Introduction
The early growth response (EGR) family comprises
four closely related zinc finger transcription factors: EGR1, EGR2,
EGR3 and EGR4. The activity of EGRs can be induced by different
extracellular stimuli, including activation, growth and
differentiation signals, tissue damage and apoptosis signals
(1). Additionally, activity can
be dynamically upregulated in response to various cellular stimuli,
including growth factors and pro-differentiation factors (2). EGR1 and EGR2 were initially
identified by screening complementary DNA libraries in mouse
fibroblasts in response to growth factor or serum stimulation,
respectively, and EGR2 exhibited functional properties similar to
those of its human homologue (3,4).
EGR3 was first described as a direct early growth-responsive gene
induced in human fibroblasts by mitogenic stimulation, while EGR4
was initially identified through screening for neurally expressed
genes and is closely associated with neuroplasticity and memory
formation (3,4). The EGR genes are located in
different chromosomal loci (5q31 for EGR1, 10q21 for EGR2, 8p21 for
EGR3 and 2p13 for EGR4), and the EGR proteins possess highly
conserved zinc finger structural domains that collectively
recognise GCG(G/T)GGGCG sequences but differ in their binding
affinities for specific nucleotides. Their isoform-specific
flanking regions and cell type-specific expression determine their
unique functions, positively and negatively regulating target gene
expression (5).
Current studies on EGR family members have focused
on EGR1, which is the most representative and extensively studied
family member. EGR1 often exhibits a dual function in various
cancer types, mostly characterised by its ability to inhibit
tumourigenesis; however, it may facilitate tumourigenesis and
tumour progression under certain conditions. For instance, EGR1 is
abnormally expressed in tumours of the urinary, digestive and
nervous systems. In gastrointestinal tumours, EGR1 is closely
associated with the pathological features of gastric cancer (GC),
and its upregulation is closely associated with enhanced
infiltration, tumour progression and poor prognosis (6); conversely, in prostate cancer, high
EGR1 expression is positively correlated with the malignancy of the
lesion, indicating its potential pro-carcinogenic role (7). Furthermore, in solid tumours,
including glioma and melanoma, EGR1 induces apoptosis, partly
through a mechanism potentially related to its regulation of cell
cycle inhibitors, including p21[wild-type p53-activated fragment
1(Waf1)/CDK-interacting protein 1 (Cip1)], indicating its
antitumour properties (8,9).
The high sequence similarity among EGR1, EGR2 and EGR3 may account
for their similar functional bidirectionality. EGR2 is an oncogenic
factor in some cancer types, and its reduced expression is
frequently associated with malignant progression. For instance, in
hepatocellular, gastric and thyroid carcinoma, the downregulation
of EGR2 expression is closely associated with enhanced cell
proliferation and reduced apoptosis (10,11). Conversely, in other cancer types,
including bladder and kidney cancer, EGR2 may be involved in
promoting malignant behaviour, with its upregulation correlated
with enhanced cell migration, invasion and metastatic potential
(12,13). Among all EGR family members, EGR1
is regulated by a variety of upstream signalling pathways and
exhibits a wider range of pro- or anticancer functions in a variety
of tumour types (14,15). EGR2 is essential in cell
differentiation and immune regulation, with its aberrations or
mutations frequently associated with malignant behaviour [such as
in chronic lymphocytic leukaemia (CLL)], and it uniquely regulates
immune cell status (16,17). In prostate cancer and
hepatocellular carcinoma (HCC), EGR3 typically acts as an
anticancer agent by inhibiting epithelial-mesenchymal transition
(EMT), promoting apoptosis and inhibiting cell proliferation
(18,19); conversely, in breast cancer, EGR3
regulates the oestrogen signalling pathway and plays a pro-cancer
role by upregulating anti-apoptotic genes including myeloid cell
leukaemia 1 (MCL1), thereby enhancing drug resistance in cancer
cells (20,21). Additionally, EGR3 is essential in
regulating the expression of inflammatory factors within the tumour
microenvironment (TME) and facilitating the differentiation of
specific tumour cells (22,23). Although EGR4 has slightly lower
sequence similarity to the other EGRs (EGR1, EGR2 and EGR3), EGR4
has anticancer activity in non-small cell lung cancer (NSCLC).
ZNF205-AS1 is an antisense transcript of the zinc finger protein
205 (ZNF205) gene, classified as a non-coding RNA. EGR4 can
upregulate ZNF205-AS1 expression, which reduces the negative
regulation of its target mRNAs (p53 pathway components) by
competing for binding to specific microRNAs (such as miR-150-5p),
which subsequently increases tumour suppressor gene expression and
promotes apoptosis. The promoting role of ZNF205 in NSCLC has not
been widely demonstrated, and thus, the current research focuses
more on the indirect regulation of other key tumour-associated
pathways through the upregulation of ZNF205-AS1 (24). Unlike EGR1, which possesses a
broader mechanistic pathway in tumours, the functions of EGR2 and
EGR3 are likely more diverse, encompassing contributions to
cellular differentiation, immune microenvironment and hormonal
regulation. Furthermore, their mutation status is often indicative
of a poor clinical prognosis (3,4,10,11). However, the functions of EGR2,
EGR3 and EGR4 in tumours are comparatively underexplored and
generally receive less attention than EGR1.
The immune system plays a critical role in tumour
onset and progression; however, tumours tend to suppress the immune
system through several immune evasion mechanisms, facilitating
their growth and metastasis. Cytotoxic T lymphocytes [cluster of
differentiation (CD)8+ T cells], natural killer (NK)
cells and dendritic cells (DCs) can exert an antitumour effect
during the immune response (25,26); however, a number of immune cells
exhibit a duality in their effects on tumours in the TME. For
instance, antitumour immune cells such as CD8+ T cells
or M1-type macrophages can kill tumour cells directly or enhance
the immune response by cytokine secretion (25). Conversely, pro-tumour immune
cells [such as regulatory T cells (Tregs), M2-type
tumour-associated macrophages (TAMs) and myeloid-derived suppressor
cells (MDSCs)] can inhibit T-cell activity and promote tumour
growth and immune escape (26).
The EGR protein family contains a deterrent structural domain (R1)
that interacts with the co-deterrent factor NGFI-A binding protein
(NAB), and specific mutations in this region are associated with
congenital hypomyelination neuropathy, with similar mutations
playing a functional role in immune-related tumours (27).
The significance of the EGR gene family in tumour
immunity is of increasing interest, especially during T-cell
depletion. EGR2 and EGR3 regulate T cell apoptosis, maintain immune
homeostasis by regulating Fas ligand (FasL) expression and play key
roles in T cell energy metabolism and tolerance responses (28). EGR1 and EGR4 modulate T-cell
function through synergistic regulation of calcium signalling
pathways (29). The EGR family
participates in the immune regulation of B cells and macrophages,
with EGR1, EGR2 and EGR3 facilitating tumour immune escape by
regulating cytokine production (30), while in macrophages, EGR2 is
implicated in M2-type polarisation, including modulating shifts in
the tumour immune response (31). In addition, the EGR family
affects tumours by modulating metabolism and differentiation.
Therefore, the present review provides an overview of the dual role
of the EGR family in tumours and the mechanisms that regulate
multiple aspects of immunity, metabolism and differentiation.
Additionally, it describes current research on relevant tumour
therapies targeting EGRs, offering new perspectives for therapies
therein.
DNA binding properties and transcriptional
regulation mechanism of EGR proteins
The DNA binding domain of each EGR protein has three
homologous zinc finger structures that recognise DNA binding sites
abundant in GC sequences (32).
Notably, in EGR1, these zinc fingers have some similarity to the
binding sites of the Sp1 transcription factor. The DNA sequence
preferentially bound by the EGR protein is 5′-GCG GGG GGC G-3′, a
structural feature that exhibits a clear rearrangement compared
with the DNA binding site of Sp1 (5′-GGG GGG CGG GG-3′). Although
Sp1, Sp3 and EGR1 do not engage with the same DNA binding sites in
the regulation of certain target genes, they share competing
binding sites in the promoter regions of some genes, including
platelet-derived growth factor (PDGF) A and B chains and adenosine
deaminase (32). This
competition for these binding sites may affect the degree of
transcriptional activation. Furthermore, the N-terminus of the EGR
protein maps to various transcriptional activation domains that are
essential for its function (32).
In EGR1, EGR2 and EGR3, a repressor domain has been
identified alongside the DNA-binding and transcriptional activation
domain. This repressor structural domain binds to the
transcriptional repressors, NAB1 and NAB2, which inhibit the
transcriptional activity of the EGR proteins by binding to them
(33,34). Therefore, even when the
transcriptional activation of EGR1 is neutralised, transcriptional
induction of the EGR1 gene may have no biological effects. NAB1 and
NAB2 interaction indicates that they regulate the function of EGR
transcription factors through negative feedback in specific cells
(33,34). Therefore, the function of EGR
transcription factors is dependent on their direct DNA-binding
ability and is regulated by interactions with NAB factors, offering
a more complex perspective for understanding EGR function across
different cellular and physiological conditions.
EGR1 facilitates several gene expression changes in
response to different cellular stimuli, including cytokines,
mitogens and oxidative stress (1-5).
The activity of EGR1 is primarily regulated by the upregulation of
its expression and post-translational modifications (2). The human EGR1 promoter has five
serum-responsive elements (SREs) and one cAMP-responsive element
(CRE) and can recruit different transcription factors to regulate
its expression across diverse cell types (35). Moreover, EGR1 self-regulates by
binding to its promoter or inducing the expression of the repressor
NAB2, forming a negative feedback loop (33,34). ELK1 is an essential component in
EGR1 regulation, activated by phosphorylation through the ERK,
c-Jun N-terminal kinase (JNK) and p38 MAPK signalling pathways and
associated with SREs (36,37). Furthermore, the PI3K/AKT pathway
regulates EGR1 expression by interfering with the DNA-binding
ability of forkhead box protein 01 through phosphorylation and
deregulating its inhibition (38). EGR1 expression in presynaptic
neurones is inhibited by the transcriptional repressor, C-terminal
binding protein 1, and is activated upon removing this repression
due to neuronal activity (39).
During ultraviolet B-induced genotoxic stress, the nuclear factor
κ-light-chain-enhancer of activated B cells (NF-κB) initiates an
apoptotic response by binding to the EGR1 promoter (40). During T-cell activation, EGR1
upregulates stromal interaction molecule 1 (STIM1), which increases
cytoplasmic Ca2+ levels, which may further promote EGR1
expression. Previous studies have demonstrated that the
Ca2+ chelator, BAPTA-AM, reduces EGR1 expression.
BAPTA-AM blocks the phosphorylated activation of transcription
factors (such as ELK1 and CREB) by Ca2+-dependent
kinases (including calcium/calmodulin-dependent protein kinase and
calcineurin) by chelating intracellular Ca2+, resulting
in the inability of these factors to bind to EGR1 promoters on SREs
and CREs, which in turn markedly reduces EGR1 transcript levels.
This implies that SREs and CREs inside the EGR1 promoter are
essential for transcriptional machinery recruitment (41,42). Additionally, in vascular smooth
muscle cells, Ca2+ signalling modulates angiotensin
II-induced EGR1 expression through calcium/calmodulin-dependent
protein kinase II and CREB, highlighting the pivotal role of
Ca2+ signalling in EGR1 regulation (43).
EGR1 function is regulated by various
post-translational modifications, including phosphorylation,
glycosylation and polymerisation. In resting cells, low
phosphorylation levels reduce EGR1 stability (44); however, casein kinase II-mediated
phosphorylation inhibits its DNA-binding ability (45), indicating that phosphorylation at
different sites may confer different functional characteristics on
EGR1. Additionally, AKT-mediated phosphorylation is a prerequisite
for EGR1 to undergo small ubiquitin-like modifier modification, an
important step in the ubiquitination and degradation of EGR1
(46). Additionally,
O-linked glycosylation of the EGR1 transactivation
structural domain is a key regulator of its activity (47).
EGR-mediated epigenetic modifications and
DNA repair mechanisms
Recently, EGRs have been shown to be deeply involved
in the epigenetic regulation of tumourigenesis and tumour
progression. EGRs affect the expression of tumour-associated genes
by regulating chromatin modification and DNA methylation, thereby
controlling key behaviours, including cell proliferation,
migration, differentiation and immune response while demonstrating
dual regulatory effects of pro- and anticancer effects on tumours
(48-67).
At the chromatin modification level, EGR1 recruits
the histone acetyltransferase, CREB-binding protein (CBP)/E1A
binding protein p300 (p300), which increases histone H3 lysine 27
acetylation (H3K27ac) levels in the promoter region of target genes
and promotes chromatin opening and transcriptional activation. For
instance, under hypoxic or inflammatory stimuli, EGR1 promotes the
binding of CBP/p300 to target gene promoters by activating the
signal transducer and activator of transcription 3 (STAT3)/NF-κB
pathway (48). This recruitment
relies on the DNA-binding activity of EGR1 and its interactions
with the structural domains of the CBP/p300 protein. Under
conditions of oxidative stress, EGR1-dependent CBP/p300 recruitment
significantly increases H3K27ac levels in the promoters of target
genes (such as Cathepsin L) and enhances chromatin accessibility,
thus facilitating transcriptional activation (48,49). This mechanism is bidirectional in
tumours; however, EGR1 promotes the proliferation of diffuse large
B-cell lymphoma cells by upregulating the myelocytomatosis oncogene
(MYC) and E2F transcription factor pathway-related genes through
the CBP/p300-H3K27ac-BRD4 axis, while simultaneously inhibiting
interferon (IFN) pathway genes by binding to the repressor NAB2,
creating a balance between pro- and anticancer effects (50,51). In addition to regulating histone
acetylation, EGR1 is regulated by histone methylation and enhancer
of zeste homologue 2, the core histone methyltransferase of the
polycomb repressive complex 2 complex, which silences the EGR1
promoter and inhibits its expression through histone H3 lysine 27
trimethylation (H3K27me3). This suppression diminishes the
activation of EGR1 oncogenes, including DNA damage-inducible 45
(GADD45) and DNA damage-inducible transcript 3 (DDIT3), and
promotes breast cancer progression (52). In addition, the DNA binding
ability of another member of the EGR family, EGR2, is correlated
with the histone methylation status of their target sites as
enrichment of H3K4me3 increases the efficiency of it binding to
target genes (53,54).
EGRs are involved in the dynamic regulation of DNA
methylation. The DNA binding ability of EGR1 is affected by the
level of methylation at the target site; its gene activation relies
on the hypomethylated state, while hypermethylation weakens its
binding efficacy and leads to gene suppression (55,56). EGR2 interacts directly with
ten-eleven translocation 2 (TET2) and facilitates 5-methylcytosine
demethylation, promoting DNA demethylation in the promoter regions
of specific genes. During monocyte differentiation, the EGR2-TET2
complex activates differentiation-associated genes (such as zinc
finger protein 36 and suppressor of cytokine signalling 3) by
dynamically regulating DNA methylation while inhibiting
tumour-associated pathways (57,58). Accordingly, the importance of
EGRs in maintaining the DNA methylation-demethylation dynamic
balance is highlighted. The EGR family also affects the
immunological milieu of the tumour, with expression levels closely
associated with tumour-infiltrating T cells and macrophages, among
others (59-67). Low expression of EGR1 results in
increased methylation of IFN pathway-related genes, inhibits the
antitumour immune response and facilitates immunological evasion in
breast cancer (59). The
stimulator of the IFN genes (STING) pathway is a central mechanism
in antitumour immunity; its activation induces the secretion of
type I IFNs, thus enhancing the maturation of DCs and T-cell
infiltration (60,61). Low EGR1 expression may inhibit
STING pathway-related genes (such as cyclic GMP-AMP synthase and
IFN-β) through methylation, resulting in the inability of immune
cells to effectively recognise tumour antigens (62,63). In immune NK T cells (iNKT),
general control non-repressible 5 (GCN5) functions as a histone
acetyltransferase, directly enhancing the transcriptional activity
of EGR2 by catalysing its acetylation. Inhibition of GCN5 (whether
through knockout or medications) markedly diminishes the
acetylation level of EGR2, impairing its capacity to effectively
activate downstream target genes, including promyelocytic leukaemia
zinc finger (PLZF), runt-related transcription factor 1 (Runx1),
interleukin-2 receptor β and T-box transcription factor (T-bet),
which hinders the differentiation and development of iNKT cells
(64,65). Additionally, GCN5 can directly
modify EGR2 through histone acetylation and through non-histone
mechanisms. For instance, GCN5 can directly acetylate specific
sites on EGR2 rather than depending on histone deacetylases in the
nucleosome remodelling and deacetylase complex (66), and this post-translational
modification enhances the ability of EGR2 to bind to the promoters
of its target genes. The acetylation status of EGR2 In iNKT cells
affects its interaction with cofactors (such as TET2), which
modulates DNA methylation dynamics and chromatin accessibility,
which subsequently affects the expression of genes, including PLZF
(67).
EGRs also have a potential role in regulating DNA
repair and maintaining genomic stability. A study has demonstrated
that EGR1 can directly bind to the promoters of several key DNA
damage response (DDR) genes in CD34+ umbilical cord
blood haematopoietic stem/progenitor cells (HSPCs) and regulate
their expression levels (50).
These EGR1 target genes are extensively implicated in cell cycle
regulation (G1/S checkpoint), DNA repair mechanisms (including the
ataxia-telangiectasia mutated/ataxia-telangiectasia and
rad3-related pathway, homologous recombination repair and
non-homologous end joining) and oxidative stress defence,
indicating that EGR1 may serve as a crucial transcriptional
regulator connecting external stimuli to the DDR network (50). In a mouse model, EGR1 knockdown
induced haematopoietic stem cell (HSC) depletion, indicating its
essential role in preserving genomic integrity and that it may be
involved in DNA repair in unstable chromosomal regions (such as 5q
deletions) by modulating repair factors, including BRCA1/2 and
radiation sensitivity 51 (61).
EGR1 functionally overlaps with classical tumour suppressors,
including p53 and phosphatase and tensin homologue (PTEN) (47,62). Under DNA damage induction, EGR1
can act in synergy with p53 to regulate repair gene expression,
including GADD45, or participate in regulating the balance between
repair and apoptosis by inhibiting PTEN and activating the AKT
pathway (47). Moreover,
oxidative stress significantly increases EGR1 expression, which may
affect the DNA repair mechanism by regulating intracellular levels
of reactive oxygen species (ROS). However, while EGR1 can increase
the need for repair through ROS signalling, over-activation can
cause aberrant expression of repair-associated genes, resulting in
genomic instability and potentially facilitating tumourigenesis and
tumour progression (63).
Accordingly, EGR1 possesses a complex regulatory function in the
DDR, and its dysregulated balance may serve as a potential
oncogenic factor.
Aberrant expression of EGR family members is closely
associated with defective DNA repair in various tumour types. For
instance, EGR1/2/3 expression is significantly lower in several
cancer types than in normal tissues (23,64,65); EGR1 downregulation increases
resistance to chemotherapy in oesophageal cancer, while EGR1
deletion in breast cancer may be correlated with genomic
instability induced by DNA replication stress (23,64). However, EGR4 is known for
facilitating tumour proliferation in colorectal cancer by
activating the inflammatory pathway [tumour necrosis factor
(TNF)-α/TNF-κB] and may indirectly exacerbate oxidative DNA damage
(65). EGR family expression
levels were significantly and negatively correlated with tumour
mutational burden, further supporting their key role in regulating
DNA repair efficacy and affecting tumour mutation accumulation
(66). Furthermore, the
functions of EGR family members are regulated by epigenetic
modifications, such as DNA methylation and histone acetylation,
which affect their ability to bind to target gene promoters,
thereby affecting the expression of DDR-related genes. Although
studies have demonstrated a multi-level association between EGR
families and DNA repair, most are still based on correlation
analysis or indirect evidence. Additional experimental approaches,
including chromatin immunoprecipitation-sequencing, gene editing
and repair function assays, are required to classify their specific
mechanisms in the DNA damage repair network.
Previous studies have demonstrated that the EGR
family is essential in tumour progression by regulating multiple
signalling pathways, including cell proliferation, apoptosis,
migration and immune response. However, ongoing studies are
clarifying that the regulatory mechanism of the EGR family in
tumours may involve deeper processes, including epigenetic
regulation and DNA repair mechanisms. Similarly, the EGR family may
further regulate the expression and function of tumour-associated
genes by affecting chromatin conformation, DNA methylation status
and DDR. This hypothesis remains unconfirmed, and more experimental
data and comprehensive molecular mechanism studies are required to
clarify the specific mechanisms.
Dual regulation of EGR in tumours
The functions of EGR family members vary across
different cancer types, exhibiting notable heterogeneity that is
dependent on the tumour type and influenced by the TME, including
hypoxia, inflammation and hormone dependence (1-5,27). The variability in the 'on/off'
mechanisms of EGRs (such as the activation or inhibition of
specific signalling pathways) across cancer types may explain the
diversity of these transcription factors in their pro- or oncogenic
roles. Typically, the function of EGR in cancer is to influence
tumour progression by regulating various cellular behaviours,
including proliferation, migration, invasion and angiogenesis.
Carcinogenic regulation of EGR
Proliferation
Cell proliferation depends on several growth
factors, and high expression of growth factors and their receptors
is an essential feature of cancer cells. High expression of EGR1
may affect tumour cell proliferation by modulating the cell cycle.
For instance, growth factor stimulation can enhance EGR1 through
oestrogen receptor β activation of the RAF/MEK1/ERK/ELK1 pathway
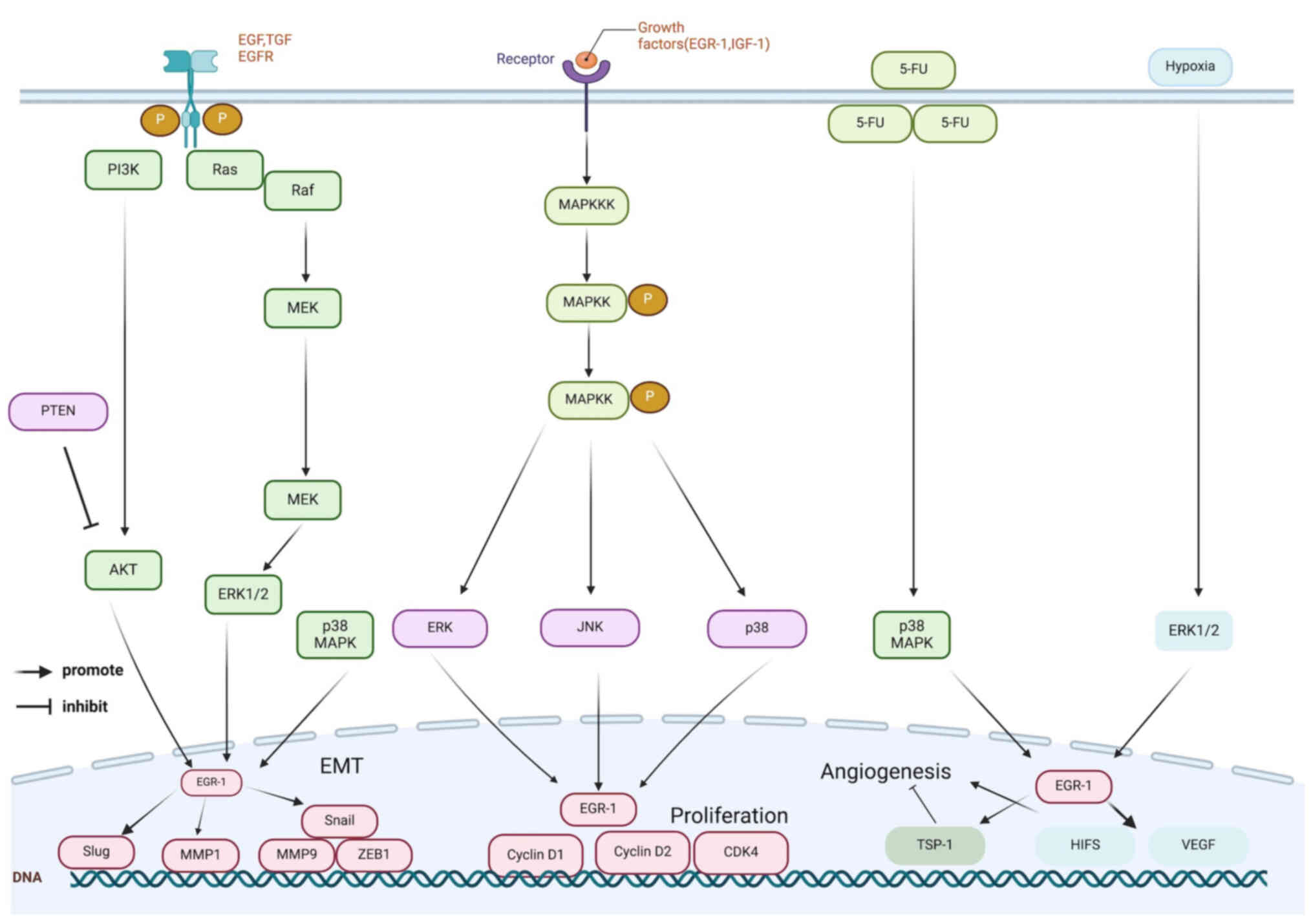
(15) (Fig. 1). Furthermore, inhibitors of the
MAPK/ERK pathway significantly reduce EGR1 expression, indicating
that EGR1 is a downstream gene within this pathway (14). Cyclin D1 is an important molecule
in cell cycle regulation, and EGR1 enhances cell proliferation by
directly binding to the cyclin D1 promoter, increasing cyclin D1
expression and facilitating the transition of cells from G1 to S
phase (67) (Fig. 1). In addition, EGR1 upregulates
other cell cycle-related proteins, including cyclin D2 and
cyclin-dependent kinase 4, further promoting tumour cell
proliferation (68). The JNK
pathway within the MAPK family enhances EGR1 expression, creating a
cyclic mechanism (69).
Additionally, insulin-like growth factor-1 and its receptor
activate the MAPK/ERK pathway, further reinforcing this cyclic
process (67) (Fig. 1).
EGR1 is a transient transcription factor whose
stability is regulated by the glycogen synthase kinase 3 β
(GSK3β)/F-box and WD repeat domain containing 7 (FBXW7) axis, which
affects cell proliferation in numerous cancer types (70). FBXW7 functions as an E3 ubiquitin
ligase for EGR1 and facilitates its ubiquitination and degradation
through GSK3β kinase activity. However, GSK3β inhibition or FBXW7
mutation prolongs the half-life and increases EGR1 levels. Under
hypoxic conditions, FBXW7 enhances the ubiquitination of EGR1,
thereby diminishing its stability (70). The downregulation of EGR1
inhibits lung cancer cell growth; however, the downregulation of
FBXW7 stimulates prostate cancer cell proliferation by stabilising
EGR1 (70). In advanced CLL,
mutations in the FBXW7 and EGR2 genes occur in similar patient
populations (16% of patients with EGR2 mutations carry both
NOTCH1/FBXW7 mutations, and FBXW7 mutations indirectly increase
their signalling activity by inhibiting NOTCH1 degradation) and are
both associated with a poorer prognosis (71). Consequently, it is possible that
both genes are involved in the same pathogenic pathway or that
their mutations collectively affect the malignant progression of
tumours.
Metastasis and invasion
EMT is a process by which epithelial cells transform
into mesenchymal cells, serving as a crucial mechanism for tumour
cell invasion and metastasis. EGR1 facilitates EMT by inducing the
expression of the epithelial cadherin (E-cadherin) transcriptional
repressors, snail family transcriptional repressor 1 (SNAIL) and
snail family transcriptional repressor 2 (SLUG), thereby promoting
tumour invasion and metastasis (72) (Fig. 1). During tumour metastasis,
mesenchymal-related genes including matrix metalloproteinases
(MMPs; such as MMP1 and MMP9), histones and zinc finger E-box
binding homeobox 1 (ZEB1) are crucial (73). EGR1 enhances the expression of
the MMP1 promoter by binding directly to it, while SNAIL
facilitates MMP9 and ZEB1 expression, thereby aiding tumour cell
invasion and metastasis (74)
(Fig. 1). Several MAPK pathways
(including ERK, JNK and p38) upregulate EGR1 and contribute to
TNF-α-induced MMP1 expression, further promoting invasion and
metastasis (75). In
hormone-independent prostate cancer, C-X-C motif chemokine ligand
(CXCL)5 enhances EGR1 transcription and increases SNAIL expression
through the RAF/MEK/ERK pathway, thereby facilitating tumour cell
EMT and metastasis. Conversely, EGR1 inactivation reduces
IL-6-associated metastasis, potentially through inhibition of the
PI3K/PTEN/AKT pathway (68).
EGR1 promotes tumour cell invasion in GC by regulating β-catenin
expression (76). In ovarian
cancer, EGF stimulates the EGR1-mediated upregulation of SLUG
through activation of the p38 MAPK, ERK1/2 and PI3K/AKT pathways,
resulting in E-cadherin inhibition and enhanced tumour metastasis
(77). In pancreatic cancer, the
EGR1-dependent p300/CBP cofactor binds to the SNAI2 promoter and
activates its transcription, subsequently inhibiting E-cadherin
expression and inducing EMT (78). In HCC, EGR1 upregulates SLUG
expression through hepatocyte growth factor-induced binding to the
SNAIL promoter or through the ERK/AKT/EGR1/SLUG pathway, thereby
promoting EMT and metastasis (79). In breast cancer, S100
calcium-binding protein A4 enhances nuclear localisation of EGR1 by
promoting its binding to input protein 7, which leads to the
EGR1-mediated downregulation of β-catenin through the
PTEN/AKT/GSK3β pathway, which in turn promotes tumour invasion and
metastasis (80).
Studies on specific metastatic sites have
demonstrated the critical role of EGR1. For instance, EGR1 has been
implicated in the metastasis of prostate cancer to bone and the
brain by regulating angiogenesis and osteoclastogenesis. EGR1
activates the NF-κB pathway by binding to TNFRSF12A (FN14) and
enhances its expression through a MEK-dependent mechanism.
Furthermore, EGR1 expression significantly reduces the number and
size of metastases and decreases vascular density and osteolytic
lesions in metastatic regions. Additionally, EGR1 regulates the
expression of numerous angiogenic and osteoclastogenic factors,
including PDGF-A, transforming growth factor β1 (TGF-β1) and
interleukin 6, which are implicated in prostate cancer metastasis
(7). Peritoneal metastasis of GC
is one of the most common forms of metastasis in advanced GC
(81). GC cells with upregulated
HOXA11 expression activate EGR1 expression in peritoneal
mesothelial cells by secreting PDGF-BB and TGF-β1, a mechanism
regulated by miR-181a-5p. EGR1 also facilitates peritoneal
mesothelial fibrosis and enhances the migration and peritoneal
dissemination of GC cells, establishing a feed-forward
amplification loop that accelerates the progression of peritoneal
metastasis (81). The role of
osteoblast protein, osteopontin (OPN; an essential glycoprotein
involved in cell signalling, immunological response and tissue
remodelling), in tumour invasion and metastasis has attracted
considerable attention (82).
OPN helps tumour cells breakthrough in situ restriction and
metastasise to other organs by facilitating cancer cell migration
and survival (82). The role of
EGR1 in cancer stem cells and metastasis in lung cancer has
received considerable attention. A previous study has demonstrated
that the transcription factor octamer-binding transcription factor
4 (Oct4) facilitates the upregulation of EGR1 by activating the
EGR1 promoter, which in turn, together with OPN, forms an
Oct4/EGR1/OPN axis and enhances the metastatic ability of lung
cancer cells (82). Lymph node
metastasis is a common feature of numerous malignant tumours and is
closely associated with cancer progression and poor prognosis.
Thyroid cancer, the most common endocrine tumour, has a high rate
of lymph node metastasis (83).
Long non-coding PTP receptor type C1 transcriptional splicing
(LNCPTCS), a tumour suppressor long non-coding RNA, has
significantly reduced expression in thyroid cancer, especially in
metastatic lymph nodes. Inflammatory cytokines, including TNF-α and
CXCL10, modulate the binding of EGR1 to the LNCPTCTS promoter,
thereby reducing LNCPTCTS expression (84).
Angiogenesis
Tumour angiogenesis is an important mechanism for
tumour growth and metastasis, and the inhibition of angiogenesis
has emerged as an effective strategy for cancer treatment. A number
of cancer cells secrete extracellular vesicles that facilitate
vascular endothelial cell migration by upregulating EGR1, hence
markedly enhancing angiogenesis (7). The rapid growth of tumours
frequently results in hypoxia in their central region, and hypoxia
is a potent angiogenesis-stimulating factor. Under hypoxic
conditions, hypoxia-inducible factor (HIF) is upregulated, leading
to increased expression of the vascular endothelial growth factor
(VEGF) protein family, which stimulates angiogenesis and promotes
tumour cell survival (85)
(Fig. 1). EGR1 expression in
prostate cancer is regulated by the inhibition of androgen receptor
signalling and PTEN deletion, facilitating angiogenesis by
upregulating HIF1 expression through activation of the HIF1
promoter under hypoxic conditions (86). In lung cancer, EGR1 directly
binds to and activates the VEGFA promoter, thereby enhancing
angiogenesis by enhancing HIF1α-dependent VEGFA expression
(87). In addition, EGR1
transcriptionally activates the angiogenic factor, C-C motif
chemokine ligand 2, which subsequently forms a positive feedback
loop through the CCR2/ERK/ELK1/EGR1 pathway (88). Programmed cell death ligand 1
(PD-L1) is abundantly distributed in the nucleus of malignant
tumours. Furthermore, nuclear nPD-L1 is an endogenous accelerator
of cancer angiogenesis; it promotes the binding of phosphorylated
STAT3 to the EGR1 promoter, resulting in EGR1-mediated angiogenic
activation (89).
EGR3 vs. EGR1 in tumour
angiogenesis
In vascular endothelial cells, the EGR transcription
factor family is essential in regulating vascular function. EGR1 is
essential in several vascular-related diseases, including
ischaemia/reperfusion-induced lung injury, atherosclerosis and
fibroblast growth factor-2 (FGF-2)-dependent angiogenesis and
tumour growth (90-92). EGR3 primarily regulates several
downstream effects of VEGF, including promoting inflammatory
responses, enhancing cell proliferation and migration in
vitro, and facilitating neovascularisation in Matrigel and
tumour growth in vivo (93). VEGF, thrombin and TNF-α rapidly
activate endothelial cells and induce high expression levels of EGR
family members, particularly EGR1 and EGR3. VEGF-induced
upregulation of EGR3 is more pronounced and persists for a longer
duration than that of EGR1 (93), whereas negative feedback pathways
are essential for the temporal regulation of endothelial cell
activation. For example, VEGF upregulates Down syndrome critical
region 1 (DSCR-1) expression through activation of the calmodulin
phosphatase/nuclear factor of activated T cells (NFAT) pathway,
while DSCR-1 subsequently inhibits NFAT activity through a negative
feedback mechanism (94,95). In endothelial cells, NFAT is
essential in the specific induction of EGR3 expression by VEGF,
while EGR3 further enhances DSCR-1 expression in VEGF-treated
endothelial cells, establishing a self-limiting regulatory circuit
that preserves the dynamic balance of the pathway by inhibiting
NFAT-dependent EGR3 activation through DSCR-1 (93). NAB1 and NAB2 synergistically
inhibit EGR1 activity in endothelial cells, with NAB acting as a
direct negative feedback inhibitor of EGR1. However, EGR3
overexpression upregulates NAB1 and NAB2 expression, while NAB2
inhibits EGR3 activity independently and may facilitate its delayed
negative feedback regulation (93). Additionally, EGR3 exhibits a dual
feedback control mechanism in response to VEGF stimulation: VEGF
induces EGR3 expression while simultaneously activating the
upregulation of NAB2 and DSCR-1, which collaboratively inhibit EGR3
activity and expression through a feedback mechanism that regulates
the functional state of endothelial cells (93). EGR3 assumes a more significant
role than EGR1 in certain specific angiogenesis and tumourigenesis,
such as VEGF-induced endothelial cell activation,
neovascularisation in Matrigel and tumour growth in
vivo.
Vasculogenic mimicry (VM) is an essential phenomenon
in tumours, characterised by the formation of capillary-like
channels composed of cancer cells within tumour tissues that are
anatomically and physiologically similar to blood vessels and are
essential in alternative angiogenesis in tumours (96). In triple-negative breast cancer
cells, EGR1 expression is upregulated and localised to the nucleus
during VM formation, while EGR1 knockdown markedly reduces VM
formation (96). Additionally,
Krüppel-like factor 4 (KLF4), a gene regulated by EGR1, exerts a
positive regulatory role in VM formation. EGR1 regulates KLF4
expression by binding to its promoter region, while the functions
of ERK and p38 kinase further affect VM formation by regulating
EGR1 and KLF4 expression (96).
Although much attention has been directed toward the role of EGR1
in VM, the impact of EGR3 on tumour angiogenesis is frequently
neglected; however, its role may surpass that of EGR1.
Anti-carcinogenic regulation
Apoptosis
Apoptosis is the process by which cells eliminate
themselves through programmed death mechanisms under physiological
conditions, and EGR1 promotes apoptosis in cancer cells through
multiple pathways. However, EGR1 can synergistically enhance
TGF-β1, fibronectin (FN), p21Waf1/Cip1 and focal adhesion kinase
(FAK) to inhibit apoptosis, thereby reducing caspase activity
(97). Additionally, EGR1
facilitates apoptosis by enhancing the PTEN-mediated downregulation
of AKT expression (98). EGR1
directly binds to the promoters of various apoptosis-inducing
factors, including BCL2-associated X (BAX), NSAID-activated gene 1
(NAG1) and PTEN, to enhance their expression (99). D-tocotrienol induces apoptosis in
pancreatic cancer cells by activating EGR1 through the JNK/c-Jun
pathway and promoting BAX expression by binding to the BAX promoter
(99,100). PTEN is a key tumour suppressor
gene whose expression is directly regulated by EGR1. A study has
demonstrated that EGR1 induces apoptosis in cancer cells by binding
to the PTEN promoter region to enhance its transcription (101). Insulin-like growth factor-II
upregulates EGR1, which subsequently increases PTEN expression;
unconjugated bilirubin regulates EGR1 through activation of the
apyrimidinic endodeoxyribonuclease 1/redox factor-1 pathway, which
affects PTEN levels; and vitamin D significantly increases PTEN
expression through synergistic interaction with the vitamin D
receptor, EGR1, and p300 PTEN expression, further inducing
apoptosis in cancer cells (101). EGR1 expression is lower in lung
and liver cancer tissues than in normal tissues (101,102). In NSCLC, EGR1 facilitates cell
cycle arrest and apoptosis through the regulation of tumour
suppressor pathways, including PTEN. Moreover, TGF-β1 downregulates
EGR1-induced EMT, while high EGR1 expression significantly inhibits
EMT through the regulation of SNAIL, SLUG and E-cadherin expression
(102). In uterine cancer,
Wilms tumour 1-associated protein (WTAP) downregulation reduces
EGR1 mRNA stability by decreasing its m6A modification, which
subsequently reduces the expression of EGR1 and the tumour
suppressor gene, PTEN, to promote tumourigenesis, while combined
WTAP, EGR1 and PTEN upregulation significantly inhibits
tumourigenicity of endometrial cancer cells and their stem cells
(103). EGR2 is essential in
mediating the anti-proliferative function of PTEN (104). In various cancer cell lines,
including two endometrial cancer cell lines (Ishikawa and HEC-1A),
one ovarian cancer cell line (SKOV3) and two colon cancer cell
lines (HCT116 and HT29), EGR2 expression significantly inhibits
cell colony formation (104). A
further study demonstrated that EGR2 induces apoptosis in cancer
cells and upregulates the transcriptional activity of the
apoptosis-related genes, BCL2 interacting protein 3-like and BAK
(105). NAG1 belongs to the
TGF-β superfamily and can prevent tumour cell growth (106). A previous study demonstrated
that EGR1 induces apoptosis by upregulating NAG1 in cancer
(106). However, NSAIDs promote
apoptotic signalling through the peroxisome proliferator-activated
receptor (PPAR)γ/EGR1/NAG1 pathway and can directly boost
EGR1-mediated NAG1 expression in a cyclo-oxygenase-2
(COX-2)-independent manner, further promoting apoptosis (107).
The TP53 gene, an anticancer gene, is closely
associated with various human tumours. The p53 protein encoded by
the gene is a tetrameric transcription factor that primarily
monitors DNA damage, preserving genome stability. Recently, the
anticancer activity of p53-deficient cells has offered new
perspectives for tumour therapy (108). p21, a major target of p53,
facilitates cell cycle regulation after DNA damage and induces the
apoptosis of cancer cells through a p53-independent mechanism after
its expression is enhanced by EGR1 (109). Mutations in the p53 gene have
been detected in numerous cancer types, and specific mutation sites
(point mutations at 156, 246, 247 and 273) exhibit a high affinity
for EGR1 activation (110).
EGR1 is essential in DNA repair and its aberrant activation
following p53 mutation may exacerbate tumour progression (111). In p53-deficient prostate cancer
cells, EGR1 promotes apoptosis by inducing TNF-α expression.
Additionally, EGR1 induces p21Waf1/Cip1 expression independently of
p53 through the ERK and JNK MAPK/ELK1/EGR1 pathways, initiating DNA
repair and enhancing apoptosis, indicating that EGR1 is a potential
therapeutic target for p53-mutant tumours. Furthermore, mutant p53
initiates the ERK1/2 pathway to mediate EGR1 upregulation, further
activating the EGR1/EGFR/ERK feedback loop (112).
Oncogene-induced senescence (OIS)
OIS is an important endogenous tumour suppressor
mechanism that induces cell cycle arrest by sensing oncogenic
signals, hence inhibiting the transformation of normal cells to
cancer cells (113,114). OIS, or apoptosis, is induced by
the activation of key anticancer pathways, including the alternate
reading frame/p53 pathway and cyclin-dependent kinase inhibitor
2A/retinoblastoma protein pathway, thereby inhibiting cancer
initiation and progression. The transcription factor,
CCAAT/enhancer-binding protein β (C/EBPβ), is essential for OIS
regulation (113-115). EGR1, EGR2 and EGR3 recognise
and transiently bind to specific sites on the C/EBPβ promoter and
are closely associated with the inducible expression of C/EBPβ.
However, the simultaneous knockdown of all three genes inhibits
C/EBPβ expression, impairing the antitumour effect of OIS (115). Accordingly, the EGR family
(namely EGR1, EGR2 and EGR3) facilitates tumour suppression through
the regulation of OIS, and its impaired function may contribute to
cancer development and progression.
Inhibition of cancer-promoting
mechanisms
The antitumour function of EGR1 involves the
inhibition of angiogenesis and invasive metastasis. The sustained
expression of EGR1 upregulates several anti-angiogenic genes,
including CXCL14, tissue inhibitor of metalloproteinase (TIMP)1,
TIMP2, TIMP3 and Fms-related receptor tyrosine kinase 1, which
inhibits tumour angiogenesis (116). In nasopharyngeal carcinoma, the
transcription factor, EGR1, binds to the promoter region of
nasopharyngeal carcinoma-associated gene 6 (NGX6) and upregulates
its expression, thereby inhibiting tumour angiogenesis and impeding
tumour progression (117).
Fluorouracil upregulates EGR1 and inhibits angiogenesis through the
p38 MAPK pathway, while EGR1 enhances thrombospondin 1 (TSP1)
expression by binding to the TSP1 promoter, further diminishing
tumour angiogenesis (117)
(Fig. 1). A colon cancer study
demonstrated that EGR1 controls the expression of Mindin by binding
to its promoter, which inhibits HIF1α and VEGFA expression and
reduces VEGFR2 phosphorylation in endothelial cells, resulting in
an anti-angiogenic effect (118). In head and neck squamous cell
carcinoma, oxytocin upregulates EGR1 through EGFR and ERK-dependent
pathways and inhibits tumour invasion and metastasis, which may be
related to E-cadherin upregulation (119). In leukaemia, thalidomide and
LY294002 upregulate EGR1, inhibit cell invasion and metastasis and
are not dependent on the PI3K/AKT pathway (120). In fibrosarcoma, EGR1
upregulation markedly inhibits tumour invasion by modulating TIMP2
expression (121).
Regulatory roles of EGR in tumour
immunity
T cells
EGR family members are essential in numerous aspects
of T cell function, including the regulation of T-cell activation,
energy metabolism and antigen-driven T cell proliferation as well
as effector function differentiation. For instance, EGR1
facilitates T helper cell type 2 (Th2) differentiation by
upregulating IL-4 expression, while EGR2 and EGR3 inhibit Th1-type
differentiation by repressing T-bet expression. Additionally, EGR4
is a key and non-redundant regulator of T-cell differentiation
(27,122-125).
EGR and regulation of T cell
function
The activation and effector functions of T
lymphocytes are regulated by EGR1 and other essential transcription
factors, including nuclear factor for activated T cells, which
collectively regulate the transcriptional processes of IL-2 and
TNF-α following T cell receptor (TCR) stimulation. The zinc finger
protein binding region (ZIP) is an important regulatory element in
the IL-2 promoter. ZIP is bound by the Sp1 transcription factor in
resting T cells and by EGR1 in activated T cells, thereby
regulating IL-2 expression (126,127). Additionally, promoter regions
containing the ZIP region and the NFAT binding element promote IL-2
and TNF-α transcription. This region) in the human IL-2 and TNF-α
promoters is a binding site for EGR1, NFAT and Sp1, which
synergistically regulate the expression of key cytokines through
heterodimer formation (126,127). EGR proteins (excluding EGR4)
typically contain a repressor binding domain regulated by NAB (NAB1
and NAB2). NAB2 regulates target gene expression by binding to
EGR1, while EGR1 concurrently regulates NAB2, forming a negative
feedback loop to balance EGR1 activity (31,32). NAB1 can effectively inhibit EGR1;
however, the role of NAB2 is more complex. NAB2 is upregulated
during T-cell activation in response to co-stimulatory signals and
collaborates with EGR1 to enhance IL-2 expression (27). IL-2 transcription is dependent on
NAB2, which is recruited to the EGR1 binding site of the IL-2
promoter upon TCR stimulation, further enhancing EGR1 activity and
facilitating IL-2 and IL-2β receptor synthesis (128). Moreover, EGR1 expression is
correlated with CD40 ligand transcription in CD4+ T
cells and collaborates with NFAT proteins to regulate CD40 ligand
expression, a process dependent on CD28 signalling. EGR1 is
significantly upregulated upon CD28 stimulation and may act as an
adaptor for TCR and CD28 signalling, facilitating full activation
of T cells and their co-stimulatory function with
antigen-presenting cells (129,130).
During tumour growth, CD8+ T cells
progressively lose their ability to release inflammatory cytokines
due to prolonged antigen exposure, accompanied by the upregulation
of inhibitory receptor expression, a condition known as T-cell
exhaustion (131-134). Although this phenomenon may
have evolved as a mechanism to mitigate immune-mediated
pathological damage, prolonged T-cell exhaustion weakens the immune
system and hence promotes tumour growth (135). Antigen-induced NFAT-dependent
regulatory mechanisms regulate the energy metabolism of
CD4+ T cells and the depletion process of
CD8+ T cells (136).
Th expression of EGR2, a key energy-regulating transcription
factor, is also induced upon TCR activation of NFAT (137). EGR2 inhibits T cell function in
response to energy requirements by directly enhancing the
expression of factors that reduce TCR signalling, including casitas
B-lineage lymphoma b (Cbl-b) (125). Certain studies have
demonstrated that the EGR2 expression level is significantly
increased in CD8+ T cells in a depleted state,
indicating that EGR2 may be crucial in the regulation of T cell
depletion (133,134). Additionally, EGR3 is more
likely to collaborate with EGR2 in regulating the tolerance
response. These two factors can bind the FasL regulatory element in
the FasL promoter and enhance the transcriptional expression of
FasL following TCR stimulation (138). EGR2 and EGR3 are essential
regulators of FasL expression, which is upregulated in
CD4+ T cells following TCR signalling and sustains
immunological homeostasis by inducing apoptosis (Fig. 2). NFAT mediates the regulation of
FasL expression through EGR2 and EGR3, which are upregulated in an
NFAT-dependent manner in CD4+ T cells in the inactivated
state. The upregulation of EGR2 and EGR3 inhibits the expression of
the T-cell activators, EGR1 and NAB2, and inhibits T-cell secretion
of IFN-γ and IL-2, while enhancing the expression of the E3
ubiquitin ligase, Cbl-b, an important mechanism for inducing T-cell
tolerance and anergic states (27,139,140) (Fig. 2). Obstructing inhibitory
receptors while simultaneously activating exhausted T-cell
responses has become one of the key targets of immunotherapy,
exhibiting significant clinical efficacy in cancer treatment
(135).
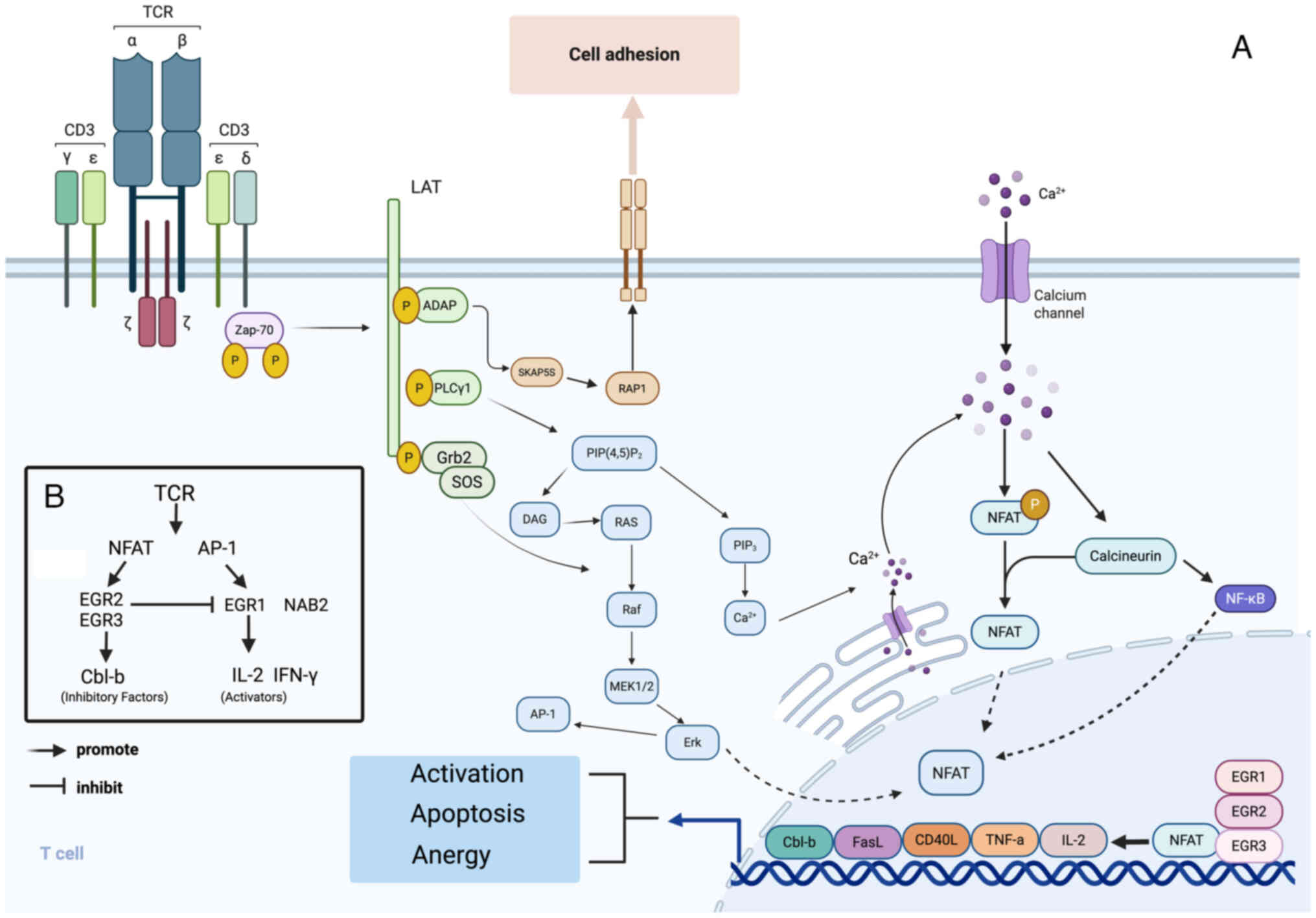 | Figure 2(A) EGR transcription factors are
essential in the functional regulation of T cells. Upon the TCR
stimulation of T cells, the transcriptional activation of EGR
factors can trigger different cell fate decisions, a process
modulated by the synergistic action of NFAT and EGR in the promoter
region. Regulators in the promoter region include cytokines (such
as IL-2 and TNF-α), co-stimulatory molecules (such as CD40L) and
several regulatory molecules, such as FasL and the ubiquitin ligase
Cbl-b, These factors do not bind directly to gene promoters; they
activate downstream signalling pathways (such as JAK-STAT, NF-κB
and MAPK) by interacting with their respective cell surface
receptors, which in turn modulate the binding of nuclear
transcription factors (including STATs, NF-κB, AP-1 and NFAT) to
bind to promoters, which ultimately affects the transcriptional
activity of genes indirectly. (B) EGR2 and EGR3 are essential
negative regulators in the NFAT pathway and play key regulatory
roles within the cell. EGR3 upregulates EGR2 expression, and
collectively, they inhibit T cell function by increasing negative
regulatory molecule expression (such as Cbl-b) and inhibiting the
expression of T-cell activators, EGR1 and NAB2. TCR, T-cell
receptor; NFAT, nuclear factor of activated T cells; JAK, Janus
kinase; STAT, signal transducer and activator of transcription;
NF-κB, nuclear factor κB; MAPK, mitogen-activated protein kinase;
AP-1, activator protein 1; Cbl-b, casitas b-lineage lymphoma
proto-oncogene-b; NAB2, NGFI-A binding protein 2. |
Functions of EGR1 and EGR4 in
store-operated calcium channel (SOCE) regulation
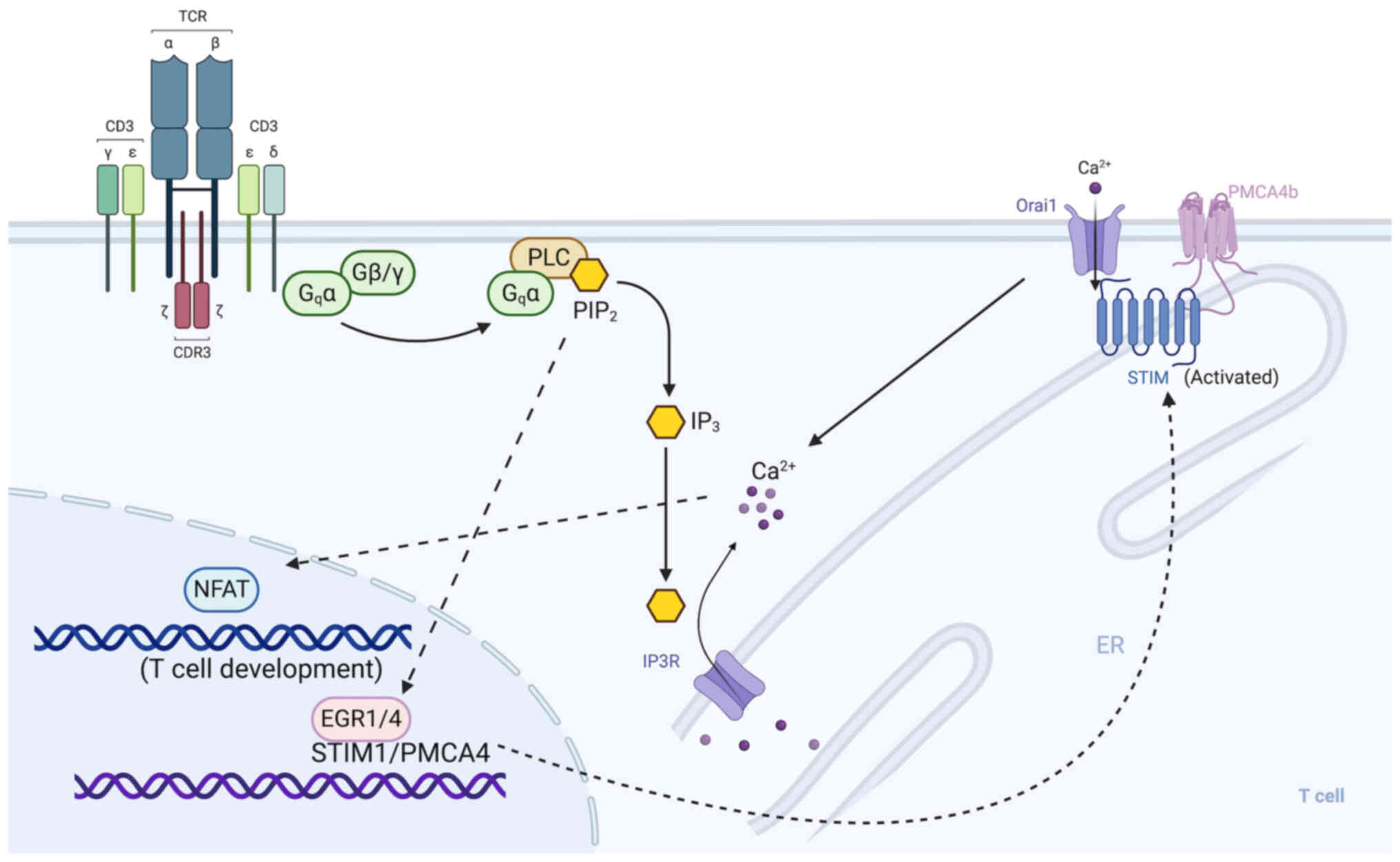
EGRs are prominent regulators of SOCE components,
with EGR1 and EGR4 capable of driving and co-regulating STIM1
expression in T cells. However, EGR2 and EGR3 lack this function.
EGR4 knockdown significantly reduced EGR1 binding to the STIM1
promoter, indicating a synergistic influence of both factors on
STIM1 expression during T-cell activation (28). The main driver of calcium
signalling during T-cell activation is TCR-mediated phospholipase C
(PLC) activity, which facilitates Orai activation by increasing the
expression of potassium channels, further amplifying the calcium
influx response (141-143). The TCR is activated upon
binding to antigenic peptides on antigen-presenting cells,
initiating a signalling cascade response. This process activates
PLCγ, which induces the release of Ca2+ from the
endoplasmic reticulum through the inositol 1,4,5-trisphosphate
receptor, while Orai1-mediated calcium influx sustains
intracellular calcium concentrations (144,145). Subsequently, the zinc finger
transcription factors, EGR1 and EGR4, are activated to modulate the
expression of Ca2+ homeostasis-associated proteins (such
as STIM1 and plasma membrane calcium ATPase 4), facilitating the
long-term maintenance of Ca2+ signalling (28). This calcium signalling
subsequently activates the calmodulin/calcineurin complex,
facilitating NFAT translocation to the nucleus, where it regulates
the expression of genes associated with T cell differentiation and
proliferation (28) (Fig. 3). However, EGR4 deficiency
disrupts the expression and function of potassium channels, causing
a persistent excessive influx of calcium ions that abnormally
activates the NFAT signalling pathway, significantly impacting
T-cell function (122).
Although SOCE relies on the upregulation of STIM1,
its functionality is significantly enhanced with a synchronous
increase in Orai1 expression. Previous studies have demonstrated
that SOCE inhibits tumour growth by facilitating apoptosis in
prostate cancer (146,147), clarifying why SOCE deficiency
may accelerate tumour progression. However, SOCE exhibits
pro-carcinogenic effects in some cases, including promoting tumour
angiogenesis by upregulating VEGF in cervical cancer and driving
activation of COX-2 (148) and
NFAT in other pathological responses (149,150). Additionally, high expression of
STIM1 and Orai1 has been associated with several cancer types,
including lung, liver and colorectal cancer (147). In prostate cancer, a negative
feedback regulation mechanism exists between Orai1 and the androgen
receptor, affecting their expression levels, while STIM1 and Orai1
expression is negatively correlated with the Gleason score
(151). In breast cancer, STIM1
and Orai1 expression is significantly associated with focal
adhesion formation; however, SOCE inhibition reduces the number of
focal adhesions while paradoxically enhancing metastasis in
vivo (152).
Repressor of EGR1 in SOCE regulation:
WT1
WT1 is a tumour suppressor gene that notably
overlaps with EGR1 in consensus targets; however, it is not a
member of the EGR family. Unlike the direct early genes of the EGR
family, WT1 expression is predominantly developmentally regulated
and generates >20 isoforms through several mechanisms, including
numerous transcription start sites, selective splicing and RNA
editing (153). The specific
splicing of exon 9 results in a lysine-threonine-serine insertion,
a modification that significantly affects the DNA binding capacity
and functional properties of WT1 (153). WT1 and EGR1 possess numerous
transcriptional targets, with WT1 typically inhibiting the gene
upregulation function of EGR1 by binding to these loci. This
disparity illustrates the significant differences between the two
in target recognition and transcriptional regulatory mechanisms
(154). In mammalian genomes,
cytosines at most CpG sites are methylated, leading to reduced EGR1
binding and downregulation of its target genes in some cases. The
zinc finger structural domains of EGR1 and WT1 exhibit similar
binding affinities to fully methylated DNA and are insensitive to
methylation (155). However,
they significantly differ in their ability to bind
5-carboxycytosine, an intermediate state of epigenetic
reprogramming. Glutamate residues in EGR1 inhibit this binding,
while glutamine residues in WT1 facilitate it (156). This binding preference may
explain the difference in target function between the two in
tumours, including breast cancer and glioma, where
5-carboxycytosine is significantly elevated and may serve as a
unique epigenetic marker. Moreover, examining the effect of
methylation on other EGR isoforms will elucidate the complex role
of the EGR family in epigenetic regulation (157,158).
In Wilms tumour cells, WT1 expression is
significantly associated with reduced STIM1 levels and SOCE
activity. EGR1 positively regulates STIM1 expression, while WT1
regulates intracellular Ca2+ homeostasis by inhibiting
STIM1 expression (159). In
addition, aberrant expression of STIM1-independent calcium channels
(such as CaV2.3) promotes Wilms tumour recurrence through the MAPK
pathway (160). Furthermore,
reduced EGR levels are significantly associated with a suboptimal
patient response to chemotherapy (161). Thus, the action of WT1 may
suppress tumours by inhibiting Ca2+ signalling, while
EGR1 upregulation and enhancement of Ca2+ signalling may
promote tumour recurrence and progression.
EGR2-mediated regulation of
progenitor-exhausted T cell differentiation
The role of EGR in differentiation is significant
in the immune escape mechanism of tumours. T cell factor 1 (TCF1)
and Slamf6-depleted progenitor T cell populations are essential in
cancer immune evasion, while EGR2 expression in these T cells
regulates their differentiation process. Sustained antigen
recognition induces high EGR2 expression in depleted and effector
CD8+ T cells, while selective EGR2 expression in
TCF1+ progenitor cells is progressively lost as these
cells transition to a more differentiated state. EGR2 maintains the
identity of progenitor-type depleted T cells and plays a vital role
in tumour immune escape by regulating transcriptional and
epigenetic networks (162).
Specifically, EGR2 works with TCF1 to preserve the self-renewal
capacity of progenitor-exhausted T cells (Tpex cells;
TCF1+PD-1+) by regulating stem cell-related
genes, including Slamf6 (163,164). Under chronic antigenic
stimulation, EGR2 prevents premature Tpex differentiation into
terminally depleted Tim-3+PD-1+ cells by
inhibiting the expression of genes associated with terminal
differentiation such as inhibitor of DNA binding 3, nuclear
receptor subfamily 4 group A member 1 and thymocyte
selection-associated high mobility group box (163,165). This regulation depends on EGR2
interaction with chromatin remodelling factors (such as
lysine-specific histone demethylase 1) to preserve progenitor cell
identity through the inhibition of epigenetic modifications,
including H3K4me2 (162,166).
In the TME, high EGR2 expression exacerbates immune escape by
enhancing an immunosuppressive phenotype while sustaining
progenitor-depleted T cell survival; EGR2 deficiency results in
accelerated functional exhaustion of CD8+
tumour-infiltrating lymphocytes and uncontrolled tumour growth
(167); however, EGR2 mitigates
immunopathological damage induced by T cell overactivation by
modulating the expression of inhibitory receptors including PD-1
and lymphocyte-activation gene 3 (168,169). This balance may determine that
therapies targeting EGR2 must be precisely regulated.
B cells
In B lymphocyte responses, EGR1 expression is
directly regulated by antigen activation through the B cell
receptor (BCR) activation of protein kinase C signalling. The
maturity level of the B cell significantly regulates BCR
signalling; in mature B cells, EGR1 expression is often correlated
with cellular proliferative and growth responses, while in immature
B cells, it may provide contrary effects (170). The expression of EGR2 and EGR3
are induced in B cells by chronic antigenic stimulation through the
NFAT pathway (170). Deletion
of EGR2 and EGR3 may result in an abnormal accumulation of B1
cells; for instance, in mouse models, EGR2 and EGR3 defective mice
exhibit significantly increased B1 cells inside the peritoneal
cavity, spleen and bloodstream, and this accumulation is closely
associated with CLL pathogenesis (16). EGR2 inhibits the differentiation
of B cells into plasma cells by modulating the expression of
specific genes (such as PR domain containing 1, with zinc
finger/B-lymphocyte-induced maturation protein 1). However, when
EGR2 mutations disrupt this regulation, B cells may undergo
aberrant proliferation and differentiation, contributing to CLL
development and progression. Further analysis has revealed that
EGR2 mutations in CLL can impair the normal function of B cells by
modifying their regulation of key target genes, hence facilitating
leukaemia onset (16). In
addition, B cells mitigate autoimmunity by secreting IL-10 and
TGF-β, which are characteristic features of regulatory B cells
(Bregs) and central mediators of their immunosuppressive effects
(171,172). IL-10 and TGF-β facilitate the
development of anti-inflammatory cells (Tregs), thus effectively
mitigating autoimmune diseases (173). Bregs are a subset of
specialised B cells with immunomodulatory roles, and hypoxic
conditions inside the TME influence their differentiation. Under
hypoxic conditions, B cells upregulate EGR1 and EGR3 expression
through activation of the MAPK pathway, which modulates the
synthesis of the downstream immunosuppressive factors, TGF-β1 and
IL-10. This pathway facilitates the conversion of B cells into
Bregs and enhances the differentiation and immunosuppressive
capabilities of Tregs through the TGF-β1, IL-10 and TNF secreted by
Bregs, promoting immune escape of tumours (29).
Macrophages
The EGR gene is activated in myeloid cells through
cytokine regulation. Preliminary studies in leukaemia cell lines
demonstrated that EGR1 expression was upregulated in response to
IL-3, granulocyte-macrophage colony-stimulating factor, granulocyte
colony-stimulating factor (G-CSF) and macrophage colony-stimulating
factor (M-CSF), although only M-CSF has been shown to induce EGR1
expression in normal bone marrow progenitor cells (174). Additionally, EGR1 and EGR2
expression is increased during monocyte differentiation in response
to phorbol 12-myristate 13-acetate stimulation. EGR1 is essential
in macrophage differentiation by rapidly responding to M-CSF
stimulation in mouse bone marrow progenitors; however, it is
insensitive to G-CSF (175). In
leukaemia cell lines and bone marrow progenitor cells, the use of
EGR1 antisense oligonucleotides inhibits macrophage
differentiation; however, it has no effect on granulocyte
differentiation (174). Further
studies have demonstrated that EGR1 overexpression in cell lines
and primary myeloid cells promotes macrophage differentiation;
however, this differentiation is counterbalanced by inhibiting
other myeloid lineages (174-176). EGR1 is not essential for
macrophage differentiation, as myeloid cells in EGR1-deficient mice
still differentiate into macrophages upon M-CSF stimulation,
indicating that other EGR proteins may substitute for EGR1 function
(175). Additionally, another
study demonstrated that PU.1 re-expression in PU.1-deficient
myeloid cells induced EGR2 upregulation and promoted macrophage
differentiation. During macrophage differentiation, EGR1 and EGR2
were upregulated in immature foetal liver myeloid progenitors or
bone marrow common myeloid progenitors, while no significant
changes were observed in EGR3 and EGR4 (176). However, subsequent a study
demonstrated that EGR1 and EGR3 expression levels were
significantly increased in myeloid cells when M-CSF induced myeloid
differentiation into macrophages, while EGR2 was weakly induced and
EGR4 could not be detected (177). EGR2, in particular, may play a
key role in monocyte differentiation into macrophages, and the
expression of this transcription factor is fine-tuned during this
process (178). Additionally,
the role of EGR1 in monocyte development is critical, especially in
enhancer regions that regulate monocyte developmental genes (such
as colony stimulating factor 1 receptor); however, differentiated
macrophages exhibit a different EGR1 binding pattern than
undifferentiated monocytes. Furthermore, in mature macrophages,
EGR1 upregulation inhibits the activation state of macrophages by
reducing cytokine secretion, inhibiting the expression of
stimulatory ligands, including CD86, and increasing the expression
of immune checkpoint molecules such as CTLA4 and PD-1 (179). Although EGR1 expression levels
are decreased in mature macrophages, especially after
lipopolysaccharide (LPS) stimulation, similar to other EGR family
members, including EGR2, they still play important regulatory roles
in mature myeloid cells (179).
Macrophages can be classified into two polarised
states: M1- and M2-like phenotypes. M1-like macrophages are induced
by Th1-associated cytokine (such as IFN-γ) and microbial products
(including LPS) and exhibit high levels of major histocompatibility
complex (MHC) class II and CD86, which efficiently stimulate
CD4+ T cells and drive a potent pro-inflammatory
response, including nitric oxide production and IL-1β, IL-6 and
TNF-α pro-inflammatory cytokines (180,181). However, M2-like macrophages are
induced by Th2-associated cytokines (such as IL-4 or IL-13),
express low levels of CD86 and typically act as promoters of tissue
repair during the resolution phase of inflammation. Characteristic
markers of M2 macrophages include arginase, found in inflammatory
zone 1 (Fizz1) and chitinase-like protein 3, and M2 macrophages
also produce anti-inflammatory cytokines, including IL-10 and
TGF-β1. M1- or M2-like phenotypes have pro-inflammatory and
anti-cancer or pro-cancer and anti-inflammatory roles, respectively
(181,182) (Fig. 4). EGR2 is regarded as a novel
M2-type marker that is essential for the selective activation of
macrophages, and its expression plays a key role in the
polarisation state of macrophages (183). EGR2 is expressed to some extent
in inactivated M0 macrophages, while it is regulated in M2-type
macrophages; however, EGR2 expression is significantly reduced in
M1-type macrophages, and its expression level remains low after
stimulation (17). EGR2 plays a
significant role in regulating M1 and M2 markers, particularly
under M2 stimuli (including IL-4 and IL-13), where the upregulation
of EGR2 promotes the expression of M2 signature markers and the
upregulation of M1 markers during M2 to M1 transition. Conversely,
EGR2 expression is downregulated under M1 stimuli (including IFNγ
and LPS), and low levels of EGR2 are associated with the
maintenance of M1 markers and weak upregulation of M2 markers,
while EGR2 knockdown results in the decreased expression of M1
markers (17). The PPARγ
transcription factor is involved in M2 polarisation, while its
upstream transcription factor, CCAAT/enhancer-binding protein
(CEBPβ), regulates the expression of markers in response to direct
stimulation of M1 and M2 phenotypes (181). Additionally, EGR2 expression is
regulated by CEBPβ, which is highly expressed and negatively
regulates EGR2 levels in M1 macrophages. CEBPβ knockdown results in
the upregulation of EGR2 expression, which affects the expression
of M1 and M2 markers. However, EGR2 positively regulates CEBPβ
expression, and EGR2 knockdown results in decreased CEBPβ levels
(182,183) (Fig. 4B). Therefore, the reciprocal
regulatory relationship between EGR2 and CEBPβ is essential for
macrophage activation and polarisation. EGR2 promotes the
expression of M1 and M2 markers by regulating CEBPβ, and this
mechanism is correlated with the ability of macrophages to respond
to inflammatory stimuli. However, low levels of EGR2 expression
indicate that macrophages are less responsive to activation
signals.
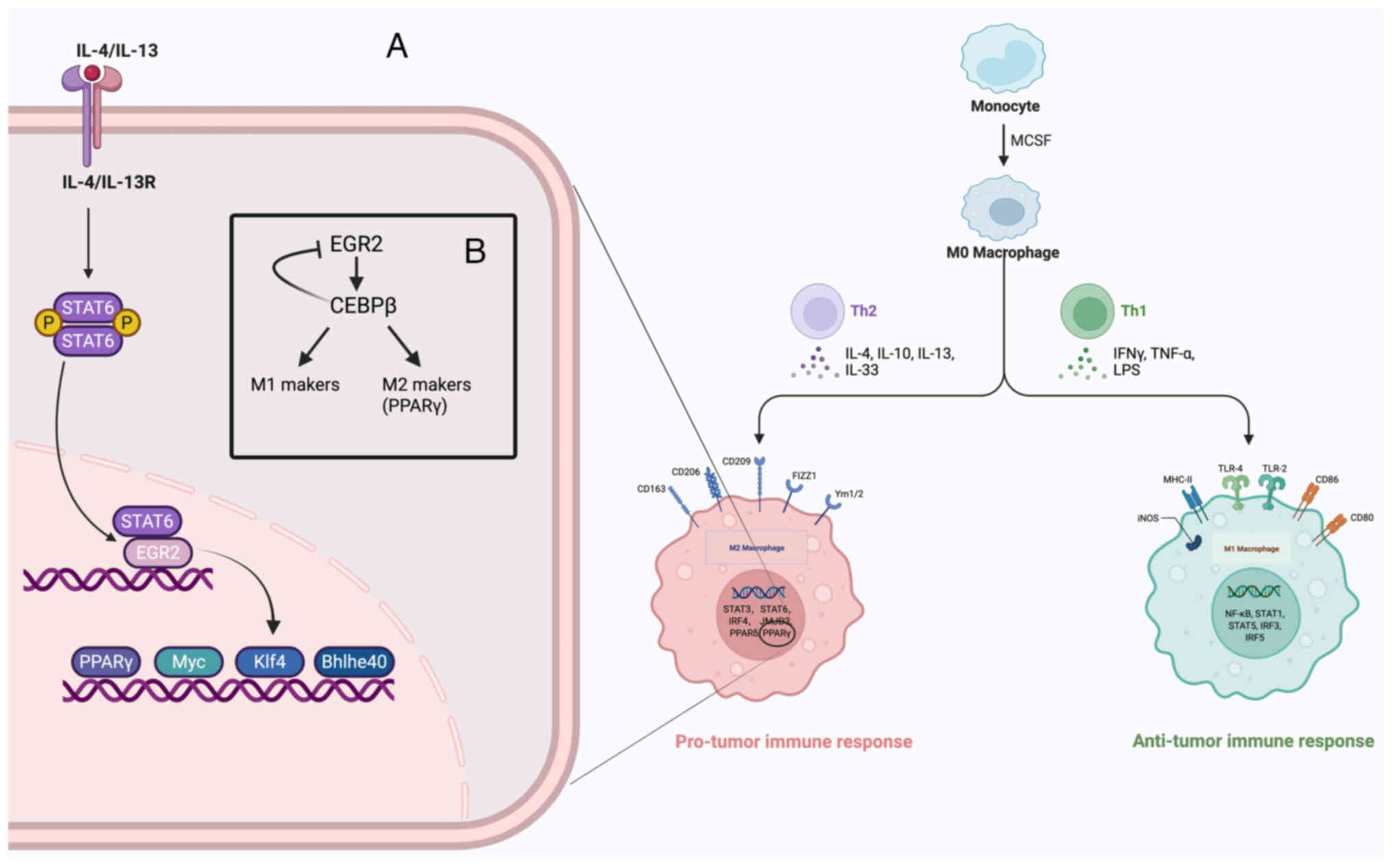 | Figure 4(A) Following IL-4 or IL-13
stimulation of macrophages, STAT6 is phosphorylated and
translocated to the nucleus, where it binds to enhancers associated
with the EGR2 regulatory site, activating EGR2 expression. EGR2
subsequently binds to de novo enhancers in its target genes
and promotes downstream effector gene activation (such as PPARγ,
MYC, Klf4 and Bhlhe40), which triggers a specific polarised
transcriptional programme. (B) In M1 and M2 type macrophages, EGR1
promotes M1 or M2 marker expression by upregulating CEBPβ, which
promotes M1 or M2 marker expression. In M1 macrophages, high levels
of CEBPβ inhibit EGR2 expression. IL, interleukin; STAT6, signal
transducer and activator of transcription 6; EGR, early growth
response; PPARγ, peroxisome proliferator-activated receptor γ; MYC,
myelocytomatosis oncogene; KLF4, Krüppel-like factor 4; BHLHE40,
basic helix-loop-helix family member e40; C/EBPβ,
CCAAT/enhancer-binding protein β. |
The selective polarisation of macrophages by IL-4
is essential for maintaining immune homeostasis in vivo.
IL-4 activates the signal transducer and activator of the
transcription 6 (STAT6) signalling pathway by binding to the IL-4
receptor (IL-4Rα). Although STAT6 activation is transient following
IL-4 stimulation, it can initiate several sustained transcriptional
changes (184) (Fig. 4A). EGR2 was identified as an
important factor in IL-4/IL-13-induced alternative activation of
macrophages, with its expression significantly upregulated in these
cells. IL-4 promotes EGR2 expression through the STAT6 pathway,
while EGR2 upregulation relies on self-regulatory mechanisms. EGR2
is essential in late enhancer activation during IL-4-mediated
processes, specifically in maintaining BRD4, the switch/sucrose
non-fermentable complex and RNA polymerase II binding to enhancers
and in regulating H3K27ac modification and chromatin accessibility
(30,75). Within 24 h of IL-4 stimulation,
EGR2 selectively occupies persistent and late enhancers,
particularly those sites that regulate the IL-4 response. After
peak STAT6 activity, EGR2 binds to these late enhancers, forming a
self-sustaining feedback loop that allows the expression of
IL-4-responsive genes to be maintained even when STAT6 is
dissociated from chromatin (30,75). EGR2 is essential in the
transcriptional regulation of IL-4-responsive genes, and almost all
IL-4-induced and repressed gene expression is dependent on EGR2
involvement (174). Unlike
STAT6, EGR2 is not directly involved in STAT6 regulation; instead,
it binds to late enhancer regions devoid of STAT6 binding sites,
which affects gene expression. EGR2 can activate and inhibit
several downstream transcription factors, including PPARγ, Klf4,
MYC and basic helix-loop-helix family member e40, which is
essential in alternative activation (185,186) (Fig. 4A). There may be synergistic
effects between EGR2 and alternative activators including PPARγ and
retinoid X receptor (RXR), especially in response to IL-4
stimulation, where PPARγ upregulation enhances the binding of RXR
heterodimers to de novo enhancers, which regulates
IL-4-responsive gene expression and facilitates the strong binding
of STAT6 in response to repeated stimulation (185-187). Additionally, EGR2 may regulate
the inhibition of LPS- or IFN-γ-induced inflammatory responses in
IL-4-treated macrophages (188,189).
NK cells
The functional inhibitory effect of the TME on NK
cells is an essential mechanism of tumour immune escape. Hypoxia,
metabolic stress and the accumulation of immunosuppressive cells
result in diminished NK cell activity, which weakens their ability
to kill tumour cells (190). In
tumours, including acute myeloid leukaemia and glioma, high EGR2
and diacylglycerol kinase α (DGKα) levels are correlated with poor
patient prognosis, while low levels of expression indicate improved
survival. In xenograft tumour models, NK cell dysfunction is
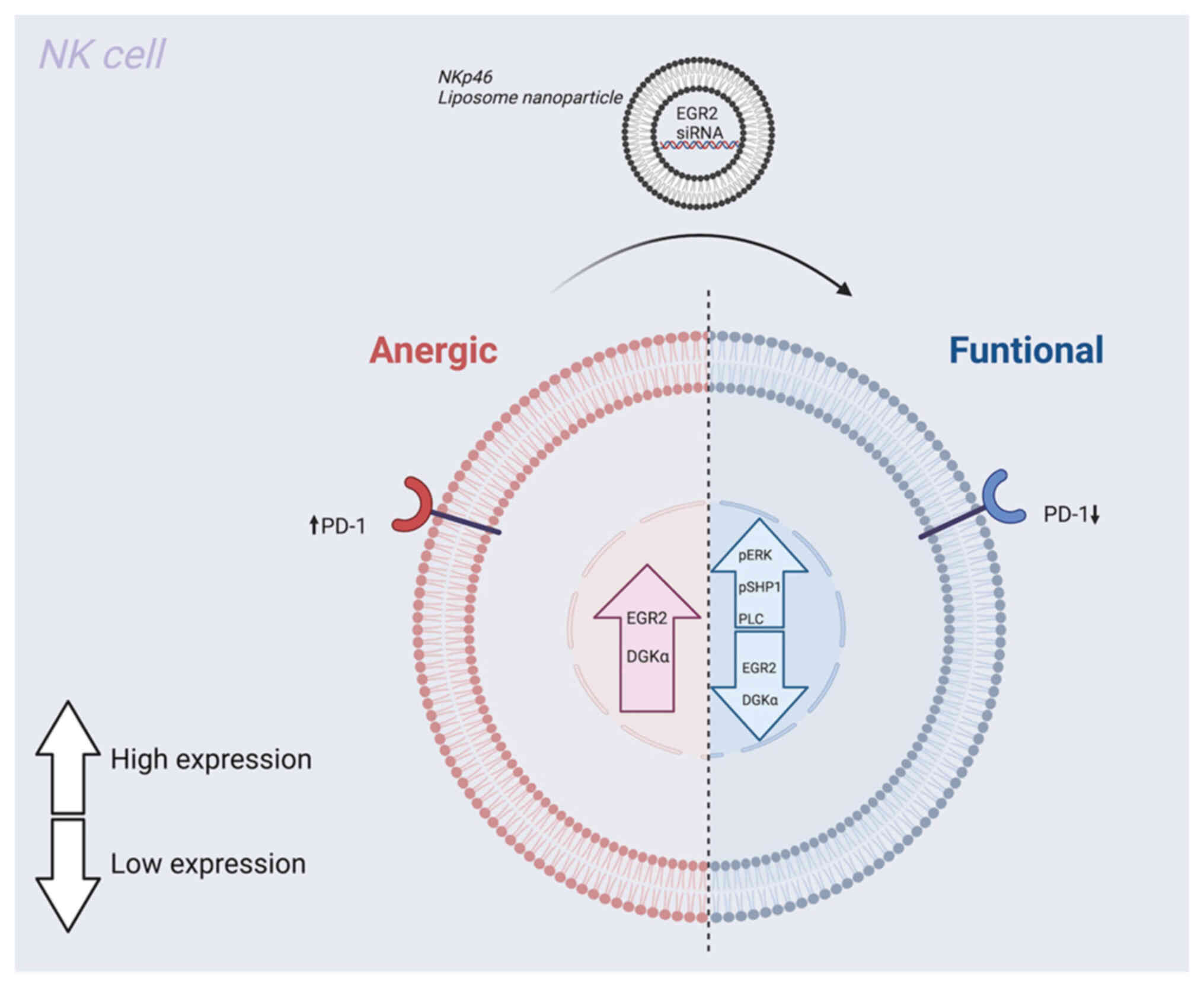
closely associated with high EGR2 and DGKα expression (190). The EGR2 and DGKα transcription
factors play key negative regulatory roles in inhibiting NK cell
function, particularly in preserving the inactive and tolerogenic
state of NK cells (Fig. 5).
Inhibiting the expression of EGR2 and DGKα markedly enhances NK
cell activity, as evidenced by an increased degranulation response,
ERK phosphorylation and intracellular calcium signalling (190). Furthermore, silencing EGR2
reduces the expression of the immune checkpoint molecule, PD-1,
further enhancing NK cell activity (190) (Fig. 6). These results indicate that
targeted modulation of the EGR2 and DGKα pathways restores NK cell
function and serves as an important therapeutic strategy to
counteract tumour immunosuppression.
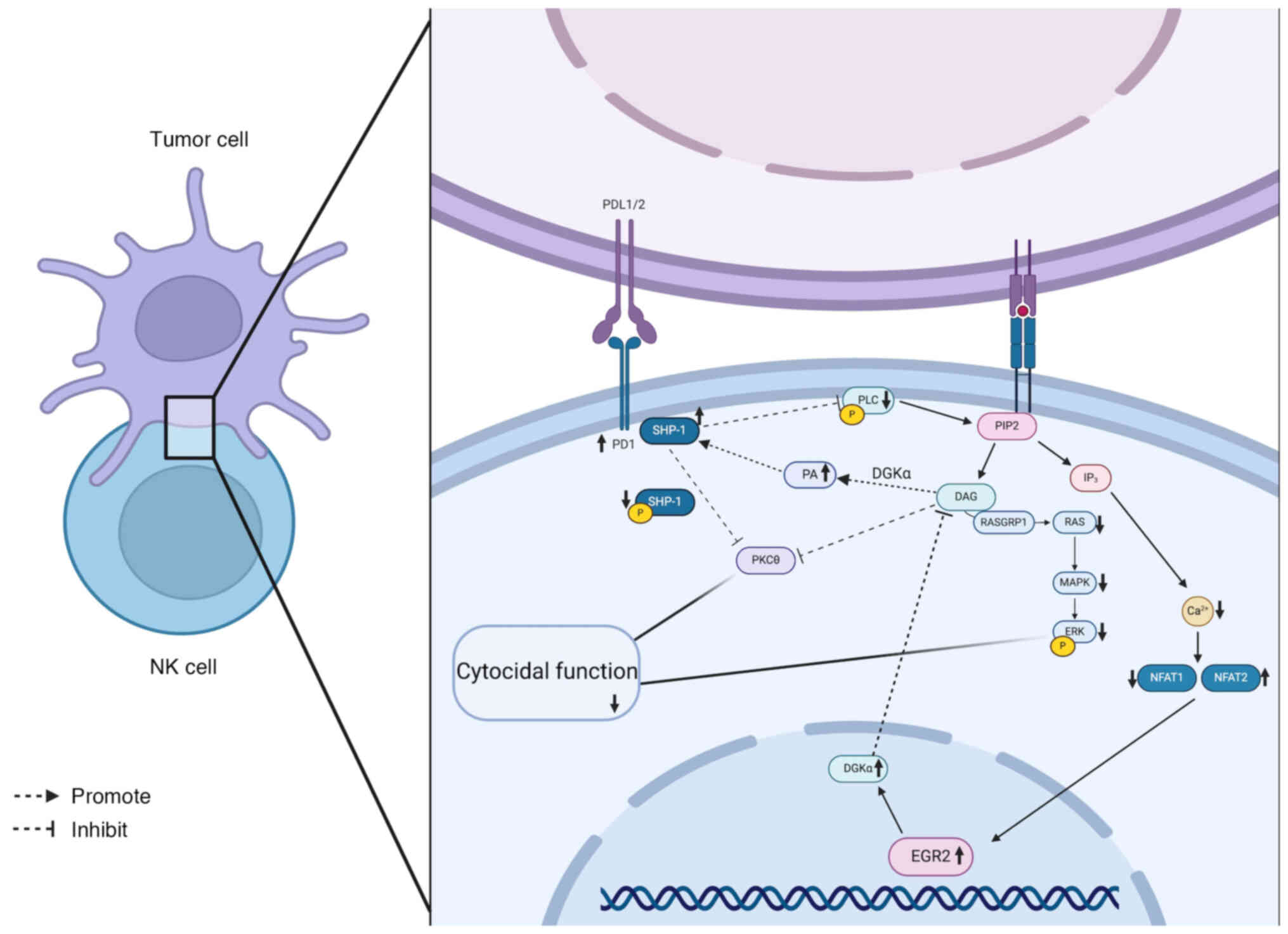 | Figure 5Schematic diagram of the signalling
pathway in anergic NK cells. In anergic NK cells, the EGR2
expression level is elevated, which subsequently triggers an
increase in DGKα. This change prompts the conversion of DAG to
phosphatidic acid, which subsequently recruits more SHP-1 to the
cell membrane. Therefore, DAG availability is limited, thereby
inhibiting protein kinase Cθ activity and preventing the regulation
of SHP-1 function. Dephosphorylation of SHP-1 further inhibits the
phosphorylation of linker for activation of T cells and PLCγ1/2,
blocking the initiation of downstream signalling. Additionally, DAG
deficiency inhibits DAG-mediated activation of the Ras/Raf/MEK/ERK
signalling pathway and IP3-mediated calcium influx. SHP-1, Src
homology 2 domain-containing phosphatase-1; PLCγ1/2, phospholipase
C γ 1/2; NK, natural killer; DGKα, diacylglycerol kinase α; DAG,
diacylglycerol; SHP-1, Src homology 2 domain-containing
phosphatase-1; PKCθ: protein kinase C θ; LAT, linker for activation
of T cells; PLCγ1/2, phospholipase C γ 1/2; IP3,
inositol 1,4,5-triphosphate. |
MDSCs and cancer-associated fibroblasts
(CAFs)
In the TME, EGR family members, especially EGR1,
have relatively complex interactions with MDSCs and CAFs (191,192). EGRs are essential in regulating
tumour immune escape, stromal remodelling and angiogenesis. EGR1
serves as an essential regulatory factor in the differentiation of
MDSCs; it facilitates the differentiation of MDSCs by directly
interacting with the promoter region of the MyoG gene while
simultaneously inhibiting their proliferation (191). This process entails a negative
regulation of the Toll-like receptor signalling pathway,
subsequently triggering the abnormal accumulation of MDSCs in the
context of chronic inflammation or chemotherapy-induced cellular
senescence [for instance, in high-risk myeloid malignancies where
del(5q) results in EGR1 haploinsufficiency] (191). In addition, EGR2 promotes
crosstalk between MDSCs and immune cells, including T cells and
macrophages, by regulating cytokine networks, including IL-8,
thereby contributing to an immunosuppressive microenvironment
(192). Additionally, EGR1 is
essential in CAFs. In placental site trophoblastic tumours, high
EGR1 expression in CAFs activates VEGFA, facilitating angiogenesis
and expediting tumour metastasis (193). In ovarian cancer, EGR1
synergistically drives the process of EMT with high mobility group
AT-hook 1, while IL-6 secreted by CAFs further enhances tumour
invasiveness through the EGR/Runx1 axis (194). In adult T-cell
leukaemia/lymphoma, a subset of CAFs display high levels of
EGFR-related transcripts, particularly EGR1 and EGR2, which
stimulate CD4+ T-cell proliferation through the
FGF7/FGF1 and PDGFA-PDGFRA/B pathways, which may drive tumour
malignant progression (195).
In colorectal cancer, EGR4 expression is significantly associated
with the abundance of CAFs, which promotes CAF activation and
extracellular matrix remodelling through activation of the
TNFα/NF-κB pathway, enhancing the proliferation and invasive
ability of cancer cells, altering the CAF phenotypes and
exacerbating the tumour-promoting effects, possibly by modulating
inflammatory factor secretion and chromatin assembly-related
pathways (65).
Regulatory role of EGR in tumour
metabolism
Glycolysis
Cancer is increasingly recognised as a metabolic
disease, and the abnormal alteration of energy metabolism in cancer
cells is one of the hallmarks of cancer (196). The Warburg (aerobic glycolysis)
effect is a major pathway of energy acquisition in cancer cells and
is regarded as an important marker of tumourigenesis (197). Among the underlying mechanisms
of the Warburg effect, TAp73, a homologue of the tumour suppressor
p53, is essential in regulating phosphofructokinase L (PFKL)
expression, which in turn promotes glycolysis and facilitates
tumour cell proliferation (198). Additionally, targeted
inhibition of PFKL is considered an effective antitumour strategy.
In HCC models, the downregulation of EGR1 expression enhances
glycolysis in tumour cells, while EGR1 overexpression binds to the
promoter region of PFKL, resulting in transcriptional repression of
PFKL and subsequent inhibition of glycolysis, reducing glucose
consumption and lactic acid production, significantly inhibiting
tumour proliferation and growth (199).
Circadian rhythms: Lipid metabolism and
glucocorticoid rhythmicity
Complex interactions exist between circadian
imbalances and cancer processes across various cancer types, and
the biological clock, an evolutionarily highly conserved mechanism,
is responsible for regulating the synchronisation of physiological
processes and behaviour with the circadian cycle (200). Molecular clocks, focused on the
transcription factors, brain and muscle arnt-like 1 (BMAL1) and
circadian locomotor output cycles kaput (CLOCK), regulate the
rhythmic expression of most protein-coding genes in the mammalian
genome, thereby affecting various key physiological activities,
including sleep, feeding, hormone secretion, metabolism and immune
function (200,201). In a healthy physiological
condition, EGR1 modulates rhythm by interacting with the feedback
loop of the biological clock. Following stimulation of EGR1 by
signals from the master clock, EGR1 binds to the promoter of the
period circadian regulator 1 (Per1) gene and activates Per1
transcription, which inhibits Per2 and reverse-Erb A (Rev-erbs)
expression. Rev-erbs repression contributes to significant rhythmic
fluctuations in BMAL1, which interacts with EGR1 to activate its
transcription, forming an EGR1/Per1/BMAL1 feedback loop that helps
to synchronise the hepatic biological clock with the master
biological clock, thereby ensuring circadian stability (202). EGR1 dysregulation in this
process can result in aberrant cell cycle regulation, thereby
facilitating cancer cell proliferation. In cancer cells, essential
clock genes, including BMAL1 and CLOCK, are commonly dysregulated
or mutated. For instance, in HCC, BMAL1 and CLOCK facilitate tumour
cell proliferation by modulating cell cycle-related factors.
Furthermore, inhibition of BMAL1 and CLOCK expression results in
increased toxicity in HCC cells, activates apoptotic pathways and
impedes normal cell cycle progression (203).
EGR1 and BMAL1/CLOCK complexes modulate the
transcription of lipid metabolism genes [including cell
death-inducing DFFA-like effector A (Cidea)], acting as a bridge
between circadian rhythms and metabolic homeostasis in peripheral
organs (202). EGR1 deficiency
or dysfunction can disrupt lipid metabolism, which affects the
energy supply and proliferation of tumour cells. Additionally, by
regulating fatty acid uptake and lipid droplet formation, EGR1 may
facilitate tumour growth and metastasis (202). During ageing, the circadian
rhythmic expression of EGR1 is modified, leading to a change in the
timing of its peak expression, which affects the expression of
lipid metabolism genes, specifically the delayed phase of Cidea
expression. Without EGR1, triglycerides accumulate in the liver due
to increased fatty acid uptake and lipid droplet formation, while
the rhythmic expression of lipid metabolism genes is disrupted,
resulting in metabolic dysfunction in the liver. Ageing leads to
decreased EGR1 expression, and the dysregulation of circadian
rhythms leads to a decoupling of the metabolic stability of
peripheral organs (the liver) from the rhythms of the master
biological clock, resulting in a decline in metabolic function
(202).
EGR3 functions as a circadian gene within the
suprachiasmatic nucleus and may exhibit a similar rhythmic
regulatory mechanism with EGR1 (203). Considering that circadian
rhythms are closely related to glucocorticoid secretion, the two
may affect glucocorticoid secretion rhythms by regulating the
expression of core biological clock genes (such as BMAL1 and Per1).
In humans, glucocorticoids exist mainly as cortisol, while in
rodents, they are primarily corticosterone; however, corticosterone
is secreted in lower amounts in humans and is more likely to
function as metabolic intermediates of cortisol (204). Rhythmic cortisol secretion is
now recognised as an important predictor of survival in lung and
breast cancer (200). The
secretion of glucocorticoids (such as cortisol and corticosterone)
is regulated by core biological clock genes (including BMAL1 and
Per1), and their rhythmic release is critical for maintaining a
stable circadian rhythm. Disruption of the secretion of these
hormones can disrupt the biological clock system, which provides
favourable conditions for tumourigenesis and tumour progression.
Additionally, disruption of glucocorticoids (such as cortisol and
corticosterone) can alter EGR1 and EGR3 expression, thereby
interfering with the normal functioning of circadian rhythms and
destabilising the biological clock, which triggers various
responses, including metabolic changes that facilitate tumour cell
development (205). However,
differences exist between cortisol and corticosterone, with
cortisol potentially regulating EGR family members (EGR1) in humans
through a more complex mechanism that encompasses a broader
transcriptional network through the dual action of the
glucocorticoid receptor (GR) and mineralocorticoid receptor (MR)
(206). The inhibition of
T-cell activation and pro-inflammatory cytokine secretion by the GR
may indirectly affect the immunoregulatory pathways in which the
EGR family is involved. For instance, the role of EGR1 in oxidative
stress and aldosterone synthesis may be inhibited by cortisol-GR
signalling, thereby facilitating tumour immune escape (207,208). However, the role of rodent
corticosterone has received less attention due to its diminished
affinity for the GR and possibly weaker regulation of the EGR
family. Corticosterone is more likely to be metabolised into its
active form by 11β-hydroxysteroid dehydrogenase or to bind to the
MR, thereby activating specific signalling pathways and indirectly
regulating EGR1-related genes (209,210). In a tongue squamous cell
carcinoma model, corticosterone promotes cancer cell migration and
invasion by downregulating EGR1, while EGR1 overexpression inhibits
these behaviours by regulating dual-specificity phosphatase 1
(211). Accordingly, the
relationship between EGR and circadian rhythms, especially the
interaction with the secretion of glucocorticoids (such as cortisol
and corticosterone), may affect cancer development, progression and
recurrence through multiple mechanisms.
Regulation of differentiation mechanisms by
EGR in tumours
The interaction between neurones and cancer cells
is one of the key processes in tumourigenesis and tumour
progression, and cancer neurogenesis has been proposed as a new
hallmark of cancer (212-214). The nervous system is
extensively distributed throughout the body; it regulates organ
development, maintains homeostasis and promotes cellular plasticity
and damage repair by innervating tissue nerves while ensuring
normal tissue function. However, the nervous system plays an
essential regulatory role in tumour growth and metastasis (215-217). Colorectal cancer stem cells
(CSCs) can differentiate into neurones, and these neurones can
synapse with nerve fibres within the tumour (218). This process is essential for
tumour onset and progression, and EGR2 serves as a key regulator of
tumour neurogenesis. EGR2 ensures the survival of CSCs and enhances
tumour progression by modulating the expression of homeobox genes,
which are master transcription factors in embryonic development,
and SRY-box (SOX) genes, which regulate neural stem cell fate
(218). In the leukaemia
environment, normal HSPCs protect themselves by assuming a
quiescent state; however, their differentiation is significantly
inhibited. EGR3 and EGR1 have been identified as key genes
regulating this suppressed state. EGR3 is highly expressed in
leukaemia bone marrow, and its ectopic upregulation induces cell
cycle arrest and diminishes the remodelling capacity of HSPCs. EGR3
deletion enhances the proliferation and implantation potential of
HSPCs and mitigates their quiescent state, indicating that EGR3 is
a potent proliferation suppressor. Similarly, EGR1 upregulation
significantly inhibits HSC function and modulates the state of
HSPCs in leukaemia (219).
Additionally, EGR3 can induce melanoma cell differentiation toward
Schwann cell-like differentiation to inhibit tumour progression,
mimicking nevus maturation, which causes significant changes in
cellular morphology, decreased motility and reduced melanin
production. These effects are achieved by modulating myelin protein
zero (MPZ) and collagen type I α 1 chain (COL1A1) expression levels
and involving SOX10-dependent and SOX10-independent regulatory
mechanisms (220).
EGRs: A family of key regulators in tumour
therapy
Combined targeted therapy and natural
compounds
A clinical trial has demonstrated that regimens
combining calcium channel blockers (CCBs) with CD20-targeted agents
yield shorter progression-free survival and overall survival times
compared with patients receiving CD20-targeted therapies alone
(221). CD20-targeted
therapies, including rituximab and obilizumab, induce cell death by
facilitating the calcium ion influx through various mechanisms,
which increases EGR1 expression. CCBs significantly inhibit EGR1
activation by preventing the influx of calcium ions, which
attenuates the efficacy of CD20-targeting drugs (221). Thus, EGR1 may be a potential
biomarker for predicting response to CD20-targeted therapy.
Natural compounds can modulate EGR1 expression,
which affects the behaviour of tumour cells. For instance,
quercetin induces apoptosis in colon cancer cells by increasing
EGR1 levels and promoting NAG1 gene expression; the EGR1 binding
site is located in the NAG1 promoter region, which is one of the
central mechanisms of this process (222). Similarly, resveratrol induces
apoptosis in cancer cells by upregulating EGR1. Resveratrol can
synergistically inhibit lung cancer cell proliferation by driving
the expression of suicide gene therapy vectors through the EGR1
promoter (223). Berberine
affects macrophage polarisation processes by regulating EGR2,
consequently modifying the composition and function of immune cells
in the TME. Macrophages transitioned from an M2-like polarised
state with reduced immunosuppressive capacity and significantly
enhanced antitumour immune responses after exposure to berberine.
For instance, in the TME, conditioned medium (CM) induced bone
marrow-derived macrophages to polarise towards M2-type TAMs,
accompanied by an upregulation of EGR2 mRNA expression levels. This
polarisation process could be inhibited by berberine treatment,
which effectively inhibited CM-induced EGR2 mRNA upregulation
(224).
Resistance to chemotherapy and
radiotherapy
Cisplatin is the primary chemotherapeutic agent for
NSCLC; however, chemoresistance significantly affects treatment
efficacy and patient prognosis. Recently, researchers have
endeavoured to explore new strategies, and the potential of natural
compounds to overcome drug resistance has attracted widespread
attention. For instance, the natural compound α-hederin, extracted
from Nigella sativa, plays an important role in reversing
cisplatin resistance in NSCLC. Specifically, α-hederin enhances the
nuclear localisation of EGR1, inhibits the transcription of
miR-96-5p and alleviates its inhibition of DDIT3, thereby
activating the DDIT3/ATF3 pathway and inducing ferroptosis
(225). Notably, EGR1 does not
act in isolation, and the synergistic effect of EGR1 and p53 in
cisplatin treatment can enhance its anticancer effect in NSCLC
(226). This finding suggests
that co-regulation of EGR1 and other key molecules may offer
additional therapeutic strategies to overcome chemotherapeutic
resistance.
In addition, the role of EGR1 extends beyond NSCLC,
with emerging studies in other types of cancer indicating its
diverse functions. For instance, in bladder cancer, EGR1 reduces
tumour invasion and gemcitabine resistance by directly inhibiting
LINC00839 expression; it regulates cancer cell migration, invasion
and drug resistance through the LINC00839/miR-142 axis and its
downstream target, SOX5 (227).
In breast cancer, medication resistance significantly impairs the
therapeutic efficacy. In tamoxifen (Tam)-sensitive and
Tam-resistant (TamR) breast cancer cell models, recombinant glial
cell line-derived neurotrophic factor (rGDNF) treatment can
activate the GDNF/RET/EGR1 signalling pathway, leading to the
binding of phosphorylated ELK1 to the EGR1 promoter, thereby
increasing EGR1 expression. Subsequently, EGR1 further binds to the
GDNF and Cyclin D1 (CCND1) promoters, forming a positive feedback
loop and upregulating CCND1 expression, driving cell proliferation
and drug resistance (228).
EGR3 is essential in TamR breast cancer. EGR3, as an estrogen
receptor α responsive factor, significantly upregulates oestrone
(E1) expression and Tam in TamR cells and promotes their
transcription by directly binding to the promoter region of the
MCL1 anti-apoptotic gene, thereby increasing cellular resistance to
apoptosis and facilitating drug resistance. Additionally, E1
significantly reduces Tam-induced apoptosis by upregulating EGR3
expression (20). This mechanism
further highlights the potential of targeting EGR in breast cancer
therapy. Gemcitabine is a key drug in the treatment of all stages
of pancreatic cancer; however, drug resistance remains a
significant challenge. EGR1 regulates multidrug resistance protein
1 (MDR1) expression in pancreatic cancer cells by binding to its
promoter, facilitating resistance to gemcitabine. EGR1 knockdown
inhibits MDR1 expression, which enhances apoptosis, attenuates
tumour proliferation and significantly improves the
chemotherapeutic efficacy of gemcitabine (229). These findings provide a new
research direction for combination therapy in pancreatic cancer. A
similar drug resistance issue has been observed in advanced thyroid
cancer, where B-raf proto-oncogene (BRAF) mutations are commonly
found in advanced thyroid cancers, such as papillary thyroid
carcinoma and anaplastic thyroid carcinoma), and a significant
increase in invasiveness is observed in BRAF inhibitor (BRAFi)
resistant thyroid cancer cells. The expression of the extracellular
matrix protein, FN, is significantly upregulated after BRAFi
treatment, which increases the invasiveness of cancer cells.
Moreover, the BRAFi-induced increase in invasiveness is dependent
upon the expression of the EGR1 transcription factor, which is
downregulated by BRAF/ERK1/2 co-inhibitory treatment, and deletion
of EGR1 inhibits the BRAFi-induced increase in invasiveness
(22). This indicates that EGR1
inhibition may be a potential strategy to mitigate BRAFi
resistance. In glioblastoma, temozolomide is a commonly employed
chemotherapeutic agent whose mechanism of action involves
inhibiting the activity of LINC00470. A previous study demonstrated
that LINC00470 activates SOX4 expression through EGR2, while
temozolomide further inhibits SOX4 expression by inhibiting the
transcriptional activation of EGR2 by LINC00470, thereby
effectively preventing glioblastoma progression (22) (Table I).
 | Table IMechanisms of EGRs in chemotherapy
and radiotherapy of various cancer types. |
Table I
Mechanisms of EGRs in chemotherapy
and radiotherapy of various cancer types.
| Cancer type | Drug/Treatment | Mechanism of
action | (Refs.) |
|---|
| Non-small cell lung
cancer | Cisplatin | α-Hederin enhances
nuclear localization of EGR1, inhibits miR-96-5p transcription,
activates the DDIT3/ATF3 pathway and induces ferroptosis. EGR1
synergizes with p53 to enhance anticancer effects. | (225,226) |
| Non-small cell lung
cancer | Radiotherapy
(IR) | IR induces ROS,
triggering lipid peroxidation and ferroptosis. miR-139 suppresses
the NRF2 signalling pathway, enhancing IR efficacy. EGR1 induces
miR-139 expression by binding to its promoter and activating
transcription. | (230) |
| Bladder cancer | Gemcitabine | EGR1 suppresses
LINC00839 expression, reducing tumour invasion and drug resistance.
Through the LINC00839/miR-142 axis, EGR1 regulates SOX5, affecting
cancer cell migration, invasion and drug resistance. | (227) |
| Breast cancer | Tamoxifen | EGR1: rGDNF
activates the GDNF/RET/EGR1 loop, increasing EGR1 expression and
forming a positive feedback loop that upregulates CCND1, promoting
cell proliferation and drug resistance. EGR3: Upregulated in
resistant cells, it directly binds to the MCL1 promoter, enhancing
its transcription, increasing anti-apoptotic capacity and
conferring drug resistance. | (20,228) |
| Pancreatic
cancer | Gemcitabine | EGR1 binds to the
MDR1 promoter to regulate its expression, promoting drug
resistance. Knockdown of EGR1 suppresses MDR1 expression, enhances
apoptosis and improves chemotherapy efficacy. | (229) |
| Thyroid Cancer | BRAFi | BRAFi upregulates
fibronectin expression, enhancing cancer cell invasiveness. EGR1, a
downregulated gene in BRAF/ERK1/2 inhibition therapy, is required
for BRAFi-induced invasiveness. Loss of EGR1 suppresses
BRAFi-induced invasiveness. | (22) |
| Glioblastoma | Temozolomide | Temozolomide
inhibits LINC00470-mediated transcriptional activation of EGR2,
further suppressing SOX4 expression and preventing tumour
progression. | (22) |
Additionally, radiotherapy, an important treatment
for NSCLC, depends on the modulation of molecular mechanisms to
improve efficacy. Ionising radiation (IR) induces lipid
peroxidation and ferroptosis in cancer cells by inducing ROS, a
process regulated by multiple factors (230). Additionally, miR-139 may be a
novel radiosensitiser in NSCLC by inhibiting the nuclear factor
erythroid 2-related factor 2 (NRF2) pathway. Furthermore, miR-139
disrupts NRF2 signalling by directly targeting c-Jun and
karyopherin α2, enhancing IR-induced lipid peroxidation and
ferroptosis. IR induces miR-139 expression through the EGR1
transcription factor, which binds to the miR-139 promoter and
activates its transcription (230) (Table I). These findings indicate that
the multifunctionality of EGRs offers new targets for improving the
efficacy of chemotherapy and radiotherapy in cancer.
Nanodelivery to reprogramme immune cell
function
The functional status of macrophages and NK cells
within the tumour immune microenvironment is essential for
antitumour therapy. Recently, research teams have explored
strategies to target the EGR family (EGR2 and EGR3) using
nanodelivery systems, offering new ideas for tumour therapy. First,
by constructing a vitamin E and sphingomyelin (VitE:SM)
nanoemulsion delivery system, the polarisation state of macrophages
can be effectively reprogrammed. Specifically, the expression of
M2-type markers (such as EGR2) was significantly decreased and the
expression of M1-type markers was significantly increased after
treatment of macrophages with this nanoemulsion, thereby converting
macrophages from M2-type to M1-type and enhancing their antitumour
activity. Additionally, TGF-βR1 inhibitor encapsulated in VitE:SM
nanoemulsions administered to tumour cells inhibited the growth of
primary tumours and significantly reduced the level of TAM
infiltration in the liver (231). Second, since targeting EGR2 can
restore the ability of NK cells, another research team effectively
administered EGR2 small interfering (si)RNA into NK cells utilising
a nanoparticle-based siRNA delivery system. The anti-tumour
function of NK cells was effectively activated using
anti-NKp46-labelled liposome nanoparticles within a 3D organ sphere
model, illustrating that this delivery system can reprogramme in
situ NK cells, thereby enhancing their antitumour activity
(Fig. 6) (190). Third, considering the role of
EGR3 in differentiation, a team of researchers designed a
therapeutic recombinant mRNA vaccine for melanoma utilising a P2A
peptide linking the CD86 and EGR3 genes, administered through lipid
nanoparticles. The vaccine significantly induced an extensive
influx of immune cells, especially T cells, and facilitated the
recruitment of NK cells and DCs. Similarly, serum concentrations of
IFN-γ and TNF-α were significantly increased in treated mice,
significantly prolonging the survival time of hormone-treated mice.
Additionally, NK cells are essential in the initial immune response
activation; after treatment, the expression levels of MPZ and
COL1A1 were significantly upregulated in tumour tissue, while the
expression of Ki-67 was reduced, indicating that the tumours
exhibited Schwann cell-like features (220).
Therefore, nanodelivery strategies targeting EGR2
and EGR3 exhibit strong immunomodulatory potential and can enhance
antitumour immune responses by reprogramming the functional state
of macrophages and NK cells. These studies provide new insights
into the role of the EGR family in tumour therapy and lay the
foundation for developing nanotechnology-based precision
immunotherapy strategies.
Future perspectives: Cutting-edge strategies
to enhance the antitumour activity of TCR-T and chimeric antigen
receptor (CAR)-T cells
Adoptive T-cell therapy has become an essential
strategy for cancer immunotherapy. Engineered TCR-T and CAR-T cell
therapies are two core immunotherapeutic platforms that precisely
direct T cells against tumour antigens and have emerged as two of
the most innovative and effective cancer treatments today. TCR-T
cell therapy is indicated for treating solid tumours and recognises
antigens on the cell membrane and intracellular tumour antigens
presented by the peptide MHC, thereby eliminating the dependence on
antigen expression on the surface of the target cells and allowing
a broader target tumour antigen spectrum (232). TCR-T cells can be activated by
a single target antigen molecule and are more sensitive to low
antigen expression levels than CAR-T cells. Upon recognition of
tumour cells, TCR-T cells are activated and secrete cytokines,
telomerase and perforin, leading to effective tumour cell
destruction (233). Previously,
the regulatory mechanisms of the EGR family in T cells were
examined, using EGR2 as an example, which is a crucial
transcription factor regulating T cell unresponsiveness (anergy)
(133,134,137). In TCR-T cell therapy, anergy is
a common problem in the TME, severely compromising the ability of T
cells to kill tumour cells (232,233). Regulating the signalling
pathway associated with EGR2 is expected to enhance the
functionality of TCR-T cells within the TME. Inhibiting TCR-T cells
from entering an unresponsive state may enhance their ability to
recognise and kill tumour cells, thereby increasing the anticancer
efficacy of TCR-T cell therapy.
CAR-T cell therapy has demonstrated good efficacy
in haematological cancer; however, drug resistance is often
encountered in treating solid tumours, and results are more
limited. CAR-T cells activate type I IFN signalling in response to
prolonged stimulation, inducing epigenetic changes and inhibiting
their antitumour effects (233,234). Knocking down the EGR2
transcription factor blocks the type I IFN-mediated inhibitory
effect and enhances early memory CAR-T cell expansion, which could
improve therapeutic efficacy against liquid and solid tumours. In
addition, IFN-β exposure reverses the protective effect against
chronic antigen-induced immune failure associated with EGR2
deficiency, indicating that EGR2 knockdown mitigates CAR-T cell
dysfunction by inhibiting the type I IFN pathway (234). A further study demonstrated
that the genetic characterisation of EGR2 is correlated with type I
IFN-induced CAR-T cell failure and shortened patient survival,
making the EGR2/type I IFN signalling pathway a new target and
strategy for solid tumour treatment (234).
At present, to the best of our knowledge, there are
no studies that systematically evaluate the feasibility of
nanoparticle-based siRNA delivery systems for the precise
introduction of EGR2 siRNA into TCR-T or CAR-T cells and their
impact on antitumour function. However, this strategy has promising
applications. For example, the use of liposomal nanoparticle
carriers to deliver EGR2 siRNA into TCR-T cells is expected to
successfully reverse the unresponsive state of T cells and enhance
their killing activity in the TME. Similarly, antibody-modified
nanoparticle systems have been used in CAR-T cells, and targeted
EGR2 inhibition is expected to improve the durable activity and
tolerability of CAR-T cells. Furthermore, in 3D tumour models,
antibody-labelled liposome nanoparticles are expected to repair
exhausted CAR-T and CAR-T cells through efficient delivery of EGR2
siRNA, and also significantly enhance their tumour recognition and
lysis. These nanotechnology-based delivery platforms demonstrate
the notable immune potential of targeting the EGR gene and open new
avenues for the next generation of personalised cancer
immunotherapies.
Conclusion
The EGR family, as key zinc finger transcription
factors, exhibits complex dual regulatory roles in tumour onset,
progression and treatment. In the present review, the multiple
functions of the EGR family in the regulation of tumour cell
proliferation, apoptosis, metastasis, angiogenesis and the
immunological milieu have been systematically summarized,
highlighting their important roles in tumour metabolism,
differentiation and neurogenesis. EGR1, the most extensively
researched member of this family, demonstrates dual pro-cancer and
anticancer functions in tumour cells and influences tumour immune
escape and the efficacy of immunotherapy by regulating the
functional status of immune cells (including T cells, B cells,
macrophages and NK cells). Additionally, the roles of EGR2, EGR3
and EGR4 in tumours, especially their unique functions in immune
regulation and metabolic regulation, have been progressively
clarified, offering new targets for tumour therapy. Based on the
regulatory mechanism of the EGR family, the present review
investigated the potential of combined targeted therapies, natural
compounds, chemotherapy, radiotherapy and nanodelivery systems in
tumour therapy, particularly focusing on the enhancement of the
antitumour efficacy of CAR-T and CAR-T cells by modulating members
of the EGR family, which represents a new strategy for overcoming
tumour drug resistance and immunosuppression. In the future,
in-depth studies on the molecular mechanisms of the EGR family in
tumours and its interaction with other signalling pathways will
facilitate the development of more precise and effective tumour
therapeutic regimens and advance the development of personalised
immunotherapy.
Availability of data and materials
Not applicable.
Authors' contributions
RG, RW, WZ, YL, YW, HW, XL and JS conceptualized
the study. RG wrote the manuscript. JS and XL reviewed the
manuscript. RG, RW, WZ, YL, YW, HW, XL and JS contributed to
manuscript editing. Data authentication is not applicable. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This research topic has been sponsored by Yancheng Science and
Technology Bureau (grant no. YCBE202421), The Special Funds for
Science Development of the Clinical Teaching Hospitals of Jiangsu
Vocational College of Medicine (grant no. 20209113). The 2021
Jiangsu Provincial Health and Health Commission Medical Research
Guidance Project (grant no. Z2021087), The Special Funds for
Science Development of the Clinical Teaching Hospitals of Jiangsu
Vocational College of Medicine (grant no. 20219105), The 2021
Yancheng Medical Science and Technology Development Plan Project
(grant no. YK2021060) and the Nantong University's 2023 Academic
Level Research Project (Special Project of Yancheng Third
Institute; grant no. YXY-Z2023008).
References
|
1
|
Beckmann AM, Matsumoto I and Wilce PA:
AP-1 and Egr DNA-binding activities are increased in rat brain
during ethanol withdrawal. J Neurochem. 69:306–314. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Skerka C, Decker EL and Zipfel PF:
Coordinate expression and distinct DNA-Binding characteristics of
the four EGR-zinc finger proteins in jurkat T lymphocytes.
Immunobiology. 198:179–191. 1997. View Article : Google Scholar
|
|
3
|
Chavrier P, Zerial M, Lemaire P, Almendral
J, Bravo R and Charnay P: A gene encoding a protein with zinc
fingers is activated during G0/G1 transition in cultured cells.
EMBO J. 7:29–35. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Joseph LJ, Le Beau MM, Jamieson GA,
Acharya S, Shows TB, Rowley JD and Sukhatme VP: Molecular cloning,
sequencing, and mapping of EGR2, a human early growth response gene
encoding a protein with 'zinc-binding finger' structure. Proc Natl
Acad Sci USA. 85:7164–7168. 1988. View Article : Google Scholar
|
|
5
|
Cao X, Mahendran R, Guy GR and Tan YH:
Detection and characterization of cellular EGR-1 binding to its
recognition site. J Biol Chem. 268:16949–16957. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Myung E, Park YL, Kim N, Chung CY, Park
HB, Park HC, Myung DS, Kim JS, Cho SB, Lee WS and Joo YE:
Expression of early growth response-1 in human gastric cancer and
its relationship with tumor cell behaviors and prognosis. Pathol
Res Pract. 209:692–699. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li L, Ameri AH, Wang S, Jansson KH, Casey
OM, Yang Q, Beshiri ML, Fang L, Lake RG, Agarwal S, et al: EGR1
regulates angiogenic and osteoclastogenic factors in prostate
cancer and promotes metastasis. Oncogene. 38:6241–6255. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schmidt K, Carroll JS, Yee E, Thomas DD,
Wert-Lamas L, Neier SC, Sheynkman G, Ritz J and Novina CD: The
lncRNA SLNCR recruits the androgen receptor to EGR1-Bound genes in
melanoma and inhibits expression of tumor suppressor p21. Cell
Reports. 27:2493–2507.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Calogero A, Arcella A, De Gregorio G,
Porcellini A, Mercola D, Liu C, Lombari V, Zani M, Giannini G,
Gagliardi FM, et al: The early growth response gene EGR-1 behaves
as a suppressor gene that is down-regulated independent of ARF/Mdm2
but not p53 alterations in fresh human gliomas. Clin Cancer Res.
7:2788–2796. 2001.PubMed/NCBI
|
|
10
|
Damm F, Mylonas E, Cosson A, Yoshida K,
Della Valle V, Mouly E, Diop M, Scourzic L, Shiraishi Y, Chiba K,
et al: Acquired initiating mutations in early hematopoietic cells
of CLL patients. Cancer Discov. 4:1088–1101. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Oakes CC, Seifert M, Assenov Y, Gu L,
Przekopowitz M, Ruppert AS, Wang Q, Imbusch CD, Serva A, Koser SD,
et al: DNA methylation dynamics during B cell maturation underlie a
continuum of disease phenotypes in chronic lymphocytic leukemia.
Nat Genet. 48:253–264. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ying Y, Ma X, Fang J, Chen S, Wang W, Li
J, Xie H, Wu J, Xie B, Liu B, et al: EGR2-mediated regulation of
m6A reader IGF2BP proteins drive RCC tumorigenesis and metastasis
via enhancing S1PR3 mRNA stabilization. Cell Death Dis. 12:7502021.
View Article : Google Scholar :
|
|
13
|
Xiao H, Huang X, Chen H, Zheng Y, Liu W,
Chen J, Wei R, Lin M, Wang Q and Zhuang W: Establishment of a SUMO
pathway related gene signature for predicting prognosis,
chemotherapy response and investigating the role of EGR2 in bladder
cancer. J Cancer. 15:3841–3856. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schwachtgen JL, Houston P, Campbell C,
Sukhatme V and Braddock M: Fluid shear stress activation of egr-1
transcription in cultured human endothelial and epithelial cells is
mediated via the extracellular signal-related kinase 1/2
mitogen-activated protein kinase pathway. J Clin Invest.
101:2540–2549. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim JH, Jeong IY, Lim YH, Lee YH and Shin
SY: Estrogen receptor β stimulates Egr-1 transcription via
MEK1/Erk/Elk-1 cascade in C6 glioma cells. BMB Rep. 44:452–457.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Masle-Farquhar E, Peters TJ, Miosge LA,
Parish IA, Weigel C, Oakes CC, Reed JH and Goodnow CC: Uncontrolled
CD21low age-associated and B1 B cell accumulation caused by failure
of an EGR2/3 tolerance checkpoint. Cell Rep. 38:1102592022.
View Article : Google Scholar
|
|
17
|
Veremeyko T, Yung AWY, Anthony DC,
Strekalova T and Ponomarev ED: Corrigendum: Early growth response
Gene-2 is essential for M1 and M2 macrophage activation and
plasticity by modulation of the transcription factor CEBPβ. Front
Immunol. 9:29232018. View Article : Google Scholar
|
|
18
|
Zhang S, Xia C, Xu C, Liu J, Zhu H, Yang
Y, Xu F, Zhao J, Chang Y and Zhao Q: Early growth response 3
inhibits growth of hepatocellular carcinoma cells via upregulation
of Fas ligand. Int J Oncol. 50:805–814. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shin SH, Kim I, Lee JE, Lee M and Park JW:
Loss of EGR3 is an independent risk factor for metastatic
progression in prostate cancer. Oncogene. 39:5839–5854. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xie Y, Han X, Yu J, Yuan M, Yan Y, Qin J,
Lan L and Wang Y: EGR3 and estrone are involved in the tamoxifen
resistance and progression of breast cancer. J Cancer Res Clin
Oncol. 149:18103–18117. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Inoue A, Omoto Y, Yamaguchi Y, Kiyama R
and Hayashi S: Transcription factor EGR3 is involved in the
estrogen-signaling pathway in breast cancer cells. J Mol
Endocrinol. 32:649–661. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hicks HM, Pozdeyev N, Sams SB, Pugazhenthi
U, Bales ES, Hofmann MC, McKenna LR and Schweppe RE: Fibronectin
contributes to a BRAF Inhibitor-driven invasive phenotype in
thyroid cancer through EGR1, which can be blocked by inhibition of
ERK1/2. Mol Cancer Res. 21:867–880. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hua Y, Wang H, Ye Z, Zheng D and Zhang X:
An integrated pan-cancer analysis of identifying biomarkers about
the EGR family genes in human carcinomas. Comput Biol Med.
148:1058892022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Joyce JA and Fearon DT: T cell exclusion,
immune privilege, and the tumor microenvironment. Science.
348:74–80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Quail DF and Joyce JA: The
microenvironmental landscape of brain tumors. Cancer Cell.
31:326–341. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
LeBlanc SE, Ward RM and Svaren J:
Neuropathy-associated Egr2 mutants disrupt cooperative activation
of myelin protein zero by Egr2 and Sox10. Mol Cell Biol.
27:3521–3529. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Collins S, Lutz MA, Zarek PE, Anders RA,
Kersh GJ and Powell JD: Opposing regulation of T cell function by
Egr-1/NAB2 and Egr-2/Egr-3. Eur J Immunol. 38:528–536. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Samakai E, Hooper R, Martin KA, Shmurak M,
Zhang Y, Kappes DJ, Tempera I and Soboloff J: Novel STIM1-dependent
control of Ca2+ clearance regulates NFAT activity during
T-cell activation. FASEB J. 30:3878–3886. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ge Y, Liu H, Huang W, Zhu H, Zong D and He
X: Immunoinhibitory effects of hypoxia-driven reprogramming of
EGR1hi and EGR3 positive B cells in the nasopharyngeal carcinoma
microenvironment. Oral Oncol. 158:1069992024. View Article : Google Scholar
|
|
30
|
Veremeyko T, Yung AWY, Anthony DC,
Strekalova T and Ponomarev ED: Early growth response Gene-2 is
essential for M1 and M2 macrophage activation and plasticity by
modulation of the transcription factor CEBPβ. Front Immunol.
9:25152018. View Article : Google Scholar
|
|
31
|
Thiel G, Müller I and Rössler OG:
Expression, signaling and function of Egr transcription factors in
pancreatic β-cells and insulin-responsive tissues. Mol Cell
Endocrinol. 388:10–19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Srinivasan R, Mager GM, Ward RM, Mayer J
and Svaren J: NAB2 Represses Transcription by Interacting with the
CHD4 Subunit of the Nucleosome Remodeling and Deacetylase (NuRD)
Complex. J Biol Chem. 281:15129–15137. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Silverman ES and Collins T: Pathways of
Egr-1-mediated gene transcription in vascular biology. Am J Pathol.
154:665–670. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gregg J and Fraizer G: Transcriptional
regulation of EGR1 by EGF and the ERK signaling pathway in prostate
cancer cells. Genes Cancer. 2:900–909. 2011. View Article : Google Scholar
|
|
35
|
Cavigelli M, Dolfi F, Claret FX and Karin
M: Induction of c-fos expression through JNK-mediated TCF/Elk-1
phosphorylation. EMBO J. 14:5957–5964. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cabodi S, Morello V, Masi A, Cicchi R,
Broggio C, Distefano P, Brunelli E, Silengo L, Pavone F, Arcangeli,
et al: Convergence of integrins and EGF receptor signaling via
PI3K/Akt/FoxO pathway in early gene Egr-1 expression. J Cell
Physiol. 218:294–303. 2009. View Article : Google Scholar
|
|
37
|
Ivanova D, Dirks A, Montenegro-Venegas C,
Schöne C, Altrock WD, Marini C, Frischknecht R, Schanze D, Zenker
M, Gundelfinger ED and Fejtova A: Synaptic activity controls
localization and function of Ct BP 1 via binding to B assoon and P
iccolo. EMBO J. 34:1056–1077. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Thyss R, Virolle V, Imbert V, Peyron JF,
Aberdam D and Virolle T: NF-κB/Egr-1/Gadd45 are sequentially
activated upon UVB irradiation to mediate epidermal cell death.
EMBO J. 24:128–137. 2005. View Article : Google Scholar
|
|
39
|
Rössler OG and Thiel G: Thrombin induces
Egr-1 expression in fibroblasts involving elevation of the
intracellular Ca2+ concentration, phosphorylation of ERK and
activation of ternary complex factor. BMC Mol Biol. 10:402009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mayer SI, Willars GB, Nishida E and Thiel
G: Elk-1, CREB, and MKP-1 regulate Egr-1 expression in
gonadotropin-releasing hormone stimulated gonadotrophs. J Cell
Biochem. 105:1267–1278. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Simo-Cheyou ER, Tan JJ, Grygorczyk R and
Srivastava AK: STIM-1 and ORAI-1 channel mediate
angiotensin-II-induced expression of Egr-1 in vascular smooth
muscle cells. J Cell Physiol. 232:3496–3509. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jain N, Mahendran R, Philp R, Guy GR, Tan
YH and Cao X: Casein Kinase II associates with Egr-1 and acts as a
negative modulator of its DNA binding and transcription activities
in NIH 3T3 Cells. J Biol Chem. 271:13530–13536. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Manente AG, Pinton G, Tavian D,
Lopez-Rodas G, Brunelli E and Moro L: Coordinated Sumoylation and
Ubiquitination Modulate EGF Induced EGR1 Expression and Stability.
PLoS One. 6:e256762011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rexach JE, Clark PM and Hsieh-Wilson LC:
Chemical approaches to understanding O-GlcNAc glycosylation in the
brain. Nat Chem Biol. 4:97–106. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cheng D, Dong Z, Lin P, Shen G and Xia Q:
Transcriptional activation of Ecdysone-responsive genes requires
H3K27 acetylation at enhancers. Int J Mol Sci. 23:107912022.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xiong YJ, Zhu Y, Liu YL, Zhao YF, Shen X,
Zuo WQ, Lin F and Liang ZQ: P300 participates in ionizing
Radiation-mediated activation of Cathepsin L by mutant p53. J
Pharmacol Exp Ther. 378:276–286. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu Z, Huang L, Zhao S, Wang J, Zhang C,
Song X, Chen Q, Du J, Yu D, Sun X, et al: Early growth Response 1
strengthens Pol-III-Directed transcription and transformed cell
proliferation by controlling PTEN/AKT signalling activity. Int J
Mol Sci. 23:49302022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang Y, Yun C, Gao B, Xu Y, Zhang Y, Wang
Y, Kong Q, Zhao F, Wang CR, Dent SYR, et al: The lysine
acetyltransferase GCN5 is required for iNKT cell development
through EGR2 acetylation. Cell Rep. 20:600–612. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Guan X, Deng H, Choi UL, Li Z, Yang Y,
Zeng J, Liu Y, Zhang X and Li G: EZH2 overexpression dampens
tumor-suppressive signals via an EGR1 silencer to drive breast
tumorigenesis. Oncogene. 39:7127–7141. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mendes K, Schmidhofer S, Minderjahn J,
Glatz D, Kiesewetter C, Raithel J, Wimmer J, Gebhard C and Rehli M:
The epigenetic pioneer EGR2 initiates DNA demethylation in
differentiating monocytes at both stable and transient binding
sites. Nat Commun. 12:15562021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Scheid R, Chen J and Zhong X: Biological
role and mechanism of chromatin readers in plants. Curr Opin Plant
Biol. 61:1020082021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hu TM, Chen SJ, Hsu SH and Cheng MC:
Functional analyses and effect of DNA methylation on the EGR1 gene
in patients with schizophrenia. Psychiatry Res. 275:276–282. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Salguero-Aranda C, Beltran-Povea A,
Postigo-Corrales F, Hitos AB, Díaz I, Caballano-Infantes E, Fraga
MF, Hmadcha A, Martín F, Soria B, et al: Pdx1 is transcriptionally
regulated by EGR-1 during nitric oxide-induced endoderm
differentiation of mouse embryonic stem cells. Int J Mol Sci.
23:39202022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fetahu IS and Taschner-Mandl S:
Neuroblastoma and the epigenome. Cancer Metastasis Rev. 40:173–189.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Saha SK, Islam SMR, Saha T, Nishat A,
Biswas PK, Gil M, Nkenyereye L, El-Sappagh S, Islam MS and Cho SG:
Prognostic role of EGR1 in breast cancer: A systematic review. BMB
Rep. 54:497–504. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gong Y, Chang C, Liu X, He Y, Wu Y, Wang S
and Zhang C: Stimulator of interferon genes signaling pathway and
its role in Anti-tumor immune therapy. Curr Pharm Des.
26:3085–3095. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yang J, Chen M, Li R, Sun Y, Ye P, Fang K,
Wang C, Shi S and Dong C: A responsive cocktail nano-strategy
breaking the immune excluded state enhances immunotherapy for
triple negative breast cancer. Nanoscale. 17:4610–4623. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yuan H, Qiu C, Wang X, Wang P, Yi L, Peng
X, Xu X, Huang W, Bai Y, Wei J, et al: Engineering semiconducting
polymeric nanoagonists potentiate cGAS-STING pathway activation and
elicit long term memory against recurrence in breast cancer. Adv
Mater. 37:e24066622025. View Article : Google Scholar
|
|
59
|
Hao M, Zhu L, Hou S, Chen S, Li X, Li K,
Zhu N, Chen S, Xue L, Ju C and Zhang C: Sensitizing tumors to
immune checkpoint blockage via STING Agonists delivered by
tumor-penetrating neutrophil cytopharmaceuticals. ACS Nano.
17:1663–1680. 2023. View Article : Google Scholar
|
|
60
|
Noritsugu K, Ito A, Nakao Y and Yoshida M:
Identification of zinc finger transcription factor EGR2 as a novel
acetylated protein. Biochem Biophys Res Commun. 489:455–459. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Stoddart A, Fernald AA, Davis EM, McNerney
ME and Le Beau MM: EGR1 haploinsufficiency confers a fitness
advantage to hematopoietic stem cells following chemotherapy. Exp
Hematol. 115:54–67. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Inoue K and Fry EA: Tumor suppression by
the EGR1, DMP1, ARF, p53, and PTEN network. Cancer Invest.
36:520–536. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sun T, Zhang Y, Zhong S, Gao F, Chen Y,
Wang B, Cai W, Zhang Z, Li W, Lu S, et al: N-n-Butyl haloperidol
iodide, a derivative of the Anti-psychotic haloperidol, antagonizes
Hypoxia/reoxygenation injury by inhibiting an Egr-1/ROS positive
feedback loop in H9c2 cells. Front Pharmacol. 9:192018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tseng YC, Shu CW, Chang HM, Lin YH, Tseng
YH, Hsu HS, Goan YG and Tseng CJ: Assessment of early growth
response 1 in tumor suppression of esophageal squamous cell
carcinoma. J Clin Med. 11:57922022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wang B, Zhang S, Wang H, Wang M, Tao Y, Ye
M, Fan Z, Wang Y and Liu L: Identification of EGR4 as a prospective
target for inhibiting tumor cell proliferation and a novel
biomarker in colorectal cancer. Cancer Gene Ther. 31:871–883. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang L, Lu J, Song Y, Bai J, Sun W, Yu J,
Cai M and Fu S: Analysis of DNA Repair-related prognostic function
and mechanism in gastric cancer. Front Cell Dev Biol.
10:8970962022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sehat B, Andersson S, Vasilcanu R, Girnita
L and Larsson O: Role of Ubiquitination in IGF-1 receptor signaling
and degradation. PLoS One. 2:e3402007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kuo P, Chen Y, Chen T, Shen K and Hsu Y:
CXCL5/ENA78 increased cell migration and epithelial-to-mesenchymal
transition of hormone-independent prostate cancer by early growth
response-1/snail signaling pathway. J Cell Physiol. 226:1224–1231.
2011. View Article : Google Scholar
|
|
69
|
Xiao D, Chinnappan D, Pestell R, Albanese
C and Weber HC: Bombesin regulates Cyclin D1 expression through the
early growth response protein Egr-1 in prostate cancer cells.
Cancer Res. 65:9934–9942. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yin L, Zhang J and Sun Y: Early growth
response-1 is a new substrate of the GSK3β-FBXW7 axis. Neoplasia.
34:1008392022. View Article : Google Scholar
|
|
71
|
Young E, Noerenberg D, Mansouri L,
Ljungström V, Frick M, Sutton LA, Blakemore SJ, Galan-Sousa J,
Plevova K, Baliakas P, et al: EGR2 mutations define a new
clinically aggressive subgroup of chronic lymphocytic leukemia.
Leukemia. 31:1547–1554. 2017. View Article : Google Scholar
|
|
72
|
Grotegut S, Von Schweinitz D, Christofori
G and Lehembre F: Hepatocyte growth factor induces cell scattering
through MAPK/Egr-1-mediated upregulation of Snail. EMBO J.
25:3534–3545. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wu WS, You RI, Cheng CC, Lee MC, Lin TY
and Hu CT: Author correction: Snail collaborates with EGR-1 and
SP-1 to directly activate transcription of MMP 9 and ZEB1. Sci Rep.
8:142262018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yeo H, Lee JY, Kim J, Ahn SS, Jeong JY,
Choi JH, Lee YH and Shin SY: Transcription factor EGR-1
transactivates the MMP1 gene promoter in response to TNFα in HaCaT
keratinocytes. BMB Rep. 53:323–328. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Walcher L, Kistenmacher AK, Suo H, Kitte
R, Dluczek S, Strauß A, Blaudszun AR, Yevsa T, Fricke S and
Kossatz-Boehlert U: Cancer stem cells-origins and biomarkers:
Perspectives for targeted personalized therapies. Front Immunol.
11:12802020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sun T, Tian H, Feng YG, Zhu YQ and Zhang
WQ: Egr-1 promotes cell proliferation and invasion by increasing
β-Catenin expression in gastric cancer. Dig Dis Sci. 58:423–430.
2012.
|
|
77
|
Cheng JC, Chang HM and Leung PC: Egr-1
mediates epidermal growth factor-induced downregulation of
E-cadherin expression via Slug in human ovarian cancer cells.
Oncogene. 32:1041–1049. 2013. View Article : Google Scholar
|
|
78
|
Wang Y, Qin C, Zhao B, Li Z, Li T, Yang X,
Zhao Y and Wang W: EGR1 induces EMT in pancreatic cancer via a
P300/SNAI2 pathway. J Transl Med. 21:2012023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chen H, Kuo T, Tseng CF, Ma JT, Yang ST,
Yen CJ, Yang CY, Sung SY and Su JL: Angiopoietin-like protein 1
antagonizes MET receptor activity to repress sorafenib resistance
and cancer stemness in hepatocellular carcinoma. Hepatology.
64:1637–1651. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yang W, Nam K, Ju J, Lee K, Oh S and Shin
I: S100A4 negatively regulates β-catenin by inducing the
Egr-1-PTEN-Akt-GSK3β degradation pathway. Cell Signal.
26:2096–2106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wang C, Ji J, Jin Y, Sun Y, Cai Q, Jiang
J, Guo L, Zhou C and Zhang J: Tumor-mesothelium HOXA11-PDGF BB/TGF
β1-miR-181a-5p-Egr1 feedforward amplifier circuity propels
mesothelial fibrosis and peritoneal metastasis of gastric cancer.
Oncogene. 43:171–188. 2024. View Article : Google Scholar
|
|
82
|
Feng YH, Su YC, Lin SF, Lin PR, Wu CL,
Tung CL, Li CF, Shieh GS and Shiau AL: Oct4 upregulates osteopontin
via Egr1 and is associated with poor outcome in human lung cancer.
BMC Cancer. 19:7912019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Raue F and Frank-Raue K: Thyroid cancer:
Risk-stratified management and individualized therapy. Clin Cancer
Res. 22:5012–5021. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Ma C, Zhang N, Wang T, Guan H, Huang Y,
Huang L, Zheng Y, Zhang L, Han L, Huo Y, et al: Inflammatory
cytokine-regulated LNCPTCTS suppresses thyroid cancer progression
via enhancing Snail nuclear export. Cancer Lett. 575:2164022023.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Brown KC, Lau JK, Dom AM, Witte TR, Luo H,
Crabtree CM, Shah YH, Shiflett BS, Marcelo AJ, Proper NA, et al:
MG624, an α7-nAChR antagonist, inhibits angiogenesis via the
Egr-1/FGF2 pathway. Angiogenesis. 15:99–114. 2012. View Article : Google Scholar
|
|
86
|
Sperandio S, Fortin J, Sasik R, Robitaille
L, Corbeil J and De Belle I: The transcription factor Egr1
regulates the HIF-1α gene during hypoxia. Mol Carcinog. 48:38–44.
2009. View Article : Google Scholar
|
|
87
|
Shimoyamada H, Yazawa T, Sato H, Okudela
K, Ishii J, Sakaeda M, Kashiwagi K, Suzuki T, Mitsui H, Woo T, et
al: Early growth Response-1 induces and enhances vascular
endothelial growth Factor-A expression in lung cancer cells. Am J
Pathol. 177:70–83. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yan J, Gao Y, Lin S, Li Y, Shi L and Kan
Q: EGR1-CCL2 feedback loop maintains Epithelial-mesenchymal
transition of Cisplatin-resistant gastric cancer cells and promotes
tumor angiogenesis. Dig Dis Sci. 67:3702–3713. 2022. View Article : Google Scholar
|
|
89
|
Yu J, Zhuang A, Gu X, Hua Y, Yang L, Ge S,
Ruan J, Chai P, Jia R and Fan X: Nuclear PD-L1 promotes
EGR1-mediated angiogenesis and accelerates tumorigenesis. Cell
Discov. 9:332023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
McCaffrey TA, Fu C, Du B, Eksinar S, Kent
KC, Bush H Jr, Kreiger K, Rosengart T, Cybulsky MI, Silverman ES
and Collins T: High-level expression of Egr-1 and Egr-1-inducible
genes in mouse and human atherosclerosis. J Clin Invest.
105:653–662. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Harja E, Bucciarelli LG, Lu Y, Stern DM,
Zou YS, Schmidt AM and Yan SF: Early growth Response-1 promotes
atherogenesis: Mice deficient in early growth Response-1 and
apolipoprotein E display decreased atherosclerosis and vascular
inflammation. Circ Res. 94:333–339. 2004. View Article : Google Scholar
|
|
92
|
Yan SF, Fujita T, Lu J, Okada K, Shan Zou
Y, Mackman N, Pinsky DJ and Stern DM: Egr-1, a master switch
coordinating upregulation of divergent gene families underlying
ischemic stress. Nat Med. 6:1355–1361. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Suehiro J, Hamakubo T, Kodama T, Aird WC
and Minami T: Vascular endothelial growth factor activation of
endothelial cells is mediated by early growth response-3. Blood.
115:2520–2532. 2010. View Article : Google Scholar :
|
|
94
|
Minami T, Miura M, Aird WC and Kodama T:
Thrombin-induced autoinhibitory factor, down syndrome critical
Region-1, attenuates NFAT-dependent vascular cell adhesion
Molecule-1 expression and inflammation in the endothelium. J Biol
Chem. 281:20503–20520. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Minami T, Horiuchi K, Miura M, Abid MR,
Takabe W, Noguchi N, Kohro T, Ge X, Aburatani H, Hamakubo T, et al:
Vascular endothelial growth Factor- and Thrombin-induced
termination factor, down syndrome critical Region-1, attenuates
endothelial cell proliferation and angiogenesis. J Biol Chem.
279:50537–50554. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Sambasivan S: Epithelial ovarian cancer:
Review article. Cancer Treat Res Commun. 33:1006292022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
De Belle I, Huang RP, Fan Y, Liu C,
Mercola D and Adamson ED: p53 and Egr-1 additively suppress
transformed growth in HT1080 cells but Egr-1 counteracts
p53-dependent apoptosis. Oncogene. 18:3633–3642. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Ryu J, Choe SS, Ryu SH, Park EY, Lee BW,
Kim TK, Ha CH and Lee SW: Paradoxical induction of growth arrest
and apoptosis by EGF via the up-regulation of PTEN by activating
Redox factor-1/Egr-1 in human lung cancer cells. Oncotarget.
8:4181–4195. 2017. View Article : Google Scholar :
|
|
99
|
Wang C, Husain K, Zhang A, Centeno BA,
Chen DT, Tong Z, Sebti SM and Malafa MP: EGR-1/Bax pathway plays a
role in vitamin E δ-tocotrienol-induced apoptosis in pancreatic
cancer cells. J Nutr Biochem. 26:797–807. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Tian X, Traub B, Shi J, Huber N, Schreiner
S, Chen G, Zhou S, Henne-Bruns D, Knippschild U and Kornmann M:
c-Jun N-terminal kinase 2 suppresses pancreatic cancer growth and
invasion and is opposed by c-Jun N-terminal kinase 1. Cancer Gene
Ther. 29:73–86. 2022. View Article : Google Scholar :
|
|
101
|
Moorehead RA, Hojilla CV, De Belle I, Wood
GA, Fata JE, Adamson ED, Watson KL, Edwards DR and Khokha R:
Insulin-like Growth Factor-II regulates PTEN expression in the
mammary gland. J Biol Chem. 278:50422–50427. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Shan LN, Song YG, Su D, Liu YL, Shi XB and
Lu SJ: Early growth response Protein-1 involves in transforming
growth factor-β1 induced Epithelial-mesenchymal transition and
inhibits migration of Non-Small-cell lung cancer cells. Asian Pac J
Cancer Prev. 16:4137–4142. 2015. View Article : Google Scholar
|
|
103
|
Wang B, Wang Y, Wang W, Wang Z, Zhang Y,
Pan X, Wen X, Leng H, Guo J and Ma XX: WTAP/IGF2BP3 mediated m6A
modification of the EGR1/PTEN axis regulates the malignant
phenotypes of endometrial cancer stem cells. J Exp Clin Cancer Res.
43:2042024. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Unoki M and Nakamura Y: Growth-suppressive
effects of BPOZ and EGR2, two genes involved in the PTEN signaling
pathway. Oncogene. 20:4457–4465. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Unoki M and Nakamura Y: EGR2 induces
apoptosis in various cancer cell lines by direct transactivation of
BNIP3L and BAK. Oncogene. 22:2172–2185. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Chen YL, Lin PC, Chen SP, Lin CC, Tsai NM,
Cheng YL, Chang WL, Lin SZ and Harn HJ: Activation of nonsteroidal
Anti-inflammatory Drug-activated Gene-1 via extracellular
Signal-regulated kinase 1/2 Mitogen-Activated protein kinase
revealed a isochaihulactone-triggered apoptotic pathway in human
lung cancer A549 cells. J Pharmacol Exp Ther. 323:746–756. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Vaish V, Piplani H, Rana C, Vaiphei K and
Sanyal SN: NSAIDs may regulate EGR-1-mediated induction of reactive
oxygen species and non-steroidal anti-inflammatory drug-induced
gene (NAG)-1 to initiate intrinsic pathway of apoptosis for the
chemoprevention of colorectal cancer. Mol Cell Biochem. 378:47–64.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Liu Y, Su Z, Tavana O and Gu W:
Understanding the complexity of p53 in a new era of tumor
suppression. Cancer Cell. 42:946–967. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Ko H, Kim JM, Kim SJ, Shim SH, Ha CH and
Chang HI: Induction of apoptosis by genipin inhibits cell
proliferation in AGS human gastric cancer cells via Egr1/p21
signaling pathway. Bioorg Med Chem Letts. 25:4191–4196. 2015.
View Article : Google Scholar
|
|
110
|
Liu J, Grogan L, Nau M, Allegra C, Chu E
and Wright J: Physical interaction between p53 and primary response
gene Egr-1. Int J Oncol. 18:863–870. 2001.PubMed/NCBI
|
|
111
|
Meng W, Yu S, Li Y, Wang H, Feng Y, Sun W,
Liu Y, Sun S and Liu H: Mutant p53 achieves function by regulating
EGR1 to induce epithelial mesenchymal transition. Tissue Cell.
90:1025102024. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ahmed MM, Sells SF, Venkatasubbarao K,
Fruitwala SM, Muthukkumar S, Harp C, Mohiuddin M and Rangnekar VM:
Ionizing Radiation-inducible apoptosis in the absence of p53 linked
to transcription factor EGR-1. J Biol Chem. 272:33056–33061. 1997.
View Article : Google Scholar
|
|
113
|
Adams PD: Healing and hurting: Molecular
mechanisms, functions, and pathologies of cellular senescence. Mol
Cell. 36:2–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Serrano M, Lin AW, McCurrach ME, Beach D
and Lowe SW: Oncogenic ras provokes premature cell senescence
associated with accumulation of p53 and p16INK4a. Cell. 88:593–602.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Salotti J, Sakchaisri K, Tourtellotte WG
and Johnson PF: An Arf-Egr-C/EBPβ pathway linked to Ras-induced
senescence and cancer. Mol Cell Biol. 35:866–883. 2015. View Article : Google Scholar :
|
|
116
|
Lucerna M, Pomyje J, Mechtcheriakova D,
Kadl A, Gruber F, Bilban M, Sobanov Y, Schabbauer G, Breuss J,
Wagner O, et al: Sustained expression of early growth response
Protein-1 blocks angiogenesis and tumor growth. Cancer Res.
66:6708–6713. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Liu M, Wang X, Peng Y, Shen S and Li G:
Egr-1 regulates the transcription of NGX6 gene through a Sp1/Egr-1
overlapping site in the promoter. BMC Mol Biol. 15:142014.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Wang LF, Liu YS, Yang B, Li P, Cheng XS,
Xiao CX, Liu JJ, Li S, Ren JL and Guleng B: The extracellular
matrix protein mindin attenuates colon cancer progression by
blocking angiogenesis via Egr-1-mediated regulation. Oncogene.
37:601–615. 2018. View Article : Google Scholar
|
|
119
|
Kim J, Kang SM, Lee HJ, Choi SY and Hong
SH: Oxytocin inhibits head and neck squamous cell carcinoma cell
migration by early growth response-1 upregulation. Anticancer
Drugs. 28:613–622. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Liu P, Li J, Lu H and Xu B: Thalidomide
inhibits leukemia cell invasion and migration by upregulation of
early growth response gene 1. Leuk Lymphoma. 50:109–113. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Huang R, Li S, Yang W, Chen L, Yao C and
Huang RP: Early growth Response-1 suppresses human fibrosarcoma
cell invasion and angiogenesis. Cancer Genomics Proteomics.
3:71–82. 2006.PubMed/NCBI
|
|
122
|
Mookerjee-Basu J, Hooper R, Gross S,
Schultz B, Go CK, Samakai E, Ladner J, Nicolas E, Tian Y, Zhou B,
et al: Suppression of Ca2+ signals by EGR 4 controls Th1
differentiation and anti-cancer immunity in vivo. EMBO Rep.
21:e489042020. View Article : Google Scholar
|
|
123
|
Zheng Y, Zha Y, Spaapen RM, Mathew R, Barr
K, Bendelac A and Gajewski TF: Egr2-dependent gene expression
profiling and ChIP-Seq reveal novel biologic targets in T cell
anergy. Mol Immunol. 55:283–291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Williams JB, Horton BL, Zheng Y, Duan Y,
Powell JD and Gajewski TF: The EGR2 targets LAG-3 and 4-1BB
describe and regulate dysfunctional antigen-specific CD8+ T cells
in the tumor microenvironment. J Exp Med. 214:381–400. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Zheng Y, Zha Y, Driessens G, Locke F and
Gajewski TF: Transcriptional regulator early growth response gene 2
(Egr2) is required for T cell anergy in vitro and in vivo. J Exp
Med. 209:2157–2163. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Crispín JC and Tsokos GC: Transcriptional
regulation of IL-2 in health and autoimmunity. Autoimmun Rev.
8:190–195. 2009. View Article : Google Scholar :
|
|
127
|
Decker EL: Early growth response proteins
(EGR) and nuclear factors of activated T cells (NFAT) form
heterodimers and regulate proinflammatory cytokine gene expression.
Nucleic Acids Res. 31:911–921. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Lin JX and Leonard WJ: The immediate-early
gene product Egr-1 regulates the human interleukin-2 receptor
beta-chain promoter through noncanonical Egr and Sp1 binding sites.
Mol Cell Biol. 17:3714–3722. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Lindgren H, Axcrona K and Leanderson T:
Regulation of transcriptional activity of the murine CD40 Ligand
promoter in response to signals through TCR and the costimulatory
molecules CD28 and CD2. J Immunol. 166:4578–4585. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Cron RQ, Bandyopadhyay R, Genin A, Brunner
M, Kersh GJ, Yin J, Finkel TH and Crow MK: Early growth Response-1
is required for CD154 transcription. J Immunol. 176:811–818. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Wherry EJ, Ha SJ, Kaech SM, Haining WN,
Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL and Ahmed
R: Molecular signature of CD8+ T cell exhaustion during chronic
viral infection. Immunity. 27:670–684. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Sen DR, Kaminski J, Barnitz RA, Kurachi M,
Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, et
al: The epigenetic landscape of T cell exhaustion. Science.
354:1165–1169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Man K, Gabriel SS, Liao Y, Gloury R,
Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt
F, Febbraio MA, et al: Transcription factor IRF4 Promotes CD8+ T
cell exhaustion and limits the development of Memory-like T cells
during chronic infection. Immunity. 47:1129–1141.e5. 2017.
View Article : Google Scholar
|
|
134
|
Doering TA, Crawford A, Angelosanto JM,
Paley MA, Ziegler CG and Wherry EJ: Network analysis reveals
centrally connected genes and pathways involved in CD8+ T cell
exhaustion versus memory. Immunity. 37:1130–1144. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Kallies A, Zehn D and Utzschneider DT:
Precursor exhausted T cells: Key to successful immunotherapy? Nat
Rev Immunol. 20:128–136. 2020. View Article : Google Scholar
|
|
136
|
Macián F, Garcı́a-Cózar F, Im SH, Horton
HF, Byrne MC and Rao A: Transcriptional mechanisms underlying
lymphocyte tolerance. Cell. 109:719–731. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Martinez GJ, Pereira RM, Äijö T, Kim EY,
Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S,
et al: The transcription factor NFAT promotes exhaustion of
activated CD8 + T Cells. Immunity. 42:265–278. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Mittelstadt PR and Ashwell JD: Cyclosporin
A-Sensitive transcription factor Egr-3 regulates fas ligand
expression. Mol Cell Biol. 18:3744–3751. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Safford M, Collins S, Lutz MA, Allen A,
Huang CT, Kowalski J, Blackford A, Horton MR, Drake C, Schwartz RH
and Powell JD: Egr-2 and Egr-3 are negative regulators of T cell
activation. Nat Immunol. 6:472–480. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Rengarajan J, Mittelstadt PR, Mages HW,
Gerth AJ, Kroczek RA, Ashwell JD and Glimcher LH: Sequential
Involvement of NFAT and Egr transcription factors in FasL
regulation. Immunity. 12:293–300. 2000. View Article : Google Scholar
|
|
141
|
Kodakandla G, Akimzhanov AM and Boehning
D: Regulatory mechanisms controlling store-operated calcium entry.
Front Physiol. 14:13302592023. View Article : Google Scholar
|
|
142
|
Kim HJ, Park S, Shin HY, Nam YR, Lam Hong
PT, Chin YW, Nam JH and Kim WK: Inhibitory effects of α-Mangostin
on T cell cytokine secretion via ORAI1 calcium channel and
K+ channels inhibition. PeerJ. 9:e109732021. View Article : Google Scholar
|
|
143
|
Srikanth S, Woo JS, Sun Z and Gwack Y:
Immunological disorders: Regulation of Ca2+ signaling in
T lymphocytes. Adv Exp Med Biol. 993:397–424. 2017. View Article : Google Scholar
|
|
144
|
Gross S, Womer L, Kappes DJ and Soboloff
J: Multifaceted control of T cell differentiation by STIM1. Trends
Biochem Sci. 48:1083–1097. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Thompson JL, Lai-Zhao Y, Stathopulos PB,
Grossfield A and Shuttleworth TJ: Phosphorylation-mediated
structural changes within the SOAR domain of stromal interaction
molecule 1 enable specific activation of distinct Orai channels. J
Biol Chem. 293:3145–3155. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Flourakis M, Lehen'kyi V, Beck B, Raphaël
M, Vandenberghe M, Abeele FV, Roudbaraki M, Lepage G, Mauroy B,
Romanin C, et al: Orai1 contributes to the establishment of an
apoptosis-resistant phenotype in prostate cancer cells. Cell Death
Dis. 1:e752010. View Article : Google Scholar
|
|
147
|
Xie J, Pan H, Yao J, Zhou Y and Han W:
SOCE and cancer: Recent progress and new perspectives. Int J
Cancer. 138:2067–2077. 2016. View Article : Google Scholar :
|
|
148
|
Im SJ, Hashimoto M, Gerner MY, Lee J,
Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al:
Defining CD8+ T cells that provide the proliferative burst after
PD-1 therapy. Nature. 537:417–421. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Armesilla AL, Lorenzo E, Gómez Del Arco P,
Martínez-Martínez S, Alfranca A and Redondo JM: Vascular
Endothelial growth factor activates nuclear factor of activated T
cells in human endothelial cells: A role for tissue factor gene
expression. Mol Cell Biol. 19:2032–2043. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Chen YF, Chiu WT, Chen YT, Lin PY, Huang
HJ, Chou CY, Chang HC, Tang MJ and Shen MR: Calcium store sensor
stromal-interaction molecule 1-dependent signaling plays an
important role in cervical cancer growth, migration, and
angiogenesis. Proc Natl Acad Sci USA. 108:15225–15230. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Xu Y, Zhang S, Niu H, Ye Y, Hu F, Chen S,
Li X, Luo X, Jiang S, Liu Y, et al: STIM1 accelerates cell
senescence in a remodeled microenvironment but enhances the
epithelial-to-mesenchymal transition in prostate cancer. Sci Rep.
5:117542015. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Yang S, Zhang JJ and Huang XY: Orai1 and
STIM1 are critical for breast tumor cell migration and metastasis.
Cancer Cell. 15:124–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Wilm B and Muñoz-Chapuli R: The role of
WT1 in Embryonic Development and Normal Organ Homeostasis. The
Wilms' Tumor (WT1) Gene. 1467. Hastie N: Springer New York; New
York, NY: pp. 23–39. 2016, View Article : Google Scholar
|
|
154
|
Madden SL, Cook DM, Morris JF, Gashler A,
Sukhatme VP and Rauscher FJ: Transcriptional repression mediated by
the WT1 wilms tumor gene product. Science. 253:1550–1553. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Zandarashvili L, White MA, Esadze A and
Iwahara J: Structural impact of complete CpG methylation within
target DNA on specific complex formation of the inducible
transcription factor Egr-1. FEBS Lett. 589:1748–1753. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Hashimoto H, Olanrewaju YO, Zheng Y,
Wilson GG, Zhang X and Cheng X: Wilms tumor protein recognizes
5-carboxylcytosine within a specific DNA sequence. Genes Dev.
28:2304–2313. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Eleftheriou M, Pascual AJ, Wheldon LM,
Perry C, Abakir A, Arora A, Johnson AD, Auer DT, Ellis IO,
Madhusudan S and Ruzov A: 5-Carboxylcytosine levels are elevated in
human breast cancers and gliomas. Clin Epigenet. 7:882015.
View Article : Google Scholar
|
|
158
|
Neri F, Incarnato D, Krepelova A, Rapelli
S, Anselmi F, Parlato C, Medana C, Dal Bello F and Oliviero S:
Single-base resolution analysis of 5-formyl and 5-carboxyl cytosine
reveals promoter DNA methylation dynamics. Cell Rep. 10:674–683.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Ritchie MF, Yue C, Zhou Y, Houghton PJ and
Soboloff J: Wilms tumor suppressor 1 (WT1) and early growth
response 1 (EGR1) are regulators of STIM1 expression. J Biol Chem.
285:10591–10596. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Natrajan R, Little SE, Reis-Filho JS, Hing
L, Messahel B, Grundy PE, Dome JS, Schneider T, Vujanic GM,
Pritchard-Jones K and Jones C: Amplification and overexpression of
CACNA1E correlates with relapse in favorable histology Wilms'
tumors. Clinical Cancer Res. 12:7284–7293. 2006. View Article : Google Scholar
|
|
161
|
Wittmann S, Wunder C, Zirn B, Furtwängler
R, Wegert J, Graf N and Gessler M: New prognostic markers revealed
by evaluation of genes correlated with clinical parameters in Wilms
tumors. Genes Chromosomes Cancer. 47:386–395. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Wagle MV, Vervoort SJ, Kelly MJ, Van Der
Byl W, Peters TJ, Martin BP, Martelotto LG, Nüssing S, Ramsbottom
KM, Torpy JR, et al: Antigen-driven EGR2 expression is required for
exhausted CD8+ T cell stability and maintenance. Nat Commun.
12:27822021. View Article : Google Scholar :
|
|
163
|
Lee J, Lee K, Bae H, Lee K, Lee S, Ma J,
Jo K, Kim I, Jee B, Kang M and Im SJ: IL-15 promotes self-renewal
of progenitor exhausted CD8 T cells during persistent antigenic
stimulation. Front Immunol. 14:11170922023. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Sun L, Ma Z, Zhao X, Tan X, Tu Y, Wang J,
Chen L, Chen Z, Chen G and Lan P: LRP11 promotes stem-like T cells
via MAPK13-mediated TCF1 phosphorylation, enhancing anti-PD1
immunotherapy. J Immunother Cancer. 12:e0083672024. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Omodho B, Miao T, Symonds ALJ, Singh R, Li
S and Wang P: Transcription factors early growth response gene
(Egr) 2 and 3 control inflammatory responses of tolerant T cells.
Immun Inflamm Dis. 6:221–233. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Liu Y, Debo B, Li M, Shi Z, Sheng W and
Shi Y: Author correction: LSD1 inhibition sustains T cell
invigoration with a durable response to PD-1 blockade. Nat Commun.
13:2632022. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Symonds ALJ, Miao T, Busharat Z, Li S and
Wang P: Egr2 and 3 maintain anti-tumour responses of exhausted
tumour infiltrating CD8 + T cells. Cancer Immunol Immunother.
72:1139–1151. 2023. View Article : Google Scholar
|
|
168
|
Symonds AL, Zheng W, Miao T, Wang H, Wang
T, Kiome R, Hou X, Li S and Wang P: Egr2 and 3 control
inflammation, but maintain homeostasis, of PD-1high
memory phenotype CD4 T cells. Life Sci Alliance. 3:e2020007662020.
View Article : Google Scholar
|
|
169
|
Miao T, Symonds ALJ, Singh R, Symonds JD,
Ogbe A, Omodho B, Zhu B, Li S and Wang P: Egr2 and 3 control
adaptive immune responses by temporally uncoupling expansion from T
cell differentiation. J Exp Med. 214:1787–1808. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Seyfert VL, McMahon SB, Glenn WD, Yellen
AJ, Sukhatme VP, Cao X and Monroe JG: Methylation of an
Immediate-early inducible gene as a mechanism for B cell tolerance
induction. Science. 250:797–800. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Natarajan P, Singh A, McNamara JT, Secor
ER, Guernsey LA, Thrall RS and Schramm CM: Regulatory B cells from
hilar lymph nodes of tolerant mice in a murine model of allergic
airway disease are CD5+, express TGF-β, and co-localize with
CD4+Foxp3+ T cells. Mucosal Immunol. 5:691–701. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Matsumoto M, Baba A, Yokota T, Nishikawa
H, Ohkawa Y, Kayama H, Kallies A, Nutt SL, Sakaguchi S, Takeda K,
et al: Interleukin-10-Producing plasmablasts exert regulatory
function in autoimmune inflammation. Immunity. 41:1040–1051. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Xie J, Shi CW, Huang HB, Yang WT, Jiang
YL, Ye LP, Zhao Q, Yang GL and Wang CF: Induction of the
IL-10-producing regulatory B cell phenotype following Trichinella
spiralis infection. Mol Immunol. 133:86–94. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Nguyen HQ, Hoffman-Liebermann B and
Liebermann DA: The zinc finger transcription factor Egr-1 is
essential for and restricts differentiation along the macrophage
lineage. Cell. 72:197–209. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Kharbanda S, Nakamura T, Stone R, Hass R,
Bernstein S, Datta R, Sukhatme VP and Kufe D: Expression of the
early growth response 1 and 2 zinc finger genes during induction of
monocytic differentiation. J Clin Invest. 88:571–577. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Laslo P, Spooner CJ, Warmflash A, Lancki
DW, Lee HJ, Sciammas R, Gantner BN, Dinner AR and Singh H:
Multilineage transcriptional priming and determination of alternate
hematopoietic cell fates. Cell. 126:755–766. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Carter JH and Tourtellotte WG: Early
growth response transcriptional regulators are dispensable for
macrophage differentiation. J Immunol. 178:3038–3047. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Pham TH, Benner C, Lichtinger M,
Schwarzfischer L, Hu Y, Andreesen R, Chen W and Rehli M: Dynamic
epigenetic enhancer signatures reveal key transcription factors
associated with monocytic differentiation states. Blood.
119:e161–e171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Trizzino M, Zucco A, Deliard S, Wang F,
Barbieri E, Veglia F, Gabrilovich D and Gardini A: EGR1 is a
gatekeeper of inflammatory enhancers in human macrophages. Sci Adv.
7:eaaz88362021. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Ponomarev ED, Shriver LP, Maresz K,
Pedras-Vasconcelos J, Verthelyi D and Dittel BN: GM-CSF production
by autoreactive T cells is required for the activation of
microglial cells and the onset of experimental autoimmune
encephalomyelitis. J Immunol. 178:39–48. 2007. View Article : Google Scholar
|
|
181
|
Murray PJ: Macrophage Polarization. Annu
Rev Physiol. 79:541–566. 2017. View Article : Google Scholar
|
|
182
|
Italiani P and Boraschi D: From monocytes
to M1/M2 macrophages: Phenotypical vs. functional differentiation.
Front Immunol. 5:5142014. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Jablonski KA, Amici SA, Webb LM,
Ruiz-Rosado JDD, Popovich PG, Partida-Sanchez S and
Guerau-de-Arellano M: Novel Markers to delineate murine M1 and M2
macrophages. PLoS One. 10:e01453422015. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Horvath A, Daniel B, Szeles L,
Cuaranta-Monroy I, Czimmerer Z, Ozgyin L, Steiner L, Kiss M,
Simandi Z, Poliska S, et al: Labelled regulatory elements are
pervasive features of the macrophage genome and are dynamically
utilized by classical and alternative polarization signals. Nucleic
Acids Res. 47:2778–2792. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Daniel B, Nagy G, Horvath A, Czimmerer Z,
Cuaranta-Monroy I, Poliska S, Hays TT, Sauer S, Francois-Deleuze J
and Nagy L: The IL-4/STAT6/PPARγ signaling axis is driving the
expansion of the RXR heterodimer cistrome, providing complex ligand
responsiveness in macrophages. Nucleic Acids Res. 46:4425–4439.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Daniel B, Nagy G, Czimmerer Z, Horvath A,
Hammers DW, Cuaranta-Monroy I, Poliska S, Tzerpos P, Kolostyak Z,
Hays TT, et al: The nuclear receptor PPARγ controls progressive
macrophage polarization as a Ligand-insensitive epigenomic ratchet
of transcriptional memory. Immunity. 49:615–626.e6. 2018.
View Article : Google Scholar
|
|
187
|
Szanto A, Balint BL, Nagy ZS, Barta E,
Dezso B, Pap A, Szeles L, Poliska S, Oros M, Evans RM, et al: STAT6
transcription factor is a facilitator of the nuclear receptor
PPARγ-regulated gene expression in macrophages and dendritic cells.
Immunity. 33:699–712. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Czimmerer Z, Daniel B, Horvath A, Rückerl
D, Nagy G, Kiss M, Peloquin M, Budai MM, Cuaranta-Monroy I, Simandi
Z, et al: The transcription factor STAT6 mediates direct repression
of inflammatory enhancers and limits activation of alternatively
polarized macrophages. Immunity. 48:75–90.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Piccolo V, Curina A, Genua M, Ghisletti S,
Simonatto M, Sabò A, Amati B, Ostuni R and Natoli G: Opposing
macrophage polarization programs show extensive epigenomic and
transcriptional cross-talk. Nat Immunol. 18:530–540. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Sabag B, Puthenveetil A, Levy M, Joseph N,
Doniger T, Yaron O, Karako-Lampert S, Lazar I, Awwad F, Ashkenazi S
and Barda-Saad M: Dysfunctional natural killer cells can be
reprogrammed to regain anti-tumor activity. EMBO J. 43:2552–2581.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Zhang W, Tong H, Zhang Z, Shao S, Liu D,
Li S and Yan Y: Transcription factor EGR1 promotes differentiation
of bovine skeletal muscle satellite cells by regulating MyoG gene
expression. J Cell Physiol. 233:350–362. 2018. View Article : Google Scholar
|
|
192
|
Wei S, Xu G, Zhao S, Zhang C, Feng Y, Yang
W, Lu R, Zhou J and Ma Y: EGR2 promotes liver cancer metastasis by
enhancing IL-8 expression through transcription regulation of PDK4
in M2 macrophages. Int Immunopharmacol. 153:1144842025. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Zhang S, Tao X, Cao Q, Feng X, Wu J, Yu H,
Yu Y, Xu C and Zhao H: lnc003875/miR-363/EGR1 regulatory network in
the carcinoma-associated fibroblasts controls the angiogenesis of
human placental site trophoblastic tumor (PSTT). Exp Cell Res.
387:1117832020. View Article : Google Scholar
|
|
194
|
Zhang Y, Sun S, Qi Y, Dai Y, Hao Y, Xin M,
Xu R, Chen H, Wu X, Liu Q, et al: Characterization of tumour
microenvironment reprogramming reveals invasion in epithelial
ovarian carcinoma. J Ovarian Res. 16:2002023. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Joo EH, Bae JH, Park J, Bang YJ, Han J,
Gulati N, Kim JI, Park CG, Park WY and Kim HJ: Deconvolution of
adult T-Cell Leukemia/lymphoma with Single-cell RNA-Seq using
frozen archived skin tissue reveals new subset of Cancer-associated
fibroblast. Front Immunol. 13:8563632022. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Seyfried TN, Flores RE, Poff AM and
D'Agostino DP: Cancer as a metabolic disease: Implications for
novel therapeutics. Carcinogenesis. 35:515–527. 2014. View Article : Google Scholar :
|
|
197
|
Ward PS and Thompson CB: Metabolic
reprogramming: A cancer hallmark even warburg did not anticipate.
Cancer Cell. 21:297–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Li L, Li L, Li W, Chen T, Bin Zou, Zhao L,
Wang H, Wang X, Xu L, Liu X, et al: TAp73-induced
phosphofructokinase-1 transcription promotes the Warburg effect and
enhances cell proliferation. Nat Commun. 9:46832018. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Pan M, Luo M, Liu L, Chen Y, Cheng Z, Wang
K, Huang L, Tang N, Qiu J, Huang A and Xia J: EGR1 suppresses HCC
growth and aerobic glycolysis by transcriptionally downregulating
PFKL. J Exp Clin Cancer Res. 43:352024. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Sulli G, Lam MTY and Panda S: Interplay
between circadian clock and cancer: New frontiers for cancer
treatment. Trends Cancer. 5:475–494. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Zhang R, Lahens NF, Ballance HI, Hughes ME
and Hogenesch JB: A circadian gene expression atlas in mammals:
Implications for biology and medicine. Proc Natl Acad Sci USA.
111:16219–16224. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Wu J, Bu D, Wang H, Shen D, Chong D, Zhang
T, Tao W, Zhao M, Zhao Y, Fang L, et al: The rhythmic coupling of
Egr-1 and Cidea regulates age-related metabolic dysfunction in the
liver of male mice. Nat Commun. 14:16342023. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Qu M, Zhang G, Qu H, Vu A, Wu R, Tsukamoto
H, Jia Z, Huang W, Lenz HJ, Rich JN and Kay SA: Circadian regulator
BMAL1::CLOCK promotes cell proliferation in hepatocellular
carcinoma by controlling apoptosis and cell cycle. Proc Natl Acad
Sci USA. 120:e22148291202023. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Spencer RL and Deak T: A users guide to
HPA axis research. Physiol Behav. 178:43–65. 2017. View Article : Google Scholar :
|
|
205
|
Wan X, Wang L, Khan MA, Peng L, Zhang K,
Sun X, Yi X, Wang Z and Chen K: Shift work promotes adipogenesis
via cortisol-dependent downregulation of EGR3-HDAC6 pathway. Cell
Death Discov. 10:1292024. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Deuter CE, Kaczmarczyk M, Hellmann-Regen
J, Kuehl LK, Wingenfeld K and Otte C: The influence of
pharmacological mineralocorticoid and glucocorticoid receptor
blockade on the cortisol response to psychological stress. Prog
Neuropsychopharmacol Biol Psychiatry. 129:1109052024. View Article : Google Scholar
|
|
207
|
Wu K, Liu Z, Liang J, Zhu Y, Wang X and Li
X: Discovery of a glucocorticoid receptor (GR) activity signature
correlates with immune cell infiltration in adrenocortical
carcinoma. J Immunother Cancer. 11:e0075282023. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Pang Y, Gong S, Tetti M, Sun Z,
Mir-Bashiri S, Bidlingmaier M, Knösel T, Wolf E, Reincke M, Kemter
E and Williams TA: EGR1 regulates oxidative stress and aldosterone
production in adrenal cells and aldosterone-producing adenomas.
Redox Biol. 80:1034982025. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Miki Y, Iwabuchi E, Takagi K, Yamazaki Y,
Shibuya Y, Tokunaga H, Shimada M, Suzuki T and Ito K: Intratumoral
cortisol associated with aromatase in the endometrial cancer
microenvironment. Pathol Res Pract. 251:1548732023. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Heyns B, Pieters R, Stander MA, Atkin SL
and Swart AC: Glucocorticoids and mineralocorticoids in hair:
Facilitating accurate diagnosis of adrenal-related endocrine
disorders. Front Endocrinol (Lausanne). 15:14480132024. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Zhou L, Shan Y, Li J, Li M, Meng Z and Guo
N: Early growth response 1 regulates dual-specificity protein
phosphatase 1 and inhibits cell migration and invasion of tongue
squamous cell carcinoma. Oncol Lett. 27:2402024. View Article : Google Scholar
|
|
212
|
Senga SS and Grose RP: Hallmarks of
cancer-the New testament. Open Biol. 11:2003582021. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Magnon C, Hall SJ, Lin J, Xue X, Gerber L,
Freedland SJ and Frenette PS: Autonomic nerve development
contributes to prostate cancer progression. Science.
341:12363612013. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Cheng Y, Sun F, D'Souza A, Dhakal B,
Pisano M, Chhabra S, Stolley M, Hari P and Janz S: Autonomic
nervous system control of multiple myeloma. Blood Rev.
46:1007412021. View Article : Google Scholar :
|
|
215
|
Tan X, Sivakumar S, Bednarsch J,
Wiltberger G, Kather JN, Niehues J, de Vos-Geelen J, Valkenburg-van
Iersel L, Kintsler S, Roeth A, et al: Nerve fibers in the tumor
microenvironment in neurotropic cancer-pancreatic cancer and
cholangiocarcinoma. Oncogene. 40:899–908. 2021. View Article : Google Scholar
|
|
216
|
Monje M, Borniger JC, D'Silva NJ, Deneen
B, Dirks PB, Fattahi F, Frenette PS, Garzia L, Gutmann DH, Hanahan
D, et al: Roadmap for the emerging field of cancer neuroscience.
Cell. 181:219–222. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Hayakawa Y, Sakitani K, Konishi M, Asfaha
S, Niikura R, Tomita H, Renz BW, Tailor Y, Macchini M, Middelhoff
M, et al: Nerve growth factor promotes gastric tumorigenesis
through aberrant cholinergic signaling. Cancer Cell. 31:21–34.
2017. View Article : Google Scholar :
|
|
218
|
Regan JL, Schumacher D, Staudte S, Steffen
A, Lesche R, Toedling J, Jourdan T, Haybaeck J, Golob-Schwarzl N,
Mumberg D, et al: Identification of a neural development gene
expression signature in colon cancer stem cells reveals a role for
EGR2 in tumorigenesis. iScience. 25:1044982022. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Cheng H and Cheng T: 'Waterloo': When
normal blood cells meet leukemia. Curr Opin Hematol. 23:304–310.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Chen CH, Chen Y, Li YN, Zhang H, Huang X,
Li YY, Li ZY, Han JX, Wu XY, Liu HJ and Sun T: EGR3 inhibits tumor
progression by inducing schwann Cell-Like differentiation. Adv Sci
(Weinh). 11:e24000662024. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Spasevska I, Matera EL, Chettab K, Ville
J, Potier-Cartereau M, Jordheim LP, Thieblemont C, Sahin D, Klein C
and Dumontet C: Calcium channel blockers impair the antitumor
activity of Anti-CD20 monoclonal antibodies by blocking EGR-1
induction. Mol Cancer Ther. 19:2371–2381. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Lim JH, Park JW, Min DS, Chang JS, Lee YH,
Park YB, Choi KS and Kwon TK: NAG-1 up-regulation mediated by EGR-1
and p53 is critical for quercetin-induced apoptosis in HCT116 colon
carcinoma cells. Apoptosis. 12:411–421. 2007. View Article : Google Scholar
|
|
223
|
Shi Q and Bhatia D: Resveratrol-responsive
CArG elements from the Egr-1 promoter for the induction of GADD45α
to arrest the G2/M transition. Suicide Gene Therapy. 1895. Düzgüneş
N: Springer New York; New York, NY: pp. 111–122. 2019, View Article : Google Scholar
|
|
224
|
Shah D, Challagundla N, Dave V, Patidar A,
Saha B, Nivsarkar M, Trivedi VB and Agrawal-Rajput R: Berberine
mediates tumor cell death by skewing tumor-associated
immunosuppressive macrophages to inflammatory macrophages.
Phytomedicine. 99:1539042022. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Han S, Yang X, Zhuang J, Zhou Q, Wang J,
Ru L, Niu F and Mao W: α-Hederin promotes ferroptosis and reverses
cisplatin chemoresistance in non-small cell lung cancer. Aging.
16:1298–1317. 2024. View Article : Google Scholar
|
|
226
|
Wang W, Li R, Chen Z, Li D, Duan Y and Cao
Z: Cisplatin-controlled p53 gene therapy for human non-small cell
lung cancer xenografts in athymic nude mice via the CArG elements.
Cancer Sci. 96:706–712. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Wang Z, Wei B and Ma S:
EGR1/LINC00839/SOX5 axis modulates migration, invasion and
Gemcitabine resistance of bladder cancer cells. Cancer Biol Ther.
24:22701062023. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Marks BA, Pipia IM, Mukai C, Horibata S,
Rice EJ, Danko CG and Coonrod SA: GDNF-RET signaling and EGR1 form
a positive feedback loop that promotes tamoxifen resistance via
cyclin D1. BMC Cancer. 23:1382023. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Yang Z, Chen F, Wei D, Chen F, Jiang H and
Qin S: EGR1 mediates MDR1 transcriptional activity regulating
gemcitabine resistance in pancreatic cancer. BMC Cancer.
24:2682024. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Lin Z, Liu Z, Pan Z, Zhang Y, Yang X, Feng
Y, Zhang R, Zeng W, Gong C and Chen J: EGR1 Promotes
Erastin-induced ferroptosis through activating Nrf2-HMOX1 signaling
pathway in breast cancer cells. J Cancer. 15:4577–4590. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Palencia-Campos A, Ruiz-Cañas L,
Abal-Sanisidro M, López-Gil JC, Batres-Ramos S, Saraiva SM, Yagüe
B, Navarro D, Alcalá S, Rubiolo JA, et al: Reprogramming
tumor-associated macrophages with lipid nanosystems reduces PDAC
tumor burden and liver metastasis. J Nanobiotechnol. 22:7952024.
View Article : Google Scholar
|
|
232
|
Mo Z, Du P, Wang G and Wang Y: The
Multi-purpose tool of tumor immunotherapy: Gene-engineered T cells.
J Cancer. 8:1690–1703. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Xu Y, Yang Z, Horan LH, Zhang P, Liu L,
Zimdahl B, Green S, Lu J, Morales JF, Barrett DM, et al: A novel
antibody-TCR (AbTCR) platform combines Fab-based antigen
recognition with gamma/delta-TCR signaling to facilitate T-cell
cytotoxicity with low cytokine release. Cell Discov. 4:622018.
View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Jung IY, Bartoszek RL, Rech AJ, Collins
SM, Ooi SK, Williams EF, Hopkins CR, Narayan V, Haas NB, Frey NV,
et al: Type I interferon signaling via the EGR2 transcriptional
regulator potentiates CAR T Cell-intrinsic dysfunction. Cancer
Discov. 13:1636–1655. 2023. View Article : Google Scholar : PubMed/NCBI
|