Introduction
Diabetes is a chronic and progressive metabolic
disorder, with an incidence rate increasing year by year (1). Diabetes can cause systemic changes,
which increase the mortality and disability rates of patients with
diabetes and it has attracted widespread attention from clinicians
and patients (2). However, once
diabetes occurs, the symptoms are generally severe and progress
rapidly, which seriously threatens the survival of patients with
diabetes (3). Diabetes
complications have always been a research hotspot and focus in the
diagnosis and treatment for researchers, including neurological
complications, microvascular complications and macrovascular
complications. Macrovascular complications mainly include
cardiovascular and cerebrovascular complications caused by
involvement of the coronary arteries, cerebrovascular vessels and
peripheral arteries, as well as diabetic foot. Microvascular
complications mainly include diabetic nephropathy and retinopathy
(4).
At present, numerous studies focus on
diabetes-related lung diseases, especially diabetes-induced lung
injury. Due to the abundant reserve of the pulmonary capillary
network, the respiratory symptoms of diabetes are not obvious in
the early stages of the disease and are easily confused with the
manifestations of cardiovascular and cerebrovascular complications,
making them easily overlooked by clinicians and patients. However,
once diabetes-related lung injury occurs, the symptoms are
generally severe and progress rapidly, posing a serious threat to
the survival of patients with diabetes (5). Diabetes-related lung injury is
mainly manifested as interstitial lung disease, including
inflammatory cell infiltration, increased collagen content,
oxidative stress, widened alveolar septa and tissue fibrosis
(6). Research shows that
significant abnormalities in lung function have been found in
patients with both type 1 and type 2 diabetes, as well as in
children and adults with diabetes (7). Studies have found that the reduced
pulmonary diffusion function in patients with diabetes is mainly
due to thickening of the alveolar basement membrane and alveolar
microvascular disease. Although pulmonary dysfunction is widely
observed in patients with diabetes, its specific mechanisms and
clinical implications require further exploration (8,9).
Diabetes-related pulmonary complications encompass heightened
susceptibility to respiratory infections and chronic conditions,
including pneumonia, asthma, pulmonary fibrosis and tuberculosis,
along with sleep-disordered breathing. A study indicates that
>50% of mortality cases among Japanese patients with diabetes
stem from these respiratory disorders (9). Another study shows that the
fibroblast growth factor (FGF) family is closely related to the
biological function of the lungs (10). Other members of the FGF family
play important roles in lung development and disease. For example,
FGF9 and FGF18 also exhibit important biological effects in
idiopathic pulmonary fibrosis (10). Endocrine members of the FGF
family, such as FGF19 and FGF21, play an anti-fibrotic role in
various organs. Research has shown that FGF19 can markedly reduce
pulmonary fibrosis in mouse models and may affect the fibrosis
process in the lungs by regulating the function of fibroblasts
(11). In addition, the FGF
signaling pathway also plays an important role in the repair and
regeneration of the lungs. Recent studies have shown that FGF4 has
an important biological role in the lungs (12,13). FGF4 belongs to the paracrine
subfamily of fibroblast growth factors (which also includes FGF5
and FGF6). It has a molecular weight of 22 kDa and is encoded by a
gene located on human chromosome 11 (11q13.3). FGF4 primarily binds
to and activates specific fibroblast growth factor receptors
(FGFRs), including FGFR1c, FGFR2c and FGFR3c. In vivo, FGF4
can be secreted by multiple organs under specific physiological
conditions, such as the liver, heart and testes. It has been
reported that FGF4 can alleviate diabetes and hyperglycemia
(14,15). However, up to now, the effect of
FGF4 on diabetic lung injury has not been elucidated.
The present study evaluated the effect of FGF4 on
high glucose-induced lung injury. In vitro, MLE-12 (a murine
alveolar epithelial cell line) and BEAS-2B (a human bronchial
epithelial cell line) were used to evaluate FGF4's protective
effect against lung cell damage induced by high glucose. It was
found that FGF4 markedly alleviated the damage caused by high
glucose to lung cells. The current study has laid a solid
foundation for further research on the effect of FGF4 on
high-glucose lung injury.
Materials and methods
Reagents and antibodies
Streptozotocin (STZ) was purchased from
MilliporeSigma. PVDF membrane, Immobilon, BCA Kit (protein
concentration determination) and hematoxylin-eosin (H&E)
staining kit were from Beijing Solarbio Science & Technology
Co., Ltd. and Masson trichrome staining solution were from Beijing
Solarbio Science & Technology Co., Ltd. Antibodies for TNF-α,
IL-6, superoxide dismutase (SOD)1, 4-Hydroxynonenal and
NFE2-related factor 2 (Nrf2) were from Abcam. Goat anti-rabbit IgG
labeled with horseradish peroxidase, goat anti-mouse IgG labeled
with horseradish peroxidase and electrochemiluminescence (ECL) were
from Beijing Solarbio Science & Technology Co., Ltd. TGF-β1 and
4-hydroxynonenal (4HNE) were from Cell Signaling Technology, Inc.
Cell culture plates, FBS, BSA and TBST buffer were from Beyotime
Institute of Biotechnology. CyclinD1, proliferating cell nuclear
antigen (PCNA), phosphorylated (p-)IκB, p-P65 and p-STAT3 were from
Proteintech Group, Inc. malondialdehyde (MDA; cat. no. A003-1-2),
SOD (cat. no. A001-3-2) and glutathione peroxidase (GPX; cat. no.
H545-1-1) were from Nanjing Jiancheng Bioengineering Institute.
Cell culture
MLE12 cells and BEAS-2B were cultured using
Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific,
Inc.) supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.). The cells were cultured in a 5%
CO2 incubator at 37°C for 24-48 h.
CCK-8 detection assay
Cells in the logarithmic growth phase (8,000 per
well) were inoculated into 96-well plates. When the cells reached
~70% confluence, they were treated with high glucose (30 mmol/l) at
different time points. After incubation for 24 h at 37°C, the
absorbance was detected using a microplate reader (OD:450 nm).
ELISA assay
The cell culture supernatant was collected,
subjected to centrifugation (2,000 × g) for 20 min at 4°C then the
supernatant was collected for subsequent detection. The reagent kit
was taken out of the refrigerator and allowed to reach room
temperature. The standard curve was prepared according to the kit's
operating instructions (TNF-α, cat. no. SEKM-0034; IL6, cat. no.
SEKM-0007; IL-1β, cat. no. SEKM-0002) and the expression levels of
inflammatory factors were finally calculated based on the results
of the microplate reader.
Cellular immunofluorescence assay
Lung cells were seeded onto 12-well cell culture
plates containing coverslips (5×104). After seeding, the
plates were transferred to a cell incubator until the cells reached
60% confluence. After FGF4 administration, the seeded cell culture
plates were removed from the incubator and the culture supernatant
was gently aspirated from each well to remove metabolites and
unabsorbed nutrients. Each well was washed to ensure removal of
non-cellular components. Fixative (4% paraformaldehyde solution)
was slowly and evenly added to ensure coverage of all cells for 20
min at 37°C. After fixation, the fixative was carefully discarded
to avoid damaging the cells. Each well was washed four times to
remove excess fixative. For cell permeabilization, Triton X-100 was
appropriately diluted with PBS to 0.5%. This permeabilization
solution was added at 200 μl per well to cover the cells and
incubated at room temperature for 10 min. After washing with PBS,
10% goat serum solution (cat. no. S9070; Beijing Solarbio Science
& Technology Co., Ltd.) was added at 200 μl per well and
incubated at room temperature for 1 h to block non-specific sites
in the wells and reduce non-specific binding of subsequent primary
antibodies to non-target proteins. After blocking, the
corresponding primary antibodies (LC3, 1:50 dilution; LAMP1, 1:60
dilution; TOM20, 1:100 dilution) were added and incubated overnight
at 4°C. After incubation, 1 ml of PBS was added to each well and
unbound primary antibodies were removed by washing to reduce
background signal. Diluted secondary antibody solution (1:500
dilution) was added for 60 min to promote the formation of
complexes between the secondary antibody and the primary
antibody-bound protein. After secondary antibody incubation, the
wells were washed four times with PBS. DAPI was used to
counterstain the cell nuclei. After secondary antibody incubation
and washing, 200 μl of DAPI solution was added to label the
cell nuclei through the binding of DAPI (0.5 μg/ml) to DNA
for 24 h at 37°C. After DAPI incubation, the cells were washed with
PBS in the dark at low speed. After mounting, the cell samples were
observed using a confocal microscope (Fv3000; Olympus
Corporation).
Measurement of mitochondrial membrane
potential (MMP)
MMP was assessed using the fluorescent dye TMRE.
Cells were seeded onto confocal dishes and incubated with 2
μg/ml TMRE at 37°C in a 5% CO2 incubator for 30
min. After washing, the cells were examined under a confocal
microscope (Fv3000; Olympus Corporation).
JC-1 staining
After treatment with FGF4, the cells were washed
three times with PBS. JC-1 staining working solution was added to
the cells and incubated in a culture incubator for 30 min at 4°C.
Upon completion of the incubation, the cells were washed three
times with PBS and the cell samples were then examined using a
(Fv3000; Olympus Corporation).
Determination of oxidative stress
indicators
The determination of MDA (cat. no. A003-1-2), SOD
(cat. no. A001-3-2) and GPX (cat. no. H545-1-1) was performed using
commercial kits according to the manufacturer's instructions. The
cell supernatant was collected and the absorbance of the sample was
measured at a 450 nm wavelength.
Knockdown of adenosine monophosphate
(AMP)-activated protein kinase (AMPK) by siRNA
The AMPK-targeting siRNA was designed and
synthesized by Shanghai GenePharma Corporation with the following
sequences: Sense strand 5'-CAGGCAUCCUCAUAAUUTT-3' and Anti-sense
strand 5'-AAUUAUAUGAGGAUGCCUGTT-3'. A scrambled control siRNA
(Sense: 5'-UUCUCCGAACGUGUCACGUdTdT-3'; Anti-sense:
5'-ACGUGACACGUUCGGAGAAdTdT-3') was used as negative control. For
transfection, 15 pmol of siRNA was pre-mixed with 1.5 μl
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) for 5 min before being added to cells. After 8 h
of incubation at 37°C, the medium was replaced with fresh complete
medium, followed by 24-48 h of culture before assessing
transfection efficiency (Fig.
S1).
Establishment of diabetic mouse
models
Male C57BL/6J mice (n=20; weight: 20-22g; 6-8 weeks
old) were purchased from Guangdong Yaokang Biotechnology Co., Ltd.
and housed at an indoor temperature of 21±2°C with a humidity of
30-70% (12-h light/dark cycle). The animals had ad libitum
access to food and water. After one week of adaptive feeding, all
experimental mice were subjected to body weight and fasting blood
glucose measurements. A type 1 diabetic model was established using
a low-dose STZ intraperitoneal injection method as reported in the
literature. A certain amount of STZ powder was dissolved in 0.1 M
sodium citrate solution (pH 4.5) to prepare a 1% STZ solution.
Prior to administration, mice in all groups were fasted for 3 h
with free access to water. Mice in the diabetes mellitus (DM)
groups received intraperitoneal injections of STZ at a dose of 50
mg/kg body weight once daily for five consecutive days, while mice
in the Ctrl groups received intraperitoneal injections of an equal
volume of 0.1 M sodium citrate solution once daily for five
consecutive days. One week after the last STZ injection, blood
glucose levels were measured from tail vein blood samples. Mice
with blood glucose levels ≥16.7 mmol/l on three separate days were
considered to have successfully developed diabetes. Mice that did
not meet this criterion received supplementary injections until
their blood glucose levels reached the required threshold. After
successfully establishing the DM model, the mice were randomly
divided into four groups (n=5 per group): The control group, DM
group, DM + FGF4 treatment group (0.5 mg/kg) and DM + FGF4
treatment group (1 mg/kg). FGF4 was administered via
intraperitoneal injection at a dose of 0.5-1 mg/kg twice a week for
a total experimental period of 15 days. At the end of the
experiment, all mice were sacrificed by anesthesia (the mice were
sacrificed before the end of the experiment due to reaching humane
endpoint criteria) to ensure no additional suffering. The criteria
(humane endpoints) established in this study also included
emergency situations that might occur during the experimental
process, including: 20% body weight loss from baseline, severe
mobility impairment, overt signs of distress and end-stage disease.
The entire experimental period was 45 days. Animal health and
behavior were monitored twice daily (morning and evening)
throughout the study (physical condition, behavioral signs).
Sacrifice was performed by cervical dislocation, under anesthesia
(sodium pentobarbital; 50 mg/kg, intraperitoneal injection).
Mortality was confirmed by sustained absence of vital signs
(respiration, heartbeat, reflexes) for 1 min. The present study was
performed in accordance with the NIH Guide for the Care and Use of
Laboratory Animals and approved by the Institutional Animal Care
and Use Committee (IACUC) of Guangdong University Affiliated
Hospital (202410-235) (https://www.gdmuah.com/kxyj/llsc.htm).
H&E staining
After fixation with 4% formaldehyde for 24 h at
25°C, tissue samples were dehydrated using alcohol of different
concentrations. The tissues were embedded in paraffin and sectioned
at 4 μm. The tissue sections were stained with hematoxylin
staining solution at room temperature for 5 min. After rinsing with
running water, the sections were treated with differentiation
solution for 30 sec. After washing, eosin staining solution was
added to stain the sections for 30 sec. The sections were immersed
in tap water for 5 min. After clearing with xylene, the sections
were mounted using neutral balsam. The tissue sections were then
observed using a light microscope.
Masson staining
Lung tissue sections (4 μm) were incubated
with Weigert iron hematoxylin staining solution for ~10 min at
25°C. The sections were then treated with alcohol differentiation
solution for 5-15 sec. After rinsing with tap water, the sections
were immersed in Masson bluing solution for 3 min to restore blue
coloration. After rinsing with tap water, the tissue sections were
stained with Ponceau fuchsin staining solution for 5-10 min.
Subsequently, the sections were rinsed with weak acid working
solution for 1 min. They were then washed with phosphomolybdic acid
solution for 1-2 min, followed by another rinse with weak acid
working solution for 1 min. The tissue sections were directly
transferred to aniline blue staining solution for 1-2 min and then
rinsed with weak acid working solution for 1 min. After dehydration
with ethanol, the sections were observed using a light microscope
(magnification, ×10-20). A total of five non-overlapping fields
were randomly selected.
Western blot analysis
Lung tissue was placed in a 2 ml EP tube, pre-cooled
tissue RIPA lysis buffer (cat. no. R0020, Solarbio) was added and
the tissue was homogenized using a tissue homogenizer. The sample
was centrifuged at 13,000 × g for 15 min at 4°C and the supernatant
was collected. Protein concentration was measured using a BCA kit.
The proteins (30 μg/lane) were separated by 4-10% SDS-PAGE.
After electrophoresis, proteins were transferred to a PVDF
membrane, which was washed three times with TBST (0.05% Tween)
solution for 10 min each. The PVDF membrane with target proteins
was blocked in 5% skimmed milk powder at room temperature for 1 h.
The membrane was then washed three times with TBST solution for 10
min each. Primary antibodies (PGC-1α, 1:3,000 dilution; TFAM,
1:2,000 dilution; β-actin, 1:4,000 dilution) diluted in 5% BSA were
added and the membrane was incubated on a shaker at 4°C overnight.
After washing three times with TBST (10 min each), secondary
antibodies (HRP-conjugated goat anti-rabbit secondary antibody at a
1:5,000 dilution) were added and incubated for 2 h. ECL luminescent
solution was prepared according to the instructions. The PVDF
membrane with target proteins was evenly coated with the
luminescent solution and placed in a fully automated
chemiluminescence imaging system for exposure. Blots were
quantified using ImageJ 1.52a software (National Institutes of
Health).
Immunohistochemistry
The tissue sections were permeabilized with 0.1%
Triton X-100 for 15 min. The sections (4 μm) were blocked
with 10% goat serum (as antigen blocking solution) for 2 h at 25°C.
Endogenous peroxidase activity was quenched by incubating the
sections with 3% hydrogen peroxide in methanol for 15 min at room
temperature. The corresponding primary antibodies solution (IL6,
cat. no. 1:EPR23819-103, 1:150 dilution; IL-1β, cat. no. EPR19147,
1:200 dilution) was then added and the slides were incubated at 4°C
for 12 h. After incubation, the sections were thoroughly washed
with PBS. Fluor594-labeled secondary antibody (cat. no. K1034G,
1:500 dilution) was added and incubated for 60 min at 37°C. After
washing, the sample was observed using a laser confocal microscope
(Fv3000; Olympus Corporation).
Statistical analysis
All experimental results are presented as mean ± SD,
SPSS 24.0 (IBM Corp.) was used for statistical analysis. All data
were tested for normality using the Shapiro-Wilk method and data
that met normal distribution were analyzed using One-way ANOVA
variance analysis. Tukey's post hoc test was used to assess the
associations among different groups. Data that did not meet normal
distribution were analyzed using the rank sum test. P<0.05 was
considered to indicate a statistically significant difference.
Results
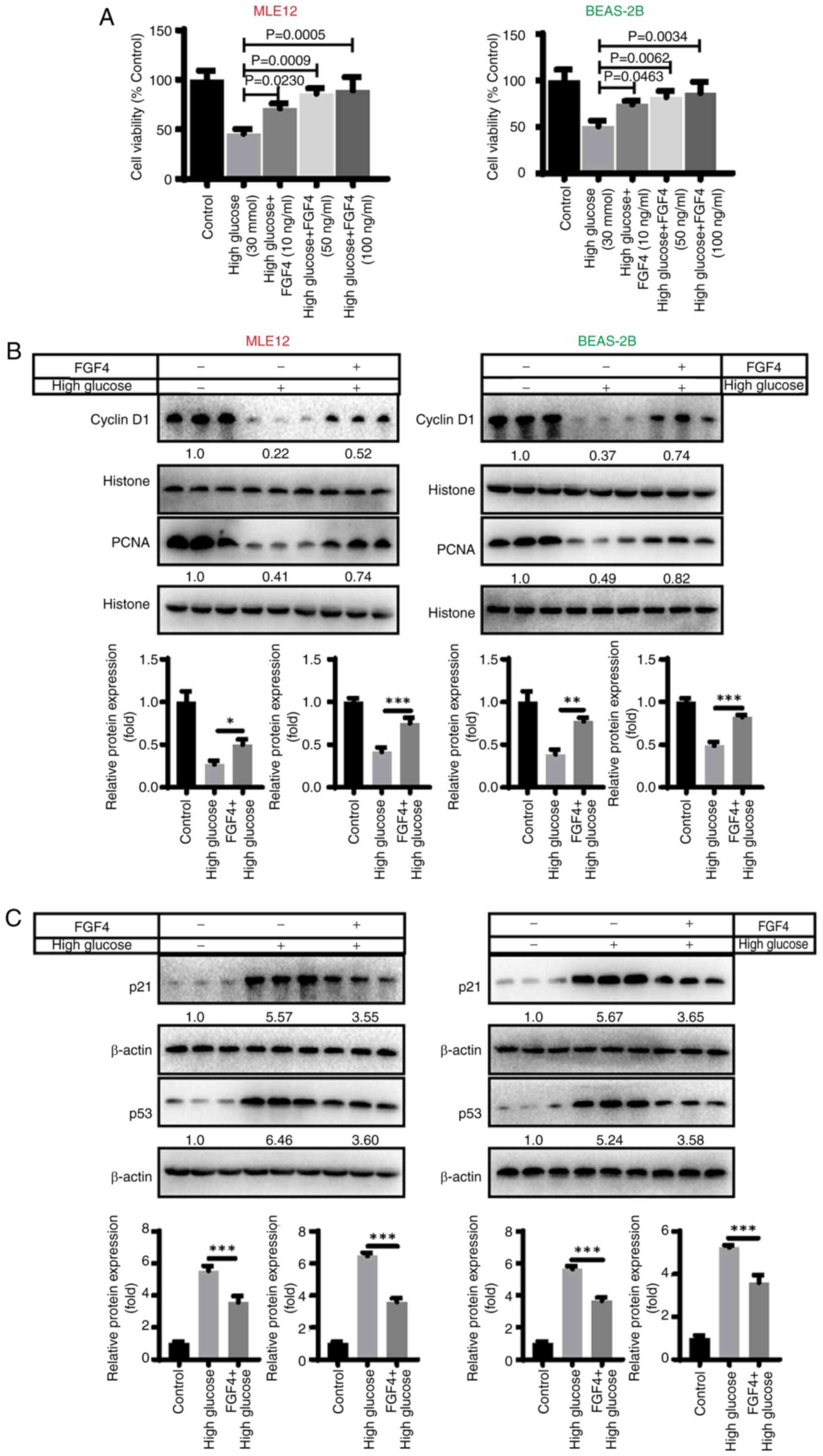
The effect of FGF4 on high
glucose-induced lung cell injury
CCK8 assays were used to detect the effect of FGF4
on high-glucose-induced lung cells. High glucose treatment [30
mmol/l; this selection of glucose concentrations is based on
reference (15)] induced a
decrease in the proliferative capacity of lung cells, indicating
that high glucose has a significant inhibitory effect on lung cell
proliferation. By contrast, when cells were treated with FGF4 (10,
50 and 100 ng/ml; the selection of these concentrations was based
on preliminary experiments), the proliferative capacity of cells
was markedly increased (Fig.
1A). In addition, the present study evaluated the effect of
FGF4 on the expression of cell cycle-related proteins. The results
illustrated that the expression of CyclinD1 and PCNA was markedly
downregulated in the high glucose group, while in the FGF4
treatment group, the expression level of CyclinD1 was markedly
upregulated (Fig. 1B).
Similarly, the expression of p21 and p53 (cell cycle inhibitors)
was markedly reduced after FGF4 treatment, implying that FGF4 can
promote cell proliferation (Fig.
1C).
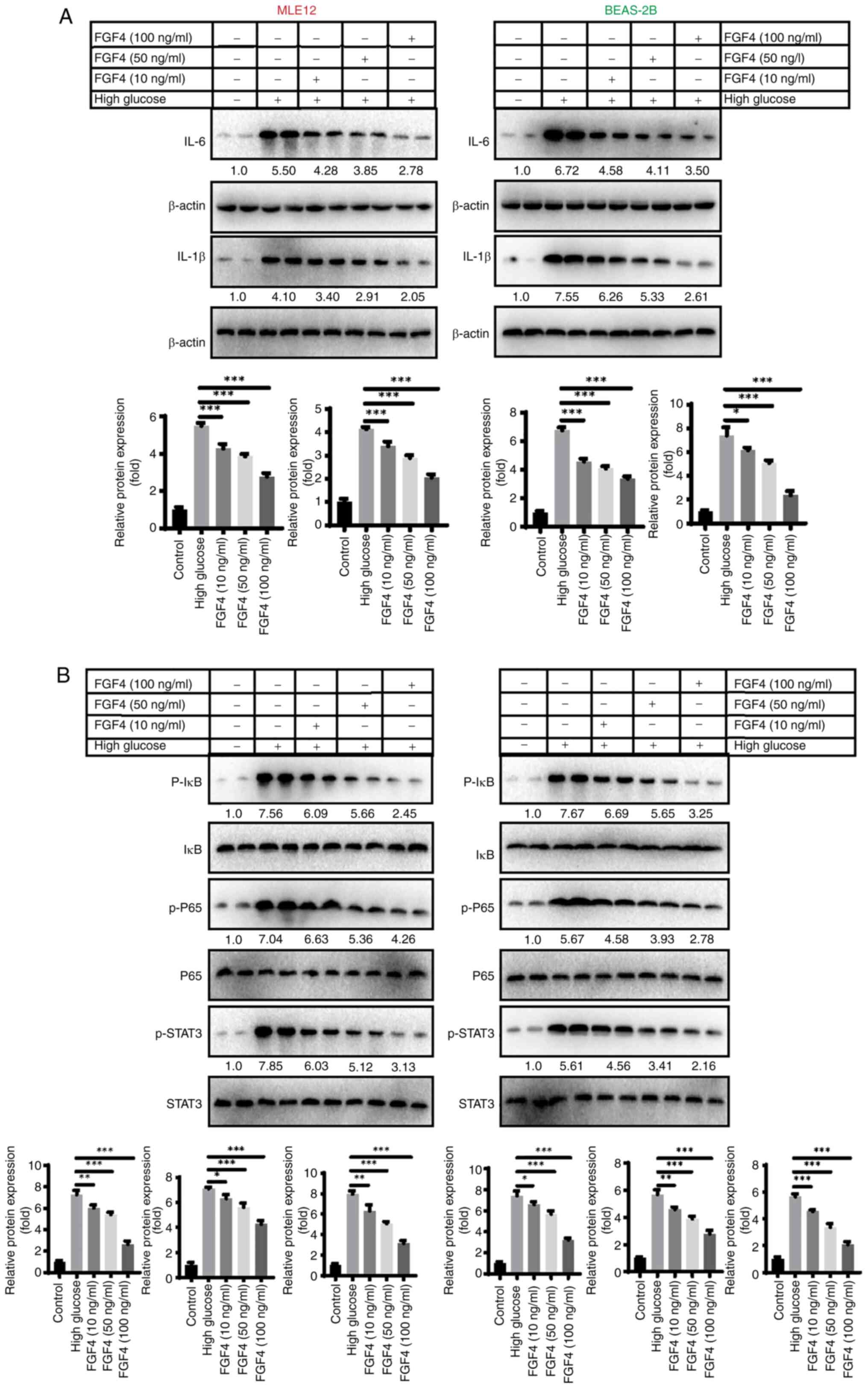
The effect of FGF4 on high
glucose-induced lung cell inflammation and oxidative stress
The present study first evaluated the effect of FGF4
on the expression of inflammatory factors in high glucose-induced
lung cells. As shown in Fig. 2A,
western blot analysis revealed that treatment with FGF4 at
concentrations of 10, 50 and 100 ng/ml led to decreased expression
levels of IL-6 and IL-1β. Additionally, the protein expression
levels of p-IκB, p-P65 and p-STAT3 were all reduced following FGF4
treatment (Fig. 2B). The present
study also assessed the effect of FGF4 on oxidative stress and the
results demonstrated that FGF4 could reduce the levels of reactive
oxygen species (ROS) and MDA, while increasing the level of SOD
(Fig. S2).
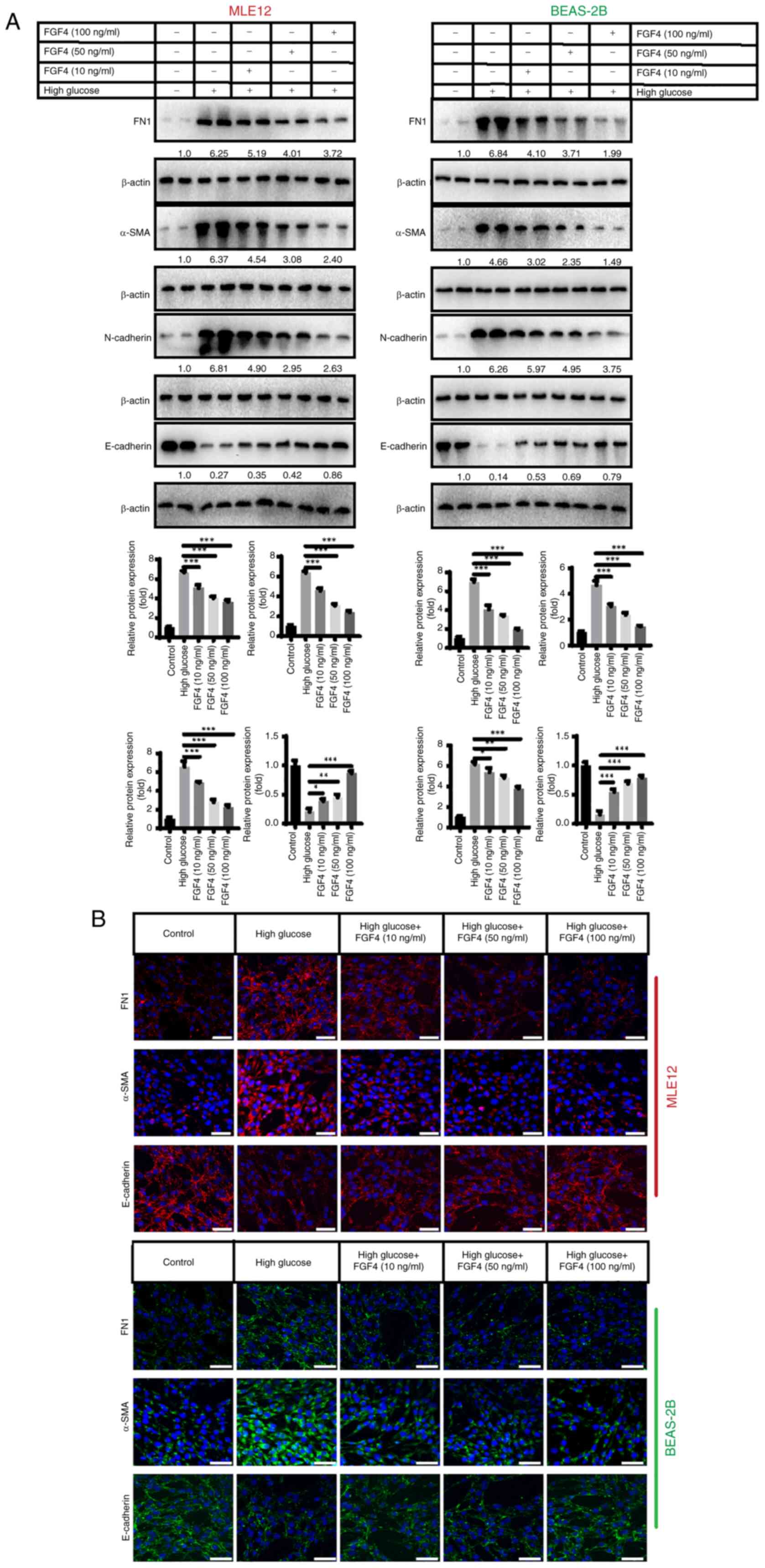
Effect of FGF4 on high glucose-induced
lung tissue fibrosis
Fig. 3 shows that
high glucose stimulation promotes lung tissue fibrosis. The present
study investigated the expression of fibronectin 1 (FN1), α-smooth
muscle actin (α-SMA) and epithelial cell markers (E-cadherin and
N-cadherin). The results revealed that in the high-glucose group,
the protein expression of FN1, α-SMA and N-cadherin was
upregulated, while E-cadherin protein expression was downregulated.
It was observed that after FGF4 treatment, the expression levels of
FN1, α-SMA and N-cadherin were also markedly decreased, while
E-cadherin expression was increased (Fig. 3A). In addition, indirect
immunofluorescence assays yielded similar results (Fig. 3B).
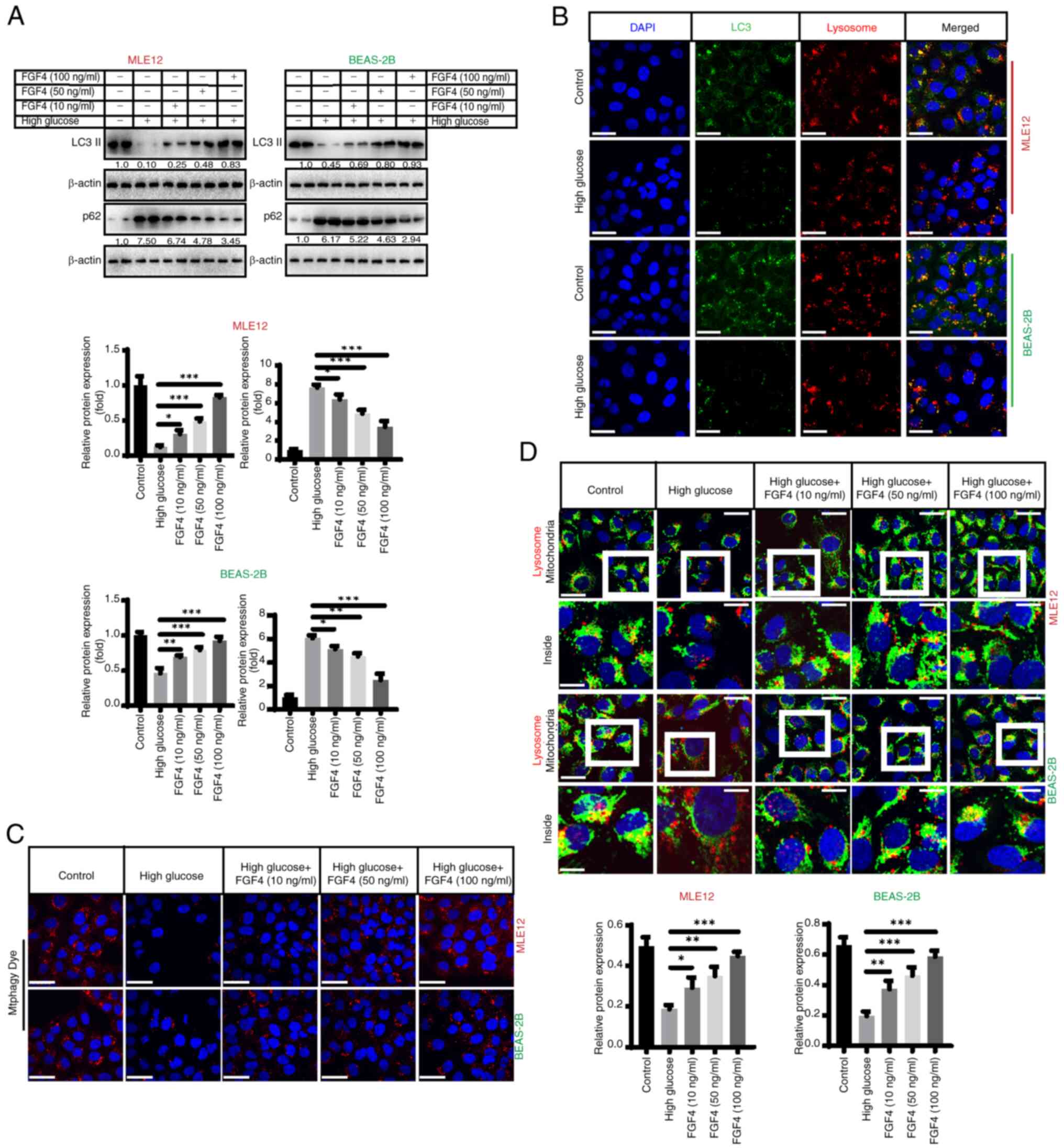
FGF4 alleviates high glucose-induced
inhibition of mitochondrial autophagy
The present study assessed the impact of high
glucose on mitochondrial autophagy. It first evaluated the effect
of high glucose on autophagy and found that the expression of
LC3-II decreased, while the expression level of p62 markedly
increased (Fig. 4A). Indirect
immunofluorescence also demonstrated that high glucose inhibited
autophagy, as the co-localization (yellow fluorescence) of LC3
(green fluorescence) and lysosomes (red fluorescence) was markedly
reduced (Fig. 4B). Furthermore,
we found that mitophagy (red fluorescence) was markedly inhibited
by high glucose treatment and FGF4 promoted autophagy when assessed
using specific fluorescent probes (Fig. 4C). Additionally, fluorescence
co-localization (yellow fluorescence) showed that mitochondrial
autophagy (lysosomes: red fluorescence; mitochondria: green
fluorescence) was partially restored following FGF4 treatment
(Fig. 4D). In addition, the
expression level of TOM20 increased in the high-glucose treatment
group, while its level markedly decreased after FGF4 treatment,
indicating enhanced mitophagy (Fig.
S4A). MMP staining with JC-1 revealed that mitochondrial
membrane potential had undergone depolarization and its level
increased after FGF4 treatment (Fig. S4B).
In general, PTEN-induced kinase 1 (PINK1)/Parkin
mediates autophagy of mitochondria with impaired membrane
potential. However, PINK1/Parkin decreased under high-glucose
treatment. After FGF4 treatment, the level of mitochondrial
autophagy was partially restored (Fig. S4C). This suggested that FGF4 may
regulate mitochondrial activity by modulating mitochondrial
biosynthesis and autophagy pathways.
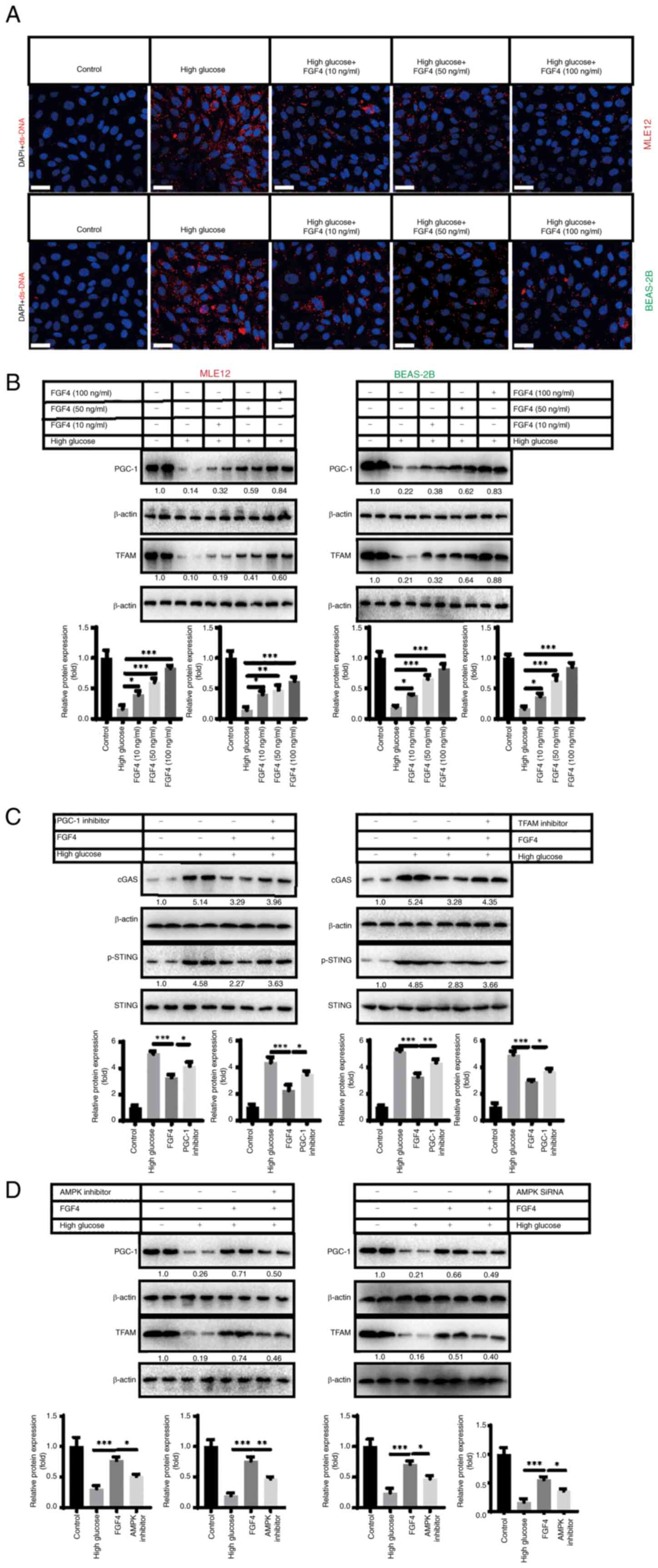
High glucose induces release of mtDNA
from lung mitochondria and activates cGAS-STING signaling
High glucose-induced injury can lead to
mitochondrial damage (16).
Therefore, the present study further focused on mitochondria as a
target and explored the potential molecular mechanisms by which
FGF4 combats high glucose-induced lung cell damage. It was
hypothesized that dysfunctional mitochondria may release
damage-associated molecular patterns (DAMPs) into the cytoplasm.
Mitochondrial DNA (mtDNA), as an important DAMP, further activates
the inflammatory response in lung cells. This may be one of the
potential molecular mechanisms underlying the current study. To
this end, the present study conducted relevant experiments to
verify this hypothesis. Using indirect immunofluorescence, it was
found that the level of mitochondrial DNA (red fluorescence) in the
cytoplasm markedly increased under high-glucose treatment, while
mtDNA levels markedly decreased following FGF4 treatment (Fig. 5A). The present study continued to
explore how FGF4 repaired mitochondria and reduced mtDNA leakage. A
previous study showed that the PGC-1-mitochondrial transcription
factor A (TFAM) signaling pathway regulates mitochondrial
biosynthesis and maintains mitochondrial function (17). The present study demonstrated
that high glucose severely inhibited the expression of PGC-1 and
TFAM, while FGF4 treatment partially restored the expression of
these molecules (Fig. 5B). When
the expression of PGC-1/TFAM was inhibited, it was found that the
alleviating effect of FGF4 on cGAS-STING signaling was partly
offset (Fig. 5C). Further
experiments showed that FGF4 activated the PGC-1/TFAM signaling
axis through AMPK: when AMPK was inhibited, the activity of the
PGC-1/TFAM signaling pathway was markedly reduced (Figs. 5D and S3). These data suggested that FGF4
regulates cGAS-STING signaling through the AMPK-PGC-1-TFAM
signaling axis.
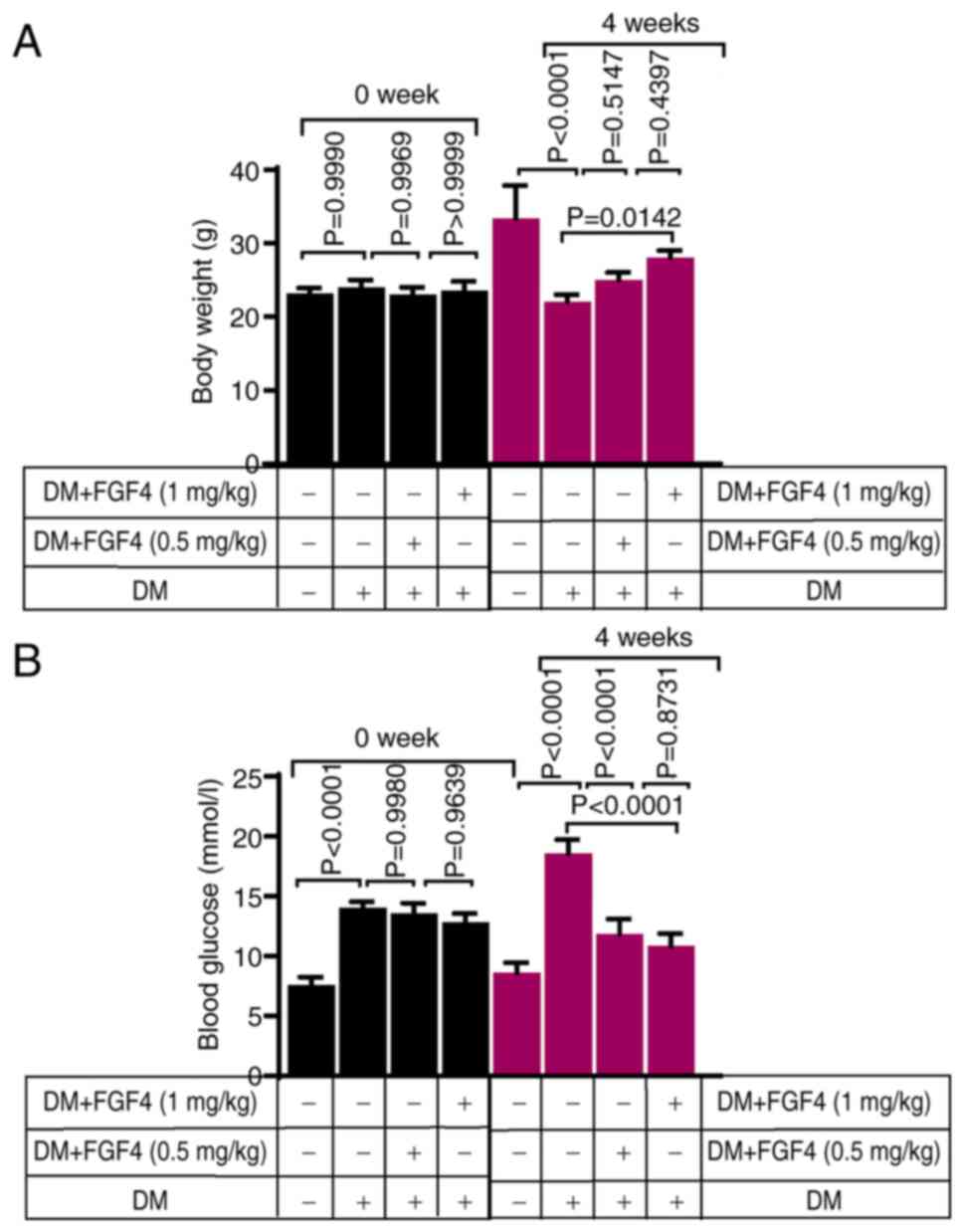
The effect of FGF4 on body weight and
blood glucose in diabetic mice
Changes in body weight of mice in each group before
and after the experiment are shown in Fig. 6A. As expected, at week 0, there
were no significant differences in body weight among the groups.
Compared with the diabetic group, the body weight of mice in the
FGF4 treatment group increased slightly, suggesting that FGF4 can
alleviate diabetes-induced weight loss. In addition, changes in
blood glucose levels in mice across all groups before and after the
experiment were evaluated. Blood glucose levels of mice in each
group before and after the experiment are shown in Fig. 6B. Prior to the experiment (week
0), blood glucose levels in the diabetic and diabetic + FGF4 groups
were markedly higher than those in the control group, indicating
successful establishment of the diabetic mouse model. At the end of
the fourth week, blood glucose levels in the FGF4 treatment group
were markedly lower than those in the diabetic group, suggesting
that FGF4 has a significant hypoglycemic effect in diabetic mice,
which is consistent with previous findings.
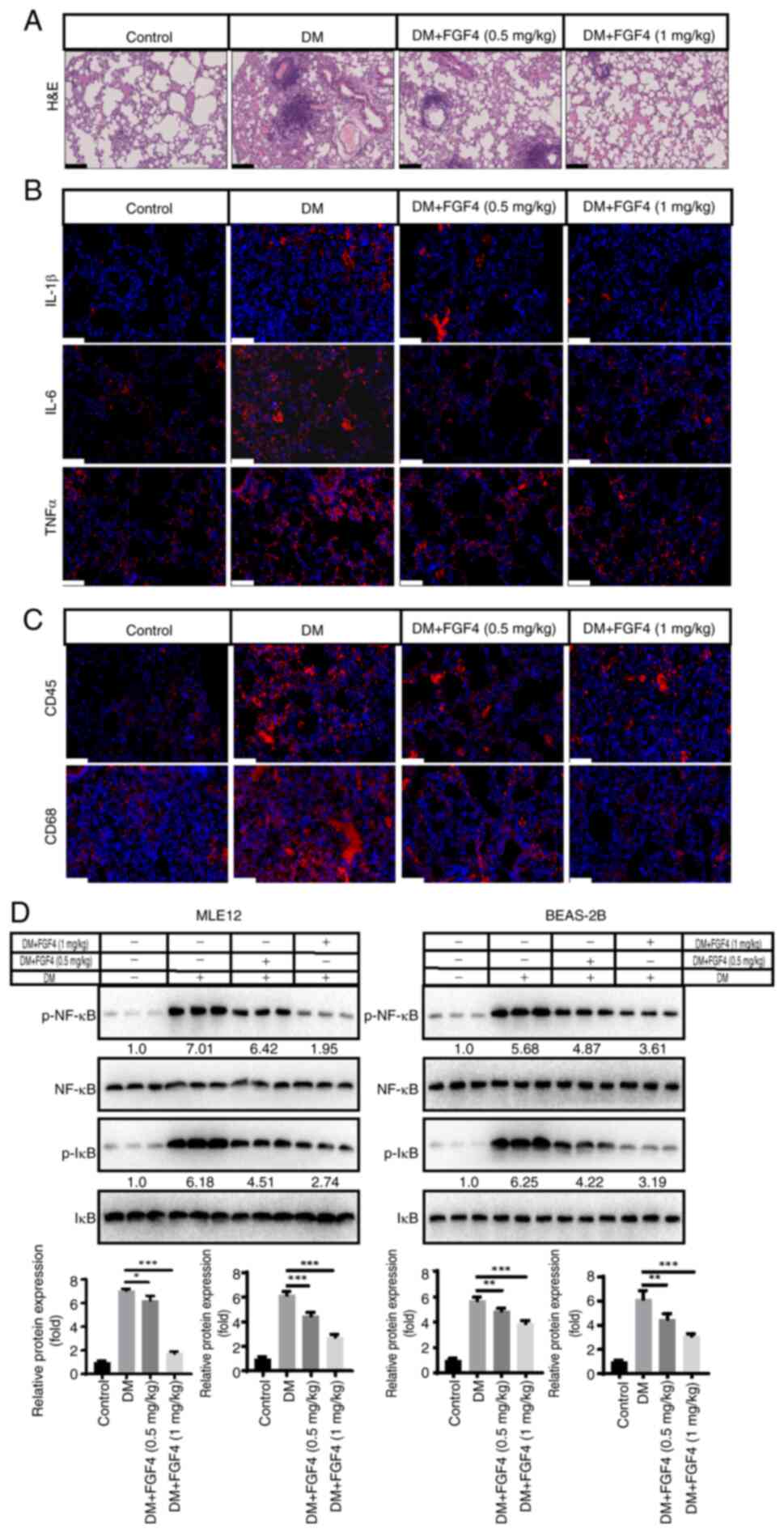
Effect of FGF4 on lung tissue
inflammation in diabetic mice
The present study evaluated the effect of FGF4 on
pathological changes in lung tissue of diabetic mice. From H&E
staining results, compared with the diabetic model group, the
alveolar structure in the control group was clear and intact, with
no obvious exudation in the alveolar cavity and no significant
widening of alveolar septa. However, in diabetic mice, the alveolar
structure was destroyed and inflammatory cell infiltration was
observed. In the FGF4 treatment group, pathological changes were
markedly alleviated compared with the DM group (Fig. 7A). To further evaluate pulmonary
inflammation in diabetic mice, we used immunohistochemistry to
detect the levels of TNF-α, IL-1β and IL-6. As shown in Fig. 7B, compared with the control
group, the expression levels of TNF-α, IL-1β and IL-6 in lung
tissues of diabetic mice were markedly higher, while their
expression levels in the FGF4 treatment group were markedly lower
than those in the diabetic group (DM). These results suggested that
FGF4 can downregulate the expression of inflammatory factors.
Furthermore, immunofluorescence showed that the infiltration of
inflammatory cells (CD45+ and CD68+ cells)
was markedly reduced (Fig. 7C).
In addition, the expression level of the inflammatory factor NF-κB
was also markedly decreased (Fig.
7D).
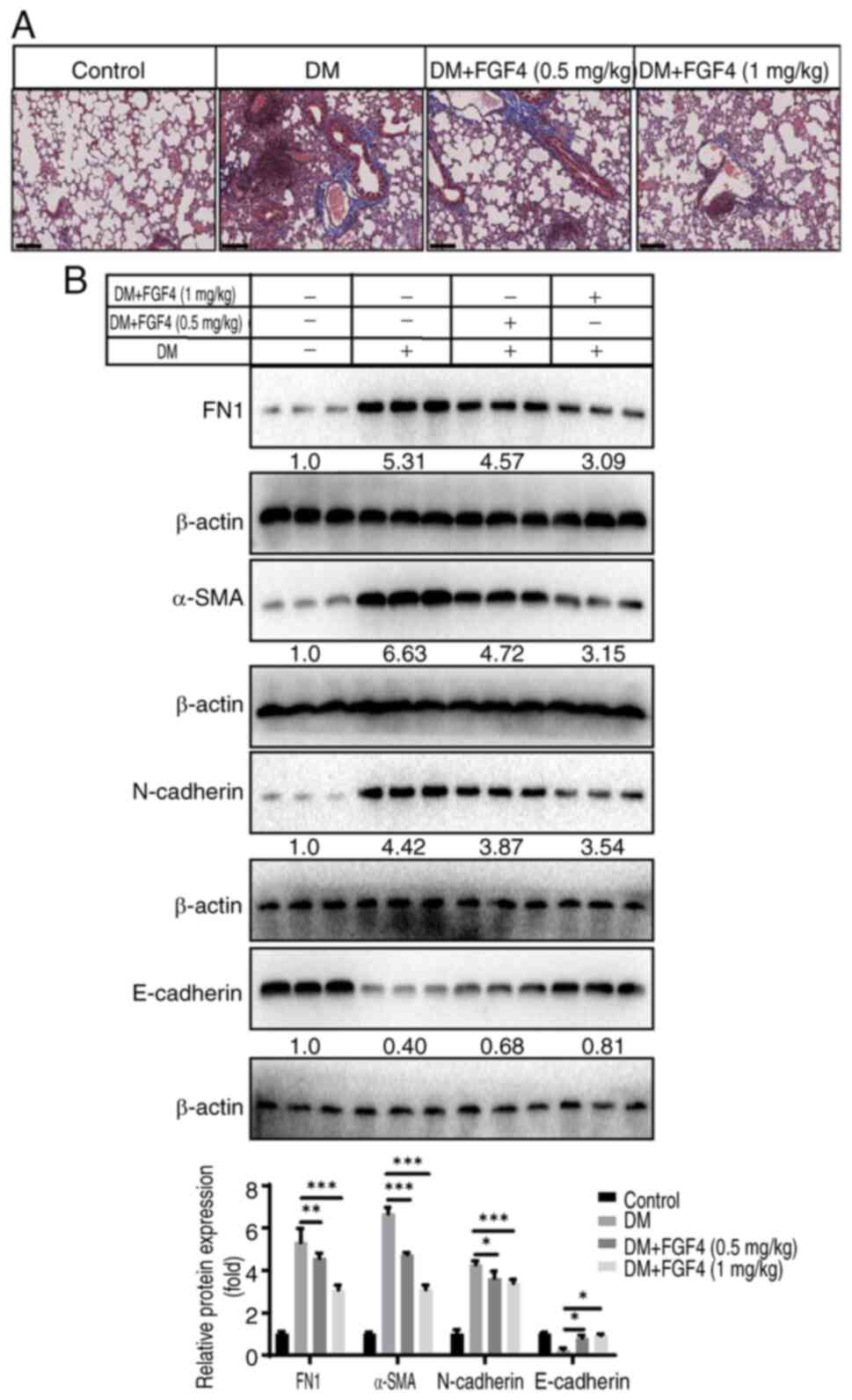
Effect of FGF4 on the degree of pulmonary
fibrosis in diabetic mice
The present study continued to evaluate the effect
of FGF4 on the fibrosis level in mouse lung tissues and performed
Masson staining: As shown in Fig.
8A, there was no significant collagen deposition in lung
tissues of the control group, while more blue-stained collagen
fibers were observed in lung tissues of the DM group. By contrast,
only a small amount of collagen fibers were observed in lung
tissues of the FGF4 treatment group. The present study also
examined the expression of FN1, α-SMA, E-cadherin and N-cadherin
proteins. The results showed that after FGF4 treatment, the
expression of FN1, α-SMA and N-cadherin was downregulated, while
the expression of E-cadherin was upregulated (Fig. 8B).
Effect of FGF4 on the
oxidative-antioxidant balance in lung tissue of diabetic mice
Given that oxidative stress is involved in the
pathogenesis of diabetes-related lung injury (1), the present study first analyzed the
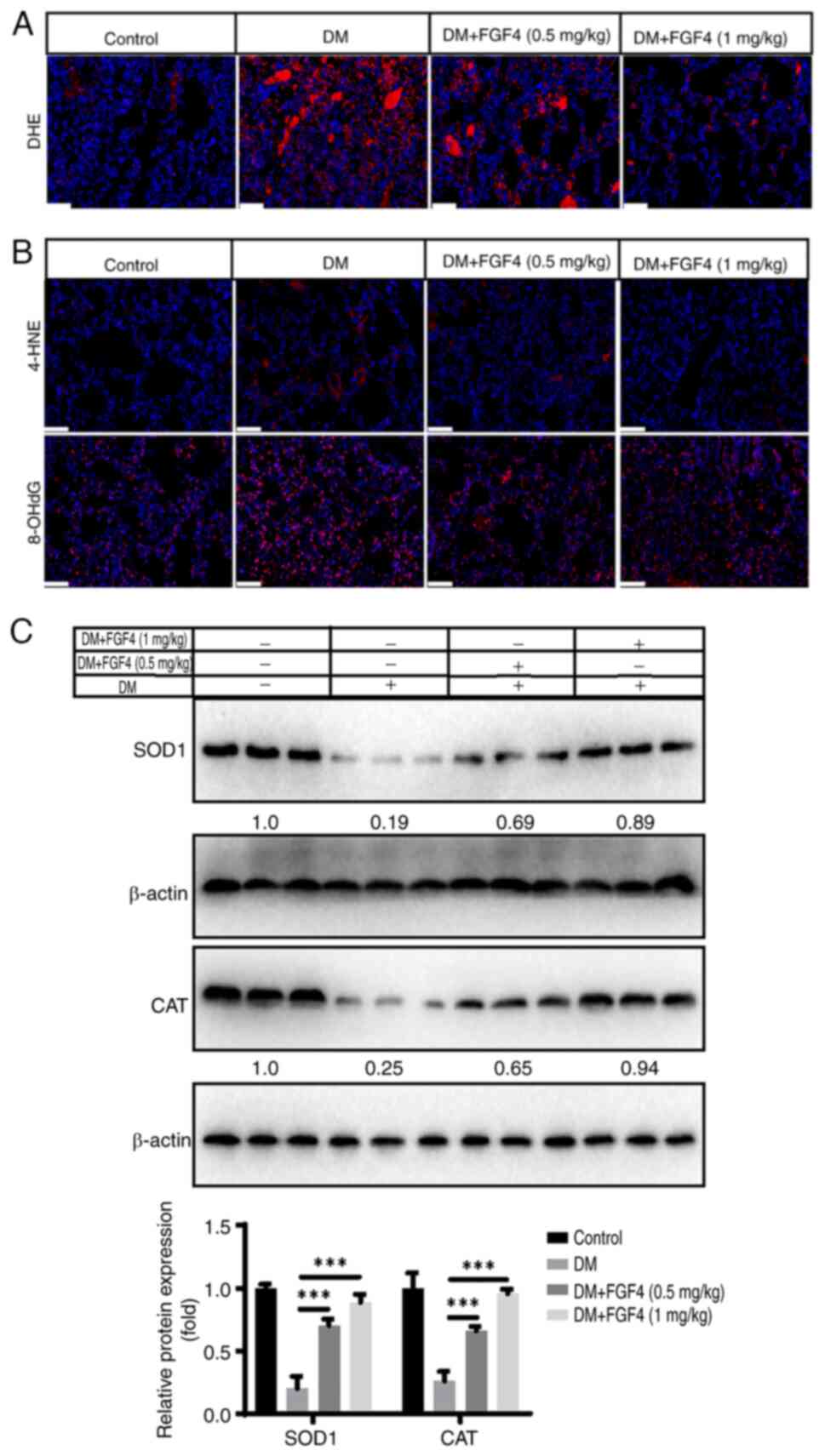
effect of FGF4 on oxidative stress. DHE staining showed that ROS
levels in lung tissue markedly decreased under FGF4 treatment
(Fig. 9A). In addition, the
present study evaluated the levels of 4-HNE and 8-OHdG. Compared
with the control group, the expression level of 4-NHE and 8-OHdG in
the diabetic group was markedly increased, while the expression
levels of 4-HNE and 8-OHdG in the FGF4 group were markedly lower
than those in the DM group. These results suggested that oxidative
stress damage occurs in the lung tissues of diabetic mice and FGF4
administration can alleviate this oxidative stress response
(Fig. 9B). Antioxidant proteins,
including SOD1 and catalase (CAT) were also evaluated. The results
showed that the expression of SOD1 and CAT in the diabetic group
was markedly reduced, whereas their expression levels were markedly
increased after FGF4 treatment (Fig.
9C).
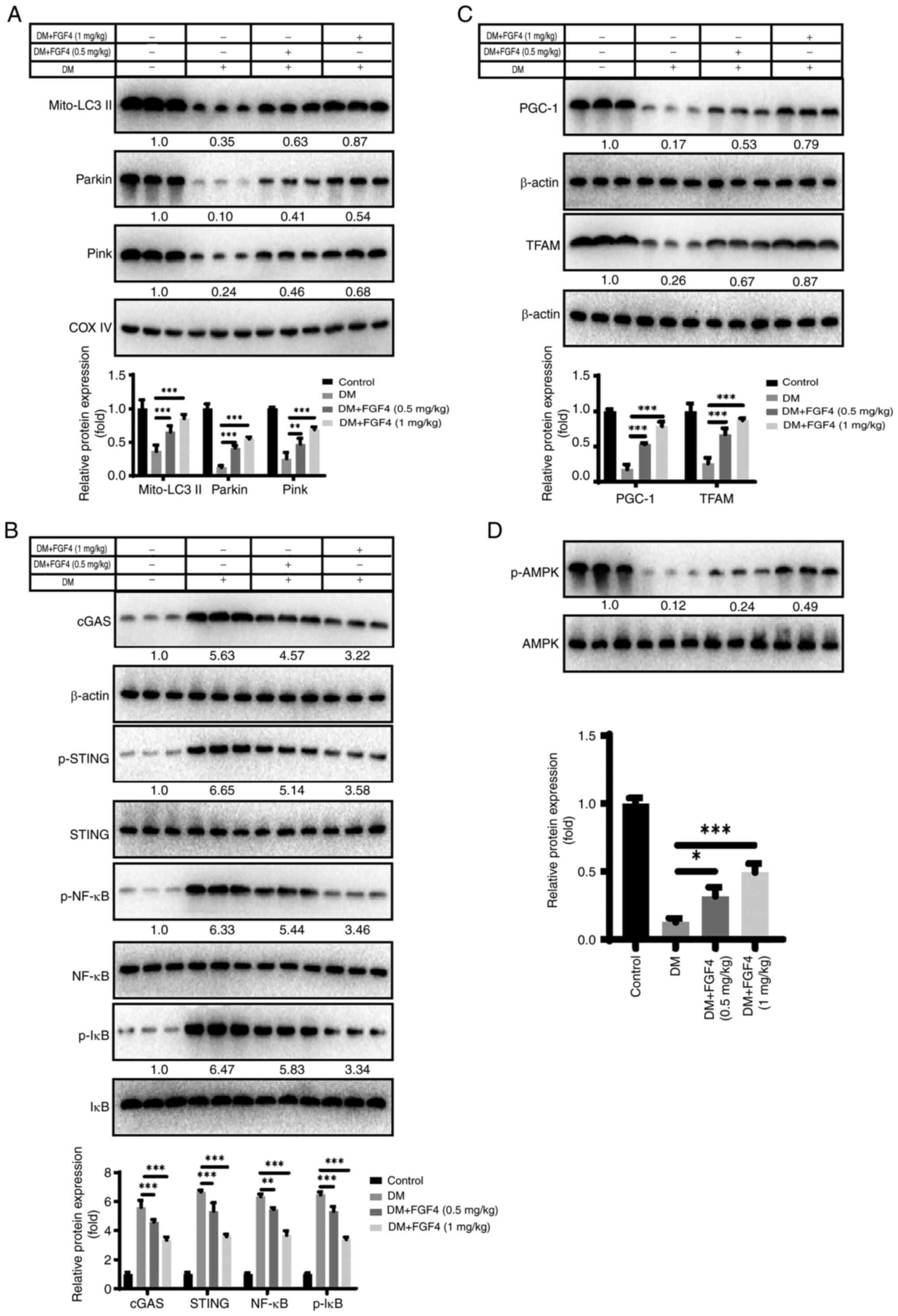
Effect of FGF4 on mitochondrial autophagy
and cGAS-STING signaling in vivo
The present study also evaluated explored the
potential molecular mechanism by which FGF4 ameliorates lung cell
injury in vivo. First, it analyzed the effect of diabetes
mellitus (DM) on mitochondrial autophagy. Western blot analysis
showed that FGF4 alleviated the inhibition of mitochondrial
autophagy induced by DM (Fig.
10A). The present study further analyzed the effect of FGF4 on
the cGAS-STING signaling pathway and the results revealed that the
activity levels of the cGAS/STING/NF-κB signaling pathway were
markedly reduced (Fig. 10B).
Similarly, FGF4 increased the expression of PGC-1 and TFAM, which
is consistent with the findings from in vitro cell models
(Fig. 10C). Additionally, FGF4
treatment increased AMPK phosphorylation in vivo (Fig. 10D).
Discussion
Diabetes can lead to lung injury and abnormal lung
function, a condition known as diabetic lung disease. Chronic and
persistent hyperglycemia stimulates the production of inflammatory
proteins, which accumulate in small blood vessels and connective
tissues. Glycation modifications of collagen and elastin in lung
tissue can induce interstitial fibrotic changes (18). In addition, diabetes-induced
systemic inflammation is another major cause of lung injury.
Dysregulation of inflammatory regulatory mechanisms can trigger
excessive inflammatory responses in the lungs, resulting in
impaired lung function (19). In
the present study, a type 1 diabetes model was established via
intraperitoneal injection of low-dose STZ. After successful
modeling, FGF4 intervention was administered to further investigate
the role and potential mechanism of FGF4 in diabetes-related lung
injury. Results showed that blood glucose levels in experimental
mice decreased and their body weight rebounded. These findings
indicated that FGF4 exhibits a protective effect against
diabetes-related lung injury in both in vitro and in
vivo models. First, to the best of the authors' knowledge, this
is the first study to reveal that FGF4 can alleviate
high-glucose-induced lung injury; Second, it identified a new
molecular mechanism by which FGF4 promotes mitophagy to mitigate
high glucose-mediated lung damage; Third, it was found that FGF4
can ameliorate lung injury, particularly pulmonary fibrosis
(another original observation).
In vitro experiments showed that FGF4 could
mitigate high glucose-induced lung cell damage and promote cell
proliferation. For instance, FGF4 increased Ki67 expression while
suppressing p21 and p53 expression.
Inflammatory responses and oxidative stress play
important roles in the pathogenesis of diabetes-related lung injury
(20). The present study first
evaluated the effect of FGF4 on lung tissue in both in vitro
and in vivo models of diabetic mice. H&E staining showed
that the alveolar structure was destroyed, the alveolar septa were
markedly widened and inflammatory cell infiltration was observed in
the widened alveolar septa, small bronchi and around blood vessels,
suggesting that diabetic mice had developed pulmonary
complications. Furthermore, the levels of the inflammatory factors
TNF-α, IL-1β and IL-6 were assessed and the results showed that
FGF4 treatment reduced the inflammatory response. The present study
also evaluated NF-κB levels and found that its expression was
markedly decreased.
Clinical and laboratory evidence supports that
oxidative stress plays an important role in the pathophysiology of
diabetes and its complications. Continuous high-glucose stimulation
can lead to the accumulation of large amounts of ROS in the lungs,
inducing oxidative stress injury and further aggravating pulmonary
inflammatory responses (21).
The coexistence of oxidative stress and inflammation is a common
cause and high-risk factor for diabetes. The present study found
that FGF4 alleviated oxidative stress levels in both in
vitro and in vivo models. Previous studies have also
shown that multiple members of the FGF family exhibit antioxidant
stress effects; for example, FGF1, FGF2 and FGF21 play important
roles in antioxidant stress and aging (10,22).
In addition, pulmonary fibrosis is another important
pathological change in diabetes-induced lung injury (23). A key mechanism promoting tissue
fibrosis is epithelial-mesenchymal transition (EMT) and animal
experiments have confirmed that inhibiting EMT can reduce the
progression of tissue fibrosis. Continuous high-glucose stimulation
can induce EMT, mainly mediated by the upregulation of
fibroblast-specific TGF-β1. The present study showed that FGF4
relieved pulmonary fibrosis in diabetic mice by inhibiting the
production of pulmonary fibrosis, as evaluated by the expression of
relevant markers.
Autophagy is a cellular degradation program involved
in responses to various stimuli (24). Thus, autophagy dysfunction may
lead to a number of pathological changes, including cancer and
immune abnormalities (25). It
is ubiquitous across organisms, involving the formation of
autophagosomes that recognize damaged or dysfunctional organelles
and macromolecules. Autophagy can be divided into different types,
such as chaperone-mediated autophagy, microautophagy and
macroautophagy. By analyzing its nature, mitochondrial autophagy
(mitophagy) is recognized as a specialized type of autophagy and
studies have found that it is closely associated with cellular
homeostasis. Based on differences in phagocytic and recognition
mechanisms, these processes can be roughly categorized into two
pathways: Ubiquitin-dependent and receptor-dependent. In the first
pathway, PINK1/Parkin is most critical; its mechanism primarily
involves the binding of damaged mitochondria to ubiquitinated outer
membrane proteins, which recruits p62. p62 then binds to LC3 on the
autophagosome membrane, promoting the encapsulation of damaged
mitochondria within autophagosomes to complete mitochondrial
degradation (26). The second
pathway relies on interactions between γ-aminobutyric acid
receptor-related proteins and LC3 on autophagosomal membranes, such
as NIX, FUNDC1 and BNIP3 located on the outer membrane of damaged
mitochondria. FUNDC1 and BNIP3 directly interact with LC3 before
being recognized by phagocytic vesicles (26).
The present study showed that under high-glucose
conditions, FGF4 markedly enhanced mitophagy levels. Specifically,
LC3 protein expression was markedly increased, while p62 protein
levels were markedly reduced. Additionally, under high-glucose
conditions, the mitochondrial membrane potential in the FGF4
treatment group was higher than that in both the empty vector group
and the high-glucose group. This finding further confirmed the
increased mitophagy levels in the FGF4-treated group.
The cGAS-STING signaling pathway and mitochondrial
autophagy play important roles in inhibiting mtDNA leakage
(27). Mitophagy is a cellular
process that maintains mitochondrial health by eliminating damaged
mitochondria. When mitochondria are damaged, mitophagy prevents the
accumulation of mtDNA in the cytoplasm, thereby avoiding the
triggering of inflammatory responses mediated by the cGAS-STING
pathway (28). In this process,
mitophagy not only optimizes mitochondrial function but also
maintains cellular homeostasis by inhibiting the cGAS-STING
signaling. By contrast, the cGAS-STING pathway triggers an immune
response upon detecting mtDNA leakage, further affecting cellular
function and survival. The present study found that FGF4 can
inhibit the activation of cGAS-STING by regulating the PGC-1α-TFAM
signaling pathway.
It is important to mention that most other FGF
family members (such as FGF21 and FGF23) have been widely reported
for their association with hyperglycemia, whereas research on FGF4
remains relatively limited. In terms of practical application
potential, the experimental results demonstrated that FGF4 holds
significant promise, as its biological activity may be stronger
compared with other FGF members. Therefore, FGF4 may be a promising
drug for treating lung damage caused by diabetes.
The current study has several limitations. First,
toxicological studies of FGF4 need to be conducted in future work.
Second, the targeting specificity of FGF4 requires improvement, as
it can distribute to various tissues and organs after entering the
bloodstream. Third, the short half-life of FGF4 needs to be
addressed in future studies, to overcome this limitation, it is
planned to employ the nano-sustained release materials as a
strategic solution to markedly prolong FGF4's therapeutic window
and enhance its bioavailability. Fourth, while the current study
employed a type 1 diabetes model, future research will use a type 2
diabetes model to evaluate the effects of FGF4. Fifth, the lack of
an insulin control group in vivo also represents one of the
limitations in the current study. Future research will focus on
developing strategies to enhance the specific targeting of FGF4 to
lung tissue, such as using lung-targeted nanomaterials.
FGF4 holds important significance in alleviating
diabetic lung injury. Patients with diabetes often develop
pulmonary complications and FGF4, as a novel long-acting
glucose-regulating factor, can not only effectively regulate blood
glucose but also alleviate pulmonary inflammation and injury
through its anti-inflammatory and antioxidant properties. This
discovery provides a new strategy for the treatment of
diabetes-related lung injury, which is expected to improve the lung
health of patients with diabetes, enhance their quality of life and
holds important clinical and scientific value.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
QF and JX participated in study design and data
collection. QW, FL, ZL, YO and KH carried out the initial analysis.
QF and JX drafted the article. JG and TW confirm the authenticity
of all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Zhanjiang Science and
Technology Plan Projects (grant nos. 2024B01314, 2025B01228 and
2022A01192), the Innovation and Entrepreneurship Training Program
for College Students of Guangdong Province (grant no.
S202510571085); and the Innovation and Entrepreneurship Training
Program for College Students of Guangdong Medical University (grant
nos. GDMUCX2024081 and GDMUCX2024304).
References
|
1
|
Banday MZ, Sameer AS and Nissar S:
Pathophysiology of diabetes: An overview. Avicenna J Med.
10:174–188. 2020. View Article : Google Scholar
|
|
2
|
Cole JB and Florez JC: Genetics of
diabetes mellitus and diabetes complications. Nat Rev Nephrol.
16:377–390. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Klein S, Gastaldelli A, Yki-Järvinen H and
Scherer PE: Why does obesity cause diabetes? Cell Metab. 34:11–20.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan JCN, Lim LL, Wareham NJ, Shaw JE,
Orchard TJ, Zhang P, Lau ESH, Eliasson B, Kong APS, Ezzati M, et
al: The lancet commission on diabetes: Using data to transform
diabetes care and patient lives. Lancet. 396:2019–2082. 2021.
View Article : Google Scholar
|
|
5
|
Goldman MD: Lung dysfunction in diabetes.
Diabetes Care. 26:1915–1918. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hsia CCW and Raskin P: Lung function
changes related to diabetes mellitus. Diabetes Technol Ther.
9(Suppl 1): S73–S82. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mameli C, Ghezzi M, Mari A, Cammi G,
Macedoni M, Redaelli FC, Calcaterra V, Zuccotti G and D'Auria E:
The diabetic lung: Insights into pulmonary changes in children and
adolescents with type 1 diabetes. Metabolites. 11:692021.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Gent R, Brackel HJ, de Vroede M and
van der Ent CK: Lung function abnormalities in children with type 1
diabetes. Respir Med. 96:976–978. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng H, Wu J, Jin Z and Yan LJ: Potential
biochemical mechanisms of lung injury in diabetes. Aging Dis.
8:7–16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang L, Zhou F, Zheng D, Wang D, Li X,
Zhao C and Huang X: FGF/FGFR signaling: From lung development to
respiratory diseases. Cytokine Growth Factor Rev. 62:94–104. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ghanem M, Archer G, Crestani B and
Mailleux AA: The endocrine FGFs axis: A systemic anti-fibrotic
response that could prevent pulmonary fibrogenesis? Pharmacol Ther.
259:1086692024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang X, Zhou L, Ye S, Liu S, Chen L, Cheng
Z, Huang Y, Wang B, Pan M, Wang D, et al: rFGF4 alleviates
lipopolysaccharide-induced acute lung injury by inhibiting the
TLR4/NF-κB signaling pathway. Int Immunopharmacol. 117:1099232023.
View Article : Google Scholar
|
|
13
|
Liu Y, Ji J, Zheng S, Wei A, Li D, Shi B,
Han X and Chen X: Senescent lung-resident mesenchymal stem cells
drive pulmonary fibrogenesis through FGF-4/FOXM1 axis. Stem Cell
Res Ther. 15:3092024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sundaram SM, Lenin RR and Janardhanan R:
FGF4 alleviates hyperglycemia in diabetes and obesity conditions.
Trends Endocrinol Metab. 34:583–585. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baumgartner-Parzer SM, Wagner L,
Pettermann M, Grillari J, Gessl A and Waldhäusl W:
High-glucose-triggered apoptosis in cultured endothelial cells.
Diabetes. 44:1323–1327. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Blake R and Trounce IA: Mitochondrial
dysfunction and complications associated with diabetes. Biochim
Biophys Acta. 1840:1404–1412. 2014. View Article : Google Scholar
|
|
17
|
Yao K, Zhang WW, Yao L, Yang S, Nie W and
Huang F: Carvedilol promotes mitochondrial biogenesis by regulating
the PGC-1/TFAM pathway in human umbilical vein endothelial cells
(HUVECs). Biochem Biophys Res Commun. 470:961–966. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Khateeb J, Fuchs E and Khamaisi M:
Diabetes and lung disease: An underestimated relationship. Rev
Diabet Stud. 15:1–15. 2019. View Article : Google Scholar :
|
|
19
|
Xiong XQ, Wang WT, Wang LR, Jin LD and Lin
LN: Diabetes increases inflammation and lung injury associated with
protective ventilation strategy in mice. Int Immunopharmacol.
13:280–283. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lontchi-Yimagou E, Sobngwi E, Matsha TE
and Kengne AP: Diabetes mellitus and inflammation. Curr Diab Rep.
13:435–444. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Forgiarini LA Jr, Kretzmann NA, Porawski
M, Dias AS and Marroni NA: Experimental diabetes mellitus:
Oxidative stress and changes in lung structure. J Bras Pneumol.
35:788–791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shimizu M and Sato R: Endocrine fibroblast
growth factors in relation to stress signaling. Cells. 11:5052022.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang J, Xue Q, Miao L and Cai L: Pulmonary
fibrosis: A possible diabetic complication. Diabetes Metab Res Rev.
27:311–317. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim J, Kim HS and Chung JH: Molecular
mechanisms of mitochondrial DNA release and activation of the
cGAS-STING pathway. Exp Mol Med. 55:510–519. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiménez-Loygorri JI, Villarejo-Zori B,
Viedma-Poyatos Á, Zapata-Muñoz J, Benítez-Fernández R, Frutos-Lisón
MD, Tomás-Barberán FA, Espín JC, Area-Gómez E, Gomez-Duran A and
Boya P: Mitophagy curtails cytosolic mtDNA-dependent activation of
cGAS/STING inflammation during aging. Nat Commun. 15:8302024.
View Article : Google Scholar : PubMed/NCBI
|