Introduction
Brain-derived neurotrophic factor precursor
(proBDNF) is a precursor protein of the neurotrophic factor family,
which has an essential role in the development and function of the
central nervous system (CNS) (1). It is generated through the cleavage
of the pro-region signal sequence by the Golgi apparatus from
pre-pro-BDNF. proBDNF consists of a 129-amino acid N-terminal
pre-structural domain and a 118-amino acid C-terminal mature
structural domain (2). Within
cells, proBDNF can be processed into mature brain-derived
neurotrophic factor (mBDNF) by furin or proprotein convertases,
while in the extracellular space, it can be converted to mBDNF by
tissue-type plasminogen activator (t-PA) or matrix
metalloproteinases (MMPs) (3).
The efficiency of cleavage and hence the ratio of pro-BDNF to
mature BDNF, is different at different stages of development.
proBDNF and mBDNF are detectable in the neonatal and adolescent
stages, whereas mBDNF predominates in adulthood (4). proBDNF is predominantly found in
CNS structures such as the dorsal horn of the spinal cord, raphe
nucleus, trigeminal nucleus of the spinal cord, hypothalamus and
amygdala; it also occurs in peripheral tissues including skin,
intestine, adrenal gland, pituitary gland, liver and skeletal
muscle. Furthermore, it is expressed in immune cells such as
monocytes/macrophages and T and B cells (5,6).
proBDNF interacts with high-affinity to the p75 neurotrophin
receptor (p75NTR), Sortilin, Sortilin-related VPS10
domain-containing receptor 2 (SorCS2), and human follistatin-like
4; by activating signaling pathways such as NF-κB, JNK, or RhoA, it
plays crucial roles in neuronal apoptosis, synaptic inhibition,
axon pruning and synapse elimination (7-11). proBDNF contributes to axonal
retraction dendritic degeneration, and long-term depression, which
is associated with cognitive impairment (CI) (11); abnormalities in proBDNF
biosynthesis may correspond to different CIs in several brain
diseases (12). For example,
elevated proBDNF and p75NTR levels in the aged hippocampus lead to
learning and memory deficits (13). There are two common polymorphisms
of proBDNF, Val66 and Met66. proBDNF Val66 inhibits long-term
potentiation (LTP) and promotes long-term depression (LTD) by
activating the glycogen synthase kinase 3β (GSK3β)-pTau signaling
pathway, which may facilitate synaptic weakening (14). The present review aims to provide
an overview of the function and application of proBDNF and to
highlight its role in the progression of various diseases.
CI
CI encompasses a spectrum of conditions
characterized by persistent and acquired cognitive deficits as the
primary clinical manifestation, resulting from abnormalities in
higher-level cognitive processing associated with learning and
memory in the brain. CI includes mild CI (MCI) and dementia, the
latter of which can lead to significant impairments in learning and
memory, accompanied by aphasia, agnosia, apraxia, visual-spatial
deficits, as well as psychological and behavioral dysfunction
(15,16). A 2020 study revealed that among
individuals aged ≥60 years old, the prevalence of MCI was 15.54%,
whereas the prevalence of dementia was 6.04% (17). The etiology of CI remains unclear
but may involve excessive CNS immune-inflammatory responses,
neuronal apoptosis, accumulation of amyloid-β (Aβ), microvascular
damage, and blood-brain barrier (BBB) dysfunction (18). Cortisol, secreted by the
hypothalamus-pituitary-adrenal axis, binds glucocorticoid receptors
in the hippocampus, amygdala and prefrontal cortex, influencing
neuronal function and cognitive processes alongside
neurotransmitters such as glutamate (19). Currently, the diagnosis of CI
largely depends on neuropsychological assessments, laboratory
biomarkers and relevant neuroimaging examinations. Treatment
strategies typically involve a comprehensive approach integrating
pharmacological therapy, physical rehabilitation, and psychological
and behavioral interventions. However, achieving satisfactory
treatment outcomes remains challenging (20). Thus, effective interventions to
prevent or slow the course of CI are critically needed.
Neurodegenerative diseases
BDNF plays a role in the development of
neurodegenerative diseases, including Parkinson's disease (PD) and
Alzheimer's disease (AD). An imbalance in the conversion of proBDNF
to mBDNF can significantly affect the progression of these diseases
by decreasing synaptic plasticity. The degeneration of neurons in
the substantia nigra, striatum and hippocampus can impede the
conversion of proBDNF to mBDNF, resulting in higher levels of
proBDNF. The concentration of proBDNF is closely associated with
the severity of neurodegenerative diseases (3). Prediabetes is associated with an
elevated risk of AD and related dementias (ADRD) (21); exercise has been shown to
mitigate this risk by modulating brain metabolism and reducing
neuroinflammation (22,23). Neuronal extracellular vesicles
(nEVs) can carry a variety of biomolecules, including proteins,
lipids and RNA. These vesicles facilitate intercellular
communication and play a role in various physiological and
pathological processes. Furthermore, nEVs can serve as indicators
of insulin signaling in the brain (24,25). Insulin regulates a wide array of
cellular functions, such as glucose metabolism, mitochondrial
activity, oxidative stress response, autophagy, synaptic plasticity
and cognitive processes. Alterations in the phosphorylation status
of signaling molecules within the insulin pathway contribute to
oxidative stress and neuroinflammation. Moreover, insulin signaling
is pivotal in maintaining neuronal health and mitigating
neurodegenerative processes (26). In a study involving 21 older
adults with prediabetes who underwent 12 sessions of 60-min cycling
training over a 2-week period, researchers observed enhanced
neuronal responsiveness to insulin, as well as increased peripheral
insulin sensitivity. The underlying mechanism may involve a
significant reduction in fasting levels of proBDNF and increased
expression of total protein kinase B (Akt) in nEVs under glucose
stimulation following short-term exercise. These changes were
closely linked to improvements in peripheral insulin sensitivity,
while the decrease in proBDNF suggested that exercise may alleviate
neuroinflammation or oxidative stress (27). However, due to limitations such
as a small sample size, absence of a control group, and
insufficient long-term monitoring of the direct preventative
effects of exercise on ADRD, further investigation is warranted to
elucidate how alterations in Akt within nEVs influence neuronal
function and cognition. Additionally, the precise mechanisms by
which proBDNF affects insulin signaling require further exploration
to clarify its relationship with brain insulin action following
exercise.
AD
AD is a progressive condition that primarily affects
memory and cognitive functions. The pathogenesis of AD involves
multiple hypotheses, including the Aβ hypothesis (28), molecular mechanisms of tau
protein pathology (29), glial
cell activation and neuroinflammation (30), insulin resistance and brain
energy metabolism (31), gut
microbiota dysbiosis (32),
pathogenic mechanisms of APOE4 (33), abnormal DNA methylation and
histone modifications (34), as
well as environmental and lifestyle factors such as sleep
disturbances (35) and air
pollution (36). Collectively,
these factors may contribute to the onset and progression of AD.
Microcystin is a potent hepatotoxin produced by certain
cyanobacteria. Its relevance to AD stems from its capacity to
induce oxidative stress, neuroinflammation and neuronal damage,
which are mechanisms central to the pathophysiology of AD.
Therefore, microcystin serves as a valuable model toxin for
investigating neurodegenerative processes (37). Research has indicated that
prolonged exposure to microcystin can markedly decrease the levels
of the t-PA enzyme, increase the expression of proBDNF, activate
the JNK pathway, and trigger neuronal apoptosis in the hippocampal
CA1 and CA2 areas. This exposure also leads to a reduction in
spinal cord density, accumulation of Aβ protein plaques, and
increased phosphorylated tau. These changes contribute to the
development of learning and memory impairments, as well as
Alzheimer's-like alterations (38). Similar to microcystin,
β-methylamino-L-alanine, a neurotoxin produced by cyanobacteria,
has been shown to induce neurotoxicity; Domoic acid, which acts as
a glutamate receptor agonist and is produced by algae, triggers
excitotoxicity and induces neuronal damage; heavy metals, such as
lead and mercury, have also been demonstrated to promote oxidative
stress and cognitive decline, thereby positioning them as
significant factors in AD research. Nevertheless, the precise
relationship between these toxins and proBDNF requires further
investigation (39,40).
The progression of AD is marked by an exceptionally
prolonged course, and it remains unclear whether this is due to
ongoing neuronal degeneration, dysregulation of associated
molecular networks, or a combination of these factors that
contribute to the disease's pathogenesis (41-43). As a result, further investigation
is necessary to elucidate the complexities of signaling pathways,
the synergistic effects between molecules, as well as the presence
of positive and negative feedback loops and their spatiotemporal
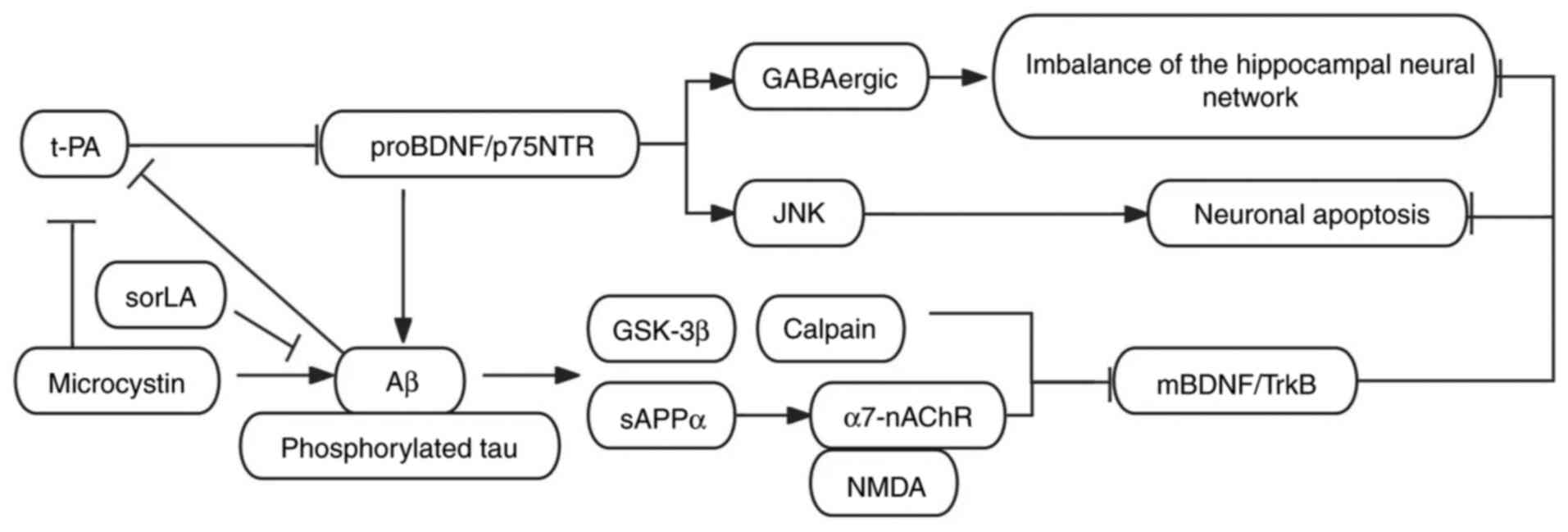
interrelationships. The p75NTR receptor is essential for proBDNF to
facilitate the accumulation of Aβ plaques and the phosphorylated
tau (44). In Aβ1-40-injected
rats and APP/PS1 transgenic mice, the ratio of proBDNF to mBDNF in
the CA1 region of the hippocampus was significantly elevated.
proBDNF inhibits GABAergic synaptic function via the p75NTR
receptor, whereas mBDNF promotes the survival of GABAergic neurons
and the expression of KCC2 through the TrkB receptor. Functional
abnormalities in the hippocampus are closely associated with
cognitive deficits, with the inhibition of GABAergic transmission
and the imbalance of the BDNF signaling pathway serving as key
pathological mechanisms. The suppression of GABAergic transmission
results in reduced synchrony of the hippocampal neural network,
thereby impairing memory encoding. Restoring GABAergic inhibitory
function can re-establish the excitation-inhibition balance of the
hippocampal neural network, rescuing cognitive function (45). Notably, another study
demonstrated that in 5xFAD and APP/PS1 transgenic mice, alongside
the progression of Aβ deposition and tau pathology in hippocampal
neurons, Aβ oligomers reduce the conversion of proBDNF to mBDNF by
inhibiting the tPA/plasmin system. Simultaneously, they activate
GSK-3β and calpain, promoting the cleavage of the TrkB receptor,
which further diminishes the neuroprotective effects of mBDNF.
Although soluble amyloid precursor protein α (sAPPα), produced via
the non-amyloidogenic pathway, indirectly enhances the mBDNF/TrkB
signaling pathway by activating α7-nicotinic acetylcholine
receptors and NMDA receptors, Aβ deposition reduces sAPPα,
weakening this protective mechanism. Additionally, the decreased
expression of sorLA in AD disrupts Aβ clearance, exacerbating the
imbalance in the proBDNF/mBDNF ratio (7). The imbalance of proBDNF/mBDNF in
the AD hippocampus mediates neurodegeneration via p75NTR and TrkB
signaling pathways. The signal transduction mechanisms of proBDNF
across AD are illustrated in Fig.
1. Furthermore, the interaction between Aβ and sAPPα
exacerbates this pathological process. Future research should
concentrate on restoring BDNF homeostasis, inhibiting proBDNF
signaling, and targeting critical nodes in sAPPα processing to
provide a novel therapeutic strategy for AD treatment.
PD
PD is the second most prevalent neurodegenerative
disorder globally, with an incidence rate exceeding 1% among
individuals aged ≥65 years old, and it is projected that the
incidence rate will double by 2030. In addition to the
characteristic motor symptoms of PD, numerous non-motor symptoms
are also present, with CI being the most prevalent and occurring at
any stage (46). The research
data indicates that the average incidence rate of MCI in patients
with PD is 25.8%, while the incidence rate of dementia is 26.3%.
Additionally, the incidence rate of dementia can escalate to 83%
within 20 years following a PD diagnosis (47). Currently, the primary focus lies
on early cognitive changes, characterized by impaired executive
function and spatial abilities, often accompanied by memory
deficits, thereby increasing the risk of early progression to
dementia (46). CI represents a
prevalent non-motor symptom of PD, spanning from MCI (PD-MCI) to
dementia (PDD); the manifestations, severity, and progression of CI
exhibit significant heterogeneity and may involve alterations in
multiple neurotransmitter systems, including dopamine,
acetylcholine, norepinephrine and serotonin, and the neuropathology
of CI in PD encompasses a range of pathological changes, such as
Lewy bodies, Aβ and neurofibrillary tangles. Additionally,
deficiencies in multiple neurotransmitters and genetic risk
factors, including α-synuclein mutations and apolipoprotein E
variants, contribute to its development (48). A systematic search was conducted
in the PsycINFO (https://www.apa.org/pubs/databases/psycinfo/), PubMed
(https://pubmed.ncbi.nlm.nih.gov/) and
Scopus (https://www.scopus.com/home.uri) databases up to April
2019, yielding 41 eligible studies (involving 7,053 patients with
PD); PD-MCI was relatively prevalent among non-demented patients
with PD, with an estimated prevalence of ~40%. The multiple-domain
subtype represents the predominant manifestation of PD-MCI and is
associated with the postural instability and gait difficulty
subtype, longer disease duration, higher levodopa equivalent daily
dose, and more severe motor symptoms. PD-MCI is further linked to a
reduced quality of life, elevated levels of depression and apathy,
and serves as a significant predictor of dementia progression
(49). Neuropsychological
assessment remains the gold standard for diagnosing CI, offering
comprehensive evaluations across various cognitive domains; only
rivastigmine has been approved by the U.S. Food and Drug
Administration for the treatment of PDD, and future treatments for
PD will necessitate further research in the standardization of
cognitive assessments, early diagnosis and intervention of CIs, as
well as the development of effective symptom management and
disease-modifying therapies, which represent significant challenges
(48). More longitudinal studies
are warranted to comprehensively investigate the risk factors and
progression of PD-MCI. Additionally, there is a need to develop
anxiety assessment tools specifically designed for patients with PD
in order to more precisely evaluate the relationship between
anxiety and PD-MCI (49).
When neurotrophic factors NT3 and NT4 interact with
p75NTR, they activate the PI3K/AKT pathway, upregulate Bcl-2, and
confer a protective effect on dopaminergic neurons. Conversely,
when proBDNF binds to p75NTR, it triggers apoptosis in dopaminergic
neurons, accelerating the progression of PD. The underlying
mechanism involves the activation of the JNK, Caspase-3, NF-κB and
RhoA pathways (50). The precise
mechanism by which proBDNF binds to the p75NTR receptor to mediate
the pro-apoptotic pathway leading to CI remains incompletely
understood, necessitating further research.
Multiple sclerosis (MS)
MS is a primary inflammatory demyelinating disease
of the CNS. CI is a common and debilitating symptom within its
broad and unpredictable spectrum of clinical manifestations,
significantly impacting the quality of life for patients (51). The prevalence of CI in MS ranges
from 20-88%, with the highest occurrence and most severe symptoms
observed in primary progressive MS. The underlying mechanism of
MS-related CI requires further elucidation (52). Research indicates that the
upregulation of proBDNF and its receptor in immune cells in
patients with MS and MS model mice is closely associated with
demyelination and CNS inflammation. This may occur through the
proBDNF/p75NTR/NF-κB pathway, which mediates the release of
inflammatory cytokines such as TNF-α, IL-β, IL-6, IL-17 and INF-γ
by peripheral immune cells. Furthermore, intraperitoneal (i.p.)
injection of an anti-proBDNF antibody has been shown to improve MS
by modulating immune system function (53).
The Vps10p-domain receptor family consists of a
group of evolutionarily conserved sorting receptors that play
essential roles in intracellular protein trafficking and signaling,
and the primary members of this family include Sortilin (SORT1),
SorLA (LR11/SORL1), SorCS1, SorCS2 and SorCS3, which are implicated
in neurodevelopment, synaptic plasticity and metabolic regulation
(54). P75NTR, a member of the
tumor necrosis factor receptor superfamily, plays a critical role
in the survival, apoptosis and synaptic plasticity of neurons
(55). The interaction between
Sortilin and p75NTR serves as a critical regulatory mechanism for
apoptosis, inflammatory responses and metabolic processes (54,56). When proBDNF binds to the
p75NTR-Sortilin complex, it activates the downstream JNK pathway,
leading to the release of cytochrome C from mitochondria and
promoting apoptosis, and this mechanism may exacerbate cell damage
in neurodegenerative and inflammatory diseases (57). It is worth noting that the
interaction between p75NTR and Sortilin may exhibit distinct
functional roles across various cell types and disease contexts
(54). The expression of proBDNF
is significantly increased in patients with MS and experimental
autoimmune encephalomyelitis (EAE) models, thereby promoting
neuronal apoptosis and inflammatory responses through the
activation of p75NTR-Sortilin receptors. Additionally, the reduced
activity of proteases, such as plasmin, may hinder the conversion
of proBDNF to mBDNF, thereby exacerbating the pathological
progression of MS (58).
Although Sortilin is highly expressed in the brain tissue of
patients with MS, its deficiency does not appear to significantly
influence disease progression in the EAE model and it has been
proposed that Sortilin predominantly engages in the innate immune
response rather than the adaptive immune response within immune
cells, thereby suggesting a limited role of Sortilin in MS
(59). A comprehensive
understanding of its specific mechanisms across a range of disease
contexts, along with the development of highly selective drugs,
will continue to be a focal point for future research
endeavors.
Depression
Pathogenesis of depression
The pathogenesis of depression includes the
neurotransmitter hypothesis, hypothalamic-pituitary-adrenal axis
hypothesis, neuroplasticity hypothesis, and immunoinflammatory
hypothesis, in which microglia and astrocytes are important
(60). It shows neuronal atrophy
and synaptic loss in the prefrontal cortex and hippocampus of
depressed patients. These changes are associated with deficits in
executive function, memory, attention, self-referential processing
bias and cognitive rigidity (61). In addition, individuals with
major depression often have significant impairments in cognitive
function, such as learning, memory, executive function, processing
speed, attention and concentration, which are underpinned by
multiple interacting neurobiological mechanisms, including
neuroinflammation. Moreover, most antidepressants have not been
specifically developed or evaluated to improve these cognitive
deficits (62). It has been
indicated that elevated proBDNF levels may result in CI, leading to
behaviors that resemble depression (63).
proBDNF regulates synaptic
plasticity
Researchers have discovered that the expression
levels of proBDNF and its receptor p75NTR are significantly
elevated in patients with severe depression and in mouse models of
depression. When mice were injected with proBDNF into both sides of
their hind legs, they displayed depressive-like behaviors within 4
weeks. Furthermore, it was observed that the density and length of
synapses in the hippocampal dentate gyrus and amygdala of the mice
were diminished, whereas treatment with a proBDNF antibody
mitigated depressive symptoms. Thus, proBDNF appears to be a key
driver of depression-like behaviors in mice, potentially through
reduced synaptic plasticity (64).
proBDNF mediates neuroinflammation and
glial cell activation
A recent study comparing the levels of related
molecules in the serum and lymphocytes of 32 patients with
depression and 20 healthy individuals found that the levels of
IL-1β and IL-10 in lymphocytes were positively correlated with the
severity of depression, while the levels of TNF-α and IL-6 were
negatively correlated. Sortilin levels were positively correlated
with IL-1β levels. Furthermore, proBDNF and p75NTR were primarily
upregulated in CD4+ and CD8+ T cells in
patients with severe depression, and their levels returned to
normal following antidepressant treatment. The
proBDNF/p75NTR/sortilin pathway may upregulate the expression
levels of IL-1β and IL-10 in patients with severe depression,
downregulate TNF-α and IL-6, and thus regulate the progression of
depression (65). In the mouse
depression model associated with periodontitis, researchers
observed that microglia and astrocytes in the hippocampus were
activated, and the expression of inflammatory factors IL-1β and
TNF-α significantly increased. The expression of occludin and
claudin5, proteins related to tight junctions, was reduced, and the
levels of IL-1β in the serum were elevated; proBDNF was found to be
involved in this process (66).
Li et al (67) found that
the deletion of SorCS2 alleviated depressive-like behaviors induced
by periodontitis in mice. Thus, it is hypothesized that the
deletion of SorCS2 downregulated proBDNF and glutamatergic signal
transduction in the hippocampus, restoring neuronal activity.
proBDNF-induced neuronal
apoptosis
Post-stroke depression (PSD) is a common
complication following a stroke, characterized by persistent
depression and diminished interest (68). Yang et al (69) established an in-vitro
model of PSD by depriving neuronal cells of oxygen and glucose and
treating them with corticosterone. It was found that proBDNF
protein levels in the prefrontal cortex and hippocampal neurons of
the PSD model group were significantly elevated compared with the
control group. proBDNF binds to the p75NTR receptor, activating the
RhoA/JNK signaling pathway, which upregulates the expression of
PSD-related proteins such as PSD-95, Syn and P-cofilin.
Simultaneously, it downregulates the expression of anti-apoptotic
proteins, including RIP2, caspase-3 and Cytochrome C, thereby
inducing neuronal apoptosis, reducing the number of mature
synapses, and influencing the development of depression. A recent
study has shown that the injection of hippocampal exosomes,
extracted and purified from a mouse model of stroke, exacerbated
depressive-like behaviors in mice. The mechanism is hypothesized to
be related to the increased expression of proBDNF and p75NTR, as
well as the insufficient expression of synaptophysin and PSD95,
caused by hippocampal exosomes from stroke (70).
Based on the aforementioned research findings, it
can be inferred that neural-immune mechanisms play a pivotal role
in the progression of depression. The proBDNF/p75NTR/sortilin
pathway shows considerable promise as a therapeutic target for
addressing CIs associated with depression, and future studies
should focus on further exploring its clinical translational
potential.
HIV-associated neurocognitive
disorders (HANDs)
HANDs refer to a spectrum of neuro-CIs linked to HIV
infection. Despite the substantial reduction in HIV-related
mortality achieved through antiretroviral therapy (ART), HANDs
continue to be prevalent complications among individuals living
with HIV (71,72). HANDs can be categorized into
three levels based on severity: Asymptomatic neuro-CI, mild
neurocognitive disorder, and HIV-associated dementia (73). The pathophysiological mechanisms
underlying the development of HANDs are multifaceted, encompassing
direct viral damage to the CNS, immune activation and inflammatory
responses, disruption of the BBB, and release of neurotoxic factors
such as glutamate, reactive oxygen species (ROS) and nitric oxide
(74,75). Treatment strategies for HANDs
include the optimization of ART, incorporation of anti-inflammatory
therapy, neuroprotective agents and application of immunomodulatory
approaches (76-78). The management of HANDs continues
to pose significant difficulties. Future research will concentrate
on early diagnosis, innovative treatment approaches, and
personalized medicine strategies to enhance the quality of life for
patients with HANDs (79).
A significant number of individuals infected with
HIV-1 develop HANDs, including spatial memory impairment and
learning difficulties, even if they are undergoing combination ART
(80). Synaptic simplification
and loss are prominent features of HANDs. The HIV-1 envelope
protein gp120 has been shown to elevate the levels of proBDNF in
the body, leading to the binding of proBDNF to p75NTR and a
subsequent reduction in synaptic plasticity (81).
Researchers have discovered that gp120 causes
intermittent memory impairment and learning deficits by disrupting
mitochondrial function and energy production. This disruption may
occur through the impairment of the glycolytic pathway at the
pyruvate level, leading to the accumulation of glycated end
products and inhibiting the cleavage of proBDNF into mBDNF. The
accumulation of proBDNF triggers the expression of Inducible cAMP
Early Repressor, which occupies cAMP response element binding sites
present on the promoter regions II and IV of the BDNF gene, thereby
altering normal synaptic plasticity (82). A cross-sectional study revealed
that among 157 HIV-infected individuals from sub-Saharan Africa who
had not undergone ART, elevated serum mBDNF levels were
significantly and positively correlated with the patients'
cognitive abilities. By contrast, increased proBDNF levels were
negatively correlated with the patients' cognitive abilities,
particularly affecting fine motor skills and speed (83). Modulating proBDNF levels could
represent a potential therapeutic strategy for ameliorating
learning and memory impairments in individuals infected with
HIV-1.
Sepsis-associated encephalopathy
(SAE)
Microglia, as an endogenous immune cell, release
both pro-inflammatory and anti-inflammatory cytokines, nitric oxide
and neurotrophic factors. The upregulation of intracellular
Ca2+ concentration in microglia is crucial for its
functionality. proBDNF binds to p75NTR and activates the targeting
transient receptor potential melastatin 7 (TRPM7) channel, leading
to a sustained elevation of intracellular Ca2+ in
microglia, thereby enhancing nitric oxide production induced by
INF-γ. proBDNF may thus play a significant role in regulating
inflammatory responses in the brain (84).
SAE refers to diffuse brain dysfunction caused by
sepsis, characterized by decreased attention and disorientation
(85). The pathogenesis of SAE
primarily involves neuroinflammation, disruption of the BBB,
impairment of cerebral vascular function, and alterations in neural
metabolism (86). Researchers
have discovered that sepsis can increase proBDNF levels, leading to
a reduction in the infiltration of cerebral meningeal
CD4+T cells. This disrupts the balance of the
pro-inflammatory and anti-inflammatory microenvironment in the
meninges, including an upregulation of pro-inflammatory factors
such as IL-1β and IL-6, as well as a downregulation of
anti-inflammatory factors including IL-4 and IL-13. Ultimately,
resulting in cognitive dysfunction (87), SAE increases the likelihood of
cognitive and memory impairment, with proBDNF potentially playing a
significant role in the pathogenesis of SAE.
Cerebral ischemia-reperfusion injury
(CIRI)
CIRI represents a complex pathological process that
ensues when blood flow is reestablished to the brain following a
period of ischemia, characterized by an absence of adequate blood
supply (88). CIRI involves a
variety of complex mechanisms, including oxidative stress and
hypoxia, which lead to mitochondrial dysfunction, resulting in the
excess release of ROS (89,90); calcium overload activates
protease, lipase and endonuclease, leading to cell injury and death
(91), mitochondrial dysfunction
(89), inflammatory responses
mediated by immune cells (92,93), and the breakdown of the BBB. This
breakdown of the BBB results in the excessive release of excitatory
neurotransmitters, resulting in calcium ion inflow. Reperfusion
triggers programmed cell death and necrotic cell death (94,95). CIRI significantly contributes to
the morbidity and mortality associated with stroke and other
ischemic events. This condition complicates treatment strategies,
as while the restoration of blood flow is essential, it may
exacerbate outcomes if not carefully managed (96). At present, the primary treatment
methods for CIRI include edaravone and N-acetylcysteine, which are
used to neutralize ROS and reduce oxidative stress; calcium channel
blockers to inhibit calcium overload; anti-inflammatory drugs such
as IL-1 receptor antagonists and TNF-α inhibitors; neuroprotective
agents to protect neurons from excitotoxicity and apoptosis; and
therapeutic hypothermia to reduce metabolic demands and alleviate
reperfusion injury (97-99). Emerging therapeutic approaches
include targeting the Wnt/β-Catenin signaling pathway for brain
ischemia-reperfusion injury (100), and TRPM channels to inhibit
oxidative stress, mitochondrial dysfunction, inflammation and
calcium overload (101).
CIRI promotes the release of inflammatory factors,
resulting in BBB damage (102).
This structural and functional impairment of the BBB can further
trigger microglial activation, neuronal apoptosis and Aβ
deposition, ultimately leading to spatial learning and memory
dysfunction (103). In the
mouse model of acute ischemic stroke, researchers observed the
infiltration of NK cells into the surrounding area of the brain
infarction. This subsequently promoted microglial activation,
resulting in an inflammatory response and neuronal damage (104). Previous research has shown that
microglia can modulate synaptic plasticity; however, the exact
mechanism by which they contribute to CIRI is not fully understood.
Subsequent studies using a mouse model of middle cerebral artery
occlusion (MCAO) have indicated that proBDNF/p75NTR may disrupt
glutamatergic synapses within 24 h of MCAO, whereas the mBDNF/TrkB
pathway could interfere with GABAergic synapses. This activation
subsequently triggers the ERK1/2 and GSK3β pathways, leading to the
phosphorylation of the postsynaptic protein Gephyrin at the Ser268
and Ser270 residues, ultimately resulting in excessive synaptic
pruning by microglia. By pharmacologically depleting microglia or
employing a multi-functional gene editing system known as
CRISPR-Cas9 to generate GphnS268A/S270A mutant mice and mice
lacking BDNF expression in their microglia, researchers were able
to reduce microglial activation within 24 h. This intervention
ensured the preservation of both glutamatergic and GABAergic
synapses, enhanced synaptic protection, reduced infarct size
following ischemia, facilitated tissue repair, and promoted neural
network reorganization after CIRI (105). In the rat photochemical
occlusion model of ischemic cerebral infarction, researchers
observed that administering an anti-proBDNF antibody at a dosage of
5 mg/kg via i.p. injection at 6 h and 3 days post-ischemia
exhibited anti-apoptotic and anti-inflammatory effects,
demonstrating improved sensory-motor function after CIRI (106); confirming that the proBDNF
antibody could reduce neuronal apoptosis and alleviate inflammatory
responses in the infarcted area. However, further verification is
required to determine whether the proBDNF antibody can improve CI.
Additionally, further larger-scale studies are required to
investigate the therapeutic effects of different doses of proBDNF
antibody to obtain the optimal dose for quicker recovery following
CIRI.
Postoperative cognitive dysfunction
(POCD)
POCD is a prevalent complication among elderly
patients undergoing anesthesia and surgery, with studies indicating
an incidence ranging from 10-54%. The incidence of POCD in patients
undergoing cardiac surgery is notably higher, with up to 80%
experiencing postoperative delirium and a subsequent incidence of
POCD within 12 months ranging from 10-40% (107,108). POCD primarily presents as
anxiety, alterations in personality, psychosis and memory
impairment. Its mechanism involves inflammation of the CNS,
oxidative stress and apoptosis (109). POCD represents a significant
neurological complication following surgery, and there is evidence
to suggest that neuroinflammation plays a pivotal role in its
pathogenesis. The age-dependent nature of the neuroinflammatory
mechanisms underlying POCD underscores the importance of
comprehending the involvement of neuroinflammation to elucidate its
underlying mechanisms and develop effective strategies for
prevention and treatment (110).
Researchers induced POCD in 16-month-old mice
through stable femoral fracture surgery under sevoflurane
anesthesia, establishing an animal model of POCD. They observed
that anesthesia and surgery led to an increase in proBDNF, which
negatively regulated synaptic function in the hippocampus and
resulted in cognitive dysfunction in aged mice. The administration
of P75NTR inhibitors and exogenous BDNF was found to mitigate the
loss of dendritic spines and long-term potentiation in the
hippocampus by altering the mBDNF/proBDNF ratio, thereby
alleviating postoperative cognitive dysfunction. Modulating the
mBDNF/proBDNF ratio may present a promising target for treating
POCD (111). Oxidative stress
injury is a key pathogenic mechanism of POCD. Studies have revealed
a close association between the integrated stress response (ISR)
and oxidative stress. In an experiment, researchers induced POCD in
male mice by performing tibial surgery at 4 months of age under
sevoflurane anesthesia and assessed cognitive function using fear
conditioning tests and Y-mazes from postoperative days 3 to 14. The
use of ISR inhibitors was found to downregulate the expression of
molecules associated with oxidative stress, such as ROS, superoxide
dismutase and malondialdehyde in the hippocampus. Additionally, it
led to decreased levels of proBDNF and increased levels of mBDNF,
thereby mitigating oxidative stress and ameliorating cognitive
dysfunction in mice (112). The
imbalance between proBDNF and mBDNF holds significant implications
for postoperative cognitive dysfunction, yet the associated
pathways and mechanisms remain incompletely elucidated. In
instances of POCD resulting from anesthesia surgery, aberrant
expression levels of proBDNF are associated with neuroimmune
inflammatory responses, a relationship requiring further validation
in future research. In the present review, the association between
proBDNF and cognitive disorders is described and is further
summarized in Table I.
 | Table ICascades between cognitive
impairment, proBDNF and downstream effector molecules. |
Table I
Cascades between cognitive
impairment, proBDNF and downstream effector molecules.
| First author,
year | Disease | proBDNF and
receptor | Downstream
molecular | Function | (Refs.) |
|---|
| Eggert et
al, 2022; Manucat-Tan et al, 2019; Bie et al,
2022 | AD |
proBDNF/p75NTR/sortilin↑ | JNK↑, KCC2↓ | Neuronal apoptosis,
tau phosphorylation, Aβ plaques | (7,44,45) |
| Ali et al,
2024 | PD |
proBDNF/p75NTR↑ | JNK, Caspase-3,
NF-κB, RhoA↑ | Neuronal
apoptosis | (50) |
| Hu et al,
2021 | MS |
proBDNF/p75NTR↑ | NF-κB, TNF-α, IL-β,
IL-6, IL-17, INF-γ↑ | Central nervous
system inflammation, demyelination | (53) |
| Al-Kuraishy et
al, 2024 | MS |
proBDNF/Sortilin1↓ | mBDNF↓, B cell
apoptosis | Weakened autoimmune
and inflammatory responses | (58) |
| Li et al,
2024 | Depression |
proBDNF/SorCS2↓ | glutamatergic↑ | restore neuronal
activities | (67) |
| Yang et al,
2024; Li et al, 2023 | MDD |
proBDNF/p75NTR/sortilin↑ | IL-1β, IL-10↑,
TNF-α, IL-6↓ | Reduce dendritic
spine plasticity | (65,66) |
| Yang et al,
2021; Huang et al, 2024 | PSD |
proBDNF/p75NTR↑ | RhoA/JNK, PSD-95,
Syn, P-cofilin↑, RIP2, caspase-3, Cytochrome C↓ | Neuronal
apoptosis | (69,70) |
| Speidell et
al, 2020; Allen et al, 2022; Michael et al,
2025 | HAND |
proBDNF/p75NTR↑ | ICER↑ | Reduce synaptic
plasticity | (81-83) |
| Mizoguchi et
al, 2021; Luo et al, 2020 | SAE | proBDNF↑ | IL-1β, IL-6↑, IL-4,
IL-13↓ | CNS
inflammation | (84,87) |
| Cramer et
al, 2022 | CIRI |
proBDNF/p75NTR↑ | ERK1/2, GSK3β↑ | Downregulate
glutamatergic dendritic spines and gephyrin scaffold stability | (105) |
Inflammatory diseases
Sepsis
Sepsis is a life-threatening condition characterized
by organ dysfunction resulting from a dysregulated response to
infection (113). Sepsis is
primarily associated with excessive inflammation; infection induces
the release of a substantial quantity of inflammatory mediators,
such as TNF-α, IL-1β and IL-6. Additionally, inflammation-induced
increases in microvascular permeability are frequently accompanied
by the activation of the coagulation system, resulting in
microthrombus formation, tissue edema and subsequent organ
dysfunction (114). Sepsis is
associated with a high mortality rate, particularly in patients
with septic shock. The prognosis is significantly influenced by
early diagnosis, timely intervention, and the underlying health
condition of the patient (115). Researchers observed that the
expression of proBDNF and p75NTR in the cortical area of
CD3+ and CD4+T cells in mesenteric lymph
nodes of septic mice was upregulated. The upregulation of proBDNF
may disrupt the immune-inflammatory microenvironment and
significantly contribute to the progression of sepsis (116). Further investigation explored
the changes in expression of proBDNF/p75NTR, derived from
lymphocytes in the peripheral blood of septic patients, and its
impact on lymphocyte differentiation. The expression of proBDNF in
the peripheral blood of septic patients was upregulated in
CD19+B cells, while the expression of p75NTR was
upregulated in CD19+B cells, CD4+T cells and
CD8+T cells. The proBDNF/p75NTR signaling pathway may
regulate lymphocyte differentiation, decrease the percentage of
CD4+ T cells and CD19+B cells, increase the
production of inflammatory factors IL-1β and IL-6, and promote the
progression of sepsis (117).
Immune-mediated inflammatory diseases
(IMIDs)
IMIDs encompass a diverse range of conditions
characterized by persistent inflammation and immune dysregulation,
affecting multiple organ systems. With >100 distinct types
identified, the global incidence of IMIDs is increasing. Future
treatment strategies for IMIDs should prioritize restoring immune
balance and investigating potential interactions between the immune
and nervous systems, rather than solely focusing on alleviating
inflammation (118). Therefore,
it is necessary to understand the pathogenic mechanism of IMIDs and
to explore novel therapeutic targets.
Rheumatoid arthritis
In the synovial tissue of the mouse model of
arthritis, the levels of proBDNF and p75NTR are elevated in
CD4+ and CD8+ T cells, and the levels of
inflammatory cytokines TNF-α, IL-1β, IL-6 and IL-10 are also
elevated. This elevation can be reversed by treatment with
methotrexate (119). The
proBDNF/p75NTR pathway is closely associated with the pathogenesis
of rheumatoid arthritis, while the expression levels of sortilin
are positively correlated with disease severity in patients.
Inhibition of the proBDNF/p75NTR pathway may lead to downregulation
of the JNK/p38MAPK signaling pathway and a reduction in the release
of inflammatory cytokines such as IL-6, IL-8 and MCP-1, thereby
alleviating joint pain in individuals with rheumatoid arthritis
(120).
Systemic lupus erythematosus (SLE)
SLE is a complex autoimmune disease, primarily
triggered by a genetic predisposition, exposure to silica and
cigarette smoke particles, ultraviolet light, EB virus infection,
or environmental factors including the use of medications including
procainamide, hydroxychloroquine and contraceptives. These factors
interact to activate both innate and specific immunity.
T-cell-mediated autoimmune responses stimulate B cells to generate
anti-ds-DNA antibodies and other autoantibodies. Hyperactive
autoimmune B cells and dysregulation of antibody-secreting cells
(ASCs) lead to the production of numerous immune complexes,
resulting in widespread tissue and organ damage (121). proBDNF and p75NTR are
significantly upregulated in ASCs of patients with SLE. Moreover,
there is a positive correlation between proBDNF levels and joint
symptoms, erythrocyte sedimentation rate, autoantibody levels, and
the degree of SLE activity. In addition, in a mouse model of lupus,
it was found that proBDNF and p75NTR were highly expressed in the
ASCs of peripheral blood and spleen. The activation of the
proBDNF/p75NTR pathway was shown to promote ASC proliferation and
differentiation, stimulate the secretion of autoantibodies as well
as the deposition of IgG complexes, and exacerbate renal injury,
thus confirming the potential pathogenic role of proBDNF in SLE
(122).
Immune dysregulation and mitochondrial
dysfunction in IMIDs
Chronic inflammation in IMIDs induces aberrant
activation of immune cells, including macrophages, T cells, and B
cells, leading to a significant increase in their metabolic
requirements, such as glycolysis and oxidative phosphorylation.
Upon binding to the p75NTR/sortilin complex, proBDNF modulates
metabolic enzyme activity, mitochondrial membrane stability, and
respiratory chain function, thereby directly or indirectly
exacerbating the pathological progression of IMIDs (57). The proBDNF/p75NTR/sortilin
complex activates the downstream JNK signaling pathway, resulting
in the activation of pro-apoptotic Bak and Bax proteins within the
Bcl-2 family. This leads to the release of cytochrome C from
mitochondria and the formation of apoptotic bodies, ultimately
culminating in the activation of caspase 3 and caspase 9, thereby
inducing apoptosis and synaptic suppression (57,123,124). proBDNF/p75NTR may mediate
neuroimmune inflammatory responses, apoptosis and mitochondrial
dysfunction, thereby facilitating the progression of MS (58). In individuals with rheumatoid
arthritis, proBDNF/p75NTR may regulate mitochondrial glucose
metabolism in fibroblast-like synoviocytes and CD4+ T
cells, leading to fibroblast-like synoviocyte proliferation,
migration, invasion and cytokine secretion that exacerbate joint
destruction. Additionally, the proBDNF/p75NTR signal can promote
the assembly of mitochondrial respiratory chain complexes in the B
cells of patients with SLE, resulting in mitochondrial damage and
dysfunction that intensify disease activity. In an animal model of
type A acute aortic dissection, upregulation of proBDNF in M2
monocytes may mediate inflammatory responses as well as induce
mitochondrial dysfunction and apoptosis within the aortic wall
(57).
In conclusion, proBDNF and its receptor p75NTR can
trigger tissue and organ damage via an exaggerated inflammatory
response. Additionally, proBDNF, p75NTR and the sortilin receptor
are capable of regulating mitochondrial function and energy
metabolism in cells, inducing apoptosis, and thereby influencing
the progression of IMIDs. The role of proBDNF in inflammatory
diseases is summarized in Table
II. However, the underlying etiology of immune dysregulation in
inflammatory diseases remains unclear. Future research should
explore proBDNF and its receptors from various angles, focusing
particularly on their role in regulating cellular metabolism and
mitochondrial function across different immune cell subpopulations,
to obtain a deeper understanding of their mechanisms of action.
| Table IICascades between inflammatory
disease, proBDNF and downstream effector molecules. |
Table II
Cascades between inflammatory
disease, proBDNF and downstream effector molecules.
| First author,
year | Disease | proBDNF and
receptor | Downstream
molecular | Function | (Refs.) |
|---|
| Wang et al,
2019; Wang et al, 2023 | Sepsis | proBDNF/p75NTR↑
CD | 4+T,
CD19+B↓, IL-1β, IL-6↑ | Excessive
inflammation | (116,117) |
| Yang et al,
2022; Farina et al, 2022 | Rheumatoid
arthritis |
proBDNF/p75NTR/sortilin↑ | TNF-α, IL-1β, IL-6,
IL-10↑, JNK/p38MAPK↑, MCP1↑ | Excessive
inflammation | (119,120) |
| Shen et al,
2022 | Systemic lupus
erythematosus |
proBDNF/p75NTR↑ | ASCs↑, IgG↑ | Excessive
inflammation | (122) |
| Li et al,
2023; Al-Kuraishy et al, 2024; Putcha et al, 2001;
Sankorrakul et al, 2021 | Mitochondrial
dysfunction |
proBDNF/p75NTR/sortilin↑ | JNK↑, Bak, Bax,
caspase3, caspase9↑ | Synaptic
inhibition, neuronal apoptosis, mitochondrial dysfunction | (57,58,123,124) |
Cancer
Tumor neutralization
The role of the nervous system in regulating tumors
is being increasingly recognized, and a recent study by Taylor
et al (125) published
in Nature has revealed that the BDNF-TrkB pathway upregulates AMPA
receptors on the membrane of astrocytoma cells. This process
increases the fluidity of Ca2+, and enhances the
strength and duration of excitatory postsynaptic currents,
augmenting synaptic connections between neurons and astrocytoma
cells, thereby amplifying the depolarization amplitude of
astrocytoma cell membranes, and promoting their proliferation
(125). Glioma cells exploit
neuroplasticity processes in a sophisticated and dynamic manner,
leveraging neural electrical activity to facilitate tumor
growth.
The role of neuro-invasion in the tumor
microenvironment as a promoter of cancer progression is being
increasingly recognized. In vitro experiments have shown
that cancer cells induce endoplasmic reticulum stress, leading to
the synthesis and release of proBDNF through an X-box binding
protein 1 (XBP1)-dependent mechanism. proBDNF mediates the
transmission of endoplasmic reticulum stress to neurons, thereby
promoting the expression of oncogene C-myc-mediated Egl-9 family
hypoxia-inducible factor 3 in tumor tissues, which in turn
stimulates tumor-associated synapse growth and tumor neurogenesis.
In in vivo tumor transplant models, endoplasmic reticulum
stress induces the expression of XBP1 and proBDNF, as well as tumor
neurogenesis. The use of a proBDNF antibody not only inhibits tumor
neurogenesis induced by endoplasmic reticulum stress but also
impedes cancer progression. Interestingly, it has been observed
that the chemotherapeutic drug 5-fluorouracil induces endoplasmic
reticulum stress and subsequent tumor neurogenesis; however, this
effect can be counteracted by using a proBDNF antibody (126). However, in a prior study, the
opposite result was reported. The expression of proBDNF and its
receptors, p75NTR and sortilin, were examined in 52 cases of human
glioma and 13 control cases using immunohistochemistry, reverse
transcription-quantitative PCR and western blot analyses. The
expression levels of proBDNF, p75NTR and sortilin were
significantly elevated in high-grade gliomas and positively
correlated with tumor malignancy. In vitro, the
proBDNF/p75NTR pathway promoted apoptosis and differentiation of C6
glioma cells while inhibiting their growth and migration (127). The bidirectional effects of
proBDNF in glioma may be attributed to the distinct signaling
pathways mediated by its precursor form and mature form (mBDNF)
through their respective receptors. proBDNF primarily activates JNK
and caspase via p75NTR binding, thereby inducing apoptosis, whereas
mBDNF activates PI3K/Akt and MAPK pathways via TrkB receptor,
promoting cell proliferation. The relative expression levels of
proBDNF/p75NTR/sortilin and mBDNF/TrkB may play a critical role in
glioma malignancy and prognosis (9,128,129). Changes in the microenvironment
of malignant glioma can modulate the signaling of various growth
factors. In the tumor microenvironment, the levels of
proBDNF-converting enzymes, such as MMPs and tPA, also influence
the balance between proBDNF and mBDNF (130-132). Furthermore, the heterogeneity
of glioma may result in differential responses of distinct cellular
subpopulations to proBDNF. Investigating the precise mechanisms
underlying the regulation of proBDNF and mBDNF expression levels in
glioma could have significant implications for therapeutic
strategies.
Clear cell renal cell carcinoma
(ccRCC)
ccRCC constitutes 70-80% of all renal cell
carcinomas and is characterized by a poor prognosis due to the lack
of effective therapeutic targets. Research has demonstrated that
p75NTR is significantly overexpressed in tumor tissues and this
increased expression is correlated with high Fuhrman nuclear
grades. proBDNF is detectable in both tumor and normal tissues,
with notably elevated levels observed in tumor samples. proBDNF
binds to p75NTR, recruiting TrkB-95 and sortilin to form a
functional complex, thereby activating the AKT/ERK signaling
pathway; activation of this pathway promotes the proliferation and
migration of ccRCC cells, as evidenced in the 786-O and ACHN cell
lines. Targeting p75NTR or proBDNF may represent a promising
strategy to inhibit ccRCC progression, particularly for patients
who exhibit resistance to TrkB inhibitors (133).
Breast cancer
Breast cancer represents a significant healthcare
challenge, with limited therapeutic options available for brain
metastases (134). Research
indicates that migrating cancer cells interact with brain
microvascular endothelial cells, thereby facilitating angiogenesis
around the metastatic tumor (135). Alhusban et al (136) investigated the interaction
between proBDNF/mBDNF in brain microvascular endothelial cells and
the breast cancer cell line MDA-MB-231. proBDNF suppressed
angiogenesis in breast cancer brain metastasis, potentially through
inhibition of signal transduction mediated by VEGF. Tamoxifen is
commonly used to treat recurrent or metastatic breast cancer and as
adjuvant therapy following early breast cancer surgery. Prolonged
use of tamoxifen downregulated the mBDNF/ERK/AKT/CREB pathway in
the female rat hippocampus, while increasing the levels of proBDNF,
leading to reduced neuroplasticity and cognitive dysfunction
(137). The primary lesion of
breast cancer may predominantly involve TrkB as the receptor
(138), leading to the
activation of distinct downstream signaling pathways by proBDNF in
different tissue locations, and as previously discussed in the
context of tumor heterogeneity, glioma and breast cancer cell lines
utilized across various studies exhibit distinct receptor
expression profiles and differential activation of signaling
pathways, thereby contributing to variations in experimental
outcomes. In addition, the effects of signaling pathways related to
proBDNF on learning and memory function warrant further
investigation.
Basal cell carcinoma (BCC)
BCC is the most common type of skin cancer. Most
cases can be controlled through local treatment, but treatment of
advanced BCC remains a challenge. Western blot and reverse
transcription-quantitative PCR analysis showed that the protein and
mRNA levels of p75NTR/proBDNF in samples from patients with BCC and
BCC cell lines (TE354.T and ASZ001) were significantly lower than
those in normal skin tissues and immortalized keratinocytes
(HaCaT). p75NTR/proBDNF activates the RIPK1/RIPK3/MLKL pathway,
significantly inhibiting the proliferation of BCC cells, inducing
apoptosis, and reducing tumor-associated macrophages, significantly
increasing the infiltration of neutrophils and CD8+T
cells, promoting M1-type macrophage polarization, and reshaping the
immune microenvironment (139).
The bidirectional effects of proBDNF are likely
attributable to its antagonistic receptor signaling pathways, the
dynamic regulation of its processing mechanisms, and the
heterogeneity inherent in the tumor microenvironment. The emerging
field of cancer neuroscience has ushered in a new era in the study
of the nervous system and tumors. proBDNF is intricately linked to
the malignant biological behaviors exhibited by various tumors
necessitating further research into elucidating the mechanisms
underlying the actions of proBDNF and its associated signaling
molecules in tumorigenesis (Table
III). Future research efforts should concentrate on elucidating
the regulatory mechanisms underlying the equilibrium between
proBDNF and mBDNF while integrating targeted therapeutic strategies
against p75NTR or TrkB receptors.
| Table IIIThe function and molecular mechanisms
of proBDNF across cancer types. |
Table III
The function and molecular mechanisms
of proBDNF across cancer types.
| First author,
year | Cancer types | proBDNF and
Receptor | Downstream
molecular | Function | (Refs.) |
|---|
| Jiang et al,
2022 | Prostate cancer
Pancreatic cancer Colon cancer | ER stress→XBP1/
proBDNF↑ | c-myc/EGLN3↑ | Neurite
outgrowth | (126) |
| Xiong et al,
2013 | Glioma |
proBDNF/p75NTR↑ | GFAP↑ | Inhibits
proliferation and migration; induce apoptosis and
differentiation | (127) |
| De la Cruz-Morcillo
et al, 2016 | Clear cell renal
cell carcinoma | ProBDNF/p75NTR/
TrkB-95/sortilin↑ | AKT/ERK↑ | Promotes
proliferation and migration | (133) |
| Klann et al,
2023 | Breast cancer | proBDNF↑ | VEGF↓ | Suppresses
angiogenesis | (137) |
| Myhre et al,
2013 | Basal cell
carcinoma |
proBDNF/p75NTR↑ | RIPK1/RIPK3/
MLKL↑ | Restore neuronal
activities | (37) |
Neurogenesis
In the adult hippocampal dentate gyrus and
subventricular zone of mature mammalian brains, ongoing
neurogenesis occurs. Here, multipotent neural stem cells can
differentiate into glial cells and neurons in response to stimuli
such as epilepsy, traumatic brain injury and ischemic stroke. These
cells develop dendrites and axons, establish synaptic contacts with
surrounding neurons, and ultimately integrate into existing neural
circuits, facilitating the repair of damaged brain tissue and the
restoration of function (140).
The activation of astrocytes is characterized by the upregulation
of proBDNF and increased metabolic activity within the cell. This
modulates neuroplasticity through the release of proBDNF and its
impact on synaptic pruning, tissue and histone modification,
apoptotic signal transduction, and the expression of genes
associated with protein processing. Consequently, this creates an
environment in the brain that facilitates neural regeneration
(141).
Neural development of the CNS is widely studied and
plays a crucial role in maintaining brain function. Notably,
neurogenesis also occurs in the peripheral nervous system.
Peripheral nerve injury (PNI) represents a relatively prevalent
neurological disorder in clinical settings, with etiologies
encompassing trauma, compression, metabolic disorders and
infections (142). The
pathophysiological mechanisms of this condition are intricate,
involving axonal damage, demyelination, inflammatory responses and
impaired regeneration. Based on the Sunderland classification
system, PNI can be categorized into grades I through V. Grade I
injury (neuropraxia) is characterized solely by myelin sheath
disruption, representing a reversible lesion with recovery
typically occurring within several weeks to months. Grade II injury
(axonotmesis) involves axonal rupture while maintaining an intact
endoneurium, enabling self-regeneration over a period of several
months. By contrast, grades III to V injuries (neurotmesis) entail
the rupture of the endoneurium, perineurium, or epineurium,
necessitating surgical intervention for repair (143). During the acute injury phase,
following axonal rupture, distal axons and myelin sheaths undergo
degeneration, with macrophages clearing debris. Schwann cells
subsequently proliferate to form Bungner bands, facilitating axonal
regeneration (144,145). However, the release of
pro-inflammatory cytokines such as TNF-α and IL-1β may exacerbate
neural damage and hinder the establishment of a regenerative
microenvironment (146,147). Additionally, scar tissue
formation may impede axonal regeneration. Concurrently,
insufficient secretion of neurotrophic factors, including NGF and
BDNF, contributes to regeneration failure (148). During chronic injury
progression, Schwann cells undergo dedifferentiation, losing their
capacity to support nerve regeneration, thereby further
compromising repair outcomes (149). A study investigated the impact
of proBDNF on peripheral sensory neurons and satellite glial cells
(SGCs) through in vitro neuron and SGC cultures (150). The proBDNF/p75NTR/sortilin
signaling pathway upregulates apoptotic factors such as Bax,
caspase-3 and p53 via the mitochondrial pathway while
downregulating anti-apoptotic factors such as Bcl-2, leading to
neuronal apoptosis following sciatic nerve transection. It also
hinders neuronal proliferation and migration. Treatment with a
proBDNF antibody inhibited mitochondrial-dependent apoptosis by
modulating the mBDNF/TrkB/PI3K/AKT and
proBDNF/p75NTR/sortilin/PI3K/AKT signaling pathways, thereby
exerting a neuroprotective effect. This presents a novel approach
to treating peripheral nerve injury (150).
The binding of mBDNF with the TrkB receptor triggers
a signal cascade associated with neuronal survival and plasticity,
whereas the interaction between proBDNF and the p75NTR/sortilin
receptor complex is involved in the apoptotic process. The relative
levels of proBDNF and mBDNF in the cellular microenvironment are
likely to play an important role in regulating astrocyte phenotype
(150). Ma et al
(151) further extracted and
purified SGCs from the Dorsal root ganglion (DRGs) of rats within
24 h of birth and cultured them ex vivo, demonstrating that
SGCs are involved in the neural development of the peripheral
nervous system. By treating the SGCs extracted from the DRGs with
proBDNF antibody, it was shown that SGCs could differentiate into
sensory neuron-like phenotypes, whereas proBDNF inhibits their
differentiation. The researchers also established a rat sciatic
nerve transection model and used proBDNF antibody to promote the
differentiation of SGCs into neuron-like phenotypes. Endogenous
proBDNF can maintain the SGC phenotype and prevent SGCs from
differentiating into neuron-like cells. After treatment with
proBDNF antiserum, the mBDNF/TrkB signal transduction may become
dominant and promote the differentiation of SGCs into neuron-like
phenotypes.
Regulation of fear memory
Fear is an emotional response of organisms to
potential threats, endowing individual life forms with the ability
to adapt to ever-changing and challenging environments. However, it
has a dual nature, as severe trauma can etch traumatic memories
into the brain, potentially resulting in post-traumatic stress
disorder (PTSD) (152). PTSD is
an anxiety and/or memory disorder developed after experiencing
disasters, domestic violence, combat-related trauma, or other
severely traumatic events (153). PTSD is intricately associated
with the excessive consolidation of fear memories and challenges in
overcoming them (154), and is
mediated by functional disruptions within the prefrontal
cortex-amygdala-hippocampus circuit (155), chronic inflammatory states
(156) and compromised neuronal
plasticity (153). The
mBDNF/TrkB signaling pathway in the amygdala plays a crucial role
in fear-related learning and memory. Synaptic plasticity is widely
considered to be the cellular mechanism underlying fear-related
memory formation, but further investigation into the fear
extinction mechanism is warranted (157). The regulation of fear memories
is crucial during the early stages of establishing the neural
circuit for fear. These memories may undergo alterations throughout
neural development, and the associated neural circuits also
experience dynamic changes (158).
Investigation into the conditioned fear model in
rats revealed that elevated levels of proBDNF in the prefrontal
cortex of young rats could destabilize retrieval-dependent fear
memories that are not strongly consolidated by activating the
p75NTR/GluN2B signaling pathway. Researchers examined θ-γ
oscillation coupling in the prefrontal cortex using EEG signals
from rats and observed an inhibitory effect of proBDNF on memory
retrieval in young rats. The experiment confirmed that proBDNF
plays a critical role in synaptic function and memory instability,
potentially facilitating the extinction of fear memories that are
difficult to reconsolidate (159). It has been demonstrated that
proBDNF plays a critical role in LTD within the lateral nucleus of
the amygdala through its interaction with p75NTR. Suppression of
p75NTR signaling leads to a reduction in LTD, and a comparable
phenomenon is observed during fear memory extinction in mice.
Furthermore, inhibition of proBDNF signaling or administration of
anti-proBDNF antibodies attenuates both LTD and fear memory
extinction. These findings suggest that the proBDNF/p75NTR
signaling pathway is essential for regulating fear extinction and
that impaired fear memory removal may be closely associated with
the pathophysiological mechanisms underlying PTSD (160).
Phosphodiesterase 4 (PDE4) is an enzyme expressed
in the dorsal hippocampus that can hydrolyze cAMP, thereby limiting
the phosphorylation of CREB by cAMP-dependent protein kinase (PKA)
and the expression of mBDNF, PKA and phosphorylated CREB can
mediate memory consolidation or decay. Researchers employed the
PDE4 inhibitor roflumilast to upregulate the cAMP/PKA signaling
pathway, thereby promoting fear extinction rather than
reconsolidation. PDE4 inhibition was found to enhance the
expression of proBDNF in the dorsal hippocampus, whereas the
expression of mBDNF remained unaffected. This suggests that proBDNF
may serve as a critical factor in the long-term modulation of fear
memory removal (161). The
proBDNF-mediated LTD mechanism via p75NTR plays a critical role in
fear extinction. Modulating the levels of proBDNF may offer novel
insights into the treatment of PTSD. It is important to highlight
that the existing research has primarily been conducted using
animal models. Future investigations will need to validate its
clinical applicability and refine the targeting strategy. Moreover,
the equilibrium between proBDNF and mBDNF within the nervous system
may significantly influence the efficacy of PTSD treatment,
necessitating careful consideration of potential adverse
effects.
proBDNF can facilitate the extinction of fear
memories and suppress synaptic transmission, potentially enhancing
individual adaptability. Cognitive and behavioral adaptability
enable individuals to adjust to changing circumstances involving
rewards and objectives. The dorsolateral striatum (DLS) is a
component of the cortico-striatal circuitry and plays a role in
regulating cognitive adaptability. Researchers observed a
significant enhancement in early-stage rat reversal learning
correct response rates following the injection of anti-cleaved
proBDNF into the DLS of rats, thereby augmenting cognitive
adaptability (162). Elevated
exogenous proBDNF enhances neural coupling associated with
flexibility, leading to the upregulation of endogenous proBDNF in
the DLS and infralimbic cortex regions. Conversely, the inhibition
of proBDNF in the infralimbic cortex region hampers cognitive
flexibility that is mediated by the DLS. These findings suggest the
involvement of both the infralimbic cortex and DLS regions in
cognitive flexibility, emphasizing the crucial role of proBDNF in
this process (163). The
precise mechanisms through which proBDNF in the infralimbic cortex
and DLS regions modulate cognitive flexibility remain unclear,
necessitating further research.
Significance of proBDNF in disease
diagnosis
Autism spectrum disorders (ASDs)
ASDs are neurodevelopmental disorders characterized
by impairments in motor function, which are one of their core
features. These disorders manifest as persistent deficits in
communication and social interaction, along with limited and
repetitive behaviors and narrow interests (164). The data indicates a significant
increase in the prevalence of ASD, which is hypothesized to be
attributed to interactions between the immune system and the
nervous system, congenital abnormalities in cell function,
neuroinflammation and the activation of microglia (165). ELISA was used to assess the
serum levels of mBDNF, proBDNF and insulin-like growth factors
(IGF-1) in 22 children aged 5-15 diagnosed with mild to moderate
ASD and 29 typically developing controls. No significant
differences in mBDNF levels were observed between the ASD and
control groups; however, proBDNF levels were notably lower in the
ASD group compared with the normal controls, whereas IGF-1 levels
were higher. Among the medicated ASD subjects, serum proBDNF levels
were also observed to be lower compared with the control group;
conversely, there was no difference in IGF-1 and mBDNF levels
between those medicated and those who were not (166). These findings appear to
corroborate the notion that proBDNF serves as a crucial biomarker
for the diagnosis and treatment of ASD. Although the aforementioned
study offers valuable insights into the roles of BDNF, proBDNF and
IGF-1 in autism, several limitations, including the small sample
size, variability in detection methods and the heterogeneity of
ASD, constrain the generalizability and reliability of the
findings. Future research should enhance the robustness of these
conclusions by establishing standardized protocols, conducting
multi-center trials, and incorporating additional biomarkers,
thereby providing more reliable foundations for the diagnosis and
treatment of autism.
Neurological disorders
Due to the absence of early diagnostic techniques
in clinical practice, PD, a neurodegenerative condition, cannot be
promptly treated, potentially resulting in disability. Therefore,
it is crucial to seek biomarkers with high sensitivity and
specificity for the early diagnosis of PD (167). A study enrolled 156
participants not receiving medication. The levels of mBDNF and
proBDNF in the serum of both the PD and control groups were
measured using ELISA. Additionally, a 1-year follow-up was
performed, during which the levels of mBDNF and proBDNF in the
serum were re-examined. The researchers compared and evaluated the
mBDNF/proBDNF ratio and mBDNF levels using ROC curve analysis both
cross-sectionally and longitudinally. The findings indicated that
the mBDNF/proBDNF ratio can function as an early biomarker for PD,
surpassing testing only for mBDNF. proBDNF exhibits superior
repeatability and individual discriminability compared with mBDNF
(168). Future research
involving larger sample sizes is necessary to evaluate the
association between proBDNF and the severity of progression,
follow-up, and response to treatment of PD. This may contribute to
a better understanding of the clinical applicability of proBDNF as
a biomarker for PD-related CI.
Researchers have investigated whether mBDNF and
proBDNF serum levels may serve as valuable biomarkers for assessing
AD. Using ELISA, they measured the levels of mBDNF and proBDNF in
the serum of 126 subjects and then assessed their correlation with
Mini-Mental State Examination (MMSE) scores. The study revealed
significant correlations among MMSE scores, proBDNF levels, and
mBDNF/proBDNF ratios. Lower serum levels of proBDNF were associated
with higher MMSE scores, suggesting that a combined assessment of
both proBDNF and the mBDNF/proBDNF ratio could be an effective
diagnostic approach for patients with AD (169).
β-hydroxybutyrate (BHB) is a small molecule
produced by the body during fat metabolism. It has various effects,
including protecting brain neurons, managing epilepsy, improving
cognitive function disorders, preventing or treating diabetes and
insulin resistance, aiding in weight loss, exhibiting antitumor
properties, delaying aging, and mitigating osteoporosis (170). Interestingly, a study involving
healthy older adults aged 65-75 revealed a positive correlation
between serum BHB and changes in proBDNF. This correlation was
found to be more stable and stronger than that with mBDNF.
Consequently, proBDNF may serve as a reliable predictor of brain
health (171).
Overactive bladder (OAB)
OAB is a syndrome characterized by urgent, frequent
and nocturnal urination in the absence of urinary tract infection
or other obvious pathological conditions. OAB can be classified
into dry OAB and wet OAB based on the presence or absence of
urgency incontinence (172).
The diagnosis of OAB primarily relies on clinically relevant
symptoms combined with self-assessment questionnaires such as the
B-SAQ, the Overactive Bladder Symptom Score (OABSS), and the
OAB-V8. It is also crucial to exclude factors such as pelvic floor
muscle damage, excessive fluid intake and urinary tract infections
before making a comprehensive judgment (173,174). The researchers discovered a
significant negative correlation between the proBDNF/mBDNF ratio
and scores on the OABSS and the Incontinence Impact Questionnaire
(IIQ-7), as well as a negative correlation with disease severity.
Compared with individual measurements of mBDNF or proBDNF, the
proBDNF/mBDNF ratio demonstrated superior diagnostic value. This
ratio could serve as an objective and repeatable test for
diagnosing and phenotypically classifying OAB (175).
Mental health conditions
Antidepressant medications can achieve their
therapeutic effects by reestablishing the equilibrium between mBDNF
and proBDNF in the brain. As depressive symptoms are ameliorated by
treatment, the levels of proBDNF decrease (176). tPA and plasminogen activator
inhibitor-1 (PAI-1) play a significant role in depression by
regulating the ratio of mBDNF to proBDNF (177). The combination of tPA, PAI-1,
and the mBDNF/proBDNF ratio may provide a useful method for the
diagnosis of major depressive disorder, and proBDNF may serve as a
more refined biomarker for assessing treatment efficacy. ELISA was
used to examine the alterations in plasma and lymphocyte levels of
mBDNF and proBDNF in patients with severe depression and bipolar
disorder, aimed at evaluating their diagnostic value. The results
confirmed that untreated individuals with severe depression and
bipolar disorder exhibited significantly lower plasma mBDNF levels.
Furthermore, antidepressant medications may influence mBDNF
concentrations. Both proBDNF and mBDNF levels were found to be
upregulated in lymphocytes. The disparity in mBDNF expression
between plasma and lymphocytes may function as a potential
biomarker for diagnosing severe depression. Specifically, a plasma
mBDNF level <12.4 ng/ml may indicate severe depression or
support a diagnosis of bipolar disorder. Additionally, plasma mBDNF
levels can act as predictors of the severity of severe depression.
However, further research is required to elucidate the implications
of elevated proBDNF levels in lymphocytes and whether the combined
values of proBDNF alongside mBDNF offer superior diagnostic
capabilities compared with mBDNF alone (178). Zwolinska et al (179) used ELISA to quantify the levels
of mBDNF, proBDNF and calcium-binding protein B (S100B) in the
serum of patients diagnosed with severe depression. As treatment
progressed, a significant reduction in serum proBDNF levels was
observed, while mBDNF and S100B levels remained relatively stable.
Consequently, it is hypothesized that decreased human serum proBDNF
levels may serve as a potential biomarker for recovery following
depression treatment. However, it is important to note that the
aforementioned study included only 31 female participants.
Therefore, further validation with a larger sample size is
necessary. These studies have certain limitations, necessitating
future large-scale clinical investigations to evaluate disease
progression and severity, follow-up protocols, and treatment
responses. Such research will enhance our understanding of the
clinical significance of proBDNF and its metabolites as biomarkers
for disease progression, diagnosis and early detection.
Utilization of proBDNF monoclonal
antibody
Researchers have conducted a series of innovative
studies focusing on proBDNF and successfully developed a humanized
monoclonal antibody against it, named McAb-proB. This antibody is
protected by independent intellectual property rights and patents.
It specifically targets peripheral immune cells, effectively
neutralizes proBDNF, and importantly, does not interfere with
endogenous BDNF in the CNS. Preclinical studies have demonstrated
that McAb-proB is efficacious in mitigating the progression of
various immune-mediated inflammatory diseases, organ damage and
inflammatory pain (180).
Applications in inflammatory pain
In the CFA-induced persistent inflammatory pain
mouse model, i.p. administration of 5 mg/kg McAb-proB 2 h post-CFA
injection was found to effectively alleviate pain within the 3-72 h
timeframe. Furthermore, in the surgical pain model, pretreatment
with McAb-proB significantly decreased pain scores. With respect to
its ability to penetrate the BBB, researchers conducted an
intracerebroventricular injection of McAb-proB. The results
indicated that direct central administration had no significant
impact on pain relief, suggesting that McAb-proB may primarily
inhibit spinal p-ERK activation via peripheral mechanisms rather
than through direct penetration of the BBB (181). Regarding pharmacokinetics,
comprehensive descriptions of the antibody's half-life, absorption,
distribution, metabolism and excretion (ADME) parameters have not
been determined to the best of our knowledge. In a separate study,
intrathecal administration of 10 µg McAb-proB was shown to
mitigate CFA-induced inflammatory pain. Intrathecal injection
circumvented the BBB directly, delivering the drug into the
vicinity of the spinal cord (182). Unfortunately, the
aforementioned study also did not provide a detailed description of
the ADME characteristics of McAb-proB. Although no significant
acute toxic reactions were reported after a single dose, further
repeated-dose and long-term toxicity studies are still needed to
evaluate its safety.
Applications for organ damage
In the MRL/lpr lupus mouse model, researchers
administered an initial i.p. injection of McAb-proB at a dose of
100 µg, followed by weekly injections of 30 µg for a
total duration of 8 weeks. The study demonstrated that McAb-proB
could induce apoptosis in CD3+B220+ cells,
reduce proteinuria and lymph node enlargement, and promote body
weight gain, while exhibiting no apparent toxicity, although, the
long-term risks associated with immunosuppression were not assessed
(183). i.p. administration of
100 µg McAb-proB in acute myocardial infarction mouse models
may inhibit the activity of MMP-9, which is associated with cardiac
repair processes. This inhibition could result in impaired cardiac
function, enlarged infarct size, and increased mortality. These
findings indicate that McAb-proB may exert pro-inflammatory side
effects in the context of cardiovascular diseases (184). The bidirectional effects of
McAb-proB in different diseases may indicate variations in the
signaling pathways of proBDNF across various disease states.
Further investigation into its pro-inflammatory/anti-inflammatory
balance mechanism, as well as validation of its safety profile,
represent the next critical steps in the study of McAb-proB as a
potential therapeutic.
Applications in IMID
In the EAE mouse model, McAb-proB was administered
i.p. at a dose of 100 µg per injection, once weekly for 3
months. The treatment was initiated either in the early phase (on
days 9, 13, and 17) or the late phase (on days 17, 21, and 25)
following EAE induction. The study demonstrated that McAb-proB
effectively modulated the immune system by suppressing the
expression of inflammatory factors in the spleen and spinal cord,
reducing the percentages of CD19+B, CD4+T,
and CD8+T cells, alleviating demyelination and
inflammatory responses, and significantly ameliorating the clinical
symptoms of EAE mice (53). In
the lipopolysaccharide-induced mouse model of septic encephalopathy
(SAE), two administration methods of McAb-proB were investigated:
i.p. injection and intrahippocampal microinjection. i.p. injection
was administered 12 post-SAE induction, using 100 µg of
McAb-proB dissolved in 0.3 ml physiological saline.
Intrahippocampal microinjection involved dissolving McAb-proB to a
concentration of 1 µg/µl, followed by stereotactic
delivery of 1 µg into the hippocampus. Both administration
routes of McAb-proB effectively restored neuronal counts and Nissl
body density, enhanced the expression of glutamate receptor 4,
N-methyl-D-aspartate receptor subunits NR1, NR2A, NR2B, and
postsynaptic density protein PSD95, thereby alleviating cognitive
dysfunction associated with SAE. Furthermore, the researchers noted
that as a macromolecule, McAb-proB typically exhibited limited
permeability across the blood. However, in the SAE model,
inflammatory damage may compromise BBB integrity, enabling partial
antibody penetration into the brain. Future studies should focus on
optimizing McAb-proB BBB penetration, such as through the use of Fc
fragment-based BBB transport vectors or the development of small
molecule modulators targeting p75NTR (185). Interestingly, in a separate
study, pretreatment via i.p. injection of 100 µg McAb-proB
was found to improve cognitive dysfunction and modulate the
distribution of immune cells in both peripheral tissues and the
meninges. By contrast, intraventricular administration of 1
µg McAb-proB did not result in any significant improvement
in cognitive function, thereby reinforcing the notion that the
peripheral effects of McAb-proB are more critical. Specifically,
i.p. injection of McAb-proB significantly ameliorated CIs and
restored the proportion of CD4+T cells in the meninges.
Conversely, direct central administration exhibited limited
efficacy, further corroborating its relatively weak capacity to
penetrate the BBB (87).
Thus, McAb-proB may be a safe and promising
candidate drug for addressing IMID, inflammatory pain, organ damage
and CI. Nevertheless, it is crucial to highlight that varying
injection sites in mouse models yield distinct effects. Given that
the action of McAb-proB is more likely to manifest within the
peripheral immune system, it remains uncertain whether it can
effectively penetrate the BBB to target the CNS and exert its
therapeutic benefits. The development of more efficient delivery
technologies is, therefore, warranted. Additionally, further
investigation is required to elucidate the detailed pharmacokinetic
parameters (for example half-life, bioavailability, clearance rate)
associated with the efficacy and safety of McAb-proB in treating
relevant diseases. Moreover, the underlying mechanisms necessitate
further exploration.
Conclusion
proBDNF, the precursor protein of BDNF, is widely
distributed in human tissues and cells. It binds to receptors such
as p75NTR, sortilin, or SorCS2 to regulate synaptic activity,
pruning, and network reorganization. The binding of proBDNF to
p75NTR can promote apoptosis, enhance hippocampal LTD, weaken
synaptic transmission, reduce neuronal plasticity, and play a
crucial role in the development and progression of depression,
cognitive dysfunction, neurogenesis, ischemia-reperfusion injury,
and inflammatory diseases. However, proBDNF also has a positive
effect on accelerating the removal of fear memories and reducing
PTSD, as well as improving cognitive flexibility. Additionally,
proBDNF acts as a biomarker, offering a reference for the early
diagnosis and prognosis of diseases. McAb-proB, a monoclonal
antibody targeting proBDNF, effectively blocks the proBDNF
signaling pathway and specifically recognizes proBDNF without
cross-reactivity to mBDNF (186). It has demonstrated substantial
therapeutic potential in various IMIDs and tissue/organ injuries,
particularly in septic-associated encephalopathy (87), inflammatory pain (181), MS (53), SLE (122) and aortic dissection (180); through the neutralization of
proBDNF activity, McAb-proB significantly mitigates disease
progression, restores immune homeostasis, and suppresses
inflammatory responses.
While previous reviews have primarily addressed the
potential mechanisms of proBDNF and its influence on the activity
of mature neurotrophic factors (1,11), the present review uniquely
focuses the relationship between proBDNF and CI, thereby offering a
more in-depth perspective on this topic. Additionally, an overview
of recent advancements in research on proBDNF in relation to
sepsis, tumors, neurogenesis, and regulation of fear memory, was
provided. The diagnostic potential of proBDNF and the proBDNF/mBDNF
ratio as biomarkers for various diseases was also explored. In
contrast to other reviews (57),
which emphasize the role of proBDNF in metabolic and mitochondrial
regulation, the present review encompasses a wider range of
perspectives. Compared with other reviews (3,4),
our study offers more actionable insights for clinical translation
and application of proBDNF monoclonal antibody.
Despite the confirmed importance of proBDNF in
basic physiology, research results remain conflicting and unclear.
The varying biological effects of proBDNF across different tumor
types are primarily attributed to the expression levels of its
receptors (p75NTR and sortilin), the characteristics of the tumor
microenvironment, and the heterogeneity of downstream signaling
pathways. Future studies should prioritize investigating the
binding affinity and signal transduction mechanisms of proBDNF with
its receptors in a range of tumor contexts, as well as elucidating
how the tumor microenvironment modulates the proBDNF signaling
pathway. Cognitive disorder diseases are the result of multiple
factors and are among the most critical global health issues.
Understanding the mechanism of proBDNF in cognitive disorders
remains a research priority for the next stage. Building on the
significant breakthroughs achieved in the treatment of IMIDs with
McAb-proB, future research will continue to explore its therapeutic
targets in cognitive disorder-related diseases and advance clinical
preliminary trials to achieve the translation of clinical research
findings.
Availability of data and materials
Not applicable.
Authors' contributions
WeiC and RC conceived the topic of study. RC and
WeiC performed the literature search and data analysis and drafted
the manuscript. DC, PL, YZ, QZ and WenC critically revised the
manuscript. Data authentication is not applicable. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Health System Scientific
Research Project of Pingshan (Shenzhen; grant no. 2024340).
References
|
1
|
Rafieva LM and Gasanov EV: Neurotrophin
propeptides: Biological functions and molecular mechanisms. Curr
Protein Pept Sci. 17:298–305. 2016. View Article : Google Scholar
|
|
2
|
Arévalo JC and Deogracias R: Mechanisms
controlling the expression and secretion of BDNF. Biomolecules.
13:7892023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang M, Xie Y and Qin D: Proteolytic
cleavage of proBDNF to mBDNF in neuropsychiatric and
neurodegenerative diseases. Brain Res Bull. 166:172–184. 2021.
View Article : Google Scholar
|
|
4
|
Yang J, Harte-Hargrove LC, Siao CJ,
Marinic T, Clarke R, Ma Q, Jing D, Lafrancois JJ, Bath KG, Mark W,
et al: proBDNF negatively regulates neuronal remodeling, synaptic
transmission, and synaptic plasticity in hippocampus. Cell Rep.
7:796–806. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Edman S, Horwath O, Van der Stede T,
Blackwood SJ, Moberg I, Stromlind H, Nordström F, Ekblom M, Katz A,
Apró W and Moberg M: Pro-brain-derived neurotrophic factor (BDNF),
but not mature BDNF, is expressed in human skeletal muscle:
Implications for exercise-induced neuroplasticity. Function (Oxf).
5:zqae52024.
|
|
6
|
Gibon J and Barker PA: Neurotrophins and
proneurotrophins: Focus on synaptic activity and plasticity in the
brain. Neuroscientist. 23:587–604. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eggert S, Kins S, Endres K and Brigadski
T: Brothers in arms: proBDNF/BDNF and sAPPα/Aβ-signaling and their
common interplay with ADAM10, TrkB, p75NTR, sortilin, and sorLA in
the progression of Alzheimer's disease. Biol Chem. 403:43–71. 2022.
View Article : Google Scholar
|
|
8
|
Hempstead BL: Deciphering proneurotrophin
actions. Handb Exp Pharmacol. 220:17–32. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Teng HK, Teng KK, Lee R, Wright S, Tevar
S, Almeida RD, Kermani P, Torkin R, Chen ZY, Lee FS, et al: ProBDNF
induces neuronal apoptosis via activation of a receptor complex of
p75NTR and sortilin. J Neurosci. 25:5455–5463. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Anastasia A, Deinhardt K, Chao MV, Will
NE, Irmady K, Lee FS, Hempstead BL and Bracken C: Val66Met
polymorphism of BDNF alters prodomain structure to induce neuronal
growth cone retraction. Nat Commun. 4:24902013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Je HS, Yang F, Ji Y, Nagappan G, Hempstead
BL and Lu B: Role of pro-brain-derived neurotrophic factor
(proBDNF) to mature BDNF conversion in activity-dependent
competition at developing neuromuscular synapses. Proc Natl Acad
Sci USA. 109:15924–15929. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Carlino D, De Vanna M and Tongiorgi E: Is
altered BDNF biosynthesis a general feature in patients with
cognitive dysfunctions? Neuroscientist. 19:345–353. 2013.
View Article : Google Scholar
|
|
13
|
Buhusi M, Etheredge C, Granholm AC and
Buhusi CV: Increased hippocampal ProBDNF contributes to memory
impairments in aged mice. Front Aging Neurosci. 9:2842017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kailainathan S, Piers TM, Yi JH, Choi S,
Fahey MS, Borger E, Gunn-Moore FJ, O'Neill L, Lever M, Whitcomb DJ,
et al: Activation of a synapse weakening pathway by human Val66 but
not Met66 pro-brain-derived neurotrophic factor (proBDNF).
Pharmacol Res. 104:97–107. 2016. View Article : Google Scholar :
|
|
15
|
Pérez Palmer N, Trejo Ortega B and Joshi
P: Cognitive impairment in older adults: Epidemiology, diagnosis,
and treatment. Psychiatr Clin North Am. 45:639–661. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ampil ER, Ong PA, Krespi Y and Yang YH: A
review of SaiLuoTong (MLC-SLT) development in vascular cognitive
impairment and dementia. Front Pharmacol. 15:13438202024.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jia L, Du Y, Chu L, Zhang Z, Li F, Lyu D,
Li Y, Li Y, Zhu M, Jiao H, et al: Prevalence, risk factors, and
management of dementia and mild cognitive impairment in adults aged
60 years or older in China: A cross-sectional study. Lancet Public
Health. 5:e661–e671. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aderinto N, Olatunji G, Abdulbasit M,
Ashinze P, Faturoti O, Ajagbe A, Ukoaka B and Aboderin G: The
impact of diabetes in cognitive impairment: A review of current
evidence and prospects for future investigations. Medicine
(Baltimore). 102:e355572023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang J, Qin TS, Bo Y, Li YJ, Liu RS, Yu
Y, Li XD, He JC, Ma AX, Tao DP, et al: The role of the intestinal
flora and its derivatives in neurocognitive disorders: A narrative
review from surgical perspective. Mol Neurobiol. 62:1404–1414.
2025. View Article : Google Scholar :
|
|
20
|
Benedict RHB, Amato MP, Deluca J and
Geurts JJG: Cognitive impairment in multiple sclerosis: Clinical
management, MRI, and therapeutic avenues. Lancet Neurol.
19:860–871. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Haroon NN, Austin PC, Shah BR, Wu J, Gill
SS and Booth GL: Risk of dementia in seniors with newly diagnosed
diabetes: A population-based study. Diabetes Care. 38:1868–1875.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ruegsegger GN, Vanderboom PM, Dasari S,
Klaus KA, Kabiraj P, McCarthy CB, Lucchinetti CF and Nair KS:
Exercise and metformin counteract altered mitochondrial function in
the insulin-resistant brain. JCI Insight. 4:e1306812019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kullmann S, Goj T, Veit R, Fritsche L,
Wagner L, Schneeweiss P, Hoene M, Hoffmann C, Machann J, Niess A,
et al: Exercise restores brain insulin sensitivity in sedentary
adults who are overweight and obese. JCI Insight. 7:e1614982022.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim B and Feldman EL: Insulin resistance
in the nervous system. Trends Endocrinol Metab. 23:133–141. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Delgado-Peraza F, Nogueras-Ortiz C,
Simonsen AH, Knight DD, Yao PJ, Goetzl EJ, Jensen CS, Høgh P,
Gottrup H, Vestergaard K, et al: Neuron-derived extracellular
vesicles in blood reveal effects of exercise in Alzheimer's
disease. Alzheimers Res Ther. 15:1562023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dahiya M, Yadav M, Goyal C and Kumar A:
Insulin resistance in Alzheimer's disease: Signalling mechanisms
and therapeutics strategies. Inflammopharmacology. 33:1817–1831.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Malin SK, Battillo DJ, Beeri MS, Mustapic
M, Delgado-Peraza F and Kapogiannis D: Two weeks of exercise alters
neuronal extracellular vesicle insulin signaling proteins and
pro-BDNF in older adults with prediabetes. Aging Cell.
24:e143692025. View Article : Google Scholar
|
|
28
|
Da Mesquita S, Papadopoulos Z, Dykstra T,
Brase L, Farias FG, Wall M, Jiang H, Kodira CD, de Lima KA, Herz J,
et al: Meningeal lymphatics affect microglia responses and anti-Aβ
immunotherapy. Nature. 593:255–260. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li P, Chen J, Wang X, Su Z, Gao M and
Huang Y: Liquid-liquid phase separation of tau: Driving forces,
regulation, and biological implications. Neurobiol Dis.
183:1061672023. View Article : Google Scholar
|
|
30
|
Wang Y, Cella M, Mallinson K, Ulrich JD,
Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S,
Zinselmeyer BH, et al: TREM2 lipid sensing sustains the microglial
response in an Alzheimer's disease model. Cell. 160:1061–1071.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kellar D and Craft S: Brain insulin
resistance in Alzheimer's disease and related disorders: mechanisms
and therapeutic approaches. Lancet Neurol. 19:758–766. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chandra S, Sisodia SS and Vassar RJ: The
gut microbiome in Alzheimer's disease: What we know and what
remains to be explored. Mol Neurodegener. 18:92023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sienski G, Narayan P, Bonner JM, Kory N,
Boland S, Arczewska AA, Ralvenius WT, Akay L, Lockshin E, He L, et
al: APOE4 disrupts intracellular lipid homeostasis in human
iPSC-derived glia. Sci Transl Med. 13:eaaz45642021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bellenguez C, Küçükali F, Jansen IE,
Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R,
Grenier-Boley B, Andrade V, et al: New insights into the genetic
etiology of Alzheimer's disease and related dementias. Nat Genet.
54:412–436. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu H, Dunnett S, Ho YS and Chang RCC: The
role of sleep deprivation and circadian rhythm disruption as risk
factors of Alzheimer's disease. Front Neuroendocrinol.
54:1007642019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hussain R, Graham U, Elder A and
Nedergaard M: Air pollution, glymphatic impairment, and Alzheimer's
disease. Trends Neurosci. 46:901–911. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Myhre O, Utkilen H, Duale N, Brunborg G
and Hofer T: Metal dyshomeostasis and inflammation in Alzheimer's
and Parkinson's diseases: Possible impact of environmental
exposures. Oxid Med Cell Longev. 2013:7269542013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang J, Chen Y, Zhang C, Xiang Z, Ding J
and Han X: Learning and memory deficits and alzheimer's
disease-like changes in mice after chronic exposure to
microcystin-LR. J Hazard Mater. 373:504–518. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Goldman SM: Environmental toxins and
Parkinson's disease. Annu Rev Pharmacol Toxicol. 54:141–164. 2014.
View Article : Google Scholar
|
|
40
|
Vasefi M, Ghaboolian-Zare E, Abedelwahab H
and Osu A: Environmental toxins and Alzheimer's disease
progression. Neurochem Int. 141:1048522020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bovis K, Davies-Branch M and Day PJR:
Dysregulated neurotransmission and the role of viruses in
Alzheimer's disease. ACS Chem Neurosci. 16:982–987. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dai L, Zou L, Meng L, Qiang G, Yan M and
Zhang Z: Cholesterol metabolism in neurodegenerative diseases:
Molecular mechanisms and therapeutic targets. Mol Neurobiol.
58:2183–2201. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Y, Zhang J, Wan J, Liu A and Sun J:
Melatonin regulates Aβ production/clearance balance and Aβ
neurotoxicity: A potential therapeutic molecule for Alzheimer's
disease. Biomed Pharmacother. 132:1108872020. View Article : Google Scholar
|
|
44
|
Manucat-Tan NB, Shen LL, Bobrovskaya L,
Al-Hawwas M, Zhou FH, Wang YJ and Zhou XF: Knockout of p75
neurotrophin receptor attenuates the hyperphosphorylation of Tau in
pR5 mouse model. Aging (Albany NY). 11:6762–6791. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bie B, Wu J, Lin F, Naguib M and Xu J:
Suppression of hippocampal GABAergic transmission impairs memory in
rodent models of Alzheimer's disease. Eur J Pharmacol.
917:1747712022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Aarsland D, Batzu L, Halliday GM, Geurtsen
GJ, Ballard C, Ray Chaudhuri K and Weintraub D: Parkinson
disease-associated cognitive impairment. Nat Rev Dis Primers.
7:472021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jellinger KA: Morphological basis of
Parkinson disease-associated cognitive impairment: An update. J
Neural Transm (Vienna). 129:977–999. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Goldman JG and Sieg E: Cognitive
impairment and dementia in Parkinson disease. Clin Geriatr Med.
36:365–377. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Baiano C, Barone P, Trojano L and
Santangelo G: Prevalence and clinical aspects of mild cognitive
impairment in Parkinson's disease: A meta-analysis. Mov Disord.
35:45–54. 2020. View Article : Google Scholar
|
|
50
|
Ali NH, Al-Kuraishy HM, Al-Gareeb AI,
Alnaaim SA, Saad HM and Batiha GES: The molecular pathway of p75
neurotrophin receptor (p75NTR) in Parkinson's disease: The way of
new inroads. Mol Neurobiol. 61:2469–2480. 2024. View Article : Google Scholar
|
|
51
|
Kazzi C, Alpitsis R, O'Brien TJ, Malpas CB
and Monif M: Cognitive and psychopathological features of
neuromyelitis optica spectrum disorder and myelin oligodendrocyte
glycoprotein antibody-associated disease: A narrative review. Mult
Scler Relat Disord. 85:1055962024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jellinger KA: Cognitive impairment in
multiple sclerosis: From phenomenology to neurobiological
mechanisms. J Neural Transm (Vienna). 131:871–899. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hu ZL, Luo C, Hurtado PR, Li H, Wang S, Hu
B, Xu JM, Liu Y, Feng SQ, Hurtado-Perez E, et al: Brain-derived
neurotrophic factor precursor in the immune system is a novel
target for treating multiple sclerosis. Theranostics. 11:715–730.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Al-Yozbaki M, Acha-Sagredo A, George A,
Liloglou T and Wilson CM: Balancing neurotrophin pathway and
sortilin function: Its role in human disease. Biochim Biophys Acta
Rev Cancer. 1874:1884292020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Vilar M: Structural characterization of
the p75 neurotrophin receptor: A stranger in the TNFR superfamily.
Vitam Horm. 104:57–87. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen C, Li J, Matye DJ, Wang Y and Li T:
Hepatocyte sortilin 1 knockout and treatment with a sortilin 1
inhibitor reduced plasma cholesterol in Western diet-fed mice. J
Lipid Res. 60:539–549. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li Q, Hu YZ, Gao S, Wang PF, Hu ZL and Dai
RP: ProBDNF and its receptors in immune-mediated inflammatory
diseases: Novel insights into the regulation of metabolism and
mitochondria. Front Immunol. 14:11553332023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Al-Kuraishy HM, Sulaiman GM, Mohammed HA,
Albukhaty S, Albuhadily AK, Al-Gareeb AI, Klionsky DJ and
Abomughaid MM: The compelling role of brain-derived neurotrophic
factor signaling in multiple sclerosis: Role of BDNF activators.
CNS Neurosci Ther. 30:e701672024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Reuter E, Weber J, Paterka M, Ploen R,
Breiderhoff T, van Horssen J, Willnow TE, Siffrin V and Zipp F:
Role of sortilin in models of autoimmune neuroinflammation. J
Immunol. 195:5762–5769. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sarno E, Moeser AJ and Robison AJ:
Neuroimmunology of depression. Adv Pharmacol. 91:259–292. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Price RB and Duman R: Neuroplasticity in
cognitive and psychological mechanisms of depression: An
integrative model. Mol Psychiatry. 25:530–543. 2020. View Article : Google Scholar :
|
|
62
|
Pan Z, Park C, Brietzke E, Zuckerman H,
Rong C, Mansur RB, Fus D, Subramaniapillai M, Lee Y and Mcintyre
RS: Cognitive impairment in major depressive disorder. CNS Spectr.
24:22–29. 2019. View Article : Google Scholar
|
|
63
|
Zhao XP, Li H and Dai RP: Neuroimmune
crosstalk through brain-derived neurotrophic factor and its
precursor pro-BDNF: New insights into mood disorders. World J
Psychiatry. 12:379–392. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lin LY, Kelliny S, Liu LC, Al-Hawwas M,
Zhou XF and Bobrovskaya L: Peripheral ProBDNF delivered by an AAV
vector to the muscle triggers depression-like behaviours in mice.
Neurotox Res. 38:626–639. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yang CR, Liang R, Liu Y, Meng FJ, Zhou F,
Zhang XY, Ning L, Wang ZQ, Liu S and Zhou XF: Upregulation of
proBDNF/p75NTR signaling in immune cells and its correlation with
inflammatory markers in patients with major depression. FASEB J.
38:e233122024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Li Y, Guan X, He Y, Jia X, Pan L, Wang Y,
Han Y, Zhao R, Yang J and Hou T: ProBDNF signaling is involved in
periodontitis-induced depression-like behavior in mouse
hippocampus. Int Immunopharmacol. 116:1097672023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Li Y, Yang Y, Guan X, Liu Z, Pan L, Wang
Y, Jia X, Yang J and Hou T: SorCS2 is involved in promoting
periodontitis-induced depression-like behaviour in mice. Oral Dis.
30:5408–5420. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Jin HJ, Pei L, Li YN, Zheng H, Yang S, Wan
Y, Mao L, Xia YP, He QW, Li M, et al: Alleviative effects of
fluoxetine on depressive-like behaviors by epigenetic regulation of
BDNF gene transcription in mouse model of post-stroke depression.
Sci Rep. 7:149262017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Yang B, Wang L, Nie Y, Wei W and Xiong W:
proBDNF expression induces apoptosis and inhibits synaptic
regeneration by regulating the RhoA-JNK pathway in an in vitro
post-stroke depression model. Transl Psychiatry. 11:5782021.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Huang S, Nie Y, Qin J, Wen M, Wang Q, Xie
F, Song F and Yang B: Hippocampal exosomes from stroke aggravate
post-stroke depression by regulating the expression of proBDNF and
p75NTR and altering spine density. Sci Rep. 14:282232024.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sanmarti M, Ibáñez L, Huertas S, Badenes
D, Dalmau D, Slevin M, Krupinski J, Popa-Wagner A and Jaen A:
Hiv-associated neurocognitive disorders. J Mol Psychiatry. 2:22014.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Saylor D, Dickens AM, Sacktor N, Haughey
N, Slusher B, Pletnikov M, Mankowski JL, Brown A, Volsky DJ and
Mcarthur JC: Hiv-associated neurocognitive disorder-pathogenesis
and prospects for treatment. Nat Rev Neurol. 12:234–248. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Antinori A, Arendt G, Becker JT, Brew BJ,
Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K,
et al: Updated research nosology for HIV-associated neurocognitive
disorders. Neurology. 69:1789–1799. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kaul M, Zheng J, Okamoto S, Gendelman HE
and Lipton SA: HIV-1 infection and AIDS: Consequences for the
central nervous system. Cell Death Differ. 12(Suppl 1): S878–S892.
2005. View Article : Google Scholar
|
|
75
|
Eggers C, Arendt G, Hahn K, Husstedt IW,
Maschke M, Neuen-Jacob E, Obermann M, Rosenkranz T, Schielke E and
Straube E; German Association of Neuro-AIDS und Neuro-Infectiology
(DGNANI): HIV-1-associated neurocognitive disorder: Epidemiology,
pathogenesis, diagnosis, and treatment. J Neurol. 264:1715–1727.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Avedissian SN, Dyavar SR, Fox HS and
Fletcher CV: Pharmacologic approaches to HIV-associated
neurocognitive disorders. Curr Opin Pharmacol. 54:102–108. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Erdos T, Masuda M and Venketaraman V:
Glutathione in HIV-associated neurocognitive disorders. Curr Issues
Mol Biol. 46:5530–5549. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Riviere-Cazaux C, Cornell J, Shen Y and
Zhou M: The role of CCR5 in HIV-associated neurocognitive
disorders. Heliyon. 8:e99502022. View Article : Google Scholar
|
|
79
|
Hou C, Wei J, Zhang H and Li H: Evolving
strategies in the diagnosis and treatment of HIV-associated
neurocognitive disorders. Rev Neurosci. Mar 6–2025.Epub ahead of
print. View Article : Google Scholar
|
|
80
|
Johnson TP and Nath A: Biotypes of
HIV-associated neurocognitive disorders based on viral and immune
pathogenesis. Curr Opin Infect Dis. 35:223–230. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Speidell A, Asuni GP, Wakulski R and
Mocchetti I: Up-regulation of the p75 neurotrophin receptor is an
essential mechanism for HIV-gp120 mediated synaptic loss in the
striatum. Brain Behav Immun. 89:371–379. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Allen CNS, Arjona SP, Santerre M, De Lucia
C, Koch WJ and Sawaya BE: Metabolic reprogramming in HIV-associated
neurocognitive disorders. Front Cell Neurosci. 16:8128872022.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Michael HU, Rapulana AM, Smit T, Xulu N,
Danaviah S, Ramlall S and Oosthuizen F: Serum mature and precursor
brain-derived neurotrophic factors and their association with
neurocognitive function in ART-Naïve adults living with HIV in
Sub-Saharan Africa. Mol Neurobiol. 62:5442–5451. 2025. View Article : Google Scholar
|
|
84
|
Mizoguchi Y, Ohgidani M, Haraguchi Y,
Murakawa-Hirachi T, Kato TA and Monji A: ProBDNF induces sustained
elevation of intracellular Ca2+ possibly mediated by
TRPM7 channels in rodent microglial cells. Glia. 69:1694–1708.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Sonneville R, de Montmollin E, Poujade J,
Garrouste-Orgeas M, Souweine B, Darmon M, Mariotte E, Argaud L,
Barbier F, Goldgran-Toledano D, et al: Potentially modifiable
factors contributing to sepsis-associated encephalopathy. Intensive
Care Med. 43:1075–1084. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Long LH, Cao WY, Xu Y and Xiang YY:
Research progress on the role of microglia in sepsis-associated
encephalopathy. Sheng Li Xue Bao. 76:289–300. 2024.In Chinese.
PubMed/NCBI
|
|
87
|
Luo RY, Luo C, Zhong F, Shen WY, Li H, Hu
ZL and Dai RP: ProBDNF promotes sepsis-associated encephalopathy in
mice by dampening the immune activity of meningeal CD4+
T cells. J Neuroinflammation. 17:1692020. View Article : Google Scholar
|
|
88
|
Huang J, Chen L, Yao ZM, Sun XR, Tong XH
and Dong SY: The role of mitochondrial dynamics in cerebral
ischemia-reperfusion injury. Biomed Pharmacother. 162:1146712023.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wu M, Gu X and Ma Z: Mitochondrial quality
control in cerebral ischemia-reperfusion injury. Mol Neurobiol.
58:5253–5271. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kumar SS and Singh D: Mitochondrial
mechanisms in cerebral ischemia-reperfusion injury: Unravelling the
intricacies. Mitochondrion. 77:1018832024. View Article : Google Scholar
|
|
91
|
Fiskum G, Murphy AN and Beal MF:
Mitochondria in neurodegeneration: Acute ischemia and chronic
neurodegenerative diseases. J Cereb Blood Flow Metab. 19:351–369.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Jurcau A and Simion A: Neuroinflammation
in cerebral ischemia and ischemia/reperfusion injuries: From
pathophysiology to therapeutic strategies. Int J Mol Sci.
23:142021. View Article : Google Scholar
|
|
93
|
Yang K, Zeng L, Ge A, Wang S, Zeng J, Yuan
X, Mei Z, Wang G and Ge J: A systematic review of the research
progress of non-coding RNA in neuroinflammation and immune
regulation in cerebral infarction/ischemia-reperfusion injury.
Front Immunol. 13:9301712022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zhang Q, Jia M, Wang Y, Wang Q and Wu J:
Cell death mechanisms in cerebral ischemia-reperfusion injury.
Neurochem Res. 47:3525–3542. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Liu X, Xie C, Wang Y, Xiang J, Chen L,
Yuan J, Chen C and Tian H: Ferritinophagy and ferroptosis in
cerebral ischemia reperfusion injury. Neurochem Res. 49:1965–1979.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Li M, Tang H, Li Z and Tang W: Emerging
treatment strategies for cerebral ischemia-reperfusion injury.
Neuroscience. 507:112–124. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ginsberg MD: Neuroprotection for ischemic
stroke: Past, present and future. Neuropharmacology. 55:363–389.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Chamorro A, Dirnagl U, Urra X and Planas
AM: Neuroprotection in acute stroke: Targeting excitotoxicity,
oxidative and nitrosative stress, and inflammation. Lancet Neurol.
15:869–881. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Xiong XY, Liu L and Yang QW: Functions and
mechanisms of microglia/macrophages in neuroinflammation and
neurogenesis after stroke. Prog Neurobiol. 142:23–44. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zhang M, Liu Q, Meng H, Duan H, Liu X, Wu
J, Gao F, Wang S, Tan R and Yuan J: Ischemia-reperfusion injury:
Molecular mechanisms and therapeutic targets. Signal Transduct
Target Ther. 9:122024. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Liu DQ, Mei W, Zhou YQ and Xi H: Targeting
TRPM channels for cerebral ischemia-reperfusion injury. Trends
Pharmacol Sci. 45:862–867. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Candelario-Jalil E, Dijkhuizen RM and
Magnus T: Neuroinflammation, stroke, blood-brain barrier
dysfunction, and imaging modalities. Stroke. 53:1473–1486. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yang C, Hawkins KE, Doré S and
Candelario-Jalil E: Neuroinflammatory mechanisms of blood-brain
barrier damage in ischemic stroke. Am J Physiol Cell Physiol.
316:C135–C153. 2019. View Article : Google Scholar :
|
|
104
|
Li S, Dou B, Shu S, Wei L, Zhu S, Ke Z and
Wang Z: Suppressing NK Cells by astragaloside IV protects against
acute ischemic stroke in mice via inhibiting STAT3. Front
Pharmacol. 12:8020472022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Cramer T, Gill R, Thirouin ZS, Vaas M,
Sampath S, Martineau F, Noya SB, Panzanelli P, Sudharshan TJJ,
Colameo D, et al: Cross-talk between GABAergic postsynapse and
microglia regulate synapse loss after brain ischemia. Sci Adv.
8:eabj1122022. View Article : Google Scholar
|
|
106
|
Rahman M, Luo H, Bobrovskaya L and Zhou
XF: Neuroprotective effects of anti-probdnf in a RAT
photothrombotic ischemic model. Neuroscience. 446:261–270. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kotekar N, Shenkar A and Nagaraj R:
Postoperative cognitive dysfunction-current preventive strategies.
Clin Interv Aging. 13:2267–2273. 2018. View Article : Google Scholar :
|
|
108
|
Yang X, Huang X, Li M, Jiang Y and Zhang
H: Identification of individuals at risk for postoperative
cognitive dysfunction (POCD). Ther Adv Neurol Disord.
15:175628642211143562022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhong Y, Zhang Y and Zhu Z: Research
progress on the association between MicroRNA and postoperative
cognitive dysfunction. Minerva Anestesiol. 90:191–199. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Luo A, Yan J, Tang X, Zhao Y, Zhou B and
Li S: Postoperative cognitive dysfunction in the aged: The
collision of neuroinflammaging with perioperative
neuroinflammation. Inflammopharmacology. 27:27–37. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Xue Z, Shui M, Lin X, Sun Y, Liu J, Wei C,
Wu A and Li T: Role of BDNF/ProBDNF imbalance in postoperative
cognitive dysfunction by modulating synaptic plasticity in aged
mice. Front Aging Neurosci. 14:7809722022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Jiang L, Dong R, Xu M, Liu Y, Xu J, Ma Z,
Xia T and Gu X: Inhibition of the integrated stress response
reverses oxidative stress damage-induced postoperative cognitive
dysfunction. Front Cell Neurosci. 16:9928692022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Hotchkiss RS, Moldawer LL, Opal SM,
Reinhart K, Turnbull IR and Vincent JL: Sepsis and septic shock.
Nat Rev Dis Primers. 2:160452016. View Article : Google Scholar
|
|
115
|
Fleischmann C, Scherag A, Adhikari NKJ,
Hartog CS, Tsaganos T, Schlattmann P, Angus DC and Reinhart K;
International Forum of Acute Care Trialists: Assessment of global
incidence and mortality of hospital-treated sepsis. Current
estimates and limitations. Am J Respir Crit Care Med. 193:259–272.
2016. View Article : Google Scholar
|
|
116
|
Wang Z, Wu JL, Zhong F, Liu Y, Yu YQ, Sun
JJ, Wang S, Li H, Zhou XF, Hu ZL and Dai RP: Upregulation of
proBDNF in the mesenteric lymph nodes in septic mice. Neurotox Res.
36:540–550. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wang S, Zeng Q, Gao H, Gao S, Dai R and Hu
Z: Expression of proBDNF/p75NTR in peripheral blood
lymphocytes of patients with sepsis and its impact on lymphocyte
differentiation. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 48:1629–1638.
2023.In English, Chinese.
|
|
118
|
Bezzio C, Della CC, Vernero M, Di Luna I,
Manes G and Saibeni S: Inflammatory bowel disease and
immune-mediated inflammatory diseases: Looking at the less frequent
associations. Therap Adv Gastroenterol. 15:175628482211153122022.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Yang CR, Ding HJ, Yu M, Zhou FH, Han CY,
Liang R, Zhang XY, Zhang XL, Meng FJ, Wang S, et al: proBDNF/p75NTR
promotes rheumatoid arthritis and inflammatory response by
activating proinflammatory cytokines. FASEB J.
36:e221802022.PubMed/NCBI
|
|
120
|
Farina L, Minnone G, Alivernini S, Caiello
I, MacDonald L, Soligo M, Manni L, Tolusso B, Coppola S, Zara E, et
al: Pro nerve growth factor and its receptor p75NTR activate
inflammatory responses in synovial fibroblasts: A novel targetable
mechanism in arthritis. Front Immunol. 13:8186302022. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Ameer MA, Chaudhry H, Mushtaq J, Khan OS,
Babar M, Hashim T, Zeb S, Tariq MA, Patlolla SR, Ali J, et al: An
overview of systemic lupus erythematosus (SLE) pathogenesis,
classification, and management. Cureus. 14:e303302022.PubMed/NCBI
|
|
122
|
Shen WY, Luo C, Hurtado PR, Liu XJ, Luo
RY, Li H, Hu ZL, Xu JM, Coulson EJ, Zhao M, et al: Up-regulation of
proBDNF/p75NTR signaling in antibody-secreting cells
drives systemic lupus erythematosus. Sci Adv. 8:eabj27972022.
View Article : Google Scholar
|
|
123
|
Putcha GV, Moulder KL, Golden JP, Bouillet
P, Adams JA, Strasser A and Johnson EM: Induction of BIM, a
proapoptotic BH3-only BCL-2 family member, is critical for neuronal
apoptosis. Neuron. 29:615–628. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Sankorrakul K, Qian L, Thangnipon W and
Coulson EJ: Is there a role for the p75 neurotrophin receptor in
mediating degeneration during oxidative stress and after hypoxia? J
Neurochem. 158:1292–1306. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Taylor KR, Barron T, Hui A, Spitzer A,
Yalçin B, Ivec AE, Geraghty AC, Hartmann GG, Arzt M, Gillespie SM,
et al: Glioma synapses recruit mechanisms of adaptive plasticity.
Nature. 623:366–374. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Jiang CC, Marsland M, Wang Y, Dowdell A,
Eden E, Gao F, Faulkner S, Jobling P, Li X, Liu L, et al: Tumor
innervation is triggered by endoplasmic reticulum stress. Oncogene.
41:586–599. 2022. View Article : Google Scholar
|
|
127
|
Xiong J, Zhou L, Yang M, Lim Y, Zhu YH, Fu
DL, Li ZW, Zhong JH, Xiao ZC and Zhou XF: ProBDNF and its receptors
are upregulated in glioma and inhibit the growth of glioma cells in
vitro. Neuro Oncol. 15:990–1007. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Fan YJ, Wu LLY, Li HY, Wang YJ and Zhou
XF: Differential effects of pro-BDNF on sensory neurons after
sciatic nerve transection in neonatal rats. Eur J Neurosci.
27:2380–2390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Koshimizu H, Hazama S, Hara T, Ogura A and
Kojima M: Distinct signaling pathways of precursor BDNF and mature
BDNF in cultured cerebellar granule neurons. Neurosci Lett.
473:229–232. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Salmaggi A, Croci D, Prina P, Cajola L,
Pollo B, Marras CE, Ciusani E, Silvani A, Boiardi A and Sciacca FL:
Production and post-surgical modification of VEGF, tPA and PAI-1 in
patients with glioma. Cancer Biol Ther. 5:204–209. 2006. View Article : Google Scholar
|
|
131
|
Nakabayashi H, Yawata T and Shimizu K:
Anti-invasive and antiangiogenic effects of MMI-166 on malignant
glioma cells. BMC Cancer. 10:3392010. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Levicar N, Nuttall RK and Lah TT:
Proteases in brain tumour progression. Acta Neurochir (Wien).
145:825–838. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
De la Cruz-Morcillo MA, Berger J,
Sánchez-Prieto R, Saada S, Naves T, Guillaudeau A, Perraud A,
Sindou P, Lacroix A, Descazeaud A, et al: p75 neurotrophin receptor
and pro-BDNF promote cell survival and migration in clear cell
renal cell carcinoma. Oncotarget. 7:34480–34497. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Corti C, Antonarelli G, Criscitiello C,
Lin NU, Carey LA, Cortés J, Poortmans P and Curigliano G: Targeting
brain metastases in breast cancer. Cancer Treat Rev.
103:1023242022. View Article : Google Scholar
|
|
135
|
Fidler IJ: The biology of brain
metastasis: Challenges for therapy. Cancer J. 21:284–293. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Alhusban L, Ayoub NM and Alhusban A:
ProBDNF is a novel mediator of the interaction between MDA-MB-231
breast cancer cells and brain microvascular endothelial cells. Curr
Mol Med. 21:914–921. 2021. View Article : Google Scholar
|
|
137
|
Klann IP, Fulco BCW and Nogueira CW:
Subchronic exposure to Tamoxifen modulates the hippocampal
BDNF/ERK/Akt/CREB pathway and impairs memory in intact female rats.
Chem Biol Interact. 382:1106152023. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Tajbakhsh A, Mokhtari-Zaer A, Rezaee M,
Afzaljavan F, Rivandi M, Hassanian SM, Ferns GA, Pasdar A and Avan
A: Therapeutic potentials of BDNF/TrkB in breast cancer; current
status and perspectives. J Cell Biochem. 118:2502–2515. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Lu Q, Qu Y, Ding Y and Kang X:
p75NTR/proBDNF modulates basal cell carcinoma (BCC) immune
microenvironment via necroptosis signaling pathway. J Immunol Res.
2021:66528462021. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Culig L, Chu X and Bohr VA: Neurogenesis
in aging and age-related neurodegenerative diseases. Ageing Res
Rev. 78:1016362022. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Bhalla M and Lee CJ: Long-term inhibition
of ODC1 in APP/PS1 mice rescues amyloid pathology and switches
astrocytes from a reactive to active state. Mol Brain. 17:32024.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Sunderland S: A classification of
peripheral nerve injuries producing loss of function. Brain.
74:491–516. 1951. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Kamble N, Shukla D and Bhat D: Peripheral
nerve injuries: Electrophysiology for the neurosurgeon. Neurol
India. 67:1419–1422. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Gordon T: Peripheral nerve regeneration
and muscle reinnervation. Int J Mol Sci. 21:86522020. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Rotshenker S: Wallerian degeneration: The
innate-immune response to traumatic nerve injury. J
Neuroinflammation. 8:1092011. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Defrancesco-Lisowitz A, Lindborg JA, Niemi
JP and Zigmond RE: The neuroimmunology of degeneration and
regeneration in the peripheral nervous system. Neuroscience.
302:174–203. 2015. View Article : Google Scholar
|
|
147
|
Cámara-Lemarroy CR, Guzmán-de la Garza FJ
and Fernández-Garza NE: Molecular inflammatory mediators in
peripheral nerve degeneration and regeneration.
Neuroimmunomodulation. 17:314–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Huang EJ and Reichardt LF: Neurotrophins:
Roles in neuronal development and function. Annu Rev Neurosci.
24:677–736. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Jessen KR and Mirsky R: The repair schwann
cell and its function in regenerating nerves. J Physiol.
594:3521–3531. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Wang X, Ma W, Wang T, Yang J, Wu Z, Liu K,
Dai Y, Zang C, Liu W, Liu J, et al: BDNF-TrkB and
proBDNF-p75NTR/Sortilin signaling pathways are involved in
mitochondria-mediated neuronal apoptosis in dorsal root ganglia
after sciatic nerve transection. CNS Neurol Disord Drug Targets.
19:66–82. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Ma W, Yang JW, Wang XB, Luo T, Zhou L,
Lagares A, Li H, Liang Z, Liu KP, Zang CH, et al: Negative
regulation by proBDNF signaling of peripheral neurogenesis in the
sensory ganglia of adult rats. Biomed Pharmacother. 144:1122732021.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Fronza MG, Ferreira BF, Pavan-Silva I,
Guimarães FS and Lisboa SF: 'NO' time in fear response: Possible
implication of nitric-oxide-related mechanisms in PTSD. Molecules.
29:892023. View Article : Google Scholar
|
|
153
|
He M, Wei JX, Mao M, Zhao GY, Tang JJ,
Feng S, Lu XM and Wang YT: Synaptic plasticity in PTSD and
associated comorbidities: The function and mechanism for
diagnostics and therapy. Curr Pharm Des. 24:4051–4059. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Andero R and Ressler KJ: Fear extinction
and BDNF: Translating animal models of PTSD to the clinic. Genes
Brain Behav. 11:503–512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Bennett MR, Hatton SN and Lagopoulos J:
Stress, trauma and PTSD: Translational insights into the core
synaptic circuitry and its modulation. Brain Struct Funct.
221:2401–2426. 2016. View Article : Google Scholar
|
|
156
|
Sagarwala R and Nasrallah HA: Changes in
inflammatory biomarkers before and after SSRI therapy in PTSD: A
review. Ann Clin Psychiatry. 31:292–297. 2019.PubMed/NCBI
|
|
157
|
Meis S, Endres T and Lessmann V:
Neurotrophin signalling in amygdala-dependent cued fear learning.
Cell Tissue Res. 382:161–172. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Beckers T and Kindt M: Memory
reconsolidation interference as an emerging treatment for emotional
disorders: Strengths, limitations, challenges, and opportunities.
Annu Rev Clin Psychol. 13:99–121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Sun W, Chen X, Mei Y, Yang Y, Li X and An
L: Prelimbic proBDNF facilitates retrieval-dependent fear memory
destabilization by regulation of synaptic and neural functions in
juvenile rats. Mol Neurobiol. 59:4179–4196. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Ma X, Vuyyuru H, Munsch T, Endres T,
Lessmann V and Meis S: ProBDNF dependence of LTD and fear
extinction learning in the amygdala of adult mice. Cereb Cortex.
32:1350–1364. 2022. View Article : Google Scholar
|
|
161
|
Machado BSJ, Cardoso NC, Raymundi AM,
Prickaerts J and Stern CAJ: Phosphodiesterase 4 inhibition after
retrieval switches the memory fate favoring extinction instead of
reconsolidation. Sci Rep. 13:203842023. View Article : Google Scholar
|
|
162
|
van der Merwe RK, Nadel JA, Copes-Finke D,
Pawelko S, Scott JS, Ghanem M, Fox M, Morehouse C, Mclaughlin R,
Maddox C, et al: Characterization of striatal dopamine projections
across striatal subregions in behavioral flexibility. Eur J
Neurosci. 58:4466–4486. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Sun W, Che H, Li J, Tang D, Liu X, Liu W
and An L: Dorsolateral striatal proBDNF improves reversal learning
by enhancing coordination of neural activity in rats. Mol
Neurobiol. 57:4642–4656. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Zheng W: Implicit motor learning in
children with autism spectrum disorder: Current approaches and
future directions. Front Psychiatry. 15:12531992024. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Hughes HK, Moreno RJ and Ashwood P: Innate
immune dysfunction and neuroinflammation in autism spectrum
disorder (ASD). Brain Behav Immun. 108:245–254. 2023. View Article : Google Scholar
|
|
166
|
Robinson-Agramonte MDLA, Michalski B,
Vidal-Martinez B, Hernández LR, Santiesteban MW and Fahnestock M:
BDNF, proBDNF and IGF-1 serum levels in naïve and medicated
subjects with autism. Sci Rep. 12:137682022. View Article : Google Scholar
|
|
167
|
Armstrong MJ and Okun MS: Diagnosis and
treatment of parkinson disease: A review. JAMA. 323:548–560. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Yi X, Yang Y, Zhao Z, Xu M, Zhang Y, Sheng
Y, Tian J and Xu Z: Serum mBDNF and ProBDNF expression levels as
diagnosis clue for early stage Parkinson's disease. Front Neurol.
12:6807652021. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Li Y, Chen J, Yu H, Ye J, Wang C and Kong
L: Serum brain-derived neurotrophic factor as diagnosis clue for
Alzheimer's disease: A cross-sectional observational study in the
elderly. Front Psychiatry. 14:11276582023. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Campisi J, Kapahi P, Lithgow GJ, Melov S,
Newman JC and Verdin E: From discoveries in ageing research to
therapeutics for healthy ageing. Nature. 571:183–192. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Norgren J, Daniilidou M, Kåreholt I, Sindi
S, Akenine U, Nordin K, Rosenborg S, Ngandu T, Kivipelto M and
Sandebring-Matton A: Serum proBDNF is associated with changes in
the ketone body β-hydroxybutyrate and shows superior repeatability
over mature BDNF: Secondary outcomes from a cross-over trial in
healthy older adults. Front Aging Neurosci. 13:7165942021.
View Article : Google Scholar
|
|
172
|
Yu WR, Jiang YH, Jhang JF and Kuo HC:
Urine biomarker could be a useful tool for differential diagnosis
of a lower urinary tract dysfunction. Tzu Chi Med J. 36:110–119.
2023.
|
|
173
|
Nitti VW, Patel A and Karram M: Diagnosis
and management of overactive bladder: A review. J Obstet Gynaecol
Res. 47:1654–1665. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Malde S, Kelly S, Saad S and Sahai A:
Case-finding tools for the diagnosis of OAB in women: A narrative
review. Neurourol Urodyn. 39:13–24. 2020. View Article : Google Scholar
|
|
175
|
Covarrubias C, Cammisotto PG, Shamout S
and Campeau L: Decrease in the ratio proBDNF/BDNF in the urine of
aging female patients with OAB. Metabolites. 13:7232023. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Gelle T, Sa mey R A, Pla nsont B, Besset
te B, Jauberteau-Marchan MO, Lalloué F and Girard M: BDNF and
pro-BDNF in serum and exosomes in major depression: Evolution after
antidepressant treatment. Prog Neuropsychopharmacol Biol
Psychiatry. 109:1102292021. View Article : Google Scholar
|
|
177
|
Yang Z, Gao C, Li Z, Jiang T, Liang Y,
Jiang T, Yu C, Yan S, Li P and Zhou L: The changes of tPA/PAI-1
system are associated with the ratio of BDNF/proBDNF in major
depressive disorder and SSRIs antidepressant treatment.
Neuroscience. 559:220–228. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Lin L, Fu XY, Zhou XF, Liu D, Bobrovskaya
L and Zhou L: Analysis of blood mature BDNF and proBDNF in mood
disorders with specific ELISA assays. J Psychiatr Res. 133:166–173.
2021. View Article : Google Scholar
|
|
179
|
Zwolińska W, Skibinska M, Słopień A and
Dmitrzak-Weglarz M: ProBDNF as an indicator of improvement among
women with depressive episodes. Metabolites. 12:3582022. View Article : Google Scholar
|
|
180
|
Shen WY, Luo C, Reinaldo HP, Hurtado-Perez
E, Luo RY, Hu ZL, Li H, Xu JM, Zhou XF and Dai RP: The regulatory
role of ProBDNF in monocyte function: Implications in Stanford
type-A aortic dissection disease. FASEB J. 34:2541–2553. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Luo C, Zhong XL, Zhou FH, Li JY, Zhou P,
Xu JM, Song B, Li CQ, Zhou XF and Dai RP: Peripheral brain derived
neurotrophic factor precursor regulates pain as an inflammatory
mediator. Sci Rep. 6:271712016. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Li H, Liu T, Sun J, Zhao S, Wang X, Luo W,
Luo R, Shen W, Luo C and Fu D: Up-regulation of
ProBDNF/p75NTR signaling in spinal cord drives
inflammatory pain in male rats. J Inflamm Res. 16:95–107. 2023.
View Article : Google Scholar :
|
|
183
|
Zha AH, Luo C, Shen WY, Fu D and Dai RP:
Systemic blockade of proBDNF inhibited the expansion and altered
the transcriptomic expression in CD3+B220+
cells in MRL/lpr lupus mice. Lupus Sci Med. 9:e0008362022.
View Article : Google Scholar
|
|
184
|
Li JN, Luo RY, Luo C, Hu ZL, Zha AH, Shen
WY, Li Q, Li H, Fu D and Dai RP: Brain-derived neurotrophic factor
precursor contributes to a proinflammatory program in
monocytes/macrophages after acute myocardial infarction. J Am Heart
Assoc. 12:e281982023. View Article : Google Scholar
|
|
185
|
Cui YH, Zhou SF, Liu Y, Wang S, Li F, Dai
RP, Hu ZL and Li CQ: Injection of anti-proBDNF attenuates
hippocampal-dependent learning and memory dysfunction in mice with
sepsis-associated encephalopathy. Front Neurosci. 15:6657572021.
View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Lim Y, Zhong JH and Zhou XF: Development
of mature BDNF-specific sandwich ELISA. J Neurochem. 134:75–85.
2015. View Article : Google Scholar : PubMed/NCBI
|