Introduction
Retinitis pigmentosa (RP) is an inherited retinal
disease (IRD) characterized by progressive degeneration of
photoreceptor cells, with a global prevalence of ~1/4,000, and it
currently has no effective cure (1-4).
X-linked RP (XLRP) is one of the most severe forms of RP. Mutations
in the retinitis pigmentosa GTPase regulator (RPGR)
gene are the main cause of XLRP and are also associated with
cone-rod dystrophy (CORD) (5-9).
The RPGR gene is located on the short arm of
the human X chromosome (Xp11.4) (3,10). RPGR proteins interact with other
proteins at the connecting cilium (CC) of photoreceptor cells to
form a complex regulatory network, which maintains the functional
stability of the CC by coordinating key processes such as vesicle
transport. Mutations in the RPGR gene lead to loss of RPGR
function. This disruption impairs the normal protein transport
system in photoreceptor cells, which subsequently disturbs
metabolic and synthetic homeostasis [such as, renewal of outer
segment (OS) disc membranes] and alters light signaling cascades.
Together, these changes ultimately lead to photoreceptor damage and
retinal structural degeneration (1,11,12).
Although considerable progress has been made in gene
therapy research on RPGR mutations worldwide and multiple
relevant animal models have been established, the key pathogenic
mechanisms and pathological features underlying RP caused by
RPGR mutations remain incompletely elucidated. Moreover, the
translation of these findings into clinical applications continues
to face key challenges. For patients with RPGR
mutation-associated retinal degeneration, the development of highly
sensitive genetic detection technologies (such as long-read
sequencing) and functional validation systems is important. A
systematic analysis of the population genetic characteristics and
the pathogenic mechanisms of RPGR gene mutations will
provide a theoretical basis for individualized diagnostic
stratification and targeted gene therapy. The present review
assesses the types of RPGR gene mutations, pathogenic
mechanisms and their effects on photoreceptor cell function
reported in recent years. In the present review, the research
progress in cross-species models (such as mouse, dog and zebrafish)
is summarized and the effectiveness of existing gene therapy
strategies [such as adeno-associated virus (AAV)-mediated gene
replacement and CRISPR-Cas9 gene editing] in preclinical models is
evaluated. Meanwhile, preclinical studies of RPGR gene therapy are
also discussed and recent clinical advances are analyzed.
Clinical and genetic features of
RPGR-associated retinopathies
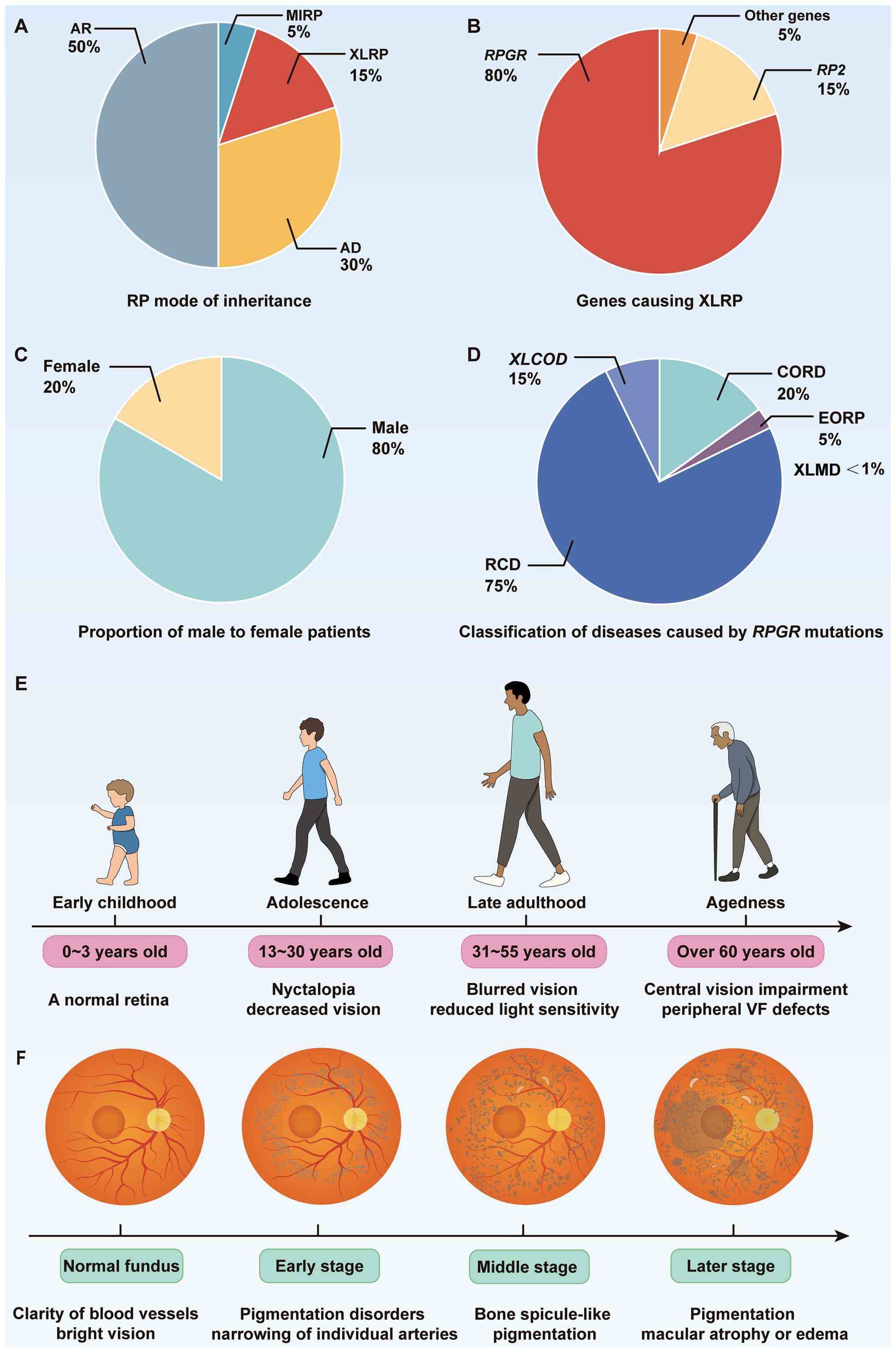
Based on the inheritance pattern, RP can be
categorized into four types: Autosomal recessive (AR) RP, autosomal
dominant (AD) RP, X-linked RP and mitochondrial RP (Fig. 1A) (13). Global statistics show that the
total number of patients with RP is >1 million (3,10,13,14). Clinical statistics show that AR
accounts for ~50% of cases, whereas AD accounts for ~30% (15,16). XLRP has a relatively low
prevalence of ~20%, but its patients are found across all age
groups (17,18). Individuals with RP show
considerable differences in disease severity and progression rates,
and this heterogeneity is determined by both the type of causative
mutation and the molecular mechanisms that the mutation mediates
(3). In addition, >100
causative genes have been identified (2,4,10,12). The majority of patients with RP
show an age-related pattern of progression. The disease initially
presents with night blindness due to rod dysfunction, followed by
rod degeneration that results in peripheral visual field (VF)
defects. As the disease progresses, secondary cone damage leads to
loss of central vision, with progressive loss of metabolic support
from the retinal pigment epithelium (RPE), ultimately leading to
blindness (1,11,12).
XLRP is a severe form of retinal ciliopathy,
characterized by the progressive degeneration of rod and cone
photoreceptor function in the retina (7). XLRP typically has a rapid onset and
progression. Some patients first experience nighttime vision loss
during adolescence, which progresses to pronounced night blindness
in early adulthood and eventually leads to central vision
impairment (19). As the disease
progresses, patients are observed to have retinal vascular
narrowing and bone spicule-like pigmentation on fundus imaging
(2,20). The risk of blindness markedly
increases in patients >50 years of age (Fig. 1). In patients with XLRP, ~70 to
80% of cases are caused by RPGR mutations, whereas
RP2 mutations account for ~10 to 15%. Notably, XLRP
resulting from RPGR mutations tends to present with a more
severe phenotype (20-22). Although both RPGR and
RP2 mutations can cause XLRP, the patterns of disease
progression are distinct. Patients with RPGR mutations
typically develop night blindness in early childhood and may
experience severe vision loss in their early 20s. In addition,
mutations in this gene have been associated with rod-cone dystrophy
(RCD), CORD, X-linked cone dystrophy (XLCOD), X-linked macular
dystrophy (XLMD) and early-onset RP (1,20,23,24). Mutations in the RPGR gene
account for 73% of X-linked CORD cases. Affected individuals
typically present with reduced visual acuity (VA), color vision
deficits, central VF defects and photophobia. They usually have a
late onset at ~40 years (25).
Clinical studies have found that vision loss
progresses relatively rapidly in these patients, with a decline in
best-corrected VA (BCVA) of ~7% per year and >60% exhibit a BCVA
of 1 logMAR or worse by the age of 50 years (26,27). There is a notable phenotypic
difference between male and female patients with
RPGR-associated XLRP. Male patients typically present with
progressive night blindness and peripheral VF reduction in early
childhood, reaching legal blindness by ~40 years of age (3,28,29). A long-term follow-up of 74 male
patients with RPGR mutations by Talib et al (24) demonstrated that the therapeutic
window for gene therapy in RPGR-associated retinal
dystrophies is relatively wide. Another 10-year study evaluating
139 male patients with RPGR-associated RP found that
binocular VA was highly associated with age, with the difference
being more pronounced in patients with worse vision (30). The study found that VA began to
decline sharply at an average age of 44 years, with the median age
at legal blindness being 45 years, consistent with previous
findings. Women who carry XLRP display a more variable phenotype,
with differing degrees of vision loss, and often exhibit color
vision deficits (31).
Some individuals with a family history of the
disease may exhibit X-linked dominant traits, and this phenotypic
variability may be associated with random or skewed X-chromosome
inactivation (XCI) (18,29,32). Fahim et al (33) found that skewed XCI of the
RPGR allele was positively associated with disease severity
in a study of 77 RPGR mutation-carrying female patients from
41 family lines, and could serve as an important predictor of
disease progression. Notably, the same RPGR gene mutation
may present with different phenotypes and severities in different
patients or even among members of the same family, suggesting that
additional factors may influence disease development. Tuekprakhon
et al (34) reported that
an 8-year-old individual who carried XLRP showed only mild
early-onset progressive CORD, although they carried the same
disease-causing mutation as their affected father. Recent studies
have also found that some female patients who carry RPGR
mutations exhibit increased levels of myopia (31,33,35-40). In a case study, Seliniotaki et
al (31) described a
4-year-old girl with no family history or other notable medical
history, but whole exome sequencing revealed a pathogenic
heterozygous stop codon variant c.212C>G (p.Ser71Ter) in the
RPGR gene, accompanied by XCI. These findings highlight the
complexity of phenotypic variation associated with the RPGR
mutation and the potential role of XCI in modulating disease
severity.
The study of RPGR-associated XLRP faces two
major problems. First, the difficulty of obtaining data from
patients at different stages of the disease course limits a
detailed understanding of pathogenic mechanisms. Second, the high
genetic heterogeneity of RPGR-associated XLRP complicates
the generalization of the results of individual studies to the
entire spectrum of the disease. This poses a challenge for studying
the pathogenic mechanisms of RPGR-associated XLRP.
Molecular features of RPGR and disease
associations
The complete sequence of the human RPGR gene
in the GRCh38 reference genome is 58,347 bp (NC_000023.11) and
includes all introns and exons. The gene produces multiple splice
variants at the transcriptional stage through extensive alternative
splicing, thereby generating functionally distinct isoforms. The
RPGR gene generates >10 alternative transcripts, of which
at least five (for example, RPGRORF15 and
RPGRex1-19) have been shown to encode functional
proteins (41). It has been
reported that RPGRex1-19 is widely expressed in a
variety of tissues, while the expression level of
RPGRORF15 is highest in retinal tissues (10).
The RPGRex1-19 transcript
(NM_000328.3) includes exons 1-19, with a full length of 3,053 bp,
encoding an 815-amino-acid protein with a molecular weight of ~90
kDa (41-42). The transcript is expressed in
several human tissues, including the testis, kidney, lung, RPE and
photoreceptors. In the retina, the primary function of this
transcript is to maintain the structural integrity and functional
stability of photoreceptor cells. The RPGRORF15
transcript (NM_001034853) consists of exons 1-14 and an open
reading frame (ORF15) derived from the variably spliced exon 15 and
intron 15. It is 4,377 bp in length, encodes a protein of 1,152
amino acids and is localized primarily to the CC of photoreceptor
cells (Fig. 2A) (34,36). The ORF15 region is a specialized
exonic region of the RPGR gene with a highly repetitive
sequence rich in purines [adenine (A) and guanine (G)]. It encodes
a region of the protein consisting of low-complexity sequences with
high glutamate (Glu) and glycine (Gly) content. The region is ~1 kb
and contains 567 amino acids. A probe targeting exons 16-19
detected an 8.9 kb band but failed to detect a 5 kb band for exons
3-10, 14 and 15 (24).
Therefore, exon ORF15 may function as a terminal exon or be
co-spliced with exons 16-19. However, ORF15 contains an internal
stop codon and does not include exons 16-19. To the best of our
knowledge, to date, no detailed studies focusing on mutations in
exons 16-19 have been reported.
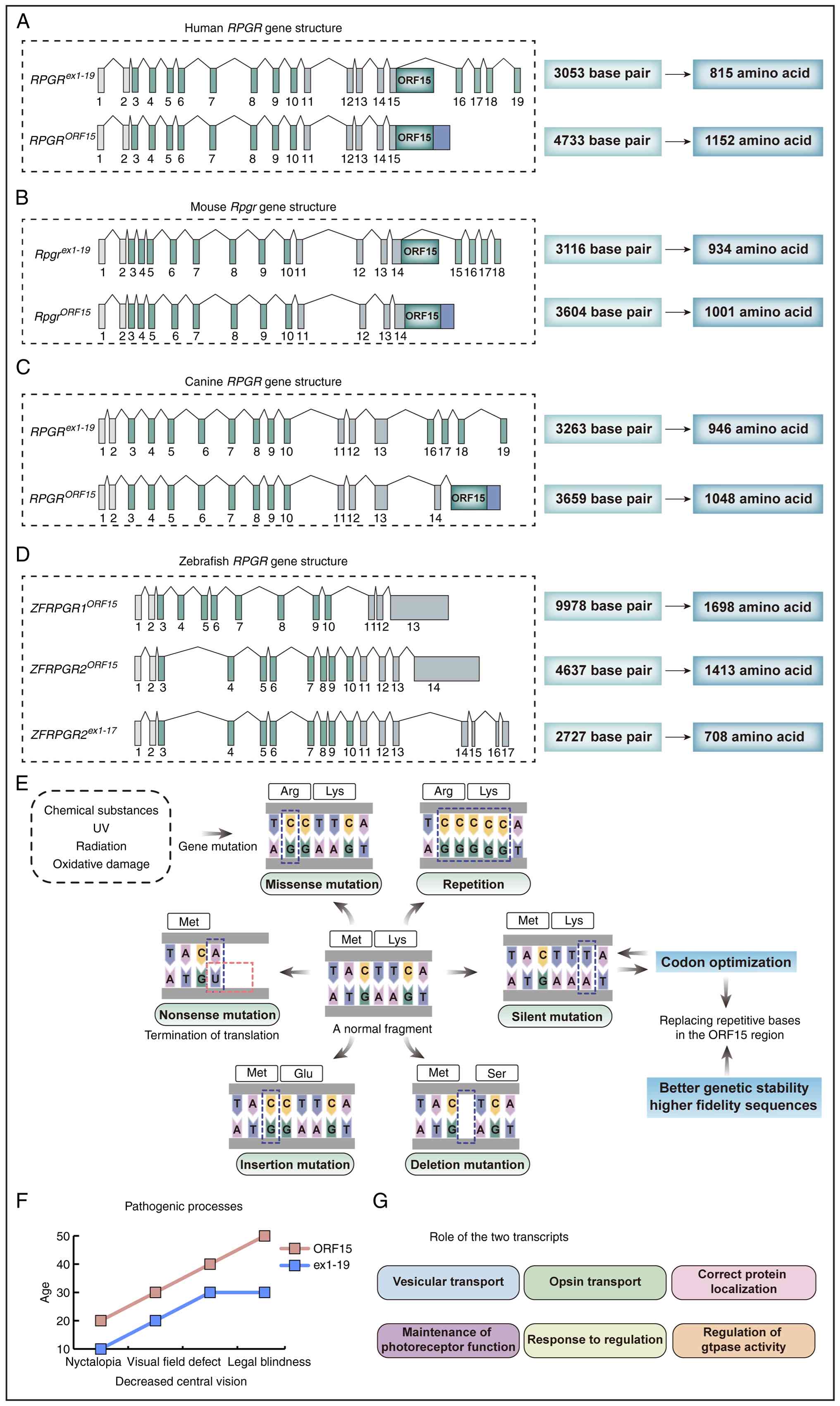 | Figure 2Information on the structure and
mutation of RPGR genes in different species. Structure of
the RPGR gene in (A) human, (B) mice, (C) canines and (D)
zebrafish, including exon-intron composition, number of base pairs
and amino acid length of the different transcripts. (E) Common
mutations in RCC1-like domains. (F) Pathogenic processes resulting
from mutations in the two transcripts leading to age-dependent
phenotypic changes. The horizontal axis represents age, and the
vertical axis reflects the severity of ocular symptoms. From left
to right, the symptoms progress from night blindness to central
vision loss, VF defects, and eventually legal blindness,
illustrating the trajectory of disease development under different
transcript mutations. (G) Role of the two transcripts in the
organism. RPGR gene, retinitis pigmentosa GTPase
regulator gene; VF, visual field. |
The exon and intron structures and mRNA expression
patterns of RPGR transcripts from different species differ
(Fig. 2B-D). For example, the
murine RpgrORF15 type (NM_001177950) is 3,604 bp
in length and encodes 1,001 amino acids, whereas the murine
Rpgrex1-19 type (NM_001177951) is 3,116 bp in
length and encodes 934 amino acids. To further assess the
evolutionary conservation among species, it is necessary to
determine the chromosomal localization of the RPGR gene.
Rpgr is located at 4.62 cM of the mouse X chromosome genetic
map, while in rats it maps to Xq12. In zebrafish, two transcripts,
rpgra and rpgrb, are located on chromosomes 9 and 11,
respectively. In dogs and rhesus monkeys, the gene is also situated
on the X chromosome, the chromosomal position most similar to that
of human RPGR.
The two major RPGR transcripts share an
identical N-terminal structure, that is, exons 1-14. Mutations
within exons 1-14 are typically associated with earlier disease
onset, faster progression and more severe visual impairment.
Patients carrying mutations in this region generally exhibit worse
visual preservation compared with those harboring ORF15 mutations
(43). Statistical analyses show
that several individuals with exon 1-14 mutations develop night
blindness and VF defects at the age of ~10 and often present with
moderate to severe RP. Missense mutations, small insertions or
deletions, and nonsense mutations are the most common types
observed (Fig. 2E). Mutations
within the ORF15 region account for ~66% of XLRP cases, making it
the most prevalent pathogenic hotspot in human XLRP (7,18,44). However, ORF15 mutations generally
exhibit a milder phenotype, with later onset and slower disease
progression (Fig. 2F). In
addition to the aforementioned mutation types, ORF15 variants may
also introduce aberrant splicing sites, thereby affecting normal
mRNA processing (10,32,44-46). Moreover, mutations in this region
have been implicated in X-linked CORD and XLMD. These phenotypic
differences highlight the complex relationship between gene
structure and function. Understanding this
structure-function-phenotype association not only provides insight
into the molecular pathology of RPGR-related diseases but
also lays the theoretical foundation for the development of
precision therapies targeting specific transcripts or mutation
types. The following section will further explore the mechanisms by
which RPGR mutations lead to photoreceptor dysfunction and
retinal degeneration.
Mechanistic insights into RPGR-associated
retinal degeneration
Photoreceptor architecture and the
functional significance of RPGR
The retina is located at the back of the eye as a
curved, multilayered structure comprising photoreceptors, bipolar
cells and ganglion cells (Fig. 3A
and B). From the outside to the inside, the retinal structure
includes the RPE, outer nuclear layer (ONL), outer plexiform layer,
inner nuclear layer (INL), inner plexiform layer, ganglion cell
layer (GCL) and nerve fiber layer, each of which contributes to the
capture and processing of light signals (47-49). Photoreceptors are responsible for
detecting light and converting it into electrical signals (50). Rods, which are primarily
distributed in the peripheral retina, contain rhodopsin and mediate
scotopic vision (51). By
contrast, cones are concentrated in the central region and contain
three distinct opsins that mediate color discrimination and fine VA
under bright light conditions (51,52). Both cell types consist of four
main components: The synaptic terminal, the inner segment (IS), the
OS and the CC linking the IS and OS (Fig. 3C). The OS is a highly specialized
primary cilium characterized by densely stacked membranous discs
that serve as the central site of phototransduction. The IS
contains organelles such as mitochondria, endoplasmic reticulum and
Golgi apparatus, which sustain the high metabolic activity required
for photoreceptor function. The CC is structurally homologous to
the transition zone of the primary cilium and is essential for the
efficient trafficking of proteins and lipids. Owing to their
exceptionally high metabolic demands, photoreceptors rely on
efficient transport of proteins synthesized in the IS through the
CC. This transport is essential to sustain their structure and
function (7). As a key component
of this intersegmental transport system, the CC requires specific
proteins to preserve its structural stability and regulate
molecular trafficking. Among these proteins, RPGR acts as a
ciliary-associated protein, carrying out a central role in
sustaining these processes.
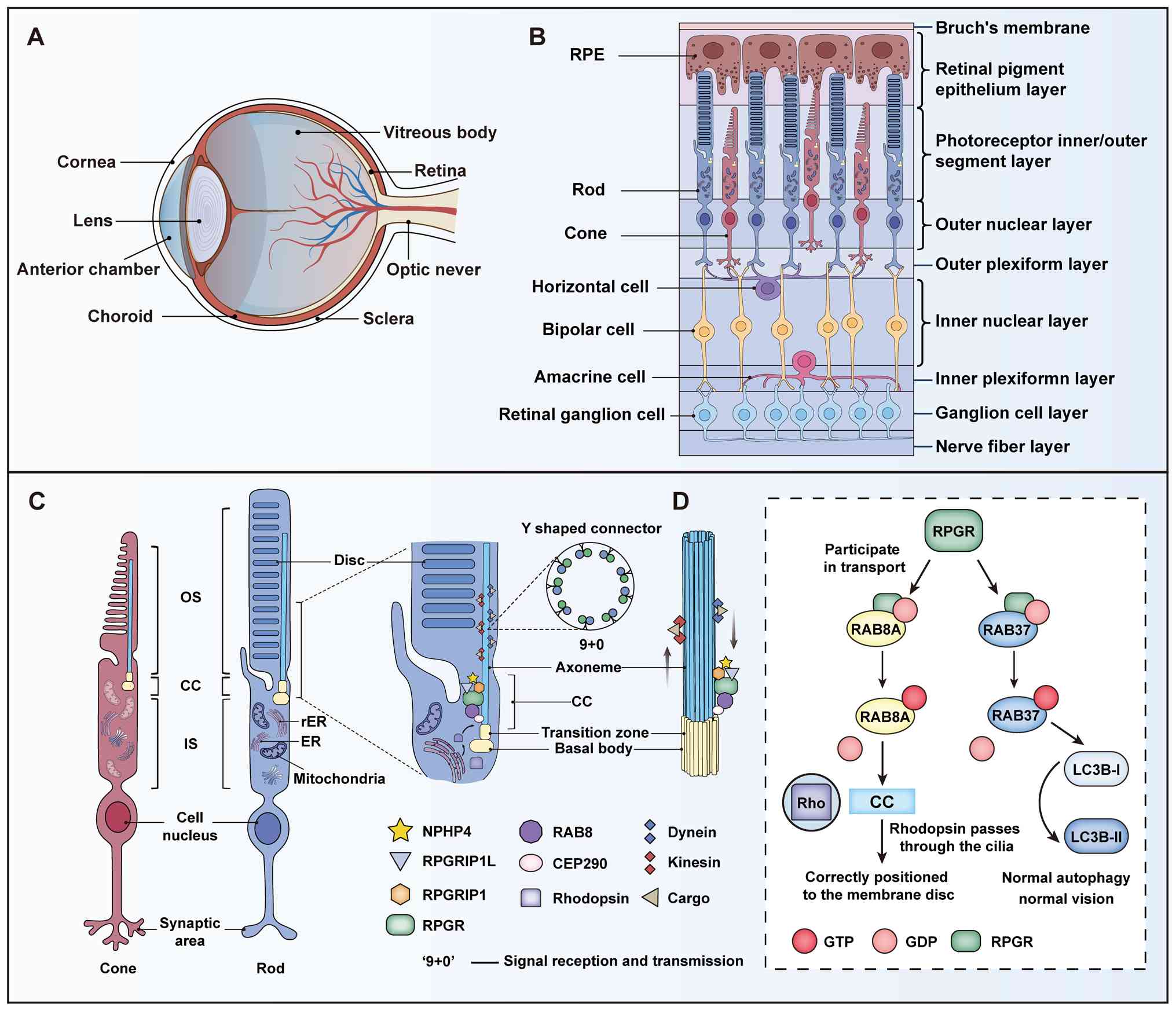 | Figure 3Structure of the eye and the role of
RPGR in photoreceptors. (A) Layered structure of the eye and
retina. (B) Retinal layers organized into three functional domains:
The support layer (BrM; RPE), the light-signal-processing layer
(OS, IS and ONL) and the neurointegrative layer (OPL, INL, IPL, GCL
and NFL). (C) Photoreceptor subcellular structures and the '9+0'
signaling axis, representing the microtubule arrangement pattern
unique to non-motile cilia. (D) Mechanisms of internal ciliary
transport involving the RPGR complex. IFT: KIF3A and Dynein mediate
bi-directional cargo transport. RAB8A participates in vesicle
transport and regulates photoreceptor OS disc membrane renewal.
Mechanisms involving RPGR, RAB8A and RAB37 in vision-related
physiological processes. RPGR participates in the transport
processes involving RAB8A and RAB37. These GTPases exert their
functions through cycling between GTP-bound and GDP-bound states.
RAB8A is involved in the correct localization of RHO to CC, thereby
maintaining normal vision. RAB37 facilitates the conversion of
LC3B-I to LC3B-II to support normal autophagy, which in turn helps
maintain vision. Key regulatory proteins: RPGR (green), RPGRIP1
(orange), NPHP4 (earthy yellow), RAB8A (bright yellow), GTP (red),
GDP (light red), RHO (purple), CC (blue), RAB37 (blue), LC3B-I
(light blue), LC3B-II (dark blue). BrM, Bruch's membrane; RPE,
retinal pigment epithelium; OS, outer segment; ONL, outer nuclear
layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL,
inner plexiform layer; GCL, ganglion cell layer; NFL, nerve fiber
layer. RPGR, retinitis pigmentosa GTPase regulator; RHO, Rhodopsin;
RPGRIP1L, RPGRIP1-like protein; RPGRIP1, RPGR-interacting protein
1; RAB8A, RAS-related protein Rab-8A; IFT, intraflagellar
transport; KIF3A, kinesin family member 3A; NPHP4, nephrocystin 4;
CC, connecting cilium; LC3B-I, microtubule-associated protein 1
light chain 3 β-I; LC3B-II, microtubule-associated protein 1 light
chain 3 β-II. |
The N-terminus of the RPGR protein contains six
complete tandem repeats, each comprising 52-54 amino acids
(32). It has structural
homology with Regulator of Chromosome Condensation 1 (RCC1),
forming RCC1-like domains (RLDs) (53). The RLDs possess nucleotide
exchange factor (GEF) activity, promoting the conversion of small
GTPases from the GDP-bound to GTP-bound forms and thereby carrying
out a key role in ciliary function and intracellular transport.
RPGR is predominantly localized in the CC between the IS and OS of
the photoreceptor (Fig. 3C)
(3,8,54,55). In the retina, the RPGR
gene primarily produces two major isoforms,
RPGRex1-19 and RPGRORF15. Both
isoforms contain identical RLDs (exons 1-14) at the N-terminus but
differ markedly in their C-terminal sequences.
RPGRex1-19 contains an isoprenylation domain that
mediates its localization to the CC. By contrast,
RPGRORF15 features a unique C-terminal ORF15 region rich
in glutamic acid and glycine residues. Early studies suggested that
RPGR might not be essential for photoreceptor development and early
function (55-58). However, subsequent studies have
confirmed that RPGR is indispensable for protein trafficking,
ciliary stability and signal maintenance during photoreceptor
maturation. It also serves as a 'longevity factor' key for
preserving their structural and functional integrity (7,48).
Disruption of RPGR-mediated protein
trafficking and proteostasis leads to photoreceptor
degeneration
RPGR carries out a key role in maintaining ciliary
function and stability, as well as in regulating opsin transport,
vesicular trafficking and proper protein localization. It forms
complexes with multiple cilia-associated proteins, including
RPGR-interacting protein 1 (RPGRIP1), RPGRIP1-like protein
(RPGRIP1L), centrosomal protein 290 (CEP290) and IQ motif
containing B1 (IQCB1) (59).
Among these, RPGRIP co-localizes with RPGR in the photoreceptor OS
and CC, contributing to structural maintenance and protein
trafficking (45,56). Distinct RPGR-associated complexes
function at different stages of molecular trafficking, and loss of
RPGR function disrupts its pivotal role within the ciliary
transport network. Under normal conditions, RPGR forms complexes
with RPGRIP1 and IQCB1 to mediate rhodopsin trafficking. When the
interaction between RPGR and either RPGRIP1 or IQCB1 is disrupted
by RPGR mutations, the severity of the associated disease is
further exacerbated (18,59).
In wild-type photoreceptors, RPGR interacts with CEP290 within the
transition zone (60). However,
in RPGR mutant mice, this interaction is altered, leading to
transition zone defects that impair molecular transport.
RPGRORF15 interacts with transition zone proteins such
as CEP290 to facilitate the transport and renewal of photoreceptor
discs in the photoreceptor OS. This interaction helps maintain the
integrity of the transition zone and the Y-link structure (Fig. 3C).
In addition to its interactions with RPGRIP1 and
IQCB1, RPGR further facilitates molecular transport and signaling
in photoreceptors by interacting proteins such as the δ subunit of
rod-specific photoreceptor cGMP phosphodiesterase (PDEδ) and
ADP-ribosylation factor-like 3 (ARL3). It is also involved in
regulating vesicular trafficking and intracellular signaling
pathways. Using a yeast two-hybrid screen, Linari et al
(61) identified that PDEδ binds
to the RLDs of RPGR. The researchers therefore proposed that RPGR
mutations may cause protein mislocalization and trafficking
defects, ultimately leading to retinal degeneration. RPGR also
associates with ARL3, linking the cell membrane to the
photoreceptor cytoskeleton and participating in specific signaling
and vesicular transport processes (62,63). A study published in 2024 reported
that RPGR functions as a GEF, facilitating the conversion of
RAS-related protein Rab-37 (RAB37) from the GDP-bound to GTP-bound
state (Fig. 3D) (64). This activation of RAB37 promotes
vesicular trafficking, particularly within the lysosome-autophagy
pathway, thereby supporting normal autophagic activity and visual
function. The role of RPGR in maintaining photoreceptor function
may therefore involve the regulation of intracellular signaling and
vesicular transport. Moreover, RPGR helps preserve protein
homeostasis and functional stability in mature photoreceptors by
modulating proteasome activity and the expression of key transport
proteins (65).
Studies have shown that RPGR mutations are
prevalent in various ciliopathies characterized by severe
photoreceptor degeneration. In a 2023 study using rpgra
mutant zebrafish, the mutation was found to cause downregulation
and mislocalization of RAS-related protein Rab-8A (RAB8A) within
the cilium, thereby disrupting the trafficking of essential
photoreceptor molecules (66).
Similarly, a 2010 study showed that RPGR knockdown in
hTERT-RPE1 cells resulted in shortened primary cilia and
mislocalization of RAB8A (67).
Proper binding of RPGR to RAB8A ensures the efficient incorporation
of rhodopsin into the OS disk membranes for light-signal capture
(Fig. 3D). Mutations in
RPGR lead to abnormal accumulation of rhodopsin in the IS or
CC, resulting in disorganization of the OS disk structure (66,67). Multiple Rpgr animal models
have confirmed that such trafficking defects are a major cause of
both rod and cone photoreceptor degeneration (68).
At the molecular level, RPGR encodes a protein
essential for the assembly and maintenance of photoreceptor cilia,
serving as a key regulator of CC integrity (55). Its N-terminal domain shares
homology with RCC1 and catalyze the conversion of Ran guanosine
diphosphate (RanGDP) to Ran guanosine triphosphate (RanGTP). Based
on this, researchers have proposed that RPGR may regulate
intraphotoreceptor protein transport through a RanGTP-dependent
mechanism. Under normal conditions, the concentration of RanGTP is
increased in the CC compared with the IS and this gradient is
considered to underlie the directionality of protein trafficking.
Loss of RPGR function may disrupt this gradient, leading to
impaired opsin transport. In 2021, Moreno-Leon et al
(7) reported that imbalanced
expression of the two major RPGR transcripts may cause
XLRP-associated ciliopathy, suggesting that transcriptional
dysregulation may contribute to the pathogenesis. Furthermore, some
studies have proposed a potential association between RPGR
mutations and primary ciliary dyskinesia, suggesting that shared
molecular mechanisms may underlie different ciliopathies (69-72). These findings provide new
insights into the ciliary aspects of XLRP and offer perspectives
for developing future therapeutic targets.
Mechanisms of RPGR gene mutations in
photoreceptor cells
Mutations in exons 1-14 of the RPGR gene
usually lead to abnormalities of the RCC1-like domain. By contrast,
mutations in the ORF15 region are more complex and can affect
multiple aspects, including DNA conformation, protein stability,
trafficking and ciliary function (73). As aforementioned, the ORF15
region represents a mutational hotspot of RPGR (Fig. 4A), with the majority of
pathogenic variants clustered between 949 and 1,047 bp (18,32,74). The majority of these are small
deletions of 1-5 bp, typically resulting in truncated or
frameshifted proteins (41).
Although such truncations may retain a modest role in genomic
stability, evidence from the XLPRA2 canine model shows that ORF15
frameshift mutations nonetheless cause severe retinal degeneration
(32,75). Subsequent studies have shown that
missense mutations disrupt the interaction of RPGR isoforms with
their endogenous partners, including Inositol
polyphosphate-5-phosphatase E (INPP5E), Phosphodiesterase 6 delta
subunit (PDE6D) and RPGRIP1L (65,76-78) The C-terminus of
RPGRex1-19 contains a prenylation site that regulates
its interaction with PDE6D, INPP5E and RPGRIP1L (Fig. 4C) (76,78). Both RPGR isoforms also
interact with CEP290 and INPP5E at the genetic and physical levels,
and this interaction is important for maintaining the function and
survival of the photoreceptor OS (7).
![Interaction network of RPGR proteins
and their functional mechanism in cilium. (A) Structural domain
characterization of RPGR protein isoforms, where blue dots indicate
sites of frameshift mutations. RPGRex1-19
contains exons 1-10, 11-14 and 16-19; its N terminus includes the
RLD, and its C terminus contains several regions of unknown
function. RPGRORF15 consists of exons 1-10, 11-14
and ORF15; it contains an acidic, glutamate-rich domain (EG-rich
domain) and a basic domain. (B) Interacting protein networks of
RPGR. Proteins directly binding to the RLDs: RPGRIP1, RPGRIP1L,
RAB8A and PDE6D. Complex-associated proteins:
Cilium-transport-related proteins (CEP290, IFT88, KIF3A, RAB11 and
γ-Tubulin); signaling-regulation-related [NPHP family proteins
(NPHP1/4/5), TTLL5, ARL2/3]; and structure-maintenance-related
(SMC1/3, SPATA7). (C) Functional pathways of RPGR in ciliary
signaling. RPGR is involved in the regulation of
phosphatidylinositol metabolism. INPP5E is isoprenylated through
its C-terminal CAAX motif and binds PDE6D to form a complex, which
ensures its proper membrane localization. ARL3 promotes the release
of PDE6D in the activated state. ARL3, in its activated state,
promotes the release of INPP5E from PDE6D and the dissociated
INPP5E is translocated to the ciliary membrane via the IFT
mechanism. ARL13B ensures the stable localization of INPP5E to the
ciliary membrane by binding to INPP5E. RPGR, retinitis pigmentosa
GTPase regulator; RLDs, RCC1-like domains; RPGRIP1,
RPGR-interacting protein 1; RPGRIP1L, RPGRIP1-like protein; RAB8A,
RAS-related protein Rab-8A; PDE6D, Phosphodiesterase 6 δ subunit,
CEP290, Centrosomal Protein 290; IFT88, Intraflagellar Transport
88; KIF3A, Kinesin Family Member 3A; RAB11, Ras-Related Protein
Rab-11; NPHP1, Nephrocystin 1; NPHP4, Nephrocystin 4; NPHP 5,
Nephrocystin 5; TTLL5, Tubulin tyrosine ligase-like family member
5; ARL2, ADP-Ribosylation Factor-Like Protein 2; ARL3,
ADP-Ribosylation Factor-Like Protein 3; ARL13B, ADP-Ribosylation
Factor-Like Protein 13B; SMC1, Structural maintenance of
chromosomes protein 1; SMC3, Structural maintenance of chromosomes
protein 3; SPATA7, Spermatogenesis-Associated Protein 7; INPP5E,
Inositol polyphosphate-5-phosphatase E.](/article_images/ijmm/57/3/ijmm-57-03-05723-g03.jpg) | Figure 4Interaction network of RPGR proteins
and their functional mechanism in cilium. (A) Structural domain
characterization of RPGR protein isoforms, where blue dots indicate
sites of frameshift mutations. RPGRex1-19
contains exons 1-10, 11-14 and 16-19; its N terminus includes the
RLD, and its C terminus contains several regions of unknown
function. RPGRORF15 consists of exons 1-10, 11-14
and ORF15; it contains an acidic, glutamate-rich domain (EG-rich
domain) and a basic domain. (B) Interacting protein networks of
RPGR. Proteins directly binding to the RLDs: RPGRIP1, RPGRIP1L,
RAB8A and PDE6D. Complex-associated proteins:
Cilium-transport-related proteins (CEP290, IFT88, KIF3A, RAB11 and
γ-Tubulin); signaling-regulation-related [NPHP family proteins
(NPHP1/4/5), TTLL5, ARL2/3]; and structure-maintenance-related
(SMC1/3, SPATA7). (C) Functional pathways of RPGR in ciliary
signaling. RPGR is involved in the regulation of
phosphatidylinositol metabolism. INPP5E is isoprenylated through
its C-terminal CAAX motif and binds PDE6D to form a complex, which
ensures its proper membrane localization. ARL3 promotes the release
of PDE6D in the activated state. ARL3, in its activated state,
promotes the release of INPP5E from PDE6D and the dissociated
INPP5E is translocated to the ciliary membrane via the IFT
mechanism. ARL13B ensures the stable localization of INPP5E to the
ciliary membrane by binding to INPP5E. RPGR, retinitis pigmentosa
GTPase regulator; RLDs, RCC1-like domains; RPGRIP1,
RPGR-interacting protein 1; RPGRIP1L, RPGRIP1-like protein; RAB8A,
RAS-related protein Rab-8A; PDE6D, Phosphodiesterase 6 δ subunit,
CEP290, Centrosomal Protein 290; IFT88, Intraflagellar Transport
88; KIF3A, Kinesin Family Member 3A; RAB11, Ras-Related Protein
Rab-11; NPHP1, Nephrocystin 1; NPHP4, Nephrocystin 4; NPHP 5,
Nephrocystin 5; TTLL5, Tubulin tyrosine ligase-like family member
5; ARL2, ADP-Ribosylation Factor-Like Protein 2; ARL3,
ADP-Ribosylation Factor-Like Protein 3; ARL13B, ADP-Ribosylation
Factor-Like Protein 13B; SMC1, Structural maintenance of
chromosomes protein 1; SMC3, Structural maintenance of chromosomes
protein 3; SPATA7, Spermatogenesis-Associated Protein 7; INPP5E,
Inositol polyphosphate-5-phosphatase E. |
Mutations within exons 1-14 and the proximal ORF15
region are generally associated with RCD, whereas mutations in the
distal ORF15 region are more often linked to CORD or XLCOD. The
genotype-phenotype association of RPGR mutations is largely
attributed to glutamylation. This post-translational modification
carries out a key role in maintaining the structural stability of
the ORF15 domain. Tubulin tyrosine ligase-like 5 (TTLL5) interacts
with the basic domain of ORF15 and serves as a key regulator of its
glutamylation. Loss of this modification leads to
RPGRORF15 dysfunction and may even cause a phenotypic
shift from RCD to CORD. In a cohort of 116 male patients, mutations
located near the C-terminal ORF15 were found to shift the disease
phenotype from rod-dominant to cone-dominant (74). Further investigations revealed
that truncations in the distal ORF15 region disrupt its interaction
with TTLL5, impair RPGR glutamylation and consequently result in
cone photoreceptor degeneration. However, the C-terminal ORF15
domain contains 11 glutamate-rich consensus motifs, and the
artificial addition of negatively charged glutamate residues in
this region may alter protein folding and stability, thereby
affecting protein-protein interactions (22). Therefore, therapeutic strategies
involving artificial modification of the ORF15 region should be
approached with caution.
Globally, the major types of RPGR mutations
identified in patients include codon deletions, duplications,
nonsense mutations and frameshift mutations (79-85). These variants can lead to
abnormal protein synthesis, structural alterations, loss of
function and disruption of protein-protein interaction networks
(Fig. 4B), thereby compromising
ciliary stability and ultimately resulting in retinal degeneration.
Studies have also revealed regional differences in the distribution
of RPGR mutations. Buraczynska et al (46) analyzed 80 unrelated patients with
XLRP and found that the majority of RPGR mutations were located
within the N-terminal RLDs. In another study, Pusch et al
(45) screened 37 European
patients with XLRP and identified mutations in the ORF15 region
that resulted in premature translation termination, thereby
affecting the structure and function of the RPGR protein (7,45). Similarly, in a cohort of 25
familial XLRP cases, both a previously reported mutation (c.3317)
and a novel deletion mutation in the ORF15 region (c.3300_3301del)
were identified. These mutations were located at the 3'end of the
exon, causing premature termination of translation and the loss of
40-50 amino acids from the RPGR C-terminus (86). All affected individuals exhibited
relatively preserved rod photoreceptor function despite these
truncations.
Different types of mutations lead to loss of RPGR
protein function, resulting in varying degrees of clinical
severity. To accurately identify pathogenic variants, the ORF15
region must be analyzed using high-throughput, robust and scalable
sequencing approaches (87).
Future studies should first aim to precisely identify mutation
sites and then develop targeted therapeutic strategies tailored to
each mutation type. RPGR-XLRP exhibits high clinical
phenotypic heterogeneity (53,88). This phenotypic heterogeneity may
arise from population differences or environmental factors
(89). Therefore, studying the
association between different mutations and their corresponding
phenotypes is important for understanding the pathogenic mechanisms
of RPGR. In addition, analyzing modifier genes that can markedly
influence RPGR mutant phenotypes is important for developing
potential therapeutic strategies (53,71).
Potential involvement of RPGR in
photoreceptor metabolic signaling via the mTOR/AMPK axis
Similar to other neurodegenerative diseases, RP
exhibits disruptions in core cellular metabolic pathways, with its
pathogenesis involving dysregulation of multi-level molecular
signaling networks. Abnormalities in multiple signaling pathways
have been shown to lead to photoreceptor loss and apoptosis. These
abnormalities include transmembrane signaling disruptions,
metabolic dysregulation, oxidative stress and apoptotic pathway
activation. Given that RPGR mutations also contribute to
photoreceptor degeneration, we hypothesized that RPGR might be
associated with these signaling pathways (Fig. 5D). Metabolic homeostasis carries
out a key regulatory role in photoreceptor degeneration (Fig. 5A-C). RPGR mutation may
disrupt the metabolic homeostasis of photoreceptors through
aberrant activation of the mTOR complex 1 (mTORC1) pathway,
accelerating their degeneration. mTOR serves as a central regulator
of cellular metabolism, primarily controlling lipid synthesis,
autophagy and cell growth through the mTORC1 complex (90-92). At the mechanistic level, we
hypothesized that the RPGR protein affects phosphatidylinositol
metabolism in rods through interactions with PDE6D and INPP5E,
leading to the degradation triggered during transport (Fig. 5E). Its deletion results in
abnormally elevated levels of phosphatidylinositol (3,4,5)-trisphosphate. This elevation in turn
activates the protein kinase B (AKT) signaling pathway, promotes
phosphorylation of tuberous sclerosis complex 2 (TSC2) and disrupts
Rheb function, resulting in continuous activation of mTORC1. This
activation not only promotes anabolic metabolism but also inhibits
autophagy and disrupts cellular metabolic homeostasis.
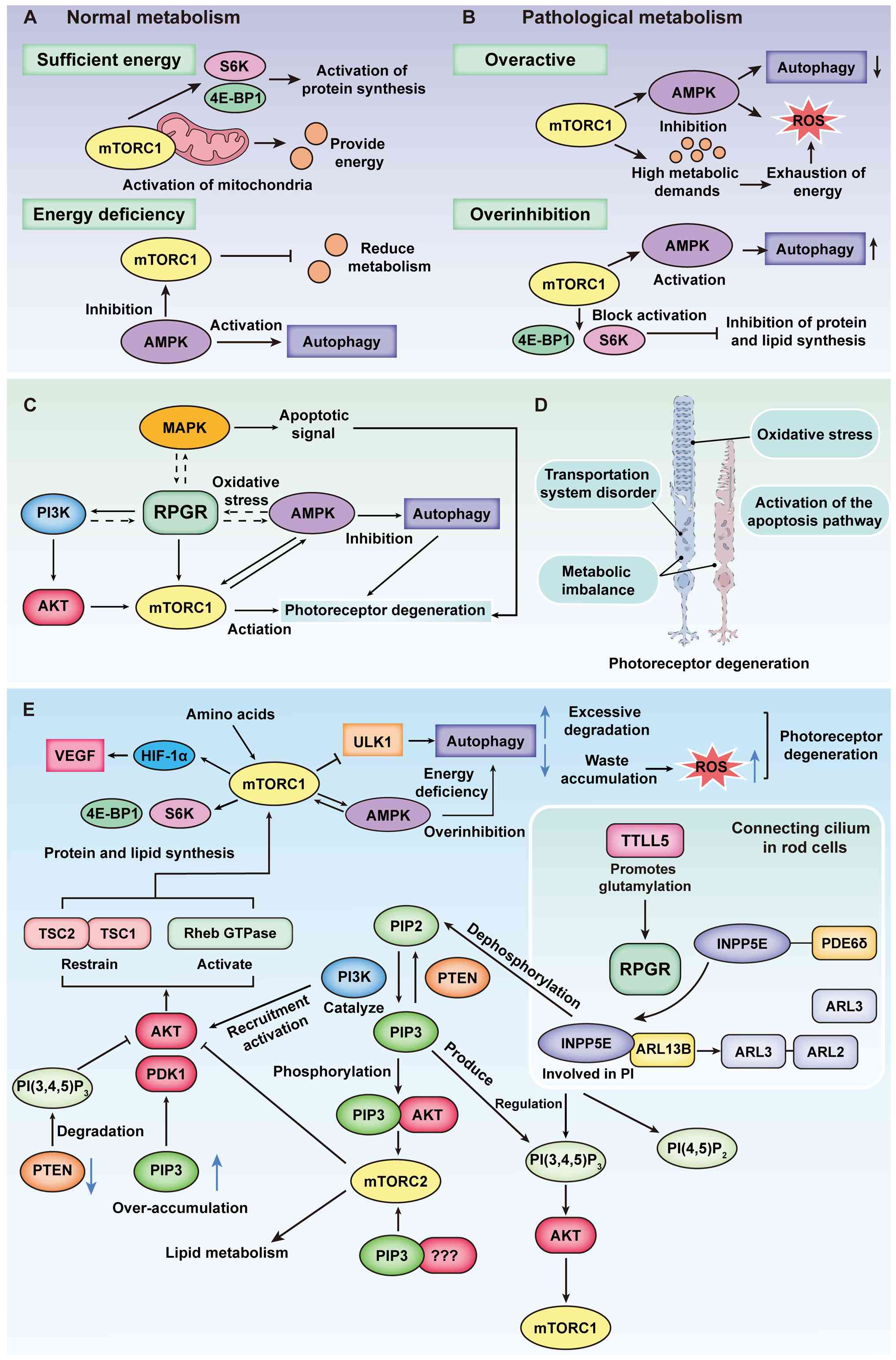 | Figure 5Impact of RPGR-associated
phosphoinositide signaling on cellular energy homeostasis. (A)
Normal metabolic conditions. In retinal cells under normal
metabolic conditions, mTORC1 is activated in the presence of
sufficient energy, promoting mitochondrial activity to sustain
cellular survival. Additionally, mTORC1 activates S6K and 4E-BP1,
thereby enhancing protein synthesis. Conversely, during energy
depletion, AMPK inhibits mTORC1, reducing metabolic activity while
simultaneously activating autophagy to provide an alternative
energy source for the cell. (B) Pathological metabolic conditions.
In pathological retinal cells, excessive activation of mTORC1
occurs when energy is abundant. Under these conditions, mTORC1
suppresses the AMPK pathway, leading to decreased autophagy and an
inability to efficiently clear intracellular waste. AMPK activity
also contributes to increased ROS levels. Meanwhile, the heightened
metabolic demand results in energy exhaustion, further triggering
ROS activation, excessive intracellular waste accumulation and
oxidative stress. By contrast, severe energy deprivation leads to
excessive inhibition of mTORC1, which blocks the activation of S6K
and 4E-BP1, thereby suppressing protein and lipid synthesis and
disrupting normal cellular metabolism. This energy-deficient state
also activates the AMPK pathway, increasing intracellular
autophagy. (C) RPGR may regulate photoreceptor degeneration through
multiple signaling pathways. Solid lines indicate pathways
supported by existing studies, such as PI3K/AKT/mTORC1 and AMPK in
autophagy regulation; dashed lines represent hypothetical
mechanisms suggesting potential involvement of MAPK and metabolic
stress pathways. (D) RPGR dysfunction leads to structural and
functional degeneration of photoreceptors. Key mechanisms include
transport defects, metabolic imbalance, oxidative stress and
activation of apoptotic pathways. (E) Indirect role of RPGR in
phosphoinositide metabolism. mTOR, in complex with mTORC1, serves
as a central regulator influenced by multiple factors, including
AMPK, the TSC1-TSC2 complex and AKT. These pathways collectively
modulate protein and lipid synthesis as well as autophagy. In
phosphoinositide metabolism, AKT is activated via the
PI3K-PIP3 pathway, while PTEN dephosphorylates
PIP3 to maintain homeostasis. PTEN dysfunction leads to
excessive PIP3 accumulation. TTLL5 facilitates the
proper glutamylation of RPGR, enabling INPP5E to function correctly
within the phosphoinositide pathway, thereby indirectly regulating
the mTOR pathway and sustaining normal cellular growth and
metabolism. mTOR, mechanistic Target of Rapamycin; mTORC1, mTOR
Complex 1; S6K, Ribosomal protein S6 kinase; 4E-BP1, Eukaryotic
Translation Initiation Factor 4E-Binding Protein 1; AMPK,
AMP-activated protein kinase signaling pathway; ROS, Reactive
oxygen species; PI3K/AKT, Phosphoinositide 3-kinase/AKT signaling
pathway; MAPK, Mitogen-activated protein kinase signaling pathway;
PI3K-PIP3, Phosphoinositide
3-kinase–phosphatidylinositol (3,4,5)-trisphosphate signaling pathway;
PTEN, Phosphatase and tensin homolog; TTLL5, Tubulin tyrosine
ligase-like family member 5; INPP5E, Inositol
polyphosphate-5-phosphatase E; TSC1, Tuberous sclerosis complex 1;
TSC2, Tuberous sclerosis complex 2. |
mTORC1 is a key kinase that regulates cell
metabolism by balancing demand with supply (93,94). Its activity is modulated by AMPK,
a central energy sensor whose impairment may cause photoreceptor
degeneration by disrupting cellular energy homeostasis (95). RPGR-mutant mice exhibit
early autophagic dysfunction, with vesicular structures
progressively accumulating in photoreceptor IS (64,96). Proteomic analysis further
revealed a notable reduction in mTOR protein expression. Notably,
Pde6b mutant mice display similar metabolic alterations,
including elevated pAKT levels (96). mTORC1 activity is modulated by
multiple upstream signals, including AMPK and various growth factor
receptors. These signals form a highly intricate feedback
regulatory network that highlights its central role in
photoreceptor degeneration. RPGR mutations likely trigger a
cascade of downstream events by disrupting photoreceptor metabolic
homeostasis (Fig. 5C). AMPK is
an energy sensor that regulates metabolic homeostasis. Upon
activation, AMPK phosphorylates TSC2 or Raptor, inhibiting protein
translation and fatty acid synthesis to maintain cellular energy
balance (97). In age-related
macular degeneration (AMD) mouse models, treatment with glucosamine
activates AMPK phosphorylation and suppresses mTORC1
phosphorylation via the AMPK/mTOR signaling pathway. This leads to
a reduction in lipofuscin-like autofluorescence in the RPE
(98). Meanwhile, in a study of
retinal autophagy homeostasis in wAMD mice, the treatment targeting
the AMPK/mTOR/hypoxia inducible factor-1α/vascular endothelial
growth factor (VEGF) and AMPK/reactive oxygen species (ROS)/heme
oxygenase-1/VEGF pathways reduced retinal damage (99). Although direct evidence of
AMPK-mTOR dysregulation in the retinas of Rpgr-KO mice is
still limited, mitochondrial stress and reduced basal respiration
observed in RpgrEx3d8 retinas indicate altered
energy states. This suggests that RPGR mutations may trigger
activation of the AMPK/mTOR pathway and decreased AMPK activity can
compromise mTORC1 inhibition, thereby promoting increased lipid and
protein synthesis. This dysregulation also impairs autophagic
clearance, exacerbating the accumulation of metabolic waste
products such as lipofuscin (Fig.
5B). Moreover, this metabolic imbalance is conserved across
species in RPGR-deficient models, as lipid droplet
accumulation has been found in the RPE of rpgra-knockout
zebrafish (66). These
pathological features suggest that RPGR mutations may
disrupt retinal metabolic homeostasis by interfering with the
AMPK/mTOR pathway. However, in Rpgr-KO mice, the activity
markers of this pathway (such as p-AMPK, p-S6K and p-4EBP1) and
potential functional interventions have not yet been evaluated,
which is essential for developing therapies targeting metabolic
dysregulation in RP.
In addition to the mTORC1 pathway, other key
signaling pathways including PI3K-AKT and MAPK are also implicated
in photoreceptor degeneration (96,100). The PI3K-AKT pathway carries out
a key role in cell survival and metabolism, and its dysregulation
may contribute to photoreceptor apoptosis by affecting downstream
targets including mTORC1. Meanwhile, the MAPK pathway, particularly
JNK and p38 kinases, is involved in pro-inflammatory responses and
stress-induced apoptosis during retinal degeneration (101-104). Although the PI3K-AKT and MAPK
pathways both carry out key roles in photoreceptor degeneration,
their functional interactions during disease progression in
RPGR-deficient models remain poorly understood. Emerging
evidence indicates that the interplay between these pathways may
generate a vicious cycle of metabolic stress, oxidative damage and
inflammation, thereby exacerbating photoreceptor cell death. Future
studies targeting these signaling cascades may uncover novel
therapeutic approaches for RPGR-associated retinal
degeneration.
Interventional therapy targeting mTOR has emerged
as a potential strategy. Rapamycin, an mTOR inhibitor, enhances
autophagic activity by upregulating beclin-1 and
microtubule-associated protein 1 light chain 3, effectively
counteracting z-VAD-induced necroptosis and markedly improving
photoreceptor survival (105).
Rapamycin markedly increases photoreceptor survival by activating
autophagy and inhibiting the ROS-apoptosis-inducing factor pathway
in the z-VAD-induced necrotic apoptosis model. However, its
therapeutic potential has not yet been systematically evaluated in
degeneration models caused by RPGR mutations. RPGR
mutations typically result in impaired protein transport and
mitochondrial stress, with hyperactivation of the mTORC1 pathway
further exacerbating degeneration. Theoretically, rapamycin could
alleviate endoplasmic reticulum stress by inhibiting mTORC1 and
restoring autophagic activity, thereby facilitating clearance of
mutant RPGR protein aggregates. It also enhances mitochondrial
autophagy to reduce ROS production, inhibiting secondary necrotic
apoptosis and helping maintain retinal microenvironmental
homeostasis. Given the key role of the mTOR signaling axis in
retinal degeneration, further exploration of the mechanisms
regulating its activity in RPGR-associated retinopathy could
provide a theoretical basis for the development of new intervention
strategies (106).
RPGR, oxidative stress and apoptotic
mechanisms in photoreceptors
Photoreceptor cells exhibit extremely high
metabolic activity and are highly sensitive to oxidative damage.
Early studies in a pig model of RP showed that following rod death,
the resulting increase in local retinal oxygen tension induces
oxidative stress, ultimately leading to secondary cone cell death
(107). Campochiaro and Mir
(108) noted that excess oxygen
can disrupt the mitochondrial electron transport chain and activate
NADPH oxidase. These processes promote the generation of superoxide
and peroxynitrite, thereby causing sustained oxidative damage to
cone cells. In RP, the gradual degeneration of cone cells following
rod cell death is mainly caused by oxidative damage. Based on the
metabolic imbalance observed in RPGR models, we hypothesize
that initial rod cell injury may trigger oxidative stress in cones,
thereby accelerating disease progression. Thus, antioxidant therapy
shows notable potential in slowing disease progression. ERG
assessments by Komeima et al (109) demonstrated that systemic
administration of antioxidants can markedly delay functional loss
in cone cells. In RD10 mice, long-term oral antioxidant treatment
using compounds such as naringenin or quercetin reduced
intracellular ROS levels, preserved retinal morphology and improved
retinal function (110).
However, the protective effects of antioxidants have not yet been
validated in other mutant models, such as Rpgr-KO mice.
Under sustained oxidative stress, photoreceptor
cells may undergo programmed cell death. The core regulatory
mechanisms involve the coordinated activation of both
caspase-dependent and mitochondrial pathways. RPGR mutations
destabilize the OS disc membranes, impairing normal mitochondrial
function. Aberrantly activated caspase-3 and caspase-9 initiate a
canonical proteolytic cascade. There is a synergistic effect
between mitochondrial damage and caspase activation, and
ligand-dependent activation of the death receptor signaling further
amplifies the process through the assembly of the death-inducing
signaling complex (DISC), triggering downstream effector caspase
cascades and ultimately accelerating photoreceptor cell death.
Notably, in 2017, Venkatesh et al (111) demonstrated through a
conditional knockout model that specific deletion of caspase-7 did
not notably affect cone survival in the RP model. This suggests
that the mechanism of secondary cone death may be independent of
the endoplasmic reticulum stress pathway and involves signaling
pathways beyond the unfolded protein response. Currently, the
specific effects of RPGR mutations on mitochondrial
homeostasis and apoptotic signaling remain to be further elucidated
in animal models.
Regardless of the initial pathogenic mechanism,
gene defects eventually lead to photoreceptor cell death.
Therefore, investigating the mechanisms underlying photoreceptor
cell death is essential. By analyzing the diverse clinical
phenotypes resulting from specific site mutations, researchers can
further explore the potential mechanisms of interaction and provide
a theoretical basis for the development of future therapeutic
targets. Additionally, biochemical analyses can help assess the
physiological and metabolic status of patients, thereby
facilitating the development of targeted therapies. Currently, the
expression pattern of RPGR protein remains incompletely understood,
but its functionally distinct isoforms, generated by alternative
splicing, are closely associated with specific roles in retinal
photoreceptors. Strategies to compensate for the loss of RPGR and
its isoforms, as well as to modulate physiological and metabolic
processes to mitigate the consequences of RPGR deficiency, may
become key directions for future research and therapy.
Preclinical research progress in RPGR
mutation
Preclinical animal models for RPGR
mutation studies
To elucidate the mechanisms underlying
RPGR-related retinal degeneration and to develop effective
gene therapy strategies, a variety of animal models have been
established to recapitulate the genetic and phenotypic features of
RPGR-associated XLRP (Fig.
6A). Mice have a short life cycle, strong reproductive capacity
and a high degree of genetic homology with humans, allowing
long-term disease progression to be observed within a relatively
short period. These characteristics make them valuable for
identifying pathogenic factors and associated signaling pathways
(13). In Rpgr mutation
studies, the RD9 mice represent one of the naturally occurring
mutants (17,112-114). In addition, researchers have
constructed mouse models carrying known mutation sites, including
the Rpgr knockout (Rpgr-KO) and conditional knockout
(Rpgr-CKO) models, using gene editing techniques to
facilitate further studies (17).
The RD9 mouse is a naturally occurring mutant model
carrying 32 bp duplication in the ORF15 region, which causes a
frameshift mutation and introduces a premature stop codon (17,115). In this model, the
RpgrORF15 transcript can be detected, but the
corresponding protein is absent, whereas the
Rpgrex1-19 protein remains detectable. At the
early stage, a mottled fundus appearance is observed, along with
mislocalization of M-opsin to the IS, perinuclear region and
synaptic terminals. As the disease progresses to the mid-stage,
rhodopsin expression decreases markedly, whereas no mislocalization
of S-opsin is observed. In the late stage, severe cell loss is
observed in the ONL, with the thickness of the photoreceptor layer
reduced by ~50% of its normal thickness (17). ERG recordings show a continuous
decline in retinal function starting as early as one month of age.
The notably reduced oscillatory potential (OP) amplitudes indicate
that both photoreceptors (OP1) and inner retinal neurons (OP2-OP4)
are affected by degenerative changes. The retinal degeneration in
this model is primarily driven by rod cell death (115). This serves as an important tool
for investigating the pathogenic mechanisms of ORF15 mutations in
XLRP and for evaluating potential therapeutic strategies.
The Rpgr-KO (Rpgr−/−)
mouse was generated on a C57BL/6 genetic background by replacing
exons 4-6 of the Rpgr gene with an antibiotic selection
cassette. This modification truncates both major transcripts
(Rpgrex1-19 and RpgrORF15) and
results in complete loss of RPGR protein expression. Although ERG
responses remain normal at postnatal day 20, some proteins that
should be localized to the OS are aberrantly distributed within the
IS (48). Müller cells exhibit
reactive gliosis, which becomes progressively more pronounced over
time. Even in the absence of overt degenerative changes, these
findings indicate that the retina has already initiated a
damage-response process. At mid-to-late stages, ONL thickness is
markedly reduced and ERG a-wave and b-wave amplitudes are decreased
by ~25 and 31%, respectively (48). Similar findings have been
reported in another study of Rpgr-KO mice, where rhodopsin
was abnormally localized to the CC, accompanied by mistrafficking
of proteins to the IS and ONL (116).
The Rpgr-CKO model was generated by deleting
the proximal promoter and exons 1-3 (3,189 bp). In this model,
nephrocystin-4 fails to localize properly to the CC, leading to
structural abnormalities in the OS. Consistent with previous
observations, mislocalization of rhodopsin and M-opsin is apparent
at early stages and is accompanied by progressive photoreceptor
degeneration. By late stages, >50% of the ONL is lost. Two new
Rpgr-KO mouse lines (L1 and L2) have been generated using
CRISPR-Cas9 technology (64).
Unlike the exons 4-6 deletion model, this version deletes exons
7-13, resulting in the deletion of 502 amino acids.
The zebrafish model is not as homologous to human
genes as the mouse model, but it has a faster growth rate and
developmental cycle, which helps researchers obtain study samples
quickly. Liu et al (66)
showed that zebrafish rpgra possesses a single transcript
homologous to human RPGRORF15. In this model,
visual dysfunction and early degenerative features can manifest as
early as 5 days post-fertilization. With increasing age,
photoreceptor OS progressively shorten, the ONL progressively thins
and OS architecture becomes disorganized, concomitant with declines
in ERG amplitudes. By mid-to-late stages, the cone OS begins to
degenerate, accompanied by substantial downregulation of retinal
phototransduction genes. Concurrently, the OS disc membranes are
disorganized and loosely stacked, with vesicular accumulation
around the cilia. Shu et al (117) identified two RPGR
homologs in zebrafish, ZFRPGR1 and ZFRPGR2, with
ZFRPGR2 regarded as the functional homolog of human
RPGR. Knockdown of ZFRPGR2 results in abnormal eye
development and retinal lamination defects, including failure of
the retina to form the normal three-layer structure (GCL, INL and
ONL) as well as underdevelopment of the photoreceptor OS, leading
to widespread retinal cell apoptosis, a phenotype distinct from
that observed in mammalian Rpgr mutant models.
Two natural mutations in the ORF15 region were
detected in canines that are collectively referred to as
progressive retinal atrophy and are homologous to human RP
(13). The loss of
photoreceptors in the naturally mutated canine model is similar to
previous RD9 models, rpgra models and RPGR ablation
in human photoreceptors (112).
In XLPRA1, a 5-nucleotide deletion in this region results in a
230-amino acid truncation at the C-terminus, but does not affect
normal photoreceptor development (32). Photoreceptors initially develop
normally and function properly, but rod degeneration initiates at
~11 months. In XLPRA2, a comparable degeneration pattern is
observed, albeit with an earlier onset, beginning at 4 weeks of age
(89).
Animal models of RPGR mutations have been
instrumental in understanding XLRP pathophysiology, identifying
early markers of the disease, evaluating the efficacy of genetic
and pharmacological treatments and elucidating the mechanisms by
which RPGR mutations drive the retinal degeneration. A
comparative overview of the strengths and limitations of these
models is presented in Table I
(13,17,55,57,66,117-132). However, these models still have
limitations: The rodent retina is dominated by rods, whereas the
human retina contains a higher proportion of cones, especially
concentrated in the central concave region, which poses challenges
for translating these findings to humans (13,116). Existing mouse models exhibit a
slower progression of retinal degeneration, which is not entirely
consistent with the more severe and rapidly progressive phenotype
of human XLRP. In terms of disease progression, histopathologic
features and molecular mechanisms, these models do not fully
recapitulate the diverse clinical manifestations observed in humans
with RPGR mutations. Due to the complexity of alternative
splicing of the RPGR gene, the expression patterns and
functions of RPGR isoforms can differ substantially between
species, thereby restricting the translational potential of
findings obtained from animal models. Existing animal models have
limited capacity to accurately replicate the systemic
manifestations of RPGR mutations, as demonstrated by
respiratory infections and hearing loss observed in some patients
(41). In translational and
therapeutic studies, judicious selection of animal models according
to research goals, disease phenotypes and experimental parameters
is important. Despite their inherent limitations, these models
offer a vital preclinical platform for testing and refining gene
therapy approaches for RPGR-related retinal
degeneration.
 | Table IComparison of different animal models
as treatment models for RPGR mutations. |
Table I
Comparison of different animal models
as treatment models for RPGR mutations.
| Models | Advantages | Disadvantages | (Refs.) |
|---|
| Mouse | i) The genetic
background is clear; ii) nature gene editing technology; iii) low
cost; iv) abundant data from existing studies. | i) Retinal
structure is very different from humans; ii) slow progression of
the RPGR mutant phenotype; iii) differences in immune
response exist. | (13,17,55,57,118,119) |
| Zebrafish | i) Transparent
embryo; ii) fast reproduction and low cost; iii) easy gene editing;
iv) retinal development: Human retina development is highly
similar. | i) Simple retinal
structure; ii) RPGR mutation phenotype may not be as
pronounced as mammals; iii) drug metabolism is different from
mammals. | (57,66,117,120,121) |
| Canine | i) Retinal
structure is close to that of humans; ii) the RPGR mutant
canine model exhibits retinal degeneration similar to that of
humans; iii) suitable for long-term studies. | i) High cost of
breeding and experimentation; ii) difficulty of gene editing; iii)
canine models face more ethical controversies compared with other
anima models. | (13,57,118,119,122,123) |
| Hog | i) The structure of
the retina is highly similar to that of humans; ii) the RPGR
mutant pig model exhibits retinal degeneration similar to that of
humans; iii) close to human size suitable for surgical eye and gene
therapy studies. | i) High breeding
and experimental costs; ii) long breeding cycle; iii) difficult for
gene editing. | (13,124,125) |
| Macaque | i) Retinal
structure almost identical to humans; ii) disease phenotypes are
closest to humans; iii) suitable for preclinical studies, with
results of gene therapy and surgical operations directly
extrapolated to humans. | i) Extremely high
cost; ii) study on apes and monkeys face serious ethical
controversies as experimental animals; iii) long breeding
cycle. | (126-132) |
Progression of treatment strategies in
RPGR-related XLRP
Early therapeutic strategies for
RPGR-related XLRP primarily focus on nutritional
supplementation aimed at slowing photoreceptor degeneration.
Vitamin A is converted into 11-cis-retinal, whereas omega-3
polyunsaturated fatty acids (such as docosahexaenoic acid, DHA)
contribute to the structural stability of the photoreceptor
membrane by integrating into the phospholipid bilayer (133-135). Such supplements are considered
to delay photoreceptor degeneration during the early stages of XLRP
(136,137). These interventions merely slow
visual deterioration and do not repair the retinal structural
damage caused by the RPGR mutation (138). Moreover, long-term DHA
supplementation may lead to side effects such as gastrointestinal
discomfort (139). To address
the limitations of such supplements, researchers have also explored
neuroprotective strategies, including supplementation with
brain-derived neurotrophic factor. These factors have shown
efficacy in conditions such as macular degeneration and glaucoma by
promoting the survival of retinal ganglion cells (RGCs) and
reducing microglia-mediated inflammation (140,141). Unlike supplement intervention
strategies, neuroprotective strategies target the secondary
neurodegenerative processes of XLRP. However, the majority of
studies of these neuroprotective strategies have been performed in
non-RPGR models (142-146), leaving their applicability to
XLRP unconfirmed. Importantly, supplements and neuroprotective
strategies are not mutually exclusive; rather, they may exert
complementary effects by targeting different pathological stages of
XLRP. Combination approaches may synergistically delay disease
progression through multiple pathways. Future studies using
RPGR mutation models are needed to systematically evaluate
the timing, dosage and interactions of such combined
strategies.
Although these interventions may help preserve
residual vision, they cannot fundamentally correct the underlying
genetic defect. By contrast, gene therapy aims to directly restore
normal RPGR function, thereby halting ongoing photoreceptor
degeneration at its source and potentially altering the natural
course of XLRP. It has emerged as a promising and potentially
transformative approach for treating IRDs, particularly those
associated with RPGR mutations (29,147-150). Because the majority of
inherited retinal disorders are monogenic, and the eye possesses
immune privilege as well as direct accessibility for observation
and intervention, a single gene therapy treatment may achieve
long-term, even lifelong, therapeutic benefits (151-153). Among various gene therapy
approaches, viral vector-mediated gene replacement represents the
primary strategy for treating RPGR-related diseases.
Gene therapy studies in Rpgr mutant
models
Despite inherent limitations, animal models remain
the primary experimental systems for evaluating the safety and
efficacy of gene therapy strategies. The therapeutic outcomes
largely depend on factors such as promoter choice, vector platform,
AAV serotype, capsid modification strategy and host immune
response.
In retinal gene therapy, the use of cell
type-specific promoters is important for achieving stable and
precise transgene expression. Researchers have compared the
transcriptional activities of several promoters in rod and cone
photoreceptors, including rhodopsin kinase (RK), human
interphotoreceptor retinoid-binding protein (hIRBP) and human RK
(hGRK1). The hGRK1 promoter exhibited more sustained expression in
rod photoreceptors, making it an optimal candidate for
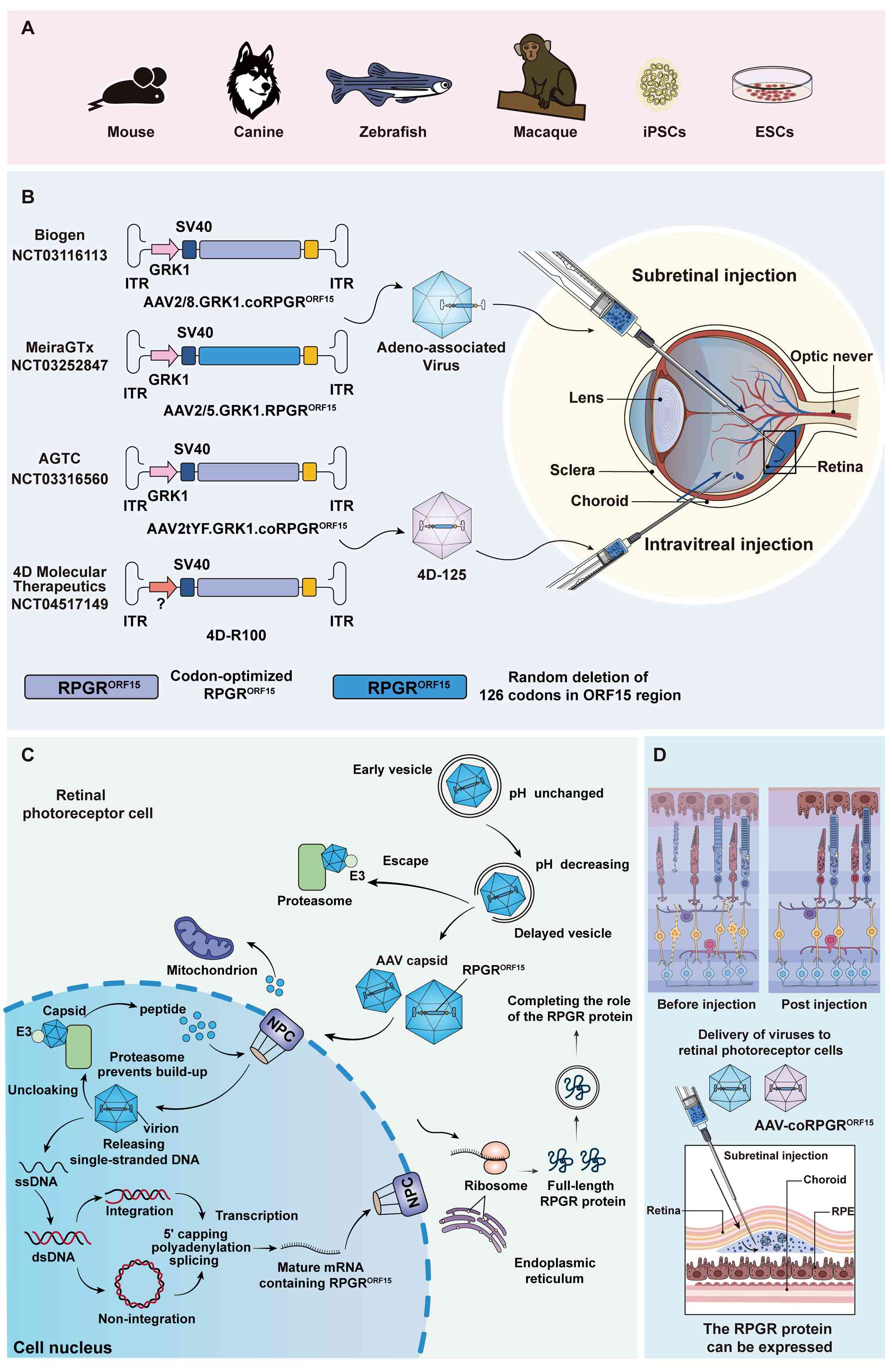
RPGR-related gene therapy (10,122). In an XLRP canine model,
delivery of human RPGRORF15 using an AAV2/5
vector driven by either the hIRBP or hGRK1 promoter successfully
preserved photoreceptor nuclei and normal retinal structure. This
treatment restored both rod and cone function and corrected
rhodopsin mislocalization (122).
In terms of vector design, continuous improvements
in recombinant AAV (rAAV) vectors have enabled targeted expression
in specific retinal cells and markedly enhanced transduction
efficiency (154-156). With a packaging capacity of
~4.8 kb, AAV vectors can readily accommodate the RPGR gene
(116,156). Because different diseases
affect retinal photoreceptors in distinct ways, vector design must
be approached with particular caution. Future optimization of AAV
vectors should focus on several key aspects: i) Selecting
appropriate and cell type-specific promoters; ii) designing safer
vectors with minimal off-target effects; iii) simplifying gene
expression regulatory mechanisms while enhancing expression
stability; iv) improving transduction efficiency alongside scalable
production; and v) systematically evaluating the durability and
safety of the therapy. Because the ORF15 region undergoes complex
post-transcriptional processing with multiple alternatively spliced
isoforms and possesses intrinsic sequence instability, maintaining
its integrity during vector production and therapeutic application
remains a key challenge (157).
Fischer et al (22)
carried out codon optimization of the Rpgr sequence and
constructed the AAV8-hRK-coRPGR system, which successfully restored
retinal function in two animal models (Rpgr−/y
and C57BL/6JRD9/Boc), markedly ameliorating retinal
degeneration phenotypes. Giacalone et al (158) used a bioinformatics approach to
introduce synonymous mutations into a highly repetitive region of
ORF15, enhancing sequence stability and expression efficiency while
maintaining amino acid sequence invariance. Hong et al
(159) demonstrated earlier
that a shortened version of RPGRORF15, with a 654
bp deletion in the repetitive region, effectively alleviated
retinal degeneration in Rpgr-KO mice. Pawlyk et al
(75) further compared the
effects of varying degrees of linker region truncation and found
that moderate truncation (removing ~33% of the sequence) preserved
protein function while markedly improving retinal morphology and
function in Rpgr-KO mice.
Currently, preclinical RPGR gene therapy studies
have primarily focused on highly efficient AAV-based delivery
systems (8,160-162). AAV is the most widely used
viral vector in clinical research on IRDs, with different serotypes
exhibiting tropism for specific tissues and cell types. For
example, AAV2 shows high affinity for RPE cells and is commonly
used for targeted delivery to RGCs (163,164). In animal models of retinal
degeneration, AAV2 has successfully restored both retinal structure
and function (165-167). AAV8, which is associated with a
reduced incidence of neutralizing antibodies and improved immune
tolerance, is considered a clinically promising alternative to
AAV2. In therapeutic studies using RPGR-mutant animal
models, AAV2, AAV5 and AAV8 are commonly employed (22,122,123). Different AAV serotypes have
distinct capsid protein structures, which influence their
transduction efficiency, tropism and immunogenicity. Therefore,
gene therapy design requires careful consideration of both serotype
selection and administration route to achieve an optimal balance
among efficacy, safety and immune tolerance (168,169). The AAV capsid is a key factor
in triggering immune responses. Once the capsid proteins are
recognized, T cells can be activated, leading to local
inflammation. Meanwhile, pre-existing neutralizing antibodies may
bind the capsid, thereby blocking vector entry into target cells
(170). Song et al
(123) evaluated the AAV2
vector in which three tyrosine residues of the capsid were
substituted with phenylalanine (AAV2tYF). The rAAV2tYF-GRK1-hRPGRco
demonstrated excellent transduction efficiency in the RPGR
mutant canine model and subsequently became a candidate vector for
clinical evaluation in the AGTC-501 study. Additionally, the
engineered AAV7m8 and modified AAV8 capsids have demonstrated
superior transduction efficiency, achieving notably higher delivery
rates in developing photoreceptor organoids compared with AAV5 and
standard AAV8 (8,171). Pavlou et al (172) developed a novel AAV capsid in
2021 that exhibited markedly enhanced delivery efficiency and lower
invasiveness across mouse, canine and non-human primate models,
confirming its cross-species applicability. These capsid
engineering strategies, which modify surface amino acids, provide a
foundation for more efficient and targeted gene delivery.
Beyond gene replacement therapy, gene-editing
approaches have also demonstrated promising therapeutic potential.
In a 2020 study, a single subretinal delivery of CRISPR-Cas9 via
AAV in Rpgr-KO mice precisely corrected the RPGR
mutation in vivo, protecting photoreceptors for ≥12 months
without detectable off-target effects (173). In the RD9 mouse model,
CRISPR-Cas9 targeted and excised the ORF15 mutation region,
allowing repair via non-homologous end joining and successfully
restoring the normal RPGR reading frame and protein expression
(113). This markedly improved
the retinal pathological phenotype, further confirming the efficacy
of gene editing for in vivo repair of RPGR mutations.
Additionally, prime editing has demonstrated a notable effect in
treating mice with RP (4,174).
Animal studies have also verified the feasibility
of RPGR gene therapy from the perspective of long-term efficacy. Wu
et al (175) conducted a
two-year dose-response study in Rpgr-KO mice, delivering
full-length RPGRORF15 via AAV8 and AAV9 vectors.
The results showed that treated mice maintained retinal structure
and function over the long term, as evidenced by higher ERG
amplitudes, a thicker photoreceptor layer and correct localization
of photoreceptor proteins (10,175). While animal models have been
invaluable in advancing RPGR gene therapy, emerging organoid models
provide promising platforms for therapeutic evaluation and
mechanistic insights, as discussed in the following section.
Application of organoid models in RPGR
gene therapy
Researchers have utilized human induced pluripotent
stem cell (hiPSC) differentiated organoid models to investigate
both the simulation and therapeutic targeting of RPGR
mutations. Organoids are highly organized three-dimensional
structures that serve dual roles in disease modeling and
therapeutic research. These models can differentiate into retinal
tissues containing all major cell types, exhibiting highly refined
structural organization and functional characteristics that closely
resemble those of native retinal tissue (8,116). Previous studies have
investigated the potential of hiPSC-derived organoids as a platform
for biomarker screening in RPGR-mutant patients (171,176,177). The first study employing
CRISPR-Cas9 to correct autologous hiPSC from patients with
RPGR mutations achieved 13% repair efficiency, and the
corrected cells could be used for subsequent transplantation
studies, underscoring the potential of precision medicine for
RPGR-related diseases (178). Similarly, a 2018 study at the
in vitro stage demonstrated that the application of
CRISPR-Cas9 to correct RPGR mutations successfully restored
the structure and electrophysiological properties of photoreceptors
(177). In addition, Sladen
et al (8) used
CRISPR-Cas9 to repair RPGR-mutated hiPSC, differentiated
them into photoreceptor cells and evaluated the effects of the AAV
delivery system, providing preliminary data to support subsequent
clinical trials (8). Another
study conducted single-cell RNA sequencing on organoids derived
from normal and RPGR-mutant hiPSCs, providing key insights
into the molecular mechanisms of RPGR-associated retinal
degeneration and informing the development of precision-targeted
gene therapy strategies (179).
Although hiPSC-derived organoids show great
potential for modeling RPGR-related diseases and testing
gene therapy strategies, they still face several challenges. The
majority of organoids have not yet developed structured
photoreceptor OS, making it difficult to fully replicate the
pathological process of cilia-associated diseases. Additionally
generating organoids is time-consuming and expensive (116,180). Although studies have attempted
to introduce antioxidants, lipid supplements and hyaluronic acid to
promote photoreceptor OS maturation, the results have been limited
(180,181). Overall, organoids cannot yet
fully replicate the in vivo microenvironment, and their
findings should therefore be interpreted cautiously when
considering clinical translation.
Delivery routes for ocular RPGR gene
therapy
In addition to the aforementioned factors
influencing gene therapy, the success of treatment largely depends
on the delivery route of the vector. The route of administration
not only affects the spatial distribution and cellular uptake
efficiency of the therapeutic gene but also directly influences the
magnitude of immune responses and the overall precision of
treatment. Therefore, the choice of injection modality is important
in RPGR gene therapy studies. Currently, the commonly used delivery
routes for ocular gene therapy include intravitreal injection,
subretinal injection and suprachoroidal injection. Table II summarizes the key features
and applicability differences of the three delivery modalities
(128,169,182-197). Intravitreal injection delivers
the AAV vector carrying the RPGR gene directly into the
vitreous cavity but may result in reduced therapeutic efficacy and
a stronger immune response due to uneven vector distribution. By
contrast, subretinal injection administers the vector into the
subretinal space between the RPE and photoreceptors, achieving a
higher concentration and more efficient transduction in target
cells. This approach is particularly suited for targeted and
precise RPGR gene therapy (Fig.
6D). Subretinal injection has also become the primary
administration route in current clinical trials for patients with
RPGR-associated RP (Fig.
6B).
| Table IIComparison of different injection
methods for ocular drug delivery. |
Table II
Comparison of different injection
methods for ocular drug delivery.
| Injection
method | Injection site | Applicable
conditions | Advantages | Challenges | (Refs.) |
|---|
| Intravitreal
injection | Inside the vitreous
cavity | Age-related macular
degeneration; diabetic retinopathy. | Fewer
procedure-related complications; low risk of immune response;
directly into the vitreous cavity; higher concentration of drugs;
considerable therapeutic effect; simple to operate and less
invasive. | Potential immune
response; high accuracy requirements; low delivery efficiency. | (128,169,182-186) |
| Subretinal
injection | Subretinal (between
the retina and retinal pigment epithelium) | Treatment of
hereditary retinal diseases. | Direct drug
delivery to target area; small injection dose; overall reduction in
side effects. | Relatively
difficult technique; moderate risk of complications; temporary
decrease in visual function; long recovery time; keep pressure
low. | (183,187-191) |
| Suprachoroidal
injection | Above the choroid
(the space between the retina and the choroid) | Macular edema;
photoreceptor loss; choroidal; neovascularization; retinal
detachment; uveitis; glaucoma; uveal melanoma. | Short-term
effectiveness; ocular tolerability; wider distribution; higher drug
utilization; less impact on the immediate period; lower
complication and infection rates; relatively minimally invasive;
high targeting; low surgical risk. | Demanding injection
techniques; patient stress; limitations of post-injection
distribution; need for continuous intraocular monitoring. | (169,192-197) |
Although ocular gene therapy offers advantages such
as localized administration, the ability to monitor treatment
effects, and using one eye as a control for the other, it still has
certain limitations. Currently, only a few gene therapy drugs for
incurable eye diseases have been approved worldwide (165). Particularly during subretinal
injection applications, several technical challenges remain,
including potential damage to the retina and macula, cataract
formation, subretinal deposit development and drug reflux into the
vitreous cavity (198). In
addition, improper technique can lead to more severe complications,
including endophthalmitis, choroidal rupture, Bruch's membrane
damage, drug misinjection into the sub-scleral cavity, retinal
detachment and even irreversible vision loss.
Emerging non-viral delivery strategies
for RPGR therapy
Owing to their high efficiency and cell-type
specificity, AAV vectors remain the preferred choice for retinal
gene delivery; however, their use may pose risks related to
inflammation and immune responses. Given these limitations and the
potential risks associated with AAV-based therapies, alternative
non-viral delivery strategies have gained increasing attention in
recent years. Non-viral vectors, with advantages in safety,
flexibility and mass production, are gradually becoming an
important complement to viral vectors in gene therapy (199-202). The use of nanoparticles for
ocular gene delivery has been continuously explored in recent
years. Engineered nanoparticles exhibit enhanced targeting
capability and reduced immunogenicity, offering great potential for
clinical translation in IRD therapy (203,204). Lipid nanoparticles, polymeric
nanoparticles (such as polylactic-co-glycolic acid) and inorganic
nanoparticles (such as cerium oxide) have demonstrated efficient
drug and gene delivery in ocular diseases such as AMD (202,205,206).
There are no published reports directly applying
non-viral vectors in Rpgr mutant animal models. Given the
therapeutic success of non-viral vectors in AMD, future research
should prioritize assessing their delivery efficiency, durability
of RPGR gene expression, and safety profiles in mutant
models (such as RD9, Rpgr-KO) to accelerate the translation
of preclinical research to clinical application.
Clinical progress of RPGR gene therapy
Clinical assessment and trial design of
RPGR gene therapy
Comprehensive and accurate assessment tools are
essential for detection of RP associated with RPGR mutations
and for evaluating the efficacy of gene therapy. Currently,
commonly used examinations include optical coherence tomography
(OCT), fundus autofluorescence (FAF), ERG, visual evoked potential,
BCVA and VF testing (37,207,208).
With the advancement of RPGR gene therapy clinical trials, the
evaluation methodology has been further refined to incorporate
multidimensional metrics such as microperimetry, contrast
sensitivity function and vision-guided mobility assessment (VMA),
thereby enabling a more comprehensive detection of subtle
treatment-induced changes (28).
The 2024 study highlighted that optimizing subject selection
criteria is important for improving the success rate of gene
therapy trials (209). In
particular, the structural integrity of the ellipsoid zone (EZ) has
been identified as a potential biomarker. Patients with RPGR
mutations who exhibit an EZ width of 600-800 μm and a
preserved 1° central VF are likely to exhibit improved response to
treatment, providing an important basis for accurate patient
stratification and efficacy prediction in clinical trials (209,210). These assessment tools are not
only suitable for dynamic follow-up across different disease
stages, but also facilitate the identification of ideal candidates
who retain central vision despite moderate vision impairment. In
addition, the stratification and evaluation system for patients
with RP also provides important reference value (35,211,212).
Current clinical studies of gene therapy for
RPGR mutation-associated XLRP primarily focus on AAV
vector-based gene replacement approaches (9,73,213). The majority of RPGR gene
therapy trials have employed the (RK/GRK1 promoter, as
aforementioned in the preclinical model section, the core
advantages of this promoter lie in two aspects: Its precise
targeting of retinal photoreceptor cells and its compatibility with
the packaging capacity limitations of AAV vectors. The main
variations among the different studies concern the viral subtypes
and the selection of coding sequences (Fig. 6B). To date, four clinical trials
have been reported, each sponsored by a different company: i)
Biogen Inc. (NCT03116113), AAV2/8.
hRK.coRPGRORF15 carrying a codon-optimized
RPGRORF15 cDNA sequence. ii) MeiraGTx
(NCT03252847), AAV2/5. hRK.RPGRORF15 without
codon optimization; iii) AGTC (NCT03316560),
AAV2tYF.GRK1.coRPGRORF15 employing an AAV2 capsid
containing a single tyrosine-to-phenylalanine (Y→F) substitution
(10); iv) 4D Molecular
Therapeutics (NCT04517149): 4D-R100-based vector carrying
codon-optimized human RPGR. R100 is an AAV capsid engineered
through directed evolution, characterized by highly efficient
targeting of retinal photoreceptor cells.
Clinical outcomes and safety of
AAV-mediated RPGR gene therapy
AAV-mediated delivery of the RPGR gene has led to
substantial advances in the treatment of retinal degenerative
diseases such as XLRP (Fig. 6C).
The Phase I/II clinical trial AGTC-501 (NCT03316560), initiated in
2017, employed rAAV2tYF-GRK1-RPGR to evaluate the safety and
preliminary efficacy of the modified capsid. The codon-optimized
human CDS coRPGRORF15 sequence overcame the
intrinsic instability associated with transgene-based therapies and
enhanced gene expression levels in mouse models, while preserving
post-translational characteristics (22). In 2020, a Phase I/II
dose-escalation trial (NCT03116113) was initiated to evaluate
codon-optimized AAV8-RPGR gene therapy for XLRP. The study met its
primary safety endpoint, with no dose-limiting toxicities (DLTs) or
serious adverse events (SAEs) observed across all dose cohorts.
These findings provide initial evidence supporting the safety and
feasibility of the codon optimization strategy (73). During the 6-month follow-up, no
DLTs or SAEs were observed in the low-dose and intermediate-dose
groups (29,61). However, the low-dose cohort
showed no improvement in visual function, likely due to
insufficient dosing and delayed intervention. The high-dose group
exhibited potentially enhanced therapeutic effects, but 77.8% of
patients developed mild intraocular inflammation, which was
alleviated by local or systemic anti-inflammatory therapy but still
affected the treatment experience and overall efficacy. By
contrast, the intermediate dose (5×1011 gp/ml) provided
the best balance between therapeutic efficacy and minimal incidence
of SAEs. Efficacy was primarily assessed based on retinal
sensitivity responses and the changes over time. In another gene
therapy trial for diabetic macular edema, several cases of vision
loss were reported due to severe delayed-onset intraocular
inflammation. However, pharmacokinetic modeling from the ADVM-022
ocular gene therapy study in monkeys provided useful reference data
for fine-tuning dosage regimens (214,215). Based on the aforementioned
clinical findings, several considerations should be emphasized in
subsequent Phase II/III studies: i) Dose selection strategies:
Optimize dosing to maximize efficacy while minimizing inflammation
risk; ii) immunosuppressive regimen optimization: Adopt more
effective anti-inflammatory strategies to improve patient
experience and treatment outcomes; iii) patient enrollment
criteria: Focus on patients with early-stage disease to enhance
therapeutic benefit.
At the 2023 Annual Meeting of the Association for
Research in Vision and Ophthalmology, MeiraGTx in collaboration
with Janssen reported on the immunogenicity of the AAV5-RPGR
(Botaretigene Sparoparvovec, a gene therapy method using AAV5) gene
therapy in the Phase I/II trial (NCT03252847) (41). At baseline, 44% of patients had
pre-existing antibodies against AAV5 and 24% had antibodies against
RPGR. Following treatment, only 13.3% of patients developed new
anti-RPGR antibodies, and no association between antibody status
and dose was observed. In this study, 96.9% of AEs were related to
the surgical procedure, and 82.2% of patients experienced at least
one treatment-emergent AE (TEAE), with no other immune-related
reactions detected. The majority of AEs were procedure-related,
including conjunctival hemorrhage (low dose, 72.7%; intermediate
dose, 64.7%; high dose, 100.0%) and transient decreases in vision
(low dose, 54.5%; intermediate dose, 47.1%; high dose, 50.0%),
which negatively affected the patient experience (41). However, these events were
generally reversible and resolved over time. Notably, one patient
in the low-dose cohort experienced retinal detachment, which was
successfully repaired surgically. At the end of the study, another
patient still had uveitis (day 33), which may suggest a potential
delayed immune response; however, longer-term follow-up is required
to determine the incidence and clinical significance of such
events.
In 2020, 4D Molecular Therapeutics initiated a
Phase I/II clinical trial (NCT04517149) to evaluate the safety and
tolerability of intravitreal injection administration of 4D-125
(216). This multicenter study
included both an observational natural history cohort and an
interventional treatment arm. The therapy utilized a proprietary
AAV capsid variant (4D-R100) carrying codon-optimized human
RPGRORF15. Updated data presented at the 2021
American Society of Retina Specialists meeting showed that, among
the eight enrolled patients, two single-eye dose levels
(3×1011 and 1×1012 vg/ml) had been tested in
preliminary experiments. The results demonstrated that 4D-125 was
well tolerated, with no DLTs or SAEs observed, and with only 25% of
patients experiencing mild, transient inflammation. These findings
supported the feasibility of administering a single-eye dose of
1×1012 vg/ml in patients with advanced XLRP. During the
intervention phase, TEAEs were generally reversible, including
transient intraocular pressure elevation within 24 h post-injection
(21%); conjunctival hemorrhage resolving within 1 week (43%); and
mild vitreous opacities resolving within 3 months (14%), all of
which resolved without medical intervention. Although the small
patient population limited the statistical power of these
observations, the therapeutic vector and dosing used in NCT04517149
exhibited a favorable safety profile. Further studies in larger
patient cohorts are required to validate the true therapeutic
potential of this approach.
Results from the AGTC-501 Phase I/II gene therapy
trial HORIZON (NCT03316560), published in 2024, showed that 78% of
patients receiving central retinal injections exhibited
improvements in visual function at 6 months, based on
microperimetry and BCVA assessments, providing preliminary
validation of the feasibility of the gene replacement strategy.
Data from a 2-year follow-up indicated that all participants
experienced at least one TEAE, with 93% considered by investigators
to be mild and self-limiting, such as transient vitreous opacities
(217). Although these SAEs
resolved spontaneously or following intervention, procedure-related
events such as retinal tears or intraocular pressure elevation
could have different implications for patients. Even transient AEs
can negatively affect patient experience and potentially impact
treatment outcomes. Of the four patients receiving the highest dose
(1.99×1012 vg/eye), three developed RPE changes, which
appeared as progressively depigmented areas on FAF imaging. These
changes were observed not only at the surgical incision site but
also beyond it, gradually enlarging over the follow-up period.
While these changes did not appear to adversely affect improvements
in retinal sensitivity for some patients over 24 months, the high
incidence (75%) and the uncertainty of potential long-term risks
led the study team to select 6.8×10¹¹ vg/eye as the maximum
tolerated dose. At this dose, half of the patients maintained
visual sensitivity improvements at a minimum of five retinal test
loci (≥7 dB improvement per locus) over 24 months (217). These findings highlight the
need to carefully consider the potential RPE toxicity of high-dose
AAV vectors in clinical gene therapy development and emphasize the
importance of extended follow-up.
In 2024, the MeiraGTx-sponsored AAV5-hRKp.RPGR
(NCT03252847) published results from its Phase I/II trial, with
primary endpoints divided into assessments of safety and efficacy
(41). The core efficacy
endpoint focused on functional vision, using VMA as the primary
measure, while secondary endpoints included static perimetry mean
retinal sensitivity (MRS) within the central 10°, dark-adapted
microperimetry MRS, BCVA and contrast sensitivity. The treatment
group showed a notable improvement in MRS in the central 10° field
of view at week 26 compared with the control group (LS mean
difference 1.96 dB). MRS in the dark-adapted microperimetry was
also notably improved at Week 26 (LS mean difference 1.06 dB). BCVA
in the treatment group remained stable at Week 26, whereas it
decreased in the control group and the treatment group continued to
maintain stability at Week 52 (LS mean change of 0.40 letters)
(218). Overall, the treatment
group demonstrated superior visual function improvement at Week 26
and AEs remained manageable, with intraocular inflammation
occurring in 22% of patients and no SAEs reported. AAV5-hRKp.RPGR
appeared to exhibit good tolerability and a low immunogenic
profile. However, the study noted that 52-week follow-up data were
insufficient to assess long-term efficacy. Despite this,
participation in this trial was not without risk for patients.
In 2022, a Phase II/III study was initiated using
AAV8-based gene therapy Cotoretigene Toliparvovec
(BIIB112/AAV8-RPGR, NCT03116113). Unfortunately, according to
published 12-month evaluations of participating patients, the study
did not meet its primary endpoint, as there was no statistically
significant difference in microperimetry response rates. Clinical
data indicated that low-dose Cotoretigene Toliparvovec reduced the
risk of inflammation, but maintained efficacy was limited, with
visual improvement declining by ~15% at 52 weeks and a 1.8-fold
increased risk of RPE atrophy (28). The primary endpoint was defined
based on VF assessments. Specifically, it measured the proportion
of patients who showed improvements of ≥7 dB in retinal sensitivity
in at least five of the 16 central loci of the study eye at 12
months post-treatment. Results showed no significant differences
among the low-dose group (37.5%), high-dose group (25.0%) and
untreated group (22.2%). Additionally, microperimetry assessments
at 52 weeks did not demonstrate statistically significant
improvements in retinal sensitivity in the treatment groups
compared with controls. Despite not achieving the primary endpoint,
the low-dose group showed trends toward improvement in mean
continuous microperimetry sensitivity and low-luminance VA (LLVA),
with ~33% of patients experiencing LLVA gains ≥15 letters,
suggesting that the therapy may be effective in some patients.
Limitations of current trials and future
directions in clinical RPGR gene therapy
These differences in therapeutic outcomes also
reflect the inherent complexity of RPGR gene therapy. Patient type
and individual variability can lead to heterogeneous responses,
highlighting the need for clearly defined enrollment criteria.
Compared with clinical trials using other AAV serotypes, AAV8
exhibits higher immunogenicity and RPE toxicity; high doses are
limited by safety concerns, while low doses may provide
insufficient efficacy. Attempts to reduce inflammation by lowering
the dose reveal the current challenge of balancing dose, efficacy
and safety, as simply reducing the dose cannot entirely mitigate
the risk of RPE atrophy. Although this study did not meet its
primary expectations, its 'failure' provides valuable insights for
subsequent vector optimization, dose adjustment, patient selection
and treatment strategies. Multiple clinical trials for Leber
Congenital Amaurosis, including NCT00999609, NCT02781480,
NCT00749957 and NCT00643747, have similarly reported ocular
immune-related AEs, such as inflammation, transient increases in
AAV neutralizing antibodies and AAV capsid-specific T cell
responses (219). These events
not only caused temporary declines in visual function but also
limited the overall benefit of retinal function improvement.
Collectively, these clinical studies demonstrate that patients
receiving AAV-mediated ocular gene therapy remain at risk of
pronounced immune responses.
Differences in clinical outcomes among vector
systems underscore the complexity of vector selection. Patients
participating in AAV5-hRKp.RPGR trials exhibited lower rates of
inflammation compared with those in AAV8-RPGR (BIIB112) trials. In
the BIIB112 high-dose group, the incidence of inflammation reached
77.8%, of which 28% were SAEs, presenting as retinal detachment. By
contrast, the high-dose group of AAV5-hRKp.RPGR exhibited a 75%
incidence of inflammation without any SAEs. Although AAV8 (XIRIUS)
and AAV5 (MeiraGTx) showed similar post-treatment inflammation
rates, their efficacy differed markedly, likely reflecting inherent
differences in tissue tropism between the vectors. Notably, both
AGTC and XIRIUS employed codon-optimized sequences; however, the
Phase II/III trial of XIRIUS did not achieve its primary endpoint,
suggesting that codon optimization alone may not be decisive for
patient outcomes in gene therapy. As previously noted, optimization
of AAV vectors extends beyond promoters and codon usage, with
capsid engineering and serotype selection being important for
enhancing stability, retinal cell targeting, transduction
efficiency and minimizing host immune responses to gene
therapy.
Regarding endpoint assessments, all clinical trials
employed AE, SAE and TEAEs to evaluate safety; however, efficacy
endpoints varied among studies. In the Phase I/II trial
NCT03116113, efficacy was assessed using BCVA, VF and MRS;
NCT03316560 evaluated efficacy with MRS, BCVA and OCT; and
NCT03252847 employed VMA, MRS and BCVA. Although safety was
achieved at each stage in the Phase I/II trials, the Phase II/III
trial of NCT03116113, which focused on VF and MRS, did not meet its
primary endpoint. These differences in efficacy assessment led to
two key issues: i) Patient outcomes across trials were not directly
comparable, making it difficult to objectively evaluate the
relative effectiveness of each treatment strategy; ii) the choice
of endpoints itself may influence whether a trial achieves its
prespecified endpoint, potentially causing treatments that appeared
effective in early studies to 'fail' in subsequent trials.
Moreover, early-stage patients retain more photoreceptors than
late-stage patients, making functional recovery easier to observe.
For the clinical development of RPGR gene therapy, establishing a
unified and standardized efficacy assessment framework is important
to facilitate translation.
A common and key challenge faced by all current
RPGR gene therapy trials is the insufficient duration of follow-up.
To date, the maximum follow-up period has been only 24 months,
which is far from adequate for evaluating the true long-term value
of gene therapy. Given that treatment decline has already been
observed in the XIRIUS and AGTC-501 trials, the efficacy of these
therapies may further diminish over time, and current clinical data
remain insufficient to predict patient outcomes beyond this period.
Maintaining durable therapeutic effects therefore remains a key
concern. Extended follow-up is required to accurately determine the
long-term disease trajectory of patients receiving these gene
therapy interventions, assess the persistence of dose-dependent
efficacy and monitor delayed toxicity risks. Only with such
longitudinal data can reliable, comparable and reproducible
clinical conclusions be drawn.
Future research should build upon monitoring and
optimizing existing gene therapy strategies by further addressing
multiple aspects of research and clinical practice. i) Vector and
therapeutic development: The creation of next-generation,
low-immunogenicity vectors, exploration of non-viral delivery
systems or combination therapies integrating anti-inflammatory and
neuroprotective strategies is needed to minimize immune responses.
Concurrently, improving surgical techniques is essential to reduce
treatment-related SAEs arising from procedural issues. ii) Patient
selection and management: More comprehensive enrollment criteria
should be established, incorporating assessments such as the EZ as
part of the screening process. iii) Post-treatment clinical
evaluation: A unified and standardized efficacy assessment
framework should be implemented to ensure consistent measurement of
outcomes. iv) Informed consent and patient counseling: Patients
should be fully informed prior to treatment regarding the need for
intensive and frequent follow-up, potential systemic side effects
from high-dose immunosuppression (for example, prednisone-induced
mood changes) and the risks of treatment failure or ocular injury
(220). Although challenges
remain in optimizing vector and delivery systems and regulatory
oversight, the continuous integration of these insights and
innovations holds promise for advancing therapeutic outcomes
(221). Systematically
addressing these issues in RPGR gene therapy clinical research and
balancing considerations across these levels is key to translating
exploratory trials into routine clinical practice. Table III summarizes both completed
clinical trials and ongoing studies with results yet to be
reported. Table IV summarizes
the main results and efficacy outcomes of completed clinical
trials.
| Table IIIClinical trials of RPGR gene therapy
for retinal degenerative diseases (data was retrieved from
ClinicalTrials.gov). |
Table III
Clinical trials of RPGR gene therapy
for retinal degenerative diseases (data was retrieved from
ClinicalTrials.gov).
| ClinicalTrials.gov ID; date | Type and phase | Patient
population | Assessment | Sponsors |
|---|
| NCT03252847
(AAV5-hRKp.RPGR); Jul 2017-Nov 2021 | Interventional,
I/II; participants, 49. | Male patients aged
≥5 years; have X-linked retinitis pigmentosa confirmed by a retinal
specialist (co-investigator or principal investigator). | Primary: Incidence
of serious adverse events within 9 weeks after administration;
secondary: Visual acuity, static visual field, questionnaires (6
months after treatment). | MeiraGTx
Limited |
| NCT03314207; Dec
2017-Feb 2022 | Observational;
participants: 14. | i) Male patients
aged ≥6 years who have mutations in exon ORF15 of the RPGR
gene; ii) willing and able to perform study procedures. iii) Signed
informed consents obtained (child assent where applicable). | Primary: Disease
progression; secondary: Visual acuity, perimetry, OCT,
electroretinography, national eye institute visual functioning
questionnaire-25 (VFQ-25) quality of life questionnaire (Day
0-Month 36). | Beacon
Therapeutics |
| NCT03349242; Dec
2017-Apr 2024 | Observational;
participants, 140. | i) Males and female
patients aged ≥5 years; ii) are able to give informed consent or
assent, with the guidance of their parent/guardian where
appropriate. | Primary: Retinal
structure; secondary: Analysis of retinal structure and function to
assess disease progression (6 years); method: Retinal sensitivity,
retinal structural detailed phenotyping, fundus autofluorescence,
visual field. | MeiraGTx
Limited |
| NCT03116113
(AAV8-RPGR); Mar 2017-Nov 2020 | Interventional,
I/II; participants: 50. | i) Male patients
aged ≥10 years; ii) having XLRP (with a mutation in the RPGR
gene) and active disease, with obvious lesions observable in the
macular area of both eyes. | Primary: Safety ≥24
months after treatment; aim: Dose-limiting toxicities,
treatment-emergent adverse events. Secondary: Visual acuity,
central retinal thickness, best-corrected visual acuity, low
luminance visual acuity, macular integrity assessment
microperimetry. | Biogen |
| NCT04868916; Jul
2021-Apr 2024 | Observational;
participants: 15. | i) Patients aged ≥5
years; ii) are able to give informed consent or assent, with the
guidance of their parent/guardian where appropriate. | Primary: Visual
function, retinal structure, retinal function; method: Visual
acuity, SD-OCT, static visual field testing. | Japan
Pharmaceutical Manufacturers Association (JPMA) |
| NCT04926129; Sep
2017-Dec 2022 | Observational;
participants: 201. | i) Patients ages ≥7
years and older; ii) are willing and able to undergo ophthalmic
examinations, as required by protocol; ii) have an ETDRS BCVA in ≥1
eye of ≥34 letters (Equivalent to Snellen ≥6/60 or 20/200; decimal
0.1; LogMAR 1.0). | Primary: Changes in
retinal sensitivity within the macula; secondary: Change from
baseline in contrast sensitivity, low luminance visual acuity,
visual function questionnaire score, RP-specific patient-reported
outcome (PRO) questionnaire, EuroQol-5 Dimension 5-level
(EQ-5D-5L), Health Utilities Index Mark 3 (HUI3), visual field
readings, microperimetry readings, multi-luminance mobility test
(MLMT) readings, SD-OCT, FAF, fundus photography, morphology of eye
as assessed by slit-lamp examination. | Biogen |
| NCT03316560
(rAAV2tYF-GRK1-RPGR) Apr 2018-Mar 2025 | Interventional,
I/II; participants: 29. | i) Male patients
aged 6-50 years of age with documented RPGR mutations; ii)
BCVA ≤78 ETDRS letters; iii) be capable of conducting visual and
retinal function tests, evaluated through OCT and have detectable
ellipsoid zone lines. | Primary: Number and
proportion of AEs, safety and efficiency; secondary: VA, SD-OCT,
quality of life questionnaire responses, visual function by
light-adapted perimetry, fundus imaging, FST and visual function by
dark-adapted full field perimetry (for subjects treated
peripherally). | Beacon
Therapeutics |
| NCT06333249
(rAAV2tYF-GRK1-RPGR); Apr 2021-Feb 2027 | Interventional, II;
participants: 14. | i) Provide written
informed consent; ii) male patients ages 8-50 years; iii) 35
letters ≤BCVA≤75 letters; iv) have detectable EZ line in both eyes
as assessed by SD-OCT and confirmed by the CRC. | Primary: Efficacy,
safety and tolerance. The difference in the proportion of
responding eyes between treated and control eyes in the low dose
group and high dose group at 12 months, as measured by MAIA
microperimetry, where response is defined as a 7dB or more
improvement in at least 5 loci. Secondary: VA; ORA-VNC mobility
test. | Beacon
Therapeutics |
| NCT06492850
(FT-002); Apr 2024-Feb 2026 | Interventional,
I/II; participants: 32. | i) Are willing and
able to follow study procedures including scheduled visits,
treatment plan, and laboratory tests, and sign a written informed
consent form; ii) Phase I: Male patients aged 18-45 years. Phase
II: Male patients aged 8-45 years old. | Primary: Evaluate
the safety and tolerance of FT-002. Incidence and severity of AEs.
Secondary: Evaluate the efficacy of FT-002. Changes in visual
sensitivity, FST and BCVA. | Frontera
Therapeutics, Inc. |
| NCT05874310
(FT-002); Feb 2023-Nov 2027 | Interventional, I;
participants: 18. | i) Are able to
follow study procedures. Male patients ages 8-45 years. ii)
confirmed with variants of RPGR. Has not received any other
gene therapy products. | Primary: Number and
proportion of AEs. Secondary: Change in visual function, retinal
structure. Method: Retinal sensitivity within the 30-degree visual
field and OCT. | Frontera
Therapeutics, Inc. |
| NCT06275620
(rAAV2tYF-GRK1-RPGR); Nov 2023-Dec 2029 | Interventional, II;
participants: 24. | i) Male patients
aged ≥12 years. ii) Have one eye previously treated with an AAV
vector-based gene therapy. iii) Have detectable baseline mean
macular sensitivity measured by MAIA microperimetry and have
detectable EZ lines. iv) Corticosteroid drugs can be used. | Primary: Incidence
of SAEs (Day 0-Month 12). Secondary: Incidence and severity of AEs.
Assess photoreceptor function under low light, FST, BCVA, LLVA, EZ
area, Ora-VNC mobility test, mobility standardized test-virtual
reality (MOST-VR) mobility course test score. | Beacon
Therapeutics |
| NCT04671433
(AAV5-hRKp.RPG); Dec 2020-Sep 2024 | Interventional,
III; participants: 97. | i) Patients aged ≥3
years. ii) Has XLRP confirmed by a retinal specialist and has a
predicted disease-causing sequence variant in RPGR confirmed
by an accredited laboratory. iii) Those who have undergone other
ocular treatments or eye surgeries are excluded. | Primary: Change
from baseline to Week 52 in VMA as measured by the ability of the
participant to navigate through a VMA maze. Secondary: Mean retinal
sensitivity within the central 10 degrees excluding scotoma,
pointwise response in full visual field, pointwise response in
worse-seeing eye in the central 30 degrees visual field, mean
retinal sensitivity within the full visual field (MRS90) in static
perimetry, the modified low luminance questionnaire (mLLQ) extreme
lighting domain score, BCVA, LLVA, ocular and non-ocular AEs,
abnormalities in laboratory assessments (from baseline to Week
52). | Johnson &
Johnson |
| NCT04312672
(AAV5-hRKp.RPGR); Jul 2017-Nov 2026 | Observational;
participants: 42. | i) Male patients
aged ≥5 years. ii) Received AAV5-hRKp.RPGR injection in MGT009
study. | Primary: Safety.
Secondary: Change in functional vision of LLQ domain scores, BCVA,
LLVA, MRS10, MRS90, pointwise response in full visual field,
functional vision of walk time in VMA. | Johnson &
Johnson |
| NCT04794101
(AAV5-hRKp.RPGR); Dec 2020-Sep 2029 | Interventional,
III; participants: 97. | i) Patients aged ≥3
years. ii) Have XLRP confirmed by a retinal specialist. | Primary: Incidence
of AEs, BCVA, number of participants with abnormalities in
laboratory assessments, LLVA. | Johnson &
Johnson |
| NCT03584165
(AAV8-RPGR); Jun 2018-Jun 2026 | Interventional,
III; participants: 330 (CHM and XLRP participants). | i) Male patients
aged ≥18 years. ii) Have received a sub-retinal injection of
BIIB112 for XLRP and have exited an antecedent study. | Primary: Incidence
of AEs, IOP, abnormal slit lamp examination, lens opacity grading,
anterior chamber and vitreous inflammation, indirect ophthalmoscopy
(up to 5 years). Secondary: Change from baseline in BCVA, VFQ-25,
VF, autofluorescence (AF), fundus photography, SD-OCT,
microperimetry. | Biogen |
| NCT04517149
(4D-125); Jun 2020-May 2029 | Interventional,
I/II; participants: 21. | i) Male patients
aged ≥12 years. ii) ≥1 eye amenable to intravitreal injection and
34 letters ≤ BCVA ≤78 letters. | Primary: Treatment.
Incidence and severity of TEAEs and SAEs. | 4D Molecular
Therapeutics |
| NCT05926583
(AAV5-hRKp.RPGR); Sep 2023-Feb 2030 | Interventional,
III; participants: 4. | i) Japanese male or
female patients aged ≥5 years. ii) Mean retinal sensitivity of
greater than or equal to ≥2 decibel (dB). | Primary: Incidence
of AEs, number of participants with abnormalities in clinical
laboratory assessments. Secondary: Change from baseline LLVA, BCVA
(Baseline-Week 52). | Johnson &
Johnson |
| NCT06646289
(AAV5-hRKp.RPGR); Oct 2024-Oct 2030 | Interventional, II;
participants: 42. | i) Male patients ≥5
years. ii) Have been treated with AAV5-hRKp.RPGR in study MGT009
and have completed or is currently enrolled in Study MGT010. iii)
Sign an informed consent form. iv) Willing to adhere to the
protocol and long-term follow-up. | Primary: Incidence
of AEs (from baseline up to 5.5 years). Change from baseline in
BCVA, LLVA (baseline, months 12, 24, 36, 48 and 60). | Johnson &
Johnson |
| NCT04850118
(rAAV2tYF-GRK1-hRPGRco); Mar 2024-Oct 2029 | Interventional,
II/III; participants: 75. | i) Have a parent,
guardian or legal representative provide written informed consent.
ii) Male patients aged 12-50 year. iii) Follow study instructions,
complete study assessments, comply with the protocol. iv) 34
letters ≤ BCVA ≤78 letters, LLVA ≤64 letters, 10< LLD letters.
Have detectable EZ line in both eyes as assessed by SD-OCT and
confirmed by the CRC. | Primary: The
proportion of participants with a ≥15 letter increase from baseline
in LLVA. Secondary: Change from baseline in LLVA, mean sensitivity
across the whole grid, as measured by MAIA microperimetry, FST.
(Day 0-Month 12). Ocular/non-ocular adverse events are collected
during the duration of the trial (Day 0-Year 5). Exclusion criteria
can be found on the official website. | Beacon
Therapeutics |
| Table IVCompleted clinical trials of RPGR
gene therapy: Main results and efficacy outcomes. |
Table IV
Completed clinical trials of RPGR
gene therapy: Main results and efficacy outcomes.
| Name of gene
product | Vector | Primary
endpoint | Main clinical
results | Limitations |
|---|
|
rAAV2tYF-GRK1-hRPGRco (AGTC-501) | i) Employing an
AAV2 capsid containing a single tyrosine-to-phenylalanine (Y→F)
substitution.
ii) Codon-optimized full-length DNA sequence. | i) HORIZON:
Incidence and severity of TEAEs and SAEs.
ii) VISTA: Proportion of participants achieving a ≥15 letter
improvement in LLVA. | i) HORIZON: It has
good safety and nodose-limiting inflammation. Early data support
the advancement of subsequent experiments.
ii) 12-24 months, retinal sensitivity functions are continuously
enhanced (≥5 loci with >7 dB improvement).
iii) 12 months: MAIA, +1.96 dB (treated), −0.39 dB (control). 24
months: MAIA, +1.63 dB (treated), −1.56 dB (control).
iv) The overall response rate was 33% in the treatment group (50%
in the highest-dose cohort) and 0% in the control group. | i) In the high-dose
cohort, 75% of patients exhibited RPE changes, accompanied by
procedure-related complications.
ii) Dose-finding studies indicated a narrow optimal dose
range.
iii) The small number of participants, with only four receiving the
high-dose injection, limited the generalizability of the
findings. |
| AAV5-hRKp.RPGR | i) Using AAV5 and
hRKp.
ii) Carrying a truncated RPGR sequence. | i) MGT009:
Incidence of SAEs within 9 weeks after administration.
ii) LUMEOS: Change from baseline to Week 52 in VMA (the ability of
the participant to navigate through a VMA maze). | i) MGT009: SAEs
occurred, including retinal detachment and uveitis.
ii) LUMEOS: At Week 52, the primary endpoint VMA data were not
statistically significant, but showed a favorable trend. However,
improvements were observed in the three secondary endpoints:
Retinal function, VA and visual function. | i) Using VMA as an
endpoint in RPE65 therapy studies may be less appropriate for XLRP,
given differences in visual field and sensitivity.
ii) Inaccurate patient stratification: Stratification based on EZ
area, age and disease duration may be insufficient, posing a risk
of mismatched therapeutic windows.
iii) Variations in surgical procedures and immunosuppression
regimens further reduce cross-trial comparability. |
| AAV8-RPGR (BIIB112,
XIRIUS) | i) Using AAV8 and
GRK1/hRKp.
ii) Carrying a codon-optimized full-length cDNA sequence. | i) Part 1: Number
of participants with DLTs and the number of participants with
TEAEs.
ii) Part 2: Percentage of study eyes with ≥7 dB improvement from
baseline at ≥5 of the 16 central loci on the 10-2 grid and the
number of participants with TEAEs. | i) XIRIUS: The
predefined primary endpoint was not met, and a dose-safety
relationship was observed, with higher doses more likely to cause
SAEs, including vision loss and noninfectious retinitis.
ii) Although the primary endpoint was not met, the secondary
endpoint assessments showed favorable trends in visual outcomes. In
the mid-dose to high-dose cohorts, both LLVA and microperimetric
sensitivity demonstrated sustained improvement over time. iii) 12M:
retinal sensitivity, 2.8 dB (treated), 0.1 dB (untreated). iv) 12M:
MAIA, + 5.1 dB (treated), −0.9 dB (untreated). | i) Using MAIA
microperimetry as an endpoint is susceptible to the effects of
fixation instability and locus registration drift.
ii) There were differences in disease stage and residual EZ area
across patient strata.
iii) Patients receiving mid-to high-dose treatment experienced more
frequent inflammatory events, which have affected the overall
therapeutic outcomes. |
| 4D-125 | i) Using 4D
-R100;
ii) Engineered from AAV2 and having a codon-optimized DNA
sequence. | i) The incidence of
TEAEs and SAEs.
ii) Patients undergoing the intervention received a single-eye
injection and were followed for 24 months to monitor safety and
tolerability. | i) All 8 patients
tolerated the treatment well, with no DLTs or SAEs reported. Only
two patients experienced mild and transient inflammation.
ii) 9 months: Retinal sensitivity, 1.65 dB (treated), 0.25 dB
(untreated). | i) The small sample
size limited the data analysis.
ii) Participants enrolled in the dose-escalation phase were all in
the late clinical stage of XLRP, with residual photoreceptor
regions and retinal sensitivity that were limited or even
unmeasurable. |
Current research consensus suggests that the key to
XLRP-RPGR gene therapy lies in extending the follow-up
period, optimizing dose and vector systems, and exploring
combination therapy strategies. Future studies should evaluate the
long-term efficacy (such as maintenance of visual function for ≥3
years) and delayed risks, such as the mutagenic potential of vector
genome integration. Dosing should also be dynamically adjusted
based on EZ biomarkers to balance short-term efficacy with
long-term safety. Additionally, the combined use of
anti-inflammatory agents, including topical corticosteroids, with
gene therapy should be investigated. This approach may help reduce
inflammation-related complications, alleviate patient anxiety and
further improve quality of life.
Conclusion and perspective
RPGR-associated RP has attracted increasing
attention, and studies on its pathogenic mechanism and gene therapy
strategies have become a major focus in the field (9,222-224). This present review summarizes
the molecular mechanisms of photoreceptor degeneration triggered by
RPGR mutations and analyzes the functions of RPGR along with
its interacting protein networks. Although the interaction networks
of RPGR have been identified, the specific function of individual
proteins and the mechanisms of their interaction with RPGR remain
to be investigated in depth. RPGR regulates signal transduction and
metabolic homeostasis in photoreceptors by maintaining the
structural integrity of the CC and its transport functions.
Mutations lead to failure of rhodopsin transport, accumulation of
metabolic waste and ROS. The survival of photoreceptors depends on
the coordinated action of multiple metabolic and signaling
regulatory mechanisms and RPGR mutations may disrupt these
pathways, thereby accelerating photoreceptor degeneration. In
addition, an imbalance in the transmembrane signaling systems may
further reduce the adaptability of photoreceptors, making them more
susceptible to apoptosis under conditions of oxidative stress or
limited nutrient availability. Animal models carrying RPGR
mutations have provided valuable insights into disease mechanisms
and carry out an important role in the development of therapeutic
strategies. Future research should further investigate the
spatiotemporal dynamics and regulatory interactions of
RPGR-related signaling pathways in animal models. Such
studies will help clarify the specific functions of RPGR proteins
within these pathways and provide a theoretical basis for the
development of targeted therapies.
In recent years, research on RPGR-associated
XLRP has progressed from efficacy validation in preclinical animal
models to the preliminary success of clinical trials. These studies
have demonstrated that some patients show pronounced improvements
in retinal sensitivity and VF after treatment, offering new hope
for gene therapy. However, treatment still faces challenges related
to dose control, immune responses and the maintenance of long-term
efficacy (219,225). Although codon optimization and
long-term immune monitoring have been implemented in clinical
studies (175,217,218,223,226), issues remain regarding the
balance between dosage and safety, management of postoperative
adverse events, delayed immune reactions and the sustained
preservation of therapeutic effects. Regarding viral vector
optimization, improvements in AAV vectors through capsid
engineering are important, and rational functional modification of
various viral vectors also represent a key strategy to enhance
their clinical utility (227,228). Beyond reducing immunogenicity,
it is also necessary to comprehensively assess vector transduction
efficiency and safety to improve overall treatment efficacy and
precision. At the clinical implementation stage, patient selection,
immune monitoring and long-term follow-up systems should be
strengthened to balance therapeutic efficacy with safety risks,
thereby laying the foundation for the standardization of subsequent
gene therapy strategies.
As ocular gene therapy technologies continue to
mature, relying solely on a single viral vector for delivery may be
insufficient to fully restore retinal function and stabilize the
retinal microenvironment. Currently, several neuroprotective agents
are undergoing Phase I/II clinical trials (such as, UBX1325,
MTP-131 and Metformin) (142).
Future research is trending toward combined approaches that
integrate gene therapy with neuroprotection. By simultaneously
correcting genetic mutations, activating cell survival signaling
pathways and upregulating neurotrophic factor expression, such
strategies hold the potential to further slow photoreceptor
degeneration and maintain microenvironmental stability. These
combination therapies enhance treatment stability and long-term
efficacy, thereby overcoming the limitations of conventional
monotherapies. However, achieving precise temporal control of gene
editing and neuroprotective signaling, verifying the safety of
co-delivery systems and establishing regulatory evaluation
frameworks for combination therapies remain key challenges for
future clinical translation.
Meanwhile, non-viral vectors have been advancing
toward the development of new platforms with low immunogenicity,
strong targeting capabilities and scalable production, including
liposomes, solid nanoparticles, polymeric nanoparticles and
polyplexes for nucleic acid delivery (229-232). These vectors are being explored
for neuroprotective strategies, offering new avenues for multimodal
treatment of retinal degenerative diseases. However,
nanoparticle-based gene delivery systems share common challenges:
Systemic accumulation can induce toxicity, making them unsuitable
for long-term use, and the degradation of nanocarriers following
nucleic acid release in vivo may generate toxic byproducts
(229,231,233,234). Injectable retinal nanoimplants
combining donor-acceptor polymers with graphene have been
developed, capable of restoring light-driven behavior, visual brain
activity and retinal function without inducing inflammation,
providing an alternative perspective for RP treatment (235). Therefore, delivery strategies
using non-viral vectors still require further optimization in terms
of material design, biodegradability and long-term safety. In the
future, integrated approaches that combine gene therapy,
neuroprotective agents and traditional treatments such as DHA may
further enhance therapeutic outcomes (Fig. 7A). However, these strategies will
require more comprehensive regulatory evaluation frameworks
(15,135).
 | Figure 7Strategies for integrated management
of ocular diseases in patients with mutated RPGR. The
program encompasses interventions (medications, nutrition and
lifestyle modifications), diagnostic tests (such as ERG),
therapeutic strategies (genetic and cellular treatments) and
preventive screening measures. (A) Interventions: Commonly used
medications, vitamin A supplementation, ophthalmic solutions, glare
avoidance and DHA intake. (B) Diagnostic assessments: ERG, BCVA, VA
and VF testing. (C) Therapeutic approaches: Treatment modalities
encompass CRISPR-Cas9-based gene editing (NHEJ and HDR), cell
transplantation (iPSCs, MSCs and ESCs), artificial vision
technologies and neurotrophic factor therapy (GDNF and BDNF). (D)
Preventive measures: Detection of pathogenic variants through NGS
and nanopore sequencing. DHA, docosahexaenoic acid; ERG,
electroretinography; BCVA, best-corrected visual acuity; VA, visual
acuity; VF, visual field; NHEJ, Non-Homologous End Joining; HDR,
Homology-Directed Repair; iPSCs, induced pluripotent stem cells;
MSCs, mesenchymal stem cells; ESCs, embryonic stem cells; GDNF,
Glial cell line-derived neurotrophic factor; BDNF, Brain-derived
neurotrophic factor; NGS, next-generation sequencing. |
Ethical and regulatory challenges remain prominent
in the clinical translation of ocular gene therapies. These
challenges encompass not only the evaluation of safety and efficacy
at the technical application level but also the protection of
patient rights and equitable access to treatment. Current
regulatory frameworks primarily focus on safety and efficacy as
core approval criteria; however, specific guidelines addressing
potential delayed inflammatory responses following treatment are
still under development. Moreover, regulatory systems vary across
countries and regions, potentially increasing the costs of clinical
translation. Ethical concerns primarily involve patient privacy
protection, informed consent and treatment equity. On one hand, a
balance must be achieved between safeguarding patient privacy and
ensuring transparency of information. On the other hand, the high
cost of therapy may exacerbate socio-economic disparities (236). Therefore, ensuring standardized
production of gene therapy products, long-term safety validation,
transparent informed consent procedures and equitable access are
important prerequisites for the standardized and sustainable
clinical translation of ocular gene therapies. Although pronounced
progress has been made in RPGR gene therapy, patient phenotype and
disease progression pattern vary. Thus, precise stratification of
patient populations is essential to implement individualized
treatment strategies. Modern technologies allow for more accurate
patient assessment and tailored therapeutic planning by analyzing
mutation sites in conjunction with genotype-expression
associations.
High-precision molecular diagnostics combined with
multidimensional analyses are required to characterize the mutation
regions of the RPGR of patients, overcoming sequencing
limitations and ensuring the reliability of clinical stratification
and therapeutic strategies. Integrating early intervention with
conventional drugs and emerging approaches such as gene editing,
gene replacement and combined neuroprotection, as well as cell
transplantation and artificial retinal implantation (Fig. 7B-D), can advance precision
medicine for RPGR mutations. These strategies are also
expected to provide new directions for the treatment of RP.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Key Research and
Development Program of China (grant no. 2024YFA1108701), the
National Natural Science Foundation of China (grant nos. 82471107
and 31970930), the Hubei Natural Science Foundation (grant nos.
2025AFB042, 2020CFA069 and 2018CFB434), the Neuroscience Team
Development Project of Wuhan University of Science and Technology
(grant nos. 1180002 and 1180030) and the Innovation and
Entrepreneurship Project of Wuhan University of Science and
Technology (grant no. JCX2024033).
Availability of data and materials
Not applicable.
Authors' contributions
YL and JQ drafted, revised the manuscript and
created figures. WZ assisted with editing. HQ and KY edited the
manuscript, supervised the study and provided funding. All authors
read and approved the final manuscript. Data authentication is not
applicable. Kai Yao-Lead contact.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang Y, Liu S, Zhai Y, Liu Y, Wan X, Wang
W, Wang F and Sun X: Identification of a novel RPGR mutation
associated with X-linked cone-rod dystrophy in a Chinese family.
BMC Ophthalmol. 21:4012021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu W, Liu S, Li P and Yao K: Retinitis
Pigmentosa: Progress in molecular pathology and biotherapeutical
strategies. Int J Mol Sci. 23:48832022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Benson MD, Mukherjee S, Agather AR, Blain
D, Cunningham D, Mays R, Sun X, Li T, Hufnagel RB, Brooks BP, et
al: RPGR: Deep phenotyping and genetic characterization with
findings specific to the 3'-end of ORF15. Invest Ophthalmol Vis
Sci. 64:192023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qin H, Zhang W, Zhang S, Feng Y, Xu W, Qi
J, Zhang Q, Xu C, Liu S, Zhang J, et al: Vision rescue via
unconstrained in vivo prime editing in degenerating neural retinas.
J Exp Med. 220:e202207762023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu YS, Pan JQ, Wan JF, Ren CY, Xu ZH, Pan
XB, Gao RN, Liu SQ, Zhang JL, Yao QH, et al: A novel missense
mutation of RPGR identified from retinitis pigmentosa affects
splicing of the ORF15 region and causes loss of transcript
heterogeneity. Biochem Biophys Res Commun. 531:172–179. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Murga-Zamalloa C, Swaroop A and Khanna H:
Multiprotein complexes of Retinitis Pigmentosa GTPase regulator
(RPGR), a ciliary protein mutated in X-linked Retinitis Pigmentosa
(XLRP). Adv Exp Med Biol. 664:105–114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moreno-Leon L, West EL, O'Hara-Wright M,
Li L, Nair R, He J, Anand M, Sahu B, Chavali VRM, Smith AJ, et al:
RPGR isoform imbalance causes ciliary defects due to exon ORF15
mutations in X-linked retinitis pigmentosa (XLRP). Hum Mol Genet.
29:3706–3716. 2021. View Article : Google Scholar :
|
|
8
|
Sladen PE, Naeem A, Adefila-Ideozu T,
Vermeule T, Busson SL, Michaelides M, Naylor S, Forbes A, Lane A
and Georgiadis A: AAV-RPGR gene therapy rescues opsin
mislocalisation in a human retinal organoid model of
RPGR-Associated X-linked retinitis pigmentosa. Int J Mol Sci.
25:18392024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Awadh Hashem S, Georgiou M, Ali RR and
Michaelides M: RPGR-Related Retinopathy: Clinical features,
molecular genetics, and gene replacement therapy. Cold Spring Harb
Perspect Med. 13:a0412802023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cehajic Kapetanovic J, McClements ME,
Martinez-Fernandez de la Camara C and MacLaren RE: Molecular
strategies for RPGR gene therapy. Genes (Basel). 10:6742019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Karuntu JS, Almushattat H, Nguyen XT,
Plomp AS, Wanders RJA, Hoyng CB, van Schooneveld MJ, Schalij-Delfos
NE, Brands MM, Leroy BP, et al: Syndromic Retinitis Pigmentosa.
Prog Retin Eye Res. 107:1013242025. View Article : Google Scholar
|
|
12
|
Hartong DT, Berson EL and Dryja TP:
Retinitis pigmentosa. Lancet. 368:1795–1809. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rivas MA and Vecino E: Animal models and
different therapies for treatment of retinitis pigmentosa. Histol
Histopathol. 24:1295–1322. 2009.PubMed/NCBI
|
|
14
|
Liu Y, Zong X, Cao W, Zhang W, Zhang N and
Yang N: Gene therapy for retinitis pigmentosa: Current challenges
and new progress. Biomolecules. 14:9032024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Napoli D, Di Marco B, Salamone G, Orsini
N, Mazziotti R and Strettoi E: Keeping the lights on: A new role
for an old drug to support cone survival in Retinitis Pigmentosa.
Prog Retin Eye Res. 109:1014032025. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ghanawi H and Koch SF: The versatile roles
of retinal pigment epithelium in the pathophysiology of retinitis
pigmentosa. Prog Retin Eye Res. 108:1013902025. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thompson DA, Khan NW, Othman MI, Chang B,
Jia L, Grahek G, Wu Z, Hiriyanna S, Nellissery J, Li T, et al: Rd9
is a naturally occurring mouse model of a common form of retinitis
pigmentosa caused by mutations in RPGR-ORF15. PLoS One.
7:e358652012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Birch DG, Cheetham JK, Daiger SP, Hoyng C,
Kay C, MacDonald IM, Pennesi ME and Sullivan LS: Overcoming the
challenges to clinical development of X-Linked retinitis pigmentosa
therapies: Proceedings of an expert panel. Transl Vis Sci Technol.
12:52023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chivers M, Li N, Pan F, Wieffer H, Slowik
R and Leartsakulpanitch J: The Burden of X-Linked retinitis
pigmentosa on patients and society: A narrative literature review.
Clinicoecon Outcomes Res. 13:565–572. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang J, Wu X, Shen D, Dong L, Jiao X,
Hejtmancik JF and Li N: Analysis of RP2 and RPGR mutations in five
X-linked Chinese families with retinitis pigmentosa. Sci Rep.
7:444652017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li L, Khan N, Hurd T, Ghosh AK, Cheng C,
Molday R, Heckenlively JR, Swaroop A and Khanna H: Ablation of the
X-linked retinitis pigmentosa 2 (Rp2) gene in mice results in opsin
mislocalization and photoreceptor degeneration. Invest Ophthalmol
Vis Sci. 54:4503–4511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fischer MD, McClements ME,
Martinez-Fernandez de la Cama ra C, Bellingrath JS, Dauletbekov D,
Ramsden SC, Hickey DG, Barnard AR and MacLaren RE: Codon-optimized
RPGR improves stability and efficacy of AAV8 gene therapy in two
mouse models of X-Linked Retinitis pigmentosa. Mol Ther.
25:1854–1865. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ebenezer ND, Michaelides M, Jenkins SA,
Audo I, Webster AR, Cheetham ME, Stockman A, Maher ER, Ainsworth
JR, Yates JR, et al: Identification of novel RPGR ORF15 mutations
in X-linked progressive cone-rod dystrophy (XLCORD) families.
Invest Ophthalmol Vis Sci. 46:1891–1898. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Talib M, van Schooneveld MJ, Thiadens AA,
Fiocco M, Wijnholds J, Florijn RJ, Schalij-Delfos NE, van Genderen
MM, Putter H, Cremers FPM, et al: Clinical and genetic
characteristics of male patients with RPGR-associated retinal
dystrophies: A Long-term follow-up study. Retina. 39:1186–1199.
2019. View Article : Google Scholar
|
|
25
|
MacLaren RE, Duncan JL, Fischer MD, Lam
BL, Meunier I, Pennesi ME, Sankila EK, Gow JA, Li J and Tsang SF:
XOLARIS: A 24-Month, prospective, natural history study of 201
participants with retinitis pigmentosa GTPase Regulator-associated
X-linked retinitis pigmentosa. Ophthalmol Sci. 5:1005952025.
View Article : Google Scholar
|
|
26
|
Birtel J, Gliem M, Mangold E, Müller PL,
Holz FG, Neuhaus C, Lenzner S, Zahnleiter D, Betz C, Eisenberger T,
et al: Next-generation sequencing identifies unexpected
genotype-phenotype correlations in patients with retinitis
pigmentosa. PLoS One. 13:e02079582018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gill JS, Georgiou M, Kalitzeos A, Moore AT
and Michaelides M: Progressive cone and cone-rod dystrophies:
Clinical features, molecular genetics and prospects for therapy. Br
J Ophthalmol. 103:711–720. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lam BL, Pennesi ME, Kay CN, Panda S, Gow
JA, Zhao G and MacLaren RE: Assessment of visual function with
cotoretigene toliparvovec in X-linked retinitis pigmentosa in the
randomized XIRIUS phase 2/3 study. Ophthalmology. 131:1083–1093.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nuzbrokh Y, Ragi SD and Tsang SH: Gene
therapy for inherited retinal diseases. Ann Transl Med. 9:12782021.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
De Silva SR, Chan HW, Agarwal A, Webster
AR, Michaelides M and Mahroo OA: Visual acuity by decade in 139
males with RPGR-Associated retinitis pigmentosa. Ophthalmol Sci.
4:1003752024. View Article : Google Scholar
|
|
31
|
Seliniotaki AK, Ververi A, Koukoula S,
Efstathiou G, Gerou S, Ziakas N and Mataftsi A: Female carrier of
RPGR mutation presenting with high myopia. Ophthalmic Genet.
45:159–163. 2024. View Article : Google Scholar
|
|
32
|
Zhang Q, Acland GM, Wu WX, Johnson JL,
Pearce-Kelling S, Tulloch B, Vervoort R, Wright AF and Aguirre GD:
Different RPGR exon ORF15 mutations in Canids provide insights into
photoreceptor cell degeneration. Hum Mol Genet. 11:993–1003. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fahim AT, Sullivan LS, Bowne SJ, Jones KD,
Wheaton DKH, Khan NW, Heckenlively JR, Jayasundera KT, Branham KH,
Andrews CA, et al: X-Chromosome inactivation is a biomarker of
clinical severity in female carriers of RPGR-Associated X-Linked
retinitis pigmentosa. Ophthalmol Retina. 4:510–520. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tuekprakhon A, Pawestri AR, Suvannaboon R,
Thongyou K, Trinavarat A and Atchaneeyasakul LO: Rare Co-occurrence
of visual snow in a female carrier with RPGR(ORF15)-Associated
retinal disorder. Front Genet. 12:7280852021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gocuk SA, Ayton LN, Edwards TL, Jolly JK
and Britten-Jones AC: Cone contrast sensitivity testing in X-linked
retinal diseases: Insights into genotype, sex and disease severity.
Ophthalmic Physiol Opt. 45:2046–2053. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nassisi M, De Bartolo G, Mohand-Said S,
Condroyer C, Antonio A, Lancelot ME, Bujakowska K, Smirnov V,
Pugliese T, Neidhardt J, et al: Retrospective natural history study
of RPGR-related Cone- and Cone-rod dystrophies while expanding the
mutation spectrum of the disease. Int J Mol Sci. 23:71892022.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tran M, Kolesnikova M, Kim AH, Kowal T,
Ning K, Mahajan VB, Tsang SH and Sun Y: Clinical characteristics of
high myopia in female carriers of pathogenic RPGR mutations: A case
series and review of the literature. Ophthalmic Genet. 44:295–303.
2023. View Article : Google Scholar :
|
|
38
|
Liu C, Sheri N and Benson MD: Inherited
retinal diseases with high myopia: A review. Genes (Basel).
16:11832025. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang L, Lai Y, Sun L, Li S and Ding X:
High myopia is common in patients with X-linked retinopathies:
Myopic maculopathy analysis. Retina. 44:117–126. 2024. View Article : Google Scholar
|
|
40
|
Branham K, Andrews CA, Milentijevic D,
Narayanan D and Jayasundera KT: Early symptoms in RPGR-associated
retinal degeneration: Can we shorten time to diagnosis in the gene
therapy era? Ophthalmic Genet. 46:569–575. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wongchaisuwat N, Amato A, Lamborn AE, Yang
P, Everett L and Pennesi ME: Retinitis pigmentosa GTPase
regulator-related retinopathy and gene therapy. Saudi J Ophthalmol.
37:276–286. 2023. View Article : Google Scholar :
|
|
42
|
Meindl A, Dry K, Herrmann K, Manson F,
Ciccodicola A, Edgar A, Carvalho MR, Achatz H, Hellebrand H, Lennon
A, et al: A gene (RPGR) with homology to the RCC1 guanine
nucleotide exchange factor is mutated in X-linked retinitis
pigmentosa (RP3). Nat Genet. 13:35–42. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zou X, Fang S, Wu S, Li H, Sun Z, Zhu T,
Wei X and Sui R: Detailed comparison of phenotype between male
patients carrying variants in exons 1-14 and ORF15 of RPGR. Exp Eye
Res. 198:1081472020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vervoort R, Lennon A, Bird AC, Tulloch B,
Axton R, Miano MG, Meindl A, Meitinger T, Ciccodicola A and Wright
AF: Mutational hot spot within a new RPGR exon in X-linked
retinitis pigmentosa. Nat Genet. 25:462–466. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pusch CM, Broghammer M, Jurklies B, Besch
D and Jacobi FK: Ten novel ORF15 mutations confirm mutational hot
spot in the RPGR gene in European patients with X-linked retinitis
pigmentosa. Hum Mutat. 20:4052002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Buraczynska M, Wu W, Fujita R, Buraczynska
K, Phelps E, Andréasson S, Bennett J, Birch DG, Fishman GA, Hoffman
DR, et al: Spectrum of mutations in the RPGR gene that are
identified in 20% of families with X-linked retinitis pigmentosa.
Am J Hum Genet. 61:1287–1292. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xu Z, Lei Y, Qin H, Zhang S, Li P and Yao
K: Sigma-1 receptor in retina: Neuroprotective effects and
potential mechanisms. Int J Mol Sci. 23:75722022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang J, Feng S, Zhang Q, Qin H, Xu C, Fu
X, Yan L, Zhao Y and Yao K: Roles of histone acetyltransferases and
deacetylases in the retinal development and diseases. Mol
Neurobiol. 60:2330–2354. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhao Y, Sun B, Fu X, Zuo Z, Qin H and Yao
K: YAP in development and disease: Navigating the regulatory
landscape from retina to brain. Biomed Pharmacother.
175:1167032024. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang S, Xu W, Liu S, Xu F, Chen X, Qin H
and Yao K: Anesthetic effects on electrophysiological responses
across the visual pathway. Sci Rep. 14:278252024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yang Z, Yan L, Zhang W, Qi J, An W and Yao
K: Dyschromatopsia: A comprehensive analysis of mechanisms and
cutting-edge treatments for color vision deficiency. Front
Neurosci. 18:12656302024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang Z, Zhang Y, Xu C, Peng A, Qin H and
Yao K: Advancements in age-related macular degeneration treatment:
From traditional anti-VEGF to emerging therapies in gene, stem
cell, and nanotechnology. Biochem Pharmacol. 236:1169022025.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Khanna H: More than meets the eye: Current
understanding of RPGR function. Adv Exp Med Biol. 1074:521–538.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yan D, Swain PK, Breuer D, Tucker RM, Wu
W, Fujita R, Rehemtulla A, Burke D and Swaroop A: Biochemical
characterization and subcellular localization of the mouse
retinitis pigmentosa GTPase regulator (mRpgr). J Biol Chem.
273:19656–19663. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hong DH, Pawlyk BS, Shang J, Sandberg MA,
Berson EL and Li T: A retinitis pigmentosa GTPase regulator
(RPGR)-deficient mouse model for X-linked retinitis pigmentosa
(RP3). Proc Natl Acad Sci USA. 97:3649–3654. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hong DH, Yue G, Adamian M and Li T:
Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein
is stably associated with the photoreceptor ciliary axoneme and
anchors RPGR to the connecting cilium. J Biol Chem.
276:12091–12099. 2001. View Article : Google Scholar
|
|
57
|
Megaw RD, Soares DC and Wright AF: RPGR:
Its role in photoreceptor physiology, human disease, and future
therapies. Exp Eye Res. 138:32–41. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dias MF, Joo K, Kemp JA, Fialho SL, da
Silva Cunha A Jr, Woo SJ and Kwon YJ: Molecular genetics and
emerging therapies for retinitis pigmentosa: Basic research and
clinical perspectives. Prog Retin Eye Res. 63:107–131. 2018.
View Article : Google Scholar
|
|
59
|
Fahim AT, Bowne SJ, Sullivan LS, Webb KD,
Williams JT, Wheaton DK, Birch DG and Daiger SP: Allelic
heterogeneity and genetic modifier loci contribute to clinical
variation in males with X-linked retinitis pigmentosa due to RPGR
mutations. PLoS One. 6:e230212011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rao KN, Zhang W, Li L, Ronquillo C, Baehr
W and Khanna H: Ciliopathy-associated protein CEP290 modifies the
severity of retinal degeneration due to loss of RPGR. Hum Mol
Genet. 25:2005–2012. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Linari M, Ueffing M, Manson F, Wright A,
Meitinger T and Becker J: The retinitis pigmentosa GTPase
regulator, RPGR, interacts with the delta subunit of rod cyclic GMP
phosphodiesterase. Proc Natl Acad Sci USA. 96:1315–1320. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Grayson C, Bartolini F, Chapple JP,
Willison KR, Bhamidipati A, Lewis SA, Luthert PJ, Hardcastle AJ,
Cowan NJ and Cheetham ME: Localization in the human retina of the
X-linked retinitis pigmentosa protein RP2, its homologue cofactor C
and the RP2 interacting protein Arl3. Hum Mol Genet. 11:3065–3074.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wätzlich D, Vetter I, Gotthardt K,
Miertzschke M, Chen YX, Wittinghofer A and Ismail S: The interplay
between RPGR, PDEδ and Arl2/3 regulate the ciliary targeting of
farnesylated cargo. EMBO Rep. 14:465–472. 2013. View Article : Google Scholar
|
|
64
|
Ying R, Li C, Li H, Zou J, Hu M, Hong Q,
Shen Y, Hou L, Cheng H and Zhou R: RPGR is a guanine nucleotide
exchange factor for the small GTPase RAB37 required for retinal
function via autophagy regulation. Cell Rep. 43:1140102024.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Rao KN, Zhang W, Li L, Anand M and Khanna
H: Prenylated retinal ciliopathy protein RPGR interacts with PDE6δ
and regulates ciliary localization of Joubert syndrome-associated
protein INPP5E. Hum Mol Genet. 25:4533–4545. 2016.
|
|
66
|
Liu X, Han S, Liu F, Yu S, Qin Y, Li J,
Jia D, Gao P, Chen X, Tang Z, et al: Retinal degeneration in rpgra
mutant zebrafish. Front Cell Dev Biol. 11:11699412023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Murga-Zamalloa CA, Atkins SJ, Peranen J,
Swaroop A and Khanna H: Interaction of retinitis pigmentosa GTPase
regulator (RPGR) with RAB8A GTPase: Implications for cilia
dysfunction and photoreceptor degeneration. Hum Mol Genet.
19:3591–3598. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lyraki R, Megaw R and Hurd T: Disease
mechanisms of X-linked retinitis pigmentosa due to RP2 and RPGR
mutations. Biochem Soc Trans. 44:1235–1244. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Baz-Redón N, Sánchez-Bellver L,
Fernández-Cancio M, Rovira-Amigo S, Burgoyne T, Ranjit R, Aquino V,
Toro-Barrios N, Carmona R, Polverino E, et al: Primary ciliary
dyskinesia and retinitis pigmentosa: Novel RPGR variant and
possible modifier gene. Cells. 13:5242024. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Moore A, Escudier E, Roger G, Tamalet A,
Pelosse B, Marlin S, Clément A, Geremek M, Delaisi B, Bridoux AM,
et al: RPGR is mutated in patients with a complex X linked
phenotype combining primary ciliary dyskinesia and retinitis
pigmentosa. J Med Genet. 43:326–333. 2006. View Article : Google Scholar
|
|
71
|
Kolkova Z, Durdik P, Holubekova V,
Durdikova A, Jesenak M and Banovcin P: Identification of a novel
RPGR mutation associated with retinitis pigmentosa and primary
ciliary dyskinesia in a Slovak family: A case report. Front
Pediatr. 12:13396642024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lucas JS, Davis SD, Omran H and Shoemark
A: Primary ciliary dyskinesia in the genomics age. Lancet Respir
Med. 8:202–216. 2020. View Article : Google Scholar
|
|
73
|
Cehajic-Kapetanovic J, Xue K,
Martinez-Fernandez de la Camara C, Nanda A, Davies A, Wood LJ,
Salvetti AP, Fischer MD, Aylward JW, Barnard AR, et al: Initial
results from a first-in-human gene therapy trial on X-linked
retinitis pigmentosa caused by mutations in RPGR. Nat Med.
26:354–359. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cehajic-Kapetanovic J, Martinez-Fernandez
de la Camara C, Birtel J, Rehman S, McClements ME, Charbel Issa P,
Lotery AJ and MacLaren RE: Impaired glutamylation of RPGR(ORF15)
underlies the cone-dominated phenotype associated with truncating
distal ORF15 variants. Proc Natl Acad Sci USA. 119:e22087071192022.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Pawlyk BS, Bulgakov OV, Sun X, Adamian M,
Shu X, Smith AJ, Berson EL, Ali RR, Khani S, Wright AF, et al:
Photoreceptor rescue by an abbreviated human RPGR gene in a murine
model of X-linked retinitis pigmentosa. Gene Ther. 23:196–204.
2016. View Article : Google Scholar :
|
|
76
|
Zhang Q, Giacalone JC, Searby C, Stone EM,
Tucker BA and Sheffield VC: Disruption of RPGR protein interaction
network is the common feature of RPGR missense variations that
cause XLRP. Proc Natl Acad Sci USA. 116:1353–1360. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Hakeem A and Yang S: Regulation of INPP5E
in ciliogenesis, development, and disease. Int J Biol Sci.
21:579–594. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Vössing C, Atigbire P, Eilers J, Markus F,
Stieger K, Song F and Neidhardt J: The major ciliary isoforms of
RPGR build different interaction complexes with INPP5E and
RPGRIP1L. Int J Mol Sci. 22:35832021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhang Z, Dai H, Wang L, Tao T, Xu J, Sun
X, Yang L and Li G: Novel mutations of RPGR in Chinese families
with X-linked retinitis pigmentosa. BMC Ophthalmol. 19:2402019.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Parmeggiani F, Barbaro V, De Nadai K,
Lavezzo E, Toppo S, Chizzolini M, Palù G, Parolin C and Di Iorio E:
Identification of novel X-linked gain-of-function RPGR-ORF15
mutation in Italian family with retinitis pigmentosa and pathologic
myopia. Sci Rep. 6:391792016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yokoyama A, Maruiwa F, Hayakawa M, Kanai
A, Vervoort R, Wright AF, Yamada K, Niikawa N and Naōi N: Three
novel mutations of the RPGR gene exon ORF15 in three Japanese
families with X-linked retinitis pigmentosa. Am J Med Genet.
104:232–238. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Andréasson S, Breuer DK, Eksandh L,
Ponjavic V, Frennesson C, Hiriyanna S, Filippova E, Yashar BM and
Swaroop A: Clinical studies of X-linked retinitis pigmentosa in
three Swedish families with newly identified mutations in the RP2
and RPGR-ORF15 genes. Ophthalmic Genet. 24:215–223. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Miano MG, Testa F, Strazzullo M, Trujillo
M, De Bernardo C, Grammatico B, Simonelli F, Mangino M, Torrente I,
Ruberto G, et al: Mutation analysis of the RPGR gene reveals novel
mutations in south European patients with X-linked retinitis
pigmentosa. Eur J Hum Genet. 7:687–694. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Hwang S, Jeon S, Yoon JM, Woo SJ, Joo K,
Choi YJ, Yoon CK, Kim M, Lee HJ, Byeon SH, et al: RPGR-associated
X-linked retinitis pigmentosa: Molecular genetics and clinical
characteristics. Am J Ophthalmol. 274:171–183. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kurata K, Hosono K, Hayashi T, Mizobuchi
K, Katagiri S, Miyamichi D, Nishina S, Sato M, Azuma N, Nakano T,
et al: X-linked retinitis pigmentosa in Japan: Clinical and genetic
findings in male patients and female carriers. Int J Mol Sci.
20:15182019. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Thiadens AA, Soerjoesing GG, Florijn RJ,
Tjiam AG, den Hollander AI, van den Born LI, Riemslag FC, Bergen AA
and Klaver CC: Clinical course of cone dystrophy caused by
mutations in the RPGR gene. Graefes Arch Clin Exp Ophthalmol.
249:1527–1535. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Bonetti G, Cozza W, Bernini A, Kaftalli J,
Mareso C, Cristofoli F, Medori MC, Colombo L, Martella S,
Staurenghi G, et al: Towards a Long-read sequencing approach for
the molecular diagnosis of RPGRORF15 genetic variants. Int J Mol
Sci. 24:168812023. View Article : Google Scholar :
|
|
88
|
Daiger SP, Sullivan LS and Bowne SJ: Genes
and mutations causing retinitis pigmentosa. Clin Genet. 84:132–141.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Liu HL, Gao FG, Wang DD, Hu FY, Xu P,
Chang Q, Xu GZ and Wu JH: Mutation analysis of the RPGR gene in a
Chinese cohort. Front Genet. 13:8501222022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Battaglioni S, Benjamin D, Wälchli M,
Maier T and Hall MN: mTOR substrate phosphorylation in growth
control. Cell. 185:1814–1836. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Liu GY and Sabatini DM: mTOR at the nexus
of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol.
21:183–203. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Marques-Ramos A and Cervantes R:
Expression of mTOR in normal and pathological conditions. Mol
Cancer. 22:1122023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Venkatesh A, Ma S and Punzo C: TSC but not
PTEN loss in starving cones of retinitis pigmentosa mice leads to
an autophagy defect and mTORC1 dissociation from the lysosome. Cell
Death Dis. 7:e22792016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Song DJ, Bao XL, Fan B and Li GY:
Mechanism of cone degeneration in retinitis pigmentosa. Cell Mol
Neurobiol. 43:1037–1048. 2023. View Article : Google Scholar
|
|
95
|
Brown EE, Lewin AS and Ash JD: AMPK may
play an important role in the retinal metabolic ecosystem. Adv Exp
Med Biol. 1185:477–481. 2019. View Article : Google Scholar
|
|
96
|
Newton F, Halachev M, Nguyen L, McKie L,
Mill P and Megaw R: Autophagy disruption and mitochondrial stress
precede photoreceptor necroptosis in multiple mouse models of
inherited retinal disorders. Nat Commun. 16:40242025. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Xu L, Kong L, Wang J and Ash JD:
Stimulation of AMPK prevents degeneration of photoreceptors and the
retinal pigment epithelium. Proc Natl Acad Sci USA.
115:10475–10480. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Chen CL, Chen YH, Liang CM, Tai MC, Lu DW
and Chen JT: Glucosamine-Induced autophagy through AMPK-mTOR
pathway attenuates Lipofuscin-like autofluorescence in human
retinal pigment epithelial cells in vitro. Int J Mol Sci.
19:14162018. View Article : Google Scholar
|
|
99
|
Liu J, Zhang Y, Xu X, Dong X, Pan Y, Sun X
and Luo Y: Ginsenoside Ro prevents endothelial injury via promoting
Epac1/AMPK-mediated mitochondria protection in early diabetic
retinopathy. Pharmacol Res. 211:1075622025. View Article : Google Scholar
|
|
100
|
Kucharska J, Del Río P, Arango-Gonzalez B,
Gorza M, Feuchtinger A, Hauck SM and Ueffing M: Cyr61 activates
retinal cells and prolongs photoreceptor survival in rd1 mouse
model of retinitis pigmentosa. J Neurochem. 130:227–240. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
German OL, Insua MF, Gentili C, Rotstein
NP and Politi LE: Docosahexaenoic acid prevents apoptosis of retina
photoreceptors by activating the ERK/MAPK pathway. J Neurochem.
98:1507–1520. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Muraleva NA and Kolosova NG: P38 MAPK
signaling in the retina: Effects of aging and Age-related macular
degeneration. Int J Mol Sci. 24:115862023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wang L, Tian Y, Li L, Cai M, Zhou X, Su W,
Hua X and Yuan X: Temporary alleviation of MAPK by arbutin
alleviates oxidative damage in the retina and ARPE-19 cells.
Heliyon. 10:e328872024. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Maugeri G, Bucolo C, Drago F, Rossi S, Di
Rosa M, Imbesi R, D'Agata V and Giunta S: Attenuation of high
glucose-induced damage in RPE cells through p38 MAPK signaling
pathway inhibition. Front Pharmacol. 12:6846802021. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ding J, Yang N, Yan Y, Wang Y, Wang X, Lu
L and Dong K: Rapamycin inhibited photoreceptor necroptosis and
protected the retina by activation of autophagy in experimental
retinal detachment. Curr Eye Res. 44:739–745. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 168:960–976. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Shen J, Yang X, Dong A, Petters RM, Peng
YW, Wong F and Campochiaro PA: Oxidative damage is a potential
cause of cone cell death in retinitis pigmentosa. J Cell Physiol.
203:457–464. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Campochiaro PA and Mir TA: The mechanism
of cone cell death in retinitis pigmentosa. Prog Retin Eye Res.
62:24–37. 2018. View Article : Google Scholar
|
|
109
|
Komeima K, Rogers BS, Lu L and Campochiaro
PA: Antioxidants reduce cone cell death in a model of retinitis
pigmentosa. Proc Natl Acad Sci USA. 103:11300–11305. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Piano I, D'Antongiovanni V, Testai L,
Calderone V and Gargini C: A nutraceutical strategy to slowing down
the progression of cone death in an animal model of retinitis
pigmentosa. Front Neurosci. 13:4612019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Venkatesh A, Cheng SY and Punzo C: Loss of
the cone-enriched caspase-7 does not affect secondary cone death in
retinitis pigmentosa. Mol Vis. 23:944–951. 2017.
|
|
112
|
Falasconi A, Biagioni M, Novelli E, Piano
I, Gargini C and Strettoi E: Retinal phenotype in the rd9 mutant
mouse, a model of X-linked RP. Front Neurosci. 13:9912019.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Gumerson JD, Alsufyani A, Yu W, Lei J, Sun
X, Dong L, Wu Z and Li T: Restoration of RPGR expression in vivo
using CRISPR/Cas9 gene editing. Gene Ther. 29:81–93. 2022.
View Article : Google Scholar :
|
|
114
|
Napoli D, Biagioni M, Billeri F, Di Marco
B, Orsini N, Novelli E and Strettoi E: Retinal pigment epithelium
remodeling in mouse models of retinitis pigmentosa. Int J Mol Sci.
22:53812021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Slijkerman RW, Song F, Astuti GD, Huynen
MA, van Wijk E, Stieger K and Collin RW: The pros and cons of
vertebrate animal models for functional and therapeutic research on
inherited retinal dystrophies. Prog Retin Eye Res. 48:137–159.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhang X, Shahani U, Reilly J and Shu X:
Disease mechanisms and neuroprotection by tauroursodeoxycholic acid
in Rpgr knockout mice. J Cell Physiol. 234:18801–18812. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Shu X, Zeng Z, Gautier P, Lennon A,
Gakovic M, Patton EE and Wright AF: Zebrafish Rpgr is required for
normal retinal development and plays a role in dynein-based
retrograde transport processes. Hum Mol Genet. 19:657–670. 2010.
View Article : Google Scholar
|
|
118
|
Tee JJ, Smith AJ, Hardcastle AJ and
Michaelides M: RPGR-associated retinopathy: Clinical features,
molecular genetics, animal models and therapeutic options. Br J
Ophthalmol. 100:1022–1027. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Mansouri V: X-Linked retinitis pigmentosa
gene therapy: Preclinical aspects. Ophthalmol Ther. 12:7–34. 2023.
View Article : Google Scholar
|
|
120
|
Raghupathy RK, McCulloch DL, Akhtar S,
Al-mubrad TM and Shu X: Zebrafish model for the genetic basis of
X-linked retinitis pigmentosa. Zebrafish. 10:62–69. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Liu F, Chen J, Yu S, Raghupathy RK, Liu X,
Qin Y, Li C, Huang M, Liao S, Wang J, et al: Knockout of RP2
decreases GRK1 and rod transducin subunits and leads to
photoreceptor degeneration in zebrafish. Hum Mol Genet.
24:4648–4659. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Beltran WA, Cideciyan AV, Lewin AS, Iwabe
S, Khanna H, Sumaroka A, Chiodo VA, Fajardo DS, Román AJ, Deng WT,
et al: Gene therapy rescues photoreceptor blindness in dogs and
paves the way for treating human X-linked retinitis pigmentosa.
Proc Natl Acad Sci USA. 109:2132–2137. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Song C, Dufour VL, Cideciyan AV, Ye GJ,
Swider M, Newmark JA, Timmers AM, Robinson PM, Knop DR, Chulay JD,
et al: Dose range finding studies with two RPGR transgenes in a
Canine model of X-Linked retinitis pigmentosa treated with
subretinal gene therapy. Hum Gene Ther. 31:743–755. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Huckenpahler AL, Carroll J, Salmon AE,
Sajdak BS, Mastey RR, Allen KP, Kaplan HJ and McCall MA:
Noninvasive imaging and correlative histology of cone photoreceptor
structure in the pig retina = Dose range finding studies with two
RPGR transgenes in a Canine model of X-Linked retinitis pigmentosa
treated with subretinal gene therapy. Transl Vis Sci Technol.
8:382019. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Mussolino C, della Corte M, Rossi S, Viola
F, Di Vicino U, Marrocco E, Neglia S, Doria M, Testa F, Giovannoni
R, et al: AAV-mediated photoreceptor transduction of the pig
cone-enriched retina. Gene Ther. 18:637–645. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Yin L, Greenberg K, Hunter JJ, Dalkara D,
Kolstad KD, Masella BD, Wolfe R, Visel M, Stone D, Libby RT, et al:
Intravitreal injection of AAV2 transduces macaque inner retina.
Invest Ophthalmol Vis Sci. 52:2775–2783. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Dacey DM: Primate retina: Cell types,
circuits and color opponency. Prog Retin Eye Res. 18:737–763. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Fujii R, Matsushita M, Itani Y, Hama A,
Natsume T and Takamatsu H: Intravitreal administration of
avacincaptad pegol in a nonhuman primate model of Dry Age-related
macular degeneration. Pharmacol Res Perspect. 13:e700522025.
View Article : Google Scholar
|
|
129
|
Jia X, Yu Z, Wu J, Hou S, Du Y, Zhu Y, Li
Z, Tu S, Zhao L, Su W, et al: Discovery of clinical manifestations
in spontaneous glaucoma suspect nonhuman primates. Ophthalmic Res.
66:1406–1416. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Ardon M, Nguyen L, Chen R, Rogers J, Stout
T, Thomasy S and Moshiri A: Onset and progression of disease in
nonhuman primates with PDE6C cone disorder. Invest Ophthalmol Vis
Sci. 65:162024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Bremmer F: Multisensory space: From
eye-movements to self-motion. J Physiol. 589:815–823. 2011.
View Article : Google Scholar :
|
|
132
|
Dominik Fischer M, Zobor D, Keliris GA,
Shao Y, Seeliger MW, Haverkamp S, Jägle H, Logothetis NK and
Smirnakis SM: Detailed functional and structural characterization
of a macular lesion in a rhesus macaque. Doc Ophthalmol.
125:179–194. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Mukherjee PK, Marcheselli VL, Serhan CN
and Bazan NG: Neuroprotectin D1: A docosahexaenoic acid-derived
docosatriene protects human retinal pigment epithelial cells from
oxidative stress. Proc Natl Acad Sci USA. 101:8491–8496. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Bazan NG: Neuroprotectin D1 (NPD1): A
DHA-derived mediator that protects brain and retina against cell
injury-induced oxidative stress. Brain Pathol. 15:159–166. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Jain N, Maguire MG, Flaxel CJ, Kim SJ,
Patel S, Smith JR, Weng CY, Kim LA and Yeh S: Dietary
supplementation for retinitis pigmentosa: A report by the american
academy of ophthalmology. Ophthalmology. 132:354–367. 2025.
View Article : Google Scholar
|
|
136
|
Mukherjee PK, Marcheselli VL, Barreiro S,
Hu J, Bok D and Bazan NG: Neurotrophins enhance retinal pigment
epithelial cell survival through neuroprotectin D1 signaling. Proc
Natl Acad Sci USA. 104:13152–13157. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Zhong M, Kawaguchi R, Kassai M and Sun H:
Retina, retinol, retinal and the natural history of vitamin A as a
light sensor. Nutrients. 4:2069–2096. 2012. View Article : Google Scholar
|
|
138
|
Hughbanks-Wheaton DK, Birch DG, Fish GE,
Spencer R, Pearson NS, Takacs A and Hoffman DR: Safety assessment
of docosahexaenoic acid in X-linked retinitis pigmentosa: The
4-year DHAX trial. Invest Ophthalmol Vis Sci. 55:4958–4966. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Hoffman DR, Hughbanks-Wheaton DK, Pearson
NS, Fish GE, Spencer R, Takacs A, Klein M, Locke KG and Birch DG:
Four-year placebo-controlled trial of docosahexaenoic acid in
X-linked retinitis pigmentosa (DHAX trial): A randomized clinical
trial. JAMA Ophthalmol. 132:866–873. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Lin JB, Murakami Y, Miller JW and Vavvas
DG: Neuroprotection for Age-related macular degeneration.
Ophthalmol Sci. 2:1001922022. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Fudalej E, Justyniarska M, Kasarełło K,
Dziedziak J, Szaflik JP and Cudnoch-Jędrzejewska A: Neuroprotective
factors of the retina and their role in promoting survival of
retinal ganglion cells: A review. Ophthalmic Res. 64:345–355. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Ou K, Li Y, Liu L, Li H, Cox K, Wu J, Liu
J and Dick AD: Recent developments of neuroprotective agents for
degenerative retinal disorders. Neural Regen Res. 17:1919–1928.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Huang K, Deng H, Wang S, Zhang F, Huang G,
Wang L, Liu J, Zhao X, Ren H, Yang G, et al: Melanin-like
nanomedicine functions as a novel RPE ferroptosis inhibitor to
ameliorate retinal degeneration and visual impairment in Dry
Age-related macular degeneration. Adv Healthc Mater.
13:e24016132024. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Wang J, Zhao J, Cui X, Mysona BA, Navneet
S, Saul A, Ahuja M, Lambert N, Gazaryan IG, Thomas B, et al: The
molecular chaperone sigma 1 receptor mediates rescue of retinal
cone photoreceptor cells via modulation of NRF2. Free Radic Biol
Med. 134:604–616. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Zou M, Ke Q, Nie Q, Qi R, Zhu X, Liu W, Hu
X, Sun Q, Fu JL, Tang X, et al: Inhibition of cGAS-STING by JQ1
alleviates oxidative stress-induced retina inflammation and
degeneration. Cell Death Differ. 29:1816–1833. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zhu X, Liu W, Tang X, Chen Y, Ge X, Ke Q,
Liang X, Gan Y, Zheng Y, Zou M, et al: The BET PROTAC inhibitor
dBET6 protects against retinal degeneration and inhibits the
cGAS-STING in response to light damage. J Neuroinflammation.
20:1192023. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Qin H, Xu W and Yao K: CRISPR-based genome
editing in disease treatment. Trends Mol Med. 29:673–674. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Dasgupta I, Flotte TR and Keeler AM:
CRISPR/Cas-dependent and Nuclease-free in vivo therapeutic gene
editing. Hum Gene Ther. 32:275–293. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Pulman J, Sahel JA and Dalkara D: New
editing tools for gene therapy in inherited retinal dystrophies.
Crispr J. 5:377–388. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Xu F, Zheng C, Xu W, Zhang S, Liu S, Chen
X and Yao K: Breaking genetic shackles: The advance of base editing
in genetic disorder treatment. Front Pharmacol. 15:13641352024.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Fu X, Feng S, Qin H, Yan L, Zheng C and
Yao K: Microglia: The breakthrough to treat neovascularization and
repair blood-retinal barrier in retinopathy. Front Mol Neurosci.
16:11002542023. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Campa C, Gallenga CE, Bolletta E and Perri
P: The role of gene therapy in the treatment of retinal diseases: A
review. Curr Gene Ther. 17:194–213. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Xu W, Zhang S, Qin H and Yao K: From bench
to bedside: Cutting-edge applications of base editing and prime
editing in precision medicine. J Transl Med. 22:11332024.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Xia X and Guo X: Adeno-associated virus
vectors for retinal gene therapy in basic research and clinical
studies. Front Med (Lausanne). 10:13100502023. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Lykken EA, Shyng C, Edwards RJ, Rozenberg
A and Gray SJ: Recent progress and considerations for AAV gene
therapies targeting the central nervous system. J Neurodev Disord.
10:162018. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Wiley LA, Boyce TM, Meyering EE, Ochoa D,
Sheehan KM, Stone EM, Mullins RF, Tucker BA and Han IC: The degree
of Adeno-Associated Virus-induced retinal inflammation varies based
on serotype and route of delivery: Intravitreal, subretinal, or
suprachoroidal. Hum Gene Ther. 34:530–539. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Puertas-Neyra K, Usategui-Martín R, Coco
RM and Fernandez-Bueno I: Intravitreal stem cell paracrine
properties as a potential neuroprotective therapy for retinal
photoreceptor neurodegenerative diseases. Neural Regen Res.
15:1631–1638. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Giacalone JC, Andorf JL, Zhang Q, Burnight
ER, Ochoa D, Reutzel AJ, Collins MM, Sheffield VC, Mullins RF, Han
IC, et al: Development of a molecularly stable gene therapy vector
for the treatment of RPGR-Associated X-Linked retinitis pigmentosa.
Hum Gene Ther. 30:967–974. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Hong DH, Pawlyk BS, Adamian M, Sandberg MA
and Li T: A single, abbreviated RPGR-ORF15 variant reconstitutes
RPGR function in vivo. Invest Ophthalmol Vis Sci. 46:435–441. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Wang JH, Zhan W, Gallagher TL and Gao G:
Recombinant adeno-associated virus as a delivery platform for
ocular gene therapy: A comprehensive review. Mol Ther.
32:4185–4207. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Trapani I, Colella P, Sommella A, Iodice
C, Cesi G, de Simone S, Marrocco E, Rossi S, Giunti M, Palfi A, et
al: Effective delivery of large genes to the retina by dual AAV
vectors. EMBO Mol Med. 6:194–211. 2014. View Article : Google Scholar :
|
|
162
|
Ford JL, Karatza E, Mody H, Nagaraja
Shastri P, Khajeh Pour S, Yang TY, Swanson M, Chao D and Devineni
D: Clinical pharmacology perspective on development of
Adeno-associated virus Vector-based retina gene therapy. Clin
Pharmacol Ther. 115:1212–1232. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Mendell JR, Al-Zaidy SA, Rodino-Klapac LR,
Goodspeed K, Gray SJ, Kay CN, Boye SL, Boye SE, George LA,
Salabarria S, et al: Current clinical applications of in vivo gene
therapy with AAVs. Mol Ther. 29:464–488. 2021. View Article : Google Scholar
|
|
164
|
Alsalloum A, Gornostal E, Mingaleva N,
Pavlov R, Kuznetsova E, Antonova E, Nadzhafova A, Kolotova D,
Kadyshev V, Mityaeva O, et al: A Comparative analysis of models for
AAV-Mediated gene therapy for inherited retinal diseases. Cells.
13:17062024. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Naldini L: Gene therapy returns to centre
stage. Nature. 526:351–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Fuller-Carter PI, Basiri H, Harvey AR and
Carvalho LS: Focused update on AAV-Based gene therapy clinical
trials for inherited retinal degeneration. BioDrugs. 34:763–781.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Narfström K, Katz ML, Bragadottir R,
Seeliger M, Boulanger A, Redmond TM, Caro L, Lai CM and Rakoczy PE:
Functional and structural recovery of the retina after gene therapy
in the RPE65 null mutation dog. Invest Ophthalmol Vis Sci.
44:1663–1672. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Smith AJ, Schlichtenbrede FC, Tschernutter
M, Bainbridge JW, Thrasher AJ and Ali RR: AAV-Mediated gene
transfer slows photoreceptor loss in the RCS rat model of retinitis
pigmentosa. Mol Ther. 8:188–195. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Kansara V, Muya L, Wan CR and Ciulla TA:
Suprachoroidal delivery of viral and nonviral gene therapy for
retinal diseases. J Ocul Pharmacol Ther. 36:384–392. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Biber J, Gandor C, Becirovic E and
Michalakis S: Retina-directed gene therapy: Achievements and
remaining challenges. Pharmacol Ther. 271:1088622025. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
McDonald A and Wijnholds J: Retinal
ciliopathies and potential gene therapies: A focus on human
iPSC-derived organoid models. Int J Mol Sci. 25:28872024.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Pavlou M, Schön C, Occelli LM, Rossi A,
Meumann N, Boyd RF, Bartoe JT, Siedlecki J, Gerhardt MJ, Babutzka
S, et al: Novel AAV capsids for intravitreal gene therapy of
photoreceptor disorders. EMBO Mol Med. 13:e133922021. View Article : Google Scholar :
|
|
173
|
Hu S, Du J, Chen N, Jia R, Zhang J, Liu X
and Yang L: In vivo CR ISPR/Cas9-mediated genome editing mitigates
photoreceptor degeneration in a mouse model of X-Linked retinitis
pigmentosa. Invest Ophthalmol Vis Sci. 61:312020. View Article : Google Scholar
|
|
174
|
Fu Y, He X, Ma L, Gao XD, Liu P, Shi H,
Chai P, Ge S, Jia R, Liu DR, et al: In vivo prime editing rescues
photoreceptor degeneration in nonsense mutant retinitis pigmentosa.
Nat Commun. 16:23942025. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Wu Z, Hiriyanna S, Qian H, Mookherjee S,
Campos MM, Gao C, Fariss R, Sieving PA, Li T, Colosi P, et al: A
long-term efficacy study of gene replacement therapy for
RPGR-associated retinal degeneration. Hum Mol Genet. 24:3956–3970.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Chahine Karam F, Loi TH, Ma A, Nash BM,
Grigg JR, Parekh D, Riley LG, Farnsworth E, Bennetts B,
Gonzalez-Cordero A, et al: Human iPSC-derived retinal organoids and
retinal pigment epithelium for novel intronic RPGR variant
assessment for therapy suitability. J Pers Med. 12:5022022.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Deng WL, Gao ML, Lei XL, Lv JN, Zhao H, He
KW, Xia XX, Li LY, Chen YC, Li YP, et al: Gene correction reverses
ciliopathy and photoreceptor loss in iPSC-Derived retinal organoids
from retinitis pigmentosa patients. Stem Cell Reports.
10:1267–1281. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Bassuk AG, Zheng A, Li Y, Tsang SH and
Mahajan VB: Precision medicine: Genetic repair of retinitis
pigmentosa in patient-derived stem cells. Sci Rep. 6:199692016.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Li T, Ma Y, Cheng Y, Zhao Y, Qiu Z, Liu H,
Zhang D, Wu J, Li J, Zhang S, et al: Single-cell transcriptomic
dataset of RPGR-associated retinitis pigmentosa patient-derived
retinal organoids. Sci Data. 11:12852024. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Lin B, McLelland BT, Aramant RB, Thomas
BB, Nistor G, Keirstead HS and Seiler MJ: Retina organoid
transplants develop photoreceptors and improve visual function in
RCS rats with RPE dysfunction. Invest Ophthalmol Vis Sci.
61:342020. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
West EL, Majumder P, Naeem A, Fernando M,
O'Hara-Wright M, Lanning E, Kloc M, Ribeiro J, Ovando-Roche P, Shum
IO, et al: Antioxidant and lipid supplementation improve the
development of photoreceptor outer segments in pluripotent stem
cell-derived retinal organoids. Stem Cell Reports. 17:775–788.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Scherbakova I, Ragi SD and Sharma T:
Ocular injection techniques for retinitis pigmentosa: Intravitreal,
subretinal, and suprachoroidal. Methods Mol Biol. 2560:375–392.
2023. View Article : Google Scholar
|
|
183
|
Kovacs KD, Ciulla TA and Kiss S:
Advancements in ocular gene therapy delivery: Vectors and
subretinal, intravitreal, and suprachoroidal techniques. Expert
Opin Biol Ther. 22:1193–1208. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Mackenbrock LHB, Auffarth GU, Albrecht M,
Naujokaitis T, Kessler LJ, Mayer CS and Khoramnia R: Anterior
segment complications following intravitreal injection. Klin Monbl
Augenheilkd. 241:917–922. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Zhao CS, Chwialkowski K, Wai KM,
Mruthyunjaya P, Rahimy E and Koo EB: Risk of cataract surgery
complications in patients with prior intravitreal injection
therapy. Am J Ophthalmol. 272:106–116. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Prado DA, Acosta-Acero M and Maldonado RS:
Gene therapy beyond luxturna: A new horizon of the treatment for
inherited retinal disease. Curr Opin Ophthalmol. 31:147–154. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Takahashi K, Morizane Y, Hisatomi T,
Tachibana T, Kimura S, Hosokawa MM, Shiode Y, Hirano M, Doi S,
Toshima S, et al: The influence of subretinal injection pressure on
the microstructure of the monkey retina. PLoS One. 13:e02099962018.
View Article : Google Scholar
|
|
188
|
Dufour VL, Cideciyan AV, Ye GJ, Song C,
Timmers A, Habecker PL, Pan W, Weinstein NM, Swider M, Durham AC,
et al: Toxicity and efficacy evaluation of an Adeno-associated
virus vector expressing Codon-optimized RPGR delivered by
subretinal injection in a Canine model of X-linked retinitis
pigmentosa. Hum Gene Ther. 31:253–267. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Qi Y, Dai X, Zhang H, He Y, Zhang Y, Han
J, Zhu P, Zhang Y, Zheng Q, Li X, et al: Trans-corneal subretinal
injection in mice and its effect on the function and morphology of
the retina. PLoS One. 10:e01365232015. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Peng Y, Tang L and Zhou Y: Subretinal
injection: A review on the novel route of therapeutic delivery for
vitreoretinal diseases. Ophthalmic Res. 58:217–226. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Tripepi D, Jalil A, Ally N, Buzzi M,
Moussa G, Rothschild PR, Rossi T, Ferrara M and Romano MR: The role
of subretinal injection in ophthalmic surgery: Therapeutic agent
delivery and other indications. Int J Mol Sci. 24:105352023.
View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Wu KY, Fujioka JK, Gholamian T, Zaharia M
and Tran SD: Suprachoroidal injection: A novel approach for
targeted drug delivery. Pharmaceuticals (Basel). 16:12412023.
View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Rahman S, Tayyab H and Siddiqui MAR:
Suprachoroidal triamcinolone acetonide injection to treat macular
edema: A Review. J Vitreoretin Dis. 8:699–708. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Han IC, Cheng JL, Burnight ER, Ralston CL,
Fick JL, Thomsen GJ, Tovar EF, Russell SR, Sohn EH, Mullins RF, et
al: Retinal tropism and transduction of Adeno-Associated virus
varies by serotype and route of delivery (Intravitreal, Subretinal,
or Suprachoroidal) in rats. Hum Gene Ther. 31:1288–1299. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Wu KY, Gao A, Giunta M and Tran SD: What's
new in ocular drug delivery: Advances in suprachoroidal injection
since 2023. Pharmaceuticals (Basel). 17:10072024. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Jung JH, Chiang B, Grossniklaus HE and
Prausnitz MR: Ocular drug delivery targeted by iontophoresis in the
suprachoroidal space using a microneedle. J Control Release.
277:14–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Gagnon NA, Hartley C and Gilger BC:
Efficacy and safety of suprachoroidal triamcinolone injection in
horses with poorly responsive equine recurrent uveitis. Vet
Ophthalmol. 24:308–312. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Lee H and Lotery A: Gene therapy for
RPE65-mediated inherited retinal dystrophy completes phase 3.
Lancet. 390:823–824. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Zu H and Gao D: Non-viral vectors in gene
therapy: Recent development, challenges, and prospects. AAPS J.
23:782021. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Wang C, Pan C, Yong H, Wang F, Bo T, Zhao
Y, Ma B, He W and Li M: Emerging non-viral vectors for gene
delivery. J Nanobiotechnology. 21:2722023. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Ren S, Wang M, Wang C, Wang Y, Sun C, Zeng
Z, Cui H and Zhao X: Application of Non-viral vectors in drug
delivery and gene therapy. Polymers (Basel). 13:33072021.
View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Chapa González C, Martínez Saráoz JV,
Roacho Pérez JA and Olivas Armendáriz I: Lipid nanoparticles for
gene therapy in ocular diseases. Daru. 31:75–82. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Carvalho C, Lemos L, Antas P and Seabra
MC: Gene therapy for inherited retinal diseases: Exploiting new
tools in genome editing and nanotechnology. Front Ophthalmol
(Lausanne). 3:12705612023. View Article : Google Scholar
|
|
204
|
Schnichels S, Simmang D, Löscher M,
Herrmann A, de Vries JW, Spitzer MS and Hurst J: Lipid-DNA
nanoparticles as Drug-delivery vehicles for the treatment of
retinal diseases. Pharmaceutics. 15:5322023. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Baig MS, Karade SK, Ahmad A, Khan MA,
Haque A, Webster TJ, Faiyazuddin M and Al-Qahtani NH: Lipid-based
nanoparticles: Innovations in ocular drug delivery. Front Mol
Biosci. 11:14219592024. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Gugleva V and Andonova V: Recent progress
of solid lipid nanoparticles and nanostructured lipid carriers as
ocular drug delivery platforms. Pharmaceuticals (Basel).
16:4742023. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Lam BL, Scholl HPN, Doub D, Sperling M,
Hashim M and Li N: A systematic literature review of disease
progression reported in RPGR-associated X-linked retinitis
pigmentosa. Retina. 44:1–9. 2024. View Article : Google Scholar
|
|
208
|
Zada M, Cornish EE, Fraser CL, Jamieson RV
and Grigg JR: Natural history and clinical biomarkers of
progression in X-linked retinitis pigmentosa: A systematic review.
Acta Ophthalmol. 99:499–510. 2021. View Article : Google Scholar
|
|
209
|
Christou EE, Josan AS, Cehajic-Kapetanovic
J and MacLaren RE: Establishing clinical trial endpoints in
selecting patients for RPGR retinal gene therapy. Transl Vis Sci
Technol. 13:182024. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Birch DG, Locke KG, Felius J, Klein M,
Wheaton DK, Hoffman DR and Hood DC: Rates of decline in regions of
the visual field defined by frequency-domain optical coherence
tomography in patients with RPGR-mediated X-linked retinitis
pigmentosa. Ophthalmology. 122:833–839. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Stephenson KAJ, Tse T, Hwang J, Kavetskyi
A, Dhanji SR, Kolawole OU, Gregory-Evans CY, Pakzad-Vaezi K, Mammo
ZN, Gregory-Evans K, et al: Quantitative choroidal analysis of
molecularly characterized retinitis pigmentosa. Invest Ophthalmol
Vis Sci. 66:112025. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Iftikhar M, Lemus M, Usmani B, Campochiaro
PA, Sahel JA, Scholl HPN and Shah SMA: Classification of disease
severity in retinitis pigmentosa. Br J Ophthalmol. 103:1595–1599.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
De Silva SR, Arno G, Robson AG, Fakin A,
Pontikos N, Mohamed MD, Bird AC, Moore AT, Michaelides M, Webster
AR, et al: The X-linked retinopathies: Physiological insights,
pathogenic mechanisms, phenotypic features and novel therapies.
Prog Retin Eye Res. 82:1008982021. View Article : Google Scholar
|
|
214
|
Ghoraba HH, Akhavanrezayat A, Karaca I,
Yavari N, Lajevardi S, Hwang J, Regenold J, Matsumiya W, Pham B,
Zaidi M, et al: Ocular gene therapy: A Literature review with
special focus on immune and inflammatory responses. Clin
Ophthalmol. 16:1753–1771. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Hugi F, Vollmer J, Renaud L and Machacek
M: A Semimechanistic ocular pharmacokinetic model for ADVM-022 gene
therapy describing the Dose-exposure relationship in monkeys and
the scaling to human. Mol Pharm. 22:4612–4623. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Martinez-Fernandez de la Camara C,
Cehajic-Kapetanovic J and MacLaren RE: Emerging gene therapy
products for RPGR-associated X-linked retinitis pigmentosa. Expert
Opin Emerg Drugs. 27:431–443. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Yang P, Birch D, Lauer A, Sisk R, Anand R,
Pennesi ME, Iannaccone A, Yaghy A, Scaria A, Jung JA, et al:
Subretinal gene therapy drug AGTC-501 for XLRP Phase 1/2
multicenter study (HORIZON): 24-month safety and efficacy results.
Am J Ophthalmol. 271:268–285. 2025. View Article : Google Scholar
|
|
218
|
Michaelides M, Besirli CG, Yang Y, DE
Guimaraes TAC, Wong SC, Huckfeldt RM, Comander JI, Sahel JA, Shah
SM, Tee JJL, et al: Phase 1/2 AAV5-hRKp.RPGR (Botaretigene
Sparoparvovec) gene therapy: Safety and efficacy in RPGR-associated
X-linked retinitis pigmentosa. Am J Ophthalmol. 267:122–134. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Ren D, Fisson S, Dalkara D and Ail D:
Immune responses to gene editing by viral and Non-viral delivery
vectors used in retinal gene therapy. Pharmaceutics. 14:19732022.
View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Casey GA, Papp KM and MacDonald IM: Ocular
gene therapy with Adeno-associated virus vectors: Current outlook
for patients and researchers. J Ophthalmic Vis Res. 15:396–399.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Butterfield GL, Reisman SJ, Iglesias N and
Gersbach CA: Gene regulation technologies for gene and cell
therapy. Mol Ther. 33:2104–2122. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Beltran WA, Cideciyan AV, Iwabe S, Swider
M, Kosyk MS, McDaid K, Martynyuk I, Ying GS, Shaffer J, Deng WT, et
al: Successful arrest of photoreceptor and vision loss expands the
therapeutic window of retinal gene therapy to later stages of
disease. Proc Natl Acad Sci USA. 112:E5844–E5853. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Beltran WA, Cideciyan AV, Boye SE, Ye GJ,
Iwabe S, Dufour VL, Marinho LF, Swider M, Kosyk MS, Sha J, et al:
Optimization of retinal gene therapy for X-Linked retinitis
pigmentosa due to RPGR mutations. Mol Ther. 25:1866–1880. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Ren M, Chen X, Gao P, Huang Y, Yu S,
Reilly J, Sun K, Han Y, Hu H, Li P, et al: RPGRORF15 mutations
disrupt lysosomal lipid metabolism in retinal pigment epithelium
cells and cause retinitis pigmentosa. Invest Ophthalmol Vis Sci.
66:612025. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Far BF, Akbari M, Habibi MA, Katavand M
and Nasseri S: CRISPR technology in disease management: An updated
review of clinical translation and therapeutic potential. Cell
Prolif. 58:e700992025. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
von Krusenstiern L, Liu J, Liao E, Gow JA,
Chen G, Ong T, Lotery AJ, Jalil A, Lam BL and MacLaren RE; XIRIUS
Part 1 Study GroupXOLARIS Study Group: Changes in retinal
sensitivity associated with cotoretigene toliparvovec in X-Linked
retinitis pigmentosa with RPGR gene variations. JAMA Ophthalmol.
141:275–283. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Ling S, Zhang X, Dai Y, Jiang Z, Zhou X,
Lu S, Qian X, Liu J, Selfjord N, Satir TM, et al: Customizable
virus-like particles deliver CRISPR-Cas9 ribonucleoprotein for
effective ocular neovascular and Huntington's disease gene therapy.
Nat Nanotechnol. 20:543–553. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Banou L, Sarrafpour S, Teng CC and Liu J:
Ocular gene therapy: An overview of viral vectors, immune
responses, and future directions. Yale J Biol Med. 97:491–503.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Jony MJ, Joshi A, Dash A and Shukla S:
Non-Viral delivery systems to transport nucleic acids for inherited
retinal disorders. Pharmaceuticals (Basel). 18:872025. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Lim Y, Campochiaro PA and Green JJ:
Suprachoroidal delivery of viral and nonviral vectors for treatment
of retinal and choroidal vascular diseases. Am J Ophthalmol.
277:518–533. 2025. View Article : Google Scholar
|
|
231
|
Wu H, Dong L, Jin S, Zhao Y and Zhu L:
Innovative gene delivery systems for retinal disease therapy.
Neural Regen Res. 21:542–552. 2026. View Article : Google Scholar
|
|
232
|
Sultana S, Yusuf M and Sharma V:
Nanovesicular drug delivery systems for rare ocular diseases:
Advances, challenges, and future directions. AAPS PharmSciTech.
26:1972025. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Adewale OB, Davids H, Cairncross L and
Roux S: Toxicological behavior of gold nanoparticles on various
models: Influence of physicochemical properties and other factors.
Int J Toxicol. 38:357–384. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Chis AA, Dobrea CM, Rus LL, Frum A,
Morgovan C, Butuca A, Totan M, Juncan AM, Gligor FG and Arseniu AM:
Dendrimers as Non-viral vectors in gene-directed enzyme prodrug
therapy. Molecules. 26:59762021. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Galluzzi F, Francia S, Cupini S, Gianiorio
T, Mantero G, DiFrancesco ML, Ravasenga T, Jasnoor, Attanasio M,
Maya-Vetencourt JF, et al: Graphene oxide increases the
phototransduction efficiency of copolymeric nanoimplants and
rescues visual functions in rat and pig models of Retinitis
pigmentosa. Nat Commun. 16:87212025. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Szabó V, Varsányi B, Barboni M, Takács Á,
Knézy K, Molnár MJ, Nagy ZZ, György B and Rivolta C: Insights into
eye genetics and recent advances in ocular gene therapy. Mol Cell
Probes. 79:1020082025. View Article : Google Scholar : PubMed/NCBI
|