Histone deacetylases (HDACs) are enzymes that
regulate gene expression by deacetylating histones, thereby
modulating their interaction with chromatin (1). HDACs play critical roles in
controlling cell proliferation, differentiation and development
(2). Among them, HDAC4 belongs
to class IIa HDACs and is exclusively expressed in
non-proliferating cells (3,4).
Studies have shown that HDAC4 is highly expressed in the heart,
brain, skeletal muscle and thymus (5,6).
CVD, a group of disorders affecting the heart or
blood vessels, poses a significant threat to human health. Common
types of CVD include heart failure (HF), myocardial infarction (MI)
and coronary artery disease (CAD) (7). Multiple studies have demonstrated
altered HDAC4 expression in CVD, suggesting its potential as a
biomarker for patients with cardiovascular conditions (8-10). Moreover, previous findings
indicate that HDAC4 contributes to the progression of CVD by
regulating processes such as cardiac hypertrophy, inflammation,
fibrosis and apoptosis (11-13).
In the present review, the biochemical properties of
HDAC4 were analyzed and an in-depth discussion of its roles and
mechanisms in CVD was provided. Additionally, factors that
influence HDAC4 expression were summarized. Finally, a summary and
outlook were added as a conclusion, aiming to provide new insights
into the potential application of HDAC4 in the treatment of
CVD.
Inflammation is a response initiated by the immune
system in reaction to infections or non-infectious tissue injury
(18). Extensive research has
confirmed that inflammation is a key contributor to the development
of CVD (19-21). Emerging evidence indicates that
HDAC4 plays a critical role in mediating inflammatory responses
associated with CVD. In vitro experiments have shown that
the inhibition of HDAC4 alleviates Ang II-induced inflammatory
responses in rat aortic endothelial cells (RAECs) (22). Another study has reported that
silencing HDAC4 reverses the TNF-α-induced expression of
inflammatory markers vascular cell adhesion molecule-1 (VCAM-1) and
phosphorylated nuclear factor kappa B (NF-κB) in smooth muscle
cells (SMCs) (23). Furthermore,
HDAC4 knockout suppresses the effect of long non-coding RNA cancer
susceptibility candidate 11 (lncRNA CASC11) in downregulating the
expression of pro-inflammatory cytokines IL-6 and IL-1β and
promoting the expression of the anti-inflammatory cytokine IL-10 in
human cardiac microvascular endothelial cells (CMECs) (24). Interestingly, a clinical study
has shown that HDAC4 expression is reduced in patients with
coronary heart disease (CHD), and it is negatively correlated with
the levels of pro-inflammatory cytokines such as TNF-α, IL-1β and
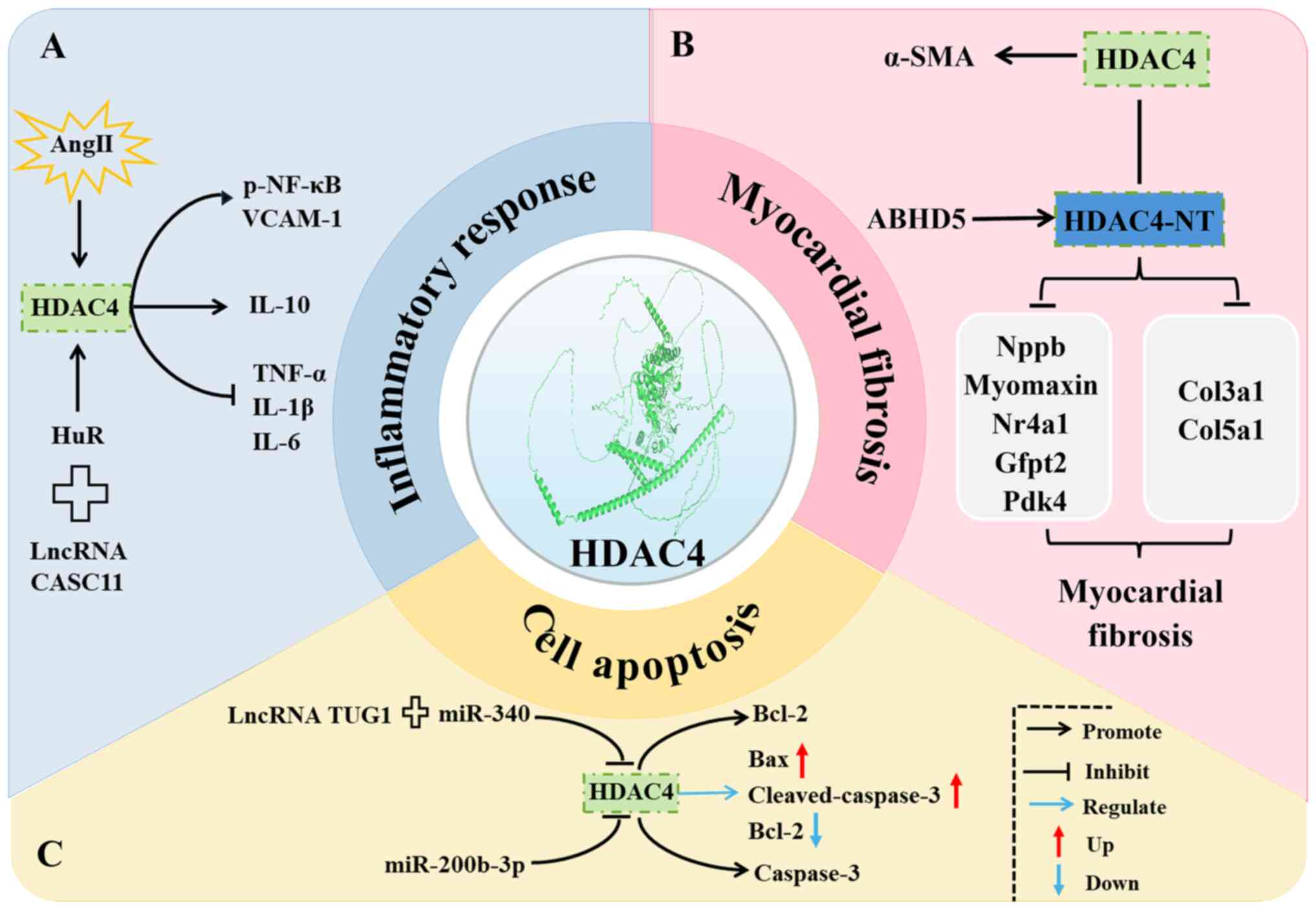
IL-6 (13) (Fig. 2A). This alteration may result
from the influence of the inflammatory microenvironment on HDAC4
expression in vivo, or it may represent a compensatory
response to the disease state in patients with CHD. Therefore,
further studies are warranted to elucidate the context-dependent
roles and underlying mechanisms of HDAC4 under different
pathological conditions.
Following myocardial injury, cardiac fibroblasts are
activated and differentiate into myofibroblasts. These
myofibroblasts exhibit proliferative and secretory properties that
promote extracellular matrix remodeling and collagen deposition,
ultimately leading to fibrotic scarring and HF (25). One study demonstrated that
inhibition of HDAC4 expression downregulates the Ang II-induced
expression of the cardiac pericyte fibrosis marker α-smooth muscle
actin (α-SMA) (26). Another
study showed that HDAC4 knockout suppresses myocardial fibrosis in
mice with MI (27). By contrast,
HDAC4 overexpression promotes myocardial fibrosis in MI mouse
models (27,28). HDAC4-NT, the N-terminal fragment
of HDAC4, has also been investigated for its potential protective
role. A study has shown that HDAC4-NT overexpression ameliorates
cardiac hypertrophy and fibrosis caused by abhydrolase
domain-containing 5 (ABHD5) deficiency. Mechanistically, this
effect may be associated with the downregulation of genes such as
natriuretic peptide B (Nppb), Myomaxin, nuclear receptor subfamily
4 group A member 1 (Nr4a1), glutamine-fructose-6-phosphate
transaminase 2 (Gfpt2), and pyruvate dehydrogenase kinase 4 (Pdk4)
(29). Similarly, another study
confirmed that HDAC4-NT suppresses the expression of
fibrosis-related genes collagen type III alpha 1 chain (Col3a1) and
Col5a1 in transverse aortic constriction (TAC)-induced models
(30) (Fig. 2B).
Apoptosis, also known as programmed cell death, is
an active process regulated by specific genes (31). It plays a crucial role in
maintaining cellular homeostasis and in the prevention and
treatment of CVD (32). Studies
have shown that HDAC4 overexpression can induce apoptosis in
cardiomyocytes (33,34). Mechanistically, this may be
related to elevated expression of the pro-apoptotic protein,
caspase-3 (33). Evidence
indicates that HDAC4 is involved in the anti-apoptotic effects of
the lncRNA CASC11 in human CMECs (24). Similarly, Wu et al
(35) reported that silencing
lncRNA taurine-upregulated gene 1 (TUG1) suppresses the expression
of pro-apoptotic markers Bcl-2-associated X protein (Bax) and
cleaved caspase-3 but enhances the expression of the anti-apoptotic
protein B-cell lymphoma 2 (Bcl-2); this effect is associated with
increased HDAC4 expression. In addition, another study has revealed
the regulatory role of HDAC4 in apoptosis; specifically, HDAC4 can
inhibit cell death induced by the overexpression of microRNA
(miR)-200b-3p (36) (Fig. 2C).
HDAC4 plays a regulatory role in inflammation,
myocardial fibrosis and apoptosis in the context of CVD (Fig. 2). These findings highlight the
significance of HDAC4 in the onset and progression of CVD. In the
following sections, the specific mechanisms by which HDAC4
functions in various types of CVD, particularly its
pathophysiological roles in common conditions such as cardiac
hypertrophy and CHD, will be further explored. A thorough
understanding of these mechanisms may offer new targets for the
treatment of these diseases.
CHD, a myocardial condition caused by
atherosclerosis (AS) of the coronary arteries, remains the leading
cause of mortality worldwide (51,52). Despite advancements in medical
interventions, current treatment options for CHD are limited and
often associated with complications. A recent study has proposed
that HDAC4 expression levels may serve as a potential predictive
biomarker for CHD (13). This
hypothesis is supported by findings showing significantly reduced
HDAC4 expression in the serum of patients diagnosed with CHD.
Moreover, HDAC4 expression has been shown to correlate with several
clinical indicators, including serum creatinine (Scr), low-density
lipoprotein cholesterol (LDL-C), C-reactive protein (CRP), and
blood glucose levels. In addition, correlation analysis revealed a
negative association between HDAC4 and inflammatory markers such as
TNF-α, IL-1β and IL-6 (13).
CMECs, which are among the most abundant cell types in the heart,
play a crucial role in regulating coronary blood flow (53-55). A study has shown that lncRNA
CASC11 can upregulate HDAC4 expression, thereby suppressing the
secretion of IL-6 and IL-1β and enhancing IL-10 expression in human
CMECs under ox-LDL stimulation (24). Knockdown of HDAC4 reverses these
effects, suggesting that HDAC4 plays a key regulatory role in the
anti-inflammatory process mediated by CASC11. Furthermore, HDAC4
was found to mediate the effects of CASC11 in suppressing apoptosis
and promoting angiogenesis (24). Bioinformatic analyses predicted
that CASC11 interacts with the RNA-binding protein human antigen R
(HuR), which binds to HDAC4, suggesting that CASC11 regulates HDAC4
expression through HuR-mediated stabilization. This mechanism
likely contributes to the protective effects of CASC11 against
ox-LDL-induced injury in CMECs (24). Collectively, these findings
suggested that HDAC4 may serve as a promising biomarker for the
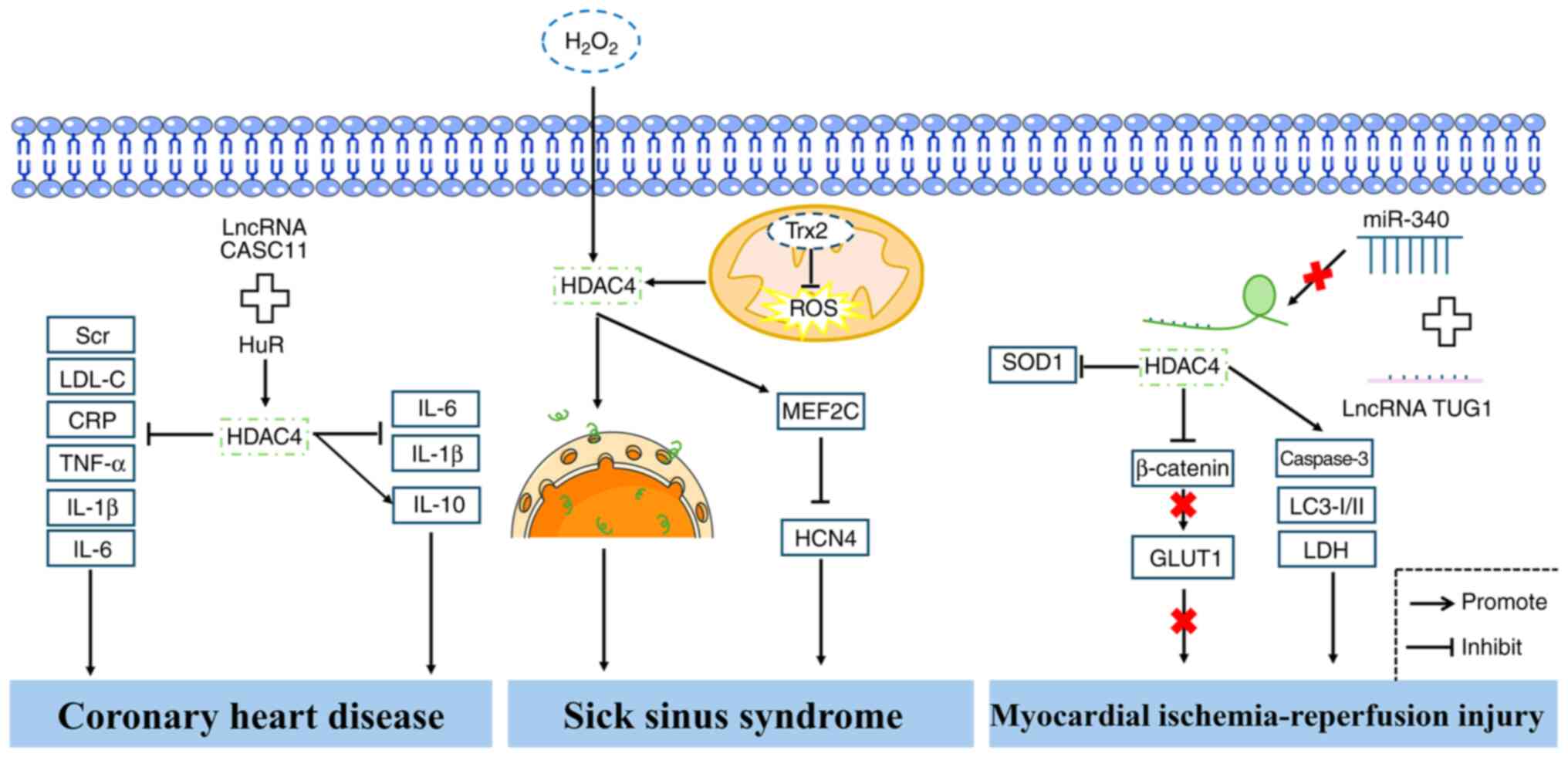
prediction and potential therapeutic targeting of CHD (Fig. 4). However, further studies are
warranted to elucidate the precise molecular mechanisms by which
HDAC4 contributes to the pathogenesis of CHD.
Preliminary studies have identified a correlation
between the development of SSS and advancing age, with the
condition primarily characterized by dysfunction of the sinoatrial
node (SAN) (56,57). Currently, treatment options for
SSS remain limited, highlighting the urgent need to explore novel
therapeutic targets. In a recent study, Zhang et al
(58) treated mouse atrial
myocytes (HL-1 cells) with hydrogen peroxide
(H2O2) to mimic oxidative stress conditions
affecting SAN pace-making function. Their results demonstrated that
H2O2 treatment leads to increased HDAC4
expression and its subsequent nuclear translocation, which in turn
contribute to impaired SAN function (58). As the central organelle for
maintaining energy metabolic homeostasis in sinoatrial node cells,
mitochondria play a critical role in regulating their
electrophysiological excitability and rhythm stability (59,60). HDAC4 has been shown to play a
crucial role in mediating the protective effects of thioredoxin-2
(Trx2) against sinus bradycardia by inhibiting mitochondrial
reactive oxygen species (ROS) production within the SAN.
Mechanistically, Trx2 deficiency can suppress the expression of
hyperpolarization-activated cyclic nucleotide-gated potassium
channel 4 via the mitochondrial ROS-HDAC4-MEF2C signaling axis,
resulting in SSS (61) (Fig. 4). These findings collectively
suggested that HDAC4 may represent a key therapeutic target for the
treatment of SSS.
MI-reperfusion injury refers to the damage inflicted
on cardiomyocytes upon the restoration of blood flow following MI,
and it has been shown to significantly affect patient prognosis
(62). An in vitro and
in vivo study demonstrated that the expression of the lncRNA
TUG1 is elevated in the hearts of ischemia/reperfusion (I/R) mice
and in cardiomyocytes subjected to hypoxia/reoxygenation (H/R).
Bioinformatic analysis predicted a direct interaction between TUG1
and miR-340, which in turn targets HDAC4. Subsequent experiments
confirmed that miR-340 overexpression reverses the upregulation of
HDAC4 induced by TUG1. Moreover, TUG1 knockdown suppresses the
H/R-induced expression of pro-apoptotic proteins Bax and cleaved
caspase-3 while enhancing the expression of the anti-apoptotic
protein Bcl-2 in cardiomyocytes. Conversely, HDAC4 overexpression
promotes apoptosis under these conditions (35). Interestingly, silencing β-catenin
reverses the upregulation of glucose transporter type 1 (GLUT1)
induced by HDAC4 knockdown, suggesting that HDAC4 negatively
regulates β-catenin, thereby promoting GLUT1 expression, as further
supported by correlation analyses. In vivo experiments
demonstrated that TUG1 knockdown improves cardiac function, reduces
infarct size, and inhibits cardiomyocyte apoptosis in I/R mice.
However, these protective effects were partially abrogated by the
additional knockdown of miR-340 or GLUT1. These findings indicated
that targeting the TUG1/miR-340/HDAC4/β-catenin/GLUT1 regulatory
axis may offer a novel therapeutic approach for MI-reperfusion
injury (35). Mitochondrial
quality control (including mitophagy, mitochondrial dynamics and
mitochondrial biogenesis) has been recognized as a critical
regulatory mechanism in the pathological progression of MI injury
(63-66). A previous study found that HDAC4
overexpression increases H9c2 cell death, enhances mitochondrial
membrane permeability transition pore activity, elevates lactate
dehydrogenase release, and upregulates cleaved caspase-3 expression
under H/R conditions (67). In
line with these observations, Zhang et al (34) reported that HDAC4 overexpression
in I/R mice exacerbates myocardial injury, as evidenced by
increased infarct size, upregulation of autophagy-related proteins
microtubule-associated protein 1A/1B-light chain 3 (LC3-I/II) and
apoptosis marker caspase-3, and downregulation of the antioxidant
protein superoxide dismutase 1 (SOD1), all of which contributed to
worsened ventricular dysfunction. Notably, treatment with the HDAC
inhibitor trichostatin A (TSA, 0.1 mg/kg) reversed these
pathological changes, suggesting that HDAC4 overexpression
aggravates I/R injury, whereas its inhibition may confer
cardioprotective effects (34)
(Fig. 4).
MI is a CVD with high lethality due to MI caused by
insufficient coronary blood supply (73). However, current treatment options
for MI remain limited, highlighting the urgent need to identify
novel therapeutic targets (74).
In vivo and in vitro studies have revealed that HDAC4
expression is elevated in MI (75,76). Overexpression of HDAC4 in MI
mouse models has been shown to result in impaired cardiac function,
enlarged cardiomyocytes, reduced vascular density, myocardial
fibrosis, and increased expression of the hypertrophic marker ANP
(28). Conversely,
transplantation of cardiac stem cells (CSCs) transfected with siRNA
targeting HDAC4 improves cardiac function in MI mice, suppresses
myocardial hypertrophy and fibrosis, and promotes CSC-derived
neovascularization and cardiomyocyte proliferation (27). A study has also shown that HDAC4
interferes with the inhibitory effect of G protein-coupled receptor
kinase 4 on the autophagy-related genes LC3-II and Beclin-1 in the
myocardium of MI mice (77). In
another study, treatment with the HDAC4 inhibitor LMK235 was found
to rescue the upregulation of SP1 and the fatty acid oxidation
marker PPARα induced by KN93, suggesting a novel therapeutic
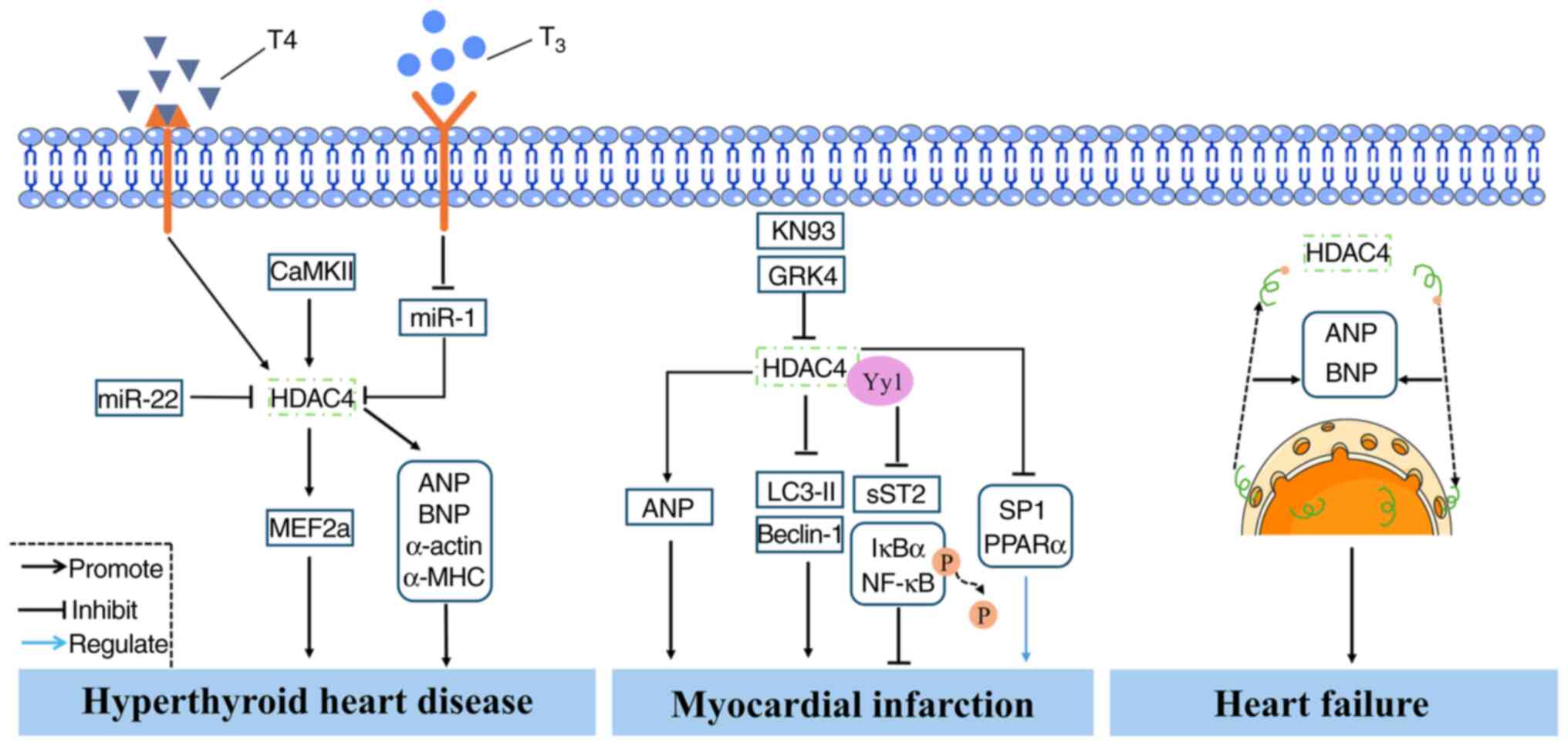
approach for MI (78).
Additionally, Asensio-Lopez et al (75) reported that HDAC4 specifically
interacts with yin-yang1 (Yy1) to suppress the expression of sST2,
thereby inhibiting cardiomyocyte hypertrophy and the activation of
phosphorylated inhibitor of nuclear factor κB α(IκBα)/NF-κB
signaling and ultimately ameliorating MI pathology (75). Current evidence indicates that
HDAC4 is upregulated in patients with MI, suggesting its potential
role as a key biomarker. Inhibition of HDAC4 expression has been
shown to improve MI outcomes by suppressing myocardial hypertrophy,
inflammation and fibrosis but promoting cardiomyocyte autophagy
(Fig. 5). Hypoxia contributes to
the development of MI (79).
However, a study has shown that hypoxia does not significantly
alter HDAC4 expression in H9c2 cells. Instead, HDAC1 expression is
increased under hypoxic conditions, indicating that relying solely
on HDAC4 expression levels as a predictive biomarker for MI may
have certain limitations (80).
HF is widely recognized as the end stage of numerous
CVDs and is associated with high morbidity and mortality rates
(81). Therefore, the prevention
and treatment of HF are of particular clinical importance. To date,
numerous studies have demonstrated that HDAC4 plays a critical role
in the pathogenesis of HF (29,82,83). Clinical evidence indicates that,
compared with non-failing hearts, the nuclear expression of HDAC4
is reduced in the cardiomyocytes of patients with HF (5,84). Notably, HDAC4 nucleocytoplasmic
shuttling has been found to be positively correlated with the
expression of ANP and BNP (5).
An animal study has reported that HDAC4 expression is elevated in
the left ventricle of rat models of HF (83). Further research has shown that
inhibition of HDAC4 expression improves cardiac function in HF mice
and enhances myocardial glucose uptake (85) (Fig. 5). Collectively, these findings
suggested that targeting HDAC4 may provide therapeutic benefits in
the treatment of HF.
A recent study indicated that hypertension affects
over one billion people worldwide (86). Although various approaches have
been developed for the management of hypertension with advances in
medical science, a definitive cure has yet to be achieved (87). A characteristic feature of
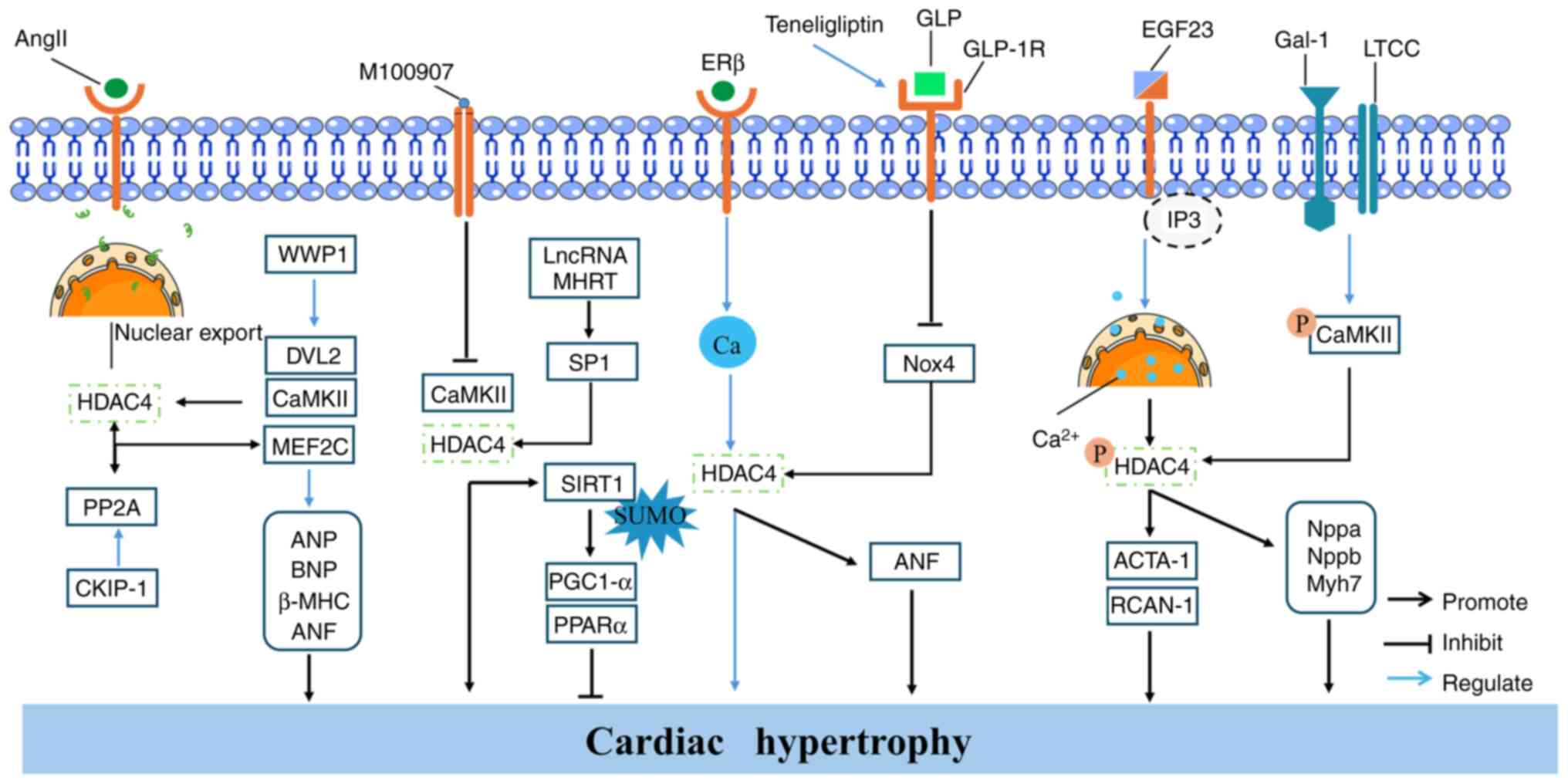
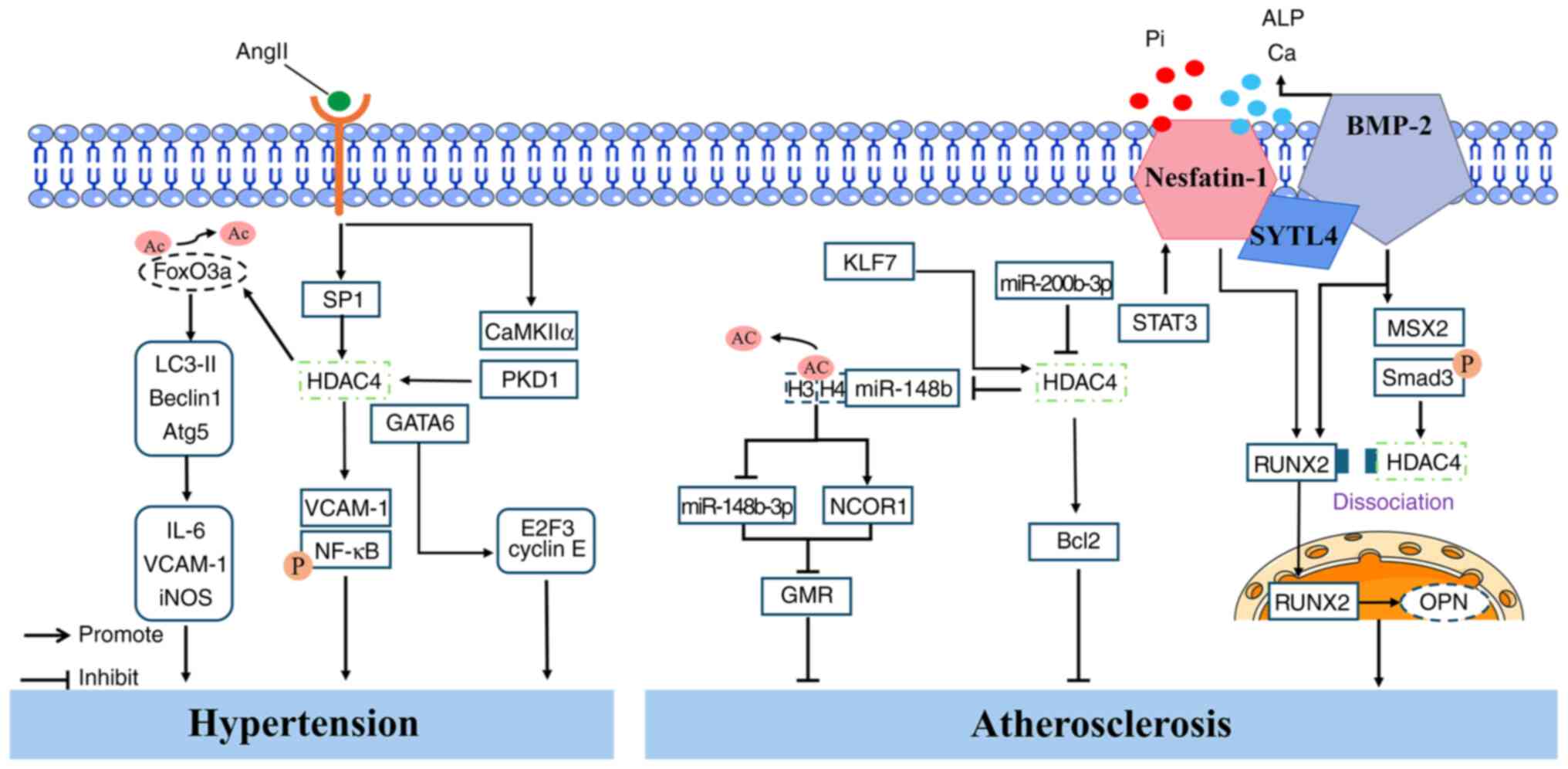
hypertension is increased vascular stiffness (88). A study has shown that AngII
treatment upregulates the expression of inflammatory cytokines
(IL-6, VCAM-1 and inducible nitric oxide synthase), autophagy
markers (LC3-II, Beclin1 and Atg5), and HDAC4 in primary RAECs and
in vivo in mice (22).
Further investigation revealed that AngII-induced autophagy
requires the involvement of endogenous forkhead box protein O3a
(FoxO3a). Treatment of RAECs with autophagy inhibitors LY294002 or
3-MA suppresses the AngII-induced expression of inflammatory
cytokines. Downregulation of HDAC4 inhibits AngII-induced vascular
inflammation and reverses the AngII-mediated reduction in FoxO3a
acetylation. These findings suggested that HDAC4 mediates the
acetylation of FoxO3a and regulates AngII-induced excessive
autophagy, thereby contributing to vascular inflammation (22). Similarly, Usui et al
(89) also demonstrated that
HDAC4 plays a key role in mediating vascular inflammation. In
animal experiments, HDAC4 expression was found to be elevated in
the mesenteric arteries of spontaneously hypertensive rats compared
with Wistar Kyoto rats. In vitro, treatment of rat
mesenteric artery SMCs with TNF-α upregulates the expression of
inflammatory markers VCAM-1 and p-NF-κB, as well as HDAC4. Notably,
silencing HDAC4 with siRNA reverses the TNF-α-induced expression of
these inflammatory markers in SMCs (23). Interestingly, HDAC4 protein
levels were found to be decreased in the aorta of hypertensive rats
but elevated in the mesenteric arteries, suggesting region-specific
differences in HDAC4 expression. This highlights the limitation of
evaluating disease progression based solely on HDAC4 levels in a
single vascular bed (23). One
study reported that MC1568, a class II HDAC inhibitor, suppresses
the AngII-induced expression of p-HDAC4S632 and
GATA-binding factor 6 (GATA6) in the kidneys and aortas of mice. An
in vitro study further showed that HDAC4 promotes the
expression of cell cycle regulatory genes E2F3 and cyclin E in
vascular smooth muscle cells (VSMCs). Notably, an endogenous
association between HDAC4 and GATA6 was confirmed in VSMCs. An
additional study demonstrated interactions among HDAC4, CaMKIIα and
PKD1 in 293T cells, which were disrupted by MC1568 treatment; thus,
MC1568 attenuates VSMC hypertrophy and proliferation by
downregulating the CaMKIIα/PKD1/HDAC4/GATA6 signaling pathway and
alleviating hypertension (90).
In summary, HDAC4 expression is elevated in multiple models of
hypertension and contributes to disease progression by promoting
vascular inflammation, VSMC hypertrophy and VSMC proliferation.
These findings underscore the potential of targeting HDAC4 as a
therapeutic strategy to delay the progression of hypertension
(Fig. 6).
AS is a chronic inflammatory disease characterized
by the accumulation of atherosclerotic plaques within the arterial
wall, resulting in reduced blood flow (91). Chen et al (92) demonstrated that HDAC4 mediates
the role of Krüppel-like factor 7 (KLF7) in AS. Specifically, KLF7
binds to the HDAC4 promoter to activate HDAC4 transcription, which
subsequently suppresses miR-148b-3p expression by reducing the
acetylation of histones H3 and H4 at the miR-148b promoter. This
suppression promotes the transcription of nuclear receptor
corepressor 1 (NCOR1), thereby inhibiting GMR in macrophages and
alleviating AS (92). AS is a
major underlying cause of CAD. A clinical study revealed that
miR-200b-3p is significantly upregulated in the epicardial adipose
tissue of patients with CAD (36). Further in vitro
experiments showed that the overexpression of miR-200b-3p
downregulates HDAC4 expression in HUVECs and reduces the expression
of the anti-apoptotic protein Bcl-2. Notably, the overexpression of
HDAC4 reverses miR-200b-3p-induced apoptosis, suggesting that HDAC4
plays a protective role against miR-200b-3p-mediated endothelial
apoptosis in AS (36). Vascular
calcification (VC) contributes to the progression of AS plaques. A
recent study reported that HDAC4 is involved in VC (93). The researchers found that
nesfatin-1 expression is elevated in patients with VS, in a vitamin
D3-induced VC mouse model, and in sodium phosphate (Pi)-treated
VSMCs. Moreover, nesfatin-1 expression was positively correlated
with the severity of VC in patients (93). In vivo experiments showed
that nesfatin-1 knockout reduces the expression of osteogenic
markers RUNX2 and bone morphogenetic protein 2 (BMP-2) in the
aortas of VC mice but enhances the expression of contractile
proteins α-SMA and SM22α. A further study revealed that BMP-2
promotes the expression of p-Smad3, HDAC4, RUNX2 and MSX2 in VSMCs;
enhances the binding of RUNX2 to the OPN promoter; increases
calcium content and alkaline phosphatase (ALP) activity; and
inhibits the formation of the HDAC4/RUNX2 complex. These effects
were reversed by nesfatin-1 knockout. Additionally, the proteasome
inhibitor MG-132 was shown to prevent BMP-2 degradation induced by
nesfatin-1 knockout. Bioinformatic analysis identified an
interaction between SYTL4 and BMP-2, and knockdown of SYTL4 also
inhibited BMP-2 ubiquitination and degradation triggered by
nesfatin-1 knockout. This reduced calcium deposition and ALP
activity under high-Pi conditions. Notably, the overexpression of
STAT3 was found to upregulate the expression of nesfatin-1, BMP-2
and RUNX2, indicating that the
STAT3/nesfatin-1/BMP-2/HDAC4/RUNX2/OPN signaling axis contributes
to VC progression (93)
(Fig. 6). In summary, these
findings highlight the potential of HDAC4 as a promising
therapeutic target in the treatment of AS.
Diabetic cardiomyopathy (DC) refers to myocardial
structural and functional impairments caused by diabetes,
independent of other traditional confounding factors (94). DC has emerged as a significant
threat to human health (95).
Catalpol, one of the major active components of Rehmannia
glutinosa, has been shown to exert cardioprotective effects. A
study has found that knockdown of HDAC4 enhances the inhibitory
effects of catalpol on the expression of pro-apoptotic proteins
caspase-3 and Bax under high-glucose conditions, while promoting
the expression of the anti-apoptotic protein Bcl-2. By contrast,
upregulation of HDAC4 expression facilitates apoptosis (33). However, another study reported
that cardiomyocyte-specific deletion of HDAC4 exacerbates cardiac
dysfunction in diabetic mice (96). These contradictory findings
suggested that the precise role of HDAC4 in DC remains
controversial and warrants further investigation.
Dilated cardiomyopathy (DCM) is a cardiac disorder
characterized by ventricular dilation and systolic dysfunction in
the absence of abnormal loading conditions (97). A clinical study reported the
elevated expression of p-HDAC4 in the hearts of patients with
end-stage DCM (98). Similarly,
increased p-HDAC4 levels have also been observed in the hearts of
cTnTR141W familial DCM mouse models (99). Notably, HDAC4 is involved in
mediating the protective effects of Dickkopf 3, which suppresses
the expression of hypertrophic marker ANF and myocardial fibrosis
in DCM mice (99).
Acute coronary syndrome (ACS) is a clinical
condition characterized by the sudden onset of acute ischemia or
necrosis of the myocardium. ACS encompasses ST-segment elevation MI
(STEMI), non-STEMI and unstable angina (104). A recent clinical study reported
the decreased expression of HDAC4 in patients with ACS, with the
lowest levels observed in those with STEMI. Furthermore, the study
found that HDAC4 expression was negatively correlated with total
cholesterol, LDL-C, CRP, cardiac troponin I, and a history of
hyperlipidemia (105). These
findings suggested that HDAC4 may serve as a predictive marker for
adverse cardiovascular events.
The peptide ligand apelin and its receptor APJ play
a critical role in regulating cardiovascular function. Homozygous
APJ knockout mice exhibit partial embryonic lethality, and
surviving embryos display cardiac and vascular defects. A further
study has shown that the apelin-APJ pathway regulates
cardiovascular function by modulating MEF2 activity through
Gα13-mediated phosphorylation of HDAC4 and HDAC5. Targeted
inhibition of HDAC4 phosphorylation and its cytoplasmic
translocation may potentially address cardiovascular defects and
embryonic lethality (106). A
study involving ventricular cardiomyocytes from mice, rabbits and
humans revealed that HDAC4 is regulated not only by CaMKII but also
by protein kinase A (PKA). Specifically, PKA was shown to regulate
the nuclear accumulation of HDAC4 through phosphorylation at the
S265/266 sites (107). This
novel finding provides new insights for the development of
therapeutic drugs or genetic interventions for CVD.
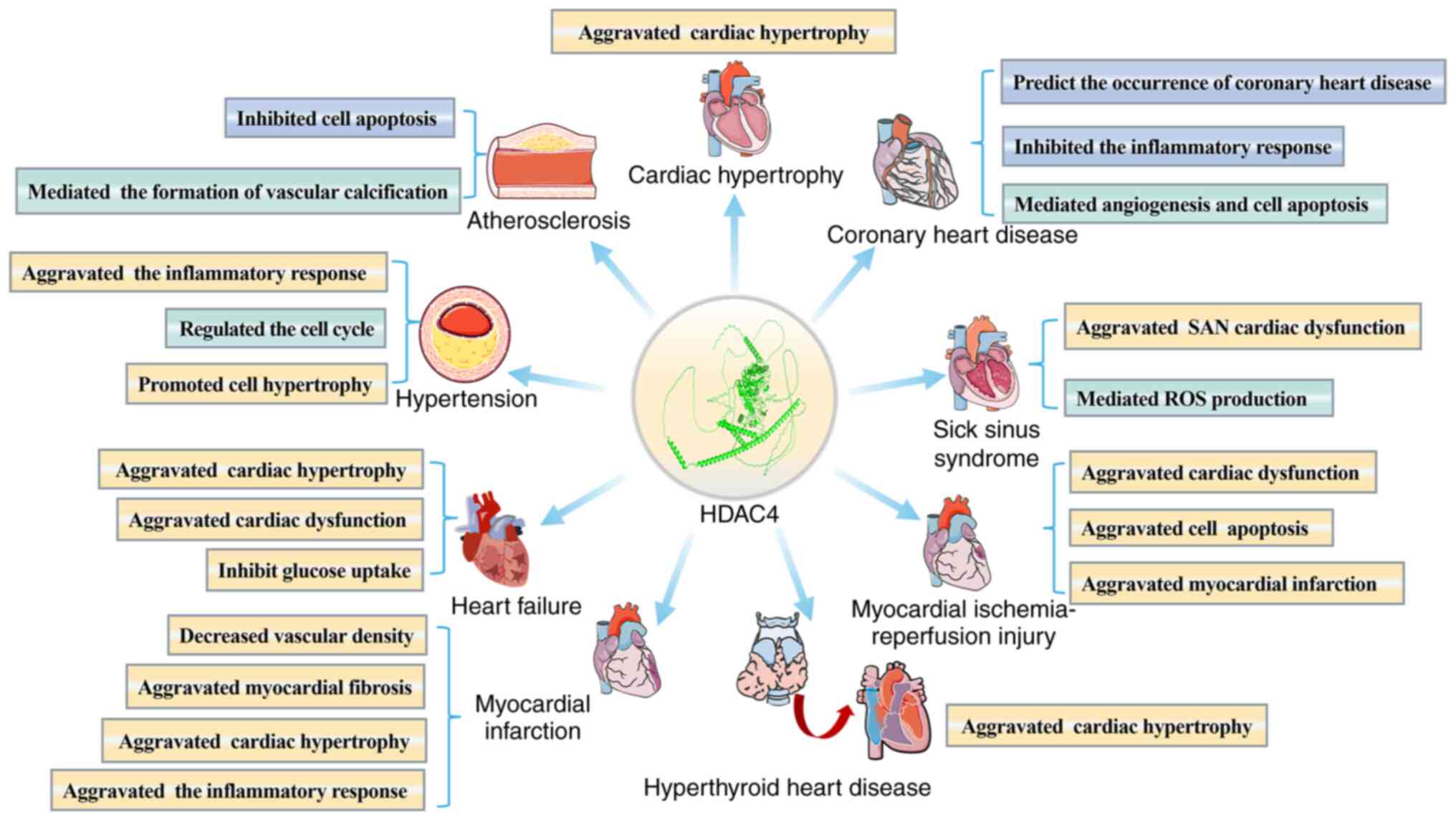
In summary, HDAC4 plays a crucial role in various
CVDs. Studies have shown that HDAC4 is directly or indirectly
involved in the onset and progression of CVD by modulating multiple
biological processes, including cell proliferation, inflammatory
responses and apoptosis. Moreover, an increasing number of studies
suggested that the regulatory role of HDAC4 in CVD may be closely
associated with mitochondrial function. As the central organelle
responsible for maintaining energy homeostasis in cardiomyocytes,
mitochondrial dysfunction is considered a major contributor to cell
death and the progression of CVD (108-111). Recent evidence has shown that
HDAC4 can modulate cardiomyocyte homeostasis by regulating the
mitochondrial permeability transition pore. These findings suggest
the existence of an as-yet undefined regulatory network between
HDAC4 and mitochondrial function, and further elucidation of this
interaction may deepen our understanding of CVD pathogenesis and
provide a theoretical basis for targeted therapeutic strategies.
However, despite numerous studies highlighting the potential role
of HDAC4 in cardiovascular pathology, research on its specific
molecular mechanisms and clinical applications remains in the
exploratory stage. Therefore, further elucidation of the factors
influencing HDAC4 expression is essential for the development of
targeted therapeutic strategies.
TSA is a commonly used hydroxamic acid-based HDAC
inhibitor. A study has shown that 20 nmol/l TSA can promote the
degradation of HDAC4 via the proteasome pathway, thereby inhibiting
H/R-induced cardiomyocyte apoptosis and LDH release (112). Yang et al (22) reported that treatment of RAECs
with 10 μM HDAC4 inhibitor tasquinimod can alleviate
AngII-induced vascular inflammation. LMK235, another hydroxamic
acid-based HDAC inhibitor, has been reported to exhibit high
selectivity for HDAC4 (113).
Chen et al (114) found
that the intraperitoneal injection of 5 mg/kg LMK235 can abolish
the protective effects of Huangqi Guizhi Wuwu decoction on
microvascular and endothelial dysfunction in diabetic mice.
However, another study has shown that LMK235 inhibits the
expression of HDAC1, HDAC2, HDAC3, HDAC4, HDAC5 and HDAC7 induced
by CaMKIIα overexpression. Thus, LMK235 may act as a broad-spectrum
HDAC inhibitor, and its specificity for HDAC4 remains uncertain
(115). MC-1568 is a commonly
used inhibitor of HDAC4 and HDAC6. Research indicates that 1
μM MC-1568 can reduce the beating rate of cardiomyocytes in
rabbit pulmonary veins, indicating its potential application in the
regulation of arrhythmias (116).
An increasing body of research indicates that
various drugs can modulate HDAC4 expression, thereby influencing
the progression of CVD. A study has shown that 1 μM
lercanidipine or 0.1 μM tacrolimus can reverse the
AngII-induced expression of cardiac hypertrophy markers ANP and
BNP, possibly by inhibiting the CaMKII-HDAC4 signaling pathway
(117). Similarly, Wang et
al (118) found that 5
μM autocamtide-2-related inhibitory peptide suppresses
ISO-induced cardiac hypertrophy by inhibiting CaMKII and HDAC4
expression. Panax quinquefolium saponin was shown to downregulate
CaMKII and HDAC4 expression in cardiomyocytes of rats subjected to
hindlimb unloading, thereby improving cardiac function (119). Liu et al (120) reported that ISO induces
hypertrophy in H9c2 cells and inhibits nuclear HDAC4 expression,
whereas 5 μmol/l isosteviol sodium (STVNa) reverses these
effects. Moreover, the addition of the Trx1 inhibitor PX-12
attenuates the effects of STVNa (120). Leucine is considered an
essential amino acid (121).
The administration of 3% leucine to HFpEF female rats has been
shown to inhibit the expression of HDAC4 in the heart, thereby
improving diastolic dysfunction (8). Quercetin, which has been
extensively studied for its cardiovascular benefits (122,123), was shown in an animal study to
alleviate cardiac hypertrophy by downregulating HDAC4 and p-HDAC4
Ser246 (123).
Additionally, 20 nM insulin-like growth factor II (IGF-II) analogue
Leu27IGF-II was found to increase the expression of CaMKIIδ,
p-HDAC4, p-HDAC5 and BNP in H9c2 cells. However, co-treatment with
Carthamus tinctorius extract inhibits the expression of
these proteins, suggesting that targeting HDAC4 holds therapeutic
potential for cardiac hypertrophy (124). An earlier study identified that
VSMCs contribute to increased vascular stiffness in hypertension
(125). Choi et al
(126) demonstrated that TMP269
inhibits class IIa HDACs (HDAC4, 5, 7, and 9) in VSMCs in a
dose-dependent manner. Similarly, panobinostat (LBH589) was found
to suppress class IIa HDAC activity. Notably, 10 μM TMP269
or LBH589 showed stronger inhibitory effects on HDAC4, HDAC5 and
HDAC9 in VSMCs compared with TSA (126). Gallic acid, a dietary phenolic
acid commonly found in edible plants (127,128), was also shown to inhibit class
IIa HDAC activity in VSMCs. Interestingly, 100 μM
sulforaphane inhibited the enzymatic activity of HDAC4, HDAC5 and
HDAC7, whereas a low concentration (1 μM) increased their
activity (126). A previous
study reported that 50 μM nifedipine downregulates
PE-induced p-HDAC4Ser632 expression and inhibits the
nuclear export of HDAC4, thereby alleviating pathological cardiac
hypertrophy (129). In
addition, Guo et al (130) discovered that aconitine (AC), a
key compound in Aconitum species, induces cardiotoxicity.
Transcriptomic sequencing and molecular docking experiments
demonstrated that AC promotes the interaction of HBB with ABHD5 and
AMPK, thereby regulating the ABHD5/AMPK/HDAC4 axis and contributing
to cardiotoxic effects (130).
Exercise, as a safe and effective multi-system
intervention, has shown beneficial effects in the prevention and
treatment of CVD (131). An
animal study demonstrated that 2 weeks of exercise increased the
expression of HDAC4-NT in the hearts of wild-type mice, whereas
mice with cardiomyocyte-specific knockout of HDAC4 exhibited
reduced exercise capacity (30).
By contrast, suppression of HDAC4 expression was found to enhance
exercise tolerance in mice with HF (85). Moreover, a recent study reported
that high-intensity interval training improves cardiac function in
HF by promoting skeletal muscle-derived meteorin-like, which
activates the AMPK-HDAC4 signaling pathway (132). These findings suggested that
exercise may regulate cardiac function via modulation of HDAC4,
offering new insights into personalized interventions for CVD.
The present review examined the role and underlying
mechanisms of HDAC4 in CVD, highlighting its regulatory potential
in key pathophysiological processes such as inflammation, fibrosis,
apoptosis and mitochondrial function. To date, an increasing body
of evidence has demonstrated the involvement of HDAC4 in the
progression of CVD, strongly suggesting that it may serve as a
promising molecular target for early intervention or personalized
therapy in the future (Fig. 7).
In cardiac pathologies such as myocardial hypertrophy, CHD, SSS,
MI-reperfusion injury, HHD, MI and HF, most studies have shown that
HDAC4 inhibition can alleviate cardiac damage by negatively
regulating cardiac function, suppressing myocardial inflammation
and fibrosis, and reducing cardiomyocyte apoptosis. In
hypertension, HDAC4 promotes vascular inflammation and hypertrophy
of VSMCs. By contrast, HDAC4 appears to play a protective role in
AS; for example, it can ameliorate AS by inhibiting
miR-200b-3p-induced apoptosis. In DC, HDAC4 exhibits dual
functions: On the one hand, its overexpression induces
cardiomyocyte apoptosis. On the other hand, HDAC4 deletion
exacerbates cardiac dysfunction in DC mice. These findings provide
a theoretical foundation for the development of HDAC4-centered
precision intervention strategies, which may facilitate a shift in
CVD treatment from late-stage symptom management to early-stage
molecular mechanism-based interventions.
However, there are still certain limitations in the
reports on HDAC4 in CVD. First, the expression patterns and
functional roles of HDAC4 in CVD induced by different pathological
stimuli remain controversial. There is a lack of comprehensive
analysis regarding its correlation with disease severity and
clinical prognosis, which warrants further investigation. Notably,
a previous study has revealed that HDAC4 inhibition can enhance
exercise capacity in mice with HF (85). Second, current research on the
safety, specificity and long-term efficacy of HDAC4-targeted
interventions remains in its early stages, limiting the feasibility
of clinical translation. Therefore, future studies should integrate
multi-omics approaches such as transcriptomics, proteomics and
metabolomics, combined with validation in large animal models and
analyses of patient-derived samples, to comprehensively assess the
mechanistic integrity and clinical translational potential of the
HDAC4 signaling pathway. Lastly, the potential role of HDAC4 in
diseases beyond the cardiovascular system warrants consideration.
In pulmonary arterial hypertension (PAH), stromal-derived factor 1
has been shown to activate the CaMKII/HDAC4 signaling pathway,
which stabilizes runt-related transcription factor 2, subsequently
promoting osteopontin expression and contributing to pulmonary
artery smooth muscle cell proliferation and vascular remodeling
(133). Simultaneously, ongoing
discussions surrounding the revised diagnostic threshold for mean
pulmonary arterial pressure have underscored the urgent need for
early molecular biomarkers in PAH (134). Thus, HDAC4 may serve as a
promising regulatory target, offering new opportunities for
mechanistic investigations and therapeutic interventions.
In summary, HDAC4 demonstrates potential as a
biomarker for monitoring CVD. Despite existing controversies,
ongoing research may confirm HDAC4 as a highly promising
therapeutic target in cardiovascular medicine.
Not applicable.
XM was responsible for conceptualization,
investigation, writing the original draft and illustration. RW was
responsible for conceptualization, investigation, illustration,
reviewing and editing the manuscript. AS was responsible for
conceptualization, reviewing and editing the manuscript. XZ was
responsible for illustration, reviewing and editing the manuscript.
JZ was responsible for investigation, reviewing and editing the
manuscript. SH was responsible for funding, supervision, reviewing
and editing the manuscript. All authors read and approved the final
version of the manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the Scientific Research Fund
Program of Shandong University of Traditional Chinese Medicine
(grant no. KYZK2024Q18) and the Undergraduate Research Training
Program of Shandong University of Traditional Chinese Medicine
(grant no. 2025054).
|
1
|
Li P, Ge J and Li H: Lysine
acetyltransferases and lysine deacetylases as targets for
cardiovascular disease. Nat Rev Cardiol. 17:96–115. 2020.
View Article : Google Scholar
|
|
2
|
Haberland M, Montgomery RL and Olson EN:
The many roles of histone deacetylases in development and
physiology: Implications for disease and therapy. Nat Rev Genet.
10:32–42. 2009. View Article : Google Scholar
|
|
3
|
Zhang D, Hu X, Henning RH and Brundel BJ:
Keeping up the balance: Role of HDACs in cardiac proteostasis and
therapeutic implications for atrial fibrillation. Cardiovasc Res.
109:519–526. 2016. View Article : Google Scholar
|
|
4
|
Backs J and Olson EN: Control of cardiac
growth by histone acetylation/deacetylation. Circ Res. 98:15–24.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hohl M, Wagner M, Reil JC, Müller SA,
Tauchnitz M, Zimmer AM, Lehmann LH, Thiel G, Böhm M, Backs J and
Maack C: HDAC4 controls histone methylation in response to elevated
cardiac load. J Clin Invest. 123:1359–1370. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Z, Qin G and Zhao TC: HDAC4:
Mechanism of regulation and biological functions. Epigenomics.
6:139–150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ouyang J, Wang H and Huang J: The role of
lactate in cardiovascular diseases. Cell Commun Signal. 21:3172023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alves PKN, Schauer A, Augstein A, Männel
A, Barthel P, Joachim D, Friedrich J, Prieto ME, Moriscot AS, Linke
A and Adams V: Leucine Supplementation improves diastolic function
in HFpEF by HDAC4 inhibition. Cells. 12:25612023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ling S, Sun Q, Li Y, Zhang L, Zhang P,
Wang X, Tian C, Li Q, Song J, Liu H, et al: CKIP-1 inhibits cardiac
hypertrophy by regulating class II histone deacetylase
phosphorylation through recruiting PP2A. Circulation.
126:3028–3040. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li J, Gao Q, Wang S, Kang Z, Li Z, Lei S,
Sun X, Zhao M, Chen X, Jiao G, et al: Sustained increased CaMKII
phosphorylation is involved in the impaired regression of
isoproterenol-induced cardiac hypertrophy in rats. J Pharmacol Sci.
144:30–42. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ginnan R, Sun LY, Schwarz JJ and Singer
HA: MEF2 is regulated by CaMKIIdelta2 and a HDAC4-HDAC5 heterodimer
in vascular smooth muscle cells. Biochem J. 444:105–114. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Berthouze-Duquesnes M, Lucas A, Sauliere
A, Sin YY, Laurent AC, Galés C, Baillie G and Lezoualc'h F:
Specific interactions between Epac1, β-arrestin2 and PDE4D5
regulate β-adrenergic receptor subtype differential effects on
cardiac hypertrophic signaling. Cell Signal. 25:970–980. 2013.
View Article : Google Scholar
|
|
13
|
Guo Z, Wu Y, Feng Q, Wang C, Wang Z, Zhu
Y, Lu X, Chen W, Yang Q and Huo Y: Circulating HDAC4 reflects lipid
profile, coronary stenosis and inflammation in coronary heart
disease patients. Biomark Med. 17:41–49. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kong Q, Hao Y, Li X, Wang X, Ji B and Wu
Y: HDAC4 in ischemic stroke: Mechanisms and therapeutic potential.
Clin Epigenetics. 10:1172018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen Z, Zhang Z, Guo L, Wei X, Zhang Y,
Wang X and Wei L: The role of histone deacetylase 4 during
chondrocyte hypertrophy and endochondral bone development. Bone
Joint Res. 9:82–89. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mathias RA, Guise AJ and Cristea IM:
Post-translational modifications regulate class IIa histone
deacetylase (HDAC) function in health and disease. Mol Cell
Proteomics. 14:456–470. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cuttini E, Goi C, Pellarin E, Vida R and
Brancolini C: HDAC4 in cancer: A multitasking platform to drive not
only epigenetic modifications. Front Mol Biosci. 10:11166602023.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Duarte LRF, Pinho V, Rezende BM and
Teixeira MM: Resolution of inflammation in acute
graft-versus-host-disease: Advances and perspectives. Biomolecules.
12:752022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liberale L, Badimon L, Montecucco F,
Luscher TF, Libby P and Camici GG: Inflammation, aging, and
cardiovascular disease: JACC review topic of the week. J Am Coll
Cardiol. 79:837–847. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cui C, Liu L, Qi Y, Han N, Xu H, Wang Z,
Shang X, Han T, Zha Y, Wei X and Wu Z: Joint association of TyG
index and high sensitivity C-reactive protein with cardiovascular
disease: A national cohort study. Cardiovasc Diabetol. 23:1562024.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fredman G and Serhan CN: Specialized
pro-resolving mediators in vascular inflammation and
atherosclerotic cardiovascular disease. Nat Rev Cardiol.
21:808–823. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang D, Xiao C, Long F, Su Z, Jia W, Qin
M, Huang M, Wu W, Suguro R, Liu X and Zhu Y: HDAC4 regulates
vascular inflammation via activation of autophagy. Cardiovasc Res.
114:1016–1028. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Usui T, Okada M, Hara Y and Yamawaki H:
Exploring calmodulin-related proteins, which mediate development of
hypertension, in vascular tissues of spontaneous hypertensive rats.
Biochem Biophys Res Commun. 405:47–51. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu K, Huang MJ, Ling S, Li YX, Cao XY,
Chen YF, Lei JM, Fu WZ and Tan BF: LncRNA CASC11 upregulation
promotes HDAC4 to alleviate oxidized low-density
lipoprotein-induced injury of cardiac microvascular endothelial
cells. Kaohsiung J Med Sci. 39:758–768. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ravassa S, Lopez B, Treibel TA, José GS,
Losada-Fuentenebro B, Tapia L, Bayés-Genís A, Díez J and González
A: Cardiac Fibrosis in heart failure: Focus on non-invasive
diagnosis and emerging therapeutic strategies. Mol Aspects Med.
93:1011942023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y, Gao F, Tang Y, Xiao J, Li C,
Ouyang Y and Hou Y: Valproic acid regulates Ang II-induced
pericyte-myofibroblast trans-differentiation via MAPK/ERK pathway.
Am J Transl Res. 10:1976–1989. 2018.PubMed/NCBI
|
|
27
|
Zhang LX, DeNicola M, Qin X, Du J, Ma J,
Zhao YT, Zhuang S, Liu PY, Wei L, Qin G, et al: Specific inhibition
of HDAC4 in cardiac progenitor cells enhances myocardial repairs.
Am J Physiol Cell Physiol. 307:C358–C372. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang LX, Du J, Zhao YT, Wang J, Zhang S,
Dubielecka PM, Wei L, Zhuang S, Qin G, Chin YE and Zhao TC:
Transgenic overexpression of active HDAC4 in the heart attenuates
cardiac function and exacerbates remodeling in infarcted
myocardium. J Appl Physiol (1985). 125:1968–1978. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jebessa ZH, Shanmukha KD, Dewenter M,
Lehmann LH, Xu C, Schreiter F, Siede D, Gong XM, Worst BC, Federico
G, et al: The lipid droplet-associated protein ABHD5 protects the
heart through proteolysis of HDAC4. Nat Metab. 1:1157–1167. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lehmann LH, Jebessa ZH, Kreusser MM,
Horsch A, He T, Kronlage M, Dewenter M, Sramek V, Oehl U,
Krebs-Haupenthal J, et al: A proteolytic fragment of histone
deacetylase 4 protects the heart from failure by regulating the
hexosamine biosynthetic pathway. Nat Med. 24:62–72. 2018.
View Article : Google Scholar
|
|
31
|
Zhan J, Wang J, Liang Y, Wang L, Huang L,
Liu S, Zeng X, Zeng E and Wang H: Apoptosis dysfunction:
Unravelling the interplay between ZBP1 activation and viral
invasion in innate immune responses. Cell Commun Signal.
22:1492024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Emdad L, Bhoopathi P, Talukdar S, Pradhan
AK, Sarkar D, Wang XY, Das SK and Fisher PB: Recent insights into
apoptosis and toxic autophagy: The roles of MDA-7/IL-24, a
multidimensional anti-cancer therapeutic. Semin Cancer Biol.
66:140–154. 2020. View Article : Google Scholar :
|
|
33
|
Zou G, Zhong W, Wu F, Wang X and Liu L:
Catalpol attenuates cardiomyocyte apoptosis in diabetic
cardiomyopathy via Neat1/miR-140-5p/HDAC4 axis. Biochimie.
165:90–99. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang L, Wang H, Zhao Y, Wang J,
Dubielecka PM, Zhuang S, Qin G, Chin YE, Kao RL and Zhao TC:
Myocyte-specific overexpressing HDAC4 promotes myocardial
ischemia/reperfusion injury. Mol Med. 24:372018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu X, Liu Y, Mo S, Wei W, Ye Z and Su Q:
LncRNA TUG1 competitively binds to miR-340 to accelerate myocardial
ischemia-reperfusion injury. FASEB J. 35:e211632021.
|
|
36
|
Zhang F, Cheng N, Du J, Zhang H and Zhang
C: MicroRNA-200b-3p promotes endothelial cell apoptosis by
targeting HDAC4 in atherosclerosis. BMC Cardiovasc Disord.
21:1722021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bazgir F, Nau J, Nakhaei-Rad S, Amin E,
Wolf MJ, Saucerman JJ, Lorenz K and Ahmadian MR: The
microenvironment of the pathogenesis of cardiac hypertrophy. Cells.
12:17802023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ago T, Liu T, Zhai P, Chen W, Li H,
Molkentin JD, Vatner SF and Sadoshima J: A redox-dependent pathway
for regulating class II HDACs and cardiac hypertrophy. Cell.
133:978–993. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Backs J, Song K, Bezprozvannaya S, Chang S
and Olson EN: CaM kinase II selectively signals to histone
deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest.
116:1853–1864. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fujioka R, Yamamoto T, Maruta A, Nakamura
Y, Tominaga N, Inamitsu M, Oda T, Kobayashi S and Yano M: Herpud1
modulates hypertrophic signals independently of calmodulin nuclear
translocation in rat myocardium-derived H9C2 cells. Biochem Biophys
Res Commun. 652:61–67. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zheng L, Wang J, Zhang R, Zhang Y, Geng J,
Cao L, Zhao X, Geng J, Du X, Hu Y and Cong H: Angiotensin II
mediates cardiomyocyte hypertrophy in atrial cardiomyopathy via
epigenetic transcriptional regulation. Comput Math Methods Med.
2022:63121002022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhao D, Zhong G, Li J, Pan J, Zhao Y, Song
H, Sun W, Jin X, Li Y, Du R, et al: Targeting E3 ubiquitin ligase
WWP1 prevents cardiac hypertrophy through destabilizing DVL2 via
inhibition of K27-linked ubiquitination. Circulation. 144:694–711.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li C, Cai X, Sun H, Bai T, Zheng X, Zhou
XW, Chen X, Gill DL, Li J and Tang XD: The deltaA isoform of
calmodulin kinase II mediates pathological cardiac hypertrophy by
interfering with the HDAC4-MEF2 signaling pathway. Biochem Biophys
Res Commun. 409:125–130. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lairez O, Cognet T, Schaak S, Calise D,
Guilbeau-Frugier C, Parini A and Mialet-Perez J: Role of serotonin
5-HT2A receptors in the development of cardiac hypertrophy in
response to aortic constriction in mice. J Neural Transm (Vienna).
120:927–935. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu MY, Yue LJ, Luo YC, Lu J, Wu GD, Sheng
SQ, Shi YQ and Dong ZX: SUMOylation of SIRT1 activating
PGC-1alpha/PPARalpha pathway mediates the protective effect of
LncRNA-MHRT in cardiac hypertrophy. Eur J Pharmacol.
930:1751552022. View Article : Google Scholar
|
|
46
|
Pedram A, Razandi M, Narayanan R, Dalton
JT, McKinsey TA and Levin ER: Estrogen regulates histone
deacetylases to prevent cardiac hypertrophy. Mol Biol Cell.
24:3805–3818. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Okabe K, Matsushima S, Ikeda S, Ikeda M,
Ishikita A, Tadokoro T, Enzan N, Yamamoto T, Sada M, Deguchi H, et
al: DPP (Dipeptidyl Peptidase)-4 inhibitor attenuates Ang II
(Angiotensin II)-induced cardiac hypertrophy via GLP (Glucagon-Like
Peptide)-1-dependent suppression of Nox (Nicotinamide Adenine
Dinucleotide Phosphate Oxidase) 4-HDAC (Histone Deacetylase) 4
pathway. Hypertension. 75:991–1001. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mhatre KN, Wakula P, Klein O, Bisping E,
Völkl J, Pieske B and Heinzel FR: Crosstalk between FGF23- and
angiotensin II-mediated Ca(2+) signaling in pathological cardiac
hypertrophy. Cell Mol Life Sci. 75:4403–4416. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fan J, Fan W, Lei J, Zhou Y, Xu H, Kapoor
I, Zhu G and Wang J: Galectin-1 attenuates cardiomyocyte
hypertrophy through splice-variant specific modulation of
CaV1.2 calcium channel. Biochim Biophys Acta Mol Basis
Dis. 1865:218–229. 2019. View Article : Google Scholar
|
|
50
|
Matsushima S, Kuroda J, Ago T, Zhai P,
Park JY, Xie LH, Tian B and Sadoshima J: Increased oxidative stress
in the nucleus caused by Nox4 mediates oxidation of HDAC4 and
cardiac hypertrophy. Circ Res. 112:651–663. 2013. View Article : Google Scholar :
|
|
51
|
Zhou P, Zhao XN, Ma YY, Tang TJ, Wang SS,
Wang L and Huang J: Virtual screening analysis of natural
flavonoids as trimethylamine (TMA)-lyase inhibitors for coronary
heart disease. J Food Biochem. 46:e143762022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shaya GE, Leucker TM, Jones SR, Martin SS
and Toth PP: Coronary heart disease risk: Low-density lipoprotein
and beyond. Trends Cardiovasc Med. 32:181–194. 2022. View Article : Google Scholar
|
|
53
|
Wang Y, Zhang J, Wang Z, Wang C and Ma D:
Endothelial-cell-mediated mechanism of coronary microvascular
dysfunction leading to heart failure with preserved ejection
fraction. Heart Fail Rev. 28:169–178. 2023. View Article : Google Scholar :
|
|
54
|
Yan P, Sun C, Ma J, Jin Z, Guo R and Yang
B: MicroRNA-128 confers protection against cardiac microvascular
endothelial cell injury in coronary heart disease via negative
regulation of IRS1. J Cell Physiol. 234:13452–13463. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang X, Zhou H and Chang X: Involvement
of mitochondrial dynamics and mitophagy in diabetic endothelial
dysfunction and cardiac microvascular injury. Arch Toxicol.
97:3023–3035. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Haqqani HM and Kalman JM: Aging and
sinoatrial node dysfunction: Musings on the not-so-funny side.
Circulation. 115:1178–1179. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mesquita T, Miguel-Dos-Santos R and
Cingolani E: Aging and sinus node dysfunction: Mechanisms and
future directions. Clin Sci (Lond). 139:577–593. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang H, Li L, Hao M, Chen K, Lu Y, Qi J,
Chen W, Ren L, Cai X, Chen C, et al: Yixin-Fumai granules improve
sick sinus syndrome in aging mice through Nrf-2/HO-1 pathway: A new
target for sick sinus syndrome. J Ethnopharmacol. 277:1142542021.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chang X, Zhou S, Liu J, Wang Y, Guan X, Wu
Q, Zhang Q, Liu Z and Liu R: Zishen Tongyang Huoxue decoction
(TYHX) alleviates sinoatrial node cell ischemia/reperfusion injury
by directing mitochondrial quality control via the VDAC1-β-tubulin
signaling axis. J Ethnopharmacol. 320:1173712024. View Article : Google Scholar
|
|
60
|
Chang X, Li Y, Liu J, Wang Y, Guan X, Wu
Q, Zhou Y, Zhang X, Chen Y, Huang Y and Liu R: β-tubulin
contributes to Tongyang Huoxue decoction-induced protection against
hypoxia/reoxygenation-induced injury of sinoatrial node cells
through SIRT1-mediated regulation of mitochondrial quality
surveillance. Phytomedicine. 108:1545022023. View Article : Google Scholar
|
|
61
|
Yang B, Huang Y, Zhang H, Huang Y, Zhou
HJ, Young L, Xiao H and Min W: Mitochondrial thioredoxin-2
maintains HCN4 expression and prevents oxidative stress-mediated
sick sinus syndrome. J Mol Cell Cardiol. 138:291–303. 2020.
View Article : Google Scholar
|
|
62
|
Chen L, Mao LS, Xue JY, Jian YH, Deng ZW,
Mazhar M, Zou Y, Liu P, Chen MT, Luo G and Liu MN: Myocardial
ischemia-reperfusion injury: The balance mechanism between
mitophagy and NLRP3 inflammasome. Life Sci. 355:1229982024.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Chang X, Zhou S, Liu J, Wang Y, Guan X, Wu
Q, Liu Z and Liu R: Zishenhuoxue decoction-induced myocardial
protection against ischemic injury through TMBIM6-VDAC1-mediated
regulation of calcium homeostasis and mitochondrial quality
surveillance. Phytomedicine. 132:1553312024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chang X, Liu R, Li R, Peng Y, Zhu P and
Zhou H: Molecular mechanisms of mitochondrial quality control in
ischemic cardiomyopathy. Int J Biol Sci. 19:426–448. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wang J, Zhuang H, Jia L, He X, Zheng S, Ji
K, Xie K, Ying T, Zhang Y, Li C and Chang X: Nuclear receptor
subfamily 4 group A member 1 promotes myocardial
ischemia/reperfusion injury through inducing mitochondrial fission
factor-mediated mitochondrial fragmentation and inhibiting FUN14
domain containing 1-depedent mitophagy. Int J Biol Sci.
20:4458–4475. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Pu X, Zhang Q, Liu J, Wang Y, Guan X, Wu
Q, Liu Z, Liu R and Chang X: Ginsenoside Rb1 ameliorates heart
failure through DUSP-1-TMBIM-6-mediated mitochondrial quality
control and gut flora interactions. Phytomedicine. 132:1558802024.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhao YT, Wang H, Zhang S, Du J, Zhuang S
and Zhao TC: Irisin ameliorates hypoxia/reoxygenation-induced
injury through modulation of histone deacetylase 4. PLoS One.
11:e01661822016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lee SY and Pearce EN: Hyperthyroidism: A
review. JAMA. 330:1472–1483. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kim HJ and McLeod DSA: Subclinical
hyperthyroidism and cardiovascular disease. Thyroid. 34:1335–1345.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Nie D, Xia C, Wang Z, Ding P, Meng Y, Liu
J, Li T, Gan T, Xuan B, Huang Y, et al: CaMKII inhibition protects
against hyperthyroid arrhythmias and adverse myocardial remodeling.
Biochem Biophys Res Commun. 615:136–142. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Diniz GP, Lino CA, Moreno CR, Senger N and
Barreto-Chaves MLM: MicroRNA-1 overexpression blunts cardiomyocyte
hypertrophy elicited by thyroid hormone. J Cell Physiol.
232:3360–3368. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Huang ZP, Chen J, Seok HY, Zhang Z,
Kataoka M, Hu X and Wang DZ: MicroRNA-22 regulates cardiac
hypertrophy and remodeling in response to stress. Circ Res.
112:1234–1243. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yang W, Lin J, Zhou J, Zheng Y, Jiang S,
He S and Li D: Innate lymphoid cells and myocardial infarction.
Front Immunol. 12:7582722021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Turkieh A, El Masri Y, Pinet F and
Dubois-Deruy E: Mitophagy regulation following myocardial
infarction. Cells. 11:1992022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Asensio-Lopez MC, Lax A, Del Palacio MJ,
Sassi Y, Hajjar RJ, Januzzi JL, Bayes-Genis A and Pascual-Figal DA:
Yin-Yang 1 transcription factor modulates ST2 expression during
adverse cardiac remodeling post-myocardial infarction. J Mol Cell
Cardiol. 130:216–233. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Lv F, Xie L, Li L and Lin J: LMK235
ameliorates inflammation and fibrosis after myocardial infarction
by inhibiting LSD1-related pathway. Sci Rep. 14:234502024.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Li L, Fu W, Gong X, Chen Z, Tang L, Yang
D, Liao Q, Xia X, Wu H, Liu C, et al: The role of G protein-coupled
receptor kinase 4 in cardiomyocyte injury after myocardial
infarction. Eur Heart J. 42:1415–1430. 2021. View Article : Google Scholar :
|
|
78
|
Zhao J, Li L, Wang X and Shen J: KN-93
promotes HDAC4 nucleus translocation to promote fatty acid
oxidation in myocardial infarction. Exp Cell Res. 438:1140502024.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hermann DM, Xin W, Bahr M, Giebel B and
Doeppner TR: Emerging roles of extracellular vesicle-associated
non-coding RNAs in hypoxia: Insights from cancer, myocardial
infarction and ischemic stroke. Theranostics. 12:5776–5802. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Li Y, Zhang Z, Zhou X, Li R, Cheng Y,
Shang B, Han Y, Liu B and Xie X: Histone deacetylase 1 inhibition
protects against hypoxia-induced swelling in H9c2 cardiomyocytes
through regulating cell stiffness. Circ J. 82:192–202. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Savarese G, Becher PM, Lund LH, Seferovic
P, Rosano GMC and Coats AJS: Global burden of heart failure: A
comprehensive and updated review of epidemiology. Cardiovasc Res.
118:3272–3287. 2023. View Article : Google Scholar
|
|
82
|
Ljubojevic-Holzer S, Herren AW, Djalinac
N, Voglhuber J, Morotti S, Holzer M, Wood BM, Abdellatif M, Matzer
I, Sacherer M, et al: CaMKIIdeltaC drives early adaptive
Ca2+ change and late eccentric cardiac hypertrophy. Circ
Res. 127:1159–1178. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lkhagva B, Lin YK, Kao YH, Chazo TF, Chung
CC, Chen SA and Chen YJ: Novel histone deacetylase inhibitor
modulates cardiac peroxisome proliferator-activated receptors and
inflammatory cytokines in heart failure. Pharmacology. 96:184–191.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Calalb MB, McKinsey TA, Newkirk S, Huynh
K, Sucharov CC and Bristow MR: Increased phosphorylation-dependent
nuclear export of class II histone deacetylases in failing human
heart. Clin Transl Sci. 2:325–332. 2009. View Article : Google Scholar
|
|
85
|
Jiang H, Jia D, Zhang B, Yang W, Dong Z,
Sun X, Cui X, Ma L, Wu J, Hu K, et al: Exercise improves cardiac
function and glucose metabolism in mice with experimental
myocardial infarction through inhibiting HDAC4 and upregulating
GLUT1 expression. Basic Res Cardiol. 115:282020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Dzau VJ and Hodgkinson CP: Precision
hypertension. Hypertension. 81:702–708. 2024. View Article : Google Scholar
|
|
87
|
Kanbay M, Copur S, Tanriover C, Ucku D and
Laffin L: Future treatments in hypertension: Can we meet the unmet
needs of patients? Eur J Intern Med. 115:18–28. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Laurent S, Alivon M, Beaussier H and
Boutouyrie P: Aortic stiffness as a tissue biomarker for predicting
future cardiovascular events in asymptomatic hypertensive subjects.
Ann Med. 44(Suppl 1): S93–S97. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Usui T, Okada M, Mizuno W, Oda M, Ide N,
Morita T, Hara Y and Yamawaki H: HDAC4 mediates development of
hypertension via vascular inflammation in spontaneous hypertensive
rats. Am J Physiol Heart Circ Physiol. 302:H1894–H1904. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kim GR, Cho SN, Kim HS, Yu SY, Choi SY,
Ryu Y, Lin MQ, Jin L, Kee HJ and Jeong MH: Histone deacetylase and
GATA-binding factor 6 regulate arterial remodeling in angiotensin
II-induced hypertension. J Hypertens. 34:2206–2219. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Saigusa R, Winkels H and Ley K: T cell
subsets and functions in atherosclerosis. Nat Rev Cardiol.
17:387–401. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Chen F, Li J, Zheng T, Chen T and Yuan Z:
KLF7 alleviates atherosclerotic lesions and inhibits glucose
metabolic reprogramming in macrophages by regulating
HDAC4/miR-148b-3p/NCOR1. Gerontology. 68:1291–1310. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhu XX, Meng XY, Chen G, Sru JB, Fu X, Xu
AJ, Liu Y, Hou XH, Qiu HB, Sun QY, et al: Nesfatin-1 enhances
vascular smooth muscle calcification through facilitating BMP-2
osteogenic signaling. Cell Commun Signal. 22:4882024. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zhao X, Liu S, Wang X, Chen Y, Pang P,
Yang Q, Lin J, Deng S, Wu S, Fan G and Wang B: Diabetic
cardiomyopathy: Clinical phenotype and practice. Front Endocrinol
(Lausanne). 13:10322682022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ma X, Mei S, Wuyun Q, Zhou L, Sun D and
Yan J: Epigenetics in diabetic cardiomyopathy. Clin Epigenetics.
16:522024. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kronlage M, Dewenter M, Grosso J, Fleming
T, Oehl U, Lehmann LH, Falcão-Pires I, Leite-Moreira AF, Volk N,
Gröne HJ, et al: O-GlcNAcylation of histone deacetylase 4 protects
the diabetic heart from failure. Circulation. 140:580–594. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Heymans S, Lakdawala NK, Tschope C and
Klingel K: Dilated cardiomyopathy: Causes, mechanisms, and current
and future treatment approaches. Lancet. 402:998–1011. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Castillero E, Ali ZA, Akashi H, Giangreco
N, Wang C, Stöhr EJ, Ji R, Zhang X, Kheysin N, Park JS, et al:
Structural and functional cardiac profile after prolonged duration
of mechanical unloading: Potential implications for myocardial
recovery. Am J Physiol Heart Circ Physiol. 315:H1463–H1476. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Lu D, Bao D, Dong W, Liu N, Zhang X, Gao
S, Ge W, Gao X and Zhang L: Dkk3 prevents familial dilated
cardiomyopathy development through Wnt pathway. Lab Invest.
96:239–248. 2016. View Article : Google Scholar
|
|
100
|
Li T, Mu N, Yin Y, Yu L and Ma H:
Targeting AMP-activated protein kinase in aging-related
cardiovascular diseases. Aging Dis. 11:967–977. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Tyrrell DJ and Goldstein DR: Ageing and
atherosclerosis: Vascular intrinsic and extrinsic factors and
potential role of IL-6. Nat Rev Cardiol. 18:58–68. 2021. View Article : Google Scholar
|
|
102
|
Saravi SS and Feinberg MW: Can removal of
zombie cells revitalize the aging cardiovascular system? Eur Heart
J. 45:867–869. 2024. View Article : Google Scholar
|
|
103
|
Shabanian K, Shabanian T, Karsai G,
Pontiggia L, Paneni F, Ruschitzka F, Beer JH and Saravi SS: AQP1
differentially orchestrates endothelial cell senescence. Redox
Biol. 76:1033172024. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bhatt DL, Lopes RD and Harrington RA:
Diagnosis and treatment of acute coronary syndromes: A review.
JAMA. 327:662–675. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Xu H, Zhang J, Jia H, Xing F and Cong H:
Serum histone deacetylase 4 longitudinal change for estimating
major adverse cardiovascular events in acute coronary syndrome
patients receiving percutaneous coronary intervention. Ir J Med
Sci. 192:2689–2696. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kang Y, Kim J, Anderson JP, Wu J, Gleim
SR, Kundu RK, McLean DL, Kim JD, Park H, Jin S, et al: Apelin-APJ
signaling is a critical regulator of endothelial MEF2 activation in
cardiovascular development. Circ Res. 113:22–31. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Helmstadter KG, Ljubojevic-Holzer S, Wood
BM, Taheri KD, Sedej S, Erickson JR, Bossuyt J and Bers DM: CaMKII
and PKA-dependent phosphorylation co-regulate nuclear localization
of HDAC4 in adult cardiomyocytes. Basic Res Cardiol. 116:112021.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Li Y, Yu J, Li R, Zhou H and Chang X: New
insights into the role of mitochondrial metabolic dysregulation and
immune infiltration in septic cardiomyopathy by integrated
bioinformatics analysis and experimental validation. Cell Mol Biol
Lett. 29:212024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Chang X, Zhang Q, Huang Y, Liu J, Wang Y,
Guan X, Wu Q, Liu Z and Liu R: Quercetin inhibits necroptosis in
cardiomyocytes after ischemia-reperfusion via
DNA-PKcs-SIRT5-orchestrated mitochondrial quality control.
Phytother Res. 38:2496–2517. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wang J, Zhuang H, Yang X, Guo Z, Zhou K,
Liu N, An Y, Chen Y, Zhang Z, Wang M, et al: Exploring the
mechanism of ferroptosis induction by sappanone A in cancer:
Insights into the mitochondrial dysfunction mediated by
NRF2/xCT/GPX4 axis. Int J Biol Sci. 20:5145–5161. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Pang B, Dong G, Pang T, Sun X, Liu X, Nie
Y and Chang X: Emerging insights into the pathogenesis and
therapeutic strategies for vascular endothelial injury-associated
diseases: Focus on mitochondrial dysfunction. Angiogenesis.
27:623–639. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Du J, Zhang L, Zhuang S, Qin GJ and Zhao
TC: HDAC4 degradation mediates HDAC inhibition-induced protective
effects against hypoxia/reoxygenation injury. J Cell Physiol.
230:1321–1331. 2015. View Article : Google Scholar :
|
|
113
|
Marek L, Hamacher A, Hansen FK, Kuna K,
Gohlke H, Kassack MU and Kurz T: Histone deacetylase (HDAC)
inhibitors with a novel connecting unit linker region reveal a
selectivity profile for HDAC4 and HDAC5 with improved activity
against chemoresistant cancer cells. J Med Chem. 56:427–436. 2013.
View Article : Google Scholar
|
|
114
|
Chen M, Cheng H, Chen X, Gu J, Su W, Cai
G, Yan Y, Wang C, Xia X, Zhang K, et al: The activation of histone
deacetylases 4 prevented endothelial dysfunction: A crucial
mechanism of HuangqiGuizhiWuwu decoction in improving
microcirculation dysfunction in diabetes. J Ethnopharmacol.
307:1162402023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Choi SY, Kee HJ, Sun S, Seok YM, Ryu Y,
Kim GR, Kee SJ, Pflieger M, Kurz T, Kassack MU and Jeong MH:
Histone deacetylase inhibitor LMK235 attenuates vascular
constriction and aortic remodelling in hypertension. J Cell Mol
Med. 23:2801–2812. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Lkhagva B, Chang SL, Chen YC, Kao YH, Lin
YK, Chiu CT, Chen SA and Chen YJ: Histone deacetylase inhibition
reduces pulmonary vein arrhythmogenesis through calcium regulation.
Int J Cardiol. 177:982–989. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Chen Y, Yuan J, Jiang G, Zhu J, Zou Y and
Lv Q: Lercanidipine attenuates angiotensin II-induced cardiomyocyte
hypertrophy by blocking calcineurin-NFAT3 and CaMKII-HDAC4
signaling. Mol Med Rep. 16:4545–4552. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Wang S, Li J, Liu Y, Zhang J, Zheng X, Sun
X, Lei S, Kang Z, Chen X, Lei M, et al: Distinct roles of
calmodulin and Ca(2+)/calmodulin-dependent protein kinase II in
isopreterenol-induced cardiac hypertrophy. Biochem Biophys Res
Commun. 526:960–966. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Sun H, Ling S, Zhao D, Li Y, Zhong G, Guo
M, Li Y, Yang L, Du J, Zhou Y, et al: Panax quinquefolium saponin
attenuates cardiac remodeling induced by simulated microgravity.
Phytomedicine. 56:83–93. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Liu F, Su H, Liu B, Mei Y, Ke Q, Sun X and
Tan W: STVNa attenuates isoproterenol-induced cardiac hypertrophy
response through the HDAC4 and Prdx2/ROS/Trx1 pathways. Int J Mol
Sci. 21:6822020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Akbay B, Omarova Z, Trofimov A, Sailike B,
Karapina O, Molnár F and Tokay T: Double-Edge effects of leucine on
cancer cells. Biomolecules. 14:14012024. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Dulf PL, Coada CA, Florea A, Moldovan R,
Baldea I, Dulf DV, Blendea D and Filip AG: Mitigating
doxorubicin-induced cardiotoxicity through quercetin intervention:
An experimental study in rats. Antioxidants (Basel). 13:10682024.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Zhang W, Zheng Y, Yan F, Dong M and Ren Y:
Research progress of quercetin in cardiovascular disease. Front
Cardiovasc Med. 10:12037132023. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Lin CY, Shibu MA, Wen R, Day CH, Chen RJ,
Kuo CH, Ho TJ, Viswanadha VP, Kuo WW and Huang CY: Leu(27)
IGF-II-induced hypertrophy in H9c2 cardiomyoblasts is ameliorated
by saffron by regulation of calcineurin/NFAT and CaMKIIδ signaling.
Environ Toxicol. 36:2475–2483. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Sehgel NL, Zhu Y, Sun Z, Trzeciakowski JP,
Hong Z, Hunter WC, Vatner DE, Meininger GA and Vatner SF: Increased
vascular smooth muscle cell stiffness: A novel mechanism for aortic
stiffness in hypertension. Am J Physiol Heart Circ Physiol.
305:H1281–H1287. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Choi SY, Kee HJ, Jin L, Ryu Y, Sun S, Kim
GR and Jeong MH: Inhibition of class IIa histone deacetylase
activity by gallic acid, sulforaphane, TMP269, and panobinostat.
Biomed Pharmacother. 101:145–154. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Xiang Z, Guan H, Zhao X, Xie Q, Xie Z, Cai
F, Dang R, Li M and Wang C: Dietary gallic acid as an antioxidant:
A review of its food industry applications, health benefits,
bioavailability, nano-delivery systems, and drug interactions. Food
Res Int. 180:1140682024. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Hadidi M, Linan-Atero R, Tarahi M,
Christodoulou MC and Aghababaei F: The potential health benefits of
gallic acid: Therapeutic and food applications. Antioxidants
(Basel). 13:10012024. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Ago T, Yang Y, Zhai P and Sadoshima J:
Nifedipine inhibits cardiac hypertrophy and left ventricular
dysfunction in response to pressure overload. J Cardiovasc Transl
Res. 3:304–313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Guo YJ, Yao JJ, Guo ZZ, Ding M, Zhang KL,
Shen QH, Li Y, Yu SF, Wan T, Xu FP, et al: HBB contributes to
individualized aconitine-induced cardiotoxicity in mice via
interfering with ABHD5/AMPK/HDAC4 axis. Acta Pharmacol Sin.
45:1224–1236. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Tucker WJ, Fegers-Wustrow I, Halle M,
Haykowsky MJ, Chung EH and Kovacic JC: Exercise for primary and
secondary prevention of cardiovascular disease: JACC focus seminar
1/4. J Am Coll Cardiol. 80:1091–1106. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Wang Y, Yuan J, Liu H, Chen J, Zou J, Zeng
X, Du L, Sun X, Xia Z, Geng Q, et al: Elevated meteorin-like
protein from high-intensity interval training improves heart
function via AMPK/HDAC4 pathway. Genes Dis. 11:1011002024.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Chen Y, Liu J, Zhang Q, Chai L, Chen H, Li
D, Wang Y, Qiu Y, Shen N, Zhang J, et al: Activation of
CaMKII/HDAC4 by SDF1 contributes to pulmonary arterial hypertension
via stabilization Runx2. Eur J Pharmacol. 970:1764832024.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Xiong C and Yang B: Revising the
hemodynamic criteria for pulmonary hypertension: A perspective from
China. J Transl Int Med. 11:1–3. 2023. View Article : Google Scholar : PubMed/NCBI
|