Mitochondria, which serve as the 'powerhouses' of
cells, play crucial roles in cellular energy metabolism,
intracellular and extracellular signaling, reactive oxygen species
(ROS) production and apoptosis signaling regulation. Within the
digestive system-an organ characterized by high turnover rates and
metabolic activity-cells demand an exceptional energy supply and
precise regulatory control. Consequently, mitochondrial dysfunction
can trigger or exacerbate digestive disorders through multiple
mechanisms (5,6). The sirtuin family comprises seven
members: SIRT1, SIRT6 and SIRT7, which reside in the nucleus;
SIRT2, which is localized to the cytoplasm; and SIRT3-5, which are
found within mitochondria (7,8).
Among these, SIRT3 functions as an NAD+-dependent
deacetylase. Within the mitochondrial matrix, it regulates energy
balance and redox status by modifying enzymes involved in metabolic
pathways, including the tricarboxylic acid (TCA) cycle, the urea
cycle, amino acid metabolism, fatty acid oxidation (FAO) and
glucose metabolism (9). In
addition to regulating material metabolism, SIRT3 acetylation also
modulates numerous mitochondrial proteins involved in metabolic
homeostasis, oxidative stress and cell survival. SIRT3 has been
demonstrated to control mitochondrial DNA repair, participate in
maintaining mitochondrial integrity and regulate apoptosis. Of
note, SIRT3 is the only sirtuin reported to influence human
lifespan (10). Studies have
indicated that SIRT3 also regulates multiple processes critical for
mitochondrial structural integrity and function, including
oxidative phosphorylation (OXPHOS), mitochondrial dynamics, the
mitochondrial unfolded protein response and mitochondrial autophagy
(7,9,11). Thus, SIRT3 plays a vital role in
maintaining mitochondrial function and homeostasis. Concurrently,
SIRT3 is highly expressed in the brain, heart, kidneys, brown
adipose tissue and liver-an organ with high oxidative capacity-in
which its core enzyme undergoes lysine acetylation (12). In the absence of SIRT3, many of
these proteins undergo hyperacetylation. Acetylated proteins induce
significant conformational changes in substrate proteins,
potentially altering the function and impairing the catalytic
activity of most mitochondrial enzymes (13). This disruption may trigger a
series of digestive system disorders (14). Therefore, this review describes
the role of SIRT3 in digestive system diseases and its potential
therapeutic targets, providing new strategies for the diagnosis and
treatment of digestive system diseases.
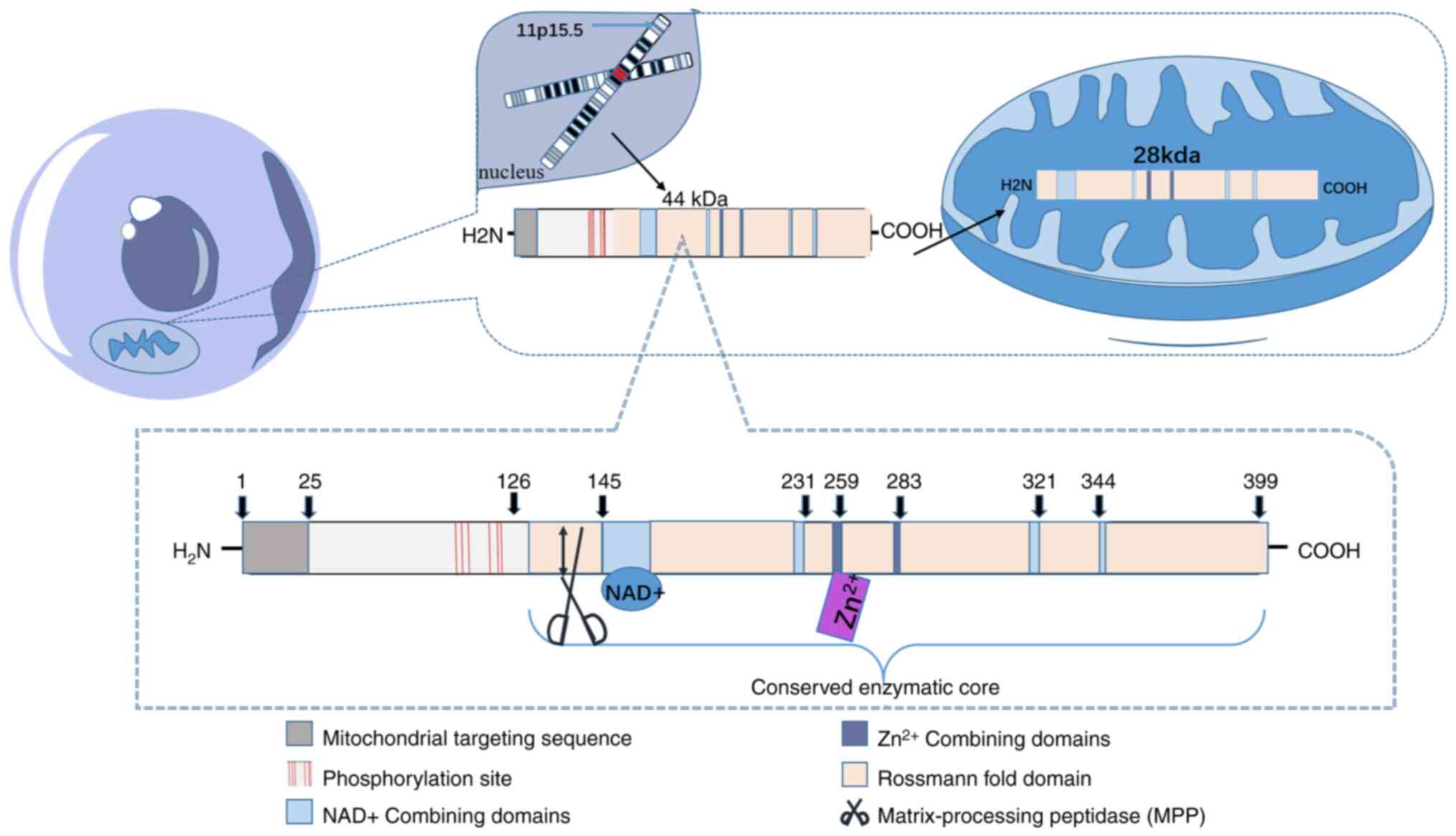
The human SIRT3 protein exists in two forms: A long
isoform and a short isoform. Both forms of SIRT3 exhibit
NAD+-dependent deacetylase activity (15). SIRT3 is transcribed in the
11p15.5 chromosomal region of the cell nucleus. The fully
transcribed, 399-residue full-length SIRT3 (FLSIRT3) constitutes
the long isoform. The mitochondrial targeting sequence located
between N-terminal residues 1-25 is crucial for subsequent
mitochondrial localization and proteolytic processing. Residues
101, 103, 105, 114, 117 and 118 can prevent substrates from
prematurely binding to the SIRT3 active site before mitochondrial
transfer (9,12). Upon localization to mitochondria
via its mitochondrial targeting sequence, FLSIRT3 is cleaved at
residue 142 by mitochondrial proline protease in the mitochondrial
matrix, yielding a short isoform with ~28 kDa of class III histone
deacetylase activity (16). The
conserved catalytic core domain positions SIRT3 as one of the most
critical deacetylases, featuring a large Rossmann fold domain that
binds NAD+ and a small domain composed of a helical
bundle and zinc-binding motif, formed by two loops extending from
the larger domain (8). The
remainder of the enzyme core comprises the binding site for SIRT3
substrates (9) (Fig. 1). Each structural feature of
SIRT3 directly governs its biological function: The cleft between
the Rossmann fold domain and the zinc finger domain forms the
substrate binding pocket. Its size and chemical properties
determine the ability of SIRT3 to recognize and bind specific
acetylated lysine residues; for instance, leucyl-tRNA synthetase 2
specifically binds to SIRT3, and NADH-ubiquinone oxidoreductase
subunit V3 specifically binds to SIRT5 (17). The mitochondrial targeting
sequence ensures its localization to the mitochondrial matrix,
while SIRT3 exclusively deacetylates substrates within
mitochondria, guaranteeing its role in regulating mitochondrial
activity. The zinc ion-binding domain not only maintains the
overall structural stability of the SIRT3 protein but also
determines the specific structures of various Sirtuins (18). The highly conserved Rossmann fold
domain within SIRT3 perfectly complements NAD+,
initiating the deacetylase reaction by hydrolyzing the cofactor
NAD+. This hydrolysis yields two molecules, nicotinamide
and ADP-ribose. The acetylated lysine residue on the substrate
protein transfers its acetyl group to ADP-ribose. At this point,
the C1'-alkylamide intermediate is converted into a bicyclic
intermediate with the assistance of the conserved His 224 residue.
This induces a nucleophilic attack by the 2'-OH group of ribose on
the amine carbon of the o-alkylamide intermediate. The intermediate
is then cleaved by an activated water molecule, ultimately forming
20-acetyl-ADP-ribose to complete deacetylation (8,9).
NAD+, as a cofactor of SIRT3, directly regulates its
activity (19). Consequently,
SIRT3 expression and activity are modulated by the
NAD+:NADH ratio. Elevated ratios-such as those during
starvation, exercise or stress-increase SIRT3 activity and
expression, whereas low ratios or aging decrease SIRT3 levels
(11,12). Thus, the structure of SIRT3 plays
a crucial role in linking cellular metabolic states to
mitochondrial functional adaptation. Alterations in SIRT3 structure
lead to reduced enzyme activity, causing cellular metabolic
reprogramming and increased genomic instability, thereby promoting
tumorigenesis (20). Similarly,
reduced SIRT3 activity diminishes mitochondrial capacity to adapt
to metabolic stress, increasing susceptibility to hepatic
steatosis, insulin resistance and oxidative damage. This
significantly increases the risk and severity of non-alcoholic
fatty liver disease (NAFLD) (21,22).
SIRT3 performs deacetylation functions because of
its conserved enzyme core. Deacetylation by SIRT3 is the most
common posttranslational modification of mitochondrial proteins,
altering the structure and functional activity of substrate
proteins via histone/lysine acetyltransferases (13). Peterson et al (23) reported that SIRT3 deacetylates
626 sites across 242 proteins, including 32 sites in 12 FAO
proteins, 27 sites in 14 proteins involved in the TCA cycle, 18
sites in 11 proteins involved in the electron transport chain
(ETC), 18 sites in 9 proteins involved in protein quality control,
and 9 sites in 6 proteins involved in ROS detoxification. In SIRT3
knockout (SIRT3-KO) cells, acetylation levels at mitochondrial
protein sites increase twofold (24). These proteins are involved in
various pathways, such as the TCA cycle, amino acid metabolism,
FAO, ETC/OXPHOS and mitochondrial dynamics. Consequently, they
regulate oxidative stress, apoptosis, energy metabolism,
inflammation, DNA damage and aging.
Studies reported 283 SIRT3-specific targets among
136 mitochondrial proteins in a SIRT3-KO mouse liver model,
particularly within fatty acid metabolism pathways. These include
acetyl-CoA synthase 2, long-chain acyl-CoA dehydrogenase (LCAD),
and 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2, as well as
the stress-responsive enzymes isocitrate dehydrogenase 2,
superoxide dismutase 2 (SOD2) and manganese superoxide dismutase
(MnSOD) (15,25). When SIRT3 is absent or its
activity is reduced, the ability of the liver to oxidize fatty
acids decreases. Large amounts of free fatty acids (FFAs) are
esterified into triglycerides (TGs) and deposited within
hepatocytes, leading to hepatic steatosis. Simultaneously, SOD2
inactivation leads to massive ROS accumulation, triggering
oxidative stress that causes hepatocyte injury, ballooning
degeneration and cell death. In digestive system cancers, SIRT3
suppresses Bax-mediated mitochondrial apoptosis pathways by
deacetylating and activating Ku70, enabling its binding to the
pro-apoptotic protein Bax (26).
Previous studies revealed that SIRT3 deficiency increases ROS
levels, activating hypoxia-inducible factor (HIF)-1α. In mice, this
leads to HIF-1α-dependent tumor growth patterns, indicating that
SIRT3 suppresses the carcinogenic effects of HIF-1α by regulating
ROS, thereby inhibiting tumor growth (27). SIRT3 deacetylation activates
pyruvate dehydrogenase, promoting pyruvate entry into the TCA cycle
and inhibiting the characteristics of aerobic glycolysis in cancer
cells (28).
Increasing evidence has indicated that SIRT3
regulates proteins extensively involved in mitochondrial reactions,
including energy production, signaling and apoptosis. When SIRT3 is
absent, numerous mitochondrial proteins involved in metabolism and
stress undergo hyperacetylation, directly contributing to the onset
and progression of various digestive system diseases (11,28,29).
NAFLD encompasses simple steatosis (SS) and
non-alcoholic steatohepatitis (NASH) and is characterized by
inflammation and ballooning degeneration with or without fibrosis.
NASH-associated cirrhosis features the formation of cirrhotic
nodules due to fibrotic septa, potentially progressing to HCC
(29,30).
Globally, the prevalence of NAFLD and NASH has
increased in tandem with that of obesity and metabolic syndrome,
indicating that these conditions are becoming serious health
threats (31). In NAFLD
pathogenesis, hepatocytes exhibit adipocyte-like functions and
maintain the balance of lipid metabolism by regulating the
production, oxidation, and transport of TGs, FFAs, cholesterol and
bile acids (32). When the
capacity of hepatocytes is exceeded, lipid metabolism disorders
lead to hepatic fat accumulation. Cells exposed to a high-fat
environment experience lipotoxicity, which impairs mitochondrial
function by activating mitochondrial defects, endoplasmic reticulum
(ER) stress and oxidative stress. Concurrently, mitochondria can
reduce cellular β-oxidation levels through multiple pathways,
including cell division, oxidative stress, autophagy and
mitochondrial quality control, thereby promoting hepatic fat
accumulation and injury, accelerating NAFLD progression and
ultimately causing a rapid decline in liver function (33). Furthermore, disruption of
mitochondrial dysfunction-mediated lipid metabolism in patients
with NAFLD leads to excessive TG accumulation (>5%) and hepatic
steatosis in hepatocytes (34).
When obesity remains uncontrolled during the SS stage, innate
immune cells within the liver-including Kupffer cells, dendritic
cells and hepatic stellate cells-become activated, leading to
progressive immune cell infiltration of the liver (35). In the liver, these immune cells
release cytokines that exacerbate the inflammatory process,
propelling hepatocytes from the SS stage into the NASH stage and
ultimately triggering fibrosis (36). Liver biopsy tissues from patients
with NAFLD showed hypermethylation of NADH dehydrogenase-6, which
correlated with disease severity (37). These changes in hypermethylation
were associated with loss of the inner mitochondrial membrane, deep
crista folding and loss of mitochondrial granules, indicating that
mitochondrial structural stability is critical for normal hepatic
physiological activity. Previous studies revealed reduced SIRT3
expression in animals fed a high-fat diet (HFD). When
SIRT3-knockout mice were fed an HFD, hepatic steatosis worsened
(25). These findings suggest
that SIRT3 plays a role in the pathogenesis of NAFLD. A further
study revealed that SIRT3 upregulates β-oxidation and ATP
production, suppresses ROS and enhances mitochondrial biogenesis
through the activation of peroxisome proliferator-activated
receptor (PPAR) gamma coactivator 1 alpha (PGC-1α) (38). Conversely, SIRT3 deficiency leads
to excessive acetylation at the K24 site of the mitochondrial
metabolic enzyme LCAD. When an acetyl group is covalently modified
at this site, the LCAD conformation changes, impairing enzyme
activity and obstructing FAO. This results in abnormal lipid
metabolism, manifested as massive accumulation of lipid droplets in
the liver (25). Furthermore, it
affects transcription coactivators involved in FAO, such as PPARα
and several target genes involved in FAO, leading to reduced
DNA-binding activity and increased acetylation of several hepatic
proteins involved in lipid uptake, ultimately resulting in NAFLD
development (39). Additionally,
SIRT3 promotes β-oxidation by driving LCAD activity and enhancing
ketone body production by promoting the deacetylation of
3-hydroxy-3-methylglutaryl-CoA synthase. In SIRT3-deficient mice,
hyperacetylation of mitochondrial FAO-related enzymes and reduced
enzyme activity lead to increased FFA levels and hepatic steatosis
(40). In SIRT3-deficient mice
fed an HFD, hepatic steatosis is exacerbated by the activation of
oxidative stress-induced nuclear factor erythroid 2-related factor
(Nrf)2, which increases the expression and protein levels of genes
involved in lipid uptake (very low-density lipoprotein receptor and
CD36) in the liver (41).
Concurrently, it reduces the activity of respiratory complexes III
and IV, exacerbating oxidative stress (42). The administration of adenovirus
expressing SIRT3 to mice alleviated obesity, insulin resistance,
hyperlipidemia, hepatic steatosis and inflammation in
SIRT3-deficient mice (43).
Currently, NAFLD treatment relies primarily on lifestyle
modifications such as weight reduction, increased exercise and
dietary control; however, the adherence to and efficacy of these
measures remain suboptimal. Identifying isoenzyme-specific SIRT
activators and their target signaling pathways to enhance hepatic
mitochondrial function may represent a novel therapeutic strategy
for NAFLD. A study revealed that Polygonum cuspidatum
glycoside (PD), a natural resveratrol precursor isolated from
Polygonum cuspidatum, activates SIRT3, which in turn
activates the deacetylase-activated transcription factor forkhead
box (FOX)O3a. FOXO3a induces the interaction between BCL2 and the
B19kDa protein-interacting protein 3 (BNIP3), which promotes
mitochondrial autophagy (44).
This herbal compound, PD, improves NAFLD liver function,
histopathology and mitochondrial function through the
aforementioned SIRT3-FOXO3-BNIP3 signaling axis and PINK1/parikin
(PRKN)-dependent mitochondrial autophagy regulatory mechanism,
PINK1 detects mitochondrial damage and recruits PRKN to the
mitochondrial surface, thereby mediating ubiquitin-tagging and
ultimately initiating the autophagy pathway for the clearance of
damaged mitochondria. demonstrating therapeutic efficacy against
NAFLD (45). PD has demonstrated
broad potential in preclinical studies, but its practical
application in clinical settings still faces several challenges
(46-48). Although the vitamin D analogue
paricalcitol is well-established in clinical treatment for
secondary hyperparathyroidism caused by chronic kidney disease, its
application in addressing liver inflammation remains in the
preclinical stage. The vitamin D analog paricalcitol reduced the
acetylation of the transcription factors FOXO3a and NF-κB in the
rat liver by increasing SIRT1 and SIRT3 expression, thereby
alleviating hepatic inflammation and oxidative stress activation
(49).
Primary liver cancer includes HCC (accounting for
75-85% of cases) and intrahepatic cholangiocarcinoma (10-15%),
along with other rare types (50). During the past decade, most
patients were diagnosed at an advanced stage when surgery and local
treatments were no longer feasible (51). Although clinical chemotherapy and
immunotherapy have demonstrated efficacy in treating HCC, its
incidence and prevalence continue to increase (52,53).
Research has indicated that HCC progression and
metastasis are associated with altered mitochondrial metabolism
(50). Mitochondrial
dysfunction, stress responses and protein aberrations in cancer
cells lead to mitochondrial defects. In damaged hepatocytes, ROS
production, metabolic reprogramming and mitochondrial hormone
responses within mitochondria drive tumor growth and metastasis.
Previous studies have indicated that when cells lack SIRT3, the
inability to deacetylate MnSOD leads to increased ROS levels,
readily causing mitochondrial metabolic abnormalities. Under
specific intracellular conditions, this triggers genetic
instability mutations, ultimately resulting in dedifferentiation
and carcinogenesis (54).
Compared with that in normal liver cells, SIRT3 expression is
downregulated in HCC cells, and this downregulation is associated
with tumor size and grade. These findings indicate that SIRT3 plays
a tumor-suppressing role in humans and suggest that it can serve as
a reliable prognostic indicator (55,56). Cancer cells primarily rely on
aerobic glycolysis for energy production, a process that also
creates favorable conditions for their survival in the
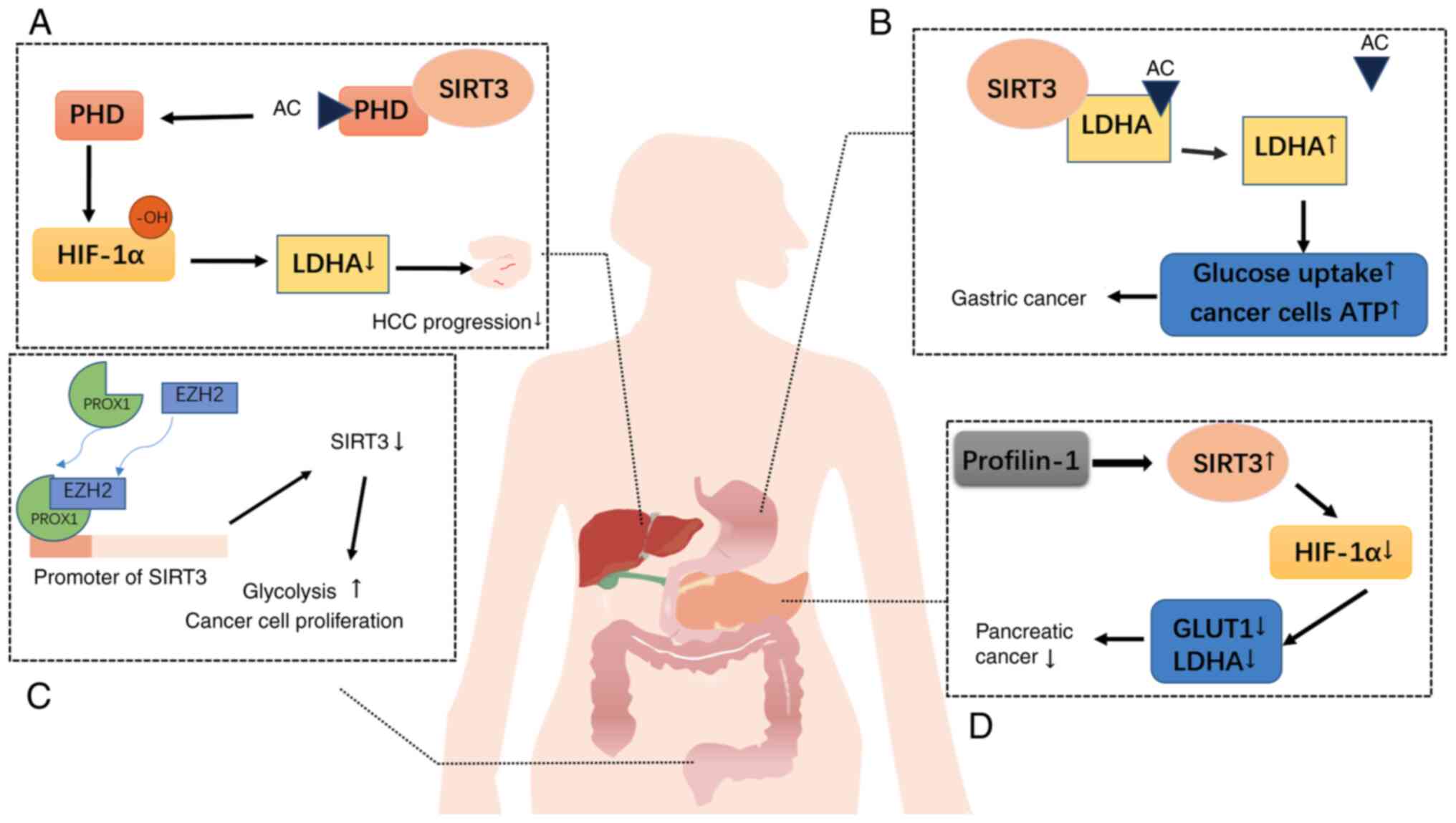
microenvironment-known as the Warburg effect (57). SIRT3 acts as a tumor suppressor
gene by promoting oxidative phosphorylation, thereby inhibiting
glycolysis and regulating metabolism to suppress the Warburg
effect. The most extensively studied mechanism involves the
inhibition of HIF-1α by SIRT3. Since many cancer cells extensively
consume glucose to produce lactate via lactate dehydrogenase A
(LDHA), which is encoded by c-Myc target genes and HIF-1α, HIF1α
has been identified as a key factor that activates glycolytic
pathways in tumors. SIRT3 modulates HIF1α activity by deacetylating
prolyl hydroxylase (PHD). Activated PHDs hydroxylate the substrate
HIF-1α, affecting its stability and preventing aerobic glycolysis
in tumors (58,59). Additionally, pyruvate
dehydrogenase complexes (PDCs) serve as SIRT3 substrates linked to
glycolysis. As an upstream deacetylase of PDCs, SIRT3 deacetylates
and activates these substrates, thereby inhibiting tumor cell
glycolysis and promoting apoptosis (60) (Fig. 2). SIRT3/SIRT4 reduces the high
expression of cyclooxygenase-2 (COX-2) in tumor tissues. Since
COX-2 impedes PINK1/PRKN-mediated mitochondrial autophagy, its
reduction enhances mitochondrial autophagy, contributing to the
early prevention of HCC (61).
As HCC is a chemoresistant cancer and its multidrug resistance
characteristics contribute to high recurrence and metastasis rates
(62). Consequently, drugs that
enhance anticancer activity are urgently needed. Given the critical
role of SIRT3 in tumors, researchers are now focusing on the
function of SIRT3 modulators in HCC. Among these, the small
molecule 7-hydroxy-3-(4'-methoxyphenyl)coumarin, a SIRT3 activator,
binds to SIRT3 to specifically increase MnSOD deacetylation and
activity (63). It also
increases sensitivity to HCC treatments such as sorafenib (55,64). Furthermore, the SIRT3 activator
resveratrol enables SIRT3 to deacetylate mitochondrial
cyclooxygenase-2 (mito-COX-2), thereby inhibiting
mito-COX-2/dintenin-1-driven mitochondrial fission, inducing cancer
apoptosis and increasing the sensitivity of HCC cells to cisplatin
chemotherapy (63). Both of
these activators are currently in preclinical trials. Although
several effective SIRT3 activators have been identified, further
research is still needed to discover and develop specific and
potent SIRT3 activators.
The gut serves as a barrier against harmful external
substances and pathogenic infections, interacting with the gut
microbiome and food antigens. Consequently, the intestinal immune
system responds to both internal and external environments to
maintain homeostasis. Disruption of this equilibrium can lead to
IBD (65). IBD is a chronic,
recurrent disease that encompasses ulcerative colitis (UC) and
Crohn's disease. Its etiology remains unclear, although it is
widely considered an autoimmune disorder in which key pathogenic
factors involve the dysregulation of T-cell subsets (66). Compared with healthy individuals,
patients with IBD exhibit increased levels of helper T cells (Th17)
and the corresponding transcription factor retinoic acid-related
orphan receptor gamma-t (RORγt). Additionally, both SIRT3 gene
transcription and protein expression are downregulated in the
colonic tissue of patients with UC (67). RORγt is a key transcription
factor for Th17 cell development. Its signaling is regulated by
signal transducer and activator of transcription 3 (STAT3), which
directly activates RORγt by modulating STAT3, thereby inducing Th17
cells (68). Thus, STAT3 plays a
crucial role in regulating the Th17/T-regulatory cell (Treg)
balance and the release of inflammation-related cytokines (69). Increased STAT3 promotes Th17-cell
differentiation, disrupting the Th17/Treg balance and triggering
inflammatory events (70).
Honokiol (HKL), an activator of SIRT3, was used to treat an
inflammatory cell model and a dextran sulfate sodium-induced mouse
colitis model. Compared with untreated mice, HKL-treated mice
exhibited reduced inflammatory cell infiltration, significantly
less structural damage and relatively lower edema levels. As a
SIRT3 activator, HKL inhibits STAT3-induced RORγt activity, reduces
the Th17 ratio and does not affect Th-cell differentiation, thereby
regulating the Th17-cell mechanism to alleviate colitis symptoms
(67). Dysregulation of
intestinal macrophages also plays a crucial role in the
pathogenesis of IBD. Macrophage NAD+ synthesis exerts
immunoregulatory effects by enhancing oxidative phosphorylation
(71). This maintains intestinal
immune homeostasis by clearing infections while preventing chronic
inflammation and inducing tissue repair. During this process,
macrophages polarize into classically activated macrophages (M1)
and alternatively activated macrophages (M2) (72). SIRT3 deacetylates glutamate
dehydrogenase 1 to promote α-ketoglutarate (α-KG) production. α-KG
accumulation not only enhances OXPHOS metabolism but also reduces
histone H3 K27 trimethylation in the nucleus, leading to the
upregulation of genes associated with M2 polarization (73). Given that IBD is a chronic,
recurrent disease imposing a significant health burden, the
discovery of the role of SIRT3 in IBD potentially reveals a
pharmacological target.
The ENS is a vast, complex and autonomous system
distinct from the central nervous system. Neurons within the ENS
regulate critical functions such as digestion, nutrient absorption
and intestinal motility. The ENS, intestinal epithelium, gut
microbiota and immune cells work in concert to ensure the
maintenance of normal intestinal function (74). ENS-related neurodegeneration is
particularly pronounced in individuals with aging and
neurodegenerative diseases. Stress from antibiotic treatment also
affects ENS function by altering the gut microbiota (75). ENS dysfunction can lead to
numerous disorders associated with motility alterations and
inflammation, including primary achalasia (76,77) and irritable bowel syndrome
(78). Owing to high energy
demands, neurotransmitter autooxidation and limited replication
capacity, ENS neurons are highly susceptible to oxidative damage
from free radicals. Oxidative stress has also been demonstrated to
alter electrophysiological properties, damage neuronal membranes
and trigger neuronal death (79). Currently, the known mechanisms of
oxidative stress in enteric neurons can be categorized into those
associated with endogenous nitrosative damage, mitochondrial
dysfunction or inflammation (79). Studies have revealed that SIRT3
protects cortical and dopaminergic neurons from oxidative stress by
regulating mitochondrial homeostasis (80). The SIRT3 activator
hexafluoromagnol activates SIRT3 and its downstream target PGC-1α
to counteract stress-induced ROS production (81). Concurrently, PGC-1α directly
enhances SIRT3 transcription to mitigate oxidative damage, thereby
promoting intestinal neuronal survival and differentiation
(82). Furthermore, SIRT3
significantly increased the density of neural networks and axons in
intestinal neurons while increasing neuronal nitric oxide synthase
and choline acetyltransferase mRNA levels. SIRT3 exerts
neuroprotective effects by inhibiting superoxide release, thereby
suppressing palmitate and lipopolysaccharide-induced neuronal
pyroptosis, which play crucial roles in promoting intestinal
neuronal survival and differentiation (83,84). Beyond regulating host energy
balance, the gut microbiota modulates numerous metabolic processes
by participating in the complex bidirectional communication system
known as gut-brain crosstalk, which is mediated by signaling
molecules such as short-chain fatty acids produced by the
microbiota (85). Short-chain
fatty acids generated by gut bacteria regulate NAD+
metabolism to influence SIRT3 activity (86). In summary, SIRT3 plays a crucial
role in maintaining normal ENS function. Investigating the role of
SIRT3 in the ENS provides direction for disease prevention and
treatment.
CRC is a malignant tumor arising from the mucosal
epithelium and glands of the large intestine. Its incidence is
increasing annually and the 5-year survival rate significantly
decreases once tumor cells infiltrate the submucosal layer.
Prognosis worsens and survival decreases upon metastasis to
extraintestinal organs (87). In
CRC, SIRT3 functions as a tumor suppressor to inhibit cancer
progression. Compared with those in adjacent normal colorectal
epithelium, the expression levels of proco-related homeobox 1
(PROX1) are significantly greater in colorectal tumor cells. As a
homologous transcription factor, PROX1 regulates cellular
differentiation and development during embryonic growth and is
positively correlated with tumor glucose metabolism (88). The mechanism involves the
recruitment of the enhancer of zeste homolog 2 (EZH2) to the SIRT3
promoter region in CRC cells by PROX1, thereby inhibiting SIRT3
transcription. This increases glucose metabolism in cancer cells to
sustain proliferation (89).
When PROX1 is highly expressed, it binds to the SIRT3 promoter
region, inhibiting SIRT3 transcription and translation and
ultimately leading to low SIRT3 expression. Knocking down PROX1
expression increases SIRT3 expression and reverses the malignant
characteristics of CRC (90)
(Fig. 2). SIRT3 regulates the
expression of PGC1α and NRF1. Reduced SIRT3 expression impairs
mitochondrial dynamics and affects the malignancy of CRC cells
(91). Of note, SIRT3 may also
promote cancer cell growth. Metabolic reprogramming is a hallmark
of cancer and is closely linked to cancer progression and
metastasis. To enable rapid cell division, cancer cells exhibit
abnormal glycolysis, glutamine catabolism and lipid synthesis
(92). Previous studies revealed
disrupted serine-to-glycine ratios in CRC, where serine/glycine
synthesis influences the tumor status by regulating cellular
antioxidant capacity (93). The
expression of serine hydroxymethyltransferase 2 (SHMT2), a key
serine/glycine interconverting enzyme, is significantly elevated in
patients with CRC and is correlated with poor survival outcomes.
Further study revealed acetylation at the K95 site of SHMT2.
Acetylated K95 disrupts the SHMT2 tetrameric conformation to
inhibit its enzymatic activity and promotes SHMT2 degradation via
the K63-ubiquitin-lysosomal pathway. This leads to reduced serine
consumption and decreased NADPH levels, thereby suppressing tumor
growth and cell proliferation (94). In the aforementioned SHMT2-K95-Ac
pathway, SIRT3 acts as a deacetylase for SHMT2, deacetylating K95.
Deacetylation of K95 fails to inhibit SHMT2 activity, leading to
SHMT2 overexpression, which promotes colorectal tumorigenesis.
These tumors are highly invasive and have poor prognosis (95). SIRT3 also deacetylates
methylenetetrahydrofolate reductase 2, increasing its activity and
promoting cell proliferation and cancer progression (96). These studies indicate that SIRT3
influences cancer cell proliferation by controlling whether
endogenous regulatory factors undergo deacetylation. In recent
years, researchers have been exploring novel therapeutic and
preventive strategies against cancer. It should not be overlooked
that dietary prevention can reduce the risk of CRC. Apigenin, a
natural antioxidant found in fruits and vegetables, reduces the
expression of SHMT2, SIRT3 and their upstream long intergenic
noncoding RNA LINC01234 when CRC cells are treated. This activity
inhibits metabolic reprogramming by regulating the
LINC01234/SIRT3/SHMT2 axis (97). Ergothioneine, a dietary betaine,
exerts anti-CRC effects by activating SIRT3 (98). Clinically, SGLT2 inhibitors such
as canagliflozin, dapagliflozin, tolvogliflozin and empagliflozin
are commonly used as hypoglycemic agents. A recent study revealed
their novel anticancer activity by reducing glucose uptake and
severely impairing cancer-specific cellular metabolism (99). In CRC, canagliflozin can be
repurposed to specifically inhibit CRC cell proliferation and
induce S-phase arrest through mechanisms of regulating metabolism,
mitochondrial function and ER stress via the SGLT2/SIRT3/dipeptidyl
peptidase-4 axis (100). It
should be noted that the aforementioned drugs remain in the
preclinical exploration phase. The findings from preclinical
studies provide crucial theoretical foundations and preliminary
scientific hypotheses for future formal clinical trials. Radiation
therapy is among the primary treatments for cancer patients.
However, prolonged radiotherapy can induce radiation resistance in
cancer cells, often leading to treatment failure and poor prognosis
in patients receiving radiotherapy (101). Research has indicated that
SIRT3 promotes radiation resistance in tumor cells by enhancing DNA
damage repair through excessive activation of mitochondrial
autophagy, which is mediated by PINK1/PRKN (102). In addition to promoting
radiation resistance during radiotherapy, SIRT3 modulates
chemotherapy resistance in CRC cells via the regulation of SOD2 and
PGC-1α during chemotherapy. SIRT3 serves as an independent
prognostic factor for colorectal cancer (99). Undoubtedly, the role of SIRT3 in
colon cancer is a double-edged sword. Whether its ultimate effect
is tumor suppression or promotion depends on a complex
decision-making network. Different colon cancer subtypes, tumor
microenvironments and tumor stages influence whether SIRT3 promotes
or inhibits tumorigenesis in CRC. Therefore, further understanding
of the underlying molecular mechanisms is warranted for the future
clinical development of SIRT3-targeted therapeutic agents.
AP is a localized inflammatory response in
pancreatic tissue caused by the activation of pancreatic enzymes
due to various etiologies. The activation of pancreatic enzymes is
a prerequisite for local inflammation in pancreatitis (100). When pancreatic cells undergo
necrosis and vascular permeability increases, the pancreas
experiences edema, necrosis and hemorrhage. Concurrently, impaired
pancreatic function affects other organs, such as through gut
microbiota dysbiosis, increased intestinal barrier permeability and
bacterial translocation (103).
Unlike conventional treatments that primarily reduce disease
severity by alleviating pancreatic burden, fecal microbiota
transplantation (FMT) achieves similar effects by mitigating tissue
damage and inflammation through the reversal of dysbiosis (104). Research has indicated that FMT
modulates the intestinal microbiota to concentrate nicotinamide
mononucleotide in the pancreas, thereby increasing pancreatic
NAD+ levels and reducing disease severity. This process
requires an NAD+-dependent enzyme for conversion. SIRT3,
an NAD+-dependent deacetylase, mitigates AP-induced
oxidative stress and inflammation by reducing the acetylation of
the mitochondrial protein peroxiredoxin-5 (PRDX5) in acinar cells
and increasing the expression of PRDX5 (105). These findings indicate that
SIRT3 exerts an inhibitory effect on AP and that its expression
levels in patients may serve as a therapeutic biomarker.
Pancreatic cancer, particularly pancreatic ductal
adenocarcinoma (PDAC), is characterized by late symptom onset,
early metastasis and rapid progression, resulting in low patient
survival rates. Therefore, identifying potential biomarkers for
patients with pancreatic cancer is critically important (106). Studies of sirtuin protein
expression profiles in PDAC have revealed that in the absence of
chemotherapy intervention, low SIRT3 expression in the tumor
cytoplasm is associated with high-grade, poorly differentiated
tumors. Low SIRT3 expression is associated with high invasiveness,
high recurrence rates and poor prognosis (107). These findings indicate that
SIRT3 functions as a tumor suppressor in pancreatic cancer. As an
enzyme that regulates macronutrient metabolism, SIRT3 redirects
cellular glycolysis toward oxidative phosphorylation and the TCA
cycle, suppressing the Warburg effect in tumor cells and reducing
proliferation (108). Research
has revealed that Profilin1 (Pfn1) expression is downregulated in
pancreatic cancer. Pfn1 fails to increase HIF1-α stability by
upregulating SIRT3, thereby exerting a growth-inhibitory function
in pancreatic cancer. HIF1α promotes metabolic reprogramming and
plays a crucial role in maintaining glycolysis in pancreatic cancer
(109,110) (Fig. 2). SIRT3 also regulates iron
metabolism in PDAC by modulating the activity of iron regulatory
protein 1 (IRP1), a key regulator of cellular iron levels that
control genes containing iron response elements (IREs) (111). SIRT3 reduces the binding
affinity of IRP1 for IREs, leading to decreased expression of
iron-related genes such as transferrin receptor and thereby
inhibiting pancreatic cancer cell proliferation (112). Furthermore, ZMAT1 belongs to a
5-member family (ZMAT1-5) in humans, in which all encoded proteins
contain zinc finger domains, functions as tumor suppressors by
triggering cell cycle arrest and apoptosis. ZMAT1 expression is
downregulated in PDAC and is associated with tumor differentiation,
tumor stage and patient survival. ZMAT1 promotes SIRT3
transcription by binding to three sites on its promoter,
subsequently upregulating p53 expression and inhibiting cancer cell
proliferation (113). SIRT3
also participates in regulating the ETC in pancreatic cancer,
specifically by modulating mitochondrial complex II (CII) activity.
Dysfunction of SIRT3 leads to decreased CII activity, causing
mitochondrial dysfunction and promoting tumor progression. Recent
research has identified a pharmacological agent, poplar
nanocapsules, that targets the ubiquinone site to restore CII
activity and SIRT3 expression, thereby inducing apoptosis and
reducing pancreatic cancer cell survival (114). Given that pancreatic cancer is
characterized by rapid growth and high resistance to
chemoradiotherapy, developing novel effective treatments targeting
its pathogenesis is critically important (115). Identifying SIRT3-regulated
pathways that drive PDAC initiation and progression is important
for identifying potential therapeutic targets. Reconstructing SIRT3
activity or activating upstream components of SIRT3-associated
oncogenic signaling suppression pathways offers a novel,
multifaceted strategy to promote cancer progression and improve
patient prognosis.
Gastric cancer ranks fourth among cancer types.
Although its global incidence has shown an overall downward trend,
long-term trends in incidence and mortality vary significantly
across countries (116).
Increasing experimental evidence has demonstrated that SIRT3
deficiency leads to increased ROS production and oxidative stress
in the body, potentially causing cellular dedifferentiation or
carcinogenesis. SIRT3 plays a crucial role in protecting cells and
preventing cancer progression during carcinogenesis and tumor
development. For instance, SIRT3 regulates MnSOD activity to reduce
ROS levels in gastric cancer cells, thereby shielding them from
oxidative stress-induced damage (117-119). However, in SIRT3-positive
gastric tumor cells, SIRT3 acts as an oncogenic factor. By
deacetylating LDHA, it enhances enzyme activity. Increased LDHA
activity increases glucose uptake and lactic acid production,
reducing mitochondrial oxygen consumption while increasing the
mitochondrial membrane potential. This increases overall ATP
production and aerobic glycolysis in cancer cells, accelerating
tumor growth. Concurrently, the expression of glycolysis-related
genes is upregulated in SIRT3-overexpressing gastric tumor cells
(119) (Fig. 2). Although the incidence and
mortality rates of gastric cancer have generally declined, it
remains among the leading causes of death worldwide (120). Decreasing gastric cancer
mortality may be achieved by developing drugs through studying the
regulatory mechanisms of SIRT3 in gastric cancer.
Gallbladder cancer (GBC) is a common malignant tumor
of the biliary tract. Early stages show no specific symptoms, but
when significant weight loss, jaundice, abdominal pain and diarrhea
occur, the disease has already progressed and is often missing the
optimal treatment window. Radical cholecystectomy is the preferred
treatment, but owing to its high degree of invasiveness and
susceptibility to peritoneal and intrahepatic metastasis, early
prevention and treatment are particularly crucial (121). SIRT3 plays a role in the
pathogenesis of GBC. Studies have revealed that SIRT3 expression is
significantly lower in GBC cells than in adjacent nontumor tissue,
and patients with low SIRT3 expression exhibit poorer overall
survival than those with high SIRT3 expression (122). As a mitochondrial deacetylase,
SIRT3 enhances ferroptosis by inhibiting AKT to increase acyl-CoA
synthetase long-chain family member 4 expression (123). When SIRT3 expression is
reduced, inhibition of AKT-dependent ferroptosis protects cancer
cells from ferroptosis. Simultaneously, activating AKT in GBC cells
induces the upregulation of epithelial-mesenchymal transition (EMT)
markers, enhancing tumor cell invasion activity and migration
capacity (124). Reduced SIRT3
expression promotes tumor cell respiration, ATP production and ROS
reduction (122,125). Thus, in GBC pathogenesis, SIRT3
induces Akt-mediated ferroptosis while inhibiting EMT, thereby
suppressing tumor progression.
As early as 2003, studies reported an association
between SIRT3 expression and lifespan (126). It protects vital human tissues
such as the heart, liver, brain and kidneys from disease through
the deacetylation of substrates. SIRT3 also participates in the
overall progression of cancer. Current research on sirtuin targets
has made significant progress. Studies on these targets can be
extended to identify sirtuin-activating compounds (STACs) or
inhibitory compounds (8). Most
STACs belong to the natural product polyphenol family, with
resveratrol being the first compound discovered to increase SIRT1
activity nearly 10-fold (127).
Compounds such as SRT1720, SRT2183 and SRT3025 have been developed
to increase sirtuin enzymatic activity, and these drugs have been
shown to limit kidney damage (128). Currently, most SIRT3 activators
are natural products. These compounds can deacetylate SIRT3 or
regulate its expression levels through various mechanisms. HKL, a
SIRT3 activator, is a natural compound that increases SIRT3
expression and deacetylase activity. In mice with cardiac
hypertrophy, it inhibits cardiac hypertrophy by regulating the AKT
and ERK1/2 pathways (82). HKL
exhibits anti-inflammatory effects in mice with enterotoxin-induced
diarrhea by promoting intestinal barrier function and regulating
intestinal epithelial apoptosis. It can also cross the blood-brain
barrier to directly act on neuronal cells in the central nervous
system (84). The novel
fluorinated synthetic HKL analog hexafluoro-HKL exhibits protective
effects against melanoma and enhances SIRT3 expression (129). Dihydromyricetin, which is
structurally similar to resveratrol, enhances SIRT3 expression
through SIRT3-mediated cell protection and inflammation resistance,
thereby treating osteoarthritis (130). In a mouse model of sulfur
mustard-induced liver injury, this natural product was shown to
exert its hepatoprotective effects via SIRT3 (131). Another natural product,
pyrroloquinoline quinone, improves hepatic metabolic disorders by
increasing SIRT3 expression (132). In the treatment of HCC,
sorafenib is a clinically used drug for treating HCC. Sorafenib has
been shown to reduce SIRT3 expression, thereby decreasing drug
sensitivity, while upregulating SIRT3 can restore HCC sensitivity
to sorafenib therapy (82).
These findings indicate that SIRT3 is a promising therapeutic
target for liver diseases. However, the dual role of SIRT3 in
cancer poses risks to its use as a cancer treatment target
(9). In addition to natural
compounds, SIRT3 activators can be developed through
high-throughput screening and structural optimization. Recent
discoveries have identified 2-APQC, a structurally selected
SIRT3-targeted small-molecule activator. By activating SIRT3, it
modulates the AKT-mTOR and TGFβ-Smad3 signaling pathways, thereby
inhibiting myocardial hypertrophy and alleviating myocardial
fibrosis (133). Unlike
traditional 'inhibitory' or 'blocking' drugs, SIRT3 activators aim
to 'restore and optimize' cellular physiological functions,
offering broad and profound application prospects. However, further
development through preclinical and clinical studies is needed to
address their pleiotropic effects and enhance their specificity.
Furthermore, achieving tissue-specific targeting remains
challenging, as other members of the sirtuin family are widely
expressed in the digestive system and regulate both tumor-promoting
and tumor-suppressing processes. Currently, allosteric activators
are gaining attention in the design of SIRT-targeting activators
(e.g., SIRT1 and -6). Further elucidation of the structure and
biological functions of SIRT3 will facilitate the development of
small-molecule activators targeting SIRT3 (9).
Compared with the development of SIRT3 activators,
the development of inhibitors within the dynamic deacetylation
process is somewhat simpler. By leveraging the peptide-binding
region within the SIRT3 crystal structure, a structure-based
approach can be used to identify novel isoform-specific inhibitors
(134). With respect to
deacetylation substrates, competitive inhibition of SIRT3
deacetylation activity can be achieved by synthesizing structural
analogs of endogenous acetylated substrates. Alternatively,
NAD+ coenzyme competitive inhibitors accelerate the
reverse reaction of deacetylation, thereby inhibiting the
deacetylation process. Tenovin-6 is a bioactive p53 activator that
was later identified as a SIRT3 inhibitor with antitumor activity.
Its mechanism of SIRT3 inhibition remains elusive, but it has been
demonstrated to act as a noncompetitive inhibitor (135). LC-0296 is a synthetic SIRT3
inhibitor whose mechanism of action is currently unknown. On the
basis of its structure, LC-0296 may act as a competitive inhibitor
of NAD+. It exhibits potent antiproliferative and
proapoptotic activity against head and neck squamous cell carcinoma
(136). As summarized in
Table I, an increasing number of
natural and synthetic compounds have been identified as SIRT3
activators or inhibitors, providing a critical resource for
developing targeted therapeutic strategies (Table I). Research on SIRT3 activators
is promising, yet their practical clinical application remains
challenging. For instance, precisely delivering drugs to specific
diseased organs while minimizing effects on other tissues is
crucial for enhancing efficacy and reducing side effects. This
precise targeting becomes particularly vital when SIRT3 expression
levels exhibit either pro- or antitumor effects across different
cancers. Careful monitoring of future research is essential,
especially in preclinical and clinical settings, to explore
SIRT3-modulating therapies for digestive system diseases.
As a member of the sirtuin family, SIRT3 influences
mitochondrial energy balance, redox homeostasis and mitochondrial
DNA repair by regulating the acetylation status of mitochondrial
proteins. The functional integrity of mitochondria is vital for
maintaining the health of digestive organs. Dysfunction of SIRT3
leads to mitochondrial disruption and serves as a common pathway
linking multiple pathophysiological processes. This review
summarizes the structure and deacetylase function of SIRT3,
highlighting its vital role in digestive system diseases. In such
diseases, loss of SIRT3 activity leads to metabolic disorders,
oxidative stress-induced accumulation of energy and substances,
cellular damage and mutations, and genomic instability. Ultimately,
this leads to tissue carcinogenesis. Notably, however, SIRT3
expression exhibits duality in certain digestive system tumors,
such as CRC, and influences chemoresistance in CRC cells.
Therefore, when SIRT3 activators are designed, combining SIRT3
modulators with chemotherapeutic agents or metabolic inhibitors
could significantly improve clinically observed chemoresistance.
Although a series of SIRT3-targeting small molecules have
demonstrated efficacy, the vast majority of SIRT3 activators remain
in preclinical stages. SIRT3 remains a largely unexplored
therapeutic target, and future research should focus on optimizing
SIRT3-targeted therapies by enhancing their specificity and
improving their efficacy when combined with conventional
treatments. Despite these existing challenges, this approach offers
novel therapeutic strategies for digestive system diseases.
Not applicable.
JL was responsible for conceptualization of the
review, methodological design, literature search, drafting of the
manuscript, and visualization of charts and graphs. QL performed
the literature review and data organization and participated in
drafting portions of the initial manuscript. SY provided resources
and performed quality assessment. XL provided resources and was
involved in formal analysis. LZ provided resources and was involved
in data curation. YW supervised the study and was responsible for
methodology. GW, JA and HJ acquired funding, performed project
administration, provided resources and supervision and were
involved in writing-review and editing. BT acquired funding and
contributed to writing-review and editing. Data authentication is
not applicable. All authors have read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81960507, 82073087
and 82160112), the Science and Technology Plan Project of Guizhou
Province (grant no. QIAN KE HE JI CHU-ZK (2024)YI BAN 323) and the
Medical Research Union Fund for High-Quality Health Development of
Guizhou Province (grant no. 2024GZYXKYJJXM0019).
|
1
|
Zhang Y, Zhao L, Gao H, Zhai J and Song Y:
Potential role of irisin in digestive system diseases. Biomed
Pharmacother. 166:1153472023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chakraborty E and Sarkar D: Emerging
therapies for hepatocellular carcinoma (HCC). Cancers (Basel).
14:27982022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Menini S, Iacobini C, Vitale M, Pesce C
and Pugliese G: Diabetes and pancreatic cancer-a dangerous liaison
relying on carbonyl stress. Cancers (Basel). 1:3132021. View Article : Google Scholar
|
|
4
|
Hu JX, Zhao CF, Chen WB, Liu QC, Li QW,
Lin YY and Gao F: Pancreatic cancer: A review of epidemiology,
trend, and risk factors. World J Gastroenterol. 27:4298–321. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Haque PS, Kapur N, Barrett TA and Theiss
AL: Mitochondrial function and gastrointestinal diseases. Nat Rev
Gastroenterol Hepatol. 21:537–555. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
LeFort KR, Rungratanawanich W and Song BJ:
Contributing roles of mitochondrial dysfunction and hepatocyte
apoptosis in liver diseases through oxidative stress,
post-translational modifications, inflammation, and intestinal
barrier dysfunction. Cell Mol Life Sci. 81:342024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou L, Pinho R, Gu Y and Radak Z: The
role of SIRT3 in exercise and aging. Cells. 11:25962022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Feldman JL, Dittenhafer-Reed KE and Denu
JM: Sirtuin catalysis and regulation. J Biol Chem. 287:42419–42427.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang J, Xiang H, Liu J, Chen Y, He RR and
Liu B: Mitochondrial sirtuin 3: New emerging biological function
and therapeutic target. Theranostics. 10:8315–8342. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grabowska W, Sikora E and Bielak-Zmijewska
A: Sirtuins, a promising target in slowing down the ageing process.
Biogerontology. 18:447–476. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Verdin E, Hirschey MD, Finley LW and
Haigis MC: Sirtuin regulation of mitochondria: Energy production,
apoptosis, and signaling. Trends Biochem Sci. 35:669–675. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mishra Y and Kaundal RK: Role of SIRT3 in
mitochondrial biology and its therapeutic implications in
neurodegenerative disorders. Drug Discov Today. 28:1035832023.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Y, Wen P, Luo J, Ding H, Cao H, He
W, Zen K, Zhou Y, Yang J and Jiang L: Sirtuin 3 regulates
mitochondrial protein acetylation and metabolism in tubular
epithelial cells during renal fibrosis. Cell Death Dis. 12:8472021.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hebert AS, Dittenhafer-Reed KE, Yu W,
Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ,
Higbee AJ, et al: Calorie restriction and SIRT3 trigger global
reprogramming of the mitochondrial protein acetylome. Mol Cell.
49:186–199. 2013. View Article : Google Scholar :
|
|
15
|
Iwahara T, Bonasio R, Narendra V and
Reinberg D: SIRT3 functions in the nucleus in the control of
stress-related gene expression. Mol Cell Biol. 32:5022–5034. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Onyango P, Celic I, McCaffery JM, Boeke JD
and Feinberg AP: SIRT3, a human SIR2 homologue, is an NAD-dependent
deacetylase localized to mitochondria. Proc Natl Acad Sci USA.
99:13653–13658. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang W, Nagasawa K, Münch C, Xu Y,
Satterstrom K, Jeong S, Hayes SD, Jedrychowski MP, Vyas FS,
Zaganjor E, et al: Mitochondrial sirtuin network reveals dynamic
SIRT3-dependent deacetylation in response to membrane
depolarization. Cell. 167:985–1000.e21. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shen H, Qi X, Hu Y, Wang Y, Zhang J, Liu Z
and Qin Z: Targeting sirtuins for cancer therapy: Epigenetics
modifications and beyond. Theranostics. 14:6726–6767. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Griffiths HBS, Williams C, King SJ and
Allison SJ: Nicotinamide adenine dinucleotide (NAD+): Essential
redox metabolite, co-substrate and an anti-cancer and anti-ageing
therapeutic target. Biochem Soc Trans. 48:733–744. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ouyang S, Zhang Q, Lou L, Zhu K, Li Z, Liu
P and Zhang X: The double-edged sword of SIRT3 in cancer and its
therapeutic applications. Front Pharmacol. 13:8715602022.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen LJ, Guo J, Zhang SX, Xu Y, Zhao Q,
Zhang W, Xiao J and Chen Y: Sirtuin3 rs28365927 functional variant
confers to the high risk of non-alcoholic fatty liver disease in
Chinese Han population. Lipids Health Dis. 20:922021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kane AE and Sinclair DA: Sirtuins and
NAD+ in the development and treatment of metabolic and
cardiovascular diseases. Circ Res. 123:868–885. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peterson BS, Campbell JE, Ilkayeva O,
Grimsrud PA, Hirschey MD and Newgard CB: Remodeling of the
acetylproteome by SIRT3 manipulation fails to affect insulin
secretion or β cell metabolism in the absence of overnutrition.
Cell Rep. 24:209–223.e6. 2018. View Article : Google Scholar
|
|
24
|
Sol EM, Wagner SA, Weinert BT, Kumar A,
Kim HS, Deng CX and Choudhary C: Proteomic investigations of lysine
acetylation identify diverse substrates of mitochondrial
deacetylase sirt3. PLoS One. 7:e505452012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hirschey MD, Shimazu T, Jing E, Grueter
CA, Collins AM, Aouizerat B, Stančáková A, Goetzman E, Lam MM,
Schwer B, et al: SIRT3 deficiency and mitochondrial protein
hyperacetylation accelerate the development of the metabolic
syndrome. Mol Cell. 44:177–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu J, Li D, Zhang T, Tong Q, Ye RD and
Lin L: SIRT3 protects hepatocytes from oxidative injury by
enhancing ROS scavenging and mitochondrial integrity. Cell Death
Dis. 8:e31582017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bell EL, Emerling BM, Ricoult SJ and
Guarente L: SirT3 suppresses hypoxia inducible factor 1α and tumor
growth by inhibiting mitochondrial ROS production. Oncogene.
30:2986–2996. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fukushi A, Kim HD, Chang YC and Kim CH:
Revisited metabolic control and reprogramming cancers by means of
the warburg effect in tumor cells. Int J Mol Sci. 23:100372022.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rinella ME, Neuschwander-Tetri BA,
Siddiqui MS, Abdelmalek MF, Caldwell S, Barb D, Kleiner DE and
Loomba R: AASLD practice guidance on the clinical assessment and
management of nonalcoholic fatty liver disease. Hepatology.
77:1797–1835. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Paternostro R and Trauner M: Current
treatment of non-alcoholic fatty liver disease. J Intern Med.
292:190–204. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grander C, Grabherr F and Tilg H:
Non-alcoholic fatty liver disease: Pathophysiological concepts and
treatment options. Cardiovasc Res. 119:1787–1798. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Di Ciaula A, Passarella S, Shanmugam H,
Noviello M, Bonfrate L, Wang DQ and Portincasa P: Nonalcoholic
fatty liver disease (NAFLD). Mitochondria as players and targets of
therapies? Int J Mol Sci. 22:53752021.
|
|
33
|
Zheng Y, Wang S, Wu J and Wang Y:
Mitochondrial metabolic dysfunction and non-alcoholic fatty liver
disease: New insights from pathogenic mechanisms to clinically
targeted therapy. J Transl Med. 21:5102023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pirola CJ, Gianotti TF, Burgueño AL,
Rey-Funes M, Loidl CF, Mallardi P, Martino JS, Castaño GO and
Sookoian S: Epigenetic modification of liver mitochondrial DNA is
associated with histological severity of nonalcoholic fatty liver
disease. Gut. 62:1356–1363. 2013. View Article : Google Scholar
|
|
35
|
Mazzoccoli G, De Cosmo S and Mazza T: The
biological clock: A pivotal hub in non-alcoholic fatty liver
disease pathogenesis. Front Physiol. 9:1932018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Polyzos SA, Kountouras J and Mantzoros CS:
Obesity and nonalcoholic fatty liver disease: From pathophysiology
to therapeutics. Metabolism. 92:82–97. 2019. View Article : Google Scholar
|
|
37
|
Ramanathan R, Ali AH and Ibdah JA:
Mitochondrial dysfunction plays central role in nonalcoholic fatty
liver disease. Int J Mol Sci. 23:72802022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kong X, Wang R, Xue Y, Liu X, Zhang H,
Chen Y, Fang F and Chang Y: Sirtuin 3, a new target of PGC-1alpha,
plays an important role in the suppression of ROS and mitochondrial
biogenesis. PLoS One. 5:e117072010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Barroso E, Rodríguez-Rodríguez R, Zarei M,
Pizarro-Degado J, Planavila A, Palomer X, Villarroya F and
Vázquez-Carrera M: SIRT3 deficiency exacerbates fatty liver by
attenuating the HIF1α-LIPIN 1 pathway and increasing CD36 through
Nrf2. Cell Commun Signal. 18:1472020. View Article : Google Scholar
|
|
40
|
Chen DD, Shi Q, Liu X, Liang DL, Wu YZ,
Fan Q, Xiao K, Chen C and Dong XP: Aberrant SENP1-SUMO-Sirt3
signaling causes the disturbances of mitochondrial deacetylation
and oxidative phosphorylation in prion-infected animal and cell
models. ACS Chem Neurosci. 14:1610–1621. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang Z, Dou X, Li S, Zhang X, Sun X, Zhou
Z and Song Z: Nuclear factor (erythroid-derived 2)-like 2
activation-induced hepatic very-low-density lipoprotein receptor
overexpression in response to oxidative stress contributes to
alcoholic liver disease in mice. Hepatology. 59:1381–1392. 2014.
View Article : Google Scholar
|
|
42
|
Green MF and Hirschey MD: SIRT3 weighs
heavily in the metabolic balance: A new role for SIRT3 in metabolic
syndrome. J Gerontol A Biol Sci Med Sci. 68:105–107. 2013.
View Article : Google Scholar
|
|
43
|
Kendrick AA, Choudhury M, Rahman SM,
McCurdy CE, Friederich M, Van Hove JL, Watson PA, Birdsey N, Bao J,
Gius D, et al: Fatty liver is associated with reduced SIRT3
activity and mitochondrial protein hyperacetylation. Biochem J.
433:505–514. 2011. View Article : Google Scholar
|
|
44
|
Lai CS, Tsai ML, Badmaev V, Jimenez M, Ho
CT and Pan MH: Xanthigen suppresses preadipocyte differentiation
and adipogenesis through down-regulation of PPARγ and C/EBPs and
modulation of SIRT-1, AMPK, and FoxO pathways. J Agric Food Chem.
60:1094–1101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
He J, Qian YC, Yin YC, Kang JR and Pan TR:
Polydatin: A potential NAFLD therapeutic drug that regulates
mitochondrial autophagy through SIRT3-FOXO3-BNIP3 and PINK1-PRKN
mechanisms-a network pharmacology and experimental investigation.
Chem Biol Interact. 398:1111102024. View Article : Google Scholar
|
|
46
|
Ren B, Kwah MX, Liu C, Ma Z, Shanmugam MK,
Ding L, Xiang X, Ho PC, Wang L, Ong PS and Goh BC: Resveratrol for
cancer therapy: Challenges and future perspectives. Cancer Lett.
515:63–72. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Karami A, Fakhri S, Kooshki L and Khan H:
Polydatin: Pharmacological mechanisms, therapeutic targets,
biological activities, and health benefits. Molecules. 27:64742022.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Imtiyaz K, Shafi M, Fakhri KU, Uroog L,
Zeya B, Anwer ST and Rizvi MMA: Polydatin: A natural compound with
multifaceted anticancer properties. J Tradit Complement Med.
15:447–466. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Malladi N, Lahamge D, Somwanshi BS, Tiwari
V, Deshmukh K, Balani JK, Chakraborty S, Alam MJ and Banerjee SK:
Paricalcitol attenuates oxidative stress and inflammatory response
in the liver of NAFLD rats by regulating FOXO3a and NFκB
acetylation. Cell Signal. 121:1112992024. View Article : Google Scholar
|
|
50
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI
|
|
51
|
Ladd AD, Duarte S, Sahin I and Zarrinpar
A: Mechanisms of drug resistance in HCC. Hepatology. 79:926–940.
2024. View Article : Google Scholar
|
|
52
|
Ahmed O and Pillai A: Hepatocellular
carcinoma: A contemporary approach to locoregional therapy. Am J
Gastroenterol. 115:1733–1736. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Vogel A, Meyer T, Sapisochin G, Salem R
and Saborowski A: Hepatocellular carcinoma. Lancet. 400:1345–1362.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang B, Qin L, Zhou CJ, Liu YL, Qian HX
and He SB: SIRT3 expression in hepatocellular carcinoma and its
impact on proliferation and invasion of hepatoma cells. Asian Pac J
Trop Med. 6:649–652. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang JX, Yi Y, Li YW, Cai XY, He HW, Ni
XC, Zhou J, Cheng YF, Jin JJ, Fan J and Qiu SJ: Down-regulation of
sirtuin 3 is associated with poor prognosis in hepatocellular
carcinoma after resection. BMC Cancer. 14:2972014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
De Matteis S, Scarpi E, Granato AM,
Vespasiani-Gentilucci U, La Barba G, Foschi FG, Bandini E, Ghetti
M, Marisi G, Cravero P, et al: Role of SIRT-3, p-mTOR and HIF-1α in
hepatocellular carcinoma patients affected by metabolic
dysfunctions and in chronic treatment with metformin. Int J Mol
Sci. 20:15032019. View Article : Google Scholar
|
|
57
|
Liu H, Li S, Liu X, Chen Y and Deng H:
SIRT3 overexpression inhibits growth of kidney tumor cells and
enhances mitochondrial biogenesis. J Proteome Res. 17:3143–3152.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Le A, Cooper CR, Gouw AM, Dinavahi R,
Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL and Dang
CV: Inhibition of lactate dehydrogenase A induces oxidative stress
and inhibits tumor progression. Proc Natl Acad Sci USA.
107:2037–2042. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kwon SM, Lee YK, Min S, Woo HG, Wang HJ
and Yoon G: Mitoribosome defect in hepatocellular carcinoma
promotes an aggressive phenotype with suppressed immune reaction.
iScience. 23:1012472020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Fan J, Shan C, Kang HB, Elf S, Xie J,
Tucker M, Gu TL, Aguiar M, Lonning S, Chen H, et al: Tyr
phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3
to regulate the pyruvate dehydrogenase complex. Mol Cell.
53:534–548. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Che L, Wu JS, Du ZB, He YQ, Yang L, Lin
JX, Lei Z, Chen XX, Guo DB, Li WG, et al: Targeting mitochondrial
COX-2 enhances chemosensitivity via Drp1-dependent remodeling of
mitochondrial dynamics in hepatocellular carcinoma. Cancers
(Basel). 14:8212022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ceballos MP, Quiroga AD and Palma NF: Role
of sirtuins in hepatocellular carcinoma progression and multidrug
resistance: Mechanistical and pharmacological perspectives. Biochem
Pharmacol. 212:1155732023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lu J, Zhang H, Chen X, Zou Y, Li J, Wang
L, Wu M, Zang J, Yu Y, Zhuang W, et al: A small molecule activator
of SIRT3 promotes deacetylation and activation of manganese
superoxide dismutase. Free Radic Biol Med. 112:287–297. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
De Matteis S, Granato AM, Napolitano R,
Molinari C, Valgiusti M, Santini D, Foschi FG, Ercolani G,
Vespasiani Gentilucci U, Faloppi L, et al: Interplay between
SIRT-3, metabolism and its tumor suppressor role in hepatocellular
carcinoma. Dig Dis Sci. 62:1872–1880. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kim YI, Ko I, Yi EJ, Kim J, Hong YR, Lee W
and Chang SY: NAD+ modulation of intestinal macrophages
renders anti-inflammatory functionality and ameliorates gut
inflammation. Biomed Pharmacother. 185:1179382025. View Article : Google Scholar
|
|
66
|
Guan Q: A comprehensive review and update
on the pathogenesis of inflammatory bowel disease. J Immunol Res.
2019:72472382019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chen X, Zhang M, Zhou F, Gu Z, Li Y, Yu T,
Peng C, Zhou L, Li X, Zhu D, et al: SIRT3 activator honokiol
inhibits Th17 cell differentiation and alleviates colitis. Inflamm
Bowel Dis. 29:1929–1940. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Nistala K and Wedderburn LR: Th17 and
regulatory T cells: Rebalancing pro- and anti-inflammatory forces
in autoimmune arthritis. Rheumatology (Oxford). 48:602–606. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Yu R, Zuo F, Ma H and Chen S:
Exopolysaccharide-producing Bifidobacterium adolescentis strains
with similar adhesion property induce differential regulation of
inflammatory immune response in Treg/Th17 axis of DSS-colitis mice.
Nutrients. 11:7822019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhang M, Zhou L, Xu Y, Yang M, Xu Y,
Komaniecki GP, Kosciuk T, Chen X, Lu X, Zou X, et al: A STAT3
palmitoylation cycle promotes TH17 differentiation and
colitis. Nature. 586:434–439. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Cros C, Margier M, Cannelle H, Charmetant
J, Hulo N, Laganier L, Grozio A and Canault M: Nicotinamide
mononucleotide administration triggers macrophages reprogramming
and alleviates inflammation during sepsis induced by experimental
peritonitis. Front Mol Biosci. 9:8950282022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Vergadi E, Ieronymaki E, Lyroni K,
Vaporidi K and Tsatsanis C: Akt signaling pathway in macrophage
activation and M1/M2 polarization. J Immunol. 198:1006–1014. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhou W, Hu G, He J, Wang T, Zuo Y, Cao Y,
Zheng Q, Tu J, Ma J, Cai R, et al: SENP1-Sirt3 signaling promotes
α-ketoglutarate production during M2 macrophage polarization. Cell
Rep. 39:1106602022. View Article : Google Scholar
|
|
74
|
Furness JB: The enteric nervous system and
neurogastroenterology. Nat Rev Gastroenterol Hepatol. 9:286–294.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Warnecke T, Schäfer KH, Claus I, Del
Tredici K and Jost WH: Gastrointestinal involvement in Parkinson's
disease: Pathophysiology, diagnosis, and management. NPJ Parkinsons
Dis. 8:312022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Gockel I, Müller M and Schumacher J:
Achalasia-a disease of unknown cause that is often diagnosed too
late. Dtsch Arztebl Int. 109:209–214. 2012.PubMed/NCBI
|
|
77
|
Niesler B, Kuerten S, Demir IE and Schäfer
KH: Disorders of the enteric nervous system-a holistic view. Nat
Rev Gastroenterol Hepatol. 18:393–410. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wood JD, Liu S, Drossman DA, Ringel Y and
Whitehead WE: Anti-enteric neuronal antibodies and the irritable
bowel syndrome. J Neurogastroenterol Motil. 18:78–85. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Brown IAM, McClain JL, Watson RE, Patel BA
and Gulbransen BD: Enteric glia mediate neuron death in colitis
through purinergic pathways that require connexin-43 and nitric
oxide. Cell Mol Gastroenterol Hepatol. 2:77–91. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Shi H, Deng HX, Gius D, Schumacker PT,
Surmeier DJ and Ma YC: Sirt3 protects dopaminergic neurons from
mitochondrial oxidative stress. Hum Mol Genet. 26:1915–1926. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhang X, Ren X, Zhang Q, Li Z, Ma S, Bao
J, Li Z, Bai X, Zheng L, Zhang Z, et al: PGC-1α/ERRα-Sirt3 pathway
regulates daergic neuronal death by directly deacetylating SOD2 and
ATP synthase β. Antioxid Redox Signal. 24:312–328. 2016. View Article : Google Scholar :
|
|
82
|
Pillai VB, Samant S, Sundaresan NR,
Raghuraman H, Kim G, Bonner MY, Arbiser JL, Walker DI, Jones DP,
Gius D and Gupta MP: Honokiol blocks and reverses cardiac
hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun.
6:66562015. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Pillai VB, Kanwal A, Fang YH, Sharp WW,
Samant S, Arbiser J and Gupta MP: Honokiol, an activator of
Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart
from doxorubicin-induced cardiomyopathy in mice. Oncotarget.
8:34082–34098. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Balasubramaniam A, Li G, Ramanathan A,
Mwangi SM, Hart CM, Arbiser JL and Srinivasan S: SIRT3 activation
promotes enteric neurons survival and differentiation. Sci Rep.
12:220762022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Sudo N: Biogenic amines: Signals between
commensal microbiota and gut physiology. Front Endocrinol
(Lausanne). 10:5042019. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Munteanu C, Onose G, Poștaru M, Turnea M,
Rotariu M and Galaction AI: Hydrogen sulfide and gut microbiota:
Their synergistic role in modulating sirtuin activity and potential
therapeutic implications for neurodegenerative diseases.
Pharmaceuticals (Basel). 17:14802024. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM
and Wallace MB: Colorectal cancer. Lancet. 394:1467–1480. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Sánchez-Aragó M, Chamorro M and Cuezva JM:
Selection of cancer cells with repressed mitochondria triggers
colon cancer progression. Carcinogenesis. 31:567–576. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ku M, Koche RP, Rheinbay E, Mendenhall EM,
Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, et al:
Genomewide analysis of PRC1 and PRC2 occupancy identifies two
classes of bivalent domains. PLoS Genet. 4:e10002422008. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Gan L, Li Q, Nie W, Zhang Y, Jiang H, Tan
C, Zhang L, Zhang J, Li Q, Hou P, et al: PROX1-mediated epigenetic
silencing of SIRT3 contributes to proliferation and glucose
metabolism in colorectal cancer. Int J Biol Sci. 19:50–65. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Torrens-Mas M, Hernández-López R, Pons DG,
Roca P, Oliver J and Sastre-Serra J: Sirtuin 3 silencing impairs
mitochondrial biogenesis and metabolism in colon cancer cells. Am J
Physiol Cell Physiol. 317:C398–C404. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metabol. 23:27–47. 2016.
View Article : Google Scholar
|
|
93
|
Amelio I, Cutruzzolá F, Antonov A,
Agostini M and Melino G: Serine and glycine metabolism in cancer.
Trends Biochem Sci. 39:191–198. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Lee GY, Haverty PM, Li L, Kljavin NM,
Bourgon R, Lee J, Stern H, Modrusan Z, Seshagiri S, Zhang Z, et al:
Comparative oncogenomics identifies PSMB4 and SHMT2 as potential
cancer driver genes. Cancer Res. 74:3114–3126. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wei Z, Song J, Wang G, Cui X, Zheng J,
Tang Y, Chen X, Li J, Cui L, Liu CY and Yu W: Deacetylation of
serine hydroxymethyl-transferase 2 by SIRT3 promotes colorectal
carcinogenesis. Nat Commun. 9:44682018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Colloca A, Balestrieri A, Anastasio C,
Balestrieri ML and D'Onofrio N: Mitochondrial sirtuins in chronic
degenerative diseases: New metabolic targets in colorectal cancer.
Int J Mol Sci. 23:32122022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Abdelmaksoud NM, Abulsoud AI, Abdelghany
TM, Elshaer SS, Rizk SM, Senousy MA and Maurice NW: Uncovering
SIRT3 and SHMT2-dependent pathways as novel targets for apigenin in
modulating colorectal cancer: In vitro and in vivo studies. Exp
Cell Res. 441:1141502024. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
D'Onofrio N, Martino E, Balestrieri A,
Mele L, Cautela D, Castaldo D and Balestrieri ML: Diet-derived
ergothioneine induces necroptosis in colorectal cancer cells by
activating the SIRT3/MLKL pathway. FEBS Lett. 596:1313–1329. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Colloca A, Donisi I, Anastasio C,
Balestrieri ML and D'Onofrio N: Metabolic alteration bridging the
prediabetic state and colorectal cancer. Cells. 13:6632024.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Anastasio C, Donisi I, Del Vecchio V,
Colloca A, Mele L, Sardu C, Marfella R, Balestrieri ML and
D'Onofrio N: SGLT2 inhibitor promotes mitochondrial dysfunction and
ER-phagy in colorectal cancer cells. Cell Mol Biol Lett. 29:802024.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Sharma A, Baker S, Duijm M, Oomen-de Hoop
E, Cornelissen R, Verhoef C, Hoogeman M and Jan Nuyttens J:
Prognostic factors for local control and survival for inoperable
pulmonary colorectal oligometastases treated with stereotactic body
radiotherapy. Radiother Oncol. 144:23–29. 2020. View Article : Google Scholar
|
|
102
|
Wei Y, Xiao G, Xu H, Sun X, Shi Y, Wang F,
Kang J, Peng J and Zhou F: Radiation resistance of cancer cells
caused by mitochondrial dysfunction depends on SIRT3-mediated
mitophagy. FEBS J. 290:3629–3645. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Frost F, Kacprowski T, Rühlemann M, Bülow
R, Kühn JP, Franke A, Heinsen FA, Pietzner M, Nauck M, Völker U, et
al: Impaired exocrine pancreatic function associates with changes
in intestinal microbiota composition and diversity.
Gastroenterology. 156:1010–1015. 2019. View Article : Google Scholar
|
|
104
|
Li X, He C, Li N, Ding L, Chen H, Wan J,
Yang X, Xia L, He W, Xiong H, et al: The interplay between the gut
microbiota and NLRP3 activation affects the severity of acute
pancreatitis in mice. Gut Microbes. 11:1774–1789. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Liu LW, Xie Y, Li GQ, Zhang T, Sui YH,
Zhao ZJ, Zhang YY, Yang WB, Geng XL, Xue DB, et al: Gut
microbiota-derived nicotinamide mononucleotide alleviates acute
pancreatitis by activating pancreatic SIRT3 signalling. Br J
Pharmacol. 180:647–666. 2023. View Article : Google Scholar
|
|
106
|
Cai J, Chen H, Lu M, Zhang Y, Lu B, You L,
Zhang T, Dai M and Zhao Y: Advances in the epidemiology of
pancreatic cancer: Trends, risk factors, screening, and prognosis.
Cancer Lett. 520:1–11. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhou Y, Cheng S, Chen S and Zhao Y:
Prognostic and clinicopathological value of SIRT3 expression in
various cancers: A systematic review and meta-analysis. Onco
Targets Ther. 11:2157–2167. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Haigis MC, Deng CX, Finley LW, Kim HS and
Gius D: SIRT3 is a mitochondrial tumor suppressor: A scientific
tale that connects aberrant cellular ROS, the Warburg effect, and
carcinogenesis. Cancer Res. 72:2468–2472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Yao W, Cai X, Liu C, Qin Y, Cheng H, Ji S,
Xu W, Wu C, Chen T, Xu J, et al: Profilin 1 potentiates apoptosis
induced by staurosporine in cancer cells. Curr Mol Med. 13:417–428.
2013.PubMed/NCBI
|
|
110
|
Yao W, Ji S, Qin Y, Yang J, Xu J, Zhang B,
Xu W, Liu J, Shi S, Liu L, et al: Profilin-1 suppresses
tumorigenicity in pancreatic cancer through regulation of the
SIRT3-HIF1α axis. Mol Cancer. 13:1872014. View Article : Google Scholar
|
|
111
|
Jeong SM, Lee J, Finley LW, Schmidt PJ,
Fleming MD and Haigis MC: SIRT3 regulates cellular iron metabolism
and cancer growth by repressing iron regulatory protein 1.
Oncogene. 34:2115–2124. 2015. View Article : Google Scholar
|
|
112
|
Chouhan S, Kumar A, Muhammad N, Usmani D
and Khan TH: Sirtuins as key regulators in pancreatic cancer:
Insights into signaling mechanisms and therapeutic implications.
Cancers (Basel). 16:40952024. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Ma Z, Li Z, Wang S, Zhou Z, Liu C, Zhuang
H, Zhou Q, Huang S, Zhang C and Hou B: ZMAT1 acts as a tumor
suppressor in pancreatic ductal adenocarcinoma by inducing
SIRT3/p53 signaling pathway. J Exp Clin Cancer Res. 41:1302022.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ragab EM, El Gamal DM, Mohamed TM and
Khamis AA: Impairment of electron transport chain and induction of
apoptosis by chrysin nanoparticles targeting succinate-ubiquinone
oxidoreductase in pancreatic and lung cancer cells. Genes Nutr.
18:42023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Shi S, Yao W, Xu J, Long J, Liu C and Yu
X: Combinational therapy: New hope for pancreatic cancer? Cancer
Lett. 317:127–135. 2012. View Article : Google Scholar
|
|
116
|
Thrift AP, Wenker TN and El-Serag HB:
Global burden of gastric cancer: Epidemiological trends, risk
factors, screening and prevention. Nat Rev Clin Oncol. 20:338–349.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Kim HS, Patel K, Muldoon-Jacobs K, Bisht
KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage
J, Owens KM, et al: SIRT3 is a mitochondria-localized tumor
suppressor required for maintenance of mitochondrial integrity and
metabolism during stress. Cancer Cell. 17:41–52. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Tao R, Coleman MC, Pennington JD, Ozden O,
Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, et
al: Sirt3-mediated deacetylation of evolutionarily conserved lysine
122 regulates MnSOD activity in response to stress. Mol Cell.
40:893–904. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Cui Y, Qin L, Wu J, Qu X, Hou C, Sun W, Li
S, Vaughan AT, Li JJ and Liu J: SIRT3 enhances glycolysis and
proliferation in SIRT3-expressing gastric cancer cells. PLoS One.
10:e01298342015. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Arnold M, Park JY, Camargo MC, Lunet N,
Forman D and Soerjomataram I: Is gastric cancer becoming a rare
disease? A global assessment of predicted incidence trends to 2035.
Gut. 69:823–829. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Roa JC, García P, Kapoor VK, Maithel SK,
Javle M and Koshiol J: Gallbladder cancer. Nat Rev Dis Primers.
8:692022. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Liu L, Li Y, Cao D, Qiu S, Li Y, Jiang C,
Bian R, Yang Y, Li L, Li X, et al: SIRT3 inhibits gallbladder
cancer by induction of AKT-dependent ferroptosis and blockade of
epithelial-mesenchymal transition. Cancer Lett. 510:93–104. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Aiello NM and Kang Y: Context-dependent
EMT programs in cancer metastasis. J Exp Med. 216:1016–1026. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Osborne B, Bentley NL, Montgomery MK and
Turner N: The role of mitochondrial sirtuins in health and disease.
Free Radic Biol Med. 100:164–174. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Rose G, Dato S, Altomare K, Bellizzi D,
Garasto S, Greco V, Passarino G, Feraco E, Mari V, Barbi C, et al:
Variability of the SIRT3 gene, human silent information regulator
Sir2 homologue, and survivorship in the elderly. Exp Gerontol.
38:1065–1070. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Sinclair DA and Guarente L: Small-molecule
allosteric activators of sirtuins. Annu Rev Pharmacol Toxicol.
54:363–380. 2014. View Article : Google Scholar :
|
|
128
|
Morigi M, Perico L and Benigni A: Sirtuins
in renal health and disease. J Am Soc Nephrol. 29:1799–1809. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Akamata K, Wei J, Bhattacharyya M, Cheresh
P, Bonner MY, Arbiser JL, Raparia K, Gupta MP, Kamp DW and Varga J:
SIRT3 is attenuated in systemic sclerosis skin and lungs, and its
pharmacologic activation mitigates organ fibrosis. Oncotarget.
7:69321–69336. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Wang J, Wang K, Huang C, Lin D, Zhou Y, Wu
Y, Tian N, Fan P, Pan X, Xu D, et al: SIRT3 activation by
dihydromyricetin suppresses chondrocytes degeneration via
maintaining mitochondrial homeostasis. Int J Biol Sci.
14:1873–1882. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zhang H, Chen Y, Pei Z, Gao H, Shi W, Sun
M, Xu Q, Zhao J, Meng W and Xiao K: Protective effects of polydatin
against sulfur mustard-induced hepatic injury. Toxicol Appl
Pharmacol. 367:1–11. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Zhang J, Meruvu S, Bedi YS, Chau J,
Arguelles A, Rucker R and Choudhury M: Pyrroloquinoline quinone
increases the expression and activity of Sirt1 and -3 genes in
HepG2 cells. Nutr Res. 35:844–849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Peng F, Liao M, Jin W, Liu W, Li Z, Fan Z,
Zou L, Chen S, Zhu L, Zhao Q, et al: 2-APQC, a small-molecule
activator of Sirtuin-3 (SIRT3), alleviates myocardial hypertrophy
and fibrosis by regulating mitochondrial homeostasis. Signal
Transduct Target Ther. 9:1332024. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Schlicker C, Boanca G, Lakshminarasimhan M
and Steegborn C: Structure-based development of novel sirtuin
inhibitors. Aging (Albany NY). 3:852–872. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Nakamura Y, Suganami A, Fukuda M, Hasan
MK, Yokochi T, Takatori A, Satoh S, Hoshino T, Tamura Y and
Nakagawara A: Identification of novel candidate compounds targeting
TrkB to induce apoptosis in neuroblastoma. Cancer Med. 3:25–35.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Alhazzazi TY, Kamarajan P, Xu Y, Ai T,
Chen L, Verdin E and Kapila YL: A novel sirtuin-3 inhibitor,
LC-0296, inhibits cell survival and proliferation, and promotes
apoptosis of head and neck cancer cells. Anticancer Res. 36:49–60.
2016.PubMed/NCBI
|