Cardiovascular diseases (CVDs) continue to be the
primary cause of death worldwide, with myocardial infarction
contributing markedly to global morbidity and mortality (1). The timely restoration of coronary
blood flow through thrombolytic therapy or percutaneous coronary
intervention (PCI) has been proven to effectively reduce infarct
size (2). However,
ischemia/reperfusion (I/R) injury induces secondary myocardial
damage, mainly affecting cardiomyocytes and cardiac microvascular
endothelial cells (CMECs). This secondary injury is characterized
by myocardial stunning, microvascular dysfunction, reperfusion
arrhythmias and lethal reperfusion injury (3). Microvascular I/R injury involves
CMEC apoptosis, microvascular spasm and impaired perfusion, all of
which exacerbate myocardial damage after reperfusion (4). Beyond conventional clinical risk
scores and left ventricular ejection fraction, microvascular
obstruction due to I/R injury has been identified as an independent
predictor of infarct size and poor prognosis (5-7).
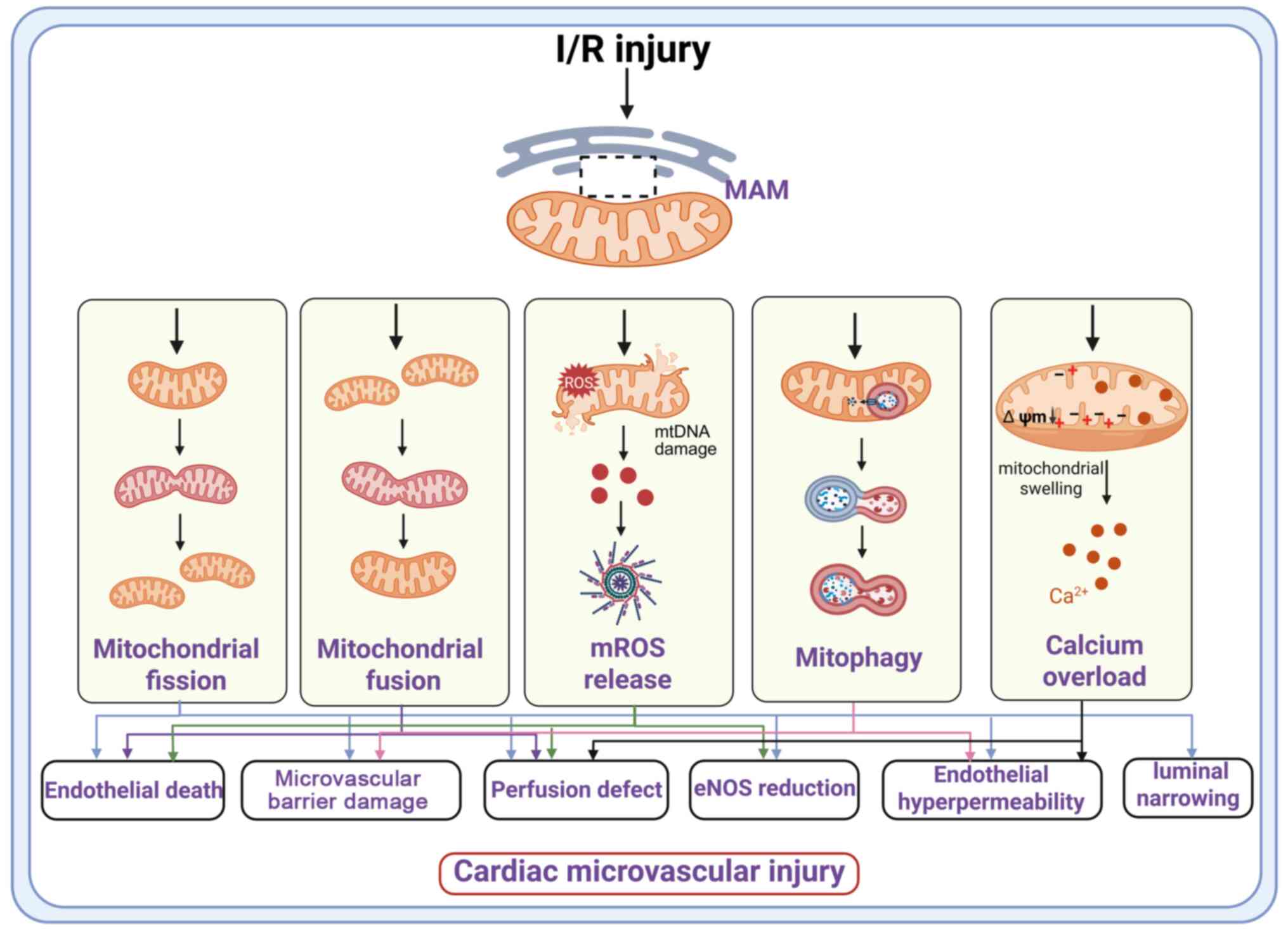
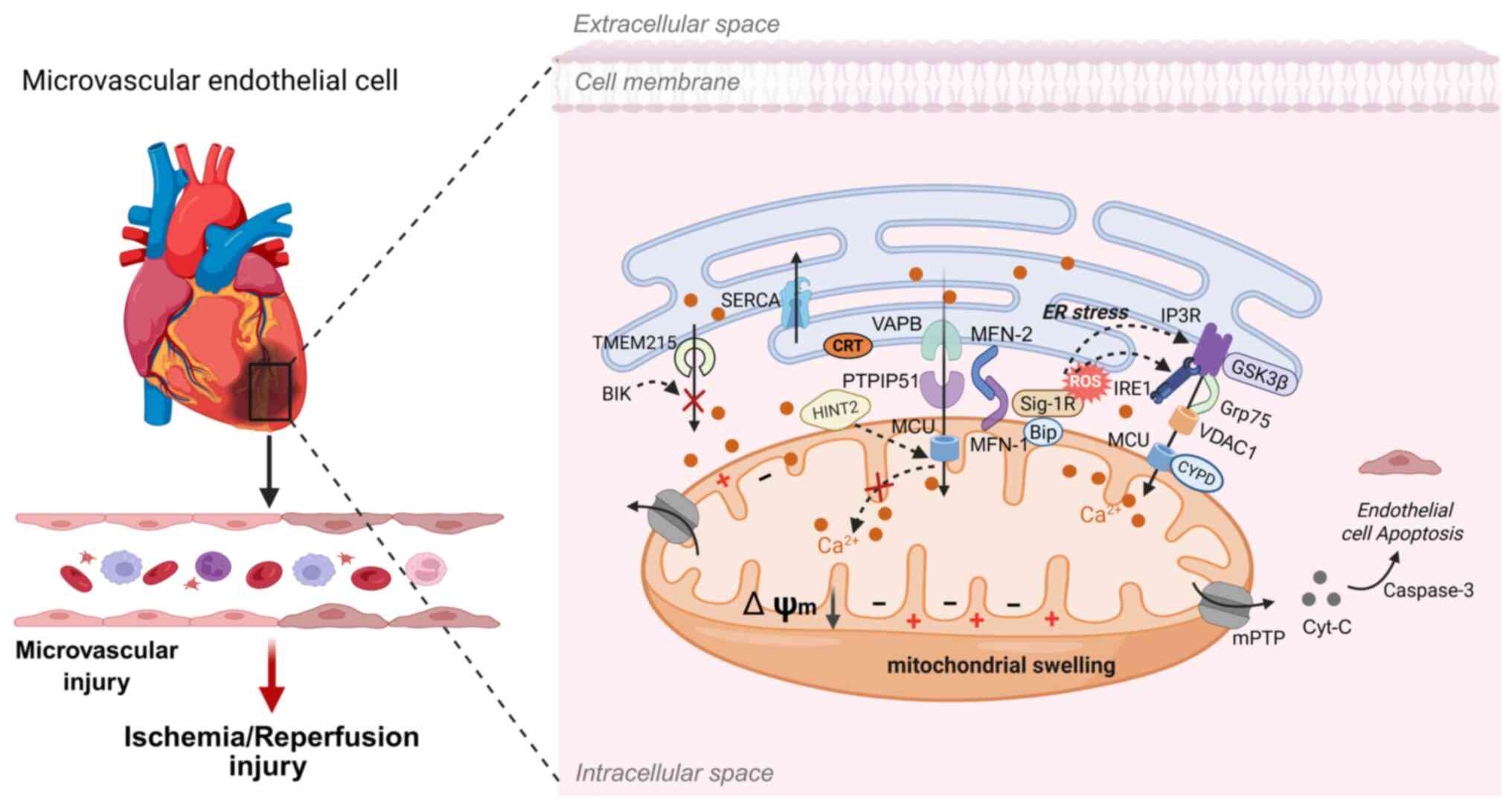
Mitochondria-associated endoplasmic reticulum
membranes (MAMs) serve as key signaling hubs involved in
fundamental cellular processes, including calcium (Ca2+)
and lipid homeostasis, mitochondrial dynamics and energy production
(8). During cardiac
microvascular I/R injury, abnormal mitochondrial dynamics and
impaired mitophagy (2,9) further exacerbate microvascular
dysfunction by compromising vascular patency, altering vascular
tone and amplifying inflammatory responses (10). Further, excessive mitochondrial
Ca2+ uptake leads to overactivation of the electron
transport chain (ETC) and excessive generation of reactive oxygen
species (ROS) (11). This
pathological cascade leads to the collapse of the mitochondrial
membrane potential and promotes the opening of the mitochondrial
permeability transition pore (mPTP) (12). Thus, mitochondrial dysfunction
represents a central mechanism in microvascular I/R injury
(13,14) (Fig. 1).
The endoplasmic reticulum (ER), the largest cellular
organelle composed of a continuous network of tubules and sheets,
plays a fundamental role in protein folding, lipid synthesis and
Ca2+ storage (15,16). Maintenance of ER proteostasis is
vital for secretory function, whereas prolonged ER stress drives
cellular dysfunction and contributes to the pathogenesis of CVD
(17). Research highlights
dynamic crosstalk between the ER and mitochondria as a critical
regulator of cellular homeostasis (18). First described by Wilhelm
Bernhard in 1956 as the connection between the ER and mitochondria
(19), MAMs were later isolated
as unique subcellular structures using subcellular isolation
techniques and density-gradient centrifugation (20). Disruption of MAM integrity
impairs ER-mitochondria communication, leading to homeostatic
imbalance and contributing to various diseases, including cancer,
neurological disorders and CVDs (21). The present review summarized
recent advances in MAM-mediated regulation of mitochondrial quality
control and Ca2+ signaling, offering valuable insights
for future investigations into the molecular mechanisms with
particular emphasis on potential therapeutic strategies.
Mitochondria are central to cellular bioenergetics
and are traditionally described as the powerhouse of the cell,
producing ATP through oxidative phosphorylation driven by the ETC
and oxygen reduction (22). As
well as ATP generation, mitochondria regulate apoptosis,
Ca2+ homeostasis, inflammation and immune responses
(23). In microvascular
endothelial cells, which primarily depend on glycolysis for energy,
mitochondria exhibit a punctate distribution pattern (24). Mitochondria consist of a double
membrane defining four compartments: the outer mitochondrial
membrane (OMM), the intermembrane space (IMS), the inner
mitochondrial membrane (IMM) and the matrix. The OMM contains
porins that mediate exchange with other organelles (25), the IMS stores key apoptotic
factors (26) and the IMM, with
its highly invaginated cristae, harbors the ETC and ATP synthase,
enabling efficient ATP synthesis (27). The IMM is characterized by its
low permeability and high cardiolipin content (28).
MAMs are dynamic contact sites where the OMM and ER
membranes are closely apposed but remain distinct (29). The distance between the two
membranes ranges from ~10 nm up to 80-100 nm (29), as identified by electron
microscopy, with smooth ER-mitochondria contacts typically closer
(10-15 nm) than rough ER contacts (20-30 nm) (30), suggesting that gap width
influences the degree of functional coupling (31). Based on the extent of
ER-mitochondria membrane contact, MAMs are categorized into three
forms: Type I (partial wrapping, ~10% OMM coverage), Type II
(extensive contact, ≤50% of the mitochondrial surface) and Type III
(complete encapsulation) (32).
Type I predominates in most cells, encompassing 10-15% of the OMM
(31).
Studies underscore the therapeutic potential of
targeting MAMs to mitigate hypoxia-induced endothelial injury
(33). Hypoxia directly damages
endothelial mitochondria, characterized by elevated mitochondrial
ROS (mROS), impaired mitophagy, reduced mitochondrial biogenesis
and mitochondrial cristae disruption (34-36). Regulatory proteins and non-coding
(nc)RNAs play key roles in this context. MARCH5, a key regulator of
mitochondrial dynamics, apoptosis and mitophagy, protects
endothelial cells against hypoxia-induced injury via Akt/eNOS
signaling (37), whereas RIPK3
exacerbates damage by upregulating 1,4,5-trisphosphate receptors
(IP3R) expression, leading to Ca2+ overload
and oxidative damage (38). The
long (l)ncRNA Malat1 preserves microvascular function after
myocardial infarction by regulating mitochondrial dynamics through
the miR-26b-5p/mitofusin-1 (MFN1) axis (39). Similarly, the compound FL3
promotes MFN1-dependent mitochondrial fusion, stabilizes
Ca2+ homeostasis and alleviates myocardial injury
(40). These findings
demonstrate that endothelial dysfunction and mitochondrial stress
are closely associated with the pathogenesis of microvascular I/R
injury. Targeted modulation of MAMs, therefore, represents a
promising therapeutic strategy for protecting endothelial integrity
and preserving cardiac function (41-43).
As a crucial component of the mitochondrial quality
control (MQC) system, mitochondrial dynamics facilitates the
selective removal of damaged organelles and renewal of functional
ones (44). Mitochondria
continuously undergo fission, fusion and degradation to preserve
endothelial homeostasis in response to various cellular cues
(45,46). The balance between these fusion
and fission processes determines mitochondrial morphology and
functionality (47-49). Enhanced fusion or reduced fission
promotes the formation of an elongated mitochondrial network,
whereas excessive fission or impaired fusion leads to mitochondrial
fragmentation (50-52).
MAMs serve as a vital regulatory hub that
coordinates mitochondrial by recruiting specific signaling
molecules and effector proteins, ensuring precise spatiotemporal
regulation of mitochondrial fission and fusion (53,54). Quantitative analyses indicates
that >80% of fission and ~60% of fusion events occur at MAMs
(55). These findings underscore
their role as central hubs that coordinate mitochondrial dynamics
and preserve endothelial homeostasis, particularly under stress
conditions.
Mitochondrial fission in cardiac microvascular
ischemia/reperfusion injury. Mitochondrial fission is markedly
upregulated during myocardial I/R injury (56). This process is primarily
initiated at MAMs, where the ER outlines the future division site
even before dynamin-related protein 1 (Drp1) recruitment (57). Drp1 subsequently accumulates at
ER-mitochondria contact sites, where its fission activity is
regulated by post-translational modifications (PTMs) such as
phosphorylation, ubiquitination and acetylation. These PTMs are
mediated by various enzymes, including cyclin-dependent kinases
(CDKs) and protein kinase A (PKA) (58-60). For example, phosphorylation of
Drp1 at Ser616 promotes its oligomerization and assembly into
constrictive rings on the OMM (61), whereas phosphorylation at Ser637
prevents Drp1 translocation to mitochondria (62). Experimental studies have shown
that inhibiting Drp1 improves microvascular endothelial function,
reduces mROS and attenuates mitochondrial fission (61).
Special attention should be given to the role of
mitochondrial fission proteins within the MAM-associated signaling
network. During the fission process, Drp1 is actively recruited to
the OMM (63). Drp1 then binds
to receptors mitochondrial dynamics protein of 49 kDa (MiD49),
mitochondrial dynamics protein of 51 kDa (MiD51), mitochondrial
fission factor (MFF) and mitochondrial fission protein 1 (Fis1),
constricting the mitochondrial membrane in a GTP-dependent manner,
thereby dividing a single mitochondrion into two separate
organelles (64,65). Immunofluorescence analysis of
Drp1 has shown that Fis1 and MFF are key determinants of both the
number and size of Drp1 puncta on mitochondria. Either MiD49 or
MiD51 can independently mediate Drp1 recruitment and promote
mitochondrial fission even in the absence of Fis1 and MFF (66). Modulation of mitochondrial
fission protein expression markedly influences mitochondrial
morphology: Suppression of MFF disrupts Drp1 foci from the OMM,
leading to elongation of the mitochondrial network, whereas MFF
overexpression facilitates Drp1 recruitment and promotes
mitochondrial fission (67). MFF
and Drp1 interact both in vitro and in vivo and
MFF-dependent mitochondrial fission occurs independently of Fis1.
In endothelial cells, MFF serves as the primary principal adaptor
for Drp1 recruitment (67).
FUN14 domain-containing protein 1 (FUNDC1), a
recently identified mitochondrial outer membrane protein, localizes
to mitochondria-ER contact sites by interacting with the ER
membrane protein calnexin under hypoxic conditions (68). During hypoxia, sustained
mitophagy disrupts the FUNDC1-calnexin interaction, exposing the
cytoplasmic loop of FUNDC1, which subsequently binds to Drp1 and
initiates mitochondrial fission (68). Furthermore, at mitochondria-ER
contact sites, inverted formin-2 (INF2) becomes activated and
promotes actin polymerization (69). Actin filament formation between
the ER and mitochondria likely generates mechanical force that
facilitates mitochondrial constriction and enhance Drp1 assembly
(70). Following fission, the
severing and depolymerizing activity of INF2 enables the rapid
clearance of actin filaments.
MFF-driven excessive fission promotes apoptosis
through a number of mechanisms. Pathological fission contributes to
mitochondrial outer membrane permeabilization (MOMP) and the
subsequent release of cytochrome c into the cytosol
(74,75), although whether fission precedes
or follows MOMP remains unresolved. Studies suggest three major
apoptotic pathways triggered by excessive mitochondrial fission.
First, fission induces double-strand breaks in mitochondrial DNA
(mtDNA), impairing transcription and replication. mtDNA damage
disrupts ETC activity, increases proton leakage and elevates ROS
generation (76,77). Second, mROS oxidize cardiolipin,
decreasing its binding affinity for cytochrome c and
facilitating its release into the cytosol, thereby activating
apoptotic signaling (78).
Third, mitochondrial fragmentation promotes the polymerization of
voltage-dependent anion channel 1 (VDAC1) and displaces hexokinase
2 (HK2) from VDAC1 (79). These
changes open the mPTP. The combined effects of oxidative stress,
MOMP and mPTP activation exacerbate cardiac microvascular I/R
injury, leading to impaired endothelial nitric oxide synthase
(eNOS) synthesis, endothelial barrier breakdown, loss of vascular
integrity, capillary obstruction and abnormal vascular permeability
(80,81). Thus, MFF-mediated mitochondrial
fission represents a key pathological mechanism in microvascular
I/R injury and represents a potential therapeutic target (78).
In summary, excessive fission serves as a key
pathological driver of I/R injury (82). Therapeutic intervention of
MAM-associated molecular pathways, therefore, represents a
promising strategy for attenuating cardiac microvascular I/R injury
by suppressing pathological mitochondrial fission. Several agents
have been shown to act through this axis. Shuangshen Ningxin
formula, a traditional Chinese medicine compound, alleviates
cardiac microvascular I/R injury by improving mitochondrial
function through the regulation of the NR4A1/MFF/Drp1 pathway
(83). Melatonin exerts
protective effects against cardiac microvascular I/R injury by
activating AMPKα and promoting inhibitory phosphorylation of Drp1
at Ser637, suppressing fission and enhancing ATP synthase activity.
It also attenuates cardiac microvascular injury by targeting the
mitochondrial fission-VDAC1-HK2-mPTP-mitophagy cascade (79). Similarly, Bax inhibitor-1 (BI1)
preserves mitochondrial integrity under I/R conditions by reducing
F-actin-mediated fission, suppressing xanthine oxidase (XO)
activity and limiting ROS production, ultimately maintaining
endothelial viability and barrier function (84). Furthermore, BI1 is also
associated with microvascular protection in I/R injury via
repressing Syk-Nox2-Drp1-mitochondrial fission pathways (85). The ROS-JNK-Drp1 pathway has been
identified as a primary mechanism driving endothelial cell injury
and mitochondrial fission during hypoxia-reoxygenation injury; its
inhibition may provide a novel therapeutic strategy to prevent
coronary no-reflow injury (86).
Empagliflozin protects against microvascular I/R injury by
suppressing mitochondrial fission via inactivation of the
DNA-PKcs/Fis1 pathway (87). By
contrast, pretreatment with 2'-hydroxycinnamaldehyde (HCA) reduces
Drp1 expression and improves microvascular function during
reperfusion (88).
MAMs exert multifaceted control over mitochondrial
fission. Resident proteins and signaling cascades at MAMs
facilitate mitochondrial constriction and Drp1 recruitment,
promoting fragmentation. Protective mechanisms suppress pro-fission
proteins or inhibit associated signaling pathways. These mechanisms
collectively reduce mROS, prevent mPTP opening and preserve
endothelial cell viability. The MAM-mitochondrial fission axis
represents a promising therapeutic target for ameliorating cardiac
microvascular I/R injury.
Mitochondrial fusion in cardiac microvascular
ischemia/reperfusion injury. Compared with mitochondrial fission,
mitochondrial fusion in endothelial cells during myocardial
infarction has been less extensively studied. However,
mitochondrial fusion is essential for mitochondrial repair and
functional recovery, as it facilitates the exchange of matrix and
membrane components, preserving mitochondrial integrity and
maintaining bioenergetic homeostasis. Mitochondrial fusion involves
the coordinated fusion of the OMM and IMM, which is regulated
primarily by MFN1/2 and optic atrophy 1 (OPA1), respectively
(56,89) (Table I). Mitofusin-2 (MFN2) is
particularly important because it not only mediates OMM fusion but
also regulates mitochondria-ER tethering at MAMs (90,91). MFN2 plays a crucial role in
maintaining normal mitochondrial functions, including fusion,
axonal transport, inter-organelle communication and mitosis
(92). Unlike MFN1, which is
confined to the mitochondrial membrane, MFN2 is distributed across
the ER, OMM and MAMs. Homotypic interactions between MFN2 molecules
on adjacent membranes, or heterotypic interactions between
ER-localized MFN2 and mitochondrial MFN1, form dimeric or
multimeric complexes that maintain an optimal distance of 10-30 nm
between the two organelles (91,93). Loss or downregulation of MFN2
enhances both structural and functional coupling between the ER and
mitochondria. Thus, MFN2 acts as a negative regulator of excessive
ER-mitochondria tethering and its reduced expression disrupts this
control, promoting aberrant and potentially harmful ER-mitochondria
crosstalk (94). OPA1 then
remodels the IMM, restoring membrane continuity and structural
integrity. This step requires the cardiolipin-binding domain of
OPA1.
Fusion of the inner and outer mitochondrial
membranes is further regulated by proteolytic processing and
ubiquitination (95). At the
IMM, long OPA1 (L-OPA1) is cleaved at sites S1 and S2 to form short
OPA1 (S-OPA1) (96,97). The AAA+ protease YME1L mediates
S2 cleavage under basal conditions, whereas the stress-responsive
metalloprotease OMA1 cleaves at S1, thereby disrupting the OPA1
isoform balance (98-100). Under physiological conditions,
a near-equivalent ratio of L-OPA1 to S-OPA1 preserves membrane
fusion activity (101,102). Pathological stimuli,
particularly loss of membrane potential, activate OMA1 (103), shifting the ratio and impairing
fusion, leading to fragmentation (104).
MFN1 and MFN2 are not only central to mitochondrial
structure but are also closely associated with angiogenic signaling
pathways in vascular endothelial cells (105). Loss of MFN1/2 disrupts fusion
and mitochondrial networks, leading to reduced membrane potential,
impaired endothelial cell survival and reduced responsiveness to
VEGF signaling, which then suppresses angiogenesis. Fusion is also
essential for maintaining protein synthesis and energy metabolism,
as well as preventing mitochondrial DNA loss (106). Despite these protective roles,
MFN2 exerts context-dependent regulatory effects on cellular
physiological and pathological processes. In cardiomyocytes, MFN2
depletion limits mPTP opening and increases tolerance to
Ca2+-induced injury, improving resistance to ischemia
during I/R (107). In other
cell types or under pathological stress, silencing or deletion of
MFN2 enhances mitochondria-ER interactions, promotes
Ca2+ transport into mitochondria and increases
susceptibility to Ca2+-induced cell death (93). These findings suggest that the
function of MFN2 varies according to cell type and disease state.
Moreover, MFN2 and OPA1 transcriptional levels are markedly reduced
during I/R injury, consistent with functional inhibition of
mitochondrial fusion. Regulation of mitochondrial function can
mitigate hypoxic-stress-induced apoptosis (108). Overexpression of the
sarcoplasmic reticulum Ca2+ pump sarco/endoplasmic
reticulum Ca2+ATPase (SERCA) in CMECs reverses MFN2/OPA1
downregulation, increases mitochondrial fusion, reduces
fragmentation and improves microvascular integrity by restoring
Ca2+ homeostasis (109).
ER stress and impaired intercellular connections are
closely associated with mitochondrial fusion-related disorders.
This pathogenic cascade is characterized by the loss of
cardiac-specific Lon peptidase 1 (LonP1), disruption of the MAM
structure, defective mitochondrial fusion and activation of the ER
unfolded protein response (UPRER) (110). These interrelated changes
contribute to metabolic reprogramming and cardiac structural
remodeling. Fusion dysfunction also activates the
nucleotide-binding oligomerization domain-, leucine-rich repeat-
and pyrin domain-containing receptor 3 (NLRP3) inflammasome,
triggering vascular inflammatory responses (111). During the early stages of I/R,
impaired fusion leads to interstitial edema and endothelial cell
swelling, further reducing microcirculatory perfusion (112). In female mouse models, I/R
injury disrupts endothelial connections, decreases connexin 43
expression and alters the balance of MMP-3 and TIMP-1 (113,114). Restoration of MFN2 expression
reverses these changes and increases endothelial progenitor cell
marker expression, including platelet endothelial cell adhesion
molecule 1 (CD31), vascular endothelial growth factor receptor 2
(VEGFR2), Fms-related tyrosine kinase 4 (FLT4) and Kinase Insert
Domain Receptor (KDR) (114).
Loss of cell junctions compromises endothelial barrier function,
increasing susceptibility to platelet and coagulation system
activation and exacerbating microvascular thrombosis (115). This mechanism impairs
myocardial perfusion during reperfusion by promoting microvascular
obstruction. Downregulation of MFN2 reduces mitochondrial
Ca2+ overload by decreasing CypD-VDAC1-IP3R1
interactions. However, MFN2 broadly supports endothelial integrity,
microvascular structure and maintenance of fusion (116). These results suggest that MFN2
exerts a dual role in cardiac microvascular I/R injury by
regulating endothelial cell metabolism, apoptosis and inflammatory
responses while preserving MAM homeostasis.
Most of these findings are derived from
loss-of-function studies using RNA interference to inhibit MFN1/2
or OPA1 expression in endothelial cells in vitro (51,117). However, the protective effects
of MFN1/2- or OPA1-mediated fusion have not been conclusively
validated in rescue experiments using transgenic mouse models or
virus-mediated overexpression. Moreover, interactions between
mitochondrial fission and fusion in cardiac microvascular I/R
injury remain poorly defined. For example, whether active induction
of fusion can directly inhibit fission is unclear. Research
suggests that the MFN2/Drp1 expression ratio may reflect the
dynamic balance between fusion and fission (118), providing a potential avenue for
further investigation; however, its clinical applicability has yet
to be established.
Oxygen is transported from the bloodstream through
endothelial cells to the surrounding perivascular tissues. Research
indicates that endothelial dysfunction can impair this process by
reducing the rate of oxygen diffusion across the arteriolar wall
(119). Under hypoxic
conditions, impaired respiration promotes excessive production of
mROS (120). The mROS are
primarily generated within the ETC of the IMM during oxidative
phosphorylation (121,122). While moderate ROS levels act as
second messengers in physiological signaling, excessive ROS induces
oxidative stress, causing endothelial senescence and cell death.
Ischemia- or inflammation-induced oxidative stress activates
mitochondria-dependent apoptotic pathways, leading to further cell
death (123,124). I/R injury is particularly
associated with ROS accumulation. Activation of XO during
reperfusion markedly increases ROS, reduces nitric oxide (NO)
bioavailability and triggers microvascular spasm and impaired
perfusion (125). Furthermore,
mitochondrial superoxide oxidizes tetrahydrobiopterin (BH4) to
dihydrobiopterin (BH2), preventing BH4 binding to eNOS. This
uncoupling of eNOS decreases NO production and exacerbates vascular
dysfunction (126).
Protective mechanisms also regulate oxidative
stress. MAMs are implicated in multiple cardiovascular disorders,
mainly by promoting oxidative stress and inflammation in cardiac
tissue (127). They represent
the only known binding platform for the NLRP3 inflammasome complex,
contributing markedly to ROS-mediated oxidative injury (128). The NLRP3 inflammasome is
composed of NLRP3, apoptosis-associated speck-like protein (ASC)
and caspase-1 (129). NLRP3 is
localized to the ER, whereas ASC is anchored on the OMM. MAMs serve
as scaffolds that facilitate NLRP3 inflammasome assembly by
enabling the interaction between NLRP3 and ASC (130). mROS are essential regulators of
NLRP3 activation. They promote dissociation of
thioredoxin-interacting protein (TXNIP) from thioredoxin (TRX),
after which TXNIP binds to NLRP3, initiating caspase-1 activation
and cleavage of pro-IL-1β and pro-IL-18 (131). This cascade drives robust
inflammatory responses. In CMECs, TXNIP-dependent NLRP3 activation
has been identified as a novel mechanism of injury during
myocardial I/R (129). The
extent of myocardial infarction and subsequent functional recovery
is strongly affected by the intensity of this inflammatory response
(132). Thus, NLRP3 activation
serves as a critical mechanistic link between mitochondrial
dysfunction and inflammation, playing a key role in microvascular
dysfunction following I/R injury (128). Experimental studies demonstrate
that early inhibition of NLRP3 inflammasome activity during
reperfusion improves cardiac function and reduces infarct size,
highlighting it as a promising therapeutic target (133).
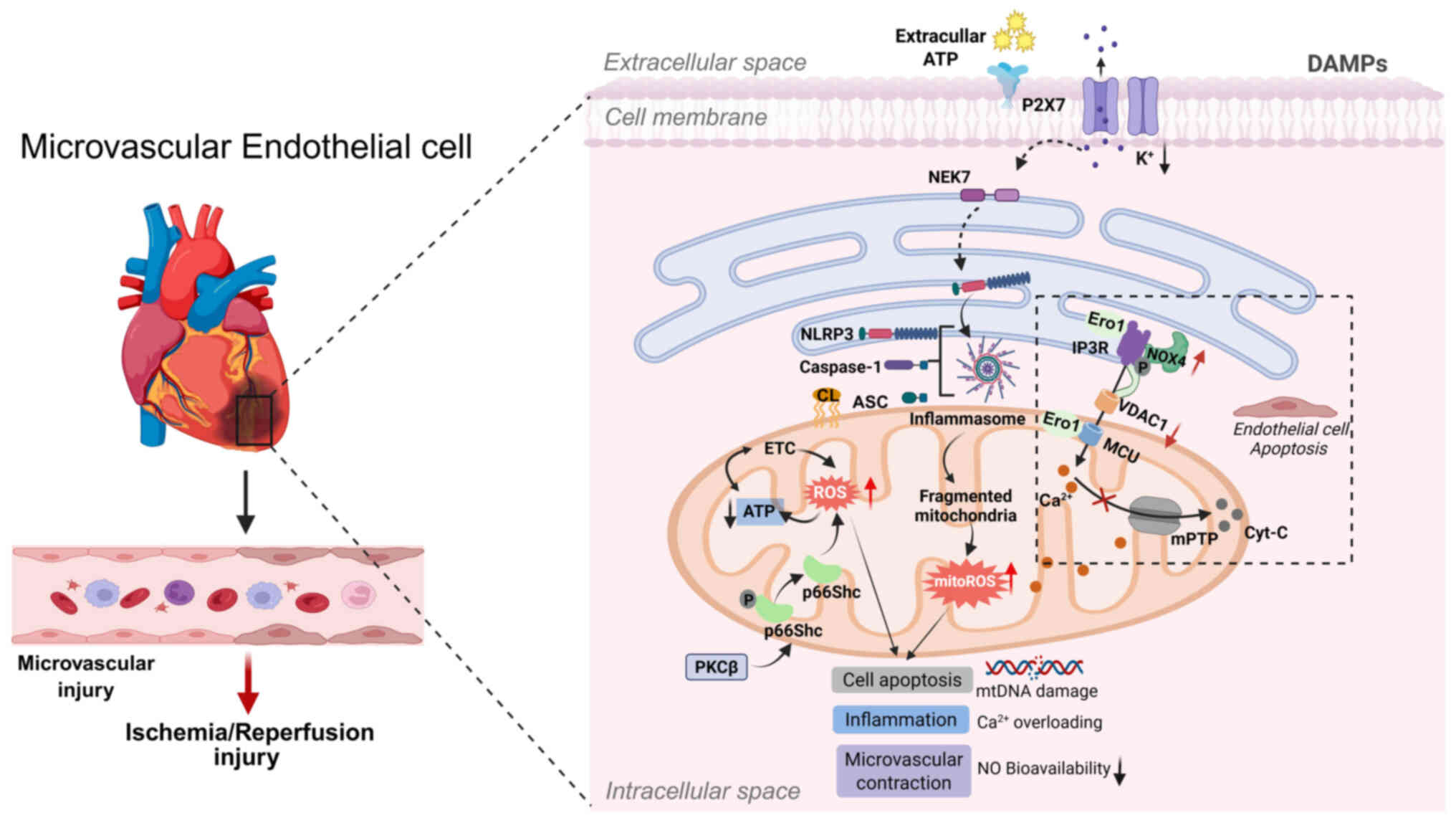
Elevated ROS levels at MAMs can trigger the release
of mtDNA and the opening of the mPTP. Within MAMs, mtDNA functions
as a damage-associated molecular pattern, initiating NLRP3
inflammasome activation and downstream inflammatory signaling
(134). During cardiac
ischemia-reperfusion, necrotic cardiomyocytes release ATP, which
binds to P2X7 receptors on neighboring non-ischemic cardiomyocytes.
This interaction induces potassium ion (K+) efflux,
lowering cytoplasmic K+ concentration. The resulting
hypokalemic state activates NIMA-related kinase 7 (NEK7), promoting
inflammasome assembly and NLRP3 activation (135). Moreover, decreased
extracellular H+ concentration increases
Na+/H+ and Na+/Ca2+
exchange, aggravating intracellular Ca2+ overload
(135). This increases
mitochondrial Ca2+ accumulation within MAMs and further
stimulates ROS production. Excess ROS induces cardiolipin release
from the IMM, enabling cardiolipin to bind NLRP3 and promote
inflammasome formation. Inhibition of two voltage-dependent anion
channel (VDAC1 and VDAC2) isoforms in the OMM markedly reduces
NLRP3 inflammasome activation, caspase-1 cleavage and IL-1β
production (127). These
findings suggest potential therapeutic targets for reducing
inflammation and improving heart function following myocardial
infarction.
The MAM serves as an important site for ROS
generation. Structural proteins located at the MAM, such as
endoplasmic reticulum oxidoreductase 1 (Ero1) (136) and 66-kDa isoform of the growth
factor adaptor Shc (p66Shc) (137), play pivotal roles in redox
signaling between mitochondria and the ER, thereby regulating ROS
production (Table I). Ero1α,
localized at the MAM (138),
facilitates Ca2+ influx by activating the mitochondrial
calcium uniporter (MCU) (139).
During early stages of ER stress, Ero1α interacts with PKR-like
endoplasmic reticulum kinase (PERK), a PKR-like ER kinase, forming
the Ero1-PERK complex that coordinates mitochondrial fusion,
enhances ER-mitochondria interactions, restores mitochondrial
bioenergetics and modulates intracellular ROS levels (140). PERK functions as a
MAM-anchoring protein, promoting the formation of MAMs through
oligomerization (141) and
transmitting ROS signals to mitochondria (142). Furthermore, the Ero1-PERK
complex reduces ER Ca2+ levels and promotes
ER-to-mitochondria Ca2+ flux (143). Conversely, the absence of PERK
impairs Ero1α-mediated IP3R oxidation, thereby
disturbing mitochondrial redox homeostasis (144). Similarly, under oxidative
stress, activated protein kinase C β (PKCβ) phosphorylates p66Shc
at Ser36, triggering its translocation to mitochondria or the MAMs,
where it contributes to ROS generation (137,145). Although ROS derived from p66Shc
may support short-term cellular repair responses (146), persistent activation can lead
to the onset and progression of various cardiovascular diseases
(147). NADPH oxidase (Nox),
particularly the Nox1, Nox2 and Nox4 isoforms (148), is a major contributor to ROS
production in myocardial I/R injury. Nox4 localizes to MAMs, where
its expression increases under stress conditions. It functions as
an endoplasmic reticulum-localized source of ROS, maintaining basal
IP3R oxidation necessary for OXPHOS and regulating redox
signaling within the ER under stress conditions (149). At these sites, Nox4 facilitates
Akt-mediated phosphorylation of IP3R, which inhibits
Ca2+ transfer to mitochondria and prevents
mPTP-dependent cell death. This localized redox signaling underlies
the protective role of Nox4 in reducing necrosis in both
cardiomyocytes and intact hearts following I/R injury (149) (Fig. 2).
A number of therapeutic strategies have been
reported to alleviate oxidative stress and protect cardiac
function. ENPP2 regulates redox balance and mitochondrial activity,
reducing hypoxia/reoxygenation injury in CMECs (150). Liproxstatin-1, a ferroptosis
inhibitor, protects the myocardium by downregulating VDAC1 and
restoring GPX4 (151). In a
pressure-overload mouse model, ginkgolide A (GA) reduced oxidative
stress and increased NO bioavailability in the heart (152). Glucagon-like peptide-1 (GLP-1)
analogs, such as liraglutide, have demonstrated cardioprotective
effects in clinical trials by lowering oxidative stress and
vascular inflammation, preventing eNOS uncoupling and preserving
endothelial function (153).
Histone deacetylase 7-derived peptides promote angiogenesis and
limit oxidative stress in hind limb ischemia, preserving
endothelial integrity (154).
Naringin (Nar) improves cardiac microvascular function, potentially
by facilitating NADH ubiquinone oxidoreductase core subunit S1
(NDUFS1) translocation to mitochondria, reducing ROS and protecting
endothelial cells (155).
Furthermore, PSS-carrying nanoparticles minimize oxidative stress
and reverse coronary microcirculatory dysfunction (156).
Regulation of ROS production and mitigation of
oxidative damage constitute the primary mechanisms by which MAMs
modulate cellular functions during oxidative stress responses
(157). Importantly,
MAM-induced mitochondrial oxidative stress does not occur in
isolation; elevated ROS levels drive a shift in mitochondrial
dynamics from fusion toward fission. These fragmented mitochondria
become major sources of apoptotic signals and excessive mROS
generation (158). Excessive
ROS stimulation further amplifies mitochondrial dysfunction,
leading to swelling, loss of cristae structure and rupture of the
outer membrane, ultimately releasing apoptosis-related proteins
into the cytoplasm and activating the apoptotic cascade (159). Further research on how
MAM-inflammasome crosstalk contributes to oxidative stress is
essential for elucidating the mechanisms underlying cardiac
microvascular I/R injury and developing effective therapeutic
strategies.
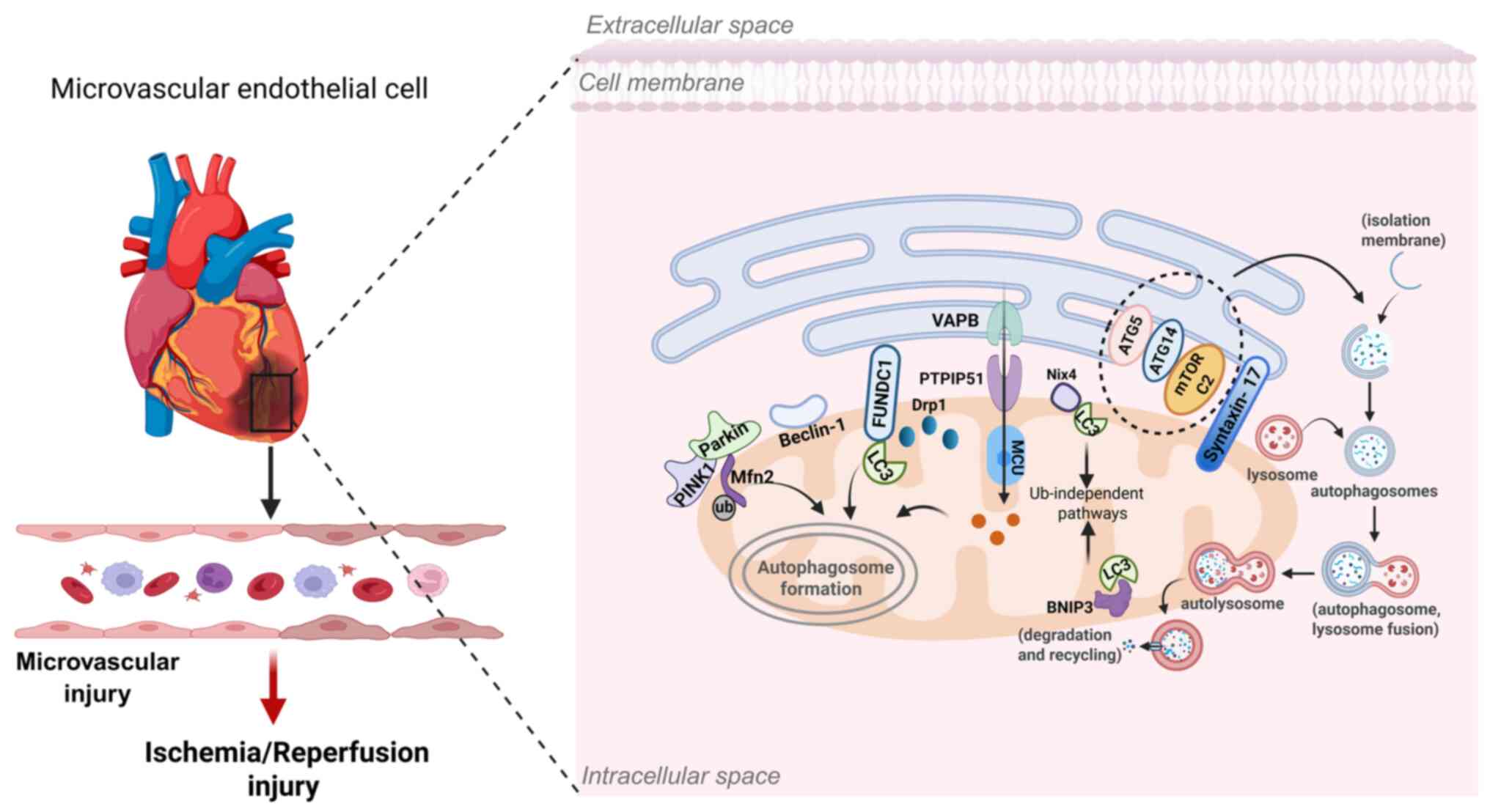
Mitophagy, the selective autophagic degradation of
damaged mitochondria, is essential for maintaining cellular
homeostasis and regulating energy metabolism. It has emerged as a
potential therapeutic target for limiting I/R injury (160) and protecting the
microvasculature (48). MAMs
play a central role in this process by functioning as specialized
subdomains where both initiation and progression of mitophagy
occur. The pathway proceeds through different stages: initiation,
phagophore elongation, autophagosome closure, lysosomal fusion and
degradation and is regulated by a set of autophagy-related genes
and proteins (161). Mitophagy
signaling depends on receptor proteins at the OMM, including
FUNDC1, BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3),
Nip3-like protein X (Nix) and mitochondrial E3 ubiquitin protein
ligase 1 (Mul1), as well as the E3 ubiquitin ligase Parkin
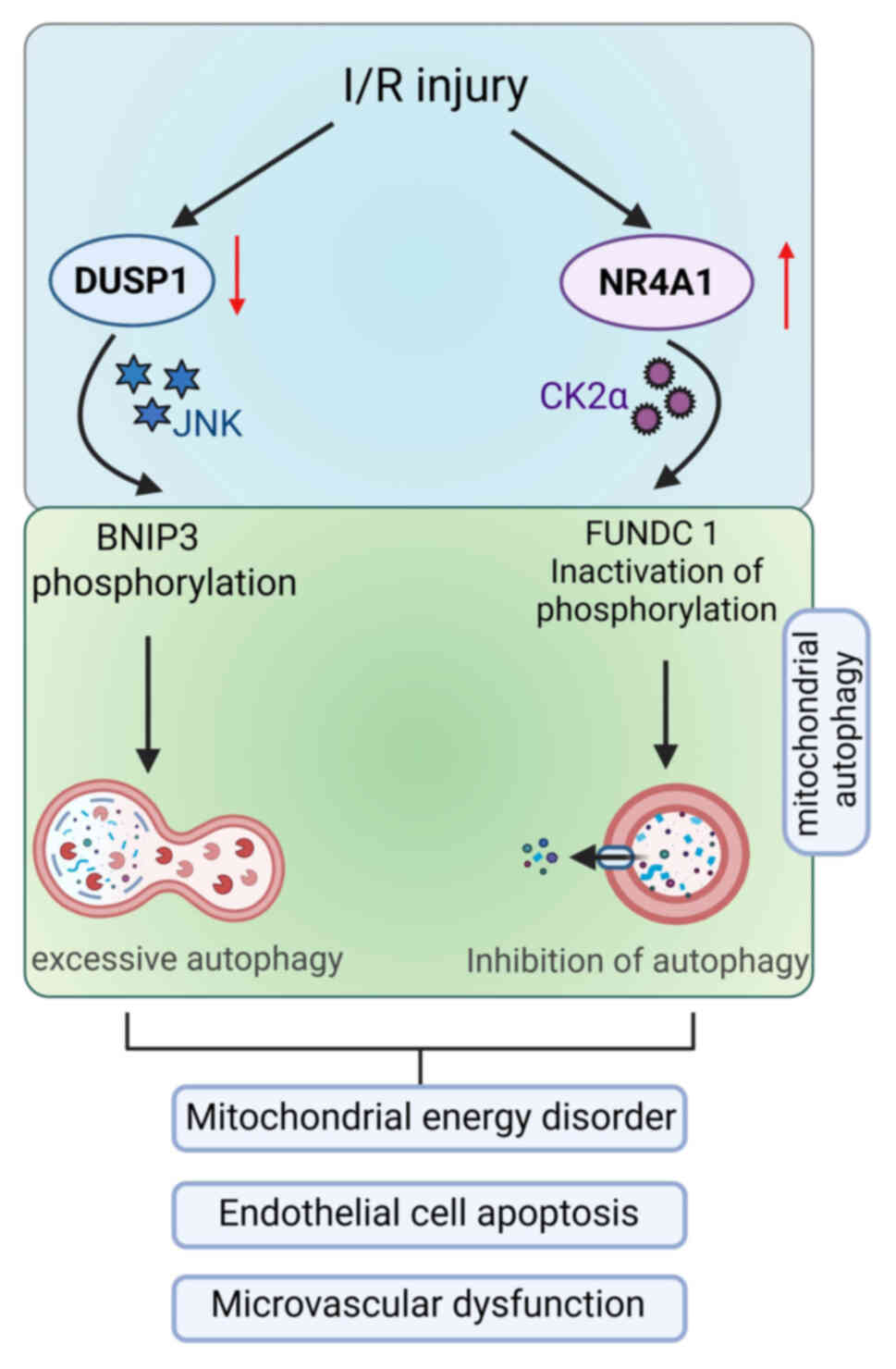
(162). Upstream regulation is
closely associated with mitochondrial dynamics. Mitochondrial
fission, often a prerequisite for mitophagy, is regulated by
proteins such as MFF, whose activity is influenced by DUSP1
(72) and NR4A1 (73). In CMECs, NR4A1 inhibits mitophagy
by suppressing FUNDC1 (73),
whereas DUSP1 promotes it through BNIP3 phosphorylation (Fig. 3).
Beclin1, which is also enriched at MAMs, enhances
autophagic flux and regulates ER-mitochondria interactions,
functioning as a protective factor against I/R injury (171). The serine residue at position
15 of Beclin1 can be phosphorylated by Unc-51-like
autophagy-activating kinase 1 (ULK1) (172), a kinase involved in mitophagy
(173) and this modification is
essential for Beclin1's association with MAMs during the mitophagic
process. Localization of Beclin1 at MAMs ensures that autophagosome
formation occurs in proximity to damaged mitochondria, facilitating
their efficient sequestration and degradation (171). Furthermore, studies have shown
that Beclin1-driven autophagy suppresses caspase-4-mediated
apoptosis, thereby protecting microvascular endothelial cells from
I/R-induced injury (174).
Crosstalk among various mitophagy adaptors
ultimately determines the net effect of mitophagy. Fission of
damaged mitochondria into smaller fragments facilitates their
elimination through mitophagy (186). Autophagy and fission occur
simultaneously, forming a coordinated response (187). Moderate mitophagy limits
excessive fragmentation induced by pathological fission (175,188,189). Thus, fission is both a
prerequisite for mitophagy activation and, when excessive, a
process that can be counterbalanced by appropriately regulated
mitophagy. Taken together, these findings underscore that mitophagy
exerts both protective and detrimental roles depending on the
signaling context. The precise contribution of specific adaptors in
endothelial cells and their interactions with fission machinery
remain unclear. Therefore, the controlled regulation of mitophagy
represents a key area of investigation for mitigating microvascular
I/R injury.
Several molecular regulators modulate this process.
Glycogen synthase kinase-3β (GSK-3β) and the Sigma-1 receptor
(Sig-1R) are key MAM-associated proteins that alleviate ER stress
and reduce Ca2+ uptake, conferring cardioprotection
during I/R (206,207). GSK-3β, an enzyme responsible
for phosphorylating and inactivating glycogen synthase (208,209), can be recruited to the MAM,
where it regulates IP3R1-mediated calcium release. This
regulation promotes mitochondrial Ca2+ accumulation and
induces the opening of the mPTP. As a result, inhibition of GSK-3β
has been suggested to exert cardioprotective effects during I/R
injury (206). The Sig-1R is a
specific chaperone localized at MAMs (207). Overexpression of Sig-1R
enhances ER-to-mitochondria Ca2+ flux and through its
interactions with ankyrin and the ER chaperone BiP. Under
physiological conditions, Sig-1R remains bound to BiP; however,
under ER stress or following ER Ca2+depletion, Sig-1R
dissociates from BiP and binds to IP3R instead. This
interaction stabilizes IP3R, preventing its degradation
and restoring efficient Ca2+ transfer from the ER to
mitochondria (207). Moreover,
Sig-1R stabilizes MAM structure through its interactions with VDAC1
and IP3R and provides cardioprotective benefits by
mitigating MAM-mediated Ca2+ overload and ER stress
(207). Under conditions of
elevated ROS, Sig-1R also interacts with reactive oxygen species,
thereby activating and stabilizing IRE1 (210).
Cyclophilin D (CypD), a key regulator of the mPTP
located in the matrix. During H/R, increased interactions within
the CypD-VDAC1-GRP75-IP3R1 super-complex drive
mitochondrial Ca2+ overload and cardiomyocyte death
(126). Genetic ablation of
CypD protects against I/R-induced necrosis by limiting this
aberrant Ca2+ transfer (211,212). Similarly, deletion of the
PPIF gene (encoding CypD), or knockdown of IP3R1
or GRP75, reduces H/R-induced Ca2+ overload and cell
death (211). Pharmacological
interventions also provide protection; melatonin attenuates
oxidative stress and reduces CMEC mortality by inhibiting
IP3R-VDAC-mediated mitochondrial Ca2+ influx,
increasing endothelial resistance to oxidative damage (77).
MAM tethering is also regulated by the OMM protein
PTPIP51 and the ER protein vesicle-associated membrane
protein-associated protein-B (VAPB), which form a structural bridge
between the two organelles (213). Overexpression of PTPIP51
strengthens this physical connection, enhances ER-mitochondria
coupling and increases mitochondrial Ca2+ uptake through
MCU activation (214,215). However, pharmacological or
genetic inhibition of MCU abolishes this effect and protects
cardiomyocytes from apoptosis (211). The ER chaperone calreticulin
(CRT), with its high Ca2+-binding capacity, also plays a
key role in buffering and storage. Upregulation of CRT restores
Ca2+ homeostasis by improving ER Ca2+
reserves (216) (Fig. 5). Collectively, MAM-associated
proteins regulate calcium transport through distinct and highly
coordinated mechanisms (Table
II).
Therapeutically, pinacidil has been shown to
preserve CRT expression, reduce endothelial Ca2+
overload and prevent mitochondrial apoptosis in CMECs, ultimately
improving microvascular density, increasing blood flow, reducing
infarct size and attenuating the no-reflow phenomenon following I/R
injury (217). Other protective
mechanisms involve ER-transmembrane proteins such as transmembrane
protein 215 (TMEM215), which protect endothelial cells by
inhibiting BCL-2-interacting killer (BIK). Since BIK promotes
ER-to-mitochondria Ca2+ transfer, its suppression by
TMEM215 reduces pathological Ca2+ flux and supports cell
survival during reperfusion (218).
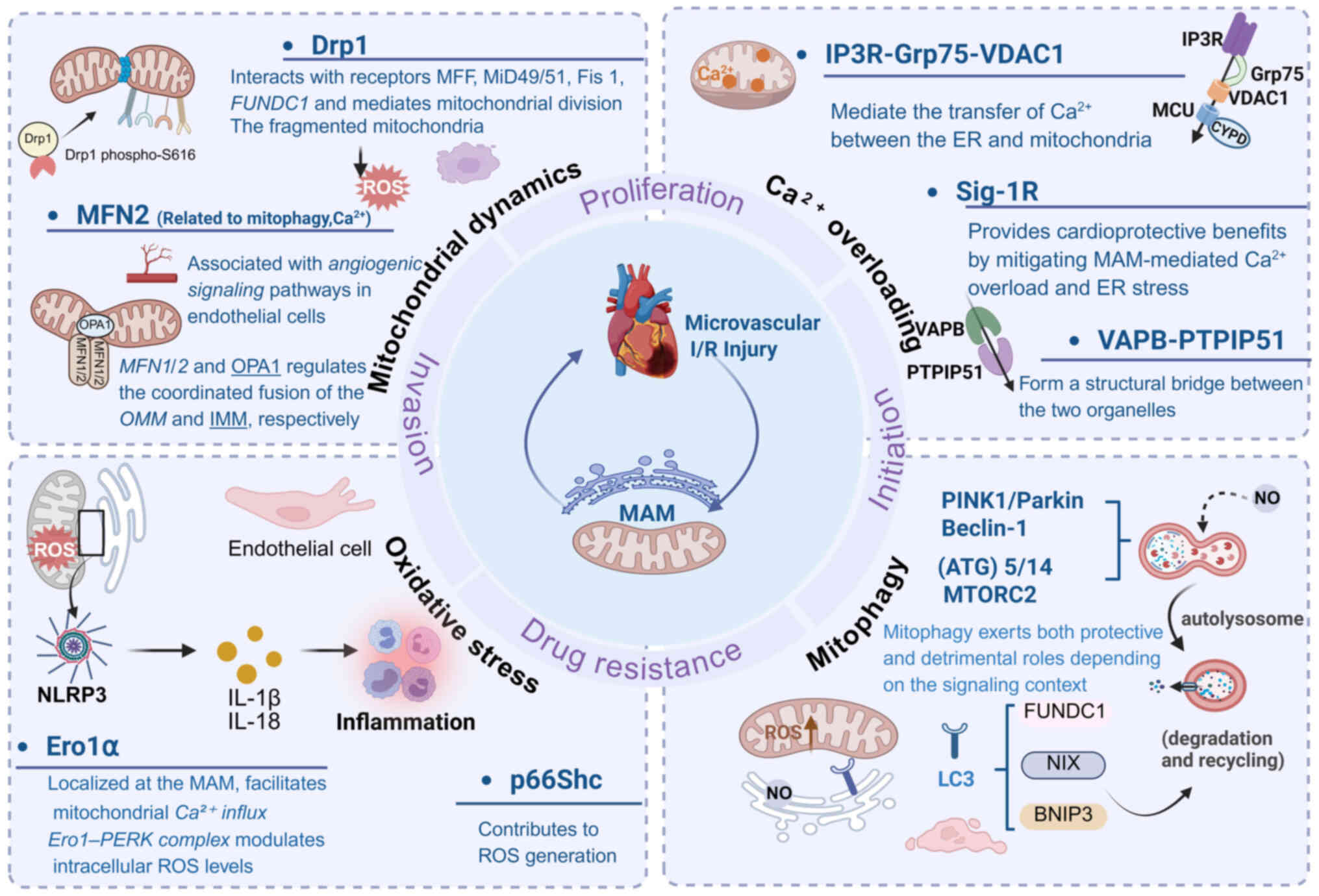
Damage to this microvascular network during I/R can
result in interstitial hemorrhage and edema (220). The MAM functions as a pivotal
signaling hub that orchestrates mitochondrial dynamics, oxidative
stress responses, mitophagy and calcium homeostasis. Importantly,
these mitochondrial alterations do not occur in isolation but are
intricately interconnected (Fig.
6). Core MAM-associated proteins such as Drp1, MFN2, FUNDC1,
Ero1α, IP3R, VDAC1 and Sig-1R play essential roles in
maintaining normal microvascular physiology. By integrating these
key MAM components, molecular mechanisms and signaling pathways,
advances in understanding cardiac microvascular I/R injury have
provided new perspectives for addressing clinical conditions such
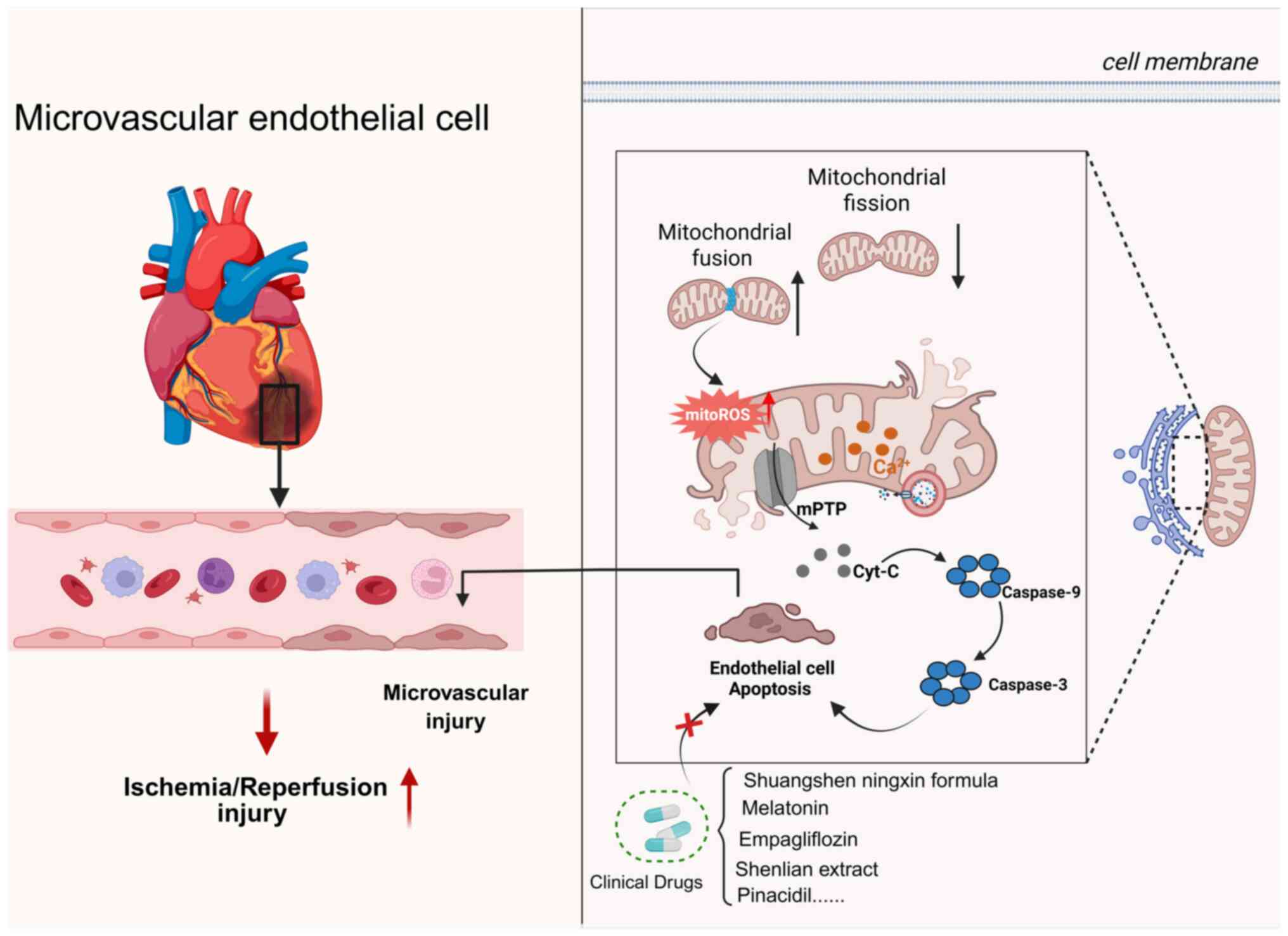
as the no-reflow phenomenon and microvascular angina (Fig. 7).
Moreover, several pharmacological agents, including
empagliflozin, Shuangshen Ningxin Pill and melatonin, have been
shown to exert protective effects against microvascular I/R injury
through MAM-related mechanisms (Table IV). However, most of the current
evidence remains confined to cellular and animal models. Future
research, focusing on the safety, controllability and efficacy of
MAM-targeted interventions at the human microvascular level may
enable the development of more effective therapeutic
strategies.
Despite these advances, the inherent complexity and
dynamic nature of MAMs present major challenges to further
investigation. Our current understanding of MAMs remains limited,
particularly regarding how their morphology, inter-organelle
distance, membrane thickness and number of contact sites influence
cellular energy metabolism and contribute to disease pathogenesis.
Beyond serving as a structural bridge between mitochondria and the
ER, the bidirectional regulatory mechanisms by which MAMs
coordinate organelle function remain incompletely understood, for
instance, the role of MAM-localized MFN2. It also remains unclear
whether MAMs show conserved structural and functional features
across different cardiovascular cell types, how their formation can
be selectively modulated for therapeutic benefit and whether they
might serve as early biomarkers of microvascular injury.
Furthermore, the temporal dynamics, spatial organization and
hierarchical signaling networks that govern MAM remodeling in
endothelial cells under stress conditions are still poorly
characterized.
Future research should aim to develop multiscale
integrative models of MAMs to elucidate the causal relationships
between their dynamic remodeling and microvascular pathological
responses. Employing advanced in vivo multi-omics and
imaging technologies, such as the Split-GFP contact sensor
(221,222) and serial block-face scanning
electron microscopy (223), may
provide deeper insights into their structural and functional
characteristics. Given the involvement of MAMs in key cellular
processes, including energy metabolism, inflammation and cell
death, their dysregulation likely contributes to the pathogenesis
of cardiovascular, metabolic and neurological disorders. Therefore,
clarifying the molecular mechanisms underlying MAM function could
not only enhance our understanding of cardiovascular
microcirculatory pathology but also reveal broadly applicable
therapeutic targets for multiple diseases. However, pharmacological
strategies targeting MAMs alone may be limited by individual
metabolic variations and comorbid conditions; thus, future
investigations should emphasize combinatorial regulatory
approaches, such as dual-target therapies that modulate both MAMs
and mitochondrial metabolism.
Not applicable.
YW and B F conceived the idea for the present
review and drafted the manuscript. YtW and ZS created the figures
and contributed to the writing of the manuscript. HY, WC and CZ
reviewed and modified the manuscript and screened literature. ZL
provided valuable guidance and analysis during the present review.
Data authentication is not applicable. All authors read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the Jilin Provincial Natural
Science Foundation (grant no. YDZJ202401052ZYTS).
|
1
|
Marin W, Marin D, Ao X and Liu Y:
Mitochondria as a therapeutic target for cardiac
ischemia-reperfusion injury (Review). Int J Mol Med. 47:485–499.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang M, Linn BS, Zhang Y and Ren J:
Mitophagy and mitochondrial integrity in cardiac
ischemia-reperfusion injury. Biochim Biophys Acta Mol Basis Dis.
1865:2293–2302. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhao BH, Ruze A, Zhao L, Li QL, Tang J,
Xiefukaiti N, Gai MT, Deng AX, Shan XF and Gao XM: The role and
mechanisms of microvascular damage in the ischemic myocardium. Cell
Mol Life Sci. 80:3412023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abbate A, Kontos MC and Biondi-Zoccai GGL:
No-reflow: The next challenge in treatment of ST-elevation acute
myocardial infarction. Eur Heart J. 29:1795–1797. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Davidson SM, Arjun S, Basalay MV, Bell RM,
Bromage DI, Bøtker HE, Carr RD, Cunningham J, Ghosh AK, Heusch G,
et al: The 10th biennial hatter cardiovascular institute workshop:
Cellular protection-evaluating new directions in the setting of
myocardial infarction, ischaemic stroke, and cardio-oncology. Basic
Res Cardiol. 113:432018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
de Waha S, Patel MR, Granger CB, Ohman EM,
Maehara A, Eitel I, Ben-Yehuda O, Jenkins P, Thiele H and Stone GW:
Relationship between microvascular obstruction and adverse events
following primary percutaneous coronary intervention for ST-segment
elevation myocardial infarction: An individual patient data pooled
analysis from seven randomized trials. Eur Heart J. 38:3502–3510.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eitel I, de Waha S, Wöhrle J, Fuernau G,
Lurz P, Pauschinger M, Desch S, Schuler G and Thiele H:
Comprehensive prognosis assessment by CMR imaging after ST-segment
elevation myocardial infarction. J Am Coll Cardiol. 64:1217–1226.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao P, Yan Z and Zhu Z:
Mitochondria-associated endoplasmic reticulum membranes in
cardiovascular diseases. Front Cell Dev Biol. 8:6042402020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuznetsov AV, Javadov S, Margreiter R,
Grimm M, Hagenbuchner J and Ausserlechner MJ: The role of
mitochondria in the mechanisms of cardiac ischemia-reperfusion
injury. Antioxidants (Basel). 8:4542019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kluge MA, Fetterman JL and Vita JA:
Mitochondria and endothelial function. Circ Res. 112:1171–1188.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bertero E, Popoiu TA and Maack C:
Mitochondrial calcium in cardiac ischemia/reperfusion injury and
cardioprotection. Basic Res Cardiol. 119:569–585. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wong R, Steenbergen C and Murphy E:
Mitochondrial permeability transition pore and calcium handling.
Methods Mol Biol. 810:235–242. 2012. View Article : Google Scholar :
|
|
13
|
Pang B, Dong G, Pang T, Sun X, Liu X, Nie
Y and Chang X: Emerging insights into the pathogenesis and
therapeutic strategies for vascular endothelial injury-associated
diseases: Focus on mitochondrial dysfunction. Angiogenesis.
27:623–639. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luo Z, Yao J, Wang Z and Xu J:
Mitochondria in endothelial cells angiogenesis and function:
Current understanding and future perspectives. J Transl Med.
21:4412023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parkkinen I, Their A, Asghar MY, Sree S,
Jokitalo E and Airavaara M: Pharmacological regulation of
endoplasmic reticulum structure and calcium dynamics: Importance
for neurodegenerative diseases. Pharmacol Rev. 75:959–978. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou X, Jiang Y, Wang Y, Fan L, Zhu Y,
Chen Y, Wang Y, Zhu Y, Wang H, Pan Z, et al: Endothelial FIS1
DeSUMOylation protects against hypoxic pulmonary hypertension. Circ
Res. 133:508–531. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ren J, Bi Y, Sowers JR, Hetz C and Zhang
Y: Endoplasmic reticulum stress and unfolded protein response in
cardiovascular diseases. Nat Rev Cardiol. 18:499–521. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lackner LL: The expanding and unexpected
functions of mitochondria contact sites. Trends Cell Biol.
29:580–590. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Copeland DE and Dalton AJ: An association
between mitochondria and the endoplasmic reticulum in cells of the
pseudobranch gland of a teleost. J Biophys Biochem Cytol.
5:393–396. 1959. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vance JE: Phospholipid synthesis in a
membrane fraction associated with mitochondria. J Biol Chem.
265:7248–7256. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ronayne CT and Latorre-Muro P: Navigating
the landscape of mitochondrial-ER communication in health and
disease. Front Mol Biosci. 11:13565002024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Anderson AJ, Jackson TD, Stroud DA and
Stojanovski D: Mitochondria-hubs for regulating cellular
biochemistry: Emerging concepts and networks. Open Biol.
9:1901262019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mishra SR, Mahapatra KK, Behera BP, Patra
S, Bhol CS, Panigrahi DP, Praharaj PP, Singh A, Patil S, Dhiman R
and Bhutia SK: Mitochondrial dysfunction as a driver of NLRP3
inflammasome activation and its modulation through mitophagy for
potential therapeutics. Int J Biochem Cell Biol. 136:1060132021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leung SWS and Shi Y: The glycolytic
process in endothelial cells and its implications. Acta Pharmacol
Sin. 43:251–259. 2022. View Article : Google Scholar :
|
|
25
|
Kühlbrandt W: Structure and function of
mitochondrial membrane protein complexes. BMC Biol. 13:892015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vázquez-Meza H, Vilchis-Landeros MM,
Vázquez-Carrada M, Uribe-Ramírez D and Matuz-Mares D: Cellular
compartmentalization, glutathione transport and its relevance in
some pathologies. Antioxidants (Basel). 12:8342023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jagtap YA, Kumar P, Kinger S, Dubey AR,
Choudhary A, Gutti RK, Gutti RK, Singh S, Jha HC, Poluri KM and
Mishra A: Disturb mitochondrial associated proteostasis:
Neurodegeneration and imperfect ageing. Front Cell Dev Biol.
11:11465642023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schiaffarino O, Valdivieso González D,
García-Pérez IM, Peñalva DA, Almendro-Vedia VG, Natale P and
López-Montero I: Mitochondrial membrane models built from native
lipid extracts: Interfacial and transport properties. Front Mol
Biosci. 9:9109362022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Giacomello M and Pellegrini L: The coming
of age of the mitochondria-ER contact: A matter of thickness. Cell
Death Differ. 23:1417–1427. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Csordás G, Renken C, Várnai P, Walter L,
Weaver D, Buttle KF, Balla T, Mannella CA and Hajnóczky G:
Structural and functional features and significance of the physical
linkage between ER and mitochondria. J Cell Biol. 174:915–921.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Filadi R, Theurey P and Pizzo P: The
endoplasmic reticulum-mitochondria coupling in health and disease:
Molecules, functions and significance. Cell Calcium. 62:1–15. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang N, Wang C, Zhao H, He Y, Lan B, Sun L
and Gao Y: The MAMs structure and its role in cell death. Cells.
10:6572021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mohan AA and Talwar P: MAM kinases:
Physiological roles, related diseases, and therapeutic
perspectives-a systematic review. Cell Mol Biol Lett. 30:352025.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Adesina SE, Kang BY, Bijli KM, Ma J, Cheng
J, Murphy TC, Michael Hart C and Sutliff RL: Targeting
mitochondrial reactive oxygen species to modulate hypoxia-induced
pulmonary hypertension. Free Radic Biol Med. 87:36–47. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Haslip M, Dostanic I, Huang Y, Zhang Y,
Russell KS, Jurczak MJ, Mannam P, Giordano F, Erzurum SC and Lee
PJ: Endothelial uncoupling protein 2 regulates mitophagy and
pulmonary hypertension during intermittent hypoxia. Arterioscler
Thromb Vasc Biol. 35:1166–1178. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ye JX, Wang SS, Ge M and Wang DJ:
Suppression of endothelial PGC-1α is associated with
hypoxia-induced endothelial dysfunction and provides a new
therapeutic target in pulmonary arterial hypertension. Am J Physiol
Lung Cell Mol Physiol. 310:L1233–L1242. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lei W, Li J, Li C, Chen L, Huang F, Xiao
D, Zhang J, Zhao J, Li G, Qu T, et al: MARCH5 restores endothelial
cell function against ischaemic/hypoxia injury via Akt/eNOS
pathway. J Cell Mol Med. 25:3182–3193. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou H, Wang J, Zhu P, Hu S and Ren J:
Ripk3 regulates cardiac microvascular reperfusion injury: The role
of IP3R-dependent calcium overload, XO-mediated oxidative stress
and F-action/filopodia-based cellular migration. Cell Signal.
45:12–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen Y, Li S, Zhang Y, Wang M, Li X, Liu
S, Xu D, Bao Y, Jia P, Wu N, et al: The lncRNA Malat1 regulates
microvascular function after myocardial infarction in mice via
miR-26b-5p/Mfn1 axis-mediated mitochondrial dynamics. Redox Biol.
41:1019102021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhong Z, Hou Y, Zhou C, Wang J, Gao L, Wu
X, Zhou G, Liu S, Xu Y and Yang W: FL3 mitigates cardiac
ischemia-reperfusion injury by promoting mitochondrial fusion to
restore calcium homeostasis. Cell Death Discov. 11:3042025.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Groschner LN, Waldeck-Weiermair M, Malli R
and Graier WF: Endothelial mitochondria-less respiration, more
integration. Pflugers Arch. 464:63–76. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang J, Toan S and Zhou H: New insights
into the role of mitochondria in cardiac microvascular
ischemia/reperfusion injury. Angiogenesis. 23:299–314. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pang B, Dong G, Pang T, Sun X, Liu X, Nie
Y and Chang X: Advances in pathogenesis and treatment of vascular
endothelial injury-related diseases mediated by mitochondrial
abnormality. Front Pharmacol. 15:14226862024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dorn GW II: Evolving concepts of
mitochondrial dynamics. Annu Rev Physiol. 81:1–17. 2019. View Article : Google Scholar
|
|
45
|
Murphy E and Steenbergen C: Mechanisms
underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang HH, Wu YJ, Tseng YM, Su CH, Hsieh CL
and Yeh HI: Mitochondrial fission protein 1 up-regulation
ameliorates senescence-related endothelial dysfunction of human
endothelial progenitor cells. Angiogenesis. 22:569–582. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu Y, Huo JL, Ren K, Pan S, Liu H, Zheng
Y, Chen J, Qiao Y, Yang Y and Feng Q: Mitochondria-associated
endoplasmic reticulum membrane (MAM): A dark horse for diabetic
cardiomyopathy treatment. Cell Death Discov. 10:1482024. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tagaya M and Arasaki K: Regulation of
mitochondrial dynamics and autophagy by the mitochondria-associated
membrane. Adv Exp Med Biol. 997:33–47. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bernal AF, Mota N, Pamplona R, Area-Gomez
E and Portero-Otin M: Hakuna MAM-Tata: Investigating the role of
mitochondrial-associated membranes in ALS. Biochim Biophys Acta Mol
Basis Dis. 1869:1667162023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chan DC: Fusion and fission: Interlinked
processes critical for mitochondrial health. Annu Rev Genet.
46:265–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen W, Zhao H and Li Y: Mitochondrial
dynamics in health and disease: Mechanisms and potential targets.
Signal Transduct Target Ther. 8:3332023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ferrier V: Mitochondrial fission in life
and death. Nat Cell Biol. 3:E2692001. View Article : Google Scholar
|
|
53
|
Gatti P, Schiavon C, Cicero J, Manor U and
Germain M: Mitochondria- and ER-associated actin are required for
mitochondrial fusion. Nat Commun. 16:4512025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hemel IMGM, Sarantidou R and Gerards M: It
takes two to tango: The essential role of ER-mitochondrial contact
sites in mitochondrial dynamics. Int J Biochem Cell Biol.
141:1061012021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Guo Y, Li D, Zhang S, Yang Y, Liu JJ, Wang
X, Liu C, Milkie DE, Moore RP, Tulu US, et al: Visualizing
intracellular organelle and cytoskeletal interactions at nanoscale
resolution on millisecond timescales. Cell. 175:1430–1442.e17.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Al Ojaimi M, Salah A and El-Hattab AW:
Mitochondrial fission and fusion: Molecular mechanisms, biological
functions, and related disorders. Membranes (Basel). 12:8932022.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Friedman JR, Lackner LL, West M,
DiBenedetto JR, Nunnari J and Voeltz GK: ER tubules mark sites of
mitochondrial division. Science. 334:358–362. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hyun HW, Min SJ and Kim JE: CDK5
inhibitors prevent astroglial apoptosis and reactive astrogliosis
by regulating PKA and DRP1 phosphorylations in the rat hippocampus.
Neurosci Res. 119:24–37. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ganesan V, Willis SD, Chang KT, Beluch S,
Cooper KF and Strich R: Cyclin C directly stimulates Drp1 GTP
affinity to mediate stress-induced mitochondrial hyperfission. Mol
Biol Cell. 30:302–311. 2019. View Article : Google Scholar :
|
|
60
|
Bravo-Sagua R, Parra V, Ortiz-Sandoval C,
Navarro-Marquez M, Rodríguez AE, Diaz-Valdivia N, Sanhueza C,
Lopez-Crisosto C, Tahbaz N, Rothermel BA, et al: Author correction:
Caveolin-1 impairs PKA-DRP1-mediated remodelling of ER-mitochondria
communication during the early phase of ER stress. Cell Death
Differ. 26:24942019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Giedt RJ, Yang C, Zweier JL, Matzavinos A
and Alevriadou BR: Mitochondrial fission in endothelial cells after
simulated ischemia/reperfusion: Role of nitric oxide and reactive
oxygen species. Free Radic Biol Med. 52:348–356. 2012. View Article : Google Scholar
|
|
62
|
Ko AR, Hyun HW, Min SJ and Kim JE: The
differential DRP1 phosphorylation and mitochondrial dynamics in the
regional specific astroglial death induced by status epilepticus.
Front Cell Neurosci. 10:1242016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Otera H, Ishihara N and Mihara K: New
insights into the function and regulation of mitochondrial fission.
Biochim Biophys Acta. 1833:1256–1268. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ding Y, Liu N, Zhang D, Guo L, Shang Q,
Liu Y, Ren G and Ma X: Mitochondria-associated endoplasmic
reticulum membranes as a therapeutic target for cardiovascular
diseases. Front Pharmacol. 15:13983812024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ilamathi HS and Germain M: ER-mitochondria
contact sites in mitochondrial DNA dynamics, maintenance, and
distribution. Int J Biochem Cell Biol. 166:1064922024. View Article : Google Scholar
|
|
66
|
Losón OC, Song Z, Chen H and Chan DC:
Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in
mitochondrial fission. Mol Biol Cell. 24:659–667. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Otera H, Wang C, Cleland MM, Setoguchi K,
Yokota S, Youle RJ and Mihara K: Mff is an essential factor for
mitochondrial recruitment of Drp1 during mitochondrial fission in
mammalian cells. J Cell Biol. 191:1141–1158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wu W, Lin C, Wu K, Jiang L, Wang X, Li W,
Zhuang H, Zhang X, Chen H, Li S, et al: FUNDC1 regulates
mitochondrial dynamics at the ER-mitochondrial contact site under
hypoxic conditions. EMBO J. 35:1368–1384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Korobova F, Ramabhadran V and Higgs HN: An
actin-dependent step in mitochondrial fission mediated by the
ER-associated formin INF2. Science. 339:464–467. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ji WK, Chakrabarti R, Fan X, Schoenfeld L,
Strack S and Higgs HN: Receptor-mediated Drp1 oligomerization on
endoplasmic reticulum. J Cell Biol. 216:4123–4139. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Li S, Li J, Li Y, Ye Q, Wang R, Liu X, Li
H, Peng D and Duan X: Regulation of NR4A1 by Taohong Siwu decoction
inhibits endothelial cell apoptosis in cerebral
ischemia-reperfusion injury. J Ethnopharmacol. 353:1202852025.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma
S, Zhu H, Ren J and Zhou H: DUSP1 alleviates cardiac
ischemia/reperfusion injury by suppressing the Mff-required
mitochondrial fission and Bnip3-related mitophagy via the JNK
pathways. Redox Biol. 14:576–587. 2018. View Article : Google Scholar
|
|
73
|
Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu
S, Ren J and Chen Y: NR4A1 aggravates the cardiac microvascular
ischemia reperfusion injury through suppressing FUNDC1-mediated
mitophagy and promoting Mff-required mitochondrial fission by CK2α.
Basic Res Cardiol. 113:232018. View Article : Google Scholar
|
|
74
|
Landes T and Martinou JC: Mitochondrial
outer membrane permeabilization during apoptosis: The role of
mitochondrial fission. Biochim Biophys Acta. 1813:540–545. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Adebayo M, Singh S, Singh AP and Dasgupta
S: Mitochondrial fusion and fission: The fine-tune balance for
cellular homeostasis. FASEB J. 35:e216202021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hu SY, Zhang Y, Zhu PJ, Zhou H and Chen
YD: Liraglutide directly protects cardiomyocytes against
reperfusion injury possibly via modulation of intracellular calcium
homeostasis. J Geriatr Cardiol. 14:57–66. 2017.PubMed/NCBI
|
|
77
|
Zhu H, Jin Q, Li Y, Ma Q, Wang J, Li D,
Zhou H and Chen Y: Melatonin protected cardiac microvascular
endothelial cells against oxidative stress injury via suppression
of IP3R-[Ca2+] c/VDAC-[Ca2+]m axis by
activation of MAPK/ERK signaling pathway. Cell Stress Chaperones.
23:101–113. 2018. View Article : Google Scholar
|
|
78
|
Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu
P, Ma Q, Tian F and Chen Y: Mff-dependent mitochondrial fission
contributes to the pathogenesis of cardiac microvasculature
ischemia/reperfusion injury via induction of mROS-mediated
cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP
opening. J Am Heart Assoc. 6:e0053282017. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q,
Jin Q, Cao F, Tian F and Chen Y: Melatonin protects cardiac
microvasculature against ischemia/reperfusion injury via
suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis.
J Pineal Res. 63:e124132017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Shenouda SM, Widlansky ME, Chen K, Xu G,
Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, et
al: Altered mitochondrial dynamics contributes to endothelial
dysfunction in diabetes mellitus. Circulation. 124:444–453. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Qu K, Yan F, Qin X, Zhang K, He W, Dong M
and Wu G: Mitochondrial dysfunction in vascular endothelial cells
and its role in atherosclerosis. Front Physiol. 13:10846042022.
View Article : Google Scholar
|
|
82
|
Ong SB, Subrayan S, Lim SY, Yellon DM,
Davidson SM and Hausenloy DJ: Inhibiting mitochondrial fission
protects the heart against ischemia/reperfusion injury.
Circulation. 121:2012–2022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Liu Z, Han X, You Y, Xin G, Li L, Gao J,
Meng H, Cao C, Liu J, Zhang Y, et al: Shuangshen ningxin formula
attenuates cardiac microvascular ischemia/reperfusion injury
through improving mitochondrial function. J Ethnopharmacol.
323:1176902024. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zhou H, Wang J, Hu S, Zhu H, Toanc S and
Ren J: BI1 alleviates cardiac microvascular ischemia-reperfusion
injury via modifying mitochondrial fission and inhibiting
XO/ROS/F-actin pathways. J Cell Physiol. 234:5056–5069. 2019.
View Article : Google Scholar
|
|
85
|
Zhou H, Shi C, Hu S, Zhu H, Ren J and Chen
Y: BI1 is associated with microvascular protection in cardiac
ischemia reperfusion injury via repressing
Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis.
21:599–615. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Chen Y, Liu C, Zhou P, Li J, Zhao X, Wang
Y, Chen R, Song L, Zhao H and Yan H: Coronary endothelium no-reflow
injury is associated with ROS-modified mitochondrial fission
through the JNK-Drp1 signaling pathway. Oxid Med Cell Longev.
2021:66995162021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zou R, Shi W, Qiu J, Zhou N, Du N, Zhou H,
Chen X and Ma L: Empagliflozin attenuates cardiac microvascular
ischemia/reperfusion injury through improving mitochondrial
homeostasis. Cardiovasc Diabetol. 21:1062022. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Cheng YH, Chiang CY, Wu CH and Chien CT:
2'-Hydroxycinnamaldehyde, a natural product from cinnamon,
alleviates ischemia/reperfusion-induced microvascular dysfunction
and oxidative damage in rats by upregulating cytosolic BAG3 and
Nrf2/HO-1. Int J Mol Sci. 25:129622024. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Mishra P, Carelli V, Manfredi G and Chan
DC: Proteolytic cleavage of Opa1 stimulates mitochondrial inner
membrane fusion and couples fusion to oxidative phosphorylation.
Cell Metab. 19:630–641. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zhao L, Zhuang J, Wang Y, Zhou D, Zhao D,
Zhu S, Pu J, Zhang H, Yin M, Zhao W, et al: Propofol ameliorates
H9c2 cells apoptosis induced by oxygen glucose deprivation and
reperfusion injury via inhibiting high levels of mitochondrial
fusion and fission. Front Pharmacol. 10:612019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
de Brito OM and Scorrano L: Mitofusin 2
tethers endoplasmic reticulum to mitochondria. Nature. 456:605–610.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Stuppia G, Rizzo F, Riboldi G, Del Bo R,
Nizzardo M, Simone C, Comi GP, Bresolin N and Corti S: MFN2-related
neuropathies: Clinical features, molecular pathogenesis and
therapeutic perspectives. J Neurol Sci. 356:7–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Filadi R, Greotti E, Turacchio G, Luini A,
Pozzan T and Pizzo P: Mitofusin 2 ablation increases endoplasmic
reticulum-mitochondria coupling. Proc Natl Acad Sci USA.
112:E2174–E2181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Prinz WA: Bridging the gap: Membrane
contact sites in signaling, metabolism, and organelle dynamics. J
Cell Biol. 205:759–769. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Yang F, Wu R, Jiang Z, Chen J, Nan J, Su
S, Zhang N, Wang C, Zhao J, Ni C, et al: Leptin increases
mitochondrial OPA1 via GSK3-mediated OMA1 ubiquitination to enhance
therapeutic effects of mesenchymal stem cell transplantation. Cell
Death Dis. 9:5562018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Sprenger HG and Langer T: The good and the
bad of mitochondrial breakups. Trends Cell Biol. 29:888–900. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Romanello V, Scalabrin M, Albiero M,
Blaauw B, Scorrano L and Sandri M: Inhibition of the fission
machinery mitigates OPA1 impairment in adult skeletal muscles.
Cells. 9:5972019. View Article : Google Scholar
|
|
98
|
Yin W, Li R, Feng X and James Kang Y: The
involvement of cytochrome c oxidase in mitochondrial fusion in
primary cultures of neonatal rat cardiomyocytes. Cardiovasc
Toxicol. 19:365–373. 2018. View Article : Google Scholar
|
|
99
|
Anderson CJ, Kahl A, Fruitman H, Qian L,
Zhou P, Manfredi G and Iadecola C: Prohibitin levels regulate OMA1
activity and turnover in neurons. Cell Death Differ. 27:1896–1906.
2020. View Article : Google Scholar :
|
|
100
|
Schulman JJ, Szczesniak LM, Bunker EN,
Nelson HA, Roe MW, Wagner LE II, Yule DI and Wojcikiewicz RJH: Bok
regulates mitochondrial fusion and morphology. Cell Death Differ.
26:2682–2694. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ding M, Liu C, Shi R, Yu M, Zeng K, Kang
J, Fu F and Mi M: Mitochondrial fusion promoter restores
mitochondrial dynamics balance and ameliorates diabetic
cardiomyopathy in an optic atrophy 1-dependent way. Acta Physiol
(Oxf). 229:e134282020. View Article : Google Scholar
|
|
102
|
Hong Y, Tak H, Kim C, Kang H, Ji E, Ahn S,
Jung M, Kim HL, Lee JH, Kim W and Lee EK: RNA binding protein HuD
contributes to β-cell dysfunction by impairing mitochondria
dynamics. Cell Death Differ. 27:1633–1643. 2020. View Article : Google Scholar
|
|
103
|
Meyer JN, Leuthner TC and Luz AL:
Mitochondrial fusion, fission, and mitochondrial toxicity.
Toxicology. 391:42–53. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Wu W, Zhao D, Shah SZA, Zhang X, Lai M,
Yang D, Wu X, Guan Z, Li J, Zhao H, et al: OPA1 overexpression
ameliorates mitochondrial cristae remodeling, mitochondrial
dysfunction, and neuronal apoptosis in prion diseases. Cell Death
Dis. 10:7102019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Lugus JJ, Ngoh GA, Bachschmid MM and Walsh
K: Mitofusins are required for angiogenic function and modulate
different signaling pathways in cultured endothelial cells. J Mol
Cell Cardiol. 51:885–893. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Sabouny R and Shutt TE: The role of
mitochondrial dynamics in mtDNA maintenance. J Cell Sci.
134:jcs2589442021. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Papanicolaou KN, Khairallah RJ, Ngoh GA,
Chikando A, Luptak I, O'Shea KM, Riley DD, Lugus JJ, Colucci WS,
Lederer WJ, et al: Mitofusin-2 maintains mitochondrial structure
and contributes to stress-induced permeability transition in
cardiac myocytes. Mol Cell Biol. 31:1309–1328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Peng C, Rao W, Zhang L, Wang K, Hui H,
Wang L, Su N, Luo P, Hao YL, Tu Y, et al: Corrigendum to 'Mitofusin
2 ameliorates hypoxia-induced apoptosis via mitochondrial function
and signaling pathways title of article' [International Journal of
Biochemistry and Cell Biology 69 (2015) 29-40]. Int J Biochem Cell
Biol. 73:1372016. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Tan Y, Mui D, Toan S, Zhu P, Li R and Zhou
H: SERCA overexpression improves mitochondrial quality control and
attenuates cardiac microvascular ischemia-reperfusion injury. Mol
Ther Nucleic Acids. 22:696–707. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Li Y, Huang D, Jia L, Shangguan F, Gong S,
Lan L, Song Z, Xu J, Yan C, Chen T, et al: LonP1 links
mitochondria-er interaction to regulate heart function. Research
(Wash D C). 6:01752023.PubMed/NCBI
|
|
111
|
Liu P, Xie Q, Wei T, Chen Y, Chen H and
Shen W: Activation of the NLRP3 inflammasome induces vascular
dysfunction in obese OLETF rats. Biochem Biophys Res Commun.
468:319–325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Jung M, Dodsworth M and Thum T:
Inflammatory cells and their non-coding RNAs as targets for
treating myocardial infarction. Basic Res Cardiol. 114:42018.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Xue J, Yan X, Yang Y, Chen M, Wu L, Gou Z,
Sun Z, Talabieke S, Zheng Y and Luo D: Connexin 43
dephosphorylation contributes to arrhythmias and cardiomyocyte
apoptosis in ischemia/reperfusion hearts. Basic Res Cardiol.
114:402019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Veeranki S and Tyagi SC: Mdivi-1 induced
acute changes in the angiogenic profile after ischemia-reperfusion
injury in female mice. Physiol Rep. 5:e132982017. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Rouault P, Guimbal S, Cornuault L,
Bourguignon C, Foussard N, Alzieu P, Choveau F, Benoist D, Chapouly
C, Gadeau AP, et al: Thrombosis in the coronary microvasculature
impairs cardiac relaxation and induces diastolic dysfunction.
Arterioscler Thromb Vasc Biol. 44:e1–e18. 2024. View Article : Google Scholar
|
|
116
|
Paillard M, Tubbs E, Thiebaut PA, Gomez L,
Fauconnier J, Da Silva CC, Teixeira G, Mewton N, Belaidi E, Durand
A, et al: Depressing mitochondria-reticulum interactions protects
cardiomyocytes from lethal hypoxia-reoxygenation injury.
Circulation. 128:1555–1565. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Hall AR, Burke N, Dongworth RK, Kalkhoran
SB, Dyson A, Vicencio JM, Dorn GW II, Yellon DM and Hausenloy DJ:
Hearts deficient in both Mfn1 and Mfn2 are protected against acute
myocardial infarction. Cell Death Dis. 7:e22382016. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Hu C, Huang Y and Li L: Drp1-dependent
mitochondrial fission plays critical roles in physiological and
pathological progresses in mammals. Int J Mol Sci. 18:1442017.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
He P, Talukder MAH and Gao F: Oxidative
stress and microvessel barrier dysfunction. Front Physiol.
11:4722020. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Ivanina AV, Nesmelova I, Leamy L, Sokolov
EP and Sokolova IM: Intermittent hypoxia leads to functional
reorganization of mitochondria and affects cellular bioenergetics
in marine molluscs. J Exp Biol. 219:1659–1674. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zhao RZ, Jiang S, Zhang L and Yu ZB:
Mitochondrial electron transport chain, ROS generation and
uncoupling (Review). Int J Mol Med. 44:3–15. 2019.PubMed/NCBI
|
|
122
|
Qing G, Huang C, Pei J and Peng B:
Alteration of cardiac energetics and mitochondrial function in
doxorubicin-induced cardiotoxicity: Molecular mechanism and
prospective implications (Review). Int J Mol Med. 56:1832025.
View Article : Google Scholar :
|
|
123
|
Potier E, Ferreira E, Meunier A, Sedel L,
Logeart-Avramoglou D and Petite H: Prolonged hypoxia concomitant
with serum deprivation induces massive human mesenchymal stem cell
death. Tissue Eng. 13:1325–1331. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Zhou H, Yang J, Xin T, Li D, Guo J, Hu S,
Zhou S, Zhang T, Zhang Y, Han T and Chen Y: Exendin-4 protects
adipose-derived mesenchymal stem cells from apoptosis induced by
hydrogen peroxide through the PI3K/Akt-Sfrp2 pathways. Free Radic
Biol Med. 77:363–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Granger DN and Kvietys PR: Reperfusion
injury and reactive oxygen species: The evolution of a concept.
Redox Biol. 9:524–551. 2015. View Article : Google Scholar
|
|
126
|
Kar S and Kavdia M: Modeling of
biopterin-dependent pathways of eNOS for nitric oxide and
superoxide production. Free Radic Biol Med. 51:1411–1427. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Giorgi C, Missiroli S, Patergnani S,
Duszynski J, Wieckowski MR and Pinton P: Mitochondria-associated
membranes: Composition, molecular mechanisms, and
physio-pathological implications. Antioxid Redox Signal.
22:995–1019. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Missiroli S, Patergnani S, Caroccia N,
Pedriali G, Perrone M, Previati M, Wieckowski MR and Giorgi C:
Mitochondria-associated membranes (MAMs) and inflammation. Cell
Death Dis. 9:3292018. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao
C, Xin C, Zhu D, Li Y, Yan W, et al: TXNIP mediates NLRP3
inflammasome activation in cardiac microvascular endothelial cells
as a novel mechanism in myocardial ischemia/reperfusion injury.
Basic Res Cardiol. 109:4152014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Ovciarikova J, Shikha S, Lacombe A,
Courjol F, McCrone R, Hussain W, Maclean A, Lemgruber L,
Martins-Duarte ES, Gissot M and Sheiner L: Two ancient membrane
pores mediate mitochondrial-nucleus membrane contact sites. J Cell
Biol. 223:e2023040752024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Yan YR, Zhang L, Lin YN, Sun XW, Ding YJ,
Li N, Li HP, Li SQ, Zhou JP and Li QY: Chronic intermittent
hypoxia-induced mitochondrial dysfunction mediates endothelial
injury via the TXNIP/NLRP3/IL-1β signaling pathway. Free Radic Biol
Med. 165:401–410. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Francisco J and Del Re DP: Inflammation in
myocardial ischemia/reperfusion injury: Underlying mechanisms and
therapeutic potential. Antioxidants (Basel). 12:19442023.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
van Hout GPJ, Bosch L, Ellenbroek GHJM, de
Haan JJ, van Solinge WW, Cooper MA, Arslan F, de Jager SC,
Robertson AAB, Pasterkamp G and Hoefer IE: The selective
NLRP3-inflammasome inhibitor MCC950 reduces infarct size and
preserves cardiac function in a pig model of myocardial infarction.
Eur Heart J. 38:828–836. 2017.
|
|
134
|
Zhang X, Zeng W, Zhang Y, Yu Q, Zeng M,
Gan J, Zhang W, Jiang X and Li H: Focus on the role of mitochondria
in NLRP3 inflammasome activation: A prospective target for the
treatment of ischemic stroke (Review). Int J Mol Med. 49:742022.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Toldo S, Mauro AG, Cutter Z and Abbate A:
Inflammasome, pyroptosis, and cytokines in myocardial
ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol.
315:H1553–H1568. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Frand AR and Kaiser CA: Ero1p oxidizes
protein disulfide isomerase in a pathway for disulfide bond
formation in the endoplasmic reticulum. Mol Cell. 4:469–477. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Giorgio M, Migliaccio E, Orsini F,
Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S,
Marcaccio M, et al: Electron transfer between cytochrome c and
p66Shc generates reactive oxygen species that trigger mitochondrial
apoptosis. Cell. 122:221–233. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Gilady SY, Bui M, Lynes EM, Benson MD,
Watts R, Vance JE and Simmen T: Ero1alpha requires oxidizing and
normoxic conditions to localize to the mitochondria-associated
membrane (MAM). Cell Stress Chaperones. 15:619–629. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Anelli T, Bergamelli L, Margittai E,
Rimessi A, Fagioli C, Malgaroli A, Pinton P, Ripamonti M, Rizzuto R
and Sitia R: Ero1α regulates Ca(2+) fluxes at the endoplasmic
reticulum-mitochondria interface (MAM). Antioxid Redox Signal.
16:1077–1087. 2012. View Article : Google Scholar
|
|
140
|
Bassot A, Chen J, Takahashi-Yamashiro K,
Yap MC, Gibhardt CS, Le GNT, Hario S, Nasu Y, Moore J, Gutiérrez T,
et al: The endoplasmic reticulum kinase PERK interacts with the
oxidoreductase ERO1 to metabolically adapt mitochondria. Cell Rep.
42:1118992023. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Muñoz JP, Ivanova S, Sánchez-Wandelmer J,
Martínez-Cristóbal P, Noguera E, Sancho A, Díaz-Ramos A,
Hernández-Alvarez MI, Sebastián D, Mauvezin C, et al: Mfn2
modulates the UPR and mitochondrial function via repression of
PERK. EMBO J. 32:2348–2361. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
van Vliet AR, Giordano F, Gerlo S, Segura
I, Van Eygen S, Molenberghs G, Rocha S, Houcine A, Derua R,
Verfaillie T, et al: The ER stress sensor PERK coordinates
ER-plasma membrane contact site formation through interaction with
filamin-A and F-actin remodeling. Mol Cell. 65:885–899.e6. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Verfaillie T, Rubio N, Garg AD, Bultynck
G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A
and Agostinis P: PERK is required at the ER-mitochondrial contact
sites to convey apoptosis after ROS-based ER stress. Cell Death
Differ. 19:1880–1891. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Li G, Mongillo M, Chin KT, Harding H, Ron
D, Marks AR and Tabas I: Role of ERO1-alpha-mediated stimulation of
inositol 1,4,5-triphosphate receptor activity in endoplasmic
reticulum stress-induced apoptosis. J Cell Biol. 186:783–792. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Pinton P, Rimessi A, Marchi S, Orsini F,
Migliaccio E, Giorgio M, Contursi C, Minucci S, Mantovani F,
Wieckowski MR, et al: Protein kinase C beta and prolyl isomerase 1
regulate mitochondrial effects of the life-span determinant p66Shc.
Science. 315:659–663. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Akhmedov A, Montecucco F, Braunersreuther
V, Camici GG, Jakob P, Reiner MF, Glanzmann M, Burger F, Paneni F,
Galan K, et al: Genetic deletion of the adaptor protein p66Shc
increases susceptibility to short-term ischaemic myocardial injury
via intracellular salvage pathways. Eur Heart J. 36:516–526a. 2015.
View Article : Google Scholar
|
|
147
|
Carreras-Sureda A, Jaña F, Urra H, Durand
S, Mortenson DE, Sagredo A, Bustos G, Hazari Y, Ramos-Fernández E,
Sassano ML, et al: Publisher correction: Non-canonical function of
IRE1α determines mitochondria-associated endoplasmic reticulum
composition to control calcium transfer and bioenergetics. Nat Cell
Biol. 21:9132019. View Article : Google Scholar
|
|
148
|
Braunersreuther V, Montecucco F, Asrih M,
Pelli G, Galan K, Frias M, Burger F, Quinderé AL, Montessuit C,
Krause KH, et al: Role of NADPH oxidase isoforms NOX1, NOX2 and
NOX4 in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol.
64:99–107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Beretta M, Santos CX, Molenaar C, Hafstad
AD, Miller CC, Revazian A, Betteridge K, Schröder K,
Streckfuß-Bömeke K, Doroshow JH, et al: Nox4 regulates
InsP3 receptor-dependent Ca2+ release into
mitochondria to promote cell survival. EMBO J. 39:e1035302020.
View Article : Google Scholar
|
|
150
|
Fang G, Shen Y and Liao D: ENPP2
alleviates hypoxia/reoxygenation injury and ferroptosis by
regulating oxidative stress and mitochondrial function in human
cardiac microvascular endothelial cells. Cell Stress Chaperones.
28:253–263. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Feng Y, Madungwe NB, Imam Aliagan AD,
Tombo N and Bopassa JC: Liproxstatin-1 protects the mouse
myocardium against ischemia/reperfusion injury by decreasing VDAC1
levels and restoring GPX4 levels. Biochem Biophys Res Commun.
520:606–611. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
You W, Wu Z, Ye F and Wu X: Ginkgolide A
protects adverse cardiac remodeling through enhancing antioxidation
and nitric oxide utilization in mice with pressure overload.
Pharmazie. 74:698–702. 2019.PubMed/NCBI
|
|
153
|
Helmstädter J, Frenis K, Filippou K, Grill
A, Dib M, Kalinovic S, Pawelke F, Kus K, Kröller-Schön S, Oelze M,
et al: Endothelial GLP-1 (glucagon-like peptide-1) receptor
mediates cardiovascular protection by liraglutide in mice with
experimental arterial hypertension. Arterioscler Thromb Vasc Biol.
40:145–158. 2020. View Article : Google Scholar :
|
|
154
|
Yang J, Moraga A, Xu J, Zhao Y, Luo P, Lao
KH, Margariti A, Zhao Q, Ding W, Wang G, et al: A histone
deacetylase 7-derived peptide promotes vascular regeneration via
facilitating 14-3-3γ phosphorylation. Stem Cells. 38:556–573. 2020.
View Article : Google Scholar
|
|
155
|
Guan X, Yang Z, Wang J, Lu W, Wang S, Yang
M, Sun P, Hu W, Yang L and Li H: Naringin attenuates myocardial
ischemia-reperfusion injury by promoting mitochondrial
translocation of NDUFS1 and suppressing cardiac microvascular
endothelial cell ferroptosis. J Nutr Biochem. 145:1100192025.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Mao Y, Hu Y, Feng W, Yu L, Li P, Cai B, Li
C and Guan H: Effects and mechanisms of PSS-loaded nanoparticles on
coronary microcirculation dysfunction in streptozotocin-induced
diabetic cardiomyopathy rats. Biomed Pharmacother. 121:1092802020.
View Article : Google Scholar
|
|
157
|
Booth DM, Enyedi B, Geiszt M, Várnai P and
Hajnóczky G: Redox nanodomains are induced by and control calcium
signaling at the ER-mitochondrial interface. Mol Cell. 63:240–248.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Ježek J, Cooper KF and Strich R: The
impact of mitochondrial fission-stimulated ROS production on
pro-apoptotic chemotherapy. Biology (Basel). 10:332021.
|
|
159
|
Li B, Yang WW, Yao BC, Chen QL, Zhao LL,
Song YQ, Jiang N and Guo ZG: Liriodendrin alleviates myocardial
ischemia-reperfusion injury via partially attenuating apoptosis,
inflammation and mitochondria damage in rats. Int J Mol Med.
55:652025. View Article : Google Scholar :
|
|
160
|
Chen Z, Liu T, Xiong L and Liu Z: Shen-fu
injection modulates HIF-1α/BNIP3-mediated mitophagy to alleviate
myocardial ischemia-reperfusion injury. Cardiovasc Toxicol.
25:898–914. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Ashrafi G and Schwarz TL: The pathways of
mitophagy for quality control and clearance of mitochondria. Cell
Death Differ. 20:31–42. 2013. View Article : Google Scholar
|
|
162
|
Puri R, Cheng XT, Lin MY, Huang N and
Sheng ZH: Mul1 restrains Parkin-mediated mitophagy in mature
neurons by maintaining ER-mitochondrial contacts. Nat Commun.
10:36452019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Martens S and Fracchiolla D: Activation
and targeting of ATG8 protein lipidation. Cell Discov. 6:232020.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Condon KJ and Sabatini DM: Nutrient
regulation of mTORC1 at a glance. J Cell Sci. 132:jcs2225702019.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Hamasaki M, Furuta N, Matsuda A, Nezu A,
Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et
al: Autophagosomes form at ER-mitochondria contact sites. Nature.
459:389–393. 2013. View Article : Google Scholar
|
|
166
|
Barazzuol L, Giamogante F, Brini M and
Calì T: PINK1/Parkin mediated mitophagy, Ca2+
signalling, and ER-mitochondria contacts in Parkinson's disease.
Int J Mol Sci. 21:17722020. View Article : Google Scholar
|
|
167
|
McLelland GL, Goiran T, Yi W, Dorval G,
Chen CX, Lauinger ND, Krahn AI, Valimehr S, Rakovic A, Rouiller I,
et al: Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent
release of ER from mitochondria to drive mitophagy. Elife.
7:e328662018. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Gelmetti V, De Rosa P, Torosantucci L,
Marini ES, Romagnoli A, Di Rienzo M, Arena G, Vignone D, Fimia GM
and Valente EM: PINK1 and BECN1 relocalize at
mitochondria-associated membranes during mitophagy and promote
ER-mitochondria tethering and autophagosome formation. Autophagy.
13:654–669. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Fu M, St-Pierre P, Shankar J, Wang PT,
Joshi B and Nabi IR: Regulation of mitophagy by the Gp78 E3
ubiquitin ligase. Mol Biol Cell. 24:1153–1162. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Zhao X, Wang Z, Wang L, Jiang T, Dong D
and Sun M: The PINK1/Parkin signaling pathway-mediated mitophagy: A
forgotten protagonist in myocardial ischemia/reperfusion injury.
Pharmacol Res. 209:1074662024. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Quiles JM, Najor RH, Gonzalez E, Jeung M,
Liang W, Burbach SM, Zumaya EA, Diao RY, Lampert MA and Gustafsson
ÅB: Deciphering functional roles and interplay between Beclin1 and
Beclin2 in autophagosome formation and mitophagy. Sci Signal.
16:eabo44572023. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Russell RC, Tian Y, Yuan H, Park HW, Chang
YY, Kim J, Kim H, Neufeld TP, Dillin A and Guan KL: ULK1 induces
autophagy by phosphorylating Beclin-1 and activating VPS34 lipid
kinase. Nat Cell Biol. 15:741–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Kundu M, Lindsten T, Yang CY, Wu J, Zhao
F, Zhang J, Selak MA, Ney PA and Thompson CB: Ulk1 plays a critical
role in the autophagic clearance of mitochondria and ribosomes
during reticulocyte maturation. Blood. 112:1493–1502. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Sun W, Lu H, Dong S, Li R, Chu Y, Wang N,
Zhao Y, Zhang Y, Wang L, Sun L and Lu D: Beclin1 controls caspase-4
inflammsome activation and pyroptosis in mouse myocardial
reperfusion-induced microvascular injury. Cell Commun Signal.
19:1072021. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Lampert MA, Orogo AM, Najor RH, Hammerling
BC, Leon LJ, Wang BJ, Kim T, Sussman MA and Gustafsson ÅB:
BNIP3L/NIX and FUNDC1-mediated mitophagy is required for
mitochondrial network remodeling during cardiac progenitor cell
differentiation. Autophagy. 15:1182–1198. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Wang C, Dai X, Wu S, Xu W, Song P, Huang K
and Zou MH: FUNDC1-dependent mitochondria-associated endoplasmic
reticulum membranes are involved in angiogenesis and
neoangiogenesis. Nat Commun. 12:26162021. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Chen G, Han Z, Feng D, Chen Y, Chen L, Wu
H, Huang L, Zhou C, Cai X, Fu C, et al: A regulatory signaling loop
comprising the PGAM5 phosphatase and CK2 controls receptor-mediated
mitophagy. Mol Cell. 54:362–377. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Wu W, Tian W, Hu Z, Chen G, Huang L, Li W,
Zhang X, Xue P, Zhou C, Liu L, et al: ULK1 translocates to
mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO
Rep. 15:566–575. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Zhang Y, Zhuang H, Liu H and Feng D:
Molecular regulations of FUNDC1 at ER-mitochondria contacts under
hypoxic stress. Contact (Thousand Oaks).
5:251525642210924872022.PubMed/NCBI
|
|
180
|
Ji H, Wang J, Muid D, Song W, Jiang Y and
Zhou H: FUNDC1 activates the mitochondrial unfolded protein
response to preserve mitochondrial quality control in cardiac
ischemia/reperfusion injury. Cell Signal. 92:1102492022. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D,
Hu S, Ren J, Cao F and Chen Y: Ripk3 induces mitochondrial
apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury.
Redox Biol. 13:498–507. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Cai C, Guo Z, Chang X, Li Z, Wu F, He J,
Cao T, Wang K, Shi N, Zhou H, et al: Empagliflozin attenuates
cardiac microvascular ischemia/reperfusion through activating the
AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox Biol. 52:1022882022.
View Article : Google Scholar
|
|
183
|
Zhou H, Li D, Zhu P, Hu S, Hu N, Ma S,
Zhang Y, Han T, Ren J, Cao F and Chen Y: Melatonin suppresses
platelet activation and function against cardiac
ischemia/reperfusion injury via PPARγ/FUNDC1/mitophagy pathways. J
Pineal Res. 63:e124382017. View Article : Google Scholar
|
|
184
|
Li JJ, Wang YJ, Wang CM, Li YJ, Yang Q,
Cai WY, Chen Y and Zhu XX: Shenlian extract decreases mitochondrial
autophagy to regulate mitochondrial function in microvascular to
alleviate coronary artery no-reflow. Phytother Res. 37:1864–1882.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Liu S, Faitg J, Tissot C, Konstantopoulos
D, Laws R, Bourdier G, Andreux PA, Davey T, Gallart-Ayala H,
Ivanisevic J, et al: Urolithin A provides cardioprotection and
mitochondrial quality enhancement preclinically and improves human
cardiovascular health biomarkers. iScience. 28:1118142025.
View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Wang J, Toan S and Zhou H: Mitochondrial
quality control in cardiac microvascular ischemia-reperfusion
injury: New insights into the mechanisms and therapeutic
potentials. Pharmacol Res. 156:1047712020. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Shirihai OS, Song M and Dorn GW II: How
mitochondrial dynamism orchestrates mitophagy. Circ Res.
116:1835–1849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Wong YC, Ysselstein D and Krainc D:
Mitochondria-lysosome contacts regulate mitochondrial fission via
RAB7 GTP hydrolysis. Nature. 554:382–386. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Wang J, Zhu P, Li R, Ren J and Zhou H:
Fundc1-dependent mitophagy is obligatory to ischemic
preconditioning-conferred renoprotection in ischemic AKI via
suppression of Drp1-mediated mitochondrial fission. Redox Biol.
30:1014152020. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Huang CH, Chiang CY, Pen RH, Tsai MS, Chen
HW, Hsu CY, Wang TD, Ma MH, Chen SC and Chen WJ: Hypothermia
treatment preserves mitochondrial integrity and viability of
cardiomyocytes after ischaemic reperfusion injury. Injury.
46:233–239. 2015. View Article : Google Scholar
|
|
191
|
Wang Z, Liu D, Varin A, Nicolas V,
Courilleau D, Mateo P, Caubere C, Rouet P, Gomez AM, Vandecasteele
G, et al: A cardiac mitochondrial cAMP signaling pathway regulates
calcium accumulation, permeability transition and cell death. Cell
Death Dis. 7:e21982016. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Jiang RQ, Li QQ and Sheng R: Mitochondria
associated ER membranes and cerebral ischemia: Molecular mechanisms
and therapeutic strategies. Pharmacol Res. 191:1067612023.
View Article : Google Scholar : PubMed/NCBI
|
|
193
|
He X, Bi XY, Lu XZ, Zhao M, Yu XJ, Sun L,
Xu M, Wier WG and Zang WJ: Reduction of mitochondria-endoplasmic
reticulum interactions by acetylcholine protects human umbilical
vein endothelial cells from hypoxia/reoxygenation injury.
Arterioscler Thromb Vasc Biol. 35:1623–1634. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X,
Huang K, Xie Z and Zou MH: Binding of FUN14 domain containing 1
with inositol 1,4,5-trisphosphate receptor in
mitochondria-associated endoplasmic reticulum membranes maintains
mitochondrial dynamics and function in hearts in vivo. Circulation.
136:2248–2266. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Li C, Ma Q, Toan S, Wang J, Zhou H and
Liang J: SERCA overexpression reduces reperfusion-mediated cardiac
microvascular damage through inhibition of the
calcium/MCU/mPTP/necroptosis signaling pathways. Redox Biol.
36:1016592020. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Yang R, Zhang X, Xing P, Zhang S, Zhang F,
Wang J, Yu J, Zhu X and Chang P: Grpel2 alleviates myocardial
ischemia/reperfusion injury by inhibiting MCU-mediated
mitochondrial calcium overload. Biochem Biophys Res Commun.
609:169–175. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Carreras-Sureda A, Jaña F, Urra H, Durand
S, Mortenson DE, Sagredo A, Bustos G, Hazari Y, Ramos-Fernández E,
Sassano ML, et al: Non-canonical function of IRE1α determines
mitochondria-associated endoplasmic reticulum composition to
control calcium transfer and bioenergetics. Nat Cell Biol.
21:755–767. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Goodman JB, Qin F, Morgan RJ, Chambers JM,
Croteau D, Siwik DA, Hobai I, Panagia M, Luptak I, Bachschmid M, et
al: Redox-Resistant SERCA [Sarco(endo)plasmic Reticulum Calcium
ATPase] attenuates oxidant-stimulated mitochondrial calcium and
apoptosis in cardiac myocytes and pressure overload-induced
myocardial failure in mice. Circulation. 142:2459–2469. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Chen LT, Xu TT, Qiu YQ, Liu NY, Ke XY,
Fang L, Yan JP and Zhu DY: Homocysteine induced a calcium-mediated
disruption of mitochondrial function and dynamics in endothelial
cells. J Biochem Mol Toxicol. 35:e227372021. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Ma L, Zou R, Shi W, Zhou N, Chen S, Zhou
H, Chen X and Wu Y: SGLT2 inhibitor dapagliflozin reduces
endothelial dysfunction and microvascular damage during cardiac
ischemia/reperfusion injury through normalizing the
XO-SERCA2-CaMKII-coffilin pathways. Theranostics. 12:5034–5050.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Hoppe UC: Mitochondrial calcium channels.
FEBS Lett. 584:1975–1981. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Israelson A, Abu-Hamad S, Zaid H, Nahon E
and Shoshan-Barmatz V: Localization of the voltage-dependent anion
channel-1 Ca2+-binding sites. Cell Calcium. 41:235–244. 2007.
View Article : Google Scholar
|
|
203
|
Szabadkai G, Bianchi K, Várnai P, De
Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T and Rizzuto
R: Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J Cell Biol. 175:901–911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Li Y, Hu H, Chu C and Yang J:
Mitochondrial calcium uniporter complex: An emerging therapeutic
target for cardiovascular diseases (Review). Int J Mol Med.
55:402025. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Xu H, Guan N, Ren YL, Wei QJ, Tao YH, Yang
GS, Liu XY, Bu DF, Zhang Y and Zhu SN:
IP3R-Grp75-VDAC1-MCU calcium regulation axis antagonists
protect podocytes from apoptosis and decrease proteinuria in an
Adriamycin nephropathy rat model. BMC Nephrol. 19:1402018.
View Article : Google Scholar
|
|
206
|
Gomez L, Thiebaut PA, Paillard M, Ducreux
S, Abrial M, Crola Da Silva C, Durand A, Alam MR, Van Coppenolle F,
Sheu SS and Ovize M: The SR/ER-mitochondria calcium crosstalk is
regulated by GSK3β during reperfusion injury. Cell Death Differ.
23:313–322. 2016. View Article : Google Scholar
|
|
207
|
Hayashi T and Su TP: Sigma-1 receptor
chaperones at the ER-mitochondrion interface regulate Ca(2+)
signaling and cell survival. Cell. 131:596–610. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Embi N, Rylatt DB and Cohen P: Glycogen
synthase kinase-3 from rabbit skeletal muscle. Separation from
cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J
Biochem. 107:519–527. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Woodgett JR and Cohen P: Multisite
phosphorylation of glycogen synthase. Molecular basis for the
substrate specificity of glycogen synthase kinase-3 and casein
kinase-II (glycogen synthase kinase-5). Biochim Biophys Acta.
788:339–347. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Thoudam T, Jeon JH, Ha CM and Lee IK: Role
of mitochondria-associated endoplasmic reticulum membrane in
inflammation-mediated metabolic diseases. Mediators Inflamm.
2016:18514202016. View Article : Google Scholar
|
|
211
|
Elrod JW and Molkentin JD: Physiologic
functions of cyclophilin D and the mitochondrial permeability
transition pore. Circ J. 77:1111–1122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Nakagawa T, Shimizu S, Watanabe T,
Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T and Tsujimoto Y:
Cyclophilin D-dependent mitochondrial permeability transition
regulates some necrotic but not apoptotic cell death. Nature.
434:652–658. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Stoica R, De Vos KJ, Paillusson S, Mueller
S, Sancho RM, Lau KF, Vizcay-Barrena G, Lin WL, Xu YF, Lewis J, et
al: ER-mitochondria associations are regulated by the VAPB-PTPIP51
interaction and are disrupted by ALS/FTD-associated TDP-43. Nat
Commun. 5:39962014. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Gomez-Suaga P, Paillusson S, Stoica R,
Noble W, Hanger DP and Miller CCJ: The ER-mitochondria tethering
complex VAPB-PTPIP51 regulates autophagy. Curr Biol. 27:371–385.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
De Vos KJ, Mórotz GM, Stoica R, Tudor EL,
Lau KF, Ackerley S, Warley A, Shaw CE and Miller CC: VAPB interacts
with the mitochondrial protein PTPIP51 to regulate calcium
homeostasis. Hum Mol Genet. 21:1299–1311. 2012. View Article : Google Scholar :
|
|
216
|
Fucikova J, Spisek R, Kroemer G and
Galluzzi L: Calreticulin and cancer. Cell Res. 31:5–16. 2021.
View Article : Google Scholar :
|
|
217
|
Liu M, Li S, Yin M, Li Y, Chen J, Chen Y,
Zhou Y, Li Q, Xu F, Dai C, et al: Pinacidil ameliorates cardiac
microvascular ischemia-reperfusion injury by inhibiting
chaperone-mediated autophagy of calreticulin. Basic Res Cardiol.
119:113–131. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Zhang J, Wang L, Xie W, Hu S, Zhou H, Zhu
P, Zhu P and Zhu H: Melatonin attenuates ER stress and
mitochondrial damage in septic cardiomyopathy: A new mechanism
involving BAP31 upregulation and MAPK-ERK pathway. J Cell Physiol.
235:2847–2856. 2020. View Article : Google Scholar
|
|
219
|
Li S, Chen J, Liu M, Chen Y, Wu Y, Li Q,
Ma T, Gao J, Xia Y, Fan M, et al: Protective effect of HINT2 on
mitochondrial function via repressing MCU complex activation
attenuates cardiac microvascular ischemia-reperfusion injury. Basic
Res Cardiol. 116:652021. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Zhou H and Toan S: Pathological roles of
mitochondrial oxidative stress and mitochondrial dynamics in
cardiac microvascular ischemia/reperfusion injury. Biomolecules.
10:852020. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Vallese F, Catoni C, Cieri D, Barazzuol L,
Ramirez O, Calore V, Bonora M, Giamogante F, Pinton P, Brini M and
Calì T: An expanded palette of improved SPLICS reporters detects
multiple organelle contacts in vitro and in vivo. Nat Commun.
11:60692020. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Calì T and Brini M: Quantification of
organelle contact sites by split-GFP-based contact site sensors
(SPLICS) in living cells. Nat Protoc. 16:5287–5308. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Marshall AG, Neikirk K, Stephens DC, Vang
L, Vue Z, Beasley HK, Crabtree A, Scudese E, Lopez EG, Shao B, et
al: Serial block face-scanning electron microscopy as a burgeoning
technology. Adv Biol (Weinh). 7:e23001392023. View Article : Google Scholar : PubMed/NCBI
|