Introduction
As reported in the Diabetes Atlas (11th Edition)
published by the International Diabetes Federation (2025), 589
million adults around the world are estimated to be living with
diabetes mellitus (DM), with forecasts suggesting that this figure
could increase to 853 million by 2050 (1). DM is a metabolic condition that
results from an interplay of genetic and environmental influences,
which can lead to inadequate insulin production and/or impaired
insulin function. The condition is primarily manifested by
dysregulation in the metabolism of carbohydrates, proteins and
fats, and clinically presents as chronic hyperglycemia (2). Until now, the full causes and
development of DM have yet to be fully understood. DM is mainly
divided into four types: Type 1 DM (T1DM), T2DM, gestational DM and
other specific diabetes types not previously enumerated. Among
these, T2DM is the most common type, representing ~90% of all
diabetes cases (3); thus, it is
the core focus of the present review. T2DM is caused by genetic
susceptibility and environmental risk factors, which lead to the
inability of β-cells to produce sufficient insulin or the poor
effectiveness of insulin (4,5).
In terms of genetic factors, T2DM exhibits a significant family
aggregation pattern. At present, a number of susceptibility genes
(such as transcription factor 7 like 2, peroxisome
proliferator-activated receptor γ and potassium inwardly rectifying
channel subfamily J member 11) have been identified, which can
affect an individual's susceptibility to the disease by regulating
processes including insulin secretion, glucose transport and lipid
metabolism (6). Among the
environmental factors, obesity, a high-calorie diet, lack of
physical activity and age are the main factors for T2DM.
Additionally, patients with hypertension or dyslipidemia have an
increased risk of developing T2DM. Moreover, T2DM can result in
chronic metabolic disorders accompanied by multi-system
complications, which may lead to the onset of eye disease, kidney
disease, cardiac disease, vascular disease and dysfunction of the
central nervous system (7).
The present treatment plan for T2DM involves a
comprehensive strategy that includes considerable lifestyle changes
along with medication interventions. Pharmacological agents mainly
consist of oral hypoglycemic medications and insulin formulations.
Oral hypoglycemics encompass various medications, including
traditional drugs such as metformin, α-glucosidase inhibitors,
glinides, sulfonylureas and thiazolidinediones, and newer agents
such as dipeptidyl peptidase-4 inhibitors. Traditional oral
hypoglycemic medications have been demonstrated to improve glycemic
management and decrease the likelihood of complications and
mortality associated with T2DM. However, their effectiveness across
various organ systems, particularly the cardiovascular and renal
systems, is restricted, and they come with specific side effects
(7). For instance, α-glucosidase
inhibitors cause various side effects including abdominal
discomfort, bloating, diarrhea, pain and flatulence (8). Hypoglycemia is the main side effect
of all sulfonylurea drugs, while minor side effects such as
headache, dizziness, nausea, hypersensitivity reactions and weight
gain are also common (9). As a
result, reliance on these medications has decreased in preference
for newer therapies, such as insulin pumps, sodium-glucose
cotransporter-2 inhibitors and glucagon-like peptide-1 (GLP-1)
receptor agonists, which have demonstrated notable efficacy.
However, the percentage of patients achieving well-controlled T2DM
has not risen as expected (10).
Insulin treatment encompasses a variety of insulin types, such as
basal insulin and premixed (or biphasic) insulin analogs. Commonly
utilized basal insulins include neutral protamine hagedorn insulin,
insulin glargine in U100 or U300 formulations and detemir (11). At the same time, lifestyle
changes, which include dietary adjustments and regular exercise,
are essential for the successful management of T2DM and its related
complications (12). However,
clinical data show that the proportion of T2DM patients with
well-controlled blood glucose has not met expectations, suggesting
that there is still room for optimization in existing treatment
regimens (13).
β-hydroxybutyric acid (β-HB) is the most abundant
ketone body (KB) in the human body, accounting for ~70% of the
circulating KBs (14). β-HB
serves as an efficient energy carrier from the liver to peripheral
tissues and it can act as a crucial alternative energy source,
especially when glucose supply for energy production is
insufficient. The maintenance of its concentration in the body
mainly relies on two pathways: i) Endogenous production when the
body is in a state of insufficient glucose supply for energy such
as prolonged starvation, ketogenic diet (KD) with a
low-carbohydrate and high-fat ratio and after strenuous exercise
(15); ii) exogenous ketone
supplement. Exogenous supplementation can rapidly increase the
concentration of β-HB in the circulation (16). Recent studies have shown that
β-HB is not merely a metabolite; it also possesses important
cellular signaling functions as it can link changes in the external
environment to cellular functions and gene expression by regulating
key intracellular pathways. Specifically, studies have confirmed
that β-HB plays an important role in the pathogenesis and
therapeutic management of diseases such as aging (17), intestinal diseases (18), liver diseases (19), septicemia (20), obesity (21) and T2DM (22). Particularly in the field of T2DM,
β-HB can participate in blood glucose regulation and insulin
resistance (IR) by influencing the physiological functions of
various organ systems including the liver (23), kidney (24) and adipose tissue (25). However, comprehensive reports
discussing and summarizing these effects are lacking.
In the present review, a comprehensive search in
major databases [PubMed (https://pubmed.ncbi.nlm.nih.gov), Google Scholar
(https://scholar.google.com) and Web of
Science (https://www.webofscience.com)] up to
June 2025 was conducted using the keywords 'β-HB', 'KB', 'β-HB and
T2DM', 'KB and T2DM', 'β-HB and T2DM complications' and 'KB and
T2DM complications'. Subsequently, the retrieved articles were
screened by reading them one by one to exclude irrelevant articles.
The present review aimed to clarify the metabolic pathways
associated with β-HB, examine its effectiveness and mechanisms of
action concerning T2DM and its related complications, to analyze
the potential value of endogenous and exogenous ketogenesis methods
in increasing β-HB levels as an adjuvant nutritional therapy for
T2DM and to provide a reference for subsequent research and
clinical translation in this field.
Properties of β-HB
Structure of β-HB
β-HB is also known as 3-hydroxybutyric acid or
D-3-hydroxybutyrate and has a molecular formula of
C4H8O3 and a molecular weight of
104 Da. β-HB has two enantiomers: D-β-HB and L-β-HB. In the human
body, endogenously produced β-HB in the liver is predominantly in
the form of D-β-HB, while L-β-HB is a byproduct generated by
certain tissues under ketotic conditions, and its proportion in
serum is typically extremely low (26). Compared with D-β-HB, the
oxidative metabolism efficiency of L-β-HB is significantly lower.
The key enzyme responsible for catalyzing the metabolism of β-HB,
namely D-β-hydroxybutyrate dehydrogenase 1 (BDH1), exhibits
stereoselectivity; it preferentially catalyzes only the reaction
between D-β-HB and acetoacetic acid (ACAC). By contrast, L-β-HB
cannot be effectively oxidized by BDH1, resulting in a longer
half-life and slower clearance rate in the blood, cells and tissues
(27). This notable difference
in metabolic utilization efficiency underscores the importance of
distinguishing between these enantiomers when considering the
signaling functions and therapeutic applications of β-HB.
Metabolic pathway of β-HB Anabolism of
β-HB
β-HB is synthesized from acetyl coenzyme (CoA),
which is derived from the β-oxidation of free fatty acids (FFAs).
This procedure is facilitated by several enzymes located in the
mitochondria of the liver (Fig.
1). The β-oxidation of FFAs yields acetyl CoA, with two acetyl
CoA molecules being combined to form acetoacetyl CoA through the
action of acetoacetyl CoA sulfatase, releasing one molecule of
coenzyme A (CoASH). Afterwards, the enzyme 3-hydroxyglutaryl-CoA
(HMG-CoA) synthase catalyzes the reaction between acetoacetyl CoA
and an additional acetyl CoA molecule, resulting in the production
of HMG-CoA and the release of another CoA molecule. HMG-CoA is then
cleaved by 3-hydroxymethylglutaryl-CoA lyase to produce ACAC and
acetyl CoA. BDH1 catalyzes the reduction of ACAC to β-HB by using
nicotinamide adenine dinucleotide (NADH), while a fraction of ACAC
is transformed into acetone. Therefore, ACAC, acetone and β-HB are
collectively referred to as KBs (28).
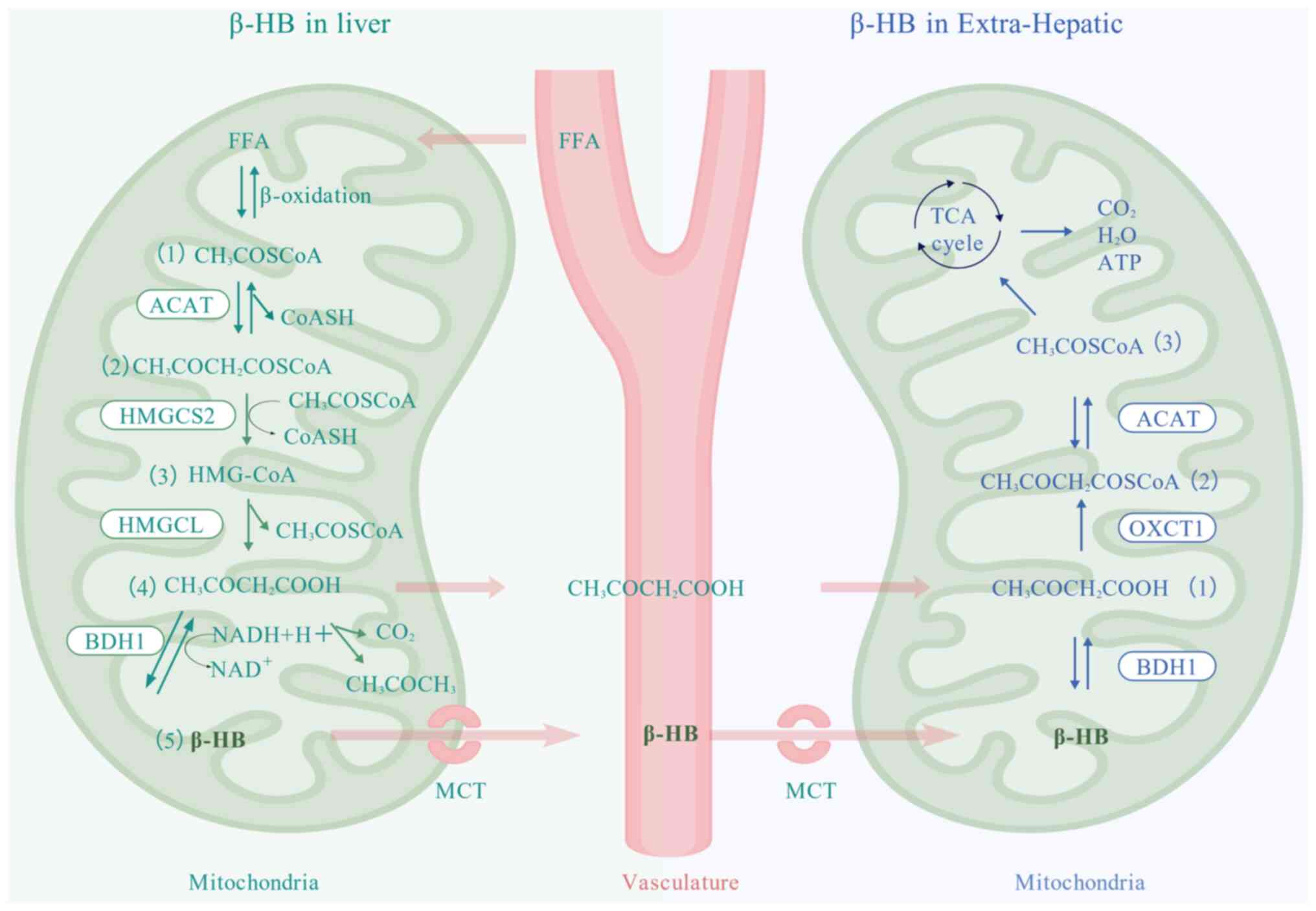 | Figure 1Production of β-HB by the liver and
the breakdown of β-HB by extrahepatic tissues. β-HB is synthesized
through the following steps: i) The β-oxidation of FFAs results in
the production of a substantial amount of acetyl CoA
(CH3COSCoA) in the mitochondria of the liver; ii) the
condensation of two molecules of acetyl CoA (CH3COSCoA)
into acetoacetyl CoA (CH3COCH2COSCoA) is
catalyzed by HMGCS2 with the release of one molecule of CoASH; iii)
the condensation of acetoacetyl CoA
(CH3COCH2COSCoA) with another molecule of
acetyl CoA (CH3COSCoA) forms HMG-CoA, catalyzed by
HMGCL, releasing an additional molecule of CoASH; iv) HMG-CoA is
then cleaved by HMG-CoA lyase to produce ACAC
(CH3COCH2COOH) and acetyl CoA; and v) The
reduction of ACAC (CH3COCH2COOH) to β-HB is
mediated by BDH1, utilizing NADH as the hydrogen donor. A minor
fraction of ACAC is converted to acetone
(CH3COCH3). β-HB is transported by MCTs into
the vasculature into the circulatory system and eventually into
extrahepatic tissues. The catabolism of β-HB: i) β-HB is
dehydrogenated to ACAC (CH3COCH2COOH) in the
mitochondria of extrahepatic tissues, which is catalyzed by BDH1;
ii) ACAC (CH3COCH2COOH) is subsequently
converted to acetoacetyl CoA (CH3COCH2COSCoA)
by OXCT1; and iii) acetoacetyl CoA (CH3COCH2COSCoA) is catalyzed by
ACAT to become acetyl CoA, which then enters the TCA cycle for
complete oxidative decomposition to CO2, H2O
and release of ATP. FFA, free fatty acids; ACAT, acetyl-CoA
acetyltransferase; HMGCS2, 3-hydroxymethylglutaryl-CoA synthase 2;
HMGCL, 3-hydroxymethylglutaryl-CoA lyase; BDH1, β-hydroxybutyrate
dehydrogenase 1; NAD, nicotinamide adenine dinucleotide; β-HB,
β-hydroxybutyric acid; TCA, tricarboxylic acid; ATP, adenosine
triphosphate; OXCT1, 3-oxoacid CoA transferase 1; MCTs,
monocarboxylate transporters; ACAC, acetoacetate; CoA, coenzyme
A. |
Traditionally, β-HB synthesis is considered to be
exclusive to the liver due to the hepatic specificity of the key
ketogenesis enzyme, HMG-CoA synthase 2 (19). However, investigations have
implicated extrahepatic organization in the production of β-HB and
other KBs including glial cells (29,30), kidney (31), pancreatic β-cells (32), retina (33,34) and tumor cells (35). However, the presence of these
processes in some of these tissues remains a subject of debate
(36).
Catabolism of β-HB
Hepatic tissues contain a robust β-HB synthase
system. However, they do not possess an enzyme system for β-HB
utilization (14). By contrast,
extrahepatic tissues exhibit a well-developed expression of enzymes
that utilize β-HB. Consequently, the β-HB that is produced in the
liver is primarily utilized by extrahepatic tissues such as the
heart, kidneys, brain and skeletal muscles (14). Within the mitochondria, β-HB is
transformed into ACAC and NADH through the action of BDH1. ACAC,
along with succinyl CoA, is converted into NADH with the help of
succinyl CoA transsulfatase [also termed as 3-oxoacid CoA
transferase 1 (OXCT1)], leading to the generation of activated
acetoacetyl CoA and succinic acid. The conversion of acetoacetyl
CoA is catalyzed by acetyl-CoA acetyltransferase, resulting in the
formation of two molecules of acetyl CoA, which subsequently enter
the tricarboxylic acid (TCA) cycle for thorough oxidation (Fig. 1) (37). In mammals, glucose, FFAs and KBs
(specifically β-HB) are sources of adenosine triphosphate (ATP).
Among these sources, β-HB is known to yield the highest amount of
ATP per oxygen atom produced (38).
Regulation of β-HB metabolism
Regulation of β-HB metabolism is mainly influenced
by factors such as satiety, fasting, carbohydrate metabolism and
the activity of specific enzymes. In the presence of satiety or
sufficient carbohydrate availability, insulin secretion rises,
which leads to a suppression of β-HB production. Conversely, during
periods of starvation or when glucose metabolism is impaired,
glucagon secretion increases, facilitating the catabolism of β-HB.
Malonyl CoA hinders the transport of fatty acyl CoA into the
mitochondria by competitively inhibiting carnitine
palmitoyltransferase, which leads to a decrease in β-oxidation of
fatty acids and a subsequent reduction in β-HB synthesis (28). Additionally, OXCT1 serves as a
crucial rate-limiting enzyme in the catabolism of β-HB, and the
liver is incapable of metabolizing β-HB due to the absence of OXCT1
(39).
Transport of β-HB
β-HB must be transported across both the plasma
membrane and the inner mitochondrial membrane. The primary
transporters responsible for β-HB passage at the plasma membrane
are the monocarboxylate transporter (MCT) family (40), while at the inner mitochondrial
membrane, the pyruvate carrier facilitates its transport (41). There are two types of MCTs:
Proton-coupled MCTs and sodium-coupled monocarboxylate transporters
(SMCTs). To date, 14 MCTs and 2 SMCTs have been characterized
(42-45). Specifically, MCT1, MCT2, MCT4,
MCT7 and SMCT1 have been identified as transporters for β-HB
(42,44,46-48).
MCT1 is expressed widely in a variety of tissues,
such as muscle, kidney, liver and heart (45). MCT2 is expressed in a more
limited set of tissues, such as the liver, kidney and testis
(49). MCT4 is mainly present in
skeletal muscle (50), while
MCT7 is detected in the liver, pancreas, skin, vas deferens and
testis, as well as other tissues (51). Another significant transporter,
SMCT1, is present in the intestine, kidney, thyroid gland and
retina (52). This distribution
of MCTs and SMCTs suggests a specialized and regulated transport
system for β-HB across different tissues.
Functions of β-HB
β-HB as an energetic substrate
Under conditions of intermittent fasting (IF),
caloric restriction (CR), KD and exercise, FFA undergoes extensive
β-oxidation in the liver, yielding a significant quantity of ACAC.
A portion of this ACAC is converted into β-HB (53). β-HB possesses physicochemical
properties such as a small molecular weight and good water
solubility (54); it can not
only be efficiently transported through the blood circulation but
also penetrates the blood-brain barrier (BBB) and the capillary
walls of muscle tissue, easily reaching extrahepatic tissues where
it is oxidized and decomposed as an energy substrate (Fig. 2). Once there, β-HB can be
metabolized and utilized, making it a crucial energy source for the
brain, heart, kidneys and skeletal muscle, among other tissues, in
times of metabolic stress (55).
Consequently, β-HB assumes a pivotal role as a compensatory energy
fuel during these periods of increased energy demand (56-58). Furthermore, the contribution of
β-HB to energy metabolism is notably amplified during the perinatal
lactation period and the neonatal phase (59).
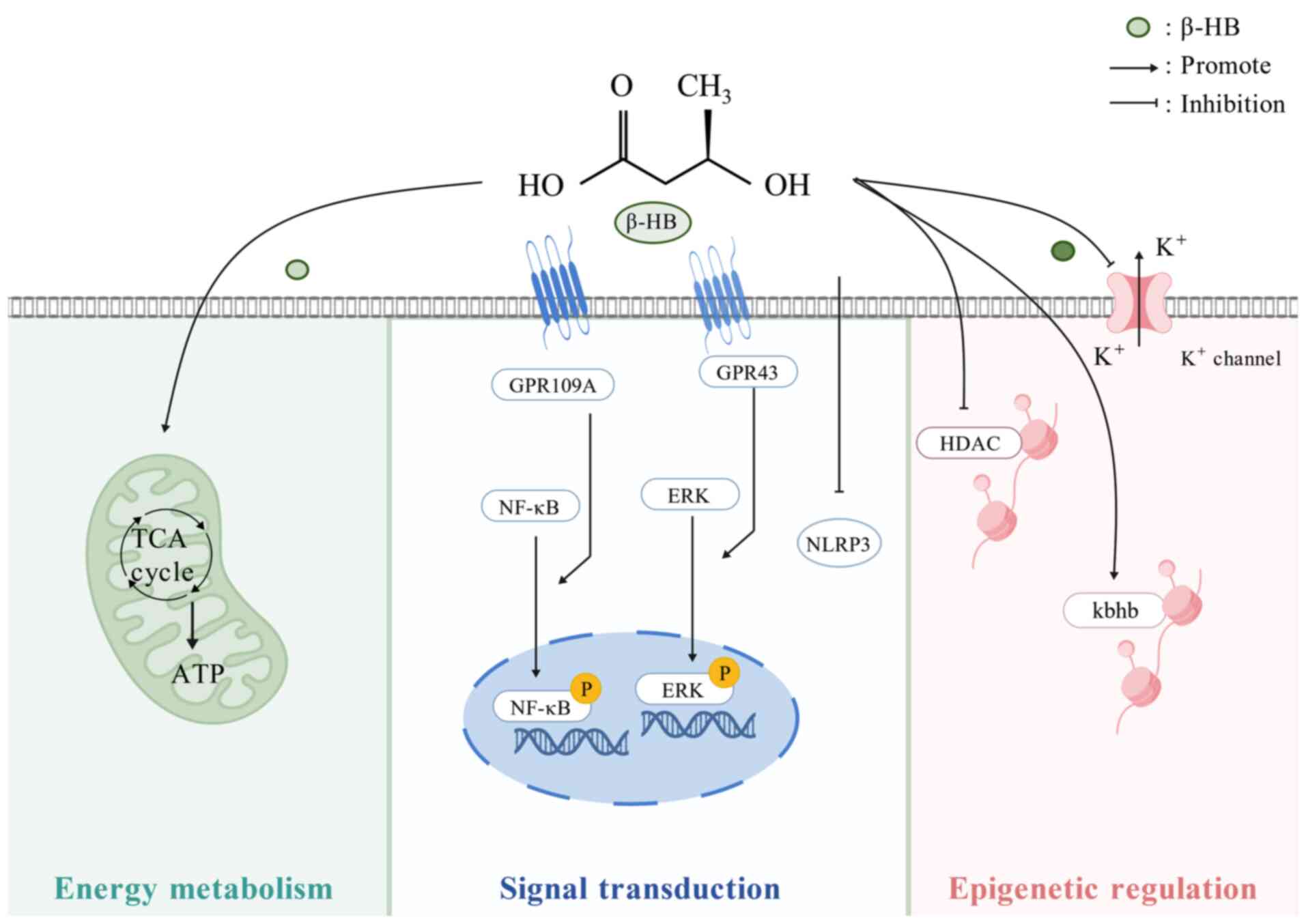 | Figure 2Biological functions of β-HB. β-HB
acts as an energy substrate to regulate metabolic reactions as a
signaling molecule that binds to the ligand of the GPCR to modulate
downstream signaling molecules and inhibits NLRP3. As an epigenetic
regulator, β-HB inhibits HDAC, promotes Kbhb and controls
K+ channels. HDAC, histone deacetylase; Kbhb, lysine
β-hydroxybutyrylation; GPCRs, G protein-coupled receptors; NF-κB:
nuclear factor κB; ERK, extracellular regulated protein kinase;
TCA, tricarboxylic acid; ATP, adenosine triphosphate; NLRP3,
NOD-like receptor family pyrin domain containing 3; β-HB,
β-hydroxybutyric acid. |
β-HB as a signaling molecule
β-HB serves as a mediator for metabolic signaling
that influences numerous cellular processes (Fig. 2). β-HB is a ligand for the G
protein-coupled receptors (GPCRs), GPR109A (also known as HM74A in
humans and PUMA-G in mice) and GPR41. GPR109A is found in
adipocytes, retinal tissue and macrophages. Within physiologically
relevant concentrations (Ki=0.7 mM), β-HB selectively stimulates
GPR109A, leading to the activation or inhibition of various
signaling pathways linked to lipid metabolism and cellular growth
(60). Moreover, GPR41, also
known as free fatty acid receptor 3, is present in sympathetic
ganglia; it can inhibit sympathetic activity in mice through the G
protein β-γ complex/phospholipase C β/MAPK signaling pathway,
thereby suppressing the overall metabolic rate (61,62). Besides GPCRs, β-HB engages
directly with ribonucleoproteins; it plays a role in histone
acetylation (63), histone
lysine β-hydroxybutyrylation (Kbhb) (64) and indirectly promotes protein
hyperacetylation (65).
Furthermore, β-HB exerts direct regulatory influences on K+
channels and neuronal vesicular glutamate transporters (66) and it inhibits inflammation
mediated by the NOD-like receptor family pyrin domain-containing 3
(NLRP3) (67).
β-HB and ketosis
Under normal circumstances, the concentration of
β-HB in human plasma and tissues is maintained at ~0.1 mM (68). Under conditions of prolonged
fasting or a KD, insufficient carbohydrate intake prompts the body
to reduce protein breakdown to maintain blood glucose levels.
Instead, it shifts to fat breakdown to produce KBs, which serve as
an alternative energy source to glucose. During this period, the KB
levels of the body increase slightly (69). Pathological states such as
obesity and DM, when the KB production of the body exceeds its
utilization capacity and accumulates, lead to ketoacidosis (KA).
Ketosis is characterized by elevated serum KB levels and is
classified into nutritional ketosis (NK) and pathological KA. The
core differences between the two lie in KB concentration, acid-base
balance status and inducing mechanisms (58,70). As a key hormone regulating
ketogenesis, insulin maintains KB homeostasis primarily through
three mechanisms: i) It inhibits lipolysis in adipose tissue,
reducing the transport of FFAs to the liver; ii) it directly
decreases the activity of enzymes involved in KB synthesis in the
liver; and iii) it enhances the efficiency of KB oxidation and
utilization in peripheral tissues such as the brain and muscles
(71).
NK
NK is an adaptive metabolic state in the body where
KBs serve as the primary energy source under specific physiological
conditions or dietary interventions. NK is typically induced by
factors such as starvation, IF, KD or prolonged exercise. The
criterion widely accepted in most studies for diagnosing NK is a
serum β-HB level ranging from 0.5 to 3 mmol/l (72). From the perspective of metabolic
effects, NK has clear physiological advantages, such as improving
insulin sensitivity and optimizing energy utilization efficiency
(73-76). Moreover, although the blood
glucose level of the body decreases slightly in this state, the
blood pH value remains within the normal range at all times. It
should be noted that the induction process of NK may be accompanied
by transient discomfort symptoms such as drowsiness and dizziness.
Additionally, long-term dietary interventions (such as strict KD)
may have potential impacts on the homeostasis of intestinal flora.
Meanwhile, due to the high requirements for dietary adherence, most
individuals find it difficult to persist with such interventions
over the long term (77). In the
ketogenic state, on one hand, the low-insulin environment allows
the liver to activate the ketogenesis pathway to supplement energy;
on the other hand, insulin can increase the concentration of
malonyl-CoA by activating acetyl-CoA carboxylase, thereby
inhibiting the activity of carnitine palmitoyl transferase 1
(CPT-1), restricting the excessive entry of fatty acids into the
mitochondria, and ultimately precisely controlling the blood ketone
concentration within the safe range of NK (78).
Diabetic ketoacidosis (DKA)
DKA is the most common acute hyperglycemic emergency
in patients with DM. Among these patients, those with T1DM are at a
high risk of developing DKA due to absolute insulin deficiency
(58). The typical clinical
features of DKA are characterized by a triad of hyperglycemia
(blood glucose ≥13.9 mmol/l), hyperketonemia (typically β-HB of
≥3.0 mM) and hyperketonuria (urinary KB test strip ≥2+), along with
electrolyte disturbances and acid-base imbalance (79,80). Due to the absolute insulin
deficiency, the inhibition of hormone-sensitive lipase is lifted,
leading to a surge in FFAs; a sharp drop in malonyl-CoA causes
excessive activation of CPT-1 and, combined with the upregulation
of ketogenic enzymes, this results in an abnormal increase in KB
production. In the state of DKA, the level of β-HB can rise to
10-20 mM (or even higher) (81).
β-HB in T2DM and its complications
T2DM arises from the interplay of two main factors:
Pancreatic β-cells exhibiting impaired insulin secretion and
tissues sensitive to insulin exhibiting an inadequate response.
Inflammation, endoplasmic reticulum stress (ERS) and
metabolic/oxidative stress have been identified as potential
contributors to β cell dysfunction or IR (82-84). Research has underscored the role
of β-HB in antioxidant, anti-inflammatory and mitochondrial
function-protective mechanisms (85). It is suggested that β-HB may
regulate the occurrence and development of T2DM through these
mechanisms. This provides a new clinical diagnosis and treatment
avenue for the early diagnosis and management of various
complications of T2DM; however, its underlying mechanism of action
still needs further exploration and clarification (Fig. 3 and Table I).
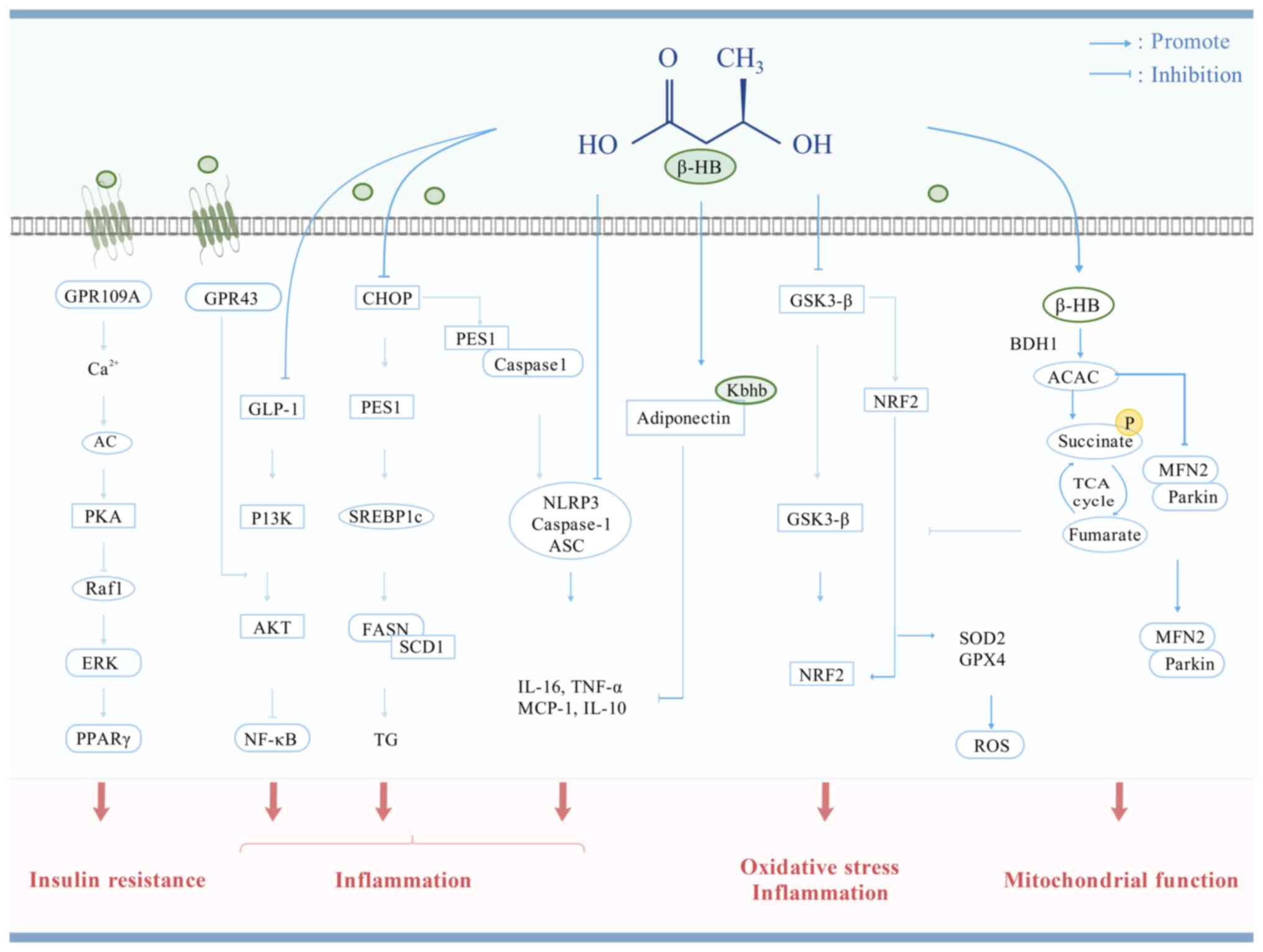 | Figure 3Possible mechanisms of β-HB against
T2DM. β-HB acts as a signaling molecule to regulate IR,
inflammation, oxidative stress, mitochondrial function and cell
apoptosis to improve T2DM. GPR, G protein-coupled receptor; AC,
adenylyl cyclase; PKA, protein kinase A; Raf1, Raf-1
proto-oncogene, serine/threonine-protein kinase; ERK, extracellular
regulated protein kinase; PPARγ, peroxisome proliferator-activated
receptor γ; GLP-1, glucagon-like peptide-1; P13K,
phosphatidylinositol-3-kinase; NF-κB: nuclear factor-κB; CHOP,
C/EBP-homologous protein; PES1, pescadillo 1; SREBP1c, sterol
regulatory element binding protein 1c; FASN, fatty acid synthase;
SCD1, stearoyl-CoA desaturase 1; TG, triglyceride; Caspase-1,
cysteinyl aspartate specific proteinase 1; NLRP3, NOD-like receptor
family pyrin domain-containing 3; TNF-α, tumor necrosis factor-α;
MCP1, monocyte chemoattractant protein 1; BDH1, β-hydroxybutyrate
dehydrogenase 1; ACAC, acetoacetate; TCA, tricarboxylic acid; MFN2,
mitofusin 2; Nrf2, nuclear factor-erythroid 2-related factor 2;
GSK3-β, glycogen synthase kinase 3-β; SOD, superoxide dismutase;
GPX4, glutathione peroxidase 4; ROS, reactive oxygen species;
β-hydroxybutyric acid; T2DM, type 2 diabetes mellitus. |
 | Table IRole of β-HB in T2DM and its
complications and related mechanisms. |
Table I
Role of β-HB in T2DM and its
complications and related mechanisms.
A, Liver
|
|---|
| Authors, year | In
vivo/in vitro | Model | Methods for
intervening in β-HB | Main findings | Mechanisms and
indicators of change | (Refs.) |
|---|
| Zhou et al,
2022 | In vivo | KKAy mice | KD | B-HB reduced the
hepatic adipose tissue inflammatory response and improved hepatic
lipid metabolism in KKAy mice. | CHOP, PES1, p300,
SREBP1c, ↓: N'-SREBP1c, FASN, SCD1, NLRP3, Caspase1,
cleaved-Caspase1, IL-1β, IL-18 | (23) |
|
| B, Kidney |
|
| Guo et al,
2023 | In vivo | HFD feeding
combined with STZ injected to induce T2DM mice | 1,3-BDO solution as
a drinking solution. | Supplementation
with 1,3-BDO reprograms energy metabolism and attenuates kidney
damage. | ↑: ATP, β-HB | (24) |
| Wan et al,
2023 | In vivo | db/db mice | Supplementation of
β-HB at 100 mM in drinking water for 6 weeks. | B-HB prevents
diabetic environmentally-induced glomerular podocyte senescence and
injury. | ↑: BDH1, Nrf2 | (95) |
| Wan et al,
2023 | In
vitro | HK-2 cells | 5 mM treatment for
48 h. | B-HB prevents
diabetic environmentally-induced glomerular podocyte senescence and
injury. | ↓: ROS, IL-1β,
IL-18; ↑: Nrf2, ACAC, succinate, fumarate | (95) |
| Fang et al,
2021 | In vivo | STZ injected | Intraperitoneal
injection of β-HB (100 mg/kg/day) every other day for 4 weeks | B-HB reduces
albuminuria, renal hypertrophy and histologic signs of DKD | ↓: Urinary protein,
p-GSK3β; ↑: Nrf2, GSK3β | (96) |
| Fang et al,
2021 | In vivo | Glomerular
podocytes treated with PA combined with TGF-β | 4 mM treatment for
48 h | B-HB enhances the
antioxidant response and ultimately attenuates podocyte
senescence. | ↓: p-GSK3β; ↑:
Nrf2, GSK3β | (96) |
|
| C, Adipose |
|
| Zhang et al,
2023 | In vivo | HFD feeding
combined with STZ injected to induce T2DM mice. | 10% 1,3-BDO
solution as a drinking solution. | B-HB reduces
fasting blood glucose levels and improves glucose tolerance and IR
in T2DM mice via HCAR2. | ↓: Raf-1, ERK1/2,
p-PPARγ; ↑: PPARγ, Ca2+, AC, cAMP, PKA. | (22) |
| Park et al,
2011 | In vivo | Rats had a 90%
Px. | 150 mg/kg of β-HB
was injected intraperitoneally twice daily into rats for 5
weeks. | Intraperitoneal
injection of β-HB decreased epididymal fat pads and serum leptin
levels. | ↓: PEPCK; ↑: IRS2,
p-AKT/AKT | (107) |
|
| D, Blood
vessel |
|
| Wang et al,
2023 | In vivo | db/db mice | KD (5%
carbohydrate, 75% fat and 20% protein). | KD inhibits the
T2DM-induced increase in PES1, which may lead to vascular
hyperpermeability in T2DM mice through the ubiquitination of
VE-cadherin. | ↓: PES1, VEGF; ↑:
VE-cadherin, Occludin | (116) |
| Wang et al,
2023 | In
vitro | MVECs intervened
with HG. | 2 mM β-HB
intervention for 24 h. | β-HB reduces the
ubiquitination of VE-cadherin promoted by PES1. | ↓: PES1, VEGF;
↑:VE-cadherin, Occludin | (116) |
|
| E, Heart |
|
| Thai et al,
2021 | In vivo | db/db mice | KE feeding 4
weeks. | KE supplementation
prevents progression toward DCM in T2DM by limiting oxidative
stress and enhancing mitochondrial quality control via
mitophagy. | ↓:
H2O2; ↑: BDH1, OXCT1, ACAT, mitochondrial
complex II, mitochondrial complex IV, mitochondrial complex V,
GPX4, Parkin, Mfn2, pro-LC3B. | (117) |
|
| F, Brain |
|
| Park et al,
2011 | In vivo | Px diabetic
rats | B-HB injection at
12 mg/h for 28 days. | β-HB central
infusion improves hypothalamic leptin and insulin signaling. | ↑: IRS2, p-AKT,
GLUT2, glucokinase, STAT3 | (130) |
|
| G, Retina |
|
| Trotta et
al, 2019 | In vivo | STZ-injected
mice | Inject twice weekly
for 10 weeks at 25, 50 and 100 mg/kg. | β-HB attenuates
retinal NLRP3 inflammatory vesicle activation markers, reduces
apoptotic cells and improves retinal permeability and
homeostasis. | ↓: NLRP3, ASC,
caspase–1, IL–1β, IL–18, p-PERK, p-IRE1, ATF6α; ↑: Connexin 43,
HCA2 | (141) |
Potential effects of β-HB on the liver in
T2DM
Effects of T2DM on the liver
The liver is essential for regulating glucose and
lipid metabolism and it is a key site for the development of IR.
Chronic increase in plasma-free FFA leads to metabolic imbalance in
the body and induces IR, which promotes FFA delivery to the liver
and hepatic fat deposition. Increased hepatic fat accumulation
results in heightened lipotoxicity, adipose tissue inflammation,
impaired mitochondrial function and ERS. Excessive deposition of
hepatic extracellular matrix forms hepatic fibrosis, which
ultimately leads to cirrhosis; T2DM hepatic fibrosis is one of its
pathological manifestations (86). It is noteworthy that the abnormal
activation of the hepatic gluconeogenesis pathway is a crucial
mechanism leading to the hyperglycemic state. This process is
regulated by key gluconeogenic enzymes and the increased activity
of these enzymes significantly promotes gluconeogenesis, thereby
exacerbating hyperglycemia (87).
Effects of β-HB on the liver
A persistent, low-grade inflammatory state is
recognized as a pivotal element of IR and metabolic disorders. Zhou
et al (23) demonstrated
that β-HB can inhibit lipid synthesis in the hepatocytes of KKAy
mice (T2DM mouse model), thereby reducing hepatic lipid
accumulation and decreasing the expression of hepatic inflammatory
factors. The mechanism may involve β-HB impairing the binding
ability of the transcription factor, C/EBP homologous protein, to
the promoters of ribosome biogenesis factor 1 (PES1), thereby
leading to downregulated expression of the PES1 protein.
Ultimately, this improves liver pathology through two key effects:
i) It inhibits the binding of PES1 to the promoters of E1A-binding
protein p300 (p300) and cysteine-aspartic acid protease 1; and ii)
it reduces p300-mediated acetylation of sterol regulatory
element-binding protein 1c (SREBP1c) and the associated
inflammatory response pathways. SREBP1c is a core transcription
factor for lipid synthesis, and the inhibition of its activity can
directly reduce hepatic triglyceride synthesis. Furthermore, the
authors observed that KD increased the levels of circulating β-HB,
suppressed hepatic PES1 expression and improved hepatic lipid
regulation, the inflammatory response, blood glucose levels and IR
in KKAy mice (23).
The accumulation of reactive oxygen species (ROS)
caused by excessive hepatic lipid deposition is a crucial driving
factor for hepatic fibrosis in T2DM (88). Xu et al (89) discovered that upregulation of
BDH1 reduces fibrosis, inflammation and apoptosis in the livers of
db/db mice, processes that are mediated by ROS. The underlying
mechanism may be that β-HB metabolism mediated by BDH1 upregulates
the production of fumarate. In turn, fumarate can inhibit the
activity of Kelch-like ECH-associated protein 1 (Keap1), thereby
activating nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2
is a core transcription factor for resisting oxidative stress; its
activation can induce the expression of antioxidant enzymes, which
in turn scavenge excessive ROS (89). BDH1 promotes the interconversion
of β-HB with ACAC (37).
Therefore, BDH1-mediated hepatic β-HB metabolism opens a new avenue
for the treatment of db/db mice.
Effects of β-HB on the kidneys in
T2DM
Effects of T2DM on the kidneys
The kidneys are one of the most susceptible target
organs in T2DM. Diabetic kidney disease (DKD) caused by T2DM has
become the leading cause of end-stage renal disease and ~40% of
T2DM patients will progress to DKD (90). Persistent hyperglycemia can
induce functional abnormalities in the glomerular feedback system
and mediate cellular damage via glucotoxicity, thereby triggering a
state of glomerular hyperfiltration. This early pathological change
can further activate a series of cascade reactions, including
metabolic disorders, hemodynamic abnormalities, ERS, inflammatory
responses and fibrotic processes, leading DKD to progress from
early functional impairment to irreversible organic lesions
(91). Patients with DKD
frequently exhibit early signs of hyperfiltration and albuminuria,
as well as glomerular and tubular lesions (92).
Effects of β-HB on the kidneys
SMCT1, as a high-affinity transporter for
monocarboxylates, is a key molecule for β-HB uptake in tissues
(93). Under the state of
hyperinsulinemia in T2DM, the protective effect of β-HB on renal
tubular epithelial cells may depend on the 'β-HB uptake and
utilization' mediated by SMCT1. The authors (24) observed a decrease in renal SMCT1
expression in patients with DKD and in mice with T2DM. Following
overexpression of SMCT1, the levels of β-HB in the serum and
kidneys of T2DM mice increased, while the urinary β-HB levels
decreased and renal energy metabolism improved. By contrast, a lack
of the SLc5α8 gene responsible for encoding SMCT1 led to structural
damage and functional impairment of the renal tubules in T2DM.
However dietary supplementation with 1,3-butanediol (1,3-BDO), a
precursor of β-HB, can ameliorate renal injury in SMCT1-knockout
mice (24). Mechanistic studies
have shown that hyperinsulinemia inhibits SMCT1, impairs the uptake
of β-HB and thereby compromises mitochondrial function and cell
survival in renal tubular epithelial cells (94). Therefore, the transport of β-HB
into renal tissues mediated by SMCT1 is a necessary process for
maintaining the mitochondrial function of renal tubules. In
addition, renal β-HB metabolism mediated by BDH1 may also provide a
new therapeutic approach for DKD. Both KD intervention and β-HB
intervention can increase the serum β-HB levels and upregulate the
renal expression of BDH1, thereby ameliorating DKD (95). Mechanistically, β-HB metabolism
mediated by BDH1 upregulates the production of fumarate, which in
turn activates Nrf2 to inhibit oxidative stress and alleviate renal
tubular injury (95). The
protective effect of β-HB on the kidney is not limited to renal
tubules; it also exerts a protective effect on glomerular cells.
Supplementation with β-HB can reduce albuminuria and alleviate
renal hypertrophy in mice with DKD (96). In vitro experiments have
confirmed that β-HB can protect glomerular podocytes from injury
and senescence under stimulation by high glucose combined with
transforming growth factor-β (TGF-β). The underlying mechanism may
be as follows: β-HB inhibits the activity of glycogen synthase
kinase 3β and the phosphorylation of Nrf2, reduces the nuclear
export of Nrf2 and increases its nuclear accumulation, thereby
enhancing the antioxidant response and delaying podocyte senescence
(96). In summary, β-HB protects
renal tubules and glomeruli in DKD from oxidative stress-induced
injury by inhibiting the nuclear translocation of Nrf2.
Effect of β-HB on adipose tissue in
T2DM
Effects of T2DM on adipose tissue
It has been indicated that ~80% of individuals with
T2DM exhibit signs of overweight or obesity (97). When adipose tissue exceeds its
normal storage capacity, it accumulates ectopically (such as in the
heart, skeletal muscle, liver and pancreas), leading to increased
visceral lipid deposition and the onset of FFA-induced toxicity,
commonly referred to as lipotoxicity (98). Lipotoxicity denotes the harmful
effects of lipid byproducts on tissues that are not composed of
fat, such as the liver, skeletal muscle, heart, kidneys and
pancreatic β-cells (99), which
are pivotal in the pathogenesis of T2DM IR (100). Furthermore, under healthy
conditions, adipokines (such as adiponectin) secreted by adipose
tissue can enhance insulin sensitivity. By contrast, the 'endocrine
dysfunction' of adipose tissue under pathological states exhibits
pathogenicity (101). The
increase in adipose tissue mass leads to low-grade inflammation by
altering the secretion of adipokines and cytokines, and the
production of these adipocytokines is negatively correlated with IR
in T2DM (102).
Effects of β-HB on adipose tissue
Zhang et al (22) employed a db/db mouse model
alongside a T2DM mouse model induced by a high-fat diet (HFD) and
treated with streptozocin to investigate how administering a 10%
aqueous solution of 1,3-BDO affected these mice. It was discovered
that β-HB binds to hydroxycarboxylic acid receptor 2 (HCAR2) and
triggers an increase in intracellular Ca2+ levels within
adipocytes. This process triggers adenylate cyclase, leading to an
increase in cyclic adenosine monophosphate (cAMP) levels, which in
turn activates protein kinase A (PKA). When activated, PKA
restrains the activity of Raf-1 proto-oncogene
serine/threonine-protein kinase (Raf1), causing a decrease in the
activity of extracellular regulated protein kinases 1/2 (ERK1/2).
This reduction ultimately inhibits the phosphorylation of
peroxisome proliferator-activated receptor γ (PPARγ) at Ser273 in
adipocytes. Such changes in the expression levels of genes
regulated by PPARγ contribute to a decrease in IR (22). As a result, β-HB regulates the
activity of ERK1/2 through the HCAR2/Ca+/cAMP/PKA/Raf1
pathway, ultimately enhancing PPARγ function through
post-translational changes and decreasing IR linked to T2DM.
Insufficient secretion of adiponectin is a core
feature of adipose tissue dysfunction in T2DM. Nishitani et
al (25) found that in KKAy
mice, the serum level of β-HB was increased, while in periovarian
adipose tissue (visceral adipose tissue), the expression of
adiponectin was decreased, the inflammatory response was enhanced,
the metabolism of the insulin signaling pathway was weakened, the
expression levels of MCT1 and MCT2 were downregulated and the
expression of OXCT1 (the enzyme responsible for KB catabolism) was
reduced. These findings suggest that abnormal β-HB metabolism is
closely associated with adipose tissue dysfunction. In in
vitro intervention experiments on 3T3-L1 adipocytes, β-HB can
significantly upregulate the mRNA and protein expression levels of
adiponectin, while inhibiting inflammatory responses (25). Further mechanistic studies have
shown that the regulatory effect of β-HB depends on a novel
epigenetic modification, Kbhb. Specifically, β-HB can induce an
increase in the Kbhb level of the histone H3 lysine 9 (H3K9) site
within the adiponectin gene promoter region in 3T3-L1 adipocytes.
This modification serves to open the chromatin structure of the
adiponectin gene, facilitate the binding of transcription factors,
and thereby activating the expression of the adiponectin gene.
Lipocalin expression has also been found to be regulated by two
epigenetic modifications (103,104) and promoter activity (105,106), and lipocalin gene expression
has been reported to be epigenetically associated with 3T3-L1
adipocytes (104). The research
findings of Nishitani et al (25) revealed that β-HB induces an
increase in Kbhb at the H3K9 site in 3T3-L1 adipocytes. Therefore,
β-HB seems to offer defense in KKAy mice by inducing epigenetic
changes in the lipocalin gene within adipocytes. β-HB can directly
regulate adiponectin gene expression through epigenetic
modifications and this mechanism provides a new perspective for
adipose tissue protection.
The positive effects of KD on managing energy
metabolism and maintaining glucose homeostasis are still a topic of
contention. A notable detail is that KD and the intraperitoneal
administration of β-HB produce distinct effects on lipid
metabolism. Park et al (107) observed that KD led to an
increase in epididymal fat pads and serum leptin levels, while
leptin-related signal transducer and activator of transcription 3
(STAT3) signaling was impaired, leading to visceral fat
accumulation in diabetic rats with 90% pancreatectomy (Px). By
contrast, intraperitoneal administration of β-HB led to decreased
testicular fat pads and serum leptin levels, partially restoring
hepatic insulin receptor expression and signaling without affecting
STAT3 signaling. This discrepancy may be attributed to the fact
that a KD inhibits hypothalamic STAT3 phosphorylation, thereby
inducing leptin resistance, whereas β-HB injection exerts no effect
on STAT3 signal transduction. Additionally, the high-fat content in
a KD leads to an increase in FFAs, which promotes fat accumulation.
Apart from β-HB, a KD contains substantial amounts of saturated
fats and trans fats; these components themselves can disrupt lipid
metabolism (such as increasing FFA levels and inducing
inflammation) (108). These
findings suggest that β-HB exerts a protective effect on lipid
metabolism, while a KD is not suitable for lipid metabolism
management in non-obese patients with T2DM. Additionally, research
has shown that the supraphysiological levels of β-HB (ranging from
15 to 50 mM) triggers the occurrence of lipofuscinosis (109), while it does not promote
lipo-browning at physiological concentrations (110).
Effects of β-HB on the cardiovascular
system in T2DM
Effects of T2DM on the cardiovascular
system
T2DM is strongly correlated with the development of
cardiovascular disease (111).
The metabolic environment characterized by hyperglycemia and
hyperlipidemia in patients with T2DM increases the risk of
developing heart disease (112). Diabetic cardiomyopathy (DCM)
refers to heart diseases complicated by or associated with DM,
including coronary atherosclerotic heart disease, DCM itself and
arrhythmias and cardiac dysfunction caused by autonomic nerve
disorders (113). The main
pathological features of DCM include cardiomyocyte hypertrophy,
myocardial fibrosis and impaired coronary microvascular perfusion.
The pathogenesis involves oxidative stress, inflammation and
impaired Ca2+ handling as well as alterations in
substrate metabolism/utilization, insulin signaling, gene
regulation, mitochondrial dysfunction, ERS, neurohumoral activation
and cardiomyocyte death (114).
However, there is currently a lack of effective therapeutic
approaches for DCM (115).
Effects of β-HB on the cardiovascular
system
β-HB, as a key metabolite of a KD, downregulates the
expression of vascular PES1, thereby inhibiting PES1-mediated
ubiquitin-dependent degradation of vascular endothelial cadherin
(VE-cadherin). Consequently, β-HB upregulates barrier-protective
proteins such as VE-cadherin and downregulates pro-leakage proteins
such as vascular endothelial growth factor (VEGF), ultimately
improving vascular hyperpermeability in T2DM mice; it also assists
in reducing fasting blood glucose (116). This finding provides a novel
'PES1/β-HB-targeted' direction for the treatment of vascular
complications in T2DM. Ketone esters (KEs) increase the circulating
level of β-HB and upregulate the expression of myocardial BDH1,
ultimately improving cardiac function in mice with T2DM. The
underlying mechanism may be associated with β-HB regulating the
expression of mitofusin 2, promoting Parkin-mediated mitophagy and
enhancing mitochondrial biogenesis. These processes collectively
optimize mitochondrial quality control and reduce the level of
oxidative stress (117).
Another study also indirectly demonstrated the myocardial
antioxidant stress effect of β-HB. Lin et al (118) observed that the expression of
BDH1 was decreased in the aorta of T2DM model mice, and systemic
overexpression of BDH1 reduced the area of atherosclerotic plaques
in T2DM. In their in vitro studies, BDH1 was found to
mitigate oxidative stress and inflammatory reactions in Raw264.7
cells (mouse macrophage cell line) by enhancing ferredoxin
metabolic flux and stimulating the Nrf2 signaling pathway. The
study by Uchihashi et al (119) further supports their
conclusion. This study showed that heart-specific overexpression of
BDH1 can also improve oxidative stress and cardiac remodeling in
heart failure induced by pressure overload. In addition, the
reduction in β-HB concentration can be considered a marker of
overall FFA oxidation (120).
Liepinsh et al (121)
found that the plasma β-HB level in Goto-Kakizaki (GK) rats was
significantly decreased, while after treatment with light phosphate
at a dose of 200 mg/kg for 4 and 8 weeks, the β-HB concentration
was further reduced. Therefore, the authors concluded that the
cardioprotective effect of light phosphate treatment in GK rats
could be explained by partial inhibition of FFA oxidation and
increased glucose metabolism.
Effects of β-HB on the brain in T2DM
Effects of T2DM on the central nervous
system
Individuals with T2DM exhibit an accelerated rate of
brain aging at ~26% faster than those without diabetes (122). The effect of diabetes on the
central nervous system has garnered considerable attention in
recent years. 'Diabetic encephalopathy (DE)' was raised by
Reske-Nielsen et al (123) to describe a central nervous
system complication associated with diabetes, characterized by
cognitive and behavioral deficits. Clinically, DE presents as
cognitive dysfunction, decision-making disorders and mood
disorders, and pathologically as structural and functional
alterations in intracranial tissues. The pathological hallmarks of
DE include gray matter, white matter, hippocampal atrophy,
compromised synaptic plasticity, glial cell dysfunction and
alterations in the structure and function of cerebral blood
vessels. The pathological mechanisms of DE encompass an imbalance
in pancreatic amyloid polypeptide homeostasis, microRNAs,
macrophage autophagy, Lipin1, advanced glycation end products
(AGEs), oxidative stress, hyperphosphorylation of Tau proteins and
intestinal homeostasis dysregulation (124,125).
Effects of β-HB on the central nervous
system in T2DM
β-HB is produced in the liver and it can traverse
the BBB to provide energy to the brain when glucose levels are low
(126). Andersen et al
(127) found that cerebral
glucose metabolism was reduced in db/db mice, whereas hippocampal
β-HB metabolism was increased. Furthermore, an enhancement in
mitochondrial oxygen consumption and the rate of ATP synthesis was
observed. This suggests that β-HB can partially compensate for
insufficient glucose metabolism by enhancing mitochondrial
oxidation, thereby maintaining the energy homeostasis of
hippocampal neurons. The entry of β-HB into nerve cells depends on
MCTs. Pierre et al (128) reported that, at 6 weeks of age,
the hippocampus of db/db mice exhibit increased levels of MCT1 and
MCT2, and that this upregulated expression of these transporter
proteins may support the utilization of KBs. MCT1 is expressed by
endothelial cells, astrocytes, oligodendrocytes and microglial
cells in the brain, whereas MCT2 is primarily expressed by neurons
(129). Thus, the augmented KB
metabolism in db/db mice may be related to the expression levels of
neuronal transporter proteins.
The hypothalamus, a pivotal regulator of energy
homeostasis, has been a subject of debate in terms of its potential
influence on energy and glucose homeostasis through central KBs.
Park et al (130)
administered β-HB at a dose of 12 μg/h into the lateral
ventricle of diabetic rats with 90% Px. After 28 days, they
observed increased β-HB levels in the hypothalamus and liver,
enhanced leptin and insulin signaling in the hypothalamus and
elevated STAT3 phosphorylation. By contrast, intraperitoneal
injection of β-HB has no effect on hypothalamic signaling in Px
rats (107). This discrepancy
may be caused by the BBB: β-HB administered via intraperitoneal
injection needs to cross the BBB through MCTs. At the cellular and
molecular levels, β-HB exhibits multifaceted neuroprotective
potential. Majrashi et al (131) demonstrated that supplementation
of β-HB at doses of 250 and 500 μM exert a neuroprotective
effect on HT22 cells (mouse hippocampal neuronal cell line). The
proliferative effect of hippocampal neurons can reduce oxidative
stress, maintain energy metabolism, improve mitochondrial function
and regulate cell apoptosis. Combined with computational
pharmacokinetic and molecular modeling analyses. This study further
confirmed the neuroprotective potential of β-HB in
cognition-related neurodegenerative diseases. The brain is one of
the organs with the highest lipid content and lipids account for
~50% of its dry weight. Dabke et al (132) simulated the effect of
endogenous β-HB production induced by a KD on the lipids of
neuronal cells. Their research showed that when HT22 cells
incubated under low-glucose conditions were treated with β-HB at 5
mM, the levels of cholesterol and phosphatidylserine decreased,
while the ratio of phospholipids to cholesterol increased.
Effects of β-HB on the retina in
T2DM
Effects of T2DM on the retina
DM is linked to various eye-related issues, such as
diabetic retinopathy (DR), cataracts, diabetic papillopathy,
glaucoma and diseases affecting the ocular surface (133). DR is a major complication of DM
that manifests as retinal microangiopathy and stands as the leading
cause of vision impairment in middle-aged individuals (134). The condition of diabetes
enhances the permeability of the blood-retinal barrier, promoting
angiogenesis in the retina (135). The development of DR is
complex, and it involves increased production of free radicals, the
stimulation of AMP-activated protein kinase/mammalian target of
rapamycin signaling pathways, the activation of the
renin-angiotensin system, engagement of TGF-β/Smad signaling, the
kinin system involving kinin-releasing enzymes, the accumulation of
AGEs and various inflammatory agents such as VEGF (136-138). The abnormal signaling pathway
of TGF-β plays a role in the development of DR. Systemic inhibition
of TGF-β signaling offers protection against obesity, diabetes and
liver fat accumulation in mice (139). However, TGF-β has been proposed
to safeguard retinal ganglion cells against oxidative harm induced
by hyperglycemia by enhancing cellular antioxidant and
neuroprotective mechanisms, such as the Nrf2/Keap1 pathway
(140).
Effects of β-HB on the retina in
T2DM
Trotta et al (141) demonstrated that diabetic mice
with heightened ERS markers [phosphorylated (p)ERK, phosphorylated
inositol requiring enzyme 1 and activating transcription factor
6α], increased NLRP3 inflammasome activity (NLRP3, apoptosis
associated speck-like protein containing a CARD and caspase-1) and
increased levels of pro-inflammatory cytokines (IL-1β and IL-18)
experienced significant reductions in these parameters following
intraperitoneal injections of 50 and 100 mg/kg β-HB. The injections
led to increased plasma and retinal β-HB levels as well as enhanced
expression of the β-HB receptor, GPR109A. Consequently, ERS
markers, NLRP3 inflammasome activation markers and pro-inflammatory
cytokine levels were significantly reduced. Moreover, retinal outer
nuclear layer cell death was diminished, effectively safeguarding
the retina from diabetic-induced damage. Therefore, β-HB may offer
protection to the retinas of diabetic mice by mitigating
inflammation and ERS through the GPR109A receptor.
Application of β-HB in the clinical setting
of T2DM
Clinical research on β-HB mainly includes three
aspects: i) The potential of β-HB as a clinical diagnostic
biomarker for T2DM; ii) the effect of endogenous ketogenesis on
T2DM; and iii) the effects of exogenous β-HB supplementation on
patients with T2DM (Table
II).
| Table IITherapeutic applications of β-HB in
T2DM. |
Table II
Therapeutic applications of β-HB in
T2DM.
A, IF
|
|---|
| Authors, year | Human subject | Intervention
method | Main findings | Mechanisms and
indicators of change | (Refs.) |
|---|
| Arnason et
al, 2017 | Patients with T2DM
(n=10) | 2 weeks, fasting
for 18-20 h daily | Short-term daily IF
may be a safe and tolerable dietary intervention for patients with
T2DM. | ↓: Body weight,
BMI, target morning blood glucose, fasting blood glucose, IR, CRP,
caloric intake | (155) |
| Nuttall et
al, 2020 | Patients with T2DM
(n=7) | 3-day IF | β-HB increases with
the duration of fasting. | ↑: β-HB,
IGFBP-1 | (156) |
| Kramer et
al, 2024 | Overweight patients
with early-stage T2DM (n=39) | 6 weeks, fasting
for 20 h every day. | IF improved β-cell
function and IR in early-stage T2DM with overweight, accompanied by
beneficial effects on obesity. | ↓: HOMA-IR, HbA1c,
body weight, waist, circumference; ↑: insulin secretion sensitivity
index 2. | (186) |
|
| B, CR |
|
| Steven et
al, 2016 | Patients with T2DM
(n=30) | 6 months of CR (43%
carbohydrates, 34% protein and 19.5% fat, with an energy intake of
624 kcal per day). | CR diet reduces
fasting blood glucose in patients with T2DM. | ↓: Body weight,
HbA1c, fasting blood glucose levels. | (157) |
| Vigili et
al, 2017 | Patients with T2DM
before and after coronary angiography (n=11) | IF for 12-17 h in
patients with T2DM. | Overnight fasting
leads to inappropriate increase in β-HB in patients with T2DM | ↑: β-HB | (158) |
|
| C, KD |
|
| Goday et al,
2016 | Patients with T2DM
(n=44) | Very low-calorie KD
(15 g protein, 4 g carbohydrates, 3 g fat and 20 μg
chromium, 0.8 g ginseng and 0.4 mg biotin). | The very
low-calorie KD is safe and well-tolerated in patients with
T2DM. | ↓: Body weight,
HbA1c, blood glucose | (160) |
| Nuttall et
al, 2020 | Patients with T2DM
(n=7) | 3-day high-fat diet
(85% fat, 15% protein, virtually carbohydrate-free). | KD leads to a
significant increase in TAGs and NEFAs, which return to initial
levels after 24 h. | ↑: TAG, NEFA | (156) |
| Merovci et
al, 2024 | Overweight/obese
patients with T2DM (n=10) | 10-day intervention
with a KD (15-25% protein, 5-10% carbohydrate and 70-80% fat). | It stimulates the
production of ATP in β-cell mitochondria and significantly enhances
the insulin secretion function. | ↑: Plasma β-HB,
insulin, and C-peptide | (161) |
|
| D, KE
supplementation |
|
| Soto-Mota et
al, 2021 | Patients with T2DM
(n=21) | Continuously for 4
weeks, take 25 ml of KEM three times a day. | Exogenous KE
supplementation induced significant reductions in all markers of
blood sugar control. | ↓: Fructosamin,
HbA1c, average daily blood glucose. | (168) |
| Falkenhain et
al, 2024 | Patients with T2DM
(n=18) | Single
supplementation of KEM at a dose of 0.3 g/kg. | β-HB inhibits
lipolysis in patients with T2DM, reduces FFA, decreases the supply
of gluconeogenic amino acids and slightly increases insulin
concentration. | ↓: NEFAs, Met, Ser;
↑: β-HB | (170) |
| Jensen et
al, 2020 | Patients with T2DM
(n=14) | Intravenous
injection Na-DL-β-HB. | KB infusion
improves working memory performance in patients with T2DM. | ↑: Working
memory | (171) |
| Baranowski et
al, 2025 | Patients with T2DM
(n=15) | Acute and
short-term (14-day) supplementation of exogenous ketone
monoester. | Ketone monoester
has no effect on plasma BDNF or cognition. | - | (172) |
| Monteyne et
al, 2024 | Patients with T2DM
(n=10) | Single
supplementation of ketone monoester at a dose of 0.5 g/kg body
weight. | β-HB delays glucose
absorption in adults with T2DM, thereby reducing postprandial
glucose concentrations. | ↓: Glucose
concentrations at 2 and 4 h post-prandially; ↑: plasma β-HB | (169) |
|
| E, KD + KE |
|
| Merovci et
al, 2024 | Overweight/obese
patients with T2DM (n=10) | 10-day intervention
with KE plus β-HB KE (8 g every 8 h). | It stimulates the
production of ATP in the mitochondria of β-cells and significantly
enhances the insulin-secreting function. | ↑: Plasma β-HB,
insulin, and C-peptide | (161) |
|
| F, Intravenous
infusion |
|
| Solis-Herrera et
al, 2025 | Patients with T2DM
complicated by heart failure (n=36) | Intravenous
infusion of β-HB at doses of 0.7, 1.6 and 3.2 mmol/l. | The myocardial
benefits of β-HB are attributed to its ability to provide
additional fuel to the heart without inhibiting MGU. | ↑: Cardiac output,
LVEF, and stroke volume | (173) |
β-HB as a potential clinical diagnostic
biomarker for T2DM
The level of β-HB in the blood may be associated
with the risk of developing T2DM. Researchers from the Netherlands
and Sweden found a positive correlation between fasting plasma β-HB
levels and the incidence of T2DM in the general population without
diabetes or impaired fasting glucose (142). However, Bae et al
(143) followed up 453 patients
with impaired fasting glucose from South Korea for 10.9 years and
found that the incidence of T2DM was lower in patients in the high
β-HB group (≥0.05 mmol/l). A previous study has shown that high
levels of KBs are a marker of glucose metabolism disorders in
prediabetes and an indicator of hyperglycemia in diabetes.
Additionally, insulin sensitivity is negatively correlated with
β-HB levels (144). The
discrepancies between the conclusions made by Szili-Torok et
al (142) and Bae et
al (143) may be related to
ethnicity, or they may stem from the key role of insulin as a KB
regulator. Sufficient insulin secretion maintains low levels of KBs
by inhibiting the expression of hormone-sensitive lipase (144). Based on the existing evidence,
β-HB may serve as a novel predictive biomarker for the risk of
developing T2DM. Additionally, β-HB may act as an early diagnostic
biomarker for T2DM. Lucidi et al (145) conducted a study on 11 patients
with T2DM and found that, compared with the normal control
subjects, the blood β-HB levels were increased in patients with
T2DM and the level of β-HB was higher in the afternoon than in the
morning in these individuals. Garcia et al (146) employed nuclear magnetic
resonance spectroscopy to measure plasma KBs in 373 patients with
T2DM. The findings indicated an increase in all three types of KBs
in patients with T2DM, with KBs levels showing a negative
correlation with IR. In addition, elevated levels of β-HB may
reduce the risk of developing complications in T2DM (146). The β-HB levels are increased in
patients with T2DM and obesity (147,148). T2DM patients with impaired
ketogenic function exhibit reduced insulin sensitivity and an
increased risk of developing metabolism-related fatty liver disease
(149). However, among patients
with early-stage T2DM, those with intact ketogenic capacity have a
lower risk of hepatic steatosis or fibrosis (150). A study by Liu et al
(151) suggests that the higher
the β-HB level, the better the renal function and the lower the
risk of DKD. β-HB may also serve as a potential predictive
biomarker for the therapeutic response in T2DM. Lee et al
(152) showed that patients
with T2DM with high initial serum β-HB levels are more likely to
achieve well-controlled hemoglobin A1C levels after 6 months of
antidiabetic therapy.
Endogenous ketosis
Clinical studies indicate that lifestyle
modifications, including dietary control and regular physical
activity, can induce a state of NK that is beneficial for T2DM
(153,154). Dietary control encompasses IF,
CR and KD. IF refers to a period during which no food is consumed
either daily or weekly. During the fasting period, there is a shift
in metabolic pathways, transitioning from hepatic glucose
metabolism to adipocyte-derived ketone metabolism. A study has
shown that IF can alleviate IR in T2DM (155). Nuttall et al (156) conducted a 3-day fasting study
on male patients with T2DM and observed that plasma β-HB levels
increased (2.233±0.2 mM), while the insulin concentration remained
unchanged. Currently available studies have not explained this
result.
CR refers to a reduction of 25-50% in total daily
caloric intake while providing adequate nutritional components such
as essential amino acid and vitamins, to ensure that malnutrition
does not occur. Steven et al (157) demonstrated that a 6-month
intervention with a very low-calorie diet led to a decrease in
fasting blood glucose levels in patients with T2DM. Vigili et
al (158) showed that the
baseline levels of β-HB increased in patients with T2DM, and after
undergoing coronary angiography and an overnight fast, their β-HB
levels increased further. Although this was a small-scale clinical
trial, it also suggests that exploring and optimizing the
preoperative fasting protocol for this group of patients is
valuable.
KD is a formula diet characterized by a high
proportion of fat, a low proportion of carbohydrates and
appropriate amounts of protein and other nutrients. When undergoing
a KD, the body metabolizes to produce increased levels of KB, which
are utilized as an energy source (159). An extremely low-calorie KD can
effectively reduce body weight in patients with T2DM and enhance
glycemic control (160). In
patients with T2DM, a HFD (85% fat, 15% protein and virtually
carbohydrate-free) caused a sudden increase in β-HB only at the 8th
h, which reached its maximum at the 10th h; however, this effect
was far less significant than that of fasting. This mechanism may
be associated with effectors other than insulin (156). Intervention with a KD can
increase the plasma β-HB concentration in obese patients with T2DM
(from a baseline of 0.22 mM to 0.44 mM during the intervention
period). By contrast, in the group receiving combined intervention
of KD and β-HB supplementation, the fasting β-HB concentration
(baseline of 0.23 mM) reached a peak of 0.57 mM at 45 min and
returned to the baseline level after 120 min. Both intervention
approaches increased plasma insulin levels and C-peptide levels;
however, they had no significant effects on blood glucose control,
lipid metabolism or insulin sensitivity in muscle, liver and
adipose tissues (161). This
effect may be attributed to the conversion of β-HB into acetyl-CoA
in β-cells. Acetyl-Coenzyme A then enters mitochondrial metabolism
to generate ATP, which provides energy for the maintenance of
β-cell function and may directly stimulate glucose-induced insulin
secretion.
Therefore, the effects of IF, CR and KD on
promoting β-HB to regulate body weight, fasting blood glucose
levels, IR and other indicators in patients with T2DM remain
inconsistent. Moreover, additional research evidence is required to
clarify whether β-HB mediates these effects and to elucidate their
underlying mechanisms. It is noteworthy that although dietary
interventions can induce the human body to enter a state of NK, the
adaptation period is extremely long and difficult to sustain
(77). An excessively strict KD
may also lead to adverse side effects (162,163).
Exogenous ketosis
Exogenous ketosis can increase the ketone levels in
the body more rapidly and to a greater extent than endogenous
ketosis (164). Current ketone
supplements are roughly categorized into two major types: Ketone
salts and ketone esters (KEs). Ketone salts consist of β-HB bound
to minerals such as sodium, potassium and magnesium, whereas KE are
ketones bonded to precursor molecules such as 1,3-BDO (165). The ketone monoester (KME)
(R)-3-hydroxybutyl-(R)-3-hydroxybutyrate is a beverage that, upon
ingestion, is metabolized by intestinal esterases into β-HB and
1,3-BDO in equal proportions. Both compounds then enter the portal
circulation where the latter is transformed into β-HB in the liver
(166). Compared with
endogenous ketosis, exogenous KB supplementation can cause a sharp
increase in blood β-HB levels without the need for prolonged
fasting or adherence to a KD (167). Long-term exogenous
supplementation of β-HB can improve blood glucose control in
patients with T2DM. Soto-Mota et al (168) found that administering 25 ml of
exogenous ketone three times a day for 4 weeks reduced the levels
of glycemic control markers in patients with T2DM. However, the
effect of a single-dose intervention on blood glucose control
remains controversial. In a study by Monteyne et al
(169), 10 patients with T2DM
were enrolled. These patients were instructed to take
(R)-3-hydroxybutyl-(R)-3-hydroxybutyrate orally at a dose of 0.5
g/kg, 30 min before meals. The results showed that the plasma β-HB
concentration in the patients increased from 0.3±0.03 to a peak of
4.3±1.2 mmol/l and their postprandial blood glucose levels were
significantly reduced. Oral administration of KME before meals can
safely induce ketosis in T2DM patients and lower postprandial blood
glucose, providing a new metabolic intervention approach for
postprandial blood glucose management in patients with T2DM.
Falkenhain et al (170)
found that 30 min after a single oral administration of 0.3 g/kg
KME, blood β-HB levels increased from 0.2±0.1 mM to 1.5±0.8 mM,
reached a peak of 1.7-1.8±0.6 mM at 60-90 min and then decreased to
0.8±0.4 mM at 180 min. Additionally, serum insulin levels
increased, while lipolysis was inhibited and gluconeogenic
precursors were reduced; however, there was no effect on fasting
blood glucose levels. This discrepancy may be related to the dose
as a relatively small oral dose was used in the study.
Intravenous injection of β-HB (0.22 g/kg/h for 120
min) have been shown to improve working memory in patients with
T2DM (age, 65±4 years) (171).
By contrast, a study by Baranowski et al (172) found that acute (0.3 g/kg) or
short-term (15 g, 14 days) oral β-HB supplementation had no
beneficial effect on the cognitive function of patients with T2DM
(age, 30-70 years). These discrepancies may be related to the
duration of β-HB supplementation, the dosage administered and the
methods used to assess cognitive function. A study by Solis-Herrera
et al (173) found that
intravenous infusion of β-HB exerts a clear 'threshold effect' on
cardiac protection in patients with T2DM complicated by heart
failure. When the plasma β-HB concentration is ≥1.6 mmol/l, β-HB
provides energy substrates for the heart without inhibiting
myocardial glucose uptake, thereby improving left ventricular
systolic function. A 2-week KE intervention was shown to increase
the circulating β-HB level by ~10-fold in patients with T2DM
complicated by heart failure with preserved ejection fraction and
was accompanied by improvements in resting and exercise hemodynamic
status (174). This indicates
that β-HB supplementation exerts a protective effect on
cardiovascular function in patients with T2DM. The pharmacology and
safety of KEs have been intensively investigated in animals
(175), healthy humans
(176) and patients with T2DM
(168). However, most ketone
salts are racemic (with mixed chirality) and L-β-HB is not a
naturally occurring substance in the human body. For ketone salts
existing in the D/L form, if the amount of L-chiral isomer is
higher than the D-chiral isomer, it tends to prolong the time
originally required to induce ketosis (177). Ketone salts have a relatively
simple manufacturing process and low cost, but they are prone to
causing gastrointestinal side effects (178). By contrast, KEs have much
greater safety and tolerability in the human body than ketone salts
(179); however, they need to
be metabolized by the liver first before being degraded into acids
for absorption.
Conclusion
With the prevalence of T2DM and its related
complications on the rise, this condition has become a pressing
global health issue that poses a notable risk to human health and
well-being. The scientific community has been diligently
researching new preventative and therapeutic approaches. Recent
findings indicate that β-HB is a predominant KB with preventive and
therapeutic potential, functioning as an energy metabolite and
signaling molecule across various pathologies, including aging,
cancer, neurological disorders and T2DM. Current clinical studies
have shown a correlation between circulating β-HB levels and the
pathological progression of T2DM, suggesting its potential utility
as a diagnostic indicator for the disease. Additionally, β-HB,
whose levels are elevated either through endogenous ketosis or
exogenous supplementation, possesses pharmacological activity. This
activity may be associated with the pathogenesis of T2DM and β-HB
thus holds promise as a potential therapeutic agent for the
prevention and treatment of T2DM. Animal and cellular studies have
explained the potential mechanisms of β-HB in the pathological
progression of T2DM. β-HB exerts its effects by modulating glucose
and lipid metabolism, safeguarding pancreatic β-cells and
mitigating IR. Additionally, it acts as a signaling molecule that
promotes cellular protein homeostasis, inhibits oxidative stress
and inflammation, alleviates ERS and regulates mitochondrial
biosynthesis, autophagy, apoptosis and other pathways to combat
T2DM and its multi-organ complications.
Limitations and future directions
At present, the optimization of pathological
diagnostic indicators for T2DM, in-depth analysis of its
pathogenesis and the development of clinical drugs still face a
number of unsolved challenges. Particularly in the research field
related to β-HB, the limitations of existing evidence have
significantly restricted its clinical translational application.
There remain numerous pending issues to be addressed regarding the
pathological diagnostic indicators of T2DM, as well as in-depth
mechanistic research and the development and application of
clinical drugs.
At the clinical level, studies on the detection of
β-HB levels in patients with T2DM are not only scarce in quantity,
but the existing studies also generally suffer from limitations
such as small sample sizes and inconsistent detection methods.
These issues result in the inability to clearly define the
threshold value for T2DM at present, making it difficult to use
β-HB as a reliable indicator for clinical diagnosis or disease
condition assessment. In the field of mechanistic research, the
exploration of the mechanism by which β-HB acts on T2DM and its
complications remains confined to in vitro cell experiments
and animal model studies. There is a lack of mechanism validation
based on human clinical samples, leading to a distinct
'translational gap' between laboratory conclusions and practical
clinical applications. Meanwhile, existing animal and cellular
studies have notable technical limitations. Most studies focus on
the detection of plasma β-HB concentration, while neglecting the
differences in the expression and distribution of β-HB in T2DM
target organs such as the liver, brain and kidneys, making it
impossible to reveal its 'organ-specific regulatory effect'.
Although current studies have demonstrated that β-HB is generally
considered to exert metabolic benefits in most organ systems of
patients with T2DM, its effect of inhibiting GLP-1 secretion in
vitro (where GLP-1 is a first-line clinical target for glucose
lowering) creates a significant contradiction (180). In GLUTag cells (mouse colonic
endocrine cells), both low-dose (0.01 mM) and high-dose (100 mM)
β-HB inhibit glucose-induced GLP-1 secretion, while intermediate
doses have no effect. In human jejunum-like monolayer cells, β-HB
at a dose of 10 mM inhibits glucose-induced GLP-1 secretion
(180). This result is somewhat
pharmacologically puzzling. Even though the local level in the
intestinal mucosa is close to 10 mM (181), 100 mM is still far beyond the
physiological concentration in vivo. However, current
experimental studies only represent results at the cellular
experimental level and the effect of β-HB on GLP-1 in the digestive
system under in vivo conditions remains unknown.
Additionally, existing experiments involve only short-term exposure
with no consideration given to the impact of time. Additionally,
Wang et al (182) used
resonance Raman scattering technology and found that
intraperitoneal injection of β-HB improves mitochondrial function
in T2DM model mice, thereby alleviating pathological phenotypes of
T2DM such as elevated blood glucose, IR, systemic inflammation and
multi-organ damage. However, from an overall perspective, the
experimental methods are relatively singular and there is a lack of
application of precise tools such as omics technologies (such as
combined metabolomic and transcriptomic analysis) and plasmid
transfection (such as for the overexpression/silencing of specific
genes). This makes it difficult to systematically analyze the
interaction between β-HB and other signaling molecules as well as
the mechanism of multi-organ crosstalk.
In terms of regulatory strategies for the source of
β-HB, the induction of endogenous ketosis (such as IF, CR, KD and
exercise intervention) can increase circulating β-HB levels,
thereby providing a potential direction for the treatment of T2DM
and its complications. However, existing studies have not fully
considered the impact of the 'time factor'. For instance, questions
such as whether long-term endogenous ketosis can trigger metabolic
adaptation in the body and the differences in the regulatory effect
of β-HB between different intervention durations (such as
short-term vs. long-term KD) remain unclear. Additionally, most
intervention strategies utilize a single dose of β-HB (such as
intraperitoneal injection of β-HB salts at a fixed concentration).
Comparative studies investigating different doses and various
administration routes (such as intraperitoneal injection vs. oral
administration) have not been conducted, making it impossible to
clarify the 'dose-effect relationship' through which β-HB exerts
its effects and the optimal administration regimen. Notably,
prolonged moderate-intensity exercise or high-intensity interval
exercise can significantly increase the oxidation of fatty acids in
muscles, thereby promoting the production of KBs in the liver.
These KBs are then oxidized as an energy source during exercise and
increase significantly during the post-exercise recovery period;
the skeletal muscles adapted to exercise training exhibit a higher
capacity to utilize KBs (70).
During exercise, the oxidation of KBs in the muscles involved in
the exercise increases (183,184). Scientific exercise, as an
effective method for ketone elevation and improvement of T2DM, has
received little attention in research. Only Wang et al
(185), using untargeted
metabolomics, found that β-HB levels were decreased in the cardiac
tissue of Sprague Dawley rats with T2DM. However, 8 weeks of
aerobic exercise increased β-HB levels and improved cardiac
function in these T2DM rats. This suggests that β-HB may be a key
target for aerobic exercise to improve cardiac function in
T2DM.
Exogenous β-HB supplementation faces practical
challenges related to dosage forms. Ketone salt preparations,
though easily accessible, may cause gastrointestinal adverse
reactions (such as nausea and diarrhea) and excessive mineral load
in the body (such as sodium and potassium overload) and KE
preparations carry potential risks of excessive L-β-HB intake,
while pharmaceutically active pure D-β-HB acid preparations are
currently scarce in market supply and expensive, making them unable
to meet the needs of basic research and clinical translation. More
notably, the mechanisms of action between endogenously induced β-HB
production and exogenous β-HB supplementation in improving T2DM are
both related and distinct. The core regulatory networks and
interactive relationships between the two remain unclear, which
limits the precise design of β-HB intervention strategies. At the
level of drug development, as a potential drug, β-HB still lacks
comprehensive basic research on its quality standardization
(including purity and impurity control), pharmacodynamics
(including duration of action and the dose-effect relationship) and
pharmacokinetics (including absorption, distribution, metabolism
and excretion patterns), which further hinders its drug development
process.
In summary, β-HB has demonstrated clear potential
in the prevention and treatment of T2DM and its complications and
is expected to become a key focus in the fields of healthcare and
pharmaceutical research in the future. However, current research
must prioritize breaking through core bottlenecks such as the
standardization of clinical detection, the clinical translation of
mechanistic research, the optimization of intervention strategies
and the development of pharmaceutical dosage forms. Through the
integration of multidisciplinary technologies and multi-center
collaboration, β-HB research can be advanced from basic research to
clinical application. Ultimately, this will provide a novel and
efficient metabolic intervention regimen for the prevention and
treatment of T2DM, contributing to the development of the human
health industry.
Availability of data and materials
Not applicable.
Authors' contributions
XYD contributed to conceptualization, funding
acquisition, project administration, original draft writing as well
as reviewing and editing the manuscript. JBW, LW, KL, HYG, MYW, QYZ
and RNH contributed to reviewing and editing the manuscript. WHW
contributed to conceptualization as well as reviewing and editing
the manuscript; WHX contributed to funding acquisition, project
administration as well as reviewing and editing the manuscript.
Data authentication is not applicable. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
β-HB
|
β-hydroxybutyric acid
|
|
KB
|
ketone body
|
|
KD
|
ketogenic diet
|
|
BDH1
|
β-hydroxybutyrate dehydrogenase 1
|
|
MCTs
|
monocarboxylate transporters
|
|
SMCTs
|
sodium-dependent MCTs
|
|
IF
|
intermittent fasting
|
|
CR
|
caloric restriction
|
|
GPCR
|
G protein-coupled receptor
|
|
Kbhb
|
lysine β-hydroxybutyrylation
|
|
KA
|
ketoacidosis
|
|
NK
|
nutritional ketosis
|
|
DKA
|
diabetic ketoacidosis
|
|
DKD
|
diabetic kidney disease
|
|
1,3-BDO
|
1,3-butanediol
|
|
HCAR2
|
hydroxycarboxylic acid receptor 2
|
|
DCM
|
diabetic cardiomyopathy
|
|
DE
|
diabetic encephalopathy
|
|
AGEs
|
advanced glycation end products
|
|
DR
|
diabetic retinopathy
|
|
KE
|
ketone esters
|
|
KME
|
ketone monoester
|
Acknowledgements
Not applicable.
Funding
This study was sponsored by the National Natural Science
Foundation of China (grant no. 32371185), Shanghai Oriental Talents
Program (Youth Project), the Shanghai Science and Technology Plan
Project (grant no. 23010504200), the Key Lab of Exercise and Health
Sciences of Ministry of Education (Shanghai University of Sport)
(grant no. 2025KF002), the Shanghai Key Lab of Human Performance
(Shanghai University of Sport) (grant no. 11DZ2261100) and the
Research and Innovation Grant for Graduate Students (Shanghai
University of Sport) (grant no. YJSCX-2024-019).
References
|
1
|
IDF Diabetes Atlas 11th Edition 2025. The
International Diabetes Federation; 2025
|
|
2
|
Karamanou M, Protogerou A, Tsoucalas G,
Androutsos G and Poulakou-Rebelakou E: Milestones in the history of
diabetes mellitus: The main contributors. World J Diabetes. 7:1–7.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zheng Y, Ley SH and Hu FB: Global
aetiology and epidemiology of type 2 diabetes mellitus and its
complications. Nat Rev Endocrinol. 14:88–98. 2018. View Article : Google Scholar
|
|
4
|
Roden M and Shulman GI: The integrative
biology of type 2 diabetes. Nature. 576:51–60. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Holst JJ and Orskov C: Incretin
hormones-an update. Scand J Clin Lab Invest Suppl. 234:75–85.
2001.
|
|
6
|
Kwak SH and Park KS: Recent progress in
genetic and epigenetic research on type 2 diabetes. Exp Mol Med.
48:e2202016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu Y, Ding Y, Tanaka Y and Zhang W: Risk
factors contributing to type 2 diabetes and recent advances in the
treatment and prevention. Int J Med Sci. 11:1185–1200. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tan MH: α-Glucosidase inhibitors in the
treatment of diabetes. Curr Opin Endocrinol Diabetes Obesity.
4:48–55. 1997. View Article : Google Scholar
|
|
9
|
Chaudhury A, Duvoor C, Reddy Dendi VS,
Kraleti S, Chada A, Ravilla R, Marco A, Shekhawat NS, Montales MT,
Kuriakose K, et al: Clinical review of antidiabetic drugs:
Implications for type 2 diabetes mellitus management. Front
Endocrinol (Lausanne). 8:62017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nauck MA, Wefers J and Meier JJ: Treatment
of type 2 diabetes: Challenges, hopes, and anticipated successes.
Lancet Diabetes Endocrinol. 9:525–544. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rosenstock J, Bajaj HS, Lingvay I and
Heller SR: Clinical perspectives on the frequency of hypoglycemia
in treat-to-target randomized controlled trials comparing basal
insulin analogs in type 2 diabetes: A narrative review. BMJ Open
Diabetes Res Care. 12:e0039302024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Malakar S, Singh SK and Usman K:
Optimizing blood pressure management in type 2 Diabetes: A
comparative investigation of One-time versus periodic lifestyle
modification counseling. Cureus. 16:e616072024.PubMed/NCBI
|
|
13
|
Pitak P, Tasai S, Kumpat N, Na Songkla P,
Fuangchan A, Krass I and Dhippayom T: The prevalence of glycemic
control in patients with type 2 diabetes treated with insulin: A
systematic review and meta-analysis. Public Health. 225:218–228.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei S, Binbin L, Yuan W, Zhong Z, Donghai
L and Caihua H: β-Hydroxybutyrate in Cardiovascular diseases: A
minor metabolite of great expectations. Front Mol Biosci.
9:8236022022. View Article : Google Scholar
|
|
15
|
Holmes E, Wilson ID and Nicholson JK:
Metabolic phenotyping in health and disease. Cell. 134:714–717.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wishart DS: Metabolomics for investigating
physiological and pathophysiological processes. Physiol Rev.
99:1819–1875. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu D, Wang L, Gao H, Wang Z, Li K, Ma X,
Zhao L and Xiao W: Aerobic exercise delays Age-related sarcopenia
in mice via alleviating imbalance in mitochondrial quality control.
Metabolites. 15:4722025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ge Z, Chen C, Chen J, Jiang Z, Chen L, Wei
Y, Chen H, He L, Zou Y, Long X, et al: Gut Microbiota-derived
3-Hydroxybutyrate blocks GPR43-mediated IL6 signaling to ameliorate
radiation proctopathy. Adv Sci (Weinh). 11:e23062172024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li K, Wang WH, Wu JB and Xiao WH:
β-hydroxybutyrate: A crucial therapeutic target for diverse liver
diseases. Biomed Pharmacother. 165:1151912023. View Article : Google Scholar
|
|
20
|
Huang M, Yu Y, Tang X, Dong R, Li X, Li F,
Jin Y, Gong S, Wang X, Zeng Z, et al: 3-Hydroxybutyrate ameliorates
sepsis-associated acute lung injury by promoting autophagy through
the activation of GPR109α in macrophages. Biochem Pharmacol.
213:1156322023. View Article : Google Scholar
|
|
21
|
Mishima M, Takeda S, Nagane M, Suzuki T,
Ogata M, Shima A, Aihara N, Kamiie J, Suzuki R, Mizugaki H, et al:
Prebiotic effect of poly-D-3-hydroxybutyrate prevents dyslipidemia
in obese mice. FASEB J. 37:e231212023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y, Li Z, Liu X, Chen X, Zhang S,
Chen Y, Chen J, Chen J, Wu F and Chen GQ: 3-Hydroxybutyrate
ameliorates insulin resistance by inhibiting PPARγ Ser273
phosphorylation in type 2 diabetic mice. Signal Transduct Target
Ther. 8:1902023. View Article : Google Scholar
|
|
23
|
Zhou J, Lu Y, Jia Y, Lu J, Jiang Z and
Chen K: Ketogenic diet ameliorates lipid dysregulation in type 2
diabetic mice by downregulating hepatic pescadillo 1. Mol Med.
28:12022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guo Z, Zhong F, Hou M, Xie J, Zhang AZ, Li
X, Li Y, Chang B and Yang J: Key enzyme in charge of ketone
reabsorption of renal tubular SMCT1 may be a new target in diabetic
kidney disease. Nephrol Dial Transplant. 38:2754–2766. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nishitani S, Fukuhara A, Shin J, Okuno Y,
Otsuki M and Shimomura I: Metabolomic and microarray analyses of
adipose tissue of dapagliflozin-treated mice, and effects of
3-hydroxybutyrate on induction of adiponectin in adipocytes. Sci
Rep. 8:88052018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Puchalska P, Nelson AB, Stagg DB and
Crawford PA: Determination of ketone bodies in biological samples
via rapid UPLC-MS/MS. Talanta. 225:1220482021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Newman JC and Verdin E: β-Hydroxybutyrate:
A signaling metabolite. Annu Rev Nutr. 37:51–76. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McGarry JD and Foster DW: Ketogenesis and
its regulation. Am J Med. 61:9–13. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guzmán M and Blázquez C: Is there an
astrocyte-neuron ketone body shuttle? Trends Endocrinol Metab.
12:169–173. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Silva B, Mantha OL, Schor J, Pascual A,
Plaçais PY, Pavlowsky A and Preat T: Glia fuel neurons with locally
synthesized ketone bodies to sustain memory under starvation. Nat
Metab. 4:213–224. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang D, Yang H, Kong X, Wang K, Mao X,
Yan X, Wang Y, Liu S, Zhang X, Li J, et al: Proteomics analysis
reveals diabetic kidney as a ketogenic organ in type 2 diabetes. Am
J Physiol Endocrinol Metab. 300:E287–E295. 2011. View Article : Google Scholar
|
|
32
|
El Azzouny M, Longacre MJ, Ansari IH,
Kennedy RT, Burant CF and MacDonald MJ: Knockdown of ATP citrate
lyase in pancreatic beta cells does not inhibit insulin secretion
or glucose flux and implicates the acetoacetate pathway in insulin
secretion. Mol Metab. 5:980–987. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Adijanto J, Du J, Moffat C, Seifert EL,
Hurle JB and Philp NJ: The retinal pigment epithelium utilizes
fatty acids for ketogenesis. J Biol Chem. 289:20570–20582. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Reyes-Reveles J, Dhingra A, Alexander D,
Bragin A, Philp NJ and Boesze-Battaglia K: Phagocytosis-dependent
ketogenesis in retinal pigment epithelium. J Biol Chem.
292:8038–8047. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Grabacka MM, Wilk A, Antonczyk A, Banks P,
Walczyk-Tytko E, Dean M, Pierzchalska M and Reiss K: Fenofibrate
induces ketone body production in melanoma and glioblastoma cells.
Front Endocrinol (Lausanne). 7:52016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Venable AH, Lee LE, Feola K, Santoyo J,
Broomfield T and Huen SC: Fasting-induced HMGCS2 expression in the
kidney does not contribute to circulating ketones. Am J Physiol
Renal Physiol. 322:F460–F467. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Puchalska P and Crawford PA:
Multi-dimensional roles of ketone bodies in fuel metabolism,
signaling, and therapeutics. Cell Metab. 25:262–284. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mudaliar S, Alloju S and Henry RR: Can a
shift in fuel energetics explain the beneficial cardiorenal
outcomes in the EMPA-REG OUTCOME study? A unifying hypothesis.
Diabetes Care. 39:1115–1122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Orii KE, Fukao T, Song XQ, Mitchell GA and
Kondo N: Liver-specific silencing of the human gene encoding
succinyl-CoA: 3-ketoacid CoA transferase. Tohoku J Exp Med.
215:227–236. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Halestrap AP: The monocarboxylate
transporter family-Structure and functional characterization. IUBMB
Life. 64:1–9. 2012. View Article : Google Scholar
|
|
41
|
Bricker DK, Taylor EB, Schell JC, Orsak T,
Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N,
et al: A mitochondrial pyruvate carrier required for pyruvate
uptake in yeast, Drosophila, and humans. Science. 337:96–100. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Halestrap AP: The SLC16 gene
family-structure, role and regulation in health and disease. Mol
Aspects Med. 34:337–349. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Halestrap AP and Meredith D: The SLC16
gene family-from monocarboxylate transporters (MCTs) to aromatic
amino acid transporters and beyond. Pflugers Arch. 447:619–628.
2004. View Article : Google Scholar
|
|
44
|
Carneiro L and Pellerin L: Monocarboxylate
transporters: New players in body weight regulation. Obes Rev.
16(Suppl 1): S55–S66. 2015. View Article : Google Scholar
|
|
45
|
Vijay N and Morris ME: Role of
monocarboxylate transporters in drug delivery to the brain. Curr
Pharm Des. 20:1487–1498. 2014. View Article : Google Scholar :
|
|
46
|
Martin PM, Gopal E, Ananth S, Zhuang L,
Itagaki S, Prasad BM, Smith SB, Prasad PD and Ganapathy V: Identity
of SMCT1 (SLC5A8) as a neuron-specific Na+-coupled transporter for
active uptake of L-lactate and ketone bodies in the brain. J
Neurochem. 98:279–288. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bröer S, Schneider HP, Bröer A, Rahman B,
Hamprecht B and Deitmer JW: Characterization of the monocarboxylate
transporter 1 expressed in Xenopus laevis oocytes by changes in
cytosolic pH. Biochem J. 333:167–174. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hugo SE, Cruz-Garcia L, Karanth S,
Anderson RM, Stainier DY and Schlegel A: A monocarboxylate
transporter required for hepatocyte secretion of ketone bodies
during fasting. Genes Dev. 26:282–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Koehler-Stec EM, Simpson IA, Vannucci SJ,
Landschulz KT and Landschulz WH: Monocarboxylate transporter
expression in mouse brain. Am J Physiol. 275:E516–E524.
1998.PubMed/NCBI
|
|
50
|
Bonen A: The expression of lactate
transporters (MCT1 and MCT4) in heart and muscle. Eur J Appl
Physiol. 86:6–11. 2001. View Article : Google Scholar
|
|
51
|
Enerson BE and Drewes LR: Molecular
features, regulation, and function of monocarboxylate transporters:
Implications for drug delivery. J Pharm Sci. 92:1531–1544. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Felmlee MA, Morse BL and Morris ME:
γ-Hydroxybutyric acid: Pharmacokinetics, pharmacodynamics, and
toxicology. AAPS J. 23:222021. View Article : Google Scholar
|
|
53
|
Costa TJ, Linder BA, Hester S, Fontes M,
Pernomian L, Wenceslau CF, Robinson AT and McCarthy CG: The janus
face of ketone bodies in hypertension. J Hypertens. 40:2111–2119.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yao A, Li Z, Lyu J, Yu L, Wei S, Xue L,
Wang H and Chen GQ: On the nutritional and therapeutic effects of
ketone body D-β-hydroxybutyrate. Appl Microbiol Biotechnol.
105:6229–6243. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang L, Chen P and Xiao W:
β-hydroxybutyrate as an Anti-aging metabolite. Nutrients.
13:34202021. View Article : Google Scholar
|
|
56
|
Balasse EO and Féry F: Ketone body
production and disposal: Effects of fasting, diabetes, and
exercise. Diabetes Metab Rev. 5:247–270. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Veech RL, Chance B, Kashiwaya Y, Lardy HA
and Cahill GF Jr: Ketone bodies, potential therapeutic uses. IUBMB
Life. 51:241–247. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Veech RL: The therapeutic implications of
ketone bodies: The effects of ketone bodies in pathological
conditions: Ketosis, ketogenic diet, redox states, insulin
resistance, and mitochondrial metabolism. Prostaglandins Leukot
Essent Fatty Acids. 70:309–319. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hawkins RA and Biebuyck JF: Ketone bodies
are selectively used by individual brain regions. Science.
205:325–327. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Taggart AK, Kero J, Gan X, Cai TQ, Cheng
K, Ippolito M, Ren N, Kaplan R, Wu K, Wu TJ, et al:
(D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the
nicotinic acid receptor PUMA-G. J Biol Chem. 280:26649–26652. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kimura I, Inoue D, Maeda T, Hara T,
Ichimura A, Miyauchi S, Kobayashi M, Hirasawa A and Tsujimoto G:
Short-chain fatty acids and ketones directly regulate sympathetic
nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl
Acad Sci USA. 108:8030–8035. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Won YJ, Lu VB, Puhl HL III and Ikeda SR:
β-Hydroxybutyrate modulates N-type calcium channels in rat
sympathetic neurons by acting as an agonist for the
G-protein-coupled receptor FFA3. J Neuroscience. 33:19314–19325.
2013. View Article : Google Scholar
|
|
63
|
Shimazu T, Hirschey MD, Newman J, He W,
Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD,
et al: Suppression of oxidative stress by β-hydroxybutyrate, an
endogenous histone deacetylase inhibitor. Science. 339:211–214.
2013. View Article : Google Scholar
|
|
64
|
Xie Z, Zhang D, Chung D, Tang Z, Huang H,
Dai L, Qi S, Li J, Colak G, Chen Y, et al: Metabolic regulation of
gene expression by histone lysine β-hydroxybutyrylation. Mol Cell.
62:194–206. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Newman JC and Verdin E: Ketone bodies as
signaling metabolites. Trends Endocrinol Metab. 25:42–52. 2014.
View Article : Google Scholar :
|
|
66
|
Lund TM, Ploug KB, Iversen A, Jensen AA
and Jansen-Olesen I: The metabolic impact of β-hydroxybutyrate on
neurotransmission: Reduced glycolysis mediates changes in calcium
responses and KATP channel receptor sensitivity. J Neurochem.
132:520–531. 2015. View Article : Google Scholar
|
|
67
|
Youm YH, Nguyen KY, Grant RW, Goldberg EL,
Bodogai M, Kim D, D'Agostino D, Planavsky N, Lupfer C, Kanneganti
TD, et al: The ketone metabolite β-hydroxybutyrate blocks NLRP3
inflammasome-mediated inflammatory disease. Nat Med. 21:263–269.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Robinson AM and Williamson DH:
Physiological roles of ketone bodies as substrates and signals in
mammalian tissues. Physiological Rev. 60:143–187. 1980. View Article : Google Scholar
|
|
69
|
Pięta A, Frączek B, Wiecek M and
Mazur-Kurach P: Impact of paleo diet on body composition,
carbohydrate and fat metabolism of professional handball players.
Nutrients. 15:41552023. View Article : Google Scholar
|
|
70
|
Evans M, Cogan KE and Egan B: Metabolism
of ketone bodies during exercise and training: Physiological basis
for exogenous supplementation. J Physiol. 595:2857–2871. 2017.
View Article : Google Scholar :
|
|
71
|
Keller U, Lustenberger M, Müller-Brand J,
Gerber PP and Stauffacher W: Human ketone body production and
utilization studied using tracer techniques: Regulation by free
fatty acids, insulin, catecholamines, and thyroid hormones.
Diabetes Metab Rev. 5:285–298. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Poff AM, Koutnik AP and Egan B:
Nutritional ketosis with ketogenic diets or exogenous ketones:
Features, convergence, and divergence. Curr Sports Med Rep.
19:251–259. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Crabtree CD, Kackley ML, Buga A, Fell B,
LaFountain RA, Hyde PN, Sapper TN, Kraemer WJ, Scandling D,
Simonetti OP and Volek JS: Comparison of ketogenic diets with and
without ketone salts versus a Low-fat diet: Liver fat responses in
overweight adults. Nutrients. 13:9662021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Di Lorenzo C, Pinto A, Ienca R, Coppola G,
Sirianni G, Di Lorenzo G, Parisi V, Serrao M, Spagnoli A, Vestri A,
et al: A Randomized Double-blind, Cross-over trial of very
Low-calorie diet in overweight migraine patients: A possible role
for ketones? Nutrients. 11:17422019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Joo NS, Lee DJ, Kim KM, Kim BT, Kim CW,
Kim KN and Kim SM: Ketonuria after fasting may be related to the
metabolic superiority. J Korean Med Sci. 25:1771–1776. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kim G, Lee SG, Lee BW, Kang ES, Cha BS,
Ferrannini E, Lee YH and Cho NH: Spontaneous ketonuria and risk of
incident diabetes: A 12 year prospective study. Diabetologia.
62:779–788. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Firman CH, Mellor DD, Unwin D and Brown A:
Does a ketogenic diet have a place within diabetes clinical
practice? review of current evidence and controversies. Diabetes
Ther. 15:77–97. 2024. View Article : Google Scholar :
|
|
78
|
Dhillon KK and Gupta S: Biochemistry,
Ketogenesis. StatPearls StatPearls Publishing Copyright © 2025.
StatPearls Publishing LLC., Treasure Island (FL) ineligible
companies. Disclosure: Sonu Gupta declares no relevant financial
relationships with ineligible companies. 2025
|
|
79
|
Dhatariya KK, Glaser NS, Codner E and
Umpierrez GE: Diabetic ketoacidosis. Nat Rev Dis Primers. 6:402020.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Umpierrez GE, Davis GM, ElSayed NA, Fadini
GP, Galindo RJ, Hirsch IB, Klonoff DC, McCoy RG, Misra S, Gabbay
RA, et al: Hyperglycemic crises in adults with diabetes: A
consensus report. Diabetes Care. 47:1257–1275. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Cahill GF Jr and Veech RL: Ketoacids? Good
medicine? Trans Am Clin Climatol Assoc. 114:149–163.
2003.PubMed/NCBI
|
|
82
|
Stumvoll M, Goldstein BJ and van Haeften
TW: Type 2 diabetes: Principles of pathogenesis and therapy.
Lancet. 365:1333–1346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Galicia-Garcia U, Benito-Vicente A, Jebari
S, Larrea-Sebal A, Siddiqi H, Uribe KB, Ostolaza H and Martín C:
Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci.
21:62752020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Christensen AA and Gannon M: The beta cell
in type 2 diabetes. Curr Diab Rep. 19:812019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Yurista SR, Chong CR, Badimon JJ, Kelly
DP, de Boer RA and Westenbrink BD: Therapeutic potential of ketone
bodies for patients with cardiovascular disease: JACC
State-of-the-Art review. J Am Coll Cardiol. 77:1660–1669. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang L, Wang H, Wu J, Ji C, Wang Y, Gu M,
Li M and Yang H: Gut microbiota and metabolomics in metabolic
dysfunction-associated fatty liver disease: Interaction, mechanism,
and therapeutic value. Front Cell Infect Microbiol. 15:16356382025.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Song K, Kong X, Xian Y, Yu Z, He M, Xiao
D, Liang D, Zhang Z, Liu T, Huang Z, et al: Roux-en-Y gastric
bypass improves liver and glucose homeostasis in Zucker diabetic
fatty rats by upregulating hepatic trefoil factor family 3 and
activating the phosphatidylinositol 3-kinase/protein kinase B
pathway. Surg Obes Relat Dis. 21:792–805. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Xiang H, Lyu Q, Chen S, Ouyang J, Xiao D,
Liu Q, Long H, Zheng X, Yang X and Lu H: PACS2/CPT1A/DHODH
signaling promotes cardiomyocyte ferroptosis in diabetic
cardiomyopathy. Cardiovasc Diabetol. 23:4322024. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Xu BT, Teng FY, Wu Q, Wan SR, Li XY, Tan
XZ, Xu Y and Jiang ZZ: Bdh1 overexpression ameliorates hepatic
injury by activation of Nrf2 in a MAFLD mouse model. Cell Death
Discov. 8:492022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Jager KJ, Kovesdy C, Langham R, Rosenberg
M, Jha V and Zoccali C: A single number for advocacy and
communication-worldwide more than 850 million individuals have
kidney diseases. Kidney Int. 96:1048–1050. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Tuttle KR, Agarwal R, Alpers CE, Bakris
GL, Brosius FC, Kolkhof P and Uribarri J: Molecular mechanisms and
therapeutic targets for diabetic kidney disease. Kidney Int.
102:248–260. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Sagoo MK and Gnudi L: Diabetic
Nephropathy: An Overview. Diabetic Nephropathy: Methods and
Protocols. Gnudi L and Long DA: Springer US; New York, NY: pp. 3–7.
2020, View Article : Google Scholar
|
|
93
|
Gopal E, Fei YJ, Sugawara M, Miyauchi S,
Zhuang L, Martin P, Smith SB, Prasad PD and Ganapathy V: Expression
of slc5a8 in kidney and its role in Na(+)-coupled transport of
lactate. J Biol Chem. 279:44522–44532. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Xie J, Zhong F, Guo Z, Li X, Wang J, Gao
Z, Chang B and Yang J: Hyperinsulinemia impairs the metabolic
switch to ketone body utilization in proximal renal tubular
epithelial cells under energy crisis via the inhibition of the
SIRT3/SMCT1 pathway. Front Endocrinol (Lausanne). 13:9608352022.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wan SR, Teng FY, Fan W, Xu BT, Li XY, Tan
XZ, Guo M, Gao CL, Zhang CX, Jiang ZZ and Xu Y: BDH1-mediated βOHB
metabolism ameliorates diabetic kidney disease by activation of
NRF2-mediated antioxidative pathway. Aging. 15:13384–13410. 2023.
View Article : Google Scholar
|
|
96
|
Fang Y, Chen B, Gong AY, Malhotra DK,
Gupta R, Dworkin LD and Gong R: The ketone body β-hydroxybutyrate
mitigates the senescence response of glomerular podocytes to
diabetic insults. Kidney Int. 100:1037–1053. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Lean ME: Obesity: Burdens of illness and
strategies for prevention or management. Drugs Today. 36:773–784.
2000. View Article : Google Scholar
|
|
98
|
Biondi G, Marrano N, Borrelli A, Rella M,
Palma G, Calderoni I, Siciliano E, Lops P, Giorgino F and
Natalicchio A: Adipose tissue secretion pattern influences β-cell
wellness in the transition from obesity to type 2 diabetes. Int J
Mol Sci. 23:55222022. View Article : Google Scholar
|
|
99
|
Schaffer JE: Lipotoxicity: When tissues
overeat. Curr Opin Lipidol. 14:281–287. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Meex RCR, Blaak EE and van Loon LJC:
Lipotoxicity plays a key role in the development of both insulin
resistance and muscle atrophy in patients with type 2 diabetes.
Obesity Rev. 20:1205–1217. 2019. View Article : Google Scholar
|
|
101
|
Kim JE, Kim JS, Jo MJ, Cho E, Ahn SY, Kwon
YJ and Ko GJ: The roles and associated mechanisms of adipokines in
development of metabolic syndrome. Molecules. 27:3342022.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Booth A, Magnuson A, Fouts J and Foster
MT: Adipose tissue: An endocrine organ playing a role in metabolic
regulation. Horm Mol Biol Clin Investig. 26:25–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Kim AY, Park YJ, Pan X, Shin KC, Kwak SH,
Bassas AF, Sallam RM, Park KS, Alfadda AA, Xu A and Kim JB:
Obesity-induced DNA hypermethylation of the adiponectin gene
mediates insulin resistance. Nat Commun. 6:75852015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Sakurai N, Mochizuki K and Goda T:
Modifications of histone H3 at lysine 9 on the adiponectin gene in
3T3-L1 adipocytes. J Nutr Sci Vitaminol (Tokyo). 55:131–138. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Iwaki M, Matsuda M, Maeda N, Funahashi T,
Matsuzawa Y, Makishima M and Shimomura I: Induction of adiponectin,
a fat-derived antidiabetic and antiatherogenic factor, by nuclear
receptors. Diabetes. 52:1655–1663. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Segawa K, Matsuda M, Fukuhara A, Morita K,
Okuno Y, Komuro R and Shimomura I: Identification of a novel distal
enhancer in human adiponectin gene. J Endocrinol. 200:107–116.
2009. View Article : Google Scholar
|
|
107
|
Park S, Kim DS, Kang S and Daily JW III: A
ketogenic diet impairs energy and glucose homeostasis by the
attenuation of hypothalamic leptin signaling and hepatic insulin
signaling in a rat model of non-obese type 2 diabetes. Exp Biol Med
(Maywood). 236:194–204. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Biesiekierska M, Strigini M, Śliwińska A,
Pirola L and Balcerczyk A: The impact of ketogenic nutrition on
obesity and metabolic health: Mechanisms and clinical implications.
Nutr Rev. 83:1957–1972. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Carrière A, Jeanson Y, Berger-Müller S,
André M, Chenouard V, Arnaud E, Barreau C, Walther R, Galinier A,
Wdziekonski B, et al: Browning of white adipose cells by
intermediate metabolites: An adaptive mechanism to alleviate redox
pressure. Diabetes. 63:3253–3265. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
de Oliveira Caminhotto R, Andreotti S,
Komino ACM, de Fatima Silva F, Antônio Laurato Sertié R, Augusto
Christoffolete M, Boltes Reis G and Lima FB: Physiological
concentrations of β-hydroxybutyrate do not promote adipocyte
browning. Life Sci. 232:1166832019. View Article : Google Scholar
|
|
111
|
Gast KB, Tjeerdema N, Stijnen T, Smit JW
and Dekkers OM: Insulin resistance and risk of incident
cardiovascular events in adults without diabetes: Meta-analysis.
PLoS One. 7:e520362012. View Article : Google Scholar
|
|
112
|
Nesto RW: Correlation between
cardiovascular disease and diabetes mellitus: Current concepts. Am
J Med. 116(Suppl 5A): 11S–22S. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Dillmann WH: Diabetic Cardiomyopathy. Circ
Res. 124:1160–1162. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ritchie RH and Abel ED: Basic mechanisms
of diabetic heart disease. Circ Res. 126:1501–1525. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Song K, Liang D, Xiao D, Kang A and Ren Y:
Role of bariatric surgery in improving diabetic cardiomyopathy:
Molecular mechanisms and therapeutic perspectives (Review). Mol Med
Rep. 30:1992024. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wang S, Zhou J, Lu J, Lin Y, Liu S and
Chen K: A ketogenic diet improves vascular hyperpermeability in
type 2 diabetic mice by downregulating vascular pescadillo1
expression. J Cell Mol Med. 27:1410–1422. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Thai PN, Miller CV, King MT, Schaefer S,
Veech RL, Chiamvimonvat N, Bers DM and Dedkova EN: Ketone Ester
D-β-Hydroxybutyrate-(R)-1,3 butanediol prevents decline in cardiac
function in type 2 diabetic mice. J Am Heart Assoc. 10:e0207292021.
View Article : Google Scholar
|
|
118
|
Lin J, Ren Q, Zhang F, Gui J, Xiang X and
Wan Q: D-β-Hydroxybutyrate dehydrogenase mitigates Diabetes-induced
atherosclerosis through the activation of Nrf2. Thromb Haemost.
123:1003–1015. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Uchihashi M, Hoshino A, Okawa Y, Ariyoshi
M, Kaimoto S, Tateishi S, Ono K, Yamanaka R, Hato D, Fushimura Y,
et al: Cardiac-Specific Bdh1 overexpression ameliorates oxidative
stress and cardiac remodeling in pressure overload-induced heart
failure. Circ Heart Fail. 10:e0044172017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
van Knegsel AT, van den Brand H, Dijkstra
J, Tamminga S and Kemp B: Effect of dietary energy source on energy
balance, production, metabolic disorders and reproduction in
lactating dairy cattle. Reprod Nutr Dev. 45:665–688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Liepinsh E, Vilskersts R, Zvejniece L,
Svalbe B, Skapare E, Kuka J, Cirule H, Grinberga S, Kalvinsh I and
Dambrova M: Protective effects of mildronate in an experimental
model of type 2 diabetes in Goto-Kakizaki rats. Br J Pharmacol.
157:1549–1556. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Antal B, McMahon LP, Sultan SF, Lithen A,
Wexler DJ, Dickerson B, Ratai EM and Mujica-Parodi LR: Type 2
diabetes mellitus accelerates brain aging and cognitive decline:
Complementary findings from UK Biobank and meta-analyses. eLife.
11:e731382022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Reske-Nielsen E, Lundbaek K, Gregersen G
and Harmsen A: Pathological changes in the central and peripheral
nervous system of young long-term diabetics. The terminal
neuro-muscular apparatus. Diabetologia. 6:98–103. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Lionetti N, Di Lago MG, Brescia T,
Bevilacqua F and Gnoni A: Diabetes and brain: Omics approaches to
study diabetic encephalopathy. Front Endocrinol. 16:15705852025.
View Article : Google Scholar
|
|
125
|
Xu Y, Huang C, Zhang Y, Li H, Yang H, Liu
M, Zhu L, Li C, Zhong Y, Tang L, et al: Diabetic encephalopathy
models: A systematic review from cells to animals. Exp Neurol.
395:1154772025. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Hein ZM, Arbain MFF, Kumar S, Mehat MZ,
Hamid HA, Che Ramli MD and Che Mohd Nassir CMN: Intermittent
fasting as a neuroprotective strategy: Gut-brain axis modulation
and metabolic reprogramming in neurodegenerative disorders.
Nutrients. 17:22662025. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Andersen JV, Christensen SK, Nissen JD and
Waagepetersen HS: Improved cerebral energetics and ketone body
metabolism in db/db mice. J Cereb Blood Flow Metab. 37:1137–1147.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Pierre K, Parent A, Jayet PY, Halestrap
AP, Scherrer U and Pellerin L: Enhanced expression of three
monocarboxylate transporter isoforms in the brain of obese mice. J
Physiol. 583:469–486. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Pellerin L, Bergersen LH, Halestrap AP and
Pierre K: Cellular and subcellular distribution of monocarboxylate
transporters in cultured brain cells and in the adult brain. J
Neurosci Res. 79:55–64. 2005. View Article : Google Scholar
|
|
130
|
Park S, Kim DS and Daily JW: Central
infusion of ketone bodies modulates body weight and hepatic insulin
sensitivity by modifying hypothalamic leptin and insulin signaling
pathways in type 2 diabetic rats. Brain Res. 1401:95–103. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Majrashi M, Altukri M, Ramesh S,
Govindarajulu M, Schwartz J, Almaghrabi M, Smith F, Thomas T,
Suppiramaniam V, Moore T, et al: β-hydroxybutyric acid attenuates
oxidative stress and improves markers of mitochondrial function in
the HT-22 hippocampal cell line. J Integr Neurosci. 20:321–329.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Dabke P, Brogden G, Naim HY and Das AM:
Ketogenic diet: Impact on cellular lipids in hippocampal murine
neurons. Nutrients. 12:38702020. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Sayin N, Kara N and Pekel G: Ocular
complications of diabetes mellitus. World J Diabetes. 6:92–108.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Jian Q, Wu Y and Zhang F: Metabolomics in
diabetic retinopathy: From potential biomarkers to molecular basis
of oxidative stress. Cells. 11:30052022. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Herat LY, Matthews VB, Rakoczy PE,
Carnagarin R and Schlaich M: Focusing on sodium glucose
cotransporter-2 and the sympathetic nervous system: Potential
impact in diabetic retinopathy. Int J Endocrinol. 2018:92541262018.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Heng LZ, Comyn O, Peto T, Tadros C, Ng E,
Sivaprasad S and Hykin PG: Diabetic retinopathy: Pathogenesis,
clinical grading, management and future developments. Diabet Med.
30:640–650. 2013. View Article : Google Scholar
|
|
137
|
Wong TY, Cheung CM, Larsen M, Sharma S and
Simó R: Diabetic retinopathy. Nat Rev Dis Primers. 2:160122016.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Saika S, Yamanaka O, Okada Y, Tanaka S,
Miyamoto T, Sumioka T, Kitano A, Shirai K and Ikeda K: TGF beta in
fibroproliferative diseases in the eye. Front Biosci (Schol Ed).
1:376–390. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
139
|
Yadav H, Quijano C, Kamaraju AK, Gavrilova
O, Malek R, Chen W, Zerfas P, Zhigang D, Wright EC, Stuelten C, et
al: Protection from obesity and diabetes by blockade of TGF-β/Smad3
signaling. Cell Metab. 14:67–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Chen HY, Ho YJ, Chou HC, Liao EC, Tsai YT,
Wei YS, Lin LH, Lin MW, Wang YS, Ko ML and Chan HL: The role of
transforming growth factor-beta in retinal ganglion cells with
hyperglycemia and oxidative stress. Int J Mol Sci. 21:64822020.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Trotta MC, Maisto R, Guida F, Boccella S,
Luongo L, Balta C, D'Amico G, Herman H, Hermenean A, Bucolo C and
D'Amico M: The activation of retinal HCA2 receptors by systemic
beta-hydroxybutyrate inhibits diabetic retinal damage through
reduction of endoplasmic reticulum stress and the NLRP3
inflammasome. PLoS One. 14:e02110052019. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Szili-Torok T, de Borst MH, Garcia E,
Gansevoort RT, Dullaart RPF, Connelly MA, Bakker SJL and Tietge
UJF: Fasting ketone bodies and incident type 2 diabetes in the
general population. Diabetes. 72:1187–1192. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Bae J, Kim YE, Jung KJ, Jee SH and Lee BW:
Association between serum beta-hydroxybutyrate levels and risk of
type 2 diabetes mellitus in patients with impaired fasting glucose.
Nutr Diabetes. 15:162025. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Mahendran Y, Vangipurapu J, Cederberg H,
Stancáková A, Pihlajamäki J, Soininen P, Kangas AJ, Paananen J,
Civelek M, Saleem NK, et al: Association of ketone body levels with
hyperglycemia and type 2 diabetes in 9,398 Finnish men. Diabetes.
62:3618–3626. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Lucidi P, Perriello G, Porcellati F,
Pampanelli S, De Fano M, Tura A, Bolli GB and Fanelli CG: Diurnal
cycling of insulin sensitivity in type 2 diabetes: Evidence for
deviation from physiology at an early stage. Diabetes.
72:1364–1373. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Garcia E, Shalaurova I, Matyus SP,
Oskardmay DN, Otvos JD, Dullaart RPF and Connelly MA: Ketone bodies
are mildly elevated in subjects with type 2 diabetes mellitus and
are inversely associated with insulin resistance as measured by the
lipoprotein insulin resistance index. J Clin Med. 9:3212020.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Gogna N, Krishna M, Oommen AM and Dorai K:
Investigating correlations in the altered metabolic profiles of
obese and diabetic subjects in a South Indian Asian population
using an NMR-based metabolomic approach. Mol Biosyst. 11:595–606.
2015. View Article : Google Scholar
|
|
148
|
Fikri AM, Smyth R, Kumar V, Al-Abadla Z,
Abusnana S and Munday MR: Pre-diagnostic biomarkers of type 2
diabetes identified in the UAE's obese national population using
targeted metabolomics. Sci Rep. 10:176162020. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Lee S, Bae J, Jo DR, Lee M, Lee YH, Kang
ES, Cha BS and Lee BW: Impaired ketogenesis is associated with
metabolic-associated fatty liver disease in subjects with type 2
diabetes. Front Endocrinol. 14:11245762023. View Article : Google Scholar
|
|
150
|
Lim K, Kang M and Park J: Association
between fasting ketonuria and advanced liver fibrosis in
non-alcoholic fatty liver disease patients without prediabetes and
diabetes mellitus. Nutrients. 13:34002021. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Liu Y, Wang J, Xu F, Zhang S, Cui S, Li Y,
Wang X, Zheng H, Li J, Kong Y, et al: A J-shaped relationship
between ketones and the risk of diabetic kidney disease in patients
with type 2 diabetes: New insights from a cross-sectional study.
Diabetes Obes Metab. 25:3317–3326. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Lee M, Cho Y, Lee YH, Kang ES, Cha BS and
Lee BW: β-hydroxybutyrate as a biomarker of β-cell function in
new-onset type 2 diabetes and its association with treatment
response at 6 months. Diabetes Metab. 49:1014272023. View Article : Google Scholar
|
|
153
|
Park SB and Yang SJ: Ketogenic diet
preserves muscle mass and strength in a mouse model of type 2
diabetes. PLoS One. 19:e02966512024. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Miller VJ, Villamena FA and Volek JS:
Nutritional Ketosis and Mitohormesis: Potential Implications for
Mitochondrial Function and Human Health. J Nutr Metab.
2018:51576452018. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Arnason TG, Bowen MW and Mansell KD:
Effects of intermittent fasting on health markers in those with
type 2 diabetes: A pilot study. World J Diabetes. 8:154–164. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Nuttall FQ, Almokayyad RM and Gannon MC:
Circulating lipids in men with type 2 diabetes following 3 days on
a carbohydrate-free diet versus 3 days of fasting. Physiological
Rep. 8:e145692020. View Article : Google Scholar
|
|
157
|
Steven S, Hollingsworth KG, Al-Mrabeh A,
Avery L, Aribisala B, Caslake M and Taylor R: Very low-calorie diet
and 6 months of weight stability in type 2 diabetes:
Pathophysiological changes in responders and nonresponders.
Diabetes Care. 39:808–815. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Vigili de Kreutzenberg S and Avogaro A:
The role of point-of-care 3-hydroxybutyrate testing in patients
with type 2 diabetes undergoing coronary angiography. J Endocrinol
Invest. 40:627–634. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Goldberg IJ, Ibrahim N, Bredefeld C, Foo
S, Lim V, Gutman D, Huggins LA and Hegele RA: Ketogenic diets, not
for everyone. J Clin Lipidol. 15:61–67. 2021. View Article : Google Scholar :
|
|
160
|
Goday A, Bellido D, Sajoux I, Crujeiras
AB, Burguera B, García-Luna PP, Oleaga A, Moreno B and Casanueva
FF: Short-term safety, tolerability and efficacy of a very
low-calorie-ketogenic diet interventional weight loss program
versus hypocaloric diet in patients with type 2 diabetes mellitus.
Nutr Diabetes. 6:e2302016. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Merovci A, Finley B, Hansis-Diarte A,
Neppala S, Abdul-Ghani MA, Cersosimo E, Triplitt C and DeFronzo RA:
Effect of weight-maintaining ketogenic diet on glycemic control and
insulin sensitivity in obese T2D subjects. BMJ Open Diabetes Res
Care. 12:e0041992024. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Durrer C, Lewis N, Wan Z, Ainslie PN,
Jenkins NT and Little JP: Short-term Low-carbohydrate high-fat diet
in healthy young males renders the endothelium susceptible to
hyperglycemia-induced damage, an exploratory analysis. Nutrients.
11:4892019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Harvey C, Schofield GM and Williden M: The
use of nutritional supplements to induce ketosis and reduce
symptoms associated with keto-induction: A narrative review. PeerJ.
6:e44882018. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Holland AM, Qazi AS, Beasley KN and
Bennett HR: Blood and cardiovascular health parameters after
supplementing with ketone salts for six weeks. J Insul Resist.
4:e472019.
|
|
165
|
Falkenhain K, Islam H and Little JP:
Exogenous ketone supplementation: An emerging tool for
physiologists with potential as a metabolic therapy. Exp Physiol.
108:177–187. 2023. View Article : Google Scholar
|
|
166
|
Suissa L, Kotchetkov P, Guigonis JM, Doche
E, Osman O, Pourcher T and Lindenthal S: Ingested ketone ester
leads to a rapid rise of Acetyl-CoA and competes with glucose
metabolism in the brain of Non-fasted mice. Int J Mol Sci.
22:5242021. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Stubbs BJ, Cox PJ, Evans RD, Santer P,
Miller JJ, Faull OK, Magor-Elliott S, Hiyama S, Stirling M and
Clarke K: On the metabolism of exogenous ketones in humans. Front
Physiol. 8:8482017. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Soto-Mota A, Norwitz NG, Evans R, Clarke K
and Barber TM: Exogenous ketosis in patients with type 2 diabetes:
Safety, tolerability and effect on glycaemic control. Endocrinol
Diabetes Metab. 4:e002642021. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Monteyne AJ, Falkenhain K, Whelehan G,
Neudorf H, Abdelrahman DR, Murton AJ, Wall BT, Stephens FB and
Little JP: A ketone monoester drink reduces postprandial blood
glucose concentrations in adults with type 2 diabetes: A randomised
controlled trial. Diabetologia. 67:1107–1113. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Falkenhain K, Oliveira BF, Islam H,
Neudorf H, Cen HH, Johnson JD, Madden K, Singer J, Walsh JJ and
Little JP: The effect of acute and 14-day exogenous ketone
supplementation on glycemic control in adults with type 2 diabetes:
Two randomized controlled trials. Am J Physiol Endocrinol Metab.
326:E61–E72. 2024. View Article : Google Scholar
|
|
171
|
Jensen NJ, Nilsson M, Ingerslev JS, Olsen
DA, Fenger M, Svart M, Møller N, Zander M, Miskowiak KW and Rungby
J: Effects of β-hydroxybutyrate on cognition in patients with type
2 diabetes. Eur J Endocrinol. 182:233–242. 2020. View Article : Google Scholar
|
|
172
|
Baranowski BJ, Oliveira BF, Falkenhain K,
Little JP, Mohammad A, Beaudette SM, Finch MS, Caldwell HG, Neudorf
H, MacPherson REK and Walsh JJ: Effect of exogenous
β-hydroxybutyrate on BDNF signaling, cognition, and amyloid
precursor protein processing in humans with T2D and
insulin-resistant rodents. Am J Physiol Cell Physiol.
328:C541–C556. 2025. View Article : Google Scholar
|
|
173
|
Solis-Herrera C, Qin Y, Honka H, Cersosimo
E, Triplitt C, Neppala S, Rajan J, Acosta FM, Moody AJ, Iozzo P, et
al: Effect of hyperketonemia on myocardial function in patients
with heart failure and type 2 diabetes. Diabetes. 74:43–52. 2025.
View Article : Google Scholar
|
|
174
|
Gopalasingam N, Berg-Hansen K, Christensen
KH, Ladefoged BT, Poulsen SH, Andersen MJ, Borlaug BA, Nielsen R,
Møller N and Wiggers H: Randomized crossover trial of 2-week ketone
ester treatment in patients with type 2 diabetes and heart failure
with preserved ejection fraction. Circulation. 150:1570–1583. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Clarke K, Tchabanenko K, Pawlosky R,
Carter E, Knight NS, Murray AJ, Cochlin LE, King MT, Wong AW,
Roberts A, et al: Oral 28-day and developmental toxicity studies of
(R)-3-hydroxybutyl (R)-3-hydroxybutyrate. Regul Toxicol Pharmacol.
63:196–208. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Clarke K, Tchabanenko K, Pawlosky R,
Carter E, Todd King M, Musa-Veloso K, Ho M, Roberts A, Robertson J,
Vanitallie TB and Veech RL: Kinetics, safety and tolerability of
(R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects.
Regul Toxicol Pharmacol. 63:401–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Engel MH and Macko SA: Isotopic evidence
for extraterrestrial non-racemic amino acids in the Murchison
meteorite. Nature. 389:265–268. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
178
|
Fischer T, Och U, Klawon I, Och T,
Grüneberg M, Fobker M, Bordewick-Dell U and Marquardt T: Effect of
a Sodium and Calcium DL-β-Hydroxybutyrate salt in healthy adults. J
Nutr Metab. 2018:98128062018. View Article : Google Scholar
|
|
179
|
Soto-Mota A, Vansant H, Evans RD and
Clarke K: Safety and tolerability of sustained exogenous ketosis
using ketone monoester drinks for 28 days in healthy adults. Regul
Toxicol Pharmacol. 109:1045062019. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Elebring E, Casselbrant A, Persson SMT,
Fändriks L and Wallenius V: βHB inhibits glucose-induced GLP-1
secretion in GLUTag and human jejunal enteroids. J Mol Endocrinol.
70:e2201152023. View Article : Google Scholar
|
|
181
|
Wallenius V, Elias E, Elebring E, Haisma
B, Casselbrant A, Larraufie P, Spak E, Reimann F, le Roux CW,
Docherty NG, et al: Suppression of enteroendocrine cell
glucagon-like peptide (GLP)-1 release by fat-induced small
intestinal ketogenesis: A mechanism targeted by Roux-en-Y gastric
bypass surgery but not by preoperative very-low-calorie diet. Gut.
69:1423–1431. 2020. View Article : Google Scholar
|
|
182
|
Wang N, Yang A, Tian X, Liao J, Yang Z,
Pan Y, Guo Y and He S: Label-free analysis of the
β-hydroxybutyricacid drug on mitochondrial redox states repairment
in type 2 diabetic mice by resonance raman scattering. Biomed
Pharmacother. 172:1163202024. View Article : Google Scholar
|
|
183
|
Féry F and Balasse EO: Response of ketone
body metabolism to exercise during transition from postabsorptive
to fasted state. Am J Physiol. 250:E495–E501. 1986.PubMed/NCBI
|
|
184
|
Féry F and Balasse EO: Effect of exercise
on the disposal of infused ketone bodies in humans. J Clin
Endocrinol Metab. 67:245–250. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Wang T, Ning M, Mo Y, Tian X, Fu Y, Laher
I and Li S: Metabolomic profiling reveals that exercise lowers
biomarkers of cardiac dysfunction in rats with type 2 diabetes.
Antioxidants (Basel). 13:11672024. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Kramer CK, Zinman B, Feig DS and
Retnakaran R: Effect of time-restricted eating on β-cell function
in adults with type 2 diabetes: A randomized cross-over trial. J
Clin Endocrinol Metab. 110:e2045–e2053. 2025. View Article : Google Scholar
|














