα-treated C8-D1A cells, thus suggesting that CEF could suppress
ferroptosis via regulating the SLC7A11/GPX4 axis in activated
astrocytes (Fig. 6G and H). In
addition, CM from TNFα-treated C8-D1A cells pre-treated with CEF
reduced cellular toxicity in SH-SY5Y cells (Fig. 6I and J). Overall, the
aforemetioned findings suggested that CEF could inhibit astrocyte
activation and protect neurons against astrocyte activation-induced
neuronal death in vitro (Fig.
7).
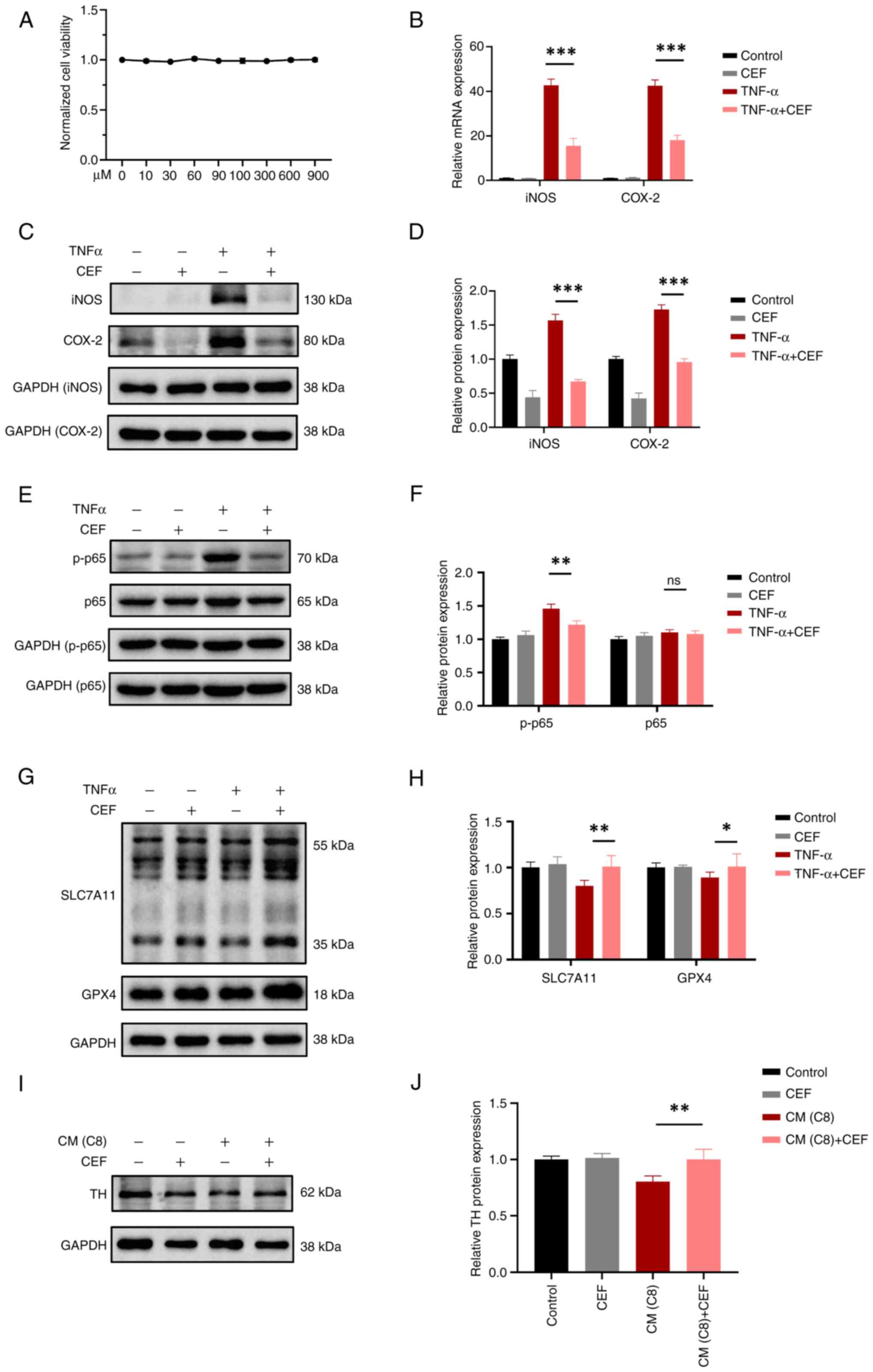 | Figure 6CEF attenuates neuronal injury via
inhibiting the activation and ferroptosis of astrocytes. (A) A Cell
Counting Kit-8 assay was performed to evaluate the effect of CEF on
cell viability. C8-D1A cells were treated with different
concentrations of CEF (0-900 μM) for 24 h. (B) C8-D1A cells
were pre-treated with 300 μM CEF for 4 h and then with 10
ng/ml TNF-α for an additional 6 h. The mRNA expression levels of
iNOS and COX-2 were measured by reverse transcription-quantitative
PCR. (C) C8-D1A cells were pre-treated with CEF for 4 h. Following
cell treatment with TNF-α (10 ng/ml) for 24 h, the protein
expression levels of iNOS and COX-2 were detected by western blot
analysis. (D) The relative expression levels of the indicated
proteins in (C) are shown. (E) C8-D1A cells were pre-treated with
CEF for 4 h. After treatment of cells with 10 ng/ml TNF-α for 15
min, the protein expression levels of p65 and p-p65 were measured
by western blot assays. (F) The relative expression levels of the
indicated proteins in (E) are presented. (G) C8-D1A cells were
pre-treated with CEF for 4 h. Following treatment with TNF-α (10
ng/ml) for 24 h, the protein expression levels of SLC7A11 and GPX4
were measured by western blot analysis. (H) The relative expression
levels of the indicated proteins in (G) are shown. (I) SH-SY5Y were
cultured in culture medium from C8-D1A cells for 24 h and the
protein expression levels of TH were detected using western blot
analysis. (J) The relative protein expression levels of TH are
presented. Data are expressed as the mean the ± SD.
*P<0.05, **P<0.01 and
***P<0.001 (n=3). CEF, ceftriaxone; iNOS, inducible
nitric oxide synthase; COX-2, cyclooxygenase-2; p-, phosphorylated;
SLC7A11, solute carrier family 7 member 11; GPX4, glutathione
peroxidase 4; TH, tyrosine hydroxylase; ns, not significant. |
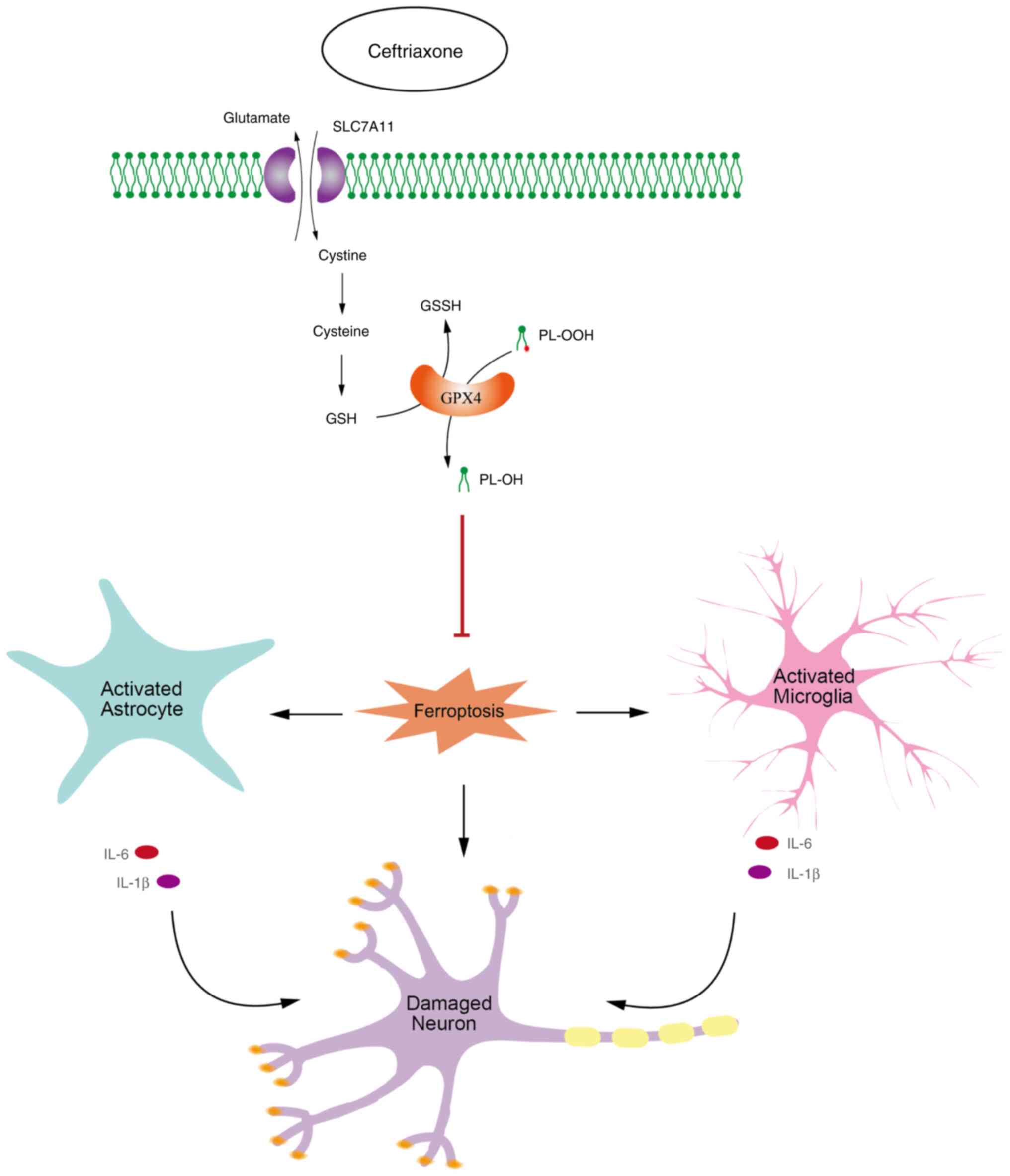 | Figure 7CEF alleviates glial cell activation
and neuronal damage via inhibiting ferroptosis. In turn, CEF can
inhibit the ferroptosis pathway via regulating the expression
levels of SLC7A11 and GPX4 in a non-cell-specific manner, thus
directly protecting dopaminergic neurons and preventing glial cell
activation, and its indirect effect on damaging neurons. CEF,
ceftriaxone; SLC7A11, solute carrier family 7 member 11; GPX4,
glutathione peroxidase 4; GSH, reduced glutathione; GSSG, oxidized
glutathione; IL, interleukin; PL-OOH, phospholipid
hydroperoxides. |
Discussion
Neuroinflammation is an early and persistent
phenomenon in PD, which is commonly driven by microglia and
astrocytes in the CNS (24).
This immune response is crucial for maintaining tissue homeostasis,
removing pathogens and recovering from damage (25). The primary feature of PD, α-syn,
directly activates microglia, thus leading to increased production
of pro-inflammatory cytokines and reactive oxygen species (ROS)
(26). It has been reported that
cytokines, such as TNFα, released from microglia, can activate
neighboring astrocytes (20). A
previous study demonstrated that persistent inflammation was
involved in α-syn aggregation, while the resulting ROS could lead
to lipid peroxidation, thus driving ferroptosis and non-apoptotic
cell death (27). It was
previously shown that glial activation-induced ferroptosis in
neurons is a key molecular mechanism in PD (11). Ferroptosis, a regulated cell
death, which is characterized by overwhelming lipid peroxidation,
is closely associated with PD pathogenesis (28). The end-product of ferroptosis,
4-hydroxy-2,3-trans-nonenal, promotes the inflammatory response by
α-syn aggregation, indicating that ferroptosis and
neuroinflammation potentiate each other in PD progression (22). Therefore, regulating inflammation
and ferroptosis could offer a potential strategy for treating PD
(28,29).
CEF, a third-generation broad-spectrum antibiotic,
has been widely used for over 30 years and is generally considered
safe (30). In 2005, Rothstein
et al (31) found that
CEF could upregulate astrocytic glutamate transporter-1 (GLT-1)
through transcriptional activation. GLT-1 regulates the majority of
extracellular glutamate, a significant excitatory neurotransmitter
in the CNS (32). Dysregulation
of glutamate homeostasis is considered as a core feature of
neuropsychiatric disorders (33). Via upregulating GLT-1, CEF could
attenuate the behavioral manifestations of various
hyper-glutamatergic brain disorders in over 100 preclinical studies
(34). In MPTP-induced PD animal
models, CEF could reverse behavioral deficits, possibly due to
GLT-1 upregulation and its antioxidant and anti-inflammatory
properties, as well as the attenuation of neuronal toxicity
mediated by BDNF restoration (6-9).
Furthermore, CEF has been recognized to upregulate the
glutamate/cystine antiporter (SLC7A11/xCT), which imports cystine
in exchange for intracellular glutamate (35-37). This exchange can promote
glutamate homeostasis and increase antioxidant activity in
astrocytes via promoting the synthesis of GSH, the principal
antioxidant in mammalian cells (38). Using GSH as the preferred
substrate, GPX4 can convert phospholipid hydroperoxides into
non-toxic lipid alcohols, while oxidizing GSH to oxidized
glutathione (39). GPX4 is
considered as the hub of lipid oxidation, ferroptosis, disease and
treatment (40). The
SLC7A11/GSH/GPX4 axis, dependent on the cystine-glutamate
antiporter system, is considered the mainstay restricting
ferroptosis (17). However,
whether ferroptosis is associated with the SLC7A11/GPX4 axis in the
neuroprotective mechanism of CEF remains poorly understood.
The current study revealed that CEF exerted
neuroprotective properties via inhibiting ferroptosis in PD. The
results showed that CEF inhibited ferroptosis in both neuronal
injury models (Fig. 2D) and
glial cell activation in vitro models (Figs. 5G and 6G). Since the term was coined in 2012,
the research field of ferroptosis has increased exponentially
(41). The underlying
pathogenesis of PD based on ferroptosis could offer potential
therapeutics for this disease (42,43). Previous studies on CEF focused on
the role of GLT-1 in PD (43-45). GLT-1 and SLC7A11 could promote
glutamate homeostasis via regulating its flow into and out of cells
(32,38). Although previous studies
demonstrated that CEF upregulated SLC7A11 in APP/PS1 Alzheimer's
disease mice and in rats exposed to ethanol (35,36), whether SLC7A11-related
ferroptosis is involved in the neuroprotective mechanism of CEF in
PD remains poorly understood. A previous in vitro study
indicated that pre-treatment of PC12 cells with 100 μM CEF
could protect them against 6-hydroxydopamine-induced injury
(10). Additionally, 100
μM CEF could alleviate MPP+-induced neurotoxicity
in astrocytes (46) and increase
SLC7A11 activity, which was associated with GSH levels in HT22
cells treated with 300 μM CEF for 7 days (37). In the present study, the
neuroprotective mechanism of CEF (300 μM) and its
association with ferroptosis was investigated. The results revealed
that CEF inhibited the ferroptosis pathway via regulating SLC7A11
expression in a non-cell-specific manner. Additionally, CEF could
block ferroptosis via regulating GPX4, the hub of ferroptosis. The
aforementioned findings highlighted the potential research and
therapeutic value of CEF in regulating ferroptosis in PD through
the SLC7A11/GPX4 axis.
Previous studies also showed that CEF downregulated
GFAP and IBA1, thus supporting its potential effect on modulating
neuroinflammation in PD rats (47). The MPTP-induced PD model is a
recognized and reproducible L-dopa-responsive lesion in the
nigrostriatal system, since it can accurately recapitulate several
of the key features of the pathophysiology of PD, including the
loss of dopaminergic neurons, mitochondrial dysfunction and motor
deficits (48,49). The present experiment was
designed based on previous literature and laboratory experience,
administering MPTP at a dose of 20-30 mg/kg by intraperitoneal
injection over 1 week (50,51). Following the MPTP injections,
behavioral tests were conducted, such as the rotating rod test and
climbing rod test, to screen for successful PD models (12,13). To ensure the precision and
reliability of our experimental data, mice of the same birthdate,
sex and strain were selected to minimize the influence of
individual differences. The present study verified that treatment
with CEF could promote neuronal protection in a PD mouse model
(Fig. 1). Furthermore, MPTP
administration could promote the production of ROS and
pro-inflammatory cytokines, which in turn could exacerbate the loss
of dopaminergic neurons and motor impairments in mice via promoting
oxidative stress and neuroinflammation (52). LPS is a component of the outer
membrane of gram-negative bacteria and is used to induce
neuroinflammation in PD models. LPS-induced inflammation has been
extensively used to study the mechanisms of neuroinflammation in
PD, since the activation of microglia can lead to the production of
inflammatory cytokines and oxidative stress, which in turn cause
dopaminergic neuron death and motor deficits in mice (53). Both the MPTP and LPS-induced PD
models are widely used due to their ability to closely mimic
several of the key features of PD. The results of present study
verified the anti-inflammatory effects of CEF in LPS-induced
inflammation models (Figs. 3 and
4A-D). Furthermore, CEF
alleviated glial cell activation through the NF-κB pathway
(Figs. 5C-F and 6C-F) and reduced glial
activation-mediated neuronal toxicity (Figs. 5I and 6I). Activated glial cells can release
pro-inflammatory cytokines and other neurotoxic agents that can
damage surrounding neuronal cells (18,25). Therefore, understanding the
crosstalk between glial cells and neurons is essential in
developing effective therapeutic interventions for these diseases
(54). In future studies, the
authors will consider conducting animal behavior tests to evaluate
the efficacy of CEF in improving behavioral anomalies in the PD
animal model. Given that the role of GLT-1 in PD has been
confirmed, the present study mainly focused on the roles of SLC7A11
and related ferroptosis in PD. The association between GLT-1 and
SLC7A11 has not been previously investigated. Other studies also
suggested that inflammation could lead to ferroptosis via the
activation of multiple inflammation-related signaling pathways. The
investigation of ferroptosis in inflammation, particularly in
microglia-mediated neuroinflammation, remains in its early stages
(55). In the present study, the
results suggested that CEF could alleviate glial cell activation
and inhibit ferroptosis in PD (Figs.
5G and 6G), thus supporting
that the association between inflammation and ferroptosis could be
a potential research focus.
In summary, the present study suggested that CEF
could alleviate glial cell activation and neuronal damage in PD via
inhibiting ferroptosis (Fig. 7).
CEF could exert a significant potential in mitigating PD pathology
and ferroptosis, positioning inflammatory signaling pathways and
driving anti-ferroptosis, thus providing a potential strategy for
the treatment of PD.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
DG conceptualized the study. YC supervised the
study. HZ, DG and YC performed the experiments. XW performed data
analysis. HZ and DG wrote the main manuscript and prepared Figs. 1, 2 and 4-6.
YC prepared Fig. 3. XW and DG
revised the manuscript. ZC and JL provided revision suggestions and
supplemented experiments. All authors read and approved the final
version of the manuscript. HZ and DG confirm the authenticity of
all raw data.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of Suzhou Institute of Biomedical Engineering and
Technology, Chinese Academy of Sciences (approval no. 2024-A50;
Suzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 82001255 and 32300799), the Natural
Science Foundation of Jiangsu (grant no. SBK20200213) and the
Suzhou Science and Technology Plan Project (grant nos. SKYD2023090,
SKYD2023091 and SKYD2023180).
References
|
1
|
Weintraub D, Aarsland D, Chaudhuri KR,
Dobkin RD, Leentjens AF, Rodriguez-Violante M and Schrag A: The
neuropsychiatry of Parkinson's disease: Advances and challenges.
Lancet Neurol. 21:89–102. 2022. View Article : Google Scholar :
|
|
2
|
Jankovic J and Tan EK: Parkinson's
disease: Etiopathogenesis and treatment. J Neurol Neurosurg
Psychiatry. 91:795–808. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dong-Chen X, Yong C, Yang X, Chen-Yu S and
Li-Hua P: Signaling pathways in Parkinson's disease: Molecular
mechanisms and therapeutic interventions. Signal Transduct Target
Ther. 8:732023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bloem BR, Okun MS and Klein C: Parkinson's
disease. Lancet. 397:2284–2303. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vijiaratnam N, Simuni T, Bandmann O,
Morris HR and Foltynie T: Progress towards therapies for disease
modification in Parkinson's disease. Lancet Neurol. 20:559–572.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hsieh MH, Meng WY, Liao WC, Weng JC, Li
HH, Su HL, Lin CL, Hung CS and Ho YJ: Ceftriaxone reverses deficits
of behavior and neurogenesis in an MPTP-induced rat model of
Parkinson's disease dementia. Brain Res Bull. 132:129–138. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hsu CY, Hung CS, Chang HM, Liao WC, Ho SC
and Ho YJ: Ceftriaxone prevents and reverses behavioral and
neuronal deficits in an MPTP-induced animal model of Parkinson's
disease dementia. Neuropharmacology. 91:43–56. 2015. View Article : Google Scholar
|
|
8
|
Bisht R, Kaur B, Gupta H and Prakash A:
Ceftriaxone mediated rescue of nigral oxidative damage and motor
deficits in MPTP model of Parkinson's disease in rats.
Neurotoxicology. 44:71–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaur B and Prakash A: Ceftriaxone
attenuates glutamate-mediated neuro-inflammation and restores BDNF
in MPTP model of Parkinson's disease in rats. Pathophysiology.
24:71–79. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ruzza P, Siligardi G, Hussain R, Marchiani
A, Islami M, Bubacco L, Delogu G, Fabbri D, Dettori MA, Sechi M, et
al: Ceftriaxone blocks the polymerization of α-synuclein and exerts
neuroprotective effects in vitro. ACS Chem Neurosci. 5:30–38. 2014.
View Article : Google Scholar
|
|
11
|
Wang ZL, Yuan L, Li W and Li JY:
Ferroptosis in Parkinson's disease: Glia-neuron crosstalk. Trends
Mol Med. 28:258–269. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu LL, Han Y, Zhang ZJ, Wang YQ, Hu YW,
Kaznacheyeva E, Ding JQ, Guo DK, Wang GH, Li B and Ren HG: Loss of
DJ-1 function contributes to Parkinson's disease pathogenesis in
mice via RACK1-mediated PKC activation and MAO-B upregulation. Acta
Pharmacol Sin. 44:1948–1961. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gu C, Zhang Y, Hu Q, Wu J, Ren H, Liu CF
and Wang G: P7C3 inhibits GSK3β activation to protect dopaminergic
neurons against neurotoxin-induced cell death in vitro and in vivo.
Cell Death Dis. 8:e28582017. View Article : Google Scholar
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Nagatsu T and Nagatsu I: Tyrosine
hydroxylase (TH), its cofactor tetrahydrobiopterin (BH4), other
catecholamine-related enzymes, and their human genes in relation to
the drug and gene therapies of Parkinson's disease (PD): Historical
overview and future prospects. J Neural Transm (Vienna).
123:1255–1278. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kordower JH, Olanow CW, Dodiya HB, Chu Y,
Beach TG, Adler CH, Halliday GM and Bartus RT: Disease duration and
the integrity of the nigrostriatal system in Parkinson's disease.
Brain. 136:2419–2431. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng J and Conrad M: The metabolic
underpinnings of ferroptosis. Cell Metab. 32:920–937. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu H, Chang Q, Sun T, He X, Wen L, An J,
Feng J and Zhao Y: Metabolic reprogramming and polarization of
microglia in Parkinson's disease: Role of inflammasome and iron.
Ageing Res Rev. 90:1020322023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Patani R, Hardingham GE and Liddelow SA:
Functional roles of reactive astrocytes in neuroinflammation and
neurodegeneration. Nat Rev Neurol. 19:395–409. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kwon HS and Koh SH: Neuroinflammation in
neurodegenerative disorders: The roles of microglia and astrocytes.
Transl Neurodegener. 9:422020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mitchell JP and Carmody RJ: NF-κB and the
transcriptional control of inflammation. Int Rev Cell Mol Biol.
335:41–84. 2018. View Article : Google Scholar
|
|
22
|
Lee J and Hyun DH: The interplay between
intracellular iron homeostasis and neuroinflammation in
neurodegenerative diseases. Antioxidants (Basel). 12:9182023.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, Wu S, Li Q, Sun H and Wang H:
Pharmacological inhibition of ferroptosis as a therapeutic target
for neurodegenerative diseases and strokes. Adv Sci (Weinh).
10:e23003252023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tansey MG, Wallings RL, Houser MC, Herrick
MK, Keating CE and Joers V: Inflammation and immune dysfunction in
Parkinson disease. Nat Rev Immunol. 22:657–673. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang W, Xiao D, Mao Q and Xia H: Role of
neuroinflammation in neurodegeneration development. Signal
Transduct Target Ther. 8:2672023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Isik S, Yeman Kiyak B, Akbayir R, Seyhali
R and Arpaci T: Microglia mediated neuroinflammation in Parkinson's
disease. Cells. 12:10122023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Angelova PR, Choi ML, Berezhnov AV,
Horrocks MH, Hughes CD, De S, Rodrigues M, Yapom R, Little D, Dolt
KS, et al: Alpha synuclein aggregation drives ferroptosis: An
interplay of iron, calcium and lipid peroxidation. Cell Death
Differ. 27:2781–2796. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shibu MA, Bharath M and Velmurugan BK:
Regulating inflammation associated ferroptosis - A treatment
strategy for Parkinson disease. Curr Med Chem. 28:6895–6914. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Grubbauer HM, Dornbusch HJ, Dittrich P,
Weippl G, Mutz I, Zobel G, Georgopoulos A and Fotter R: Ceftriaxone
monotherapy for bacterial meningitis in children. Chemotherapy.
36:441–447. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rothstein JD, Patel S, Regan MR, Haenggeli
C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung
DS, et al: Beta-lactam antibiotics offer neuroprotection by
increasing glutamate transporter expression. Nature. 433:73–77.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pajarillo E, Rizor A, Lee J, Aschner M and
Lee E: The role of astrocytic glutamate transporters GLT-1 and
GLAST in neurological disorders: Potential targets for
neurotherapeutics. Neuropharmacology. 161:1075592019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Malik AR and Willnow TE: Excitatory amino
acid transporters in physiology and disorders of the central
nervous system. Int J Mol Sci. 20:56712019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abulseoud OA, Alasmari F, Hussein AM and
Sari Y: Ceftriaxone as a novel therapeutic agent for
hyperglutamatergic states: Bridging the gap between preclinical
results and clinical translation. Front Neurosci. 16:8410362022.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gao J, Liu L, Liu C, Fan S, Liu L, Liu S,
Xian XH and Li WB: GLT-1 knockdown inhibits ceftriaxone-mediated
improvements on cognitive deficits, and GLT-1 and xCT expression
and activity in APP/PS1 AD mice. Front Aging Neurosci.
12:5807722020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rao PSS, Saternos H, Goodwani S and Sari
Y: Effects of ceftriaxone on GLT1 isoforms, xCT and associated
signaling pathways in P rats exposed to ethanol. Psychopharmacology
(Berl). 232:2333–2342. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lewerenz J, Albrecht P, Tien ML, Henke N,
Karumbayaram S, Kornblum HI, Wiedau-Pazos M, Schubert D, Maher P
and Methner A: Induction of Nrf2 and xCT are involved in the action
of the neuroprotective antibiotic ceftriaxone in vitro. J
Neurochem. 111:332–343. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Patel SA, Warren BA, Rhoderick JF and
Bridges RJ: Differentiation of substrate and non-substrate
inhibitors of transport system xc(-): An obligate exchanger of
L-glutamate and L-cystine. Neuropharmacology. 46:273–284. 2004.
View Article : Google Scholar
|
|
39
|
Dar NJ, John U, Bano N, Khan S and Bhat
SA: Oxytosis/ferroptosis in neurodegeneration: The underlying role
of master regulator glutathione peroxidase 4 (GPX4). Mol Neurobiol.
61:1507–1526. 2024. View Article : Google Scholar
|
|
40
|
Liu Y, Wan Y, Jiang Y, Zhang L and Cheng
W: GPX4: The hub of lipid oxidation, ferroptosis, disease and
treatment. Biochim Biophys Acta Rev Cancer. 1878:1888902023.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sun S, Shen J, Jiang J, Wang F and Min J:
Targeting ferroptosis opens new avenues for the development of
novel therapeutics. Signal Transduct Target Ther. 8:3722023.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Y, Lv MN and Zhao WJ: Research on
ferroptosis as a therapeutic target for the treatment of
neurodegenerative diseases. Ageing Res Rev. 91:1020352023.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang X, Wu K, Ye XY, Xie T, Zhang P,
Blass BE and Bai R: Novel druggable mechanism of Parkinson's
disease: Potential therapeutics and underlying pathogenesis based
on ferroptosis. Med Res Rev. 43:872–896. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Smaga I, Fierro D, Mesa J, Filip M and
Knackstedt LA: Molecular changes evoked by the beta-lactam
antibiotic ceftriaxone across rodent models of substance use
disorder and neurological disease. Neurosci Biobehav Rev.
115:116–130. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ritter K, Somnuke P, Hu L, Griemert EV and
Schäfer MKE: Current state of neuroprotective therapy using
antibiotics in human traumatic brain injury and animal models. BMC
Neurosci. 25:102024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang Y, Zhang X and Qu S: Ceftriaxone
protects astrocytes from MPP(+) via suppression of NF-κB/JNK/c-Jun
signaling. Mol Neurobiol. 52:78–92. 2015. View Article : Google Scholar
|
|
47
|
Zhou X, Lu J, Wei K, Wei J, Tian P, Yue M,
Wang Y, Hong D, Li F, Wang B, et al: Neuroprotective effect of
ceftriaxone on MPTP-induced parkinson's disease mouse model by
regulating inflammation and intestinal microbiota. Oxid Med Cell
Longev. 2021:94245822021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tieu K: A guide to neurotoxic animal
models of Parkinson's disease. Cold Spring Harb Perspect Med.
1:a0093162011. View Article : Google Scholar
|
|
49
|
Mao Q, Qin WZ, Zhang A and Ye N: Recent
advances in dopaminergic strategies for the treatment of
Parkinson's disease. Acta Pharmacol Sin. 41:471–482. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jackson-Lewis V and Przedborski S:
Protocol for the MPTP mouse model of Parkinson's disease. Nat
Protoc. 2:141–151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Narmashiri A, Abbaszadeh M and Ghazizadeh
A: The effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
(MPTP) on the cognitive and motor functions in rodents: A
systematic review and meta-analysis. Neurosci Biobehav Rev.
140:1047922022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ge J, Lin H, Yang J, Li Q, Zhou J, Qin Z
and Wu F: TP53-induced glycolysis and apoptosis regulator (TIGAR)
ameliorates lysosomal damage in the 1-methyl-4-phenyl-1, 2, 3,
6-tetrahydropyridine-mediated mouse model of Parkinson's disease.
Toxicol Lett. 339:60–69. 2021. View Article : Google Scholar
|
|
53
|
Guo DK, Zhu Y, Sun HY, Xu XY, Zhang S, Hao
ZB, Wang GH, Mu CC and Ren HG: Pharmacological activation of
REV-ERBα represses LPS-induced microglial activation through the
NF-κB pathway. Acta Pharmacol Sin. 40:26–34. 2019. View Article : Google Scholar
|
|
54
|
He J, Zhu G, Wang G and Zhang F: Oxidative
stress and neuroinflammation potentiate each other to promote
progression of dopamine neurodegeneration. Oxid Med Cell Longev.
2020:61375212020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mohan S, Alhazmi HA, Hassani R, Khuwaja G,
Maheshkumar VP, Aldahish A and Chidambaram K: Role of ferroptosis
pathways in neuroinflammation and neurological disorders: From
pathogenesis to treatment. Heliyon. 10:e247862024. View Article : Google Scholar : PubMed/NCBI
|