The pathogenesis of heart failure is highly complex.
The reported molecular mechanisms of heart failure mainly include
the dysregulation of disease-related genes, noncoding RNAs, calcium
ion homeostasis, mitochondrial homeostasis, cell apoptosis,
extracellular matrix (ECM) remodeling, oxidative stress, the
inflammatory response and the neuroendocrine system (11-22). Cardiac hypertrophy-related and
fibrosis-related genes are associated with the occurrence and
development of heart failure (23,24). The overexpression of cardiac
hypertrophy-related genes, such as myosin binding protein C
(MYBPC3) and myosin heavy chain 7 (MYH7), can lead to
myocardial systolic and diastolic dysfunction (25,26). The expression of fibrosis-related
genes such as transforming growth factor β1 (TGFB1) and
actin α2 (ACTA2) can cause the activation and proliferation
of myofibroblasts and the inflammatory response (27,28). The dysregulation of calcium ion
homeostasis in myocardial cells also plays an important role in
heart failure (17,18,29,30). The abnormal expression of
microRNAs (miRNAs) is also associated with heart failure (14,15,31). miRNA-1 and miRNA-21 can regulate
the expression of myocardial function-related genes and their
dysregulation can disrupt the metabolism, apoptosis and contraction
of myocardial cells (31-33).
Mitochondrial dysfunction can disrupt the energy supply to
myocardial cells, leading to impaired myocardial function (18,34). In addition, increased myocardial
cell apoptosis and necroptosis are important risk factors for heart
failure (19,35,36). Cardiomyocyte necroptosis can lead
to cell swelling, cell membrane rupture and intracellular contents
overflow, which together triggers inflammatory responses and heart
failure (36-38). Previous studies have shown that
inflammatory factors such as interleukin-1 (IL-1), IL-6, tumor
necrosis factor-α (TNF-α) and excessive reactive oxygen species
(ROS) can activate the intracellular apoptotic signaling pathway,
which promotes myocardial cell death and heart failure (28,39-42). Inflammatory responses are closely
linked with the occurrence and development of heart failure
(43). The release of
inflammatory factors such as TNF-α and IL-6 can promote an
inflammatory cascade, which results in myocardial cell damage and
cardiac fibrosis (21,43). Oxidative stress induced by
excessive ROS production can cause myocardial damage and
dysfunction, which leads to heart failure (20,41). Neuroendocrine system
overactivation can also lead to heart failure (22). The abnormal activation of the
renin-angiotensin-aldosterone system (RAAS) can increase
angiotensin II (Ang II) levels, thus promoting myocardial
remodeling (22,44). However, there are few effective
preventive targets and therapeutic methods for treating heart
failure due to its complex molecular mechanisms. Further studies on
the detailed mechanisms of heart failure are highly important for
the diagnosis, treatment and prevention of heart failure.
The present study reviewed recent research progress
on the pathogenesis of heart failure, providing more references for
further studies on the molecular mechanisms of heart failure and
contributing to the development of potential therapeutic targets
for heart failure.
Arrhythmia has a high mortality rate of ~10-15%; it
is characterized by abnormalities in the frequency, rhythm, origin,
conduction velocity and sequence of cardiac impulses (45-47). Arrhythmia can lead to
insufficient heart pumping, damage the blood supply to various
tissues and organs and result in dizziness, fatigue, amaurosis and
syncope (46,48). Furthermore, long-term or severe
arrhythmia can promote myocardial oxygen consumption, which
gradually leads to myocardial remodeling and heart failure
(49,50). It is important to investigate and
reveal the pathogenesis of arrhythmia to prevent heart failure
(51). However, the mechanisms
by which arrhythmia causes heart failure are complex and remain to
be fully elucidated.
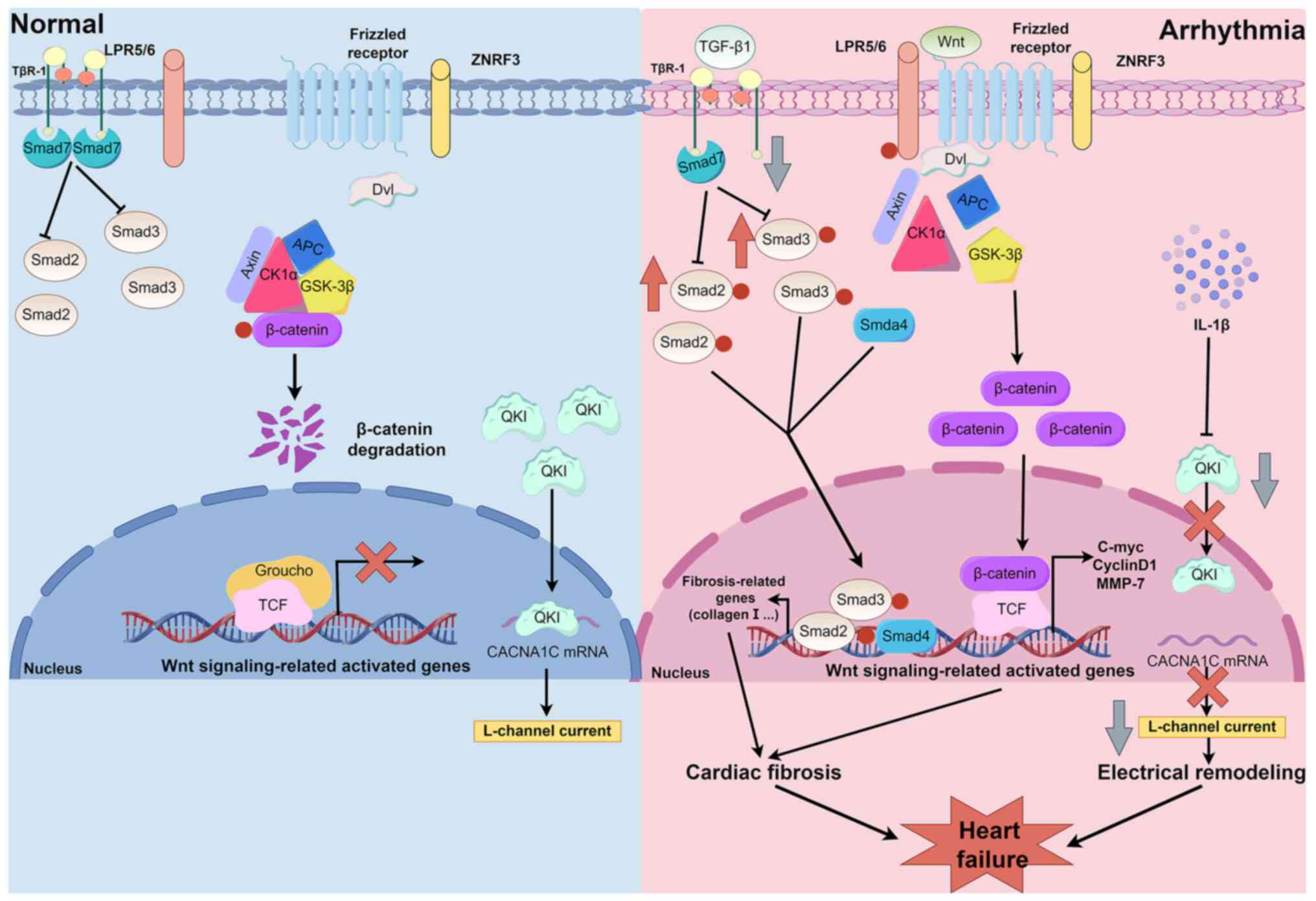
Arrhythmia can result in heart failure through
TGF-β1/Smad signaling, Wnt/β-catenin signaling and IL-1β secretion
(Fig. 1) (52-54). The activation of TGF-β1/Smad
signaling can cause cardiac fibrosis in atrial fibrillation, which
leads to structural and functional damage to the heart and to heart
failure (52). Smad2 and Smad3
are receptor-regulated Smads (R-Smads) that can be phosphorylated
and activated by type I receptors of TGF-β (55,56). In addition, Smad4 is a
comodulator Smad and Smad7 is a type of inhibitory Smad that can
compete with R-Smads for binding to type I receptors of TGF-β and
inhibit the phosphorylation of R-Smads (56,57). TGF-β1 expression is upregulated
in arrhythmias; this increase decreases Smad7, weakens the
competitiveness of Smad7 and promotes the activation of Smad2 and
Smad3 (58,59). Activated Smad2 and Smad3 can form
a complex with Smad4, and the complex can be transferred to the
nucleus, upregulate the transcription of fibrosis-related genes,
including collagen type I α1 chain (COL1A1), COL3A1,
fibronectin 1 and ACTA2, and ultimately result in cardiac
fibrosis (58,60-62). Another pathway is the classical
Wnt pathway (53,63). Without Wnt stimulation, the
'destruction complex', which is composed of Axin, adenomatous
polyposis coli protein, glycogen synthase kinase 3β and casein
kinase 1α, is active in vivo (64). β-catenin is phosphorylated by the
'destruction complex', and phosphorylated β-catenin is
ubiquitinated and then degraded by proteasomes, resulting in a low
protein level of β-catenin in the cytoplasm (63,64). Once Wnt ligands bind to the
Frizzled receptor and low-density lipoprotein receptor-related
protein 5/6, the 'destruction complex' can be disrupted and
scattered (65), protecting
β-catenin from being phosphorylated and degraded, and thus
promoting β-catenin accumulation (53,64). The accumulated β-catenin then
enters the cell nucleus, interacts with T-cell factor/lymphoid
enhancer factor to form a transcription activation complex, and
upregulates the transcription of Wnt signaling-related genes,
including ACTA2, connective tissue growth factor,
COL1A1 and phosphoribosyl anthranilate isomerase 1, thus
driving myocardial fibrosis (64,65). In the case of atrial
fibrillation, the first and second pathways are abnormally
activated, leading to myocardial fibrosis (66-69). Furthermore, as atrial
fibrillation persists and atrial fibrosis progresses, the burden on
the heart continues to increase, resulting in heart failure
(69-71). In the third pathway,
proinflammatory macrophages can induce atrial electrical remodeling
by secreting IL-1β, which decreases the protein level of the atrial
myocyte fibrillation protein Quaking (QKI) (54,72). Atrial fibrillation can induce
proinflammatory macrophage polarization and IL-1β secretion from
macrophages to downregulate QKI expression (72); it can reduce the binding of QKI
and calcium voltage-gated channel subunit α1 C (CACNA1C) mRNA in
atrial myocytes, which decreases the protein level of CACNA1C and
L-type calcium currents (72,73). Ultimately, atrial fibrillation
can induce electrical remodeling and affect cardiac function, which
can cause heart failure in the long term (72-74).
There are numerous reported arrhythmia-targeted
drugs, genes and surgical treatments for preventing heart failure
(Table I) (75-81). In clinical practice,
antiarrhythmic drugs include Class I, II, III and IV drugs
(75,76). Class I sodium channel blockers
can be divided into moderate sodium channel blockers (e.g.,
quinidine), mild blockers (e.g., lidocaine) and significant
blockers (e.g., propafenone) (75,76); they can reduce the autonomy of
myocardial cells to different degrees (75,76). Class II drugs, which are
β-blockers (e.g., propranolol), can competitively block β
adrenergic receptors to reduce myocardial autonomy; they are mainly
used to treat sympathetic nervous system excitation-related
arrhythmias (75,76). Class III drugs, which are
potassium channel blockers (e.g., amiodarone), can prolong the
action potential and refractory period of myocardial cells by
blocking potassium channels; they are commonly used to treat
structural heart disease-related arrhythmias (75,76). Class IV drugs, which are calcium
channel blockers (e.g., verapamil), can act mainly on L-type
calcium channels in myocardial cells and vascular smooth muscle
cells (VSMCs), inhibit calcium ion influx and reduce the autonomy
of the sinoatrial node (75,76); these drugs can improve myocardial
electrophysiological stability and inhibit the myocardial damage
induced by arrhythmia, which ultimately prevents heart failure
(75,76). It has been reported that the
insulin-like hormone relaxin can reverse TGFβ-induced cardiac
fibrosis and increase the conduction velocity of atrial action to
suppress arrhythmias (77,78). In addition, numerous genes have
been reported to play important roles in the development of
arrhythmia and may be prospective targets for preventing heart
failure (74,79-81). MicroRNA (miR)-210-3p, a miRNA
within extracellular vesicles derived from atrial myocytes,
promotes atrial fibrosis by targeting the glycerol-3-phosphate
dehydrogenase 1-like protein/phosphatidylinositol 3-kinase/AKT
pathway. Therefore, miR-210-3p inhibitors can prevent Ang
II-induced AF occurrence and persistence in rats, alleviate atrial
fibrosis and prevent the development of heart failure (79). QKI can act as a prospective
target for inhibiting electrical remodeling and heart failure
(75) Surgical treatments
include catheter ablation and left-ventricular assisted devices
(80,81). Catheter ablation involves the use
of radiofrequency currents, cryogenic energy or other energy
sources to generate high or low temperatures locally, causing
coagulation necrosis or cryogenic damage to abnormal myocardial
tissue that leads to arrhythmia, thereby disrupting the conduction
pathway or origin point of abnormal electrical activity, restoring
normal cardiac rhythm, improving cardiac rhythm and pumping
function, and thus playing a certain therapeutic role in heart
failure (80). Left ventricular
assistance devices use mechanical assistance to help the left
ventricle pump blood out, increase cardiac output and improve
systemic blood circulation; they are effective for treating
arrhythmia and heart failure (81).
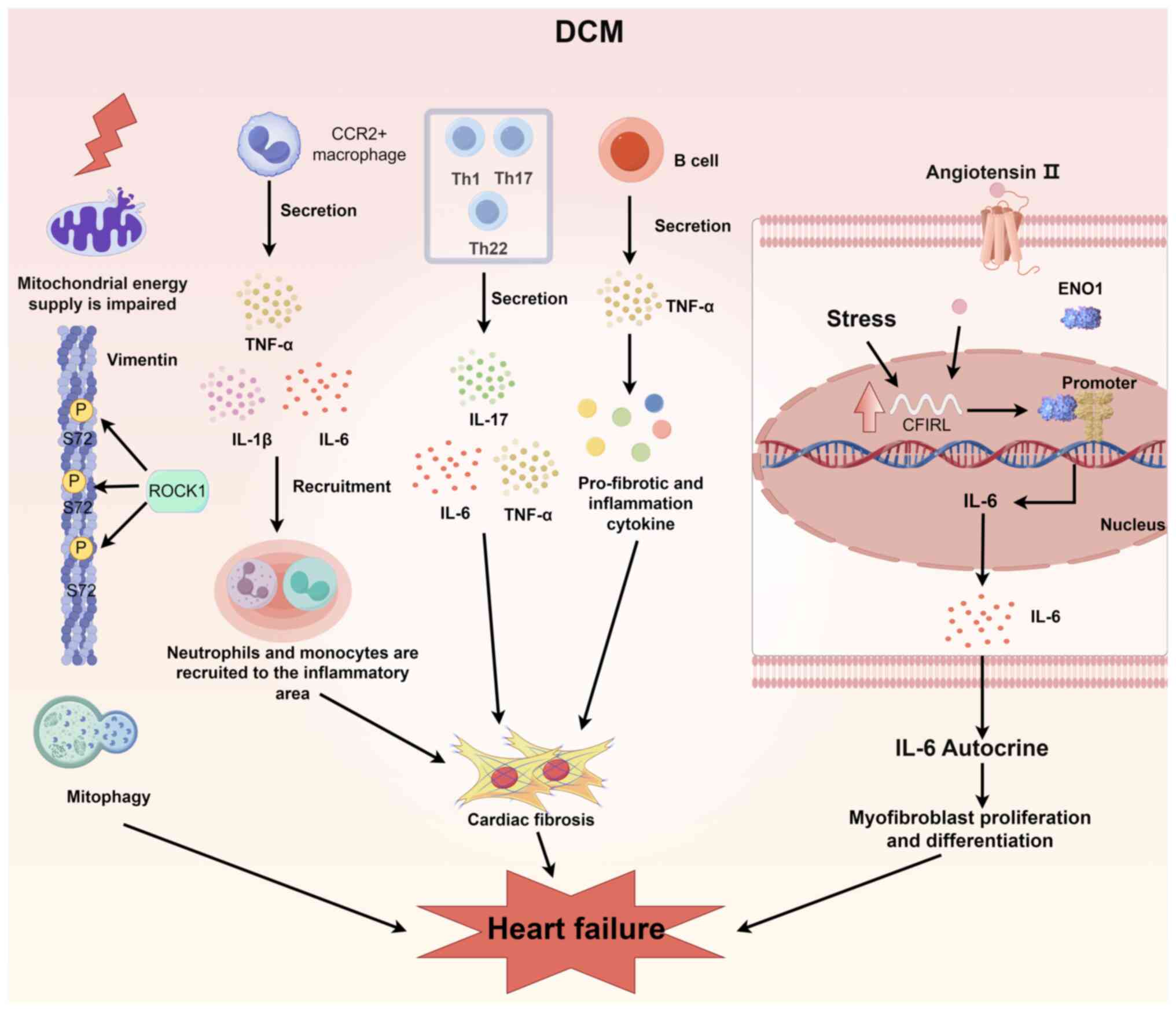
DCM can lead to heart failure through the activation
of the rho-associated coiled-coil containing protein kinase 1
(ROCK1)-vimentin (VIM) pathway, an inflammatory cascade initiated
by immune cells and calcium-independent receptor for α-latrotoxin
(CFIRL)-mediated cardiac remodeling (Fig. 2) (85-87). The first pathway involves
cytoskeleton regulation and mitochondrial autophagy (85). In DCM, mitochondrial dysfunction
can activate the ROCK1-VIM pathway, leading to mitophagy (86). VIM can interact directly with
mitochondria (85). Abnormally
activated ROCK1 can accelerate the transport speed of damaged
mitochondria by phosphorylating S72 of VIM, leading to the
substantial accumulation of damaged mitochondria and significantly
enhancing mitophagy (85,88).
These effects result in insufficient mitochondrial energy supply
and severely impaired myocardial contractility, causing heart
failure (85). The second
pathway involves immune cells and the inflammatory cascade
(86,89). In DCM, immune cell activation
leads to the development of associated inflammation (86). Among cardiomyocytes are
macrophages, monocytes, B cells, T cells, natural killer (NK)
cells, dendritic cells and granulocytes, and these immune cells
secrete a variety of fibrotic mediators, such as cytokines, growth
factors and stromal cell proteins, which play important roles in
the occurrence and progression of myocardial fibrosis (86). Monocytes and macrophages can be
transformed into myofibroblasts under the stimulation of various
cytokines, resulting in the secretion of inflammatory mediators and
fibrotic growth factors. Myeloid differentiation protein-2 can
induce the proinflammatory state of monocytes in patients with DCM
through toll-like receptor 4/nuclear factor-κB signaling, resulting
in the secretion of monocyte chemotactic protein-1 to recruit
monocytes to the site of inflammation to promote DCM progression
and, concurrently, induce the expression of adhesion factors and
the secretion of IL-6 and IL-1β by monocytes (86,89). Elevated levels of IL-6 and IL-1β
in patients with DCM may lead to cardiomyocyte apoptosis and
impaired systolic function of the heart (89). Cardiac macrophages can be divided
into two types, C-C motif chemokine receptor 2 (CCR)2+
and CCR2−, on the basis of the protein level of CCR2
(86). CCR2+
macrophages are involved in cardiac fibrosis and inflammatory
responses, whereas CCR2− macrophages mediate tissue
repair (86). CCR2+
macrophages in DCM can recruit monocytes and neutrophils to the
inflammatory area by secreting inflammatory cytokines and
chemokines, which promote the inflammatory response (86). B cells can secrete a variety of
cytokines, including proinflammatory agents (e.g., TNF-α) and
anti-inflammatory molecules (e.g., IL-1) (87). Patients with DCM have increased
TNF-α and decreased IL-1 secreted by B cells, which impairs their
anti-inflammatory ability (86).
T cells can be divided into T helper 1 (Th1) cells, Th22 cells,
Th17 cells, T follicular helper cells and regulatory T cells and
are involved in cardiac inflammation and injury, which play crucial
roles in the development of DCM (86). NK cells are important
anti-inflammatory cells and their depletion can cause DCM and heart
failure (86). The inflammatory
cascade caused by various immune cells can eventually lead to
cardiac fibrosis and heart failure (86). The third pathway is
CFIRL-mediated cardiac remodeling (87). In DCM, significantly upregulated
CFIRL expression stimulated by pressure overload or Ang II can
recruit enolase 1 to the nucleus to form a transcription activation
complex that promotes IL6 gene transcription (87). The abnormal upregulation of IL-6
expression mediated by CFIRL has an autocrine effect, directly
promoting the proliferation and differentiation of myofibroblasts
and indirectly stimulating cardiac hypertrophy, which ultimately
leads to heart failure (87).
Previous studies have reported numerous DCM-targeted
drugs, genes and surgical treatments for preventing heart failure
(Table I) (83,86,87,90-98). Immunotherapy is an important
intervention approach for preventing DCM and heart failure
(86,90). Immunotherapy can regulate the
immune system and suppress the myocardial damage caused by
autoimmune reactions (86); it
includes immunosuppressive therapy, intravenous immunoglobulin and
immunoadsorption (86). During
the clinical trial stage, immunosuppressive therapy (e.g.,
prednisolone and azathioprine) can reduce the antibody immune
response by inhibiting the activity of immune response-related
cells such as T cells, B cells and macrophages (86). Intravenous immunoglobulin can
recognize and bind to dysregulated antibodies in patients, which
reduces the attack and damage caused by dysregulated antibodies on
myocardial cells (86). In
immune adsorption therapy, extracorporeal circulation is used to
selectively remove dysregulated antibodies, immune complexes and
other related immune active substances from the blood using an
immune adsorption column (86,90). Angiotensin-converting enzyme
inhibitors (ACEIs) can effectively prevent cardiac fibrosis in DCM
and the development of heart failure (91). β-blockers can reverse left
ventricular dilation, which protects cardiac function and heart
failure (92,93). Mineralocorticoid receptor
antagonists, which can increase the ejection fraction in and reduce
the mortality rate of patients with heart failure, are used to
treat DCM (83,94). In addition, certain genes have
been reported to play important roles in DCM and may be prospective
targets for preventing heart failure (87,95). DCM is a typical hereditary
cardiomyopathy and gene therapy can be used to replace diseased
genes (95). The dysregulation
of sarcomere-related genes [e.g., titin, troponin T2
(TNNT2) and TNNC1] is the main cause of DCM (95). By replacing these dysregulated
genes, DCM can be used to intervene in the development of heart
failure (95). A significant
increase in CFIRL can mediate cardiac remodeling; therefore, CFIRL
can serve as a potential therapeutic target (87). For surgical treatment,
nonischemic DCM can be treated with a left ventricular assist
device to reverse left ventricular remodeling and prevent
progression to heart failure (96,97). Intra-aortic balloon pump
catheters are used as a bridge to heart transplantation and a
transitional therapy for patients with advanced heart failure
(98).
Over the past 30 years, ~8.5 million individuals
have died worldwide from hypertension (99). Hypertension is characterized by
increased pressure overload of the heart and its long-term
development can further enlarge the left ventricle, increase
myocardial oxygen consumption, induce weakened myocardial
contractility and reduce cardiac output, which eventually results
in heart failure (100). It is
essential to study the molecular mechanisms of hypertension and
intervene in its development to prevent heart failure.
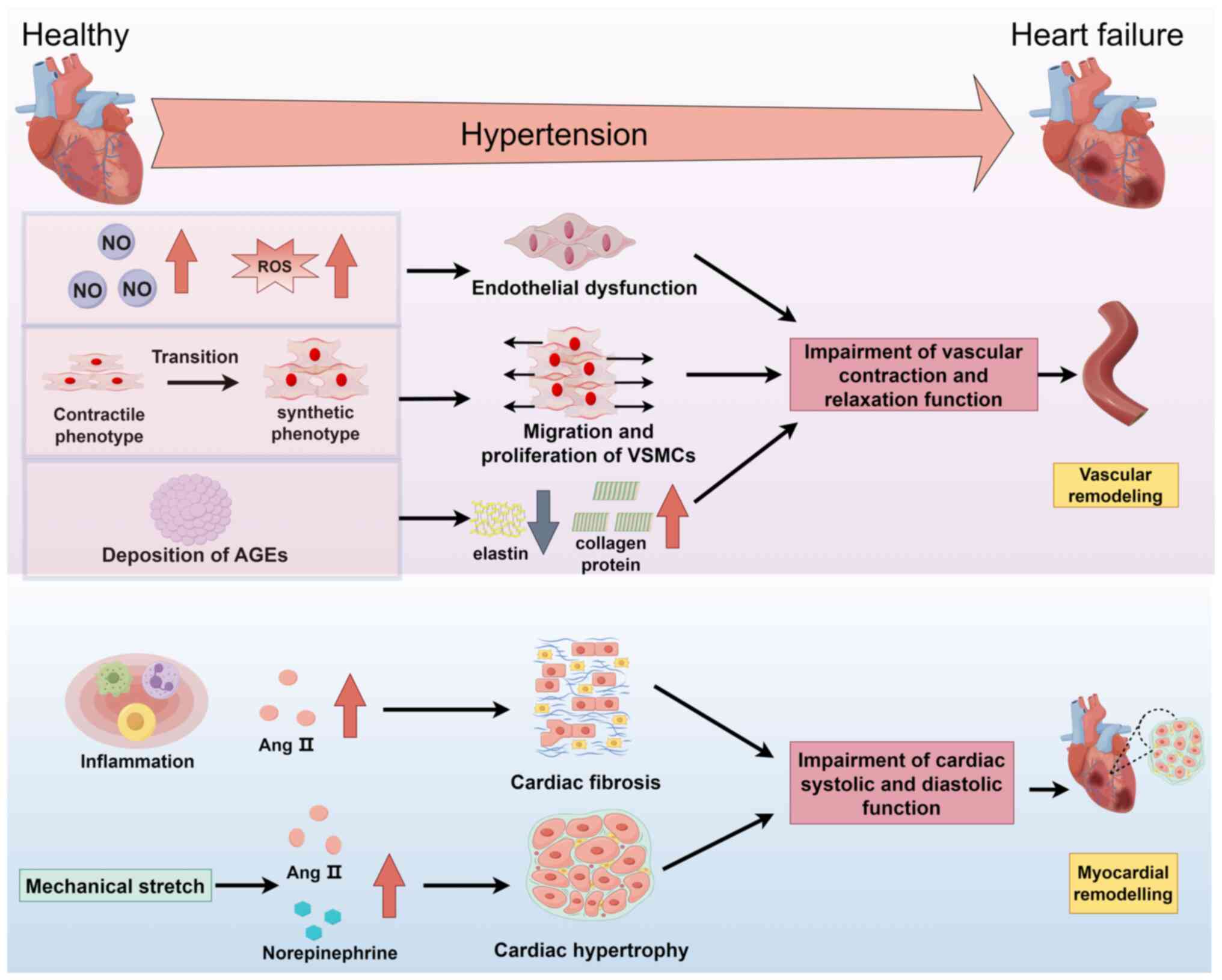
Vascular remodeling is an important pathway through
which hypertension leads to heart failure (101). Vascular remodeling is a type of
pathological change that is difficult to reverse (101). Key events in vascular
remodeling include endothelial cell (EC) dysfunction, the migration
and proliferation of VSMCs and changes in the collagen ECM
(Fig. 3) (101,102). Hypertension is an important
risk factor for EC dysfunction (102). EC dysfunction resulting from
insufficient nitric oxide production and increased levels of ROS in
blood vessels can lead to a decreased vasodilation capacity, which
causes the blood vessels to narrow and blood pressure to continue
to rise, eventually leading to vascular remodeling and heart
failure (102,103). Hypertension is closely related
to the contractile function of VSMCs (101). The phenotypic conversion of
VSMCs from a contractile to a synthetic phenotype causes vascular
calcification and the proliferation and migration of VSMCs due to
the overproduction of Ang II in patients with hypertension, which
leads to impaired vascular contraction function and heart failure
(99,101,104). Alterations in the ECM are
currently irreversible pathological changes in hypertension
(101). The dysregulated
synthesis of the ECM, such as increased collagen and decreased
elastin synthesis, is caused by the activation of matrix
metalloproteinases and the deposition of advanced glycation end
products in hypertension, which can increase the hardness and
weaken the elasticity of the vascular wall (101).
Numerous hypertension-targeted drugs for preventing
heart failure have been reported (Table I) (115-120). The acetyltransferase p300
inhibitor L002 can prevent the progression of heart failure by
reversing hypertension-induced myocardial fibrosis and left
ventricular hypertrophy (115).
Lipoprotein-associated phospholipase A2 plays a key role in
hypertensive cardiac fibrosis, and its inhibitor, darapladib, can
prevent Ang II-induced cardiac remodeling and inflammation
(116). Voltage-gated L-type
Ca2+ channel blockersincluding dihydropyridines (e.g.,
amlodipine), phenylalkylamines (e.g., verapamil) and
benzothiazepines (e.g., diltiazem), are mainly used to treat
hypertension (117). The ROS
produced by nicotinamide adenine dinucleotide phosphate oxidase
(NOX) activation can induce the dysfunction of other oxidase
systems, which leads to a vicious cycle and exacerbates
cardiovascular tissue damage (118). Novel NOX inhibitors, such as
GKT137831 and GSK2795039, have increased selectivity and
specificity for alleviating oxidative stress (118). In a mouse model, NOX inhibitors
have been shown to have inhibitory effects on ROS production, which
implies that these inhibitors have the ability to suppress cardiac
remodeling and heart failure (118). In addition, there are several
clinically recommended antihypertensive drugs, including the RAAS
blockers, ACEIs, angiotensin receptor blockers, calcium channel
blockers and thiazides or thiazide diuretics (119,120).
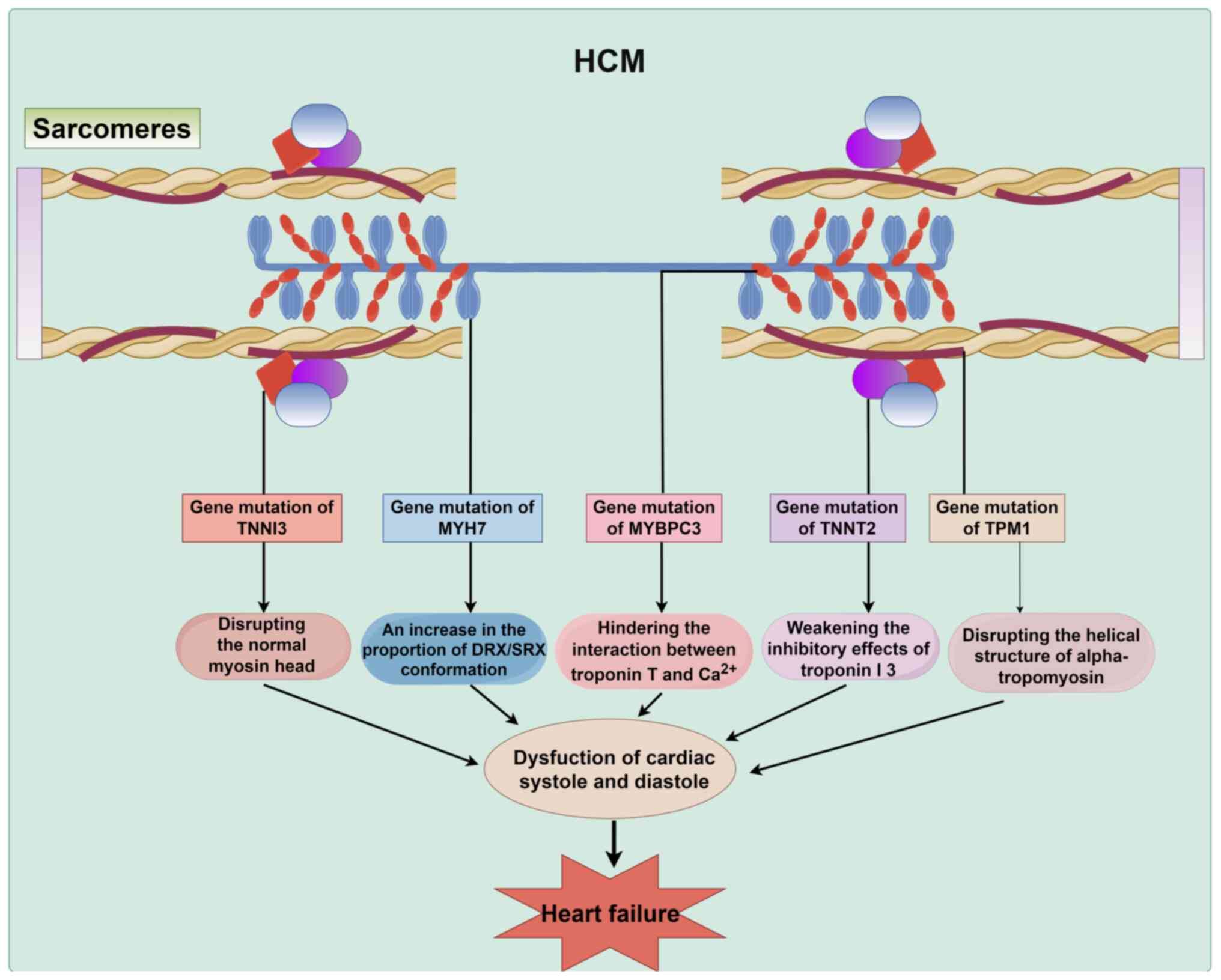
The prevalence of HCM, which is influenced by
genetic factors and sarcomere mutations, is 1:200 (121,122). HCM is characterized by
myocardial hypertrophy, especially asymmetric thickening of the
ventricular septum (123). This
abnormal myocardial hypertrophy can reduce the size of the heart
chambers and severely impair cardiac diastolic function, which
leads to hemodynamic dysregulation (123). Long-term hemodynamic
dysregulations can gradually fatigue the heart and eventually
contribute to heart failure (124). It is important to investigate
the molecular mechanisms of HCM further, to identify more potential
targets for developing therapeutic options for heart failure.
There are numerous reported HCM-targeted drugs,
genes and surgical treatments for preventing heart failure
(Table I) (130-133). In clinical practice, the
β-adrenergic antagonists verapamil and disopyramide are mainly used
to treat HCM (130). Sarcomere
contractile inhibitors can prevent heart failure by reducing
myofilament sensitivity to calcium ions or directly inhibiting
myosin to weaken myocardial contractile function (131). For instance, aficamten can
decrease myosin ATPase activity and the interaction of myosin with
actin, which reduces cardiac contractility (131); it has been reported to exhibit
an inhibitory effect on cardiac contractility and good
pharmacokinetic properties in healthy animals and HCM models
(132). Currently, phase III
clinical trials are underway (132). Gene therapy is highly important
for treating HCM (133).
Exploring various genetic mutation sites to identify exon
methylation and miRNA levels in HCM is beneficial for the
development of gene-targeted therapy methods (133). For instance, in male patients
with HCM with MYBPC3 gene GAGT deletion, the successful
repair of germline mutations was achieved, which improved
homologous-directed repair efficiency without off-target events
(133). Alcohol septal ablation
and surgical myectomy are the primary surgical treatments used
(131).
Until 2019, MI accounted for 49.2% of deaths caused
by CVD; additionally, MI has attracted widespread attention because
of its high mortality rate (134). MI is characterized by
myocardial ischemia due to atherosclerosis, which impairs cardiac
contraction and relaxation and ultimately leads to heart failure
(135,136). Studying the detailed mechanisms
of MI will be beneficial for the development of potential treatment
approaches for heart failure (137).
MI can result in heart failure through the
regulation of cell death, including apoptosis, Ang II-induced
necroptosis, mitochondrial homeostasis and Ca2+
transients (138-146). The first major pathway is cell
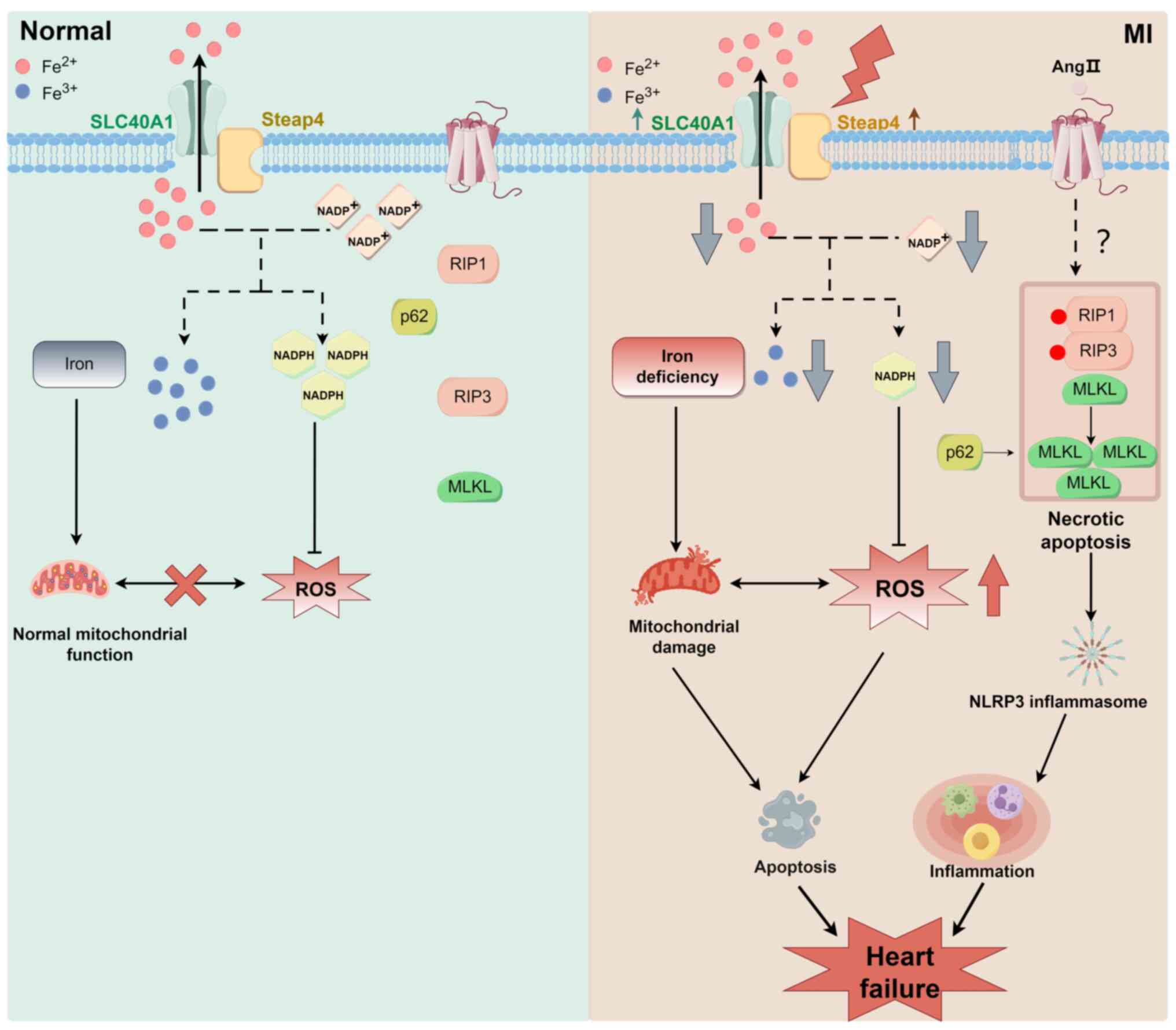
death (Fig. 5). Previous studies
have shown that solute carrier family 40 member 1 and steap family
member 4 expression is upregulated in MI, which promotes iron
efflux (138). More divalent
iron ions are transported extracellularly, which causes iron
deficiency in cells and myocardial mitochondrial dysfunction
because electron transfer within mitochondria requires significant
amounts of iron ions (138,139). Myocardial mitochondrial
dysfunction leads to insufficient energy generation, disrupts the
intracellular redox balance (e.g., NADPH/NADH levels), and promotes
the production of ROS and oxidative stress, which ultimately
stimulates myocardial cell apoptosis (Fig. 5) (138,140). In addition, Ang II-induced
necroptosis plays an important role in the transition from MI to
heart failure (Fig. 5) (141). In acute MI, Ang II levels are
significantly elevated. Ang II is one of the main neurohumoral
factors resulting in the development of heart failure (141). Ang II is involved in myocardial
cell necroptosis by promoting the release of humoral factors from
cardiac fibroblasts (141).
Necroptosis promotes cell rupture and the release of intracellular
contents, such as damage-associated molecular patterns and lactate
dehydrogenase, which stimulate an inflammatory response and
ultimately trigger heart failure (141,142). In MI, mitochondrial
dysfunction, mitochondrial morphological swelling and reduced
cristae can lead to energy metabolism disorders and an insufficient
cellular energy supply (143).
Dysfunction of the mitochondrial electron transport chain in MI can
result in the excessive production of ROS (143). Excessive ROS can attack
mitochondrial membranes, proteins and DNA, which causes
mitochondrial damage (143).
The opening of the mitochondrial permeability transition pore in MI
can result in abnormalities in the mitochondrial membrane
potential, the cessation of ATP synthesis and the release of
proinflammatory factors (e.g., cytochrome C) into the cytoplasm,
which initiates cell apoptosis (143). Calcium transients are another
important mechanism of MI (144). Changes in the calcium
concentration significantly affect myocardial cells (144,145). A decrease in the amplitude of
calcium transients can weaken the contractility of myocardial cells
and affect the blood pumping function of the heart (145,146). A prolonged duration of calcium
transients can hinder the reduction in the intracellular calcium
concentration during diastole, which impairs diastolic function and
limits cardiac filling (17,145).
There are numerous reported MI-targeted drugs and
surgical treatments for preventing heart failure (Table I) (147-153). In clinical practice,
MI-targeted drugs include ACEIs (e.g., ramipril and perindopril),
P2Y12 receptor inhibitors (e.g., clopidogrel) and statins (e.g.,
atorvastatin, simvastatin and rosuvastatin) (147). ACEIs can improve endothelial
function and reduce peripheral vascular resistance in the heart
(147). They can reduce blood
clots and the burden on the heart to intervene in heart failure
(148). P2Y12 receptor
antagonists can inhibit platelet aggregation to reduce thrombosis
and prevent myocardial ischemic necrosis and heart failure
(149). Dual antiplatelet
therapy comprises aspirin and a P2Y receptor antagonist, which has
always been used to treat acute MI (149); it can protect patients with
acute MI from developing heart failure by reducing the degree of
systemic atherothrombosis (149,150). Statins can lower blood lipid
levels, particularly the synthesis of low-density lipoprotein
cholesterol, which reduces blood lipid levels and prevents plaque
formation (151). In stem cell
therapy, differentiated new cardiomyocytes can be used to replace
cardiomyocytes damaged during MI, thus inhibiting cell apoptosis
and heart failure (152).
Coronary intervention can effectively clear up occluded coronary
arteries, which increases myocardial blood supply and prevents
heart failure (153).
Other CVDs, including doxorubicin-induced
cardiomyopathy, iron overload cardiomyopathy, sepsis
cardiomyopathy, viral myocarditis and diabetic cardiomyopathy, can
also develop into heart failure at the end stage (154-162). Doxorubicin-induced
cardiomyopathy can result in heart failure through oxidative stress
injury, apoptosis and mitochondrial damage (154,155). The development of inflammation,
cardiac hypertrophy and fibrosis are the main pathways through
which iron overload cardiomyopathy leads to heart failure (156,157). heart failure is caused
primarily by inflammatory responses in sepsis cardiomyopathy
(158). In viral myocarditis,
heart failure mainly results from direct injury and immune-mediated
injury pathways (159,160). Diabetic cardiomyopathy can
result in heart failure through autonomic dysfunction, cardiac
hypertrophy and fibrosis (161,162). However, the detailed mechanisms
of the abovementioned CVDs remain to be elucidated.
The five-year mortality rate of patients with heart
failure is estimated to be ~50% worldwide (163). The pathological mechanisms of
heart failure include cardiac fibrosis, cardiac hypertrophy,
cardiomyocyte death, cardiac sarcomere disorders, mitochondrial
dysfunction and vascular remodeling (110,132,143,164-172). Intervention approaches for
suppressing the progression of heart failure are available at both
the experimental and clinical stages. However, these approaches
have many challenges and limitations, including off-target effects,
side effects and poor translation of basic research into clinical
practice (173). Gene
therapies, which mainly consist of gene editing technology and
noncoding nucleotide acid methods, can be used to prevent and treat
heart failure (174,175). Gene editing technology can be
used to replace dysregulated sarcomere proteins by targeting
related genes (174).
CRISPR-Cas9 may be used as 'gene scissors' that can remove
erroneous gene fragments and restore related normal genes. The
RNA-guided adeno-associated virus 9 delivery of effective Cas9
nucleases can inactivate pathogenic mutant genes in HCM, thereby
preventing the progression of heart failure (176). In addition, the discovery and
application of regulatory RNA have attracted increasing attention
in medical research. Ambrose and Rufkun were awarded the Nobel
Prize in Physiology or Medicine in 2024 for their discovery of the
miRNA-Lin-14 (177). miR-133, a
type of noncoding nucleotide acid, can alleviate heart failure by
downregulating the expression of proapoptotic proteins such as
caspases-3 and Bax and reducing cardiomyocyte apoptosis (175). However, the main technical
problem and challenge are off-target risks, which may cause other
genetic abnormalities (174).
Drug therapies for heart failure have side effects, including dry
cough caused by ACEIs, slow movement and low blood pressure caused
by β-blockers, and electrolyte imbalance caused by diuretics
(178-180). In addition, it has been
reported that human fibroblasts can be reprogrammed into induced
cardiac-like myocytes to regenerate cardiomyocytes and prevent MI
in the experimental stage; however, further studies are needed to
promote their translation into clinical practice (152).
There are reported methods and strategies to
address the abovementioned issues. Nanotechnology can improve the
precise targeting and reduce off-target effects; it can be used to
package and deliver drugs to the target site of related CVDs
accurately, which thereby reduces off-target effects (181,182). The integration of traditional
Chinese and Western medicine can diminish drug side effects
(183-189). Previous studies have shown that
Platycodon grandiflorum and Flos Farfarae can alleviate
the dry cough caused by ACEIs, and that ginseng can ameliorate the
bradycardia and hypotension caused by β-blockers (184-187). Specific traditional Chinese
medicine active components, such as glycyrrhizic acid and
ginsenoside, can protect patients from the adverse effects of
Western medicine (188,189). In addition, clinical
translation requires more in-depth clinical trials.
Current cutting-edge research technologies, such as
superresolution fluorescence microscopy, cryo-electron microscopy
and photoelectric coupling technology, can promote research on
heart failure and lay the foundation for drug development (190-192). In addition, nanotechnology and
noncoding RNA technology can improve the precise targeting of
disease treatments (176,182). The present review provides
references for basic research on heart failure and lays a
theoretical foundation for the development of therapeutic
drugs.
Not applicable.
All of the authors contributed to the manuscript.
SG, YH and ZR drafted the manuscript, and LL, ZY, LW, XY, ML and XG
performed the literature search. SG, YH, LL, ZY, LW, XY, ML and XG
edited the manuscript. ZR conceived the study and edited and
finalized the manuscript. All authors have read and confirmed the
final version of the manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
The authors thank Dr Zhanhong Ren (Hubei Key
Laboratory of Diabetes and Angiopathy, Xianning Medical College,
Hubei University of Science and Technology, Xianning, China) for
contributing ideas and editing the manuscript and Miss Yingqing Hu,
Miss Li Ling, Mr. Zhuangzhuang Yang, Miss Luxuan Wan (School of
Basic Medical Sciences, Xianning Medical College, Hubei University
of Science and Technology, Xianning, China) and Miss Shuang Guo, Dr
Xiaosong Yang, Dr Min Lei and Dr Xiying Guo (Hubei Key Laboratory
of Diabetes and Angiopathy, Xianning Medical College, Hubei
University of Science and Technology, Xianning, China) for editing
the manuscript. Figures were created and designed by Figdraw.
This study was jointly supported by Hubei Provincial Natural
Science Foundation and Xianning of China (grant no. 2025AFD407),
the Special Project on Diabetes and Angiopathy (grant no.
2024TNB04), the Scientific Research and Innovation Team of Hubei
University of Science and Technology (grant no. 2022T01) and the
Horizontal Scientific Research Project of Hubei University of
Science and Technology (grant no. 2024HX132).
|
1
|
Ketabi M, Andishgar A, Fereidouni Z, Sani
MM, Abdollahi A, Vali M, Alkamel A and Tabrizi R: Predicting the
risk of mortality and rehospitalization in heart failure patients:
A retrospective cohort study by machine learning approach. Clin
Cardiol. 47:e242392024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu X and Xu Z; Association between
phenotypic age the risk of mortality in patients with heart
failure: A retrospective cohort study. Clin Cardiol. 47:e243212024.
View Article : Google Scholar
|
|
3
|
Pratley R, Guan X, Moro RJ and do Lago R:
Chapter 1: The burden of heart failure. Am J Med. 137(2S): S3–S8.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mulugeta H, Sinclair PM and Wilson A:
Prevalence of depression and its association with health-related
quality of life in people with heart failure in low- and
middle-income countries: A systematic review and meta-analysis.
PLoS One. 18:e02831462023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shahim B, Kapelios CJ, Savarese G and Lund
LH: Global public health burden of heart failure: An updated
review. Card Fail Rev. 9:e112023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Emmons-Bell S, Johnson C and Roth G:
Prevalence, incidence and survival of heart failure: A systematic
review. Heart. 108:1351–1360. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Caturano A, Vetrano E, Galiero R,
Salvatore T, Docimo G, Epifani R, Alfano M, Sardu C, Marfella R,
Rinaldi L and Sasso FC: Cardiac hypertrophy: From
pathophysiological mechanisms to heart failure development. Rev
Cardiovasc Med. 23:1652022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Castiglione V, Aimo A, Vergaro G, Saccaro
L, Passino C and Emdin M: Biomarkers for the diagnosis and
management of heart failure. Heart Fail Rev. 27:625–643. 2022.
View Article : Google Scholar :
|
|
9
|
Bozkurt B, Coats AJS, Tsutsui H,
Abdelhamid CM, Adamopoulos S, Albert N, Anker SD, Atherton J, Böhm
M, Butler J, et al: Universal definition and classification of
heart failure: A report of the heart failure society of America,
heart failure association of the European society of cardiology,
Japanese heart failure society and writing committee of the
universal definition of heart failure: Endorsed by the Canadian
heart failure society, heart failure association of India, cardiac
society of Australia and New Zealand, and Chinese heart failure
association. Eur J Heart Fail. 23:352–380. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Triposkiadis F, Xanthopoulos A, Parissis
J, Butler J and Farmakis D: Pathogenesis of chronic heart failure:
Cardiovascular aging, risk factors, comorbidities, and disease
modifiers. Heart Fail Rev. 27:337–344. 2022. View Article : Google Scholar
|
|
11
|
Huang K, Wu H, Xu X, Wu L, Li Q and Han L:
Identification of TGF-β-related genes in cardiac hypertrophy and
heart failure based on single cell RNA sequencing. Aging (Albany
NY). 15:7187–7218. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gu JJ, Du TJ, Zhang LN, Zhou J, Gu X and
Zhu Y: Identification of ferroptosis-related genes in heart failure
induced by transverse aortic constriction. J Inflamm Res.
16:4899–4912. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marian AJ: Molecular genetic basis of
hypertrophic cardiomyopathy. Circ Res. 128:1533–1553. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Madè A, Bibi A, Garcia-Manteiga JM,
Tascini AS, Piella SN, Tikhomirov R, Voellenkle C, Gaetano C,
Leszek P, Castelvecchio S, et al: circRNA-miRNA-mRNA deregulated
network in ischemic heart failure patients. Cells. 12:25782023.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wong LL, Wang J, Liew OW, Richards AM and
Chen YT: MicroRNA and heart failure. Int J Mol Sci. 17:5022016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park JH and Kho C: MicroRNAs and calcium
signaling in heart disease. Int J Mol Sci. 22:105822021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Saad NS, Mashali MA, Repas SJ and Janssen
PML: Altering calcium sensitivity in heart failure: A crossroads of
disease etiology and therapeutic innovation. Int J Mol Sci.
24:175772023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hinton A Jr, Claypool SM, Neikirk K, Senoo
N, Wanjalla CN, Kirabo A and Williams CR: Mitochondrial structure
and function in human heart failure. Circ Res. 135:372–396. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xue P, Liu Y, Wang H, Huang J and Luo M:
miRNA-103-3p-Hlf regulates apoptosis and autophagy by targeting
hepatic leukaemia factor in heart failure. ESC Heart Fail.
10:3038–3045. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pagan LU, Gomes MJ, Martinez PF and Okoshi
MP: Oxidative stress and heart failure: Mechanisms, signalling
pathways, and therapeutics. Oxid Med Cell Longev. 2022:98295052022.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hanna A and Frangogiannis NG: Inflammatory
cytokines and chemokines as therapeutic targets in heart failure.
Cardiovasc Drugs Ther. 34:849–863. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Manolis AA, Manolis TA and Manolis AS:
Neurohumoral activation in heart failure. Int J Mol Sci.
24:154722023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cappola TP and Burke MF: Studying Heart
failure through the lens of gene regulation. Circ Heart Fail.
13:e0076472020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Levin MG, Tsao NL, Singhal P, Liu C, Vy
HMT, Paranjpe I, Backman JD, Bellomo TR, Bone WP, Biddinger KJ, et
al: Genome-wide association and multi-trait analyses characterize
the common genetic architecture of heart failure. Nat Commun.
13:69142022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tudurachi BS, Zăvoi A, Leonte A, Țăpoi L,
Ureche C, Bîrgoan SG, Chiuariu T, Anghel L, Radu R, Sascău RA and
Stătescu C: An update on MYBPC3 gene mutation in hypertrophic
cardiomyopathy. Int J Mol Sci. 24:105102023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park J, Packard EA, Levin MG, Judy RL;
Regeneron Genetics Center; Damrauer SM, Day SM, Ritchie MD and
Rader DJ: A genome-first approach to rare variants in hypertrophic
cardiomyopathy genes MYBPC3 and MYH7 in a medical biobank. Hum Mol
Genet. 31:827–837. 2022. View Article : Google Scholar :
|
|
27
|
Akboua H, Eghbalzadeh K, Keser U, Wahlers
T and Paunel-Görgülü A: Impaired non-canonical transforming growth
factor-β signalling prevents profibrotic phenotypes in cultured
peptidylarginine deiminase 4-deficient murine cardiac fibroblasts.
J Cell Mol Med. 25:9674–9684. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qian L, Xu H, Yuan R, Yun W and Ma Y:
Formononetin ameliorates isoproterenol induced cardiac fibrosis
through improving mitochondrial dysfunction. Biomed Pharmacother.
170:1160002024. View Article : Google Scholar
|
|
29
|
Roe AT, Frisk M and Louch WE: Targeting
cardiomyocyte Ca2+ homeostasis in heart failure. Curr Pharm Des.
21:431–448. 2015. View Article : Google Scholar :
|
|
30
|
Shah K, Seeley S, Schulz C, Fisher J and
Gururaja Rao S: Calcium channels in the heart: Disease states and
drugs. Cells. 11:9432022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Al-Hayali MA, Sozer V, Durmus S, Erdenen
F, Altunoglu E, Gelisgen R, Atukeren P, Atak PG and Uzun H:
Clinical value of circulating microribonucleic acids miR-1 and
miR-21 in evaluating the diagnosis of acute heart failure in
asymptomatic type 2 diabetic patients. Biomolecules. 9:1932019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vilella-Figuerola A, Gallinat A, Escate R,
Mirabet S, Padró T and Badimon L: Systems biology in chronic heart
failure-identification of potential miRNA regulators. Int J Mol
Sci. 23:152262022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu R, Wu J, Yang CJ, Kang L, Ji YY, Li C,
Ding ZW and Zou YZ: A circRNA-miRNA-mRNA network analysis
underlying pathogenesis of human heart failure. J Geriatr Cardiol.
20:350–360. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gallo G, Rubattu S and Volpe M:
Mitochondrial dysfunction in heart failure: From Pathophysiological
mechanisms to therapeutic opportunities. Int J Mol Sci.
25:26672024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
van Empel VPM, Bertrand ATA, Hofstra L,
Crijns HJ, Doevendans PA and De Windt LJ: Myocyte apoptosis in
heart failure. Cardiovasc Res. 67:21–29. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo X, Chen Y and Liu Q: Necroptosis in
heart disease: Molecular mechanisms and therapeutic implications. J
Mol Cell Cardiol. 169:74–83. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kang Y and Wang Q: Potential therapeutic
value of necroptosis inhibitor for the treatment of COVID-19. Eur J
Med Res. 27:2832022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lu LQ, Tian J, Luo XJ and Peng J:
Targeting the pathways of regulated necrosis: A potential strategy
for alleviation of cardio-cerebrovascular injury. Cell Mol Life
Sci. 78:63–78. 2021. View Article : Google Scholar
|
|
39
|
Zhang H and Dhalla NS: The role of
pro-inflammatory cytokines in the pathogenesis of cardiovascular
disease. Int J Mol Sci. 25:10822024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bishopric NH, Andreka P, Slepak T and
Webster KA: Molecular mechanisms of apoptosis in the cardiac
myocyte. Curr Opin Pharmacol. 1:141–150. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tsutsui H, Kinugawa S and Matsushima S:
Oxidative stress and heart failure. Am J Physiol Heart Circ
Physiol. 301:H2181–H2190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Khoynezhad A, Jalali Z and Tortolani AJ: A
synopsis of research in cardiac apoptosis and its application to
congestive heart failure. Tex Heart Inst J. 34:352–359.
2007.PubMed/NCBI
|
|
43
|
Paulus WJ and Zile MR: From systemic
inflammation to myocardial fibrosis: The heart failure with
preserved ejection fraction paradigm revisited. Circ Res.
128:1451–1467. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kjaer A and Hesse B: Heart failure and
neuroendocrine activation: Diagnostic, prognostic and therapeutic
perspectives. Clin Physiol. 21:661–672. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Blackwell DJ, Schmeckpeper J and Knollmann
BC: Animal models to study cardiac arrhythmias. Circ Res.
130:1926–1964. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Reißmann B, Rottner L, Rillig A and
Metzner A: Cardiac arrhythmia. MMW Fortschr Med. 163:62–71. 2021.In
German. View Article : Google Scholar
|
|
47
|
Fu DG: Cardiac arrhythmias: Diagnosis,
symptoms, and treatments. Cell Biochem Biophys. 73:291–296. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Paludan-Müller C, Vad OB, Stampe NK,
Diederichsen SZ, Andreasen L, Monfort LM, Fosbøl EL, Køber L,
Torp-Pedersen C, Svendsen JH and Olesen MS: Atrial fibrillation:
Age at diagnosis, incident cardiovascular events, and mortality.
Eur Heart J. 45:2119–2129. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu X, Zhang W, Luo J, Shi W, Zhang X, Li
Z, Qin X, Liu B and Wei Y: TRIM21 deficiency protects against
atrial inflammation and remodeling post myocardial infarction by
attenuating oxidative stress. Redox Biol. 62:1026792023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bossuyt J, Borst JM, Verberckmoes M,
Bailey LRJ, Bers DM and Hegyi B: Protein kinase D1 regulates
cardiac hypertrophy, potassium channel remodeling, and arrhythmias
in heart failure. J Am Heart Assoc. 11:e0275732022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Piccini JP: Atrial fibrillation and heart
failure: Too much talk and not enough action. JACC Clin
Electrophysiol. 9:581–582. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang H, Chen Y, Zhao S, Wang X, Lu K and
Xiao H: Effect of Sox9 on TGF-β1-mediated atrial fibrosis. Acta
Biochim Biophys Sin (Shanghai). 53:1450–1458. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wolke C, Antileo E and Lendeckel U: WNT
signaling in atrial fibrillation. Exp Biol Med (Maywood).
246:1112–1120. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Huang M, Huiskes FG, de Groot NMS and
Brundel BJJM: The role of immune cells driving electropathology and
atrial fibrillation. Cells. 13:3112021. View Article : Google Scholar
|
|
55
|
Yokoyama T, Kuga T, Itoh Y, Otake S, Omata
C, Saitoh M and Miyazawa K: Smad2Δexon3 and Smad3 have distinct
properties in signal transmission leading to TGF-β-induced cell
motility. J Biol Chem. 299:1028202023. View Article : Google Scholar
|
|
56
|
Miyazawa K, Itoh Y, Fu H and Miyazono K:
Receptor-activated transcription factors and beyond: Multiple modes
of Smad2/3-dependent transmission of TGF-β signaling. J Biol Chem.
300:1072562024. View Article : Google Scholar
|
|
57
|
Hu HH, Chen DQ, Wang YN, Feng YL, Cao G,
Vaziri ND and Zhao YY: New insights into TGF-β/Smad signaling in
tissue fibrosis. Chem Biol Interact. 292:76–83. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gao S, Li X, Jiang Q, Liang Q, Zhang F, Li
S, Zhang R, Luan J, Zhu J, Gu X, et al: PKM2 promotes pulmonary
fibrosis by stabilizing TGF-β1 receptor I and enhancing TGF-β1
signaling. Sci Adv. 8:eabo09872022. View Article : Google Scholar
|
|
59
|
Stolfi C, Troncone E, Marafini I and
Monteleone G: Role of TGF-beta and Smad7 in gut inflammation,
fibrosis and cancer. Biomolecules. 11:172020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Li X, Zhu F, Meng W, Zhang F, Hong J,
Zhang G and Wang F: CYP2J2/EET reduces vulnerability to atrial
fibrillation in chronic pressure overload mice. J Cell Mol Med.
24:862–874. 2020. View Article : Google Scholar
|
|
62
|
Wang D, Wang X, Yang T, Tian H, Su Y and
Wang Q: Long non-coding RNA Dancr affects myocardial fibrosis in
atrial fibrillation mice via the MicroRNA-146b-5p/Smad5 axis. Acta
Cardiol Sin. 39:841–853. 2023.PubMed/NCBI
|
|
63
|
Tian L, Wang Y and Jang YY: Wnt signaling
in biliary development, proliferation, and fibrosis. Exp Biol Med
(Maywood). 247:360–367. 2022. View Article : Google Scholar
|
|
64
|
Somanader DVN, Zhao P, Widdop RE and
Samuel CS: The involvement of the Wnt/β-catenin signaling cascade
in fibrosis progression and its therapeutic targeting by relaxin.
Biochem Pharmacol. 223:1161302024. View Article : Google Scholar
|
|
65
|
Roberts JD, Murphy NP, Hamilton RM,
Lubbers ER, James CA, Kline CF, Gollob MH, Krahn AD, Sturm AC, Musa
H, et al: Ankyrin-B dysfunction predisposes to arrhythmogenic
cardiomyopathy and is amenable to therapy. J Clin Invest.
129:3171–3184. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lorenzon A, Calore M, Poloni G, De Windt
LJ, Braghetta P and Rampazzo A: Wnt/β-catenin pathway in
arrhythmogenic cardiomyopathy. Oncotarget. 8:60640–60655. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lv X, Li J, Hu Y, Wang S, Yang C, Li C and
Zhong G: Overexpression of miR-27b-3p targeting Wnt3a regulates the
signaling pathway of Wnt/β-catenin and attenuates atrial fibrosis
in rats with atrial fibrillation. Oxid Med Cell Longev.
2019:57037642019. View Article : Google Scholar
|
|
68
|
Lai YJ, Tsai FC, Chang GJ, Chang SH, Huang
CC, Chen WJ and Yeh YH: miR-181b targets semaphorin 3A to mediate
TGF-β-induced endothelial-mesenchymal transition related to atrial
fibrillation. J Clin Invest. 132:e1425482022. View Article : Google Scholar
|
|
69
|
Zhang Z, Li L, Hu Z, Zhou L, Zhang Z,
Xiong Y and Yao Y: Causal effects between atrial fibrillation and
heart failure: Evidence from a bidirectional Mendelian
randomization study. BMC Med Genomics. 16:1872023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sagris M, Vardas EP, Theofilis P,
Antonopoulos AS, Oikonomou E and Tousoulis D: Atrial fibrillation:
Pathogenesis, predisposing factors, and genetics. Int J Mol Sci.
23:62021. View Article : Google Scholar
|
|
71
|
Husti Z, Varró A and Baczkó I:
Arrhythmogenic remodeling in the failing heart. Cells. 10:32032021.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Sun Z, Zhou D, Xie X, Wang S, Wang Z, Zhao
W, Xu H and Zheng L: Cross-talk between macrophages and atrial
myocytes in atrial fibrillation. Basic Res Cardiol. 111:632016.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Huiskes FG, Creemers EE and Brundel BJJM:
Dissecting the molecular mechanisms driving electropathology in
atrial fibrillation: Deployment of RNA sequencing and
transcriptomic analyses. Cells. 12:22422023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Maury P, Sanchis K, Djouadi K, Cariou E,
Delasnerie H, Boveda S, Fournier P, Itier R, Mondoly P,
Voglimacci-Stephan opoli Q, et al: Catheter ablation of atrial
arrhythmias in cardiac amyloidosis: Impact on heart failure and
mortality. PLoS One. 19:e03017532024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Lei M, Wu L, Terrar DA and Huang CL:
Modernized classification of cardiac antiarrhythmic drugs.
Circulation. 138:1879–1896. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Mankad P and Kalahasty G: Antiarrhythmic
drugs: Risks and benefits. Med Clin North Am. 103:821–834. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Romero G, Martin B, Gabris B and Salama G:
Relaxin suppresses atrial fibrillation, reverses fibrosis and
reduces inflammation in aged hearts. Biochem Pharmacol.
227:1164072024. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Martin B, Gabris B, Barakat AF, Henry BL,
Giannini M, Reddy RP, Wang X, Romero G and Salama G: Relaxin
reverses maladaptive remodeling of the aged heart through
Wnt-signaling. Sci Rep. 9:185452019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hao H, Yan S, Zhao X, Han X, Fang N, Zhang
Y, Dai C, Li W, Yu H, Gao Y, et al: Atrial myocyte-derived exosomal
microRNA contributes to atrial fibrosis in atrial fibrillation. J
Transl Med. 20:4072022. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Benali K, Khairy P, Hammache N, Petzl A,
Da Costa A, Verma A, Andrade JG and Macle L: Procedure-related
complications of catheter ablation for atrial fibrillation. J Am
Coll Cardiol. 81:2089–2099. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhang Z, Xiao Y, Dai Y, Lin Q and Liu Q:
Device therapy for patients with atrial fibrillation and heart
failure with preserved ejection fraction. Heart Fail Rev.
29:417–430. 2024. View Article : Google Scholar :
|
|
82
|
Li S, Wang Y, Yang W, Zhou D, Zhuang B, Xu
J, He J, Yin G, Fan X, Wu W, et al: Cardiac MRI risk stratification
for dilated cardiomyopathy with left ventricular ejection fraction
of 35% or higher. Radiology. 306:e2130592023. View Article : Google Scholar
|
|
83
|
Schultheiss HP, Fairweather D, Caforio
ALP, Escher F, Hershberger RE, Lipshultz SE, Liu PP, Matsumori A,
Mazzanti A, McMurray J and Priori SG: Dilated cardiomyopathy. Nat
Rev Dis Primers. 5:322019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Orphanou N, Papatheodorou E and
Anastasakis A: Dilated cardiomyopathy in the era of precision
medicine: Latest concepts and developments. Heart Fail Rev.
27:1173–1191. 2022. View Article : Google Scholar
|
|
85
|
Liu M, Zhai L, Yang Z, Li S, Liu T, Chen
A, Wang L, Li Y, Li R, Li C, et al: Integrative proteomic analysis
reveals the cytoskeleton regulation and mitophagy difference
between ischemic cardiomyopathy and dilated cardiomyopathy. Mol
Cell Proteomics. 22:1006672023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang E, Zhou R, Li T, Hua Y, Zhou K, Li Y,
Luo S and An Q: The molecular role of immune cells in dilated
cardiomyopathy. Medicina (Kaunas). 59:12462023. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yuan S, Zhang X, Zhan J, Xie R, Fan J, Dai
B, Zhao Y, Yin Z, Liu Q, Wang DW, et al: Fibroblast-localized
lncRNA CFIRL promotes cardiac fibrosis and dysfunction in dilated
cardiomyopathy. Sci China Life Sci. 67:1155–1169. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Mado K, Chekulayev V, Shevchuk I, Puurand
M, Tepp K and Kaambre T: On the role of tubulin, plectin, desmin,
and vimentin in the regulation of mitochondrial energy fluxes in
muscle cells. Am J Physiol Cell Physiol. 316:C657–C667. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zhang J, Cheng L, Li Z, Li H, Liu Y, Zhan
H, Xu H, Huang Y, Feng F and Li Y: Immune cells and related
cytokines in dilated cardiomyopathy. Biomed Pharmacother.
171:1161592024. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Cavusoglu Y, Tahmazov S, Murat S and Akay
OM: Immunoadsorption therapy in refractory heart failure patients
with dilated cardiomyopathy: A potential therapeutic option. Rev
Assoc Med Bras (1992). 69:90–96. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zhong G, Chen C, Wu S, Chen J, Han Y, Zhu
Q, Xu M, Nie Q and Wang L: Mechanism of angiotensin-converting
enzyme inhibitors in the treatment of dilated cardiomyopathy based
on a protein interaction network and molecular docking. Cardiovasc
Diagn Ther. 13:534–549. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Tong X, Shen L, Zhou X, Wang Y, Chang S
and Lu S: Comparative efficacy of different drugs for the treatment
of dilated cardiomyopathy: A systematic review and network
meta-analysis. Drugs R D. 23:197–210. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Jiao M, Wang X, Liang Y, Yang Y, Gu Y,
Wang Z, Lv Z and Jin M: Effect of β-blocker therapy on the level of
soluble ST2 protein in pediatric dilated cardiomyopathy. Medicina
(Kaunas). 58:13392022. View Article : Google Scholar
|
|
94
|
Ferreira JP, Butler J, Zannad F,
Filippatos G, Schueler E, Steubl D, Zeller C, Januzzi JL, Pocock S,
Packer M and Anker SD: Mineralocorticoid receptor antagonists and
empagliflozin in patients with heart failure and preserved ejection
fraction. J Am Coll Cardiol. 79:1129–1137. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Eldemire R, Mestroni L and Taylor MRG:
Genetics of dilated cardiomyopathy. Annu Rev Med. 75:417–426. 2024.
View Article : Google Scholar :
|
|
96
|
Jordan E, Peterson L, Ai T, Asatryan B,
Bronicki L, Brown E, Celeghin R, Edwards M, Fan J, Ingles J, et al:
Evidence-based assessment of genes in dilated cardiomyopathy.
Circulation. 144:7–19. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Koga-Ikuta A, Fukushima S, Ishibashi-Ueda
H, Tadokoro N, Kakuta T, Watanabe T, Fukushima N, Suzuki K, Fukui T
and Fujita T: Immunocompetent cells in durable ventricular assist
device-implanted non-ischaemic dilated cardiomyopathy. Gen Thorac
Cardiovasc Surg. 70:685–693. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Dziekiewicz M, Banaszewski M, Kuć M and
Stępińska J: Intra-aortic balloon pump catheter insertion using a
novel left external iliac artery approach in a case of chronic
heart failure due to dilated cardiomyopathy. Am J Case Rep.
20:1826–1829. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhang X, Wei R, Wang X, Zhang W, Li M, Ni
T, Weng W and Li Q: The neutrophil-to-lymphocyte ratio is
associated with all-cause and cardiovascular mortality among
individuals with hypertension. Cardiovasc Diabetol. 23:1172024.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Messerli FH, Rimoldi SF and Bangalore S:
The transition from hypertension to heart failure: Contemporary
update. JACC Heart Fail. 5:543–551. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ma J, Li Y, Yang X, Liu K, Zhang X, Zuo X,
Ye R, Wang Z, Shi R, Meng Q and Chen X: Signaling pathways in
vascular function and hypertension: Molecular mechanisms and
therapeutic interventions. Signal Transduct Target Ther. 8:1682023.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Konukoglu D and Uzun H: Endothelial
dysfunction and hypertension. Hypertension: From basic research to
clinical practice. 511–540. 2017.
|
|
103
|
Watson T, Goon PKY and Lip GYH:
Endothelial progenitor cells, endothelial dysfunction,
inflammation, and oxidative stress in hypertension. Antioxid Redox
Signal. 10:1079–1088. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhu N, Guo ZF, Kazama K, Yi B, Tongmuang
N, Yao H, Yang R, Zhang C, Qin Y, Han L and Sun J: Epigenetic
regulation of vascular smooth muscle cell phenotypic switch and
neointimal formation by PRMT5. Cardiovasc Res. 119:2244–2255. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Liard JF and Peters G: Role of the
retention of water and sodium in two types of experimental
renovascular hypertension in the rat. Pflugers Arch. 344:93–108.
1973. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Hu Y, Lin L, Zhang L, Li Y, Cui X, Lu M,
Zhang Z, Guan X, Zhang M, Hao J, et al: Identification of
circulating plasma proteins as a mediator of hypertension-driven
cardiac remodeling: A mediation mendelian randomization study.
Hypertension. 81:1132–1144. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wang M, Han X, Yu T, Wang M, Luo W, Zou C,
Li X, Li G, Wu G, Wang Y and Liang G: OTUD1 promotes pathological
cardiac remodeling and heart failure by targeting STAT3 in
cardiomyocytes. Theranostics. 13:2263–2280. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Holopainen T, Räsänen M, Anisimov A,
Tuomainen T, Zheng W, Tvorogov D, Hulmi JJ, Andersson LC, Cenni B,
Tavi P, et al: Endothelial Bmx tyrosine kinase activity is
essential for myocardial hypertrophy and remodeling. Proc Natl Acad
Sci USA. 112:13063–13068. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Li L, Fu W, Gong X, Chen Z, Tang L, Yang
D, Liao Q, Xia X, Wu H, Liu C, et al: The role of G protein-coupled
receptor kinase 4 in cardiomyocyte injury after myocardial
infarction. Eur Heart J. 42:1415–1430. 2021. View Article : Google Scholar :
|
|
110
|
Bazgir F, Nau J, Nakhaei-Rad S, Amin E,
Wolf MJ, Saucerman JJ, Lorenz K and Ahmadian MR: The
microenvironment of the pathogenesis of cardiac hypertrophy. Cells.
12:17802023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Bertaud A, Joshkon A, Heim X, Bachelier R,
Bardin N, Leroyer AS and Blot-Chabaud M: Signaling pathways and
potential therapeutic strategies in cardiac fibrosis. Int J Mol
Sci. 24:17562023. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zhang W, Wang Q, Feng Y, Chen X, Yang L,
Xu M, Wang X, Li W, Niu X and Gao D: MicroRNA-26a protects the
heart against hypertension-induced myocardial fibrosis. J Am Heart
Assoc. 9:e0179702020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Failer T, Amponsah-Offeh M, Neuwirth A,
Kourtzelis I, Subramanian P, Mirtschink P, Peitzsch M, Matschke K,
Tugtekin SM, Kajikawa T, et al: Developmental endothelial locus-1
protects from hypertension-induced cardiovascular remodeling via
immunomodulation. J Clin Invest. 132:e1261552022. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Rai R, Sun T, Ramirez V, Lux E, Eren M,
Vaughan DE and Ghosh AK: Acetyltransferase p300 inhibitor reverses
hypertension-induced cardiac fibrosis. J Cell Mol Med.
23:3026–3031. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Lv SL, Zeng ZF, Gan WQ, Wang WQ, Li TG,
Hou YF, Yan Z, Zhang RX and Yang M: Lp-PLA2 inhibition prevents Ang
II-induced cardiac inflammation and fibrosis by blocking macrophage
NLRP3 inflammasome activation. Acta Pharmacol Sin. 42:2016–2032.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Johnson MT, Gudlur A, Zhang X, Xin P,
Emrich SM, Yoast RE, Courjaret R, Nwokonko RM, Li W, Hempel N, et
al: L-type Ca2+ channel blockers promote vascular
remodeling through activation of STIM proteins. Proc Natl Acad Sci
USA. 117:17369–17380. 2020. View Article : Google Scholar
|
|
118
|
Zhang Y, Murugesan P, Huang K and Cai H:
NADPH oxidases and oxidase crosstalk in cardiovascular diseases:
novel therapeutic targets. Nat Rev Cardiol. 17:170–194. 2020.
View Article : Google Scholar
|
|
119
|
Nardoianni G, Pala B, Scoccia A, Volpe M,
Barbato E and Tocci G: Systematic review article: New drug
strategies for treating resistant hypertension-the importance of a
mechanistic, personalized approach. High Blood Press Cardiovasc
Prev. 31:99–112. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Zhang ZY, Yu YL, Asayama K, Hansen TW,
Maestre GE and Staessen JA: Starting antihypertensive drug
treatment with combination therapy: Controversies in
hypertension-con side of the argument. Hypertension. 77:788–798.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Maron BJ, Desai MY, Nishimura RA, Spirito
P, Rakowski H, Towbin JA, Dearani JA, Rowin EJ, Maron MS and
Sherrid MV: Management of hypertrophic cardiomyopathy: JACC
state-of-the-art review. J Am Coll Cardiol. 79:390–414. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Maron BA, Wang RS, Carnethon MR, Rowin EJ,
Loscalzo J, Maron BJ and Maron MS: What causes hypertrophic
cardiomyopathy? Am J Cardiol. 179:74–82. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Cirino AL, Channaoui N and Ho C:
Nonsyndromic hypertrophic cardiomyopathy overview.
GeneReviews® [Internet]. Adam MP, Feldman J, Mirzaa GM,
Pagon RA, Wallace SE and Amemiya A: University of Washington;
Seattle, WA: 1993
|
|
124
|
Bazan SGZ, Oliveira GO, Silveira CFDSMPD,
Reis FM, Malagutte KNDS, Tinasi LSN, Bazan R, Hueb JC and Okoshi K:
Hypertrophic cardiomyopathy: A review. Arq Bras Cardiol.
115:927–935. 2020.In English, Portuguese. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Homburger JR, Green EM, Caleshu C, Sunitha
MS, Taylor RE, Ruppel KM, Metpally RP, Colan SD, Michels M, Day SM,
et al: Multidimensional structure-function relationships in human
β-cardiac myosin from population-scale genetic variation. Proc Natl
Acad Sci USA. 113:6701–6706. 2016. View Article : Google Scholar
|
|
126
|
Lin LR, Hu XQ, Lu LH, Dai JZ, Lin NN, Wang
RH, Xie ZX and Chen XM: MicroRNA expression profiles in familial
hypertrophic cardiomyopathy with myosin-binding protein C3 (MYBPC3)
gene mutations. BMC Cardiovasc Disord. 22:2782022. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Parbhudayal RY, Harms HJ, Michels M, van
Rossum AC, Germans T and van der Velden J: Increased myocardial
oxygen consumption precedes contractile dysfunction in hypertrophic
cardiomyopathy caused by pathogenic TNNT2 gene variants. J Am Heart
Assoc. 9:e0153162020. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Fahed AC, Nemer G, Bitar FF, Arnaout S,
Abchee AB, Batrawi M, Khalil A, Abou Hassan OK, DePalma SR,
McDonough B, et al: Founder mutation in N terminus of cardiac
troponin i causes malignant hypertrophic cardiomyopathy. Circ Genom
Precis Med. 13:444–452. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Schulz EM, Wilder T, Chowdhury SA, Sheikh
HN, Wolska BM, Solaro RJ and Wieczorek DF: Decreasing tropomyosin
phosphorylation rescues tropomyosin-induced familial hypertrophic
cardiomyopathy. J Biol Chem. 288:28925–28935. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Wijnker PJM and van der Velden J:
Mutation-specific pathology and treatment of hypertrophic
cardiomyopathy in patients, mouse models and human engineered heart
tissue. Biochim Biophys Acta Mol Basis Dis. 1866:1657742020.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Enriquez AD and Goldman ME: Management of
hypertrophic cardiomyopathy. Ann Glob Health. 80:35–45. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Lehman SJ, Crocini C and Leinwand LA:
Targeting the sarcomere in inherited cardiomyopathies. Nat Rev
Cardiol. 19:353–363. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Pradeep R, Akram A, Proute MC, Kothur NR,
Georgiou P, Serhiyenia T, Shi W, Kerolos ME and Mostafa JA:
Understanding the genetic and molecular basis of familial
hypertrophic cardiomyopathy and the current trends in gene therapy
for its management. Cureus. 13:e175482021.PubMed/NCBI
|
|
134
|
Liu T, Hao Y, Zhang Z, Zhou H, Peng S,
Zhang D, Li K, Chen Y and Chen M: Advanced cardiac patches for the
treatment of myocardial infarction. Circulation. 149:2002–2020.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Golforoush P, Yellon DM and Davidson SM:
Mouse models of atherosclerosis and their suitability for the study
of myocardial infarction. Basic Res Cardiol. 115:732020. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Jiang H, Fang T and Cheng Z: Mechanism of
heart failure after myocardial infarction. J Int Med Res.
51:30006052312025732023. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Jenča D, Melenovský V, Stehlik J, Staněk
V, Kettner J, Kautzner J, Adámková V and Wohlfahrt P: Heart failure
after myocardial infarction: Incidence and predictors. ESC Heart
Fail. 8:222–237. 2021. View Article : Google Scholar
|
|
138
|
Feng R, Wang D, Li T, Liu X, Peng T, Liu
M, Ren G, Xu H, Luo H, Lu D, et al: Elevated SLC40A1 impairs
cardiac function and exacerbates mitochondrial dysfunction,
oxidative stress, and apoptosis in ischemic myocardia. Int J Biol
Sci. 20:414–432. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Dharmakumar R, Nair AR, Kumar A and
Francis J: Myocardial infarction and the fine balance of iron. JACC
Basic Transl Sci. 6:581–583. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Zhao WK, Zhou Y, Xu TT and Wu Q:
Ferroptosis: Opportunities and challenges in myocardial
ischemia-reperfusion injury. Oxid Med Cell Longev.
2021:99296872021. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Marunouchi T, Onda S, Kurasawa M and
Tanonaka K: Angiotensin II is involved in MLKL activation during
the development of heart failure following myocardial infarction in
rats. Biol Pharm Bull. 47:809–817. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Santagostino SF, Assenmacher CA, Tarrant
JC, Adedeji AO and Radaelli E: Mechanisms of regulated cell death:
Current perspectives. Vet Pathol. 58:596–623. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Li A, Gao M, Liu B, Qin Y, Chen L, Liu H,
Wu H and Gong G: Mitochondrial autophagy: Molecular mechanisms and
implications for cardiovascular disease. Cell Death Dis.
13:4442022. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Held PH, Yusuf S and Furberg CD: Calcium
channel blockers in acute myocardial infarction and unstable
angina: An overview. BMJ. 299:1187–1192. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Makarewich CA, Zhang H, Davis J, Correll
RN, Trappanese DM, Hoffman NE, Troupes CD, Berretta RM, Kubo H,
Madesh M, et al: Transient receptor potential channels contribute
to pathological structural and functional remodeling after
myocardial infarction. Circ Res. 115:567–580. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Cui T, Liu W, Yu C, Ren J, Li Y, Shi X, Li
Q and Zhang J: Protective effects of allicin on acute myocardial
infarction in rats via hydrogen sulfide-mediated regulation of
coronary arterial vasomotor function and myocardial calcium
transport. Front Pharmacol. 12:7522442022. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Pietrzykowski Ł, Michalski P, Kosobucka A,
Kasprzak M, Fabiszak T, Stolarek W, Siller-Matula JM and Kubica A:
Medication adherence and its determinants in patients after
myocardial infarction. Sci Rep. 10:120282020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Heart Outcomes Prevention Evaluation Study
Investigators; Yusuf S, Sleight P, Pogue J, Bosch J, Davies R and
Dagenais G: Effects of an angiotensin-converting-enzyme inhibitor,
ramipril, on cardiovascular events in high-risk patients. N Engl J
Med. 342:145–153. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Khan SU, Singh M, Valavoor S, Khan MU,
Lone AN, Khan MZ, Khan MS, Mani P, Kapadia SR, Michos ED, et al:
Dual antiplatelet therapy after percutaneous coronary intervention
and drug-eluting stents: A systematic review and network
meta-analysis. Circulation. 142:1425–1436. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Yandrapalli S, Andries G, Gupta S, Dajani
AR and Aronow WS: Investigational drugs for the treatment of acute
myocardial infarction: Focus on antiplatelet and anticoagulant
agents. Expert Opin Investig Drugs. 28:223–234. 2019. View Article : Google Scholar
|
|
151
|
AIM-HIGH Investigators; Boden WE,
Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P,
Koprowicz K, McBride R, Teo K and Weintraub W: Niacin in patients
with low HDL cholesterol levels receiving intensive statin therapy.
N Engl J Med. 365:2255–2267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Tsai IT and Sun CK: Stem cell therapy
against ischemic heart disease. Int J Mol Sci. 25:37782024.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Broch K, Anstensrud AK, Woxholt S, Sharma
K, Tøllefsen IM, Bendz B, Aakhus S, Ueland T, Amundsen BH, Damås
JK, et al: Randomized trial of interleukin-6 receptor inhibition in
patients with acute ST-segment elevation myocardial infarction. J
Am Coll Cardiol. 77:1845–1855. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Shi S, Chen Y, Luo Z, Nie G and Dai Y:
Role of oxidative stress and inflammation-related signaling
pathways in doxorubicin-induced cardiomyopathy. Cell Commun Signal.
21:612023. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Ye X, Li Y, Lv B, Qiu B, Zhang S, Peng H,
Kong W, Tang C, Huang Y, Du J and Jin H: Endogenous hydrogen
sulfide persulfidates caspase-3 at cysteine 163 to inhibit
doxorubicin-induced cardiomyocyte apoptosis. Oxid Med Cell Longev.
2022:61537722022. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Gammella E, Recalcati S, Rybinska I,
Buratti P and Cairo G: Iron-induced damage in cardiomyopathy:
Oxidative-dependent and independent mechanisms. Oxid Med Cell
Longev. 2015:2301822015. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Cheng CF and Lian WS: Prooxidant
mechanisms in iron overload cardiomyopathy. Biomed Res Int.
2013:7405732013. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Fatmi A, Saadi W, Beltrán-García J,
García-Giménez JL and Pallardó FV: The endothelial glycocalyx and
neonatal sepsis. Int J Mol Sci. 24:3642022. View Article : Google Scholar
|
|
159
|
Carthy CM, Yang D, Anderson DR, Wilson JE
and McManus BM: Myocarditis as systemic disease: New perspectives
on pathogenesis. Clin Exp Pharmacol Physiol. 24:997–1003. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Lasrado N and Reddy J: An overview of the
immune mechanisms of viral myocarditis. Rev Med Virol. 30:1–14.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Dimitropoulos G, Tahrani AA and Stevens
MJ: Cardiac autonomic neuropathy in patients with diabetes
mellitus. World J Diabetes. 5:17–39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Lin S, Dai S, Lin J, Liang X, Wang W,
Huang W, Ye B and Hong X: Oridonin relieves angiotensin II-induced
cardiac remodeling via inhibiting GSDMD-mediated inflammation.
Cardiovasc Ther. 2022:31679592022. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Chen S, Huang Z, Liang Y, Zhao X,
Aobuliksimu X, Wang B, He Y, Kang Y, Huang H, Li Q, et al:
Five-year mortality of heart failure with preserved, mildly
reduced, and reduced ejection fraction in a 4880 Chinese cohort.
ESC Heart Fail. 9:2336–2347. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Liu M, López de Juan Abad B and Cheng K:
Cardiac fibrosis: Myofibroblast-mediated pathological regulation
and drug delivery strategies. Adv Drug Deliv Rev. 173:504–519.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Ravassa S, López B, Treibel TA, San José
G, Losada-Fuentenebro B, Tapia L, Bayés-Genís A, Díez J and
González A: Cardiac Fibrosis in heart failure: Focus on
non-invasive diagnosis and emerging therapeutic strategies. Mol
Aspects Med. 93:1011942023. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Martin TG, Juarros MA and Leinwand LA:
Regression of cardiac hypertrophy in health and disease: Mechanisms
and therapeutic potential. Nat Rev Cardiol. 20:347–363. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol
Cancer. 14:482015. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Dong Y, Liu C, Zhao Y, Ponnusamy M, Li P
and Wang K: Role of noncoding RNAs in regulation of cardiac cell
death and cardiovascular diseases. Cell Mol Life Sci. 75:291–300.
2018. View Article : Google Scholar
|
|
169
|
Cai K, Jiang H, Zou Y, Song C, Cao K, Chen
S, Wu Y, Zhang Z, Geng D, Zhang N, et al: Programmed death of
cardiomyocytes in cardiovascular disease and new therapeutic
approaches. Pharmacol Res. 206:1072812024. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Chiong M, Wang ZV, Pedrozo Z, Cao DJ,
Troncoso R, Ibacache M, Criollo A, Nemchenko A, Hill JA and
Lavandero S: Cardiomyocyte death: Mechanisms and translational
implications. Cell Death Dis. 2:e2442011. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Topriceanu CC, Pereira AC, Moon JC, Captur
G and Ho CY: Meta-analysis of penetrance and systematic review on
transition to disease in genetic hypertrophic cardiomyopathy.
Circulation. 149:107–123. 2024. View Article : Google Scholar :
|
|
172
|
Ye C, Zheng F, Wu N, Zhu GQ and Li XZ:
Extracellular vesicles in vascular remodeling. Acta Pharmacol Sin.
43:2191–2201. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Jordan-Rios A, Cannatà A, Bromage D and
McDonagh T: Challenges in the implementation of medical therapy in
heart failure. JACC Heart Fail. 11:607–609. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Pacesa M, Pelea O and Jinek M: Past,
present, and future of CRISPR genome editing technologies. Cell.
187:1076–1100. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Xiao Y, Zhao J, Tuazon JP, Borlongan CV
and Yu G: MicroRNA-133a and myocardial infarction. Cell Transplant.
28:831–838. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Reichart D, Newby GA, Wakimoto H, Lun M,
Gorham JM, Curran JJ, Raguram A, DeLaughter DM, Conner DA,
Marsiglia JDC, et al: Efficient in vivo genome editing prevents
hypertrophic cardiomyopathy in mice. Nat Med. 29:412–421. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Morris KV and Mattick JS: The rise of
regulatory RNA. Nat Rev Genet. 15:423–437. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Shim JS, Song WJ and Morice AH:
Drug-induced cough. Physiol Res. 69(Suppl 1): S81–S92. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Wong GW and Wright JM: Blood pressure
lowering efficacy of nonselective beta-blockers for primary
hypertension. Cochrane Database Syst Rev. 2014:CD0074522024.
|
|
180
|
Cheng CJ, Rodan AR and Huang CL: Emerging
targets of diuretic therapy. Clin Pharmacol Ther. 102:420–435.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Haley KE, Almas T, Shoar S, Shaikh S,
Azhar M, Cheema FH and Hameed A: The role of anti-inflammatory
drugs and nanoparticle-based drug delivery models in the management
of ischemia-induced heart failure. Biomed Pharmacother.
142:1120142021. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Pala R, Anju VT, Dyavaiah M, Busi S and
Nauli SM: Nanoparticle-mediated drug delivery for the treatment of
cardiovascular diseases. Int J Nanomedicine. 15:3741–3769. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Qiu R, Han S, Wei X, Zhong C, Li M, Hu J,
Wang P, Zhao C, Chen J and Shang H: Development of a core outcome
set for the benefits and adverse events of acute heart failure in
clinical trials of traditional Chinese medicine and western
medicine: A study protocol. Front Med (Lausanne). 8:6770682021.
View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Chen X, Ma Y, Li J, Yao L, Gui M, Lu B,
Zhou X, Wang M and Fu D: The efficacy of ginseng-containing
traditional Chinese medicine in patients with acute decompensated
heart failure: A systematic review and meta-analysis. Front
Pharmacol. 13:10830012023. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Ji MY, Bo A, Yang M, Xu JF, Jiang LL, Zhou
BC and Li MH: The pharmacological effects and health benefits of
platycodon grandiflorus-a medicine food homology species. Foods.
9:1422020. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Tang MM, Zhao ST, Li RQ and Hou W:
Therapeutic mechanisms of ginseng in coronary heart disease. Front
Pharmacol. 14:12710292023. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Yang L, Jiang H, Wang S, Hou A, Man W,
Zhang J, Guo X, Yang B, Kuang H and Wang Q: Discovering the major
antitussive, expectorant, and anti-inflammatory bioactive
constituents in Tussilago farfara L. based on the spectrum-effect
relationship combined with chemometrics. Molecules. 25:6202020.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Tan D, Tseng HHL, Zhong Z, Wang S, Vong CT
and Wang Y: Glycyrrhizic acid and its derivatives: Promising
candidates for the management of type 2 diabetes mellitus and its
complications. Int J Mol Sci. 23:109882022. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Feng Q, Ling L, Yuan H, Guo Z and Ma J:
Ginsenoside Rd: A promising target for ischemia-reperfusion injury
therapy (A mini review). Biomed Pharmacother. 171:1161112024.
View Article : Google Scholar : PubMed/NCBI
|
|
190
|
MacRitchie N and Maffia P: Light sheet
fluorescence microscopy for quantitative three-dimensional imaging
of vascular remodelling. Cardiovasc Res. 117:348–350. 2021.
View Article : Google Scholar :
|
|
191
|
Yu B, Lu Q, Li J, Cheng X, Hu H, Li Y, Che
T, Hua Y, Jiang H, Zhang Y, et al: Cryo-EM structure of human HCN3
channel and its regulation by cAMP. J Biol Chem. 300:1072882024.
View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Li H, Zhang X, Wang F, Zhou L, Yin Z, Fan
J, Nie X, Wang P, Fu XD, Chen C and Wang DW: MicroRNA-21 lowers
blood pressure in spontaneous hypertensive rats by upregulating
mitochondrial translation. Circulation. 134:734–751. 2016.
View Article : Google Scholar : PubMed/NCBI
|