Introduction
Ferroptosis, first described by Dixon in 2012 as an
iron-dependent regulated form of cell death, has emerged as a
critical determinant of cellular fate across diverse physiological
and pathological contexts (1).
This unique mode of cell death is primarily driven by phospholipid
peroxidation and iron overload, extending from decades of research
that recognized the cytotoxic consequences of iron and lipid
peroxidation (2,3). The elucidation of antioxidant
defense systems in ferroptosis regulation has advanced our
integrated understanding of the interplay between iron metabolism
and oxidative stress in regulated cell death. Emerging evidence
further indicates that ferroptosis significantly contributes to
tumor suppression, immune regulation and the maintenance of
metabolic homeostasis (4).
Concurrently, lactate has undergone a profound
conceptual transformation from a metabolic waste product to a
sophisticated signaling molecule. Once considered merely a
metabolic waste product associated with hypoxic stress and
detrimental effects, subsequent research has revealed that lactate
is actively produced and utilized even under aerobic conditions
(5). The lactate shuttle
hypothesis further revealed its pivotal roles in oxidative
substrate transport and cellular signaling via monocarboxylate
transporters (MCTs), leading to its recognition as a potent
signaling metabolite, particularly through its receptor GPR81
(6-8). subsequent discoveries have revealed
the epigenetic functions of lactate through histone lysine
lactylation (Kla), a post-translational modification analogous to
acetylation and succinylation (9,10). This modification impacts
transcriptional regulation and extends to non-histone proteins,
thereby regulating enzyme activities (11,12). However, a paradox has emerged:
Lactate exhibits opposing effects on ferroptosis, promoting cell
death in certain contexts while conferring protection in others,
particularly when comparing tumor vs. normal cells. To the best of
our knowledge, no relevant articles currently summarize and explain
these contradictory observations.
The present review elucidates the molecular
mechanisms underlying the lactate-ferroptosis axis, examining how
lactate influences ferroptosis through lactylation-independent and
-dependent pathways that modulate iron homeostasis, lipid
metabolism, redox balance and the immune response. Meanwhile, the
'dual role' refers to the context-dependent, bidirectional
regulation of ferroptosis by lactate. In non-tumor tissues, lactate
tends to promote ferroptosis, potentially accelerating disease
progression. Conversely, in tumor microenvironments, lactate tends
to inhibit ferroptosis, facilitating cancer cell survival and
therapeutic resistance. Furthermore, the present review highlights
the key contextual determinants that may dictate the divergent
roles of lactate. Understanding these context-specific mechanisms
promises new therapeutic strategies targeting a broad spectrum of
diseases ranging from cancer to neurodegeneration.
Unless otherwise specified, the term 'lactate'
throughout the present review refers to L-lactate, the predominant
enantiomer produced by mammalian lactate dehydrogenase (LDH). By
contrast, D-lactate, though present in trace amounts from bacterial
metabolism and certain metabolic disorders, has not yet been
systematically investigated in ferroptosis contexts.
Ferroptosis: Molecular mechanisms
Ferroptosis is mediated by multiple mechanisms
including lipid peroxidation, iron overload and dysfunction of the
antioxidant system (Fig. 1). A
hallmark of ferroptotic initiation and execution is the upregulated
peroxidation of phospholipid-bound polyunsaturated fatty acids
(PUFAs) within cellular membranes, a process facilitated by labile
iron pools (LIPs) and amplified through autocatalytic radical
reactions (13,14). In this context, redox-active
iron, principally existing as ferrous (Fe2+) ions,
serves as a critical cofactor, driving Fenton reactions that
generate reactive oxygen species (ROS). These ROS abstract hydrogen
atoms from bis-allylic positions within PUFA chains, producing
lipid radicals that react with molecular oxygen to yield lipid
peroxyl radicals. This initiates a self-perpetuating cascade of
membrane lipid peroxidation, ultimately compromising membrane
integrity and triggering cell death (14).
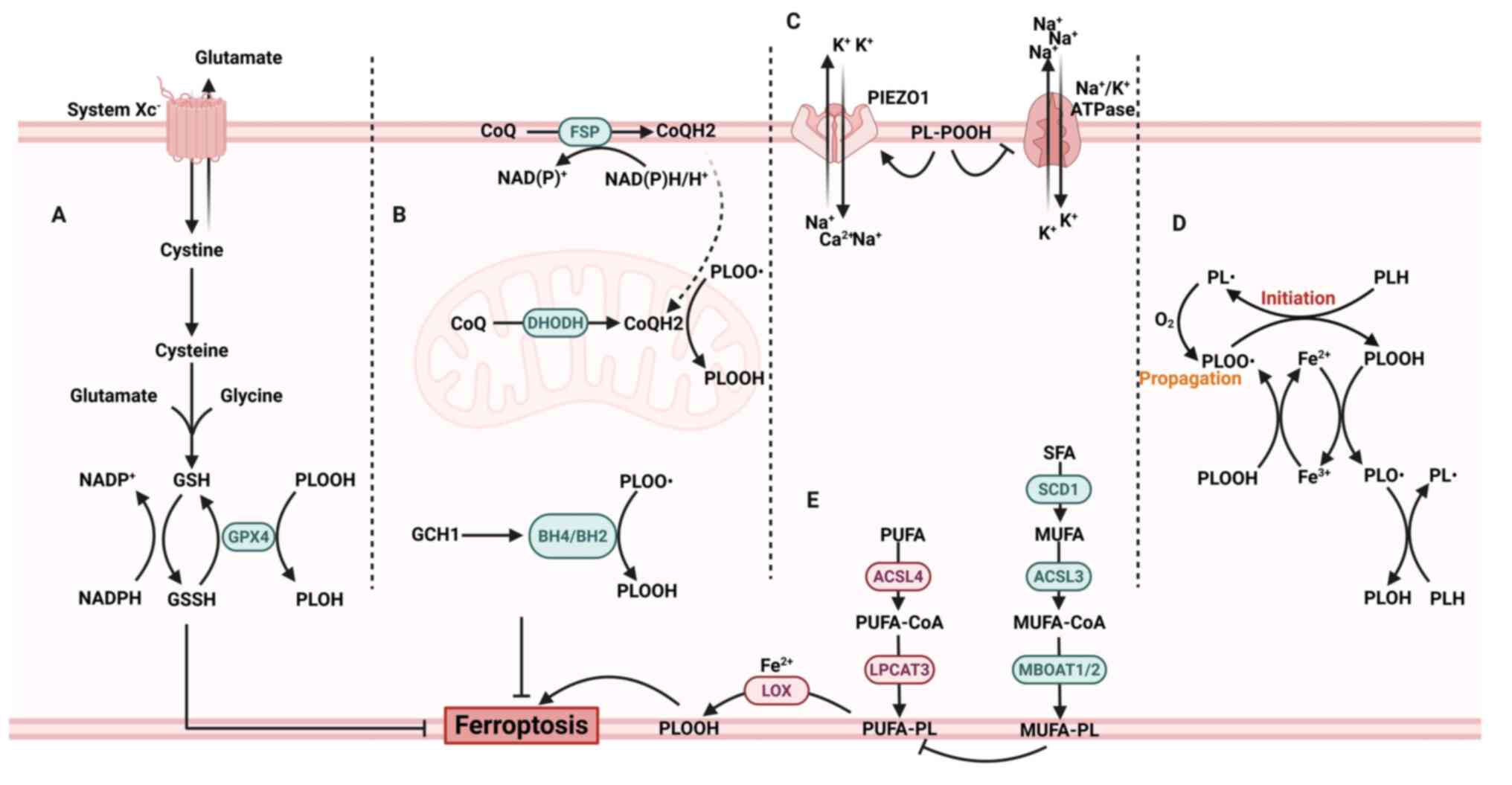 | Figure 1Molecular mechanism of ferroptosis.
(A) The canonical ferroptosis-regulating axis involves cystine
uptake via system Xc−, GSH biosynthesis and
GPX4-mediated reduction of PLOOH into their corresponding alcohols
(PLOH). NADPH provides electrons for recycling GSSG. (B) The
FSP1/CoQ10, DHODH/CoQ10 and GCH1/BH4/BH2 system serves as parallel
pathways to inhibit lipid peroxidation and ferroptosis. (C) The
peroxidation of phospholipids in the plasma membrane activates
PIEZO1, leading to the influx of Ca2+ and
Na+. This process, combined with the inactivation of the
Na+/K+ ATPase, results in the efflux of
K+. (D) The initiation and propagation of PLOOH form a
chain reaction with positive feedback. (E) PUFA and MUFA metabolic
pathways promote and inhibit lipid peroxidation, respectively. GSH,
glutathione; PLOOH, phospholipid hydroperoxides; PLOH, phospholipid
alcohol; GSSG, oxidized glutathione; FSP1, ferroptosis suppressor
protein 1; CoQ10, coenzyme Q10; DHODH, dihydroorotate
dehydrogenase; GCH1, GTP cyclohydrolase 1; BH4,
tetrahydrobiopterin; BH2, dihydrobiopterin; PIEZO1, piezo-type
mechanosensitive ion channel component 1; PUFA, polyunsaturated
fatty acid; MUFA, monounsaturated fatty acid; PL, phospholipid;
SFA, saturated fatty acid. Created with BioRender.com. |
This process of lipid peroxidation is regulated by a
complex network of metabolic and enzymatic regulators. Glutathione
peroxidase 4 (GPX4) plays a central role in counteracting
ferroptosis by reducing membrane lipid hydroperoxides to their
corresponding alcohols, utilizing glutathione (GSH) as a reducing
substrate (15). Perturbation of
this axis, either through direct GPX4 inhibition or GSH depletion
via impaired cystine import (for example, via system Xc−
inhibition), markedly sensitizes cells to ferroptotic death
(1,16). In parallel, ferroptosis
suppressor protein 1 (FSP1)/ubiquinol (CoQH2), dihydroorotate
dehydrogenase/CoQH2 and GTP cyclohydrolase 1
(GCH1)/tetrahydrobiopterin have been identified as independent
systems that scavenge free radicals to exert their antioxidative
effects and suppress ferroptosis (17-21).
The execution of the Fenton response is dependent on
iron availability. Cellular iron metabolism is exquisitely
regulated, with transferrin-mediated uptake, ferritin-based storage
and transferrin-transferrin receptor (TFRC)-mediated export
collectively maintaining intracellular iron homeostasis (22). Perturbations that expand the LIP,
whether through increased iron import, mobilization from stores or
diminished export, potentiate ferroptosis by providing increased
substrate levels for lipid peroxidation reactions (1,23). In addition, the membrane
susceptibility to ferroptotic damage is modulated by its lipidomic
composition, with phospholipids enriched in PUFAs, particularly
arachidonic acid (AA) and adrenic acid (AdA), being especially
prone to peroxidation, thereby rendering membrane vulnerability to
ferroptosis (24).
Lactate metabolism and regulation
Recently, a growing body of evidence has
demonstrated that lactate is not merely a metabolic by-product but
also serves as a key energy source and a critical signaling
molecule involved in memory formation, neuroprotection, modulation
of inflammatory responses, wound healing, ischemic injury repair,
as well as tumor growth and metastasis (5,25-27). In this section, a comprehensive
overview of the metabolic pathways of lactate is presented and its
specific mechanistic roles, with particular emphasis on its dual
role in regulating ferroptosis, are examined (Fig. 2).
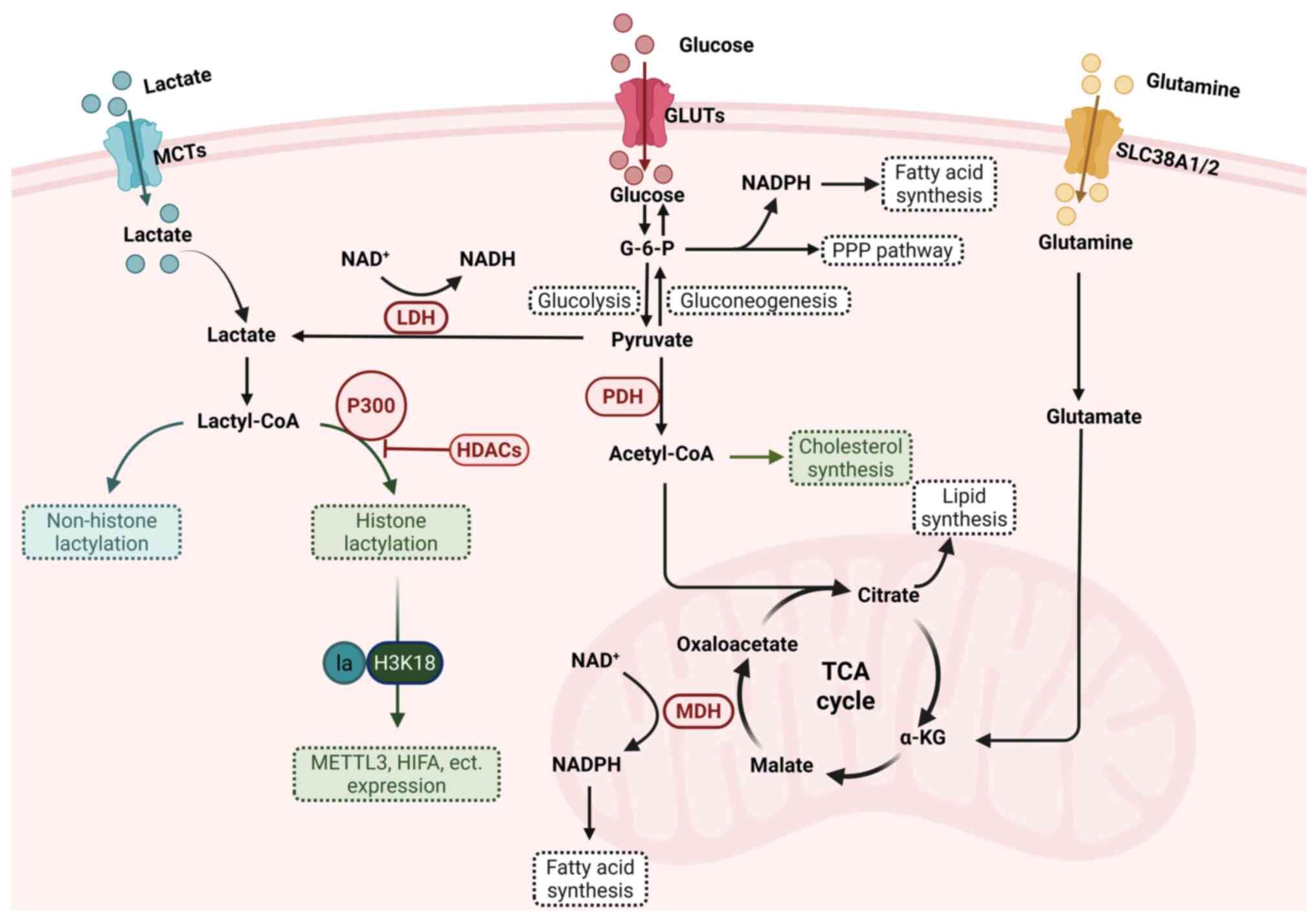 | Figure 2Lactate metabolism, lactylation and
the pathways involved in cells. Lactate is transported into cells
via MCTs and is generated through glycolysis or glutamine
decomposition in the cytoplasm. Once oxidized to pyruvate, pyruvate
can be metabolized through two major pathways: i) Entering
mitochondria for metabolism via the TCA cycle; or i) being
converted to glucose via gluconeogenesis. Intermediate products of
glycolysis and gluconeogenesis contribute to NADPH production
through the PPP. Malate can be converted to oxaloacetate via MDH,
generating NADPH, which supports fatty acid synthesis and GSSG
recycling. Citrate, another key metabolite, serves as a precursor
for lipid synthesis. Additionally, lactate can be converted into
lactyl-CoA, facilitating the lactylation of histone and non-histone
proteins, linking metabolism to epigenetic regulation. MCTs,
monocarboxylate transporters; TCA cycle, tricarboxylic acid cycle;
PPP, pentose phosphate pathway; MDH, malate dehydrogenase; GSSG,
oxidized glutathione; GLUTs, glucose transporters; LDH, lactate
dehydrogenase; HDACs, histone deacetylases; PDH, pyruvate
dehydrogenase; METTL3, methyltransferase like 3; HIFA,
hypoxia-inducible factor α; MDH, malate dehydrogenase; SLC38a1/2,
solute carrier family 38 member a 1/2. Created with BioRender.com. |
Lactate biosynthesis and metabolism
When cellular energy demands exceed the capacity of
aerobic metabolism, as occurs during intense exercise or infection,
lactate is generated via glycolysis to serve as an alternative
energy source. Under hypoxic conditions, cytoplasmic glucose is
metabolized into pyruvate through a series of enzymatic reactions.
However, instead of being transported into mitochondria for
oxidative metabolism, pyruvate is converted into lactate by LDHA,
coupled with NADH/NAD+ interconversion (5). This reaction is reversible: Under
sufficient oxygen availability, lactate can be reconverted into
pyruvate by LDHB, which is then subsequently oxidized to acetyl-CoA
by pyruvate dehydrogenase (PDH) and enters the tricarboxylic acid
cycle (TCA) for efficient energy production (5). Furthermore, an electrochemical
gradient driving ATP synthesis is created as electrons shuttle
through NAD+/NADH and FAD/FADH2 to the
electron transport chain (28).
Studies have highlighted that the conversion of
glucose to lactate by cells constitutes a tightly regulated
metabolic state, which may confer advantages during periods of
heightened biosynthetic demand (29,30). By channeling excess pyruvate
toward lactate production, proliferating cells effectively prevent
cytosolic NADH accumulation and prevent excessive ATP generation.
This regulation ensures the continuation of cytosolic glucose
metabolism without feedback inhibition from mitochondrial ATP
overproduction. Furthermore, glucose-6-phosphate derived from
glycolysis can be diverted into branching metabolic pathways, such
as the pentose phosphate pathway (PPP), where it is partially
oxidized to generate NADPH (28). This NADPH serves as a critical
reducing equivalent for fatty acid synthesis and other anabolic
processes. Additionally, isotope tracing studies have demonstrated
that lactate functions as a major fuel in the TCA cycle, where
13C-lactate labeled TCA intermediates in every tissue of
the body, even in tumors (31,32). However, the accumulation of
lactate carries significant risks to the human body. Elevated serum
lactate levels can result in lactic acidosis, a condition that
poses a greater physiological threat compared with other metabolic
intermediates (33).
In addition to glycolysis, other biochemical
pathways, such as glutamine metabolism, contribute significantly to
lactate production in vivo particularly in cancer cells
(34). Under the regulation of
the oncogene c-Myc, glutamine is metabolized through the TCA cycle
to generate pyruvate, which is subsequently converted into lactate
by LDH (35). This process
provides an alternative and critical source of lactate in rapidly
proliferating cells, supporting energy production and
biosynthesis.
Hypoxia-induced lactate accumulation
Hypoxia represents a hallmark of pathological
conditions, including tumors, infections, ischemia-reperfusion
injury (IRI) and inflammation (36); it occurs when cells experience
reduced oxygen availability and activate an adaptive response to
cope with it. In response to reduced oxygen availability, cells
activate adaptive signaling cascades mediated by hypoxia-inducible
factor-1α (HIF-1α). Under this condition, HIF-1α becomes stabilized
and subsequently translocates to the nucleus, where it binds to
hypoxia-responsive elements within target genes, thereby promoting
metabolic programming, notably the upregulation of anaerobic
glycolysis (37,38). Under hypoxic conditions, the
reduced oxygen availability diminishes the activity of prolyl
hydroxylase domain enzymes (PHDs), which typically employ oxygen
and α-ketoglutarate as substrates to hydroxylate HIF-1α, thereby
targeting it for degradation. Consequently, the decreased PHD
activity leads to HIF-1α stabilization (39).
Cellular adaptation to hypoxia involves an enhanced
glycolytic flux, primarily mediated by HIF-dependent
transcriptional upregulation of genes encoding glucose transporters
and glycolytic enzymes. This metabolic shift is accompanied by an
active suppression of mitochondrial pyruvate oxidation and
respiratory activity (40-43). These biochemical adaptations
result in a metabolic reprogramming that promotes lactate
production and accumulation, thereby reinforcing the hypoxic
cellular response.
LDHA/B
LDHA and LDHB constitute the subunits of the
catalytically active LDH enzyme, which has long been recognized for
its pivotal role in ATP generation and energy homeostasis under
both anaerobic and aerobic glycolytic conditions (44). LDHA possesses a higher affinity
for pyruvate and preferentially catalyzes its reduction to lactate,
thereby sustaining anaerobic glycolysis. By contrast, LDHB
catalyzes the reverse reaction (oxidizing lactate to pyruvate)
which subsequently fuels mitochondrial oxidative phosphorylation
(44). However, accumulating
evidence suggests that all LDH isoforms retain the inherent
capacity to mediate pyruvate-to-lactate conversion accompanied by
NAD+ regeneration, and that LDHA and LDHB can
functionally compensate for each other under metabolic stress
(44,45). By regulating the
NAD+/NADH ratio, mitochondrial function and ROS levels,
LDH isoforms indirectly modulate ferroptosis susceptibility. For
instance, LDHA has been demonstrated to promote tumor cell survival
by mitigating oxidative stress, whereas LDHB deficiency induces
mitochondrial dysfunction and oxidative damage, ultimately leading
to neurodegeneration in the adult mouse brain (46,47). More recently, non-canonical roles
of LDHA and LDHB have been identified, with both isoforms
contributing to ferroptosis resistance in IRI and cancer through
mechanisms associated with GPX4 activity or GSH availability
(48,49).
MCTs and GPR81/HCAR1
MCTs serve as the principal mediators of lactate
transport across cell membranes. By coupling lactate translocation
with proton co-transport, these transporters help maintain
acid-base balance and cellular metabolic stability (7). For instance, inhibition of MCT1
disrupts lactate homeostasis and impairs both glycolytic flux and
GSH biosynthesis in MYC-driven cancer, resulting in reduced glucose
uptake and depletion of ATP, NADPH and GSH (50). Furthermore, extracellular lactate
must enter cells through MCTs before contributing to lactylation
reactions, which have been demonstrated to modulate ferroptosis
through multiple mechanisms (51).
GPR81/HCAR1 is a cell-surface G protein-coupled
receptor that recognizes lactate as its endogenous ligand and
coordinates metabolic signaling across diverse tissues (8). In adipocytes, GPR81 acts
synergistically with insulin to lower intracellular cyclic
adenosine monophosphate (cAMP) levels, thereby suppressing
postprandial lipolysis (52).
Beyond adipose tissue, GPR81 is abundantly expressed in skeletal
muscle, kidney, brain, heart and various cancer types (8). Elevated extracellular lactate, a
defining feature of the tumor microenvironment, predicts poor
outcomes, and high GPR81 expression correlates with poorer survival
(8,53,54). Analogous to its role in
adipocytes, GPR81 activation in cancer cells decreases
intracellular cAMP and suppresses proteinkinase A (PKA) activity,
thereby modulating lipid remodeling (55,56).
Elevated lactate concentrations within the tumor
microenvironment can activate GPR81 receptors located on the
cytoplasmic membrane of cells, subsequently facilitating
MCT1-mediated lactate uptake (57,58). Through this pathway, lactate
disrupts AMPK signaling, which in turn downregulates sterol
regulatory element-binding protein 1 (SREBP1) and its downstream
target stearoyl-CoA desaturase 1 (SCD1). Consequently, cells
produce more anti-ferroptotic monounsaturated fatty acids (MUFAs),
thereby suppressing lipid peroxidation. Simultaneously, long-chain
acyl-CoA synthetase 4 (ACSL4) expression decreases, reducing the
availability of oxidizable PUFAs, although the exact mechanism
underlying this remains unclear. Notably, blocking lactate
transport by inhibiting MCT1 or GPR81 promotes ferroptosis
primarily through alterations in lipid metabolism rather than
through conventional ferroptosis regulators, as evidenced by
unchanged GPX4 and FSP1 levels (57). Additionally, other research has
demonstrated that intracellular lactate accumulation following MCT
inhibition may lead to end-product inhibition of LDH, thereby
impairing NAD+ regeneration capacity (59). This sustained disruption of
glycolysis can result in the depletion of ATP, NADPH and GSH
(50). These findings suggest
that therapeutic strategies targeting the function of MCTs could
prove effective against both oxidative and hypoxic tumors.
Dual roles of lactate in ferroptosis
Accumulating evidence indicates that lactate
metabolism modulates ferroptosis through both
lactylation-independent and dependent pathways. Notably, this
regulation exhibits context-dependent effects. The
lactylation-independent mechanisms, including the iron handling,
lipid remodeling, redox regulation and immune responses in
ferroptosis, are summarized in this section. Additionally, evidence
supporting lactylation-dependent mechanisms has been compiled.
Lactate regulates iron homeostasis in
ferroptosis
Lactate-hepcidin axis in
ferroptosis
Clinical and experimental evidence has consistently
demonstrated a robust association between hyperlactatemia and
anemia, indicating a functional interconnection between lactate
metabolism and systemic iron homeostasis (60). For instance, in endurance
athletes, repeated bouts of exercise induces transient elevations
in plasma lactate concentrations, which have been correlated with
the development of iron-restrictive anemia in 10-15% of
individuals, particularly among those engaging in >10 h of
training per week (61).
Hepcidin, a peptide hormone predominantly synthesized by
hepatocytes, regulates iron homeostasis by binding to ferroportin
(FPN), the sole identified cellular iron exporter. This interaction
triggers FPN internalization and degradation, consequently
restricting iron efflux and leading to the elevation of
intracellular iron pools. Concurrently, it limits systemic iron
availability by reducing intestinal iron absorption, ultimately
resulting in decreased serum iron concentrations (62) (Fig. 3).
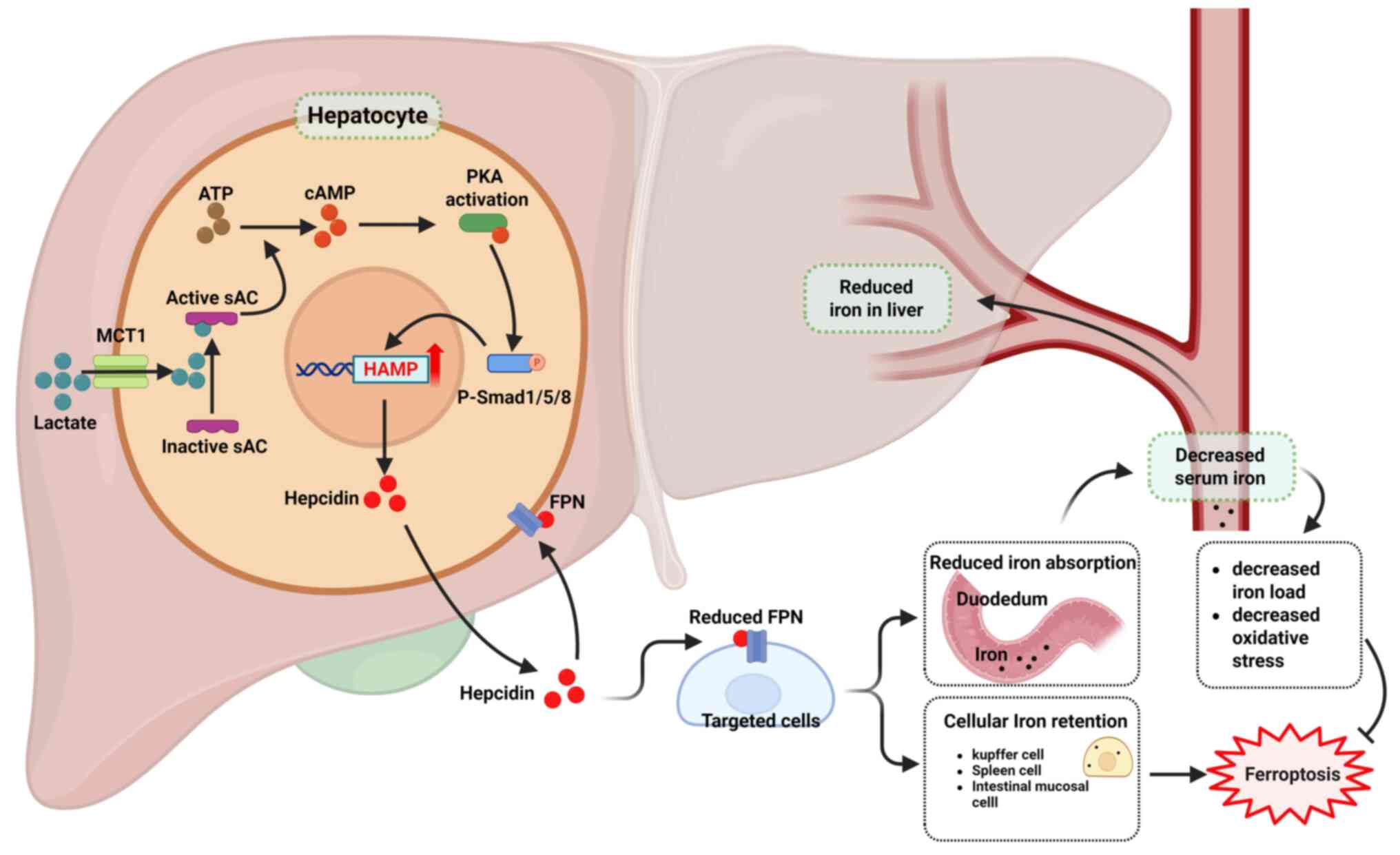 | Figure 3Lactate-hepcidin axis regulates iron
homeostasis. Lactate enters cells via MCT1 and binds sAC,
triggering its conversion of ATP to cAMP. The cAMP activates PKA,
which enhances SMAD signaling. Activated SMAD upregulates HAMP
transcription, increasing hepcidin, the master regulator of iron
homeostasis. Hepcidin then binds FPN, causing its internalization
and degradation, which promotes cellular iron retention and lowers
circulating iron. MCT1, monocarboxylate transporter 1; sAC, soluble
adenylyl cyclase; cAMP, cyclic adenosine monophosphate; PKA,
protein kinase A; HAMP, human hepcidin gene; FPN, ferroportin 1.
Created with BioRender.com. |
Liu et al (63) recently elucidated the molecular
mechanism by which lactate regulates hepcidin expression and,
consequently, systemic iron homeostasis. Their findings revealed
that lactate, transported into cells via MCT1, directly interacts
with soluble adenylyl cyclase, resulting in elevated cAMP levels.
This, in turn, activates the PKA-Smad1/5/8 signaling cascade,
ultimately upregulating hepcidin transcription (63). Further investigation demonstrated
that lactate administration in mice induced hepcidin expression,
leading to reduced FPN levels. This resulted in increased splenic
iron sequestration, diminished duodenal iron absorption and,
consequently, decreased serum and tissue iron content accompanied
by attenuation of oxidative stress (63,64). Although tissue-specific
differences in hepcidin sensitivity, along with variations in
critical thresholds, time kinetics and the compensatory capability
of cellular antioxidant defense systems, may modulate iron
accumulation and ferroptotic outcomes, existing evidence supports a
pro-ferroptotic role of hepcidin in Kupffer cells, neurons and
hepatocytes (65-67).
Although current evidence supports a sophisticated
mechanistic framework linking lactate-mediated hepcidin induction
to ferroptosis, the tissue- and cell-type-specific effects of
elevated lactate levels induced by diverse physiological and
pathophysiological processes on iron homeostasis and ferroptosis
throughout the body warrant further investigation.
Lactate regulates iron via the
TFRC
Iron can be imported into the cell via the TFRC
system. In this process, Fe3+-transferrin complexes are
internalized through TFRC and eventually trafficked to and
liberated within the acidic environment of the lysosome mediated by
nuclear receptor coactivator 4 (NCOA4) (2). TFRC silencing and NCOA4 disruption
reduce ferroptotic sensitivity by restricting iron retrieval from
ferritin and thereby limiting the LIP (68,69). Lactylation of histones or
non-histone proteins involved in iron autophagy-related pathways
can modulate iron handling, thereby regulating ferroptosis
(70-72). Notably, this regulatory effect
appears to be context-dependent; for instance, H3K14 lactylation
has been demonstrated to promote ferroptosis in endothelial cells
exposed to lipopolysaccharide (LPS) by transcriptionally
upregulating the TFRC (73).
Conversely, lactylation of lysine-specific demethylase 1 (LSD1) in
melanoma promotes its interaction with Fos-like antigen 1 (FosL1),
resulting in repression of TFRC-mediated iron uptake and conferring
resistance to ferroptosis (70).
However, at present, there is no direct evidence demonstrating that
histone lactylation regulates the TFRC in tumor cells. Notably,
ferroptotic susceptibility is determined not only by iron overload
but also by the lipid ratio and the capacity of antioxidant defense
systems.
Lactate-lipid remodeling in
ferroptosis
Beyond its established roles in energy metabolism
and immune modulation within the tumor microenvironment, lactate
serves as a crucial metabolic substrate that supports the TCA cycle
in major organs under physiological conditions (31). Tracer studies employing
13C-labeled lactate have demonstrated robust
incorporation of lactate-derived carbon into TCA intermediates
across a wide array of tissues, encompassing both healthy and
malignant cells (31,32).
Mechanistically, lactate contributes to the
intracellular acetyl-CoA pool through its conversion to pyruvate,
followed by subsequent metabolism (74). Acetyl-CoA carboxylase (ACC) then
catalyzes the rate-limiting carboxylation of acetyl-CoA to
malonyl-CoA, a critical intermediate in fatty acid biosynthesis
(75). Fatty acid synthase
subsequently utilizes malonyl-CoA to synthesize palmitic acid
(C16:0), which can be elongated to stearic acid (C18:0) through the
action of elongation-of-very-long-chain-fatty acids 6 (Fig. 4). SCD1 then introduces a double
bond at the Δ9 position, converting these saturated fatty acids to
palmitoleic and oleic acids, respectively. Malonyl-CoA also plays a
vital role in PUFA synthesis, where dietary linoleic acid undergoes
sequential desaturation and elongation reactions catalyzed by fatty
acid desaturase (FADS)2, ELOVL5 and FADS1, leading to the formation
of AA (C20:4, ω-6), which can be further elongated by ELOVL2/4 to
generate AdA (C22:4, ω-6) (76).
These interconnected pathways highlight the pivotal role of
ACC-derived malonyl-CoA in fatty acid metabolism, with ACC
inhibition potentially attenuating lipid peroxidation and
ferroptosis (77-79). Additionally, lactate metabolism
enhances cellular NADPH availability through a glucose-sparing
mechanism: When cells use lactate as their primary fuel source,
glucose is redirected towards the PPP, which generates NADPH
essential for fatty acid biosynthesis (28).
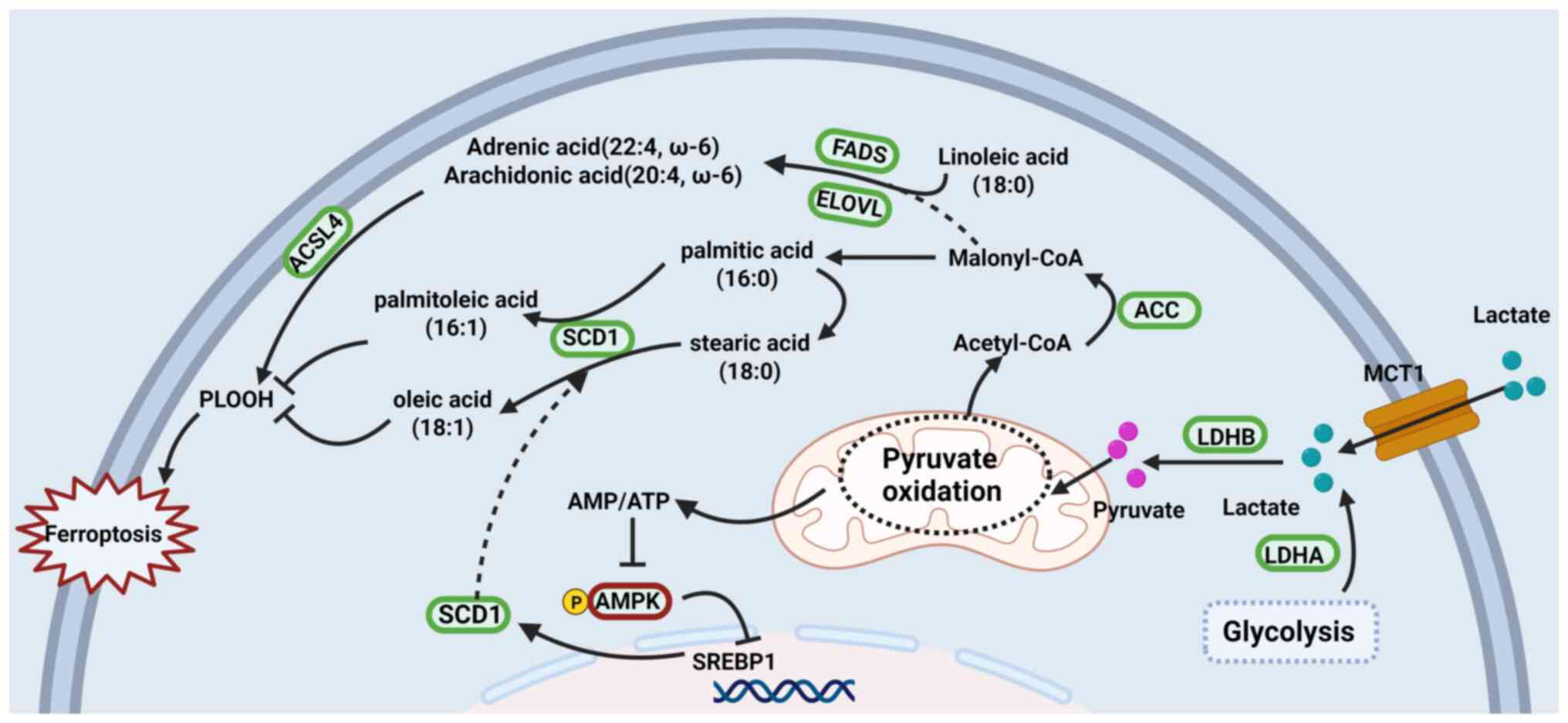 | Figure 4Lactate promotes lipid remodeling in
cells. Lactate is taken up via MCT1 and converted by LDH to
pyruvate and acetyl-CoA, which fuels de novo lipogenesis
through ACC, FASN, SCD1, FADS and ELOVL under the control of the
AMPK-SREBP1 axis. The resulting balance between MUFAs and PUFAs,
and their ACSL4-dependent incorporation into membranes, governs
lipid peroxidation and cellular susceptibility to ferroptotic cell
death. MCT1, monocarboxylate transporter 1; LDH, lactate
dehydrogenase; ACC, acetyl-CoA carboxylase; FASN, fatty acid
synthase; SCD1, stearyl-CoA desaturase 1; FADS, fatty acid
desaturases; ELOVL, elongation of very long-chain fatty acid; AMPK,
AMP-activated protein kinase; SREBP1, sterol regulatory
element-binding protein 1; MUFAs, monounsaturated fatty acids;
PUFAs, polyunsaturated fatty acids; ACSL4, acyl-CoA synthetase
long-chain family member 4. Created with BioRender.com. |
A study by Zhao et al (57) demonstrated that lactate-enriched
hepatocellular carcinoma (HCC) cells exhibit enhanced resistance to
ferroptotic damage induced by RAS-selective lethal 3 (RSL3) and
Erastin. The authors elucidated a mechanism wherein MCT1-mediated
lactate uptake promotes ATP production, leading to AMPK
deactivation and subsequent upregulation of SREBP1 and SCD1,
enhancing the production of MUFAs that confer protection against
ferroptosis (57). Corroborating
these findings, Yang et al (58) demonstrated that lactate-induced
alterations in SCD1/ACSL4 expression and ferroptosis resistance are
correlated with lactate production levels in esophageal squamous
cell carcinoma (ESCC). The conservation of this lactate/SCD1
regulatory axis across diverse tissue types has been substantiated
by multiple studies (80,81).
ACSL4 is an enzyme that esterifies CoA into specific PUFAs, such as
AA and AdA, contributing to ferroptosis execution by triggering
phospholipid peroxidation. In sepsis, lactate promotes ferroptosis
via GPR81-mediated upregulation of methyltransferase like 3
(METTL3), which may mediate ACSL4 mRNA stability via
N6-methyladenosine (m6A) modification in pulmonary epithelial cells
(82).
Although regulating the MUFA/PUFA ratio by lactate
through the SCD1/ASCL4 pathway does affect membrane lipid
peroxidation sensitivity, this protective effect against
ferroptosis may be more highly dependent on the state of the
intracellular antioxidant system (83).
Lactate-redox regulation in
ferroptosis
Disruptions in redox balance, whether toward
excessive oxidation or reduction, are generally deleterious to
cellular function. Lactate acts as a redox buffer by modulating the
NAD(P)H/NAD(P)+ ratio and serves as a signaling molecule
that regulates the activity of antioxidant enzymes involved in ROS
metabolism.
NADH/NAD+ balance in
lactate-ferroptosis crosstalk
The interconversion of lactate and pyruvate,
catalyzed by LDHA/B, is closely coupled to the NAD+/NADH
redox pair, thereby modulating cellular redox balance in a
context-dependent manner (84,85). Elevated lactate concentrations
have been demonstrated to increase the NADH/NAD+ ratio,
leading to the inhibition of key glycolytic enzymes such as
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and
phosphoglycerate dehydrogenase, ultimately suppressing both
glycolysis and mitochondrial respiration (85). When the cellular demand for
NAD+ to sustain oxidation exceeds the rate of ATP
turnover, particularly in cells exhibiting active aerobic
glycolysis, NAD+ regeneration becomes a limiting factor
under conditions of compromised mitochondrial respiration. In such
circumstances, cells preferentially rely on glycolysis, resulting
in an elevated NAD+/NADH ratio. Under these conditions,
activation of PDH facilitates pyruvate oxidation, attenuates
lactate accumulation and restores the NAD+/NADH
equilibrium (86). Conversely,
when lactate is oxidized to pyruvate and enters mitochondrial
oxidative pathways, it can indirectly elevate ROS production
through electron leakage from the respiratory chain (87-89). In adipocytes, lactate exposure
transiently elevates the NADH/NAD+ ratio, which
normalizes after 24 h due to elevated NAD+ levels
associated with increased mitochondrial membrane potential and
enhanced ROS production (90).
Similarly, Jia et al (91) reported that elevated neuronal
lactate uptake augments ROS production and mitochondrial oxidative
metabolism, thereby disrupting the balance between ROS generation
and detoxification, impairing ATP synthesis and ultimately leading
to peripheral axonal degeneration. The mitochondrial enzyme
nicotinamide nucleotide transhydrogenase further modulates redox
status by catalyzing the reversible conversion of NADH and
NADP+ into NAD+ and NADPH, the latter serving
as a crucial factor for GSH reductase-mediated recycling of reduced
GSH (92). Unlike observations
in normal cells, lactate uptake in melanoma cells via MCT1 elevates
NADH, NADPH, GPX4 and FSP1 levels, thereby conferring resistance to
ferroptosis (93). The net
effect of lactate on redox status is governed by the balance
between reducing equivalents of NADH and ROS production, which may
differ between normal and cancer cells due to variations in
oxidative phosphorylation activity and NADH-processing pathways.
This apparent contradiction may reflect a critical evolutionary
adaptation in which cancer cells reprogram lactate metabolism to
attain dual benefits (enhanced metabolic flexibility coupled with
augmented oxidative stress resistance) thereby facilitating
survival under adverse microenvironmental conditions.
NADPH/NADP+ balance in
lactate-ferroptosis
Within glucose metabolism, glucose-6-phosphate
generated either through glycolysis or via gluconeogenesis from
lactate can enter the PPP, where it is partially oxidized to
generate NADPH (94,95). Moreover, lactate metabolism
contributes to NADPH production through TCA cycle-linked pathways,
particularly those catalyzed by malic enzyme 1 (ME1) and isocitrate
dehydrogenase 1 (IDH1) (94,96). Similar to NADH, NADPH fulfills
dual roles in cellular redox regulation: It is indispensable for
antioxidant defense by facilitating GSH reduction yet
simultaneously serves as a substrate for NADPH oxidases that
generate ROS production (97).
Under glucose-deprived conditions, the knockout of IDH1 or ME1
significantly reduces the NADPH/NADP+ and GSH/oxidized
GSH ratios, leading to elevated ROS levels and increased cell death
in cancer cells (94).
Conversely, excessive NADPH accumulation can induce reductive
stress, leading to the upregulation of NADPH oxidase 4 through
activation of the PI3K/Akt signaling pathway, thereby promoting ROS
generation, as observed in osteoarthritis (96).
Collectively, cellular redox homeostasis is
maintained by a delicate equilibrium between NAD(P)H-dependent
antioxidant defense mechanisms and NAD(P)H-mediated reductive
stress. By engaging in NAD(P)H/NAD(P)+ redox cycling,
lactate functions as a dynamic redox buffer that modulates cellular
responses to oxidative and reductive stress.
Antioxidant enzymes in
lactate-ferroptosis
Under physiological conditions, lactate indirectly
regulates cellular antioxidant systems through modulation of the
NAD(P)H/NAD(P)+ redox balance. Specifically,
uncontrolled elevated intracellular lactate concentrations disrupt
the NADH/NAD+ ratio, thereby impairing the activity of
key glycolytic enzymes, particularly GAPDH due to NAD+
depletion (50,57). This metabolic perturbation,
coupled with lactate-mediated feedback inhibition of
phosphofructokinase-1, ultimately impairs cellular ATP homeostasis
and compromises GSH biosynthesis, the predominant intracellular
antioxidant (50). GPX4 plays a
pivotal role in counteracting ferroptosis. A recent investigation
has demonstrated that intracellular lactate accumulation under
ischemic conditions can inactivate the AMPK/GPX4 axis to promote
myocardial ferroptosis (98).
Conversely, emerging evidence indicates that lactate can
paradoxically potentiate antioxidant defense mechanisms through
alternative signaling cascades. Metabolically reprogrammed lactate
has been demonstrated to augment GPX4 expression and confer
resistance to ferroptosis via activation of the p38-serum
glucocorticoid-regulated kinase 1 signaling axis. This pathway
mitigates GPX4 ubiquitination and subsequent degradation in
non-small cell lung cancer (NSCLC) cells (99). Furthermore, an additional study
has elucidated that lactate activates antioxidant defense and
pro-survival pathways, including the unfolded protein response and
nuclear factor erythroid 2-related factor 2 (NRF2) signaling
cascades, by inducing mild oxidative stress in neuroblastoma cells
(100). Notably, lactate has
also been shown to alleviate oxidative stress-induced cell death
through autophagy activation in retinal pigment epithelial cells
(101).
Lactate-ferroptosis axis in
immunometabolism
The immunomodulatory properties of lactate
significantly influence ferroptosis susceptibility within both
inflammatory conditions and the tumor microenvironment, carrying
notable therapeutic relevance (102,103).
Inflammatory response and sepsis
Sepsis-induced metabolic reprogramming drives
increased lactate accumulation and systemic oxidative stress,
triggering multiple cell death pathways in both immune and
parenchymal cells (82,104). In septic lung injury, elevated
lactate levels exacerbate alveolar epithelial cell ferroptosis
through lactylated histone H3 lysine 18 (H3K18la)-mediated
upregulation of METTL3, which enhances m6A modification of ACSL4,
ultimately contributing to the development of acute respiratory
distress syndrome (82). Lactate
further augments neutrophil functions, including chemotaxis,
phagocytosis, oxidative burst and neutrophil extracellular trap
formation, via energy provision and PI3K/Akt signaling, which may
exacerbate tissue cell ferroptosis (105). Nevertheless, lactate
simultaneously exerts immunomodulatory effects in sepsis; it
suppresses LPS-induced pro-inflammatory cytokine production in
macrophages and promotes M2 polarization through MCTs and HIF-1α
activation (106). Moreover,
lactate-induced histone lactylation drives macrophages toward a
reparative phenotype characterized by reduced pro-inflammatory
cytokine expression, thereby potentially constraining excessive
inflammation (107,108). This dual role of lactate in
modulating ferroptosis and inflammation may account for the
inconsistent therapeutic outcomes associated with hypertonic sodium
lactate administration in sepsis, as these effects appear to be
highly dependent on factors such as timing of intervention, dosage
and the specific experimental animal models employed (104,109).
Tumor immunity context
The lactate-ferroptosis axis influences anti-tumor
immunity through multifaceted mechanisms that reshape the tumor
microenvironment. Lactate accumulation within the tumor
microenvironment suppresses effector T cell proliferation and
cytotoxic activity via GPR81-mediated signaling and metabolic
competition, while simultaneously polarizing tumor-associated
macrophages toward an immunosuppressive M2 phenotype that promotes
tumor progression (8). Notably,
ferroptosis induction in cancer cells can trigger immunogenic cell
death (ICD), leading to the release of damage-associated molecular
patterns that activate dendritic cells and stimulate antitumor T
cell responses (110). However,
lactate-mediated ferroptosis resistance through SCD1 upregulation
and enhanced antioxidant capacity diminishes this immunogenic
potential, effectively creating an immune-evasive tumor phenotype
(57,58). Recent evidence demonstrates that
targeting the lactate-ferroptosis axis can reprogram the
immunosuppressive tumor microenvironment. Specifically, the
combination of MCT4 inhibition with ferroptosis inducers not only
depletes lactate accumulation but also enhances CD8+ T
cell infiltration and ferroptosis-driven ICD, thereby
synergistically improving checkpoint blockade efficacy (111). This immunometabolic
reprogramming represents a promising approach to convert 'cold'
tumors into 'hot' tumors that are more responsive to
immunotherapy.
Epigenetic regulation via protein
lactylation
Kla was initially identified as an enzymatically
catalyzed post-translational modification, wherein lactyl groups
derived from lactate are covalently attached to lysine residues
(9). Originally identified on
histones, Kla has been indicated to accumulate at gene promoters in
response to diverse stimuli, including hypoxia, interferon-γ, LPS
exposure and bacterial infections. This modification directly
modulates transcriptional activity and gene expression, thereby
establishing a mechanistic link between cellular metabolism and
transcriptional regulation (107,108). Subsequent investigations have
expanded the functional repertoire of Kla, demonstrating its
presence on non-histone proteins, particularly metabolic enzymes
(11,12). The lactylation of these enzymes
regulates cellular metabolism by modulating enzymatic activity,
notably through feedback mechanisms that regulate glycolytic flux
(108,112). However, the precise enzymatic
machinery responsible for Kla deposition and removal remains
incompletely characterized and the relative contributions of
enzymatic vs. non-enzymatic lactylation pathways warrant further
clarification.
Histone lactylation
Nuclear Kla is predominantly observed at H3K18, with
p300-mediated H3K18la at gene promoters serving as a key
determinant of transcriptional regulation. For instance, H3K18la
enrichment at the METTL3 promoter upregulates METTL3 expression,
subsequently augmenting m6A modification of ACSL4 mRNA (82). This cascade stabilizes ACSL4
transcripts, elevates ACSL4 protein levels and promotes
mitochondrial ROS accumulation, ultimately driving ferroptosis in
alveolar epithelial cells during sepsis (82). Furthermore, H3K18la facilitates
ACSL4 expression through direct promoter engagement and activation
of the HIF-1α signaling pathway (113,114). Under hypoxic conditions,
elevated lactate levels stimulate H3K18la at the HIF-1α promoter,
leading to upregulation of HIF-1α expression (115). This signaling cascade
subsequently elevates ACSL4 expression, driving lipid peroxidation
and ferroptosis through the ACSL4/lysophosphatidylcholine
acyltransferase 3/arachidonate lipoxygenase 15 axis, as
demonstrated in both in vitro and in vivo models of
severe acute pancreatitis (114). Paradoxically, Kla modifications
can also confer cytoprotective effects in specific cancer contexts.
In triple-negative breast cancer, cancer-associated fibroblasts
exhibit elevated H3K18la levels, which enhance zinc finger protein
64 expression and subsequently activate the transcription of GCH1
and FTH. This cascade facilitates iron sequestration and protects
cells from doxorubicin-induced ferroptosis (71). Similarly, in colorectal cancer
stem cells, p300-mediated H4K12la upregulates glutamate-cysteine
ligase catalytic subunit (GCLC), thereby expanding the GSH pool and
conferring ferroptosis resistance (116). Furthermore, H3K18la enhances
the transcriptional activity of NFS1 cysteine desulfurase, a
cysteine desulfurase essential for iron-sulfur cluster
biosynthesis, consequently reducing the susceptibility of HCC to
ferroptosis following microwave ablation (117). These findings underscore the
notable tissue-specific and context-dependent nature of histone Kla
function. However, the molecular determinants underlying this
functional dichotomy remain poorly understood, and the systematic
frameworks capable of predicting whether Kla will promote or
suppress ferroptosis within specific cellular contexts are still
lacking.
Non-histone lactylation
Beyond its role in chromatin regulation, Kla also
influences the activity and stability of various non-histone
proteins, including key metabolic and RNA-modifying enzymes that
regulate ferroptosis. Notably, lactate-primed lysine
acetyltransferase 8 directly lactylates mitochondrial
phosphoenolpyruvate carboxykinase 2 at K100, thereby enhancing its
kinase activity and reprogramming mitochondrial fatty-acid
synthesis to promote ferroptotic processes (118). Additionally, Kla of METTL3
stabilizes the protein and promotes m6A modifications on ACSL4 and
TFRC transcripts, thereby accelerating ferroptosis in PC12 cells
(72). In the context of
Alzheimer's disease, reduced lactylation of tau at K677 inhibits
ferroptosis by disrupting ferritinophagy, resulting in dysregulated
iron metabolism and increased resistance to cell death (119). Similarly, decreased lactylation
of malate dehydrogenase 2 at K241, coupled with reduced lactate
production, leads to elevated levels of GSH and GPX4, thereby
alleviating ferroptosis and improving mitochondrial function in
myocardial IRI (120). By
contrast, lactylation of LSD1 in melanoma promotes its interaction
with FosL1, resulting in repression of TFRC-mediated iron uptake
and thereby conferring resistance to ferroptosis (70). Similarly, lactylation of NOP2/Sun
RNA methyltransferase 2 enhances its catalytic activity and
stabilizes GCLC mRNA via m5C modifications, leading to increased
intracellular GSH levels and ferroptosis resistance in gastric
cancer cells (121). These
findings underscore the versatility of Kla as a regulatory
mechanism capable of either promoting or inhibiting ferroptotic
cell death through modulation of enzymatic activity.
The bidirectional influence of Kla on ferroptosis is
of significant therapeutic interest. In cancer, Kla-driven
modulation of ferroptosis has been implicated in developing
resistance to chemotherapy and radiation. For example, evodiamine
has been found to inhibit histone lactylation at the HIF-1α
promoter, suppressing angiogenesis and programmed death-ligand 1
expression while inducing ferroptosis in prostate cancer cells
(122). This highlights the
potential of targeting Kla 'writers', 'erasers' or 'readers' to
selectively induce ferroptosis in cancer cells, thereby enhancing
the efficacy of cancer therapies while minimizing damage to normal
tissues. Conversely, Kla manipulation could also hold promise in
treating degenerative diseases and ischemic injury by reducing
ferroptotic damage (123).
However, the clinical translation of Kla-targeted therapies faces
notable challenges, including the lack of specific inhibitors,
potential off-target effects and the complex tissue-specific
functions of lactylation.
In summary, Kla represents a critical metabolic
regulator integrating cellular metabolic status with ferroptotic
outcomes. Although the context-dependent, bidirectional modulation
of ferroptosis by Kla presents promising therapeutic avenues,
notable barriers remain between current mechanistic insights and
clinical implementation. This underscores the need for more
rigorous investigations into tissue-specific regulatory networks
and the development of targeted therapeutic strategies.
Putative context-dependent mechanisms
The seemingly contradictory roles of lactate in
ferroptosis (its capacity to both promote and inhibit this
regulated form of cell death) highlight the complex interplay among
cellular metabolism, redox homeostasis and iron regulation. In the
previous section, the factors that determine whether lactate
functions in a pro-ferroptotic or anti-ferroptotic manner within
specific cellular contexts were systematically examined (Fig. 5). In pathological conditions such
as sepsis (73,82), neurodegeneration (91,119), osteoarthritis (96), pancreatitis (114), IRI (120), intracerebral hemorrhage
(72) and adipocytes browning
(90), lactate promotes
ferroptosis. Conversely, in various malignancies, including HCC
(57,117), ESCC (58), melanoma (70), NSCLC (99), neuroblastoma (100), breast cancer (71), colorectal cancer (116), gastric cancer (121) and prostate cancer (122), lactate suppresses ferroptosis.
Table I summarizes recent in
vitro studies examining the role of lactate in ferroptosis
regulation. In this section, three potential mechanisms underlying
this context-dependent regulation are discussed: Differences in
metabolic enzyme expression, pH homeostatic capacity and
antioxidant defense systems (Fig.
6).
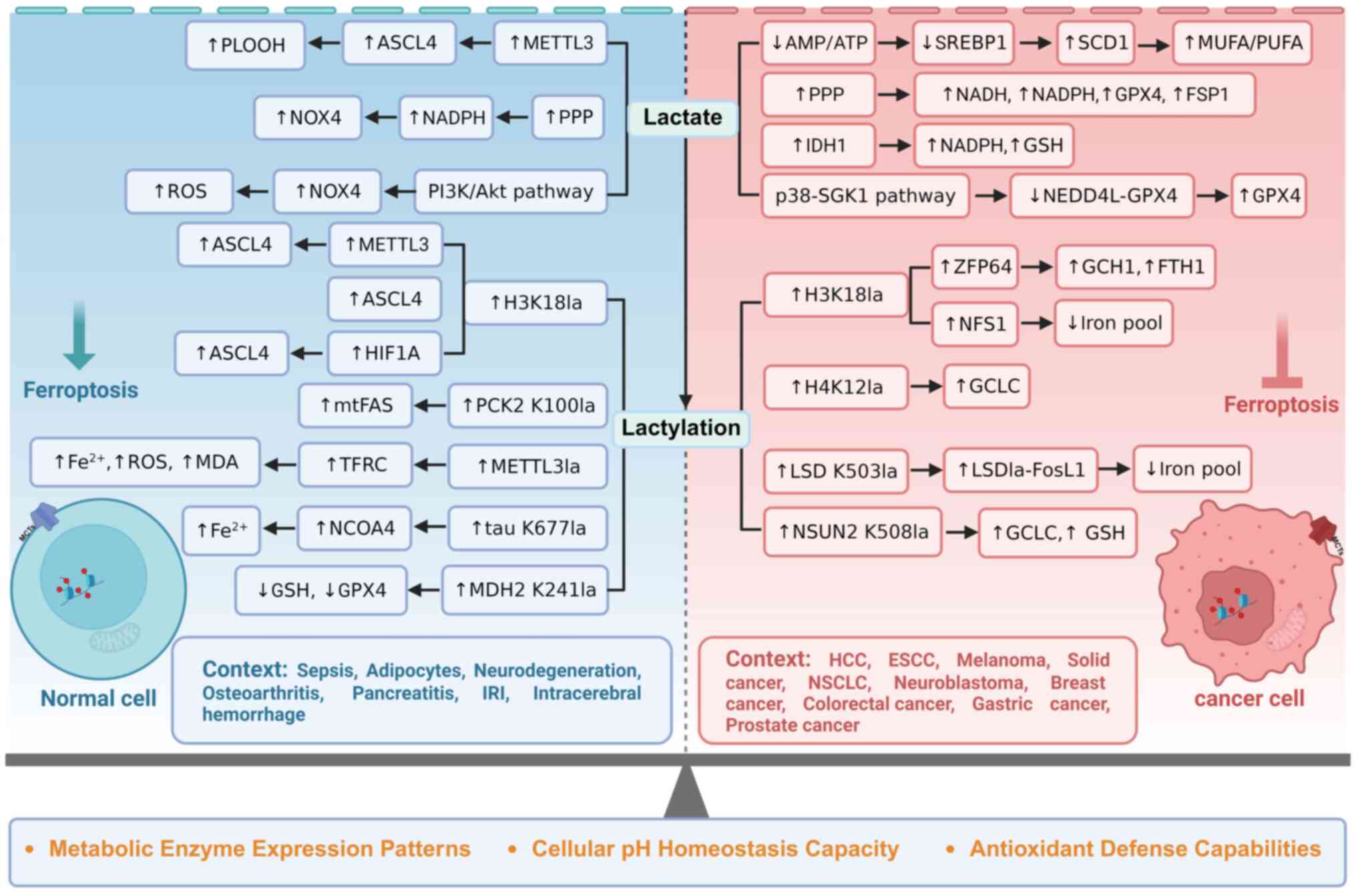 | Figure 5Dual roles of lactate and lactylation
in ferroptosis regulation across cellular contexts. Lactate and
lactylation exhibit contrasting regulatory effects on ferroptosis
in normal vs. tumor cells through both lactylation-dependent and
-independent mechanisms. In lactylation-independent pathways,
lactate modulates cellular metabolism and redox homeostasis via
MCTs and GPR81 receptor signaling, with contrasting outcomes
observed between normal and malignant cell populations. In
lactylation-dependent pathways, lactate similarly demonstrates
contrasting effects in normal vs. tumor cell contexts through
post-translational protein modifications. PLOOH, phospholipid
hydroperoxides; ACSL4, acyl-CoA synthetase long-chain family member
4; METTL3, methyltransferase like 3; NOX4, NADPH oxidase 4; PPP,
pentose phosphate pathway; ROS, reactive oxygen species; SREBP1,
sterol regulatory element-binding protein 1; SCD1, stearoyl-CoA
desaturase 1; MUFAs, monounsaturated fatty acids; PUFAs,
polyunsaturated fatty acids; GPX4, glutathione peroxidase 4; FSP1,
ferroptosis suppressor protein 1; IDH1, isocitrate dehydrogenase 1;
GSH, glutathione; SGK1, serum- and glucocorticoid-inducible kinase
1; NEDD4L, neural precursor cell expressed developmental
downregulated protein; PCK2, phosphoenolpyruvate carboxykinase 2;
mtFAS, mitochondrial fatty acid synthesis; TFRC,
transferrin-transferrin receptor; MDA, malondialdehyde; NCOA4,
nuclear receptor coactivator 4; MDH2, malate dehydrogenase 2;
ZFP64, zinc finger protein 64; GCH1, GTP cyclohydrolase 1; FTH1,
ferritin heavy chain 1; NFS1, NFS1 cysteine desulfurase; GCLC,
glutamate-cysteine ligase catalytic subunit; LSD1, lysine-specific
demethylase 1; FosL1, Fos-like antigen 1; NSUN2, NOP2/Sun RNA
methyltransferase family member 2; IRI, ischemia-reperfusion
injury; HCC, hepatocellular carcinoma; ESCC, esophageal squamous
cell carcinoma; NSCLC, non-small cell lung cancer; MCT (1,4),
monocarboxylate transporter (1,4);
GPR81, G protein-coupled receptor 81. Created with BioRender.com. |
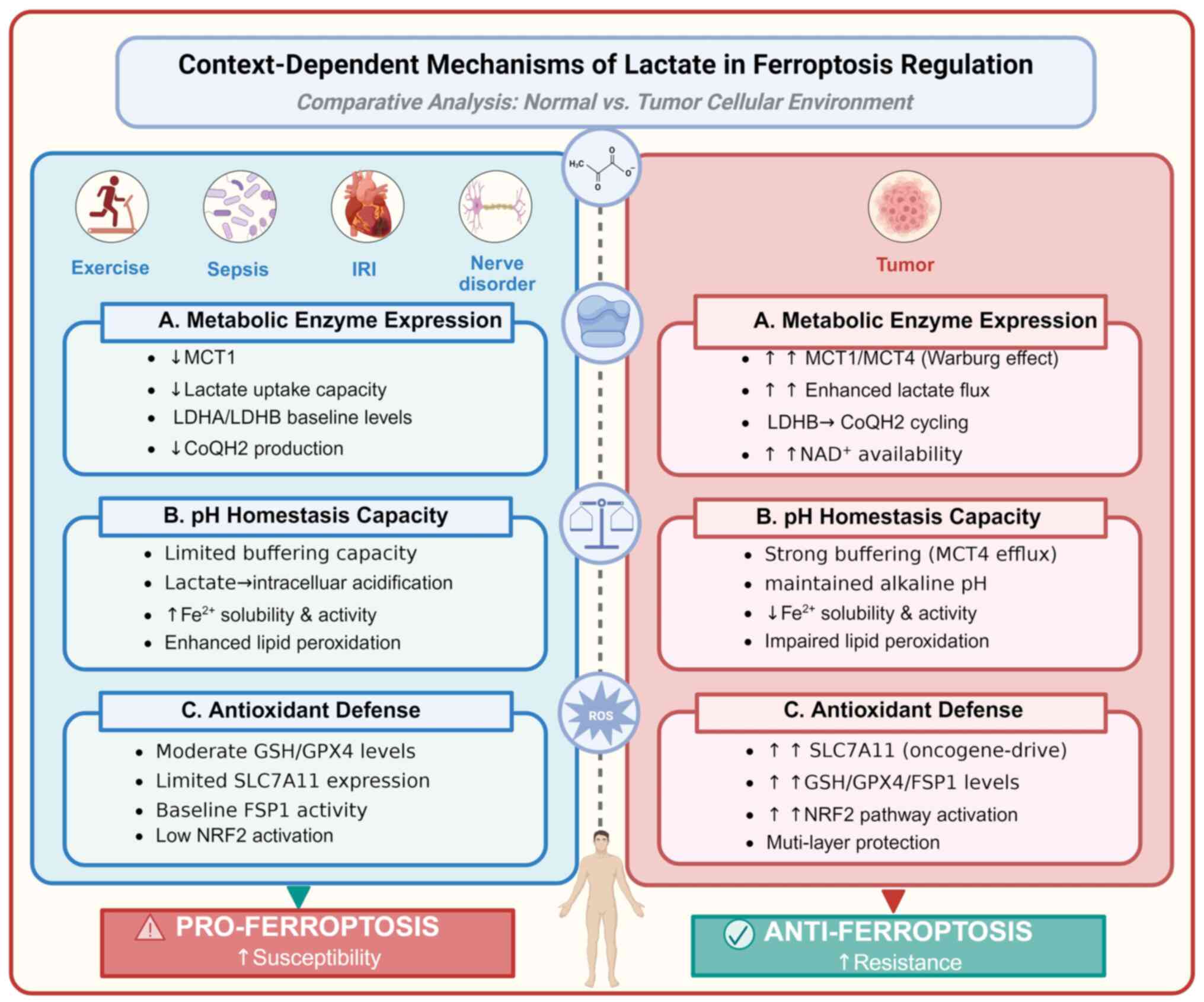 | Figure 6Context-dependent mechanisms of
lactate in ferroptosis regulation. The context-dependent mechanisms
of lactate in ferroptosis regulation include: (A) Differential
metabolic enzyme expression profiles, (B) distinct cellular pH
homeostasis capacities and (C) varying antioxidant defense
capabilities. These mechanisms may underlie the distinct effects of
the context-dependent lactate-ferroptosis axis. IRI,
ischemia-reperfusion injury; MCT (1,4),
monocarboxylate transporter (1,4);
LDHA/B, lactate dehydrogenase A/B; CoQH2, ubiquinol; GSH,
glutathione; GPX4, glutathione peroxidase 4; SLC7A11,
cystine/glutamate antiporter solute carrier family 7 member 11;
FSP1, ferroptosis suppressor protein 1; NRF2, nuclear factor
erythroid 2-related factor 2. Created with BioRender.com. |
 | Table IIn vitro evidence of lactate
regulating ferroptosis. |
Table I
In vitro evidence of lactate
regulating ferroptosis.
| Authors, year | Cell type | Lactate
concentration | Phenotype | Mechanism | Disease
context | Reduce/promote
ferroptosis | (Refs.) |
|---|
| Zhao et al,
2020 | Hep3B and
Huh-7 | 20 mM | MUFAs↑ | SCD1↑ | Hepatocellular
carcinoma | Reduce | (57) |
| Yang et al,
2024 | EC109 | 20 μM | MUFAs↑ | SCD1↑ | Esophageal
cancer | Reduce | (58) |
| Jia et al,
2021 | Primary spinal and
DRG neuron | 1 and 10 mM | mtROS↑ | Alters energy
metabolism | Neurodegenerative
disease | Promote | (91) |
| Huang et al,
2023 | Chondrocyte | 10, 20 and 40
mM | ROS↑ | NADPH/NOX4,
GPR81/NOX4 | Osteoarthritis | Promote | (96) |
| Lin et al,
2022 | COMM-SUS | 10 and 30 mM | NADPH↑, NADH↑ | PPP activation | Melanoma | Reduce | (93) |
| Bauzá-Thorbrügge
et al, 2023 | Adipocyte | 25 mM | ROS↑ | NADH↑ | Adipose tissue | Promote | (90) |
| Cheng et al,
2023 | H1229 and A549 | 5, 10, 15 and 25
mM | GPX4↑ | Inhibits GPX4
ubiquitination by inactivating the E3 ubiquitin ligase NEDD4L | NSCLC | Reduce | (99) |
| Tauffenberger et
al, 2019 | SH-SY5Y | 20 mM | UPR↑, NRF↑ | ROS burst | Neuroblastoma | Reduce | (100) |
| Zou et al,
2023 | ARPE-19 | 20 mM | ROS↓ | Activate autophagy
pathway | A cell line derived
from the retina | Reduce | (101) |
| Wu et al,
2024 | MLE12 | 10 mM | GPX4↓,
GSH/GSSG↓, |
GPR81/H3K18la/METTL3/ACSL4 | Mouse lung
epithelial cells in sepsis | Promote | (82) |
| Zhang et al,
2025 | AR42J | 5, 10, 15 and 25
mM | ACSL4↑, LPCAT3↑,
ALOX15↑ | H3K18la in HIF-1α
promoter | Pancreatitis | Promote | (114) |
| Zhang et al,
2025 | DOX-resistant
TNBC | 25 mM | GCH1↑, FTH1↑ | H3K18la in ZFP64
promoter | TNBC | Reduce | (71) |
| Deng et al,
2025 | LoVo, SW-620 and
HCT-116 | 5, 10 and 15
mM | ROS↓, MDA↓,
GPX4↑ | GCLC↑ | Colorectal
cancer | Reduce | (116) |
| Huang et al,
2025 | MHCC-97H and
Huh7. | 5 and 10 mM | ROS↓, free
iron↓ | H3K18la in NFS1
promoter | Hepatocellular
carcinoma | Reduce | (117) |
| Yuan et al,
2025 | THLE2 | 5, 10 and 20
mM | GPX4↓, COX2↑,
TFRC↑ | Lactylation in PCK2
(K100) induces mtFAS remodeling, potentiation of oxidative
phosphorylation and the tricarboxylic acid cycle | Human liver
epithelial cell line | Promote | (118) |
| She et al,
2024 | H9c2 | 10, 20 and 50
mM | GSH↓, GPX4↓, MDA↑,
iron↑ | Lactylation in MDH2
(K241) | Cardiomyocyte cell
lines from the rat heart | Promote | (120) |
| Niu et al,
2025 | HEK293T and
MKN45 | 10 and 20 mM | GSH↑, Lipid
perioxidation↓ | Lactylation in
NSUN2 (K508) stabilizes GCLC mRNA via promoting m5C
modification | Gastric cancer | Reduce | (121) |
| Yu et al,
2023 | DU145 | 10 mM | ROS↓, iron↓ | H3K18la in HIF-1α
promoter | Prostate
cancer | Reduce | (122) |
Metabolic enzyme expression patterns
Cancer cells characterized by the Warburg effect
demonstrate elevated expression of MCT1 and MCT4, which promote
enhanced lactate uptake and consequent metabolic reprogramming.
Notably, identical lactate concentrations exert diametrically
contrasting effects on ferroptosis susceptibility in malignant lung
cancer cells compared with their normal epithelial counterparts,
highlighting fundamental differences in lactate metabolism between
these cellular contexts (82,99). Although LDHA has been extensively
characterized as a tumor survival factor through its role in
mitigating ROS, targeted depletion via small interfering RNA or
pharmacological inhibition (GSK2837808A or R-GNE-140) fails to
sensitize A549 cells to ferroptosis inducers, including RSL3 or
erastin (49). This observation
necessitates the existence of LDHA-independent anti-ferroptosis
mechanisms. In KRAS-driven NSCLC, LDHB has been identified as a key
regulator of GSH-dependent ferroptosis resistance through STAT1
signaling, although the precise molecular mechanisms underlying
LDHB-mediated STAT1 activation remain incompletely elucidated
(49). A recent investigation
propose that LDHB mediates a complex three-step reaction cycle,
wherein reducing equivalents are transferred from lactate to
reduced CoQH2, with NAD+ serving as a cycling cofactor
(124). The markedly elevated
NAD+ concentrations observed in tumor cells compared
with normal tissues may provide enhanced substrate availability for
LDHB-mediated lactate oxidation cycles (125). Given that CoQH2 serves as an
alternative anti-ferroptosis system operating in parallel with
GPX4, the differential metabolic environments between malignant and
normal cells likely contribute to the divergent effects of lactate
on ferroptosis sensitivity observed across these cellular contexts.
Nevertheless, this proposed mechanism lacks direct biochemical
validation and relies heavily on circumstantial evidence from
functional studies. Despite these limitations, this finding
provides an important avenue for subsequent research.
Cellular pH homeostasis capacity
The differential expression of MCTs between normal
and malignant cells provides a mechanistic framework for
understanding lactate-mediated modulation of ferroptosis
susceptibility. Normal cells predominantly express MCT1, which
confers a relatively limited capacity for lactate transport
(126). By contrast, cancer
cells typically co-express MCT1 and MCT4, facilitating highly
efficient lactate efflux that preserves intracellular pH
homeostasis even within the acidic tumor microenvironment (127). This enhanced acid-extruding
capacity enables tumor cells to sustain intracellular pH at neutral
or slightly alkaline levels, potentially exceeding those observed
in normal cells under comparable conditions (128). The pH dependence of
Fe2+-catalyzed lipid peroxidation constitutes a critical
determinant of ferroptotic sensitivity, proceeding efficiently
under acidic conditions but being markedly impaired at neutral or
basic pH due to reduced Fe2+ solubility. Consequently,
the efficacy of ferroptosis-inducing therapies may be limited in
alkaline cytoplasmic environments (129). However, the proposed
lactate-acidification-ferroptosis axis warrants careful evaluation
in light of emerging contradictory evidence. An early study by
Jackson and Halestrap (130)
documented lactate-induced intracellular acidification in rat
hepatocytes, whereas Bozzo et al (131) observed only modest pH
reductions following 5 mM lactate treatment in mouse neurons. These
findings suggest that normal cells possess notable buffering
capacity against lactate-induced acidification, challenging the
assumption that lactate uniformly acidifies the cytoplasm across
diverse cell types. Moreover, LDHA-mediated lactate accumulation
has been reported to promote ferroptosis resistance in a
pH-dependent manner in tumors, a mechanism that may involve
inhibition of Piezo1 in an acidic environment (132-134). Accordingly, the mechanistic
link between lactate-mediated pH changes and iron-dependent
ferroptosis remains inadequately characterized.
Antioxidant defense capabilities
Tumor cells have developed intricate adaptive
mechanisms to evade ferroptosis, a key tumor-suppressive process.
In response to the heightened oxidative stress associated with
malignant transformation, cancer cells activate a comprehensive
antioxidant defense pathway that, paradoxically, shields them from
ferroptotic cell death (4). At
the core of this protective adaptation lies the upregulation of
cystine/glutamate antiporter solute carrier family 7 member 11
(SLC7A11), driven by the inactivation of key tumor suppressors
including TP53, BRCA1 associated protein 1 and alternate reading
frame (16,135). This dysregulation fundamentally
alters the cellular redox balance, conferring ferroptosis
resistance while simultaneously promoting tumor proliferation and
survival. The oncogenic KRAS signaling cascade further amplifies
this effect by directly upregulating SLC7A11 expression, thereby
establishing a robust anti-ferroptotic defense that is particularly
pronounced in lung adenocarcinoma progression (136). Beyond cystine import, the
ferroptosis evasion machinery encompasses the upregulation of
critical antioxidant enzymes, notably GSH and GPX4, which are
consistently upregulated across multiple tumor types (137,138). Complementing these classical
antioxidant systems, cancer cells also exploit radical-trapping
antioxidant mechanisms mediated by FSP1 and GCH1, both of which are
frequently upregulated in diverse malignancies and markedly
contribute to ferroptosis resistance (21,139). Furthermore, NRF2 activation, a
hallmark of numerous cancer types, serves as both a driver of tumor
progression and a coordinator of therapy resistance. Through its
transcriptional control of ferroptosis-regulatory components,
including SLC7A11, GPX4 and FSP1, NRF2 creates a unified resistance
program that simultaneously promotes tumor progression and confers
therapeutic resistance (140).
Notably, this mechanism of ferroptosis evasion may
be closely associated with altered lactate metabolism in tumor
cells. While lactate has been reported to promote ferroptosis in
normal cellular contexts by modulating iron homeostasis and lipid
peroxidation, tumor cells appear to exploit lactate signaling
pathways to reinforce their anti-ferroptotic defenses. This
metabolic rewiring suggests that the Warburg effect and ferroptosis
evasion may be mechanistically linked, representing complementary
adaptive strategies that collectively support malignant
transformation and tumor progression.
Crosstalk between the lactate-ferroptosis
axis and other cell death modalities
The lactate-ferroptosis axis does not function
independently but rather integrates with other forms of programmed
cell death, constituting a complex regulatory network that
modulates therapeutic outcomes. Growing evidence suggests that
lactate metabolism exerts regulatory control over multiple cell
death modalities, with the dominant pathway contingent upon the
specific cellular context and magnitude of stress stimuli.
Lactate-mediated coordination of
ferroptosis and apoptosis
In cancer cells, increased lactate concentrations
have been reported to suppress apoptosis through HIF-1α-mediated
upregulation of anti-apoptotic proteins, including BCL-2 and
survivin, while concurrently promoting ferroptosis resistance via
SCD1 upregulation (58,141). This coordinated suppression of
apoptotic and ferroptotic pathways confers a metabolic survival
advantage under stress conditions. Conversely, in certain
therapeutic contexts, lactate depletion strategies have been
demonstrated to induce a synergistic activation of both apoptotic
and ferroptotic pathways. For instance, MCT1 inhibition in
glycolytic tumors induces a metabolic catastrophe that triggers
caspase-dependent apoptosis alongside GSH depletion-mediated
ferroptosis, thereby yielding enhanced antitumor efficacy relative
to the activation of either pathway alone (142). A study has demonstrated that
GPX4 inhibition can simultaneously activate the caspase-8-mediated
apoptotic pathway and ferroptosis, suggesting shared upstream
signaling mechanisms (143).
Furthermore, the tumor suppressor p53 serves as a critical node
connecting these pathways as it can promote ferroptosis via SLC7A11
repression while simultaneously regulating apoptotic gene
expression (144).
Autophagy as a double-edged sword in
lactate-ferroptosis
The interplay between lactate, autophagy and
ferroptosis is notably intricate. Lactate has been reported to
induce protective autophagy in retinal pigment epithelial cells,
mitigating oxidative stress and cell death through AMPK activation
(101,145). However, in the context of
ferroptosis, selective autophagy pathways such as ferritinophagy
(autophagic degradation of ferritin) and lipophagy (degradation of
lipid droplets) can paradoxically facilitate ferroptosis by
elevating the LIP and releasing PUFAs for peroxidation (146,147). Studies have demonstrated that
lactate-induced autophagy activation can dictate cellular fate
between survival and ferroptotic death based on the iron-handling
capacity and antioxidant reserve of specific cell types (101,119,148). In Alzheimer's disease,
diminished tau lactylation suppresses ferritinophagy, resulting in
iron accumulation and altered susceptibility to ferroptosis,
highlighting the complex interplay among lactate metabolism,
autophagy and iron homeostasis (119). Additionally, clockophagy
(selective degradation of aryl hydrocarbon receptor nuclear
translocator-like protein 1, a core clock gene) has been reported
to enhance ferroptosis sensitivity by disrupting the circadian
regulation of lipid metabolism and antioxidant defenses (149).
Therapeutic approaches in
lactate-ferroptosis
As the role of lactate in disease progression
becomes increasingly evident, strategies aimed at modulating
lactate metabolism are receiving growing attention in conditions
including neurodegenerative disorders, IRI, sepsis and cancer.
These approaches encompass systemic administration of
lactate-enriched solutions to provide metabolic support, as well as
interventions aimed at inhibiting lactate production or transport
as well as promoting its depletion to disrupt metabolic symbiosis
in tumors, highlighting the dual therapeutic potential of lactate
(150,151).
Lactate in therapeutic contexts
Neurodegenerative diseases and
IRI
Increasing evidence implicates ferroptosis in the
pathogenesis of major neurodegenerative disorders, including
Alzheimer's disease, Parkinson's disease, Huntington's disease,
multiple sclerosis and amyotrophic lateral sclerosis (152). In these chronic conditions,
persistent elevations in lactate are associated with oxidative
stress and disrupted iron homeostasis, collectively promoting
ferroptotic neuronal death (153-155). Similarly, lactate accumulation
during prolonged ischemia or late reperfusion has detrimental
effects. Sun et al (98)
demonstrated that intracellular lactate overload under prolonged
ischemia inactivates the AMPK/NRF2/GPX4 protective axis, thereby
promoting myocardial ferroptosis and exacerbating cardiac injury.
By contrast, lactate exhibits striking neuroprotective properties
in acute ischemic stroke. Intraventricular administration following
reperfusion markedly reduces infarct volume and ameliorates
neurological deficits (156).
This protective action is attributed to the role of lactate as an
alternative energy substrate, whereby its conversion to pyruvate
enables mitochondrial oxidation once oxygen is restored (157). However, lactate accumulated
during the ischemic phase itself, when oxygen tension is absent,
cannot fuel oxidative metabolism. Instead, it drives protein
lactylation in ischemic tissues, a post-translational modification
that exacerbates cellular injury (158).
The neuroprotective effects of exogenous lactate
are highly dependent on both dose and timing. Following
oxygen-glucose deprivation, 4 mM lactate markedly attenuates
hippocampal neuronal death, whereas 20 mM exerts neurotoxic effects
(159). Optimal neuroprotection
is observed at ~10 mM, with concentrations below this threshold
insufficient to alleviate the metabolic crisis associated with
cerebral ischemia (160).
Similarly, perfusion of isolated mouse hearts with 20 mmol/l
L-lactate for only 15 min at reperfusion onset reduced infarct size
from 44.74 to 21.46% (161).
Clinical and preclinical studies suggest that lactate-enriched
solutions confer notable benefits in traumatic brain injury and
myocardial ischemia, including reductions in cognitive deficits,
improvements in cerebral blood flow and attenuation of reperfusion
injuries through mechanisms such as anti-inflammatory effects and
provision of alternative energy substrates (156,162). Moreover, lactate-enriched
solutions exhibit promising effects by producing a positive
inotropic response in both healthy individuals and patients with
acute heart failure, and they may further mitigate reperfusion
injuries following myocardial ischemia (163-165). Collectively, these findings
underscore the context-dependent duality of lactate: It acts as a
metabolic burden in chronic neurodegeneration and hypoxic
conditions; however, it serves as a therapeutic asset during
reperfusion when oxidative capacity is restored.
Sepsis
Elevated serum lactate levels serve as a prognostic
indicator in sepsis, with higher concentrations correlating with
increased mortality (166). A
mechanistic study has further implicated lactate in
ferroptosis-mediated lung injury during sepsis progression
(82). However, Besnier et
al (104) reported that
hypertonic sodium lactate solutions protected against cardiac
dysfunction, microcirculatory impairment and vascular leakage in
septic animals, while simultaneously attenuating inflammation and
promoting ketogenesis. This paradox, wherein lactate exhibits
context-dependent anti-inflammatory effects while promoting
ferroptosis, represents a significant challenge for its clinical
translation.
Targeting lactate therapy in tumor
Lactate production inhibition
Given its role in catalyzing the conversion of
pyruvate to lactate in glycolytic cancer cells and the observation
that LDHA deficiency results in only relatively mild symptoms, such
as exertional myopathy, LDHA represents a pivotal and safe
therapeutic target (150,167). Inhibitors, including gossypol
(AT-101) and its derivative FX-11, have demonstrated efficacy in
preclinical and early clinical studies, while other molecules, such
as galloflavin, oxamate, quinoline derivatives and
N-hydroxyindole-based compounds, demonstrate potential in
suppressing tumor progression (168-173). Despite these promising
findings, challenges, including isoform-specific expression (LDHA
vs. LDHB) and tumor metabolic plasticity, have limited their
broader application (174).
Targeting other metabolic enzymes, including hexokinase 2 and PDH
kinase 1, using agents such as 2-deoxy-D-glucose and
dichloroacetate (DCA), has demonstrated potential in reducing
lactate production and suppressing tumor growth (175,176).
Nanomedicines, owing to their nanoscale size and
multi-functional design, facilitate precise tumor targeting,
controlled drug release and enhanced bioavailability, thereby
offering innovative solutions to modulate tumor lactate metabolism
and amplify antitumor efficacy (150). For example, Zhang et al
(177) developed PMVL, a
lonidamine-loaded nanoplatform that enhances ferroptosis and immune
activation through dual inhibition of glycolysis and the PPP,
effectively reducing lactate production.
Lactate clearance enhancement
Lactate oxidase provides a direct strategy by
catalyzing lactate oxidation to pyruvate and
H2O2, reducing lactate levels, exacerbating
tumor hypoxia and increasing oxidative stress (150). This approach enhances antitumor
immune responses and activates hypoxia-sensitive prodrugs. A recent
study reported that an engineered biohybrid of DH5α Escherichia
coli with hypoxia-inducible lactate oxidase and iron-doped
zeolitic imidazolate framework-8 nanoparticles enables targeted
lactate depletion, immune activation and ferroptosis, significantly
inhibiting tumor growth and metastasis (178). However, systemic administration
of lactate oxidase carries the risk of off-target
H2O2 toxicity, underscoring the need for
tumor-specific delivery systems to improve safety and therapeutic
outcomes.
MCT-mediated transport inhibition
Inhibiting MCT-mediated lactate transport offers
another avenue to disrupt tumor metabolic symbiosis. MCT1
inhibition compels cancer cells to compete for glucose, thereby
inducing apoptosis in hypoxic cancer cells, while MCT4 inhibition
triggers intracellular acidosis under hypoxic conditions (142). Early inhibitors
(hydroxycinnamate and lonidamine) lacked isoform selectivity,
limiting their clinical potential (179,180). However, next-generation
inhibitors, such as AZD3965, AR-C155858, SR13800 and VB124,
demonstrate improved specificity, with AZD3965 progressing to phase
I trials (111,181-183). Additionally, CD147-targeting
therapies, including anti-CD147 antibodies, modulate MCT1/4 surface
expression, although off-target effects remain a concern (184). Certain statin drugs demonstrate
MCT4 inhibitory activity, with lipophilic statins exhibiting
greater potency compared with their hydrophilic counterparts
(185,186). Recently, Chen et al
(187) developed a folic
acid-decorated, manganese dioxide-coated mesoporous silica
nanoparticle to co-deliver fluvastatin sodium (MCT4 inhibitor) and
metformin, effectively targeting tumor lactate metabolism by
promoting lactate production and inhibiting lactate efflux, thereby
exacerbating intracellular acidosis and inducing cancer cell death.
This nanomedicine demonstrated enhanced antitumor efficacy,
suppressed tumor cell migration and suppressed metastasis by
disrupting the MCT4-mediated lactate shuttling in breast cancer
models. Targeting lactate metabolism has therefore emerged as a
promising therapeutic approach in oncology, and the aforementioned
strategies are summarized in Table
II.
| Table IITargeting lactate in tumor
therapeutic strategies. |
Table II
Targeting lactate in tumor
therapeutic strategies.
A, Targeting
lactate production
|
|---|
| Targeted drugs | Mechanism | Research
development | (Refs.) |
|---|
| AT-101 | Inhibits LDH | Phase Ⅱ clinical
trials | (168) |
| FX-11 | Inhibits LDHA | Preclinical
research | (169) |
| Oxamate | Inhibits LDHA | Preclinical
research | (170) |
| Galloflavin | Inhibits LDHA | Preclinical
research | (171) |
| LDHA short
interfering RNA | Suppresses LDHA
expression | Preclinical
research | (204) |
|
N-hydroxyindole-based compounds | Inhibits LDHA
activity | Preclinical
research | (172) |
| Quinoline
derivatives | Inhibits LDHA | Preclinical
research | (173) |
| 2-DG | Inhibits HK2 | Phase I clinical
trials | (175) |
| DCA | Inhibits PDK1 | Preclinical
research | (176) |
|
| B, Targeting
lactate transport |
|
| AZD3965 | Blocks MCT1 | Phase I clinical
trials | (181) |
| AR-C155858 | Blocks MCT1 | Preclinical
research | (182) |
| SR13800 | Blocks MCT1 | Preclinical
research | (183) |
| VB124 | Inhibits MCT4 | Preclinical
research | (111) |
| Fluvastatin
Sodium | Inhibits MCT4 | Preclinical
research | (185) |
| Lonidamine | Inhibits MCT
activity | Preclinical/early
clinical research | (179,180) |
|
| C, Enhancing
lactate depletion |
|
| Lactate
oxidase | Catalyzes lactate
oxidation | Preclinical
research | (205) |
| Biohybrid Materials
(e.g., iron-doped ZIF-8 nanoparticles) | Enables targeted
lactate depletion via lactate oxidase delivery | Animal models | (178) |
Targeting lactate therapy in
neurodegenerative diseases, IRI and sepsis
Therapeutic approaches aimed at modulating lactate
metabolism have demonstrated some potential in regulating
ferroptosis in neurodegenerative diseases, IRI and sepsis. However,
their effectiveness is critically context-dependent, varying
according to the metabolic characteristics of the specific tissues
and cell types (48,188,189). Approaches that suppress lactate
production, such as using PDK inhibitors (DCA and thiamine), have
been demonstrated to alleviate organ damage and mitigate
ferroptosis in sepsis by reversing the pathological Warburg
effect-like state, thereby improving mitochondrial function and
reducing ROS accumulation (189,190). Conversely, in myocardial IRI,
activation of LDHA has been found to phosphorylate and stabilize
the ferroptosis-suppressing enzyme GPX4 via its kinase activity,
suggesting that direct inhibition of LDHA could be detrimental
(48).
Enhancing lactate clearance has also emerged as a
promising strategy in sepsis, as increased lactate elimination has
been positively correlated with improved clinical outcomes in
patients with sepsis (191).
Beyond merely correcting metabolic acidosis, this approach also
restricts lactate-driven histone lactylation, which influences the
epigenetic regulation of TFRC expression and ferroptosis
susceptibility (73,192). By contrast, controlled
utilization of lactate, such as lactate post-conditioning, has been
demonstrated to exert neuroprotective effects in cerebral IRI
(156).
Inhibition of MCT-mediated lactate shuttling has
demonstrated cell-type-specific therapeutic potential. For
instance, blocking lactate efflux via MCT4, using agents such as
VB124, can redirect pyruvate toward mitochondrial oxidation,
thereby conferring cardioprotection in myocardial IRI (193). Meanwhile, MCT1 inhibitors,
including AZD3965, have been revealed to regulate immune responses
and significantly reduce mortality in septic mice by promoting
neutrophil apoptosis (194).
However, MCT1-mediated lactate transport remains crucial for
sustaining energy homeostasis and preserving tissue health in
neurons and pulmonary epithelial cells under comparable conditions
(195,196).
In summary, modulation of lactate metabolism
represents a promising avenue for managing ferroptosis in disease.
Nevertheless, future translational studies must focus on developing
drugs with high tissue specificity and isoform-selective activity
to account for the metabolic duality inherent to lactate
biology.
Conclusions and perspectives
Regulated cell death and metabolic homeostasis
constitute fundamental determinants of cellular development and
growth. In the present review, comprehensive analysis revealed a
particularly intriguing phenomenon: Lactate exhibits diametrically
contrasting effects on ferroptosis, promoting cell death in normal
cells while conferring protection in tumor cells. This apparent
paradox, largely attributed to context-dependent mechanisms, has
remained incompletely understood.
Several critical research gaps remain to be
addressed. The molecular mechanism underlying lactate-induced
downregulation of ACSL4 via MCT1 remains incompletely elucidated,
although accumulating evidence suggests a potential involvement of
the lactate-induced STAT3 signaling pathway (197,198). Notably, MCT1 blockade in HCC
cells promotes ferroptosis without observable alterations in GPX4
and FSP1 expression levels, while lactate uptake in melanoma cells
via MCT1 elevates NADH, NADPH, GPX4 and FSP1. This paradox may be
explained by enzymatic dysfunction rather than transcriptional
downregulation. In neurodegenerative contexts, the notable
metabolic differences between immature and mature neurons indicate
that the lactate-ferroptosis axis during neurogenesis is largely
unexplored, with potential implications for neurodevelopmental
disorders (199-201). Similarly, in IRI and sepsis,
studies lack systematic investigation of temporal exposure windows,
a critical limitation given that prolonged lactate exposure may
trigger adaptive responses or cumulative toxic effects. While Wu
et al (202) recently
addressed lactate-regulated ferroptosis in cancer contexts and
acknowledged the context-dependent nature of this phenomenon, their
analysis did not elucidate the mechanistic determinants underlying
this differential regulation. The present review addresses this
knowledge gap by identifying three key context-dependent mechanisms
governing the dual role of lactate: Metabolic enzyme expression
patterns, pH homeostasis capacity and antioxidant defense
capabilities. Additionally, the present review explored the
therapeutic potential of lactate in non-tumor diseases and
elucidated its crosstalk with other cell death pathways. Although
lactylation modifications have been demonstrated to regulate
ferroptosis across diverse experimental models, it remains unclear
whether these mechanisms are restricted to the specific disease
contexts and models studied or can be universally extended to all
cellular systems.
The therapeutic implications are profound yet
complex. Strategies aimed at disrupting lactate-mediated
ferroptosis resistance in tumors while reducing lactate-mediated
ferroptosis in sepsis may offer selective therapeutic windows.
However, in IRI, the dual nature of lactate, providing a protective
effect through energy supply while simultaneously promoting
ferroptotic damage, poses significant challenges for its clinical
application. In the nervous system, interventions harnessing the
neuroprotective properties of lactate while mitigating
pro-ferroptotic effects offer novel approaches (203).
Future research directions must prioritize three
key directions: First, investigating the triggering factors of
lactate-ferroptosis interactions across diverse biological contexts
to enable the design of tissue-specific lactate-targeting
therapeutic interventions; second, developing spatiotemporally
resolved analytical techniques to identify predictive biomarkers
that can distinguish between different ferroptotic responses; and
third, leveraging advanced imaging technologies, metabolomics
approaches and novel biosensors to provide essential tools for
tracking lactate-ferroptosis dynamics in vivo.
In conclusion, the lactate-ferroptosis network
represents a sophisticated cellular regulatory mechanism that
integrates metabolic status with cell fate determination.
Translating these mechanistic insights into targeted therapeutic
strategies across diverse disease contexts, from cancer to
neurodegeneration, constitutes a pivotal future challenge,
requiring interdisciplinary collaboration and innovative
experimental approaches.
Availability of data and materials
Not applicable.
Authors' contributions
QY and HY discussed the structure of the article
and completed the manuscript; YK, JH and LY provided technical and
material support and completed the manuscript. XL provided
suggestions for revision. All authors read and approved the final
version of the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, artificial
intelligence tools (Claude 4.5 Sonnet) to improve the readability
and language of the manuscript or to generate images, and
subsequently, the authors revised and edited the content produced
by the artificial intelligence tools as necessary, taking full
responsibility for the ultimate content of the present
manuscript.
Acknowledgements
Not applicable.
Funding
This work was supported by National Natural Science Foundation
of China (grant no. 824B2063).
References
|
1
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dixon SJ and Olzmann JA: The cell biology
of ferroptosis. Nat Rev Mol Cell Biol. 25:424–442. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mishima E, Nakamura T, Doll S, Proneth B,
Fedorova M, Pratt DA, Friedmann Angeli JP, Dixon SJ, Wahida A and
Conrad M: Recommendations for robust and reproducible research on
ferroptosis. Nat Rev Mol Cell Biol. 26:615–630. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou Q, Meng Y, Li D, Yao L, Le J, Liu Y,
Sun Y, Zeng F, Chen X and Deng G: Ferroptosis in cancer: From
molecular mechanisms to therapeutic strategies. Signal Transduct
Target Ther. 9:552024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li X, Yang Y, Zhang B, Lin X, Fu X, An Y,
Zou Y, Wang JX, Wang Z and Yu T: Lactate metabolism in human health
and disease. Signal Transduct Target Ther. 7:3052022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brooks GA: The science and translation of
lactate shuttle theory. Cell Metab. 27:757–785. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Felmlee MA, Jones RS, Rodriguez-Cruz V,
Follman KE and Morris ME: Monocarboxylate transporters (SLC16):
Function, regulation, and role in health and disease. Pharmacol
Rev. 72:466–485. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brown TP and Ganapathy V: Lactate/GPR81
signaling and proton motive force in cancer: Role in angiogenesis,
immune escape, nutrition, and Warburg phenomenon. Pharmacol Ther.
206:1074512020. View Article : Google Scholar
|
|
9
|
Zhang D, Tang Z, Huang H, Zhou G, Cui C,
Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al: Metabolic
regulation of gene expression by histone lactylation. Nature.
574:575–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hua S, Jeong HN, Dimapasoc LM, Kang I, Han
C, Choi JS, Lebrilla CB and An HJ: Isomer-specific LC/MS and
LC/MS/MS profiling of the mouse serum N-glycome revealing a number
of novel sialylated N-glycans. Anal Chem. 85:4636–4643. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li H, Sun L, Gao P and Hu H: Lactylation
in cancer: Current understanding and challenges. Cancer Cell.
42:1803–1807. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu YQ, Yang Q and He GW:
Post-translational acylation of proteins in cardiac hypertrophy.
Nat Rev Cardiol. 22:944–960. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Conrad M and Pratt DA: The chemical basis
of ferroptosis. Nat Chem Biol. 15:1137–1147. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liang D, Minikes AM and Jiang X:
Ferroptosis at the intersection of lipid metabolism and cellular
signaling. Mol Cell. 82:2215–2227. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Badgley MA, Kremer DM, Maurer HC,
DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J,
Firl CEM, et al: Cysteine depletion induces pancreatic tumor
ferroptosis in mice. Science. 368:85–89. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bersuker K, Hendricks JM, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Doll S, Freitas FP, Shah R, Aldrovandi M,
da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius
E, Scheel CH, et al: FSP1 is a glutathione-independent ferroptosis
suppressor. Nature. 575:693–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee
H, Koppula P, Wu S, Zhuang L, Fang B, et al: DHODH-mediated
ferroptosis defence is a targetable vulnerability in cancer.
Nature. 593:586–590. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang D, Liang W, Huo D, Wang H, Wang Y,
Cong C, Zhang C, Yan S, Gao M, Su X, et al: SPY1 inhibits neuronal
ferroptosis in amyotrophic lateral sclerosis by reducing lipid
peroxidation through regulation of GCH1 and TFR1. Cell Death
Differ. 30:369–382. 2023. View Article : Google Scholar :
|
|
21
|
Kraft VAN, Bezjian CT, Pfeiffer S,
Ringelstetter L, Müller C, Zandkarimi F, Merl-Pham J, Bao X,
Anastasov N, Kössl J, et al: GTP cyclohydrolase
1/tetrahydrobiopterin counteract ferroptosis through lipid
remodeling. ACS Cent Sci. 6:41–53. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kappler A, Bryce C, Mansor M, Lueder U,
Byrne JM and Swanner ED: An evolving view on biogeochemical cycling
of iron. Nat Rev Microbiol. 19:360–374. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cañeque T, Baron L, Müller S, Carmona A,
Colombeau L, Versini A, Solier S, Gaillet C, Sindikubwabo F,
Sampaio JL, et al: Activation of lysosomal iron triggers
ferroptosis in cancer. Nature. 642:492–500. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Magtanong L, Ko PJ and Dixon SJ: Emerging
roles for lipids in non-apoptotic cell death. Cell Death Differ.
23:1099–1109. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schwörer S, Vardhana SA and Thompson CB:
Cancer metabolism drives a stromal regenerative response. Cell
Metab. 29:576–591. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Won W, Bhalla M, Lee JH and Lee CJ:
Astrocytes as key regulators of neural signaling in health and
disease. Annu Rev Neurosci. 48:251–276. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Certo M, Tsai CH, Pucino V, Ho PC and
Mauro C: Lactate modulation of immune responses in inflammatory
versus tumour microenvironments. Nat Rev Immunol. 21:151–161. 2021.
View Article : Google Scholar
|
|
28
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metab. 23:27–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rabinowitz JD and Enerbäck S: Lactate: The
ugly duckling of energy metabolism. Nat Metab. 2:566–571. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Y and Patti GJ: The Warburg effect: A
signature of mitochondrial overload. Trends Cell Biol.
33:1014–1020. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hui S, Ghergurovich JM, Morscher RJ, Jang
C, Teng X, Lu W, Esparza LA, Reya T, Zhan L, Yanxiang Guo J, et al:
Glucose feeds the TCA cycle via circulating lactate. Nature.
551:115–118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Faubert B, Li KY, Cai L, Hensley CT, Kim
J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al:
Lactate metabolism in human lung tumors. Cell. 171:358–371.e9.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Y, Huang Y, Yang J, Zhou FQ, Zhao L
and Zhou H: Pyruvate is a prospective alkalizer to correct hypoxic
lactic acidosis. Mil Med Res. 5:132018.PubMed/NCBI
|
|
34
|
DeBerardinis RJ, Mancuso A, Daikhin E,
Nissim I, Yudkoff M, Wehrli S and Thompson CB: Beyond aerobic
glycolysis: Transformed cells can engage in glutamine metabolism
that exceeds the requirement for protein and nucleotide synthesis.
Proc Natl Acad Sci USA. 104:19345–19350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ivashkiv LB: The hypoxia-lactate axis
tempers inflammation. Nat Rev Immunol. 20:85–86. 2020. View Article : Google Scholar :
|
|
37
|
Taylor CT and Scholz CC: The effect of HIF
on metabolism and immunity. Nat Rev Nephrol. 18:573–587. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang GL and Semenza GL: General
involvement of hypoxia-inducible factor 1 in transcriptional
response to hypoxia. Proc Natl Acad Sci USA. 90:4304–4308. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Silagi ES, Schipani E, Shapiro IM and
Risbud MV: The role of HIF proteins in maintaining the metabolic
health of the intervertebral disc. Nat Rev Rheumatol. 17:426–439.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Iyer NV, Kotch LE, Agani F, Leung SW,
Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY
and Semenza GL: Cellular and developmental control of O2
homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev.
12:149–162. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Seagroves TN, Ryan HE, Lu H, Wouters BG,
Knapp M, Thibault P, Laderoute K and Johnson RS: Transcription
factor HIF-1 is a necessary mediator of the pasteur effect in
mammalian cells. Mol Cell Biol. 21:3436–3444. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Papandreou I, Cairns RA, Fontana L, Lim AL
and Denko NC: HIF-1 mediates adaptation to hypoxia by actively
downregulating mitochondrial oxygen consumption. Cell Metab.
3:187–197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kim JW, Tchernyshyov I, Semenza GL and
Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase
kinase: A metabolic switch required for cellular adaptation to
hypoxia. Cell Metab. 3:177–185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Claps G, Faouzi S, Quidville V, Chehade F,
Shen S, Vagner S and Robert C: The multiple roles of LDH in cancer.
Nat Rev Clin Oncol. 19:749–762. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ždralević M, Brand A, Di Ianni L, Dettmer
K, Reinders J, Singer K, Peter K, Schnell A, Bruss C, Decking SM,
et al: Double genetic disruption of lactate dehydrogenases A and B
is required to ablate the 'Warburg effect' restricting tumor growth
to oxidative metabolism. J Biol Chem. 293:15947–15961. 2018.
View Article : Google Scholar
|
|
46
|
Kim EY, Chung TW, Han CW, Park SY, Park
KH, Jang SB and Ha KT: A novel lactate dehydrogenase inhibitor,
1-(Phenylseleno)-4-(Trifluoromethyl) benzene, suppresses tumor
growth through apoptotic cell death. Sci Rep. 9:39692019.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Park JS, Saeed K, Jo MH, Kim MW, Lee HJ,
Park CB, Lee G and Kim MO: LDHB deficiency promotes mitochondrial
dysfunction mediated oxidative stress and neurodegeneration in
adult mouse brain. Antioxidants (Basel). 11:2612022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yin M, Li S, Liu M, Zhu W, Chen Y, Qiu W,
Li Q, Li Y, Chen J, Zhou Y, et al: GUCY1A1-LDHA axis suppresses
ferroptosis in cardiac ischemia-reperfusion injury. Circ Res.
137:986–1005. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhao L, Deng H, Zhang J, Zamboni N, Yang
H, Gao Y, Yang Z, Xu D, Zhong H, van Geest G, et al: Lactate
dehydrogenase B noncanonically promotes ferroptosis defense in
KRAS-driven lung cancer. Cell Death Differ. 32:632–645. 2025.
View Article : Google Scholar :
|
|
50
|
Doherty JR, Yang C, Scott KE, Cameron MD,
Fallahi M, Li W, Hall MA, Amelio AL, Mishra JK, Li F, et al:
Blocking lactate export by inhibiting the Myc target MCT1 disables
glycolysis and glutathione synthesis. Cancer Res. 74:908–920. 2014.
View Article : Google Scholar :
|
|
51
|
Wang N, Wang W, Wang X, Mang G, Chen J,
Yan X, Tong Z, Yang Q, Wang M, Chen L, et al: Histone lactylation
boosts reparative gene activation post-myocardial infarction. Circ
Res. 131:893–908. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ahmed K, Tunaru S, Tang C, Müller M, Gille
A, Sassmann A, Hanson J and Offermanns S: An autocrine lactate loop
mediates insulin-dependent inhibition of lipolysis through GPR81.
Cell Metab. 11:311–319. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Roland CL, Arumugam T, Deng D, Liu SH,
Philip B, Gomez S, Burns WR, Ramachandran V, Wang H,
Cruz-Monserrate Z and Logsdon CD: Cell surface lactate receptor
GPR81 is crucial for cancer cell survival. Cancer Res.
74:5301–5310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lee YJ, Shin KJ, Park SA, Park KS, Park S,
Heo K, Seo YK, Noh DY, Ryu SH and Suh PG: G-protein-coupled
receptor 81 promotes a malignant phenotype in breast cancer through
angiogenic factor secretion. Oncotarget. 7:70898–70911. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Feng J, Yang H, Zhang Y, Wei H, Zhu Z, Zhu
B, Yang M, Cao W, Wang L and Wu Z: Tumor cell-derived lactate
induces TAZ-dependent upregulation of PD-L1 through GPR81 in human
lung cancer cells. Oncogene. 36:5829–5839. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Luo M, Zhu J, Ren J, Tong Y, Wang L, Ma S
and Wang J: Lactate increases tumor malignancy by promoting tumor
small extracellular vesicles production via the
GPR81-cAMP-PKA-HIF-1α axis. Front Oncol. 12:10365432022. View Article : Google Scholar
|
|
57
|
Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z,
Cai K, Zhao Y and Luo Z: HCAR1/MCT1 regulates tumor ferroptosis
through the lactate-mediated AMPK-SCD1 activity and its therapeutic
implications. Cell Rep. 33:1084872020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang P, Li H, Sun M, Guo X, Liao Y, Hu M,
Ye P and Liu R: Zinc deficiency drives ferroptosis resistance by
lactate production in esophageal squamous cell carcinoma. Free
Radic Biol Med. 213:512–522. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Benjamin D, Robay D, Hindupur SK, Pohlmann
J, Colombi M, El-Shemerly MY, Maira SM, Moroni C, Lane HA and Hall
MN: Dual inhibition of the lactate transporters MCT1 and MCT4 Is
synthetic lethal with metformin due to NAD+ depletion in cancer
cells. Cell Rep. 25:3047–3058.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang S, Liu W, Ganz T and Liu S:
Exploring the relationship between hyperlactatemia and anemia.
Trends Endocrinol Metab. 35:300–307. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sim M, Garvican-Lewis LA, Cox GR, Govus A,
McKay AKA, Stellingwerff T and Peeling P: Iron considerations for
the athlete: A narrative review. Eur J Appl Physiol. 119:1463–1478.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Nemeth E, Tuttle MS, Powelson J, Vaughn
MB, Donovan A, Ward DM, Ganz T and Kaplan J: Hepcidin regulates
cellular iron efflux by binding to ferroportin and inducing its
internalization. Science. 306:2090–2093. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu W, Zhang S, Li Q, Wu Y, Jia X, Feng W,
Li Z, Shi Y, Hou Q, Ma J, et al: Lactate modulates iron metabolism
by binding soluble adenylyl cyclase. Cell Metab. 35:1597–1612.e6.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu W, Wu Y, Wei H, Ma J, Feng W, Yang Q,
Zhang S, Ganz T and Liu S: Lactate administration improves
laboratory parameters in murine models of iron overload. Blood.
143:1045–1049. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang J, Wang Y, Fan M, Guan Y, Zhang W,
Huang F, Zhang Z, Li X, Yuan B, Liu W, et al: Reactive oxygen
species regulation by NCF1 governs ferroptosis susceptibility of
Kupffer cells to MASH. Cell Metab. 36:1745–1763.e6. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Davaanyam D, Lee H, Seol SI, Oh SA, Kim SW
and Lee JK: HMGB1 induces hepcidin upregulation in astrocytes and
causes an acute iron surge and subsequent ferroptosis in the
postischemic brain. Exp Mol Med. 55:2402–2416. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Davaanyam D, Seol SI, Oh SA, Lee H and Lee
JK: Hepatocyte activation and liver injury following cerebral
ischemia promote HMGB1-mediated hepcidin upregulation in
hepatocytes and regulation of systemic iron levels. Exp Mol Med.
56:2171–2183. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Li A, Gong Z, Long Y, Li Y, Liu C, Lu X,
Li Q, He X, Lu H, Wu K, et al: Lactylation of LSD1 is an acquired
epigenetic vulnerability of BRAFi/MEKi-resistant melanoma. Dev
Cell. 60:1974–1990.e11. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhang K, Guo L, Li X, Hu Y and Luo N:
Cancer-associated fibroblasts promote doxorubicin resistance in
triple-negative breast cancer through enhancing ZFP64 histone
lactylation to regulate ferroptosis. J Transl Med. 23:2472025.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang L, Wang X, Che W, Zhou S and Feng Y:
METTL3 silenced inhibited the ferroptosis development via
regulating the TFRC levels in the intracerebral hemorrhage
progression. Brain Res. 1811:1483732023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Gong F, Zheng X, Xu W, Xie R, Liu W, Pei
L, Zhong M, Shi W, Qu H, Mao E, et al: H3K14la drives endothelial
dysfunction in sepsis-induced ARDS by promoting
SLC40A1/transferrin-mediated ferroptosis. MedComm. 6:e700492025.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Martinez-Outschoorn UE, Peiris-Pagés M,
Pestell RG, Sotgia F and Lisanti MP: Cancer metabolism: A
therapeutic perspective. Nat Rev Clin Oncol. 14:11–31. 2017.
View Article : Google Scholar
|
|
75
|
Zheng J and Conrad M: Ferroptosis: When
metabolism meets cell death. Physiol Rev. 105:651–706. 2025.
View Article : Google Scholar
|
|
76
|
de Kivit S, Mensink M, Kostidis S, Derks
RJE, Zaal EA, Heijink M, Verleng LJ, de Vries E, Schrama E,
Blomberg N, et al: Immune suppression by human thymus-derived
effector Tregs relies on glucose/lactate-fueled fatty acid
synthesis. Cell Rep. 43:1146812024. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Li C, Dong X, Du W, Shi X, Chen K, Zhang W
and Gao M: LKB1-AMPK axis negatively regulates ferroptosis by
inhibiting fatty acid synthesis. Signal Transduct Target Ther.
5:1872020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Song X, Liu J, Kuang F, Chen X, Zeh HJ
III, Kang R, Kroemer G, Xie Y and Tang D: PDK4 dictates metabolic
resistance to ferroptosis by suppressing pyruvate oxidation and
fatty acid synthesis. Cell Rep. 34:1087672021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sondermeijer BM, Battjes S, van Dijk TH,
Ackermans MT, Serlie MJ, Nieuwdorp M, Groen AK, Dallinga-Thie GM
and Stroes ES: Lactate increases hepatic secretion of
VLDL-triglycerides in humans. Atherosclerosis. 228:443–450. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Fan Z, Ye M, Liu D, Zhou W, Zeng T, He S
and Li Y: Lactate drives the ESM1-SCD1 axis to inhibit the
antitumor CD8+ T-cell response by activating the
Wnt/β-catenin pathway in ovarian cancer cells and inducing
cisplatin resistance. Int Immunopharmacol. 137:1124612024.
View Article : Google Scholar
|
|
82
|
Wu D, Spencer CB, Ortoga L, Zhang H and
Miao C: Histone lactylation-regulated METTL3 promotes ferroptosis
via m6A-modification on ACSL4 in sepsis-associated lung injury.
Redox Biol. 74:1031942024. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Pope LE and Dixon SJ: Regulation of
ferroptosis by lipid metabolism. Trends Cell Biol. 33:1077–1087.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tilton WM, Seaman C, Carriero D and
Piomelli S: Regulation of glycolysis in the erythrocyte: Role of
the lactate/pyruvate and NAD/NADH ratios. J Lab Clin Med.
118:146–152. 1991.PubMed/NCBI
|
|
85
|
Quinn WJ III, Jiao J, TeSlaa T, Stadanlick
J, Wang Z, Wang L, Akimova T, Angelin A, Schäfer PM, Cully MD, et
al: Lactate limits T cell proliferation via the NAD(H) redox state.
Cell Rep. 33:1085002020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Luengo A, Li Z, Gui DY, Sullivan LB,
Zagorulya M, Do BT, Ferreira R, Naamati A, Ali A, Lewis CA, et al:
Increased demand for NAD+ relative to ATP drives aerobic
glycolysis. Mol Cell. 81:691–707.e6. 2021. View Article : Google Scholar
|
|
87
|
Corkey BE and Deeney JT: The redox
communication network as a regulator of metabolism. Front Physiol.
11:5677962020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yang S and Lian G: ROS and diseases: Role
in metabolism and energy supply. Mol Cell Biochem. 467:1–12. 2020.
View Article : Google Scholar :
|
|
89
|
Young A, Oldford C and Mailloux RJ:
Lactate dehydrogenase supports lactate oxidation in mitochondria
isolated from different mouse tissues. Redox Biol. 28:1013392020.
View Article : Google Scholar
|
|
90
|
Bauzá-Thorbrügge M, Peris E, Zamani S,
Micallef P, Paul A, Bartesaghi S, Benrick A and Wernstedt Asterholm
I: NRF2 is essential for adaptative browning of white adipocytes.
Redox Biol. 68:1029512023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Jia L, Liao M, Mou A, Zheng Q, Yang W, Yu
Z, Cui Y, Xia X, Qin Y, Chen M and Xiao B: Rheb-regulated
mitochondrial pyruvate metabolism of Schwann cells linked to axon
stability. Dev Cell. 56:2980–2994.e6. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Rydström J: Mitochondrial NADPH,
transhydrogenase and disease. Biochim Biophys Acta. 1757:721–726.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Lin W, Lu X, Yang H, Huang L, Huang W,
Tang Y, Liu S, Wang H and Zhang Y: Metabolic heterogeneity protects
metastatic mucosal melanomas cells from ferroptosis. Int J Mol Med.
50:1242022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ying M, You D, Zhu X, Cai L, Zeng S and Hu
X: Lactate and glutamine support NADPH generation in cancer cells
under glucose deprived conditions. Redox Biol. 46:1020652021.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zhang W, Guo C, Jiang K, Ying M and Hu X:
Quantification of lactate from various metabolic pathways and
quantification issues of lactate isotopologues and isotopmers. Sci
Rep. 7:84892017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Huang YF, Wang G, Ding L, Bai ZR, Leng Y,
Tian JW, Zhang JZ, Li YQ, Ahmad, Qin YH, et al: Lactate-upregulated
NADPH-dependent NOX4 expression via HCAR1/PI3K pathway contributes
to ROS-induced osteoarthritis chondrocyte damage. Redox Biol.
67:1028672023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Taylor JP and Tse HM: The role of NADPH
oxidases in infectious and inflammatory diseases. Redox Biol.
48:1021592021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Sun F, He Y, Yang Z, Xu G, Wang R, Juan Z
and Sun X: Propofol pretreatment inhibits ferroptosis and
alleviates myocardial ischemia-reperfusion injury through the
SLC16A13-AMPK-GPX4 pathway. Biomed Pharmacother. 179:1173452024.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Cheng F, Dou J, Yang Y, Sun S, Chen R,
Zhang Z, Wei H, Li J and Wu Z: Drug-induced lactate confers
ferroptosis resistance via p38-SGK1-NEDD4L-dependent upregulation
of GPX4 in NSCLC cells. Cell Death Discov. 9:1652023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Tauffenberger A, Fiumelli H, Almustafa S
and Magistretti PJ: Lactate and pyruvate promote oxidative stress
resistance through hormetic ROS signaling. Cell Death Dis.
10:6532019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zou GP, Wang T, Xiao JX, Wang XY, Jiang
LP, Tou FF, Chen ZP, Qu XH and Han XJ: Lactate protects against
oxidative stress-induced retinal degeneration by activating
autophagy. Free Radic Biol Med. 194:209–219. 2023. View Article : Google Scholar
|
|
102
|
Ma M, Zhang Y, Pu K and Tang W:
Nanomaterial-enabled metabolic reprogramming strategies for
boosting antitumor immunity. Chem Soc Rev. 54:653–714. 2025.
View Article : Google Scholar
|
|
103
|
Lei G, Zhuang L and Gan B: Targeting
ferroptosis as a vulnerability in cancer. Nat Rev Cancer.
22:381–396. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Besnier E, Coquerel D, Kouadri G, Clavier
T, Favory R, Duburcq T, Lesur O, Bekri S, Richard V, Mulder P and
Tamion F: Hypertonic sodium lactate improves microcirculation,
cardiac function, and inflammation in a rat model of sepsis. Crit
Care. 24:3542020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zeng M, Niu Y, Huang J and Deng L:
Advances in neutrophil extracellular traps and ferroptosis in
sepsis-induced cardiomyopathy. Front Immunol. 16:15903132025.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhang T, Chen L, Kueth G, Shao E, Wang X,
Ha T, Williams DL, Li C, Fan M and Yang K: Lactate's impact on
immune cells in sepsis: Unraveling the complex interplay. Front
Immunol. 15:14834002024. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Irizarry-Caro RA, McDaniel MM, Overcast
GR, Jain VG, Troutman TD and Pasare C: TLR signaling adapter BCAP
regulates inflammatory to reparatory macrophage transition by
promoting histone lactylation. Proc Natl Acad Sci USA.
117:30628–30638. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Hu Y, He Z, Li Z, Wang Y, Wu N, Sun H,
Zhou Z, Hu Q and Cong X: Lactylation: The novel histone
modification influence on gene expression, protein function, and
disease. Clin Epigenetics. 16:722024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Su F, Xie K, He X, Orbegozo D, Hosokawa K,
Post EH, Donadello K, Taccone FS, Creteur J and Vincent JL: The
harmful effects of hypertonic sodium lactate administration in
hyperdynamic septic shock. Shock. 46:663–671. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Tang R, Xu J, Zhang B, Liu J, Liang C, Hua
J, Meng Q, Yu X and Shi S: Ferroptosis, necroptosis, and pyroptosis
in anticancer immunity. J Hematol Oncol. 13:1102020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Fang Y, Liu W, Tang Z, Ji X, Zhou Y, Song
S, Tian M, Tao C, Huang R, Zhu G, et al: Monocarboxylate
transporter 4 inhibition potentiates hepatocellular carcinoma
immunotherapy through enhancing T cell infiltration and immune
attack. Hepatology. 77:109–123. 2023. View Article : Google Scholar
|
|
112
|
Hagihara H, Shoji H, Otabi H, Toyoda A,
Katoh K, Namihira M and Miyakawa T: Protein lactylation induced by
neural excitation. Cell Rep. 37:1098202021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Sun K, Shi Y, Yan C, Wang S, Han L, Li F,
Xu X, Wang Y, Sun J, Kang Z and Shi J: Glycolysis-derived lactate
induces ACSL4 expression and lactylation to activate ferroptosis
during intervertebral disc degeneration. Adv Sci (Weinh).
12:e24161492025. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zhang T, Huang X, Feng S and Shao H:
Lactate-dependent HIF1A transcriptional activation exacerbates
severe acute pancreatitis through the ACSL4/LPCAT3/ALOX15 pathway
induced ferroptosis. J Cell Biochem. 126:e306872025. View Article : Google Scholar
|
|
115
|
Wu Q, You L, Nepovimova E, Heger Z, Wu W,
Kuca K and Adam V: Hypoxia-inducible factors: Master regulators of
hypoxic tumor immune escape. J Hematol Oncol. 15:772022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Deng J, Li Y, Yin L, Liu S, Li Y, Liao W,
Mu L, Luo X and Qin J: Histone lactylation enhances GCLC expression
and thus promotes chemoresistance of colorectal cancer stem cells
through inhibiting ferroptosis. Cell Death Dis. 16:1932025.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Huang J, Xie H, Li J, Huang X, Cai Y, Yang
R, Yang D, Bao W, Zhou Y, Li T and Lu Q: Histone lactylation drives
liver cancer metastasis by facilitating NSF1-mediated ferroptosis
resistance after microwave ablation. Redox Biol. 81:1035532025.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Yuan J, Yang M, Wu Z, Wu J, Zheng K, Wang
J, Zeng Q, Chen M, Lv T, Shi Y, et al: The lactate-primed KAT8-PCK2
axis exacerbates hepatic ferroptosis during ischemia/reperfusion
injury by reprogramming OXSM-dependent mitochondrial fatty acid
synthesis. Adv Sci (Weinh). 12:e24141412025. View Article : Google Scholar
|
|
119
|
An X, He J, Xie P, Li C, Xia M, Guo D, Bi
B, Wu G, Xu J, Yu W and Ren Z: The effect of tau K677 lactylation
on ferritinophagy and ferroptosis in Alzheimer's disease. Free
Radic Biol Med. 224:685–706. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
She H, Hu Y, Zhao G, Du Y, Wu Y, Chen W,
Li Y, Wang Y, Tan L, Zhou Y, et al: Dexmedetomidine ameliorates
myocardial ischemia-reperfusion injury by inhibiting MDH2
lactylation via regulating metabolic reprogramming. Adv Sci
(Weinh). 11:e24094992024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Niu K, Chen Z, Li M, Ma G, Deng Y, Zhang
J, Wei D, Wang J and Zhao Y: NSUN2 lactylation drives cancer cell
resistance to ferroptosis through enhancing GCLC-dependent
glutathione synthesis. Redox Biol. 79:1034792025. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Yu Y, Huang X, Liang C and Zhang P:
Evodiamine impairs HIF1A histone lactylation to inhibit
Sema3A-mediated angiogenesis and PD-L1 by inducing ferroptosis in
prostate cancer. Eur J Pharmacol. 957:1760072023. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Xiong J, Ge X, Pan D, Zhu Y, Zhou Y, Gao
Y, Wang H, Wang X, Gu Y, Ye W, et al: Metabolic reprogramming in
astrocytes prevents neuronal death through a UCHL1/PFKFB3/H4K8la
positive feedback loop. Cell Death Differ. 32:1214–1230. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Deng H, Zhao L, Ge H, Gao Y, Fu Y, Lin Y,
Masoodi M, Losmanova T, Medová M, Ott J, et al: Ubiquinol-mediated
suppression of mitochondria-associated ferroptosis is a targetable
function of lactate dehydrogenase B in cancer. Nat Commun.
16:25972025. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Moreira JD, Hamraz M, Abolhassani M, Bigan
E, Pérès S, Paulevé L, Nogueira ML, Steyaert JM and Schwartz L: The
redox status of cancer cells supports mechanisms behind the warburg
effect. Metabolites. 6:332016. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Halestrap AP and Wilson MC: The
monocarboxylate transporter family-role and regulation. IUBMB Life.
64:109–119. 2012. View Article : Google Scholar
|
|
127
|
Singh M, Afonso J, Sharma D, Gupta R and
Kumar V, Rani R, Baltazar F and Kumar V: Targeting monocarboxylate
transporters (MCTs) in cancer: How close are we to the clinics?
Semin Cancer Biol. 90:1–14. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Hosonuma M and Yoshimura K: Association
between pH regulation of the tumor microenvironment and
immunological state. Front Oncol. 13:11755632023. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Wang X, Zhao Y, Hu Y, Fei Y, Zhao Y, Xue
C, Cai K, Li M and Luo Z: Activatable biomineralized nanoplatform
remodels the intracellular environment of multidrug-resistant
tumors for enhanced ferroptosis/apoptosis therapy. Small.
17:e21022692021. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Jackson VN and Halestrap AP: The kinetics,
substrate, and inhibitor specificity of the monocarboxylate
(lactate) transporter of rat liver cells determined using the
fluorescent intracellular pH indicator,
2',7'-bis(carboxyethyl)-5(6)-carboxyfluorescein. J Biol Chem.
271:861–868. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Bozzo L, Puyal J and Chatton JY: Lactate
modulates the activity of primary cortical neurons through a
receptor-mediated pathway. PLoS One. 8:e717212013. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Yang Z, Su W, Wei X, Qu S, Zhao D, Zhou J,
Wang Y, Guan Q, Qin C, Xiang J, et al: HIF-1α drives resistance to
ferroptosis in solid tumors by promoting lactate production and
activating SLC1A1. Cell Rep. 42:1129452023. View Article : Google Scholar
|
|
133
|
Bae C, Sachs F and Gottlieb PA:
Protonation of the human PIEZO1 ion channel stabilizes
inactivation. J Biol Chem. 290:5167–5173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Jin C, Zhang DP, Lin Z, Lin YZ, Shi YF,
Dong XY, Jin MQ, Song FQ, Du ST, Feng YZ, et al: Piezo1-mediated
ferroptosis delays wound healing in aging mice by regulating the
transcriptional activity of SLC7A11 through activating
transcription factor 3. Research (Wash D C). 8:07182025.PubMed/NCBI
|
|
135
|
Koppula P, Zhang Y, Zhuang L and Gan B:
Amino acid transporter SLC7A11/xCT at the crossroads of regulating
redox homeostasis and nutrient dependency of cancer. Cancer Commun
(Lond). 38:122018.PubMed/NCBI
|
|
136
|
Hu K, Li K, Lv J, Feng J, Chen J, Wu H,
Cheng F, Jiang W, Wang J, Pei H, et al: Suppression of the
SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant
lung adenocarcinoma. J Clin Invest. 130:1752–1766. 2020. View Article : Google Scholar :
|
|
137
|
Harris IS, Treloar AE, Inoue S, Sasaki M,
Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA,
et al: Glutathione and thioredoxin antioxidant pathways synergize
to drive cancer initiation and progression. Cancer Cell.
27:211–222. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Zhang L, Hobeika CS, Khabibullin D, Yu D,
Filippakis H, Alchoueiry M, Tang Y, Lam HC, Tsvetkov P, Georgiou G,
et al: Hypersensitivity to ferroptosis in chromophobe RCC is
mediated by a glutathione metabolic dependency and cystine import
via solute carrier family 7 member 11. Proc Natl Acad Sci USA.
119:e21228401192022. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Koppula P, Lei G, Zhang Y, Yan Y, Mao C,
Kondiparthi L, Shi J, Liu X, Horbath A, Das M, et al: A targetable
CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1
inactive lung cancers. Nat Commun. 13:22062022. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Rojo de la Vega M, Chapman E and Zhang DD:
NRF2 and the hallmarks of cancer. Cancer Cell. 34:21–43. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Rawat SG, Tiwari RK, Jaiswara PK, Gupta
VK, Sonker P, Vishvakarma NK, Kumar S, Pathak C, Gautam V and Kumar
A: Phosphodiesterase 5 inhibitor sildenafil potentiates the
antitumor activity of cisplatin by ROS-mediated apoptosis: A role
of deregulated glucose metabolism. Apoptosis. 27:606–618. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Kobayashi M, Narumi K, Furugen A and Iseki
K: Transport function, regulation, and biology of human
monocarboxylate transporter 1 (hMCT1) and 4 (hMCT4). Pharmacol
Ther. 226:1078622021. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Ding Y, Chen X, Liu C, Ge W, Wang Q, Hao
X, Wang M, Chen Y and Zhang Q: Identification of a small molecule
as inducer of ferroptosis and apoptosis through ubiquitination of
GPX4 in triple negative breast cancer cells. J Hematol Oncol.
14:192021. View Article : Google Scholar
|
|
144
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Zhou L, Mo Y, Zhang H, Zhang M, Xu J and
Liang S: Role of AMPK-regulated autophagy in retinal pigment
epithelial cell homeostasis: A review. Medicine (Baltimore).
103:e389082024. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Chen X, Yu C, Kang R, Kroemer G and Tang
D: Cellular degradation systems in ferroptosis. Cell Death Differ.
28:1135–1148. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Zhou B, Liu J, Kang R, Klionsky DJ,
Kroemer G and Tang D: Ferroptosis is a type of autophagy-dependent
cell death. Semin Cancer Biol. 66:89–100. 2020. View Article : Google Scholar
|
|
149
|
Yang M, Chen P, Liu J, Zhu S, Kroemer G,
Klionsky DJ, Lotze MT, Zeh HJ, Kang R and Tang D: Clockophagy is a
novel selective autophagy process favoring ferroptosis. Sci Adv.
5:eaaw22382019. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Chen J, Zhu Y, Wu C and Shi J: Engineering
lactate-modulating nanomedicines for cancer therapy. Chem Soc Rev.
52:973–1000. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Annoni F, Peluso L, Gouvêa Bogossian E,
Creteur J, Zanier ER and Taccone FS: Brain protection after anoxic
brain injury: Is lactate supplementation helpful? Cells.
10:17142021. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Fei Y and Ding Y: The role of ferroptosis
in neurodegenerative diseases. Front Cell Neurosci. 18:14759342024.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Pan RY, He L, Zhang J, Liu X, Liao Y, Gao
J, Liao Y, Yan Y, Li Q, Zhou X, et al: Positive feedback regulation
of microglial glucose metabolism by histone H4 lysine 12
lactylation in Alzheimer's disease. Cell Metab. 34:634–648.e6.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Devos D, Labreuche J, Rascol O, Corvol JC,
Duhamel A, Guyon Delannoy P, Poewe W, Compta Y, Pavese N, Růžička
E, et al: Trial of deferiprone in Parkinson's disease. N Engl J
Med. 387:2045–2055. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Chen L, Shen Q, Liu Y, Zhang Y, Sun L, Ma
X, Song N and Xie J: Homeostasis and metabolism of iron and other
metal ions in neurodegenerative diseases. Signal Transduct Target
Ther. 10:312025. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Berthet C, Lei H, Thevenet J, Gruetter R,
Magistretti PJ and Hirt L: Neuroprotective role of lactate after
cerebral ischemia. J Cereb Blood Flow Metab. 29:1780–1789. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Cerina M, Levers M, Keller JM and Frega M:
Neuroprotective role of lactate in a human in vitro model of the
ischemic penumbra. Sci Rep. 14:79732024. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Xiong XY, Pan XR, Luo XX, Wang YF, Zhang
XX, Yang SH, Zhong ZQ, Liu C, Chen Q, Wang PF, et al:
Astrocyte-derived lactate aggravates brain injury of ischemic
stroke in mice by promoting the formation of protein lactylation.
Theranostics. 14:4297–4317. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Cai M, Wang H, Song H, Yang R, Wang L, Xue
X, Sun W and Hu J: Lactate Is answerable for brain function and
treating brain diseases: Energy substrates and signal molecule.
Front Nutr. 9:8009012022. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Liu Y, Hu P, Cheng H, Xu F and Ye Y: The
impact of glycolysis on ischemic stroke: From molecular mechanisms
to clinical applications. Front Neurol. 16:15143942025. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Cler M, Perez-Amodio S, Valls-Lacalle L,
Martinez E, Barba I, Ganse GFS, Engel E and Rodriguez-Sinovas A:
Abstract 4146509: L-lactic acid reduces infarct size after ischemia
in isolated mouse hearts through acidosis, MCT1-mediated uptake,
and metabolic reprogramming. Circulation. 150(Suppl1):
A41465092024. View Article : Google Scholar
|
|
162
|
Berthet C, Castillo X, Magistretti PJ and
Hirt L: New evidence of neuroprotection by lactate after transient
focal cerebral ischaemia: Extended benefit after
intracerebroventricular injection and efficacy of intravenous
administration. Cerebrovasc Dis. 34:329–335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Nalos M, Kholodniak E, Smith L, Orde S,
Ting I, Slama M, Seppelt I, McLean AS and Huang S: The comparative
effects of 3% saline and 0.5M sodium lactate on cardiac function: A
randomised, crossover study in volunteers. Crit Care Resusc.
20:124–130. 2018.PubMed/NCBI
|
|
164
|
Nalos M, Leverve X, Huang S, Weisbrodt L,
Parkin R, Seppelt I, Ting I and Mclean A: Half-molar sodium lactate
infusion improves cardiac performance in acute heart failure: A
pilot randomised controlled clinical trial. Crit Care. 18:R482014.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Koyama T: Lactated Ringer's solution for
preventing myocardial reperfusion injury. Int J Cardiol Heart Vasc.
15:1–8. 2017.PubMed/NCBI
|
|
166
|
Nolt B, Tu F, Wang X, Ha T, Winter R,
Williams DL and Li C: Lactate and immunosuppression in sepsis.
Shock. 49:120–125. 2018. View Article : Google Scholar :
|
|
167
|
Doherty JR and Cleveland JL: Targeting
lactate metabolism for cancer therapeutics. J Clin Invest.
123:3685–3692. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Sonpavde G, Matveev V, Burke JM, Caton JR,
Fleming MT, Hutson TE, Galsky MD, Berry WR, Karlov P, Holmlund JT,
et al: Randomized phase II trial of docetaxel plus prednisone in
combination with placebo or AT-101, an oral small molecule Bcl-2
family antagonist, as first-line therapy for metastatic
castration-resistant prostate cancer. Ann Oncol. 23:1803–1808.
2012. View Article : Google Scholar
|
|
169
|
Wu J, Gu X, Zhang J, Mi Z, He Z, Dong Y,
Ge W, Ghimire K, Rong P, Wang W and Ma X: 4-OI protects MIN6 cells
from oxidative stress injury by reducing LDHA-mediated ROS
generation. Biomolecules. 12:12362022. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Zhao Z, Han F, Yang S, Wu J and Zhan W:
Oxamate-mediated inhibition of lactate dehydrogenase induces
protective autophagy in gastric cancer cells: Involvement of the
Akt-mTOR signaling pathway. Cancer Lett. 358:17–26. 2015.
View Article : Google Scholar
|
|
171
|
Farabegoli F, Vettraino M, Manerba M,
Fiume L, Roberti M and Di Stefano G: Galloflavin, a new lactate
dehydrogenase inhibitor, induces the death of human breast cancer
cells with different glycolytic attitude by affecting distinct
signaling pathways. Eur J Pharm Sci. 47:729–738. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Granchi C, Roy S, Giacomelli C, Macchia M,
Tuccinardi T, Martinelli A, Lanza M, Betti L, Giannaccini G,
Lucacchini A, et al: Discovery of N-hydroxyindole-based inhibitors
of human lactate dehydrogenase isoform A (LDH-A) as starvation
agents against cancer cells. J Med Chem. 54:1599–1612. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Laganá G, Barreca D, Calderaro A and
Bellocco E: Lactate dehydrogenase inhibition: Biochemical relevance
and therapeutical potential. Curr Med Chem. 26:3242–3252. 2019.
View Article : Google Scholar
|
|
174
|
Ippolito L, Morandi A, Giannoni E and
Chiarugi P: Lactate: A metabolic driver in the tumour landscape.
Trends Biochem Sci. 44:153–166. 2019. View Article : Google Scholar
|
|
175
|
Raez LE, Papadopoulos K, Ricart AD,
Chiorean EG, Dipaola RS, Stein MN, Rocha Lima CM, Schlesselman JJ,
Tolba K, Langmuir VK, et al: A phase I dose-escalation trial of
2-deoxy-D-glucose alone or combined with docetaxel in patients with
advanced solid tumors. Cancer Chemother Pharmacol. 71:523–530.
2013. View Article : Google Scholar
|
|
176
|
Sutendra G and Michelakis ED: Pyruvate
dehydrogenase kinase as a novel therapeutic target in oncology.
Front Oncol. 3:382013. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Zhang Y, Du X, He Z, Gao S, Ye L, Ji J,
Yang X and Zhai G: A vanadium-based nanoplatform synergizing
ferroptotic-like therapy with glucose metabolism intervention for
enhanced cancer cell death and antitumor immunity. ACS Nano.
17:11537–11556. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Li F, Zhu P, Zheng B, Lu Z, Fang C, Fu Y
and Li X: A customized biohybrid presenting cascade responses to
tumor microenvironment. Adv Mater. 36:e24049012024. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Deng X, Zhu Y, Dai Z, Liu Q, Song Z, Liu
T, Huang Y and Chen H: A bimetallic nanomodulator to reverse
immunosuppression via sonodynamic-ferroptosis and lactate
metabolism modulation. Small. 20:e24045802024. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Nancolas B, Guo L, Zhou R, Nath K, Nelson
DS, Leeper DB, Blair IA, Glickson JD and Halestrap AP: The
anti-tumour agent lonidamine is a potent inhibitor of the
mitochondrial pyruvate carrier and plasma membrane monocarboxylate
transporters. Biochem J. 473:929–936. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Halford S, Veal GJ, Wedge SR, Payne GS,
Bacon CM, Sloan P, Dragoni I, Heinzmann K, Potter S, Salisbury BM,
et al: A phase I dose-escalation study of AZD3965, an oral
monocarboxylate transporter 1 inhibitor, in patients with advanced
cancer. Clin Cancer Res. 29:1429–1439. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Puri S and Juvale K: Monocarboxylate
transporter 1 and 4 inhibitors as potential therapeutics for
treating solid tumours: A review with structure-activity
relationship insights. Eur J Med Chem. 199:1123932020. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Khan A, Valli E, Lam H, Scott DA, Murray
J, Hanssen KM, Eden G, Gamble LD, Pandher R, Flemming CL, et al:
Targeting metabolic activity in high-risk neuroblastoma through
Monocarboxylate transporter 1 (MCT1) inhibition. Oncogene.
39:3555–3570. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Kirk P, Wilson MC, Heddle C, Brown MH,
Barclay AN and Halestrap AP: CD147 is tightly associated with
lactate transporters MCT1 and MCT4 and facilitates their cell
surface expression. EMBO J. 19:3896–3904. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Zhu J, Cai H, Xu C, Wang W, Song X, Li B,
Shen Y and Dong X: Acidity-responsive nanoreactors destructed
'Warburg effect' for toxic-acidosis and starvation synergistic
therapy. Small. 19:e23040582023. View Article : Google Scholar
|
|
186
|
Kobayashi M, Otsuka Y, Itagaki S, Hirano T
and Iseki K: Inhibitory effects of statins on human monocarboxylate
transporter 4. Int J Pharm. 317:19–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Chen ZX, Liu MD, Guo DK, Zou MZ, Wang SB,
Cheng H, Zhong Z and Zhang XZ: A MSN-based tumor-targeted
nanoplatform to interfere with lactate metabolism to induce tumor
cell acidosis for tumor suppression and anti-metastasis. Nanoscale.
12:2966–2972. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Minhas PS, Jones JR, Latif-Hernandez A,
Sugiura Y, Durairaj AS, Wang Q, Mhatre SD, Uenaka T, Crapser J,
Conley T, et al: Restoring hippocampal glucose metabolism rescues
cognition across Alzheimer's disease pathologies. Science.
385:eabm61312024. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Zeng Z, Huang Q, Mao L, Wu J, An S, Chen Z
and Zhang W: The pyruvate dehydrogenase complex in sepsis:
Metabolic regulation and targeted therapy. Front Nutr.
8:7831642021. View Article : Google Scholar :
|
|
190
|
Ryoo SM and Kim WY: Clinical applications
of lactate testing in patients with sepsis and septic shock. J
Emerg Crit Care Med. 2:142018. View Article : Google Scholar
|
|
191
|
Wei Y, Zhuang J, Li J, Wang Z, Wang J,
Zhang X and Leng J: Lactate trajectories and outcomes in patients
with sepsis in the intensive care unit: Group-based trajectory
modeling. Front Public Health. 13:16102202025. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Liu S, Yang T, Jiang Q, Zhang L, Shi X,
Liu X and Li X: Lactate and lactylation in sepsis: A comprehensive
review. J Inflamm Res. 17:4405–4417. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Visker JR, Cluntun AA, Velasco-Silva JN,
Eberhardt DR, Cedeño-Rosario L, Shankar TS, Hamouche R, Ling J,
Kwak H, Hillas JY, et al: Enhancing mitochondrial pyruvate
metabolism ameliorates ischemic reperfusion injury in the heart.
JCI Insight. 9:e1809062024. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Fei M, Zhang H, Meng F, An G, Tang J, Tong
J, Xiong L, Liu Q and Li C: Enhanced lactate accumulation
upregulates PD-L1 expression to delay neutrophil apoptosis in
sepsis. VIEW. 5:202300532024. View Article : Google Scholar
|
|
195
|
Suzuki A, Stern SA, Bozdagi O, Huntley GW,
Walker RH, Magistretti PJ and Alberini CM: Astrocyte-neuron lactate
transport is required for long-term memory formation. Cell.
144:810–823. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Lottes RG, Newton DA, Spyropoulos DD and
Baatz JE: Lactate as substrate for mitochondrial respiration in
alveolar epithelial type II cells. Am J Physiol Lung Cell Mol
Physiol. 308:L953–L961. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Brown CW, Amante JJ, Goel HL and Mercurio
AM: The α6β4 integrin promotes resistance to ferroptosis. J Cell
Biol. 216:4287–4297. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Pucino V, Certo M, Bulusu V, Cucchi D,
Goldmann K, Pontarini E, Haas R, Smith J, Headland SE, Blighe K, et
al: Lactate buildup at the site of chronic inflammation promotes
disease by inducing CD4+ T cell metabolic rewiring. Cell
Metab. 30:1055–1074.e8. 2019. View Article : Google Scholar
|
|
199
|
Lei P, Walker T and Ayton S:
Neuroferroptosis in health and diseases. Nat Rev Neurosci.
26:497–511. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Zheng X, Boyer L, Jin M, Mertens J, Kim Y,
Ma L, Ma L, Hamm M, Gage FH and Hunter T: Metabolic reprogramming
during neuronal differentiation from aerobic glycolysis to neuronal
oxidative phosphorylation. Elife. 5:e133742016. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Bittar PG, Charnay Y, Pellerin L, Bouras C
and Magistretti PJ: Selective distribution of lactate dehydrogenase
isoenzymes in neurons and astrocytes of human brain. J Cereb Blood
Flow Metab. 16:1079–1089. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Wu N, Wei X, Yu S, Yang L and Zhang X:
Lactate in ferroptosis regulation: A new perspective on tumor
progression and therapy. Pharmacol Res. 218:1078412025. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Kennedy L, Glesaaen ER, Palibrk V, Pannone
M, Wang W, Al-Jabri A, Suganthan R, Meyer N, Austbø ML, Lin X, et
al: Lactate receptor HCAR1 regulates neurogenesis and microglia
activation after neonatal hypoxia-ischemia. Elife. 11:e764512022.
View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Hu L, Huang S, Chen G, Li B, Li T, Lin M,
Huang Y, Xiao Z, Shuai X and Su Z: Nanodrugs incorporating LDHA
siRNA inhibit M2-like polarization of TAMs and amplify autophagy to
assist oxaliplatin chemotherapy against colorectal cancer. ACS Appl
Mater Interfaces. 14:31625–31633. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Lu H, Liang B, Hu A, Zhou H, Jia C, Aji A,
Chen Q, Ma Y, Cui W, Jiang L and Dong J: Engineered biomimetic
cancer cell membrane nanosystems trigger gas-immunometabolic
therapy for spinal-metastasized tumors. Adv Mater. 37:e24126552025.
View Article : Google Scholar
|














