Introduction
Myocardial fibrosis is a common end-pathologic
phenotype in a variety of cardiomyopathies that is characterized by
pathological dilatation of the myocardial interstitium and the
abnormal remodeling of extracellular matrix (ECM) components
(1). This ECM remodeling
involves not only an imbalance in the ratio of type I/III collagen,
but an abnormal cross-linking of fibronectin and disruption of the
laminin network, thereby leading to an increase in the elastic
modulus of myocardial tissue. In the context of acute myocardial
injury, fibrosis represents an evolutionarily conserved mechanism
of trauma repair. When the investigation of the repair response
moved beyond its spatial and temporal constraints, fibrosis
research shifted to pathological processes: A sustained activation
of the TGF-β/Smad pathway was shown to lead to a reduced
mitochondrial oxidative phosphorylation capacity in abnormally
proliferating myofibroblasts. These cells were found to depend on
glycolysis to maintain a high state of collagen synthesis and an
excessive deposition of ECM was shown to result in a marked
increase in ventricular stiffness and chronic, deregulated or
excessively aggressive fibrotic responses in these tissues were
observed to disrupt the structural framework of the heart's
structural elements, thereby impeding tissue regeneration and
leading to dysfunction (2).
The primary mechanism responsible for cardiac
fibrosis is the process of overactivation and proliferation of
cardiac fibroblasts (CFs). Activated CFs, also known as
myofibroblasts, are the major contributors to ECM production, and
are the primary effector cells of cardiac fibrosis (3,4).
The abnormal activation and proliferation of CFs underlie the cell
biology of cardiac fibrosis. Under physiological conditions,
resident CFs exist in a relatively quiescent state and are
primarily responsible for maintaining ECM homeostasis, cardiac
structure and mediating electrophysiological conduction. However,
under conditions of cardiac injury, such as myocardial ischemic
injury and myocardial infarction, myofibroblasts secrete more ECM,
leading to cardiac fibrosis. The fibrotic process is further
exacerbated by sustained activation of the TGF-β/Smad signaling
pathway. In previous studies, the binding of TGF-β1 to the TβRII
receptor was shown to trigger the phosphorylation of Smad2/3,
leading to their subsequent nuclear translocation (5-7).
This process directly activates collagen synthesis genes,
ultimately leading to a cascade of amplified fibrotic signals. In
addition, previous studies have demonstrated that this pathological
remodeling inhibits the migration of endogenous cardiac stem cells
through the physical barrier effect of migration, ultimately
leading to diastolic dysfunction and arrhythmias (8,9).
The chronological application of therapeutic interventions, which
promote moderate ECM deposition in the acute phase and inhibit
pathological remodeling in the chronic phase, has provided novel
therapeutic paradigms. Therefore, in order to design further new
targets and therapeutic strategies, a deeper understanding of the
processes of normal and pathological tissue remodeling is
required.
Recent advances in fibrosis research have focused on
elucidating the molecular mechanisms underlying cardiac
fibrogenesis, with particular emphasis on identifying potential
therapeutic targets. Among these, METTL3 has emerged as a key
regulatory enzyme in the fibrotic process (10). As was demonstrated in a
previously published study (11), METTL3, as the core catalytic
component of the methyltransferase complex, performs a crucial role
in mediating RNA modifications that influence fibrotic pathways.
Despite the advances that have been made in understanding the
processes of myocardial fibrosis, however, the therapeutic
landscape for myocardial fibrosis remains limited, highlighting the
urgent need for an even deeper understanding of its molecular
mechanisms. Currently, the most intensively studied RNA
modifications are N6-methyladenosine
(m6A) and 7-methylguanosine (m7G)
modifications, and their specific mechanisms of action in
myocardial fibrosis have been partly elucidated. Advancements in
this field of research will be crucial for the development of
effective therapeutic strategies against this debilitating
condition.
Types and functions of RNA
modifications
RNA modifications can be categorized into three main
groups, namely chemical motif modifications, nucleotide editing and
dynamic processing, and other regulatory modifications. The present
review focused on the types of RNA modifications in the first two
categories. Among the chemical motif modifications, methylation
modifications include m6A, methyladenosine
(m1A), 5-methylcytosine (m5C) and
m7G; acetylation modifications are mainly focused on
N4-acetylcytidine (ac4C); and
heterodimerization is mainly for pseudouridylation (Ψ), which
specifically modifies the U34 position of transfer RNA (tRNA)s.
Nucleotide editing and dynamic processing-type modifications
include adenosine-to-inosine (A-to-I) base substitutions and
uridylation in nucleotide addition (Fig. 1).
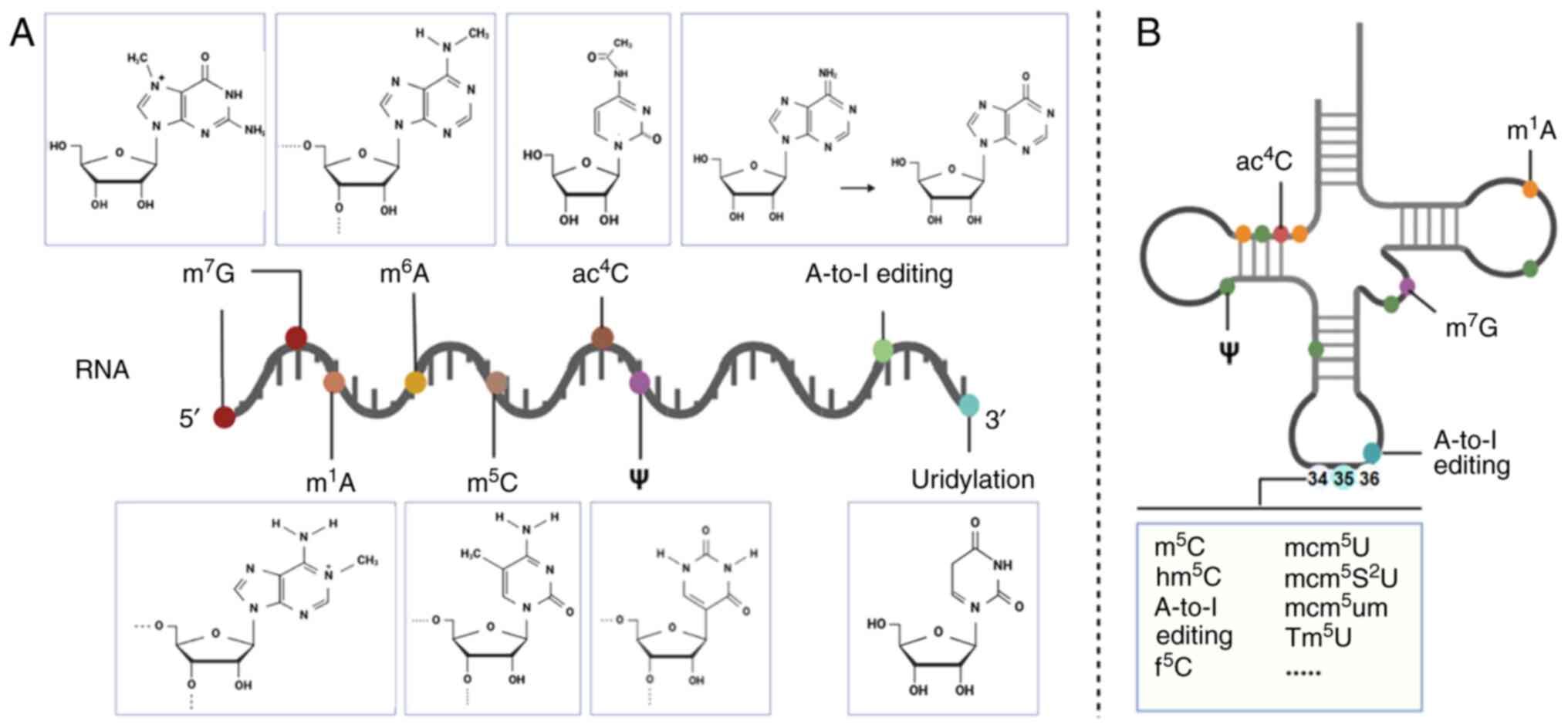 | Figure 1A graphical depiction of the chemical
modifications occurring in RNA within mammalian cells is provided.
(A) The chemical structures of various RNA modifications, such as
involving mRNAs and microRNAs, as well as (B) Transfer RNAs, are
shown. Specifically, the modification at area 34 of the tRNA
regulates swing pairing, encompassing such as m5C, hm5C, and
others. A-to-I, adenine-to-inosine; m5C,
5-methylcytosine; m6A,
N6-methyladenosine; m1A,
1-methyladenosine, m7G, 7-methylguanosine;
ac4C, N4-acetylcytidine. |
m6A modifications
Since the discovery of nucleic acid methylation in
HeLa cells in the 1970s, m6A and m7G have
gradually entered the research landscape as important
epitranscriptome markers (12,13). After half a century of intensive
research, m6A has now been shown to be the most abundant
type of chemical modification in mammalian mRNAs, and its dynamic
and reversible modification properties have a key role in the
regulation of gene expression. In 2012, researchers achieved the
first systematic resolution of m6A modification at the
whole transcriptome level in human and mouse through developing
m6A-seq, a high-throughput sequencing technology
(14). This groundbreaking study
revealed that the m6A locus exhibits a remarkable
spatial distribution feature, namely, that it is mainly enriched in
two functional regions in the transcript: One is the long internal
exonic region of the coding region of the gene, whereas the other
is the 3'-untranslated region (3'-UTR) close to the translation
termination codon. Notably, Meyer et al (15) further revealed, through a finer
localization analysis, that ~70% of the m6A sites in the
mammalian genome are densely distributed in the 3'-UTR interval
50-200 nucleotides (nt) downstream of the stop codon (16). This remarkable regiospecificity
suggests that m6A may be involved in regulating the
accessibility of microRNA (miRNA)-binding sites through either
spatial site-blocking effects or recruitment of specific reading
proteins. This discovery provides important clues towards
elucidating the precise molecular mechanism via which
m6A modification regulates post-transcriptional
processes through RNA-protein interaction networks.
The molecular architecture and functional regulatory
mechanisms of the m6A methyltransferase complex have
both been gradually elucidated. The complex comprises METTL3 and
METTL14 as the core catalytic subunits. Previous studies have shown
that METTL3 directly mediates S-adenosylmethionine (SAM)-dependent
methyl transfer reactions through its catalytic domain (17,18), whereas METTL14, though it was
previously shown not to be catalytically active (16), was responsible for substrate
recognition and spatial localization through its superior
RNA-binding ability (19-21).
The heterodimer formed by the two subunits not only constitutes the
catalytic core of the methylation reaction, but its structural
complementarity was shown to markedly enhance the thermodynamic
stability of the complex (22).
Wilms tumor 1-associated protein, which has been
identified as a key regulatory component of the complex, was found
to be not enzymatically active (23). Previous studies revealed that it
is able to regulate the methylation process through two key
mechanisms: First, it was shown to specifically interact with the
METTL3/METTL14 heterodimer, thereby promoting the aggregation of
the complex in nuclear speckles (19,23); and second, as a splicing factor,
it was shown to direct the complexes to target specific sites of
pre-mRNA through a phase separation mechanism (19,24). This dynamic regulatory network
ensures the spatiotemporal specific deposition of m6A
modifications on transcripts.
In addition to the core components, earlier studies
revealed that the m6A methylation system comprises
multiple classes of cofactors with distinct regulatory roles. For
example, Vir-like M6A methyltransferase associated/m6A
methyltransferase KIAA1429 was previously shown to mediate
co-localization of the complex with RNA polymerase II through
acting as a molecular scaffold (25). Similarly, the m6A
regulator, RNA binding motif protein 15/15B, has been shown to
enhance the complex's affinity for specific RNA motifs through its
RNA recognition motifs (26.) In addition, the E3 ubiquitin-protein
ligase Hakai and zinc finger CCCH-type containing 13 were reported
to stabilize the subcellular localization of the complex via
deubiquitination modifications (27,28). These findings collectively
demonstrate that the modular assembly of these cofactors enables
the methyltransferase complexes to dynamically respond to cellular
signals, thereby facilitating the precise regulation of
m6A modifications (29).
In terms of substrate profile, the m6A
modification catalyzed by METTL3 has a broad RNA-type specificity,
covering classical RNA molecules such as mRNAs, tRNAs and ribosomal
RNA (rRNA)s, as well as regulatory RNAs, such as miRNA precursors
and long-stranded non-coding RNAs (lncRNAs) (30). Notably, among mammalian mRNAs,
m6A modifications not only constitute the most abundant
post-transcriptional type of modification (31), but its dynamic and reversible
nature makes it a key regulatory node in the remodeling of gene
expression networks in pathological processes, including those of
cardiovascular diseases. This property has enabled METTL3 to
gradually become an important research target for the study both of
disease mechanisms and of the development of targeted
therapies.
The functional realization of m6A
modifications relies on the dynamic recognition of their
methylation sites via specific reading proteins. Structural and
mechanistic analyses have classified m6A reading
proteins into three main categories. The first class comprises the
canonical YT521-B homology (YTH) domain family (YTHDF1-3 and
YTHDC1-2), where conserved aromatic residues within their
hydrophobic binding pockets were shown to mediate multiple aspects
of mRNA metabolism through the recognition of methylated adenosine
residues (32,33). The second category comprises a
family of non-classical RNA-binding proteins, including
insulin-like growth factor 2 mRNA-binding proteins and
heterogeneous nuclear ribonucleoproteins. Although these proteins
lack the YTH domain, structural studies have demonstrated that
their RNA recognition motifs, or K-homology domains, selectively
bind m6A-modified sites to regulate RNA stability and
subcellular localization. The third class of m6A reading proteins
is defined by translation-regulating factors, such as eukaryotic
initiation factor 3 (eIF3). Key studies have shown that eIF3 is
able to recognize m6A modifications within mRNA 5'-UTRs
through its hydrophobic binding pocket, thereby promoting ribosomal
preinitiation complex assembly to exert upstream translational
control, while concurrently influencing mRNA stability and
localization (32,33). Demethylases, including AlkB
homolog 5 (ALKBH5) and fat mass and obesity-associated protein
(FTO), were demonstrated to reverse m6A modifications
through enzymatic erasure mechanisms, as evidenced by biochemical
and structural studies (34)
(Fig. 2).
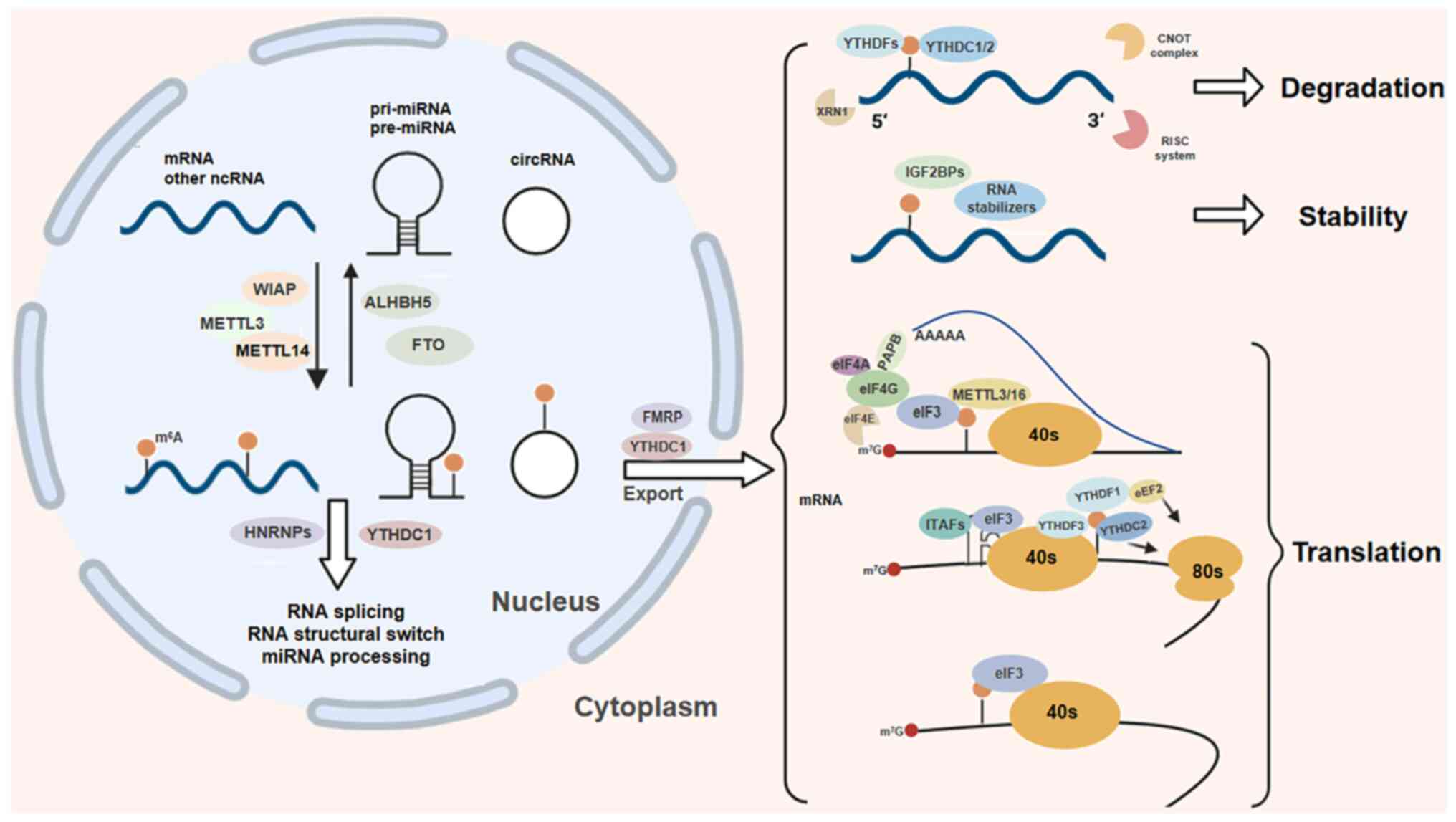 | Figure 2The biological functions of
m6A modification are diverse. This reversible process
involves 'writers' such as METTL3 and METTL14, and 'erasers' such
as FTO and ALKBH5 that remove the modification. The 'readers', such
as those with the YTH domain recognize and bind to
m6A-modified RNA, thereby influencing RNA processing and
its transport to the cytoplasm. Ultimately, m6A
modulation controls RNA translation, stability and degradation
within the cytoplasm. m6A,
N6-methyladenosine; METTL,
methyltransferase-like; FTO, fat mass and obesity-associated;
ALKBH5, AlkB homolog 5; YTH, YT521-B homology. |
m1A modifications
The biological functions of m¹A, a key modification
in the epigenetic regulation of RNA, have been gradually unveiled
with the advance of research techniques. While earlier studies
focused on the distribution of m¹A in fungal tRNAs and rRNAs, a
breakthrough discovery based on the methylated RNA
immunoprecipitation sequencing (MeRIP-seq) technique showed that
m¹A modifications are also present in the coding region (CDS) and
the 5'-UTR region of mammalian mRNAs and that the degree of their
enrichment is markedly positively associated with the number of
variable translation initiation sites (35). However, Grozhik et al
(35), by performing chemometric
analysis, showed that the abundance of m¹A modifications in mRNAs
is markedly lower compared with other RNA types, suggesting that
they may exist as precise markers for specific functional sites,
rather than as global modifications.
At the methylation regulatory level, members of the
tRNA methyltransferase (TRMT) family have been shown to exhibit a
subcellular functional division. TRMT61B, which was identified as
the first human m1A methyltransferase, primarily
catalyzes modifications of cytoplasmic tRNAs and rRNAs (36,37), whereas TRMT10C specifically
targets mitochondrial tRNAs and was further demonstrated to
maintain mitochondrial homeostasis through the regulation of energy
metabolism (38). Previous
studies have established that the AlkB homolog (ALKBH) family
enzymes precisely coordinate RNA demethylation dynamics; for
example, ALKBH3 promotes functional tRNA fragment generation via
erasing tRNA methylation, and actively mediates
m1A-dependent transcript degradation through its
demethylase activity (39,40). Another study (41) showed that ALKBH1 is able to
enhance the pool of translationally active tRNAs through
selectively removing m¹A modifications at position 58 of tRNAs,
thereby increasing their stability. Moreover, ALKBH7 was
characterized as a mitochondrial pre-tRNA-specific demethylase that
participates in cellular energy metabolism reprogramming (39). Furthermore, FTO not only
catalyzes m6A demethylation, but it also recognizes
stem-loop structures that enable it to demethylate tRNA m¹A
modifications, thereby markedly boosting nucleocytoplasmic protein
translation flux (42). In skin
scar research, staphylococcal nuclease and tudor domain containing
1 (SND1) was identified as an m¹A reader protein that both enhances
RNA stability and drives fibroblast proliferation and collagen
deposition in pathological scarring. However, its role in
cardiovascular diseases remains poorly understood (43). From the perspective of the
functions of SND1 and the existing mechanisms of cardiac fibrosis,
future studies should explore the role of SND1 in the progression
of cardiac fibrosis by investigating its effect on cardiac
fibrosis-associated signaling pathways (such as the TGF-β signaling
pathway), gene expression regulation and cellular metabolism.
Notably, m¹A modifications have been shown to
exhibit significant environmental stress sensitivity. Under serum
starvation or oxidative stress
(H2O2-stimulated) conditions, the abundance
of m¹A modification sites in mRNAs was found to be markedly
increased (39), whereas glucose
deprivation led to a decrease in m¹A tRNA levels through the
inhibition of TRMT enzyme activity, an effect that could be
reversed by knockdown of the ALKBH1 gene (41). This dynamic modification pattern
suggests that m¹A may act as a metabolic stress receptor that helps
cells to adapt to micro-environmental changes via modulating
translational reprogramming. For example, under energetic stress
conditions, dynamic changes in mitochondrial m¹A modification would
enable rapid regulation of oxidative phosphorylation activity by
influencing the translational efficiency of the respiratory chain
complex mRNA (Fig. 3).
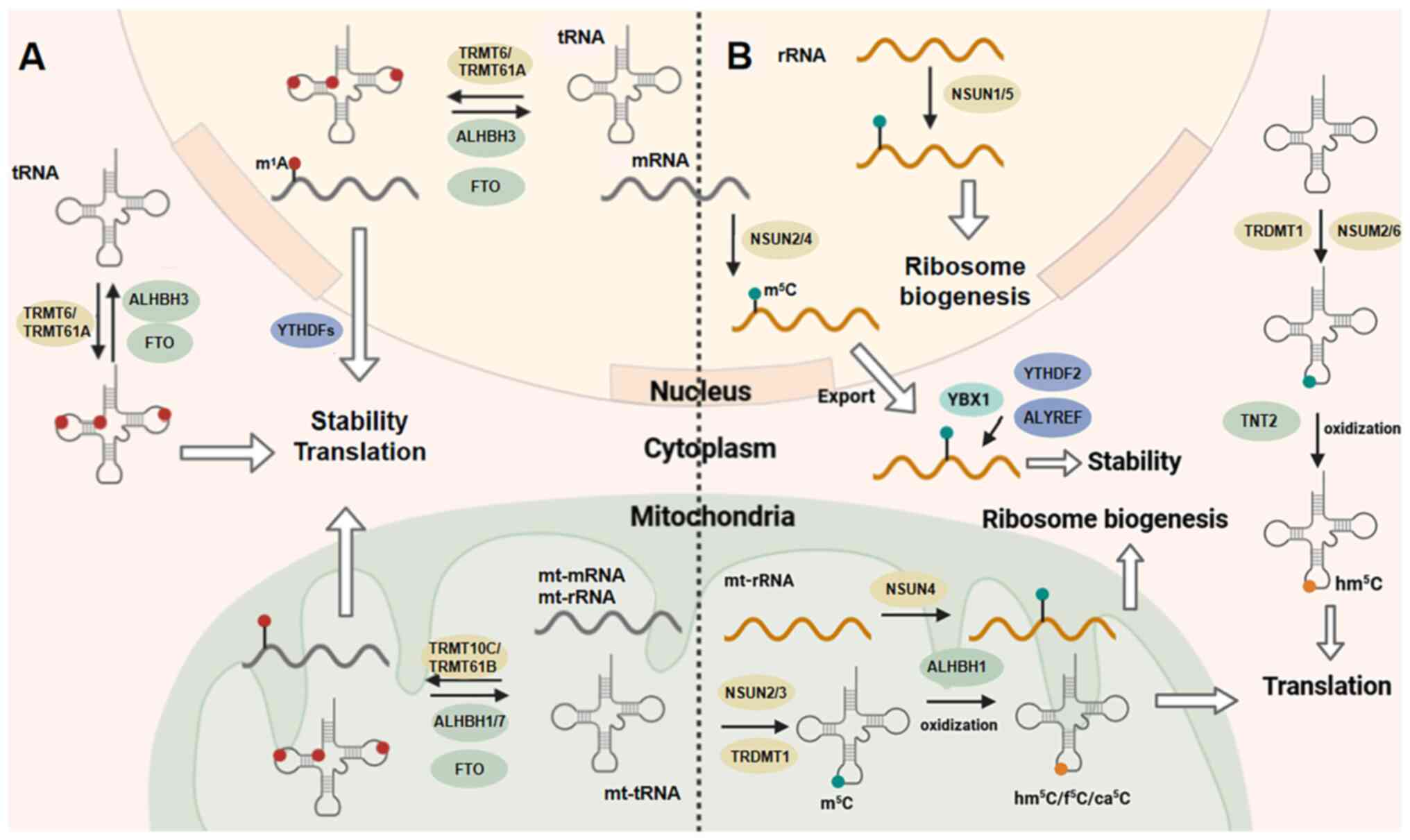 | Figure 3m1A and m5C
modifications. (A) Within the cell nucleus, mRNAs and tRNAs undergo
methylation by enzymes including TRMT6 and TRMT61A. Specifically,
the methylation of m1A on mt-tRNA, rRNA and mRNA is
catalyzed by TRMT10C/TRMT61B. The enzymes FTO and ALKBH1/3/7 are
responsible for controlling the demethylation process.
Functionally, m1A employs various mechanisms to regulate
translation and RNA stability, with YTHDF serving as a potential
'reader' of m1A modifications. (B) Aspartic acid tRNA is a specific
target for methylation by the enzyme TRDMT1. The methyltransferases
NSUN1-6 function as 'writers' of m5C modifications in RNA,
influencing RNA nuclear export, metabolism, and function. Various
proteins, including ALYREF, YBX1 and YTHDF2, have been reported to
serve as 'readers' of m5C modifications. Additionally, TNT2, ALKBH1
are responsible for catalyzing the oxidation of m5C rather than its
elimination. m1A, 1-methyladenosine; m5C,
5-methylcytosine; Mt-mitochondrial; ALKBH1, AlkB homolog 1; YTH,
YT521-B homology; FTO, fat mass and obesity-associated; ALKBH5,
AlkB homolog 5; YTH, YT521-B homology; rRNA, ribosomal RNA. |
m5C modifications
As a key contributor to RNA epigenetic modification,
the biological significance of m5C has undergone a
cognitive revolution from DNA-specific modification to widespread
regulation by RNA. during the early period of its investigation,
m5C was considered to exist only in DNA due to
limitations in the technologies available for detection, although
given the breakthroughs that have been made in high-throughput
sequencing and mass spectrometric techniques, researchers have
discovered that it is widely distributed in eukaryotic RNAs, and is
particularly enriched in high abundance in the anticodon loops of
tRNAs and in conserved regions of rRNAs (44). m5C deposition has been
shown to be mediated by the NOL1/NOP2/SUN structural domain (NSUN)
family, in concert with TRDMT1. In the tRNA modification system,
NSUN2 was identified to catalyze the methylation of both miRNA
precursors and mRNAs (45,46), whereas NSUN3, NSUN6 and TRDMT1
were shown to form a ternary system targeting tRNA-specific sites,
respectively (45,47-49); moreover, functional analyses
revealed that, in the ribosomal RNA (rRNA) modification system,
NSUN1 and NSUN5 regulate the structural maturation of rRNAs,
whereas NSUN4 participates in respiratory chain complex assembly
through mitochondrial rRNA/mRNA methylation (50-53).
A previous study also revealed diversified
m5C recognition mechanisms; namely, the YTH
domain-containing family protein 2 (YTHDF2)-mediated degradation of
m5C-methylated mRNAs through its YTH domain, the Y
box-binding protein 1 (YBX1)-mediated stabilization of
m5C-modified transcripts via cold shock domain binding,
and the functioning of Aly/REF export factor (ALYREF) as a nuclear
export factor that facilitates the cytoplasmic translocation of
m5C-marked mRNAs through specific RNA-protein
interactions (54). Notably, the
mechanism of dynamic regulation of m5C remains
controversial, given that its reversibility has not yet been
clarified (unlike that of m6A/m¹A) and this stability
implies that m5C modifications have a unique role in
long-term episodic memory regulation. Currently, the existing
experimental evidence has established that m5C
modifications are involved in organ growth, initiation of various
pathological conditions, and developmental processes (47,54-57). Taken together, these findings
have not only expanded the regulatory dimensions of RNA epigenetic
inheritance, but have also provided novel perspectives for the
continuing study of disease mechanisms and targeted therapies.
m7G modifications
Since the discovery of m7G modification
in the mammalian mRNA 5'-cap structure in the 1970s (58,59), its biological function is now
realized to extend from classical mRNA stability regulation to
multilevel gene expression networks. Genome-wide mapping has
further revealed that, beyond mRNA cap structures, m7G
is distributed across non-coding RNAs, including rRNAs, tRNAs and
miRNAs, and that this dynamic distribution pattern suggests
pervasive roles in post-transcriptional regulation (60).
Another previously published study (61) demonstrated that precise
m7G deposition relies on synergistic multi-enzyme
complexes; namely, that the mammalian mRNA cap methyltransferase
complex, RNMT-RAM, specifically catalyzes m7G
modifications in 5'-cap structures by spatially coupling
mRNA-capping enzymes into functional post-transcriptional
processing modules. In addition, the WBSCR22-TRMT112 complex
utilizes the SAM-binding structural domain to catalyze guanosine
methylation at position 1,639 of 18S rRNA, a modification that
served as a critical checkpoint for the maturation of the small
subunit of the ribosome (60)
and the METTL1-WDR4 complex has been shown to exhibit
multi-functional catalytic properties that are responsible for
mRNA, tRNA and miRNA-associated m7G modifications
(62-64).
At the level of functional regulation,
m7G coordinates RNA metabolism and translation through
multiple mechanisms, including prolongation of the mRNA half-life
of cap-structured m7G via the inhibition of 5'→3'
nucleic acid exonuclease activity (65-67), whereas internal m7G
modifications have been shown to promote RNA nucleocytoplasmic
translocation by binding to translocation factors. During
translation, METTL1-mediated modification of internal
m7G mRNA enhances the translation initiation efficiency
by reconfiguring the ribosome-binding interface, whereas the
m7G modification of tRNA led to an enhancement of the
decoding efficiency by optimizing the precision of codon-anti-codon
pairing (63,68). Non-coding RNA maturation is also
regulated by m7G; for example, the m7G
modification of 18S rRNA was identified to be a quality checkpoint
for ribosome biogenesis (60),
whereas m7G modification of miRNA precursors was shown
to regulate the abundance of mature miRNAs by altering the
accessibility of the cleavage site of Drosha, a ribonuclease III
enzyme that performs a crucial role in the biogenesis of
miRNAs.
A cryo-electron microscopy study revealed that the
METTL1-WDR4 complex achieves substrate-specific switching through
conformational changes: When the complex bound to tRNA, the WD40
structural domain of WDR4 was found to induce active-pocket
deformation, which led to a precise recognition of RAGGU motifs
(69). Abnormalities in this
dynamic catalytic mechanism are closely associated with
neurodevelopmental disorders and tumor metastasis; for example,
METTL1 deletion led to tRNA fragmentation, with the consequent
activation of the p53-dependent apoptotic pathway (63,68). In glioblastoma, METTL1
overexpression was shown to promote tumor cell invasion by
enhancing the methylation of oncogenic miRNA (for example, miR-21)
precursors (64). Taken
together, these findings have not only shed light on the molecular
regulatory network of m7G modification, but they also
may provide novel ideas for precision therapy targeting RNA
methylation.
ac4C modifications
As the only known RNA acetylation modification, the
biology of ac4C has undergone a cognitive leap from
prokaryotic to eukaryotic systems. Whereas early studies focused on
the ac4C modification of bacterial tRNA anticodon stem
loops and conserved regions of fungal rRNAs (70), the discovery of
N-acetyltransferase 10 (NAT10) revealed the widespread
presence of this modification in mammalian RNAs. NAT10 catalyzes,
through ATP-dependent acetyltransferase activity, the formation of
ac4C in a conserved region of the 18S rRNA site (namely,
at position 1,842; ac4C1842). Further structural
analysis by cryo-electron microscopy revealed that
ac4C1842 serves as a molecular checkpoint for the
assembly of the small subunit of the ribosome through stabilizing
the helix 69 conformation of the 18S rRNA; its absence led to
aberrant processing of the rRNA precursor (70).
In the dynamic regulation of tRNAs, the deletion of
NAT10 was shown to lead to a decrease in the overall acetylation
level of tRNAs in human colon cancer cells, thereby markedly
affecting their structural stability (69). Of particular note,
ac4C modification close to the tRNA anticodon swing site
led to an increase in ribosome decoding efficiency via the
optimization of codon-anticodon geometry matching (71). Subsequent mRNA-level studies
identified that ac4C in the CDS region prolonged the
half-life of transcripts by resisting 3'→5' exonuclease activity,
whereas ac4C in the 5'-UTR region facilitated the
translation initiation of upstream open reading frames (ORFs)
through a 'leaky scanning' mechanism, via which the efficiency of
the main ORFs was enhanced (71,72). Furthermore, in esophageal
squamous cell carcinoma, NAT10 was found to specifically catalyze
the acetylation modification of the lncRNA CTC-490G23.2, and this
modification drove tumor progression (73).
Physiologically, NAT10 promotes the osteogenic
differentiation of bone marrow mesenchymal stem cells by
stabilizing runt-related transcription factor 2 mRNA, which thereby
established its critical role in bone metabolic homeostasis
(74). At the pathomechanistic
level, NAT10 has been shown to be abnormally highly expressed in
malignant tumors such as hepatocellular carcinoma and breast cancer
and its stability is enhanced by the acetylation of pro-oncogene
mRNAs (for example, Myc and Cyclin D1). Additionally, specific
alterations were observed in the serum exosomal ac4C
modification profiles of patients with esophageal carcinoma,
thereby suggesting that it has potential as a liquid biopsy marker
(73,75-79). For therapeutic applications,
ac4C-modified synthetic mRNAs have been demonstrated to
have unique advantages; specifically, repeated dosing was enabled
through reducing Toll-like receptor 7/8-mediated immunogenicity and
a 3-fold extension of the shelf-life of mRNA vaccines at ambient
temperature was achieved by enhancing their thermal stability
(80). These breakthroughs
provide new directions for RNA acetylation-based precision medicine
(Fig. 4).
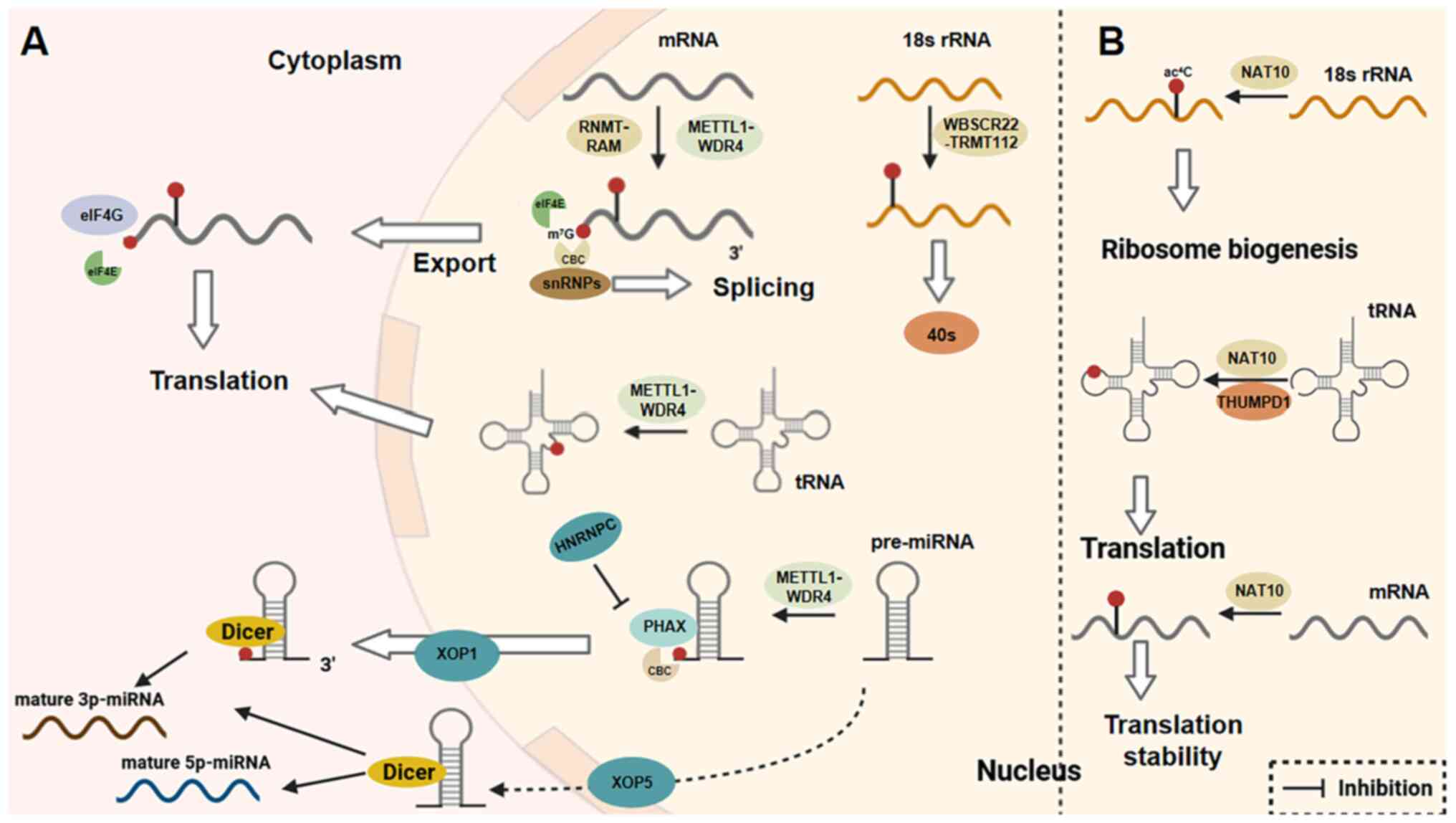 | Figure 4m7G and ac4C
modifications. (A) In the nucleus, METTL1-WDR4 serves as the
'reader' for m7G modifications in mRNAs, pre-miRNAs and
tRNAs. WBSCR22-TRMT112 is responsible for reading 18S rRNA. The
mammalian mRNA cap methyltransferase complex, RNMT-RAM, on the
other hand, primarily targets mRNA for m7G modification.
m7G has various roles in enhancing translation rates, in
accelerating RNA export, in contributing to ribosome synthesis and
in facilitating the maturation of miRNAs. (B) Within the nucleus,
NAT10 and THUMPD1 primarily function as enzymes that catalyze
ac4C modifications on rRNAs, tRNAs and mRNAs. These
modifications are associated with translation efficiency, ribosome
biosynthesis, and mRNA stability, respectively. METTL,
methyltransferase-like; tRNAs m7G, 7-methylguanosine;
ac4C, N4-acetylcytidine; miRNA,
microRNA; rRNA, ribosomal RNA; NAT10, N-acetyltransferase
10; tRNA, transfer RNA. |
Uridylation modifications
Uridylation, a typical representative of RNA
3'-end-tail modification, causes predominantly an enrichment in
short poly(A)-tailed or U-tailed RNA molecules (81,82). Its catalytic system consists of
the terminal uridylyl transferase (TUT, or TUTase) and terminal
nucleotidyl transferase (TENT) families working in concert: TUT4/7
(ZCCHC11/6) was found to specifically recognize the 3'-overhanging
structure of mature miRNAs, mediating uridylation modifications of
precursor miRNAs, such as pre-let-7 (83,84). In addition, TENT2 was shown to
regulate the loading efficiency of Argonaute proteins through
selectively catalyzing the addition of uridine to the 3'-end of
mature miRNAs (83), and TENT5C
was shown to maintain mRNA stability via antagonizing the
de-adenylation enzyme CCR4-NOT complex (85).
At the level of dynamic metabolic regulation,
uridylation and de-adenylation form a precise balance: When poly(A)
binding protein cytoplasmic 1 (PABPC1) bound to long poly(A) tails,
it was found to inhibit TUTase activity through spatial
site-blocking effects, whereas, in the short poly(A) tail state,
PABPC1 was dissociated from the TUTase, thereby initiating the
process of uridylation (85-87). This antagonistic mode coexists
with a synergistic mode: The miRNA miR-1 was shown to promote
de-adenylation through recruiting CCR4, while enhancing
TUT4-mediated uridylation, thereby creating a positive feedback
loop of mRNA decay (82,85). Mammalian cells have been found to
clear uridylated RNA species through a dual pathway: 5'→3'
degradation is initiated by XRN1 exonuclease, which specifically
recognizes the TUT4/7-extended U-tail (88), whereas 3'→5' degradation is
mediated via DIS3-like exonuclease 2 (DIS3L2), which recognizes the
uridylation marker through the S1 structural domain. Exosomes
remove uridylated mRNAs through the polyadenylate-binding nuclear
protein 1 (PABPN1)-DIS3L2 axis, and the exosome also degrades
miRNAs (89-91). RNA-binding proteins, such as
human-antigen R, antagonize the degradative activity of DIS3L2 by
binding to uridine-enriched elements to form protective
ribonucleoprotein particles, and this dynamic equilibrium
determines the fate of uridylated RNAs (92-95).
In miRNA biosynthesis, uridylation also serves a
dual regulatory role: In terms of inhibitory modifications, the
RNA-binding protein Lin28 was shown to recruit TUT4 to the GGAG
motif of pre-let-7 and other pre-miRNAs, where the addition of an
oligo-uridine tail (+UUU) was found to block Dicer cleavage
(84). The operation of a
similar mechanism was observed in the inhibition of miR-191
processing (96). In terms of
mature miRNA functional modulation, uridylated miR-105 was shown to
reprogram its target profile by altering seed sequence
complementarity (97), whereas,
in another study (98),
TUT7-mediated pre-miRNA uridylation was found to promote the
differential loading of Argonaute-2 (Fig. 5).
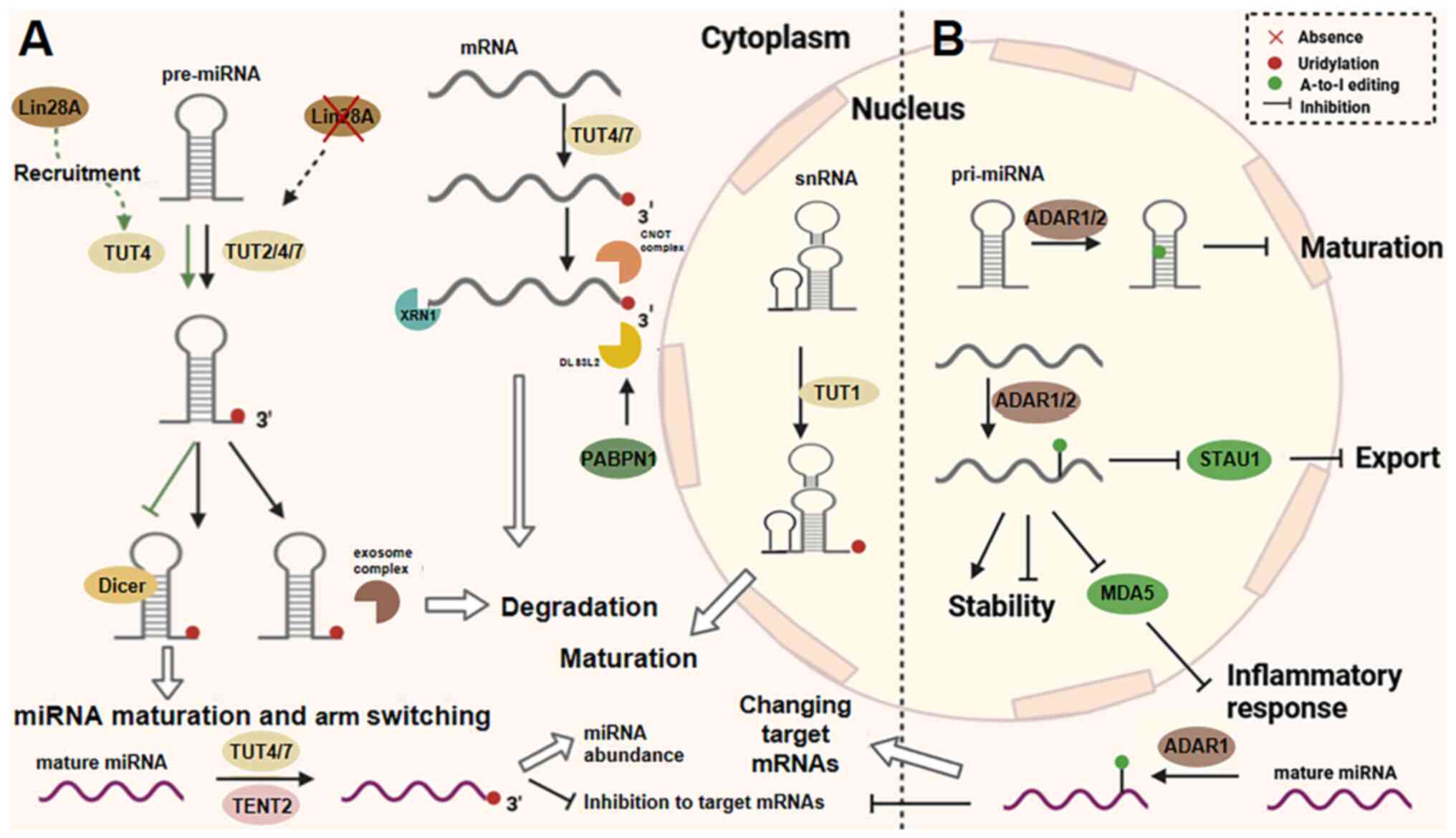 | Figure 5Uridylation and A-to-I editing. (A)
In the cytoplasm, TUT2, TUT4 and TUT7 are responsible for the
addition of uridylates to pre-miRNAs, fully mature miRNAs and
mRNAs, whereas TUT1 is responsible for the uridylation of snRNAs in
the nucleus. The maturation of miRNAs is regulated by TENT2.
Uridylation is important in the maturation of miRNAs, snRNAs, the
activity of miRNAs, decay. (B) ADAR1 and ADAR2 function as the
'writer' of A-to-I editing for mRNAs, mature miRNAs and pri-miRNAs.
ADAR1/2 affects not only mRNA stability and production, but also
miRNA maturation and function. A-to-I, adenine-to-inosine; TUT,
terminal uridylyl transferase; miRNA, microRNA; snRNA, small
nuclear RNA; ADAR, adenosine deaminase acting on RNA. |
A-to-I RNA editing modifications
The adenosine deaminase acting on RNA (ADAR) family
has been demonstrated to remodel RNA function through A-to-I
editing, and its sites of action are frequently co-localized with
Alu elements and long interspersed nuclear elements
retrotransposons in the genome (99,100). Although ADAR1 and ADAR2 share
catalytic structural domains, they display significant functional
heterogeneity: ADAR1 preferentially recognizes the
quasi-double-stranded regions formed by Alu elements through its
double-stranded RNA-binding domain (101), whereas ADAR2 targets
neuron-specific RNA substrates by virtue of its N-terminal nuclear
localization signal (102).
This difference in substrate selectivity establishes the functional
division of labor between the two ADAR family members in the
cell.
RNA editing regulates gene expression through a
mechanism that operates on multiple levels. At the level of
transcript remodeling, ADAR-mediated selective splicing has been
shown to alter protein heterodimer composition by retaining
specific exons; for example, A-to-I editing of miR-34a led to the
alteration of its seed sequence and a loss of inhibitory function
for target mRNAs (103),
whereas RNA secondary structure remodeling was found to block
Staufen double-stranded RNA binding protein 1-mediated RNA nuclear
export (101). At the level of
protein function regulation, the RNA editing of antizyme inhibitor
1 mRNA was shown to induce its nucleoplasmic shuttling function,
thereby regulating polyamine metabolic homeostasis, whereas
arginine codon (AGA) to termination codon (UGA) editing triggered
nonsense-mediated mRNA degradation (104-108).
During cancer progression, the ADAR family has been
shown to exhibit a 'double-edged sword' effect: ADAR1 inhibits
melanoma differentiation-associated gene 5 (MDA5) from recognizing
endogenous double-stranded RNAs via editing Alu elements, thereby
enabling cells to evade immune surveillance (100,109-111), whereas its p150 isoform
promotes tumor metastasis through the editing of circular RNAs
(112-114). On the other hand, ADAR2 was
shown to restore oncogene expression, exerting a tumor-suppressive
role (112,113).
Subtype-specific regulation has been demonstrated to
further refine the functional network: For example, cytoplasmic
ADAR1-p110 regulates mature miRNA function, whereas nuclear
ADAR1-p150 maintains embryonic stem cell pluripotency. In cardiac
tissues, ADAR2 was shown to protect ADAR1 targets from over-editing
through competitive binding (114) and, moreover, a hierarchical
regulatory mechanism for editing activity was demonstrated in a
different study (115).
In inflammatory regulation, ADAR1 has been shown to
maintain immune homeostasis through three mechanisms: First,
through editing endogenous double-stranded RNA to block
MDA5-mediated intrinsic immune recognition; second, by moderately
activating the protein kinase R (PKR)-Z-DNA/RNA binding protein 1
axis to induce adaptive stress responses; and thirdly, by editing
viral RNA mimicry via its p150 isoform to prevent type I interferon
storms (100,109-111). Disruption of this dynamic
equilibrium makes an important contribution to autoimmune diseases
and viral infections, thereby providing novel targets for
immunomodulatory therapy.
Ψ modifications
Ψ, a very abundant post-transcriptional modification
in RNA, occurs via the action of pseudouridine synthetase (PUS),
catalyzing the isomerization of uridine. Its unique C-C glycosidic
bond (compared with the C-N bond of uridine) results in both a
higher thermodynamic stability of, and irreversible changes to, RNA
molecules, also serving as a consistent marker of the
epitranscriptome (116-118). The catalytic pathways may be
divided into two categories, first, the RNA-independent pathway, in
which PUS family members (for example, PUS1) directly recognize
RNA-specific structures (such as the T-arm deletion conformation of
tRNAs) to accomplish site-specific modifications through conserved
catalytic structural domains (119,120); and second, the RNA-dependent
pathway, which is mediated via the box H/ACA-type small nucleolar
ribonucleoprotein complexes, in which the protein Dyskerin acts as
the core catalytic subunit to direct the Ψ-modification of rRNA and
small nuclear RNA (snRNA) (121-123).
Mammalian PUS family members exhibit a fine
subcellular division of labor: In the cytoplasmic system, PUS10 has
been shown to specifically catalyze Ψ-modification at tRNA
positions 54/55 (124,125), whereas PUS1 regulates
transcript stability by recognizing mRNA stem-loop structures
(119,126). In the mitochondrion, TruB
pseudouridine synthase family member 1 (TRUB1) maintains the
Ψ-modification at tRNA position 55, and its absence was found to
lead to tRNA conformational disorder, which resulted in triggering
the defective assembly of the respiratory chain complex (127). In the nucleoplasmic system,
PUS3 targets tRNA modifications at positions 38/39 (128,129), and PUS7 was found to recognize
the UGUAR motif to regulate both small nucleolar RNA (snoRNA)
processing and pre-mRNA splicing (120,130). Ψ also has an important role in
the control of translation. Pseudouridylation of tRNAs has been
reported to regulate translation; in addition, the
pseudouridylation of rRNAs also affects mRNA translation. In 293T
cells, the presence of Ψ in mRNA codons was shown to increase
protein production and to accelerate the rate of recognition of
associated tRNAs (131).
The Ψ-modification has been shown to regulate the
translation process via a mechanism that operates on multiple
levels. Ψ in the mRNA CDS region forms a 'ribosomal speed bump'
that enhances the translation efficiency of rare codons through
prolonging the ribosomal retention time, whereas the Ψ-modification
of termination codons (namely, changing UGA to ΨGA) results in an
inhibition of the recognition of transcripts by the eukaryotic
release factor eRF1, resulting in an ≤15% prolongation of
readthrough translation (132,133). Under stress conditions,
Ψ-modification has also been shown to maintain the eIF2A
non-phosphorylated state by inhibiting PKR activation, resulting in
an increased efficiency of translation initiation (134). In addition, Ψ has been shown to
be involved in the global regulation of RNA metabolism: The RNA
pseudouridine synthase D4 (RPUSD4)/PUS1/PUS7-mediated
Ψ-modification of the pre-mRNA splice site was demonstrated to
alter the efficiency of splicing processing, and it was proposed
that this could be explained by its facilitation of RNA-binding
protein binding (135)
(Fig. 6).
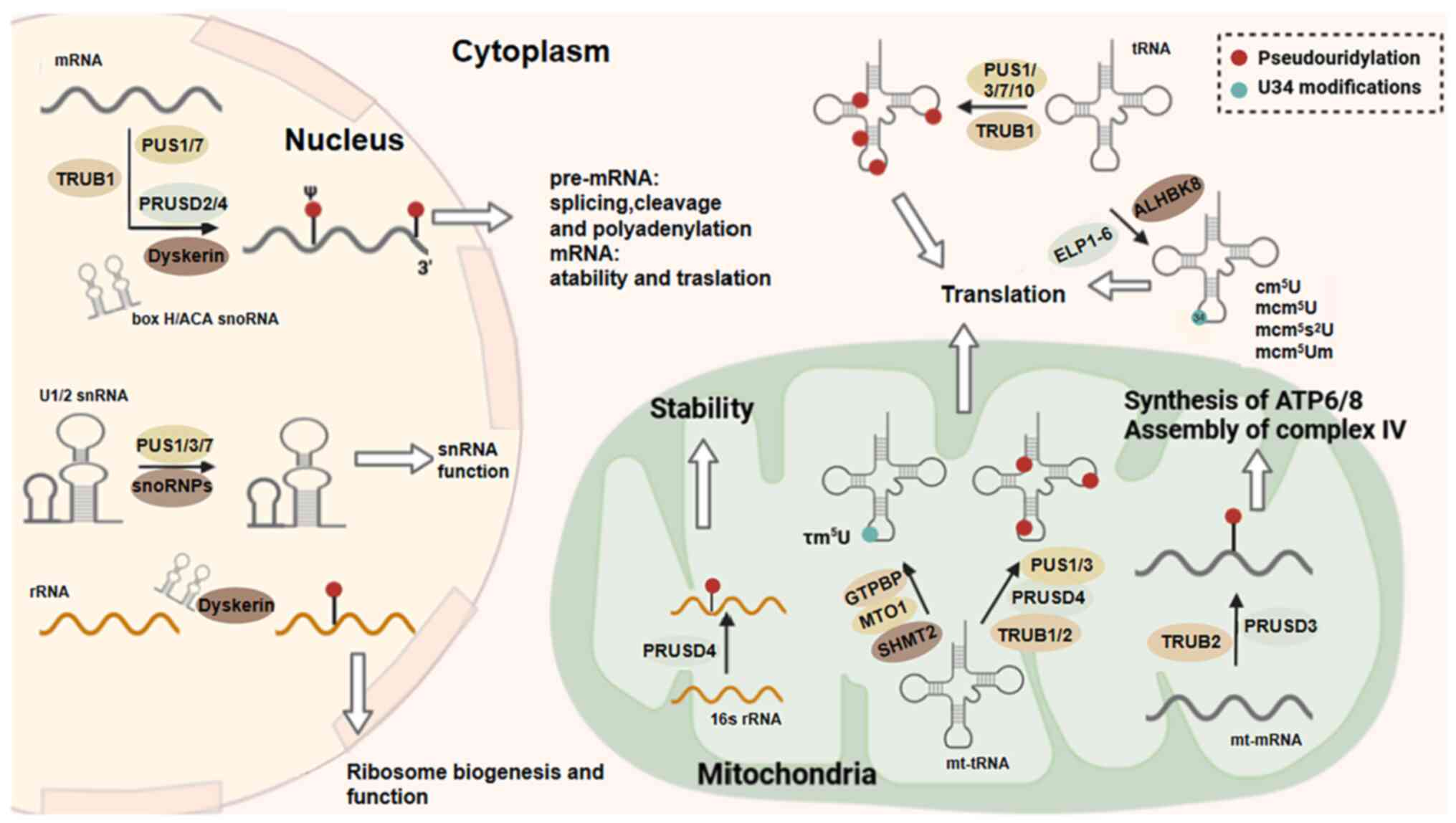 | Figure 6Pseudouridylation and U34
modification. Pseudouridylation and U34 modification are important
processes that occur on tRNAs. In mammals, the RNA
pseudouridylation 'writers' primarily include PUS1/3/7/10,
PRUSD2-4, TRUB1/2 and Dyskerin. The catalysis of pseudouridylation
by Dyskerin relies on H/ACA snoRNA. From a functional standpoint,
pseudouridylation contributes to RNA processing, stability and
functionality. The translation of tRNA is markedly affected by
alterations at the U34 position, such as cm5U, τm5U, MCM5U, MCM5s2U
and MCM5Um. MCM5U and its derivatives are formed when ALKBH8
methylates cm5U, which is catalyzed by the ELP1-6 complex. The τm5U
modification in mt-tRNA is catalyzed by GTP binding protein 3 or
mitochondrial translation optimization 1 homolog, whereas SHMT2
provides the starting material for methyl synthesis. PUS,
pseudouridine synthetase; tRNA, transfer RNA; TRUB1/2, TruB
pseudouridine synthase family member 1/2; snoRNA, small nucleolar
RNA; SHMT2, serine hydroxymethyltransferase 2. |
At the pathological level, oxidative stress has
been shown to induce an increase in Ψ sites within the mRNA CDS
region of HeLa cells, a dynamic modification that assists tumor
cells in adapting to the microenvironment through stabilizing key
transcripts such as hypoxia-inducible factor-1α (HIF-1α) (132). For example, the Ψ-modification
of HIF-1α mRNA promotes tumor cell survival under hypoxic
conditions via inhibiting nuclease degradation and enhancing the
glycolytic pathway activity mediated by HIF-1α. The discovery of
this epitranscriptomic buffering mechanism has provided novel ideas
for targeting RNA modifications in cancer therapy.
Modifications of U34 on tRNA
The chemical modification of U34 is a central
regulatory element for codon-decoding precision. In a couple of
previously published studies, U34 is able to extend the tRNA
recognition of synonymous codons through non-classical base pairing
(for example, the G-U wobble), a dynamic pairing mechanism that
fulfills a critical role in maintaining a balance between
translation rate and fidelity that is especially indispensable in
decoding NNN-AA-type codons, and an absence of U34 modifications
led to the accumulation of misfolded proteins, disruptions in
unfolded protein response processes and impaired translation
elongation (136,137) (Fig. 6).
Role of RNA modification in myocardial
fibrosis
Methylation in myocardial fibrosis
m6A in myocardial fibrosis
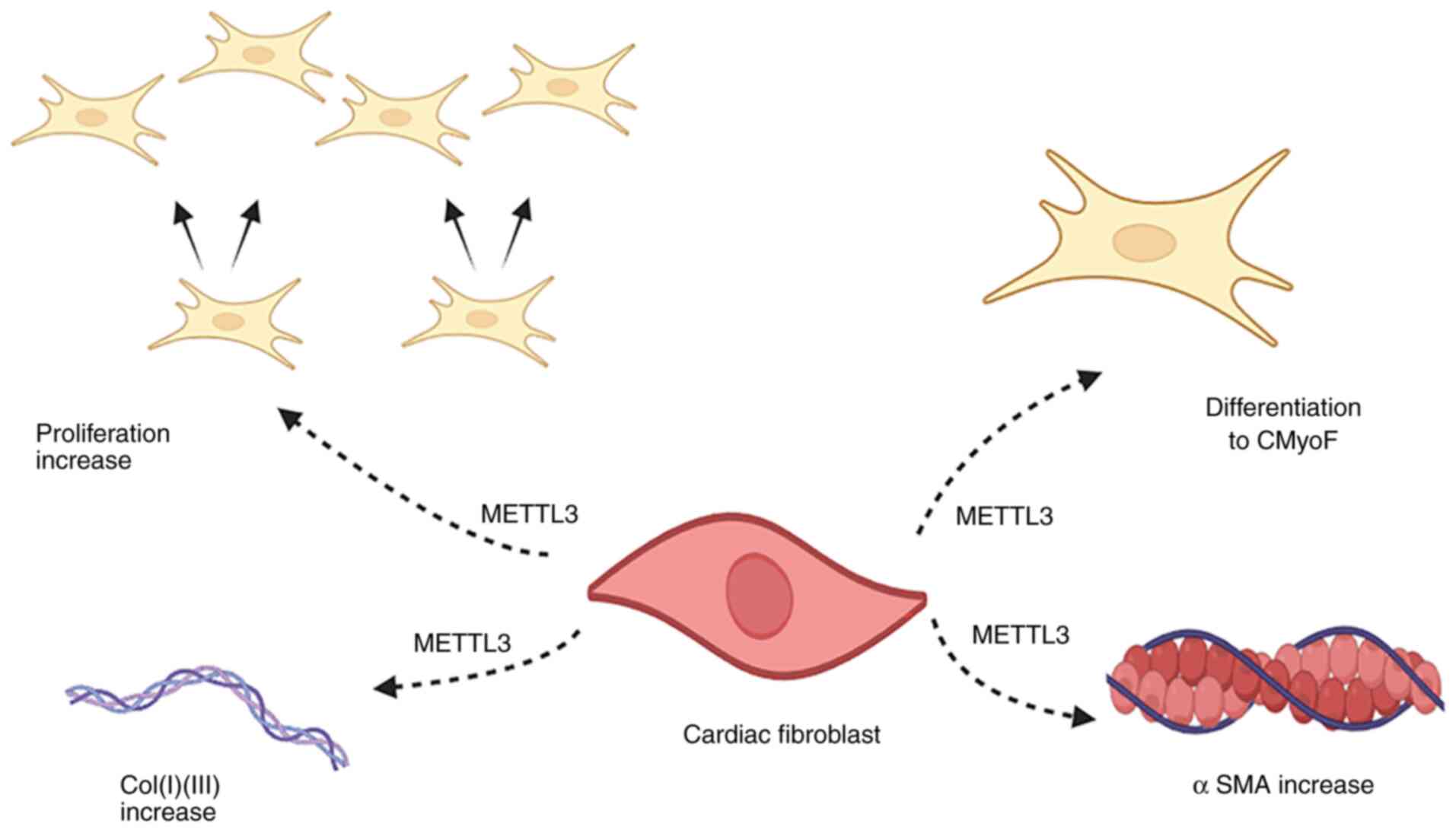
Research has shown that METTL3 is a key driver of
myocardial fibrosis, which regulates pro-fibrotic gene expression
through m6A modification to drive fibroblast activation and the
excessive deposition of ECM. Overexpression of METTL3 markedly
promotes the transdifferentiation of CFs into myofibroblasts, both
through upregulating the expression of type I/III collagen and
mesenchymal fibrosis markers and through activating the
TGF-β1-Mad2/3 signaling pathway (10) (Fig. 7). In addition, m6A modifications
weaken double-stranded RNA stability by triggering a significant
conformational shift of the RNA double strand to a hairpin
structure (for example, in the case of oligonucleotide 19/21),
which directly affects the ability to interact with recognition
proteins, such as FTO, ALKBH5 and YTHDF2. FTO and ALKBH5 were found
to be able to recognize the RNA double strand through m6A-induced
RNA conformational changes, thereby enabling them to distinguish
among substrates of highly similar sequences with dynamic
selectivity. Contrary to the currently held consensus view, the
GG(m6A)CU conserved motif was found not to be essential
for FTO/ALKBH5, whereas METTL3/YTHDF2 was strictly dependent on it.
This demonstrated that the m6A modification itself could
regulate demethylase substrate selectivity through dynamic changes
in RNA structure, challenging the traditional 'sequence motif
determinism' hypothesis, and providing a novel perspective for our
understanding of the functional complexity of the m6A
epitranscriptome (34).
The extracellular matrix glycoprotein tenascin C
(TNC) has been identified as a key downstream target of METTL3 in
recent years. A previous study showed that METTL3 is able to
enhance the mRNA stability of TNC by increasing its m6A
modification level (namely, through increasing the number of
methylated sites), leading to an elevated level of TNC protein
expression, which, in turn, exacerbates cardiomyocyte apoptosis and
fibrosis through the integrin αvβ3 signaling pathway (138). Overexpression of METTL3 has
also been shown to promote the m6A modification of
fibrogenic genes [for example, collagen, type I, α 1 (COL1A1) and
collagen, type III, α 1 (COL3A1)] through YTHDF2-dependent
mechanisms, which, in turn, promotes cardiomyocyte apoptosis and
fibrosis, leading to increased collagen deposition (139). In addition, it has been shown
that the RNA-methylated reading protein YTHDF2 inhibits excessive
mitochondrial autophagy via recognizing the m6A
modification site on BCL2/adenovirus E1B 19 kDa protein-interacting
protein 3 (BNIP3) mRNA and promoting its degradation. In myocardial
ischemia-reperfusion (I/R) injury, due to an absence of YTHDF2 led
to an accumulation of BNIP3 protein, which resulted in the
induction of excessive mitochondrial autophagy and myocardial cell
death; on the other hand, overexpression of YTHDF2 was shown to
reduce the level of BNIP3, leading to a decrease in the area of
myocardial infarction (140).
In exploring pathway synergies, the mRNA stability of METTL3 was
found to be upregulated through m6A modification (with
the consequent prolongation of its half-life), leading to increased
insulin-like growth factor binding protein 3 protein expression.
This molecule formed a synergy with TGF-β signaling to promote
α-smooth muscle actin (α-SMA) expression, thereby markedly
increasing the ability of CFs to migrate and to synthesize collagen
(141). By contrast with
previous findings on METTL3 overexpression, however, inhibition of
this protein was found to be associated with the downregulation of
fibrotic markers and decreased activity of CFs. In myocardial
fibrosis, m6A regulates programmed cell death, and
cardiomyocyte apoptosis is mediated via an upregulation of METTL3.
Elevated m6A levels could promote the transcriptional
degradation of autophagy-associated genes, which ultimately
exacerbate cardiac injury and fibrosis (142).
Although METTL3 has been shown to regulate
pre-METTL3 pre-fibrotic pathways through m6A
modifications, such as the TGF-β/HIF-1α/reactive oxygen species
(ROS) signaling pathway, identifying its therapeutic targets still
presents a major challenge: First, there is a need to clarify the
METTL3 pre-repair (early stage of injury) and pre-fibrotic
(chronic) processes, and second, there is the need to analyze the
synergistic reading proteins, such as those involved in the YTHDF2
and IGF2BP1 regulatory network. Small-molecule inhibitors targeting
METTL3 have entered preclinical studies and are able to markedly
reduce fibrotic areas by selectively inhibiting methyltransferase
activity. These advances will provide novel ideas for enabling us
to reverse pathological cardiac remodeling, although researchers
need to be wary of the risk of the genomic instability that may be
caused by overall m6A inhibition.
m7G in myocardial fibrosis
The dysregulation of m7G modification is
associated with a variety of diseases; however, the role of METTL1
in cardiac fibrosis remains poorly understood. One study (143) identified the critical
regulatory role of METTL1-mediated RNA m7G methylation
in myocardial fibrosis, and the underlying molecular mechanism.
Multidimensional experimental validation studies have shown that,
in fibrotic myocardial tissues from patients with myocardial
infarction and also on the basis of TGF-β1-induced in vitro
models, myocardial fibroblasts exhibit upregulated METTL1
expression levels (which are elevated compared with controls) and a
significant increase in the abundance of transcriptome-wide
m7G methylation (143). In a tamoxifen-induced,
fibroblast-specific METTL1-knockout mouse model (Col1a2-CreERT;
METTL1flox constructed using the Cre-loxP system), cardiac function
indices were found to be markedly improved following myocardial
infarction and histopathological analyses further revealed a
decreased interstitial collagen volume fraction and reduced
transformation rates of α-SMA-positive myofibroblasts. Moreover,
mechanistic studies demonstrated that METTL1, as a key component of
the m7G methylation system, could regulate the
TGF-β/Smad3 signaling pathway (including Smad7 and Smurf2) without
affecting fibroblast gene stability but by impacting mRNA
translation efficiency, thereby promoting an excessive deposition
of ECM proteins (namely, Col1a1, Col3a1 and fibronectin).
Single-cell sequencing analysis revealed that the deletion of
METTL1 specifically inhibited the differentiation of a fibroblast
subpopulation towards a pro-fibrotic phenotype, whereas no
significant effects were exerted on cardiomyocytes or on other
cardiac resident cells. This conclusion not only established for
the first time a direct link between RNA epigenetic modifications
and cardiac fibrosis, but it also provided a theoretical and
experimental basis for the development of novel anti-fibrotic
therapies targeting the METTL1-m7G axis.
Acetylation in myocardial
fibrosis
Recent studies (144,145) have shown that the RNA
acetyltransferase NAT10 dynamically regulates the cardiac fibrotic
process through ac4C modification. In neonatal CFs,
NAT10, through ac4C modification, increased the mRNA
stability and translational efficiency of angiomotin-like protein 1
(Amotl1), facilitating the interaction of the Amotl1 protein with
Yes-associated protein 1 (YAP), and driving YAP nuclear
translocation. This process led to a significant promotion of CF
proliferation (increased rate) and transdifferentiation of the CFs
into myofibroblasts (upregulation of α-SMA expression) through
activation of the Hippo/YAP signaling pathway. Consequently,
experiments wherein Amotl1 gene silencing was combined with the
YAP-selective inhibitor verticibufungin revealed an effective
blockade of the fibrotic phenotype induced by NAT10 overexpression,
which consequently led to reduced collagen deposition.
EGR3, an early growth response factor, promotes
myocardial fibrosis by activating the TGF-β/Smad3 signaling
pathway, which thereby promotes ECM remodeling and exacerbates
myocardial fibrosis (as denoted by an increased fibrotic area). The
lncRNA tsr007330 was shown to mediate ac4C modification
of EGR3 mRNA through recruiting NAT10, leading to an increased
expression of EGR3 protein, which, in turn, exacerbated the
condition. These findings confirmed the centrality of NAT10 in
post-myocardial infarction fibrosis, also revealing the
pathological significance of the non-coding
RNA-ac4C-EGR3 axis as a novel regulatory cascade
reaction (146).
ac4C modifications enhance fibrotic gene
expression through transcriptome reprogramming, additionally
fulfilling a key role in chromatin accessibility remodeling to
activate key signaling pathways (for example, the Hippo/YAP and
TGF-β/Smad signaling pathways). Small-molecule inhibitors targeting
NAT10 (for example, remodeling proteins) have been shown to have
preclinical potential, given their ability to markedly reduce
ac4C modification levels and to improve cardiac
function, although aspects of their tissue-specific delivery and
long-term safety have yet to be evaluated in depth.
Role of RNA modification in other
cardiovascular diseases
Hypertension
The pathogenesis of hypertension, as the leading
preventable risk factor for cardiovascular disease worldwide,
involves a complex interaction of environmental and genetic factors
and is multiply regulated by the immune response, oxidative stress,
sympathetic nerves and the renin-angiotensin system. Studies have
revealed that RNA epigenetic modifications have a key role in blood
pressure homeostasis through regulating the function of vascular
smooth muscle cells (147,148).
ADAR2-mediated RNA editing presents an important
mechanism for maintaining vascular function. This enzyme regulates
the contractile properties of vascular smooth muscle cells by
specifically editing the fine filament protein A (FLNA) precursor
mRNA. When ADAR2 activity becomes aberrant, conformational changes
of the FLNA protein lead to an imbalance in the regulation of
vascular tone, which is closely associated with elevated diastolic
blood pressure. This finding revealed the precise regulatory role
of RNA editing in vascular dynamics (149).
Genetic polymorphisms in the FTO gene, an
m6A demethylase, have been found to be associated with
susceptibility to hypertension. Although the exact mechanism has
not yet been fully elucidated, this regulation at the epigenetic
level has provided novel perspectives to explain the genetic
predisposition to hypertension (150).
The RNA-binding protein HuR also acts as a
molecular hub of multiple modification sites, and participates in
the blood pressure regulatory network through binding
m6A, uridylated and A-to-I-modified transcripts. Reduced
levels of its expression in aortic tissues are associated with the
progression of hypertension and the creation of animal models has
confirmed that HuR deficiency triggers vasodilatory dysfunction and
compensatory myocardial hypertrophy. Mechanistic studies have shown
that HuR maintains the homeostasis of the NO/cGMP signaling pathway
by stabilizing soluble guanylate cyclase and foveolar protein 1
mRNA (151,152).
It should be noted that these regulatory mechanisms
do not exist in isolation. For example, HuR may indirectly affect
the editing efficiency of FLNA by regulating the mRNA stability of
ADAR2, whereas FTO-mediated m6A modification may
interfere with the ability of HuR to bind to target RNAs, forming a
multilevel epistatic regulatory network. This cross-talk property
highlights the systems biology significance of RNA modifications in
blood pressure regulation, and also provides a theoretical basis
for the development of novel antihypertensive strategies targeting
the epitranscriptome.
Atherosclerosis and coronary heart
disease
Atherosclerosis, as an inflammation-driven vascular
lesion, is characterized by lipid deposition and fibrous plaque
formation as its core pathology, which ultimately leads to coronary
artery stenosis and thrombotic events (153). Studies have shown that
m6A modification is heavily involved in the disease
process through regulating vascular inflammation and endothelial
function. In patients with coronary artery disease and in models of
atherosclerosis, the expression levels of the methyltransferases
METTL3 and METTL14 are markedly upregulated (154-156) and their mediated m6A
modifications promote plaque development through a dual mechanism:
First, by enhancing monocyte-endothelial cell adhesion and
activating the NF-κB/IL-6 signaling pathway, which drives the
inflammatory cascade; and second, by inhibiting the mRNA stability
of MyD88 in macrophages, which hinders M2-type polarization and
exacerbates the inflammatory microenvironment within the plaque.
Notably, knockdown of METTL14 leads to a marked reduction in the
macrophage inflammatory phenotype via inhibiting the NF-κB pathway
(156), whereas the
METTL3-m6A-YTHDF2 signaling axis further amplifies
monocyte/macrophage inflammatory responses by regulating the
metabolism of oxidized low-density lipoprotein (157). In addition, the synergistic
interaction of METTL3/14 with ADAR1 regulates vascular
calcification and neovascularization, suggesting their involvement
in a complex mechanism underlying plaque stability and ischemic
compensation (158,159).
In the progression of coronary heart disease, the
dynamic balance of RNA modifications exerts a key regulatory role
in cardiac function. A previous study specifically on
cardiomyocytes showed that the downregulation of FTO expression
under hypoxic conditions leads to an increase in m6A
levels and that FTO overexpression modulates both intracellular
calcium homeostasis and improved myocardial contractile function
through stabilizing ATP2A2 mRNA (encoding the SERCA2a protein)
(160). By contrast, ALKBH5
overexpression was found to inhibit cardiomyocyte proliferation and
to reduce cardiac function through the m6A-YTHDF1-YAP
signaling pathway, revealing a dual role for demethylases in
myocardial repair. In an acute myocardial infarction model,
endothelial cell-derived extracellular vesicles were shown to carry
the METTL3-m6A-HNRNPA2B1 complex, thereby exacerbating
cardiomyocyte apoptosis and dysfunction through the upregulation of
miR-503 (160).
The mechanism of I/R injury, a serious complication
of coronary heart disease, has been demonstrated to be closely
associated with the m6A modification network. In a
myocardial I/R model, METTL3 overexpression was found to promote
the binding of heterogeneous nuclear ribonucleoprotein D to
m6A-modified TFEB mRNA, to inhibit autophagy, and to
induce apoptosis in cardiomyocytes. TFEB inhibits METTL3 stability,
and activates ALKBH5 transcription through a negative feedback
loop, forming a dynamic regulatory balance (160). Gene intervention experiments
confirmed that the myocardium-specific knockdown of METTL14 led to
a reduction in the expression of atrial natriuretic peptide (ANP),
a marker of heart failure, and also a significant improvement in
cardiac function following I/R injury (160). These findings not only revealed
the multilevel regulatory role of RNA modification in coronary
artery disease, but also provided a theoretical basis for
therapeutic strategies that are aimed at targeting the
m6A network (for example, METTL3 inhibitors or FTO
activators). Furthermore, m7G RNA modification exerts
dual roles through dynamic regulation via the methyltransferase
METTL1/WDR4 complex. A previous study (161) showed that, during the early
reperfusion phase (1-3 h), AKT-dependent phosphorylation activates
METTL1, thereby transiently enhancing the translation efficiency of
protective genes (for example, HIF-1α) to maintain metabolic energy
homeostasis. However, in the middle-to-late reperfusion phase
(>6 h), the sustained overexpression of METTL1 drives excessive
m7G modification. This serves to stabilize the mRNAs
encoding the pro-apoptotic factor caspase-3, and the pro-fibrotic
factor TGF-β1, thereby triggering oxidative stress bursts and
mitochondrial dysfunction. Mechanistic analyses have demonstrated
the core process involving the METTL1-mediated serine and arginine
rich splicing factor 9 (SRSF9)/nuclear factor of activated T-cells,
cytoplasmic 4 (NFATc4) signaling axis, where m7G
modification enhances the RNA stability of splicing factor SRSF9,
facilitating the alternative splicing of NFATc4 and nuclear
translocation, thereby activating the calcineurin pathway. This
cascade ultimately leads to pathological cardiomyocyte hypertrophy,
collagen deposition and cardiac functional deterioration. Targeting
METTL1 has also been shown to be a strategy to markedly mitigate
cardiac injury: Myocardial-specific knockout models exhibited a 40%
reduction in infarct size, with improved cardiac function (ejection
fraction and fractional shortening), whereas delivery strategies
(for example, ROS-responsive carriers) that block METTL1 activity
may offer novel translational avenues (162).
Cardiomyopathy
Primary cardiomyopathies encompass various types of
hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy, and
their pathogeneses are closely associated with genetic variation.
In HCM ~70% of cases were shown to be driven by mutations in the
genes of myosin-binding protein C and β-myosin heavy chain (MYH7)
(163), whereas mutations in
other key genes, such as α-myosin heavy chain, troponin T and
myosin ganglionic protein, may also lead to structural disorders of
the cardiac myocardium (164,165). Abnormal mitochondrial function,
specifically mitochondrial dysfunction in the myocardium, fulfills
an important role in the progression of cardiomyopathy, and
mutations in the GTP-binding protein 3 or mitochondrial translation
optimization 1 homolog genes were shown to elicit an impaired
assembly of respiratory chain complexes through impairing the
5-taurinomethyluridine (τm5U) modification of mitochondrial tRNAs,
ultimately leading to HCM (166). In a dilated cardiomyopathy
(DCM) model, specific knockdown of the pentatricopeptide repeat
domain 1 gene disrupted ribosomal large subunit assembly through
interfering with the Ψ-modification at position 2509 of 16S rRNA,
which had the effect of triggering defects in mitochondrial energy
metabolism with dysregulated mTOR signaling (167). In addition, conditional
knockdown experiments of YTHDC1 and ADAR1 confirmed that the
deletion of RNA-binding proteins could induce DCM through aberrant
m6A modification (168) and dysregulated RNA editing
(110), respectively.
Unlike genetic lesions, secondary cardiomyopathies
(for example, diabetic cardiomyopathy and chemotherapy
cardiotoxicity) are primarily driven by acquired factors. In
diabetic cardiomyopathy, METTL14-induced hypermethylation of the
lncRNA TINCR was found to enhance the degradation of TINCR via
YTHDF2, which consequently reduced the inhibitory effect of TINCR
on NLRP3 inflammasome targets. Furthermore, the lncRNA Airn was
shown to stabilize p53 mRNA through m6A modification, also
inhibiting the ubiquitin-mediated disruption of eIF2C, thereby
attenuating myocardial fibrosis in diabetic cardiomyopathy
(169,170). In adriamycin-induced
cardiotoxicity, METTL14 was shown to exacerbate myocardial injury
by promoting iron death (specifically, through increasing the
levels of lipid peroxidation) through modulation of the
m6A modification of genes involved in glutathione
metabolism (171).
The aforementioned studies have revealed the
centrality of RNA epitope modification in cardiomyopathy: First,
METTL14 inhibitors may inhibit inflammatory vesicle activation
through restoring TINCR expression; subsequently, the
m6A-reading function, targeting YTHDC1, may correct
metabolic derangements in the cardiomyocytes; and lastly, ADAR1
activators may both repair RNA-editing aberrations and improve
myocardial electrophysiological stability. However, resolution of
the tissue-specific delivery systems with dynamic modification
profiles remains a key challenge for clinical translation.
Cardiac hypertrophy
Cardiac hypertrophy, as a compensatory response of
the myocardium to stress load, can be categorized into two types,
namely physiological and pathological, which exhibit significant
differences in their epitranscriptome regulatory mechanisms.
Physiological hypertrophy (for example, that induced by exercise)
was shown to be accompanied by a decrease in the overall
m6A levels of myocardial mRNA, and this was potentially
associated with the downregulation of METTL14 expression (160,172). By contrast, the molecular
mechanism of pathological hypertrophy (for example, that induced by
pressure overload), a major trigger of heart failure, involves a
complex dysregulation of the dynamic balance of m6A
modification. It is caused by a variety of cardiac diseases,
including both heart failure with maintained ejection fraction and
heart failure with reduced ejection fraction. In the aortic arch
constriction model, the myocardium-specific knockdown of FTO was
shown to exacerbate ejection fraction-reduced heart failure
(173). On the other band, FTO
overexpression was demonstrated to inhibit the expression of
pathologic hypertrophy-associated genes, such as ANP and B-type
natriuretic peptide, through demethylation, resulting in a
reduction in the cardiomyocyte cross-sectional area (174). This bidirectional regulatory
effect suggests that FTO fulfills a key role in maintaining the
fine balance between myocardial adaptation and pathological
remodeling.
METTL3 functions as an m6A
methyltransferase in an environment-dependent manner: Its
overexpression in the basal state enhances cardiac compensatory
function, although the myocardium-specific knockdown of METTL3 was
found to accelerate the progression of heart failure under pressure
overload conditions. Mechanistic studies have shown that the
Piwi-interacting RNA CHAPIR promotes pathological ventricular
hypertrophy by binding to and inhibiting METTL3 methylation
activity, thereby leading to hypomethylation of the mRNA of the
ADP-ribosyltransferase PARP10, and blocking the YTHDF2-mediated
degradation pathway (175,176). Notably, the
m6A-reading protein YTHDF2, whose expression is
upregulated in heart failure, was shown to promote the cardiac
hypertrophic phenotype through stabilizing myosin-7 (MYH7) mRNA
(namely, prolonging its half-life), thereby creating a vicious
circle (176).
Taken together, these findings have revealed a dual
role for the m6A modification network: Its first role is
to regulate myocardial compensation and dystrophic transition
through the balance of FTO-METTL3, and its second role is to drive
sustained progression of the hypertrophic phenotype through the
YTHDF2-MYH7 axis. Targeting this regulatory network (for example,
through the development of METTL3 mutagenic activators or YTHDF2
inhibitors) may provide possible novel strategies, although for
this to occur, technical challenges presented by matters such as
tissue-specific delivery and dynamic modification monitoring will
need to be addressed.
Summary and future outlook
The RNA epitranscriptional regulatory network of
myocardial fibrosis is associated with both breakthrough
therapeutic opportunities and complex biological challenges.
Methylation reactions (for example, m6A and
m7G) and ac4C serve as core modification
mechanisms that drive fibrotic progression through precisely
regulating fibroblast activation and extracellular matrix
deposition. METTL3 silencing markedly reduces the levels of
fibrosis markers such as collagen, revealing its potential as a
drug target; however, the potential toxicity of the molecule to
cardiomyocytes presents an obstacle to its clinical application: A
paradox that highlights the critical importance of cell-specific
effects. By way of contrast, the METTL1-m7G axis has
unique properties: In fibrotic tissues, from patients with
myocardial infarction and in TGF-β1-induced in vitro models,
METTL1 is specifically overexpressed in cardiac fibroblasts and is
accompanied by elevated levels of transcriptome-wide m7G
methylation; knockdown of METTL1 not only improves cardiac
function, but it also reduces collagen deposition and
α-SMA-positive myofibroblast transformation, more critically,
without affecting cardiomyocyte function. This selectivity stems
from the fact that METTL1 specifically promotes the excessive
deposition of ECM proteins by finely regulating mRNA translation
efficiency, rather than the stability of key genes of the TGF-β
pathway (or example, Smad7/Smurf2) through m7G
modification.
Acetylation modifications amplify fibrotic signals
through mechanisms at multiple levels in concert with methylation
networks. NAT10, as an RNA acetyltransferase, dynamically regulates
cardiac fibrosis through ac4C epigenetic modifications:
It enhances Amotl1 mRNA stability and translational efficiency,
promotes the binding of Amotl1 protein to YAP, and activates the
Hippo/YAP signaling pathway, ultimately accelerating fibroblast
proliferation and transdifferentiation. Notably, silencing of the
Amotl1 gene combined with YAP-specific inhibitors has been shown to
effectively block the fibrotic phenotype induced by NAT10
overexpression, and the synergistic effect of EGR3 with the
TGF-β/Smad3 pathway, and the multilayered regulation of fibrosis by
NAT10, further established its centrality to post-infarction
fibrosis. These findings reveal a hierarchical modification network
architecture: The METTL3-m6A modification initiates the
TGF-β transcriptional cascade, the NAT10-ac4C
modification stabilizes key effectors (for example, EGR3 mRNA), and
the METTL1-m7G modification enhances the translational
efficiency of downstream genes. Altogether, the three types of
modification form a self-amplifying pathogenic cycle (Fig. 8).
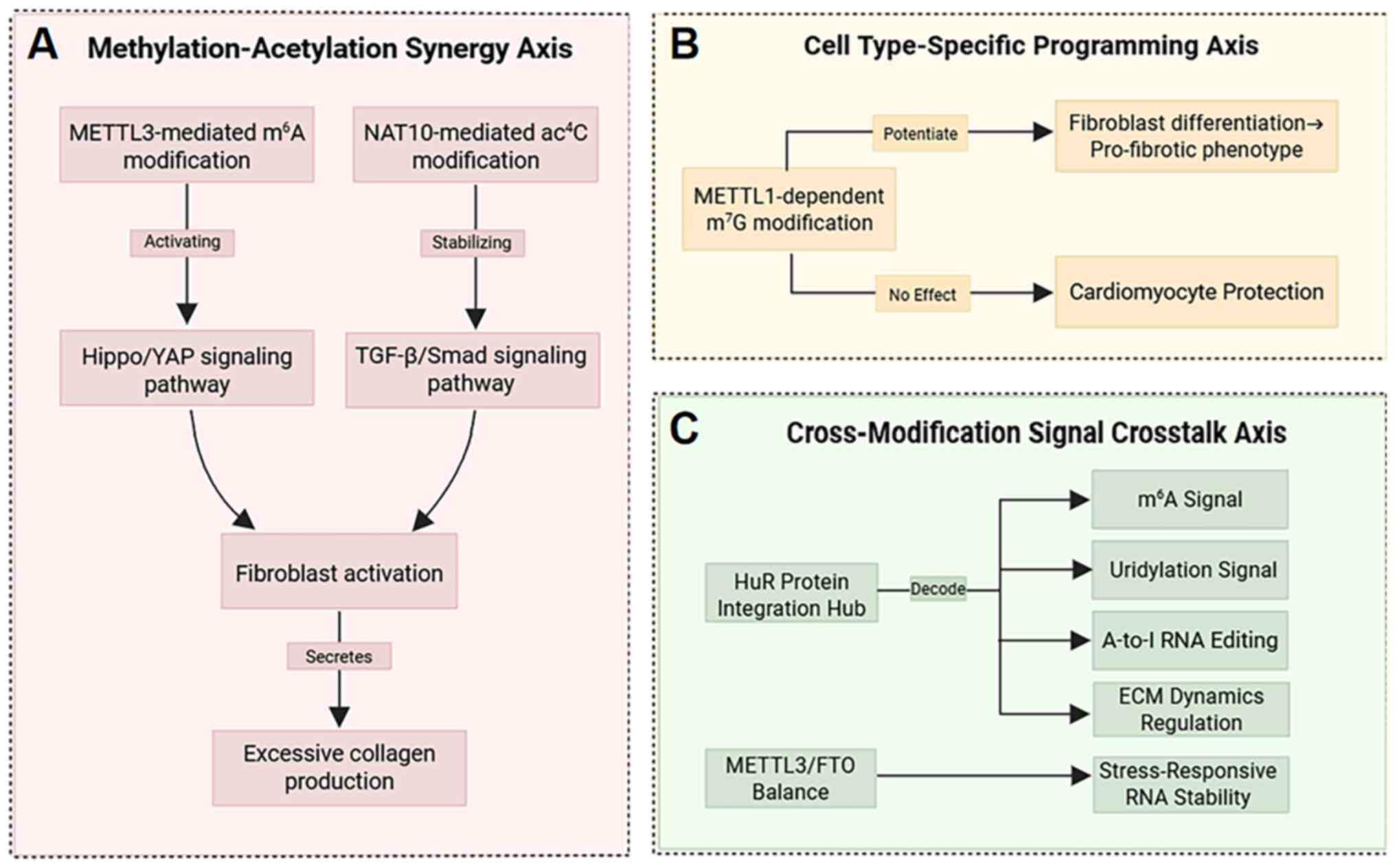 | Figure 8The three-axis regulatory model is
shown: (A) Methylation-acetylation synergy, where METTL3-mediated
m6A modifications and NAT10-driven ac4C
modifications co-operatively target the Hippo/YAP and TGF-β/Smad
signaling pathways, exacerbating fibroblast activation and
excessive collagen production. (B) Cell-type-specific programming
revealed by single-cell analysis, as exemplified by
METTL1-catalyzed m7G modifications selectively promoting
fibroblast differentiation into pro-fibrotic phenotypes while
protecting cardiomyocytes. (C) Cross-modification crosstalk,
illustrated by the HuR protein integrating m6A,
uridylation, and A-to-I editing signals to regulate extracellular
matrix dynamics, while the METTL3/FTO balance influences
stress-responsive RNA stability. Collectively, these three axes
drive the pathological phenotype of myocardial fibrosis,
characterized by excessive collagen deposition, increased
ventricular stiffness and diastolic dysfunction. METTL1/3,
methyltransferase-like 1/3; m6A,
N6-methyladenosine; NAT10, N-acetyltransferase
10; YAP, Yes-associated protein; A-to-I, adenine-to-inosine; FTO,
fat mass and obesity-associated. |
These post-transcriptional modifications have been
demonstrated to affect cell function and RNA metabolism, to
regulate gene expression, and to influence both organ development
and cell differentiation, ultimately determining the cell's fate.
Through these chemical changes, cells have an improved ability to
adapt to internal and external stimuli (39,41,160). Innovative solutions are
required to circumvent the core bottlenecks faced by translational
medicine: The dual role of METTL3 in both the early repair of
injury and the chronic fibrosis stage requires the development of
spatiotemporal regulation strategies, such as ROS-responsive
nanocarriers for precise drug delivery of myocardial infarction.
The fibroblast specificity of METTL1 offers opportunities for
spatial targeting, which can be accomplished using the Col1a2
promoter-driven CRISPR-Cas9 system for cell-type specific
interventions. Future research should focus on three frontiers:
First, resolving the precise modification sites of METTL1 on
Smad7/Smurf2 mRNA and elucidating the underlying mechanism of
interaction with the translation machinery; second, exploring the
network interactions of m7G dynamic modifications with
other nodes of the TGF-β pathway; and third, developing
METTL1-specific small-molecule inhibitors and a serum exosome
modification fingerprinting diagnostic system. Current antifibrotic
treatments are still in the developmental stages; although RNA
modification networks have opened new therapeutic windows, existing
strategies are mostly limited to symptom control. Fundamental
breakthroughs are required for the integration of
epitranscriptomics, non-coding RNA and signaling pathway research;
for example, the development of CRISPR-Cas13d-mediated editing of
the Amotl1 mRNA ac4C locus, or patient stratification
(high m7G/ac4C ratio populations) to achieve
precision intervention. Ultimately, successful translation of
primary research results to the clinic will depend on our ability
to harness both cell-specific targeting (for example,
fibroblast-restricted METTL1 inhibition) and critical node
breakthroughs to move epitranscriptional regulation from the
mechanistic discovery mode to finding clinical solutions.
Availability of data and materials
Not applicable.
Authors' contributions
KW conceived the present study and provided
suggestions for the revision of the manuscript. XW, RW and XC wrote
the manuscript and made the figures. Data authentication is not
applicable. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, a generative
AI/AI-assisted technology, namely DeepSeek, was used to improve the
readability and language of the manuscript.
Acknowledgements
Not applicable.
Funding
The present study was supported by Qingdao Science and
Technology Benefiting the People Demonstration Project (grant no.
24-1-8-smjk-7-nsh), Major Basic Research Projects in Shandong
Province (grant no. ZR2024ZD46) and Taishan Scholar Distinguished
Expert.
References
|
1
|
Liu M, López de Juan Abad B and Cheng K:
Cardiac fibrosis: Myofibroblast-mediated pathological regulation
and drug delivery strategies. Adv Drug Deliv Rev. 173:504–519.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Talman V and Ruskoaho H: Cardiac fibrosis
in myocardial infarction-from repair and remodeling to
regeneration. Cell Tissue Res. 365:563–581. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ivey MJ and Tallquist MD: Defining the
cardiac fibroblast. Circ J. 80:2269–2276. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kendall RT and Feghali-Bostwick CA:
Fibroblasts in fibrosis: Novel roles and mediators. Front
Pharmacol. 5:1232014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tallquist MD and Molkentin JD: Redefining
the identity of cardiac fibroblasts. Nat Rev Cardiol. 14:484–491.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pellman J, Zhang J and Sheikh F:
Myocyte-fibroblast communication in cardiac fibrosis and
arrhythmias: Mechanisms and model systems. J Mol Cell Cardiol.
94:22–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hinz B: Myofibroblasts. Exp Eye Res.
142:56–70. 2016. View Article : Google Scholar
|
|
8
|
Thomas TP and Grisanti LA: The dynamic
interplay between cardiac inflammation and fibrosis. Front Physiol.
11:5290752020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hinderer S and Schenke-Layland K: Cardiac
fibrosis-A short review of causes and therapeutic strategies. Adv
Drug Deliv Rev. 146:77–82. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li T, Zhuang Y, Yang W, Xie Y, Shang W, Su
S, Dong X, Wu J, Jiang W, Zhou Y, et al: Silencing of METTL3
attenuates cardiac fibrosis induced by myocardial infarction via
inhibiting the activation of cardiac fibroblasts. FASEB J.
35:e211622021. View Article : Google Scholar
|
|
11
|
Huang W, Chen TQ, Fang K, Zeng ZC, Ye H
and Chen YQ: N6-methyladenosine methyltransferases: Functions,
regulation, and clinical potential. J Hematol Oncol. 14:1172021.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wei CM, Gershowitz A and Moss B:
Methylated nucleotides block 5' terminus of HeLa cell messenger
RNA. Cell. 4:379–386. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Furuichi Y, Morgan M, Shatkin AJ, Jelinek
W, Salditt-Georgieff M and Darnell JE: Methylated, blocked 5
termini in HeLa cell mRNA. Proc Natl Acad Sci USA. 72:1904–1908.
1975. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dominissini D, Moshitch-Moshkovitz S,
Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K,
Jacob-Hirsch J, Amariglio N, Kupiec M, et al: Topology of the human
and mouse m6A RNA methylomes revealed by m6A-seq. Nature.
485:201–284. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meyer KD, Saletore Y, Zumbo P, Elemento O,
Mason CE and Jaffrey SR: Comprehensive analysis of mRNA methylation
reveals enrichment in 3' UTRs and near stop codons. Cell.
149:1635–1646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zaccara S, Ries RJ and Jaffrey SR:
Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell
Biol. 20:608–624. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bokar JA, Shambaugh ME, Polayes D, Matera
AG and Rottman FM: Purification and cDNA cloning of the
AdoMet-binding subunit of the human mRNA
(N-6-adenosine)-methyltransferase. RNA. 3:1233–1247.
1997.PubMed/NCBI
|
|
18
|
Lin Z, Hsu PJ, Xing X, Fang J, Lu Z, Zou
Q, Zhang KJ, Zhang X, Zhou Y, Zhang T, et al:
Mettl3-/Mettl14-mediated mRNA N6-methyladenosine modulates murine
spermatogenesis. Cell Res. 27:1216–1230. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ping XL, Sun BF, Wang L, Xiao W, Yang X,
Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al: Mammalian WTAP is
a regulatory subunit of the RNA N6-methyladenosine
methyltransferase. Cell Res. 24:177–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang P, Doxtader KA and Nam Y: Structural
basis for cooperative function of Mettl3 and Mettl14
methyltransferases. Mol Cell. 63:306–317. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, Feng J, Xue Y, Guan Z, Zhang D,
Liu Z, Gong Z, Wang Q, Huang J, Tang C, et al: Structural basis of
N6-adenosine methylation by the METTL3-METTL14 complex. Nature.
534:575–578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sledz P and Jinek M: Structural insights
into the molecular mechanism of the m6A writer complex. Elife.
5:e184342016. View Article : Google Scholar :
|
|
23
|
Liu JZ, Yue Y, Han D, Wang X, Fu Y, Zhang
L, Jia G, Yu M, Lu Z, Deng X, et al: A METTL3-METTL14 complex
mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem
Biol. 10:93–95. 2014. View Article : Google Scholar :
|
|
24
|
Huang Q, Mo J, Liao Z, Chen X and Zhang B:
The RNA m6A writer WTAP in diseases: Structure, roles, and
mechanisms. Cell Death Dis. 13:8522022. View Article : Google Scholar :
|
|
25
|
Yue YA, Liu J, Cui X, Cao J, Luo G, Zhang
Z, Cheng T, Gao M, Shu X, Ma H, et al: VIRMA mediates preferential
m6A mRNA methylation in 3'UTR and near stop codon and associates
with alternative polyadenylation. Cell Discov. 4:102018. View Article : Google Scholar
|
|
26
|
Patil DP, Chen CK, Pickering BF, Chow A,
Jackson C, Guttman M and Jaffrey SR: m6A RNA methylation promotes
XIST-mediated transcriptional repression. Nature. 537:369–373.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Zhang L, Ren H, Ma L, Guo J, Mao
D, Lu Z, Lu L and Yan D: Role of Hakai in m6A modification pathway
in Drosophila. Nat Commun. 12:21592021. View Article : Google Scholar :
|
|
28
|
Knuckles P, Lence T, Haussmann IU, Jacob
D, Kreim N, Carl SH, Masiello I, Hares T, Villaseñor R, Hess D, et
al: Zc3h13/Flacc is required for adenosine methylation by bridging
the mRNA-binding factor Rbm15/Spenito to the m6A machinery
component Wtap/Fl(2)d. Genes Dev. 32:415–429. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang XL, Liu B, Nie Z, Duan L, Xiong Q,
Jin Z, Yang C and Chen Y: The role of m6A modification in the
biological functions and diseases. Signal Transduct Target Ther.
6:742021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Du Y, Hou G, Zhang H, Dou J, He J, Guo Y,
Li L, Chen R, Wang Y, Deng R, et al: SUMOylation of the m6A-RNA
methyltransferase METTL3 modulates its function. Nucleic Acids Res.
46:5195–5208. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu Z, Lv B, Qin Y and Zhang B: Emerging
roles and mechanism of m6A methylation in cardiometabolic diseases.
Cells. 11:11012022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liao S, Sun H and Xu C: YTH Domain: A
family of N6-methyladenosine (m6A) Readers. Genomics Proteomics
Bioinformatics. 16:99–107. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schöller E, Weichmann F, Treiber T, Ringle
S, Treiber N, Flatley A, Feederle R, Bruckmann A and Meister G:
Interactions, localization, and phosphorylation of the m6A
generating METTL3-METTL14-WTAP complex. RNA. 24:499–512. 2018.
View Article : Google Scholar
|
|
34
|
Zou S, Toh JD, Wong KH, Gao YG, Hong W and
Woon EC: N6-Methyladenosine: A conformational marker that regulates
the substrate specificity of human demethylases FTO and ALKBH5. Sci
Rep. 6:256772016. View Article : Google Scholar :
|
|
35
|
Grozhik AV, Olarerin-George AO, Sindelar
M, Li X, Gross SS and Jaffrey SR: Antibody cross-reactivity
accounts for widespread appearance of m1A in 5'UTRs. Nat Commun.
10:51262019. View Article : Google Scholar
|
|
36
|
Bar-Yaacov D, Frumkin I, Yashiro Y, Chujo
T, Ishigami Y, Chemla Y, Blumberg A, Schlesinger O, Bieri P, Greber
B, et al: Mitochondrial 16S rRNA is methylated by tRNA
methyltransferase TRMT61B in all vertebrates. PLoS Biol.
14:e10025572016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chujo T and Suzuki T: Trmt61B is a
methyltransferase responsible for 1-methyladenosine at position 58
of human mitochondrial tRNAs. RNA. 18:2269–2276. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vilardo E, Nachbagauer C, Buzet A,
Taschner A, Holzmann J and Rossmanith W: A subcomplex of human
mitochondrial RNase P is a bifunctional methyltransferase-extensive
moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res.
40:11583–11593. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li X, Xiong X, Wang K, Wang L, Shu X, Ma S
and Yi C: Transcriptome-wide mapping reveals reversible and dynamic
N(1)-methyladenosine methylome. Nat Chem Biol. 12:311–316. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen Z, Qi M, Shen B, Luo G, Wu Y, Li J,
Lu Z, Zheng Z, Dai Q and Wang H: Transfer RNA demethylase ALKBH3
promotes cancer progression via induction of tRNA-derived small
RNAs. Nucleic Acids Res. 47:2533–2545. 2019. View Article : Google Scholar :
|
|
41
|
Liu F, Clark W, Luo G, Wang X, Fu Y, Wei
J, Wang X, Hao Z, Dai Q, Zheng G, et al: ALKBH1-Mediated tRNA
demethylation regulates translation. Cell. 167:816–828.e16. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wei JB, Liu F, Lu Z, Fei Q, Ai Y, He PC,
Shi H, Cui X, Su R, Klungland A, et al: Differential m6A, m6Am, and
m1A Demethylation Mediated by FTO in the cell nucleus and
cytoplasm. Mol Cell. 71:973–985.e5. 2018. View Article : Google Scholar
|
|
43
|
Qin G, Sun Y-W, Guo Y-D, Li J-W, Liang C,
Feng H and Yong S: SND1 recognize the methylation sites of TINCR to
promote growth of keloid fibroblasts. Med J Chin PLA. 46:1068–1076.
2021.In Chinese.
|
|
44
|
Guo GQ, Pan K, Fang S, Ye L, Tong X, Wang
Z, Xue X and Zhang H: Advances in mRNA 5-methylcytosine
modifications: Detection, effectors, biological functions, and
clinical relevance. Mol Ther Nucleic Acids. 26:575–593. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Squires JE, Patel HR, Nousch M, Sibbritt
T, Humphreys DT, Parker BJ, Suter CM and Preiss T: Widespread
occurrence of 5-methylcytosine in human coding and non-coding RNA.
Nucleic Acids Res. 40:5023–5033. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Huang T, Chen W, Liu J, Gu N and Zhang R:
Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat
Struct Mol Biol. 26:380–388. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Van Haute L, Dietmann S, Kremer L, Hussain
S, Pearce SF, Powell CA, Rorbach J, Lantaff R, Blanco S, Sauer S,
et al: Deficient methylation and formylation of mt-tRNA (Met)
wobble cytosine in a patient carrying mutations in NSUN3. Nat
Commun. 7:120392016. View Article : Google Scholar
|
|
48
|
Liu RJ, Long T, Li J, Li H and Wang ED:
Structural basis for substrate binding and catalytic mechanism of a
human RNA:m5C methyltransferase NSun6. Nucleic Acids Res.
45:6684–6697. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huang ZX, Li J, Xiong QP, Li H, Wang ED
and Liu RJ: Position 34 of tRNA is a discriminative element for
m5C38 modification by human DNMT2. Nucleic Acids Res.
49:13045–13061. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Janin M, Ortiz-Barahona V, de Moura MC,
Martínez-Cardús A, Llinàs-Arias P, Soler M, Nachmani D, Pelletier
J, Schumann U, Calleja-Cervantes ME, et al: Epigenetic loss of
RNA-methyltransferase NSUN5 in glioma targets ribosomes to drive a
stress adaptive translational program. Acta Neuropathol.
138:1053–1074. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liao H, Gaur A, McConie H, Shekar A, Wang
K, Chang JT, Breton G and Denicourt C: Human NOP2/NSUN1 regulates
ribosome biogenesis through non-catalytic complex formation with
box C/D snoRNPs. Nucleic Acids Res. 50:10695–10716. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yang L, Ren Z, Yan S, Zhao L, Liu J, Zhao
L, Li Z, Ye S, Liu A, Li X, et al: Nsun4 and Mettl3 mediated
translational reprogramming of Sox9 promotes BMSC chondrogenic
differentiation. Commun Biol. 5:4952022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Spåhr H, Habermann B, Gustafsson CM,
Larsson NG and Hallberg BM: Structure of the human MTERF4-NSUN4
protein complex that regulates mitochondrial ribosome biogenesis.
Proc Natl Acad Sci USA. 109:15253–15258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dai X, Gonzalez G, Li L, Li J, You C, Miao
W, Hu J, Fu L, Zhao Y, Li R, et al: YTHDF2 Binds to
5-Methylcytosine in RNA and modulates the maturation of ribosomal
RNA. Anal Chem. 92:1346–1354. 2020. View Article : Google Scholar :
|
|
55
|
Xue C, Gu X, Zheng Q, Shi Q, Yuan X, Su Y,
Jia J, Jiang J, Lu J and Li L: ALYREF mediates RNA m5C modification
to promote hepatocellular carcinoma progression. Signal Transduct
Target Ther. 8:1302023. View Article : Google Scholar :
|
|
56
|
Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu
Y, Li Z, Li X, Zhao K, Wang C, et al: Tet2 promotes pathogen
infection-induced myelopoiesis through mRNA oxidation. Nature.
554:123–127. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yang WL, Qiu W, Zhang T, Xu K, Gu ZJ, Zhou
Y, Xu HJ, Yang ZZ, Shen B, Zhao YL, et al: Nsun2 coupling with
RoRγt shapes the fate of Th17 cells and promotes colitis. Nat
Commun. 14:8632023. View Article : Google Scholar
|
|
58
|
Muthukrishnan S, Both GW, Furuichi Y and
Shatkin AJ: 5'-Terminal 7-methylguanosine in eukaryotic mRNA is
required for translation. Nature. 255:33–37. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Adams JM and Cory S: Modified nucleosides
and bizarre 5'-termini in mouse myeloma mRNA. Nature. 255:28–33.
1975. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Létoquarta J, Huvelle E, Wacheul L,
Bourgeois G, Zorbas C, Graille M, Heurgué-Hamard V and Lafontaine
DL: Structural and functional studies of Bud23-Trm112 reveal 18S
rRNA N7-G1575 methylation occurs on late 40S precursor ribosomes.
Proc Natl Acad Sci USA. 111:E5518–E5526. 2014.
|
|
61
|
Aregger M, Kaskar A, Varshney D,
Fernandez-Sanchez ME, Inesta-Vaquera FA, Weidlich S and Cowling VH:
CDK1-Cyclin B1 activates RNMT, coordinating mRNA Cap methylation
with G1 phase transcription. Mol Cell. 61:734–746. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Pandolfini L, Barbieri I, Bannister AJ,
Hendrick A, Andrews B, Webster N, Murat P, Mach P, Brandi R, Robson
SC, et al: METTL1 Promotes let-7 MicroRNA processing via m7G
methylation. Mol Cell. 74:1278–1290.e9. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Dai ZH, Liu H, Liao J, Huang C, Ren X, Zhu
W, Zhu S, Peng B, Li S, Lai J, et al: N7-Methylguanosine tRNA
modification enhances oncogenic mRNA translation and promotes
intrahepatic cholangiocarcinoma progression. Mol Cell.
81:3339–3355.e8. 2021. View Article : Google Scholar
|
|
64
|
Zhang LS, Liu C, Ma H, Dai Q, Sun HL, Luo
G, Zhang Z, Zhang L, Hu L, Dong X and He C: Transcriptome-wide
mapping of internal N7-methylguanosine methylome in mammalian mRNA.
Mol Cell. 74:1304–1316.e8. 2019. View Article : Google Scholar
|
|
65
|
Furuichi Y, LaFiandra A and Shatkin AJ:
5'-Terminal structure and mRNA stability. Nature. 266:235–239.
1977. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Konarska MM, Padgett RA and Sharp PA:
Recognition of cap structure in splicing in vitro of mRNA
precursors. Cell. 38:731–736. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hsu CL and Stevens A: Yeast-cells lacking
5'-3' exoribonuclease-1 contain mRNA species that are poly(A)
deficient and partially lack the 5' cap structure. Mol Cell Biol.
13:4826–4835. 1993.PubMed/NCBI
|
|
68
|
Ma JY, Han H, Huang Y, Yang C, Zheng S,
Cai T, Bi J, Huang X, Liu R, Huang L, et al: METTL1/WDR4-mediated
m7G tRNA modifications and m7G codon usage promote mRNA translation
and lung cancer progression. Mol Ther. 29:3422–3435. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lin S, Liu Q, Lelyveld VS, Choe J, Szostak
JW and Gregory RI: Mettl1/Wdr4-Mediated m7G tRNA methylome is
required for normal mRNA translation and embryonic stem cell
self-renewal and differentiation. Mol Cell. 71:244–255.e5. 2018.
View Article : Google Scholar :
|
|
70
|
Ito S, Horikawa S and Suzuki T, Kawauchi
H, Tanaka Y and Suzuki T and Suzuki T: Human NAT10 Is an
ATP-dependent RNA acetyltransferase responsible for
N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J Biol
Chem. 289:35724–35730. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Arango D, Sturgill D, Alhusaini N, Dillman
AA, Sweet TJ, Hanson G, Hosogane M, Sinclair WR, Nanan KK, Mandler
MD, et al: Acetylation of cytidine in mRNA promotes translation
efficiency. Cell. 175:1872–1886.e24. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Arango D, Sturgill D, Yang R, Kanai T,
Bauer P, Roy J, Wang Z, Hosogane M, Schiffers S and Oberdoerffer S:
Direct epitranscriptomic regulation of mammalian translation
initiation through N4-acetylcytidine. Mol Cell. 82:2797–2814.e11.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yu XM, Li SJ, Yao ZT, Xu JJ, Zheng CC, Liu
ZC, Ding PB, Jiang ZL, Wei X, Zhao LP, et al: N4-acetylcytidine
modification of lncRNA CTC-490G23.2 promotes cancer metastasis
through interacting with PTBP1 to increase CD44 alternative
splicing. Oncogene. 42:1101–1116. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yang W, Li HY, Wu YF, Mi RJ, Liu WZ, Shen
X, Lu YX, Jiang YH, Ma MJ and Shen HY: ac4C acetylation of RUNX2
catalyzed by NAT10 spurs osteogenesis of BMSCs and prevents
ovariectomy-induced bone loss. Mol Ther Nucleic Acids. 26:135–147.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang GP, Zhang M, Zhang Y, Xie Y, Zou J,
Zhong J, Zheng Z, Zhou X, Zheng Y, Chen B and Liu C: NAT10-mediated
mRNA N4-acetylcytidine modification promotes bladder cancer
progression. Clin Transl Med. 12:e7382022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Feng ZY, Li K, Qin K, Liang J, Shi M, Ma
Y, Zhao S, Liang H, Han D, Shen B, et al: Retraction Note: The
LINC00623/NAT10 signaling axis promotes pancreatic cancer
progression by remodeling ac4C modification of mRNA. J Hematol
Oncol. 16:1092023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zheng X, Wang Q, Zhou Y, Zhang D, Geng Y,
Hu W, Wu C, Shi Y and Jiang J: N-acetyltransferase 10 promotes
colon cancer progression by inhibiting ferroptosis through
N4-acetylation and stabilization of ferroptosis suppressor protein
1 (FSP1) mRNA. Cancer Commun (Lond). 42:1347–1366. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Jin C, Wang T, Zhang D, Yang P, Zhang C,
Peng W, Jin K, Wang L, Zhou J, Peng C, et al: Acetyltransferase
NAT10 regulates the Wnt/β-catenin signaling pathway to promote
colorectal cancer progression via ac4C acetylation of KIF23 mRNA. J
Exp Clin Cancer Res. 41:3452022. View Article : Google Scholar
|
|
79
|
Liao L, He Y, Li SJ, Yu XM, Liu ZC, Liang
YY, Yang H, Yang J, Zhang GG, Deng CM, et al: Lysine
2-hydroxyisobutyrylation of NAT10 promotes cancer metastasis in an
ac4C-dependent manner. Cell Res. 33:355–371. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Nance KD, Gamage ST, Alam MM, Yang A, Levy
MJ, Link CN, Florens L, Washburn MP, Gu S, Oppenheim JJ and Meier
JL: Cytidine acetylation yields a hypoinflammatory synthetic
messenger RNA. Cell Chem Biol. 29:312–320.e7. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chang H, Lim J, Ha M and Kim VN: TAIL-seq:
Genome-wide determination of poly(A) tail length and 3' end
modifications. Mol Cell. 53:1044–1052. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lim J, Ha M, Chang H, Kwon SC, Simanshu
DK, Patel DJ and Kim VN: Uridylation by TUT4 and TUT7 marks mRNA
for degradation. Cell. 159:1365–1376. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Yang A, Bofill-De Ros X, Stanton R, Shao
TJ, Villanueva P and Gu S: TENT2, TUT4, and TUT7 selectively
regulate miRNA sequence and abundance. Nat Commun. 13:52602022.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Heo I, Joo C, Kim YK, Ha M, Yoon MJ, Cho
J, Yeom KH, Han J and Kim VN: TUT4 in Concert with Lin28 suppresses
MicroRNA biogenesis through Pre-MicroRNA Uridylation. Cell.
138:696–708. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Yi H, Park J, Ha M, Lim J, Chang H and Kim
VN: PABP Cooperates with the CCR4-NOT complex to promote mRNA
deadenylation and block precocious decay. Mol Cell.
70:1081–1088.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Mullen TE and Marzluff WF: Degradation of
histone mRNA requires oligouridylation followed by decapping and
simultaneous degradation of the mRNA both 5' to 3' and 3' to 5'.
Genes Dev. 22:50–65. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Schmidt MJ, West S and Norbury CJ: The
human cytoplasmic RNA terminal U-transferase ZCCHC11 targets
histone mRNAs for degradation. RNA. 17:39–44. 2011. View Article : Google Scholar :
|
|
88
|
Malecki M, Viegas SC, Carneiro T, Golik P,
Dressaire C, Ferreira MG and Arraiano CM: The exoribonuclease
Dis3L2 defines a novel eukaryotic RNA degradation pathway. EMBO J.
32:1842–1854. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ustianenko D, Pasulka J, Feketova Z,
Bednarik L, Zigackova D, Fortova A, Zavolan M and Vanacova S:
TUT-DIS3L2 is a mammalian surveillance pathway for aberrant
structured non-coding RNAs. EMBO J. 35:2179–2191. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Koppers-Lalic D, Hackenberg M, Bijnsdorp
IV, van Eijndhoven MAJ, Sadek P, Sie D, Zini N, Middeldorp JM,
Ylstra B, de Menezes RX, et al: Nontemplated nucleotide additions
distinguish the small RNA composition in cells from exosomes. Cell
Rep. 8:1649–1658. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Liu X, Zheng Q, Vrettos N, Maragkakis M,
Alexiou P, Gregory BD and Mourelatos Z: A MicroRNA precursor
surveillance system in quality control of MicroRNA synthesis. Mol
Cell. 55:868–879. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ripin N, Boudet J, Duszczyk MM, Hinniger
A, Faller M, Krepl M, Gadi A, Schneider RJ, Šponer J, Meisner-Kober
NC and Allain FH: Molecular basis for AU-rich element recognition
and dimerization by the HuR C-terminal RRM. Proc Natl Acad Sci USA.
116:2935–2944. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Loh XY, Sun QY, Ding LW, Mayakonda A,
Venkatachalam N, Yeo MS, Silva TC, Xiao JF, Doan NB, Said JW, et
al: RNA-binding protein ZFP36L1 suppresses hypoxia and cell-cycle
signaling. Cancer Res. 80:219–233. 2020. View Article : Google Scholar
|
|
94
|
Rataj F, Planel S, Denis J, Roelants C,
Filhol O, Guyon L, Feige JJ and Cherradi N: Targeting AU-rich
element-mediated mRNA decay with a truncated active form of the
zinc-finger protein TIS11b/BRF1 impairs major hallmarks of mammary
tumorigenesis. Oncogene. 38:5174–5190. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Gu L, Wang H, Wang J, Guo Y, Tang Y, Mao
Y, Chen L, Lou H and Ji G: Reconstitution of HuR-Inhibited CUGBP1
expression protects cardiomyocytes from acute myocardial
infarction-induced injury. Antioxid Redox Signal. 27:1013–1026.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Rau F, Freyermuth F, Fugier C, Villemin
JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, et
al: Misregulation of miR-1 processing is associated with heart
defects in myotonic dystrophy. Nat Struct Mol Biol. 18:840–845.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Heo I, Ha M, Lim J, Yoon MJ, Park JE, Kwon
SC, Chang H and Kim VN: Mono-Uridylation of Pre-MicroRNA as a key
step in the biogenesis of group II let-7 MicroRNAs. Cell.
151:521–532. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Ansari MY, Khan NM, Ahmad N, Green J,
Novak K and Haqqi TM: Genetic Inactivation of ZCCHC6 suppresses
interleukin-6 expression and reduces the severity of experimental
osteoarthritis in mice. Arthritis Rheumatol. 71:583–593. 2019.
View Article : Google Scholar :
|
|
99
|
Nishikura K: A-to-I editing of coding and
non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 17:83–96. 2016.
View Article : Google Scholar :
|
|
100
|
Chung HC, Calis JJA, Wu X, Sun T, Yu Y,
Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR and
Rice CM: Human ADAR1 prevents endogenous RNA from triggering
translational shutdown. Cell. 172:811–824.e14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Yang CC, Chen YT, Chang YF, Liu H, Kuo YP,
Shih CT, Liao WC, Chen HW, Tsai WS and Tan BC: ADAR1-mediated 3'
UTR editing and expression control of antiapoptosis genes
fine-tunes cellular apoptosis response. Cell Death Dis.
8:e28332017. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Chalk AM, Taylor S, Heraud-Farlow JE and
Walkley CR: The majority of A-to-I RNA editing is not required for
mammalian homeostasis. Genome Biol. 20:2682019. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wu X, Wang L, Wang K, Li J, Chen R, Wu X,
Ni G, Liu C, Das S, Sluijter JPG, et al: ADAR2 increases in
exercised heart and protects against myocardial infarction and
doxorubicin-induced cardiotoxicity. Mol Ther. 30:400–414. 2022.
View Article : Google Scholar :
|
|
104
|
Chen J, Liu HF, Qiao LB, Wang FB, Wang L,
Lin Y and Liu J: Global RNA editing identification and
characterization during human pluripotent-to-cardiomyocyte
differentiation. Mol Ther Nucleic Acids. 26:879–891. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wang FJ, He J, Liu S, Gao A, Yang L, Sun
G, Ding W, Li CY, Gou F, He M, et al: A comprehensive RNA editome
reveals that edited Azin1 partners with DDX1 to enable
hematopoietic stem cell differentiation. Blood. 138:1939–1952.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Jiang L, Hao Y, Shao C, Wu Q, Prager BC,
Gimple RC, Sulli G, Kim LJ, Zhang G, Qiu Z, et al: ADAR1-mediated
RNA editing links ganglioside catabolism to glioblastoma stem cell
maintenance. J Clin Invest. 132:e1433972022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Solomon O, Di Segni A, Cesarkas K, Porath
HT, Marcu-Malina V, Mizrahi O, Stern-Ginossar N, Kol N,
Farage-Barhom S, Glick-Saar E, et al: RNA editing by ADAR1 leads to
context-dependent transcriptome-wide changes in RNA secondary
structure. Nat Commun. 8:14402017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Kokot KE, Kneuer JM, John D, Rebs S,
Möbius-Winkler MN, Erbe S, Müller M, Andritschke M, Gaul S, Sheikh
BN, et al: Reduction of A-to-I RNA editing in the failing human
heart regulates formation of circular RNAs. Basic Res Cardiol.
117:322022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Liddicoat BJ, Piskol R, Chalk AM,
Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH and Walkley
CR: RNA EDITING RNA editing by ADAR1 prevents MDA5 sensing of
endogenous dsRNA as nonself. Science. 349:1115–1120. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Garcia-Gonzalez C, Dieterich C, Maroli G,
Wiesnet M, Wietelmann A, Li X, Yuan X, Graumann J, Stellos K, Kubin
T, et al: ADAR1 prevents autoinflammatory processes in the heart
mediated by IRF7. Circ Res. 131:580–597. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
de Reuver R, Verdonck S, Dierick E,
Nemegeer J, Hessmann E, Ahmad S, Jans M, Blancke G, Van
Nieuwerburgh F, Botzki A, et al: ADAR1 prevents autoinflammation by
suppressing spontaneous ZBP1 activation. Nature. 607:784–789. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Chan TH, Lin CH, Qi L, Fei J, Li Y, Yong
KJ, Liu M, Song Y, Chow RK, Ng VH, et al: A disrupted RNA editing
balance mediated by ADARs (Adenosine DeAminases that act on RNA) in
human hepatocellular carcinoma. Gut. 63:832–843. 2014. View Article : Google Scholar
|
|
113
|
Chan TH, Qamra A, Tan KT, Guo J, Yang H,
Qi L, Lin JS, Ng VH, Song Y, Hong H, et al: ADAR-Mediated RNA
editing predicts progression and prognosis of gastric cancer.
Gastroenterology. 151:637–650.e10. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Shen H, An O, Ren X, Song Y, Tang SJ, Ke
XY, Han J, Tay DJT, Ng VHE, Molias FB, et al: ADARs act as potent
regulators of circular transcriptome in cancer. Nat Commun.
13:15082022. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Tan MH, Li Q, Shanmugam R, Piskol R,
Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et
al: Dynamic landscape and regulation of RNA editing in mammals.
Nature. 550:249–254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Karijolich J, Yi CQ and Yu YT:
Transcriptome-wide dynamics of RNA pseudouridylation. Nat Rev Mol
Cell Biol. 16:581–585. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Davis DR: Stabilization of RNA stacking by
pseudouridine. Nucleic Acids Res. 23:5020–5026. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zhao BS and He C: Pseudouridine in a new
era of RNA modifications. Cell Res. 25:153–154. 2015. View Article : Google Scholar :
|
|
119
|
Li X, Zhu P, Ma S, Song J, Bai J, Sun F
and Yi C: Chemical pulldown reveals dynamic pseudouridylation of
the mammalian transcriptome. Nat Chem Biol. 11:592–597. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Guegueniat J, Halabelian L, Zeng H, Dong
A, Li Y, Wu H, Arrowsmith CH and Kothe U: The human pseudouridine
synthase PUS7 recognizes RNA with an extended multi-domain binding
surface. Nucleic Acids Res. 49:11810–11822. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Ni J, Tien AL and Fournier MJ: Small
nucleolar RNAs direct site-specific synthesis of pseudouridine in
ribosomal RNA. Cell. 89:565–573. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Ruggero D, Grisendi S, Piazza F, Rego E,
Mari F, Rao PH, Cordon-Cardo C and Pandolfi PP: Dyskeratosis
congenita and cancer in mice deficient in ribosomal RNA
modification. Science. 299:259–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Li L and Ye K: Crystal structure of an
H/ACA box ribonucleoprotein particle. Nature. 443:302–307. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
McCleverty CJ, Hornsby M, Spraggon G and
Kreusch A: Crystal structure of human Pus10, a novel pseudouridine
synthase. J Mol Biol. 373:1243–1254. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Song J, Zhuang Y, Zhu C, Meng H, Lu B, Xie
B, Peng J, Li M and Yi C: Differential roles of human PUS10 in
miRNA processing and tRNA pseudouridylation. Nat Chem Biol.
16:160–169. 2020. View Article : Google Scholar
|
|
126
|
Bykhovskaya Y, Casas K, Mengesha E, Inbal
A and Fischel-Ghodsian N: Missense mutation in pseudouridine
synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic
anemia (MLASA). Am J Hum Genet. 74:1303–1308. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Jia Z, Meng F, Chen H, Zhu G, Li X, He Y,
Zhang L, He X, Zhan H, Chen M, et al: Human TRUB1 is a highly
conserved pseudouridine synthase responsible for the formation of
ψ55 in mitochondrial tRNAAsn, tRNAGln, tRNAGlu and tRNAPro. Nucleic
Acids Res. 50:9368–9381. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Shaheen R, Han L, Faqeih E, Ewida N,
Alobeid E, Phizicky EM and Alkuraya FS: A homozygous truncating
mutation in PUS3 expands the role of tRNA modification in normal
cognition. Hum Genet. 135:707–713. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Lin TY, Smigiel R, Kuzniewska B,
Chmielewska JJ, Kosińska J, Biela M, Biela A, Kościelniak A, Dobosz
D, Laczmanska I, et al: Destabilization of mutated human PUS3
protein causes intellectual disability. Hum Mutat. 43:2063–2078.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Cui Q, Yin K, Zhang X, Ye P, Chen X, Chao
J, Meng H, Wei J, Roeth D, Li L, et al: Targeting PUS7 suppresses
tRNA pseudouridylation and glioblastoma tumorigenesis. Nat Cancer.
2:932–949. 2021. View Article : Google Scholar
|
|
131
|
Eyler DE, Franco MK, Batool Z, Wu MZ,
Dubuke ML, Dobosz-Bartoszek M, Jones JD, Polikanov YS, Roy B and
Koutmou KS: Pseudouridinylation of mRNA coding sequences alters
translation. Proc Natl Acad Sci USA. 116:23068–23074. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Dai Q, Zhang LS, Sun HL, Pajdzik K, Yang
L, Ye C, Ju CW, Liu S, Wang Y, Zheng Z, et al: Quantitative
sequencing using BID-seq uncovers abundant pseudouridines in
mammalian mRNA at base resolution. Nat Biotechnol. 41:344–354.
2023. View Article : Google Scholar :
|
|
133
|
Karijolich J and Yu YT: Converting
nonsense codons into sense codons by targeted pseudouridylation.
Nature. 474:395–398. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Anderson BR, Muramatsu H, Nallagatla SR,
Bevilacqua PC, Sansing LH, Weissman D and Karikó K: Incorporation
of pseudouridine into mRNA enhances translation by diminishing PKR
activation. Nucleic Acids Res. 38:5884–5892. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Martinez NM, Su A, Burns MC, Nussbacher
JK, Schaening C, Sathe S, Yeo GW and Gilbert WV: Pseudouridine
synthases modify human pre-mRNA co-transcriptionally and affect
pre-mRNA processing. Mol Cell. 82:645–659.e9. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Rapino F, Zhou Z, Roncero Sanchez AM,
Joiret M, Seca C, El Hachem N, Valenti G, Latini S, Shostak K,
Geris L, et al: Wobble tRNA modification and hydrophilic amino acid
patterns dictate protein fate. Nat Commun. 12:21702021. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Rosu A, El Hachem N, Rapino F,
Rouault-Pierre K, Jorssen J, Somja J, Ramery E, Thiry M, Nguyen L,
Jacquemyn M, et al: Loss of tRNA-modifying enzyme Elp3 activates a
p53-dependent antitumor checkpoint in hematopoiesis. J Exp Med.
218:e202006622021. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Cheng H, Li L, Xue J, Ma J and Ge J: TNC
accelerates hypoxia-induced cardiac injury in a METTL3-dependent
manner. Genes (Basel). 14:5912023. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Zhuang Y, Li T, Hu X, Xie Y, Pei X, Wang
C, Li Y, Liu J, Tian Z, Zhang X, et al: MetBil as a novel molecular
regulator in ischemia-induced cardiac fibrosis via METTL3-mediated
m6A modification. FASEB J. 37:e227972023. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Cai X, Zou P, Hong L, Chen Y, Zhan Y, Liu
Y and Shao L: RNA methylation reading protein YTHDF2 relieves
myocardial ischemia-reperfusion injury by downregulating BNIP3 via
m6A modification. Hum Cell. 36:1948–1964. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Ding JF, Sun H, Song K, Zhou Y, Tu B, Shi
KH, Lu D, Xu SS and Tao H: IGFBP3 epigenetic promotion induced by
METTL3 boosts cardiac fibroblast activation and fibrosis. Eur J
Pharmacol. 942:1754942023. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Liu ZY, You QY, Liu ZY, Lin LC, Yang JJ
and Tao H: m6A control programmed cell death in cardiac fibrosis.
Life Sci. 353:1229222024. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wang L, Zhou J, Kong L, Ying G, Sha J, Yi
D, Zeng J, Xiong W and Wen T: Fibroblast-specific knockout of
METTL1 attenuates myocardial infarction-induced cardiac fibrosis.
Life Sci. 329:1219262023. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Kurian L and Brandes RP: RNA modification
that breaks the heart: RNA Acetylase Nat10 promotes fibrosis. Circ
Res. 133:1003–1005. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Wang XX, Zhao YM, Zhang QY, Zhao JX, Yin
DH, Zhang ZZ, Jin XY, Li SN, Ji HY, Chen HY, et al: Acetylcytidine
modification of Amotl1 by N-acetyltransferase 10 contributes to
cardiac fibrotic expansion in mice after myocardial infarction.
Acta Pharmacol Sin. 45:1425–1437. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Hao Y, Li B, Yin F and Liu W: tRNA-derived
small RNA (tsr007330) regulates myocardial fibrosis after
myocardial infarction through NAT10-mediated ac4C acetylation of
EGR3 mRNA. Biochim Biophys Acta Mol Basis Dis. 1870:1672672024.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Zhou B, Perel P, Mensah GA and Ezzati M:
Global epidemiology, health burden and effective interventions for
elevated blood pressure and hypertension. Nat Rev Cardiol.
18:785–802. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Touyz RM, Alves-Lopes R, Rios FJ, Camargo
LL, Anagnostopoulou A, Arner A and Montezano AC: Vascular smooth
muscle contraction in hypertension. Cardiovasc Res. 114:529–539.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Jain M, Mann TD, Stulić M, Rao SP, Kirsch
A, Pullirsch D, Strobl X, Rath C, Reissig L, Moreth K, et al: RNA
editing of Filamin A pre-mRNA regulates vascular contraction and
diastolic blood pressure. EMBO J. 37:e948132018. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Marcadenti A, Fuchs FD, Matte U, Sperb F,
Moreira LB and Fuchs SC: Effects of RS9939906 and MC4R RS17782313
on obesity, type 2 diabetes mellitus and blood pressure in patients
with hypertension. Cardiovasc Diabetol. 12:1032013. View Article : Google Scholar
|
|
151
|
Liu S, Jiang X, Lu H, Xing M, Qiao Y,
Zhang C and Zhang W: HuR (Human Antigen R) regulates the
contraction of vascular smooth muscle and maintains blood pressure.
Arterioscler Thromb Vasc Biol. 40:943–957. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Klöss S, Rodenbach D, Bordel R and Mülsch
A: Human-antigen R (HuR) expression in hypertension-:
Downregulation of the mRNA stabilizing protein HuR in genetic
hypertension. Hypertension. 45:1200–1206. 2005. View Article : Google Scholar
|
|
153
|
Botros M, Fadah K and Mukherjee D: The
role of inflammatory response in the development of
atherosclerosis, myocardial infarction, and remodeling. Vessel
Plus. 8:312024.
|
|
154
|
Chien CS, Li JY, Chien Y, Wang ML,
Yarmishyn AA, Tsai PH, Juan CC, Nguyen P, Cheng HM, Huo TI, et al:
METTL3-dependent N6-methyladenosine RNA modification mediates the
atherogenic inflammatory cascades in vascular endothelium. Proc
Natl Acad Sci USA. 118:e20250701182021. View Article : Google Scholar :
|
|
155
|
Jian D, Wang Y, Jian L, Tang H, Rao L,
Chen K, Jia Z, Zhang W, Liu Y, Chen X, et al: METTL14 aggravates
endothelial inflammation and atherosclerosis by increasing FOXO1
N6-methyladeosine modifications. Theranostics. 10:8939–8956. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Zheng Y, Li Y, Ran X, Wang D, Zheng X,
Zhang M, Yu B, Sun Y and Wu J: Mettl14 mediates the inflammatory
response of macrophages in atherosclerosis through the NF-κB/IL-6
signaling pathway. Cell Mol Life Sci. 79:3112022. View Article : Google Scholar
|
|
157
|
Sun ZW, Chen W, Wang Z, Wang S, Zan J,
Zheng L and Zhao W: Matr3 reshapes m6A modification complex to
alleviate macrophage inflammation during atherosclerosis. Clin
Immunol. 245:1091762022. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Chen J, Ning Y, Zhang H, Song N, Gu Y, Shi
Y, Cai J, Ding X and Zhang X: METTL14-dependent m6A regulates
vascular calcification induced by indoxyl sulfate. Life Sci.
239:1170342019. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Zhou T, Han D, Liu J, Shi J, Zhu P, Wang Y
and Dong N: Factors influencing osteogenic differentiation of human
aortic valve interstitial cells. J Thorac Cardiovasc Surg.
161:e163–e185. 2021. View Article : Google Scholar
|
|
160
|
Wang L, Wang J, Yu P, Feng J, Xu GE, Zhao
X, Wang T, Lehmann HI, Li G, Sluijter JPG and Xiao J: METTL14 is
required for exercise-induced cardiac hypertrophy and protects
against myocardial ischemia-reperfusion injury. Nat Commun.
13:67622022. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Wang C, Hou X, Guan Q, Zhou H, Zhou L, Liu
L, Liu J, Li F, Li W and Liu H: RNA modification in cardiovascular
disease: Implications for therapeutic interventions. Signal
Transduct Target Ther. 8:4122023. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Yu S, Sun Z, Ju T, Liu Y, Mei Z, Wang C,
Qu Z, Li N, Wu F, Liu K, et al: The m7G Methyltransferase Mettl1
drives cardiac hypertrophy by regulating SRSF9-mediated splicing of
NFATc4. Adv Sci (Weinh). 11:e23087692024. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Maron MS and Maron BJ: Hypertrophic
cardiomyopathy-Authors reply. Lancet. 381:1457–1458. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Schultheiss HP, Fairweather D, Caforio
ALP, Escher F, Hershberger RE, Lipshultz SE, Liu PP, Matsumori A,
Mazzanti A, McMurray J and Priori SG: Dilated cardiomyopathy. Nat
Rev Dis Primers. 5:322019. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Muchtar E, Blauwet LA and Gertz MA:
Restrictive cardiomyopathy: Genetics, pathogenesis, clinical
manifestations, diagnosis, and therapy. Circ Res. 121:819–837.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Ghezzi D, Baruffini E, Haack TB,
Invernizzi F, Melchionda L, Dallabona C, Strom TM, Parini R,
Burlina AB, Meitinger T, et al: Mutations of the mitochondrial-tRNA
Modifier MTO1 cause hypertrophic cardiomyopathy and lactic
acidosis. Am J Hum Genet. 90:1079–1087. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Perks KL, Rossetti G, Kuznetsova I, Hughes
LA, Ermer JA, Ferreira N, Busch JD, Rudler DL, Spahr H, Schöndorf
T, et al: PTCD1 Is Required for 16S rRNA maturation complex
stability and mitochondrial ribosome assembly. Cell Rep.
23:127–142. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Gao S, Sun H, Chen K, Gu X, Chen H, Jiang
L, Chen L, Zhang S, Liu Y, Shi D, et al: Depletion of m6A reader
protein YTHDC1 induces dilated cardiomyopathy by abnormal splicing
of Titin. J Cell Mol Med. 25:10879–10891. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Meng L, Lin H, Huang X, Weng J, Peng F and
Wu S: METTL14 suppresses pyroptosis and diabetic cardiomyopathy by
downregulating TINCR lncRNA. Cell Death Dis. 13:382022. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Peng T, Liu M, Hu L, Guo D, Wang D, Qi B,
Ren G, Hu C, Zhang F, Chun HJ, et al: LncRNA Airn alleviates
diabetic cardiac fibrosis by inhibiting activation of cardiac
fibroblasts via a m6A-IMP2-p53 axis. Biol Direct. 17:322022.
View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Zhuang S, Ma Y, Zeng Y, Lu C, Yang F,
Jiang N, Ge J, Ju H, Zhong C, Wang J, et al: METTL14 promotes
doxorubicin-induced cardiomyocyte ferroptosis by regulating the
KCNQ1OT1-miR-7-5p-TFRC axis. Cell Biol Toxicol. 39:1015–1035. 2023.
View Article : Google Scholar
|
|
172
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Berulava T, Buchholz E, Elerdashvili V,
Pena T, Islam MR, Lbik D, Mohamed BA, Renner A, von Lewinski D,
Sacherer M, et al: Changes in m6A RNA methylation contribute to
heart failure progression by modulating translation. Eur J Heart
Fail. 22:54–66. 2020. View Article : Google Scholar
|
|
174
|
Zhang B, Jiang H, Wu J, Cai Y, Dong Z,
Zhao Y, Hu Q, Hu K, Sun A and Ge J: m6A demethylase FTO attenuates
cardiac dysfunction by regulating glucose uptake and glycolysis in
mice with pressure overload-induced heart failure. Signal Transduct
Target Ther. 6:3772021. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Dorn LE, Lasman L, Chen J, Xu X, Hund TJ,
Medvedovic M, Hanna JH, van Berlo JH and Accornero F: The
N6-Methyladenosine mRNA Methylase METTL3 controls cardiac
homeostasis and hypertrophy. Circulation. 139:533–545. 2019.
View Article : Google Scholar :
|
|
176
|
Gao XQ, Zhang YH, Liu F, Ponnusamy M, Zhao
XM, Zhou LY, Zhai M, Liu CY, Li XM, Wang M, et al: The piRNA CHAPIR
regulates cardiac hypertrophy by controlling METTL3-dependent
N6-methyladenosine methylation of Parp10 mRNA. Nat Cell Biol.
22:1319–1331. 2020. View Article : Google Scholar : PubMed/NCBI
|