Introduction
Diabetic nephropathy (DN), one of the most
devastating microvascular complications of diabetes mellitus,
manifests initially as microalbuminuria and progressively evolves
into end-stage renal disease through characteristic glomerulopathic
changes (1). This trajectory
underscores the critical need to elucidate its molecular
pathogenesis and develop targeted interventions for early renal
preservation. Previous evidence implicates glomerular podocytes as
primary targets in diabetic renal injury, where their structural
and functional integrity governs glomerular filtration barrier
competence (2). Notably, the
high mitochondrial density in these specialized epithelial cells,
which rely predominantly on oxidative phosphorylation for energy
homeostasis, renders them particularly vulnerable to metabolic
disturbances. Consequently, podocyte mitochondrial dysfunction is
increasingly recognized as a pivotal driver of DN progression,
involving disrupted energy metabolism, exacerbated oxidative
stress, and impaired organelle quality control mechanisms (3).
Emerging evidence indicates that complement system
activation, particularly through the C3a/C3aR axis, is involved in
progressive DN (4,5). Substantial studies demonstrate that
C3aR blockade reduces podocyte mitochondrial dysfunction and
oxidative stress while alleviating renal damage in DN rodent models
(6-8). Mitophagy, a selective autophagic
process essential for cellular homeostasis, eliminates damaged
mitochondria via lysosomal degradation (9). In early diabetes, renal cells
exhibit enhanced clearance of glucose-damaged mitochondria.
However, disease progression coincides with increased mitochondrial
fission, heightened oxidative stress and defective mitophagy. This
failure to eliminate compromised mitochondria permits damaging
factor release (reactive oxygen species), thus driving DN
advancement (10). In both
high-glucose environments and DN models, a decrease in the
expression of key proteins involved in mitophagy, such as PINK1,
parkin and microtubule-associated protein 1A/1B-light chain 3B
(LC3B), has been observed in podocytes. Conversely, the expression
of P62, an indicator of impaired autophagy, significantly increases
(11-13). These findings suggest that
dysfunction in podocyte mitophagy may play a crucial role in the
pathogenesis of DN.
Despite evidence linking C3a/C3aR to DN, the
mechanisms underlying its role in podocyte mitophagy remain
unclear. While our prior clinical and animal data demonstrated
local complement C3 activation and reduced mitophagy in DN, the
direct regulatory involvement of C3a/C3aR in mitophagy
downregulation remains undefined. The current study aimed to
evaluate C3a/C3aR effects on glomerular podocytes utilizing both
human podocyte cultures and db/db murine models. Markedly increased
renal C3 and C3aR expression was observed in diabetic mice and high
glucose (HG)-exposed podocytes. Importantly, a novel regulatory
role for C3a/C3aR was identify in maintaining podocyte
mitochondrial homeostasis through PI3K/AKT/FoxO1 signaling-mediated
mitochondrial biogenesis and mitophagy. These findings suggest that
targeting C3aR to preserve mitochondrial function and cellular
bioenergetics upstream of cellular damage represents a promising
therapeutic strategy for DN.
Materials and methods
Gene expression profiling data and
preprocessing
Transcriptomic profiles of human kidney podocytes
were retrieved from the Gene Expression Omnibus (accession no.
GSE47183), comprising 122 samples across eight pathological
categories, including DN (n=14), focal segmental glomerulosclerosis
(FSGS, n=23), FSGS with minimal change disease (MCD, n=6),
membranous glomerulonephritis (n=21), MCD (n=15), rapidly
progressive glomerulonephritis (RPGN, n=23), thin membrane disease
(TMD, n=3), and tumor nephrectomy controls (n=17) (14,15), The dataset was normalized using
Robust Multichip Average standardization with the affy package
(v1.78.0; Bioconductor Release 3.18, https://bioconductor.org). After normalization, the
platform file was used to map each probe to Entrenz Gene ID. If a
probe maps to multiple genes or does not map to any genes, the
expression value of the probe is deleted. If multiple probes map to
the same gene, the average value of these probes is taken as the
expression value of the gene. The normalized data was visualized
using pheatmap (Bioconductor pheatmap package).
Animals and reagents
Male C57BLKS/JGpt wild-type (wt/wt, 8-week-old,
20-25 g, n=10) and db/db mice (8-week-old, 45-55 g, n=20) were
procured from GemPharmatech Co., Ltd. All procedures were conducted
in accordance with the NIH Guide for the Care and Use of Laboratory
Animals (16) and approved by
the Institutional Animal Care and Use Committee of Fujian Medical
University (approval no. IACUC FJMU 2023-Y-1033; Fuzhou, China).
Animals were maintained under standardized conditions (12/12-h
light/dark cycle, 22±2°C, 60% humidity) with ad libitum access to
food and water. Following a 7-day acclimatization period, wt/wt
mice served as non-diabetic controls (NC, n=6). Diabetic db/db mice
were randomly allocated into two experimental groups (n=6/group):
i) A C3aR antagonist (C3aRA)-treated group, which received
intraperitoneal injections of SB290157 (10 mg/kg; cat. no.
HY-101502A/CS-6852; MedChemExpress) every 48 h (17); and ii) a vehicle group, which
received equivalent volumes of 5% DMSO in sterile saline. SB290157
was freshly reconstituted in vehicle solution (5% DMSO in 0.9%
NaCl) under aseptic conditions. All solutions were filtered through
0.22-μm membranes prior to administration. After 8 weeks of
intervention, 24-h urine was collected from all mice using a
metabolic cage (Fig. S1).
Euthanasia was performed by intraperitoneal injection of 2% sodium
pentobarbital at a dose of 100 mg/kg. Next, bilateral kidneys were
excised, the renal capsule was removed, and the kidneys were
sectioned by sagittal. Kidney tissue was either fixed with 4%
paraformaldehyde at 4°C for 24 h or rapidly frozen in liquid
nitrogen.
Measurement of serum creatinine, urinary
albumin/creatinine ratio (UACR), 24-h urinary total protein (UTP)
and cell-culture medium C3a level
Metabolic cages were used to collect 24 h urine
samples. The contents of serum creatinine, 24-h UTP and UACR were
measured using an automatic biochemical analyzer (Beckman Coulter,
Inc.). Cell-culture medium C3a levels were measured by using ELISA
kits (Human Complement C3a; cat. no. USEA387Hu; Wuhan Cloud-Clone
Corp.), according to the manufacturer's protocol.
Histological analysis
Kidney sections were deparaffinized and rehydrated
and were then stained with hematoxylin and eosin (H&E), and
Periodic Acid-Schiff (PAS).
Cell culture
An immortalized human podocyte cell line was donated
by Professor Moin A. Saleem (Bristol University, UK) and was
retained and donated by the Nephrology Laboratory of Wuhan
University. Cells were cultured in low-glucose Roswell Park
Memorial Institute (RPMI)-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) with 10% fetal bovine serum (FBS; Gibco; Thermo
Fisher Scientific, Inc.). The human immortalized podocytes used in
the present study were of passages 16-23. The podocytes were
propagated at 33°C and treated with interferon
[insulin-Transferrin-Selenium (ITS)-G; 10 U/ml; Invitrogen; Thermo
Fisher Scientific, Inc.]. Next, cells were differentiated without
ITS-G at 37°C for 7 days. For further evaluation, the podocytes
were stimulated with HG (30 mM glucose) and mannitol (24.5 mM
mannitol + 5.5 mM glucose) containing 1% FBS with or without
incubation with C3a or C3aRA (SB290157) for 24 h.
Application of small interfering RNA
(siRNA)
A duplex siRNA was designed to target human C3aR
(NCBI: NM 004054.4; https://www.ncbi.nlm.nih.gov/nuccore/?term=NM+004054.4).
The target sequence is GCUUCAAACAACCUCUAAU (1746-1764 bp) and the
siRNA nucleotide sequences for C3aR were r(GCCUCAAACAACCUCUAAU)
dTdT (Sense) and r(AUUAGAGGUUGUUUGAGGC) dGdG (Antisense). A duplex
siRNA was designed to target human PINK1 (NCBI: NM 032409.3;
https://www.ncbi.nlm.nih.gov/nuccore/?term=NM+032409.3).
The target sequence is GCTGGAGGAGTATCTGATA (1,449-1,467 bp) and the
siRNA nucleotide sequences for PINK1 were r(GCUGGAGGAGUAUCUGAUA)
dTdT (Sense) and r(UAUCAGAUACUCCUCCAGC) dGdG (Antisense).
Meanwhile, Negative control (NC) siRNA with no homology to
mammalian genomes served as negative control, which nucleotide
sequences were r (UUCUCCGAACGUGUCACGU) dTdT (Sense) and r
(ACGUGACACGUUCGGAGAA) dTdT (Antisense). All siRNAs were synthesized
by Shanghai Hanheng Biotechnology Co., Ltd. For transfection, cells
were seeded at 60% confluency in antibiotic-free medium and
transfected with 50 nM siRNA using RNAFit™ Transfection Reagent
(cat. no. HB-RF-1000; Shanghai Hanheng Biotechnology Co., Ltd.).
Cells were harvested 48 h post-transfection for downstream
analysis, with protein expression verification performed by western
blotting (Fig. S2).
TdT-mediated dUTP nick-end labeling
(TUNEL) assay
Paraffin-embedded kidney sections and cell slides
were processed with a TUNEL assay kit (Dalian Meilun Biology
Technology Co., Ltd.) to detect apoptosis. Paraffin-embedded renal
sections were deparaffinized using xylene and rehydrated with
ethanol. Cells were fixed with 4% paraformaldehyde at 37°C for 10
min. The deparaffinized sections and fixed cell slides were
incubated first with 20 μg/ml DNase-free proteinase K for 30
min at room temperature and then with TdT reaction mix for 60 min
at 37°C in the dark. Next, nuclei were counterstained with DAPI
(0.001 mg/ml). Images were acquired with a fluorescence microscope.
The apoptotic cells were identified and quantified by counting the
number of positive cells in three fields per group.
Immunohistochemistry
Xylene and ethanol were used to deparaffinize and
dehydrate renal sections, respectively, and citrate buffer was used
for antigen retrieval. The sections were incubated with antibodies
against C3 (1:5,000; Santa Cruz Biotechnology, Inc.), C3aR
(1:5,000; Santa Cruz Biotechnology, Inc.), Mnf2 (1:1,000;
Proteintech Group, Inc.) or PINK1 (1:1,000; Proteintech Group,
Inc.) at room temperature for 1 h. Next, the sections were stained
with an enhanced polymer detection system (Beijing Zhongshan
Jinqiao Biotechnology Co., Ltd.) according to the manufacturer's
instructions. Diaminobenzidine was used as an HRP-specific
substrate.
Immunofluorescence
Renal tissue sections and cell slides were washed
with PBS, permeabilized with PBS containing 0.3% Triton X-100 and
blocked with 5% BSA (Gibco; Thermo Fisher Scientific, Inc.) at 37°C
for 30 min. Next, samples were incubated with a primary antibody
overnight at 4°C. The following antibodies were employed: Mouse
anti-TOMM20 (1:100; cat. no. 11802-1-AP; Proteintech Group, Inc.),
rabbit anti-synaptopodin (1:200; cat. no. ab224491; Abcam) and
guinea pig anti-nephrin (1:200; cat. no. GP-N2; ProGen). After PBS
washing, sections were incubated for 1 h at room temperature with
goat anti-guinea pig, goat anti-mouse, or goat anti-rabbit
secondary antibodies conjugated with Alexa Fluor™ 488 or 594,
followed by treatment with DAPI (0.001 mg/ml) for 10 min at room
temperature. Quantification of the TOMM20/nephrin toward staining
was performed in 10-15 glomeruli per section using ImageJ 1.40g
(National Institutes of Health), and data were expressed as a
percentage of the TOMM20/nephrin co-staining area (yellow) on the
total glomerular TOMM20 area (red). Samples were examined under a
confocal inverted laser microscope (LSM 510 Meta; Carl Zeiss
AG).
Preparation of C3a solution
Human complement C3a (cat. no. 204881; 50 μg;
Merck KGaA) was diluted to a concentration of 5-10 M using sterile
PBS at a pH of 7.2. The resulting solution was aliquoted into 50
μl portions and stored at −80°C. This solution was utilized
for extrinsic interventions on podocytes at different time points
and concentrations.
Cytoskeleton visualization
Cells were fixed with 4% paraformaldehyde at 37°C
for 15 min, permeabilized with PBS containing 0.3% Triton X-100,
blocked with 5% BSA for 30 min and stained with 500 nM Alexa Fluor™
488 phalloidin (Invitrogen; Thermo Fisher Scientific, Inc.) for 30
min and DAPI (0.001 mg/ml) for 15 min. The cytoskeleton was
visualized in the composite image of the F-actin (phalloidin,
green). The average intensity of F-actin was measured using ImageJ
1.40g software.
Double labeling GFP/LC3B-DsRed/Mito
adenovirus mitophagy assay
A mixture of 20 μl DsRed-Mito (red)
adenovirus stock solution (1x1010 PFU/ml) + 20 μl
GFP-LC3B (green) adenovirus stock solution (1x1010
PFU/ml) + 160 μl RPMI-1640 medium was prepared to make a
final volume of 200 μl adenovirus mix (1x109
PFU/ml). Podocytes were washed with PBS three times for 3 min each,
and 250 μl RPMI-1640 medium was added to each well. Next, 15
μl of the adenovirus mix (optimal MOI=100, Fig. S3) was added to each well, and
after 4 h of infection, an additional 250 μl RPMI-1640
medium was added. Next, the virus-containing medium was aspirated,
and complete medium was added for 12-h incubation. Subsequently,
cells were fixed, their nuclei were stained, and images were
captured. By utilizing the dual fluorescence adenovirus system
(GFP/LC3B-DsRed/Mito), the dynamic process of mitophagy could be
precisely tracked in real-time tracked. Quantification of
GFP-LC3B (green)-associated Mito-DsRed (red) staining was
performed using ImageJ 1.40g software. Analysis included
determining the total number of LC3-positive points and the
percentage of Mito-LC3B overlap among LC3-positive points. Each
experiment involved analyzing at least ≥5 cells.
Western blotting
Proteins were extracted from kidney tissues and
podocytes using RIPA lysis buffer (cat. no. P0013B; Beyotime
Institute of Biotechnology) containing a protease inhibitor
cocktail (cat. no. P1005; Beyotime Institute of Biotechnology) and
a phosphatase inhibitor cocktail (cat. no. P1082; Beyotime
Institute of Biotechnology). Tissue homogenates were centrifuged at
12,000 × g for 15 min at 4°C, the protein concentrations of
supernatants were determined by using a BCA protein assay kit (cat.
no. P0009; Beyotime Institute of Biotechnology) and stored at
−80°C. Equal amounts of proteins (40 μg) were loaded per
lane on a 10% SDS-PAGE gel and transferred to polyvinylidene
difluoride (PVDF) membranes (MilliporeSigma), which were blocked
with 5% non-fat milk for 2 h at room temperature. Next, the
membranes were incubated with primary antibody against β actin
(1:2,000; cat. no. A1978; MilliporeSigma), C3 (1:500; cat. no.
sc-28294; Santa Cruz Biotechnology, Inc.), C3aR (1:500; cat. no.
sc-53738; Santa Cruz Biotechnology, Inc.), parkin (1:1,000; cat.
no. 14060-1-AP; Proteintech Group, Inc.), PINK1 (1:500; cat. no.
23274-1-AP; Proteintech Group, Inc.), TOMM20 (1:500; cat. no.
11802-1-AP; Proteintech Group, Inc.), LC3B (1:1,000; cat. no.
ab192890; Abcam), ZO 1 (1:1,000; cat. no. ab96587; Abcam),
synaptopodin (1:1,000; cat. no. ab224491; Abcam), podocin (1:1,000;
cat. no. ab50339; Abcam), PI3K [p85α (54 + 85 kDa); 1:1,000; cat.
no. MA5-14942; Invitrogen; Thermo Fisher Scientific, Inc.),
phosphorylated (p)-AKT (1:1,000; cat. no. MA5-14952;
Invitrogen; Thermo Fisher Scientific, Inc.) and FoxO1 (1:1,000;
cat. no. MA5-17151; Invitrogen; Thermo Fisher Scientific, Inc.) at
4°C overnight. Blots were incubated with Goat anti-Rabbit IgG (H+L)
HRP conjugate (cat. no. ab6721; Abcam) or Goat anti-mouse IgG (H+L)
HRP conjugate (cat. no. ab6789; Abcam) at 1:10,000 dilution in 5%
non-fat milk/TBST for 1 h at 25°C. Images were analyzed in ImageJ
(v1.54f; National Institutes of Health) using the Gel Analyzer
tool. Rolling ball radius (50 pixels) was applied for background
subtraction.
Transmission electron microscopy
(TEM)
Mouse kidneys were fixed in 2.5% glutaraldehyde and
0.05 M sodium phosphate buffer (Ph 7.2) overnight at 4°C. Samples
were embedded in epoxy resin after being washed with 0.1 M
cacodylate buffer and saline, followed by a gradual series of
ethanol dehydration. Sections (60-90 nm thickness) were then
incubated on copper grids, contrasted with aqueous uranyl acetate
and lead citrate, and analyzed. Images were recorded with a TEM
(H7500; Hitachi, Ltd.).
Statistical analysis
All data are expressed as the mean ± standard error
of the mean. Comparisons between two groups were performed using a
paired Student's t test. For multiple groups, the data were
analyzed by one- or two way ANOVA, followed by Bonferroni post hoc
tests, where appropriate. P<0.05 was considered to indicate a
statistically significant difference. All statistical analyses were
performed using GraphPad Prism 8.3.0 software (GraphPad;
Dotmatics).
Results
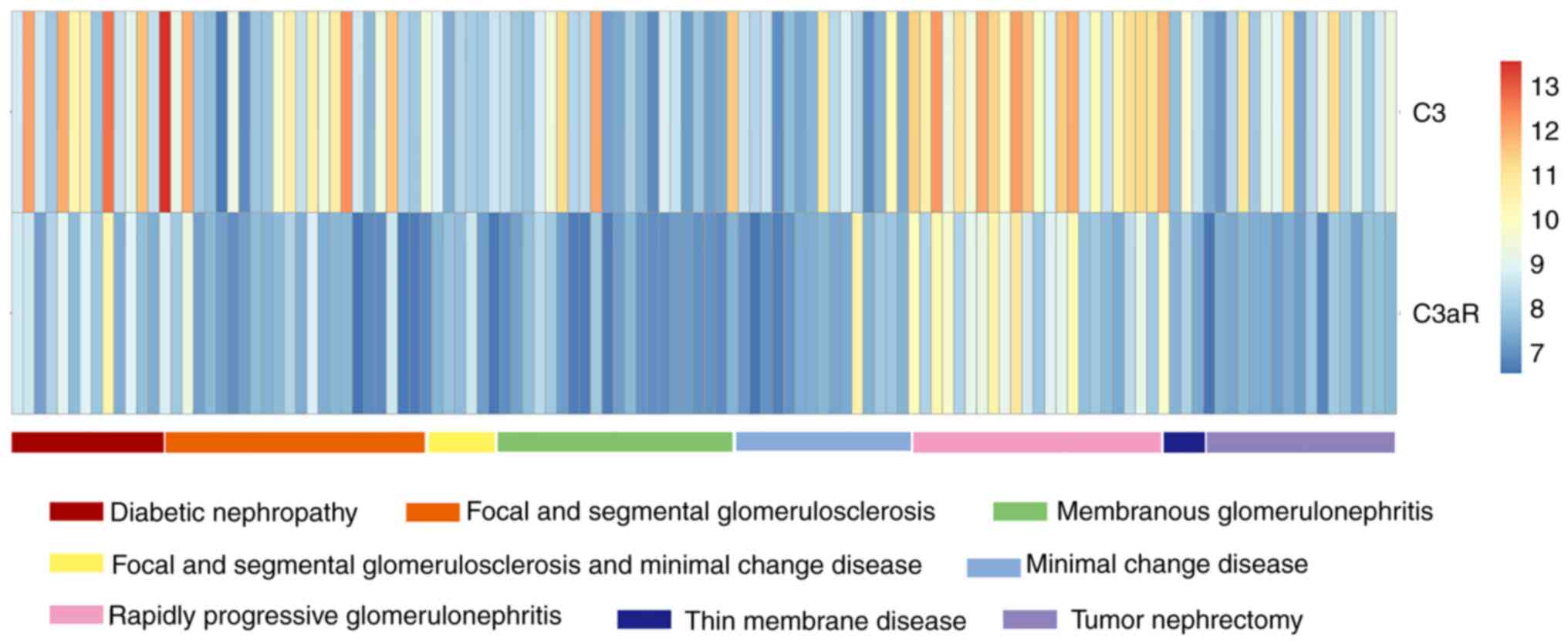
Expression of C3 and C3aR in different
kidney-related diseases
The results of the present study were analyzed by
comparing the expression levels of the same gene in different
kidney-related diseases. As shown in Fig. 1, the expression levels of C3 and
C3aR were relatively high in RPGN and DN, compared with the other
kidney-related diseases, indicating that the activation of
complement C3 may be closely related to the occurrence and
development of DN.
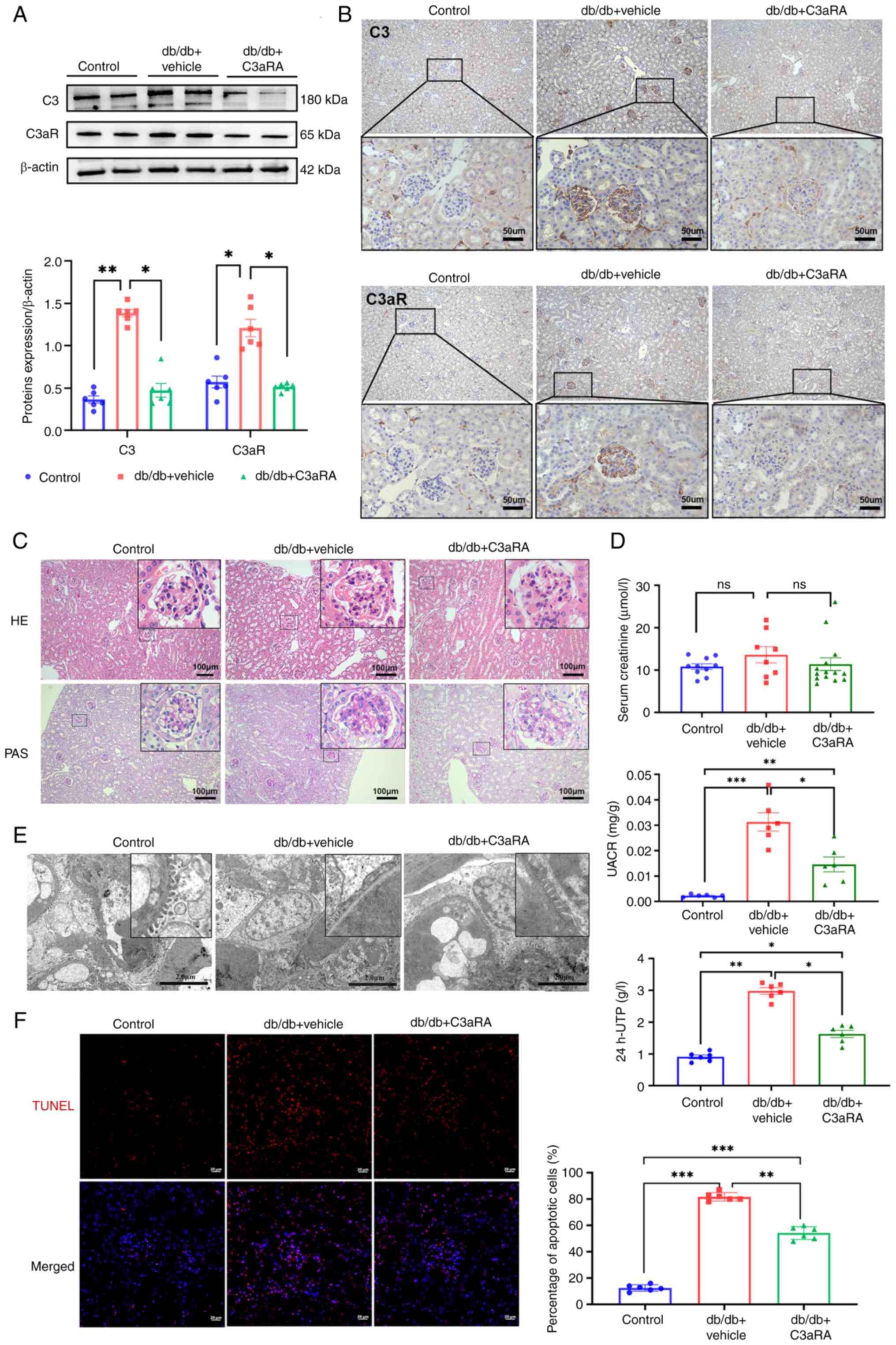
Inhibiting C3aR shows promise in
ameliorating renal damage in DN mice
Further validation was conducted in vivo in a
DN model (db/db mice). Although serum C3a levels showed no
significant difference (Fig.
S4), renal C3 and C3aR protein expression was significantly
increased in DN mice versus non-diabetic controls (Fig. 2A). Glomerular-localized
C3/C3aR proteins levels (Fig.
2B) were substantially reduced following SB290157 treatment.
Histological analysis revealed that db/db mice exhibited glomerular
hypertrophy, mesangial hyperplasia, increased cellularity and
matrix expansion (H&E and PAS staining; Fig. 2C). C3aRA intervention attenuated
mesangial matrix deposition without exacerbating cellular
proliferation. Biochemical assessments demonstrated significantly
elevated UACR, 24 h-UTP, blood glucose levels, total cholesterol,
or triglyceride levels and kidney/body weight ratios (Table SI) in db/db mice versus
controls, despite non-significant differences in serum creatinine.
Electron microscopy confirmed diffuse podocyte foot process
effacement with increased renal apoptosis. C3aRA treatment
significantly reduced body weight (Fig. S5), UACR and 24 h-UTP, restored
organized foot process architecture, and diminished apoptosis
(Fig. 2D-F). Collectively, these
data indicated that C3aR antagonism ameliorates proteinuria
and histopathological damage in experimental DN.
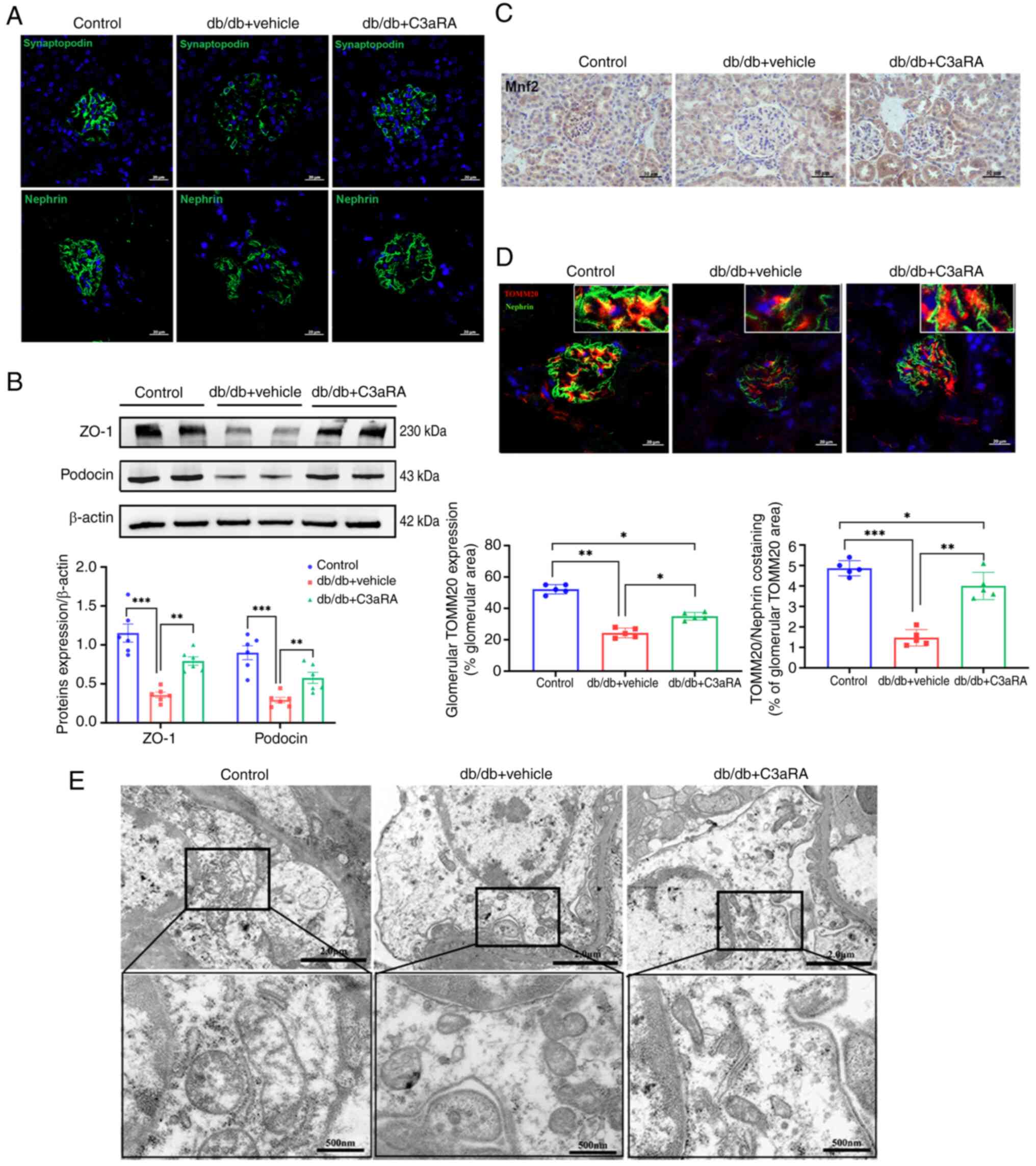
C3aR inhibition ameliorates podocyte
dysfunction and mitochondrial injury in diabetic mice
Accumulating evidence has identified podocyte injury
as a critical pathogenic mechanism in DN progression (11,18). Immunofluorescence analysis
demonstrated significantly diminished fluorescence intensity and
disrupted cytoskeletal architecture in db/db podocytes. C3aRA
intervention upregulated synaptopodin and nephrin expression in DN
models (Fig. 3A). Protein
quantification revealed marked downregulation of the glomerular
filtration barrier components ZO-1 and podocin in db/db renal
tissues versus controls, and these effects were reversed by C3aR
blockade (Fig. 3B).
The mitochondrial integrity regulators Mfn2 (fusion
protein) and TOMM20 (a translocase of the outer mitochondrial
membrane) exhibited significantly reduced glomerular deposition in
db/db mice (Fig. 3C and D).
Ultrastructural analysis confirmed mitochondrial swelling, matrix
disorganization and cristae fragmentation (Fig. 3E). C3aRA treatment normalized
Mfn2 and TOMM20 expression (Fig. 3C
and D) and restored mitochondrial morphology (Fig. 3E), indicating significant
attenuation of podocyte mitochondrial damage.
Comparative analysis of mitophagy and
signaling pathway alterations in DN
Western blot analysis of renal tissue homogenates
revealed significantly reduced expression of the mitophagy markers
LC3B-II/LC3B-I, PINK1 and parkin in DN model mice versus controls
(Fig. 4B). Immunohistochemistry
demonstrated diminished PINK1 deposition in both glomerular and
tubular compartments of db/db mice, with pronounced reduction in
glomeruli (Fig. 4A). Notably,
intraperitoneal administration of SB290157 partially reversed these
alterations, significantly elevating LC3B-II/LC3B-I, PINK1 and
parkin protein levels (Fig. 4B)
while enhancing glomerular PINK1 deposition (Fig. 4A). These findings indicated that
C3aRA mitigates impaired autophagic flux and enhances
PINK1-mediated mitophagy in diabetic kidneys.
It was observed in previous experiments by the
authors that p-AKT was activated in renal tissues of db/db mice,
while t-AKT expression levels remained stable (Fig. S6A). Further immunoblotting
demonstrated PI3K/AKT pathway activation in diabetic renal tissue,
characterized by increased PI3K expression and p-AKT, concomitant
with FoxO1 suppression (Fig.
4C). C3aR antagonism significantly attenuated PI3K/p-AKT
upregulation while restoring FoxO1 expression (Fig. 4C), indicating that modulation of
this signaling axis contributes to the therapeutic effect.
HG and C3a induce podocyte injury and
suppress mitophagy in vitro
The C3/C3aR axis is implicated in impaired podocyte
mitophagy in DN. To investigate this mechanistically, hyperglycemic
injury was modeled in vitro by using conditionally
immortalized podocytes. Podocytes were cultured for 24 h under
normal glucose (NG, 5.5 mM glucose), HG (30 mM glucose) or mannitol
high osmotic control (MG, 24.5 mM mannitol + 5.5 mM glucose)
conditions. HG-exposed podocytes exhibited F-actin disassembly with
reduced fluorescence intensity versus NG/MG controls (Fig. 5A), which was accompanied by
significant downregulation of synaptopodin and podocin (Fig. 5B). C3aR expression significantly
increased under HG conditions (Fig.
5D). ELISA revealed time-dependent C3a accumulation in
supernatants, with HG cultures showing accelerated generation (≤96
h) despite comparable cellular C3 expression across groups
(Fig. 5C). Mitophagy markers
(LC3B-II/LC3B-I, PINK1 and parkin) were significantly suppressed in
HG-treated podocytes (Fig. 5E),
confirming complement axis activation and mitophagy impairment.
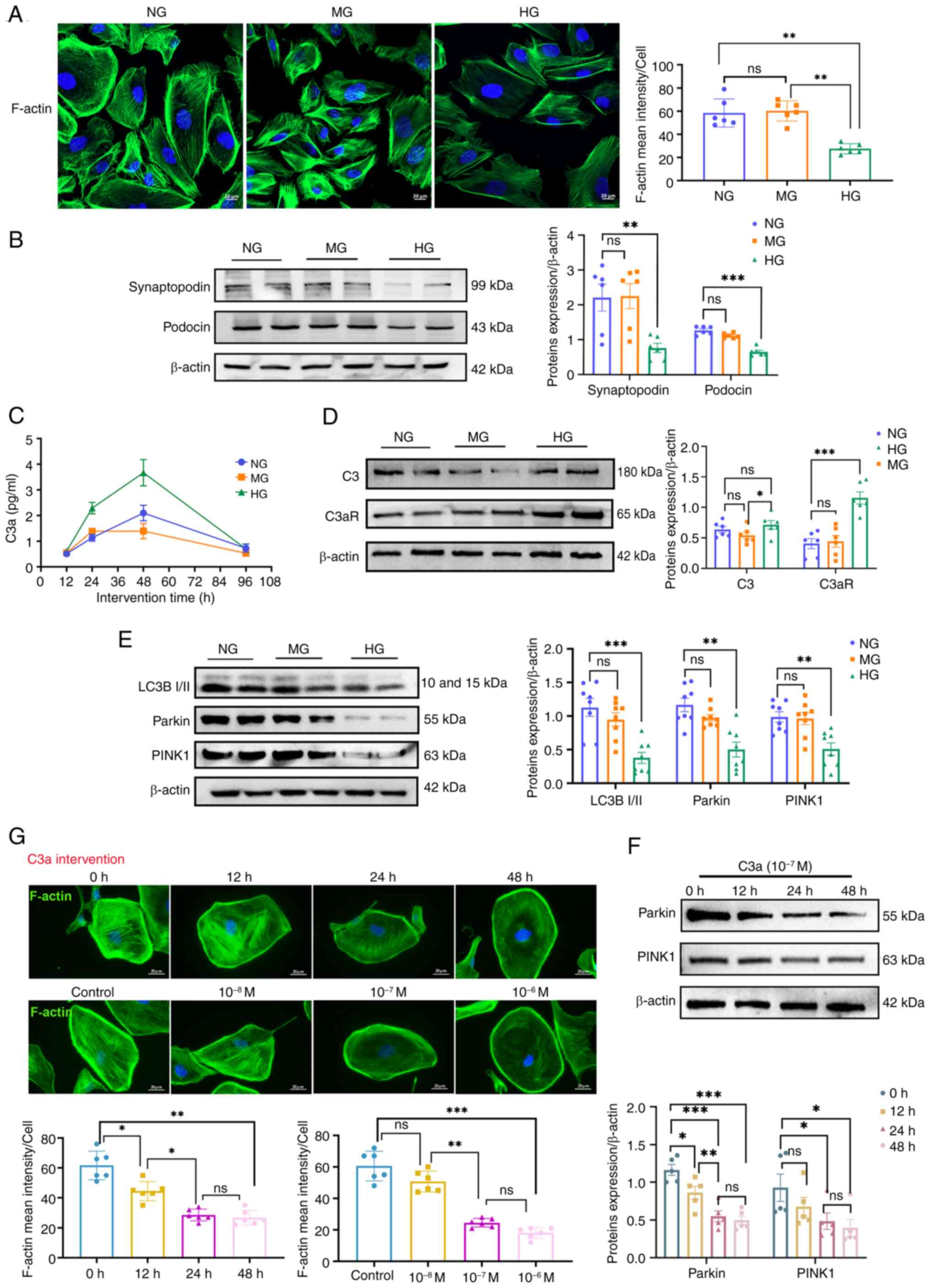 | Figure 5Effects of HG and C3a on podocyte
damage and mitophagy. Normal glucose refers to 5.5 mM glucose,
while the mannitol high osmotic control group was subjected to 24.5
mM mannitol + 5.5 mM glucose, and HG represents the intervention
group (30 mM glucose). (A) Representative images (left) and
quantification (right) of podocyte cytoskeleton, with F-actin
(green) stained using phalloidin (n=6). Scale bar, 20 μm.
(B) Protein levels of synaptopodin and podocin in podocytes (n=6).
Corresponding histograms are shown on the bottom panel of
representative protein bands. (C) ELISA detection of C3a levels in
podocytes at different time points (n=6). (D) Protein levels of C3
and C3aR in podocytes (n=6). Corresponding histograms are shown on
the right panel of representative protein bands. (E) Protein levels
of LC3B I/II, parkin and PINK1 in podocytes (n=8). Corresponding
histograms are shown on the bottom panel of representative protein
bands. (F) Protein levels of parkin and PINK1 in podocytes (n=5).
Corresponding histograms are shown on the right panel of
representative protein bands. (G) Representative images and
quantification (bottom) of podocyte cytoskeleton induced by
different times and concentrations of C3a (10−7 M for
12, 24 and 48 h; or 10−8, 10−7 and
10−6 M for 24 h) (n=6). Scale bar, 20 μm.
*P<0.05, **P<0.01 and
***P<0.001. ns, no statistically significant
difference; HG, high glucose. |
Complementing these findings, direct C3a exposure
(10−7 M) induced dose- and time-dependent cytoskeletal
disorganization, which was most pronounced at 24 h (Fig. 5G). Concomitant reductions in
PINK1 and parkin expression demonstrated C3a's causal role in
mitophagy suppression (Fig. 5F),
establishing its direct contribution to podocyte injury.
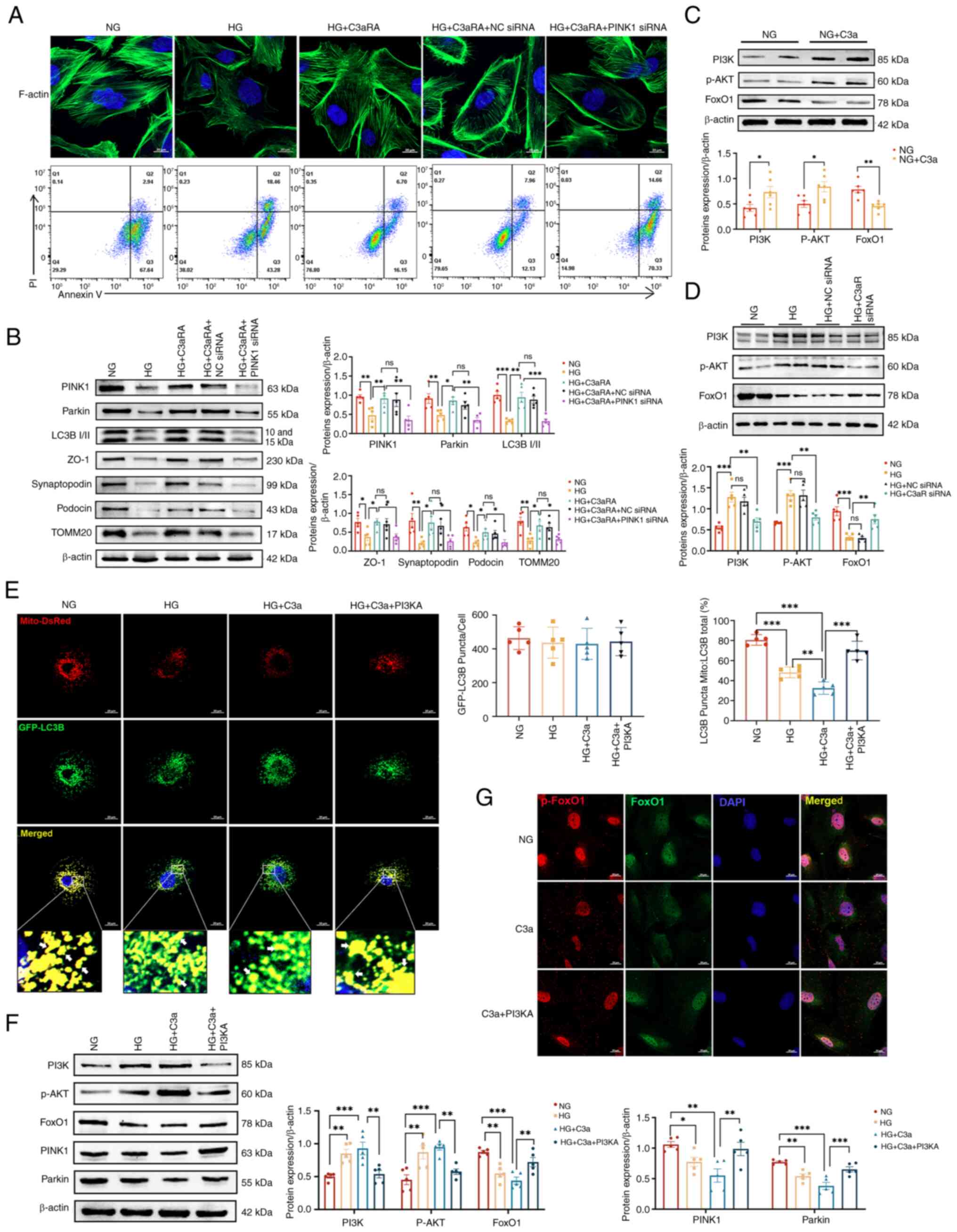
C3aR knockdown ameliorates HG-induced
podocyte injury and mitophagy suppression
To mechanistically validate the involvement of the
C3a/C3aR axis in podocyte injury and mitophagy impairment under
hyperglycemic conditions, siRNA-mediated C3aR downregulation was
employed in cultured podocytes. Comparative analysis revealed that
both HG-exposed and HG + NC siRNA groups exhibited fragmented
F-actin microfilaments with cytoskeletal disorganization, elevated
apoptotic indices, mitochondrial swelling with cristae
fragmentation, and minimal autolysosome formation, which
collectively indicate profound mitochondrial damage and suppressed
autophagic flux (Fig. 6A-C). By
contrast, C3aR-knockdown podocytes under HG conditions demonstrated
restored F-actin bundling with increased filament density, reduced
apoptosis, preserved mitochondrial ultrastructure (characterized by
mild swelling and intact cristae), decreased proportions of damaged
mitochondria and a significant increase in autophagosome-engulfed
mitochondria.
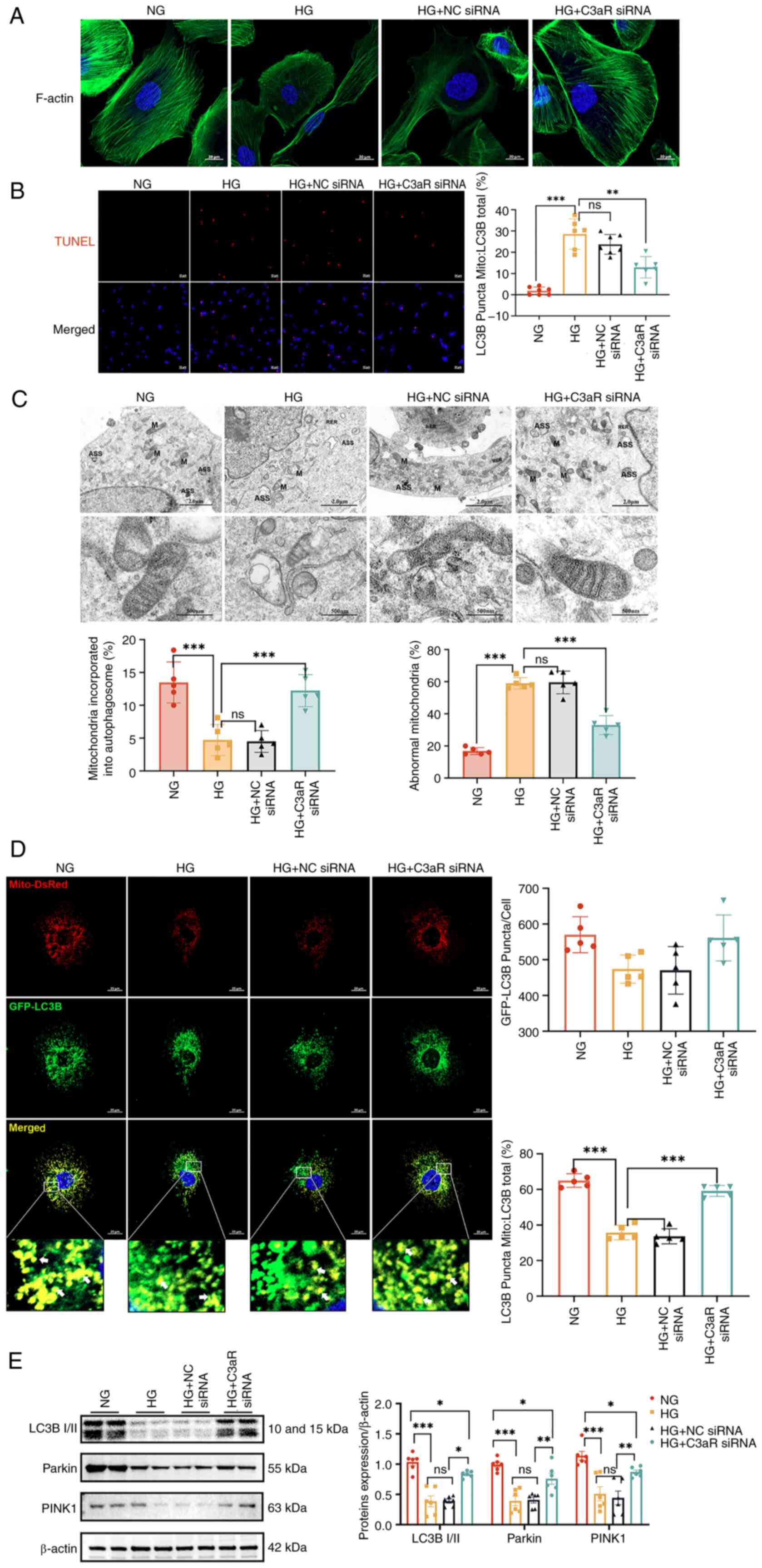 | Figure 6Inhibition of C3aR improves
HG-induced podocyte and mitochondrial autophagic damage. (A)
Changes in cell cytoskeleton after blocking C3aR in a HG
environment. Scale bar, 20 μm. (B) Immunofluorescence images
of podocytes in each group using a TdT-mediated dUTP nick-end
labeling assay. Scale bar, 50 μm. The lower panel presents
the statistical analysis of apoptosis ratios in podocytes for each
group (n=6). (C) Transmission electron microscopy examination of
mitochondrial morphology. Scale bar, 2.0 μm and 500 nm. The
statistical charts on the right side of the electron microscopy
images represent the percentages of damaged mitochondria and
mitochondria enveloped by autophagosomes (n=5). (D) Transfection of
immortalized human podocytes with adenovirus GFP-LC3B (green) and
DsRed-Mito (red). Scale bar, 20 μm. The statistical charts
on the right side represent the number of GFP-LC3B-positive puncta
per cell and the proportion of LC3B spots on mitochondria (Mito) to
total LC3B. Quantification of GFP-LC3B (green)-associated
Mito-DsRed (red) staining intensity normalized by GFP-LC3B area
(n=5). (E) Protein levels of LC3B I/II, parkin and PINK1 in
podocytes (n=6). Corresponding histograms are shown on the right
panel of representative protein bands. *P<0.05,
**P<0.01 and ***P<0.001. ns, no
statistically significant difference; HG, high glucose; NG, normal
glucose; siRNA, small interfering RNA; NC, negative control. |
For dynamic quantification of mitophagic flux,
adenoviral transduction of GFP-LC3B/DsRed-Mito reporters revealed
significantly fewer mitochondrial LC3+ puncta in the HG
and HG + NC siRNA groups versus normoglycemic controls (P<0.01),
indicative of impaired mitophagosome biogenesis. This deficit was
reversed by C3aR knockdown, which increased mitochondrial
LC3+ puncta by 2.1-fold (Fig. 6D). Immunoblot analysis further
confirmed that C3aR silencing significantly upregulated the
LC3B-II/LC3B-I ratio (1.8-fold, P<0.001), as well as PINK1
(1.5-fold, P<0.01), and parkin (1.7-fold, P<0.001) expression
levels in HG-treated podocytes (Fig.
6E), establishing that C3aR blockade restores mitophagic
activity in DN.
C3a/C3aR axis suppresses mitophagy via
PI3K/AKT/FoxO1 signaling in hyperglycemia-induced podocyte
injury
To delineate the role of C3a/C3aR in diabetic
podocyte dysfunction, PINK1 knockdown was combined with C3aRA.
HG-exposed podocytes exhibited cytoskeletal disorganization and
elevated apoptosis, and these phenotypes partially ameliorated by
C3aRA. Crucially, PINK1-silenced podocytes (HG + C3aRA + PINK1
siRNA) displayed exacerbated F-actin fragmentation with reduced
fluorescence intensity and increased apoptosis compared with HG +
C3aRA and HG + C3aRA + NC siRNA controls (Fig. 7A). Immunoblot analysis confirmed
that PINK1 knockdown significantly reduced mitophagy markers
(PINK1, parkin and LC3B-II/LC3B-I ratio) and diminished the levels
of key podocyte integrity proteins (ZO-1, synaptopodin and podocin)
and of the mitochondrial translocase TOMM20 (P<0.05).
Importantly, PINK1 silencing attenuated C3aRA-mediated protection
against HG-induced injury, indicating that C3a/C3aR induces
podocyte damage through PINK1-mediated mitophagy impairment
(Fig. 7B). Following
pretreatment of podocytes with HG and HG + C3a, activation of p-AKT
was observed (Fig. S6B).
Mechanistically, C3a stimulation significantly enhanced PI3K/AKT
phosphorylation while suppressing FoxO1 expression versus
normoglycemic controls (Fig.
7C). Both HG exposure and C3aR knockdown similarly activated
PI3K/AKT signaling and reduced FoxO1, while C3aR inhibition
reversed these effects (Fig.
7D), consistent with the aforementioned in vivo data.
Under HG conditions with C3a overexpression, treatment with
LY294002 (a PI3K inhibitor; cat. no. HY-10108; MedChemExpress),
which was dissolved in DMSO and applied to podocytes at a final
concentration of 20 μM for 24 h prior to protein extraction,
reduced PI3K/AKT phosphorylation, restored FoxO1 expression and
increased the number of mitochondrial LC3+ puncta (by
2.3-fold, P<0.001) with elevated mitophagic flux (Fig. 7E and F). C3a induced FoxO1
nuclear export through enhanced phosphorylation, a process that was
significantly attenuated by LY294002 (Fig. 7G). These results indicated that
C3a/C3aR mediates hyperglycemia-induced podocytopathy by
suppressing mitophagy through PI3K/AKT-dependent inhibition of
FoxO1-PINK1 signaling.
Discussion
Increasing evidence suggests that the complement
system and its downstream components contribute to the development
of DN (19). The kidneys, in a
diabetic state, are continuously exposed to metabolic and
hemodynamic stress, leading to cellular damage and activation of
innate immune responses, including the complement system (20), with complement C3 serving as the
central hub in the complement cascade (21). Previous studies have indicated
that patients and animals with DN exhibit renal glomerular C3
deposition (4,22). In the present study, a markedly
higher expression of C3 and C3aR was observed in podocytes of
patients with DN compared with those of patients with other
kidney-related diseases. This suggests that complement system
activation, particularly the C3a/C3aR axis, is closely associated
with HG-induced kidney damage and proteinuria. Based on clinical
problems, db/db mice were employed to simulate the process of renal
damage in clinical DN, which is a spontaneous diabetic mouse model
with leptin receptor gene deficiency. Compared with the STZ-induced
type 2 diabetes mouse model, db/db mice showed obesity and more
obvious renal damage. In addition, the occurrence of diabetes in
db/db mice is more similar to that in clinical patients with type 2
diabetes because there is no artificial interference (23,24).
In the current study, db/db mice exhibited C3
deposition and elevated C3aR levels, accompanied with podocyte
loss, increased levels of proteinuria and renal tissue damage.
Podocyte apoptosis can result in podocyte loss and damage,
exacerbating the severity of proteinuria in patients with DN
(25,26). Previous literature has reported
that the C3a/C3aR axis in podocytes can initiate autocrine
IL-1β/IL-1R1 signaling, downregulating nephrin expression and
leading to rearrangement of the actin cytoskeleton (27). Similarly, in the present study,
blocking C3aR increased the expression of the podocyte functional
proteins nephrin and synaptopodin in DN mice glomeruli, and
improved podocyte cytoskeleton alignmen in HG. This suggests the
involvement of the C3a/C3aR axis in podocyte damage in DN, which is
primarily manifested in cytoskeleton disruption and downregulation
of nephrin expression, with C3aRAs potentially contributing to the
maintenance of podocyte homeostasis.
Podocytes exhibit high dynamism, thus necessitating
substantial energy to maintain the normal organization of the
cytoskeleton and podocyte foot process remodeling (28). Impaired mitophagy is considered a
hallmark of human DN and rodent models of DN (29). Currently, the PINK1/parkin
pathway is considered one of the most crucial mediators in the
process of mitophagy (30). The
present study confirmed a decrease in PINK1/parkin-mediated
mitophagy levels in the db/db mice and in podocytes under HG
conditions in vitro. Similarly, reduced PINK1/parkin
mitophagy has been reported in HK-2 cells under HG conditions,
STZ-induced DN models (12,31), proximal tubular cells (32), glomerular mesangial cells
(33), podocytes, and db/db
mouse models of DN (34),
aligning with the current findings. By contrast, certain previous
studies have reported abnormal activation of PINK1/parkin-mediated
mitophagy in db/db mice (35,36). The db/db mice model effectively
recapitulates early-stage DN manifestations, including
characteristic glomerular pathology, podocyte injury and the
albuminuric phase, but fails to develop progressive renal fibrosis
or end-stage renal disease (ESRD) features typically observed in
advanced human DN, primarily due to its constrained 24-week
observation window, species-specific attenuation of fibrotic
pathways, and absence of sustained glomerulosclerosis that would
culminate in functional renal failure, thereby limiting its
translational relevance for late-stage DN therapeutic interventions
requiring fibrosis or ESRD endpoints. The db/db mice model remains
the gold standard for initiating events in DN but necessitates
complementary approaches for disease culmination studies. In
subsequent studies, therapeutic strategies targeting fibrosis/ESRD
reversal must be validated using complementary models (for example,
uninephrectomized db/db mice or DBA/2J-STZ mice). These seemingly
disparate results may be attributed to different stages of DN. In
the early stages of DN, the number of damaged mitochondria
increases, and compensatory mitophagy is enhanced to eliminate
these mitochondria. As DN progresses to a certain stage,
compensatory mitophagy becomes insufficient to clear an adequate
number of damaged mitochondria, resulting in a decompensated state
and a decrease in mitophagy levels (37). Since there were no genetic
background differences in the animals used in these studies,
further research is needed to explain these conflicting results
observed in the activation pattern of PINK1/parkin-mediated
mitophagy.
Numerous previous studies, including one conducted
by our group, have reported the key role of C3a/C3aR in
accelerating apoptosis. CRP interacts with the C3a/C3aR axis to
promote the process of DN through podocyte autophagy (38), and the treatment of DN mice with
a C3aRA enhanced podocyte density and preserved their phenotype,
limiting proteinuria and glomerular injury (8). Overall, this suggests that the
C3a/C3aR axis plays an important role in diabetic podocyte injury,
which may involve podocyte autophagy. The current study also found
that C3aR blockade protected podocytes and mitochondria from damage
in DN, as evidence by the corresponding changes in mitophagy levels
during the process. The majority of previous studies have shown
significant changes in the expression of mitophagy-related genes in
the entire lysates of kidney tissues or cells, with little
localization within mitochondria. Although the C3a/C3aR axis has
been demonstrated to affect mitochondrial biogenesis, including
impairing PPARα/CPT-1α-mediated renal tubular mitochondrial fatty
acid oxidation (39), there is
currently no relevant research on the involvement of the C3a/C3aR
axis in podocyte mitophagy. In the present study, Mito-LC3B
adenovirus was utilized for exogenous transfection of immortalized
human podocytes, and the co-localization assessment of mitochondria
and lysosomes helped further elucidate the role of
PINK1/parkin-mediated mitophagy in C3a-induced podocyte damage
under HG conditions. The results revealed a significant decrease in
mitophagy flux in podocytes cultured in a HG environment compared
with the NG control group. Inhibition of C3aR improved the reduced
mitophagy induced by HG, confirming that the C3a/C3aR axis
downregulates mitophagy levels in podocytes, participating in the
process of podocyte damage. While initially characterized as a C3aR
antagonist, subsequent studies have demonstrated that SB290157
inhibits C5aR1 (Ki≈1 μM) at higher concentrations, and C5aR1
similarly mediates inflammation and podocyte injury in DN.
Moreover, SB290157 exhibits concentration-dependent (>μM)
activation of PAR1 and PAR2, both expressed in renal cells. To
mitigate these potential off-target effects, our experimental
design rigorously maintained concentrations ≤0.5 μM.
Furthermore, complementary C3aR siRNA experiments in cellular
models confirmed that the pharmacological profile of SB290157
persisted under these constrained conditions.
The present study delineates a novel mechanistic
link between the C3a/C3aR axis and podocyte mitophagy dysregulation
in DN, centrally orchestrated by the PI3K/AKT/FoxO1 signaling
pathway. Numerous previous studies have reported the activation of
the PI3K/AKT pathway in DN animal models (40-43), and that inhibiting the PI3K/AKT
pathway could enhance podocyte autophagy levels and reduce
proteinuria (44,45). In the present study, increased
expression of PI3K/AKT signaling molecules was observed in the
renal tissues of db/db mice and in podocytes cultured in a HG
environment in vitro. Blocking C3aR expression inhibited the
PI3K/AKT signaling pathway. Inhibition of PI3K/AKT signaling
prevents phosphorylation of FoxO1, enabling its nuclear
translocation, thereby regulating gene expression associated with
cell death, proliferation, differentiation, cellular metabolism,
and oxidative stress (46,47). Previous studies have established
that PI3K/AKT signaling exacerbates insulin resistance in diabetes
by inhibiting FOXO-driven metabolic adaptations (48,49). FoxO1 overexpression prevents
podocyte damage and improves the progression of DN (50). The current study uncovers that
C3a, a complement effector, acts as an upstream trigger of PI3K/AKT
in podocytes. In diabetic environments, C3aR binding activates
PI3K/AKT, which subsequently phosphorylates and inactivates FoxO1.
This redirects FoxO1 from the nucleus to the cytoplasm, repressing
the transcription of mitophagy genes (such as PINK1 and parkin).
The current findings support the observations that C3aR blockade
restores mitochondrial integrity, mechanistically linking
complement to FoxO1-driven redox and metabolic homeostasis.
However, in the present study, only PI3K inhibitors were used, and
the expression changes of the downstream signal molecules AKT and
FoxO1 were indirectly detected. Their direct role and existence in
the nucleus or cytoplasm are not yet clear; thus, further studies
are required in the future.
Additionally, a limitation of the present study is
that the exclusive focus on complement C3 and C3aR, lacking the
assessment of complement fragment C3a levels. Although a
significant increase in C3a under HG conditions was detected in
vitro in cultured podocytes, no noticeable differences in
circulating C3a expression were observed in the mouse experiments.
Considering the possibility of localized activation of complement
C3a in the kidney, future studies should construct models to
measure C3a levels in urine or locally collected blood from the
kidneys.
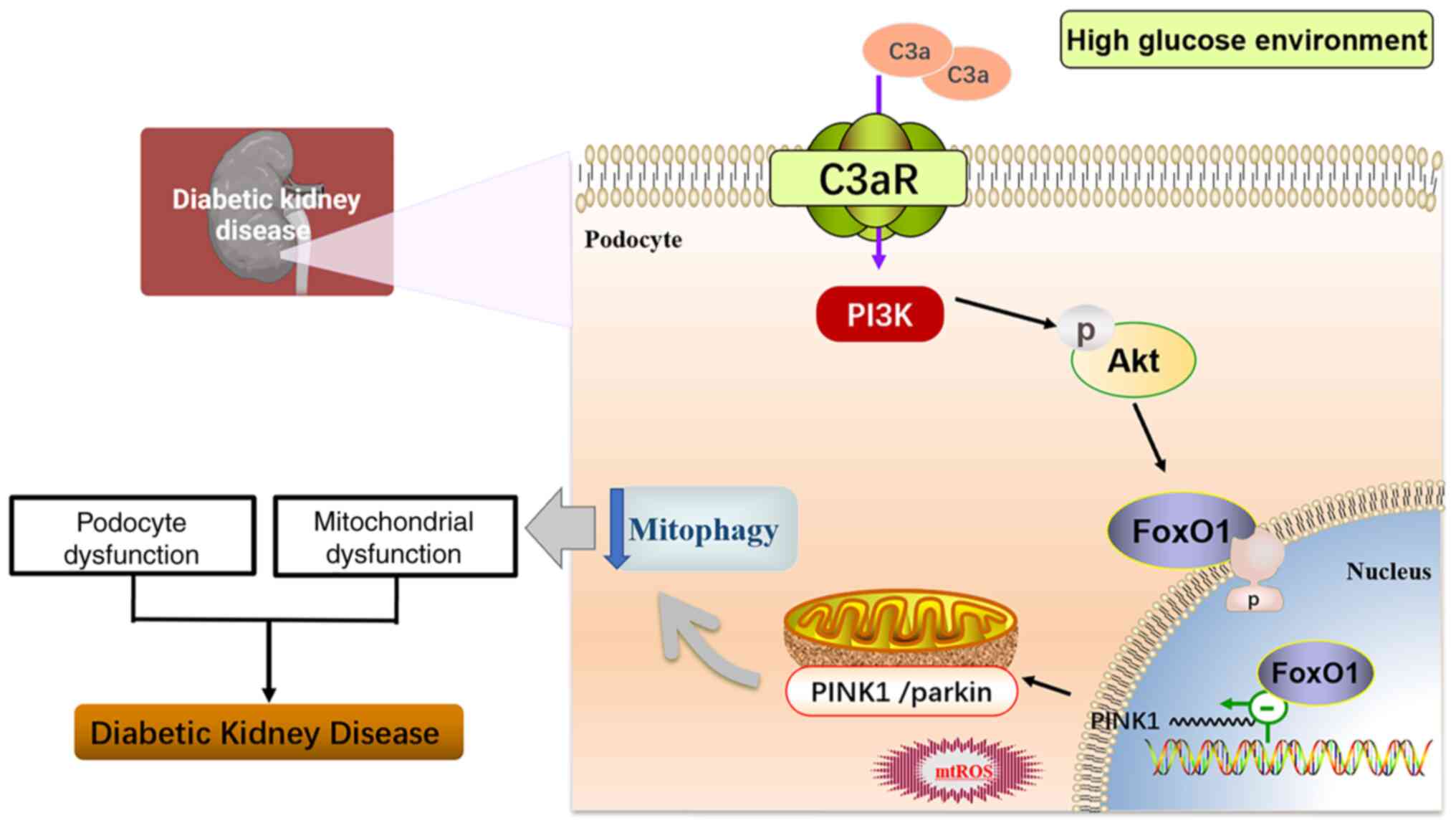
In summary (Fig.
8), the present study demonstrated for the first time that the
C3a/C3aR axis regulates the PI3K/AKT/FoxO1 signaling pathway, and
then leads to diabetic renal injury by inhibiting podocyte
mitophagy, suggesting that blocking C3aR may be an innovative
therapeutic strategy for treating patients with DN.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
MW and JW conceived and designed the study. MW, XW,
SR, TZ and JZ performed the experiments. JC, XW, SR, EL, DY and KN
analyzed the data. MW was a major contributor in writing the
manuscript. JW and MW assume overall responsibility for the
manuscript. JW and JC confirm the authenticity of all the raw data.
JW and JC was responsible for supervision, funding acquisition and
interpretation of data. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
All procedures were conducted in accordance with the
NIH Guide for the Care and Use of Laboratory Animals and approved
by the Institutional Animal Care and Use Committee of Fujian
Medical University (approval no. IACUC FJMU 2023-Y-1033; Fuzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
The authors acknowledge Professor Liangdi Xie from
the Fujian Hypertension Research Institute, The First Affiliated
Hospital of Fujian Medical University for his providing the
experimental site and instruments. Additionally, the authors would
like to thank Mr Changshen Xu and Ms Guili Lian from the Fujian
Hypertension Research Institute, The First Affiliated Hospital of
Fujian Medical University for their technical assistance and
research resources. The authors also thank the Public Technology
Platform, The First Affiliated Hospital of Fujian Medical
University for its technical support.
Funding
The present study was supported by the Fujian Provincial Health
Technology Project (grant no. 2021CXA018), the National Natural
Science Foundation of China (grant nos. 82470733 and 82401848).
References
|
1
|
Ma J, Yiu WH and Tang SCW: Complement
anaphylatoxins: Potential therapeutic target for diabetic kidney
disease. Diabet Med. 42:e154272025. View Article : Google Scholar :
|
|
2
|
Li X, Zhao S, Xie J, Li M, Tong S, Ma J,
Yang R, Zhao Q, Zhang J and Xu A: Targeting the NF-κB p65-MMP28
axis: Wogonoside as a novel therapeutic agent for attenuating
podocyte injury in diabetic nephropathy. Phytomedicine.
138:1564062025. View Article : Google Scholar
|
|
3
|
Wang T, Chen Y, Liu Z, Zhou J, Li N, Shan
Y and He Y: Long noncoding RNA Glis2 regulates podocyte
mitochondrial dysfunction and apoptosis in diabetic nephropathy via
sponging miR-328-5p. J Cell Mol Med. 28:e182042024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Woroniecka KI, Park ASD, Mohtat D, Thomas
DB, Pullman JM and Susztak K: Transcriptome analysis of human
diabetic kidney disease. Diabetes. 60:2354–2369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wehner H, Höhn D, Faix-Schade U, Huber H
and Walzer P: Glomerular changes in mice with spontaneous
hereditary diabetes. Lab Invest. 27:331–340. 1972.PubMed/NCBI
|
|
6
|
Li L, Chen L, Zang J, Tang X, Liu Y, Zhang
J, Bai L, Yin Q, Lu Y, Cheng J, et al: C3a and C5a receptor
antagonists ameliorate endothelial-myofibroblast transition via the
Wnt/β-catenin signaling pathway in diabetic kidney disease.
Metabolism. 64:597–610. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li L, Yin Q, Tang X, Bai L, Zhang J, Gou
S, Zhu H, Cheng J, Fu P and Liu F: C3a receptor antagonist
ameliorates inflammatory and fibrotic signals in type 2 diabetic
nephropathy by suppressing the activation of TGF-β/smad3 and IKBα
pathway. PLoS One. 9:e1136392014. View Article : Google Scholar
|
|
8
|
Morigi M, Perico L, Corna D, Locatelli M,
Cassis P, Carminati CE, Bolognini S, Zoja C, Remuzzi G, Benigni A
and Buelli S: C3a receptor blockade protects podocytes from injury
in diabetic nephropathy. JCI Insight. 5:e1318492020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma K, Chen G, Li W, Kepp O, Zhu Y and Chen
Q: Mitophagy, mitochondrial homeostasis, and cell fate. Front Cell
Dev Biol. 8:4672020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stanigut AM, Tuta L, Pana C, Alexandrescu
L, Suceveanu A, Blebea NM and Vacaroiu IA: Autophagy and mitophagy
in diabetic kidney disease-a literature review. Int J Mol Sci.
26:8062025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tagawa A, Yasuda M, Kume S, Yamahara K,
Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Asanuma K,
et al: Impaired podocyte autophagy exacerbates proteinuria in
diabetic nephropathy. Diabetes. 65:755–767. 2016. View Article : Google Scholar
|
|
12
|
Zhou D, Zhou M, Wang Z, Fu Y, Jia M, Wang
X, Liu M, Zhang Y, Sun Y, Zhou Y, et al: Progranulin alleviates
podocyte injury via regulating CAMKK/AMPK-mediated autophagy under
diabetic conditions. J Mol Med (Berl). 97:1507–1520. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou D, Zhou M, Wang Z, Fu Y, Jia M, Wang
X, Liu M, Zhang Y, Sun Y, Lu Y, et al: PGRN acts as a novel
regulator of mitochondrial homeostasis by facilitating mitophagy
and mitochondrial biogenesis to prevent podocyte injury in diabetic
nephropathy. Cell Death Dis. 10:5242019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Martini S, Nair V, Keller BJ, Eichinger F,
Hawkins JJ, Randolph A, Böger CA, Gadegbeku CA, Fox CS, Cohen CD,
et al: Integrative biology identifies shared transcriptional
networks in CKD. J Am Soc Nephrol. 25:2559–2572. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ju W, Greene CS, Eichinger F, Nair V,
Hodgin JB, Bitzer M, Lee YS, Zhu Q, Kehata M, Li M, et al: Defining
cell-type specificity at the transcriptional level in human
disease. Genome Res. 23:1862–1873. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
National Research Council Committee for
the Update of the Guide for the C. and A. Use of Laboratory. The
National Academies Collection: Reports funded by National
Institutes of Health, in Guide for the Care and Use of Laboratory
Animals. National Academies Press. Copyright© 2011. National
Academy of Sciences; Washington, DC: 2011
|
|
17
|
Ames RS, Lee D, Foley JJ, Jurewicz AJ,
Tornetta MA, Bautsch W, Settmacher B, Klos A, Erhard KF, Cousins
RD, et al: Identification of a selective nonpeptide antagonist of
the anaphylatoxin C3a receptor that demonstrates antiinflammatory
activity in animal models. J Immunol. 166:6341–6348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yasuda-Yamahara M, Kume S, Tagawa A,
Maegawa H and Uzu T: Emerging role of podocyte autophagy in the
progression of diabetic nephropathy. Autophagy. 11:2385–2386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Flyvbjerg A: The role of the complement
system in diabetic nephropathy. Nat Rev Nephrol. 13:311–318. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tesch GH: Diabetic nephropathy-is this an
immune disorder? Clin Sci (Lond). 131:2183–2199. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sahu A and Lambris JD: Structure and
biology of complement protein C3, a connecting link between innate
and acquired immunity. Immunol Rev. 180:35–48. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kelly KJ, Liu Y, Zhang J and Dominguez JH:
Renal C3 complement component: Feed forward to diabetic kidney
disease. Am J Nephrol. 41:48–56. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Racine KC, Iglesias-Carres L, Herring JA,
Wieland KL, Ellsworth PN, Tessem JS, Ferruzzi MG, Kay CD and
Neilson AP: The high-fat diet and low-dose streptozotocin type-2
diabetes model induces hyperinsulinemia and insulin resistance in
male but not female C57BL/6J mice. Nutr Res. 131:135–146. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang L, Zhou R, Li G, Zhang X, Li Y, Shen
Y and Fang J: Multi-omics characterization of diabetic nephropathy
in the db/db mouse model of type 2 diabetes. Comput Struct
Biotechnol J. 27:3399–3409. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin J, Shi Y, Gong J, Zhao L, Li Y, He Q
and Huang H: Exosome secreted from adipose-derived stem cells
attenuates diabetic nephropathy by promoting autophagy flux and
inhibiting apoptosis in podocyte. Stem Cell Res Ther. 10:952019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yuen DA, Stead BE, Zhang Y, White KE,
Kabir MG, Thai K, Advani SL, Connelly KA, Takano T, Zhu L, et al:
eNOS deficiency predisposes podocytes to injury in diabetes. J Am
Soc Nephrol. 23:1810–1823. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Angeletti A, Cantarelli C, Petrosyan A,
Andrighetto S, Budge K, D'Agati VD, Hartzell S, Malvi D, Donadei C,
Thurman JM, et al: Loss of decay-accelerating factor triggers
podocyte injury and glomerulosclerosis. J Exp Med.
217:e201916992020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Galvan DL, Green NH and Danesh FR: The
hallmarks of mitochondrial dysfunction in chronic kidney disease.
Kidney Int. 92:1051–1057. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen K, Dai H, Yuan J, Chen J, Lin L,
Zhang W, Wang L, Zhang J, Li K and He Y: Optineurin-mediated
mitophagy protects renal tubular epithelial cells against
accelerated senescence in diabetic nephropathy. Cell Death Dis.
9:1052018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nguyen TN, Padman BS and Lazarou M:
Deciphering the molecular signals of PINK1/parkin mitophagy. Trends
Cell Biol. 26:733–744. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li W, Du M, Wang Q, Ma X, Wu L, Guo F, Ji
H, Huang F and Qin G: FoxO1 promotes mitophagy in the podocytes of
diabetic male mice via the PINK1/parkin pathway. Endocrinology.
158:2155–2167. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao Y and Sun M: Metformin rescues Parkin
protein expression and mitophagy in high glucose-challenged human
renal epithelial cells by inhibiting NF-κB via PP2A activation.
Life Sci. 246:1173822020. View Article : Google Scholar
|
|
33
|
Yi X, Yan W, Guo T, Liu N, Wang Z, Shang
J, Wei X, Cui X, Sun Y, Ren S and Chen L: Erythropoietin mitigates
diabetic nephropathy by restoring PINK1/Parkin-mediated mitophagy.
Front Pharmacol. 13:8830572022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sun J, Zhu H, Wang X, Gao Q, Li Z and
Huang H: CoQ10 ameliorates mitochondrial dysfunction in diabetic
nephropathy through mitophagy. J Endocrinol. 240:445–465. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu X, Wang W, Song G, Wei X, Zeng Y, Han
P, Wang D, Shao M, Wu J, Sun H, et al: Astragaloside IV ameliorates
diabetic nephropathy by modulating the mitochondrial quality
control network. PLoS One. 12:e01825582017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu X, Lu J, Liu S, Huang D, Chen M, Xiong
G and Li S: Huangqi-Danshen decoction alleviates diabetic
nephropathy in db/db mice by inhibiting PINK1/Parkin-mediated
mitophagy. Am J Transl Res. 12:989–998. 2020.PubMed/NCBI
|
|
37
|
Yang M, Li C, Yang S, Xiao Y, Chen W, Gao
P, Jiang N, Xiong S, Wei L, Zhang Q, et al: Mitophagy: A novel
therapeutic target for treating DN. Curr Med Chem. 28:2717–2728.
2021. View Article : Google Scholar
|
|
38
|
Zhang L, Li W, Gong M, Zhang Z, Xue X, Mao
J, Zhang H, Li S, Liu X, Wu F, et al: C-reactive protein inhibits
C3a/C3aR-dependent podocyte autophagy in favor of diabetic kidney
disease. FASEB J. 36:e223322022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang C, Wang Z, Xu J, Ma H, Jin K, Xu T,
Pan X, Feng X and Zhang W: C3aR antagonist alleviates C3a induced
tubular profibrotic phenotype transition via restoring PPARα/CPT-1α
mediated mitochondrial fatty acid oxidation in renin-dependent
hypertension. Front Biosci (Landmark Ed). 28:2382023. View Article : Google Scholar
|
|
40
|
Chen Y, Zheng YF, Lin XH, Zhang JP, Lin F
and Shi H: Dendrobium mixture attenuates renal damage in rats with
diabetic nephropathy by inhibiting the PI3K/Akt/mTOR pathway. Mol
Med Rep. 24:5902021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ma Z, Liu Y, Li C, Zhang Y and Lin N:
Repurposing a clinically approved prescription Colquhounia root
tablet to treat diabetic kidney disease via suppressing
PI3K/AKT/NF-kB activation. Chin Med. 17:22022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dong R, Zhang X, Liu Y, Zhao T, Sun Z, Liu
P, Xiang Q, Xiong J, Du X, Yang X, et al: Rutin alleviates EndMT by
restoring autophagy through inhibiting HDAC1 via PI3K/AKT/mTOR
pathway in diabetic kidney disease. Phytomedicine. 112:1547002023.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang Y, Yang S, Cui X, Yang J, Zheng M,
Jia J, Han F, Yang X, Wang J, Guo Z, et al: Hyperinsulinemia can
cause kidney disease in the IGT stage of OLETF rats via the
INS/IRS-1/PI3-K/Akt signaling pathway. J Diabetes Res.
2019:47097152019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zheng D, Tao M, Liang X, Li Y, Jin J and
He Q: p66Shc regulates podocyte autophagy in high glucose
environment through the Notch-PTEN-PI3K/Akt/mTOR pathway. Histol
Histopathol. 35:405–415. 2020.
|
|
45
|
Wang X, Jiang L, Liu XQ, Huang YB, Wang
AL, Zeng HX, Gao L, Zhu QJ, Xia LL and Wu YG: Paeoniflorin binds to
VEGFR2 to restore autophagy and inhibit apoptosis for podocyte
protection in diabetic kidney disease through PI3K-AKT signaling
pathway. Phytomedicine. 106:1544002022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen Y, Li Z, Li H, Su W, Xie Y, Pan Y,
Chen X and Liang D: Apremilast regulates the Teff/Treg balance to
ameliorate uveitis via PI3K/AKT/FoxO1 signaling pathway. Front
Immunol. 11:5816732020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Miao Z, Liu Y, Xu Y, Bu J and Yang Q:
Oxaloacetate promotes the transition from glycolysis to
gluconeogenesis through the Akt-FoxO1 and JNK/c-Jun-FoxO1 axes and
inhibits the survival of liver cancer cells. Int Immunopharmacol.
161:1150512025. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xu Z, Liu K, Zhang G, Yang F, He Y, Nan W,
Li Y and Lin J: Transcriptome analysis reveals that the injection
of mesenchymal stem cells remodels extracellular matrix and
complement components of the brain through PI3K/AKT/FOXO1 signaling
pathway in a neuroinflammation mouse model. Genomics.
117:1110332025. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Deng A, Wang Y, Huang K, Xie P, Mo P, Liu
F, Chen J, Chen K, Wang Y and Xiao B: Artichoke (Cynara scolymus
L.) water extract alleviates palmitate-induced insulin resistance
in HepG2 hepatocytes via the activation of IRS1/PI3K/AKT/FoxO1 and
GSK-3β signaling pathway. BMC Complement Med Ther. 23:4602023.
View Article : Google Scholar
|
|
50
|
Cosenso-Martin LN, Takaoka LY and
Vilela-Martin JF: Randomized study comparing vildagliptin vs
glibenclamide on glucose variability and endothelial function in
patients with type 2 diabetes mellitus and hypertension. Diabetes
Metab Syndr Obes. 13:3221–3229. 2020. View Article : Google Scholar : PubMed/NCBI
|















![Expression levels of mitophagy and
related pathway proteins in mouse renal tissues. (A)
Immunohistochemical analysis of the mitophagy-specific protein
PINK1. Magnification, x100. (B) Protein levels of LC3B I/II, parkin
and PINK1 in kidney tissues (n=6). Corresponding histograms are
shown on the right panel of representative protein bands. (C)
Protein levels of PI3K [PI3-kinase p85α (54 + 85 kDa)],
phosphorylated-AKT and FoxO1 in kidney tissues (n=6). Corresponding
histograms are shown on the right panel of representative protein
bands. *P<0.05, **P<0.01 and
***P<0.001. p-, phosphorylated.](/article_images/ijmm/56/6/ijmm-56-06-05664-g03.jpg)