Introduction
Quinazolinone and its derivatives are important
heterocyclic compounds commonly found in various bioactive
molecules (1). They represent a
major class of biologically active compounds, with the
quinazolinone nucleus attracting substantial attention due to its
broad pharmacological activities (2). Studies have shown that
quinazolinone and its derivatives possess diverse biological
effects, including anti-HIV (3),
anti-tumor (4), anti-bacterial
(5), anti-inflammatory (6), anti-malarial (7), anti-convulsant (8), anti-diabetic (9), anti-oxidant (10), dihydrofolate reductase inhibition
(11) and kinase inhibitory
activity (12).
Chemical scaffold of quinazolinone
compounds
Heterocyclic compounds represent an important class
of organic chemicals. These compounds contain one or more
heteroatoms, such as nitrogen, oxygen or sulfur, along with carbon
atoms, forming a ring structure. Their properties are largely
influenced by the number and type of heteroatoms within the ring
(13). The cyclic structure
strongly affects the compound's characteristics, which are
primarily determined by the heteroatoms present.
The term 'cyclic' refers to the presence of at least
one ring, whereas 'hetero' indicates that non-carbon atoms, or
heteroatoms, are part of that ring (14). In general, heterocyclic compounds
resemble cyclic organic compounds composed only of carbon atoms.
However, the inclusion of heteroatoms imparts distinct physical and
chemical properties, differentiating them from their all-carbon
counterparts (15). These
compounds are highly versatile in organic chemistry and play a key
role in medicinal chemistry. They are widely used in developing
bioactive synthetic compounds, pharmaceutical agents and synthetic
intermediates (16). In the
pharmaceutical industry, heterocyclic structures are particularly
common, with >60% of top-selling drugs containing at least one
heterocyclic ring (17).
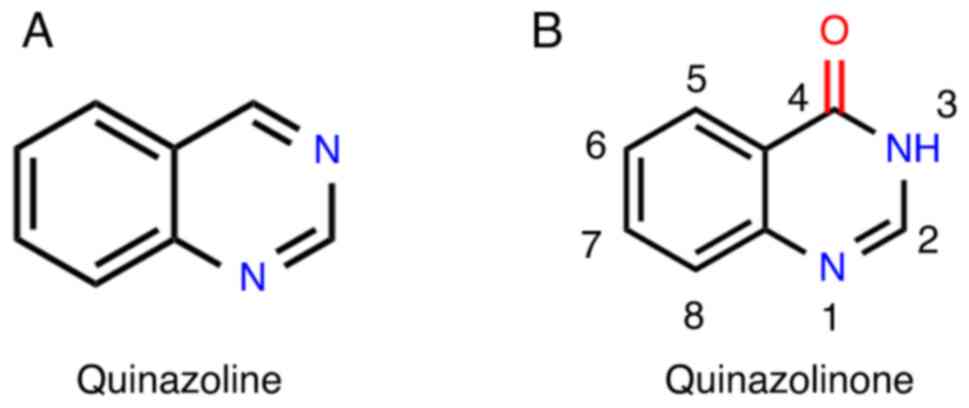
The basic structure of quinazolinone consists of a
quinazoline ring and a ketone group. This bicyclic compound
contains a pyrimidine ring fused with a benzene ring at the 4 and 8
positions (18). The quinazoline
ring comprises six carbon atoms and two nitrogen atoms located at
the 1 and 4 positions (19)
(Fig. 1). As an oxidized form of
quinazoline, quinazolinone retains the nitrogen atoms and is
categorized as a nitrogen-containing heterocyclic compound.
Quinazolinone forms the core structure of nearly 150 naturally
occurring alkaloids found in various plant families, as well as in
animals and microorganisms (20), including compounds such as
glycyrrhizin, quinazolinone, deoxycannabinoid ketone and
rutaecarpine (21). The first
quinazoline alkaloid, vasicine, was isolated in 1888 from the
Indian medicinal plant Adhatoda vasica and continues to
demonstrate significant pharmacological value (22). In recent years, studies have
shown that vasicine and its derivatives have potential
anti-inflammatory, antioxidant, and anti-Alzheimer's disease
effects (23). For instance,
vasicine has been shown to alleviate atopic dermatitis induced by
2,4-dinitrochlorobenzene in BALB/c mice by inhibiting pro-Th 2 and
Th 2 cytokines in both serum and skin tissues (24). Additionally, vasicine inhibits
acetylcholinesterase (AChE) by specifically binding to the AChE
catalytic site, such as Trp 84 and Ser 200, positioning it as a
promising lead compound for Alzheimer's disease treatment (25). Furthermore, in a rat model of
lung injury, in vitro assays of lung tissue homogenates
revealed that vasicine inhibits lipid peroxidation triggered by
excessive reactive oxygen species (ROS), while simultaneously
restoring the activity of the endogenous antioxidant enzyme system
(26). Thus, it is both a
historical milestone in drug development and a starting point for
research on new drugs. Later, vasicine and related quinazolinone
alkaloids such as vasicinone and deoxyvasicinone were also
identified in plants within the Acanthaceae family (27). Numerous additional natural
products containing quinazoline or quinazolinone structures have
since been isolated, analyzed and synthesized. The first
quinazolinone compound was synthesized in the late 1860s by
reacting anthranilic acid with cyanogen to produce
2-cyanoquinazolinone (28). This
compound is a key precursor in quinazolinone chemistry, with its
cyano group providing synthetic flexibility, making it a central
molecule for constructing structurally diverse and complex
quinazolinone derivatives. Systematic modification of its cyano
group and the quinazolinone ring-particularly at the N-3 and
C-6/C-7 positions-can efficiently generate a large library of
structurally diverse compounds for high-throughput screening and
structure-activity relationship (SAR) studies, supporting the
discovery of new lead compounds and optimization of drug activity
(29,30). It plays a pivotal role as a
synthetic building block in drug discovery, particularly in the
development of kinase inhibitors and related compounds. The unique
structural features of quinazolinone, particularly its fused
benzene and pyrimidine rings, increase the possibility and
flexibility of structural modification (31). In addition, the carbonyl group at
the 4-position is a strong electron-withdrawing group, markedly
reducing the electron density of the quinazolinone ring and
facilitating nucleophilic substitution reactions (32). A molecular docking study revealed
that certain quinazolinone derivatives can form strong hydrogen
bonds with LYS 630 and HIS 775 in topoisomerase II and stack π-π
interactions with DT15, supporting the compound's ability to
inhibit DNA replication and repair (33). Furthermore, the quinazolinone
core adapts to the hydrophobic channel of cyclooxygenase-2, where
the carbonyl group can form hydrogen bonds with Arg 121 and Tyr
356, thereby inhibiting enzyme activity (34). These properties enhance
selectivity and biological activity during interactions with
biomolecules, making quinazolinone a key scaffold in drug
development (35).
Quinazolinones can be classified by structural
features into six main groups: Those substituted at the 2- and/or
3-positions, simple 2-substituted quinazolin-4-ones and
quinazolinones fused with pyrrole, pyrroloquinoline, piperidine or
piperazine ring systems (36).
They can also be categorized by the position of the keto or oxo
group into 2(1H)-quinazolinones, 4(3H)-quinazolinones and
2,4(1H,3H)-quinazolinediones (37). Among these, 4(3H)-quinazolinones
are the most common and often serve as intermediates or natural
products in proposed biosynthetic pathways (38).
The pharmacological properties of
quinazolinone-based compounds can be optimized by modifying their
central structure with various functional groups. For instance,
introducing alkyl (39),
hydrazone (40,41) or other substituents at different
positions on the ring can significantly influence target
interactions and therapeutic efficacy. This structural adaptability
has made quinazolinones and their derivatives a central focus in
research on anti-tumor, anti-bacterial and anti-inflammatory drug
development.
Biological activities of quinazolinone
compounds
In synthetic and medicinal chemistry, the
quinazoline scaffold has attracted considerable interest due to its
simple synthesis, versatile reactivity and broad pharmacological
potential (42). Several
quinazolinone-derived drugs, such as Idelalisib and Canertinib,
demonstrate the therapeutic potential of this class in treating
hematological malignancies and other cancers. In addition, prazosin
is used to treat hypertension, Albaconazole for fungal infections,
Balaglitazone for diabetes and Dictyoquinazol A as both a
neuroprotective agent and a glutamate receptor antagonist (43). Over the past two decades, >20
drugs containing a quinazoline or quinazolinone core structure have
been approved by the Food and Drug Administration (FDA) for
anti-tumor use. A prominent example is Dacomitinib, approved in
2018 for the treatment of non-small-cell lung carcinoma. These
agents act through various mechanisms to inhibit cancer cell
growth, mainly by targeting kinases, tubulin, kinesin spindle
proteins and other related molecules (44).
Natural quinazolinone compounds, derived from
plants, animals and microorganisms and widely used in traditional
medicine, often have complex structures that have been refined
through natural selection and evolutionary processes to produce
unique biological activities (45). Quinazolinone alkaloids are
present in the traditional Chinese medicinal herb Dichroae Radix,
forming the core structure of febrifugine and isofebrifugine. These
compounds have been used as antimalarial agents in China for
>2,000 years (46). In recent
years, increasing evidence has confirmed the significant anti-tumor
activity of natural quinazolinone compounds, including their
ability to overcome tumor resistance (47). Several derivatives based on the
quinazolinone core nucleus, such as Bouchardatine, Luotonin F and
Isofebrifugine, have been widely used in cancer treatment,
underscoring the broad distribution and biological importance of
this class of compounds in nature (43,48).
By contrast, the structural design of chemically
synthesized quinazolinone-based drugs often involves modifications
to the natural structure, with researchers selectively altering
specific molecular sites (such as positions 2, 4, 6, 7 and 8)
through chemical functionalization (49). These modifications can markedly
influence pharmacological activity by introducing various
functional groups, thereby optimizing drug efficacy, selectivity,
pharmacological properties, toxicity, stability and
pharmacokinetics. The choice between natural and synthetic
quinazolinone drugs in therapy depends on their origins,
pharmacodynamics and development strategies. Natural quinazolinones
are better suited for chronic diseases that require multi-target
synergy and low toxicity, such as asthma and chronic inflammation.
By contrast, synthetic derivatives are more appropriate for
conditions requiring targeted precision, adjustable structure and
large-scale production, such as cancer therapy, anti-diabetic
treatments and central nervous system disorders (50-52).
Although quinazolinones display a wide range of
biological activities, this review focuses specifically on their
anti-tumor properties, particularly the mechanisms through which
they induce tumor cell death. It summarizes key quinazolinone
derivatives known to induce cell death and aims to provide insights
for designing structures that help elucidate the mechanisms
involved in tumor cell death.
Quinazolinone and its derivatives in
inducing cell death
Apoptosis
Apoptosis is a gene-regulated, self-controlled and
orderly form of cell death that maintains internal homeostasis
(53). It is regulated by
multiple genes, many of which are highly conserved across species.
Apoptosis enables multicellular organisms to remove damaged or
unnecessary cells, playing a critical role in tissue remodeling
during embryonic development and in maintaining tissue stability
throughout life (54). There are
two major apoptotic pathways: Intrinsic and extrinsic (55).
The intrinsic pathway is mainly triggered by
internal signals such as cellular stress, DNA damage or oxidative
stress. It involves the release of pro-apoptotic factors, including
cytochrome c, from mitochondria, which activate caspases that
execute cell death. Proteins of the Bcl-2 family are central
regulators of this pathway, controlling mitochondrial membrane
permeability (56). The
extrinsic pathway is activated by external stimuli through
membrane-bound death receptors, the most well-known being Fas and
tumor necrosis factor (TNF) receptors. Ligand binding to these
receptors activates a signaling cascade, including caspases, that
leads to apoptosis (57).
Over the past decade, most quinazolinone derivatives
have been shown to induce apoptosis through intrinsic pathways,
including mitochondrial and endoplasmic reticulum (ER) stress
pathways (Table I, Fig. 2). Regarding targeting the
intrinsic pathway, Liang et al (58) synthesized four novel series of
quinazolinone-based compounds by adding alkynyl functional groups
at the C8 position. Among them, compound 9b showed the highest
affinity for PI3Kγ and induced apoptosis in leukemic cells by
simultaneously modulating the PI3K-AKT and MAPK signaling pathways.
Similarly, Kim et al (59) modified substituents at the C-5/6
positions and optimized side-chain positioning within a hydrophobic
binding region to develop a selective PI3Kδ inhibitor. This
compound suppressed the PI3K pathway in SUDHL-5 and MOLT-4 cell
models, reduced phosphorylation of AKT, S6 and eukaryotic
translation initiation factor 4E-binding protein 1, and triggered
apoptosis in malignant cells.
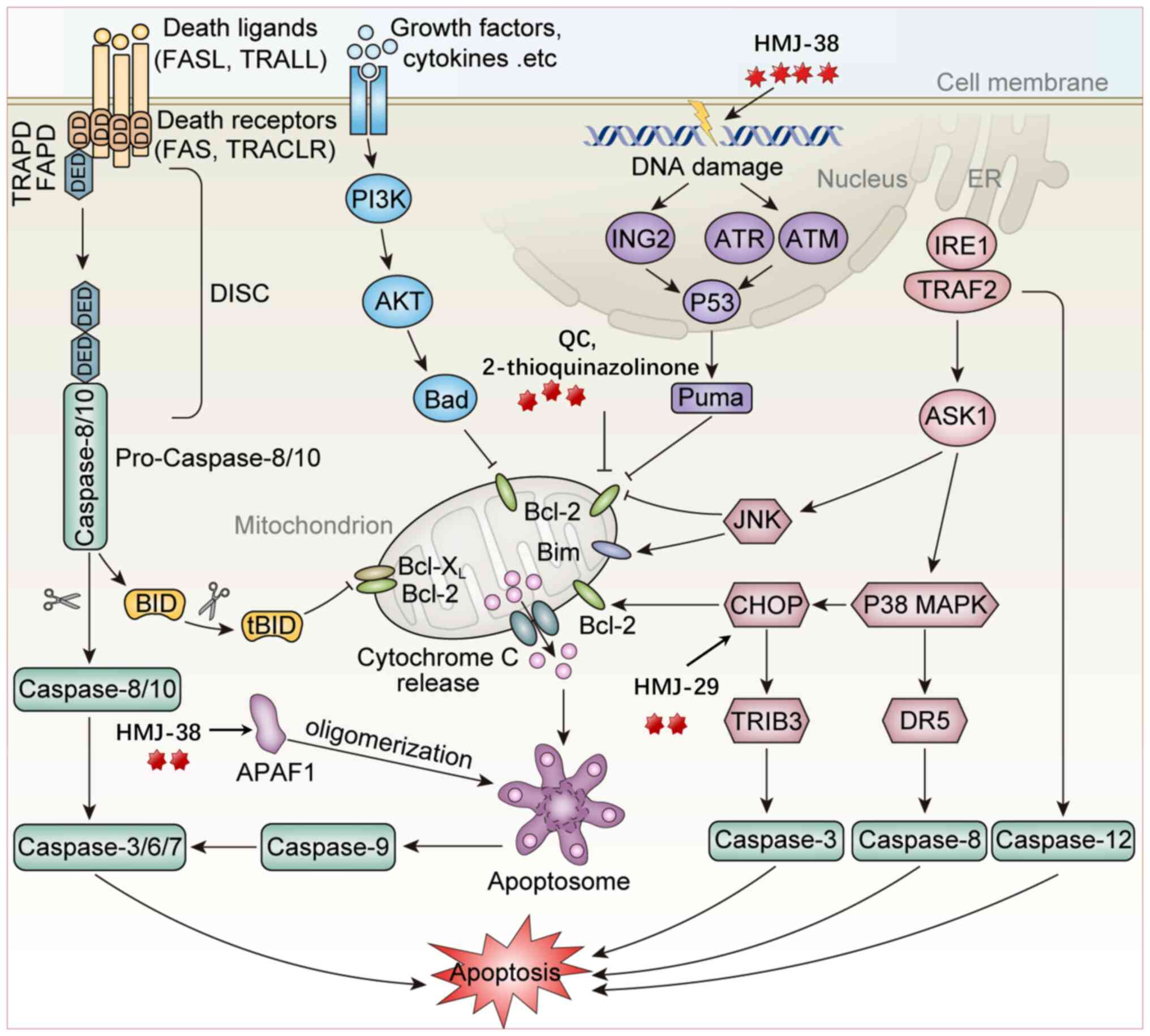 | Figure 2Mechanism of quazolinone compounds
inducing cell death through apoptosis. i) In the extrinsic pathway,
the death ligand-receptor interaction triggers apoptotic signaling
and forms the DISC complex, activating the Caspase-8/-3 cascade,
leading to cell apoptosis (HMJ-38 induces apoptosis through the
Fas/Death receptor-Caspase-8 pathway and is regulated by p53/ATM
signaling). ii) In the intrinsic pathway, quinazolinones
downregulate Bcl-2/Bcl-xL and upregulate Bax/Bad, thereby promoting
cytochrome c release, Apaf-1 apoptosome formation and
Caspase-9/Caspase-3 cascade activation (QC suppresses Bcl-2,
promotes the translocation of Bax into the mitochondria, releases
Cyt C and activates Caspase-9. MITC-12 upregulates the Bax/Bcl-2
ratio and increases Caspase-3 expression. The analogs synthesized
by Madbouly et al (61)
induce apoptosis by promoting Caspase-3 and PARP-1 cleavage
(compound 5k increases Bad and Bax, reduces Bcl-2 and Bcl-xL and
enhances pro-apoptotic signals). iii) In the DNA damage pathway,
ATM/ATR/p53 signaling induces Puma expression to enhance
mitochondrial apoptosis (one of the mechanisms of HMJ-38 is to
induce the p53/ATM signaling pathway through DNA damage, thereby
activating the death receptor pathway Caspase-8). iv) ER stress
activates the IRE1-TRAF2-ASK1 signaling pathway, which in turn
activates the JNK and p38 MAPK pathways, induces CHOP expression,
promotes the upregulation of TRIB3 and DR5 and subsequently
activates Caspase-3, Caspase-8 and Caspase-12, thereby inducing
apoptosis (MJ-29 activates key markers of ER stress by increasing
the protein levels of calpain 1 and CHOP). ER, endoplasmic
reticulum; DISC, death-inducing signaling complex; DED, death
effector domain; BID, BH3 interacting domain death agonist; APAF1,
apoptotic peptidase activating factor 1; ING2, inhibitor of growth
family member 2; Puma, P53 upregulated modulator of apoptosis; Bim,
Bcl-2 interacting mediator of cell death; Bad, BCL2-associated
agonist of cell death; JNK, c-Jun N-terminal kinase; IRE1,
immunoglobulin-regulated enhancer 1; TRAF2, TNF receptor associated
factor 2; ASK1, apoptosis signal-regulating kinase 1; CHOP, C/EBP
homologous protein; TRIB3, Tribbles pseudokinase 3; DR5, death
receptor 5; ATM, Ataxia-telangiectasia mutated proteins; ATR,
Ataxia telangiectasia mutated and Rad3 related; caspase, cysteinyl
aspartate specific proteinase. |
 | Table IApoptosis pathway changes following
quinazolinone derivative treatment. |
Table I
Apoptosis pathway changes following
quinazolinone derivative treatment.
| Authors, year | Cancer type | Chemical name | Affected molecule
or pathway | (Refs.) |
|---|
| Liang et al,
2023 | Leukemia |
3-(1-((9H-purin-6-yl)amino)ethyl)-8-((4-methoxyphenyl)
ethynyl)-2-phenylisoquinolin-1(2H)-one | PI3K-AKT↓
p-p38, p-ERK↑ | (58) |
| Kim et al,
2021 | Hematologic
malignancies |
(S)-2-(((7H-Purin-6-yl)amino)
(cyclopropyl)methyl)-5-fluoro-3-phenylquinazolin-4(3H)-one | p-AKT,
p-S6↓
4EBP1↓ | (59) |
| Wani et al,
2016 | Colon cancer |
3-(3-((E)-3-(4-hydroxy-3-methoxyphenyl)-2propenoyl)phenyl)-2-methyl-3,4-dihydro-4-quinazolinone | PI3K/AKT/mTOR
signaling pathway; Bcl-2/Bax↓, Caspase-9,3↑, PARP-1↑ | (60) |
| Madbouly et
al, 2022 | Epidermoid
carcinoma, fibrosarcoma |
(E)-2-((4-Cinnamoylphenoxy)methyl)-3-(4-fluorophenyl)-quinazolin-4(3H)-one |
Caspase-3↑
PARP↓ | (61) |
| Xie et al,
2023 | Glioma |
6,8-Dibromo-2-thio-3-(4-rhamnosylphenylmethyl)-2,3-dihydroquinazolin-4-one |
Caspase-3↑
Bax/Bcl-2↑ | (62) |
| Qiu et al,
2020 | Lung cancer |
2-(Benzylthio)-6-(3-(dimethylamino)propoxy)-3-(furan-2ylmethyl)quinazolin-4(3H)-one | Bad, Bax↑
Bcl-2, Bcl-xl↓ | (63) |
| El-shafey et
al, 2020 | Breast cancer |
3-(4-(((3-Benzyl-6-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)benzoic
acid | ROS accumulation,
Bax/Bcl-2↑
Caspase-6,7,9↑ | (64) |
| Hour et al,
2023 and Chiang et al, 2013 | Pancreatic
cancer |
2-(3'-Methoxyphenyl)-6-pyrrolidinyl-4-quinazolinone | p53/ATM signaling
pathway; Fas/CD95 and death receptor-modulated extrinsic caspase
signaling | (65,66) |
| Lu et al,
2012 | Murine
leukemia |
6-Pyrrolidinyl-2-(2-hydroxyphenyl)-4-quinazolinone | Calpain 1, CHOP and
p-eIF2α↑ | (67) |
Wani et al (60) developed a new
quinazolinonechalcone derivative,
3-(3-((E)-3-(4-hydroxy-3-methoxyphenyl)-2-propenoyl)phenyl)-2-methyl-3,4
dihydro-4-quinazolinone (QC). QC suppressed the mitochondrial
anti-apoptotic protein Bcl-2. At the same time, it promoted the
translocation of Bax from the cytoplasm to the mitochondria. Bax
forms oligomers on the mitochondria, leading to an increase in
mitochondrial membrane permeability. This results in the release of
cytochrome C from the mitochondria into the cytoplasm. Cytochrome C
binds to apoptotic protease activating factor 1 (Apaf-1) to form an
apoptosome, which recruits and activates caspase-9. Activated
caspase-9 then further activates downstream caspases, including
caspase-3, caspase-6 and caspase-7, ultimately triggering cell
apoptosis (60). Similarly,
Madbouly et al (61)
synthesized a related quinazolinone-chalcone compound
[(E)-2-((4-acetylphenoxy) methyl)-3-phenylquinazolin-4(3H)-one],
which induced apoptosis by promoting caspase-3 and poly(ADP-ribose)
polymerase 1 (PARP-1) cleavage in A431 carcinoma cells. Some
quinazolinone derivatives regulate apoptosis by modulating Bcl-2
family proteins. Xie et al (62) synthesized a series of MITC
[4-((α-L-rhamnose oxy)benzyl)] quinazolinone derivatives. Among
them, MITC-12 induced apoptosis in U251 cells by increasing
caspase-3 expression and elevating the Bax/Bcl-2 ratio. Qiu et
al (63) introduced alkoxy
substituents and showed that compound 5k induced
concentration-dependent apoptosis in HepG2 cells by increasing
pro-apoptotic proteins Bad and Bax and decreasing anti-apoptotic
proteins Bcl-2 and Bcl-xl. El-Shafey et al (64) demonstrated that novel compounds
with a 2-thioquinazolinone scaffold triggered mitochondrial
apoptosis by increasing ROS accumulation, elevating the Bax/Bcl-2
ratio and activating caspases 6, 7 and 9.
A smaller group of quinazolinone derivatives induce
apoptosis via the extrinsic pathway. HMJ-38 [2-(3'-methoxy
phenyl)-6-pyrrolidinyl-4-quinazolinone], a quinazolinone
derivative, inhibits tubulin polymerization. Hour et al
(65) showed that HMJ-38 induces
both autophagy and apoptosis in gemcitabine-resistant pancreatic
cancer cells, with the recruitment of pro-caspase-9 to the
apoptosome by the Apaf-1 complex activating caspase-9
auto-catalytically, thereby enhancing apoptosis through subsequent
activation of caspase-3, caspase-6 and caspase-7. Chiang et
al (66) reported that
HMJ-38 induces apoptosis in human umbilical vein endothelial cells
through ROS generation and activation of the Fas/death
receptor-mediated caspase-8 pathway, regulated by p53/ATM
signaling. In WEHI-3 cells, MJ-29 activates key markers of ER
stress by increasing the protein levels of calpain 1 and C/EBP
homologous protein (CHOP), playing an important role in regulating
cell apoptosis (67).
Ferroptosis
Ferroptosis is a regulated form of cell death first
identified in cancer cells with oncogenic Ras mutations. It is
characterized by iron-dependent lipid peroxidation (68,69). Unlike classical forms of cell
death such as apoptosis and necrosis, ferroptosis involves
intracellular iron accumulation and ROS generation. These processes
lead to polyunsaturated fatty acid peroxidation in cell membranes,
resulting in membrane damage and cell death (70). Ferroptosis is regulated by
multiple metabolic pathways, including those involved in redox
balance, iron metabolism, mitochondrial function, and amino acid,
lipid and carbohydrate metabolism. Several disease-related
signaling pathways also contribute to its regulation (71). The three primary pathways that
control ferroptosis are the system Xc−/glutathione (GSH)
peroxidase 4 (GPX4) axis, lipid metabolism and iron metabolism
(72). In addition to small
molecules and drugs, external stressors such as heat, cold, hypoxia
and radiation can also induce ferroptosis (73) (Table II, Fig. 3).
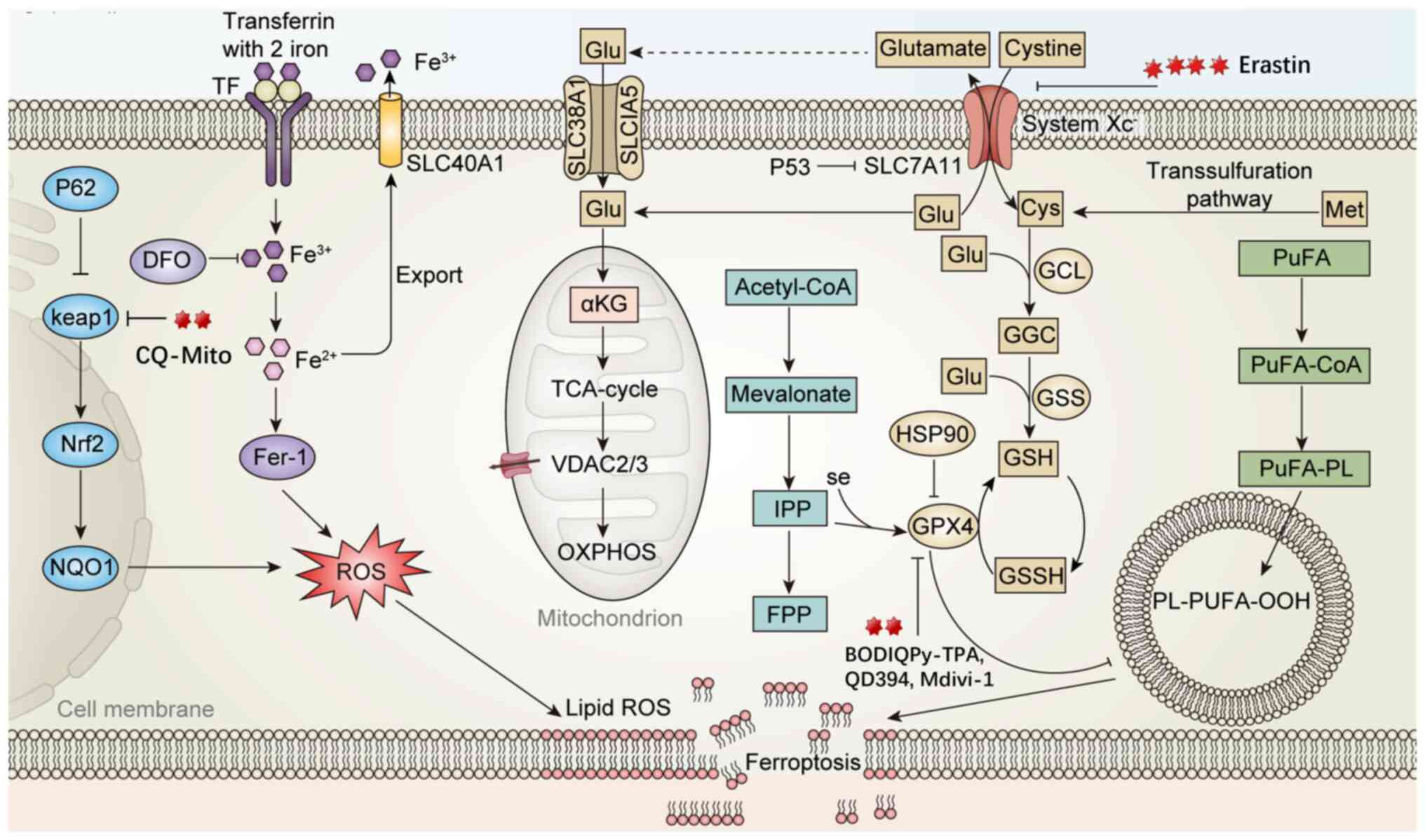 | Figure 3Mechanism of quazolinone compounds
inducing cell death through ferroptosis. i) System Xc−
is responsible for importing extracellular cysteine into cells for
GSH synthesis, maintaining GPX4 activity to clear lipid peroxides
PL-PUFA-OOH. When cysteine uptake is blocked or GSH is depleted,
GPX4 becomes inactive, leading to the accumulation of lipid
peroxides and triggering ferroptosis (Erastin inhibits SLC7A11,
prevents Cys entry, reduces GSH synthesis, weakens GPX4 activity
and enhances lipid peroxidation, thereby inducing ferroptosis;
BODIQPy-TPA directly acts on the GPX4/GSH axis, inhibits GPX4,
depletes GSH, blocks lipid peroxide clearance and promotes
ferroptosis). ii) After transferrin-bound Fe3+ enters
the cell, it is reduced to Fe2+, promoting ROS
generation and inducing lipid peroxidation. The Keap1-Nrf2
signaling axis regulates the downstream antioxidant gene NQO1,
which helps buffer ferroptosis stress to a certain extent (CQ-Mito
generates ROS under light, inhibits GPX4, reduces Keap1 and
activates the Nrf2 antioxidant pathway). iii) PUFA is acylated to
form PuFA-CoA, and under the influence of iron and ROS, lipid
peroxides PL-PUFA-OOH are generated, which are direct molecular
effectors of ferroptosis. ROS, reactive oxygen species; GSH,
glutathione; TF, transcription factors; GCL, glutamate cysteine
ligase; GGC, gamma-glutamylcysteine; GSS, glutathione synthetase;
SLC40A1, solute carrier family 40 member 1; system Xc−,
cystine/glutamate antiporter system; αKG, α-ketoglutaric acid;
TCA-cycle, tricarboxylic acid cycle; VDAC, voltage-dependent anion
channel; OXPHOS, oxidative phosphorylation; PuFA, polyunsaturated
fatty acid; CoA, coenzyme A; Cys, cysteine; Met, methionine;
PuFA-PL, polyunsaturated-fatty-acid-containing phospholipids; IPP,
Isopentenyl pyrophosphate; FPP, farnesyl pyrophosphate; HSP90, heat
shock protein 90; GPX4, glutathione peroxidase 4; DFO,
deferoxamine; Keap1, Kelch-like ECH-associated protein 1; Nrf2,
nuclear factor erythroid 2-related factor 2; NQO1, quinone
oxidoreductase. |
| Table IIFerroptosis pathway changes following
quinazolinone derivative treatment. |
Table II
Ferroptosis pathway changes following
quinazolinone derivative treatment.
| Authors, year | Cancer type | Chemical name | Affected molecule
or pathway | (Refs.) |
|---|
| Yang et al,
2020 | Melanoma |
2-(1-(4-(2-(4-chlorophenoxy)acetyl)
piperazin-1-yl)ethyl)-3-(2-ethoxyphenyl) quinazolin-4(3H)-one | FOXM1-Nedd4-VDAC2/3
negative feedback loop | (76) |
| Sun et al,
2020 | Gastric cancer |
2-(1-(4-(2-(4-chlorophenoxy)acetyl)
piperazin-1-yl)ethyl)-3-(2-ethoxyphenyl) quinazolin-4(3H)-one | System
Xc−↓
Block of the uptake of cystine, resulting in the accumulation of
ROS | (77) |
| Li et al,
2021 | Endometrial
cancer |
2-(1-(4-(2-(4-chlorophenoxy)acetyl)
piperazin-1-yl)ethyl)-3-(2-ethoxyphenyl) quinazolin-4(3H)-one | ROS↑, FPN↓ | (78) |
| Huang et al,
2018 | Lung cancer |
2-(1-(4-(2-(4-chlorophenoxy)acetyl)
piperazin-1-yl)ethyl)-3-(2-ethoxyphenyl) quinazolin-4(3H)-one | ROS↑, p53↑, p-p53↑,
p21, Bax↑, SLC7A11↓ | (79) |
| Zhao et al,
2024 | Liver cancer |
N-(2-(4-(((2-(7-(diethylamino)-2-oxo-2H-chromen-3-yl)-4-oxo-3,4-dihydroquinazolin-6-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)ethyl)-4-methylbenzenesulfonamide | Gpx4↓, Nrf2↑,
Keap1↓ | (83) |
| Xing et al,
2024 | Melanoma |
(E)-2-(4-(diphenylamino)styryl)-6-fluoro-6-methyl-5λ4,6λ4-pyrido(1',2':1,5)
(1,3,2)
diazaborolo(4,3-b)quinazolin-8(6H)-one | GPX4-GSH-cysteine
axis | (84) |
| Huang et al,
2023 | Liver damage |
3-(2,4-Dichloro-5-methoxyphenyl)-2,
3-dihydro-2-thioxo-4(1H)-quinazolinone | p-Drp1↓, MDA↓, GSH,
GPX4↑ | (85) |
| Nakamura et
al, 2023 | Liver disease |
N-(4-(2-methyl-4-oxoquinazolin-3(4H)-yl)
phenyl)-2-(3,4,5-trimethoxyphenyl)acetamide | FSP1↑, combination
with iron death inducers promotes apoptosis | (87) |
| Hu et al,
2020 | Pancreatic
cancer |
6-(4-(4-Methylpiperazin-1-yl)phenylamino)
quinazoline-5,8-dione | ROS↑,
GSH/GSSG↓
lipid peroxidation↑ | (90) |
Erastin, a piperazinyl-quinazolinone compound, is a
well-known ferroptosis inducer (74). It inhibits the system
Xc−, leading to reduced intracellular GSH levels and
impaired GPX4 function. This results in increased lipid
peroxidation, mitochondrial damage and other ferroptotic features
(75). Erastin remains in the
preclinical stage and is not yet marketed. Although it induces
ferroptosis in various tumor cells, its therapeutic effect is
limited by a feedback mechanism involving degradation of
voltage-dependent anion channel (VDAC)2 and VDAC3. In melanoma
cells, Erastin activates forkhead box M1, which transcriptionally
upregulates NEDD4 E3 ubiquitin protein ligase, leading to VDAC2/3
ubiquitination and proteasomal degradation. This reduces
intracellular ROS levels and modulates ferroptosis. Silencing Nedd4
restores VDAC2/3 levels and enhances cancer cell sensitivity to
Erastin (76). Furthermore,
Erastin induces mitochondrial dysfunction and ROS accumulation in
HGC-27 gastric cancer and endometrial stromal cells, thereby
promoting ferroptosis and suppressing malignancy (77,78). This process is also marked by
iron accumulation and decreased ferroportin expression. Huang et
al (79) found that Erastin
induces both ferroptosis and apoptosis in A549 lung cancer cells by
increasing ROS and activating p53. Erastin may also enhance the
therapeutic effects of programmed cell death 1/programmed cell
death ligand 1 inhibitors by influencing tumor-associated
macrophage polarization (80).
In another study, aspirin induced ferroptosis in HepG2 and Huh7
cells, and its effect was enhanced by Erastin co-treatment
(81,82).
Organelle-targeted photosensitizers (PSs) are
promising in enhancing photodynamic therapy, as they generate ROS
under light exposure in the presence of oxygen. Zhao et al
(83) developed a series of PSs
based on the coumarin-quinazolinone (CQ) structure. The
mitochondria-targeted CQ-Mito compound inhibits GPX4 upon light
exposure, induces lipid peroxidation and activates Nrf2 while
reducing Keap1 expression, which normally deubiquitinates Nrf2.
Treatment with the ferroptosis inhibitor Fer-1 restores
ferroptosis-related protein expression, confirming the
light-induced ferroptotic effect of CQ-Mito (83). Additionally,
BODIQPy-triphenylamine (BODIQPy-TPA), a lipophilic
quinazolinone-based probe, can directly induce lipid peroxidation
upon light exposure. In B16 and HepG2 cells, BODIQPy-TPA triggers
ferroptosis by inhibiting GPX4, depleting GSH, and promoting
cystine starvation, thereby activating the GPX4-GSH-cysteine axis
(84). It also emits strong
near-infrared fluorescence in live cells, making it useful for
real-time imaging of the ferroptosis process.
3-(2,4-Dichloro-5-methoxyphenyl)-2,3-dihydro-2-thioxo-4(1H)-quinazolinone
(Mdivi-1) is a quinazolinone derivative and a selective inhibitor
of dynamin-related protein 1 (Drp1). In cold stress-induced liver
injury, Mdivi-1 reduces malondialdehyde levels, increases GSH and
GPX4 levels, inhibits mitochondrial fission and effectively
mitigates ferroptosis (85). It
also decreases ferrous ion and lipid peroxide levels. In models of
PM2.5 exposure, Mdivi-1 significantly reduces the expression of
acyl-CoA synthetase long chain family member 4, ferritin, ferritin
heavy chain 1 and ferritin light chain 1, while increasing GPX4
protein levels, thereby inhibiting ferroptosis (86).
Ferroptosis suppressor protein 1 (FSP1), in
conjunction with mitochondrial-derived ubiquinone, exogenous
vitamin K and NAD(P)H/H+ as electron donors, has been
identified as a second ferroptosis-suppressing system (87). A recent study indicated that
3-phenyl-quinazolinone, an FSP1 inhibitor, promotes FSP1
aggregation in tumors. This aggregation synergizes with ferroptosis
inducers to enhance iron-dependent cell death and suppress tumor
progression in vivo (88).
In the search for new therapies for uveal melanoma,
coumarin-quinazolinone-phenylboronic acid has been developed as a
prodrug to induce ferroptosis by targeting elevated ROS and
tyrosinase levels in melanoma cells (89). QD394, a quinazolinone-dione
compound, promotes lipid peroxidation and destabilizes GPX4
protein, thereby triggering GPX4-mediated ferroptosis (90).
Autophagy
Autophagy is a conserved cellular process that plays
an essential role in maintaining homeostasis by degrading damaged
or unnecessary organelles and proteins (91). It begins with the formation of
autophagosomes, which enclose the targeted materials and deliver
them to lysosomes for degradation. The term 'autophagy-dependent
cell death (ADCD)' was introduced in 2018 by the Nomenclature
Committee on Cell Death (92).
ADCD is a regulated form of cell death characterized by the
activation of autophagy markers, such as enhanced degradation of
autophagosomal substrates and lipidation of
microtubule-associated-proteinlight-chain-3 (LC3) (93). During autophagy, LC3 is converted
from the cytoplasmic form LC3-I to the membrane-associated form
LC3-II, and the LC3-II/LC3-I ratio is widely used as a marker of
autophagy activity (94).
Autophagy and apoptosis may act interchangeably under certain
conditions. Excessive autophagy, referred to as 'autophagic death',
can lead to cell death resembling apoptosis. These two processes
are regulated by overlapping signaling pathways, including mTOR,
Bcl-2 and Beclin-1 (95).
Quinazolinone compounds modulate the initiation and progression of
cell autophagy (Table III,
Fig. 4).
![Mechanism of action of Quinazolinone
compounds in pyroptosis, autophagy and cellular senescence
pathways. i) In the senescence pathway, telomere shortening and
limited telomerase function activate the p53/p21/p16 pathway,
causing G1 phase arrest and cellular senescence
3-(2-(hydroxymethyl) phenyl)-2-methylquinazolin-4(3H)-ones
upregulate the expression of TRF1, POT1 and p53/p21/p16, and
inhibit telomerase, inducing cells to enter a senescence
phenotype]. ii) In the pyroptosis pathway, NLRP3 assembles with ASC
and procaspase-1 to form the inflammasome, activating caspase-1,
which mediates the maturation of IL-1β and IL-18, and induces
pyroptosis through GSDMD, forming membrane pores. Quinazolinone
derivatives can inhibit this process by blocking NLRP3 inflammasome
activation and the release of inflammatory factors (Mdivi-1
inhibits NLRP3 inflammasome activation, reduces the activation of
NLRP3, ASC, Caspase-1 and the level of GSDMD-NT, and decreases the
release of IL-1β and IL-18). iii) In the autophagy pathway, ATG
family proteins mediate phagophore nucleation and extension. LC3 is
modified by ATG4 and ATG7, transforming into LC3-II, which promotes
autophagosome formation and fusion with lysosomes for substrate
degradation (DQQ and HF promote LC3-II generation, enhance
ATG5-ATG12 complex formation and promote autophagy. MJ-33 initiates
autophagy by activating ATG proteins during vesicle nucleation but
reduces the LC3/LC3-II ratio and increases p62 levels, inhibiting
autophagy). NLRP3, NOD-like receptor thermal protein domain
associated protein 3; ASC, apoptosis-associated speck-like protein
containing a CARD; GSDMD, gasdermin D; ATG, autophagy related gene;
LC3, microtubule-associated-proteinlight-chain-3; NIX, NIP3-like
protein X; TIN2, Terf1 interacting nuclear factor 2; RAP1,
Ras-proximate-1; TRF1, telomeric repeat binding factor 2; POT1,
protection of telomeres 1.](/article_images/ijmm/56/6/ijmm-56-06-05646-g03.jpg) | Figure 4Mechanism of action of Quinazolinone
compounds in pyroptosis, autophagy and cellular senescence
pathways. i) In the senescence pathway, telomere shortening and
limited telomerase function activate the p53/p21/p16 pathway,
causing G1 phase arrest and cellular senescence
3-(2-(hydroxymethyl) phenyl)-2-methylquinazolin-4(3H)-ones
upregulate the expression of TRF1, POT1 and p53/p21/p16, and
inhibit telomerase, inducing cells to enter a senescence
phenotype]. ii) In the pyroptosis pathway, NLRP3 assembles with ASC
and procaspase-1 to form the inflammasome, activating caspase-1,
which mediates the maturation of IL-1β and IL-18, and induces
pyroptosis through GSDMD, forming membrane pores. Quinazolinone
derivatives can inhibit this process by blocking NLRP3 inflammasome
activation and the release of inflammatory factors (Mdivi-1
inhibits NLRP3 inflammasome activation, reduces the activation of
NLRP3, ASC, Caspase-1 and the level of GSDMD-NT, and decreases the
release of IL-1β and IL-18). iii) In the autophagy pathway, ATG
family proteins mediate phagophore nucleation and extension. LC3 is
modified by ATG4 and ATG7, transforming into LC3-II, which promotes
autophagosome formation and fusion with lysosomes for substrate
degradation (DQQ and HF promote LC3-II generation, enhance
ATG5-ATG12 complex formation and promote autophagy. MJ-33 initiates
autophagy by activating ATG proteins during vesicle nucleation but
reduces the LC3/LC3-II ratio and increases p62 levels, inhibiting
autophagy). NLRP3, NOD-like receptor thermal protein domain
associated protein 3; ASC, apoptosis-associated speck-like protein
containing a CARD; GSDMD, gasdermin D; ATG, autophagy related gene;
LC3, microtubule-associated-proteinlight-chain-3; NIX, NIP3-like
protein X; TIN2, Terf1 interacting nuclear factor 2; RAP1,
Ras-proximate-1; TRF1, telomeric repeat binding factor 2; POT1,
protection of telomeres 1. |
| Table IIIAutophagy pathway changes following
quinazolinone derivative treatment. |
Table III
Autophagy pathway changes following
quinazolinone derivative treatment.
| Authors, year | Cancer type | Chemical name | Affected molecule
or pathway | (Refs.) |
|---|
| Kumar et al,
2014 | Leukemia |
2,3-Dihydro-2-(quinoline-5-yl)
quinazolin-4(1H)-one | AVOs↑, beclin1↑,
ATG7↑, ATG5↑, LC3-II↑ | (96) |
| Sharma et
al, 2022 | Lung cancer |
7,8,9,10-Tetrahydroazepino (2,1-b)
quinazolin-12 (6H)-one | AVOs↑ | (97) |
| Xia et al,
2017 | Breast cancer |
7-bromo-6-chloro-3-(3-(3-hydroxypiperidin-2-yl)propyl)quinazolin-4(3H)-one | LC3↑, ATG5-ATG12
complex↑, SQSTM1↓, STMN1↓, p53↓ | (98) |
| Ha et al,
2021 | Colorectal
cancer |
2-(3-Ethoxyphenyl)-6-pyrrolidinylquinazolinone | ATG5, ATG7, ATG12,
ATG16↑
p62↑, LC3/LC3-II↓ | (99) |
| ElZahabi et
al, 2021 | Breast cancer |
3-Amino-6,8-dibromo-2-methylquinazolin-4(3H)-one | EGFR↓, PI3K, AKT,
mTOR↓ | (100) |
Kumar et al (96) investigated a novel quinazolinone
derivative, 2,3-dihydro-2-(quinoline-5-yl)quinazolin-4(1H)-one
(DQQ). DQQ induced the formation of acidic vacuolar organelles
(AVOs) in MOLT-4 cells and significantly increased the expression
of autophagy-related proteins, including LC3-II, autophagy-related
7 (ATG7), ATG5 and Beclin-1 (96). Another study showed that a fused
quinazolinone compound [compound 6, based on
7,8,9,10-tetrahydroazepino (2,1-b) quinazolin-12(6H)-one] increased
AVO formation, as detected by acridine orange staining, indicating
enhanced autophagic flux (97).
Halofuginone (HF), a quinazolinone alkaloid derived from Dichroa
febrifuga, promoted autophagy while inhibiting stathmin 1 and
p53 expression and activity. HF increased LC3 expression, promoted
ATG5-ATG12 complex formation and reduced sequestosome 1 expression
in MCF-7 cells (98).
Certain quinazolinone derivatives can also inhibit
autophagy. For instance, MJ-33 initiates autophagy at the vesicle
nucleation stage by activating ATG proteins but decreases the
LC3/LC3-II ratio and increases p62 levels, suggesting suppression
of autophagic flux. In HT-29/5FUR cells, this suppression enhances
MJ-33-induced apoptosis (99).
ElZahabi et al (100)
designed a series of quinazolin-4-one derivatives, among which
compound 7 reduced autophagy, leading to increased apoptosis and
cancer cell death.
Senescence
Cellular senescence is generally an irreversible
arrest of cell proliferation in damaged normal cells that have
exited the cell cycle. It is associated with widespread
macromolecular alterations and a secretory phenotype linked to
chronic inflammation (101).
Senescent cells show characteristic morphological changes,
including flattened cell shape, vacuolization, cytoplasmic
granularity and organelle abnormalities (102). Key features of senescence
include persistent cell cycle arrest, altered transcriptional
activity, a pro-inflammatory secretome, accumulated macromolecular
damage and metabolic dysregulation (103). Increasing evidence indicates a
complex relationship between aging and cancer (Table IV, Fig. 4). Initially, senescence functions
as a protective mechanism by limiting abnormal cell proliferation
and reducing cancer risk. However, with aging, immune decline and
accumulated DNA damage may contribute to cancer development
(104,105).
| Table IVSenescence, necrosis and pyroptosis
pathway changes following quinazolinone derivative treatment. |
Table IV
Senescence, necrosis and pyroptosis
pathway changes following quinazolinone derivative treatment.
| Authors, year | Cancer type | Chemical name | Affected molecule
or pathway | (Refs.) |
|---|
| Kamal et al,
2013 | Breast cancer,
non-small cell lung cancer, pancreatic cancer, colorectal
cancer | A series of
3-diarylethyne quinazolinone compounds | p53, p21,
p16↑
TRF1, POT1↑
SKP2, TRF2 and tankyrase protein levels↓ | (106) |
| Venkatesh et
al, 2015 | Cervical
cancer |
2-Methyl-3-(2-((4-phenyl-1H-1,2,3-Triazol-1-yl)methyl)phenyl)quinazolin-4(3H)-one | p53, p21↑
HDAC-1,2,3,4↓ | (107) |
| Shams et
al,2011 | Kidney damage |
4(3H)-Quinazolinone-2-propyl-2-phenylethyl
and 4(3H)quinazolinone-2-ethyl-2-phenylethyl | By metabolization
and production of active metabolites and free oxygen radicals, cell
membrane and organelles such as mitochondria and peroxisome are
damaged and necrosis appears in renal tubule cells | (112) |
| Piamsiri et
al, 2024 | Acute myocardial
infarction |
3-(2,4-Dichloro-5-methoxyphenyl)-2,3-dihydro-2-thioxo-4(1H)-quinazolinone | NLRP3,
GSDMD-NT↑ | (113) |
| Li et al,
2021 | Atopic
dermatitis |
3-(2,4-Dichloro-5-methoxyphenyl)-2,3-dihydro-2-thioxo-4(1H)-quinazolinone | NLRP3,
ASC↓
Cleavage of Caspase-1↓
GSDMD-NT↓
IL-1β, IL-18↓ | (114) |
| Liu et al,
2020 | Acute kidney
injury |
3-(2,4-Dichloro-5-methoxyphenyl)-2,
3-dihydro-2-thioxo-4(1H)-quinazolinone | NLRP3↓
IL-1β, IL-18↓ | (116) |
Kamal et al (106) developed a series of
3-diarylacetylene quinazolinone compounds that activate p53, p21,
p16, telomeric repeat binding factor 1 (TRF1) and protection of
telomeres 1 (POT1) proteins. These compounds exhibited significant
telomerase inhibitory activity, suggesting their potential as
senescence inducers. Similarly, certain 3-(2-(hydroxymethyl)
phenyl)-2-methylquinazolin-4(3H)-ones induced a senescence
phenotype in HeLa cells (107).
The application of quinazolinone and its derivatives in aging and
cancer therapy remains an area of active investigation.
Necrosis and pyroptosis
Necrosis is an abnormal form of cell or tissue death
caused by external factors such as trauma, hypoxia, infection or
injury. Receptor-interacting protein kinase 1 has been identified
as a key regulator of necroptosis, a programmed form of necrosis
activated by stimuli such as TNF, TNF superfamily member 10 (also
known as TRAIL), lipopolysaccharide, oxidative stress or DNA damage
(108). During necrosis,
membrane breakdown leads to the release of cytoplasmic contents
into the extracellular environment (109).
Pyroptosis is a regulated form of cell death
mediated by gasdermins and characterized by continuous cell
swelling, plasma membrane rupture and the release of intracellular
contents that trigger inflammatory and immune responses (110). It is initiated by inflammasome
activation in response to various triggers (111). Pyroptosis is involved in
cancer, neurodegenerative diseases and ischemia-reperfusion injury.
Research on the effects of quinazolinone derivatives in necrosis
and pyroptosis is still at an early stage and the specific
mechanisms remain elusive. The available literature is limited.
Quinazolinone compounds can induce cell death through necrosis and
pyroptosis (Table IV, Fig. 4).
In BALB/c mice, administration of two new
quinazolinone compounds, 4(3H)-quinazolinone-2-propyl-2-phenylethyl
and 4(3H)-quinazolinone-2-ethyl-2-phenylethyl, caused abnormal
kidney function. Quinazolinones can affect sulfhydryl groups,
disrupt protein structures, generate reactive metabolites and free
radicals, and damage organelles such as tubular membranes,
mitochondria and peroxisomes, ultimately leading to cell necrosis
(112).
Mdivi-1, a quinazolinone-derived compound, has been
shown to reduce necroptosis- and pyroptosis-related protein
expression, including NLR family pyrin domain containing 3 (NLRP3)
and gasdermin D N-terminus (GSDMD-NT), in cardiomyocytes of rats
with myocardial infarction, thereby improving cardiac function in
post-MI rats (113). Li et
al (114) were the first to
study the effect of Mdivi-1 on primary human keratinocytes in an
in vitro model of atopic dermatitis-related inflammation
induced by an inflammatory cocktail. Their results showed that
Mdivi-1 inhibited NLRP3 inflammasome activation and pyroptosis, as
indicated by reduced levels of NLRP3, apoptosis-associated
speck-like protein containing a CARD, cleaved caspase-1, GSDMD-NT,
and mature interleukins IL-1β and IL-18 in keratinocytes. In
cellular and animal models of septic acute kidney injury, Mdivi-1
suppressed NLRP3-driven pyroptosis and improved mitochondrial
function (115,116). Further research is needed to
fully understand the roles and mechanisms of quinazolinone and its
derivatives in necrosis and pyroptosis.
Application of quinazolinone derivatives in
cancer therapy
Quinazolinone and its derivatives have attracted
considerable attention in recent years for their ability to induce
cell death in various cancer types, representing a promising
therapeutic approach. As single agents, these compounds act through
multiple mechanisms, including apoptosis, autophagy and
ferroptosis. To enhance therapeutic efficacy and address challenges
such as drug resistance and toxicity, increasing efforts have
focused on combining quinazolinone derivatives with other
pharmacological agents. This strategy not only amplifies their
anti-tumor effects but also enables more targeted interventions,
potentially reducing adverse effects. The following section
discusses the clinical prospectssss of quinazolinone and its
derivatives in combination therapy, emphasizing their synergistic
effects when used with conventional chemotherapeutic agents and
molecular targeted therapies.
Combination with platinum-based
drugs
Studies have shown that co-treatment with cisplatin
and Mdivi-1 synergistically enhances apoptotic cell death in both
intrinsically and acquired cisplatin-resistant tumor cells,
including MDA-MB-231 breast cancer, H1299 non-small cell lung
cancer, glioblastoma, melanoma, cholangiocarcinoma and A2780cis
cisplatin-resistant ovarian cancer cells (117,118). In A2780cis cells, Mdivi-1
enhances cisplatin sensitivity through a Drp1-independent pathway.
The combination increases DNA damage, upregulates
phorbol-12-myristate-13-acetate-induced protein 1 and disrupts
mitochondrial function, ultimately triggering Bax- and
Bak-independent mitochondrial outer membrane permeabilization and
activating the mitochondrial apoptotic pathway (117). Tusskorn et al (118) demonstrated that Mdivi-1
increases the sensitivity of cholangiocarcinoma cells to cisplatin
by promoting oxidative stress and autophagosome formation, thereby
inducing cell death through mitochondrial pathways. Furthermore,
Mdivi-1 protects against cisplatin-induced hair cell death in
zebrafish by modulating mitochondrial dynamics, suggesting its
potential for reducing ototoxicity. Further studies in mammalian
models are required to clarify the underlying protective mechanisms
(119).
Combination with 5-fluorouracil
(5-FU)
MJ-33, a quinazolinone derivative, exhibits
anti-tumor activity by reducing the viability of 5-FU-resistant
HT-29 colon cancer cells. It induces apoptosis and autophagy
through the AKT/mTOR signaling pathway, offering potential
therapeutic value in 5-FU-resistant colorectal cancer (CRC)
(76).
Lai et al (120) reported that
2-(3-fluorophenyl)-6-morpholinoquinazolin-4(3H)-one induces mitotic
arrest, subsequently leading to apoptosis. This compound also
synergizes with 5-FU to enhance cytotoxic effects against oral
squamous cell carcinoma (OSCC).
Combination with gemcitabine
HMJ-38 promotes both autophagy and apoptosis in
MIA-RG100 pancreatic cancer cells and demonstrates cytotoxic
effects against gemcitabine-resistant cells. The mechanism involves
activation of the EGFR-AKT-mTOR signaling pathway, which induces
autophagy, and the mitochondrial pathway, which facilitates
apoptosis (65). These findings
provide new perspectives for overcoming gemcitabine resistance in
pancreatic cancer. Another quinazolinone derivative, QD232,
suppresses the growth of gemcitabine-resistant MIA PaCa-2 cells by
inhibiting STAT3 and Src phosphorylation, offering a potential
strategy for treating drug-resistant pancreatic cancer associated
with STAT3 activation (121).
Combination with temozolomide
Targeting the catalytic domain of PARP-1, a series
of quinazolin-2,4(1H,3H)-dione derivatives have been synthesized as
PARP-1 inhibitors. Zhou et al (122) developed novel compounds
containing a 3-amino-pyrrolidine group, which enhanced the
cytotoxic effects of temozolomide in the MX-1 xenograft breast
cancer model. Similarly, 1-substituted benzyl
quinazoline-2,4(1H,3H)-dione derivatives demonstrated comparable
efficacy (123). Another study
confirmed that the 2-propionyl-3H-quinazoline-4-one scaffold acts
as a novel PARP-1 inhibitor, exhibiting synergistic effects with
temozolomide in MX1 cells (124).
Combination with paclitaxel
Ucleotide binding oligomerization domain containing
1 (NOD1) and NOD2 receptors, which contain nucleotide-binding
oligomerization domains, are emerging as potential immune
checkpoints. In a study by Ma et al (125), a quinazolinone derivative
[6-(3-chlorophenyl)-3-(2-(3,3-difluoropiperidin-1-yl)-2-oxoethyl)-4-oxo-N-(3-(4-(trifluoromethyl)phenoxy)propyl)-3,4-dihydro
quinazoline-7-carboxamide] was designed as a dual antagonist of
NOD1/2. By inhibiting NOD1/2-mediated NF-κB and MAPK signaling
pathways, this compound enhanced the response of B16
melanoma-bearing mice to paclitaxel therapy (125).
Quinazolinone derivatives approved by the
FDA or under clinical investigation
Idelalisib
Idelalisib (Zydelig™; CAL-101; Gilead Sciences),
with the chemical name
5-fluoro-3-phenyl-2-((1S)-1-(9H-purin-6-yl-amino)propyl)quinazolin-4(3H)-one,
has the molecular formula
C22H18FN7O and functions as a
lipid kinase inhibitor, specifically targeting the p110δ isoform of
class I phosphatidylinositol-3 kinase (PI3Kδ) (126). Idelalisib inhibits PI3Kδ by
competitively binding to the ATP-binding site of the p110δ subunit.
This inhibition disrupts key PI3Kδ signaling functions, including
chemokine secretion, cell migration and receptor-driven kinase
phosphorylation, ultimately reducing cell survival and promoting
apoptosis (127,128).
Idelalisib has shown clinical efficacy in indolent
B-cell non-Hodgkin lymphoma and was approved by the FDA in 2014 for
monotherapy in patients with chronic lymphocytic leukemia (CLL),
follicular lymphoma and small lymphocytic lymphoma. It is also
approved in combination with rituximab for the treatment of CLL
(129,130). In patients with previously
treated indolent non-Hodgkin lymphoma, idelalisib as monotherapy
achieved an objective response rate (ORR) of 57%, with a median
duration of response (mDOR) of 12.5 months and a median
progression-free survival (PFS) of 20.3 months (131,132). When combined with rituximab,
idelalisib further improved overall response rates and 12-month
overall survival (OS) in patients with CLL (133).
Despite its clinical benefits, idelalisib carries a
black box warning from the FDA due to the risk of severe or fatal
adverse events, including hepatotoxicity, diarrhea, colitis,
pneumonia and intestinal perforation (134). Patients who developed colitis
or transaminitis after idelalisib treatment exhibited elevated
plasma chemokine levels and reduced T-regulatory cell (Treg)
populations, suggesting that Treg depletion may contribute to the
adverse effects of p110δ inhibition by releasing the suppression of
cytotoxic T cells (135). In a
Phase III clinical trial, the combination of idelalisib and
rituximab improved PFS in patients with relapsed/refractory CLL
from 11.1 to 20.8 months; however, long-term follow-up revealed a
5-year overall survival rate of only 40%, indicating disease
progression in certain patients and suggesting the development of
resistance (136,137). In CLL, resistance to idelalisib
is closely associated with the activation of insulin-like growth
factor 1 receptor (IGF1R). Specifically, the upregulation of IGF1R
enhances the MAPK signaling pathway, bypassing PI3Kδ inhibition and
thereby promoting tumor cell survival (138). To overcome Idelalisib
resistance, researchers have explored various combination therapy
strategies. For instance, idelalisib combined with bortezomib
blocks drug resistance properties of Epstein-Barr virus-related
B-cell origin cancer cells via regulation of NF-κB (139). Nevertheless, further
investigation into the molecular mechanisms of resistance is
necessary to develop more precise therapeutic strategies.
Ispinesib
Ispinesib, a quinazolinone-derived compound, is a
selective inhibitor of kinesin spindle protein (KSP) and functions
as an allosteric modulator of KSP motor ATPase activity. By
inhibiting KSP, ispinesib disrupts mitotic spindle formation,
leading to cell cycle arrest and apoptosis (140). Developed by Cytokinetics,
ispinesib entered clinical trials in 2004 for multiple indications,
becoming the first potent and selective KSP inhibitor to undergo
clinical testing in human diseases (20). According to data from the
National Institutes of Health, ispinesib has been evaluated as
monotherapy in 13 Phase I/II clinical trials for various cancer
types, including relapsed renal cell cancer (RCC), breast cancer,
recurrent or metastatic head and neck squamous cell carcinoma
(HNSCC), ovarian cancer (OC), hepatocellular carcinoma (HCC) and
CRC, with the best responses observed in breast cancer and OC
(141,142). As a combination therapy,
ispinesib has been tested in three clinical trials: With docetaxel
for HCC, with capecitabine for HNSCC and with carboplatin for
metastatic or recurrent malignant melanoma (142,143).
In clinical studies of breast cancer, Ispinesib has
shown some antitumor activity, with an ORR of 9% (144). However, its ORR was relatively
poor in other tumor types. In a phase II clinical trial for
advanced RCC, no complete responses or partial responses were
observed, and only 6 patients had stable disease (145). A phase II trial of Ispinesib
was conducted in patients with advanced HCC who had not undergone
chemotherapy. At the 8-week evaluation, 46% of patients had stable
disease as the best response, with an mDOR of 3.9 months and a
median time to tumor progression of 1.61 months (146). No significant objective
response was achieved. The most common adverse reactions associated
with ispinesib treatment include neutropenia, anemia, elevated
alanine aminotransferase and aspartate aminotransferase, and
diarrhea, while no neuropathy, mucositis or alopecia have been
observed (147).
In glioblastoma, resistance to ispinesib is closely
associated with the activation of STAT3. STAT3 mediates
anti-apoptotic and metabolic effects through dual phosphorylation
by Src and EGFR, thereby promoting drug resistance that can be
reversed by simultaneously inhibiting Src and EGFR (148). In addition, drug efflux
mediated by P-glycoprotein (P-gp) can pump ispinesib out of the
cells, reducing its intracellular concentration and leading to drug
resistance, which can be overcome by inhibiting P-gp activity to
enhance the cytotoxicity of ispinesib (149). Clinical trials have reported a
favorable safety profile, with no significant neurotoxicity,
alopecia or gastrointestinal toxicity. The most common adverse
event was reversible neutropenia (150). Ispinesib is currently in Phase
I/II clinical development and further studies on its mechanisms and
optimized clinical protocols may establish it as an effective
anti-tumor therapy.
Nolatrexed
Nolatrexed
[2-amino-6-methyl-5-(4-pyridylthio)-4(3H)-quinazolinone] is a
thymidylate synthase inhibitor primarily developed for the
treatment of HCC. In patients with unresectable HCC, nolatrexed
achieved a median overall survival of 22.3 weeks, with an ORR of
1.4% and a median PFS of 12 weeks (151). Initially developed by Agouron
Pharmaceuticals in the US, nolatrexed advanced to Phase II/III
trials for liver cancer and head and neck tumors through a
partnership with Roche. In 1999, Agouron transferred global
oncology rights for nolatrexed to Eximias Pharmaceutical Company,
which currently leads global development of nolatrexed. Ongoing
Phase II clinical trials are being conducted for CRC, HNSCC,
prostate cancer (PCA), pancreatic cancer and lung cancer in
countries including the US, UK, Canada, Italy and South Africa.
Phase III trials for liver cancer are also in progress (152,153).
Nolatrexed, a 4(3H)-quinazolinone derivative, has
shown promising efficacy in clinical trials, exhibiting typical
antimetabolite-related side effects such as short duration of
action and low toxicity, along with mucositis, vomiting, diarrhea
and thrombocytopenia (151).
Thymidylate synthase (TS) is the primary target of nolatrexed and
its overexpression is one of the key mechanisms of resistance. In
CRC cells with p53 mutations, both TS mRNA and protein expression
levels are significantly elevated, thereby increasing the cells'
resistance to nolatrexed (154). Furthermore, being lipophilic,
nolatrexed passively diffuses into cells without the need for
specific membrane transport proteins. Since inhibition of
transporter-mediated uptake is a mechanism of tumor cell
resistance, this property may allow nolatrexed to be effective
against drug-resistant tumors and reduce the likelihood of
resistance development (155).
Although granted orphan drug status by the European Medicines
Agency, it was not approved by the FDA in 2005 (20).
Halofuginone
Halofuginone is a synthetic derivative of a
quinazolinone alkaloid with the chemical name
7-bromo-6-chloro-3-(3-(3-hydroxy-2-piperidinyl)-2-oxopropyl)-4(3H)-quinazolinone
(156). It was developed by
Collgard Biopharmaceuticals and received FDA approval in 2000 as an
orphan drug for the treatment of scleroderma (20). Halofuginone inhibits
epithelial-to-mesenchymal transition in tumor cells, reducing
migration and invasiveness. It also selectively suppresses Th17
cell differentiation and inhibits Smad3 phosphorylation, a key
downstream mediator of the TGF-β pathway. These effects reduce
tumor-associated fibrosis and enhance immune surveillance (157,158). As a broad-spectrum anti-tumor
agent, it has demonstrated efficacy against Pancreatic
adenocarcinoma, CRC, breast cancer and lung cancer (159). Halofuginone has demonstrated
promising results in a Phase II study for recurrent superficial
transitional cell carcinoma of the bladder. In addition, a
sustained-release formulation of fluorofebrin is under Phase II
evaluation for safety, tolerability and pharmacokinetics in
patients with Duchenne muscular dystrophy (20).
Up to date, clinical investigations of halofuginone
have primarily focused on exploring its safety and tolerability
rather than establishing overall therapeutic efficacy, and no
specific data on ORR, PFS or DOR have yet been reported.
Nonetheless, its antitumor potential has been demonstrated in
several experimental models. For instance, in anaplastic thyroid
carcinoma (ATC), halofuginone significantly suppresses ATC cell
proliferation and tumor growth by inhibiting the enzyme-prolyl-tRNA
synthetase-activating transcription factor 4-collagen type I
signaling axis (160). In OSCC,
halofuginone suppressed collagen synthesis and myofibroblast
activation, thereby attenuating tumor invasiveness and growth
(161). Common adverse effects
include nausea, vomiting, a possible increased risk of bleeding,
and decreased red and white blood cell counts (162). A study suggested that
halofuginone can overcome drug resistance in cancer therapy with a
relatively low risk of inducing resistance. For instance, it
restores sensitivity to EGFR-tyrosine kinase inhibitors by
targeting phosphoserine aminotransferase 1 downregulation (163), reverses paclitaxel resistance
in basal-like breast cancer by modulating the BRCA1/TGF-β signaling
axis (164) and serves as a
potential therapeutic agent for 5-FU-resistant CRC by targeting
microRNA-132-3p in vitro (165).
Febrifugine
Febrifugine is the active antimalarial compound
isolated from the traditional Chinese herb Chang Shan, which has
been used for centuries to treat malaria-induced fevers (166). Febrifugine also demonstrates
anti-tumor activity and has shown potential therapeutic effects of
PCA and bladder cancer (BLCA), likely through inhibition of focal
adhesion kinase (167-169). It also suppresses
steroidogenesis and promotes apoptosis, contributing to its
anti-tumor effects by inhibiting DNA synthesis (170). The effectiveness of febrifugine
in clinical treatment is currently focused on basic research and
animal models, with no specific data reported directly on clinical
patients. A related study revealed its anticancer efficacy. In the
BLCA model, febrifugine effectively inhibited the proliferation of
T24 and SW780 BLCA cells, with IC50 values of 0.02 and
0.018 μM, respectively, and demonstrated good anticancer
effects by reducing steroidogenesis and promoting apoptosis
(168). It was also shown to
enhance the anticancer effects of cisplatin in vivo
(171). Due to significant side
effects, there has been no further research on febrifugine for a
long period of time and there is limited literature or clinical
data that discuss its resistance in different cancer types in
detail. Some potential mechanisms of resistance may include cancer
cells rejecting febrifugine, mutations in target genes or cancer
cells altering their sensitivity to the drug through metabolic
pathways. The specific mechanisms of resistance may be gradually
revealed in future studies. Common adverse effects include
dizziness, dry mouth and persistent vomiting (172,173). A summary of the aforementioned
quinazolinone derivatives is presented in Table V.
| Table VQuinazolinone derivatives approved or
under clinical investigation in cancer therapy. |
Table V
Quinazolinone derivatives approved or
under clinical investigation in cancer therapy.
SAR of anticancer quinazolinone
compounds
SAR studies of quinazolinone derivatives reveal a
close association between molecular structure and biological
activity. Structural optimization and the introduction of various
substituents can effectively modulate anti-tumor activity,
pharmacokinetic stability and targeting specificity. The
substitution sites of quinazolinone are shown in Fig. 1.
The 2- and 3-positions are key sites for SAR
studies, as they directly influence selectivity, potency and
cellular activity. In anticancer compounds, substituents at
positions 2 and 3 are predominantly sulfur ethers and aryl ketones,
as seen in the investigational drug ispinesib. Electron-withdrawing
groups at the meta position, such as halogens and trifluoromethyl,
generally enhance inhibitory activity against EGFR, as exemplified
by the quinazoline-based drugs gefitinib and erlotinib, which
contain −Cl and −CF3 groups (174).
Substituents at the 6- and 7-positions typically
target the solvent-exposed regions or entry channels of the
ATP-binding pocket (175).
Modifications at these positions significantly affect solubility,
membrane permeability and pharmacokinetics. Halogen atoms are
frequently introduced at these positions to increase lipophilicity
and stability, thereby improving membrane penetration and
bioactivity, as observed with -Cl and -Br in halofuginone (176). However, methoxy groups
generally have the opposite effect, reducing the reactivity of the
ring toward various reactions (50). The 6,7-dimethoxyquinazolinone
derivative forms an additional hydrogen bond with Thr918 of VEGFR2
via the oxygen atom of the methoxy group. A molecular docking study
showed that it can strongly bind to the hydrophobic site of VEGFR2
kinase, making it a potent VEGFR2 inhibitor (174). Furthermore, Kurogi et al
(177) found from
structure-activity relationship studies that introducing methoxy
groups at the 6- and 7-positions of quinazolinone enhanced its
hypolipidemic activity. Therefore, the addition or removal of such
groups in vivo can serve as a tool for modulating the
toxicity of quinazoline derivatives.
Substitution at the 5-position is relatively
uncommon, as introducing substituents at this site may increase
steric hindrance. However, it shows specificity for cytotoxic
agents, such as the protein kinase inhibitor idelalisib and the
dihydrofolate reductase inhibitor nolatrexed (20). These positions can also serve as
linkers for synthesizing dual-target inhibitors. In the study on
RAF kinase inhibitors, Huestis et al (178) designed the 5-fluoro-substituted
quinazolinone compound GNE-0749. By masking the adjacent polar NH
group, they enhanced the molecule's interaction with RAF kinase and
significantly improved its solubility and permeability, thereby
endowing it with properties suitable for oral administration
(178). Table VI summarizes the SAR of several
common quinazolinone derivatives. Fig. 5 shows the specific structure of
these substituents.
| Table VISubstitution at different positions
of several common quinazolinone compounds. |
Table VI
Substitution at different positions
of several common quinazolinone compounds.
| Drug | R1 | R2 | R3 | R4 | R5 | R6 | R7 |
|---|
| Idelalisib | H |
C8H10N5 | Benzene | H | F | H | H |
| Ispinesib | H |
C15H23N2O | Benzene | H | H | H | Cl |
| Nolatrexed | H | NH2 | H | H |
C5H4NS | H | H |
| Halofuginone | H | H |
C8H16NO | H | H | Cl | Br |
| Febrifugine | H | H |
C8H14NO2 | H | H | H | H |
| Mdivi-1 | H | S |
C7H5Cl2O | H | H | H | H |
| Erastin | H |
C14H18ClN2O2 | Ethoxybenzene | H | H | H | H |
| HMJ-38 | H |
C7H7O | H | H | H | Pyrrolidine | H |
| MJ-33 | H |
C8H9O | H | H | H | Pyrrolidine | H |
Discussion
This review examined the structure, applications
and mechanisms of cell death induced by quinazolinone and its
derivatives. Quinazolinone is a multifunctional heterocyclic
compound that has attracted considerable attention for its
promising biological activity (179). Quinazolinone derivatives act
through multiple cell death pathways, including classical
apoptosis, autophagy and ferroptosis associated with metabolic
stress and damage, as well as inflammatory responses triggered by
senescence, necrosis and pyroptosis. Importantly, quinazolinone and
its derivatives offer distinct advantages in overcoming tumor
resistance and immune evasion. Cancer cells often develop
resistance to chemotherapy through mechanisms such as drug efflux
and altered drug metabolism (180,181). Quinazolinone derivatives have
demonstrated the ability to counteract some of these mechanisms by
sensitizing cancer cells to chemotherapeutic agents. Several
quinazolinone-based compounds have received FDA approval for cancer
treatment, while others have shown encouraging efficacy in
preclinical studies and in Phase I and II clinical trials (182). Despite these advances, further
rigorous clinical studies are needed to fully evaluate the safety,
efficacy and optimal dosing strategies of quinazolinone
derivatives.
Although quinazolinone and its derivatives show
considerable potential in cancer therapy, several challenges and
unresolved issues remain. First, the chemical synthesis of
quinazolinones still relies heavily on high temperatures and harsh
reaction conditions, and generates numerous by-products (183). A team from South China Normal
University developed a covalent organic framework, TAPP-Cu-An,
which enables efficient dehydrogenative cross-coupling reactions
under mild conditions (room temperature and light exposure),
facilitating the photochemical synthesis of 4-quinazolinone
(184). Furthermore, a team
from Guilin University of Technology developed a 4-DPAIPN catalyst,
using acetonitrile as the solvent and blue LED light irradiation,
achieving a metal-free synthesis of tetracyclic quinazolinones and
avoiding the use of precious metals (185). Second, although it is
established that these compounds act through multiple cell death
pathways, the interactions and regulatory mechanisms among these
pathways are not yet fully understood. Future research should
investigate the crosstalk between these pathways and determine how
quinazolinone modulates them in different cancer types to optimize
its anti-tumor effects. Most current studies have examined
combinations of quinazolinone derivatives with chemotherapy agents;
however, their synergistic potential with immunotherapy also
warrants exploration (4,186). Finally, most quinazolinone
derivatives suffer from poor water solubility, low oral
bioavailability and short half-life, which limit their therapeutic
efficacy in vivo (187).
Future research should consider the use of nanocarriers (such as
nanoparticles, liposomes and polymeric microparticles) to
encapsulate the drug, improve solubility in aqueous solutions and
enable targeted delivery, thereby maximizing therapeutic potential
(188). Additionally, prodrug
strategies can be employed, incorporating solubilizing moieties
such as phosphate esters or polyethylene glycol (PEG) groups, such
as PEG-400, to improve bioavailability (189).
Quinazolinone molecules can interact with
fluorescent dyes through covalent coupling, electrostatic
adsorption, or coordination bonding. These interactions allow
real-time tracking of drug delivery in vivo, enable
observation of drug absorption, transport and dynamic changes at
the cellular level, and can be applied in fluorescent imaging of
tumors (190). With advances in
synthetic chemistry and drug design, structural modifications of
quinazolinone compounds could further enhance specificity,
selectivity and therapeutic efficacy. Designing new quinazolinone
derivatives and investigating their roles in cancer treatment will
be critical for advancing this field. By optimizing molecular
structures to improve selectivity and cytotoxicity toward cancer
cells, significant progress in therapeutic outcomes may be
achieved.
Availability of data and materials
Not applicable.
Authors' contributions
JL and YuY wrote the manuscript. YuY conceived and
supervised the study. XK and YaY revised the manuscript. LW and QL
finalized the figures and checked the grammar of the text. All
authors contributed to the article and approved the submitted
version. Data authentication is not applicable. All authors have
read and agreed to the submitted version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Natural Science
Foundation of China (grant no. 82374273), the Natural Science
Foundation of Henan (grant no. 252300420155), the Key Scientific
Research Project of Higher Education of Henan Province (grant no.
25CY031), the Innovation Project of Graduate Students at Xinxiang
Medical University (grant no. YJSCX202417Y) and the National
College Students' Innovation and Entrepreneurship Training Program
(grant nos. 202310472034 and 202410472002).
References
|
1
|
Tiwary BK, Pradhan K, Nanda AK and
Chakraborty R: Implication of quinazoline-4(3H)-ones in medicinal
chemistry: A brief review. J Chem Biol Ther. 1:1042016.
|
|
2
|
Rakesh KP, Manukumar HM and Gowda DC:
Schiff's bases of quinazolinone derivatives: Synthesis and SAR
studies of a novel series of potential anti-inflammatory and
antioxidants. Bioorg Med Chem Lett. 25:1072–1077. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sulthana MT, Chitra K, Alagarsamy V,
Saravanan G and Solomon VR: Anti-HIV and antibacterial activities
of novel 2-(3-Substituted-4-oxo-3, 4-dihydroquinazolin-2-yl)-2,
3-dihydrophthalazine-1,4-diones. Russ J Bioorg Chem. 47:112–121.
2021. View Article : Google Scholar
|
|
4
|
Salfi R, Hakim F, Bhikshapathi D and Khan
A: Anticancer evaluation of novel quinazolinone acetamides:
Synthesis and characterization. Anticancer Agents Med Chem.
22:926–932. 2022. View Article : Google Scholar
|
|
5
|
Gatadi S, Lakshmi TV and Nanduri S:
4(3H)-Quinazolinone derivatives: Promising antibacterial drug
leads. Eur J Med Chem. 170:157–172. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zayed MF: Medicinal Chemistry of
quinazolines as analgesic and anti-inflammatory agents.
ChemEngineering. 6:942022. View Article : Google Scholar
|
|
7
|
Mhetre UV, Haval NB, Bondle GM, Rathod SS,
Choudhari PB, Kumari J, Sriram D and Haval KP: Design, synthesis
and molecular docking study of novel triazole-quinazolinone hybrids
as antimalarial and antitubercular agents. Bioorg Med Chem Lett.
108:1298002024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yaduwanshi PS, Singh S, Sahapuriya P,
Dubey P, Thakur J and Yadav S: Synthesis of some noval qunazolinone
derivatives for their anticonvulsant activity. Orient J Chem.
40:369–373. 2024. View Article : Google Scholar
|
|
9
|
Khalifa MM, Sakr HM, Ibrahim A, Mansour AM
and Ayyad RR: Design and synthesis of new benzylidene-quinazolinone
hybrids as potential anti-diabetic agents: In vitro α-glucosidase
inhibition, and docking studies. J Mol Struct. 1250:1317682022.
View Article : Google Scholar
|
|
10
|
Soliman AM, Karam HM, Mekkawy MH and
Ghorab MM: Antioxidant activity of novel quinazolinones bearing
sulfonamide: Potential radiomodulatory effects on liver tissues via
NF-κB/PON1 pathway. Eur J Med Chem. 197:1123332020. View Article : Google Scholar
|
|
11
|
Osman EO, Emam SH, Sonousi A, Kandil MM,
Abdou AM and Hassan RA: Design, synthesis, anticancer, and
antibacterial evaluation of some quinazolinone-based derivatives as
DHFR inhibitors. Drug Dev Res. 84:888–906. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
El-Karim SS, Syam YM, El Kerdawy AM and
Abdel-Mohsen HT: Rational design and synthesis of novel
quinazolinone N-acetohydrazides as type II multi-kinase inhibitors
and potential anticancer agents. Bioorg Chem. 142:1069202024.
View Article : Google Scholar
|
|
13
|
Kabir E and Uzzaman M: A review on
biological and medicinal impact of heterocyclic compounds. Results
Chem. 4:1006062022. View Article : Google Scholar
|
|
14
|
Almulla AF: A review: Biological
importance of heterocyclic compounds. Der Pharma Chemica.
9:141–147. 2017.
|
|
15
|
Arora P, Arora V, Lamba HS and Wadhwa D:
Importance of heterocyclic chemistry:A review. Int J Pharm Sci Res.
3:2947–2954. 2012.
|
|
16
|
Kumar B, Babu NJ and Chowhan RL:
Sustainable synthesis of highly diastereoselective &
fluorescent active spirooxindoles catalyzed by copper oxide
nanoparticle immobilized on microcrystalline cellulose. Appl
Organomet Chem. 36: View Article : Google Scholar : 2022. View Article : Google Scholar
|
|
17
|
Khan I, Ibrar A, Abbas N and Saeed A:
Recent advances in the structural library of functionalized
quinazoline and quinazolinone scaffolds: Synthetic approaches and
multifarious applications. Eur J Med Chem. 76:193–244. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Borah B, Swain S, Patat M and Chowhan LR:
Recent advances and prospects in the organocatalytic synthesis of
quinazolinones. Front Chem. 10:9910262022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Faisal M and Saeed A: Chemical insights
into the synthetic chemistry of quinazolines: Recent advances.
Front Chem. 8:5947172021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alsibaee AM, Al-Yousef HM and Al-Salem HS:
Quinazolinones, the winning horse in drug discovery. Molecules.
28:9782023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sharma PC, Kaur G, Pahwa R, Sharma A and
Rajak H: Quinazolinone analogs as potential therapeutic agents.
Curr Med Chem. 18:4786–4812. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Khandelwal P, Wadhwani BD, Rao RS, Mali D,
Vyas P, Kumar T and Nair R: Exploring the pharmacological and
chemical aspects of pyrrolo-quinazoline derivatives in Adhatoda
vasica. Heliyon. 10:e257272024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Padmanabhan D, Manimekalai R,
Senthil-Nathan S, Suganthi M and Palanisamy S: Biosynthesis,
therapeutic characteristics, origin and strategies to improve the
yield of vasicine in plants. Vegetos. 186:2025.
|
|
24
|
Zhang Y, Du W, Zhu D, Li M, Qu L, Rao G,
Lin Y, Tong X, Sun Y and Huang F: Vasicine alleviates
2,4-dinitrochlorobenzene-induced atopic dermatitis and passive
cutaneous anaphylaxis in BALB/c mice. Clin Immunol. 244:1091022022.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ali SK, Hamed AR, Soltan MM, El-Halawany
AM, Hegazy UM and Hussein AA: Kinetics and molecular docking of
vasicine from Adhatoda vasica: An acetylcholinesterase inhibitor
for Alzheimer's disease. S Afr J Bot. 104:118–124. 2016. View Article : Google Scholar
|
|
26
|
Srinivasarao D, Jayarraj IA, Jayraaj R and
Prabha ML: A study on antioxidant and anti-inflammatory activity of
vasicine against lung damage in rats. Indian J Allergy Asthma
Immunol. 20:1–7. 2006.
|
|
27
|
Eguchi S: Quinazoline alkaloids and
related chemistry. Bioactive Heterocycles I. 6:113–156. 2006.
View Article : Google Scholar
|
|
28
|
Ghosh P, Ganguly B and Das S: C-H
functionalization of quinazolinones by transition metal catalysis.
Org Biomol Chem. 18:4497–4518. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rakesh KP, Darshini N, Shubhavathi T and
Mallesha N: Biological applications of quinazolinone analogues: A
review. Org Med Chem Int J. 2:41–45. 2017.
|
|
30
|
Kalogirou AS, Kourtellaris A and Koutentis
PA: Synthesis of 2-Cyanoquinazolin-4-ones from
3',5'-Dichloro-1H-spiro
(quinazoline-2,4'-[1,2,6]thiadiazin)-4(3H)-ones. ChemistrySelect.
5:1884–1889. 2020. View Article : Google Scholar
|
|
31
|
Zeng R, Huang C, Wang J, Zhong Y, Fang Q,
Xiao S, Nie X, Chen S and Peng D: Synthesis, crystal structure, and
antifungal activity of quinazolinone derivatives. Crystals.
13:12542023. View Article : Google Scholar
|
|
32
|
Ghoneim MM, Abdelgawad MA, Elkanzi NAA,
Parambi DGT, Alsalahat I, Farouk A and Bakr RB: A literature review
on pharmacological aspects, docking studies, and synthetic
approaches of quinazoline and quinazolinone derivatives. Arch Pharm
(Weinheim). 357:e24000572024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Haneen DSA, Abdalha AA, Alkhatib MM, Kamal
M, Youssef ASA, Abou-Elmagd WSI and Samir SS: Synthesis,
comprehensive in silico studies, and cytotoxicity evaluation of
novel quinazolinone derivatives as potential anticancer agents. Sci
Rep. 15:236972025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Borik RM and Hussein MA: A novel
quinazoline-4-one derivatives as a promising cytokine inhibitors:
Synthesis, molecular docking, and structure-activity relationship.
Curr Pharm Biotechnol. 23:1179–1203. 2022. View Article : Google Scholar
|
|
35
|
Samotrueva MA, Starikova AA, Bashkina OA,
Tsibizova AA, Borisov AV, Merezhkina DV, Tyurenkov IN and Ozerov
AA: Biochemical basis of the antimicrobial activity of
quinazolinone derivatives in the light of insights into the
features of the chemical structure and ways of binding to target
molecules. A review. Dokl Chem. 510:107–129. 2023. View Article : Google Scholar
|
|
36
|
Mhaske SB and Argade NP: The chemistry of
recently isolated naturally occurring quinazolinone alkaloids.
Tetrahedron. 62:9787–9826. 2006. View Article : Google Scholar
|
|
37
|
Mahato AK, Srivastava B and Shanthi CN:
Chemistry structure activity relationship and biological activity
of quinazoline-4(3H)-one derivatives. Inventi Rapid: MedChem.
1:0976–7541. 2011.
|
|
38
|
Asif M: Chemical characteristics,
synthetic methods, and biological potential of quinazoline and
quinazolinone derivatives. Int J Med Chem. 2014:1–27. 2014.
|
|
39
|
Garofalo A, Goossens L, Baldeyrou B,
Lemoine A, Ravez S, Six P, David-Cordonnier MH, Bonte JP, Depreux
P, Lansiaux A and Goossens JF: Design, synthesis, and DNA-binding
of N-Alkyl(anilino)quinazoline derivatives. J Med Chem.
53:8089–8103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
El-Malah A, Malebari AM, Khayyat AN,
Mohammad KA, Gineinah MM and Mahmoud Z: Design, synthesis, and
antiproliferative activities of novel
substitutedhydrazone/triazolo-linked quinazoline derivatives. J Mol
Struct. 1306:1378222024. View Article : Google Scholar
|
|
41
|
Haghighijoo Z, Firuzi O, Hemmateenejad B,
Emami S, Edraki N and Miri R: Synthesis and biological evaluation
of quinazolinone-based hydrazones with potential use in Alzheimer's
disease. Bioorg Chem. 74:126–133. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Khan I, Ibrar A, Ahmed W and Saeed A:
Synthetic approaches, functionalization and therapeutic potential
of quinazoline and quinazolinone skeletons: The advances continue.
Eur J Med Chem. 90:124–169. 2015. View Article : Google Scholar
|
|
43
|
Kaur J, Kaur S, Muskan, Kaur N, Kumar V
and Anand A: Unveiling the therapeutic potential of quinazolinone
derivatives in cancer treatment: A comprehensive exploration.
ChemistrySelect. 9:e2024013662024. View Article : Google Scholar
|
|
44
|
Haider K, Das S, Joseph A and Yar MS: An
appraisal of anticancer activity with structure-activity
relationship of quinazoline and quinazolinone analogues through
EGFR and VEGFR inhibition: A review. Drug Dev Res. 83:859–890.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Reddy MM and Sivaramakrishna A: Remarkably
flexible quinazolinones-synthesis and biological applications. J
Heterocycl Chem. 57:942–954. 2019. View Article : Google Scholar
|
|
46
|
He D, Wang M, Zhao S, Shu Y, Zeng H, Xiao
C, Lu C and Liu Y: Pharmaceutical prospects of naturally occurring
quinazolinone and its derivatives. Fitoterapia. 119:136–149. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shankar GM, Alex VV, Nisthul AA, Bava SV,
Sundaram S, Retnakumari AP, Chittalakkottu S and Anto RJ:
Pre-clinical evidences for the efficacy of tryptanthrin as a potent
suppressor of skin cancer. Cell Prolif. 53:e127102020. View Article : Google Scholar
|
|
48
|
Li H, Fu G and Zhong W: Natural
quinazolinones: From a treasure house to promising anticancer
leads. Eur J Med Chem. 245:1149152023. View Article : Google Scholar
|
|
49
|
Kushwaha N, Sahu A, Mishra J, Soni A and
Dorwal D: An insight on the prospect of quinazoline and
quinazolinone derivatives as anti-tubercular agents. Curr Org
Synth. 20:838–869. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jaiswal S, Verma K, Srivastva A, Arya N,
Dwivedi J and Sharma S: Green synthetic and pharmacological
developments in the hybrid quinazolinone moiety: An updated review.
Curr Top Med Chem. 25:493–532. 2025. View Article : Google Scholar
|
|
51
|
Wu C, Wang Y, Yang F, Shi W, Wang Z, He L,
He Y and Shen J: Synthesis and biological evaluation of
five-atom-linker-based arylpiperazine derivatives with an atypical
antipsychotic profile. ChemMedChem. 14:2042–2051. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wei M, Chai WM, Wang R, Yang Q, Deng Z and
Peng Y: Quinazolinone derivatives: Synthesis and comparison of
inhibitory mechanisms on alpha-glucosidase. Bioorg Med Chem.
25:1303–1308. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Obeng E: Apoptosis (programmed cell death)
and its signals-A review. Braz J Biol. 81:1133–1143. 2021.
View Article : Google Scholar
|
|
54
|
Nair P, Lu M, Petersen S and Ashkenazi A:
Apoptosis initiation through the cell-extrinsic pathway. Methods
Enzymol. 544:99–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lossi L: The concept of intrinsic versus
extrinsic apoptosis. Biochem J. 479:357–384. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hongmei Z: Extrinsic and intrinsic
apoptosis signal pathway review. Apoptosis and Medicine. 2012.
View Article : Google Scholar
|
|
57
|
Ashkenazi A: Targeting the extrinsic
apoptosis pathway in cancer. Cytokine Growth Factor Rev.
19:325–331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liang Y, Zheng Y, Yang J, Ke J and Cheng
K: Design, synthesis and bioactivity evaluation of a series of
quinazolinone derivatives as potent PI3Kγ antagonist. Bioorg Med
Chem. 84:1172612023. View Article : Google Scholar
|
|
59
|
Kim YS, Cheon MG, Boggu PR, Koh SY, Park
GM, Kim G, Park SH, Park SL, Lee CW, Kim JW and Jung YH: Synthesis
and biological evaluation of novel purinyl quinazolinone
derivatives as PI3Kδ-specific inhibitors for the treatment of
hematologic malignancies. Bioorg Med Chem. 45:1163122021.
View Article : Google Scholar
|
|
60
|
Wani ZA, Guru SK, Rao AVS, Sharma S,
Mahajan G, Behl A, Kumar A, Sharma PR, Kamal A, Bhushan S and
Mondhe DM: A novel quinazolinone chalcone derivative induces
mitochondrial dependent apoptosis and inhibits PI3K/Akt/mTOR
signaling pathway in human colon cancer HCT-116 cells. Food Chem
Toxicol. 87:1–11. 2016. View Article : Google Scholar
|
|
61
|
Madbouly EA, Lashine ESM, Al-Karmalawy AA,
Sebaiy MM, Pratsinis H, Kletsas D and Metwally K: Design and
synthesis of novel quinazolinone-chalcone hybrids as potential
apoptotic candidates targeting caspase-3 and PARP-1:In vitro,
molecular docking, and SAR studies. New J Chem. 46:22013–22029.
2022. View Article : Google Scholar
|
|
62
|
Xie J, Yang MR, Hu X, Hong ZS, Bai YY,
Sheng J, Tian Y and Shi CY: Moringa oleifera Lam. Isothiocyanate
quinazolinone derivatives inhibit U251 glioma cell proliferation
through cell cycle regulation and apoptosis induction. Int J Mol
Sci. 24:113762023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Qiu J, Zhou Q, Zhang Y, Guan M, Li X, Zou
Y, Huang X, Zhao Y, Chen W and Gu X: Discovery of novel
quinazolinone derivatives as potential anti-HBV and anti-HCC
agents. Eur J Med Chem. 205:1125812020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
El-Shafey HW, Gomaa RM, El-Messery SM and
Goda FE: Synthetic approaches, anticancer potential, HSP90
inhibition, multitarget evaluation, molecular modeling and
apoptosis mechanistic study of thioquinazolinone skeleton:
Promising antibreast cancer agent. Bioorg Chem. 101:1039872020.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hour MJ, Tsai FJ, Lai IL, Tsao JW, Chiang
JH, Chiu YJ, Lu HF, Juan YN, Yang JS and Tsai SC: Efficacy of
HMJ-38, a new quinazolinone analogue, against the
gemcitabine-resistant MIA-PaCa-2 pancreatic cancer cells.
Biomedicine (Taipei). 13:20–31. 2023. View Article : Google Scholar
|
|
66
|
Chiang JH, Yang JS, Lu CC, Hour MJ, Chang
SJ, Lee TH and Chung JG: Newly synthesized quinazolinone HMJ-38
suppresses angiogenetic responses and triggers human umbilical vein
endothelial cell apoptosis through p53-modulated Fas/death receptor
signaling. Toxicol Appl Pharmacol. 269:150–162. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lu CC, Yang JS, Chiang JH, Hour MJ, Lin
KL, Lin JJ, Huang WW, Tsuzuki M, Lee TH and Chung JG: Novel
quinazolinone MJ-29 triggers endoplasmic reticulum stress and
intrinsic apoptosis in murine leukemia WEHI-3 cells and inhibits
leukemic mice. PLoS One. 7:e368312012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Pope LE and Dixon SJ: Regulation of
ferroptosis by lipid metabolism. Trends Cell Biol. 33:1077–1087.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Dixon SJ and Olzmann JA: The cell biology
of ferroptosis. Nat Rev Mol Cell Biol. 25:424–442. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang M, Guo M, Gao Y, Wu C, Pan X and
Huang Z: Mechanisms and therapeutic targets of ferroptosis:
Implications for nanomedicine design. J Pharm Anal. 14:1009602024.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tang D and Kroemer G: Ferroptosis. Curr
Biol. 30:R1292–R1297. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yuan H, Li X, Zhang X, Kang R and Tang D:
Identification of ACSL4 as a biomarker and contributor of
ferroptosis. Biochem Biophys Res Commun. 478:1338–1343. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Liang X, Long L, Guan F, Xu Z and Huang H:
Research status and potential applications of circRNAs affecting
colorectal cancer by regulating ferroptosis. Life Sci.
352:1228702024. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yang Y, Luo M, Zhang K, Zhang J, Gao T,
Connell DO, Yao F, Mu C, Cai B, Shang Y and Chen W: Nedd4
ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in
melanoma. Nat Commun. 11:4332020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Sun Y, Deng R and Zhang C: Erastin induces
apoptotic and ferroptotic cell death by inducing ROS accumulation
by causing mitochondrial dysfunction in gastric cancer cell HGC-27.
Mol Med Rep. 22:2826–2832. 2020.PubMed/NCBI
|
|
78
|
Li Y, Zeng X, Lu D, Yin M, Shan M and Gao
Y: Erastin induces ferroptosis via ferroportin-mediated iron
accumulation in endometriosis. Hum Reprod. 36:951–964. 2021.
View Article : Google Scholar
|
|
79
|
Huang C, Yang M, Deng J, Li P, Su W and
Jiang R: Upregulation and activation of p53 by erastin-induced
reactive oxygen species contribute to cytotoxic and cytostatic
effects in A549 lung cancer cells. Oncol Rep. 40:2363–2370.
2018.PubMed/NCBI
|
|
80
|
Badgley MA, Kremer DM, Maurer HC,
DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J,
Firl CEM, et al: Cysteine depletion induces pancreatic tumor
ferroptosis in mice. Science. 368:85–89. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Jiang Y and Sun M: SLC7A11: The Achilles
heel of tumor? Front Immunol. 15:14388072024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R
and Tang D: Activation of the p62-Keap1-NRF2 pathway protects
against ferroptosis in hepatocellular carcinoma cells. Hepatology.
63:173–184. 2016. View Article : Google Scholar
|
|
83
|
Zhao X, Wang T, Shang F, Yan J, Jiang M,
Zou X, Li G, Song Z and Huang J: Coumarin-Quinazolinone based
photosensitizers: Mitochondria and endoplasmic reticulum targeting
for enhanced phototherapy via different cell death pathways. Eur J
Med Chem. 280:1169902024. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Xing Z, Yan J, Miao Y, Ruan Y, Yao H, Zhou
Y, Tang Y, Li G, Song Z, Peng Y and Huang J: Endoplasmic
reticulum-targeting quinazolinone-based lipophilic probe for
specific photoinduced ferroptosis and its induced lipid dynamic
regulation. J Med Chem. 67:1900–1913. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Huang Y, Xiong K, Wang A, Wang Z, Cui Q,
Xie H, Yang T, Fan X, Jiang W, Tan X and Huang Q: Cold stress
causes liver damage by inducing ferroptosis through the p38
MAPK/Drp1 pathway. Cryobiology. 113:1045632023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Li X, Ran Q, He X, Peng D, Xiong A, Jiang
M, Zhang L, Wang J, Bai L, Liu S, et al: HO-1 upregulation promotes
mitophagy-dependent ferroptosis in PM2.5-exposed hippocampal
neurons. Ecotoxicol Environ Saf. 277:1163142024. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Nakamura T, Hipp C, Dias Mourão A,
Borggräfe J, Aldrovandi M, Henkelmann B, Wanninger J, Mishima E,
Lytton E, Emler D, et al: Phase separation of FSP1 promotes
ferroptosis. Nature. 619:371–377. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wu Z, Zhu Y, Liu W, Balasubramanian B, Xu
X, Yao J and Lei X: Ferroptosis in liver disease: Natural active
compounds and therapeutic implications. Antioxidants (Basel).
13:3522024. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Balushi KA, Hadhrami AA, Balushi HA,
Lawati AA and Das S: Tebentafusp as a promising drug for the
treatment of uveal melanoma. Curr Drug Targets. 25:149–157. 2024.
View Article : Google Scholar
|
|
90
|
Hu S, Sechi M, Singh PK, Dai L, McCann S,
Sun D, Ljungman M and Neamati N: A novel redox modulator induces a
GPX4-mediated cell death that is dependent on iron and reactive
oxygen species. Eur J Med Chem. 63:9838–9855. 2020. View Article : Google Scholar
|
|
91
|
Cheng Z: The FoxO-autophagy axis in health
and disease. Trends Endocrinol Metab. 30:658–671. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Buzun K, Gornowicz A, Lesyk R, Bielawski K
and Bielawska A: Autophagy modulators in cancer therapy. Int J Mol
Sci. 22:58042021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zhang B and Liu L: Autophagy is a
double-edged sword in the therapy of colorectal cancer. Oncol Lett.
21:3782021. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Gump JM and Thorburn A: Autophagy and
apoptosis: What is the connection? Trends Cell Biol. 21:387–392.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kumar S, Guru SK, Pathania AS, Mupparapu
N, Kumar A, Malik F, Bharate SB, Ahmed QN, Vishwakarma RA and
Bhushan S: A novel quinazolinone derivative induces cytochrome c
interdependent apoptosis and autophagy in human leukemia MOLT-4
cells. Toxicol Rep. 1:1013–1025. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Sharma R, Chatterjee E, Mathew J, Sharma
S, Rao NV, Pan CH, Lee SB, Dhingra A, Grewal AS, Liou JP, et al:
Accommodation of ring C expanded deoxyvasicinone in the HDAC
inhibitory pharmacophore culminates into a tractable anti-lung
cancer agent and pH-responsive nanocarrier. Eur J Med Chem.
240:1146022022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Xia X, Wang L, Zhang X, Wang S, Lei L,
Cheng L, Xu Y, Sun Y, Hang B, Zhang G, et al: Halofuginone-induced
autophagy suppresses the migration and invasion of MCF-7 cells via
regulation of STMN1 and p53. J Cell Biochem. 119:4009–4020. 2018.
View Article : Google Scholar
|
|
99
|
Ha HA, Chiang JH, Tsai FJ, Bau DT, Juan
YN, Lo YH, Hour MJ and Yang JS: Novel quinazolinone MJ-33 induces
AKT/mTOR-mediated autophagy-associated apoptosis in 5FU-resistant
colorectal cancer cells. Oncol Rep. 45:680–692. 2020. View Article : Google Scholar
|
|
100
|
ElZahabi HSA, Nafie MS, Osman D, Elghazawy
NH, Soliman DH, El-Helby AAH and Arafa RK: Design, synthesis and
evaluation of new quinazolin-4-one derivatives as apoptotic
enhancers and autophagy inhibitors with potent antitumor activity.
Eur J Med Chem. 222:1136092021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Roger L, Tomas F and Gire V: Mechanisms
and regulation of cellular senescence. Int J Mol Sci. 22:131732021.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Huang W, Hickson LJ, Eirin A, Kirkland JL
and Lerman LO: Cellular senescence: The good, the bad and the
unknown. Nat Rev Nephrol. 18:611–627. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
de Magalhães JP: Cellular senescence in
normal physiology. Science. 384:1300–1301. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Schmitt CA, Wang B and Demaria M:
Senescence and cancer-role and therapeutic opportunities. Nat Rev
Clin Oncol. 19:619–636. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wang L, Lankhorst L and Bernards R:
Exploiting senescence for the treatment of cancer. Nat Rev Cancer.
22:340–355. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kamal A, Sultana F, Ramaiah MJ, Srikanth
YVV, Viswanath A, Bharathi EV, Nayak R, Pushpavalli SNCVL, Srinivas
C and Pal-Bhadra M: 3-Diarylethyne quinazolinones: A new class of
senescence inducers. Med Chem Comm. 4:575–581. 2013. View Article : Google Scholar
|
|
107
|
Venkatesh R, Ramaiah MJ, Gaikwad HK,
Janardhan S, Bantu R, Nagarapu L, Sastry GN, Ganesh AR and Bhadra
M: Luotonin-A based quinazolinones cause apoptosis and senescence
via HDAC inhibition and activation of tumor suppressor proteins in
HeLa cells. Eur J Med Chem. 94:87–101. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Proskuryakov SY and Gabai VL: Mechanisms
of tumor cell necrosis. Curr Pharm Des. 16:56–58. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Szabó C: Mechanisms of cell necrosis. Crit
Care Med. 33:530–534. 2005. View Article : Google Scholar
|
|
110
|
Loveless R, Bloomquist R and Teng Y:
Pyroptosis at the forefront of anticancer immunity. J Exp Clin
Cancer Res. 40:2642021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Wei X, Xie F, Zhou X, Wu Y, Yan H, Liu T,
Huang J, Wang F, Zhou F and Zhang L: Role of pyroptosis in
inflammation and cancer. Cell Mol Immunol. 19:971–992. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Shams LM, Minaei-Tehrani D, Gholipour H
and Nohehkhan M: Effects of quinazolinones on Balb/C mice embryonic
livers. Indian J Exp Biol. 49:183–190. 2011.PubMed/NCBI
|
|
113
|
Piamsiri C, Maneechote C, Jinawong K,
Arunsak B, Chunchai T, Nawara W, Kerdphoo S, Chattipakorn SC and
Chattipakorn N: Chronic mitochondrial dynamic-targeted therapy
alleviates left ventricular dysfunction by reducing multiple
programmed cell death in post-myocardial infarction rats. Eur J
Pharmacol. 977:1767362024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Li L, Mu Z, Liu P, Wang Y, Yang F and Han
X: Mdivi-1 alleviates atopic dermatitis through the inhibition of
NLRP3 inflammasome. Exp Dermatol. 30:1734–1744. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Tanriover C, Copur S, Ucku D, Cakir AB,
Hasbal NB, Soler MJ and Kanbay M: The mitochondrion: A promising
target for kidney disease. Pharmaceutics. 15:5702023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Liu R, Wang SC, Li M, Ma XH, Jia XN, Bu Y,
Sun L and Yu KJ: An inhibitor of DRP1 (Mdivi-1) alleviates
LPS-induced septic AKI by inhibiting NLRP3 inflammasome activation.
Biomed Res Int. 2020:1–11. 2020.
|
|
117
|
Qian W, Wang J, Roginskaya V, McDermott
LA, Edwards RP, Stolz DB, Llambi F, Green DR and Van Houten B:
Novel combination of mitochondrial division inhibitor 1 (mdivi-1)
and platinum agents produces synergistic pro-apoptotic effect in
drug resistant tumor cells. Oncotarget. 5:4180–4194. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Tusskorn O, Khunluck T, Prawan A,
Senggunprai L and Kukongviriyapan V: Mitochondrial division
inhibitor-1 potentiates cisplatin-induced apoptosis via the
mitochondrial death pathway in cholangiocarcinoma cells. Biomed
Pharmacother. 111:109–118. 2019. View Article : Google Scholar
|
|
119
|
Vargo JW, Walker SN, Gopal SR, Deshmukh
AR, McDermott BM Jr, Alagramam KN and Stepanyan R: Inhibition of
mitochondrial division attenuates cisplatin-induced toxicity in the
neuromast hair cells. Front Cell Neurosci. 11:3932017. View Article : Google Scholar
|
|
120
|
Lai KC, Chia YT, Yih LH, Lu YL, Chang ST,
Hong ZX, Chen TL and Hour MJ: Antitumor effects of the novel
quinazolinone Holu-12: Induction of mitotic arrest and apoptosis in
human oral squamous cell carcinoma CAL27 cells. Anticancer Res.
41:259–268. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Pathania D, Kuang Y, Sechi M and Neamati
N: Mechanisms underlying the cytotoxicity of a novel
quinazolinedione-based redox modulator, QD232, in pancreatic cancer
cells. Br J Pharmacol. 172:50–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Zhou J, Ji M, Yao H, Cao R, Zhao H, Wang
X, Chen X and Xu B: Discovery of quinazoline-2,4(1H,3H)-dione
derivatives as novel PARP-1/2 inhibitors: Design, synthesis and
their antitumor activity. Org Biomol Chem. 16:3189–3202. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Zhou Q, Ji M, Zhou J, Jin J, Xue N, Chen
J, Xu B and Chen X: Poly (ADP-ribose) polymerases inhibitor,
Zj6413, as a potential therapeutic agent against breast cancer.
Biochem Pharmacol. 107:29–40. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Giannini G, Battistuzzi G, Vesci L,
Milazzo FM, De Paolis F, Barbarino M, Guglielmi MB, Carollo V,
Gallo G, Artali R and Dallavalle S: Novel PARP-1 inhibitors based
on a 2-propanoyl-3H-quinazolin-4-one scaffold. Bioorg Med Chem
Lett. 24:462–466. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Ma Y, Yang J, Wei X, Pei Y, Ye J, Li X, Si
G, Tian J, Dong Y and Liu G: Nonpeptidic quinazolinone derivatives
as dual nucleotide-binding oligomerization domain-like receptor 1/2
antagonists for adjuvant cancer chemotherapy. Eur J Med Chem.
207:1127232020. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Smolewski P and Rydygier D: Efficacy and
safety of idelalisib for the treatment of indolent B-cell
malignancies. Expert Opin Pharmacother. 21:1915–1926. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Miller BW, Przepiorka D, de Claro RA, Lee
K, Nie L, Simpson N, Gudi R, Saber H, Shord S, Bullock J, et al:
FDA approval: Idelalisib monotherapy for the treatment of patients
with follicular lymphoma and small lymphocytic Lymphoma. Clin
Cancer Res. 21:1525–1529. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Merli M, Passamonti F and Arcaini L: The
double significance of idelalisib immune-related toxicity. Leuk
Lymphoma. 62:2815–2817. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Liu K, Li D, Zheng W, Shi M, Chen Y, Tang
M, Yang T, Zhao M, Deng D, Zhang C, et al: Discovery, optimization,
and evaluation of quinazolinone derivatives with novel linkers as
orally efficacious phosphoinositide-3-kinase delta inhibitors for
treatment of inflammatory diseases. J Med Chem. 64:8951–8970. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Yu M, Chen J, Xu Z, Yang B, He Q, Luo P,
Yan H and Yang X: Development and safety of PI3K inhibitors in
cancer. Arch Toxicol. 97:635–650. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Ramanathan S, Jin F, Sharma S and Kearney
BP: Clinical pharmacokinetic and pharmacodynamic profile of
idelalisib. Clin Pharmacokinet. 55:33–45. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Wiese W, Barczuk J, Racinska O, Siwecka N,
Rozpedek-Kaminska W, Slupianek A, Sierpinski R and Majsterek I:
PI3K/Akt/mTOR signaling pathway in blood malignancies-new
therapeutic possibilities. Cancers (Basel). 15:52972023. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Zhu J, Wang P, Shehu AI, Lu J, Bi H and Ma
X: Identification of novel pathways in idelalisib metabolism and
bioactivation. Chem Res Toxicol. 31:548–555. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Graf SA and Gopal AK: Idelalisib for the
treatment of non-Hodgkin lymphoma. Expert Opin Pharmacother.
17:265–274. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Lin X, Zhang Y, Huang H, Zhuang W and Wu
L: Post-marketing safety concern of PI3K inhibitors in the cancer
therapies: An 8-year disproportionality analysis from the FDA
adverse event reporting system. Expert Opin Drug Saf. 24:1–12.
2024.
|
|
136
|
Hus I, Puła B and Robak T: PI3K inhibitors
for the treatment of chronic lymphocytic leukemia: Current status
and future perspectives. Cancers (Basel). 14:15712022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Zirlik K and Veelken H: Idelalisib. Recent
Results Cancer Res. 212:243–264. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Scheffold A, Jebaraj BMC, Tausch E,
Bloehdorn J, Ghia P, Yahiaoui A, Dolnik A, Blätte TJ, Bullinger L,
Dheenadayalan RP, et al: IGF1R as druggable target mediating PI3K-δ
inhibitor resistance in a murine model of chronic lymphocytic
leukemia. Blood. 134:534–547. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Park GB, Chung YH, Jeong JY and Kim D: A
p110δ-specific inhibitor combined with bortezomib blocks drug
resistance properties of EBV-related B cell origin cancer cells via
regulation of NF-κB. Int J Oncol. 50:1711–1720. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Alossaimi MA, Riadi Y, Alnuwaybit GN, Md
S, Alkreathy HM, Elekhnawy E, Geesi MH, Alqahtani SM and Afzal O:
Design, synthesis, molecular docking, and in vitro studies of
2-mercaptoquinazolin-4(3H)-ones as potential anti-breast cancer
agents. Saudi Pharm J. 32:1019712024. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Škubník J, Jurášek M, Ruml T and Rimpelová
S: Mitotic poisons in research and medicine. Molecules.
25:46322020. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Shahin R and Aljamal S: Kinesin spindle
protein inhibitors in cancer: From high throughput screening to
novel therapeutic strategies. Future Sci OA. 8:FSO7782022.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Lee CW, Bélanger K, Rao SC, Petrella TM,
Tozer RG, Wood L, Savage KJ, Eisenhauer EA, Synold TW, Wainman N
and Seymour L: A phase II study of ispinesib (SB-715992) in
patients with metastatic or recurrent malignant melanoma: A
national cancer institute of Canada clinical trials group trial.
Invest New Drugs. 26:249–255. 2008. View Article : Google Scholar
|
|
144
|
Purcell JW, Davis J, Reddy M, Martin S,
Samayoa K, Vo H, Thomsen K, Bean P, Kuo WL, Ziyad S, et al:
Activity of the kinesin spindle protein inhibitor ispinesib
(SB-715992) in models of breast cancer. Clin Cancer Res.
16:566–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Lee RT, Beekman KE, Hussain M, Davis NB,
Clark JI, Thomas SP, Nichols KF and Stadler WM: A University of
Chicago consortium phase II trial of SB-715992 in advanced renal
cell cancer. Clin Genitourin Cancer. 6:21–24. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Murase Y, Ono H, Ogawa K, Yoshioka R,
Ishikawa Y, Ueda H, Akahoshi K, Ban D, Kudo A, Tanaka S and Tanabe
M: Inhibitor library screening identifies ispinesib as a new
potential chemotherapeutic agent for pancreatic cancers. Cancer
Sci. 112:4641–4654. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Gomez HL, Philco M, Pimentel P, Kiyan M,
Monsalvo ML, Conlan MG, Saikali KG, Chen MM, Seroogy JJ, Wolff AA
and Escandon RD: Phase I dose-escalation and pharmacokinetic study
of ispinesib, a kinesin spindle protein inhibitor, administered on
days 1 and 15 of a 28-day schedule in patients with no prior
treatment for advanced breast cancer. Anticancer Drugs. 23:335–341.
2012. View Article : Google Scholar
|
|
148
|
Kenchappa RS, Dovas A, Argenziano MG,
Meyer CT, Stopfer LE, Banu MA, Pereira B, Griffith J, Mohammad A,
Talele S, et al: Activation of STAT3 through combined SRC and EGFR
signaling drives resistance to a mitotic kinesin inhibitor in
glioblastoma. Cell Rep. 39:1109912022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Ansbro MR, Shukla S, Ambudkar SV, Yuspa SH
and Li L: Screening compounds with a novel high-throughput
ABCB1-mediated efflux assay identifies drugs with known therapeutic
targets at risk for multidrug resistance interference. PLoS One.
8:e603342013. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Purcell JW, Davis J, Reddy M, Martin S,
Samayoa K, Vo H, Thomsen K, Bean P, Kuo WL, Ziyad S, et al:
Activity of the kinesin spindle protein inhibitor ispinesib
(SB-715992) in models of breast cancer. Clin Cancer Res.
16:566–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Gish RG, Porta C, Lazar L, Ruff P, Feld R,
Croitoru A, Feun L, Jeziorski K, Leighton J, Gallo J and Kennealey
GT: Phase III randomized controlled trial comparing the survival of
patients with unresectable hepatocellular carcinoma treated with
nolatrexed or doxorubicin. J Clin Oncol. 25:3069–3075. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Lord R, Suddle A and Ross PJ: Emerging
strategies in the treatment of advanced hepatocellular carcinoma:
The role of targeted therapies. Int J Clin Pract. 65:182–188. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Gish RG, Porta C, Lazar L, Ruff P, Feld R,
Croitoru A, Feun L, Jeziorski K, Leighton J, Knox J and Kennealey
GT: Phase III randomized controlled trial comparing the survival of
patients with unresectable hepatocellular carcinoma treated with
nolatrexed or doxorubicin. J Clin Oncol. 25:3069–3075. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Giovannetti E, Backus HH, Wouters D,
Ferreira CG, van Houten VM, Brakenhoff RH, Poupon MF, Azzarello A,
Pinedo HM and Peters GJ: Changes in the status of p53 affect drug
sensitivity to thymidylate synthase (TS) inhibitors by altering TS
levels. Br J Cancer. 96:769–775. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Van Triest B, Pinedo HM, Giaccone G and
Peters GJ: Downstream molecular determinants of response to
5-fluorouracil and antifolate thymidylate synthase inhibitors. Ann
Oncol. 11:385–391. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Hasan RU, Mian SS, Arfi S, Begum B, Verma
S, Ahmad R, Hussain I and Asif M: Study of pharmacologically active
drugs containing quinazoline pharmacophore: A brief overview. J Adv
Zool. 45:1166–1184. 2024.
|
|
157
|
Han Y, Liu S, Zhu J, Liu P, Meng Z, Li Y,
Li S, Fan F, Zhang M and Liu H: Experimental study on the
inhibitory effect of Halofuginone on NSCLC. European Journal of
Pharmacology. 2024.
|
|
158
|
Chen Y, Liu W and Wang P, Hou H, Liu N,
Gong L, Wang Y, Ji K, Zhao L and Wang P: Halofuginone inhibits
radiotherapy-induced epithelial mesenchymal transition in lung
cancer. Oncotarget. 7:71341–71352. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Zuo R, Guo X, Song X, Gao X, Zhang J,
Jiang S, Adam V, Kuca K, Wu W and Guo D: New uses of halofuginone
to treat cancer. J Pharm Anal. 15:1010802024. View Article : Google Scholar
|
|
160
|
Mi L, Liu J, Zhang Y, Su A, Tang M, Xing
Z, He T, Wei T, Li Z and Wu W: The EPRS-ATF4-COLI pathway axis is a
potential target for anaplastic thyroid carcinoma therapy.
Phytomedicine. 129:1556702024. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Zhao H, Li R, Chen Y, Yang X and Shang Z:
Stromal nicotinamide N-methyltransferase orchestrates the crosstalk
between fibroblasts and tumour cells in oral squamous cell
carcinoma: Evidence from patient-derived assembled organoids.
Oncogene. 42:1166–1180. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Panda H, Suzuki M, Naito M, Saito R, Wen
H, Baird L, Uruno A, Miyata K and Yamamoto M: Halofuginone micelle
nanoparticles eradicate Nrf2-activated lung adenocarcinoma without
systemic toxicity. Free Radic Biol Med. 187:92–104. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Li Y, Liu P, Liu S, Zhu J, Han Y, Jiang Z,
Tang D, Meng Z, Li S, Zhang M, et al: Halofuginone targets
Serine/Glycine synthesis to reverse epidermal growth factor
receptor tyrosine Kinase inhibitor resistance in lung
adenocarcinoma. Phytomedicine. 143:1567882025. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Zhang L, Yue G, Lu Y and Tang J: BRCA1 as
a target for attenuating paclitaxel resistance by Halofuginone
treatment in basal-like breast cancer. J Funct Foods.
118:1062452024. View Article : Google Scholar
|
|
165
|
Wang C, Zhu JB, Yan YY, Zhang W, Gong XJ,
Wang X and Wang XL: Halofuginone inhibits tumorigenic progression
of 5-FU-resistant human colorectal cancer HCT-15/FU cells by
targeting miR-132-3p in vitro. Oncol Lett. 20:3852020. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Zhu S, Wang J, Chandrashekar G, Smith E,
Liu X and Zhang Y: Synthesis and evaluation of 4-quinazolinone
compounds as potential antimalarial agents. Eur J Med Chem.
45:3864–3869. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Zuo R, Zhang J, Song X, Hu S, Gao X, Wang
J, Ji H, Ji C, Peng L, Si H, et al: Encapsulating halofuginone
hydrobromide in TPGS polymeric micelles enhances efficacy against
triple-negative breast cancer cells. Int J Nanomedicine.
16:1587–1600. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Chen J, Fan S, Guo J, Yang J, Pan L and
Xia Y: Discovery of anticancer function of Febrifugine: Inhibition
of cell proliferation, induction of apoptosis and suppression
steroid synthesis in bladder cancer cells. Toxicol Appl Pharmacol.
484:1168782024. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Lin F, Wang R, Du J, Wen C, Wang X, Jin Y
and Shao L: Pharmacokinetics and bioavailability of febrifugine in
rat plasma determined by UPLC-MS/MS. Acta Chromatogr. 37:
View Article : Google Scholar : 2024.
|
|
170
|
Chen J, Fan S, Guo J, Yang J, Pan L and
Xia Y: Discovery of anticancer function of Febrifugine: Inhibition
of cell proliferation, induction of apoptosis and suppression
steroid synthesis in bladder cancer cells. Toxicol Appl Pharmacol.
484:1168782024. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Zhang J, Xu HX, Zhu JQ, Dou YX, Xian YF
and Lin ZX: Natural Nrf2 inhibitors: A review of their potential
for cancer treatment. Int J Biol Sci. 19:3029–3041. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Zhu J, Liu S, Liu P, Zhai K and Liu H:
Traditional medicine meets modern science: Halofuginone's role in
combating autoimmune diseases. J Nat Med. 79:1017–1029. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Hasan RU, Mian SS, Arfi S, Begum B, Verma
S, Ahmad R, Hussain I and Asif M: Study of pharmacologically active
drugs containing quinazoline pharmacophore: A brief overview. J Adv
Zool. 45:1166–1184. 2024.
|
|
174
|
Haider K, Das S, Joseph A and Yar MS: An
appraisal of anticancer activity with structure-activity
relationship of quinazoline and quinazolinone analogues through
EGFR and VEGFR inhibition: A review. Drug Dev Res. 83:859–890.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Jiang X, Yu J, Zhou Z, Kongsted J, Song Y,
Pannecouque C, De Clercq E, Kang D, Poongavanam V, Liu X and Zhan
P: Molecular design opportunities presented by solvent-exposed
regions of target proteins. Med Res Rev. 39:2194–2238. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
El-Sayed NNE, Al-Otaibi TM, Barakat A,
Almarhoon ZM, Hassan MZ, Al-Zaben MI, Krayem N, Masand VH and Bacha
AB: Synthesis and biological evaluation of some New
3-Aryl-2-thioxo-2,3-dihydroquinazolin-4(1H)-ones and
3-Aryl-2-(benzylthio)quinazolin-4(3H)-ones as antioxidants; COX-2,
LDHA, α-Glucosidase and α-amylase inhibitors; and anti-colon
carcinoma and apoptosis-inducing agents. Pharmaceuticals (Basel).
16:13922023. View Article : Google Scholar
|
|
177
|
Kurogi Y, Inoue Y, Tsutsumi K, Nakamura S,
Nagao K, Yoshitsugu H and Tsuda Y: Synthesis and hypolipidemic
activities of novel 2-[4-[diethoxyphosphoryl)methyl]phenyl]
quinazolines and 4(3H)-quinazolinones. J Med Chem. 39:1433–1437.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Huestis MP, Dela Cruz D, DiPasquale AG,
Durk MR, Eigenbrot C, Gibbons P, Gobbi A, Hunsaker TL, La H, Leung
DH, et al: Targeting KRAS mutant cancers via combination treatment:
Discovery of a 5-Fluoro-4-(3H)-quinazolinone Aryl urea pan-RAF
kinase inhibitor. J Med Chem. 64:3940–3955. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Rezaeinasab R, Jafari E and Khodarahmi G:
Quinazolinone-based hybrids with diverse biological activities: A
mini-review. J Res Med Sci. 27:682022. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Lv L, Maimaitiming M, Xia S, Yang J, Zhang
T, Wang Y, Li X, Pinchuk I, Wang P, Wang CY and Liu Z: MR2938
relieves DSS-induced colitis in mice through inhibiting NF-κB
signaling and improving epithelial barrier. Mar Life Sci Technol.
18: View Article : Google Scholar : 2025.
|
|
181
|
Upadhyay R, Tandel P and Patel AB:
Halogen-based quinazolin-4(3H)-one derivatives as MCF-7 breast
cancer inhibitors: Current developments and structure-activity
relationship. Arch Pharm (Weinheim). 358:e24007402024. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Wahan SK, Sharma B and Chawla PA:
Medicinal perspective of quinazolinone derivatives: Recent
developments and structure-activity relationship studies. Curr Top
Med Chem. 59:239–257. 2021.
|
|
183
|
Kumar P, Tomar V, Joshi RK and Nemiwal M:
Nanocatalyzed synthetic approach for quinazoline and quinazolinone
derivatives: A review (2015-present). Synth Commun. 52:795–826.
2022. View Article : Google Scholar
|
|
184
|
Chen Y, Sun SN, Chen XH, Chen ML, Lin JM,
Niu Q, Li SL, Liu J and Lan YQ: Predesign of covalent-organic
frameworks for efficient photocatalytic dehydrogenative
cross-coupling reaction. Adv Mater. 37:e24136382025. View Article : Google Scholar
|
|
185
|
Huang FP, Qin WJ, Pan XY, Yang K, Wang K
and Teng QH: Visible-Light-Induced chemodivergent synthesis of
tetracyclic quinazolinones and 3-iminoisoindoliones via the
substrate control strategy. J Org Chem. 89:4395–4405. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Kavitha K, Srinivasan N and Harbabu Y:
Review of quinazolinone scaffold as anticancer agents. World J
Pharm Res. 7:434–454. 2018.
|
|
187
|
Wang Q, Pan Y, Luo H, Zhang Y, Gao F, Wang
J and Zheng J: Novel approaches for the solid-phase synthesis of
dihydroquinazoline-2(1H)-one derivatives and biological evaluation
as potential anticancer agents. Molecules. 27:85772022. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Aljohani AKB, Almadani SA, Alsulaimany M,
Aljuhani N, Samman WA, Al-Shareef AH, Alghamdi R, Tayeb SM, Alharbi
HY, Aljohani MS, et al: Nano-carrier, design, synthesis, in silico
ADMET, anti-proliferative assessments and docking of
[1,2,4]triazolo[4,3-a]quinoxalines as Topo-II inhibitors and DNA
intercalators. Naunyn Schmiedebergs Arch Pharmacol. 11: View Article : Google Scholar : 2025.
|
|
189
|
Udayasree N, Haridasyam RB, Palabindela R,
Krishna TM and Narsimha S: One-pot synthesis, anticancer, EGFR and
caspases assays of novel fused
[1,2,3]triazolo-pyrrolo[2,1-b]quinazolinones. J Mol Struct.
1320:1395702025. View Article : Google Scholar
|
|
190
|
Zhu MS, Zhang G, Xu YJ, Sun R and Ge JF:
Conjugated structures based on quinazolinones and their application
in fluorescent labeling. Org Biomol Chem. 21:1992–2000. 2023.
View Article : Google Scholar : PubMed/NCBI
|