Introduction
Sepsis is defined as a life-threatening systemic
inflammatory response syndrome caused by a dysregulated host
response to infection, contributing to a considerable global
mortality rate (as of 2017, ~48.9 million cases of sepsis were
recorded globally, with 11 million reported sepsis-related
mortalities, accounting for 19.7% of all global mortalities)
(1). The initial event of sepsis
is the recognition of microbe-derived pathogen-associated molecular
patterns or endogenous damage-associated molecular patterns by host
pattern recognition receptors (2). This recognition triggers
intracellular signal transduction pathways. Under physiological
conditions, immune cells interact via this signaling network,
inducing an innate immune response to eliminate pathogens and
maintain cellular stability. However, certain host signaling
pathways in sepsis become abnormally activated, leading to an
explosive release of cytokines and alarmins (3). These inflammatory mediators
markedly upregulate the expression of inflammation-related genes
and act as key effector molecules in driving excessive systemic
inflammatory responses and subsequent immunosuppression. This
process further promotes the massive release of inflammatory
mediators such as cytokines and chemokines, ultimately culminating
in a cytokine storm (4,5).
The liver carries out a key role as a defensive
barrier that protects the body against potential threats (6). It can be activated by microbes or
alarm signals, resulting in the release of pro-inflammatory
cytokines and the recruitment of immune cells to assist in
microbial clearance. Additionally, the intricate immune
surveillance mechanisms of the liver, supported by its strategic
anatomical positioning, enable it to collaborate with both
parenchymal and non-parenchymal cells to eliminate bacteria from
the bloodstream and regulate the inflammatory and immune responses.
This collaboration mitigates the risk of bacteremia and serves as a
notable barrier against microbial invasion (7). However, the liver is frequently
targeted by dysregulation of inflammation. Immune cells are
susceptible to various substances. These substances include
antigens, endotoxins, danger signals and microorganisms circulating
in the bloodstream. Excessive stimulation may cause these cells to
become abnormally activated and may also make them undergo
apoptosis which disrupts immune function and contributes to
inflammation dysregulation (8,9).
Therefore, prompt intervention targeting liver injury during sepsis
management is key to effectively mitigating inflammatory responses
and cytokine storms, thereby protecting against further organ
damage and improving patient outcomes.
Radioprotective 105 (RP105), also known as CD180, is
a type I transmembrane protein characterized by an extracellular
domain rich in leucine-rich repeats (LRRs) and a short cytoplasmic
tail (10). Analogous to the
Toll protein in Drosophila, RP105 was the first mammalian molecule
to exhibit similarity to Toll through its extracellular LRRs.
Lipopolysaccharide (LPS) is the primary component of the cell wall
of gram-negative bacteria. It is also a key virulence factor. It
causes infections such as sepsis and septic shock. During sepsis
progression, LPS binds directly to pattern recognition receptors on
immune cell surfaces. This binding activates the Toll-like Receptor
4 (TLR4) inflammatory signaling pathway. This activation triggers a
release of inflammatory cytokines, resulting in an 'inflammatory
cytokine storm' that can damage tissues and organs. As a key
negative regulator of the TLR4 signaling pathway and an essential
modulator of the immune system (11,12), RP105 prevents the excessive
production of pro-inflammatory cytokines by inhibiting TLR4
mediated pro-inflammatory signals, serving as a protective strategy
against exaggerated inflammatory responses. Consequently, RP105
carries out a key role in regulating immune response and
controlling inflammation during sepsis.
Although numerous studies have reported the
anti-inflammatory effects of RP105 in various disease models
(13-16), its effect on sepsis-induced liver
injury remains poorly documented. Moreover, the molecular
mechanisms underlying the protective role of RP105, particularly
whether it modulates intracellular cytokine signaling pathways,
have not been elucidated. Therefore, the present study aimed to
investigate the function of RP105 in sepsis-induced hepatic injury
using a CLP mouse model.
Materials and methods
Mice and treatments
Wild-type (WT) mice and RP105 knockout (KO) mice
[total number, 24 (WT, 12; KO, 12); male C57BL/6 mice; 8-12 weeks
old; weighing 20-25 g] were purchased from GemPharmatech Co. Ltd.
After purchase, the mice were raised at the Animal Experiment
Center of Zhongnan Hospital of Wuhan University (Wuhan, China).
Mice were kept in a breeding environment with a room temperature of
20-25°C, humidity 50-70% and a 12:12 h light-dark cycle. All mice
had free access to standard animal food and water. All experimental
procedures were conducted in accordance with the National
Institutes of Health Guidelines for the Care and Use of Laboratory
Animals (17). Every effort was
made to minimize the suffering of the mice used. Before the
official experiment begins, the mice undergo an adaptive feeding
period and are provided with sterilized clean drinking water.
Genotypes of the knockout mice were confirmed using PCR. WT and KO
mice were randomly assigned to different treatment groups (n=6 for
single-variable animal experiments following previous studies that
also used six animals per group to ensure reproducibility and
statistical reliability) (18,19), data collection and analysis was
carried out under blinded conditions.
Anesthesia protocol
C57BL/6 mice were anesthetized by inhalation of 5%
isoflurane for induction, followed by maintenance with 2-2.5%
isoflurane. The animals were positioned in a prone position on a
37°C thermoregulated heating pad (Harvard Apparatus) with
continuous monitoring of rectal temperature maintained at
36.5±0.5°C.
Animal procedures
In this experiment, the duration of the laparotomy
procedure for cecal ligation and puncture (CLP) is 10 min; the
modeling cycle of the CLP model (the time from the completion of
the modeling procedure to the end of the experiment) is 24 h.
Following anesthesia, a 1 cm midline laparotomy was carried out on
the left side of the xiphoid to expose the cecum. The cecum was
then ligated at 75% of the distance between the distal pole and the
base using 4-0 silk suture, followed by two through-and-through
punctures with a 25-gauge needle near the distal end. The cecum was
subsequently repositioned and the abdominal cavity was closed in
two layers with 5-0 sutures. Postoperatively, mice received 1 ml of
pre-warmed sterile saline via subcutaneous injection for fluid
resuscitation and were maintained at 37°C for thermal support.
Control mice underwent identical surgical
procedures, including cecal exteriorization, but without ligation
or puncture. The monitored parameters included body temperature,
mobility, food/water intake and pain-related behaviors, with
assessments conducted hourly. Terminal blood samples (1 ml) were
collected from the inferior vena cava, and liver tissues were
harvested. All procedures were performed under deep anesthesia
immediately prior to euthanasia. The serum obtained by
centrifugation (3,000 × g for 10 min at 4°C) and liver tissues were
stored at −80°C for future experimental use.
Euthanasia procedure
Mice were euthanized by 5% isoflurane overdose until
respiratory arrest (~5 min), with mortality confirmed via cardiac
cessation, absent corneal reflex and fixed pupil dilation (per AVMA
Guidelines 2020) (20).
Cell culture and LPS-induced model
Both Raw264.7 cells (cat. no. YC-C020) and the RP105
(alias, CD180) knockout cell line (cat. no. YKO-M041) were
commercially obtained from Ubigene). In the present study,
CRISPR/Cas9 technology was used for gene KO, with the negative
control (NC) group consisting of untransfected WT cells. The RP015
KO cell line was generated by precisely deleting a 382-base pair
fragment (completely removing exon 3) in clone C7 (the KO cell line
was produced by Ubigene). All cells were cultured in a humidified
incubator (Thermo Fisher Scientific, Inc.) maintained at 37°C and
5% CO2 in high-glucose Dulbecco's Modified Eagle Medium
(cat. no. G4511; Wuhan Servicebio Technology Co., Ltd.)
supplemented with 10% heat-inactivated fetal bovine serum (cat. no.
A5256701; Gibco; Thermo Fisher Scientific, Inc.) For the
experiment, the cultures were placed in the aforementioned medium
overnight (at 37°C with 5% CO2) and 1 μg/ml LPS
(InvivoGen) was administered when the cells reached 80-90%
confluence, with treatment for 24 h.
H&E
After washing the excised liver tissues, which were
stored at −80°C, with sterile normal saline, they were fixed in 4%
buffered paraformaldehyde at room temperature for 24 h, then
embedded in paraffin for subsequent processing. All
paraffin-embedded sections were cut to 4-μm thickness,
stained with H&E at room temperature. After dewaxing and
rehydration (Sections were sequentially dehydrated through a graded
ethanol series (100, 95, 80, 70%) and finally immersed in distilled
water for 5 min to achieve rehydration), stain the sections were
incubated with hematoxylin for 5 min, then eosin for 15 min, both
at room temperature, followed by observation and imaging using a
light microscope. Two pathologists, blinded to the experimental
groups, randomly selected three different fields of view to
evaluate the liver injury of each animal. The liver injury score
was used to assess the characteristics of sepsis-induced liver
injury, and the pathological scoring was carried out according to
previous studies (21,22). The degrees of inflammatory
infiltration, thrombosis and necrosis were graded separately (0 was
defined as absent and four as severe). The total score of liver
histological scoring was expressed as the sum of the scores of each
parameter, with a maximum of 12 points.
Biochemical analysis
Serum samples stored at −80°C were thawed at room
temperature for 30 min. After complete thawing, they were
centrifuged at low speed (750 × g for 30 sec at 4°C), gently mixed
by vortexing and air bubbles in the tubes were thoroughly aspirated
to prevent interference with the experimental results.
Subsequently, standardized processing and streamlined detection
were conducted by professional medical technicians using a fully
automated biochemical analyzer. Serum levels of alanine
aminotransferase (ALT) and aspartate aminotransferase (AST) were
measured using the BS-2000 automatic biochemical analyzer (Shenzhen
Mindray Bio-Medical Electronics Co., Ltd.). All detection
procedures were carried out at the Experimental Center of Zhongnan
Hospital, Wuhan University (Wuhan, China).
Reverse transcription quantitative PCR
(RT-qPCR)
Total RNA was extracted from liver tissues of the
mice using the Trizol kit (cat. no. R0016; Beyotime Institute of
Biotechnology), followed by reverse transcription of the RNA into
cDNA using the Hifair® V one-step RT-gDNA digestion
SuperMix kit (cat. no. 11142ES10; Shanghai Yeasen Biotechnology
Co., Ltd.). A total of 1 μg RNA was used for cDNA
conversion. The reverse transcription procedure was carried out as
follows: Initial primer annealing at 30°C for 5 min, followed by
cDNA synthesis at 55°C for 30 min and final enzyme inactivation at
85°C for 5 min. qPCR was carried out using SYBR Green dye (Biosharp
Life Sciences). The qPCR amplification protocol consisted of an
initial denaturation step at 95°C for 5 min, followed by 40 cycles
of denaturation at 95°C for 10 sec and annealing/extension at 60°C
for 30 sec with fluorescence signal acquisition. Relative gene
expression levels were determined using the 2-ΔΔCq
(23) method and normalized to
the β-actin reference gene. The primers for the target genes are
listed in Table I.
 | Table IPrimer sequences of the target
genes. |
Table I
Primer sequences of the target
genes.
| Gene | Forward primer
sequence (5'-3') | Reverse primer
sequence (5'-3') |
|---|
| IL-1β C |
ACCTCACAAGCAGAGCACAAG |
GAAACAGTCCAGCCCATACTTTAGG |
| IL-6 |
CTTCTTGGGACTGATGCTGGTGAC |
TCTGTTGGGAGTGGTATCCTCTGTG |
| TNF-α |
AAGACACCATGAGCACAGAAAGC |
GCCACAAGCAGGAATGAGAAGAG |
| RP105 G |
ACCAACTCACTTCAGACA |
TGACTAAGAGCCTCAATGC |
| β-Actin |
CAGCAAGCAGGAGTACGATGAGTC |
CAGTAACAGTCCGCCTAGAAGCAC |
Immunohistochemistry (IHC)
Liver tissue was first fixed in 4% paraformaldehyde
for 24 h (at room temperature), embedded in paraffin and sliced
into sections of 4-μm thickness. These sections were
dewaxed, rehydrated for 30 min at room temperature, blocked with 3%
BSA for 30 min at 37°C, and finally incubated overnight at 4°C with
primary antibodies: anti-RP105 (1:1,000; cat. no. ab184956; Abcam)
and anti-Caspase3 (1:200; cat. no. 19677-1-AP; Proteintech Group,
Inc.). The slides were then incubated with Goat Anti-Rabbit IgG
H&L (1:1,000; cat. no. ab205718; Abcam) at 37°C for 1 h.
Immunoreactivity was visualized using 3,3-diaminobenzidine
tetrahydrochloride, which produces a brown precipitate at the
antigen site. Subsequently, the cell nuclei were re-counterstained
with Mayer's Hematoxylin Stain Solution (cat. no. G1080; Beijing
Solarbio Science & Technology Co., Ltd.) at room temperature
for 1 min, the slides were dehydrated and cells were observed and
imaged under a light microscope.
Immunofluorescence (IF)
IF followed a protocol similar to that reported
previously (24). The sections
were prepared following the same protocol as used for IHC; they
were then incubated (overnight at 4°C) with anti-myeloperoxidase
(MPO; 1:100; cat. no. 22225-1-AP; Wuhan Sanying Biotechnology),
anti-F4/80 (1:5,000; cat. no. ab300421; Abcam) and anti-suppressor
of cytokine signaling 2 (SOCS2; 1:1,000; cat. no. A9190; ABclonal
Biotech Co., Ltd.). After washing to remove the unbound antibody,
the slides were incubated with a secondary antibody (1:1,000; cat.
no. ab205718; Abcam) conjugated to Alexa Fluor 594 (cat. no.
ab150080; Abcam) or XFD488 tyramide reagent (100 μl was
applied to each sample and they were incubated for 5-10 min at room
temperature; cat. no. 11070; AAT Bioquest, Inc.). Next, nuclei were
stained with DAPI. Finally, the slides were observed and imaged
under a fluorescence microscope.
Western blotting (WB)
Precooled RIPA lysis buffer (Wuhan Servicebio
Technology Co., Ltd.) was added to a sterile centrifuge tube, along
with grinding beads. Frozen liver tissue (30-50 mg) was added to a
centrifuge tube, followed by grinding to obtain a homogenate (the
entire operation was carried out on ice). After grinding, the
centrifuge tube was transferred to a refrigerated centrifuge and
centrifuged at 12,000 × g for 15 min at 4°C. After centrifugation,
the supernatant was carefully aspirated, which served as the
protein extract. The total protein concentration was determined
using the BCA method. For electrophoresis, 30 μg of protein
per lane was loaded for animal tissue samples, and 50 μg of
protein per lane was loaded for cell samples. After separating
proteins with different molecular weights using SDS-PAGE (10 and
12%), the proteins were electrically transferred onto a PVDF
membrane. Following a 2-h blocking treatment of the membrane with
5% BSA solution, the membrane was incubated overnight with a
specific primary antibody at 4°C. Subsequently, followed by
labeling with Goat Anti-Rabbit IgG H&L (1:5,000, cat. no.
AS014; ABclonal Biotech Co., Ltd.). Finally, the protein bands on
the membrane were visualized using ECL (Biosharp Life Sciences).
Briefly, the membranes were incubated overnight at 4°C with the
following corresponding primary antibodies: Anti-RP105 (1:1,000,
cat. no. ab184956; Abcam), anti-SOCS2 (1:1,000; cat. no. A9190;
ABclonal Biotech Co., Ltd.), anti-JAK2 (1:1,000, cat. no A11497;
ABclonal Biotech Co., Ltd.), BCL-2 (1:1,000; cat. no. 26593-1-AP;
Wuhan Sanying Biotechnology), anti-BAX (1:1,000; cat. no. T40051;
Abmart Pharmaceutical Technology Co., Ltd.), anti-p53 upregulated
modulator of apoptosis (PUMA; 1:1,000, cat. no. A3752; ABclonal
Biotech Co., Ltd.), anti-Growth Arrest and DNA Damage-inducible α
(GADD45A; 1:500, cat. no. A13487; ABclonal Biotech Co., Ltd.),
anti-phosphorylated-JAK2 (p-JAK2; 1:2,000; cat. no. 4406; Cell
Signaling Technology, Inc.), anti-STAT3 (1:1,000; cat. no. 30835;
Cell Signaling Technology, Inc.), anti-phosphorylated (p-STAT3)
(1:2,000, cat. no. 9145; Cell Signaling Technology, Inc.) and
anti-β-actin (1:50,000, cat. no. AC026; ABclonal Biotech Co., Ltd.)
antibodies.
RNA sequencing (RNA-seq)
RNA was extracted from the animal tissues of the
four groups, using Trizol which was the same as in RT-qPCR
experiment. RNA integrity was evaluated by using the RNA Nano 6000
assay kit from Bioanalyzer After confirming RNA quality, mRNA was
enriched from total RNA using poly-T oligonucleotide-attached
magnetic beads (cat. no. N401-01/02; Vazyme Biotech Co., Ltd.). The
purified mRNA was then fragmented and used for cDNA synthesis.
First-strand cDNA was generated with random hexamer primers and
M-MuLV Reverse Transcriptase (RNase H-) (Fast RNA-seq
Lib Prep Kit V2; cat. no. RK20306; ABclonal Biotech Co., Ltd.),
followed by second-strand synthesis using the same kit. Adapter
sequences were ligated to construct the sequencing library.
High-throughput sequencing was performed on an Illumina NovaSeq
platform using the NovaSeq X Plus 25B Reagent Kit (300 cycles; cat.
no. 20104706; Illumina Inc.) to generate 150 bp paired-end reads.
The final library was loaded at a concentration of 80-120 pM.
Following construction, libraries were quantified (Qubit 2.0
Fluorometer; Thermo Fisher Scientific, Inc.), diluted to 1.5
ng/μl, assessed for insert size (Agilent 2100 Bioanalyzer;
Agilent Technologies, Inc.) and finally quantified for effective
concentration (>1.5 nM) by quantitative PCR. After obtaining the
RNA-seq gene expression matrix, differential expression analysis
was carried out using the DESeq2 R package (1.20.0; https://github.com/mikelove/DESeq2). The
CLP-treated WT group (CLP group) and CLP-treated RP105 knockout
group (RP105 KO + CLP group) were selected for differential gene
comparison analysis, aiming to explore the molecular mechanism by
which RP105 exerts its effects on mice in the CLP model.
Differentially expressed genes (DEGs) were screened by two
simultaneous criteria: Absolute log2 fold change (log2FC)≥1
(ensuring biologically significant expression changes) and adjusted
P-value (Padj) <0.05 (controlling false positives and ensuring
statistical reliability). Raw sequencing data generated in the
present study are available in the GEO database (dataset no.
GSE298411; https://www.ncbi.nlm.nih.gov/gds/?term=GSE298411).
Co-immunoprecipitation (Co-IP)
After thorough grinding, the mouse liver tissue
(30-50 mg) was lysed with 200 μl immunoprecipitation lysis
buffer on ice for 30 min using immunoprecipitation lysis buffer
(cat. no. BK0004-02; Boyi Bio ACE.). The lysate was then subjected
to centrifugation at 4 × g for 10 min at 4°C. The resulting
supernatant, which contained the protein sample, was mixed with
SOCS2 antibody (1:1,000; A9190; ABclonal Biotech Co., Ltd.) and
incubated overnight at 4°C. Next, 20 μl of Protein A/G
agarose beads were added to the antibody-protein mixture and
incubated for an additional 1 h at room temperature. The complexes
were then separated using magnetic bead-based isolation, followed
by elution and denaturation procedures according to the
manufacturer's instructions (cat. no. BK0004-02; Boyi Bio ACE) and
analyzed by WB.
Flow cytometry
Fresh Raw246.7 cells were washed with 1 ml
pre-cooled PBS at 4°C by pipetting, followed by centrifugation at
1,000-1,500 × g for 5 min at 4°C to collect the cells. The washing
step was repeated twice. A volume of 100 μl 1X binding
buffer (cat. no. G1511-50T; Wuhan Servicebio Technology Co., Ltd.)
working solution was added to the cell pellet to resuspend the
cells. Cells were then mixed thoroughly with 5 μl of Annexin
V-FITC and 5 μl of Propidium Iodide. The mixture was
incubated in the dark at room temperature for 15 min. After
staining and incubation, 400 μl 1X Binding Buffer working
solution was added to each tube, mixed well and samples were
analyzed using a flow cytometer (CytoFLEX; Beckman Coulter, Inc.).
Data were analyzed using FlowJo software (V10.8; BD
Biosciences).
Statistical analysis
Statistical analysis was carried out using GraphPad
Prism 8.0 software (Dotmatics). All data are presented as the mean
± standard deviation. Differences between groups were analyzed
using two-way ANOVA. P<0.05 was considered to indicate a
statistically significant.
Results
Knockout of RP105 exacerbates hepatic
inflammation and injury in the CLP model
To clarify the specific effect of RP105 on the liver
during sepsis, a septic mouse model was induced using CLP and was
used to evaluate the functional status of hepatocytes in the liver.
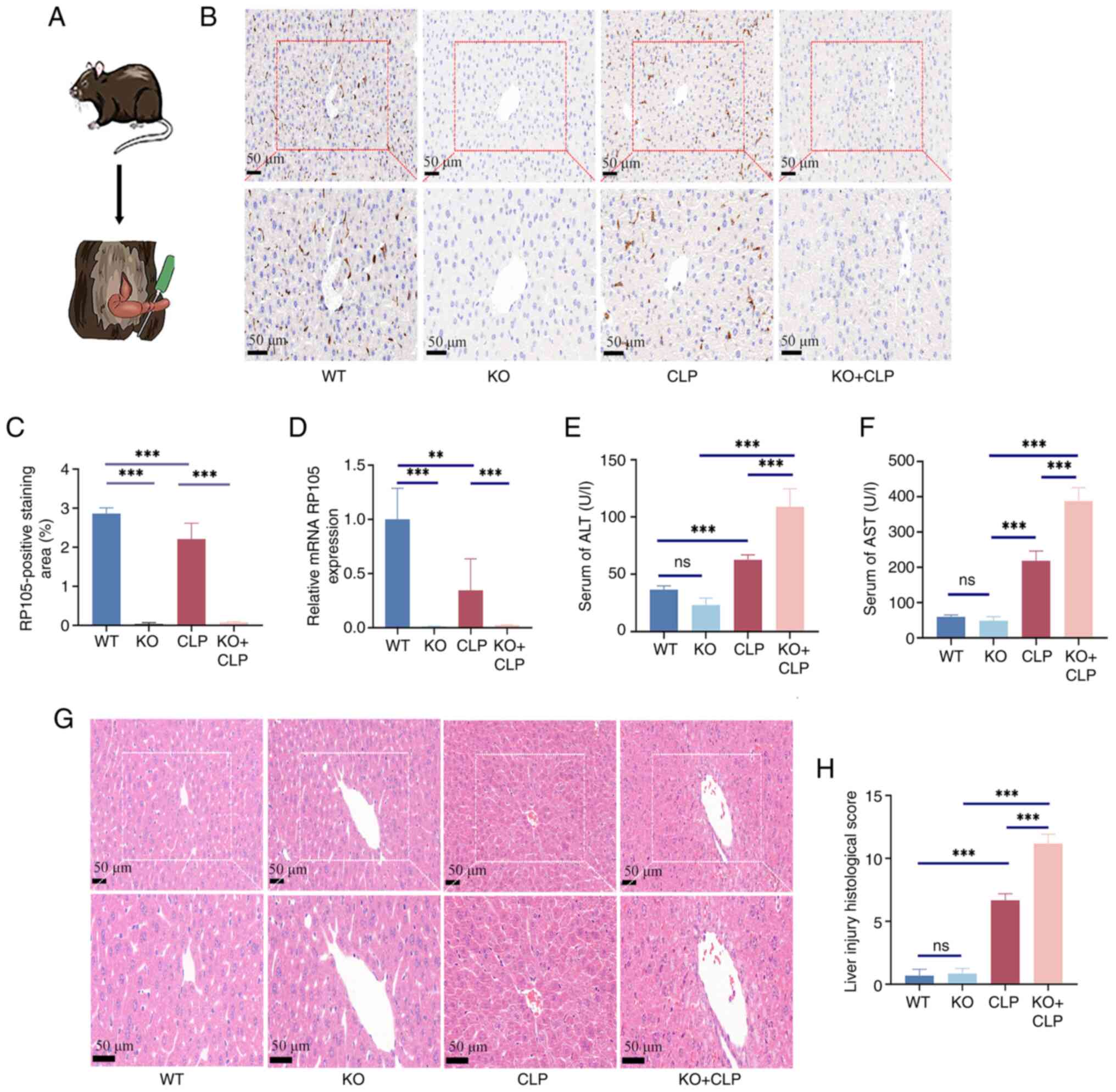
A schematic diagram of the animal experiment is shown in Fig. 1A. The knockout efficiency was
verified in the liver of RP105 KO mice using IHC and qPCR (Fig. 1B-D). Examination of serum ALT and
AST levels, indicators of liver function damage, revealed that ALT
and AST levels were significantly increased in RP105 KO + CLP mice
(ALT, 109.0±15.66; AST, 387.7±37.92) compared with the CLP group
(ALT, 62.7±4.20; AST, 218.1±27.96; Fig. 1E and F). Moreover, the
observations revealed significant pathological changes in the liver
of RP105 KO + CLP mice, including severe edema, sinusoidal
congestion and pronounced hepatocellular necrosis. By contrast,
mice in the CLP group exhibited only mild to moderate edema and
sinusoidal congestion (Fig. 1G and
H). These findings suggest that RP105 carries out a key role in
maintaining normal liver structure and function during sepsis.
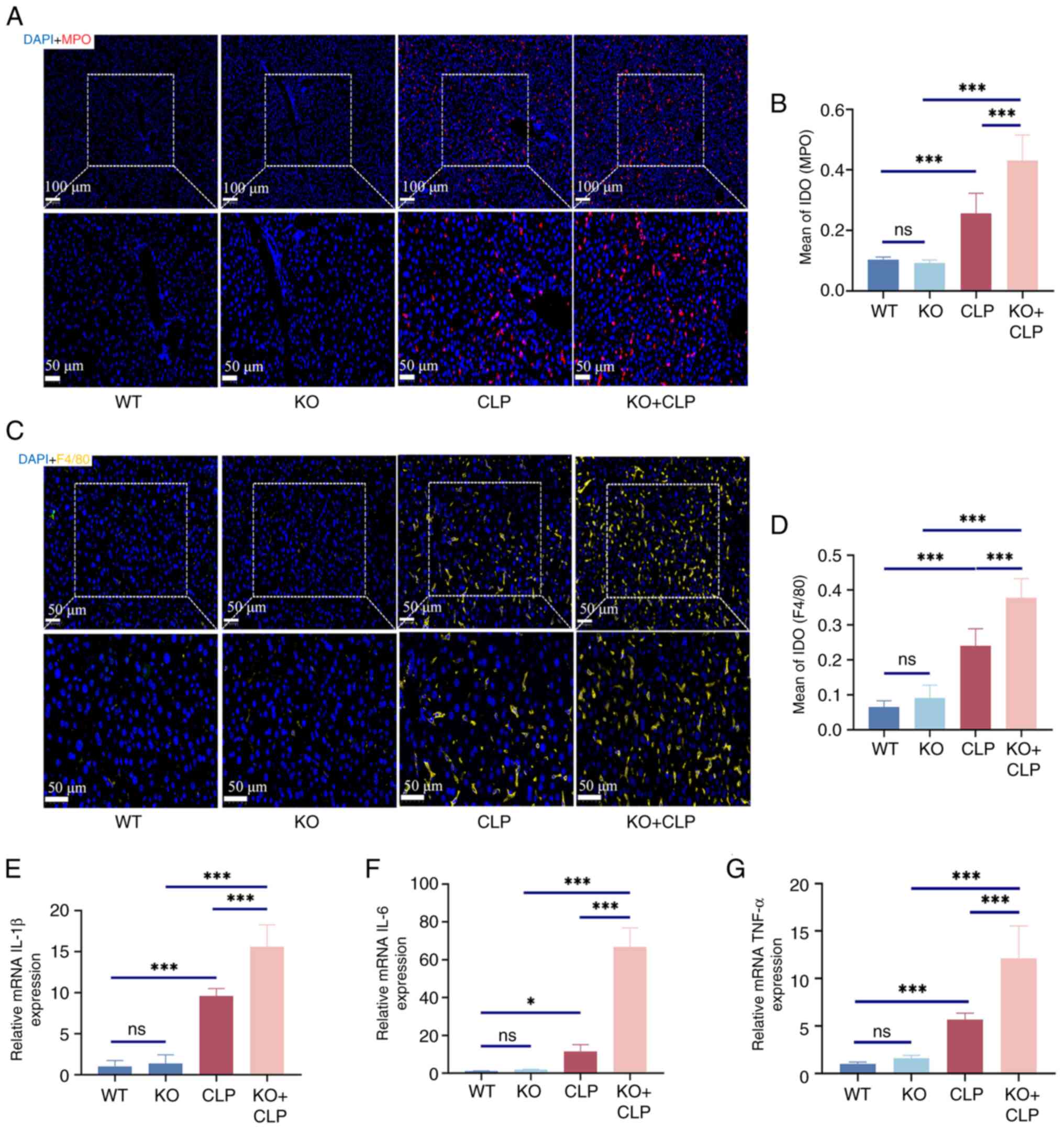
To further demonstrate the role of RP105 in septic
liver injury, additional analysis of inflammatory cell infiltration
was conducted by measuring classic markers of inflammation, MPO and
F4/80. Compared with the CLP group (MPO, 0.2562±0.0663; F4/80,
0.2408±0.0487), a significant increase in the expression levels of
these markers was observed in the liver tissues of RP105 KO mice
(MPO, 0.4306±0.0847; F4/80, 0.3776±0.0552), confirming the
intensified infiltration of inflammatory cells in the liver
(Fig. 2A-D). Furthermore, qPCR
technology was used to assess the expression levels of key
pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α. The
results indicated that, compared with the CLP group (IL-1β,
9.59±0.92; IL-6, 11.51±3.54; TNF-α, 5.70±0.66), RP105 KO + CLP mice
(IL-1β, 15.61±2.66; IL-6, 66.84±9.99; TNF-α, 12.12±3.42) exhibited
significantly elevated levels of pro-inflammatory cytokines
(Fig. 2E-G). These finding
highlights the key role of RP105 in regulating the balance between
inflammatory responses.
Knockout of RP105 aggravates hepatocyte
apoptosis in the CLP model
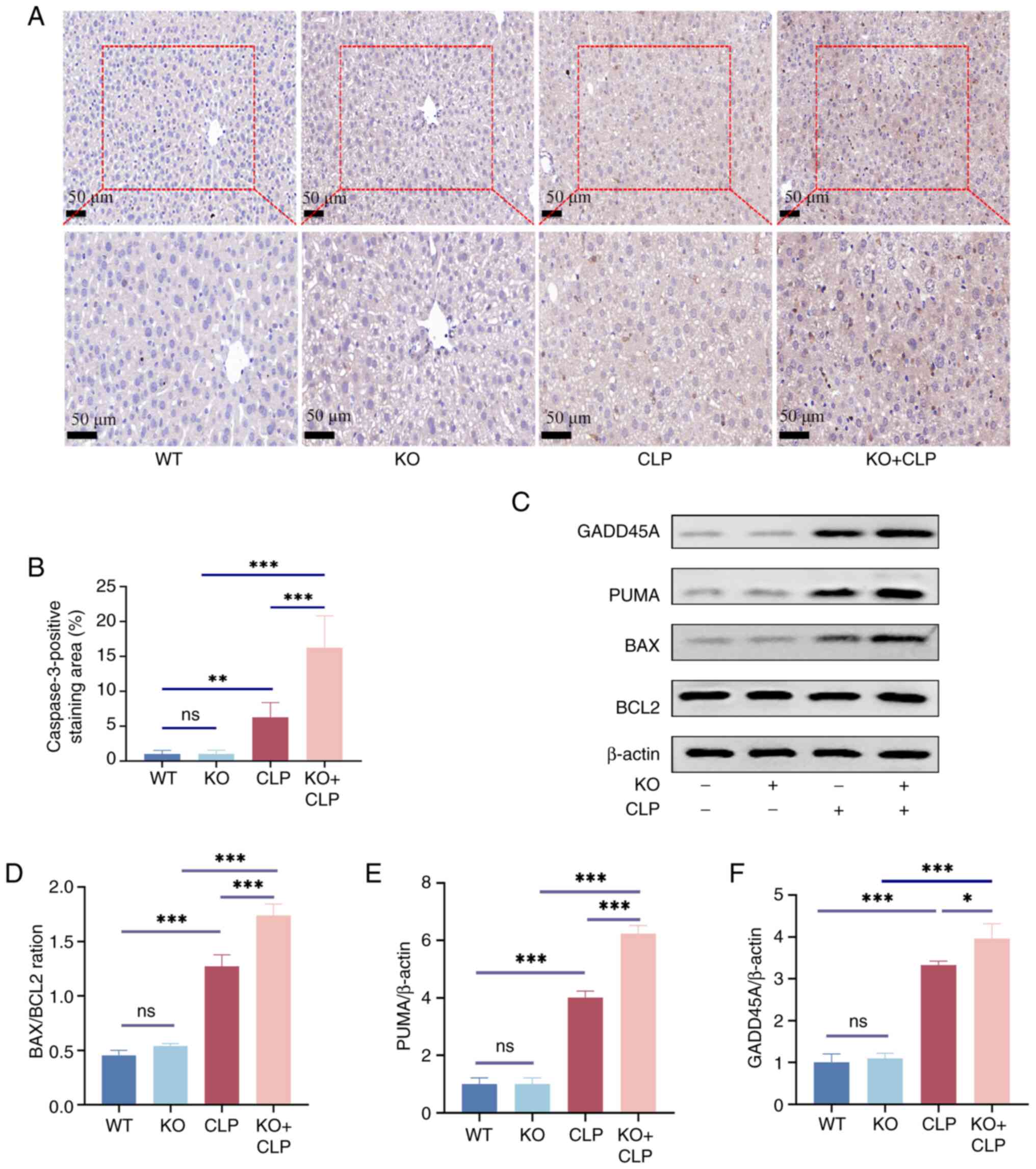
Accordingly, the level of apoptosis in the mouse
liver was measured. Analysis of the IHC experiment revealed that,
compared with the CLP group, there was enhanced expression of
Caspase3 in the RP105 KO + CLP group (Fig. 3A and B). Furthermore, compared to
the CLP group, the ratios of the apoptosis-related proteins
GADD45A, PUMA and BAX to BCL2 were significantly upregulated in the
RP105 KO + CLP group (Fig.
3C-F), further confirming the activation of the apoptotic
signaling pathway. These findings emphasize that, in the context of
sepsis, the absence of RP105 exacerbates apoptosis in the mouse
liver, highlighting the protective role of RP105 against hepatic
damage in an inflammatory condition.
RP105 exhibits the ability to interact
with SOCS2
Following RP105 knockout, the liver function
impairment, inflammatory response and apoptosis observed in the
mouse CLP model were exacerbated. To explore the potential
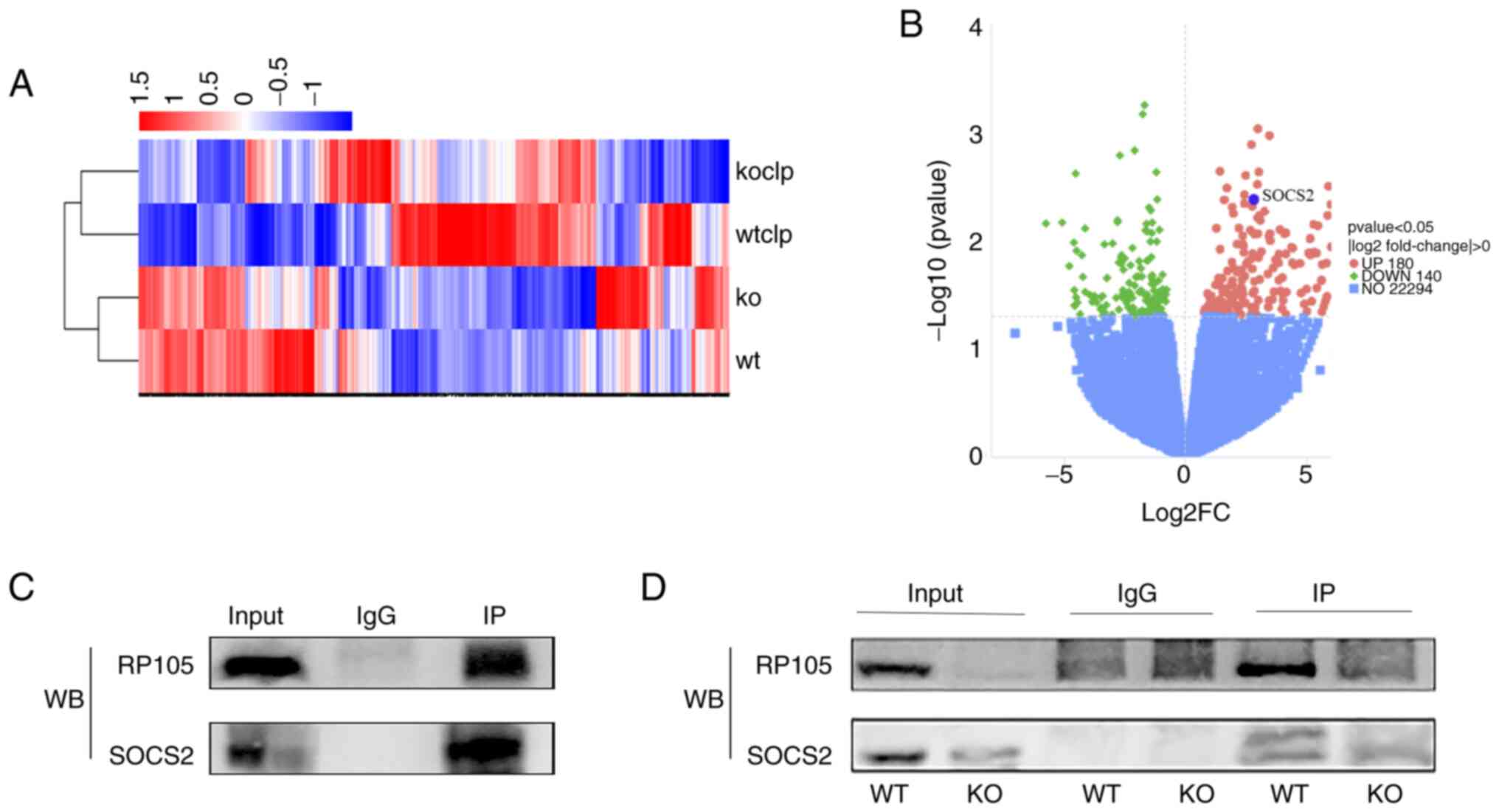
mechanisms of RP105 in this model, three samples from each of the
two groups (CLP and RP105 KO + CLP groups) were randomly selected
and RNA-seq analysis was carried out. To visually represent these
DEGs, heatmaps were created for clustering analysis (Fig. 4A). The volcano plot revealed 320
DEGs between the CLP and RP105 KO + CLP groups, with 180
upregulated and 140 downregulated genes (Fig. 4B). Notably, significant
upregulation of SOCS2 gene expression was observed within the
upregulated gene set in CLP model mice compared with the RP105 KO +
CLP group (Fig. 4A and B), which
warrants further investigation. Current evidence indicates that
SOCS2 carried out a key anti-inflammatory regulatory role. For
instance, SOCS2 suppresses inflammation and apoptosis during
nonalcoholic steatohepatitis progression by restricting NF-κB
activation in macrophages (25).
More importantly, studies have identified SOCS2 as a feedback
inhibitor of TLR-induced activation, capable of specifically
suppressing TLR signaling pathway activation (26,27), a mechanism associated with the
immune function of RP105 (15,28,29). Although RNA-seq analysis
identified multiple DEGs, pathway enrichment analysis and
literature review revealed that other upregulated DEGs revealed
weaker direct associations with inflammatory regulation. Therefore,
SOCS2 was ultimately selected as the core target for subsequent
mechanistic investigations.
To further explore the interplay between RP105 and
SOCS2, Co-IP was carried out. This experiment revealed that SOCS2
specifically binds to RP105 in mouse tissue samples, providing
preliminary evidence for a direct interaction between the two
proteins (Fig. 4C). To
strengthen this conclusion, a comparative Co-IP experiment was
carried out using liver samples from RP105 KO and WT mice. This
approach enabled the direct assessment of the effect of RP105
deficiency on the binding capacity of SOCS2. The experimental
results revealed that the specific binding between SOCS2 and RP105,
observed in the WT mouse liver, completely disappeared following
RP105 knockout (Fig. 4D). This
finding not only corroborates the initial results but also suggests
the existence of an interaction between RP105 and SOCS2.
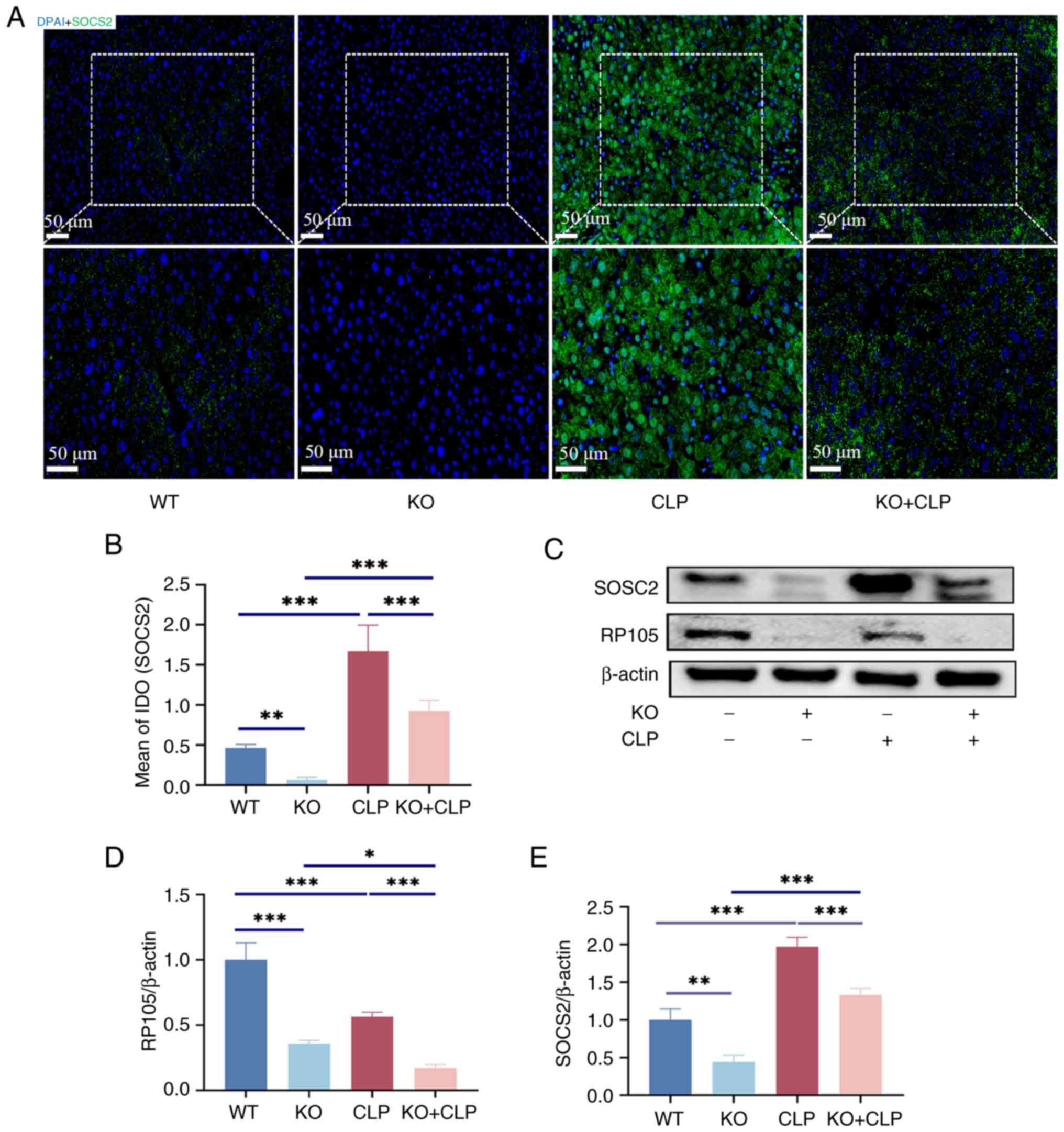
Reduction of SOCS2 expression in sepsis
liver by knockdown of RP105
To validate the RNA-seq analysis, SOCS2 protein
expression levels across four groups: WT, RP105 KO, CLP and RP105
KO + CLP groups as assessed. IHC and WB was employed to provide
direct evidence of the differential expression of SOCS2 under
various treatment conditions (Fig.
5A-E), not only to gain a deeper understanding of the
subcellular localization of the SOCS2 protein, but also to observe
potential alterations in its intracellular distribution pattern
under conditions of sepsis and RP105 knockout. After CLP surgery,
SOCS2 expression was significantly increased in the liver of mice.
Although RP105 expression was reduced in the CLP group when
compared with the WT group, SOCS2 expression was still markedly
elevated, likely due to the activation of SOCS2 expression by
CLP-induced inflammation. However, in the RP105 KO + CLP group,
SOCS2 expression was significantly suppressed. Despite this, SOCS2
expression in the RP105 KO + CLP group was still increased compared
with that in the knockout group (Fig. 5A-E), suggesting that in the
inflammatory environment, SOCS2 expression is not only regulated by
RP105 but may also be influenced by other complex signaling
pathways.
RP105 mediates SOCS2 expression to
hepatic inflammation and injury in the CLP model through the
JAK2/STAT3 pathway
In a murine model of sepsis, RNA-seq analysis
revealed significant differential expression of SOCS2 between RP105
knockout mice and wild-type controls (Fig. 4A and B). SOCS2 serves as a
negative feedback regulator within the complex regulatory network
of cytokine signaling. The classic negative feedback regulatory
signaling pathway involves the activation of the JAK2/STAT3
pathway. Building upon this finding, in-depth functional validation
of the canonical SOCS2-mediated JAK2/STAT3 signaling pathway was
conducted.
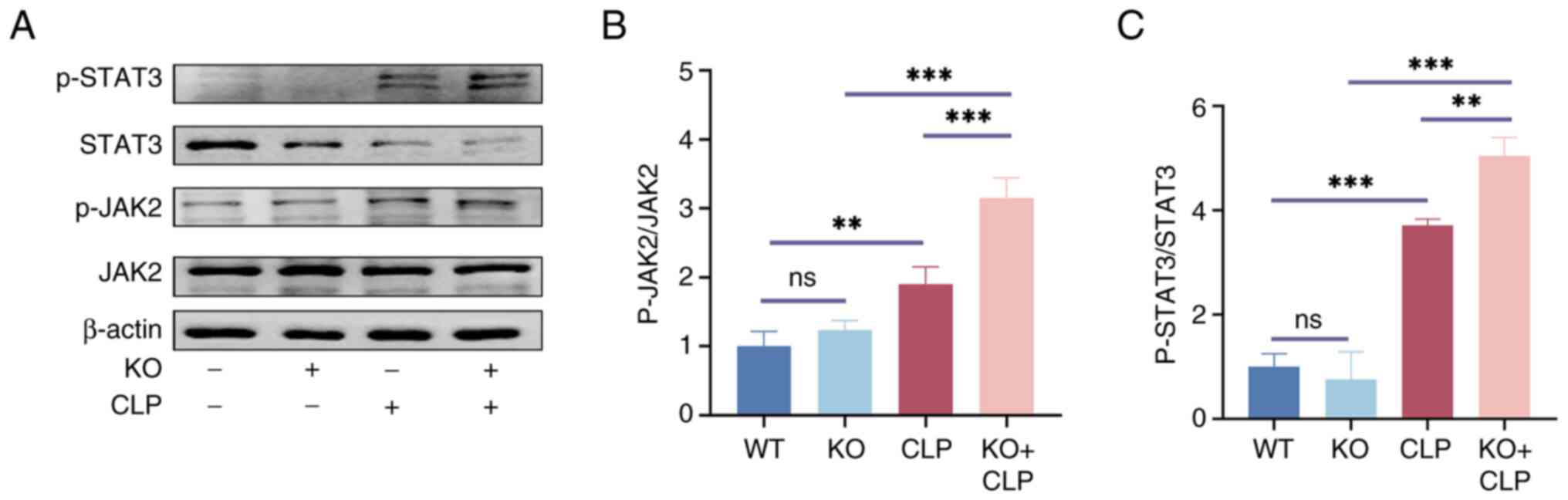
To confirm the function of RP105 and its role in
sepsis-induced liver injury through inactivation of the JAK2/STAT3
pathway via the upregulation of SOCS2, WB experiments were
conducted on liver tissues from the four groups to accurately
assess the expression levels of JAK2, p-JAK2, STAT3 and p-STAT3. WB
indicated that, in the absence of CLP treatment, there were no
significant differences in the basal expression levels of JAK2 and
STAT3 among the groups. However, a remarkable change was observed
following CLP surgery: In the RP105 KO + CLP group, the
phosphorylation levels of JAK2 and STAT3 were significantly
elevated compared with the CLP group (Fig. 6A-C).
RP105 regulates the inf lammatory
response of LPS-induced RAW264.7 macrophages by modulating the
SOCS2/JAK2/STAT3 pathway
To verify in vitro whether RP105 can
ameliorate the inflammatory response in sepsis by regulating the
SOCS2/JAK2/STAT3 pathway, the expression of relevant proteins in
RAW264.7 cells after LPS stimulation were assessed. Flow cytometry
analysis of the cellular model revealed that, compared with normal
cells, RAW264.7 macrophages with RP105 knocked out exhibited a
significant increase in apoptosis rate after LPS stimulation
(Fig. 7A and B). WB revealed
that the changes in protein expression within this pathway were
similar to those observed at the animal level (Fig. 7C-G).
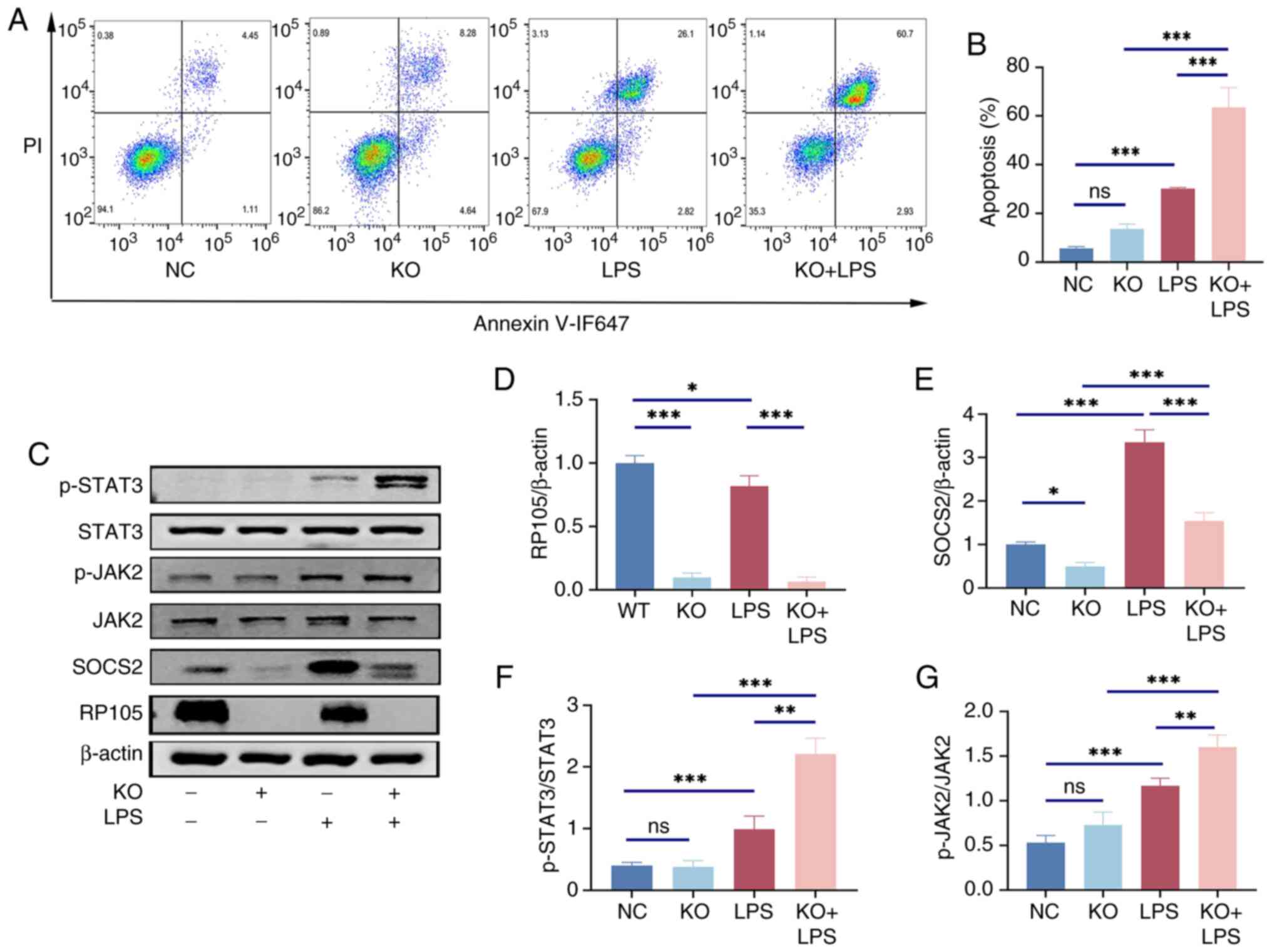 | Figure 7RAW264.7 cells with KO of RP105 gene
expression exhibits exacerbated damage from LPS under the influence
of the JAK2/STAT3 signaling pathway. (A) Representative flow
cytometric images and (B) analysis of apoptosis after LPS treatment
in RAW264.7 cells. (C) Representative western blotting images and
analysis of (D) RP105/β-actin, (E) SOCS2/β-actin, (F) p-JAK2/JAK2
and (G) p-STAT3/STAT3. ns, P>0.05, *P<0.05,
**P<0.01 and ***P<0.001. LPS,
lipopolysaccharide; NC, negative control; KO, knockout; RP105,
radioprotective 105; SOCS2, suppressor of cytokine signaling 2. |
Discussion
Sepsis is a systemic inflammatory response syndrome
triggered by pathogenic microorganisms, such as bacteria (30). When these pathogens invade the
body, they release toxins that activate the immune system of the
host. Excessive release of inflammatory mediators and cytokines
leads to a systemic inflammatory response, which can result in
multiple organ dysfunction (31). Impaired liver function is closely
associated with the prognosis of sepsis and serves as a robust
independent predictor of mortality (32). Understanding the changes in liver
function during sepsis is key to improving the survival rates of
patients with sepsis. The present study selected CLP as an animal
model for sepsis to simulate the septic environment in vivo,
as it is widely regarded as the gold standard for sepsis research
(33-35).
RP105 was initially identified as a surface marker
of B cells in mice and humans (36). It belongs to a class of type I
transmembrane receptors and exhibits substantial structural
similarity to TLRs. Similar to TLR4, RP105 contains abundant LRRs
(10), which are common
structural domains found in several proteins. The LRR domain
carries out a key role in protein-protein interactions through its
specific three-dimensional structure (37), facilitating the recognition and
activation of various immune responses and interactions with
exogenous pathogens (38). By
sensing extracellular stimuli, RP105 acts as a bridge to transmit
signals to the intracellular effectors. As a TLR homolog, RP105
carries out a key role in the negative regulation of inflammation.
Although the beneficial effects of RP105 have been well established
in TLR4-dependent contexts, recent data have also highlighted its
roles in TLR4-independent pathways (19,39,40). RP105 possesses pathways
independent of TLR4 signal transduction and can physically interact
with the PI3K pathway, carrying out a significant role in improving
myocardial ischemia-reperfusion injury through the PI3K/AKT pathway
(41).
Building on the established role of RP105 in the
anti-inflammatory process (14),
the potential of RP105 as a therapeutic target for modulating
septic liver injury was investigated. The study revealed that RP105
is key for protecting liver function. Compared with the control
group, RP105 knockout mice exhibited aggravated liver dysfunction
after CLP treatment, accompanied by relevant histopathological
changes and a more intense inflammatory response. This is
consistent with previous studies that have emphasized the role of
RP105 as a negative regulator of inflammation (13,42-44). Further RNA-Seq results revealed
that among the DEGs, SOCS2 was downregulated in RP105 KO mice.
Co-IP experiments demonstrated that SOCS2 specifically binds to
RP105 in mouse tissues. This was confirmed by comparative Co-IP
using liver samples from RP105 KO and WT mice: The SOCS2-RP105
binding observed in WT liver disappeared after RP105 knockout,
suggesting an interaction.
To validate the results of RNA-seq analysis, an
in-depth assessment of SOCS2 protein expression levels across four
experimental groups was conducted: WT, RP105 KO, CLP and RP105 KO +
CLP. Analysis revealed an apparent paradox in SOCS2 expression:
While CLP surgery alone induced a significant upregulation of SOCS2
in the liver, its expression was markedly reduced in RP105 KO + CLP
mice. This finding suggests that the induction of SOCS2 during
sepsis may require the presence of RP105. One possible explanation
is that under inflammatory stress, RP105 facilitates SOCS2
expression, possibly through a direct interaction, as supported by
the Co-IP results demonstrating specific binding between RP105 and
SOCS2. In the absence of RP105, this regulatory axis may be
disrupted, leading to an insufficient upregulation of SOCS2 in
response to the strong inflammatory stimulus induced by CLP.
Consequently, as shown by the increased phosphorylation of JAK2 and
STAT3 in RP105 KO + CLP mice, the JAK2/STAT3 signaling pathway
becomes hyperactivated, further aggravating liver inflammation and
injury. Together, these data suggest that RP105 is important not
only for maintaining hepatic homeostasis during sepsis but also for
ensuring appropriate SOCS2 induction to restrain cytokine-mediated
damage. Notably, despite the suppression of SOCS2 expression in the
CLP group with RP105 knockout, SOCS2 expression was still increased
in the RP105 KO + CLP group. This finding suggests that in an
inflammatory environment, SOCS2 expression is not only regulated by
RP105 but also is influenced by other complex pathways. For
instance, inflammatory cytokines such as IL-1β may induce SOCS2
expression through alternative signaling pathways (45).
Additionally, in the CLP group, despite a decrease
in RP105 expression compared with the WT group, SOCS2 expression
significantly increased. This may be due to the inflammatory
environment triggered by CLP surgery activating SOCS2 expression,
which may override the regulatory effect of RP105 on SOCS2.
However, this also raises the possibility that RP105 is not the
sole upstream regulator of SOCS2 and inflammatory cues such as
IL-1β or IL-6 may independently enhance SOCS2 expression through
STAT-dependent transcription. Such a compensatory induction, though
present, appears insufficient to suppress downstream signaling in
the absence of RP105. In summary, the present study not only
validates the results of RNA-seq analysis but also uncovers the
complex regulatory mechanisms of SOCS2 in inflammatory responses
and the pivotal role of RP105 in this process. These observations
suggest that the RP105-SOCS2 interaction may serve as a regulatory
checkpoint that constrains excessive activation of the JAK2/STAT3
axis during septic liver injury. The disruption of this checkpoint
in RP105-deficient conditions may contribute to signal
amplification, cytokine storm and hepatocyte apoptosis. These
findings provide new insights into the understanding of the
inflammatory responses and the search for potential therapeutic
targets.
SOCS2, operates within classic feedback loops by
targeting the degradation of signaling intermediates, thereby
blocking further signal transduction (46). Imbalances in these proteins can
lead to a wide range of pathological changes, highlighting the
essential role of SOCS2 in regulating cell fate and inflammatory
processes (47). As a pivotal
signaling protein, SOCS2 exhibits diverse functions across various
cell types (48,49), underscoring its importance.
Notably, SOCS2 carries out a key role in modulating immune
responses and inflammatory pathways, which are vital for
maintaining organismal health (50). Previous research (25) has reported that SOCS2 in
macrophages exerts anti-inflammatory and anti-apoptotic effects by
modulating the NF-κB signaling pathway, while also attenuating
inflammation through the inhibition of the inflammasome signaling
pathway. Additionally, SOCS2 has been revealed to be vital in
balancing immune responses and protecting against oxidative stress
during acetaminophen-induced acute liver injury, acting as a key
regulatory factor in liver immune responses following acetaminophen
therapy (51).
SOCS2 serves as a negative feedback regulator within
the complex regulatory network of cytokine signaling. By
interacting precisely with core molecules in the signaling complex,
it effectively blocks further extension of the signaling chain,
achieving negative feedback regulation of the cytokine signaling
pathway. A classic example of this regulatory mechanism is the
activation of the JAK/STAT pathway. During inflammation, SOCS2
forms complexes with key proteins in the JAK/STAT pathway, thereby
effectively inhibiting the amplification and transmission of
inflammatory signals. This ensures timely termination and precise
regulation of cellular responses to inflammatory factors, thus
inhibiting inflammatory reactions. We hypothesize that the
JAK2/STAT3 pathway carries out a key role in protecting against
liver injury due to sepsis mediated by RP105 through SOCS2. In the
present study, we found that the phosphorylation levels of
JAK2/STAT3 in the liver tissues of RP105 KO mice subjected to the
CLP model and in RAW264.7 macrophages treated with LPS were both
increased compared with those in the control groups. This
consistent activation across in vivo and in vitro
systems suggests that SOCS2 carries out a non-redundant role in
restraining this pathway, and its suppression in the absence of
RP105 may permit uncontrolled signaling propagation. These findings
suggest that RP105 mitigates the progression of sepsis-induced
liver injury through the inactivation of the JAK2/STAT3 pathway
mediated by SOCS2.
Based on the aforementioned research findings, we
hypothesize that intervention methods targeting the enhancement of
the RP105-SOCS2 interaction can be designed to evaluate their
intervention effects in the early stage of septic liver injury.
Additionally, through prospective cohort studies, the levels of
RP105 and SOCS2 in patients with sepsis at the time of admission
can be detected, the associations between these levels and liver
injury grading as well as treatment efficacy can be analyzed, and a
combined prediction model based on their expression levels can be
established, which will provide reference indicators for early
clinical intervention.
While the present study provides insights into the
role of RP105 in sepsis pathogenesis using murine models, it is
essential to recognize the inherent limitations in translating
these findings to human diseases due to fundamental differences in
immune regulation and pathophysiology between species. Although
murine models are indispensable for mechanistic investigations,
they cannot fully replicate the complexity of human sepsis, as
evidenced by significant interspecies variations in TLR signaling
pathways, cytokine networks and cellular immune responses.
RP105 protects against septic liver injury by
binding SOCS2 to inhibit JAK2/STAT3 signaling, thereby attenuating
inflammation and apoptosis. The study is the first to demonstrate
the RP105-SOCS2 interaction in septic liver injury, revealing the
RP105/SOCS2 axis as a potential therapeutic target.
Availability of data and materials
The data generated in the present study may be found
in the GEO database under accession number GSE298411 or at the
following URL: https://www.ncbi.nlm.nih.gov/gds/?term=GSE298411.
Authors' contributions
QD designed cell and animal experiments, curated
data, developed methodology, conducted formal analysis and wrote
the original draft. HD and QY curated data, conducted formal and
software analysis and developed methodology. RC, ZF and JX
collected the information and revised, conducted analyses using
software tools and finalized the manuscript. HP and QX proposed the
preliminary idea and design for the present study and reviewed and
revised the manuscript. QD and QX confirm the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee for Laboratory Animal Welfare of the First Affiliated
Hospital of Nanchang University (approval no.
CDYFY-IACUC-202407QR198).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 82460131 and 82060122) and Natural
Science Foundation of JiangXi province (grant no.
20224ACB206027).
References
|
1
|
Rudd KE, Johnson SC, Agesa KM, Shackelford
KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer
S, et al: Global, regional, and national sepsis incidence and
mortality, 1990-2017: Analysis for the global burden of disease
study. Lancet. 395:200–211. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Takeuchi O and Akira S: Pattern
recognition receptors and inflammation. Cell. 140:805–820. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Raymond SL, Holden DC, Mira JC, Stortz JA,
Loftus TJ, Mohr AM, Moldawer LL, Moore FA, Larson SD and Efron PA:
Microbial recognition and danger signals in sepsis and trauma.
Biochim Biophys Acta Mol Basis Dis. 1863:2564–2573. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fajgenbaum DC and June CH: Cytokine storm.
N Engl J Med. 383:2255–2273. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chousterman BG, Swirski FK and Weber GF:
Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol.
39:517–528. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Protzer U, Maini MK and Knolle PA: Living
in the liver: Hepatic infections. Nat Rev Immunol. 12:201–213.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heymann F and Tacke F: Immunology in the
liver-from homeostasis to disease. Nat Rev Gastroenterol Hepatol.
13:88–110. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yan J and Li S and Li S: The role of the
liver in sepsis. Int Rev Immunol. 33:498–510. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Elmi AN and Kwo PY: The liver in sepsis.
Clin Liver Dis. 29:453–467. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miyake K, Yamashita Y, Ogata M, Sudo T and
Kimoto M: RP105, a novel B cell surface molecule implicated in B
cell activation, is a member of the leucine-rich repeat protein
family. J Immunol. 154:3333–3340. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Divanovic S, Trompette A, Atabani SF,
Madan R, Golenbock DT, Visintin A, Finberg RW, Tarakhovsky A, Vogel
SN, Belkaid Y, et al: Negative regulation of Toll-like receptor 4
signaling by the Toll-like receptor homolog RP105. Nat Immunol.
6:571–578. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schultz TE and Blumenthal A: The
RP105/MD-1 complex: Molecular signaling mechanisms and
pathophysiological implications. J Leukoc Biol. 101:183–192. 2017.
View Article : Google Scholar
|
|
13
|
Yang J, Yang C, Yang J, Ding J, Li X, Yu
Q, Guo X, Fan Z and Wang H: RP105 alleviates myocardial ischemia
reperfusion injury via inhibiting TLR4/TRIF signaling pathways. Int
J Mol Med. 41:3287–3295. 2018.PubMed/NCBI
|
|
14
|
Fan Z, Pathak JL and Ge L: The potential
role of RP105 in regulation of inflammation and osteoclastogenesis
during inflammatory diseases. Front Cell Dev Biol. 9:7132542021.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu J, Zhang Y, Shi L, Xia Y, Zha H, Li H
and Song Z: RP105 protects against ischemic and septic acute kidney
injury via suppressing TLR4/NF-κB signaling pathways. Int
Immunopharmacol. 109:1089042022. View Article : Google Scholar
|
|
16
|
Wezel A, de Vries MR, Maassen JM, Kip P,
Peters EA, Karper JC, Kuiper J, Bot I and Quax PHA: Deficiency of
the TLR4 analogue RP105 aggravates vein graft disease by inducing a
pro-inflammatory response. Sci Rep. 6:242482016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen F, Xu W, Tang M, Tian Y, Shu Y, He X,
Zhou L, Liu Q, Zhu Q, Lu X, et al: hnRNPA2B1 deacetylation by SIRT6
restrains local transcription and safeguards genome stability. Cell
Death Differ. 32:382–396. 2025. View Article : Google Scholar
|
|
18
|
Li T, Sun H, Li Y, Su L, Jiang J, Liu Y,
Jiang N, Huang R, Zhang J and Peng Z: Downregulation of macrophage
migration inhibitory factor attenuates NLRP3 inflammasome mediated
pyroptosis in sepsis-induced AKI. Cell Death Discov. 8:612022.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Duo H, Yang Y, Luo J, Cao Y, Liu Q, Zhang
J, Du S, You J, Zhang G, Ye Q and Pan H: Modulatory role of
radioprotective 105 in mitigating oxidative stress and ferroptosis
via the HO-1/SLC7A11/GPX4 axis in sepsis-mediated renal injury.
Cell Death Discov. 11:2902025. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kollias NS, Hess WJ, Johnson CL, Murphy M
and Golab G: A literature review on current practices, knowledge,
and viewpoints on pentobarbital euthanasia performed by
veterinarians and animal remains disposal in the United States. J
Am Vet Med Assoc. 261:733–738. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gong S, Yan Z, Liu Z, Niu M, Fang H, Li N,
Huang C, Li L, Chen G, Luo H, et al: Intestinal microbiota mediates
the susceptibility to polymicrobial sepsis-induced liver injury by
granisetron generation in mice. Hepatology. 69:1751–1767. 2019.
View Article : Google Scholar
|
|
22
|
Liang H, Song H, Zhang X, Song G, Wang Y,
Ding X, Duan X, Li L, Sun T and Kan Q: Metformin attenuated
sepsis-related liver injury by modulating gut microbiota. Emerg
Microbes Infect. 11:815–828. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Zhong X, Xiao Q, Liu Z, Wang W, Lai CH,
Yang W, Yue P, Ye Q and Xiao J: TAK242 suppresses the TLR4
signaling pathway and ameliorates DCD liver IRI in rats. Mol Med
Rep. 20:2101–2110. 2019.PubMed/NCBI
|
|
25
|
Li S, Han S, Jin K, Yu T, Chen H, Zhou X,
Tan Z and Zhang G: SOCS2 suppresses inflammation and apoptosis
during NASH progression through limiting NF-κB activation in
macrophages. Int J Biol Sci. 17:4165–4175. 2021. View Article : Google Scholar :
|
|
26
|
Posselt G, Schwarz H, Duschl A and
Horejs-Hoeck J: Suppressor of cytokine signaling 2 is a feedback
inhibitor of TLR-induced activation in human monocyte-derived
dendritic cells. J Immunol. 187:2875–2884. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baetz A, Frey M, Heeg K and Dalpke AH:
Suppressor of cytokine signaling (SOCS) proteins indirectly
regulate toll-like receptor signaling in innate immune cells. J
Biol Chem. 279:54708–54715. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang J, Zeng P, Yang J and Fan ZX: The
role of RP105 in cardiovascular disease through regulating TLR4 and
PI3K signaling pathways. Curr Med Sci. 39:185–189. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Divanovic S, Trompette A, Petiniot LK,
Allen JL, Flick LM, Belkaid Y, Madan R, Haky JJ and Karp CL:
Regulation of TLR4 signaling and the host interface with pathogens
and danger: The role of RP105. J Leukoc Biol. 82:265–271. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cecconi M, Evans L, Levy M and Rhodes A:
Sepsis and septic shock. Lancet. 392:75–87. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Recknagel P, Gonnert FA, Westermann M,
Lambeck S, Lupp A, Rudiger A, Dyson A, Carré JE, Kortgen A, Krafft
C, et al: Liver dysfunction and phosphatidylinositol-3-kinase
signalling in early sepsis: Experimental studies in rodent models
of peritonitis. PLoS Med. 9:e10013382012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bastarache JA and Matthay MA: Cecal
ligation model of sepsis in mice: New insights. Crit Care Med.
41:356–357. 2013. View Article : Google Scholar :
|
|
34
|
Dejager L, Pinheiro I, Dejonckheere E and
Libert C: Cecal ligation and puncture: The gold standard model for
polymicrobial sepsis? Trends Microbiol. 19:198–208. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rittirsch D, Huber-Lang MS, Flierl MA and
Ward PA: Immunodesign of experimental sepsis by cecal ligation and
puncture. Nat Protoc. 4:31–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Miyake K, Yamashita Y, Hitoshi Y, Takatsu
K and Kimoto M: Murine B cell proliferation and protection from
apoptosis with an antibody against a 105-kD molecule:
Unresponsiveness of X-linked immunodeficient B cells. J Exp Med.
180:1217–1224. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Miura Y, Shimazu R, Miyake K, Akashi S,
Ogata H, Yamashita Y, Narisawa Y and Kimoto M: RP105 is associated
with MD-1 and transmits an activation signal in human B cells.
Blood. 92:2815–2822. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ishii A, Matsuo A, Sawa H, Tsujita T,
Shida K, Matsumoto M and Seya T: Lamprey TLRs with properties
distinct from those of the variable lymphocyte receptors. J
Immunol. 178:397–406. 2007. View Article : Google Scholar
|
|
39
|
Yang J, Zhai Y, Huang C, Xiang Z, Liu H,
Wu J, Huang Y, Liu L, Li W, Wang W, et al: RP105 attenuates
ischemia/reperfusion-induced oxidative stress in the myocardium via
activation of the Lyn/Syk/STAT3 signaling pathway. Inflammation.
47:1371–1385. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guo X, Hu S, Liu JJ, Huang L, Zhong P, Fan
ZX, Ye P and Chen MH: Piperine protects against pyroptosis in
myocardial ischaemia/reperfusion injury by regulating the
miR-383/RP105/AKT signalling pathway. J Cell Mol Med. 25:244–258.
2021. View Article : Google Scholar
|
|
41
|
Guo X, Jiang H and Chen J: RP105-PI3K-Akt
axis: A potential therapeutic approach for ameliorating myocardial
ischemia/reperfusion injury. Int J Cardiol. 206:95–96. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu B, Zhang N, Liu Z, Fu Y, Feng S, Wang
S, Cao Y, Li D, Liang D, Li F, et al: RP105 involved in activation
of mouse macrophages via TLR2 and TLR4 signaling. Mol Cell Biochem.
378:183–193. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang W and Yang J, He C and Yang J: RP105
plays a cardioprotective role in myocardial ischemia reperfusion
injury by regulating the Toll-like receptor 2/4 signaling pathways.
Mol Med Rep. 22:1373–1381. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sun Y, Liu L, Yuan J, Sun Q, Wang N and
Wang Y: RP105 protects PC12 cells from oxygen-glucose
deprivation/reoxygenation injury via activation of the PI3K/AKT
signaling pathway. Int J Mol Med. 41:3081–3089. 2018.PubMed/NCBI
|
|
45
|
Sarajlic M, Neuper T, Föhrenbach Quiroz
KT, Michelini S, Vetter J, Schaller S and Horejs-Hoeck J: IL-1β
induces SOCS2 expression in human dendritic cells. Int J Mol Sci.
20:59312019. View Article : Google Scholar
|
|
46
|
Krebs DL and Hilton DJ: SOCS proteins:
Negative regulators of cytokine signaling. Stem Cells. 19:378–387.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Elliott J and Johnston JA: SOCS: Role in
inflammation, allergy and homeostasis. Trends Immunol. 25:434–440.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Keating N and Nicholson SE: SOCS-mediated
immunomodulation of natural killer cells. Cytokine. 118:64–70.
2019. View Article : Google Scholar
|
|
49
|
Hu J, Winqvist O, Flores-Morales A,
Wikström AC and Norstedt G: SOCS2 influences LPS induced human
monocyte-derived dendritic cell maturation. PLoS One. 4:e71782009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang D, Pan A, Gu J, Liao R, Chen X and
Xu Z: Upregulation of miR-144-3p alleviates Doxorubicin-induced
heart failure and cardiomyocytes apoptosis via SOCS2/PI3K/AKT axis.
Chem Biol Drug Des. 101:24–39. 2023. View Article : Google Scholar
|
|
51
|
Monti-Rocha R, Cramer A, Gaio Leite P,
Antunes MM, Pereira RVS, Barroso A, Queiroz-Junior CM, David BA,
Teixeira MM, Menezes GB and Machado FS: SOCS2 is critical for the
balancing of immune response and oxidate stress protecting against
acetaminophen-induced acute liver injury. Front Immunol.
9:31342018. View Article : Google Scholar
|