As a sentinel of the immune system, macrophages are
characterized by the robust phagocytic capacity to play a pivotal
role in pathogen clearance and tissue homeostasis (1). Based on their developmental
origins, macrophages are categorized into hematopoietic
monocyte-derived macrophages and embryonic-derived tissue-resident
macrophages (2).
Hematopoietic-derived macrophages originate from CD34+
hematopoietic stem cells (HSCs) in the bone marrow, progressing
through granulocyte-monocyte progenitors and monocytic stages,
before entering systemic circulation (3). Under specific microenvironmental
signals, such as inflammatory mediators and chemokines, circulating
monocytes are recruited to tissues and subsequently differentiate
into functionally specialized macrophages. By contrast,
embryonic-derived tissue-resident macrophages originate from yolk
sac precursors during organogenesis (4-9),
including microglia in the central nervous system (10), Kupffer cells in the liver
(11) and Langerhans cells in
the skin (12). These
developmentally distinct populations differ fundamentally from
their hematopoietic counterparts in both ontogeny and functional
specialization (2,6,8,13-16).
Macrophages exhibit extraordinary plasticity and
functional heterogeneity, dynamically adapting their polarization
states in response to microenvironmental stimuli (17). The classical paradigm classifies
macrophages into pro-inflammatory M1 and
anti-inflammatory/reparative M2 phenotypes (18-20). M1 macrophages, activated by
interferon (IFN)-γ and lipopolysaccharide (LPS) (21,22), amplify inflammatory responses and
resist pathogenic infection through the production of IL-1β, IL-6,
IL-12, TNF-α and chemokines (23-26). While being critical for pathogen
containment, sustained M1 activation leads to chronic inflammation
and tissue destruction. Conversely, M2 macrophages polarized by
IL-4/IL-13 secrete immunosuppressive cytokines (such as IL-10 and
TGF-β) to alleviate inflammation and promote tissue regeneration
through growth factor-mediated extracellular matrix remodeling and
angiogenesis (27). However,
their immunosuppressive function may facilitate tumor progression
by establishing pro-tumorigenic microenvironments. Importantly, the
M1/M2 classification is considered an oversimplification of
macrophage functional spectrum. Under pathophysiological
conditions, macrophages exhibit a continuous spectrum of
polarization from M1 to M2 phenotypes, rather than discrete binary
states (28-31). Tissue macrophages frequently
exhibit hybrid phenotypes with overlapping M1-M2 characteristics,
reflecting their capacity to adapt to dynamic microenvironmental
changes. To further clarify this conceptual framework, it is
essential to distinguish between the processes of macrophage
activation and polarization. Macrophage activation refers to the
early process by which macrophages sense external stimuli through
receptor-mediated signaling pathways, initiating intracellular
cascades that endow them with enhanced responsiveness and effector
potential. Macrophage activation represents a transition from
resting state to functionally poised state. By contrast, macrophage
polarization, dictated by specific cytokine milieu and
environmental cues, denotes a more specialized process in which
activated macrophages differentiate along a continuous spectrum
into distinct functional phenotypes (32,33). Therefore, activation represents
the initiation of macrophage responsiveness, whereas polarization
describes the directional specification that follows.
Through coordinated phagocytosis, antigen
presentation, cytokine secretion and crosstalk with immune and
non-immune cells, macrophages play a central role in regulating
inflammatory responses, host defense and tissue homeostasis.
Dysregulated macrophage function underlies numerous pathologies.
For instance, macrophages take up lipids from the blood to
transform into foam cells, thereby promoting plaque formation and
atherosclerotic progression (34-39). Obesity-associated metabolic
disorders are featured by the macrophage-mediated chronic low-grade
inflammation, which triggers the development of insulin resistance
and type 2 diabetes (40-47).
In certain cases, M2 macrophages may promote tumor cell growth,
metastasis, invasion and drug resistance by secreting
immunosuppressive factors and enhancing tumor angiogenesis through
cytokines (such as VEGF) (48-52). As highly plastic innate immune
cells derived from myeloid precursors, the differentiation of
macrophage is orchestrated by the macrophage colony-stimulating
factor (M-CSF, also known as CSF1) (53-55). This process is fundamentally
governed by the transcription factor purine-rich U-box binding
protein 1, which drives macrophage lineage differentiation through
transcriptional activation of the M-CSF receptor CSF1R (56-58). NF-κB and STAT1 pathways drive
classical M1 polarization (59,60), while alternative M2 polarization
is mediated by JAK1/3-STAT6 signaling and nuclear receptor
peroxisome proliferator-activated receptor gamma (PPARγ) (61-65). Collectively, these observations
underscore that macrophage differentiation, activation and
polarization are governed by intricate transcriptional networks.
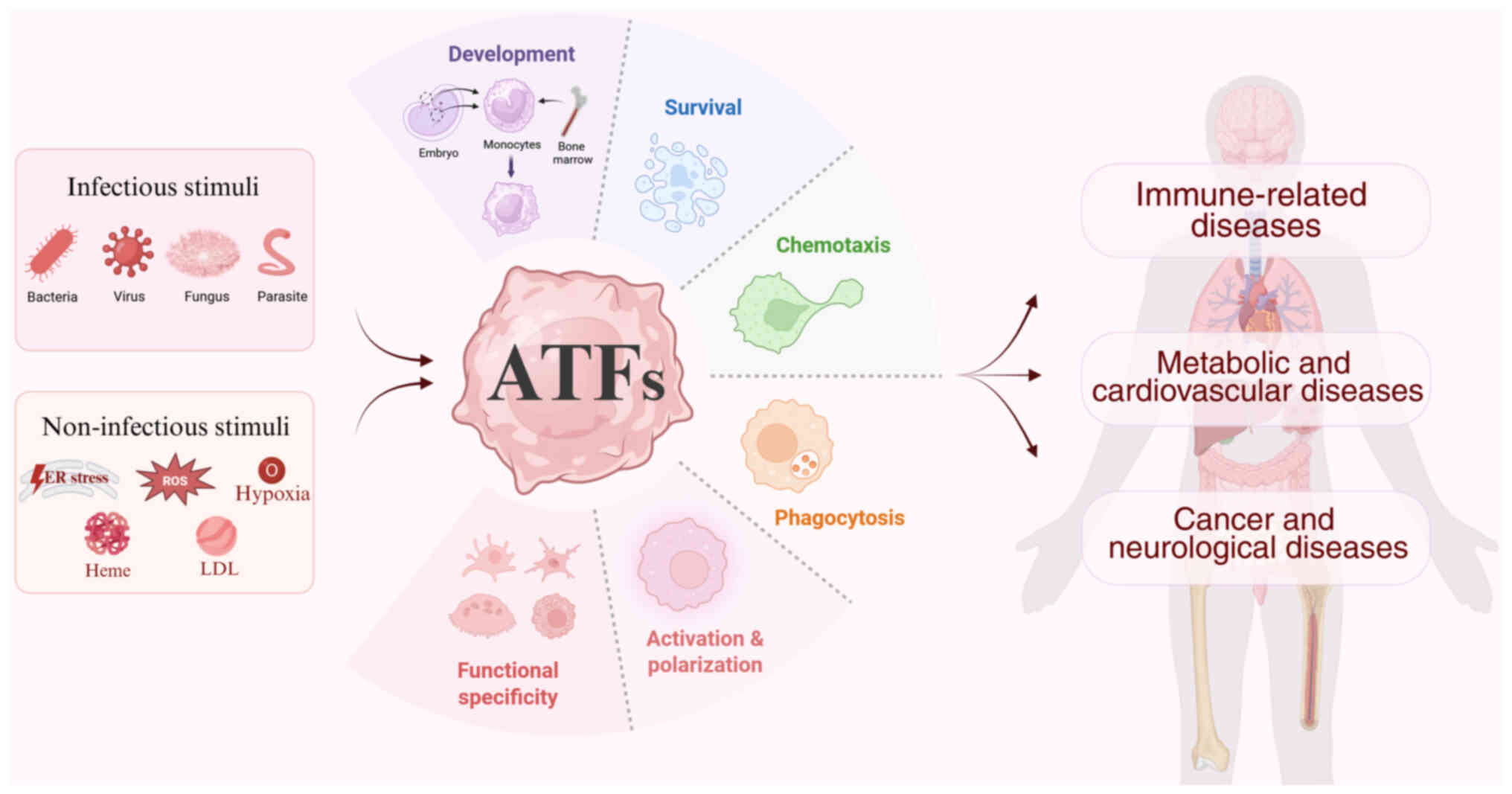
Among these, the activating transcription factors (ATFs) have
emerged as pivotal integrators of immune and metabolic cues,
orchestrating macrophage responses under diverse physiological and
pathological conditions. The current review sought to summarize the
recent findings on how activated ATFs regulate macrophage
development, survival, migration, phagocytosis, activation,
cytokine secretion, polarization and their involvement in immune,
metabolic, cardiovascular, neurological disorders and cancer, with
a particular emphasis on their therapeutic potential (Fig. 1).
The ATF family, comprising seven members (ATF1-7),
was initially characterized in 1987, as a group of transcriptional
factors involved in the regulation of gene expression across
diverse biological contexts (66). All ATF members share a highly
conserved basic leucine zipper (bZIP) domain comprising a basic
region responsible for DNA binding and a leucine zipper structure
that mediates dimerization (67,68). ATFs are activated by upstream
signals and exert transcriptional activities by forming either
homodimers or heterodimers with other bZIP transcription factors
such as AP-1 or C/EBP family members. ATF1 commonly forms
heterodimers with cAMP response element-binding protein (CREB) or
cAMP response element modulator (CREM) family members to bind to
DNA and regulate target gene transcription (69-72). ATF2 dimerizes with c-Jun to form
a canonical AP-1 complex (73,74), while its structurally related
homolog ATF7 could interact with ATF2 to generate functional
heterodimers (75-77). ATF3 cooperates with bZIP proteins
including c-Jun, JunB, or C/EBP to regulate responsive genes
(78,79). ATF4 integrates stress and
metabolic signals through heterodimerization with members of the
C/EBP family or with nuclear factor erythroid 2-related factor 2
(NRF2) to coordinate adaptive transcriptional programs (80-82). Similarly, ATF5 forms functional
heterodimers with C/EBPγ (83).
ATF6, however, undergoes a unique activation process. At steady
state, ATF6 resides in the endoplasmic reticulum (ER) membrane as a
monomer, dimer, or oligomer. Upon the challenge of ER stress, ATF6
translocates into the Golgi apparatus, where it is cleaved by
S1P/S2P proteases to release its N-terminal fragment (50 kDa) that
enters the nucleus. The nuclear p50 ATF6 forms homodimers or
heterodimers with its homolog ATF6β (84) and both of them can associate with
nuclear transcription factor Y (NF-Y) to form transcriptional
complexes (85). In addition,
ATF6 has been shown to heterodimerize with XBP1s, thereby
broadening its target gene repertoire (86).
The specific dimerization partners of each ATF
member largely determine its DNA-binding specificity and
transcriptional outcomes. Accordingly, ATF1 and ATF5 predominantly
recognize canonical CRE sequences (70,87), whereas the binding preferences of
ATF2, ATF3 and ATF7 are largely determined by their dimerization
partners (88-91). Once they form homodimers or
heterodimers with the CREB family members, they preferentially bind
to the CRE consensus sequence. By contrast, heterodimerization with
AP-1 family proteins, such as c-Jun, redirects their binding toward
AP-1 sites (74,78,92-94). Moreover, ATF3 and ATF4 can
recognize C/EBP-ATF response elements (CARE), to composite motifs
containing both CCAAT-box and CRE characteristics (95). Under stress conditions, the
C/EBPγ-ATF4 heterodimer constitutes the predominant form to bind to
the CARE motifs (81), whereas
ATF4 can also form a complex with C/EBPβ to regulate
differentiation-related gene expression (80). When ATF4 forms heterodimers with
NRF2, the complex can recognize both antioxidant response elements
(AREs) and ATF/CRE motifs, thereby synergistically activating
antioxidant and detoxifying gene transcription (96). Likewise, ATF6α/ATF6β
heterodimers, as well as ATF6α homodimers, associate with NF-Y to
recognize ER stress response elements, which comprise a 5'-CCAAT-3'
core, a GC-rich spacer and a terminal 5'-CCACG-3' motif (97-101). Within this composite motif, the
CCAAT box serves as the NF-Y binding site, whereas ATF6 recognizes
and binds to the terminal CCACG half-site to initiate transcription
of target genes (85,102). Notably, CRE sequences vary
among genes, often exhibiting one or two base substitutions and
even half-CRE sites (such as TGACG or CGTCA) can be recognized by
ATFs, suggesting that while the core CRE motif is evolutionarily
conserved, a certain degree of sequence flexibility is tolerated
(103).
The activation and function of the ATF family are
governed by multi-layered regulatory mechanisms operating across
epigenetic, transcriptional and post-translational dimensions.
Epigenetic control involves DNA methylation (such as ATF3 silencing
through promoter hypermethylation) (104), dynamic histone modifications
and mRNA m6A modifications. Transcriptionally, factors, such as
SP1/MYC and p53, modulate ATF activity via promoter binding
(104). Post-translationally,
numerous ATFs are activated through phosphorylation (PKA or MAPK)
(105), while others undergo
regulated proteolysis (such as cleavage of ATF6) or
ubiquitin-mediated turnover (106), all of which influence their
stability, localization and activity. Specifically, under stress
conditions, JNK/p38 MAPK phosphorylates ATF2 to enhance its
transcriptional activity (107), while ER stress activates ATF4
and ATF6 via the unfolded protein response (UPR) to restore
proteostasis (106,108). Moreover, ATFs could also be
regulated by signals such as IL-6, LPS or growth factors (109-111), thereby integrating immune and
metabolic cues into coordinated transcriptional responses. Once
activated, ATFs execute their transcriptional regulatory functions
primarily through the recruitment of a wide range of
transcriptional cofactors and chromatin-modifying complexes that
sculpt gene expression programs. Among them, ATF family members
recruit diverse histone-modifying enzymes and chromatin-remodeling
complexes to mediate epigenetic regulation. For example,
phosphorylated ATF1 recruits the histone acetyltransferase CBP/p300
to enhance local H3/H4 acetylation and chromatin accessibility
(112,113). Similarly, following LPS
stimulation, ATF2 recruits histone deacetylase 1 (HDAC1) to remove
repressive acetylation marks, thereby facilitating target gene
transcription (114). However,
activated ATF3 recruits HDAC1 onto promoters of proinflammatory
genes to erase permissive acetylation marks and repress gene
transcription, implying the complex nature of ATF-related
epigenetic regulation (115,116). Upon the insult of ER stress,
ATF6α recruits the mediator complex together with multiple histone
acetyltransferase assemblies, including SAGA and ATAC, to promoters
such as HSPA5, to increase local histone acetylation level
(117,118). ATF7 persistently recruits the
histone H3K9 methyltransferase G9a in resting macrophages to
deposit the repressive H3K9me2 mark, which silences the
transcription of innate immune genes and maintain the quiescent
state (119). Moreover, ATF6α
recruits arginine methyltransferases such as PRMT1, to methylate
histone arginine residues at target genes, by which it remodels
chromatin architecture and regulates stress-responsive gene
expression (120). Moreover,
some ATF members can directly or indirectly recruit ATP-dependent
chromatin-remodeling complexes, altering nucleosome positioning and
conformation to reorganize the chromatin landscape (112).
ATFs play a crucial role in various biological
processes. For instance, ATF family members regulate antioxidant
gene expression during ER stress and oxidative stress responses,
serving as a critical regulator in stress adaptation (121-123). In glucose and lipid metabolism,
ATF proteins modulate the expression of multiple metabolic genes to
maintain glucose and lipid homeostasis (111,124). Furthermore, ATF members
participate in immune responses by regulating inflammatory cascades
and immune evasion (125-128), the balance between autophagy
and apoptosis (129) and the
process of cell senescence (126). Notably, perturbation of ATF
signaling networks demonstrates strong association with tumor
(130), metabolic disorders
(121), neurodegenerative
diseases (121) and
immune-related diseases (124,131). Such dynamic nature warrants
ATFs as a crucial hub for developing targeted therapeutic
strategies.
ATFs exert multidimensional regulatory effect on
macrophage development and functional homeostasis through
regulating the proliferation, differentiation and metabolic
pathways. During macrophage development, ATF4 serves as a master
regulator to govern monocytes differentiation to gut-resident
macrophages (gMac). In septic models, ATF4 depletion in
Ly6C+ monocyte precursors (P1 cells) disrupts gMac (P4
cell) differentiation coupled with aberrant proliferation and
increased apoptosis of P1 cells, leading to exacerbated intestinal
barrier dysfunction (132).
ATF2 orchestrates monocyte differentiation through modulating the
expression of phosphatase PPM1A by binding to its promoter
(133). In atherosclerosis, the
accumulation of macrophages is markedly reduced in plaques of mice
overexpressing ATF3. ATF3 overexpression suppresses the PI3K/AKT
signaling pathway, by which it downregulates the expression of
matrix metalloproteinases (MMP-2/MMP-9) and reduces
macrophage-mediated extracellular matrix degradation, coupled with
plaque stabilization (134).
Notably, the anti-proliferative effect of ATF3 persists throughout
the macrophage lifespan by suppressing cell cycle genes
(Mem2 and Cdk2) and inhibiting the Clec4e-Csf1 axis
(135,136).
Regarding macrophage senescence and cell death, ATF4
is identified as the core responder to hypoxic stress and it
rapidly accumulates in macrophage nuclei within 1-h of hypoxic
challenge to activate adaptive gene networks to sustain cell
viability (137). ATF3 exhibits
anti-senescence properties in Pseudomonas aeruginosa
infection models. Macrophages deficient in ATF3 exhibit
elevated ROS levels and accelerated senescence, while ectopic ATF3
overexpression partially reverses these phenotypes (138). Additionally, in LPS-stimulated
microglia, ATF2 exacerbates inflammatory pyroptosis by upregulating
IL-1β, NLRP3 and key pyroptotic effector molecules such as GSDMDC1
and Caspase-1 (139).
ATF family members essentially regulate macrophage
chemotaxis and phagocytosis by modulating chemokine expression and
cytoskeletal remodeling. LPS induces phosphorylation of ATF2 in
alveolar epithelial cells, which subsequently upregulates
macrophage inflammatory protein-2 (MIP-2) to promote macrophage
recruitment (140).
Additionally, LPS enhances macrophage chemotactic capacity
via ATF3, which binds to the CRE/AP-1 motif within the
promoter of Regulated upon Activation, Normal T-cell Expressed and
Secreted (RANTES) (141). Notably, ATF3 also regulates
macrophage migration through other mechanisms. Transwell assays
suggest that ATF3-overexpressing macrophages exhibit markedly
enhanced migration under MCP-1 treatment (142,143). Mechanistically, ATF3 suppresses
gelsolin expression and F-actin de-polymerization to promote
cytoskeleton reorganization (144). On the other hand, ATF3
activates the Wnt/β-catenin signaling pathway to upregulate
extracellular matrix protein tenascin C, which subsequently
reinforces macrophage migration (142). These findings elucidate the
molecular basis by which ATF family members, ATF3 in particular,
integrate chemokine networks and cytoskeletal dynamics to precisely
orchestrate macrophage migration.
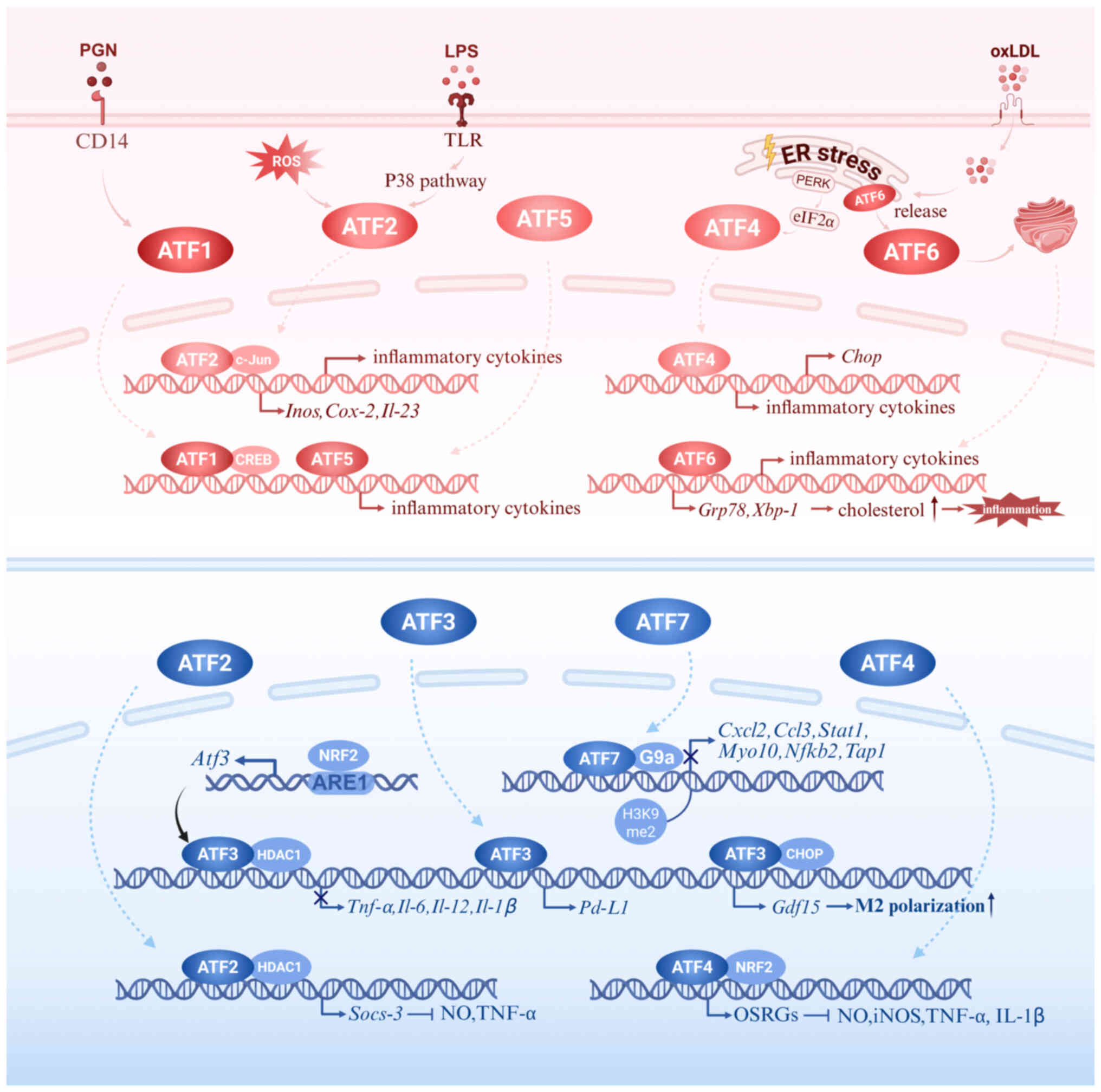
Macrophage activation and cytokine secretion are
orchestrated by ATFs that integrate microbial, metabolic and
stress-derived signals, into coordinated transcriptional programs.
ATF1, ATF5 and ATF6 predominantly act as pro-inflammatory drivers,
while ATF3 and ATF7 serve as negative feedback regulators.
Intriguingly, ATF2 and ATF4 exhibit context-dependent dual
functions, capable of both amplifying and restraining inflammatory
responses depending on the microenvironmental cues. The upstream
stimuli, signaling connections and differential impacts on cytokine
expression are summarized in Fig.
2 and Table I, which
together outline the overall regulatory network and highlight the
synergistic and antagonistic interactions among ATF members. The
dynamic equilibrium among these ATF subsets ultimately determines
the intensity and persistence of macrophage activation and cytokine
production.
ATF family members regulate macrophage
pro-inflammatory activation through multiple pathways. ATF1,
activated via sensing bacterial peptidoglycan (PGN), drives
pro-inflammatory cytokine transcription either in its
phosphorylated form or as a dimer with CREB (153). ATF5 directly amplifies
inflammatory responses by upregulating TNF-α, IL-1β and IL-6
expression (154). ATF6, a key
regulatory factor in ER stress (155), is essential for initiating the
UPR triggered by pattern recognition receptors such as
nucleotide-binding oligomerization domain-containing protein 2
(NOD2) (106). Upon the
challenge of oxLDL, ATF6 dissociates from the ER membrane, moves
into the Golgi apparatus, where it is cleaved into its active form
(p50-ATF6), which subsequently translocates into the nucleus to
regulate target genes, such as GRP78 and XBP-1 (156). Activated ATF6 stimulates the
expression of genes involved in cholesterol biosynthesis, but
represses the genes associated with cholesterol efflux (Abca1,
Abcg1, Lxrα), to drive cholesterol accumulation (157). ATF6 directly binds to the
Tnf-α promoter to enhance its transcriptional activity,
thereby activating the NF-κB and TNF signaling pathways and driving
macrophages to secrete pro-inflammatory cytokines, such as TNFα,
IL-1β and IL-6 (156). In liver
Kupffer cells, ATF6 also mediates the pro-inflammatory enhancement
of TLR4 response and cytokine production (158). Markedly, ubiquitination at K152
residue of ATF6 is essential for its antimicrobial function.
Mutations at this site (ATF6-K152A) exhibit defective bacterial
uptake, coupled with reduced ROS generation, diminished LC3II/ATG5
expression and impaired intracellular pathogen clearance (159). Together, these findings
establish ATF1, ATF5 and ATF6 as central mediators that translate
microbial and metabolic stress into potent inflammatory cytokine
programs.
In sharp contrast, ATF3 and ATF7 largely hinder the
activation of macrophages. Under physiological condition, ATF7 acts
as an epigenetic repressor by recruiting histone H3K9
dimethyltransferase G9a to the promoters of activation-related
genes (such as Cxcl2, Ccl3, Stat1,
Myo10, Nfkb2 and Tap1), thereby maintaining
macrophage at the resting state. However, LPS stimulation induces
p38-dependent phosphorylation of ATF7, prompting ATF7 dissociation
from chromatin, which leads to a significant reduction of H3K9me2
levels at target genes and the unleashing of transcriptional
repression. Such epigenetic change persists for weeks, which
sustains low H3K9me2 levels and high transcriptional activity to
confer long-term immune memory (119). Similarly, ATF3 functions as an
essential effector in curbing excessive macrophage activation.
First, ATF3 inhibits the phosphorylation of JNK, ERK and p38 MAPK,
thereby reducing the production of TNF-α, IL-6, IL-1β and IL-12β
induced by LPS, ROS, or Mycoplasma pneumoniae infection
(125,138,160-162). Second, NRF2 binds to the
antioxidant response elements (ARE1) within the Atf3
promoter to initiate Atf3 transcription (161). Activated ATF3 then recruits
HDAC1 to pro-inflammatory gene promoters (143,163), thereby reducing H3/H4 histone
acetylation (138,164) and counteracting Rel/NF-κB
transcriptional activity (115,164,165). This epigenetic pathway is
further reinforced in Clec4e-mediated response, in which ATF3
inhibits the downstream NF-κB/JNK pathway and reduces TNF-α and
CCL2 secretion (136).
Moreover, ATF3 also promotes GDF15 expression to foster the
adaptation of macrophages to metabolic stress (125). Collectively, by counteracting
the effects of ATF1, 2, 4, 5 and 6, ATF3 and ATF7 serve as critical
rheostats in maintaining macrophage homeostasis.
ATF2 and ATF4 display dual regulatory activities
that depend on the nature, strength and duration of environmental
stimuli. As a key regulator in Toll-like receptor (TLR) signaling,
ATF2 undergoes p38 MAPK-dependent phosphorylation at Thr71
(107,166), through which it promotes the
expression of pro-inflammatory mediators, such as inducible nitric
oxide synthase (iNOS), cyclooxygenase-2 (COX-2), TNF-α, IL-6 and
IL-1β (167-172). This process is further
amplified by the formation of AP-1 complex with c-JUN, which
enhances the production of nitric oxide (NO) and prostaglandin
E2 (PGE2) (173). Notably, ATF2 is highly
expressed in M1 macrophages within the white adipose tissue from
obese mice, where its phosphorylation is induced by ROS and LPS
(139) and sustains the
pro-inflammatory phenotype by suppressing the expression of
anti-inflammatory ATF3 (174).
Furthermore, ATF2 forms heterodimers with HDAC1 to induce
Socs-3 transcription to negatively regulate TLR4-mediated
inflammation (118),
demonstrating its dual regulatory nature. Under metabolic stress,
such as cholesterol accumulation, ATF4 is activated via the
protein kinase R-like ER kinase (PERK)-eukaryotic translation
initiation factor 2 alpha (eIF2α) pathway. During transient stress,
ATF4 promotes macrophage survival, whereas persistent stress
induces the expression of C/EBP homologous protein (CHOP),
triggering macrophage apoptosis along with the release of
inflammatory mediators (175,176). In obese microenvironments, ATF4
acts as a metabolic stress sensor, transcriptionally upregulating
protein disulfide isomerase a3 (PDIA3) to drive TNF-α, IL-6 and
chemokine (C-C motif) ligand 2 (CCL2) production, thereby
intensifying adipose tissue inflammation (177). The dual regulatory role of ATF4
is further reflected in its ability to influence antioxidant
defenses. Knockdown of ATF4 reduces NRF2 activity and HO-1
expression, leading to attenuated antioxidant defenses and
increased NO production (82).
Overall, ATF2 and ATF4 act as context-dependent signal integrators
that fine-tune macrophage activation, coordinating inflammatory,
oxidative and metabolic pathways to define the amplitude and
persistence of cytokine responses.
ATFs exert multidimensional roles in macrophage
polarization via metabolic reprogramming and signal transduction.
ATF1 drives macrophage polarization toward the antioxidant 'Mhem'
phenotype through coordinated regulation of iron and lipid
metabolism. This involves the induction of HO-1, which promotes the
degradation of heme into iron and antioxidant metabolites
(biliverdin and bilirubin) to help mitigate oxidative stress.
Furthermore, ATF1 activates liver X receptor-β (LXR-β) to augment
the expression of cholesterol efflux proteins ABCA1 and ApoE, by
which it reduces lipid accumulation and prevents foam cell
formation (178).
Phosphorylated ATF1 further improves macrophage adaptation to
intraplaque hemorrhage, thus maintaining the homeostasis under
dural iron-lipid stress (178).
ATF2 deficiency (THP-ΔATF2) induces abnormal macrophage morphology,
whereas ATF2 overexpression (THP-ATF2) triggers a rounded,
flattened morphology resembling M1 macrophages, along with elevated
expression of MHC class II, IL-1β and interferon-γ-induced protein
10 (133). Mechanistically,
ATF2 drives M1 polarization by enhancing glycolytic flux.
Metabolomic profiling confirmed that those ATF2-overexpressing
macrophages recapitulate the classical M1 metabolic signatures
(139).
ATF3 also plays a pivotal role in macrophage
polarization. Overexpression of ATF3 suppresses the differentiation
of M1 (pro-inflammatory) macrophage, as evidenced by the decreased
expression of M1 markers such as TNF-α and CD11c in adipose
tissues, thereby attenuating local inflammation (179). In atherosclerotic lesions, ATF3
mediated inhibition of M1 polarization reduces inflammatory
responses and lowers the risk of plaque rupture. In tumor
microenvironments, cyclophosphamide (CTX)-induced pro-tumor
macrophage traits are reversed in ATF3-knockout mice, shifting
macrophages toward anti-tumor phenotypes (147). ATF3 also enhances M2
(anti-inflammatory) macrophage program through indirect pathways.
ATF3 suppresses the Nrf2/ARE signaling pathway, leading to the
death of renal tubular epithelial cells (RTECs). The resulting
apoptotic RTECs release miR-1306-5p-containing exosomes, which in
turn inhibit M1 activation while enhance M2 polarization (180). In placental accreta spectrum
(PAS) lesions, upregulated ATF3 facilitates M2 macrophage
polarization by enhancing the expression of PD-L1, which interacts
with PD-1 to strengthen the immunosuppressive microenvironment
(181). Conversely, during
receptor activator of nuclear factor-κB ligand (RANKL)-induced
osteoclastogenesis, ATF3 overexpression promotes osteoclast
formation and activity. ATF3 directly binds to the promoters of key
transcription factors c-Fos and NFATc1, to drive
macrophage-to-osteoclast differentiation. However, as a
pro-inflammatory ATF member, ATF4 could otherwise promote M2
program (122,182). Upon hypoxic insult, ATF4
upregulates the expression of M-CSF in hemangioma stem cells
(HemSCs), to facilitate M2 macrophage polarization (183). ATF4 also activates RANKL
signaling by promoting NFATc1 expression, synergistically
regulating the differentiation of osteoclasts (184,185). Furthermore, ATF4 drives the
transition of vascular smooth muscle cells (SMCs) into foam cells,
as evidenced by the upregulation of macrophage markers such as
Cd68 and Lgals3 (186).
Under various pathological conditions, macrophages
exhibit disease-specific functional plasticity. Emerging evidence
indicates that members of the ATF family in macrophages display
distinct and highly disease-specific activation patterns rather
than a uniform stress response. Overall, ATF1, ATF2, ATF4, ATF5 and
ATF6 are persistently upregulated in most inflammatory, metabolic
and stress-related diseases, acting primarily as transcriptional
activators that drive pro-inflammatory programs or adaptive stress
responses. By contrast, ATF3 and ATF7 are predominantly induced as
negative feedback regulators to restrain excessive inflammation or
mediate innate immune memory. Importantly, the direction and
magnitude of these expression changes are strongly
context-dependent. For instance, ATF1 is markedly upregulated
during sepsis and atherosclerosis but suppressed in tissue
hemorrhage (150,153,178). In addition, ATF3 is rapidly
induced during the early hyperinflammatory phase of sepsis to
attenuate cytokine storm, whereas its sustained overexpression at
later stage contributes to immunosuppression (142,161,162,187). Furthermore, ATF4 is
downregulated during early acute sepsis, but is elevated under
chronic metabolic stress (132,177,182,186). These observations suggest that
ATF activation is finely orchestrated by disease-specific
microenvironments through the selective engagement of distinct
upstream signaling pathways. A detailed summary of ATF expression
dynamics in various diseases and their pathophysiological
consequences, along with related signaling pathways, is presented
in Table II.
In infectious and immune-related diseases,
macrophages situate at the first line of defense (1,188). During the early inflammatory
phase, they rapidly recognize and phagocytose pathogens, release
pro-inflammatory cytokines and recruit other immune cells to the
infection sites, thereby amplifying host defense (189-191). However, excessive or prolonged
activation can lead to systemic inflammatory syndromes, such as
sepsis and cause secondary tissue injury (191,192). As inflammation resolves,
macrophages adopt a reparative phenotype that clears apoptotic
cells and produces anti-inflammatory mediators to promote tissue
repair (193). Moreover,
macrophages can acquire innate immune memory, known as trained
immunity, which enables them to respond more effectively to
secondary challenges through enhanced cytokine production and
antimicrobial activity (194).
By contrast, excessive suppression of macrophage activation impairs
pathogen clearance and increases vulnerability to secondary
infections, thereby disturbing immune homeostasis (195,196). In chronic inflammatory
conditions, persistent low-grade activation or failure to
transition into a reparative state drive sustained cytokine
production, tissue remodeling and fibrosis, all of which predispose
to disease progression (197).
During bacterial infection, ATF3 exhibits pathogen-specific
regulatory effect. Specifically, ATF3 enhances macrophage clearance
of Staphylococcus aureus by directly binding to the
promoters of antimicrobial peptide genes, such as Reg3β and
S100A8/9, to bolster host defense (198). Conversely, during
Pseudomonas aeruginosa or uropathogenic E. coli
infection, ATF3 attenuates host resistance by downregulating
pro-inflammatory factors (TNF-α and IL-6) and upregulating IL-10
(138). Following
Leishmania infection, ATF3 expression reaches the highest
after 1-h of infection, which establishes an anti-inflammatory
environment in favor of parasite survival (163). ATF4 enhances macrophage
antioxidant capacity by promoting HO-1 expression to limit NO
production, thus creating an environment conducive to Leishmania
amazonensis survival (82).
By contrast, ATF7 contributes to innate immune memory by modulating
STAT1/NF-κB-dependent inflammatory networks (such as CXCL9/10),
thus enhancing macrophage-mediated responses against bacteria and
fungi (119). In antiviral
immunity, ATF2 activates the p38 MAPK/ATF2/AP-1 axis to promote the
secretion of interferon-stimulated genes and IFN-β, thereby
enhancing the resistance to vesicular stomatitis virus,
Newcastle disease virus and herpes simplex virus
(105). However, ATF3 may
compromise murine norovirus clearance by suppressing type I
interferon expression or impairing Mx1 activity (125). During Aspergillus
fumigatus infection, ATF4 is upregulated via the
TLR4/LOX-1/MAPK pathway, which modulates corneal macrophage barrier
function and disease severity (199).
In acute inflammatory diseases, ATFs exhibit a
dynamic and balanced role. Hyperactivation of ATF1 induces
excessive pro-inflammatory cytokine release from macrophages,
contributing to systemic inflammatory response syndrome (SIRS) or
septic shock (153). ATF3 plays
a complex part at different septic stages. In LPS-induced septic
models, ATF3 reduces TNF-α, IL-6 and IL-1β levels in plasma,
potentially mitigating early-stage hyperinflammation (162,187). Additionally, ATF3 promotes
macrophage polarization towards the M2 phenotype, enhances the
expression of anti-inflammatory factors such as Arg-1 and PPARγ,
but suppresses M1 markers (such as iNOS and TNF-α), thereby
alleviating inflammatory responses and facilitating tissue repair
(142). However, during the
sepsis-associated immunosuppression phase, persistent ATF3
overexpression impairs immune responses and increases
susceptibility to secondary infections (161). During septic progression,
reduced ATF4 expression in P1 cells leads to diminished gMacs,
which promotes bacterial translocation across the epithelial
barrier and worsens systemic inflammation (132). ATF5 contributes to
sepsis-associated liver injury by promoting the accumulation of
inflammatory macrophages (CD45+ CD11b+
Ly6C+) through an NF-κB-dependent pathway, leading to
the release of inflammatory cytokines (154). Excessive activation of ATF1 is
also associated with the progression of SIRS and sepsis by
enhancing macrophage cytokine secretion (153).
In chronic inflammatory diseases, ATF family
members influence macrophage polarization and amplify inflammatory
signaling, contributing to disease pathology. In LPS-induced acute
lung injury, ATF2 exacerbates pulmonary inflammation by
upregulating MIP-2 to further recruit macrophages (140), while ATF6 intensifies
macrophage activation and cytokine release via ER stress
signaling (200). ATF2
activation also amplifies TLR signaling cascades and exacerbating
chronic inflammation (107,170). Moreover, upon LPS stimulation,
ATF2 promotes IL-23 secretion in macrophages, which then induces
pathogenic Th17 cell expansion in autoimmune disorders, such as
multiple sclerosis, inflammatory bowel disease and rheumatoid
arthritis (201). In
organ-specific inflammatory injuries, ATF3 exhibits bidirectional
regulatory properties. ATF3 attenuates renal ischemia-reperfusion
injury (IRI) by inhibiting TLR4/NF-κB pathway (165). However, IRI-induced ATF3
overexpression in RTECs exacerbates ferroptosis and promotes the
release of exosomes carrying miR-1306-5p. These exosomes, upon
uptake by macrophages, drive M2 polarization and accelerate
interstitial fibrosis (180).
In alcohol-induced immunosuppression, ATF3 upregulation leads to
enhanced Kupffer cell tolerance to LPS and reduced TNF-α
production, impairing pathogen clearance and exacerbating alcoholic
liver disease (143). In
dextran sulfate sodium (DSS)-induced colitis, activation of the
ATF4-CHOP pathway drives excessive release of inflammatory
cytokines, intensifying intestinal inflammation and epithelial
damage (176). It was also
noted that ATF6 deficiency impairs macrophage antibacterial
responses, worsening intestinal inflammation (106). During hepatic
ischemia-reperfusion injury, ATF6 aggravates tissue damage by
enhancing NF-κB activation and suppressing the anti-inflammatory
Akt-GSK3β signaling pathway in Kupffer cells (158). However, during early acute
liver injury, ATF6 activation via ER stress promotes IL-1α
production, activating HSCs and driving liver fibrosis (159). ATF6 deficiency in microglia
attenuates inflammation in autoimmune encephalomyelitis by
promoting NF-κB p65 degradation (202).
In metabolic disorders, adipose tissue macrophages
produce pro-inflammatory cytokines including TNF-α, IL-6 and IL-1β
to impair insulin signaling and disrupt adipocyte homeostasis,
thereby contributing to insulin resistance and metabolic syndrome
(203-207). ATF4 exacerbates chronic
inflammation in adipose tissue, driving the progression of
metabolic syndrome and type 2 diabetes mellitus (177,182). By contrast, ATF3 mitigates
obesity-associated inflammation by suppressing TLR4-mediated
macrophage activation. Therefore, mice deficient in ATF3
exhibit aggravated obesity-related inflammation and insulin
resistance, underscoring its protective role in maintaining
metabolic homeostasis (162).
Conversely, ATF2 activation intensifies macrophage M1 program
within adipose tissue by perpetuating an 'inflammation-hypoxia-ROS'
vicious cycle, worsening insulin resistance and metabolic syndrome
(173). Additionally, ATF2
aggravates inflammation and metabolic disturbances in white adipose
tissue by suppressing ATF3 expression, further enhancing M1 program
(139).
In atherosclerosis, macrophages take up OxLDL to
form foam cells and orchestrate inflammatory and reparative
responses within vascular lesions, thereby playing a pivotal role
in plaque formation and progression (28,34,36). ATFs influence disease progression
by regulating macrophage lipid metabolism and plaque stability.
ATF3 exerts an atheroprotective effect through multiple mechanisms.
ATF3 suppresses cholesterol 25-hydroxylase to reduce oxidized
cholesterol accumulation and promotes macrophage RCT, thereby
limiting foam cell formation (148,208). Furthermore, ATF3 stabilizes
plaque structure by inhibiting macrophage apoptosis and the release
of inflammatory mediators, lowering the risk of myocardial
infarction and stroke (134).
ATF3 deficiency impairs ABCA1-mediated cholesterol efflux,
aggravating oxLDL-induced lipid deposition (136). Moreover, ATF1 induces Mhem
macrophage polarization and activates the HO-1/LXR-β pathway to
enhance cholesterol efflux and iron chelation, thereby delaying
plaque progression and rupture (150,178). By contrast, ATF6 activation
positively correlates with the severity of atherosclerotic lesion
and accelerates plaque development by facilitating cholesterol
accumulation (157). ATF4
drives necrotic core expansion and plaque destabilization
via switching an SMC phenotype ('transdifferentiated SMCs'
which express both macrophage and fibroblast markers) and inducing
CHOP-dependent cell apoptosis (186). As a downstream effector of PKCθ
signaling, ATF2 upregulates CD36 to accelerate foam cell formation
and plaque progression. PKCθ-deficient mice exhibit blunted ATF2
activation, along with reduced CD36 levels and markedly smaller
plaque areas (149). Moreover,
ATF1 deficiency impairs the segregation of iron and lipid within
macrophages, resulting in their colocalization, which disrupts iron
metabolic homeostasis, delays hematoma clearance following tissue
hemorrhage and exacerbates oxidative stress-related damage
(150). Collectively, ATFs
spatiotemporally modulate macrophage inflammatory state, thus
influencing the pathological progression of metabolic and
cardiovascular diseases.
Macrophages are major immune infiltrates that
regulate angiogenesis, immune suppression, antigen presentation and
metastatic dissemination in the tumor microenvironment (1,209). In bone lesions, macrophages
further contribute to osteoclast differentiation and tumor-induced
bone destruction (210-212). ATF4 regulates tumor-associated
macrophage polarization and promotes the differentiation of
monocyte-macrophage precursors into osteoclasts to enhance bone
resorption. In parallel, ATF4 exacerbates inflammation through the
NF-κB signaling axis, further accelerating cancer-associated bone
destruction (184). Unlike
ATF4, ATF3 exhibits functional heterogeneity in tumor
immunomodulation. Tumor-derived IL-1β upregulates ATF3 in bone
marrow hematopoietic stem cells, driving myeloid precursor
expansion and increasing peripheral CD11b+ myeloid
cells, including TAMs, tumor-associated macrophages. In a breast
cancer model, ATF3 deficiency inhibits monocyte-macrophage
differentiation, while its overexpression serves as an early
biomarker distinguishing malignant lesions (213). Furthermore, ATF3 activates
immune checkpoint pathways to amplify CD14+
immunosuppressive macrophages in PAS lesions, exacerbating
pathological progression (181).
In neurological diseases, macrophages and microglia
not only phagocytose neuronal debris but also release
neuroregulatory substances and inflammatory mediators, thereby
modulating neuronal survival and degeneration (214,215). ATF2 promotes neuroinflammatory
cascades in multiple sclerosis, Alzheimer's disease (AD) and
Parkinson's disease by promoting pyroptosis in macrophages and
microglia (139). Additionally,
ATF4 is the core transcription factor for AD microglial cells to
integrate stress response, by inducing them to enter the neurotoxic
state and mediating neurodegeneration through lipid secretion
(216). Furthermore, ATF4
synergizes with HIF-1α under hypoxic conditions in infantile
hemangioma (IH) proliferative phases, driving M2 macrophage
infiltration to fuel lesion expansion (183).
Targeting ATFs in macrophages offers a promising
therapeutic strategy, as these key transcription factors
orchestrate diverse macrophage functional states, thereby enabling
precise modulation of disease processes (Table III). Currently, therapeutic
strategies targeting ATFs can be broadly divided into three
categories: Direct intervention, which employs small-molecule
agonists or inhibitors and gene-editing approaches to directly
modulate ATF expression or activity; indirect modulation, which
targets upstream signaling pathways or downstream effector
molecules to influence ATF-dependent regulatory networks; and
emerging innovative techniques, including proteolysis-targeting
chimera (PROTAC)-mediated protein degradation, peptide or
peptidomimetic modulators and molecular glue technologies, which
offer more efficient and specific manipulation of ATF
functions.
Blocking the function of ATFs through small
inhibitors, short interference (si)RNA, or gene-editing
technologies offers a strategic approach to precisely control
disease states driven by macrophages. Targeted inhibition of ATF1
reduces osteoclastogenesis by suppressing the miR-214-5p/ITGA7
axis, suggesting a novel direction for osteoporosis treatment
(217). The traditional herbal
medicine, HangAmDan-B (HAD-B), reduces LPS-induced NO and
PGE2 production in macrophages by blocking ATF2
phosphorylation, consequently limiting tumor-promoting activity of
TAMs in gastric and colorectal cancers (166). Ginsenoside Rc markedly reduces
LPS-induced TNF-α release by suppressing the p38/ATF2 pathway,
presenting a candidate strategy for rheumatoid arthritis and other
inflammatory diseases (168).
Moreover, the traditional compound formula Qingfei Paidu Decoction
and its active component wogonoside, inhibit ATF2 phosphorylation
and enhance its ubiquitin-mediated degradation, curbing
macrophage-mediated inflammation and showing potential as adjunct
therapies for coronavirus pneumonia and colitis (169). Moreover, extracts from
Vaccinium oldhamii stems reduce ATF2 nuclear accumulation,
which blocks MAPK signaling pathway activation and limits
macrophage-driven osteoclast differentiation to mitigate bone
resorption (218). Exosomes
derived from adipose-derived stem cells (ADSCs) or mesenchymal stem
cells attenuate ATF2 expression, resulting in reduced NF-κB
activation, decreased ROS generation and diminished macrophage
infiltration, thereby mitigating vascular dysfunction (219). Similarly, perfluorocarbon (PFC)
could reduce inflammatory cell infiltration and alleviate acute
lung injury by decreasing ATF2 activity in macrophages (140). The dynamic modulation of ATF3
requires consideration of disease progression. During the early
hyperinflammatory phase of sepsis, induction of ATF3 expression in
macrophages may counteract cytokine storms, while its inhibition
during the immunosuppressive phase may help mitigate secondary
infection risk (138). ATF4
suppression also requires meticulous evaluation due to its
dualistic effect. The application of small-molecule inhibitors or
siRNA-mediated knockdown of ATF4 has been shown to attenuate M-CSF
secretion and limit M2 macrophage infiltration (183), which in turn impedes the
progression of infantile hemangioma while simultaneously
counteracts the pro-tumorigenic properties of TAMs (122). Inhibiting ATF4 can also
mitigate Leishmania infection (82), though excessive inhibition might
compromise the antifungal defense. Therefore, therapies targeting
ATF4 must balance the anti-inflammatory outcomes with host immune
protection (199). Naringenin,
a flavanone compound found in grapefruits and other citrus fruits,
could reduce ATF6 nuclear translocation and activity, by which it
decreases the expression of ER stress markers while enhances the
expression of cholesterol efflux genes (ABCA1, ABCG1 and LXRα).
This action promotes macrophage cholesterol efflux, reducing
atherosclerotic plaque formation (157). Furthermore, siRNA-mediated ATF6
knockdown alleviates macrophage-driven inflammation and fibrosis,
highlighting its potential for treating chronic inflammatory
disorders (159).
By contrast, activating or overexpressing ATFs
through gene-editing technologies (such as adenoviral vectors) and
small-molecule agonists offer an approach to enhance
anti-inflammatory, anti-infectious and tissue-repairing functions
of macrophages. Developing small-molecule drugs or gene therapies
to enhance ATF1 activity presents a potential intervention for
atherosclerosis by inducing the protective macrophage subset
(178). ATF2 activation
inhibits viral replication, with its agonists serving as potential
therapeutics for chronic viral infections (167). High-salt diets have also been
proposed as a strategy to enhance antiviral immunity via
ATF2-mediated pathways in macrophages (105). Targeted activation or
overexpression of ATF3 exhibited a promising therapeutic potential
in inflammatory diseases, infections and metabolic disorders.
Gene-editing technologies, such as lentiviral and adenoviral
deliveries (Ad/ATF3) of ATF3, alleviate excessive inflammation
following Mycoplasma pneumoniae infection (160), protect renal tubular cells from
inflammatory damage (165) and
reduce the risk of atherosclerosis development (134). ATF3 overexpression further
diminishes LPS-induced pro-inflammatory cytokine release, thus
improving the outcomes in chronic inflammatory diseases (138,164). In bacterial infections,
enhancing ATF3 activity optimizes immune responses and promotes
macrophage activation, migration and bactericidal activity, coupled
with increased host clearance of Staphylococcus aureus
(144). Across multiple disease
models, berberine and metformin have been demonstrated to enhance
ATF3 expression (162,220) and attenuates LPS-induced
production of pro-inflammatory cytokines from macrophages (164). In particular, metformin exerts
its anti-inflammatory effects by engaging the AMPK/ATF3 signaling
axis, through which it potently manages inflammation associated
with type 2 diabetes and obesity (187). ATF4 may represent a novel
strategy for treating sepsis-related intestinal injury (142). Enhancing ATF4 expression
through pharmacological or gene therapies help the restoration of
monocyte differentiation into gMacs, thereby strengthening
intestinal barrier function, reducing bacterial translocation and
mitigating sepsis-induced inflammatory damage (132). In type 2 diabetes, upregulating
ATF4 can improve insulin resistance by promoting M2 macrophage
polarization (182). Targeting
ATF7-mediated epigenetic modifications offers a strategy to refine
vaccine adjuvant efficacy, by promoting macrophage activation and
memory formation, ultimately advancing the development of more
durable and effective vaccine-induced protection (119).
However, the clinical translation of strategies
that directly targeting ATFs faces multiple inherent challenges.
The first obstacle lies in the intrinsic 'undruggability' of these
transcription factors. The interaction surfaces of ATFs with DNA or
partner proteins are typically broad and shallow, lacking
well-defined pockets that allow high-affinity binding of small
molecules, thereby rendering the development of conventional
inhibitors or agonists extremely difficult. Second, substantial
structural similarity exists among ATF family members, particularly
within their DNA-binding domains. As a result, small molecules
targeting ATFs may suffer from poor subtype selectivity,
potentially leading to off-target toxicity. Therefore, developing
compounds capable of precisely distinguishing individual ATF
isoforms or specific ATF homo- and heterodimers remains a
formidable challenge, which warrants further intensive
investigations (221).
Given the challenges associated with direct
targeting, indirect modulation of upstream signaling pathways or
downstream effector molecules have emerged as an attractive
alternative strategy. Studies show that PERK inhibitors (such as
integrated stress response inhibitor (ISRIB)) suppress the
PERK-eIF2α-ATF4 pathway, thereby reducing foam cell formation and
alleviating atherosclerotic plaque burden (186). The small molecule Limonin
blocks the PERK-ATF4-CHOP axis to attenuate inflammatory cytokine
release (186). Moreover,
drugs, such as FTY720, modulate ATF4 expression via histone
deacetylase 4 (HDAC4), thus reducing osteoclastic activity in
inflammatory bone diseases (184). For ATF2, multiple upstream
intervention strategies have been explored, including p38 MAPK
inhibitors (SB203580) and JNK inhibitors (SP600125), which reduce
inflammation by suppressing ATF2 phosphorylation (166). Additionally, PAR1 antagonists
(such as SCH79797) or PKCθ inhibitors decrease ATF2 activation to
improve insulin sensitivity (149). Novel compounds, such as
bis(5-methyl)2-furylmethane and Javamide-II, reduce inflammatory
mediator production by suppressing the p38 MAPK/ATF2 pathway.
Particularly, Javamide-II manifests lower systemic toxicity
compared with BFNM due to its selective inhibition of specific
inflammatory cytokines without affecting TNF-α or IL-1β (107,170). For ATF1, anti-CD14 monoclonal
antibodies (MY4) showed potential as an adjunctive therapy for
sepsis by inhibiting PGN-induced ATF1 activation (153). Similarly, AMPK agonists (such
as AICAR) induce ATF3 expression and offer therapeutic benefits for
metabolic disorders (162).
Conversely, the NRF2 inhibitor, trigonelline hydrochloride,
markedly reduces parasitic loads in the liver and spleen by
diminishing ATF3 expression (163). Additionally, NF-κB inhibitors
[such as dehydroxy-methylepoxy-quinomicin (DHMEQ)] alleviate
sepsis-associated liver damage by inhibiting ATF5-mediated
pro-inflammatory macrophage differentiation (154).
Nevertheless, indirect modulation strategies also
present substantial limitations. The fundamental drawback of such
approaches lies in their lack of specificity. Upstream kinases or
signaling pathways targeted by these interventions are often
ubiquitously expressed across multiple cell types, leading to
severe off-target effects and systemic toxicity. Second, the high
redundancy of intracellular signaling networks frequently triggers
compensatory activation of alternative pathways, thereby
diminishing therapeutic efficacy or even resulting in treatment
failure. Moreover, the regulatory effects of pathway-level
interventions tend to attenuate along the signaling cascade and are
highly dependent on the disease microenvironment, rendering the
ultimate modulation of specific ATFs indirect and unpredictable.
Finally, this relatively 'coarse-grained' mode of regulation is
insufficient to precisely govern the bidirectional functions of ATF
members or their heterodimeric interactions, limiting its ability
to achieve fine-tuned control over their specific physiological and
pathological activities.
To overcome the aforementioned obstacles,
researchers have explored intervention strategies such as PROTACs
and molecular glues, aiming to target traditionally 'undruggable'
molecules through novel mechanisms of action. The advent of PROTAC
technology has provided a new avenue for addressing such targets
(222-225). A PROTAC molecule contains two
functional domains, one binds to the target protein and the other
recruits an E3 ubiquitin ligase, thereby tagging the target for
selective proteasomal degradation (226,227). Although no highly potent
small-molecule ligands have yet been reported for ATF proteins,
researchers are creatively adapting the PROTAC concept for
transcription factor degradation. One proposed generalizable
strategy involves using DNA fragments recognized by transcription
factors as targeting elements, which are linked to E3 ligase
ligands to induce ubiquitination and subsequent degradation of
transcription factor-DNA complexes (228). For example, the antioxidant
response factor NRF2, a bZIP transcription factor that
heterodimerizes with small Maf proteins to bind to the AREs, can be
targeted by a chimeric molecule termed 'ARE-PROTAC', which
successfully induced simultaneous degradation of the NRF2-MafG
complex and synergistically suppressed Nrf2 signaling (229). This design suggests that
DNA-binding factors, such as ATF/CREB, DNA mimetics or DNA-derived
small molecules, could serve as decoy binding modules to achieve
targeted degradation via the PROTAC mechanism. Unlike
conventional inhibitors that merely block protein activity, this
approach eliminates the target protein itself, offering more
complete and durable inhibition while potentially alleviating
feedback activation caused by persistent protein accumulation.
Nevertheless, applying this technology to ATF targeting remains
challenging. It requires the discovery of small, high-affinity
ligands specific for ATF proteins and the relatively large
molecular weight of PROTACs limits their intracellular delivery,
especially into macrophages. To address this issue, recent studies
have explored macrophage-targeted nanocarrier systems utilizing
surface receptors such as citrate or scavenger receptors, enabling
PROTAC accumulation and selective degradation at the inflammatory
or tumor sites (230,231).
Building on this concept, molecular glues have
emerged as another promising and simplified strategy for targeted
protein degradation. Molecular glues are typically single small
molecules that 'glue' the target protein and an E3 ubiquitin
ligase, thereby promoting their interaction and inducing target
degradation (232). Unlike
PROTACs, molecular glues do not require a linker and can directly
induce contact between transcription factors and E3 ligases,
offering a simpler architecture and easier optimization. The
classic example is lenalidomide, which remodels the
substrate-binding surface of the CRBN E3 ligase, enabling selective
recognition and degradation of the transcription factors IKZF1 and
IKZF3 (233). This mechanism
holds particular interest for targeting ATF family members. First,
molecular glues generally have much smaller molecular weights than
PROTACs, conferring superior membrane permeability and drug-like
properties, thereby facilitating macrophage penetration. Second,
their activity does not depend on identifying traditional
ligand-binding pockets, which is advantageous for transcription
factors lacking well-defined active sites. Nevertheless, challenges
remain, including the difficulty of identifying suitable adaptor
molecules and the dependency on specific E3 ligases, which may
restrict the target spectrum.
In addition, peptide mimetics and interfering
peptides offer new avenues for disrupting transcription factor
activity. Researchers have developed a dominant-negative form of
ATF5 (dn-ATF5) by deleting its N-terminal activation domain while
retaining the bZIP dimerization and DNA-binding regions and further
conjugated it to an HIV-TAT sequence to generate a cell-permeable
synthetic peptide (CP-dn-ATF5) (234,235). This peptide can get into the
nucleus and compete with endogenous ATF5 for partner proteins or
DNA-binding sites, thereby blocking its transcriptional activity.
The advantages of peptide-based therapies lie in their high
specificity and flexible design. With advances in peptide drug
modification techniques, such as cyclization, D-amino acid
substitution and nanocarrier encapsulation, peptide-based ATF
inhibitors are expected to become an important complement to
small-molecule strategies.
Of note, enhancing the spatiotemporal specificity
of ATF-targeting interventions is critical for improving
therapeutic efficacy and safety. Advanced delivery systems are
under development to achieve tissue- or cell-specific targeting,
ensuring that drugs could be selectively delivered into macrophages
in affected tissues (236).
Encouragingly, advances in nanocarrier and bioengineering
technologies now enable the precise delivery of therapeutic agents
to macrophages, allowing for selective modulation of ATF-associated
pathways without affecting other cell types. For instance, in
atherosclerosis models, macrophage-targeted nanoparticles have been
utilized to deliver drugs directly to macrophages within the
plaques (237). These
nanoparticles substantially improved the pharmacokinetic profiles
of the encapsulated drugs, enhancing their accumulation at the
lesion site and promoting the therapeutic efficacy. Similarly, in
viral pneumonia models, lipid nanoparticles conjugated with
macrophage-specific antibodies (such as anti-F4/80) have been used
to deliver siRNA to lung macrophages, effectively silenced upstream
inflammatory molecules such as TAK1 (238). These findings underscore the
feasibility of macrophage-targeted delivery for therapeutic
interventions and highlight the substantial potential of
integrating delivery technologies into ATF-targeted therapies.
Therefore, innovative targeted technologies coupled with advanced
delivery systems, might overcome the barrier of ATFs-targeted
therapies and accelerate their clinical translation.
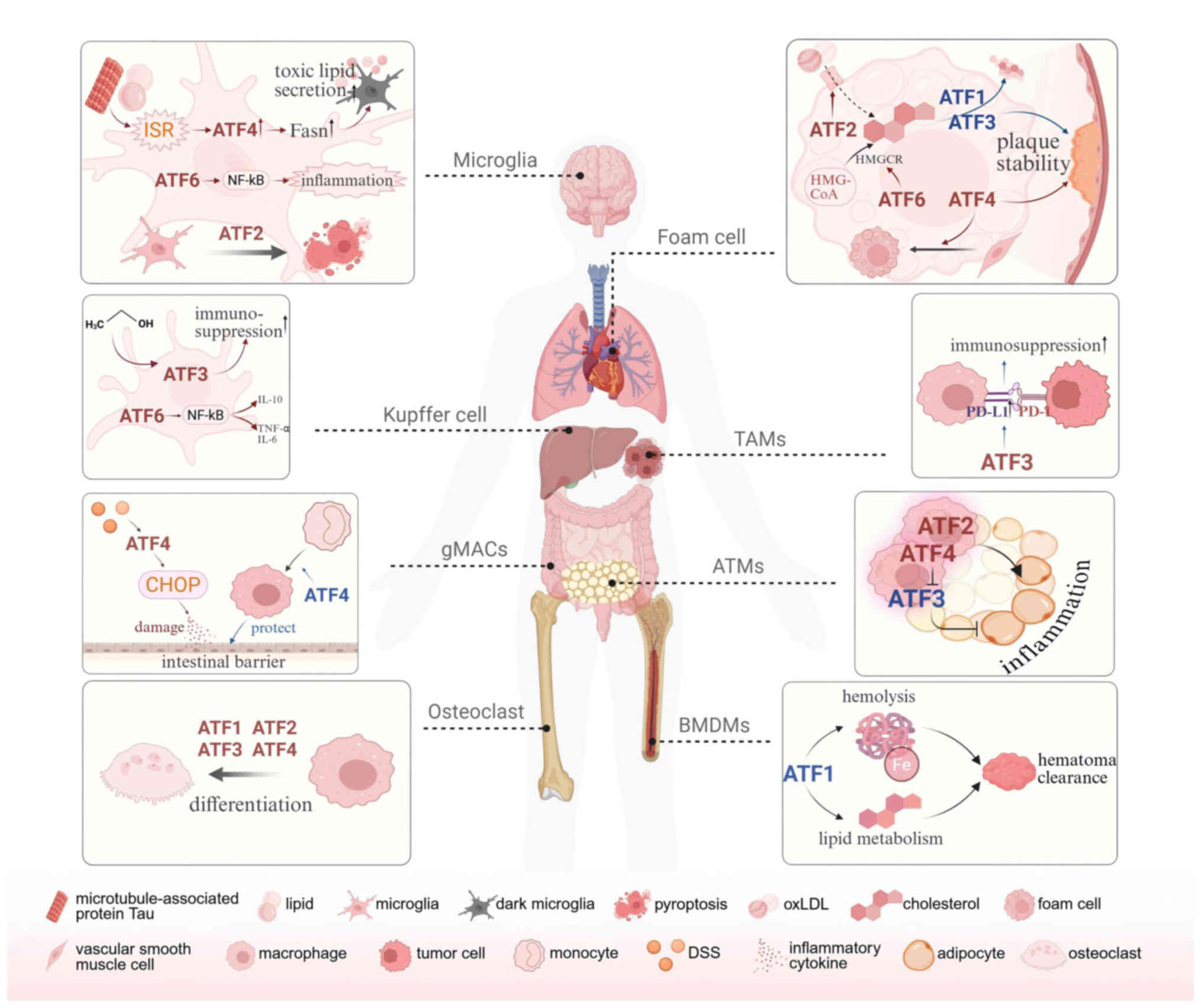
By integrating mechanistic insights and
disease-specific evidence, this review delineated how ATFs
orchestrate macrophage development, survival, migration,
phagocytosis, activation, cytokine secretion and polarization and
how these regulatory axes converge to shape macrophage-driven
pathology in infectious, inflammatory, metabolic, oncologic and
neurodegenerative settings. Through transcriptional control of
macrophage-expressed genes across diverse tissues and organs, ATFs
are not only implicated in the initiation and progression of
multiple disorders but also emerged as nodal regulators integrating
pathophysiological signals with immune and metabolic reprogramming
(Fig. 3). Of note, ATFs exhibit
complex context-dependent versatility in macrophage-mediated immune
responses. Mechanistically, the effector outcomes of ATFs largely
hinge on the variety of stimuli and upstream signaling pathways in
macrophages. For instance, TLR2 activation by Gram-positive
bacteria induces moderate inflammation, during which ATF3 promotes
antimicrobial gene expression, whereas LPS-TLR4 signaling from
Gram-negative bacteria elicits strong NF-κB and IRF3 activation
that rapidly induces ATF3 as a negative feedback regulator to
suppress excessive cytokine production (115,144,161,239-242). In addition, the intensity and
duration of stimulation further shape ATF activity. During early
sepsis, ATF3 alleviates hyperinflammation and promotes tissue
recovery, whereas its sustained overexpression contributes to
late-phase immunosuppression and secondary infections (142,161,162,187). Transient stress activates ATF4
via the PERK-eIF2α pathway, upregulating adaptive and
survival-related genes to enhance macrophage resilience; however,
prolonged stress leads to persistent ATF4-CHOP signaling, promoting
apoptosis and pro-inflammatory mediator release (175,176). Similarly, ATF6 activation under
acute ER stress (such as triggered by pattern-recognition
receptors) induces chaperone and autophagy programs that facilitate
pathogen clearance (106,155,159). By contrast, during chronic
metabolic stress, such as obesity or hyperlipidemia, sustained ATF6
activation increases cholesterol biosynthesis, suppresses
cholesterol efflux, promotes foam cell formation and amplifies
inflammatory responses (156,157). In addition, as bZIP
transcription factors, ATF family members exert highly
context-dependent effects determined by their interacting partners
and chromatin accessibility. Microenvironmental factors, such as
oxidative stress, metabolic stress and ER stress, not only regulate
ATF activation levels but also influence their combination with
c-Jun, CHOP, C/EBP, or HDAC1, thereby switching their
transcriptional output between activation and repression. Upon
inflammatory stimulation, ATF2 forms an AP-1 complex with c-Jun to
transcribe Inos and Cox-2 expression, promoting NO
and PGE2 production (156,168-173), while during inflammatory
resolution stage, ATF2 recruits HDAC1 to transcribe Socs3
and suppresses excessive TLR4 signaling (162). ATF3 similarly shifts its
binding preference. Under mild stimulation, ATF3 partners with
c-Jun or CHOP to activate antimicrobial and metabolic genes, but
under strong LPS-TLR4 signaling, ATF3 recruits HDAC1 to repress
NF-κB target genes (115,138,143,163-165). The functional direction of ATF4
also depends on its binding partners. ATF4-NRF2 heterodimers drive
adaptive metabolism and cell survival (82), while heterodimers formed with
CHOP mediate inflammation and cell apoptosis (175,176). Likewise, ATF6 enhances
cytoprotective UPR genes such as Grp78 and Xbp1
during acute stress, but under chronic lipotoxicity, it
preferentially targets inflammatory and lipid metabolic genes to
amplify macrophage inflammation (156). Moreover, changes in chromatin
accessibility under infection or metabolic stress might reshape
cis-element exposure, determining the preferential binding sites of
ATFs and their downstream transcriptional outcomes. Collectively,
these mechanisms highlight ATFs as crucial 'rheostats' that balance
inflammatory signaling, metabolic adaptation and stress responses
in macrophages. However, this functional duality also underscores
the caution in therapeutic targeting: suppressing ATFs in
pathogenic conditions must not compromise their protective roles
and activating them to resolve inflammation should avoid inducing
immunosuppression. A deeper understanding of the context-dependent
mechanisms of ATF signaling is be essentially required.
It is noteworthy that alternative splicing may
represent another critical but insufficiently investigated layer of
ATF modulation. Except for ATF4, most ATF family members possess
alternatively spliced isoforms, although their roles in macrophages
remain largely unknown. Distinct ATF1 isoforms differ in
DNA-binding affinity and subnuclear localization (243), whereas multiple ATF2 variants,
including the human isoforms ATF2-sm and ATF2 (SV5) as well as the
murine variant Atf2(Δ8,9) (244-246), exhibit reduced transcriptional
activity and may alter gene expression by modifying
nuclear-cytoplasmic trafficking or by interfering with canonical
ATF2/AP-1 dimerization. Moreover, a truncated ATF3 isoform (ΔZip)
lacking the leucine-zipper domain antagonizes the transcriptional
repressive function mediated by full-length ATF3 (247), while cytoplasmic ATF7-4 acts as
a molecular 'decoy' to sequester upstream kinases responsible for
ATF7/ATF2 phosphorylation (77).
These findings suggest that alternative splicing may establish
negative-feedback circuits and intermolecular counter-regulatory
balances responsible for the precise modulation of ATF function.
Although their implications in macrophages remain to be defined,
exploring ATF splicing isoforms represents an important direction
for elucidating the dynamic regulatory mechanisms by which ATFs
regulate immune homeostasis and stress responses.
The central involvement of ATFs in
macrophage-driven pathologies positions them as promising
therapeutic targets and biomarkers. On the one hand, ATF expression
dynamics serve as diagnostic or prognostic indicators. Rapid ATF3
upregulation (8-fold within 24 h) in silicosis macrophages is an
early diagnostic biomarker for silicosis detection (248). Additionally, due to its
plaque-stabilizing effects on atherosclerosis, ATF3 expression aids
in the identification of high-risk patients and guides personalized
treatments (134). On the other
hand, single-target interventions may be limited by compensatory
mechanisms, necessitating the need for multi-target synergistic
approaches for the regulation of macrophage functionality. Lastly,
ATF-targeting strategies such as siRNA interference, gene editing,
or more advanced protein degradation technologies such as PROTACs,
all face common challenges related to in vivo delivery
efficiency, tissue and cell specificity, immunogenicity and the
durability of their effects. Although these interventions have
shown remarkable efficacy in preclinical models, extensive studies
are still required to validate their safety, efficacy and
controllability in complex physiological settings and to further
optimize targeted delivery systems to facilitate clinical
translation.
Not applicable.
YCL conceived the study, wrote the manuscript and
prepared the figures. JWZ, QJC and XTL curated data and collected
references. SJR and FS organized the tables. SWL, CLY and CYW
reviewed and revised the manuscript. Data authentication is not
applicable. All authors approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the Noncommunicable Chronic
Diseases-National Science and Technology Major Project
(2024ZD0531400,2023ZD0507302), the National Key R&D Program of
China (2022YFA0806101), the National Natural Science Foundation of
China (81920108009, 82130023, 82570968, 82200923), the Research and
Innovative Team Project for Scientific Breakthroughs at Shanxi
Bethune Hospital (2024AOXIANG03), the Continuous Funding Program
for High-Level Research Achievements at Shanxi Bethune Hospital
(2024GSPYJ10 and 2024GSPYJ13) and the IGP Funding from QBRI, Hamad
Bin Khalifa University.
|
1
|
Chen S, Saeed AFUH, Liu Q, Jiang Q, Xu H,
Xiao GG, Rao L and Duo Y: Macrophages in immunoregulation and
therapeutics. Signal Transduct Target Ther. 8:2072023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mass E, Ballesteros I, Farlik M,
Halbritter F, Günther P, Crozet L, Jacome-Galarza CE, Händler K,
Klughammer J, Kobayashi Y, et al: Specification of tissue-resident
macrophages during organogenesis. Science. 353:aaf42382016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuznetsova T, Prange KHM, Glass CK and de
Winther MPJ: Transcriptional and epigenetic regulation of
macrophages in atherosclerosis. Nat Rev Cardiol. 17:216–228. 2020.
View Article : Google Scholar :
|
|
4
|
Ajami B, Bennett JL, Krieger C, Tetzlaff W
and Rossi FM: Local self-renewal can sustain CNS microglia
maintenance and function throughout adult life. Nat Neurosci.
10:1538–1543. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bian Z, Gong Y, Huang T, Lee CZW, Bian L,
Bai Z, Shi H, Zeng Y, Liu C, He J, et al: Deciphering human
macrophage development at single-cell resolution. Nature.
582:571–576. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gomez Perdiguero E, Klapproth K, Schulz C,
Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF,
Geissmann F and Rodewald HR: Tissue-resident macrophages originate
from yolk-sac-derived erythro-myeloid progenitors. Nature.
518:547–551. 2015. View Article : Google Scholar
|
|
7
|
Guan F, Wang R, Yi Z, Luo P, Liu W, Xie Y,
Liu Z, Xia Z, Zhang H and Cheng Q: Tissue macrophages: Origin,
heterogenity, biological functions, diseases and therapeutic
targets. Signal Transduct Target Ther. 10:932025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hashimoto D, Chow A, Noizat C, Teo P,
Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, et al:
Tissue-resident macrophages self-maintain locally throughout adult
life with minimal contribution from circulating monocytes.
Immunity. 38:792–804. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rosmus DD, Koch J, Hausmann A, Chiot A,
Arnhold F, Masuda T, Kierdorf K, Hansen SM, Kuhrt H, Fröba J, et
al: Redefining the ontogeny of hyalocytes as yolk sac-derived
tissue-resident macrophages of the vitreous body. J
Neuroinflammation. 21:1682024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Masuda T, Amann L, Monaco G, Sankowski R,
Staszewski O, Krueger M, Del Gaudio F, He L, Paterson N, Nent E, et
al: Specification of CNS macrophage subsets occurs postnatally in
defined niches. Nature. 604:740–748. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kazankov K, Jorgensen SMD, Thomsen KL,
Møller HJ, Vilstrup H, George J, Schuppan D and Grønbæk H: The role
of macrophages in nonalcoholic fatty liver disease and nonalcoholic
steatohepatitis. Nat Rev Gastroenterol Hepatol. 16:145–159. 2019.
View Article : Google Scholar
|
|
12
|
Appios A, Davies J, Sirvent S, Henderson
S, Trzebanski S, Schroth J, Law ML, Carvalho IB, Pinto MM, Carvalho
C, et al: Convergent evolution of monocyte differentiation in adult
skin instructs Langerhans cell identity. Sci Immunol.
9:eadp03442024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hassnain Waqas SF, Noble A, Hoang AC,
Ampem G, Popp M, Strauß S, Guille M and Röszer T: Adipose tissue
macrophages develop from bone marrow-independent progenitors in
Xenopus laevis and mouse. J Leukoc Biol. 102:845–855. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Perdiguero EG and Geissmann F: The
development and maintenance of resident macrophages. Nat Immunol.
17:2–8. 2016. View Article : Google Scholar :
|
|
15
|
Sakai M, Troutman TD, Seidman JS, Ouyang
Z, Spann NJ, Abe Y, Ego KM, Bruni CM, Deng Z, Schlachetzki JCM, et
al: Liver-derived signals sequentially reprogram myeloid enhancers
to initiate and maintain kupffer cell identity. Immunity.
51:655–670 e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yona S, Kim KW, Wolf Y, Mildner A, Varol
D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et
al: Fate mapping reveals origins and dynamics of monocytes and
tissue macrophages under homeostasis. Immunity. 38:79–91. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murray PJ and Wynn TA: Protective and
pathogenic functions of macrophage subsets. Nat Rev Immunol.
11:723–737. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martinez FO and Gordon S: The M1 and M2
paradigm of macrophage activation: time for reassessment.
F1000Prime Rep. 6:132014. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mills CD: Anatomy of a discovery: m1 and
m2 macrophages. Front Immunol. 6:2122015. View Article : Google Scholar :
|
|
20
|
Mills CD, Kincaid K, Alt JM, Heilman MJ
and Hill AM: M-1/M-2 macrophages and the Th1/Th2 paradigm. J
Immunol. 164:6166–6173. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bosco MC: Macrophage polarization:
Reaching across the aisle? J Allergy Clin Immunol. 143:1348–1350.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Locati M, Curtale G and Mantovani A:
Diversity, Mechanisms, and Significance of Macrophage Plasticity.
Annu Rev Pathol. 15:123–147. 2020. View Article : Google Scholar
|
|
23
|
Ivashkiv LB: Epigenetic regulation of
macrophage polarization and function. Trends Immunol. 34:216–223.
2013. View Article : Google Scholar :
|
|
24
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shapouri-Moghaddam A, Mohammadian S,
Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi
A, Afshari JT and Sahebkar A: Macrophage plasticity, polarization,
and function in health and disease. J Cell Physiol. 233:6425–6440.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dong T, Chen X, Xu H, Song Y, Wang H, Gao
Y, Wang J, Du R, Lou H and Dong T: Mitochondrial metabolism
mediated macrophage polarization in chronic lung diseases.
Pharmacol Ther. 239:1082082022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gordon S and Martinez FO: Alternative
activation of macrophages: Mechanism and functions. Immunity.
32:593–604. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Colin S, Chinetti-Gbaguidi G and Staels B:
Macrophage phenotypes in atherosclerosis. Immunol Rev. 262:153–166.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ruytinx P, Proost P, Van Damme J and
Struyf S: Chemokine-induced macrophage polarization in inflammatory
conditions. Front Immunol. 9:19302018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xue J, Schmidt SV, Sander J, Draffehn A,
Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L,
et al: Transcriptome-based network analysis reveals a spectrum
model of human macrophage activation. Immunity. 40:274–288. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan L, Wang J, Cai X, Liou YC, Shen HM,
Hao J, Huang C, Luo G and He W: Macrophage plasticity: Signaling
pathways, tissue repair, and regeneration. MedComm (2020).
5:e6582024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Murray PJ, Allen JE, Biswas SK, Fisher EA,
Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence
T, et al: Macrophage activation and polarization: nomenclature and
experimental guidelines. Immunity. 41:14–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen X, Tang J, Shuai W, Meng J, Feng J
and Han Z: Macrophage polarization and its role in the pathogenesis
of acute lung injury/acute respiratory distress syndrome. Inflamm
Res. 69:883–895. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen R, Zhang H, Tang B, Luo Y, Yang Y,
Zhong X, Chen S, Xu X, Huang S and Liu C: Macrophages in
cardiovascular diseases: Molecular mechanisms and therapeutic
targets. Signal Transduct Target Ther. 9:1302024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Frangogiannis NG: Cardiac fibrosis: Cell
biological mechanisms, molecular pathways and therapeutic
opportunities. Mol Aspects Med. 65:70–99. 2019. View Article : Google Scholar
|
|
36
|
Hou P, Fang J, Liu Z, Shi Y, Agostini M,
Bernassola F, Bove P, Candi E, Rovella V, Sica G, et al: Macrophage
polarization and metabolism in atherosclerosis. Cell Death Dis.
14:6912023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lopez B, Ravassa S, Moreno MU, José GS,
Beaumont J, González A and Díez J: Diffuse myocardial fibrosis:
Mechanisms, diagnosis and therapeutic approaches. Nat Rev Cardiol.
18:479–498. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Moore KJ and Tabas I: Macrophages in the
pathogenesis of atherosclerosis. Cell. 145:341–355. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Y, Xin W, Hu X, Wang H, Ye X, Xu C,
Nan Y, Wu Z, Ju D and Fan J: Inhibition of Hedgehog signaling
ameliorates foam cell formation by promoting autophagy in early
atherosclerosis. Cell Death Dis. 14:7402023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Byles V, Covarrubias AJ, Ben-Sahra I,
Lamming DW, Sabatini DM, Manning BD and Horng T: The TSC-mTOR
pathway regulates macrophage polarization. Nat Commun. 4:28342013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Castoldi A, Naffah de Souza C, Camara NO
and Moraes-Vieira PM: The macrophage switch in obesity development.
Front Immunol. 6:6372016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chawla A, Nguyen KD and Goh YP:
Macrophage-mediated inflammation in metabolic disease. Nat Rev
Immunol. 11:738–749. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Duan H, Jing L, Xiang J, Ju C, Wu Z, Liu
J, Ma X, Chen X, Liu Z, Feng J and Yan X: CD146 Associates with
Gp130 to Control a macrophage pro-inflammatory program that
regulates the metabolic response to obesity. Adv Sci (Weinh).
9:e21037192022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jiang H, Westerterp M, Wang C, Zhu Y and
Ai D: Macrophage mTORC1 disruption reduces inflammation and insulin
resistance in obese mice. Diabetologia. 57:2393–2404. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lu X, Kong X, Wu H, Hao J, Li S, Gu Z,
Zeng X, Shen Y, Wang S, Chen J, et al: UBE2M-mediated neddylation
of TRIM21 regulates obesity-induced inflammation and metabolic
disorders. Cell Metab. 35:1390–1405 e8. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lumeng CN, Bodzin JL and Saltiel AR:
Obesity induces a phenotypic switch in adipose tissue macrophage
polarization. J Clin Invest. 117:175–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lumeng CN, Deyoung SM, Bodzin JL and
Saltiel AR: Increased inflammatory properties of adipose tissue
macrophages recruited during diet-induced obesity. Diabetes.
56:16–23. 2007. View Article : Google Scholar
|
|
48
|
Cassetta L and Pollard JW: Targeting
macrophages: Therapeutic approaches in cancer. Nat Rev Drug Discov.
17:887–904. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
De Palma M, Biziato D and Petrova TV:
Microenvironmental regulation of tumour angiogenesis. Nat Rev
Cancer. 17:457–474. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Orecchioni M, Ghosheh Y, Pramod AB and Ley
K: Macrophage polarization: Different gene signatures in M1(LPS+)
vs. Classically and M2(LPS-) vs. alternatively activated
macrophages. Front Immunol. 10:10842019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pombo Antunes AR, Scheyltjens I, Lodi F,
Messiaen J, Antoranz A, Duerinck J, Kancheva D, Martens L, De
Vlaminck K, Van Hove H, et al: Single-cell profiling of myeloid
cells in glioblastoma across species and disease stage reveals
macrophage competition and specialization. Nat Neurosci.
24:595–610. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ruffell B, Chang-Strachan D, Chan V,
Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS and
Coussens LM: Macrophage IL-10 blocks CD8+ T cell-dependent
responses to chemotherapy by suppressing IL-12 expression in
intratumoral dendritic cells. Cancer Cell. 26:623–637. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Inoue K, Qin Y, Xia Y, Han J, Yuan R, Sun
J, Xu R, Jiang JX, Greenblatt MB and Zhao B: Bone marrow
Adipoq-lineage progenitors are a major cellular source of M-CSF
that dominates bone marrow macrophage development,
osteoclastogenesis, and bone mass. Elife. 12:e821182023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kulkarni A, Chandrasekar V, Natarajan SK,
Ramesh A, Pandey P, Nirgud J, Bhatnagar H, Ashok D, Ajay AK and
Sengupta S: A designer self-assembled supramolecule amplifies
macrophage immune responses against aggressive cancer. Nat Biomed
Eng. 2:589–599. 2018. View Article : Google Scholar
|
|
55
|
Sathi GA, Farahat M, Hara ES, Taketa H,
Nagatsuka H, Kuboki T and Matsumoto T: MCSF orchestrates branching
morphogenesis in developing submandibular gland tissue. J Cell Sci.
130:1559–1569. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Deng Y, Wang H, Liu X, Yuan H, Xu J, de
Thé H, Zhou J and Zhu J: Zbtb14 regulates monocyte and macrophage
development through inhibiting pu.1 expression in zebrafish. Elife.
11:e807602022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Solomon LA, Podder S, He J,
Jackson-Chornenki NL, Gibson K, Ziliotto RG, Rhee J and DeKoter RP:
Coordination of myeloid differentiation with reduced cell cycle
progression by PU.1 induction of MicroRNAs targeting cell cycle
regulators and lipid anabolism. Mol Cell Biol. 37:e00013–17. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tu J, Chen W, Fang Y, Han D, Chen Y, Jiang
H, Tan X, Xu Z, Wu X, Wang H, et al: PU.1 promotes development of
rheumatoid arthritis via repressing FLT3 in macrophages and
fibroblast-like synoviocytes. Ann Rheum Dis. 82:198–211. 2023.
View Article : Google Scholar
|
|
59
|
Xiao X, Li JX, Li HH and Teng F: ACE2
alleviates sepsis-induced cardiomyopathy through inhibiting M1
macrophage via NF-ĸB/STAT1 signals. Cell Biol Toxicol. 40:822024.
View Article : Google Scholar
|
|
60
|
Jin C, Zhang F, Luo H, Li B, Jiang X,
Pirozzi CJ, Liang C and Zhang M: The CCL5/CCR5/SHP2 axis sustains
Stat1 phosphorylation and activates NF-ĸB signaling promoting M1
macrophage polarization and exacerbating chronic prostatic
inflammation. Cell Commun Signal. 22:5842024. View Article : Google Scholar
|
|
61
|
Wu Q, Li B, Li J and Sun S, Yuan J and Sun
S: Cancer-associated adipocytes as immunomodulators in cancer.
Biomark Res. 9:22021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Gu L, Larson Casey JL, Andrabi SA, Lee JH,
Meza-Perez S, Randall TD and Carter AB: Mitochondrial calcium
uniporter regulates PGC-1α expression to mediate metabolic
reprogramming in pulmonary fibrosis. Redox Biol. 26:1013072019.
View Article : Google Scholar
|
|
63
|
Yu T, Gan S, Zhu Q, Dai D, Li N, Wang H,
Chen X, Hou D, Wang Y, Pan Q, et al: Modulation of M2 macrophage
polarization by the crosstalk between Stat6 and Trim24. Nat Commun.
10:43532019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Deng C, Huo M, Chu H, Zhuang X, Deng G, Li
W, Wei H, Zeng L, He Y, Liu H, et al: Exosome circATP8A1 induces
macrophage M2 polarization by regulating the miR-1-3p/STAT6 axis to
promote gastric cancer progression. Mol Cancer. 23:492024.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Choi JY, Seok HJ, Lee DH, Lee E, Kim TJ,
Bae S, Shin I and Bae IH: Tumor-derived miR-6794-5p enhances cancer
growth by promoting M2 macrophage polarization. Cell Commun Signal.
22:1902024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lee KA, Hai TY, SivaRaman L, Thimmappaya
B, Hurst HC, Jones NC and Green MR: A cellular protein, activating
transcription factor, activates transcription of multiple
E1A-inducible adenovirus early promoters. Proc Natl Acad Sci USA.
84:8355–8359. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hai T and Hartman MG: The molecular
biology and nomenclature of the activating transcription
factor/cAMP responsive element binding family of transcription
factors: Activating transcription factor proteins and homeostasis.
Gene. 273:1–11. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Vallejo M, Ron D, Miller CP and Habener
JF: C/ATF, a member of the activating transcription factor family
of DNA-binding proteins, dimerizes with CAAT/enhancer-binding
proteins and directs their binding to cAMP response elements. Proc
Natl Acad Sci USA. 90:4679–4683. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Montminy MR and Bilezikjian LM: Binding of
a nuclear protein to the cyclic-AMP response element of the
somatostatin gene. Nature. 328:175–178. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Moller E, Praz V, Rajendran S, Dong R,
Cauderay A, Xing YH, Lee L, Fusco C, Broye LC, Cironi L, et al:
EWSR1-ATF1 dependent 3D connectivity regulates oncogenic and
differentiation programs in clear cell sarcoma. Nat Commun.
13:22672022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hai TW, Liu F, Allegretto EA, Karin M and
Green MR: A family of immunologically related transcription factors
that includes multiple forms of ATF and AP-1. Genes Dev.
2:1216–1226. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Foulkes NS, Borrelli E and Sassone-Corsi
P: CREM gene: Use of alternative DNA-binding domains generates
multiple antagonists of cAMP-induced transcription. Cell.
64:739–749. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Liu H, Deng X, Shyu YJ, Li JJ, Taparowsky
EJ and Hu CD: Mutual regulation of c-Jun and ATF2 by
transcriptional activation and subcellular localization. EMBO J.
25:1058–1069. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
De Cesare D, Vallone D, Caracciolo A,
Sassone-Corsi P, Nerlov C and Verde P: Heterodimerization of c-Jun
with ATF-2 and c-Fos is required for positive and negative
regulation of the human urokinase enhancer. Oncogene. 11:365–376.
1995.PubMed/NCBI
|
|
75
|
Sun MH, Jiang WJ, Li XH, Lee SH, Heo G,
Zhou D, Choi JS, Kim KS, Lv W and Cui XS: ATF7-dependent epigenetic
changes induced by high temperature during early porcine embryonic
development. Cell Prolif. 56:e133522023. View Article : Google Scholar
|
|
76
|
Liu Y, Maekawa T, Yoshida K, Muratani M,
Chatton B and Ishii S: The transcription factor ATF7 controls
adipocyte differentiation and thermogenic gene programming.
iScience. 13:98–112. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Diring J, Camuzeaux B, Donzeau M, Vigneron
M, Rosa-Calatrava M, Kedinger C and Chatton B: A cytoplasmic
negative regulator isoform of ATF7 impairs ATF7 and ATF2
phosphorylation and transcriptional activity. PLoS One.
6:e233512011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Tsujino H, Kondo E, Fukuoka T, Dai Y,
Tokunaga A, Miki K, Yonenobu K, Ochi T and Noguchi K: Activating
transcription factor 3 (ATF3) induction by axotomy in sensory and
moto-neurons: A novel neuronal marker of nerve injury. Mol Cell
Neurosci. 15:170–182. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Rodriguez-Martinez JA, Reinke AW,
Bhimsaria D, Keating AE and Ansari AZ: Combinatorial bZIP dimers
display complex DNA-binding specificity landscapes. Elife.
6:e192722017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Tsushima H, Okazaki K, Ishihara K,
Ushijima T and Iwamoto Y: CCAAT/enhancer-binding protein β promotes
receptor activator of nuclear factor-kappa-B ligand (RANKL)
expression and osteoclast formation in the synovium in rheumatoid
arthritis. Arthritis Res Ther. 17:312015. View Article : Google Scholar
|
|
81
|
Huggins CJ, Mayekar MK, Martin N, Saylor
KL, Gonit M, Jailwala P, Kasoji M, Haines DC, Quiñones OA and
Johnson PF: C/EBPү is a critical regulator of cellular stress
response networks through heterodimerization with ATF4. Mol Cell
Biol. 36:693–713. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Dias-Teixeira KL, Calegari-Silva TC,
Medina JM, Vivarini ÁC, Cavalcanti Á, Teteo N, Santana AKM, Real F,
Gomes CM, Pereira RMS, et al: Emerging role for the
PERK/eIF2alpha/ATF4 in human cutaneous leishmaniasis. Sci Rep.
7:170742017. View Article : Google Scholar
|
|
83
|
Newman JR and Keating AE: Comprehensive
identification of human bZIP interactions with coiled-coil arrays.
Science. 300:2097–2101. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Glembotski CC, Arrieta A, Blackwood EA and
Stauffer WT: ATF6 as a nodal regulator of proteostasis in the
heart. Front Physiol. 11:2672020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Yoshida H, Okada T, Haze K, Yanagi H, Yura
T, Negishi M and Mori K: Endoplasmic reticulum stress-induced
formation of transcription factor complex ERSF including NF-Y (CBF)
and activating transcription factors 6alpha and 6beta that
activates the mammalian unfolded protein response. Mol Cell Biol.
21:1239–1248. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Yamamoto K, Sato T, Matsui T, Sato M,
Okada T, Yoshida H, Harada A and Mori K: Transcriptional induction
of mammalian ER quality control proteins is mediated by single or
combined action of ATF6alpha and XBP1. Dev Cell. 13:365–376. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hao Q, Zhao X, Zhang Y, Dong Z, Hu T and
Chen P: Targeting overexpressed activating transcription factor 1
(ATF1) inhibits proliferation and migration and enhances
sensitivity to paclitaxel in esophageal cancer cells. Med Sci Monit
Basic Res. 23:304–312. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Meijer BJ, Giugliano FP, Baan B, van der
Meer JHM, Meisner S, van Roest M, Koelink PJ, de Boer RJ, Jones N,
Breitwieser W, et al: ATF2 and ATF7 are critical mediators of
intestinal epithelial repair. Cell Mol Gastroenterol Hepatol.
10:23–42. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Maekawa T, Kim S, Nakai D, Takagi T, Ogura
H, Yamada K, Chatton B and Ishii S: Social isolation stress induces
ATF-7 phosphorylation and impairs silencing of the 5-HT 5B receptor
gene. EMBO J. 29:196–208. 2010. View Article : Google Scholar :
|
|
90
|
Gozdecka M and Breitwieser W: The roles of
ATF2 (activating transcription factor 2) in tumorigenesis. Biochem
Soc Trans. 40:230–234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Chen M, Liu Y, Yang Y, Qiu Y, Wang Z, Li X
and Zhang W: Emerging roles of activating transcription factor
(ATF) family members in tumourigenesis and immunity: Implications
in cancer immunotherapy. Genes Dis. 9:981–999. 2021. View Article : Google Scholar
|
|
92
|
Yan C and Boyd DD: ATF3 regulates the
stability of p53: A link to cancer. Cell Cycle. 5:926–929. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Hai T, Wolfgang CD, Marsee DK, Allen AE
and Sivaprasad U: ATF3 and stress responses. Gene Expr. 7:321–335.
1999.PubMed/NCBI
|
|
94
|
Giannoudis A, Malki MI, Rudraraju B,
Mohhamed H, Menon S, Liloglou T, Ali S, Carroll JS and Palmieri C:
Activating transcription factor-2 (ATF2) is a key determinant of
resistance to endocrine treatment in an in vitro model of breast
cancer. Breast Cancer Res. 22:1262020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Fawcett TW, Martindale JL, Guyton KZ, Hai
T and Holbrook NJ: Complexes containing activating transcription
factor (ATF)/cAMP-responsive-element-binding protein (CREB)
interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF
composite site to regulate Gadd153 expression during the stress
response. Biochem J. 339:135–141. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Shahriari-Felordi M, Alikhani HK,
Hashemian SR, Hassan M and Vosough M: Mini review ATF4 and GRP78 as
novel molecular targets in ER-Stress modulation for critical
COVID-19 patients. Mol Biol Rep. 49:1545–1549. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yoshida H, Okada T, Haze K, Yanagi H, Yura
T, Negishi M and Mori K: ATF6 activated by proteolysis binds in the
presence of NF-Y (CBF) directly to the cis-acting element
responsible for the mammalian unfolded protein response. Mol Cell
Biol. 20:6755–6767. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Thuerauf DJ, Morrison LE, Hoover H and
Glembotski CC: Coordination of ATF6-mediated transcription and ATF6
degradation by a domain that is shared with the viral transcription
factor, VP16. J Biol Chem. 277:20734–20739. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Thuerauf DJ, Arnold ND, Zechner D, Hanford
DS, DeMartin KM, McDonough PM, Prywes R and Glembotski CC: p38
Mitogen-activated protein kinase mediates the transcriptional
induction of the atrial natriuretic factor gene through a serum
response element. A potential role for the transcription factor
ATF6. J Biol Chem. 273:20636–20643. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Luo R, Lu JF, Hu Q and Maity SN: CBF/NF-Y
controls endoplasmic reticulum stress induced transcription through
recruitment of both ATF6(N) and TBP. J Cell Biochem. 104:1708–1723.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Kokame K, Kato H and Miyata T:
Identification of ERSE-II, a new cis-acting element responsible for
the ATF6-dependent mammalian unfolded protein response. J Biol
Chem. 276:9199–9205. 2001. View Article : Google Scholar
|
|
102
|
Haze K, Yoshida H, Yanagi H, Yura T and
Mori K: Mammalian transcription factor ATF6 is synthesized as a
transmembrane protein and activated by proteolysis in response to
endoplasmic reticulum stress. Mol Biol Cell. 10:3787–3799. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Niwano K, Arai M, Tomaru K, Uchiyama T,
Ohyama Y and Kurabayashi M: Transcriptional stimulation of the eNOS
gene by the stable prostacyclin analogue beraprost is mediated
through cAMP-responsive element in vascular endothelial cells:
Close link between PGI2 signal and NO pathways. Circ Res.
93:523–530. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Xiao X, Chen M, Sang Y, Xue J, Jiang K,
Chen Y, Zhang L, Yu S, Lv W, Li Y, et al: Methylation-mediated
silencing of ATF3 promotes thyroid cancer progression by regulating
prognostic genes in the MAPK and PI3K/AKT pathways. Thyroid.
33:1441–1454. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zhang WC, Du LJ, Zheng XJ, Chen XQ, Shi C,
Chen BY, Sun XN, Li C, Zhang YY, Liu Y, et al: Elevated sodium
chloride drives type I interferon signaling in macrophages and
increases antiviral resistance. J Biol Chem. 293:1030–1039. 2018.
View Article : Google Scholar :
|
|
106
|
Ranjan K, Hedl M, Sinha S, Zhang X and
Abraham C: Ubiquitination of ATF6 by disease-associated RNF186
promotes the innate receptor-induced unfolded protein response. J
Clin Invest. 131:e1454722021. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Udompong S, Mankhong S, Jaratjaroonphong J
and Srisook K: Involvement of p38 MAPK and ATF-2 signaling pathway
in anti-inflammatory effect of a novel compound
bis[(5-methyl)2-furyl] (4-nitrophenyl)methane on
lipopolysaccharide-stimulated macrophages. Int Immunopharmacol.
50:6–13. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Herrema H, Guan D, Choi JW, Feng X,
Salazar Hernandez MA, Faruk F, Auen T, Boudett E, Tao R, Chun H and
Ozcan U: FKBP11 rewires UPR signaling to promote glucose
homeostasis in type 2 diabetes and obesity. Cell Metab.
34:1004–1022 e8. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhou S, Zhong Z, Huang P, Xiang B, Li X,
Dong H, Zhang G, Wu Y and Li P: IL-6/STAT3 induced neuron apoptosis
in hypoxia by downregulating ATF6 expression. Front Physiol.
12:7299252021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Kwon JW, Kwon HK, Shin HJ, Choi YM, Anwar
MA and Choi S: Activating transcription factor 3 represses
inflammatory responses by binding to the p65 subunit of NF-ĸB. Sci
Rep. 5:144702015. View Article : Google Scholar
|
|
111
|
Cui A, Ding D and Li Y: Regulation of
hepatic metabolism and cell growth by the ATF/CREB family of
transcription factors. Diabetes. 70:653–664. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Takii R, Fujimoto M, Tan K, Takaki E,
Hayashida N, Nakato R, Shirahige K and akai A: ATF1 modulates the
heat shock response by regulating the stress-inducible heat shock
factor 1 transcription complex. Mol Cell Biol. 35:11–25. 2015.
View Article : Google Scholar :
|
|
113
|
Karanam B, Wang L, Wang D, Liu X,
Marmorstein R, Cotter R and Cole PA: Multiple roles for acetylation
in the interaction of p300 HAT with ATF-2. Biochemistry.
46:8207–8216. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Nguyen HCB, Adlanmerini M, Hauck AK and
Lazar MA: Dichotomous engagement of HDAC3 activity governs
inflammatory responses. Nature. 584:286–290. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Gilchrist M, Thorsson V, Li B, Rust AG,
Korb M, Roach JC, Kennedy K, Hai T, Bolouri H and Aderem A: Systems
biology approaches identify ATF3 as a negative regulator of
Toll-like receptor 4. Nature. 441:173–178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Chen BP, Liang G, Whelan J and Hai T: ATF3
and ATF3 delta Zip. Transcriptional repression versus activation by
alternatively spliced isoforms. J Biol Chem. 269:15819–15826. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Sela D, Chen L, Martin-Brown S, Washburn
MP, Florens L, Conaway JW and Conaway RC: Endoplasmic reticulum
stress-responsive transcription factor ATF6alpha directs
recruitment of the mediator of RNA polymerase II transcription and
multiple histone acetyltransferase complexes. J Biol Chem.
287:23035–23045. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Hirose N, Maekawa T, Shinagawa T and Ishii
S: ATF-2 regulates lipopolysaccharide-induced transcription in
macrophage cells. Biochem Biophys Res Commun. 385:72–77. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Yoshida K, Maekawa T, Zhu Y,
Renard-Guillet C, Chatton B, Inoue K, Uchiyama T, Ishibashi K,
Yamada T, Ohno N, et al: The transcription factor ATF7 mediates
lipopolysaccharide-induced epigenetic changes in macrophages
involved in innate immunological memory. Nat Immunol. 16:1034–1043.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Baumeister P, Luo S, Skarnes WC, Sui G,
Seto E, Shi Y and Lee AS: Endoplasmic reticulum stress induction of
the Grp78/BiP promoter: Activating mechanisms mediated by YY1 and
its interactive chromatin modifiers. Mol Cell Biol. 25:4529–4540.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Ma J, Liu Y, Valladolid-Acebes I,
Recio-López P, Peng G, Li J, Berggren PO, Juntti-Berggren L and
Tong N: ATF5 is a regulator of ER stress and β-cell apoptosis in
different mouse models of genetic- and diet-induced obesity and
diabetes mellitus. Cell Signal. 102:1105352023. View Article : Google Scholar
|
|
122
|
Raines LN, Zhao H, Wang Y, Chen HY,
Gallart-Ayala H, Hsueh PC, Cao W, Koh Y, Alamonte-Loya A, Liu PS,
et al: PERK is a critical metabolic hub for immunosuppressive
function in macrophages. Nat Immunol. 23:431–445. 2022. View Article : Google Scholar : PubMed/NCBI
Wang Y, Zhang X, Fu Y, Fu D, Zhen D, Xing
A, Chen Y, Gong G and Wei C: 1, 8-cineole protects against
ISO-induced heart failure by inhibiting oxidative stress and ER
stress in vitro and in vivo. Eur J Pharmacol. 910:1744722021.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Hu S, Li R, Gong D, Hu P, Xu J, Ai Y, Zhao
X, Hu C, Xu M, Liu C, et al: Atf3-mediated metabolic reprogramming
in hepatic macrophage orchestrates metabolic dysfunction-associated
steatohepatitis. Sci Adv. 10:eado31412024. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Brocard M, Lu J, Hall B, Borah K,
Moller-Levet C, Georgana I, Sorgeloos F, Beste DJV, Goodfellow IG
and Locker N: Murine norovirus infection results in
anti-inflammatory response downstream of amino acid depletion in
macrophages. J Virol. 95:e01134212021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Huang Y, Ge MX, Li YH, Li JL, Yu Q, Xiao
FH, Ao HS, Yang LQ, Li J, He Y and Kong QP: Longevity-associated
transcription factor ATF7 promotes healthspan by suppressing
cellular senescence and systematic inflammation. Aging Dis.
14:1374–1389. 2023.PubMed/NCBI
|
|
126
|
Liu H, Kuang X, Zhang Y, Ye Y, Li J, Liang
L, Xie Z, Weng L, Guo J, Li H, et al: ADORA1 inhibition promotes
tumor immune evasion by regulating the ATF3-PD-L1 axis. Cancer
Cell. 37:324–339 e8. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Liu T, Wen Z, Shao L, Cui Y, Tang X, Miao
H, Shi J, Jiang L, Feng S, Zhao Y, et al: ATF4 knockdown in
macrophage impairs glycolysis and mediates immune tolerance by
targeting HK2 and HIF-1alpha ubiquitination in sepsis. Clin
Immunol. 254:1096982023. View Article : Google Scholar
|
|
128
|
Zhang Q, Liu G, Liu R, Liu J, Zeng X, Ren
D, Yan X and Yuan X: Dual role of endoplasmic reticulum
stress-ATF-6 activation in autophagy and apoptosis induced by
cyclic stretch in myoblast. Apoptosis. 28:796–809. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Dai W, Hong L, Xiao W, Zhang L, Sha W, Yu
Z, Liu X, Liu S, Xiao Y, Yang P, et al: The ATF2/miR-3913-5p/CREB5
axis is involved in the cell proliferation and metastasis of
colorectal cancer. Commun Biol. 6:10262023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Wang B, Zhang J, Liu X, Chai Q, Lu X, Yao
X, Yang Z, Sun L, Johnson SF, Schwartz RC and Zheng YH: Protein
disulfide isomerases (PDIs) negatively regulate ebolavirus
structural glycoprotein expression in the endoplasmic reticulum
(ER) via the autophagy-lysosomal pathway. Autophagy. 18:2350–2367.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Wen Z, Xiong X, Chen D, Shao L, Tang X,
Shen X, Zhang S, Huang S, Zhang L, Chen Y, et al: Activating
transcription factor 4 protects mice against sepsis-induced
intestinal injury by regulating gut-resident macrophages
differentiation. Chin Med J (Engl). 135:2585–2595. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Rajabalee N, Siushansian H, Weerapura M,
Berton S, Berbatovci F, Hooks B, Geoffrion M, Yang D, Harper ME,
Rayner K, et al: ATF2 orchestrates macrophage differentiation and
activation to promote antibacterial responses. J Leukoc Biol.
114:280–298. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Qin W, Yang H, Liu G, Bai R, Bian Y, Yang
Z and Xiao C: Activating transcription factor 3 is a potential
target and a new biomarker for the prognosis of atherosclerosis.
Hum Cell. 34:49–59. 2021. View Article : Google Scholar
|
|
134
|
Nilsson R, Bajic VB, Suzuki H, di Bernardo
D, Björkegren J, Katayama S, Reid JF, Sweet MJ, Gariboldi M,
Carninci P, et al: Transcriptional network dynamics in macrophage
activation. Genomics. 88:133–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Clement M, Basatemur G, Masters L, Baker
L, Bruneval P, Iwawaki T, Kneilling M, Yamasaki S, Goodall J and
Mallat Z: Necrotic cell sensor clec4e promotes a proatherogenic
macrophage phenotype through activation of the unfolded protein
response. Circulation. 134:1039–1051. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Elbarghati L, Murdoch C and Lewis CE:
Effects of hypoxia on transcription factor expression in human
monocytes and macrophages. Immunobiology. 213:899–908. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Zhao Q, Luo YF, Tian M, Xiao YL, Cai HR
and Li H: Activating transcription factor 3 involved in Pseudomonas
aeruginosa PAO1-induced macrophage senescence. Mol Immunol.
133:122–127. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Li M, Lu H, Wang X, Duan C, Zhu X, Zhang
Y, Ge X, Ji F, Wang X, Su J and Zhang D: Pyruvate kinase M2 (PKM2)
interacts with activating transcription factor 2 (ATF2) to bridge
glycolysis and pyroptosis in microglia. Mol Immunol. 140:250–266.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Hu Y, Li CS, Li YQ, Liang Y, Cao L and
Chen LA: Perfluorocarbon inhibits lipopolysaccharide-induced
macrophage inflammatory protein-2 expression and activation of
ATF-2 and c-Jun in A549 pulmonary epithelial cells. Cell Mol Biol
(Noisy-le-grand). 62:18–24. 2016.PubMed/NCBI
|
|
140
|
Boehlk S, Fessele S, Mojaat A, Miyamoto
NG, Werner T, Nelson EL, Schlöndorff D and Nelson PJ: ATF and Jun
transcription factors, acting through an Ets/CRE promoter module,
mediate lipopolysaccharide inducibility of the chemokine RANTES in
monocytic Mono Mac 6 cells. Eur J Immunol. 30:1102–1112. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Sha H, Zhang D, Zhang Y, Wen Y and Wang Y:
ATF3 promotes migration and M1/M2 polarization of macrophages by
activating tenascin-C via Wnt/β-catenin pathway. Mol Med Rep.
16:3641–3647. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Hu C, Meng X, Huang C, Shen C and Li J:
Frontline Science: ATF3 is responsible for the inhibition of TNF-α
release and the impaired migration of acute ethanol-exposed
monocytes and macrophages. J Leukoc Biol. 101:633–642. 2017.
View Article : Google Scholar
|
|
143
|
Du Y, Ma Z, Zheng J, Huang S, Yang X, Song
Y, Dong D, Shi L and Xu D: ATF3 positively regulates antibacterial
immunity by modulating macrophage killing and migration functions.
Front Immunol. 13:8395022022. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Mylvaganam S, Freeman SA and Grinstein S:
The cytoskeleton in phagocytosis and macropinocytosis. Curr Biol.
31:R619–R632. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Taka S, Gazouli M, Politis PK, Pappa KI
and Anagnou NP: Transcription factor ATF-3 regulates allele
variation phenotypes of the human SLC11A1 gene. Mol Biol Rep.
40:2263–2271. 2013. View Article : Google Scholar
|
|
146
|
Middleton JD, Fehlman J, Sivakumar S,
Stover DG and Hai T: Stress-inducible gene Atf3 dictates a
dichotomous macrophage activity in chemotherapy-enhanced lung
colonization. Int J Mol Sci. 22:73562021. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Xu Y, Li Y, Jadhav K, Pan X, Zhu Y, Hu S,
Chen S, Chen L, Tang Y, Wang HH, et al: Hepatocyte ATF3 protects
against atherosclerosis by regulating HDL and bile acid metabolism.
Nat Metab. 3:59–74. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Raghavan S, Singh NK, Gali S, Mani AM and
Rao GN: Protein Kinase ctheta via activating transcription factor
2-mediated CD36 expression and foam cell formation of Ly6C(hi)
cells contributes to atherosclerosis. Circulation. 138:2395–2412.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Seneviratne A, Han Y, Wong E, Walter ERH,
Jiang L, Cave L, Long NJ, Carling D, Mason JC, Haskard DO and Boyle
JJ: Hematoma resolution in vivo is directed by activating
transcription factor 1. Circ Res. 127:928–944. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Illouz M, Leclercq LD, Dessenne C, Hatfull
G, Daher W and Kremer L and Guérardel Y: Multiple Mycobacterium
abscessus O-acetyltransferases influence glycopeptidolipid
structure and colony morphotype. J Biol Chem. 299:1049792023.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Perez-Arques C, Navarro-Mendoza MI, Murcia
L, Lax C, Martínez-García P, Heitman J, Nicolás FE and Garre V:
Mucor circinelloides Thrives inside the phagosome through an
Atf-Mediated germination pathway. mBio. 10:e02765–18. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Gupta D, Wang Q, Vinson C and Dziarski R:
Bacterial peptidoglycan induces CD14-dependent activation of
transcription factors CREB/ATF and AP-1. J Biol Chem.
274:14012–14020. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Shi R, Wang J, Zhang Z, Leng Y and Chen
AF: ASGR1 promotes liver injury in sepsis by modulating
monocyte-to-macrophage differentiation via NF-ĸB/ATF5 pathway. Life
Sci. 315:1213392023. View Article : Google Scholar
|
|
154
|
Abe JI, Ko KA, Kotla S, Wang Y,
Paez-Mayorga J, Shin IJ, Imanishi M, Vu HT, Tao Y, Leiva-Juarez MM,
et al: MAGI1 as a link between endothelial activation and ER stress
drives atherosclerosis. JCI Insight. 4:e1255702019. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Jin Z, Xu H, Zhao W, Zhang K, Wu S, Shu C,
Zhu L, Wang Y, Wang L, Zhang H and Yan B: Macrophage ATF6
accelerates corticotomy-assisted orthodontic tooth movement through
promoting Tnfα transcription. Int J Oral Sci. 17:282025. View Article : Google Scholar
|
|
156
|
Xu X, Lei T, Li W and Ou H: Enhanced
cellular cholesterol efflux by naringenin is mediated through
inhibiting endoplasmic reticulum stress - ATF6 activity in
macrophages. Biochim Biophys Acta Mol Cell Biol Lipids.
1864:1472–1482. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Rao J, Yue S, Fu Y, Zhu J, Wang X,
Busuttil RW, Kupiec-Weglinski JW, Lu L and Zhai Y: ATF6 mediates a
pro-inflammatory synergy between ER stress and TLR activation in
the pathogenesis of liver ischemia-reperfusion injury. Am J
Transplant. 14:1552–1561. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Wang Q, Zhu X, Li Z, Feng M and Liu X:
ATF6 promotes liver fibrogenesis by regulating macrophage-derived
interleukin-1alpha expression. Cell Immunol. 367:1044012021.
View Article : Google Scholar
|
|
159
|
Wang J, Cheng W, Wang Z, Xin L and Zhang
W: ATF3 inhibits the inflammation induced by Mycoplasma pneumonia
in vitro and in vivo. Pediatr Pulmonol. 52:1163–1170. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Hoetzenecker W, Echtenacher B, Guenova E,
Hoetzenecker K, Woelbing F, Brück J, Teske A, Valtcheva N, Fuchs K,
Kneilling M, et al: ROS-induced ATF3 causes susceptibility to
secondary infections during sepsis-associated immunosuppression.
Nat Med. 18:128–134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Bae YA and Cheon HG: Activating
transcription factor-3 induction is involved in the
anti-inflammatory action of berberine in RAW264.7 murine
macrophages. Korean J Physiol Pharmacol. 20:415–424. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Saha S, Roy S, Dutta A, Jana K and Ukil A:
Leishmania donovani targets host transcription factor NRF2 to
activate antioxidant enzyme HO-1 and transcriptional repressor ATF3
for establishing infection. Infect Immun. 89:e00764202021.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Qian L, Zhao Y, Guo L, Li S and Wu X:
Activating transcription factor 3 (ATF3) protects against
lipopolysaccharide-induced acute lung injury via inhibiting the
expression of TL1A. J Cell Physiol. 232:3727–3734. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Cheng CF and Lin H: Acute kidney injury
and the potential for ATF3-regulated epigenetic therapy. Toxicol
Mech Methods. 21:362–366. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Yu T, Moh SH, Kim SB, Yang Y, Kim E, Lee
YW, Cho CK, Kim KH, Yoo BC, Cho JY and Yoo HS: HangAmDan-B, an
ethnomedicinal herbal mixture, suppresses inflammatory responses by
inhibiting Syk/NF-ĸB and JNK/ATF-2 pathways. J Med Food. 16:56–65.
2013. View Article : Google Scholar :
|
|
166
|
Zhang C, He H, Wang L, Zhang N, Huang H,
Xiong Q, Yan Y, Wu N, Ren H, Han H, et al: Virus-triggered ATP
release limits viral replication through facilitating IFN-β
Production in a P2X7-Dependent manner. J Immunol. 199:1372–1381.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Yu T, Yang Y, Kwak YS, Song GG, Kim MY,
Rhee MH and Cho JY: Ginsenoside Rc from Panax ginseng exerts
anti-inflammatory activity by targeting TANK-binding kinase
1/interferon regulatory factor-3 and p38/ATF-2. J Ginseng Res.
41:127–133. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Xu X, Xia J, Zhao S, Wang Q, Ge G, Xu F,
Liu X, Zhang W and Yang Y: Qing-Fei-Pai-Du decoction and wogonoside
exert anti-inflammatory action through down-regulating USP14 to
promote the degradation of activating transcription factor 2. FASEB
J. 35:e218702021. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Park JB, Peters R, Pham Q and Wang TTY:
Javamide-II Inhibits IL-6 without Significant Impact on TNF-alpha
and IL-1beta in macrophage-like cells. Biomedicines. 8:1382020.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Kim SM, Park EJ and Lee HJ: Nuciferine
attenuates lipopolysaccharide-stimulated inflammatory responses by
inhibiting p38 MAPK/ATF2 signaling pathways. Inflammopharmacology.
30:2373–2383. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Endale M, Kim TH, Kwak YS, Kim NM, Kim SH,
Cho JY, Yun BS and Rhee MH: Torilin inhibits inflammation by
limiting TAK1-mediated MAP kinase and NF-ĸB activation. Mediators
Inflamm. 2017:72509682017. View Article : Google Scholar
|
|
172
|
Miyata Y, Fukuhara A, Otsuki M and
Shimomura I: Expression of activating transcription factor 2 in
inflammatory macrophages in obese adipose tissue. Obesity (Silver
Spring). 21:731–736. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Hu Y, Gu J, Wang Y, Lin J, Yu H, Yang F,
Wu S, Yin J, Lv H, Ji X and Wang S: Promotion effect of EGCG on the
raised expression of IL-23 through the signaling of
STAT3-BATF2-c-JUN/ATF2. J Agric Food Chem. 69:7898–7909. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Sukhorukov VN, Khotina VA, Bagheri Ekta M,
Ivanova EA, Sobenin IA and Orekhov AN: Endoplasmic reticulum stress
in macrophages: The vicious circle of lipid accumulation and
pro-inflammatory response. Biomedicines. 8:2102020. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Song C, Chen J, Li X, Yang R, Cao X, Zhou
L, Zhou Y, Ying H, Zhang Q and Sun Y: Limonin ameliorates dextran
sulfate sodium-induced chronic colitis in mice by inhibiting
PERK-ATF4-CHOP pathway of ER stress and NF-kappaB signaling. Int
Immunopharmacol. 90:1071612021. View Article : Google Scholar
|
|
176
|
Luo JH, Wang FX, Zhao JW, Yang CL, Rong
SJ, Lu WY, Chen QJ, Zhou Q, Xiao J, Wang YN, et al: PDIA3 defines a
novel subset of adipose macrophages to exacerbate the development
of obesity and metabolic disorders. Cell Metab. 36:2262–2280 e5.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Boyle JJ, Johns M, Kampfer T, Nguyen AT,
Game L, Schaer DJ, Mason JC and Haskard DO: Activating
transcription factor 1 directs Mhem atheroprotective macrophages
through coordinated iron handling and foam cell protection. Circ
Res. 110:20–33. 2012. View Article : Google Scholar
|
|
178
|
Suganami T, Yuan X, Shimoda Y,
Uchio-Yamada K, Nakagawa N, Shirakawa I, Usami T, Tsukahara T,
Nakayama K, Miyamoto Y, et al: Activating transcription factor 3
constitutes a negative feedback mechanism that attenuates saturated
Fatty acid/toll-like receptor 4 signaling and macrophage activation
in obese adipose tissue. Circ Res. 105:25–32. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Tang Q, Xie J, Wang Y, Dong C and Sun Q:
Exosomes secreted by ATF3/Nrf2-mediated ferroptotic renal tubular
epithelial cells promote M1/M2 ratio imbalance inducing renal
interstitial fibrosis following ischemia and reperfusion injury.
Front Immunol. 16:15105002025. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Bartels HC, Hameed S, Young C, Nabhan M,
Downey P, Curran KM, McCormack J, Fabre A, Kolch W, Zhernovkov V
and Brennan DJ: Spatial proteomics and transcriptomics of the
maternal-fetal interface in placenta accreta spectrum. Transl Res.
274:67–80. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Iwasaki Y, Suganami T, Hachiya R,
Shirakawa I, Kim-Saijo M, Tanaka M, Hamaguchi M, Takai-Igarashi T,
Nakai M, Miyamoto Y and Ogawa Y: Activating transcription factor 4
links metabolic stress to interleukin-6 expression in macrophages.
Diabetes. 63:152–161. 2014. View Article : Google Scholar
|
|
182
|
Xia HF, Zhu JY, Wang JN, Ren JG, Cai Y,
Wang FQ, Zhang W, Chen G, Zhao YF and Zhao JH: Association of ATF4
expression with tissue hypoxia and M2 macrophage infiltration in
infantile hemangioma. J Histochem Cytochem. 65:285–294. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Chen C, Zong S, Wang Z, Yang R, Guo Y,
Wang Y, Chen X, Li Y and Wang S: FTY720 Attenuates LPS-Induced
inflammatory bone loss by inhibiting osteoclastogenesis via the
NF-kappaB and HDAC4/ATF pathways. J Immunol Res. 2023:85716492023.
View Article : Google Scholar
|
|
184
|
Baek K, Park HJ, Baek JH and Kim HR:
Isoproterenol increases RANKL expression in a ATF4/NFATc1-dependent
manner in mouse osteoblastic cells. Int J Mol Sci. 18:22042017.
View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Chattopadhyay A, Kwartler CS, Kaw K, Li Y,
Kaw A, Chen J, LeMaire SA, Shen YH and Milewicz DM:
Cholesterol-induced phenotypic modulation of smooth muscle cells to
macrophage/fibroblast-like cells is driven by an unfolded protein
response. Arterioscler Thromb Vasc Biol. 41:302–316. 2021.
View Article : Google Scholar
|
|
186
|
Kim J, Kwak HJ, Cha JY, Jeong YS, Rhee SD,
Kim KR and Cheon HG: Metformin suppresses lipopolysaccharide
(LPS)-induced inflammatory response in murine macrophages via
activating transcription factor-3 (ATF-3) induction. J Biol Chem.
289:23246–23255. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Luo M, Zhao F, Cheng H, Su M and Wang Y:
Macrophage polarization: An important role in inflammatory
diseases. Front Immunol. 15:13529462024. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Xiang M, Li P, Yue X, Liu L, Wang L, Sun
N, Wang K, Zhang Y and Wang H: Dysregulated macrophage immunity in
Helicobacter pylori infection: unveiling mechanistic insights and
therapeutic implications. Front Immunol. 16:16367682025. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Fu YL and Harrison RE: Microbial
phagocytic receptors and their potential involvement in cytokine
induction in macrophages. Front Immunol. 12:6620632021. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Arango Duque G and Descoteaux A:
Macrophage cytokines: Involvement in immunity and infectious
diseases. Front Immunol. 5:4912014. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Lv K and Liang Q: Macrophages in
sepsis-induced acute lung injury: exosomal modulation and
therapeutic potential. Front Immunol. 15:15180082025. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
A-Gonzalez N, Quintana JA, Garcia-Silva S,
Mazariegos M, González de la Aleja A, Nicolás-Ávila JA, Walter W,
Adrover JM, Crainiciuc G, Kuchroo VK, et al: Phagocytosis imprints
heterogeneity in tissue-resident macrophages. J Exp Med.
214:1281–1296. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Ochando J, Mulder WJM, Madsen JC, Netea MG
and Duivenvoorden R: Trained immunity-basic concepts and
contributions to immunopathology. Nat Rev Nephrol. 19:23–37. 2023.
View Article : Google Scholar
|
|
194
|
Nathan C and Ding A: Nonresolving
inflammation. Cell. 140:871–882. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Cheng Y, Marion TN, Cao X, Wang W and Cao
Y: Park 7: A novel therapeutic target for macrophages in
sepsis-induced immunosuppression. Front Immunol. 9:26322018.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Jiang Y, Cai R, Huang Y, Zhu L, Xiao L,
Wang C and Wang L: Macrophages in organ fibrosis: From pathogenesis
to therapeutic targets. Cell Death Discov. 10:4872024. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Ahmad S, Zaki A, Manda K, Mohan A and Syed
MA: Vitamin-D ameliorates sepsis-induced acute lung injury via
augmenting miR-149-5p and downregulating ER stress. J Nutr Biochem.
110:1091302022. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Zhang S, Meng P, Liu G, Liu K and Che C:
ATF4 involvement in TLR4 and LOX-1-induced host inflammatory
response to aspergillus fumigatus keratitis. J Ophthalmol.
2018:58302022018.
|
|
199
|
Li R, Ren T, Zeng J and Xu H: ALCAM
deficiency alleviates LPS-Induced acute lung injury by inhibiting
inflammatory response. Inflammation. 46:688–699. 2023. View Article : Google Scholar
|
|
200
|
Al-Salleeh F and Petro TM: Promoter
analysis reveals critical roles for SMAD-3 and ATF-2 in expression
of IL-23 p19 in macrophages. J Immunol. 181:4523–4533. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Ta HM, Le TM, Ishii H, Takarada-Iemata M,
Hattori T, Hashida K, Yamamoto Y, Mori K, Takahashi R, Kitao Y and
Hori O: Atf6α deficiency suppresses microglial activation and
ameliorates pathology of experimental autoimmune encephalomyelitis.
J Neurochem. 139:1124–1137. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Wellen KE and Hotamisligil GS:
Obesity-induced inflammatory changes in adipose tissue. J Clin
Invest. 112:1785–1788. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Olefsky JM and Glass CK: Macrophages,
inflammation, and insulin resistance. Annu Rev Physiol. 72:219–246.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Liang W, Qi Y, Yi H, Mao C, Meng Q, Wang H
and Zheng C: The roles of adipose tissue macrophages in human
disease. Front Immunol. 13:9087492022. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Guria S, Hoory A, Das S, Chattopadhyay D
and Mukherjee S: Adipose tissue macrophages and their role in
obesity-associated insulin resistance: An overview of the complex
dynamics at play. Biosci Rep. 43:BSR202202002023. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Bai Y and Sun Q: Macrophage recruitment in
obese adipose tissue. Obes Rev. 16:127–136. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
De Nardo D, Labzin LI, Kono H, Seki R,
Schmidt SV, Beyer M, Xu D, Zimmer S, Lahrmann C, Schildberg FA, et
al: High-density lipoprotein mediates anti-inflammatory
reprogramming of macrophages via the transcriptional regulator
ATF3. Nat Immunol. 15:152–160. 2014. View Article : Google Scholar
|
|
208
|
Xu J, Ding L, Mei J, Hu Y, Kong X, Dai S,
Bu T, Xiao Q and Ding K: Dual roles and therapeutic targeting of
tumor-associated macrophages in tumor microenvironments. Signal
Transduct Target Ther. 10:2682025. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Takayanagi H: Osteoimmunology: Shared
mechanisms and crosstalk between the immune and bone systems. Nat
Rev Immunol. 7:292–304. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Sun Y, Li J, Xie X, Gu F, Sui Z, Zhang K
and Yu T: Macrophage-osteoclast associations: Origin, polarization,
and subgroups. Front Immunol. 12:7780782021. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Nakashima T, Hayashi M and Takayanagi H:
New insights into osteoclastogenic signaling mechanisms. Trends
Endocrinol Metab. 23:582–590. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Perrone M, Chiodoni C, Lecchi M, Botti L,
Bassani B, Piva A, Jachetti E, Milani M, Lecis D, Tagliabue E, et
al: ATF3 reprograms the bone marrow niche in response to early
breast cancer transformation. Cancer Res. 83:117–129. 2023.
View Article : Google Scholar :
|
|
213
|
Gao C, Jiang J, Tan Y and Chen S:
Microglia in neurodegenerative diseases: Mechanism and potential
therapeutic targets. Signal Transduct Target Ther. 8:3592023.
View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Fleiss B, Van Steenwinckel J, Bokobza C, I
KS, Ross-Munro E and Gressens P: Microglia-mediated
neurodegeneration in perinatal brain injuries. Biomolecules.
11:992021. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Flury A, Aljayousi L, Park HJ, Khakpour M,
Mechler J, Aziz S, McGrath JD, Deme P, Sandberg C, González Ibáñez
F, et al: A neurodegenerative cellular stress response linked to
dark microglia and toxic lipid secretion. Neuron. 113:554–571 e14.
2025. View Article : Google Scholar :
|
|
216
|
Liu LL, Xiao YS, Huang WM, Liu S, Huang
LX, Zhong JH, Jia P and Liu WY: ATF1/miR-214-5p/ITGA7 axis promotes
osteoclastogenesis to alter OVX-induced bone absorption. Mol Med.
28:562022. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Kim HN, Baek JK, Park SB, Kim JD, Son HJ,
Park GH, Eo HJ, Park JH, Jung HS and Jeong JB: Anti-inflammatory
effect of Vaccinium oldhamii stems through inhibition of NF-kappaB
and MAPK/ATF2 signaling activation in LPS-stimulated RAW264.7
cells. BMC Complement Altern Med. 19:2912019. View Article : Google Scholar
|
|
218
|
Comarita IK, Vilcu A, Constantin A,
Procopciuc A, Safciuc F, Alexandru N, Dragan E, Nemecz M, Filippi
A, Chiţoiu L, et al: Therapeutic potential of stem cell-derived
extracellular vesicles on atherosclerosis-induced vascular
dysfunction and its key molecular players. Front Cell Dev Biol.
10:8171802022. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Ho HH, Antoniv TT, Ji JD and Ivashkiv LB:
Lipopolysaccharide-induced expression of matrix metalloproteinases
in human monocytes is suppressed by IFN-gamma via superinduction of
ATF-3 and suppression of AP-1. J Immunol. 181:5089–5097. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Ishida M, Ueki M, Morishita J, Ueno M,
Shiozawa S and Maekawa N: T-5224, a selective inhibitor of
c-Fos/activator protein-1, improves survival by inhibiting serum
high mobility group box-1 in lethal lipopolysaccharide-induced
acute kidney injury model. J Intensive Care. 3:492015. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Kubryn N, Fijalkowski L, Nowaczyk J, Jamil
A and Nowaczyk A: PROTAC technology as a new tool for modern
pharmacotherapy. Molecules. 30:21232025. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Hu Z and Crews CM: Recent developments in
PROTAC-mediated protein degradation: From bench to clinic.
Chembiochem. 23:e2021002702022. View Article : Google Scholar :
|
|
223
|
Bekes M, Langley DR and Crews CM: PROTAC
targeted protein degraders: The past is prologue. Nat Rev Drug
Discov. 21:181–200. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Ai M, Ma H, He J, Xu F, Ming Y, Ye Z,
Zheng Q, Luo D, Yang K, Li J, et al: Targeting oncogenic
transcriptional factor c-myc by oligonucleotide PROTAC for the
treatment of hepatocellular carcinoma. Eur J Med Chem.
280:1169782024. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Simpson LM, Glennie L, Brewer A, Zhao JF,
Crooks J, Shpiro N and Sapkota GP: Target protein localization and
its impact on PROTAC-mediated degradation. Cell Chem Biol.
29:1482–1504 e7. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Liu Z, Hu M, Yang Y, Du C, Zhou H, Liu C,
Chen Y, Fan L, Ma H, Gong Y and Xie Y: An overview of PROTACs: A
promising drug discovery paradigm. Mol Biomed. 3:462022. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Liu J, Chen H, Kaniskan HU, Xie L, Chen X,
Jin J and Wei W: TF-PROTACs enable targeted degradation of
transcription factors. J Am Chem Soc. 143:8902–8910. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Ji J, Ma S, Zhu Y, Zhao J, Tong Y, You Q
and Jiang Z: ARE-PROTACs enable co-degradation of an Nrf2-MafG
heterodimer. J Med Chem. 66:6070–6081. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Wang D, Liu Y, Chen Y, Dai C, Hu W, Han J,
Li Z, Yin F, Zhang Y and Shi C: EGFR targeted liposomal PROTAC
assisted with epigenetic regulation as an efficient strategy for
osimertinib-resistant lung cancer therapy. Adv Sci (Weinh).
12:e101972025. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Chen S, Chen E, Su J, Gong Y, Tang S, Qin
A, Shen A, Tang S and Zhang L: Magnetically navigated nano-PROTAC
ameliorates acute lung injury. J Nanobiotechnology. 23:6222025.
View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Zhao L, Zhao J, Zhong K, Tong A and Jia D:
Targeted protein degradation: Mechanisms, strategies and
application. Signal Transduct Target Ther. 7:1132022. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Kronke J, Udeshi ND, Narla A, Grauman P,
Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al:
Lenalidomide causes selective degradation of IKZF1 and IKZF3 in
multiple myeloma cells. Science. 343:301–305. 2014. View Article : Google Scholar :
|
|
233
|
Greene LA, Zhou Q, Siegelin MD and
Angelastro JM: Targeting transcription factors ATF5, CEBPB and
CEBPD with cell-penetrating peptides to treat brain and other
cancers. Cells. 12:5812023. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Sun X, Angelastro JM, Merino D, Zhou Q,
Siegelin MD and Greene LA: Dominant-negative ATF5 rapidly depletes
survivin in tumor cells. Cell Death Dis. 10:7092019. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Rui Y, Eppler HB, Yanes AA and Jewell CM:
Tissue-targeted drug delivery strategies to promote
antigen-specific immune tolerance. Adv Healthc Mater.
12:e22022382023. View Article : Google Scholar
|
|
236
|
Chen W, Schilperoort M, Cao Y, Shi J,
Tabas I and Tao W: Macrophage-targeted nanomedicine for the
diagnosis and treatment of atherosclerosis. Nat Rev Cardiol.
19:228–249. 2022. View Article : Google Scholar
|
|
237
|
Zhao G, Xue L, Geisler HC, Xu J, Li X,
Mitchell MJ and Vaughan AE: Precision treatment of viral pneumonia
through macrophage-targeted lipid nanoparticle delivery. Proc Natl
Acad Sci USA. 121:e23147471212024. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Hai T, Wolford CC and Chang YS: ATF3, a
hub of the cellular adaptive-response network, in the pathogenesis
of diseases: Is modulation of inflammation a unifying component?
Gene Expr. 15:1–11. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
239
|
Nguyen CT, Kim EH, Luong TT, Pyo S and
Rhee DK: ATF3 confers resistance to pneumococcal infection through
positive regulation of cytokine production. J Infect Dis.
210:1745–1754. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Bekeredjian-Ding I, Stein C and Uebele J:
The innate immune response against staphylococcus aureus. Curr Top
Microbiol Immunol. 409:385–418. 2017.
|
|
241
|
Nguyen CT, Kim EH, Luong TT, Pyo S and
Rhee DK: TLR4 mediates pneumolysin-induced ATF3 expression through
the JNK/p38 pathway in Streptococcus pneumoniae-infected RAW 264.7
cells. Mol Cells. 38:58–64. 2015. View Article : Google Scholar :
|
|
242
|
Guo B, Stein JL, van Wijnen AJ and Stein
GS: ATF1 and CREB trans-activate a cell cycle regulated histone H4
gene at a distal nuclear matrix associated promoter element.
Biochemistry. 36:14447–14455. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Watson G, Ronai ZA and Lau E: ATF2, a
paradigm of the multifaceted regulation of transcription factors in
biology and disease. Pharmacol Res. 119:347–357. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Lau E and Ronai ZA: ATF2 - at the
crossroad of nuclear and cytosolic functions. J Cell Sci.
125:2815–2824. 2012.PubMed/NCBI
|
|
245
|
Claps G, Cheli Y, Zhang T, Scortegagna M,
Lau E, Kim H, Qi J, Li JL, James B, Dzung A, et al: A
transcriptionally inactive ATF2 variant drives melanomagenesis.
Cell Rep. 15:1884–1892. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Hashimoto Y, Zhang C, Kawauchi J, Imoto I,
Adachi MT, Inazawa J, Amagasa T, Hai T and Kitajima S: An
alternatively spliced isoform of transcriptional repressor ATF3 and
its induction by stress stimuli. Nucleic Acids Res. 0:2398–2406.
2002. View Article : Google Scholar
|
|
247
|
Chan JYW, Tsui JCC, Law PTW, So WKW, Leung
DYP, Sham MMK, Tsui SKW and Chan CWH: RNA-Seq revealed
ATF3-regulated inflammation induced by silica. Toxicology.
393:34–41. 2018. View Article : Google Scholar
|
|
248
|
Jeong BC, Kim JH, Kim K, Kim I, Seong S
and Kim N: ATF3 modulates calcium signaling in osteoclast
differentiation and activity by associating with c-Fos and NFATc1
proteins. Bone. 95:33–40. 2017. View Article : Google Scholar
|