Introduction
Heart failure (HF), one of the end-stage
manifestations of cardiovascular disease has an estimated
prevalence of ≥56 million individuals and a 5-year survival rate
following diagnosis remaining >50%. Consequently, it has been
recognized as an important issue in public health and a major cause
of morbidity and mortality worldwide (1,2).
HF not only considerably affects the quality of life of patients,
but may also lead to serious complications and can be
life-threatening (3). Despite
improvements in the guidelines for the treatment of HF, and the
development of new drugs and clinical studies, such as intravenous
recombinant human Neuregulin-1 and vericiguat (4,5),
the management of this condition remains challenging, especially in
terms of prognosis (6).
Previous studies have revealed that oxidative
stress, mitochondrial dysfunction, calcium regulation and cell
death are associated with the onset and progression of HF (7-10). Notably, pyroptosis, a mode of
programmed cell death, has been shown to be associated with HF
(11). Pyroptosis is mainly
mediated by gasdermin D (GSDMD), which causes cell membrane rupture
through the generation of GSDMD N-terminal fragments (GSDMD-NT)
with pore-forming activity, resulting in the release of
inflammatory factors, and exacerbating cell death and tissue damage
(12). A recent study revealed
that GSDMD-NT perforates the mitochondrial membrane in pyroptosis,
leading to mitochondrial damage and inducing reactive oxygen
species (ROS) release. ROS further amplifies GSDMD binding and
mitochondrial damage in pyroptosis (13). Excessive release of ROS can
activate the NLR family pyrin domain-containing protein 3 (NLRP3)
inflammasome in a positive cascade reaction with
thioredoxin-interacting protein (TXNIP), thereby mediating the
occurrence of pyroptosis (14).
Nuclear factor erythroid 2-related factor (Nrf2) is a key regulator
of the antioxidant response and can regulate oxidative stress by
eliminating ROS and inhibiting TXNIP expression (15,16).
Calycosin (CA) is a natural flavonoid compound that
has been demonstrated to exert various biological effects,
including anti-inflammatory, antioxidant and pro-angiogenic
properties (17). CA has a broad
application prospect in the treatment of cardiovascular diseases,
and studies have revealed that CA can improve myocardial oxidative
stress (18), attenuate
myocardial fibrosis and cardiac dysfunction after myocardial
infarction, and inhibit HF after acute infarction (19,20). Previous studies have revealed
that CA inhibits pyroptosis and ameliorates myocardial injury by
inhibiting the activation of the NLRP3 inflammasome (21,22). In addition, CA may upregulate
Nrf2 expression, and ameliorate myocardial oxidative stress and
mitochondrial damage, thereby protecting the myocardium (23). Therefore, the present study
further explored the therapeutic mechanism of CA based on the
Nrf2/ROS/TXNIP pathway-mediated mitochondrial damage and pyroptosis
in HF.
Materials and methods
Reagents
CA was purchased from Shanghai Yuanye Biotechnology
Co., Ltd. Captopril (CAP; CAP purchase specification: 25 mg;
national drug approval number: H44021595) was used as the positive
control and was purchased from Guangdong Pidi Pharmaceutical Co.,
Ltd. Detection kit information was as follows: Rat N-terminal pro
B-type natriuretic peptide (NT-proBNP) ELISA Kit (cat. no.
CSB-E08752r) was purchased from Cusabio Technology, LLC; lactate
dehydrogenase (LDH) ELISA kit (cat. no. SEB864Ra) was purchased
from CLOUD-CLONE CORP, CCC; malondialdehyde (MDA) assay kit (cat.
no. A003-1), superoxide dismutase (SOD) assay kit (cat. no.
A001-3-2) and microplate method total glutathione/glutathione
disulfide (T-GSH/GSSG) assay kit (cat. no. A061-1-1) were purchased
from Nanjing Jiancheng Bioengineering Institute. LDH cytotoxicity
colorimetric assay kit (cat. no. E-BC-K771-M) was purchased from
Elabscience. Antibody information was as follows: Nrf2 (cat. no.
80593-1-RR), apoptosis-associated speck-like protein containing a
CARD (ASC; cat. no. 67494-1-Ig), IL-1β (cat. no. 29530-1-AP),
cytochrome c (cat. no. 66264-1-Ig) and β-actin (cat. no.
66009-1-Ig) antibodies were purchased from Proteintech Group, Inc.;
TXNIP (cat. no. A9342), NLRP3 (cat. no. A5652), IL-18 (cat. no.
A23076) and GSDMD (full length and N terminal; cat. no. A24476)
antibodies were purchased from ABclonal Biotech Co., Ltd.;
Caspase-1 (pro- and cleaved-Caspase-1) (cat. no. ab286125), COX IV
(cat. no. ab16056), Goat Anti-rabbit IgG HRP (cat. no. ab150077)
and Goat Anti-Mouse IgG HRP (cat. no. ab205719) antibodies were
purchased from Abcam. Mitochondrial membrane potential assay kit
JC-1 (cat. no. C2006), ROS Assay kit (cat. no. S0033S) and MitoSOX
Red assay kit (cat. no. S0061S) were purchased from Beyotime
Institute of Biotechnology. Masson Tricolor Staining kit (cat. no.
G1006) was purchased from Wuhan Servicebio Technology Co., Ltd.
Cell mitochondrial stress test kit (cat. no. 103015-100),
glycolytic stress test kit (cat. no. 103020-100) and Seahorse XF
base medium (cat. no. 102353-100) were purchased from Agilent
Technologies, Inc.
Establishment and treatment of rats
A total of 50 8-week-old male Wistar rats (weight,
180-220 g) were purchased from Beijing Vital River Laboratory
Animal Technology Co., Ltd. The rats were housed in the
Experimental Center of the Affiliated Hospital of Shandong
University of Traditional Chinese Medicine (Jinan, China) under
standard conditions (12-h light/dark cycle with free access to
water and food). The animal experimental procedures were approved
by the Experimental Animal Management Committee and the Ethics
Committee of the Affiliated Hospital of Shandong University of
Traditional Chinese Medicine. The animal experiments strictly
followed the guidelines of the Ethics Committee.
Rats were randomly divided into five groups: Sham,
HF, CA low-dose (CA-L), CA high-dose (CA-H) and CAP group
(n=10/group). In the present study, the HF model was constructed 4
weeks after ligation of the left anterior descending (LAD) artery
in rats. Specifically, rats were anesthetized using 2-3% isoflurane
(3% induction and 2% maintenance) based on an isoflurane delivery
system. The left chest area of the rats was fully exposed under
aseptic conditions. The left chest area of the rats was shaved and
disinfected to ensure aseptic conditions. A minimally invasive open
thoracotomy was performed in the fourth intercostal space and the
LAD artery was permanently ligated using a 6-0 suture. After
ligation and squeezing out the gas from the chest cavity, the
muscle and skin were sutured at the surgical site layer by layer.
The Sham group underwent the same surgical procedures, but the
sutures were only passed through the LAD artery without ligation.
After 4 weeks, echocardiography was used to determine whether the
HF rat model was successfully constructed. CA was dissolved in DMSO
to prepare a 60 mg/ml stock solution, which was then diluted to the
appropriate working concentration using saline containing 20%
Sulfobutylether-β-Cyclodextrin (SBE-β-CD, Proteintech Group, Inc.)
according to experimental requirements. Subsequently, CA-L and CA-H
rats received intraperitoneal injections of 15 and 30 mg/kg CA,
respectively, daily for 4 weeks (24). The CAP group rats received 10
mg/kg CAP administered orally by gavage daily for 4 weeks. The Sham
and HF groups received intraperitoneal injections of 5 ml/kg saline
containing 20% SBE-β-CD daily for 4 weeks. Following
echocardiography and cardiac puncture for blood sampling, the rats
were euthanized through CO2 asphyxia (40% vol/min) for 5
min and death was confirmed by the absence of respiration and
heartbeat. Following euthanasia, rat hearts were immediately
collected. Due to limited tissue available from each rat, the
sample sizes used in each experiment varied. The following criteria
were established as humane endpoints: i) Weight loss >20% of the
initial body weight; ii) prolonged feeding impairment or mobility
deficits; iii) rats exhibited signs of depression accompanied by
hypothermia when not under anesthesia.
Echocardiography
At 4 weeks after surgery and treatment,
echocardiography was carried out on all rats to assess cardiac
function, in order to verify the construction of the HF model and
the therapeutic effect of CA. Specifically, after anesthetizing
rats with 2-3% isoflurane (3% induction and 2% maintenance) based
on an isoflurane delivery system, 2D long-axis images were recorded
using a portable Mindray M5 digital ultrasound scanner (Shenzhen
Mindray Bio-Medical Electronics Co., Ltd.). During three cardiac
cycles, left ventricular ejection fraction (LVEF) and fractional
shortening (FS) were measured at the myocardial level using the
M-mode.
Histological analyses
A total of three fresh rat myocardial tissue samples
in each group were fixed in 4% paraformaldehyde (cat. no.
G1101-500ML, Wuhan Servicebio Technology Co., Ltd.) at room
temperature for 24 h according to the instructions of the
manufacturer that supplied the paraformaldehyde, embedded in
paraffin and cut into 4-μm sections. Sections were stained
at room temperature with hematoxylin solution for 5 min, followed
by 15 sec in eosin solution for H&E staining. Sections were
stained at room temperature with Masson dye solution set at room
temperature according to the manufacturer's instructions for
Masson's trichrome staining. Images were obtained and
histopathological status was assessed using a digital section
scanner (WS-10; Zhiyue Medical Technology (Jiangsu) Co., Ltd.).
Masson's trichrome staining results were analyzed using ImageJ
1.52v software (National Institutes of Health).
Transmission electron microscopy
(TEM)
Three left ventricular myocardial tissue samples in
each group were rinsed three times in pre-cooled PBS buffer, cut
into 1-2 mm3 pieces and fixed in 2.5% glutaraldehyde at
4°C for 24 h. The myocardial tissues were subsequently dehydrated
in a graded ethanol series, transitioned with propylene oxide and
embedded in Epon812 epoxy resin at 60°C for 48 h. Ultrathin
sections (70 nm) were cut using an ultramicrotome, collected on
200-mesh copper grids and dual-stained with 2% uranyl acetate and
lead citrate, both at room temperature for 15 min. Sections were
examined by TEM, focusing on mitochondrial morphology, cristae
integrity and myofibril arrangement.
Measurement of NT-proBNP, T-GSH/GSSG,
MDA, SOD and LDH
After anesthetizing the rats with isoflurane to
carry out echocardiography, 1 ml blood was collected through
cardiac puncture. Rat serum was isolated from blood by centrifuging
at 1,500 × g for 10 min at 4°C. NT-proBNP and LDH levels in the
serum samples were assessed at an absorbance of 450 nm according to
the standard procedure of the assay kits. Following cardiac
puncture for blood sampling, all rats were euthanized, and their
hearts were immediately collected. After isolation of five rat
myocardial tissue samples, the levels of T-GSH/GSSG, SOD and MDA in
myocardial tissues were detected using the assay kits, according to
the standard procedures of the test kits. Absorbance was measured
at 405, 450 and 532 nm using a microplate reader to detect
T-GSH/GSSG, SOD and MDA levels, respectively. Post-treatment, LDH
levels in cells were assessed at an absorbance of 450 nm according
to the standard procedure of the assay kits.
Immunohistochemistry staining
The paraffin-embedded myocardial tissue sections
were subjected for thermally repaired antigen using citrate buffer
(pH 6.0) at 95-100°C. Following natural cooling, they were washed
three times with PBS. Hydrogen peroxide (3%) solution was used to
inactivate endogenous enzymes. Subsequently, the sections were
incubated with Nrf2 antibody (1:500) at 4°C overnight, then
sections were incubated with the Goat Anti-rabbit IgG HRP secondary
antibody (1:1,000) for 1 h at room temperature. The sections were
immersed in diaminobenzidine, counterstained with hematoxylin and
finally imaged using a digital section scanner (WS-10; Zhiyue
Medical Technology (Jiangsu) Co., Ltd.). The results were analyzed
using ImageJ 1.52v software (National Institutes of Health).
Cell culture and treatments
Rat H9c2 cardiomyocytes were purchased from The Cell
Bank of Type Culture Collection of The Chinese Academy of Sciences
and cultured in DMEM (Pricella Biotechnology) supplemented with 10%
fetal bovine serum (Vazyme Biotech Co., Ltd.) and 1%
penicillin/streptomycin at 37°C with 5% CO2. The present
study employed a hypoxia-reoxygenation (H/R) model to simulate
myocardial ischemia-reperfusion injury. CA was dissolved in DMSO to
prepare a 0.1 M stock solution, which was then diluted to the
appropriate working concentration according to experimental
requirements. After model establishment, the experimental groups
were treated with CA solutions at different concentrations (10, 20
and 30 μM). Meanwhile, control and model groups were
established and treated with an equivalent concentration of DMSO
(0.03%) solution to eliminate solvent interference.
The method used to establish the H/R model for
cardiomyocytes was as follows: First, cultured cardiomyocytes were
placed in a hypoxia chamber with gas conditions adjusted to 1%
O2, 5% CO2 and 94% N2, and
incubated at 37°C for 12 h to simulate a hypoxic environment. After
the hypoxia period, the cells were transferred to a normoxic
incubator with gas conditions of 20% O2, 5%
CO2 and 75% N2, and incubated at 37°C for 24
h to simulate the reoxygenation process. Throughout the experiment,
the temperature, humidity and gas concentration within the
incubator were strictly controlled, and cell status was regularly
monitored to ensure the reproducibility and stability of the model
(25).
Cell transfection [small interfering RNA
(siRNA)-Nrf2]
Cell transfection was performed before H/R and/or CA
treatment. Pooled siRNAs targeting Nrf2 (#1, #2 and #3) were used
to effectively silence Nrf2 and eliminate off-target effects. The
siRNA sequences were as follows: Nrf2#1, forward
5'-GGAAGUCUUCAGCAUGUUATT-3' and reverse
5'-UAACAUGCUGAAGACUUCCTT-3'; Nrf2#2, forward
5'-CGAGAAGUGUUUGACUUUATT-3' and reverse
5'-UAAAGUCAAACACUUCUCGTT-3'; Nrf2#3, forward
5'-GGGUUCAGUGACUCGGAAATT-3' and reverse
5'-UUUCCGAGUCACUGAACCCTT-3'; non-targeting negative control (NC),
forward 5'-UUCUCCGAACGUGUCACGUTT-3' and reverse
5'-ACGUGACACGUUCGGAGAATT-3' (Beijing Tsingke Biotech Co., Ltd.).
H9c2 cells were seeded in 6-well plates at 1×105
cells/well and were treated at 37°C with 5% CO2 until
they reached 70-80% confluence. The 6-well plate for transfection
was prepared by discarding the medium, washing with PBS and adding
1.5 ml Opti-MEM (Invitrogen; Thermo Fisher Scientific, Inc.) per
well. For each experimental group, 1.5-ml Eppendorf tubes were
labeled and 250 μl Opti-MEM with 5 μl Lipofectamine™
3000 (Invitrogen; Thermo Fisher Scientific, Inc.) was added, mixed
by pipetting and incubated at room temperature for 5 min.
Similarly, labeled tubes were prepared with 250 μl Opti-MEM
and 8 μl siRNA (concentration, 20 μM), mixed by
pipetting. The siRNA mixture was combined with the corresponding
Lipofectamine 3000 mixture, mixed thoroughly and incubated at room
temperature for 20 min. The mixture was then added to the 6-well
plate and the medium was replaced after 8 h at room temperature.
Cell functional assays were conducted 48 h later.
Cell Counting Kit-8 (CCK-8) assay
A H9c2 cell suspension was seeded in a 96-well plate
at 100 μl/well and incubated at 37°C with 5% CO2
for 24 h to allow cell attachment and proliferation. Different
concentrations of CA (0, 5, 10, 20, 30, 40 and 50 μM) were
added to each well, followed by an additional 24-h incubation. For
evaluating the protective effect of CA on H/R-induced cardiomyocyte
injury, cells were subjected to CA treatment following H/R
treatment; otherwise, this step was omitted for assessing the
direct impact of CA on cell viability. After incubation, 10
μl CCK-8 solution (Vazyme Biotech Co, Ltd.) was added to
each well, gently mixed and incubated for 2 h. The absorbance (OD
value) of each well was measured at 450 nm using a microplate
reader and cell viability was calculated to evaluate the effects of
different CA concentrations on H9c2 cell viability.
ROS detection
To evaluate intracellular ROS levels, a commercially
available ROS assay kit (cat. no. S0033S; Beyotime Institute of
Biotechnology) was employed. H9c2 cells were seeded in a 6-well
plate and cultured until they reached 70-80% confluence.
Post-treatment, the cell suspension was collected and transferred
to centrifuge tubes, followed by centrifugation at 200 × g at room
temperature for 5 min to pellet the cells, after which the
supernatant was carefully removed. The fluorescent probe
2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA) was diluted at
a ratio of 1:1,000 in serum-free medium to achieve a final
concentration of 10 μM and 1 ml of the diluted solution was
added to each tube. The cells were then incubated at 37°C for 20
min to facilitate the uptake of DCFH-DA. After incubation, the
cells were washed twice with serum-free medium, with centrifugation
at 200 × g at room temperature for 5 min after each wash. Finally,
the cell pellets were resuspended in 200 μl serum-free
culture medium and ROS levels were quantified using flow cytometry
(C6 Plus; Becton, Dickinson and Company) and observed under a
fluorescence microscope. The data were analyzed using FlowJo 10.6.2
(BD Biosciences).
Mitochondrial ROS staining
H9c2 cells inoculated in 6-well plates were
collected. Detection of mitochondrial ROS levels was performed
using the MitoSOX Red assay kit according to the manufacturer's
instructions. Cells were washed with PBS and were then examined
under a fluorescence microscope.
Immunofluorescence staining
H9c2 cells were seeded on coverslips in 24-well
plates and grown to 70% confluence before treatment.
Post-treatment, the plates were washed twice with PBS. Fixation was
carried out with 200 μl immunol staining fix solution
(Beyotime Institute of Biotechnology) per well for 20 min at room
temperature, followed by two washes with PBS. Permeabilization was
carried out with 200 μl immunostaining permeabilization
buffer with Triton X-100 (Beyotime Institute of Biotechnology) for
10 min at room temperature, and the cells were washed twice with
PBS. Blocking was achieved with 200 μl 5% BSA for 1 h at
room temperature, followed by two washes with PBS. A Nrf2 primary
antibody (1:500) was then added (200 μl/well) and incubated
overnight at 4°C, followed by two washes with PBS. Subsequently, a
secondary antibody Goat Anti-rabbit IgG HRP (1:500) was added (200
μl/well) and incubated overnight at 4°C or for 90 min at
room temperature, followed by two washes with PBS. DAPI staining
was carried out (200 μl/well) for 10 min at room temperature
and the samples were then washed twice with PBS. Finally, cells
were mounted with antifade medium and images were captured under a
fluorescence microscope.
Annexin V-FITC/PI staining
Annexin V-FITC/PI detection kit (Vazyme Biotech Co,
Ltd.) was used to detect cell death. H9c2 cells were seeded in
6-well plates at 1×105 cells/well and were cultured at
37°C with 5% CO2 until they reached 70-80% confluence.
Post-treatment, the cells were collected and adherent cells were
digested using EDTA-free trypsin for 2 min. Cells were then stained
with 5 μl Annexin V-FITC and 5 μl PI staining
solution, incubated in the dark at room temperature for 10 min and
mixed with 400 μl 1× Binding Buffer. Stained cells were
analyzed by flow cytometry (C6 Plus, Becton, Dickinson and Company)
within 1 h and the data were analyzed using FlowJo 10.6.2 (BD
Biosciences).
Mitochondrial membrane potential assay
with JC-1
Cells were seeded in 6-well plates and cultured
until treatment was completed. The JC-1 working solution was
prepared according to the standard procedure of the JC-1 staining
kit. Cells were incubated with JC-1 working solution at 37°C for 20
min and analyzed using flow cytometry.
Cellular energy metabolism analysis
H9c2 cells were seeded in 6-well plates at
1×105 cells/well and were cultured at 37°C with 5%
CO2 until they reached 70-80% confluence. Detection of
oxygen consumption rate (OCR) and extracellular acidification rate
(ECAR) was performed using the cell mitochondrial stress test kit
and glycolytic stress test kit, following the manufacturer's
instructions. Post-treatment, cells were incubated with Seahorse XF
Base medium at 37°C, followed by the sequential addition of
oligomycin, FCCP [trifluoromethoxy carbonylcyanide phenylhydrazone,
Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone], rotenone
and antimycin A. The Seahorse analyzer [XF96; Agilent Technologies
(China) Co., Ltd.] was used to measure OCR and ECAR of the
cells.
Western blotting
Three rat myocardial tissue samples in each group
were used for Western blotting. Proteins were extracted from
cardiac tissues and cells using RIPA buffer (Wuhan Servicebio
Technology Co., Ltd.) and PMSF (Beyotime Institute of
Biotechnology). The extraction of mitochondrial proteins from cells
was carried out using the mitochondrial isolation and protein
extraction kit (Proteintech Group, Inc.). Protein concentrations
were quantified using a BCA protein assay kit and samples were
adjusted with loading buffer to ensure equal protein loading. A
total of 20 μg protein was then separated by SDS-PAGE on 10%
gels and transferred onto a PVDF membrane. To minimize non-specific
binding, the membrane was blocked with 5% skim milk at room
temperature for 1 h. Subsequently, the membrane was incubated at
the appropriate dilutions overnight at 4°C with the primary
antibodies: Nrf2 (1:1,000), TXNIP (1:1,000), NLRP3 (1:1,000), ASC
(1:5,000), Caspase-1 (1:500), GSDMD (1:1,000), IL-1β (1:1,000),
IL-18 (1:1,000), COX IV (1:2,000) and β-actin (1:50,000). Unbound
primary antibodies were removed by washing the membrane with PBS.
The membrane was then incubated with Goat Anti-rabbit IgG HRP and
Goat Anti-Mouse IgG HRP secondary antibodies (1:10,000) at room
temperature for 1 h to facilitate antibody-antigen complex
formation. Excess secondary antibody was removed by additional PBS
washes. For detection, the membrane was incubated with
electrochemiluminescence substrate solution (Vazyme Biotech Co,
Ltd.) for 3 min in the dark before imaging. β-actin and COX IV were
used as loading controls for total protein and mitochondrial
protein expression, respectively. Band intensities were
semi-quantified using ImageJ 1.52v software (National Institutes of
Health), with β-actin/COX IV serving as the internal control for
normalization. The relative expression levels of the target
proteins were calculated as the ratio of the target protein band
density to the β-actin/COX IV band density.
Statistical analysis
Data analysis was performed using SPSS Statistics
27.0 (IBM Corp.) and GraphPad Prism 10 software (Dotmatics). All
in vitro experimental data are presented from three
independent experiments. Continuous variables were presented as
mean ± SD. Group comparisons were conducted using one-way analysis
of variance followed by Dunnett's post hoc test. Two-way analysis
of variance and post hoc tests were used to compare two variables
(siRNA and treatment). P<0.05 was considered to indicate a
statistically significant difference.
Results
CA effectively improves cardiac function
and mitochondrial damage in HF-induced rats
Echocardiography was used to assess cardiac function
in rats (Fig. 1A) and the
results revealed that LVEF and FS were significantly reduced in the
HF group compared with those in the Sham group. By contrast,
treatment with both doses of CA and with CAP exhibited improved
LVEF and FS (Fig. 1D). H&E
and Masson's trichrome staining further revealed that the
myocardial tissues of HF rats exhibited notable inflammatory
infiltration, cellular loss, disarrangement and interstitial
fibrosis, whereas CA and CAP treatment ameliorated the severity of
myocardial lesions (Fig. 1B, C and
E). In addition, CA and CAP treatment effectively reduced
levels of NT-pro BNP in HF-induced rats (Fig. 1F).
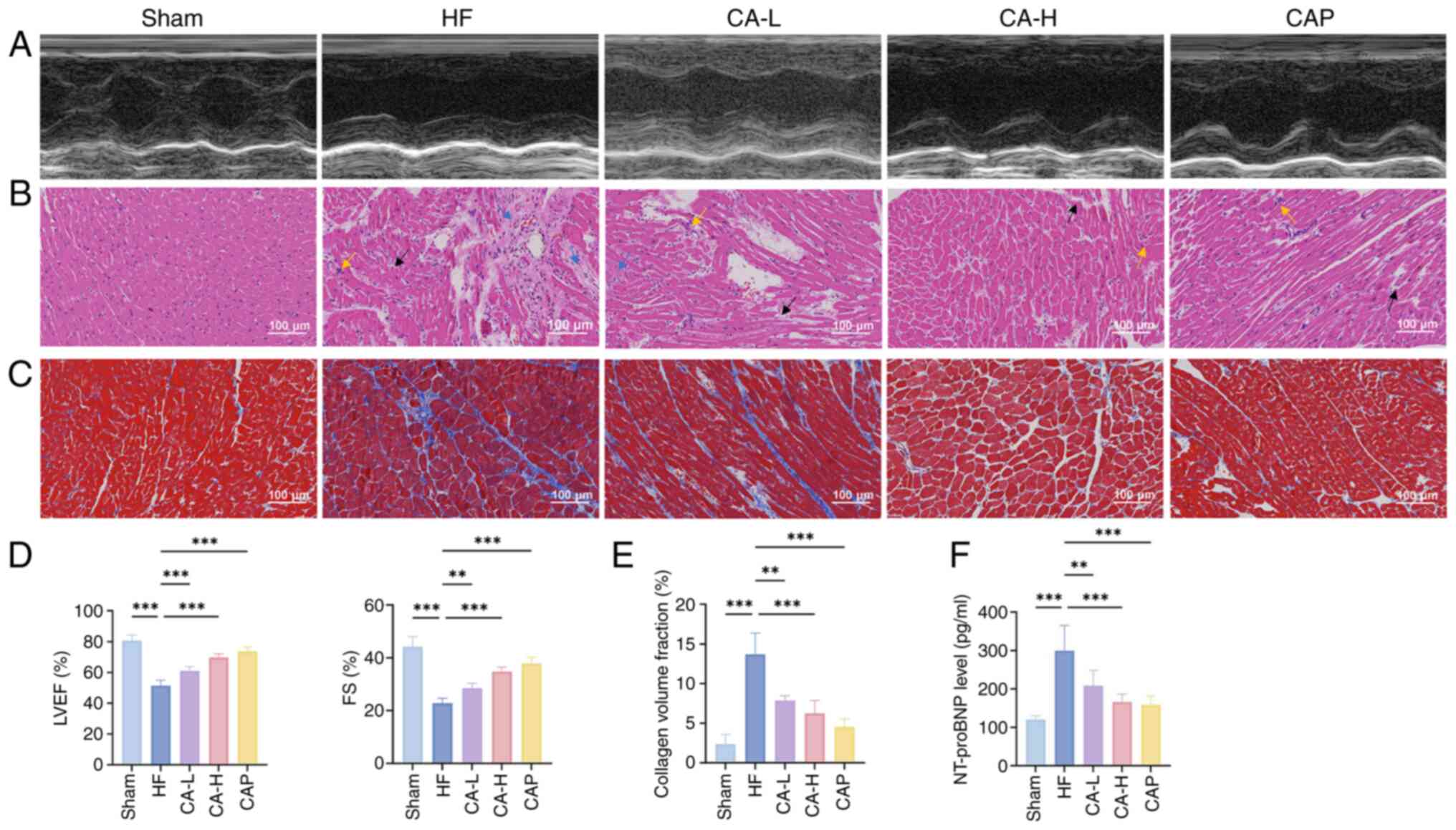 | Figure 1CA effectively improves cardiac
function in HF. (A) Representative echocardiography, (B) H&E
staining and (C) Masson's trichrome staining images of each group.
In H&E staining, the yellow arrows indicate inflammatory
infiltration, the black arrows indicate loss of myocardial cells
and the blue arrows indicate interstitial fibrosis. (D) Statistical
analysis of LVEF and FS of rats in each group, as determined by
echocardiography (n=5). (E) Quantitative analysis of Masson's
trichrome staining (n=3). (F) Serum NT-proBNP levels in rats of
each group (n=5). Data are presented as the mean ± SD,
**P<0.01, ***P<0.001. CA, calycosin;
HF, heart failure; LVEF, left ventricular ejection fraction; FS,
fractional shortening; NT-proBNP, N-terminal pro B-type natriuretic
peptide; CA-L, calycosin low-dose; CA-H, calycosin high-dose; CAP,
captopril. |
TEM was used to observe mitochondrial morphology in
the HF-induced rat myocardium. In addition, the oxidative
stress-related indicators LDH, MDA, SOD and T-GSH/GSSG were
measured to assess the levels of oxidative stress in the HF-induced
rat myocardium. TEM revealed that in the Sham group, the
mitochondria were neatly arranged, the membrane structure was
intact and the cristae were densely structured (Fig. 2A). In the HF group, mitochondrial
arrangement was disorganized, cristae were broken, mitochondria
were swollen and the membrane structure was damaged. By contrast,
CA and CAP treatment resulted in an improvement in mitochondrial
swelling, cristae breakage and relative membrane structural
integrity. In addition, rats with HF exhibited elevated levels of
LDH and MDA, and decreased levels of SOD and T-GSH/GSSG, indicating
significant oxidative stress in HF, which was significantly
prevented by CA and CAP treatment (Fig. 2B).
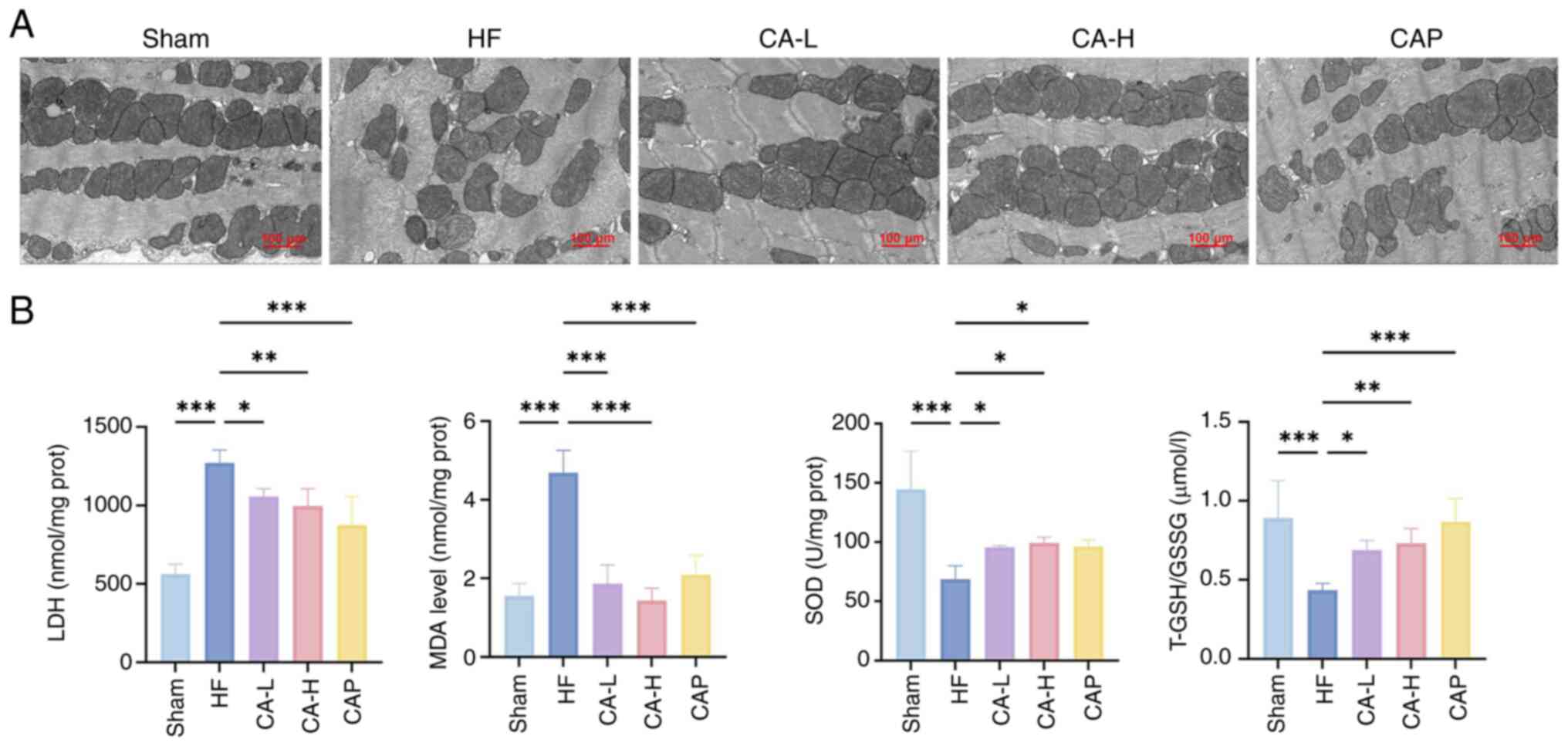 | Figure 2CA effectively improves mitochondrial
damage and cardiac oxidative stress in HF. (A) Representative
transmission electron microscopy images of rats in each group. (B)
Levels of oxidative stress-related indicators (n=5). Data are
presented as the mean ± SD, *P<0.05,
**P<0.01, ***P<0.001. CA, calycosin;
HF, heart failure; CA-L, calycosin low-dose; CA-H, calycosin
high-dose; CAP, captopril; LDH, lactate dehydrogenase; MDA,
malondialdehyde; SOD, superoxide dismutase; GSH, glutathione; GSSG,
GSH disulfide. |
CA inhibits HF-induced pyroptosis via the
Nrf2/ROS/TXNIP pathway
To further explore the mechanism of CA in the
treatment of HF, the present study measured the expression of
proteins in the Nrf2/ROS/TXNIP pathway and related pyroptosis
proteins. Immunohistochemistry staining indicated that Nrf2
expression was upregulated in the HF group compared with the Sham
group, and was further increased following CA treatment (Fig. 3A). Western blotting results
further confirmed that compared with in the Sham group, Nrf2 and
TXNIP expression levels were upregulated in HF-induced rats. CA
treatment further upregulated the expression levels of Nrf2 and
inhibited the expression of TXNIP (Fig. 3B and C). In HF-induced rats, the
expression of related pyroptosis proteins and inflammatory
cytokines was upregulated, specifically, the NLRP3 inflammasome was
activated, and the expression levels of NLRP3, ASC, pro- and
cleaved-caspase-1, GSDMD, GSDMD-NT (Fig. 3D and E), IL-18 and IL-1β
(Fig. 3F and G) were elevated.
By contrast, CA treatment significantly downregulated the
expression of these pyroptosis-related proteins and inflammatory
cytokines.
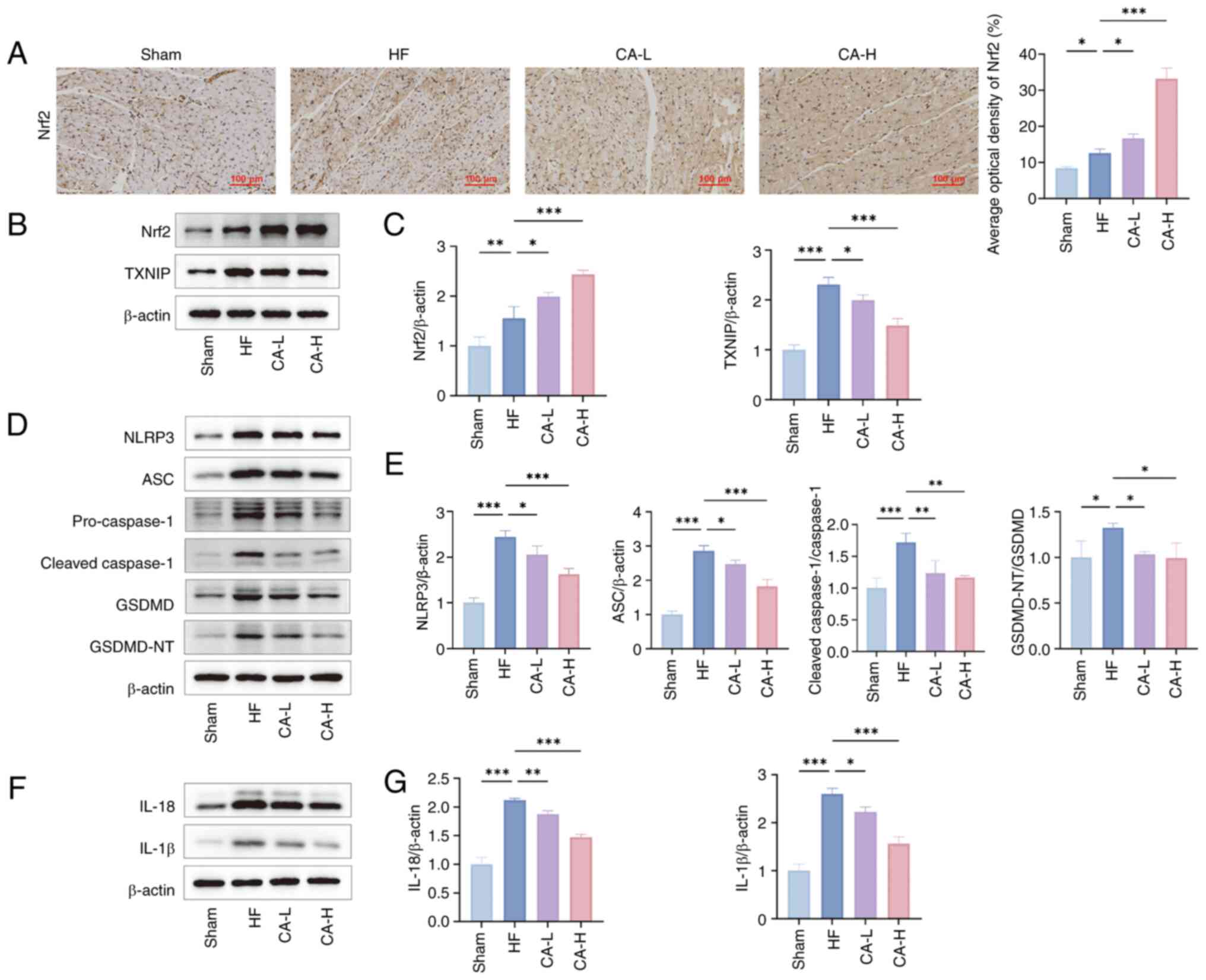 | Figure 3CA improves pyroptosis in HF through
the Nrf2/ROS/TXNIP pathway. (A) Representative Nrf2
immunohistochemistry images of each group and their quantitative
analysis (n=3). (B) Representative images of the expression levels
of Nrf2 and TXNIP in myocardial tissue of Wistar rats assessed by
western blotting. (C) Semi-quantitative analysis of Nrf2 and TXNIP
expressions (n=3). (D) Representative images of the expression
levels of NLRP3, ASC, pro- and cleaved-caspase-1, GSDMD, GSDMD-NT
in myocardial tissue of Wistar rats by western blotting. (E)
Semi-quantitative analysis of NLRP3, ASC, pro- and
cleaved-caspase-1, GSDMD, GSDMD-NT expressions (n=3). (F)
Representative images of the expression levels of IL-18 and IL-1β
in myocardial tissue of Wistar rats by western blotting. (G)
Semi-quantitative analysis of IL-18 and IL-1β expressions (n=3).
Data are presented as the mean ± SD, *P<0.05,
**P<0.01, ***P<0.001. CA, calycosin;
HF, heart failure; Nrf2, nuclear factor erythroid 2-related factor;
ROS, reactive oxygen species; TXNIP, thioredoxin-interacting
protein; CA-L, calycosin low-dose; CA-H, calycosin high-dose; ASC,
apoptosis-associated speck-like protein containing a CARD; GSDMD,
gasdermin D; GSDMD-NT, GSDMD N-terminal fragments. |
CA protects cardiomyocytes and inhibits
pyroptosis via the Nrf2/ROS/TXNIP pathway
To further validate the potential cardioprotective
effects and therapeutic mechanisms of CA, further experiments were
conducted in H/R-treated H9c2 cells. The CCK-8 assay was used to
examine the biosafety of CA in H9c2 cells and the results showed
that CA was cytotoxic at concentrations ≥40 μM (Fig. 4A). Subsequently, the H/R model
was constructed and the cardioprotective effects of CA were
evaluated through the CCK-8 assay and by measuring LDH levels. The
results showed that H/R treatment significantly reduced cell
viability and promoted LDH release, whereas CA treatment
dose-dependently reversed this effect (Fig. 4B).
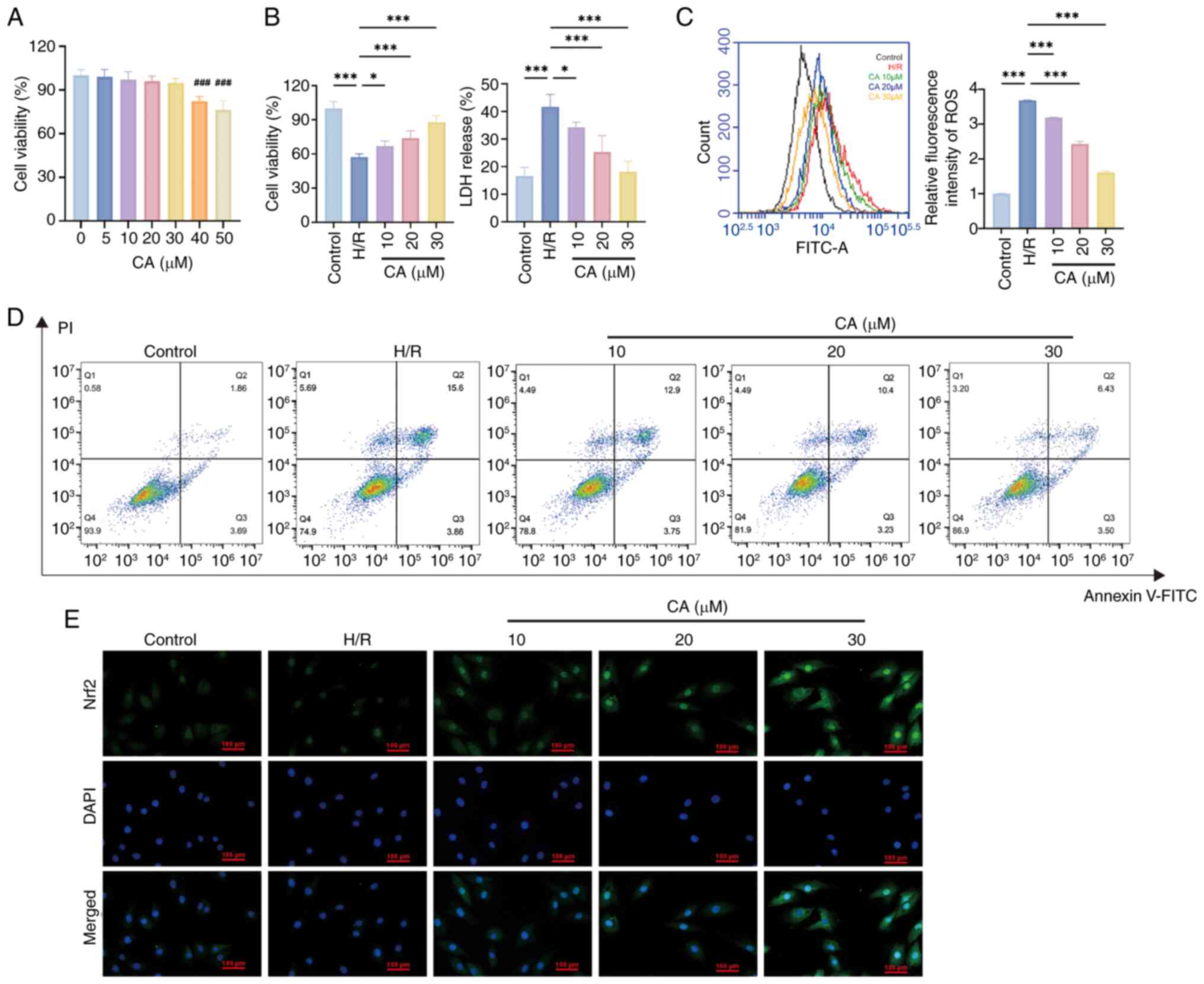 | Figure 4CA protects cardiomyocytes and
inhibits pyroptosis. (A) Detection of CA cytotoxicity using a Cell
Counting Kit-8 assay (n=3). ###P<0.001, compared with
0, 5, 10, 20, 30 μM groups. (B) Cardiomyocyte protective
effects of CA (n=3). (C) Flow cytometric analysis of cell death
with Annexin V-FITC/PI staining. (D) Immunofluorescence staining of
cardiomyocytes for Nrf2. (E) Detection of intracellular ROS levels
in cardiomyocytes (n=3). Data are presented as the mean ± SD,
*P<0.05, ***P<0.001. CA, calycosin;
Nrf2, nuclear factor erythroid 2-related factor; ROS, reactive
oxygen species; H/R, hypoxia-reperfusion; LDH, lactate
dehydrogenase. |
During pyroptosis, GSDMD-NT can form membrane pores
in the plasma membrane and mitochondrial membrane, disrupting
membrane integrity and thereby leading to mitochondrial damage and
cell death, which was observed in the present study by Annexin V/PI
staining, with an increase in Annexin V+/PI+
cells detected in response to H/R, while CA treatment ameliorated
this increase in Annexin V+/PI+ cells (Fig. 4C). Immunofluorescence staining of
cardiomyocytes for Nrf2 revealed that Nrf2 levels were increased by
H/R treatment and further increased by CA treatment, and that CA
promoted Nrf2 translocation to the nucleus (Fig. 4D). Furthermore, detection of
intracellular ROS levels revealed elevated ROS release in response
to H/R treatment, whereas CA treatment significantly reduced ROS
release (Fig. 4E). Western
blotting further indicated that H/R treatment upregulated the
expression levels of Nrf2, TXNIP, activated the NLRP3 inflammasome
and increased the expression levels of NLRP3, ASC, pro- and
cleaved-caspase-1, GSDMD, GSDMD-NT, IL-18 and IL-1β. CA treatment
further upregulated, the expression of Nrf2, inhibited the
expression of TXNIP (Fig. 5A)
and suppressed the expression and release of NLRP3, ASC, pro- and
cleaved-caspase-1, GSDMD, GSDMD-NT (Fig. 5C), IL-18 and IL-1β (Fig. 5B).
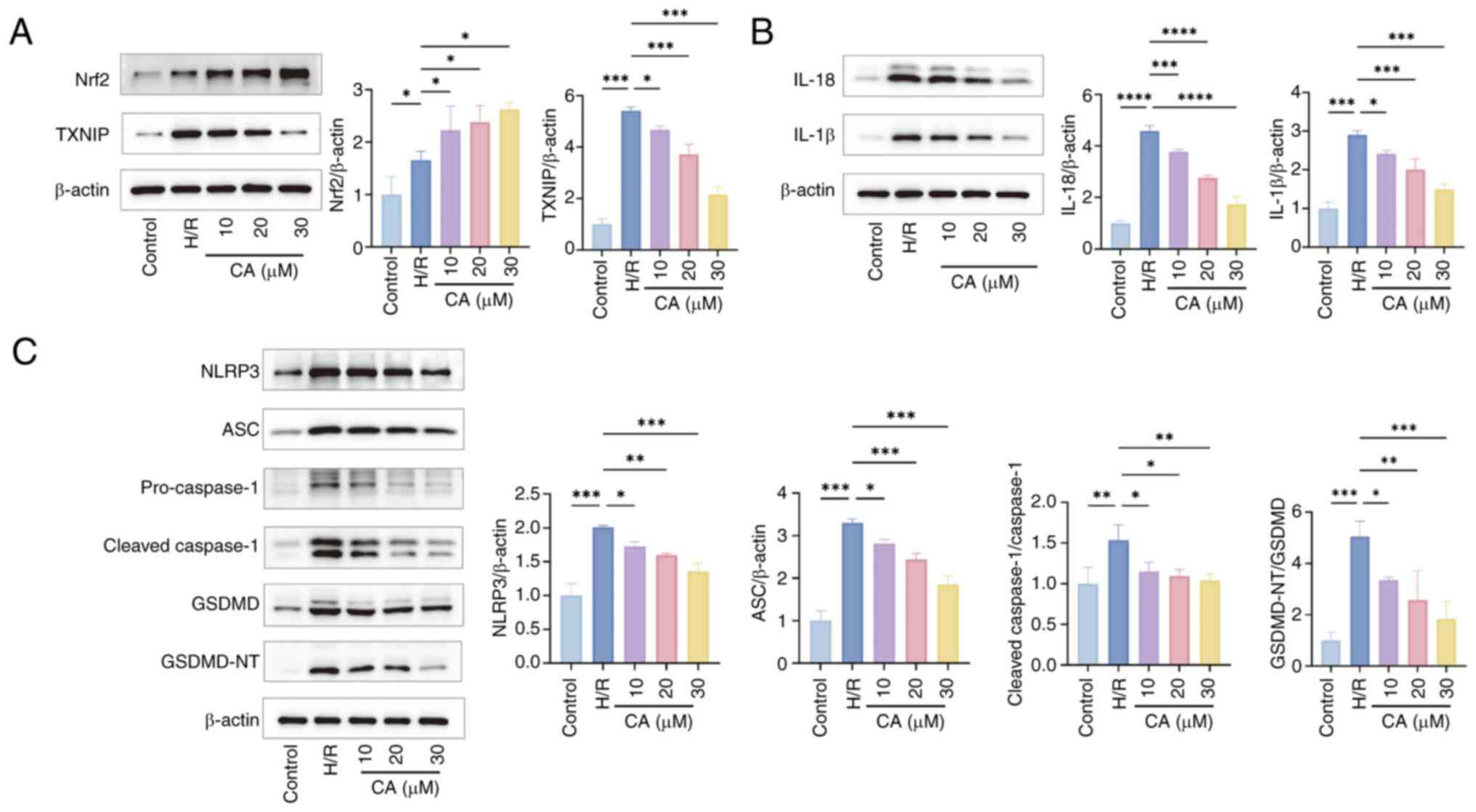 | Figure 5CA inhibits pyroptosis via the
Nrf2/reactive oxygen species/TXNIP pathway (n=3). (A)
Representative images of the expression levels and
semi-quantitative analysis of Nrf2 and TXNIP in cells by western
blotting (n=3). (B) and semi-quantitative analysis and
semi-quantitative analysis of IL-18 and IL-1β in cells by western
blotting and (n=3). (C) and semi-quantitative analysis and
semi-quantitative analysis of levels of NLRP3, ASC, pro- and
cleaved-caspase-1, GSDMD, GSDMD-NT in cells by western blotting
(n=3). Data are presented as the mean ± SD, *P<0.05,
**P<0.01, ***P<0.001. CA, calycosin;
Nrf2, nuclear factor erythroid 2-related factor; TXNIP,
thioredoxin-interacting protein; H/R, hypoxia-reperfusion, ASC,
apoptosis-associated speck-like protein containing a CARD; GSDMD,
gasdermin D; GSDMD-NT, GSDMD N-terminal fragments; NLRP3, NLR
family pyrin domain-containing protein 3. |
CA improves mitochondrial damage in
cardiomyocytes
Since CA protects cardiomyocytes in a dose-dependent
manner, CA 30 μM was used in the present study to further
investigate the improvement effect on mitochondrial damage. Flow
cytometry revealed that H/R treatment significantly decreased
mitochondrial membrane potential, while CA treatment prevented this
change (Fig. 6A). Furthermore,
ROS and mitochondrial ROS staining of the cells revealed an
increase in intracellular and mitochondrial ROS with H/R treatment,
which was improved by CA treatment (Fig. 6B and C). The ECAR and OCR were
used to assess mitochondrial glucose metabolism and oxidative
phosphorylation levels. As shown in Fig. 6D, H/R treatment promoted
glycolysis and inhibited mitochondrial respiration, while CA
treatment ameliorated this mitochondrial damage. In addition,
western blotting results indicated that H/R treatment upregulated
GSDMD expression levels, promoted GSDMD-NT activation and
upregulated cytochrome c expression levels in the
mitochondria; by contrast, CA treatment downregulated their
expression and activation (Fig.
6E).
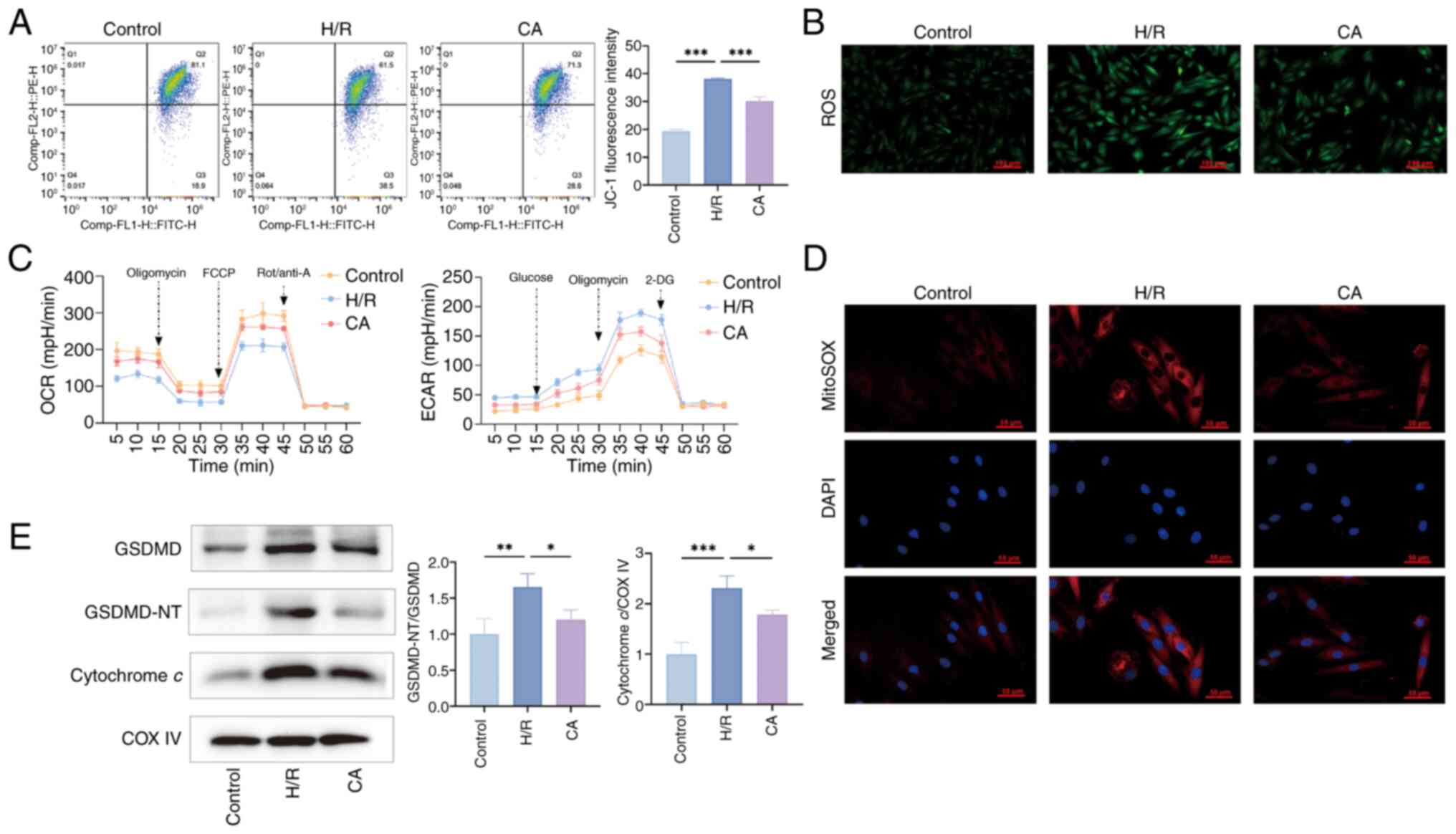 | Figure 6CA improves mitochondrial damage in
cardiomyocytes. (A) Flow cytometric analysis of JC-1 staining was
used to detect changes in mitochondrial membrane potential (n=3).
(B) Representative intracellular ROS staining images of each group.
(C) Representative mitochondrial ROS staining images of each group.
(D) ECAR and OCR of cardiomyocytes were assessed (n=3). (E)
Expression levels of GSDMD, GSDMD-NT and cytochrome c
proteins were examined by western blotting (n=3). COX IV was used
as a loading control in mitochondria. Data are presented as the
mean ± SD, *P<0.05, **P<0.01,
***P<0.001. CA, calycosin; ROS, reactive oxygen
species; ECAR, extracellular acidification rate; OCR, oxygen
consumption rate; FCCP, Trifluoromethoxy carbonylcyanide
phenylhydrazone, Carbonyl cyanide
4-(trifluoromethoxy)phenylhydrazone; Rot/anti-A, rotenone/antimycin
A; GSDMD, gasdermin D; GSDMD-NT, GSDMD N-terminal fragments; H/R,
hypoxia-reperfusion. |
Silencing Nrf2 inhibits the therapeutic
effects of CA
To further validate the therapeutic mechanism of CA,
Nrf2 expression was knocked down using siRNA in H9c2 cells. After
verifying the efficiency of Nrf2 silencing (Fig. 7A), detection of intracellular ROS
levels confirmed that Nrf2 silencing significantly upregulated
intracellular ROS levels compared with Si-NC group, further
suggesting that CA exerts antioxidant effects through Nrf2
(Fig. 7B). Western blotting
results revealed that compared with the Si-NC group, Si-Nrf2
significantly reduced Nrf2 protein expression levels. Compared with
the Si-NC group, Nrf2 silencing in the Si-Nrf2 group significantly
upregulated the expression levels of TXNIP, activated NLRP3
inflammasome, and increased the expression levels of NLRP3, ASC,
pro- and cleaved-caspase-1, GSDMD, GSDMD-NT, IL-18 and IL-1β,
further confirming that Nrf2 silencing reversed the inhibitory
effect of CA on pyroptosis (Fig.
7C-E).
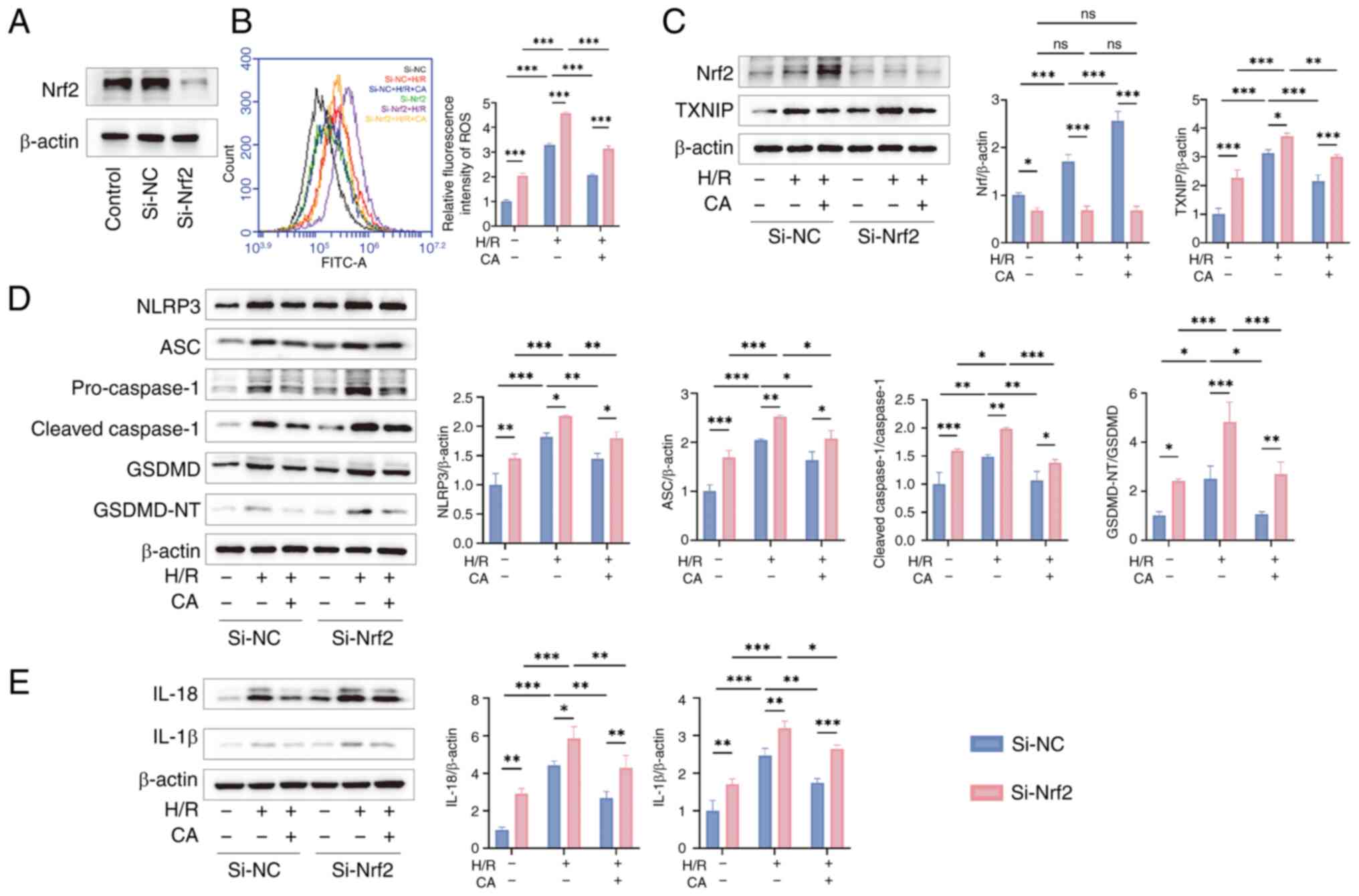 | Figure 7Silencing Nrf2 inhibits the
therapeutic effects of CA. (A) Transfection efficiency of si-Nrf2.
(B) Detection of intracellular ROS levels in each group (n=3). (C)
Representative images and semi-quantitative analysis of Nrf2 and
TXNIP in cells by western blotting (n=3). (D) Representative images
and semi-quantitative analysis of NLRP3, ASC, pro- and
cleaved-caspase-1, GSDMD, GSDMD-NT in cells by western blotting
(n=3). (E) Representative images and semi-quantitative analysis
levels of IL-18 and IL-1β in cells by western blotting (n=3). Data
are presented as the mean ± SD, *P<0.05,
**P<0.01, ***P<0.001, ns, no
significance. si, small interfering; CA, calycosin; Nrf2, nuclear
factor erythroid 2-related factor; ROS, reactive oxygen species;
TXNIP, thioredoxin-interacting protein; ASC, apoptosis-associated
speck-like protein containing a CARD; NLRP3, NLR family pyrin
domain-containing protein 3; GSDMD, gasdermin D; GSDMD-NT, GSDMD
N-terminal fragments; H/R, hypoxia-reperfusion; NC, negative
control. |
Discussion
There is a consensus that inflammation, oxidative
stress, mitochondrial dysfunction and cell death serve a central
role in the progression of HF (7,8,26). However, the specific mechanisms
of pyroptosis and effective interventions in HF remain unclear,
especially since previous studies have revealed that pyroptosis
also mediates mitochondrial damage in cells (13,27). To the best of our knowledge, the
present study is the first to systematically investigate the
therapeutic mechanism of CA to attenuate mitochondrial damage and
pyroptosis in HF via the Nrf2/ROS/TXNIP pathway, which not only
demonstrated the cardioprotective effects of CA, but also confirmed
the complex interactions between oxidative stress, mitochondrial
damage and pyroptosis. Therefore, the present study proposes the
Nrf2/ROS/TXNIP pathway as a potentially promising therapeutic
target for treating mitochondrial damage and pyroptosis in HF.
The natural flavonoid compound CA is the main
bioactive chemical in Radix Astragali, and has been shown to exert
anti-HF therapeutic effects (17-20). Previous studies have demonstrated
that CA regulates oxidative stress by modulating Nrf2 expression
(28,29), but the specific mechanism remains
to be investigated. Previous studies have demonstrated that CA
inhibits NLRP3 inflammasome activation-mediated pyroptosis and
ameliorates myocardial injury (21,22). However, these studies mainly
focused on phenotypes and downstream effector proteins, while the
effects on upstream signaling pathways were insufficiently studied,
resulting in an incomplete understanding of the mechanism of CA
treatment. Meanwhile, another study revealed that CA upregulates
Nrf2 expression levels, and alleviates myocardial oxidative stress
and mitochondrial damage, thereby protecting the myocardium
(21), warranting further
investigation of the effect of CA on Nrf2 regulation and pyroptosis
in the failing myocardium. The present study indicated that CA
improved cardiac function, attenuated cardiac oxidative stress, and
prevented myocardial fibrosis and inflammatory injury in HF via the
Nrf2/ROS/TXNIP pathway, demonstrating the complex interactions
between oxidative stress, mitochondrial damage and pyroptosis.
Nrf2 is a key regulator of cellular defense
mechanisms, particularly regulating cellular antioxidant responses
under oxidative stress conditions (15). During oxidative stress, Nrf2
dissociates from Kelch-like ECH-associated protein 1 (Keap1),
translocates to the nucleus and activates the transcription of
antioxidant-responsive genes, enhancing ROS cellular scavenging
(30). TXNIP is a negative
regulator downstream of Nrf2/ROS. The activation of Nrf2 reduces
ROS levels, thereby inhibiting ROS-mediated dissociation of TXNIP
and TRX, and consequently attenuating TXNIP expression (31,32). Downregulation of TXNIP weakens
the interaction between TXNIP and NLRP3, thereby suppressing NLRP3
inflammasome activation and ultimately attenuating pyroptosis and
inflammatory responses (33,34). Therefore, Nrf2 is a key factor
linking oxidative stress and NLRP3 inflammasome activation.
Considerable oxidative stress is present in HF (8), resulting in elevated Nrf2 levels,
but this elevation is limited (35). Therefore, although Nrf2
expression is elevated, it is insufficient to completely counteract
the complex inflammatory response and oxidative stress, therefore
TXNIP expression remains high in HF (36-38). The present study has found that
both Nrf2 and TXNIP expression were upregulated in HF, consistent
with previous research findings. In the present study, CA treatment
was shown to improve oxidative stress indicators, such as LDH, MDA,
SOD and T-GSH/GSSG, in HF-induced rats. Western blotting and flow
cytometry results also confirmed that CA treatment upregulated Nrf2
expression, decreased ROS levels, and inhibited the expression of
TXNIP and corresponding pyroptosis proteins. Nrf2
immunofluorescence experiments revealed that CA treatment elevated
Nrf2 expression levels and confirmed its nuclear translocation.
These results highlight the role of Nrf2 in the treatment of HF
with CA.
To further confirm the central role of Nrf2 in the
anti-HF effects of CA, the present study used siRNA to silence
Nrf2. The results revealed that compared to the Si-NC group,
silencing Nrf2 markedly reduced the protective effects of CA,
including a significant increase in ROS levels and the expression
of downstream molecules. However, the natural compound CA has
multi-pathway, multi-target therapeutic advantages and its
therapeutic mechanism may involve the synergistic action of
multiple signaling pathways. Studies have revealed that CA can
improve myocardial fibrosis and cardiac dysfunction through the
TGFBR1 signaling pathway and can also inhibit inflammation and
fibrosis in HF-induced rats through the AKT-IKK/STAT3 axis
(19,20).
Pyroptosis, a form of programmed cell death mediated
by GSDMD, has been reported to serve a key role in the pathological
process of HF, exacerbating inflammation and tissue damage
(11). Specifically, GSDMD is
activated by specific signals and undergoes a conformational change
into GSDMD-NT, which perforates the cell membrane, leading to
leakage of cellular contents and cellular swelling, ultimately
triggering cell rupture and death (12,39). The significant inflammatory
response and oxidative stress present in HF can activate multiple
pathways, such as the NF-κB signaling pathway that promotes GSDMD
and Caspase-1 transcription and synthesis. The in vivo and
in vitro results further confirmed that CA effectively
suppressed this synthesis. Furthermore, it inhibited NLRP3
inflammasome activation, thereby suppressing the further cleavage
and activation of GSDMD and Caspase-1, suggesting that CA inhibits
pyroptosis in failing cardiomyocytes. Mitochondrial dysfunction is
involved in a variety of pathological mechanisms such as disturbed
energy metabolism, dysregulation of calcium homeostasis,
inflammatory response and cell death, and has emerged as a key
target for HF (40). A recent
study confirmed that activation of GSDMD-NT during pyroptosis leads
not only to perforated rupture of cellular membranes, but also to
perforation of mitochondrial membranes, which in turn leads to
mitochondrial damage and release of ROS (13). Pyroptosis driven by GSDMD not
only leads to myocardial damage but also amplifies mitochondrial
damage and promotes the process toward HF. The crosstalk between
pyroptosis and mitochondrial dysfunction underscores the complexity
of HF pathophysiology. In the present study, CA treatment not only
attenuated pyroptosis but also ameliorated mitochondrial damage.
TEM examination of HF rats revealed that CA improved mitochondrial
ultrastructure and in vitro cellular experiments further
demonstrated that CA treatment downregulated the expression of
mitochondrial GSDMD-NT and improved mitochondrial membrane
potential, mitochondrial ROS levels and mitochondrial respiration,
which suggests that CA exerts its cardioprotective effects through
multiple pathways (Fig. 8).
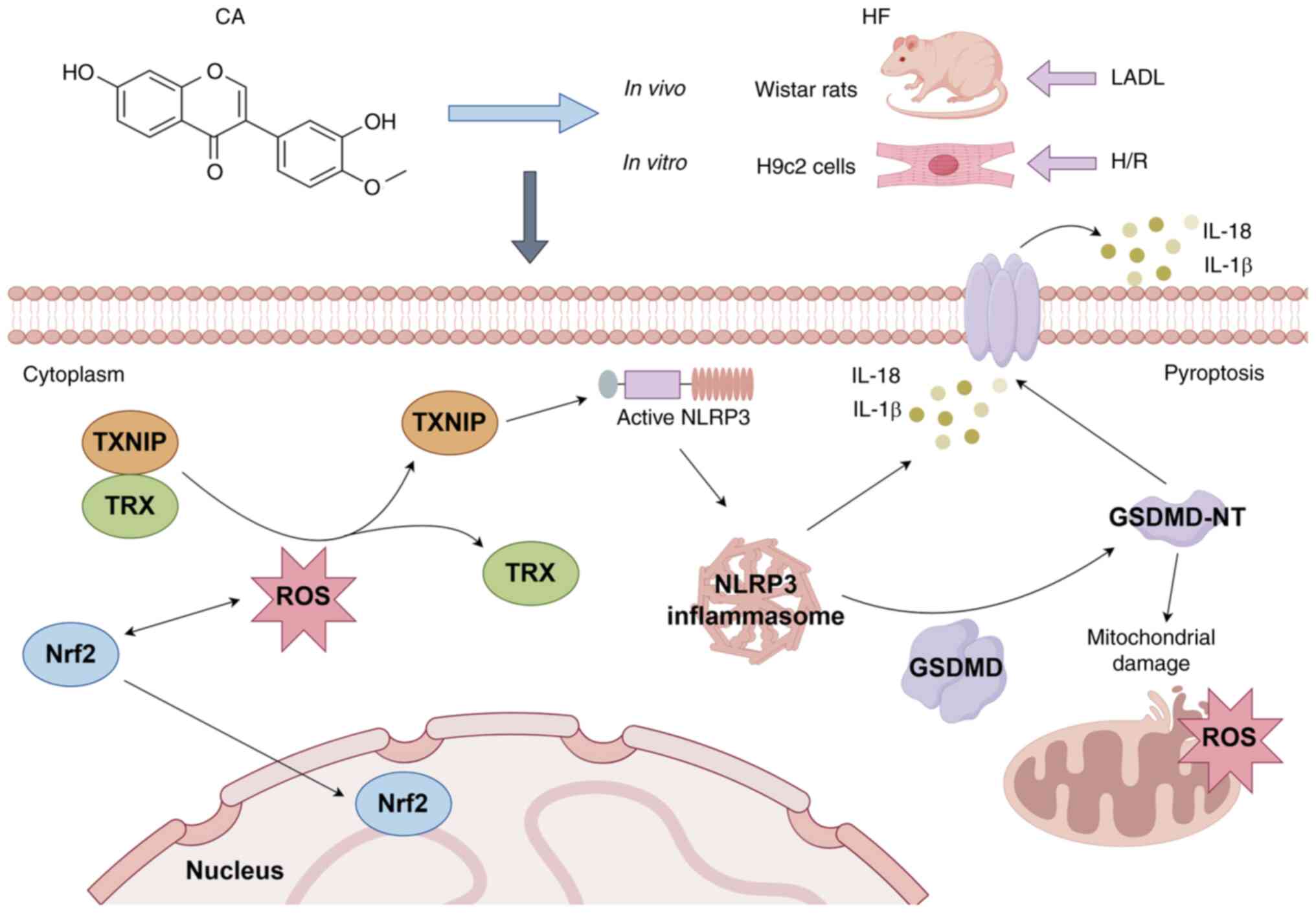 | Figure 8Molecular mechanisms of CA treatment
for HF. CA promotes Nrf2 expression and its nuclear translocation,
thereby alleviating mitochondrial damage and pyroptosis in HF via
the Nrf2/ROS/TXNIP pathway. This figure was generated by FigDraw.
CA, calycosin; HF, heart failure; LADL, left anterior descending
ligation; Nrf2, nuclear factor erythroid 2-related factor; ROS,
reactive oxygen species; TRX, thioredoxin; TXNIP,
thioredoxin-interacting protein; H/R, hypoxia-reoxygenation; NLRP3,
NLR family pyrin domain-containing protein 3; GSDMD, gasdermin D;
GSDMD-NT, GSDMD N-terminal fragments. |
Natural compounds have demonstrated promising
potential in the treatment of HF due to their multi-targeting
properties, low side effects, and excellent anti-inflammatory and
antioxidant effects (41-43).
Compared with other similar natural compounds that modulate Nrf2,
such as sulforaphane and curcumin, CA has unique advantages and
preclinical therapeutic potential. Sulforaphane has antioxidant and
anti-inflammatory properties and has been shown to improve cardiac
function and prevent worsening of HF by regulating Nrf2 expression
levels (44). However, CA has a
unique advantage because of its ability to inhibit pyroptosis and
regulate the process of cell death in cardiomyocytes (21,22). Curcumin has antioxidant,
anti-apoptotic and anti-inflammatory properties, and has been shown
to improve HF by ameliorating ventricular hypertrophy and
inhibiting myocardial fibrosis (45,46). However, the low stability of
curcumin limits its further clinical translation (47). By contrast, CA has improved
stability and may be a more suitable choice for the clinical
treatment of HF.
The present study used LAD ligation to establish the
HF model and demonstrated that CA improved cardiac function and
myocardial damage, suppressed myocardial oxidative stress and
improved mitochondrial ultrastructure in HF-induced rats. However,
the LAD ligation model is widely used to study ischemic HF, as it
simulates the pathophysiological characteristics of human ischemic
HF by inducing local myocardial ischemia (48), which may not reflect the
pathological changes and cardiac remodeling specific to
non-ischemic HF. In H9c2 cells, the present study used the H/R
model to simulate HF and further confirmed the therapeutic
mechanism of CA against HF. However, the H/R model simulates
myocardial ischemia-reperfusion injury, which captures the key
initiating mechanisms associated with ischemia-reperfusion stress
injury, but cannot fully reflect the complex, chronic adaptive and
maladaptive processes of chronic HF, whether of ischemic or
non-ischemic origin (49).
Notably, several limitations should be acknowledged.
Firstly, the LAD ligation model primarily simulates ischemic HF,
and the H/R model in H9c2 cells, although capable of capturing the
key mechanisms of myocardial ischemia-reperfusion stress injury,
may not fully reflect the pathophysiology of chronic HF. The
detection of ROS data at a single time point limits dynamic
understanding of oxidative stress in HF, therefore, this should be
further investigated using multiple time points. Furthermore, H9c2
cells are widely used in studies related to cardiovascular disease
(50-52), but they cannot fully represent
the physiological characteristics of primary cardiomyocytes. Future
studies should use primary cells to provide an improved simulation
of the pathophysiological progression of HF. Secondly, the present
study showed that CA inhibits pyroptosis in HF and improves
mitochondrial damage, possibly disrupting the crosstalk between
mitochondrial damage and pyroptosis. However, the exact mechanism
remains to be further validated through experiments such as
co-immunoprecipitation or proximity ligation assays to determine
the interaction between GSDMD-NT and mitochondria or using
cytosolic fractions to confirm mitochondrial outer membrane
permeabilization. Nrf2 silencing eliminated the protective effect
of CA; however, it remains unclear as to whether the effects of CA
are directly mediated by Nrf2 or through upstream/downstream
interactions. Therefore, additional validation is needed, including
experiments such as Nrf2 overexpression rescue after Nrf2
knockdown, or TXNIP knockout and Keap1-Nrf2 interaction after Nrf2
knockdown to confirm the specificity of Nrf2 and causality.
Finally, the present study is a preclinical basic study, which is a
preliminary exploration of the pharmacological efficacy of CA. The
clinical translational application of CA requires further
evaluation of aspects such as pharmacokinetics (bioavailability,
tissue distribution and metabolic stability) and safety. The sample
size of some experiments in the present study was also relatively
small; although consistent trends were observed in repeated
measurements, a larger sample size would enhance the accuracy and
scientific validity of the present study.
To the best of our knowledge, the present study
demonstrated, for the first time, that CA possibly disrupts the
crosstalk between pyroptosis-mitochondrial damage in HF via the
Nrf2/ROS/TXNIP pathway. CA may attenuate the activation of upstream
triggers of pyroptosis, ROS overproduction and downregulate the
downstream actuator protein, GSDMD-NT, thereby ameliorating cardiac
function and myocardial injury in HF. However, additional
validation is still required to further confirm the specificity of
Nrf2 in CA treatment.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HJY, YDY, YTX and YL conceived the study. HJY, QCH
and HY carried out the experiments. YDY and XJL analyzed the data.
HJY and QCH drafted the manuscript. YTX and YL reviewed and revised
the manuscript. YTX and YL confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The animal experiment was reviewed and approved by
the Experimental Animal Management Committee and the Ethics
Committee of the Affiliated Hospital of Shandong University of
Traditional Chinese Medicine (approval no.
SDSZYYAWE20231031001).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
ASC
|
apoptosis-associated speck-like
protein containing a CARD
|
|
CA
|
calycosin
|
|
CA-L
|
CA low-dose
|
|
CA-H
|
CA high-dose
|
|
CAP
|
captopril
|
|
CCK-8
|
Cell Counting Kit-8
|
|
ECAR
|
extracellular acidification rate
|
|
FCCP
|
Trifluoromethoxy carbonylcyanide
phenylhydrazone, Carbonyl cyanide
4-(trifluoromethoxy)phenylhydrazone
|
|
FS
|
fractional shortening
|
|
GSDMD
|
gasdermin D
|
|
GSDMD-NT
|
GSDMD N-terminal fragments
|
|
HF
|
heart failure
|
|
H/R
|
hypoxia-reoxygenation
|
|
Keap1
|
Kelch-like ECH-associated protein
1
|
|
LAD
|
left anterior descending
|
|
LDH
|
lactate dehydrogenase
|
|
LVEF
|
left ventricular ejection fraction
|
|
MDA
|
malondialdehyde
|
|
NLRP3
|
NLR family pyrin domain-containing
protein 3
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor
|
|
NT-proBNP
|
N-terminal pro B-type natriuretic
peptide
|
|
OCR
|
oxygen consumption rate
|
|
ROS
|
reactive oxygen species
|
|
SOD
|
superoxide dismutase
|
|
TEM
|
transmission electron microscopy
|
|
T-GSH/GSSG
|
total glutathione/glutathione
disulfide
|
|
TRX
|
thioredoxin
|
|
TXNIP
|
thioredoxin-interacting protein
|
Acknowledgements
Not applicable.
Funding
This work was supported by the Natural Science Foundation of
Shandong Province (grant no. ZR2023MH053).
References
|
1
|
Tsao CW, Aday AW, Almarzooq ZI, Anderson
CAM, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK,
Buxton AE, et al: Heart disease and stroke statistics-2023 update:
A report from the American heart association. Circulation.
147:e93–e621. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khan MS, Shahid I, Bennis A, Rakisheva A,
Metra M and Butler J: Global epidemiology of heart failure. Nat Rev
Cardiol. 21:717–734. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bozkurt B, Fonarow GC, Goldberg LR, Guglin
M, Josephson RA, Forman DE, Lin G, Lindenfeld J, O'Connor C,
Panjrath G, et al: Cardiac rehabilitation for patients with heart
failure: JACC expert panel. J Am Coll Cardiol. 77:1454–1469. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zannad F, O'Connor CM, Butler J, McMullan
CJ, Anstrom KJ, Barash I, Bonaca MP, Borentain M, Corda S, Gates D,
et al: Vericiguat for patients with heart failure and reduced
ejection fraction across the risk spectrum: An individual
participant data analysis of the VICTORIA and VICTOR trials.
Lancet. August 30–2025.Epub ahead of print. View Article : Google Scholar
|
|
5
|
Cools JMT, Goovaerts BK, Feyen E, Van den
Bogaert S, Fu Y, Civati C, Van Fraeyenhove J, Tubeeckx MRL, Ott J,
Nguyen L, et al: Small-molecule-induced ERBB4 activation to treat
heart failure. Nat Commun. 16:5762025. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rossignol P, Hernandez AF, Solomon SD and
Zannad F: Heart failure drug treatment. Lancet. 393:1034–1044.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brown DA, Perry JB, Allen ME, Sabbah HN,
Stauffer BL, Shaikh SR, Cleland JG, Colucci WS, Butler J, Voors AA,
et al: Expert consensus document: Mitochondrial function as a
therapeutic target in heart failure. Nat Rev Cardiol. 14:238–250.
2017. View Article : Google Scholar
|
|
8
|
van der Pol A, van Gilst WH, Voors AA and
van der Meer P: Treating oxidative stress in heart failure: Past,
present and future. Eur J Heart Fail. 21:425–435. 2019. View Article : Google Scholar
|
|
9
|
Dridi H, Kushnir A, Zalk R, Yuan Q,
Melville Z and Marks AR: Intracellular calcium leak in heart
failure and atrial fibrillation: A unifying mechanism and
therapeutic target. Nat Rev Cardiol. 17:732–747. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xiang Q, Yi X, Zhu XH, Wei X and Jiang DS:
Regulated cell death in myocardial ischemia-reperfusion injury.
Trends Endocrinol Metab. 35:219–234. 2024. View Article : Google Scholar
|
|
11
|
Zhang Z, Yang Z, Wang S, Wang X and Mao J:
Overview of pyroptosis mechanism and in-depth analysis of
cardiomyocyte pyroptosis mediated by NF-κB pathway in heart
failure. Biomed Pharmacother. 179:1173672024. View Article : Google Scholar
|
|
12
|
Shi J, Zhao Y, Wang K, Shi X, Wang Y,
Huang H, Zhuang Y, Cai T, Wang F and Shao F: Cleavage of GSDMD by
inflammatory caspases determines pyroptotic cell death. Nature.
526:660–665. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miao R, Jiang C, Chang WY, Zhang H, An J,
Ho F, Chen P, Zhang H, Junqueira C, Amgalan D, et al: Gasdermin D
permeabilization of mitochondrial inner and outer membranes
accelerates and enhances pyroptosis. Immunity. 56:2523–2541.e8.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou R, Tardivel A, Thorens B, Choi I and
Tschopp J: Thioredoxin-interacting protein links oxidative stress
to inflammasome activation. Nat Immunol. 11:136–140. 2010.
View Article : Google Scholar
|
|
15
|
Morgenstern C, Lastres-Becker I,
Demirdöğen BC, Costa VM, Daiber A, Foresti R, Motterlini R,
Kalyoncu S, Arioz BI, Genc S, et al: Biomarkers of NRF2 signalling:
Current status and future challenges. Redox Biol. 72:1031342024.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sies H and Jones DP: Reactive oxygen
species (ROS) as pleiotropic physiological signalling agents. Nat
Rev Mol Cell Biol. 21:363–383. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deng M, Chen H, Long J, Song J, Xie L and
Li X: Calycosin: A review of its pharmacological effects and
application prospects. Expert Rev Anti Infect Ther. 19:911–925.
2021. View Article : Google Scholar
|
|
18
|
Ding WJ, Chen GH, Deng SH, Zeng KF, Lin
KL, Deng B, Zhang SW, Tan ZB, Xu YC, Chen S, et al: Calycosin
protects against oxidative stress-induced cardiomyocyte apoptosis
by activating aldehyde dehydrogenase 2. Phytother Res. 37:35–49.
2023. View Article : Google Scholar
|
|
19
|
Wang X, Li W, Zhang Y, Sun Q, Cao J, Tan
N, Yang S, Lu L, Zhang Q, Wei P, et al: Calycosin as a Novel PI3K
activator reduces inflammation and fibrosis in heart failure
through AKT-IKK/STAT3 axis. Front Pharmacol. 13:8280612022.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen G, Xu H, Xu T, Ding W, Zhang G, Hua
Y, Wu Y, Han X, Xie L, Liu B and Zhou Y: Calycosin reduces
myocardial fibrosis and improves cardiac function in
post-myocardial infarction mice by suppressing TGFBR1 signaling
pathways. Phytomedicine. 104:1542772022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang L, Fan C, Jiao HC, Zhang Q, Jiang
YH, Cui J, Liu Y, Jiang YH, Zhang J, Yang MQ, et al: Calycosin
alleviates doxorubicin-induced cardiotoxicity and pyroptosis by
inhibiting NLRP3 inflammasome activation. Oxid Med Cell Longev.
2022:17338342022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yuan HJ, Han QC, Yu H, Yu YD, Liu XJ, Xue
YT and Li Y: Calycosin treats acute myocardial infarction via NLRP3
inflammasome: Bioinformatics, network pharmacology and experimental
validation. Eur J Pharmacol. 997:1776212025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han Q, Shi J, Yu Y, Yuan H, Guo Y, Liu X,
Xue Y and Li Y: Calycosin alleviates ferroptosis and attenuates
doxorubicin-induced myocardial injury via the Nrf2/SLC7A11/GPX4
signaling pathway. Front Pharmacol. 15:14977332024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu S, Huang P, Yang J, Du H, Wan H and He
Y: Calycosin alleviates cerebral ischemia/reperfusion injury by
repressing autophagy via STAT3/FOXO3a signaling pathway.
Phytomedicine. 115:1548452023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiang Q, Chen X, Gong K, Xu Z, Chen L and
Zhang F: M6a demethylase FTO regulates the oxidative stress,
mitochondrial biogenesis of cardiomyocytes and PGC-1a stability in
myocardial ischemia-reperfusion injury. Redox Rep. 30:24548922025.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Del Re DP, Amgalan D, Linkermann A, Liu Q
and Kitsis RN: Fundamental mechanisms of regulated cell death and
implications for heart disease. Physiol Rev. 99:1765–1817. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Evavold CL, Hafner-Bratkovič I, Devant P,
D'Andrea JM, Ngwa EM, Boršić E, Doench JG, LaFleur MW, Sharpe AH,
Thiagarajah JR and Kagan JC: Control of gasdermin D oligomerization
and pyroptosis by the ragulator-Rag-mTORC1 pathway. Cell.
184:4495–4511.e19. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qi XM, Zhang WZ, Zuo YQ, Qiao YB, Zhang
YL, Ren JH and Li QS: Nrf2/NRF1 signaling activation and crosstalk
amplify mitochondrial biogenesis in the treatment of
triptolide-induced cardiotoxicity using calycosin. Cell Biol
Toxicol. 41:22024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu CY, Day CH, Kuo CH, Wang TF, Ho TJ, Lai
PF, Chen RJ, Yao CH, Viswanadha VP, Kuo WW and Huang CY: Calycosin
alleviates H2 O2-induced astrocyte injury by
restricting oxidative stress through the Akt/Nrf2/HO-1 signaling
pathway. Environ Toxicol. 37:858–867. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kobayashi EH, Suzuki T, Funayama R,
Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi
H, Nakayama K and Yamamoto M: Nrf2 suppresses macrophage
inflammatory response by blocking proinflammatory cytokine
transcription. Nat Commun. 7:116242016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang J, Li X, Han X, Liu R and Fang J:
Targeting the thioredoxin system for cancer therapy. Trends
Pharmacol Sci. 38:794–808. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen Y, Cao X, Pan B, Du H, Li B, Yang X,
Chen X, Wang X, Zhou T, Qin A, et al: Verapamil attenuates
intervertebral disc degeneration by suppressing ROS overproduction
and pyroptosis via targeting the Nrf2/TXNIP/NLRP3 axis in four-week
puncture-induced rat models both in vivo and in vitro. Int
Immunopharmacol. 123:1107892023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Choi EH and Park SJ: TXNIP: A key protein
in the cellular stress response pathway and a potential therapeutic
target. Exp Mol Med. 55:1348–1356. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abderrazak A, Syrovets T, Couchie D, El
Hadri K, Friguet B, Simmet T and Rouis M: NLRP3 inflammasome: From
a danger signal sensor to a regulatory node of oxidative stress and
inflammatory diseases. Redox Biol. 4:296–307. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lu Y, An L, Taylor MRG and Chen QM: Nrf2
signaling in heart failure: Expression of Nrf2, Keap1, antioxidant,
and detoxification genes in dilated or ischemic cardiomyopathy.
Physiol Genomics. 54:115–127. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang B, Jin Y, Liu J, Liu Q, Shen Y, Zuo S
and Yu Y: EP1 activation inhibits doxorubicin-cardiomyocyte
ferroptosis via Nrf2. Redox Biol. 65:1028252023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou P, Yang L, Li R, Yin Y, Xie G, Liu X,
Shi L, Tao K and Zhang P: IRG1/itaconate alleviates acute liver
injury in septic mice by suppressing NLRP3 expression and its
mediated macrophage pyroptosis via regulation of the Nrf2 pathway.
Int Immunopharmacol. 135:1122772024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhan Y, Xu D, Tian Y, Qu X, Sheng M, Lin
Y, Ke M, Jiang L, Xia Q, Kaldas FM, et al: Novel role of macrophage
TXNIP-mediated CYLD-NRF2-OASL1 axis in stress-induced liver
inflammation and cell death. JHEP Rep. 4:1005322022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Devant P and Kagan JC: Molecular
mechanisms of gasdermin D pore-forming activity. Nat Immunol.
24:1064–1075. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu C, Zhang Z, Zhang W and Liu X:
Mitochondrial dysfunction and mitochondrial therapies in heart
failure. Pharmacol Res. 175:1060382022. View Article : Google Scholar
|
|
41
|
Sunagawa Y, Iwashimizu S, Ono M, Mochizuki
S, Iwashita K, Sato R, Shimizu S, Funamoto M, Shimizu K,
Hamabe-Horiike T, et al: The citrus flavonoid nobiletin prevents
the development of doxorubicin-induced heart failure by inhibiting
apoptosis. J Pharmacol Sci. 158:84–94. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wu Y, Huang X, He Y, Chang J, Fang X, Kang
P, Feng N, Liu R, Xiao P, Shi D, et al: Mechanism of puerarin
alleviating myocardial remodeling through NSUN2-mediated m5C
methylation modification. Phytomedicine. 143:1568492025. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sun SN, Liu X, Chen XL, Liang SL, Li J,
Liao HL, Fang HC, Ni SH, Li Y, Lu L, et al: Calycosin alleviates
myocardial fibrosis after myocardial infarction by restoring fatty
acid metabolism homeostasis through inhibiting FAP. Phytomedicine.
145:1570452025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bai Y, Chen Q, Sun YP, Wang X, Lv L, Zhang
LP, Liu JS, Zhao S and Wang XL: Sulforaphane protection against the
development of doxorubicin-induced chronic heart failure is
associated with Nrf2 upregulation. Cardiovasc Ther. 35:2017.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jiang S, Han J, Li T, Xin Z, Ma Z, Di W,
Hu W, Gong B, Di S, Wang D and Yang Y: Curcumin as a potential
protective compound against cardiac diseases. Pharmacol Res.
119:373–383. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Saeidinia A, Keihanian F, Butler AE,
Bagheri RK, Atkin SL and Sahebkar A: Curcumin in heart failure: A
choice for complementary therapy? Pharmacol Res. 131:112–119. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pan Y, Zhu G, Wang Y, Cai L, Cai Y, Hu J,
Li Y, Yan Y, Wang Z, Li X, et al: Attenuation of
high-glucose-induced inflammatory response by a novel curcumin
derivative B06 contributes to its protection from diabetic
pathogenic changes in rat kidney and heart. J Nutr Biochem.
24:146–155. 2013. View Article : Google Scholar
|
|
48
|
Grilo GA, Munoz J Jr, Lee DH, Hossain S,
Ma Y, Kain V, Lindsey ML and Halade GV: Macro- and microinjury
define the heart failure progression after permanent coronary
ligation or ischemia-reperfusion in young healthy mice. Am J
Physiol Heart Circ Physiol. 329:H521–H533. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Heusch G: Molecular basis of
cardioprotection: Signal transduction in ischemic pre-, post-, and
remote conditioning. Circ Res. 116:674–699. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Laudette M, Lindbom M, Cinato M, Bergh PO,
Skålén K, Arif M, Miljanovic A, Czuba T, Perkins R, Smith JG, et
al: PCSK9 regulates cardiac mitochondrial cholesterol by promoting
TSPO degradation. Circ Res. 136:924–942. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Khodade VS, Liu Q, Zhang C, Keceli G,
Paolocci N and Toscano JP: Arylsulfonothioates: Thiol-activated
donors of hydropersulfides which are excreted to maintain cellular
redox homeostasis or retained to counter oxidative stress. J Am
Chem Soc. 147:7765–7776. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao ST, Qiu ZC, Xu ZQ, Tao ED, Qiu RB,
Peng HZ, Zhou LF, Zeng RY, Lai SQ and Wan L: Curcumin attenuates
myocardial ischemia-reperfusion-induced autophagy-dependent
ferroptosis via Sirt1/AKT/FoxO3a signaling. Int J Mol Med.
55:512025. View Article : Google Scholar :
|














