The liver orchestrates its vital functions through
intricate cellular crosstalk among diverse cell populations
(1). Hepatic stellate cells
(HSCs), a specialized lineage of mesenchymal cells, are
strategically positioned within the perisinusoidal space (space of
Disse), forming critical anatomical interfaces between liver
sinusoidal endothelial cells (LSECs) and hepatocyte cords (2-4).
As key constituents of the non-parenchymal cell (NPC) compartment
of the liver, comprising approximately one-third of NPCs in
homeostasis (5,6), HSCs functionally interact with
resident macrophages, LSECs, Kupffer cells (KCs), portal
fibroblasts and recruited immune cells to regulate hepatic
pathophysiology (1,7). This cellular interplay positions
HSCs as central orchestrators of tissue responses during both liver
injury and regeneration (8,9).
In a quiescent state, HSCs maintain hepatic
homeostasis through vitamin A (VitA) storage and metabolism,
immunomodulatory signaling, and the paracrine secretion of
cytokines, growth factors and apolipoproteins (6,10,11). Following liver injury,
mesenchymal cell activation drives pathogenic extracellular matrix
(ECM) deposition, with transdifferentiated myofibroblasts serving
as the principal ECM producers (12,13). While multiple cellular sources,
including portal fibroblasts and bone marrow-derived progenitors,
contribute to the myofibroblast pool (14,15), lineage-tracing studies
unequivocally identify HSCs as the dominant progenitors in the
majority of liver fibrogenic contexts (16,17).
Conventional paradigms have portrayed HSCs as a
functionally homogeneous population uniformly transitioning into
profibrotic myofibroblasts (18,19). Acute injury triggers HSC
activation through paracrine signals from neighboring cells,
initiating transient myofibroblastic differentiation to support
ECM-mediated tissue repair (6).
By contrast, chronic injury leads to sustained HSC activation,
resulting in pathological ECM accumulation and architectural
distortion (20). Emerging
evidence indicates that HSCs exhibit dynamic activation states
beyond simple binary (quiescent vs. activated) classification.
While activated HSCs (aHSCs) predominantly drive fibrogenesis,
specific subpopulations demonstrate paradoxical anti-fibrotic or
hepatoprotective functions (21). This functional diversity
underscores the inherent heterogeneity and phenotypic plasticity of
HSCs across disease phases.
The resolution of single-cell RNA sequencing
(scRNA-seq) has revolutionized our understanding of mesenchymal
cell diversity (22,23). High-dimensional analyses reveal
spatially and temporally restricted HSC subpopulations in both
healthy and diseased livers, each exhibiting unique transcriptional
programs and functional specializations during fibrogenesis,
immunomodulation and tissue regeneration (24). These discoveries not only
redefine HSC biology, but also unveil novel therapeutic
opportunities for the precision targeting of pathogenic HSC
subsets, while preserving their reparative functions (25,26).
The present review synthesizes current insights into
HSC heterogeneity, delineates context-dependent phenotypic switches
driven by niche-specific cues and metabolic reprogramming, and
discusses emerging strategies that can be used to therapeutically
target HSC subpopulations in liver diseases.
By integrating marker-based analyses, single-cell
sequencing and spatial transcriptomic techniques, the multilayered
heterogeneity of HSCs across transcriptional, functional and
spatial dimensions has been revealed (27). Collectively, these perspectives
converge to portray HSCs as context-dependent regulators whose
identities vary with analytical scale and tissue niche (Table I). Single-cell transcriptomic
profiling and lineage tracing have resolved a long-standing
controversy, establishing quiescent HSCs (qHSCs) as progenitors of
injury-induced myofibroblasts across liver diseases (28,29). Pseudotime analyses have revealed
a dynamic continuum in which transitioning HSCs undergo
transcriptional priming, fate bifurcation and context-dependent
functional specialization during fibrogenesis (17). Crucially, this plasticity is
spatially constrained by hepatic zonation, with periportal HSCs
more responsive to inflammatory signals and pericentral HSCs more
attuned to metabolic stress (17,30). Disease etiology further imprints
epigenetic memory on aHSCs, reinforcing pathological feedback
loops. Such multidimensional adaptability positions HSCs as
biomechanical integrators that decode parenchymal damage patterns
into spatially calibrated fibrotic responses, offering therapeutic
entry points for precision anti-fibrotic strategies (Table II).
Previous studies have broadly categorized qHSCs as a
single transcriptional cluster characterized by VitA storage and
homeostatic functions (19,31-33). These cells are molecularly
defined by the high expression of lecithin retinol acyltransferase
(LRAT), a canonical marker of VitA metabolism, alongside
quiescence-associated genes, such as peroxisome
proliferator-activated receptor γ (PPARγ) and adipogenic genes,
such as perilipin 2 and glial fibrillary acidic protein (GFAP),
that enforce their dormant state (22,34). The maintenance of this inactive
phenotype further involves stellate-specific markers, such as Reln
and Ecm1, which regulate ECM interactions and signaling quiescence
(35,36). Transcriptomically, qHSCs marked
by nerve growth factor receptor (Ngfr), LRAT, and ADAMTS-like
protein 2 (Adamtsl2) serve as the precursor pool for aHSCs,
predominantly functioning as VitA reservoirs and homeostatic
regulators in uninjured livers (17).
Under physiological conditions, the liver lobule is
divided into three functional zones, namely the periportal (zone
1), midlobular (zone 2) and pericentral (zone 3). Each zone is
characterized by distinct oxygen tension, metabolite concentrations
and signaling molecules (37).
Building on this spatial framework, recent scRNA-seq and spatial
transcriptomic analyses have redefined zonally restricted qHSC
subpopulations. qHSCs bifurcate into portal vein-associated HSC
(PaHSC, Ngfrhigh) and central vein-associated HSC
(CaHSC, Adamtsl2high) subpopulations (30,38,39). This zonation aligns with lobular
gradients of oxygen, nutrients and signaling molecules, shaping HSC
functional identities with PaHSCs primed for pro-fibrotic
activation, while CaHSCs are more associated with detoxification
(26).
Notably, a pre-specified qHSC subtype, termed
1-HSCs, has recently been identified in zone 1 of the healthy liver
lobule (40). Although
functionally quiescent under homeostatic conditions, 1-HSCs are
spatially localized and poised for activation upon injury. Unlike
conventional myofibroblast precursors, 1-HSCs primarily orchestrate
sinusoidal capillarization in response to damage, revealing a
reparative trajectory distinct from classical fibrogenesis
(40). The identification of
1-HSCs further reinforces the notion that HSCs constitute a
spatially organized ecosystem, wherein certain subpopulations are
preconfigured for specialized responses despite appearing
phenotypically dormant in steady-state conditions. These
discoveries collectively illustrate that even in homeostasis, HSCs
exist as a functionally partitioned ecosystem, their phenotypic
diversity spatially encoded and primed for context-dependent
responses.
Notably, the functional zonation of HSCs has
identified VitA-enriched and VitA-deficient HSC subsets occupying
distinct lobular niches, with VitA droplet size and distribution
exhibiting zonation-dependent patterns (6,41). While PaHSCs display heightened
VitA storage with desmin expression (42), VitA-poor HSCs near central veins
may act as 'first responders' to injury, transitioning more rapidly
into activated states (43).
Transcriptomic stratification in human livers
further reveals a dichotomy within HSC subpopulations. Glypican
3-expressing HSC1 localize to portal-central vascular regions and
is enriched in elastic fiber-related genes, consistent with
structural maintenance roles. By contrast, dopamine
beta-hydroxylase -high HSC2 distribute diffusely along sinusoids,
displaying antigen presentation pathways suggestive of
immunomodulatory potential (44). Furthermore, developmental
research has identified embryonic HSCs transiently express collagen
type I alpha 1 chain (Col1a1) alongside GFAP and Desmin, but lack α
smooth muscle actin [α-SMA; also known as actin alpha 2 (Acta2)],
mirroring qHSC signatures, while retaining the plasticity required
for later activation (45).
The injury-activated transformation of HSCs reveals
a dynamic spectrum of phenotypic states in which qHSC populations
undergo significant changes that drive their differentiation and
mobilization in response to tissue damage (17). However, despite this
activation-driven depletion, a resilient subset of qHSCs persists
across various disease models. These residual qHSC pools exhibit
unique phenotypic and functional adaptations that enable their
survival and potential contribution to tissue repair and
regeneration (30,46-48). For instance, in carbon
tetrachloride (CCl4)-induced liver fibrosis, a distinct
subset of stress-adapted qHSCs emerges, characterized by the
expression of specific markers, such as betaine-homocysteine
S-methyltransferase and fatty acid binding protein (Fabp)1
(49). This subset appears to
represent a metabolically reprogrammed survival state by altering
their metabolic pathways to adapt to the fibrotic environment,
thereby preserving a reservoir capable of contributing to
regeneration (17,49).
The myofibroblastic aHSCs conventionally
characterized by Acta2, Col1a1 and tissue inhibitor of
metalloproteinases 1 (Timp1) signatures (17,50-52), have been further delineated along
the transitional activation gradients, revealing discrete clusters
from quiescent reservoirs to fully differentiated matrix-producing
myofibroblasts (53). For
example, in early-stage non-alcoholic steatohepatitis (NASH), qHSCs
undergo partial activation, contributing to low-grade fibrosis
through controlled ECM deposition (30). As the disease progresses, a
subset of HSCs acquires a highly profibrogenic phenotype, driven by
pro-inflammatory signals from macrophages and damaged hepatocytes
(30). Indeed, scRNA-seq
indicates that HSC activation encompasses a spectrum of cell states
rather than a uniform transition, with distinct subsets exhibiting
transcriptional programs related to collagen synthesis, immune
modulation, or matrix remodeling (27,54). Additionally, zonally restricted
HSC activation patterns are observed, with PaHSCs exhibiting
heightened responsiveness to cholestatic injury, whereas CaHSCs
dominate in metabolic stress-induced fibrosis (55).
Notably, previous studies have uncovered a third
'inactive' HSC state (iHSCs) across various disease models in both
human and mouse livers. These cells are transcriptionally distinct
from classical qHSCs, exhibiting a low expression of fibrogenic
genes and the upregulation of quiescence-associated genes, but not
adipogenic genes (30,45). As an intermediate population,
iHSCs are closer to qHSCs than to aHSCs, although they do not fully
revert to qHSCs and remain poised for swift reactivation if
fibrogenic stimuli recur (45).
However, compared with terminally differentiated aHSCs, they retain
partial quiescence features with reversible traits, making them a
promising therapeutic target because restoring iHSCs to a quiescent
state is easier than deactivating fully activated myofibroblasts.
This concept has been functionally validated using in vivo
fate-mapping experiments in fibrotic regression models (45).
While α-SMA remains a canonical activation marker,
its restricted expression to discrete HSC subsets in fibrotic
niches underscores the existence of heterogeneous myofibroblast
differentiation states (35,54). A breakthrough came with the
identification of syndecan-4 as a pan-activation marker universally
expressed in diverse injury contexts, mechanistically linked to HSC
migration through integrin signaling and cytoskeletal remodeling
(56).
Of note, HSC activation dynamics differ based on the
underlying pathogenic stimulus. For example, NASH models induce
lipid-associated activation signatures characterized by Fabp4 and
perilipin 2 (Plin2) expression, while cholestatic injury promotes
Twist1-driven ductular reaction phenotypes (32,57,58). Within similar pathologies,
spatially restricted activation programs emerge in liver disease,
with peri-injury HSCs becoming proliferative [platelet-derived
growth factor (PDGF)Rβhigh], while periportal HSCs
adopting inflammatory [C-C motif chemokine ligand 2
(Ccl2)-expressing] states (21).
This phenotypic mosaicism reflects microenvironmental instructive
signals ranging from transforming growth factor β (TGF-β) gradients
to mechanical stiffness thresholds.
The single-cell transcriptomic analyses of fibrotic
liver models has revealed dynamic HSC activation states, ranging
from quiescent VitA-storing cells to fully differentiated
collagen-producing myofibroblasts. The activation cascade begins
with early-responsive HSCs
(Cdc20high/Birc5high) that proliferate
rapidly upon injury, creating an expansion pool for downstream
differentiation (38,55). These cells give rise to a
matrix-remodeling HSC population [Acta2+/regulator of
G-protein signaling 5 (Rgs5)+], which serves as central
executors of fibrogenesis by secreting provisional ECM components,
while simultaneously regulating neighboring HSC fate through
paracrine signaling (55). A
critical transitional Adamtsl2+/Alcam+
subpopulation emerges at this stage, exhibiting hybrid
quiescent-activated features and functioning as a pivotal decision
point (49). It is capable of
either progressing toward terminal metabolically-committed HSCs or
potentially reverting toward quiescence under appropriate signals
such as PPARγ agonists (34,59).
Spatiotemporal analyses further demonstrate
model-specific activation patterns, with CCl4-induced
injury favoring VitA+ HSCs that retain metabolic
capacity, while bile duct ligation models promote VitA-depleted
progenitor-like populations (45,60,61). Functional specialization occurs
through distinct phases, including inflammatory-phase [C-X-C motif
chemokine ligand 5 (Cxcl5)+/serum amyloid A3
(Saa3)+], migratory-phase [Spp1+/matrix
metalloproteinase (Mmp)+] and ECM-producing (C-type
lectin domain family 3 member B-positive) hybrid cells, with
vascular zonation dictating myofibroblast origins, as evidenced by
transcription factor 21 (Tcf21)+ CaHSCs driving
CCl4-mediated fibrosis vs. portal Tcf21+ populations
dominating cholestatic injury (17,22,29).
Human cirrhosis exhibits an amplified, yet conserved
differentiation trajectory progressing from
RGS5+/FABP4+ transitional HSCs to fibulin
(FBLN)1+/COL1A1+ terminal myofibroblasts,
with spatial transcriptomics identifying periostin-positive aHSCs
as primary fibrogenic effectors alongside rare RGS5+
subsets potentially regulating inflammatory niches (55,62,63). The complexity of HSC
heterogeneity is further compounded by aging through the emergence
of distinct senescent subpopulations that perpetuate fibrotic
microenvironments via senescence-associated secretory phenotype
(SASP) mediated signaling, highlighting the multi-faceted nature of
HSC biology in liver fibrosis pathogenesis across different injury
models and disease stages.
Recent advances in single-cell technologies have
revealed both conserved and disease-specific patterns of HSC
activation across various liver pathologies. A core transitional
pathway (HSC1-HSC4), marked by sequential surface marker changes,
emerges in both non-alcoholic fatty liver disease (NAFLD) and
CCl4-induced injury, demonstrating conserved mechanisms
in the progression from quiescence to myofibroblastic states
(35,54). Notably, this shared activation
trajectory coexists with disease-specific adaptations.
Particularly, in NAFLD, a unique endothelial-chimeric protein
tyrosine phosphatase receptor type B (Ptprb)+/lumican
(Lum)+ subset suggests vascular niche-dependent
reprogramming (35), while
alcoholic hepatitis features a multifunctional
Lrat+/Fbln2+ population that simultaneously
drives fibrosis through collagen deposition, modulates immunity and
sustains proliferation through programmed death-ligand 1 (PD-L1)
and autocrine PDGFRβ signaling (61).
The NASH microenvironment further diversifies HSC
functionality, where different subpopulations cooperate to promote
disease progression. While Timp1+ cells initiate matrix
remodeling, interferon regulatory factor (Irf)7+ subsets
recruit inflammatory macrophages, Cd36+ populations
perpetuate steatosis and cyclin-dependent kinase 1
(Cdk1)+ clusters expand the myofibroblast pool (30). This functional specialization is
evolutionarily conserved, as human NASH biopsies similarly exhibit
ACTA2+ subsets mediating reparative ductular reactions,
while retinol binding protein 1-positive populations directly drive
fibrogenesis (64). The
transition to hepatocellular carcinoma (HCC) reveals an additional
layer of complexity, with the balance between tumor-suppressive
cytokine-producing Cxcl9+ HSCs and pro-carcinogenic
Col1a1+ myofibroblastic subsets determining clinical
outcomes (65,66). Spatial transcriptomics of HCC
further reveal region-specific HSC subsets, with COL1A1-high HSC1
supporting tumor growth through matrix-rich desmoplasia, whereas
ACTA2-high HSC2 adopts a contractile, pro-invasive phenotype
(67). These findings
collectively demonstrate how HSC heterogeneity creates distinct
pathological niches, with disease progression depending on both
conserved activation pathways and context-dependent adaptations
that emerge in specific etiologies.
Across diverse hepatotoxic agents, a core fibrogenic
program emerges through the consistent appearance of
collagen-producing myofibroblast subsets
(Acta2+/Col1a1+), suggesting a fundamental
response pathway to parenchymal damage (29). However, the specific
manifestation of HSC activation varies significantly with toxicant
type, creating distinct pathological microenvironments.
Glyphosate exposure induces a characteristic
four-subpopulation response that includes not only the expected
fibrogenic clusters but also prominently features an inflammatory
IL-6+/Ccl6+ subset, indicating strong
immunomodulatory effects (68).
Similarly, triclosan triggers ECM-producing and proliferative
populations, while uniquely generating migratory
chemokine-expressing clusters (Cxcl2+/Ccl2+),
potentially facilitating immune cell recruitment (69). The paradoxical effects of
triptolide are particularly noteworthy, as it drives simultaneous
pro-inflammatory [NOD-like receptor family pyrin domain containing
3 (NLRP3) activation] and reparative
(Acta2+/Myh11+) HSC states, reflecting its
complex therapeutic-toxic duality (46,70).
Acute liver failure models reveal another dimension
of HSC plasticity, where fibrotic collagen-producing populations
coexist with specialized Acta2+ repair-promoting subsets
that orchestrate macrophage polarization through STAT6 signaling
(71,72). This reparative population appears
particularly prominent in acetaminophen and thioacetamide models,
suggesting its potential role in acute injury resolution (48). The universal presence of cycling
HSC populations, marked by Mki67/Cdk1 or Ccnb1 across all models,
underscores the fundamental need for cellular expansion in injury
response, while the varying proportions of fibrotic, inflammatory
and reparative subpopulations reflect toxicant-specific
microenvironmental programming (48).
Collectively, these findings demonstrate that while
HSCs maintain core response modules to hepatic injury, their
activation spectrum adjusts in a compound-specific manner, forming
distinct cellular ecosystems that influence disease progression and
recovery potential. The balance between these conserved and
adaptive responses likely determines the ultimate pathological
outcome, providing novel targets for tailored therapeutic
interventions based on injury etiology.
The HSC response during liver regeneration following
partial hepatectomy (PH) exhibits a distinct activation spectrum
compared with pathological fibrosis, with specialized
subpopulations orchestrating regenerative rather than fibrotic
programs. The predominant weakly activated cluster
(Acta2low/Lrat+) represents a novel
adaptation to regenerative demands, maintaining quiescence markers
while likely serving as a rapidly mobilizable reserve pool, a
feature rarely observed in chronic injury settings where HSCs
typically progress rapidly to full activation (73).
A PH-specific proliferative population emerges as a
hallmark of regenerative HSC activation, co-expressing mitotic
regulators (Cdk1/DNA topoisomerase II alpha) alongside regenerative
cytokines (73). This dual
functionality suggests an elegant coupling of self-renewal with
paracrine support for hepatocyte proliferation that is
conspicuously absent in the majority of injury models, where
proliferating HSCs primarily contribute to fibrogenesis rather than
regeneration (30,68,69).
Computational modeling reveals a sophisticated
division of labor among regenerating HSCs, with pro-regenerative
subsets [hepatocyte growth factor
(Hgf)high/Vegfahigh/Col1a1low]
actively suppressing fibrogenic programs to prioritize growth
factor production, reflecting reprogramming that prevents the
scarring typical of pathological responses (47). Notably, the discovery of a
transitional 'mixed' state
(Col1a1high/Hgfhigh) introduces a previously
unrecognized layer of regulation, where these cells appear to
function as biological rheostats that use Yes-associated protein
(YAP)/transcriptional coactivator with PDZ-binding motif (TAZ)
mechanosensing to dynamically adjust the fibro-reparative balance
according to microenvironmental demands (47).
The progression from aHSCs to senescent phenotypes
represents a critical adaptation in chronic liver disease, with
cells exiting the cell cycle, yet remaining metabolically active
and shaping disease outcomes context-dependently (74). In fibrotic environments, aHSCs
transition into senescent HSCs (sHSCs) marked by
TP53/CDKN1A-mediated cell cycle arrests yet paradoxically acquire
pro-tumorigenic potential through SASP components, including
inflammatory cytokines and matrix-remodeling proteases (18,75-77). Early metabolic dysfunction adds
further complexity to this transition, generating
Mrc1+/Slc9a9+ sHSCs that evolve into
inflammatory-senescent hybrids
(Cxcl10+/Ccl5+), linking steatosis injury to
progressive microenvironmental dysfunction (78).
The functional duality of sHSCs becomes particularly
evident in HCC, where early TP53high sHSCs suppress
tumor growth via CXCL9-driven immune recruitment, while persistent
NASH-derived sHSCs promote malignancy through TGFβ1/PDGFA-induced
epithelial-mesenchymal transition (79-81). Contrastingly, in liver
regeneration, transient sHSCs [interleukin
(IL)-6+/CXCL2+] enhance hepatocyte
proliferation, illustrating how senescence duration and niche
signals determine beneficial vs. detrimental outcomes (82).
Parallel to senescence, aHSCs may also adopt
inactivated states during injury resolution (34). These cells downregulate
fibrogenic markers and partially regain quiescence-associated genes
but retain epigenetic scars of activation, such as loss of lipid
droplets (45). NASH regression
models have identified a specialized
CXCL1+/GABRA3+ iHSC subset that balances
reduced ECM production with heightened sensitivity to reactivation
(30), while PH induces
metabolically reprogrammed COL1A1low iHSCs that persist
in peri-sinusoidal niches (73).
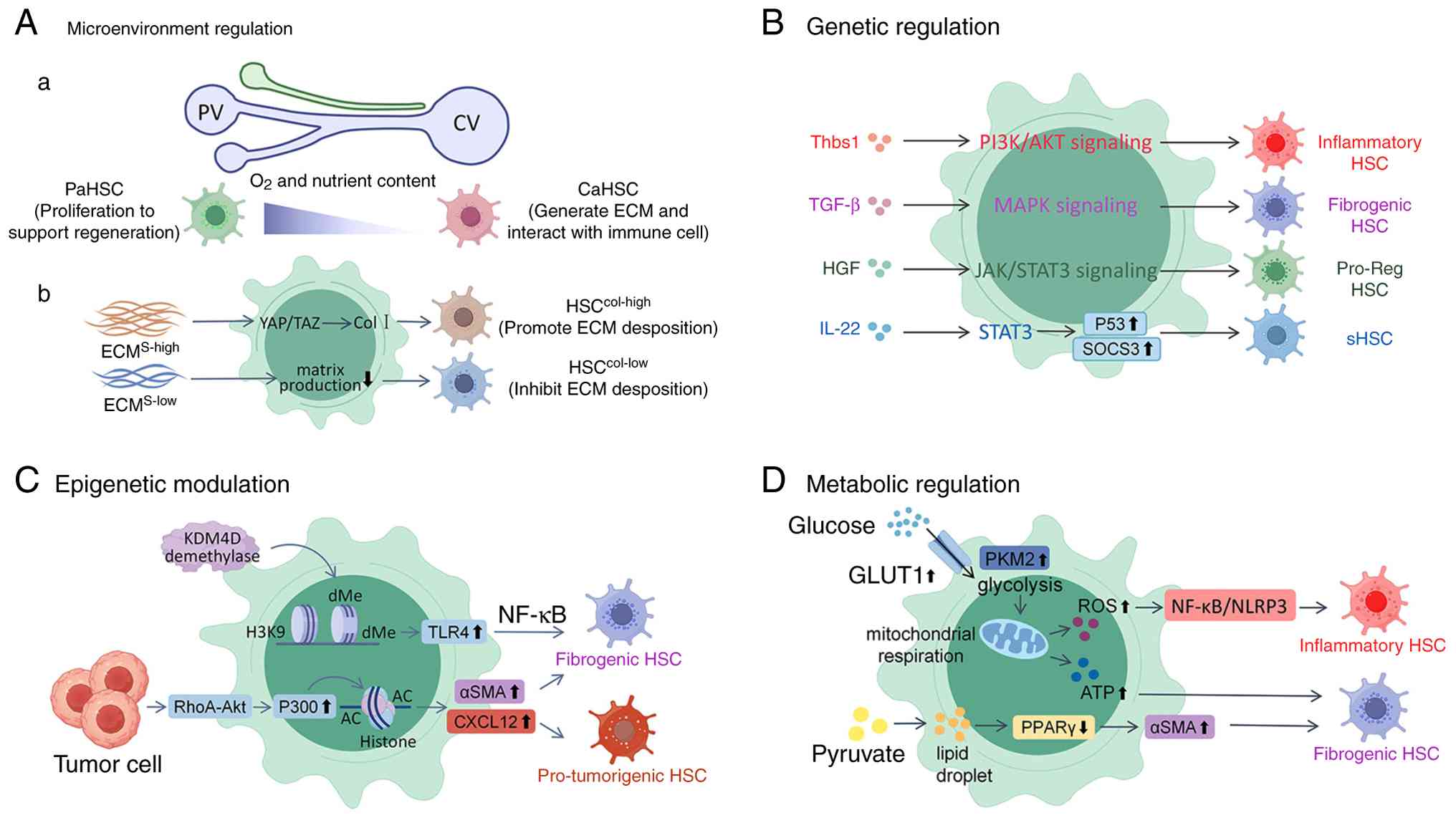
HSC heterogeneity largely arises from
microenvironmental regulation, genetic or epigenetic modulation,
and metabolic reprogramming (Fig.
1). Within the liver lobule, the spatial distribution of HSCs
establishes functional diversity driven by zonation-specific
gradients of oxygen, metabolites and signaling molecules (37,83). CaHSCs, residing in hypoxic and
metabolite-rich zones, acquire pro-fibrogenic traits through
hypoxia-inducible and CYP450-dependent pathways (37,84). By contrast, PaHSCs near the
oxygen- and nutrient-rich portal triad serve as a proliferative
reservoir during regeneration (85). Midlobular HSCs exhibit
intermediate phenotypes balancing ECM remodeling and immune
modulation (85).
Biomechanical heterogeneity further amplifies these
zonal responses. Uneven ECM deposition generates stiffness
gradients that differentially activate mechanotransduction
pathways, YAP/TAZ signaling predominates in stiff pericentral
regions to promote fibrosis, while softer periportal areas favor
HSC migration and proliferation via integrin-dependent cues
(49,86,87).
Spatial specialization is also shaped by paracrine
and juxtacrine interactions with neighboring cells (88). Pericentral LSECs secrete Hedgehog
ligands and pericentral KCs produce TGF-β to polarize HSCs toward a
fibrogenic phenotype, whereas periportal LSECs release nitric oxide
and periportal KCs secrete IL-10 to suppress excessive
proliferation and support reparative functions (20,89-91). Hepatocytes also contribute to
this zonation: Oxidative stress products from pericentral
hepatocytes activate adjacent HSCs, while periportal hepatocytes
provide pro-regenerative signals supporting proliferation (92,93).
HSCs further shape immune-metabolic niches through
crosstalk with infiltrating immune cells. Pericentral subsets
recruit monocytes via CCL2 and CXCL5 to exacerbate inflammation,
whereas periportal HSCs modulate lipid antigen presentation and
perpetuate steatoinflammation in NASH (22,30,55). These interactions integrate local
damage-associated molecular patterns with systemic immune inputs,
forming spatially organized regulatory networks that determine HSC
behavior across disease contexts.
Emerging single-cell and spatial transcriptomic
technologies have validated these mechanisms, revealing
zonation-specific ligand-receptor interactions between HSCs and
neighboring cell types and identifying spatially restricted
activation markers. Collectively, these findings demonstrate that
HSC heterogeneity represents an adaptive response to
microenvironmental demands rather than random variation (37,85). Understanding such spatial
organization provides therapeutic opportunities, selectively
targeting pericentral HSCs to mitigate fibrosis or modulating
periportal subsets to enhance regeneration. The future integration
of spatial omics and functional mapping will further refine
precision interventions for liver diseases.
The activation of HSCs involves a tightly regulated
transcriptional program that governs phenotypic transitions between
quiescent, activated, and inactivated/senescent states. In their
quiescent state, HSCs maintain lipid droplets and express
adipogenic transcription factors (TFs), such as C/EBPα and PPARγ,
which suppress fibrogenic genes such as Col1a1 and α-SMA (34,55,94). Upon liver injury,
lineage-determining TFs, including E26 transformation-specific
transcription factor ½ (ETS1/2), GATA binding protein 4/6 (GATA4/6)
and IRF1/2, which maintain quiescent phenotype in qHSCs, are
repressed during activation, leading to the disruption of
quiescence-associated genes such as PPARγ (34,59,95). During fibrosis resolution, PPARγ
is re-expressed in iHSCs, collaborating with GATA6 to restore
quiescence, highlighting the potential of GATA6 and PPARγ agonists
as a promising target in anti-fibrotic therapy (34,59).
Similarly, in NASH models, ETS1 has been shown to
preserve the quiescent identity of HSCs by suppressing
pro-fibrogenic gene programs and maintaining lipid storage
capacity, while IRF1 marks aHSCs and promotes inflammatory cytokine
production and collagen deposition during the progression of NASH
(30). Furthermore, the genetic
ablation of GATA6 in HSCs disrupts their ability to revert to a
quiescent state post-injury, as GATA6-deficient cells fail to
upregulate lipid-droplet-associated proteins, such as PLIN2 and
downregulate α-SMA expression, directly linking this TF to HSC
deactivation and metabolic reprogramming (30). These findings underscore a
TF-centric regulatory hierarchy governing HSC phenotypic
plasticity.
The hierarchical integration of upstream signaling
pathways dictates the fate of HSCs through metabolic and mechanical
checkpoints. TGF-β and PDGF function as dominant fibrogenic
drivers. TGF-β activates SMAD3 to enforce myofibroblast
transdifferentiation (20,96), while PDGFRβ signaling licenses
proliferative expansion via mechanistic target of rapamycin
(mTOR)-dependent glycolytic reprogramming (97). Conversely, Wnt pathway
modulators, such as Dickkopf-related protein 1 exert
context-dependent control (98).
While transient Wnt inhibition restores lipid storage by activating
PPARγ-dependent lipogenic genes, chronic Wnt activation promotes
senescence evasion via p21 suppression (99,100). Notably, telomerase-positive
HSCs exemplify this regulatory plasticity, coupling TERT-mediated
replicative longevity with retinol-responsive metabolic switching,
as retinol uptake is reported to reactivate RXRα-driven lipid
droplet biogenesis, enabling reversion to quiescence despite
persistent activation cues (101,102).
Emerging evidence reveals that the induction of
senescence in HSC operates through interconnected tumor suppressor
networks, which integrate inflammatory, metabolic, and mechanical
signals to enforce growth arrest and restrict ECM overproduction.
For example, IL-22 signaling initiates a senescence cascade through
the STAT3-mediated upregulation of suppressor of cytokine signaling
3 and TP53, driving HSC into growth arrest (103). This process is reinforced by
the parallel activation of the p16/Rb pathway, which stabilizes the
senescent phenotype by blocking cell cycle progression (75). sHSCs undergo dual transcriptional
reprogramming. They suppress ECM-producing genes while enhancing
immune surveillance molecules, such as major histocompatibility
complex class II and PD-L1 (104,105). Notably, HSCs lacking both TP53
and INK4a/ARF evade senescence entirely, leading to
hyperproliferation and aggravated fibrotic responses, highlighting
the cooperative action of these tumor suppressor pathways in
constraining pathological HSC activation (75). Concurrently, the loss of
YAP-mediated mechanosensing disrupts cytoskeletal tension, inducing
p21-dependent quiescence against aberrant activation (106). These overlapping pathways
collectively indicate a 'senescence barrier' to suppress HSC-driven
matrix deposition.
Of note, the integrity of this barrier determines
pathological outcomes. Compromised TP53 or p16/Rb signaling
dismantles senescence enforcement, releasing HSCs from growth
constraints and permitting their transition into aggressive
profibrotic effectors that functionally mirror tumorigenic stromal
cells. Thus, this senescence-mediated barrier underscores the
evolutionary conservation of tumor suppressor networks in fibrosis
containment, where senescence acts not only as an anti-aging
mechanism, but also as a spatial regulator ensuring fidelity of
tissue repair.
The phenotypic plasticity of HSCs during fibrosis
is also governed by coordinated epigenetic reprogramming involving
DNA methylation, histone modifications, and non-coding RNA
networks, which collectively translate microenvironmental stimuli
into transcriptional outputs (107). Genome-wide methylation
profiling reveals activation-state-specific 5-methylcytosine
patterns, particularly at pericentromeric regions, with
hypermethylation silencing anti-fibrotic loci, such as PPARγ and
hypomethylation enabling pro-fibrotic drivers, such as spondin 2
via Wnt/β-catenin activation (108-110). The pharmacological inhibition
of DNA methyltransferases (DNMTs) counteracts this shift, restoring
quiescence by suppressing TGF-β receptor signaling (111).
Complementing these DNA-centric modifications,
histone post-translational dynamics have been shown to reinforce
fibrogenic commitment. For example, histone deacetylases (HDACs)
remove H3K27 acetylation at ACTA2 promoters to sustain
myofibroblast identity (112),
while mechanical cues from fibrotic ECM stiffening trigger nuclear
accumulation of p300, amplifying histone acetylation at fibrotic
loci to lock HSCs into self-reinforcing activation (86,87,113). Concurrently, H3K9me3 deposition
at senescence-associated genes and H3K9me2 demethylation at TLR4
enhancers link chromatin states to inflammatory fibrosis (114,115).
Non-coding RNAs further integrate metabolic and
epigenetic regulation. The age-dependent decline of geromiRs
derepresses IL-6/tumor necrosis factor α (TNF-α), fueling
inflammation and pre-metastatic niches (116), while miR-23a and miR-195
coordinate lipid metabolism with DNA methylation patterns during
phenotype switching (117).
Moreover, the hypermethylation of SAD1/UNC84 domain protein 2
perturbs nuclear lamina dynamics, destabilizing the genome and
driving chronic pathogenic activation (118).
This multilayered epigenetic plasticity positions
HSCs as microenvironmental rheostats, dynamically calibrating
fibrogenic responses. Targeting nodal regulators, such as DNMTs in
early fibrosis or p300 in established scarring, could selectively
disrupt pathological subsets, while preserving reparative
functions.
The phenotypic diversity of HSCs stems from their
marked metabolic plasticity, where dynamic shifts in glucose, lipid
and mitochondrial metabolism govern their activation states.
Following liver injury, HSCs undergo a metabolic reprogramming
characterized by enhanced glucose transporter type 1-mediated
glucose uptake and pyruvate kinase M2-driven glycolytic flux,
fueling mitochondrial oxidative phosphorylation for myofibroblast
differentiation (119). This
transition is accompanied by reactive oxygen species accumulation
that activates NF-κB/NLRP3 inflammasome pathways, driving
pro-inflammatory polarization (120). Notably, although aHSCs were
traditionally considered lipid-depleted, recent evidence indicates
they maintain active lipid metabolism. Mechanistically, the
metabolic reprogramming redirects pyruvate toward lipid synthesis,
leading to lipid droplet reformation, a paradoxical feature that
actually sustains fibrotic activity through PPARγ suppression and
TGF-β activation (121).
Mitochondrial dynamics play a pivotal role in
regulating the fate of HSCs. Studies have indicated that qHSCs
maintain fusion-dominant states that support efficient β-oxidation,
while aHSCs exhibit fission-prone mitochondria that generate
oncometabolites such as succinate and 2-hydroxyglutarate (122-124). These metabolites stabilize
hypoxia-inducible factor-1α and promote angiogenic secretomes
(120).
In the tumor microenvironment, a metabolic
cross-talk emerges between cancer cells and HSCs (125). Malignant cells export lactate
through monocarboxylate transporter 4, creating an acidic
microenvironment that reprograms neighboring HSCs (126,127). This acid adaptation triggers
the release of MMP9 from HSCs, thereby fostering a permissive
environment for metastatic spread (128,129).
The complex interplay between ammonia metabolism
and HSC biology reveals an intriguing paradox. Disrupted
ureagenesis in injured livers results in the accumulation of
nitrogenous metabolites, which paradoxically elicit two opposing
outcomes in HSCs (130). While
promoting fibrogenic activation through mitochondrial stress, these
metabolites simultaneously induce growth arrest through
p53/p21-mediated senescence (131-133). This dual effect creates a
biological checkpoint that limits uncontrolled HSC expansion while
permitting controlled ECM deposition during tissue repair.
While notable progress has been made in
understanding HSC metabolism, several fundamental questions persist
about what drives their functional diversity. Currently available
research has largely illuminated the metabolic changes occurring
during initial activation; however, the exact mechanisms through
which distinct HSC subpopulations, particularly those with
immunomodulatory vs. pro-angiogenic properties, establish and
maintain their unique metabolic identities, remain to be fully
elucidated. Moreover, critical gaps remain in the knowledge of how
nutrient-sensing mechanisms interface with epigenetic regulation to
control HSC subset specification. The potential involvement of
NAD+-dependent metabolic sensors in coordinating
mitochondrial function across different HSC activation states also
warrants further investigation (134).
Resolving these unanswered questions could pave the
way for more sophisticated therapeutic strategies. Such approaches
would ideally target disease-promoting metabolic pathways in aHSCs,
while sparing their beneficial roles in liver repair and
regeneration. This precision targeting represents a crucial next
step in developing effective anti-fibrotic treatments that maintain
the innate regenerative capacity of the liver.
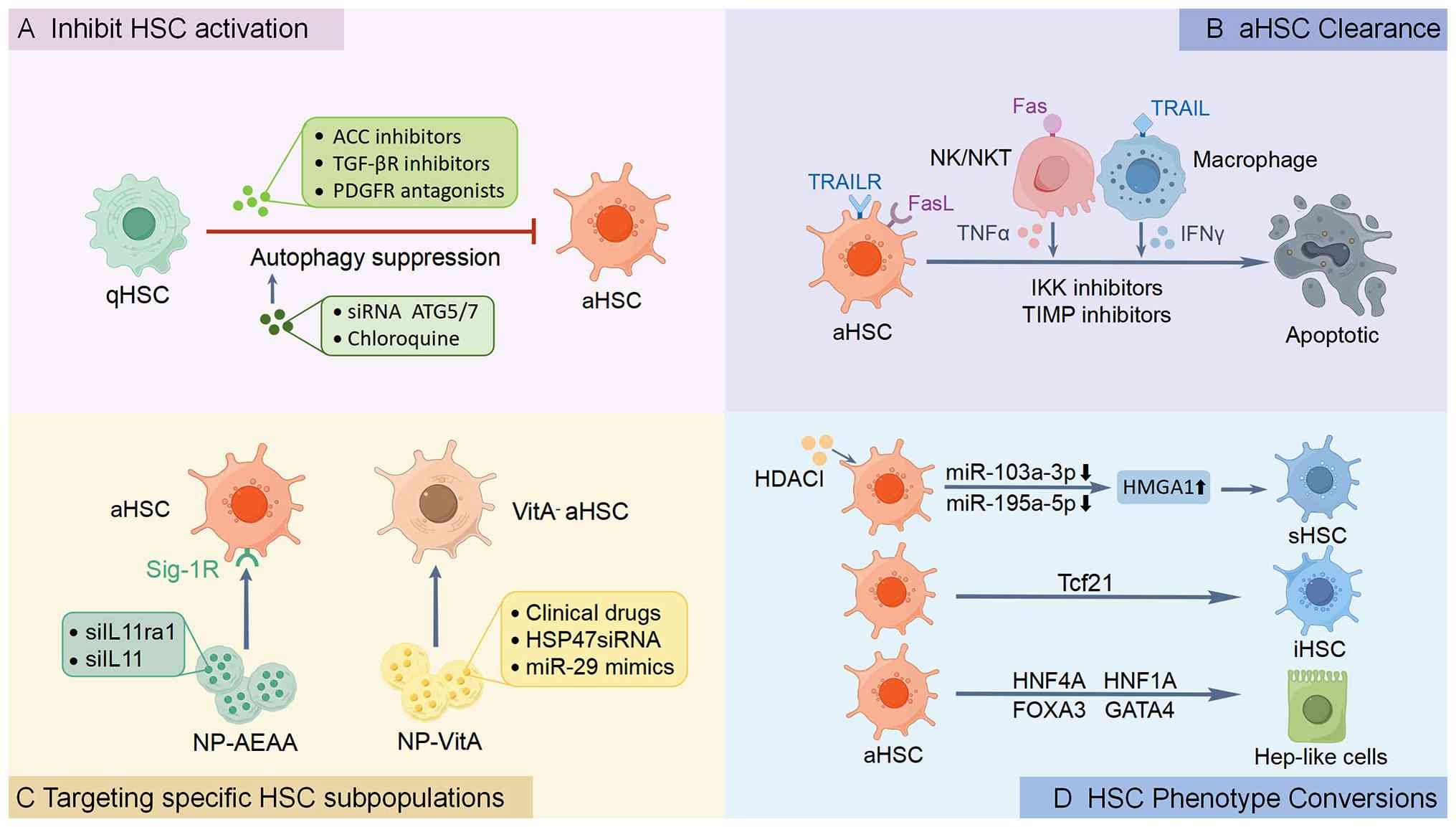
Current approaches to modulate HSC activity focus
on four primary mechanisms: i) The interruption of activation
signaling cascades; ii) the selective induction of aHSC apoptosis;
iii) targeting specific HSC subsets with defined pathogenic roles;
and iv) reprogramming aHSCs toward quiescent or alternative
functional phenotypes (Fig. 2).
Single-cell transcriptomic studies have revolutionized the
understanding of HSC heterogeneity, identifying distinct
pro-fibrotic and pro-regenerative subpopulations that coexist in
injured livers (135). These
findings support the development of precision therapies capable of
selectively targeting pathological subsets while sparing
regenerative populations.
HSC inactivation has emerged as a cornerstone in
the treatment of liver fibrosis and cirrhosis, aiming to halt or
reverse the pathological accumulation of ECM in chronic liver
diseases. HSC activation is orchestrated by an intricate signaling
network integrating paracrine cues and metabolic reprogramming
(136), including TGF-β/Smad3
(137,138), Wnt/β-catenin (139) and Akt/mTOR (22,140). Among these pathways, TGF-β
signaling enhances glycolysis and de novo lipogenesis, both of
which are indispensable for the activation of HSCs. This process
can be disrupted by Acetyl-CoA carboxylase inhibitors, such as
firsocostat, which has exhibited potential in reducing steatosis
and fibrosis in preclinical models and in patients with NASH,
although further investigations are warranted (NCT02856555)
(119,141). The inhibition of stearoyl-CoA
desaturase-1 by aramchol has been shown to attenuate HSC activation
and fibrogenesis independently of steatosis reduction, highlighting
the lipogenic contribution to HSC plasticity (142,143). Likewise, thyroid hormone
receptor agonists exert direct anti-fibrotic effects by suppressing
profibrogenic gene expression in HSCs and improving fibrosis
through coordinated metabolic and stellate cell-mediated mechanisms
(144,145). Glucagon-like peptide-1 receptor
agonists, primarily known for alleviating steatosis and insulin
resistance, further attenuate fibrosis when combined with the
ATP-citrate lyase inhibitor, bempedoic acid, in mice with NASH,
revealing a synergistic link between hormonal and metabolic
signaling in HSC inactivation (146,147).
Beyond metabolic reprogramming, anti-fibrotic
strategies also target canonical signaling pathways that drive HSC
activation. Inhibitors such as galunisertib, a TGF-β receptor I
kinase inhibitor, have been shown to exert anti-fibrotic effects in
preclinical models, although clinical trials have revealed limited
efficacy in advanced-stag cirrhosis, possibly due to the
compensatory mechanisms or off-target effects (NCT01246986)
(Table III) (148,149). Similarly, PDGF signaling,
critical for HSC proliferation, can be blocked by tyrosine kinase
inhibitors, such as imatinib or receptor antagonists; however,
challenges in specificity persist (150,151). In addition, probiotics such as
Mutaflor® inhibit HSC activation and fibrogenic
signaling in NAFLD/NASH models via the Hedgehog and Hippo pathways,
underscoring gut-liver crosstalk as a complementary anti-fibrotic
target (152).
Emerging evidence highlights the critical role of
epigenetic dysregulation in maintaining HSC activation. DNMT
inhibitors (DNMTIs, e.g., 5-azacytidine) and HDAC inhibitors
(HDACIs, e.g., vorinostat) have demonstrated anti-fibrotic effects
by remodeling chromatin architecture and suppressing collagen
expression in aHSCs (153,154). The therapeutic potential of
targeting non-coding RNAs is particularly promising, with microRNA
(miRNA/miR)-29 family members showing efficacy in restoring
epigenetic homeostasis and attenuating fibrogenesis in preclinical
models (155).
Building upon the epigenetic mechanisms discussed
earlier, autophagy emerges as another critical regulator of HSC
behavior during liver fibrosis progression. In chronic liver
injury, dysregulated autophagy plays a dual role in modulating HSC
activation, survival and fibrogenic activity (156). During the early stages of HSC
activation, autophagy serves as a crucial survival mechanism by
providing energy substrates to sustain proliferation and collagen
production under conditions of metabolic stress (156). This pro-fibrotic function is
supported by preclinical evidence showing that pharmacological
inhibitors such as chloroquine or genetic ablation of
autophagy-related genes such as autophagy-related gene 5/7
significantly attenuate collagen deposition and HSC activation in
rodent models (157,158).
However, the association between autophagy and
fibrosis becomes more complex in advanced stages of disease.
Paradoxically, excessive autophagic flux can trigger HSC senescence
through p53/p21 pathway activation, leading to cell cycle arrest
and reduced fibrogenic output (159,160). This dual nature has inspired
innovative therapeutic strategies, including combination approaches
that synergize autophagy inhibitors with pro-apoptotic agents to
enhance HSC clearance.
The clearance of aHSCs is orchestrated through
complex cellular crosstalk within the liver microenvironment, where
multiple cell types collectively determine the fate of HSCs
(161). LSECs play a dual
regulatory role, maintaining HSC quiescence under physiological
conditions through mediators including nitric oxide and hedgehog
inhibitors (162); yet,
transforming into pro-fibrotic stimulators upon injury-induced
capillarization (162,163). This phenotypic switch
highlights the therapeutic potential of vascular normalization
strategies to restore LSEC function and mitigate fibrogenesis
(163). The immune compartment
further modulates HSC clearance through coordinated actions.
Natural killer (NK) cells induce activated HSC apoptosis via
interferon γ secretion and death receptor engagement (20,164,165), while macrophage subsets
differentially regulate fibrosis progression (166). For example, pro-inflammatory M1
macrophages promote HSC death through TNF-α signaling and
potentiate NK cell cytotoxicity (167,168), whereas specialized restorative
macrophages drive fibrosis resolution by secreting MMPs to degrade
the ECM (169).
Pharmacological interventions targeting HSC
clearance have emerged through multiple approaches, including IKK
inhibitors and TIMP antagonists that rebalance NF-κB signaling and
MMPs activity (170,171), as well as drug repurposing
strategies exemplified by the antiviral agent tenofovir disoproxil
fumarate which suppresses HSC survival through PI3K/Akt/mTOR
pathway inhibition (172).
While the selective depletion of aHSCs accelerates fibrosis
resolution (106,173), excessive elimination risks
compromising hepatic function through reduced liver mass and
exacerbated inflammation (26,174). These observations underscore
the need for precision therapeutics capable of discriminating
between pathogenic and reparative HSC subpopulations (175).
The advent of single-cell omics technologies has
unmasked the functional diversity of HSC subpopulations, paving the
way for precision interventions that selectively neutralize
fibrosis-driving subsets, while preserving homeostatic or
regenerative HSCs. Clinically validated targets include
VitA-depleted aHSCs, which can be selectively addressed using
VitA-coupled liposomal nanoparticles (NPs). For instance,
BMS-986263, a lipid NP encapsulating siRNA against heat shock
protein 47 associated with collagen production, utilizes VitA to
target aHSCs in fibrotic livers (176). In a phase II trial
(NCT03420768), BMS-986263 reduced collagen production in patients
with advanced hepatic fibrosis, demonstrating proof-of-concept for
HSC-specific delivery (177).
Preclinically, VitA-coupled NPs loaded with miR-29 mimics restored
miR-29, a key anti-fibrotic miRNA in aHSCs of
CCl4-treated mice, reversing fibrosis without
hepatotoxicity (155,178). Another clinical-stage
candidate, nanoparticle aminoethyl anisamide (NP-AEAA), utilizes
aminoethyl anisamide surface modifications to selectively target
sigma-1 receptors that are upregulated on aHSCs in NASH
progression. In mice with diet-induced NASH, NP-AEAA delivering
siRNA against IL-11 reduced liver inflammation and fibrosis by
~50%, with ongoing optimization for first-in-human trials (179).
Furthermore, receptor-specific NP systems have
demonstrated significant potential for precision anti-fibrotic
therapy. The IGF2 E12-C21 peptide-conjugated NP platform
selectively binds IGF2R that are markedly upregulated during HSC
transdifferentiation. In bile duct ligation models, this targeted
delivery system enhanced fibrosis resolution through the
preferential transport of anti-fibrotic compounds such as
pentoxifylline to aHSC populations (180).
Complementary genetic approaches have further
advanced the ability to precisely manipulate HSC activity.
Preclinical studies utilizing GFAP-thymidine kinase transgenic
models achieved the selective elimination of proliferating HSCs
through ganciclovir administration, yielding a 70% reduction in
fibrosis, while preserving qHSC pools and maintaining hepatic
regenerative capacity (51,181). The parallel development of
inducible genetic systems, such as PDGFRβ-CreERT2, has enabled
temporal control over collagen-producing HSC ablation via tamoxifen
induction, demonstrating the feasibility of promoter-specific
interventions (182).
These approaches collectively represent significant
advances in cell-type-specific therapeutic strategies, offering
improved specificity compared to conventional broad-acting
anti-fibrotics.
Nevertheless, the translation of HSC-targeted
therapies from bench to bedside has encountered substantial
challenges that underscore the complexity of liver fibrosis
pathogenesis. The discouraging clinical performance of simtuzumab,
a monoclonal antibody targeting lysyl oxidase like 2, in phase II
cirrhosis trials revealed critical limitations in therapeutic
specificity and the adaptive capacity of the liver for compensatory
ECM remodeling (183).
Similarly, the TGF-β receptor inhibitor, galunisertib, despite
exhibiting robust efficacy in preclinical models, demonstrated only
modest clinical benefits in patients with advanced-stage fibrosis
(184). These clinical
experiences have catalyzed the development of more sophisticated
therapeutic strategies designed to overcome these limitations.
Emerging solutions currently focus on
dual-targeting approaches that combine VitA conjugates with PDGFRβ
ligands to better address HSC heterogeneity, as well as
microenvironment-responsive carriers that utilize fibrotic
niche-specific enzymes like MMPs for spatially controlled drug
release (185). Notably, recent
breakthroughs in gene-editing platforms, particularly CRISPR-Cas9
targeting aHSC-specific phosphorylation sites of A-kinase anchoring
protein 12, have demonstrated notable efficacy in preclinical
fibrosis models through precise silencing of profibrotic pathways
(186).
The notable plasticity of HSCs presents multiple
therapeutic avenues for combating liver fibrosis, while promoting
regeneration. Research has revealed that ~50% of aHSCs avoid
apoptosis during fibrosis resolution, instead undergoing phenotypic
reversion characterized by downregulation of fibrogenic markers and
acquisition of a quiescent-like state resistant to reactivation
(34,45). This plasticity has been
conclusively demonstrated through transplantation studies where
human aHSCs engrafted in immunodeficient mice regained
lipid-storing capacity and other quiescent cell features (34). PPARγ activation plays a central
role by restoring lipogenic programs that promote HSC inactivation
(34,187). Equally critical is Tcf21, a TF
that simultaneously suppresses profibrotic pathways, while
activating quiescence-associated genes in both cellular and animal
models (188). Complementary to
these reversion pathways, the targeted induction of senescence
through p53 activation has been shown to exert significant
anti-fibrotic effects (189-191).
Emerging evidence demonstrates that targeted
epigenetic modulation can effectively redirect aHSC fate toward
beneficial phenotypes. The small molecule, CM272, a dual inhibitor
of G9a histone methyltransferase and DNMT1, drives aHSC conversion
into adipocyte-like cells by demethylating and reactivating the
PPARγ promoter, resulting in the significant attenuation of
fibrosis in preclinical NASH models (10,192). The therapeutic potential of
this approach is further supported by the ability of CM272 to
simultaneously suppress pro-fibrotic signaling pathways. while
restoring metabolic functions characteristic of qHSCs (193).
HDACIs represent another class of epigenetic
modifiers with significant anti-fibrotic effects. Valproic acid
(VPA), a clinically approved HDACI, mediates aHSC senescence
through miRNA-dependent mechanisms. By downregulating miR-103a-3p
and miR-195-5p, VPA promotes the expression of high mobility group
AT-hook 1, a critical driver of cellular senescence (194). This epigenetic-miRNA cascade
not only induces growth arrest, but also shifts aHSCs toward an
anti-fibrotic secretome phenotype characterized by enhanced MMP
activity and reduced collagen production (22,195).
Notably, the complete lineage reprogramming of
aHSCs has been achieved through TF overexpression. The
combinatorial expression of forkhead box A3 (FOXA3), GATA4,
hepatocyte nuclear factor (HNF)1A and HNF4A converts aHSCs into
functional hepatocyte-like cells in vitro, which upon
transplantation, can repopulate damaged liver parenchyma and
ameliorate fibrosis (196,197). This direct reprogramming
approach bypasses pluripotent intermediates, while maintaining the
epigenetic memory of hepatic identity, providing potential
advantages for regenerative applications. Mechanistically, FOXA3, a
canonical pioneer factor, initiates this process by binding to
compacted chromatin at hepatocyte-specific enhancers and displacing
linker histone H1, thereby initiating local chromatin opening
(198). GATA4 acts
synergistically with FOXA3, and together they prime the epigenetic
landscape for the core hepatocyte regulators (197). Subsequently, HNF4A binds to
these accessible regulatory elements to activate a comprehensive
suite of genes defining hepatocyte identity, including those
governing metabolism and synthetic function (199). HNF1A reinforces this program by
stabilizing the transcriptional network and activating HNF4A
expression, creating a positive feedback loop that locks in the
hepatocyte fate (197).
Concurrently, the expression of the aHSC genetic program is
effectively silenced, as evidenced by the downregulation of key
markers including α-SMA and COL1A1. The reprogramming process also
suppresses critical aHSC maintenance pathways, such as YAP/TAZ
signaling, which is essential for maintaining the myofibroblastic
phenotype (200,201). This combinatorial action of the
four factors thereby orchestrates a complete phenotypic conversion,
overriding the fibrogenic identity of aHSCs to re-establish
functional hepatocyte properties in vivo.
The advent of single-cell and spatial
transcriptomics has established HSC heterogeneity and plasticity as
foundational to liver fibrogenesis and resolution. Given the
recurrent failure of broad-spectrum anti-fibrotics, future
therapeutic breakthroughs must leverage precision strategies
capable of distinguishing pathogenic from reparative HSC
subpopulations. Achieving this will require combinatorial
approaches, such as cell-specific NP delivery or temporally
controlled epigenetic reprogramming, to therapeutically guide HSC
fate decisions, thereby selectively inhibiting fibrosis while
actively promoting regeneration.
Not applicable.
DY conceived and supervised the study. DY and CG
wrote the manuscript. GC and HJ contributed to the literature
collection and the revision of the manuscript. HZ and YC assisted
with the literature search, figure preparation and revision of the
manuscript. KZ and DY finalized the manuscript and made the
conceptual evaluation of the manuscript. All authors have read and
approved the final manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by grants from the Key
Laboratory Open Project Foundation of Jiangsu University (no.
8161280002), Science and Technology Plan (Apply Basic Research) of
Changzhou City (no. CJ20230002), Science and Technology Plan (Apply
Basic Research) of Changzhou City (no. CJ20241041), the Soft
Science Research Program of Zhenjiang City (no. RK2025040), the
Natural Science Foundation of China (no. 82100666) and the Young
Scientists Initiative Foundation of Jiangsu University.
|
1
|
Campana L, Esser H, Huch M and Forbes S:
Liver regeneration and inflammation: From fundamental science to
clinical applications. Nat Rev Mol Cell Biol. 22:608–624. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wake K: 'Sternzellen' in the liver.
Perisinusoidal cells with special reference to storage of vitamin
A. Am J Anat. 132:429–462. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cogliati B, Yashaswini CN, Wang S, Sia D
and Friedman SL: Friend or foe? The elusive role of hepatic
stellate cells in liver cancer. Nat Rev Gastroenterol Hepatol.
20:647–661. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kamm DR and McCommis KS: Hepatic stellate
cells in physiology and pathology. J Physiol. 600:1825–1837. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen L, Ye X, Yang L, Zhao J, You J and
Feng Y: Linking fatty liver diseases to hepatocellular carcinoma by
hepatic stellate cells. J Natl Cancer Cent. 4:25–35.
2024.PubMed/NCBI
|
|
6
|
Friedman SL: Hepatic stellate cells:
Protean, multifunctional, and enigmatic cells of the liver. Physiol
Rev. 88:125–172. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang T, Gu Z, Feng J, Shan J, Qian C and
Zhuang N: Non-parenchymal cells: Key targets for modulating chronic
liver disease. Front Immunol. 16:15767392025. View Article : Google Scholar
|
|
8
|
Shu W, Yang M, Yang J, Lin S, Wei X and Xu
X: Cellular crosstalk during liver regeneration: unity in
diversity. Cell Commun Signal. 20:1172022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Trefts E, Gannon M and Wasserman DH: The
liver. Curr Biol. 27:R1147–R1151. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Carter JK and Friedman SL: Hepatic
stellate cell-immune interactions in NASH. Front Endocrinol
(Lausanne). 13:8679402022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xia M, Li J, Martinez Aguilar LM, Wang J,
Trillos Almanza MC, Li Y, Buist-Homan M and Moshage H: Arctigenin
attenuates hepatic stellate cell activation via endoplasmic
reticulum-associated degradation (ERAD)-Mediated restoration of
lipid homeostasis. J Agric Food Chem. 73:13918–13933. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kisseleva T: The origin of fibrogenic
myofibroblasts in fibrotic liver. Hepatology. 65:1039–1043. 2017.
View Article : Google Scholar
|
|
13
|
Wiering L, Subramanian P and Hammerich L:
Hepatic stellate cells: Dictating outcome in nonalcoholic fatty
liver disease. Cell Mol Gastroenterol Hepatol. 15:1277–1292. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Acharya P, Chouhan K, Weiskirchen S and
Weiskirchen R: Cellular mechanisms of liver fibrosis. Front
Pharmacol. 12:6716402021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nishio T, Hu R, Koyama Y, Liang S,
Rosenthal SB, Yamamoto G, Karin D, Baglieri J, Ma HY, Xu J, et al:
Activated hepatic stellate cells and portal fibroblasts contribute
to cholestatic liver fibrosis in MDR2 knockout mice. J Hepatol.
71:573–585. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim HY, Sakane S, Eguileor A, Carvalho
Gontijo Weber R, Lee W, Liu X, Lam K, Ishizuka K, Rosenthal SB,
Diggle K, et al: The origin and fate of liver myofibroblasts. Cell
Mol Gastroenterol Hepatol. 17:93–106. 2024. View Article : Google Scholar
|
|
17
|
Yang W, He H, Wang T, Su N, Zhang F, Jiang
K, Zhu J, Zhang C, Niu K, Wang L, et al: Single-Cell transcriptomic
analysis reveals a hepatic stellate cell-activation roadmap and
myofibroblast origin during liver fibrosis in mice. Hepatology.
74:2774–2790. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hernandez-Gea V and Friedman SL:
Pathogenesis of liver fibrosis. Annu Rev Pathol. 6:425–456. 2011.
View Article : Google Scholar
|
|
19
|
Krenkel O, Hundertmark J, Ritz TP,
Weiskirchen R and Tacke F: Single Cell RNA sequencing identifies
subsets of hepatic stellate cells and myofibroblasts in liver
fibrosis. Cells. 8:5032019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsuchida T and Friedman SL: Mechanisms of
hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol.
14:397–411. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ramachandran P, Dobie R, Wilson-Kanamori
JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice
M, Marwick JA, et al: Resolving the fibrotic niche of human liver
cirrhosis at single-cell level. Nature. 575:512–518. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cheng S, Zou Y, Zhang M, Bai S, Tao K, Wu
J, Shi Y, Wu Y, Lu Y, He K, et al: Single-cell RNA sequencing
reveals the heterogeneity and intercellular communication of
hepatic stellate cells and macrophages during liver fibrosis.
MedComm (2020). 4:e3782023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Deng G, Liang X, Pan Y, Luo Y, Luo Z, He
S, Huang S, Chen Z, Wang J and Fang S: Single-cell transcriptomic
analysis of different liver fibrosis models: Elucidating molecular
distinctions and commonalities. Biomedicines. 13:17882025.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang ZY, Keogh A, Waldt A, Cuttat R, Neri
M, Zhu S, Schuierer S, Ruchti A, Crochemore C, Knehr J, et al:
Single-cell and bulk transcriptomics of the liver reveals potential
targets of NASH with fibrosis. Sci Rep. 11:193962021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Peyser R, MacDonnell S, Gao Y, Cheng L,
Kim Y, Kaplan T, Ruan Q, Wei Y, Ni M, Adler C, et al: Defining the
activated fibroblast population in lung fibrosis using single-cell
sequencing. Am J Respir Cell Mol Biol. 61:74–85. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sugimoto A, Saito Y, Wang G, Sun Q, Yin C,
Lee KH, Geng Y, Rajbhandari P, Hernandez C, Steffani M, et al:
Hepatic stellate cells control liver zonation, size and functions
via R-spondin 3. Nature. 640:752–761. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Geng Y and Schwabe RF: Hepatic stellate
cell heterogeneity: Functional aspects and therapeutic
implications. Hepatology. May 8–2025.Epub ahead of print.
View Article : Google Scholar
|
|
28
|
Mederacke I, Hsu CC, Troeger JS, Huebener
P, Mu X, Dapito DH, Pradere JP and Schwabe RF: Fate tracing reveals
hepatic stellate cells as dominant contributors to liver fibrosis
independent of its aetiology. Nat Commun. 4:28232013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang SS, Tang XT, Lin M, Yuan J, Peng YJ,
Yin X, Shang G, Ge G, Ren Z and Zhou BO: Perivenous stellate cells
are the main source of myofibroblasts and cancer-associated
fibroblasts formed after chronic liver injuries. Hepatology.
74:1578–1594. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rosenthal SB, Liu X, Ganguly S, Dhar D,
Pasillas MP, Ricciardelli E, Li RZ, Troutman TD, Kisseleva T, Glass
CK and Brenner DA: Heterogeneity of HSCs in a mouse model of NASH.
Hepatology. 74:667–685. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
MacParland SA, Liu JC, Ma XZ, Innes BT,
Bartczak AM, Gage BK, Manuel J, Khuu N, Echeverri J, Linares I, et
al: Single cell RNA sequencing of human liver reveals distinct
intrahepatic macrophage populations. Nat Commun. 9:43832018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xiong X, Kuang H, Ansari S, Liu T, Gong J,
Wang S, Zhao XY, Ji Y, Li C, Guo L, et al: Landscape of
intercellular crosstalk in healthy and NASH liver revealed by
single-cell secretome gene analysis. Mol Cell. 75:644–660.e5. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lotto J, Drissler S, Cullum R, Wei W,
Setty M, Bell EM, Boutet SC, Nowotschin S, Kuo YY, Garg V, et al:
Single-cell transcriptomics reveals early emergence of liver
parenchymal and non-parenchymal cell lineages. Cell. 183:702–716
e14. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu X, Xu J, Rosenthal S, Zhang LJ,
McCubbin R, Meshgin N, Shang L, Koyama Y, Ma HY, Sharma S, et al:
Identification of lineage-specific transcription factors that
prevent activation of hepatic stellate cells and promote fibrosis
resolution. Gastroenterology. 158:1728–1744.e14. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Su Q, Kim SY, Adewale F, Zhou Y, Aldler C,
Ni M, Wei Y, Burczynski ME, Atwal GS, Sleeman MW, et al:
Single-cell RNA transcriptome landscape of hepatocytes and
non-parenchymal cells in healthy and NAFLD mouse liver. iScience.
24:1032332021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fan W, Liu T, Chen W, Hammad S, Longerich
T, Hausser I, Fu Y, Li N, He Y, Liu C, et al: ECM1 prevents
activation of transforming growth factor β, hepatic stellate cells,
and fibrogenesis in mice. Gastroenterology. 157:1352–1367.e13.
2019. View Article : Google Scholar
|
|
37
|
Ben-Moshe S and Itzkovitz S: Spatial
heterogeneity in the mammalian liver. Nat Rev Gastroenterol
Hepatol. 16:395–410. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dobie R, Wilson-Kanamori JR, Henderson
BEP, Smith JR, Matchett KP, Portman JR, Wallenborg K, Picelli S,
Zagorska A, Pendem SV, et al: Single-cell transcriptomics uncovers
zonation of function in the mesenchyme during liver fibrosis. Cell
Rep. 29:1832–1847.e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Watson BR, Paul B, Rahman RU,
Amir-Zilberstein L, Segerstolpe Å, Epstein ET, Murphy S,
Geistlinger L, Lee T, Shih A, et al: Spatial transcriptomics of
healthy and fibrotic human liver at single-cell resolution. Nat
Commun. 16:3192025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Khan MA, Fischer J, Harrer L, Schwiering
F, Groneberg D and Friebe A: Hepatic stellate cells in zone 1
engage in capillarization rather than myofibroblast formation in
murine liver fibrosis. Sci Rep. 14:188402024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ramm GA, Britton RS, O'Neill R, Blaner WS
and Bacon BR: Vitamin A-poor lipocytes: A novel desmin-negative
lipocyte subpopulation, which can be activated to myofibroblasts.
Am J Physiol. 269(4 Pt 1): G532–G541. 1995.PubMed/NCBI
|
|
42
|
Ballardini G, Groff P, Badiali de Giorgi
L, Schuppan D and Bianchi FB: Ito cell heterogeneity:
Desmin-negative Ito cells in normal rat liver. Hepatology.
19:440–446. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
D'Ambrosio DN, Walewski JL, Clugston RD,
Berk PD, Rippe RA and Blaner WS: Distinct populations of hepatic
stellate cells in the mouse liver have different capacities for
retinoid and lipid storage. PLoS One. 6:e249932011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Payen VL, Lavergne A, Alevra Sarika N,
Colonval M, Karim L, Deckers M, Najimi M, Coppieters W, Charloteaux
B, Sokal EM and El Taghdouini A: Single-cell RNA sequencing of
human liver reveals hepatic stellate cell heterogeneity. JHEP Rep.
3:1002782021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kisseleva T, Cong M, Paik Y, Scholten D,
Jiang C, Benner C, Iwaisako K, Moore-Morris T, Scott B, Tsukamoto
H, et al: Myofibroblasts revert to an inactive phenotype during
regression of liver fibrosis. Proc Natl Acad Sci USA.
109:9448–9453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Guo Q, Wu J, Wang Q, Huang Y, Chen L, Gong
J, Du M, Cheng G, Lu T, Zhao M, et al: Single-cell transcriptome
analysis uncovers underlying mechanisms of acute liver injury
induced by tripterygium glycosides tablet in mice. J Pharm Anal.
13:908–925. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cook D, Achanta S, Hoek JB, Ogunnaike BA
and Vadigepalli R: Cellular network modeling and single cell gene
expression analysis reveals novel hepatic stellate cell phenotypes
controlling liver regeneration dynamics. BMC Syst Biol. 12:862018.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kolodziejczyk AA, Federici S, Zmora N,
Mohapatra G, Dori-Bachash M, Hornstein S, Leshem A, Reuveni D,
Zigmond E, Tobar A, et al: Acute liver failure is regulated by MYC-
and microbiome-dependent programs. Nat Med. 26:1899–1911. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kostallari E, Wei B, Sicard D, Li J,
Cooper SA, Gao J, Dehankar M, Li Y, Cao S, Yin M, et al: Stiffness
is associated with hepatic stellate cell heterogeneity during liver
fibrosis. Am J Physiol Gastrointest Liver Physiol. 322:G234–G246.
2022. View Article : Google Scholar :
|
|
50
|
Andrews TS, Atif J, Liu JC, Perciani CT,
Ma XZ, Thoeni C, Slyper M, Eraslan G, Segerstolpe A, Manuel J, et
al: Single-cell, single-nucleus, and spatial RNA sequencing of the
human liver identifies cholangiocyte and mesenchymal heterogeneity.
Hepatol Commun. 6:821–840. 2022. View Article : Google Scholar
|
|
51
|
Wang S, Li K, Pickholz E, Dobie R,
Matchett KP, Henderson NC, Carrico C, Driver I, Borch Jensen M,
Chen L, et al: An autocrine signaling circuit in hepatic stellate
cells underlies advanced fibrosis in nonalcoholic steatohepatitis.
Sci Transl Med. 15:d39492023. View Article : Google Scholar
|
|
52
|
He W, Huang C, Shi X, Wu M, Li H, Liu Q,
Zhang X, Zhao Y and Li X: Single-cell transcriptomics of hepatic
stellate cells uncover crucial pathways and key regulators involved
in non-alcoholic steatohepatitis. Endocr Connect. 12:e2205022023.
View Article : Google Scholar :
|
|
53
|
Cavalli M, Diamanti K, Pan G, Spalinskas
R, Kumar C, Deshmukh AS, Mann M, Sahlén P, Komorowski J and
Wadelius C: A multi-omics approach to liver diseases: Integration
of single nuclei transcriptomics with proteomics and HiCap bulk
data in human liver. OMICS. 24:180–194. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Terkelsen MK, Bendixen SM, Hansen D, Scott
EAH, Moeller AF, Nielsen R, Mandrup S, Schlosser A, Andersen TL,
Sorensen GL, et al: Transcriptional dynamics of hepatic
sinusoid-associated cells after liver injury. Hepatology.
72:2119–2133. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang W, Conway SJ, Liu Y, Snider P, Chen
H, Gao H, Liu Y, Isidan K, Lopez KJ, Campana G, et al:
Heterogeneity of hepatic stellate cells in fibrogenesis of the
liver: Insights from single-cell transcriptomic analysis in liver
injury. Cells. 10:21292021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yin L, Qi Y, Xu Y, Xu L, Han X, Tao X,
Song S and Peng J: Dioscin Inhibits HSC-T6 cell migration via
adjusting SDC-4 Expression: Insights from iTRAQ-Based quantitative
proteomics. Front Pharmacol. 8:6652017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Krenkel O, Puengel T, Govaere O, Abdallah
AT, Mossanen JC, Kohlhepp M, Liepelt A, Lefebvre E, Luedde T,
Hellerbrand C, et al: Therapeutic inhibition of inflammatory
monocyte recruitment reduces steatohepatitis and liver fibrosis.
Hepatology. 67:1270–1283. 2018. View Article : Google Scholar
|
|
58
|
Nevi L, Costantini D, Safarikia S, Di
Matteo S, Melandro F, Berloco PB and Cardinale V:
Cholest-4,6-Dien-3-One promote epithelial-to-mesenchymal transition
(EMT) in biliary tree stem/progenitor cell cultures in vitro.
Cells. 8:14432019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wang Y, Nakajima T, Gonzalez FJ and Tanaka
N: PPARs as metabolic regulators in the liver: Lessons from
liver-specific PPAR-Null mice. Int J Mol Sci. 21:20612020.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Katsumata LW, Miyajima A and Itoh T:
Portal fibroblasts marked by the surface antigen Thy1 contribute to
fibrosis in mouse models of cholestatic liver injury. Hepatol
Commun. 1:198–214. 2017. View Article : Google Scholar
|
|
61
|
Balog S, Fujiwara R, Pan SQ, El-Baradie
KB, Choi HY, Sinha S, Yang Q, Asahina K, Chen Y, Li M, et al:
Emergence of highly profibrotic and proinflammatory Lrat+Fbln2+ HSC
subpopulation in alcoholic hepatitis. Hepatology. 78:212–224. 2023.
View Article : Google Scholar
|
|
62
|
Li X, Wang Q, Ai L and Cheng K: Unraveling
the activation process and core driver genes of HSCs during
cirrhosis by single-cell transcriptome. Exp Biol Med (Maywood).
248:1414–1424. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Chung BK, Ogaard J, Reims HM, Karlsen TH
and Melum E: Spatial transcriptomics identifies enriched gene
expression and cell types in human liver fibrosis. Hepatol Commun.
6:2538–2550. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Fred RG, Steen PJ, Thompson JJ, Lee J,
Timshel PN, Stender S, Opseth Rygg M, Gluud LL, Bjerregaard
Kristiansen V, Bendtsen F, et al: Single-cell transcriptome and
cell type-specific molecular pathways of human non-alcoholic
steatohepatitis. Sci Rep. 12:134842022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wang Z, Zhao Z, Xia Y, Cai Z, Wang C, Shen
Y, Liu R, Qin H, Jia J and Yuan G: Potential biomarkers in the
fibrosis progression of nonalcoholic steatohepatitis (NASH). J
Endocrinol Invest. 45:1379–1392. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Filliol A, Saito Y, Nair A, Dapito DH, Yu
LX, Ravichandra A, Bhattacharjee S, Affo S, Fujiwara N, Su H, et
al: Opposing roles of hepatic stellate cell subpopulations in
hepatocarcinogenesis. Nature. 610:356–365. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yu Y, Li Y, Zhou L, Cheng X and Gong Z:
Hepatic stellate cells promote hepatocellular carcinoma development
by regulating histone lactylation: Novel insights from single-cell
RNA sequencing and spatial transcriptomics analyses. Cancer Lett.
604:2172432024. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wu J, Sun X, Wu C, Hong X, Xie L, Shi Z,
Zhao L, Du Q, Xiao W, Sun J and Wang J: Single-cell transcriptome
analysis reveals liver injury induced by glyphosate in mice. Cell
Mol Biol Lett. 28:112023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bai YM, Yang F, Luo P, Xie LL, Chen JH,
Guan YD, Zhou HC, Xu TF, Hao HW, Chen B, et al: Single-cell
transcriptomic dissection of the cellular and molecular events
underlying the triclosan-induced liver fibrosis in mice. Mil Med
Res. 10:72023.PubMed/NCBI
|
|
70
|
Hu S, Tang B, Lu C, Wang S, Wu L, Lei Y,
Tang L, Zhu H, Wang D and Yang S: Lactobacillus rhamnosus GG
ameliorates triptolide-induced liver injury through modulation of
the bile acid-FXR axis. Pharmacol Res. 206:1072752024. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yang AT, Kim YO, Yan XZ, Abe H, Aslam M,
Park KS, Zhao XY, Jia JD, Klein T, You H and Schuppan D: Fibroblast
activation protein activates macrophages and promotes parenchymal
liver inflammation and fibrosis. Cell Mol Gastroenterol Hepatol.
15:841–867. 2023. View Article : Google Scholar :
|
|
72
|
Li Y, Sheng Q, Zhang C, Han C, Bai H, Lai
P, Fan Y, Ding Y and Dou X: STAT6 up-regulation amplifies M2
macrophage anti-inflammatory capacity through mesenchymal stem
cells. Int Immunopharmacol. 91:1072662021. View Article : Google Scholar
|
|
73
|
Kimura Y, Koyama Y, Taura K, Kudoh A,
Echizen K, Nakamura D, Li X, Nam NH, Uemoto Y, Nishio T, et al:
Characterization and role of collagen gene expressing hepatic cells
following partial hepatectomy in mice. Hepatology. 77:443–455.
2023. View Article : Google Scholar
|
|
74
|
Kumar S, Duan Q, Wu R, Harris EN and Su Q:
Pathophysiological communication between hepatocytes and
non-parenchymal cells in liver injury from NAFLD to liver fibrosis.
Adv Drug Deliv Rev. 176:1138692021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Krizhanovsky V, Yon M, Dickins RA, Hearn
S, Simon J, Miething C, Yee H, Zender L and Lowe SW: Senescence of
activated stellate cells limits liver fibrosis. Cell. 134:657–667.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bernard M, Yang B, Migneault F, Turgeon J,
Dieudé M, Olivier MA, Cardin GB, El-Diwany M, Underwood K, Rodier F
and Hébert MJ: Autophagy drives fibroblast senescence through
MTORC2 regulation. Autophagy. 16:2004–2016. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Yoshimoto S, Loo TM, Atarashi K, Kanda H,
Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et
al: Obesity-induced gut microbial metabolite promotes liver cancer
through senescence secretome. Nature. 499:97–101. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yashaswini CN, Qin T, Bhattacharya D, Amor
C, Lowe S, Lujambio A, Wang S and Friedman SL: Phenotypes and
ontogeny of senescent hepatic stellate cells in metabolic
dysfunction-associated steatohepatitis. J Hepatol. 81:207–217.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Devarbhavi H, Asrani SK, Arab JP, Nartey
YA, Pose E and Kamath PS: Global burden of liver disease: 2023
update. J Hepatol. 79:516–537. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhou Y, Zhang L, Ma Y, Xie L, Yang YY, Jin
C, Chen H, Zhou Y, Song GQ, Ding J and Wu J: Secretome of senescent
hepatic stellate cells favors malignant transformation from
nonalcoholic steatohepatitis-fibrotic progression to hepatocellular
carcinoma. Theranostics. 13:4430–4448. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lujambio A, Akkari L, Simon J, Grace D,
Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA,
Krizhanovsky V and Lowe SW: Non-cell-autonomous tumor suppression
by p53. Cell. 153:449–460. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Cheng N, Kim KH and Lau LF: Senescent
hepatic stellate cells promote liver regeneration through IL-6 and
ligands of CXCR2. JCI Insight. 7:e1582072022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Ramachandran P, Matchett KP, Dobie R,
Wilson-Kanamori JR and Henderson NC: Single-cell technologies in
hepatology: New insights into liver biology and disease
pathogenesis. Nat Rev Gastroenterol Hepatol. 17:457–472. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Xu J, Ma H, Liang S, Sun M, Karin G,
Koyama Y, Hu R, Quehenberger O, Davidson NO, Dennis EA, et al: The
role of human cytochrome P450 2E1 in liver inflammation and
fibrosis. Hepatol Commun. 1:1043–1057. 2017. View Article : Google Scholar
|
|
85
|
Ben-Moshe S, Veg T, Manco R, Dan S,
Papinutti D, Lifshitz A, Kolodziejczyk AA, Bahar Halpern K, Elinav
E and Itzkovitz S: The spatiotemporal program of zonal liver
regeneration following acute injury. Cell Stem Cell.
29:973–989.e10. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Jain I, Brougham-Cook A and Underhill GH:
Effect of distinct ECM microenvironments on the genome-wide
chromatin accessibility and gene expression responses of hepatic
stellate cells. Acta Biomater. 167:278–292. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Brougham-Cook A, Jain I, Kukla DA, Masood
F, Kimmel H, Ryoo H, Khetani SR and Underhill GH: High throughput
interrogation of human liver stellate cells reveals
microenvironmental regulation of phenotype. Acta Biomater.
138:240–253. 2022. View Article : Google Scholar :
|
|
88
|
Cai X, Wang J, Wang J, Zhou Q, Yang B, He
Q and Weng Q: Intercellular crosstalk of hepatic stellate cells in
liver fibrosis: New insights into therapy. Pharmacol Res.
155:1047202020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Li W, Chang N and Li L: Heterogeneity and
function of kupffer cells in liver injury. Front Immunol.
13:9408672022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Dai Q, Ain Q, Seth N, Rooney M and
Zipprich A: Liver sinusoidal endothelial cells: Friend or foe in
metabolic dysfunction-associated steatotic liver disease/metabolic
dysfunction-associated steatohepatitis. Dig Liver Dis. 57:493–503.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wang Z, Du K, Jin N, Tang B and Zhang W:
Macrophage in liver fibrosis: Identities and mechanisms. Int
Immunopharmacol. 120:1103572023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Walesky CM, Kolb KE, Winston CL, Henderson
J, Kruft B, Fleming I, Ko S, Monga SP, Mueller F, Apte U, et al:
Functional compensation precedes recovery of tissue mass following
acute liver injury. Nat Commun. 11:57852020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Lee YA, Wallace MC and Friedman SL:
Pathobiology of liver fibrosis: A translational success story. Gut
2015. 64:830–841. 2015.
|
|
94
|
Wang L, Feng J, Deng Y, Yang Q, Wei Q, Ye
D, Rong X and Guo J: CCAAT/enhancer-binding proteins in fibrosis:
Complex roles beyond conventional understanding. Research (Wash
DC). 2022:98916892022.
|
|
95
|
Lee W, Liu X, Rosenthal SB, Miciano C,
Sakane S, Hokutan K, Dhar D, Kim HY, Brenner DA and Kisseleva T:
ETS1 suppresses hepatic stellate cell activation and liver
fibrosis. JCI Insight. Nov 4–2025.Epub ahead of print. View Article : Google Scholar
|
|
96
|
Altamirano-Barrera A, Barranco-Fragoso B
and Mendez-Sanchez N: Management strategies for liver fibrosis. Ann
Hepatol. 16:48–56. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Guan Y, Fang Z, Hu A, Roberts S, Wang M,
Ren W, Johansson PK, Heilshorn SC, Enejder A and Peltz G: Live-cell
imaging of human liver fibrosis using hepatic micro-organoids. JCI
Insight. 10:e1870992024. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Duspara K, Bojanic K, Pejic JI, Kuna L,
Kolaric TO, Nincevic V, Smolic R, Vcev A, Glasnovic M, Curcic IB
and Smolic M: Targeting the Wnt signaling pathway in liver fibrosis
for drug options: An update. J Clin Transl Hepatol. 9:960–971.
2021.PubMed/NCBI
|
|
99
|
Cheng JH, She H, Han YP, Wang J, Xiong S,
Asahina K and Tsukamoto H: Wnt antagonism inhibits hepatic stellate
cell activation and liver fibrosis. Am J Physiol Gastrointest Liver
Physiol. 294:G39–G49. 2008. View Article : Google Scholar
|
|
100
|
Lv X, Liu C, Liu S, Li Y, Wang W, Li K,
Hua F, Cui B, Zhang X, Yu J, et al: The cell cycle inhibitor P21
promotes the development of pulmonary fibrosis by suppressing lung
alveolar regeneration. Acta Pharm Sin B. 12:735–746. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Xu R, Zhang L, Pan H and Zhang Y: Retinoid
X receptor heterodimers in hepatic function: Structural insights
and therapeutic potential. Front Pharmacol. 15:14646552024.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Friedman SL and Weiskirchen R: Working
with immortalized hepatic stellate cell lines. Methods Mol Biol.
2669:129–162. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Kong X, Feng D, Wang H, Hong F, Bertola A,
Wang FS and Gao B: Interleukin-22 induces hepatic stellate cell
senescence and restricts liver fibrosis in mice. Hepatology.
56:1150–1159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Wang TW, Johmura Y, Suzuki N, Omori S,
Migita T, Yamaguchi K, Hatakeyama S, Yamazaki S, Shimizu E, Imoto
S, et al: Blocking PD-L1-PD-1 improves senescence surveillance and
ageing phenotypes. Nature. 611:358–364. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Akkiz H, Gieseler RK and Canbay A: Liver
fibrosis: From basic science towards clinical progress, focusing on
the central role of hepatic stellate cells. Int J Mol Sci.
25:78732024. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Du K, Maeso-Diaz R, Oh SH, Wang E, Chen T,
Pan C, Xiang K, Dutta RK, Wang XF, Chi JT and Diehl AM: Targeting
YAP-mediated HSC death susceptibility and senescence for treatment
of liver fibrosis. Hepatology. 77:1998–2015. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Liu R, Li Y, Zheng Q, Ding M, Zhou H and
Li X: Epigenetic modification in liver fibrosis: Promising
therapeutic direction with significant challenges ahead. Acta Pharm
Sin B. 14:1009–1029. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Page A, Paoli P, Moran Salvador E, White
S, French J and Mann J: Hepatic stellate cell transdifferentiation
involves genome-wide remodeling of the DNA methylation landscape. J
Hepatol. 64:661–673. 2016. View Article : Google Scholar :
|
|
109
|
Li N, Liu S, Zhang Y, Yu L, Hu Y, Wu T,
Fang M and Xu Y: Transcriptional activation of matricellular
protein spondin2 (SPON2) by BRG1 in vascular endothelial cells
promotes macrophage chemotaxis. Front Cell Dev Biol. 8:7942020.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Gotze S, Schumacher EC, Kordes C and
Häussinger D: Epigenetic changes during Hepatic stellate cell
activation. PLoS One. 10:e1287452015. View Article : Google Scholar
|
|
111
|
Dewidar B, Meyer C, Dooley S and
Meindl-Beinker AN: TGF-β in hepatic stellate cell activation and
liver fibrogenesis-updated 2019. Cells. 8:14192019. View Article : Google Scholar
|
|
112
|
Huang YH, Yang YL and Wang FS: The role of
miR-29a in the regulation, function, and signaling of liver
fibrosis. Int J Mol Sci. 19:18892018. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Dou C, Liu Z, Tu K, Zhang H, Chen C,
Yaqoob U, Wang Y, Wen J, van Deursen J, Sicard D, et al: P300
acetyltransferase mediates stiffness-induced activation of hepatic
stellate cells into tumor-promoting myofibroblasts.
Gastroenterology. 154:2209–2221.e14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Paluvai H, Di Giorgio E and Brancolini C:
The histone code of senescence. Cells. 9:4662020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Dong F, Jiang S, Li J, Wang Y, Zhu L,
Huang Y, Jiang X, Hu X, Zhou Q, Zhang Z and Bao Z: The histone
demethylase KDM4D promotes hepatic fibrogenesis by modulating
Toll-like receptor 4 signaling pathway. EBioMedicine. 39:472–483.
2019. View Article : Google Scholar :
|
|
116
|
Gerovska D, Garcia-Gallastegi P, Crende O,
Márquez J, Larrinaga G, Unzurrunzaga M, Araúzo-Bravo MJ and Badiola
I: GeromiRs are downregulated in the tumor microenvironment during
colon cancer colonization of the liver in a murine metastasis
model. Int J Mol Sci. 22:48192021. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Gholizadeh M, Szelag-Pieniek S, Post M,
Kurzawski M, Prieto J, Argemi J, Drozdzik M and Kaderali L:
Identifying differentially expressed MicroRNAs, target genes, and
key pathways deregulated in patients with liver diseases. Int J Mol
Sci. 21:73682020. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Chen X, Li WX, Chen Y, Li XF, Li HD, Huang
HM, Bu FT, Pan XY, Yang Y, Huang C, et al: Suppression of SUN2 by
DNA methylation is associated with HSCs activation and hepatic
fibrosis. Cell Death Dis. 9:10212018. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Bates J, Vijayakumar A, Ghoshal S,
Marchand B, Yi S, Kornyeyev D, Zagorska A, Hollenback D, Walker K,
Liu K, et al: Acetyl-CoA carboxylase inhibition disrupts metabolic
reprogramming during hepatic stellate cell activation. J Hepatol.
73:896–905. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Horn P and Tacke F: Metabolic
reprogramming in liver fibrosis. Cell Metab. 36:1439–1455. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
McCommis KS, Hodges WT, Brunt EM,
Nalbantoglu I, McDonald WG, Holley C, Fujiwara H, Schaffer JE,
Colca JR and Finck BN: Targeting the mitochondrial pyruvate carrier
attenuates fibrosis in a mouse model of nonalcoholic
steatohepatitis. Hepatology. 65:1543–1556. 2017. View Article : Google Scholar
|
|
122
|
Benedicto AM, Lucantoni F, Fuster-Martinez
I, Diaz-Pozo P, Dorcaratto D, Muñoz-Forner E, Victor VM, Esplugues
JV, Blas-García A and Apostolova N: Interference with mitochondrial
function as part of the antifibrogenic effect of Rilpivirine: A
step towards novel targets in hepatic stellate cell activation.
Biomed Pharmacother. 178:1172062024. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Smith-Cortinez N, van Eunen K, Heegsma J,
Serna-Salas SA, Sydor S, Bechmann LP, Moshage H, Bakker BM and
Faber KN: Simultaneous induction of glycolysis and oxidative
phosphorylation during activation of hepatic stellate cells reveals
novel mitochondrial targets to treat liver fibrosis. Cells.
9:24562020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Cho EH: Succinate as a regulator of
hepatic stellate cells in liver fibrosis. Front Endocrinol
(Lausanne). 9:4552018. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Sinha S, Aizawa S, Nakano Y, Rialdi A,
Choi HY, Shrestha R, Pan SQ, Chen Y, Li M, Kapelanski-Lamoureux A,
et al: Hepatic stellate cell stearoyl co-A desaturase activates
leukotriene B4 receptor 2 - β-catenin cascade to promote liver
tumorigenesis. Nat Commun. 14:26512023. View Article : Google Scholar
|
|
126
|
Li J, Yao S, Zimny S, Koob D, Jin H,
Wimmer R, Denk G, Tuo B and Hohenester S: The acidic
microenvironment in the perisinusoidal space critically determines
bile salt-induced activation of hepatic stellate cells. Commun
Biol. 7:15912024. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Yasukawa K, Shimada S, Akiyama Y, Taniai
T, Igarashi Y, Tsukihara S, Tanji Y, Umemura K, Kamachi A, Nara A,
et al: ACVR2A attenuation impacts lactate production and
hyperglycolytic conditions attracting regulatory T cells in
hepatocellular carcinoma. Cell Rep Med. 6:1020382025. View Article : Google Scholar
|
|
128
|
Kikuchi K and Tsukamoto H: Stearoyl-CoA
desaturase and tumorigenesis. Chem Biol Interact. 316:1089172020.
View Article : Google Scholar :
|
|
129
|
Tian XP, Wang CY, Jin XH, Li M, Wang FW,
Huang WJ, Yun JP, Xu RH, Cai QQ and Xie D: Acidic microenvironment
up-regulates exosomal miR-21 and miR-10b in early-stage
hepatocellular carcinoma to promote cancer cell proliferation and
metastasis. Theranostics. 9:1965–1979. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Gallego-Duran R, Ampuero J, Pastor-Ramirez
H, Álvarez-Amor L, Del Campo JA, Maya-Miles D, Montero-Vallejo R,
Cárdenas-García A, Pareja MJ, Gato-Zambrano S, et al: Liver injury
in non-alcoholic fatty liver disease is associated with urea cycle
enzyme dysregulation. Sci Rep. 12:34182022. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Wu B, Feng J, Guo J, Wang J, Xiu G and Xu
J, Ning K, Ling B, Fu Q and Xu J: ADSCs-derived exosomes ameliorate
hepatic fibrosis by suppressing stellate cell activation and
remodeling hepatocellular glutamine synthetase-mediated glutamine
and ammonia homeostasis. Stem Cell Res Ther. 13:4942022. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Li L, Mao Y, Zhao L, Li L, Wu J, Zhao M,
Du W, Yu L and Jiang P: p53 regulation of ammonia metabolism
through urea cycle controls polyamine biosynthesis. Nature.
567:253–256. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Thomsen KL, Eriksen PL, Kerbert AJ, De
Chiara F, Jalan R and Vilstrup H: Role of ammonia in NAFLD: An
unusual suspect. JHEP Rep. 5:1007802023. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Zheng W, Guan F, Xu G, Yu Y, Xiao J and
Huang X: FAT10 silencing prevents liver fibrosis through regulating
SIRT1 expression in hepatic stellate cells. Int J Med Sci.
20:557–565. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
He J, Deng C, Krall L and Shan Z:
ScRNA-seq and ST-seq in liver research. Cell Regen. 12:112023.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Gilgenkrantz H, Mallat A, Moreau R and
Lotersztajn S: Targeting cell-intrinsic metabolism for antifibrotic
therapy. J Hepatol. 74:1442–1454. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Sun Y, Chen X, Chen L, Bao B, Li C and
Zhou Y: MFAP2 promotes HSCs activation through FBN1/TGF-β/Smad3
pathway. J Cell Mol Med. 27:3235–3246. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Li HM, Zhang JY, Wang XQ, Ye LT, Ren B,
Leng YH, Zhang JX, Yang Y, Jiang Q, Feng LL, et al: Therapeutic
potential of PDA@CeO2 in suppressing hepatic stellate cell
activation and preventing liver fibrosis. Int J Nanomedicine.
20:9073–9091. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Li C, Zhan Y, Zhang R, Tao Q, Lang Z and
Zheng J: 20(S)-Protopanaxadiol suppresses hepatic stellate cell
activation via WIF1 demethylation-mediated inactivation of the
Wnt/β-catenin pathway. J Ginseng Res. 47:515–523. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Zhang Z, Shang J, Yang Q, Dai Z, Liang Y,
Lai C, Feng T, Zhong D, Zou H, Sun L, et al: Exosomes derived from
human adipose mesenchymal stem cells ameliorate hepatic fibrosis by
inhibiting PI3K/Akt/mTOR pathway and remodeling choline metabolism.
J Nanobiotechnology. 21:292023. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Loomba R, Kayali Z, Noureddin M, Ruane P,
Lawitz EJ, Bennett M, Wang L, Harting E, Tarrant JM, McColgan BJ,
et al: GS-0976 reduces hepatic steatosis and fibrosis markers in
patients with nonalcoholic fatty liver disease. Gastroenterology.
155:1463–1473.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Bhattacharya D, Basta B, Mato JM, Craig A,
Fernández-Ramos D, Lopitz-Otsoa F, Tsvirkun D, Hayardeny L, Chandar
V, Schwartz RE, et al: Aramchol downregulates stearoyl
CoA-desaturase 1 in hepatic stellate cells to attenuate cellular
fibrogenesis. JHEP Rep. 3:1002372021. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Ratziu V, de Guevara L, Safadi R, Poordad
F, Fuster F, Flores-Figueroa J, Arrese M, Fracanzani AL, Ben Bashat
D, Lackner K, et al: Aramchol in patients with nonalcoholic
steatohepatitis: A randomized, double-blind, placebo-controlled
phase 2b trial. Nat Med. 27:1825–1835. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Tacke F, Puengel T, Loomba R and Friedman
SL: An integrated view of anti-inflammatory and antifibrotic
targets for the treatment of NASH. J Hepatol. 79:552–566. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Harrison SA, Bashir MR, Guy CD, Zhou R,
Moylan CA, Frias JP, Alkhouri N, Bansal MB, Baum S,
Neuschwander-Tetri BA, et al: Resmetirom (MGL-3196) for the
treatment of non-alcoholic steatohepatitis: A multicentre,
randomised, double-blind, placebo-controlled, phase 2 trial.
Lancet. 394:2012–2024. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Yabut JM and Drucker DJ: Glucagon-like
Peptide-1 Receptor-based therapeutics for metabolic liver disease.
Endocr Rev. 44:14–32. 2023. View Article : Google Scholar
|
|
147
|
Desjardins EM, Wu J, Lavoie DCT, Ahmadi E,
Townsend LK, Morrow MR, Wang D, Tsakiridis EE, Batchuluun B,
Fayyazi R, et al: Combination of an ACLY inhibitor with a GLP-1R
agonist exerts additive benefits on nonalcoholic steatohepatitis
and hepatic fibrosis in mice. Cell Rep Med. 4:1011932023.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Giannelli G, Santoro A, Kelley RK, Gane E,
Paradis V, Cleverly A, Smith C, Estrem ST, Man M, Wang S, et al:
Biomarkers and overall survival in patients with advanced
hepatocellular carcinoma treated with TGF-βRI inhibitor
galunisertib. PLoS One. 15:e2222592020. View Article : Google Scholar
|
|
149
|
Faivre S, Santoro A, Kelley RK, Gane E,
Costentin CE, Gueorguieva I, Smith C, Cleverly A, Lahn MM, Raymond
E, et al: Novel transforming growth factor beta receptor I kinase
inhibitor galunisertib (LY2157299) in advanced hepatocellular
carcinoma. Liver Int. 39:1468–1477. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Brauer NR, Kempen AL, Hernandez D and
Sintim HO: Non-kinase off-target inhibitory activities of
clinically-relevant kinase inhibitors. Eur J Med Chem.
275:1165402024. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Pesce A, Ciurleo R, Bramanti A, Armeli
Iapichino EC, Petralia MC, Magro GG, Fagone P, Bramanti P,
Nicoletti F and Mangano K: Effects of combined admistration of
imatinib and sorafenib in a murine model of liver fibrosis.
Molecules. 25:43102020. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Hany NM, Eissa S, Basyouni M, Hasanin AH,
Aboul-Ela YM, Elmagd NMA, Montasser IF, Ali MA, Skipp PJ and
Matboli M: Modulation of hepatic stellate cells by Mutaflor((R))
probiotic in non-alcoholic fatty liver disease management. J Transl
Med. 20:3422022. View Article : Google Scholar
|
|
153
|
Habash NW, Sehrawat TS, Shah VH and Cao S:
Epigenetics of alcohol-related liver diseases. JHEP Rep.
4:1004662022. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Pan XY, You HM, Wang L, Bi YH, Yang Y,
Meng HW, Meng XM, Ma TT, Huang C and Li J: Methylation of RCAN1.4
mediated by DNMT1 and DNMT3b enhances hepatic stellate cell
activation and liver fibrogenesis through Calcineurin/NFAT3
signaling. Theranostics. 9:4308–4323. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Wang X, He Y, Mackowiak B and Gao B:
MicroRNAs as regulators, biomarkers and therapeutic targets in
liver diseases. Gut. 70:784–795. 2021. View Article : Google Scholar
|
|
156
|
Lucantoni F, Martinez-Cerezuela A,
Gruevska A, Moragrega ÁB, Víctor VM, Esplugues JV, Blas-García A
and Apostolova N: Understanding the implication of autophagy in the
activation of hepatic stellate cells in liver fibrosis: Are we
there yet? J Pathol. 254:216–228. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Le TV, Phan-Thi HT, Huynh-Thi MX, Dang TM,
Holterman AXL, Grassi G, Nguyen-Luu TU and Truong NH: Autophagy
inhibitor chloroquine downmodulates hepatic stellate cell
activation and liver damage in bile-duct-ligated mice. Cells.
12:10252023. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Yu X, Elfimova N, Muller M, Bachurski D,
Koitzsch U, Drebber U, Mahabir E, Hansen HP, Friedman SL, Klein S,
et al: Autophagy-related activation of hepatic stellate cells
reduces cellular miR-29a by promoting its vesicular secretion. Cell
Mol Gastroenterol Hepatol. 13:1701–1716. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Zhang Z, Yao Z, Zhao S, Shao J, Chen A,
Zhang F and Zheng S: Interaction between autophagy and senescence
is required for dihydroartemisinin to alleviate liver fibrosis.
Cell Death Dis. 8:e28862017. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Bao YN, Yang Q, Shen XL, Yu WK, Zhou L,
Zhu QR, Shan QY, Wang ZC and Cao G: Targeting tumor suppressor p53
for organ fibrosis therapy. Cell Death Dis. 15:3362024. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Yan M, Cui Y and Xiang Q: Metabolism of
hepatic stellate cells in chronic liver diseases: Emerging
molecular and therapeutic interventions. Theranostics.
15:1715–1740. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
He S, Luo Y, Ma W, Wang X, Yan C, Hao W,
Fang Y, Su H, Lai B, Liu J, et al: Endothelial POFUT1 controls
injury-induced liver fibrosis by repressing fibrinogen synthesis. J
Hepatol. 81:135–148. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Kisseleva T and Brenner D: Molecular and
cellular mechanisms of liver fibrosis and its regression. Nat Rev
Gastroenterol Hepatol. 18:151–166. 2021. View Article : Google Scholar
|
|
164
|
Maretti-Mira AC, Salomon MP, Hsu AM, Dara
L and Golden-Mason L: Etiology of end-stage liver cirrhosis impacts
hepatic natural killer cell heterogenicity. Front Immunol.
14:11370342023. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Xu F, Gao Y, Li T, Jiang T, Wu X, Yu Z,
Zhang J, Hu Y and Cao J: Single-cell sequencing reveals the
heterogeneity of hepatic natural killer cells and identifies the
cytotoxic natural killer subset in schistosomiasis mice. Int J Mol
Sci. 26:32112025. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Ma X, Qiu J, Zou S, Tan L and Miao T: The
role of macrophages in liver fibrosis: Composition, heterogeneity,
and therapeutic strategies. Front Immunol. 15:14942502024.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Wang WM, Xu XS and Miao CM: Kupffer
cell-derived TNF-α triggers the apoptosis of hepatic stellate cells
through TNF-R1/Caspase 8 due to ER stress. Biomed Res Int.
2020:80356712020. View Article : Google Scholar
|
|
168
|
Jia K, Ma Z, Zhang Y, Xie K, Li J, Wu J,
Qu J, Li F and Li X: Picroside II promotes HSC apoptosis and
inhibits the cholestatic liver fibrosis in Mdr2(-/-) mice by
polarizing M1 macrophages and balancing immune responses. Chin J
Nat Med. 22:582–598. 2024.PubMed/NCBI
|
|
169
|
Li Y, Zhang Y, Pan G, Xiang LX, Luo DC and
Shao JZ: Occurrences and functions of Ly6C(hi) and Ly6C(lo)
macrophages in health and disease. Front Immunol. 13:9016722022.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Zhou Z, Qi J, Zhao J, Lim CW, Kim JW and
Kim B: Dual TBK1/IKKε inhibitor amlexanox attenuates the severity
of hepatotoxin-induced liver fibrosis and biliary fibrosis in mice.
J Cell Mol Med. 24:1383–1398. 2020. View Article : Google Scholar
|
|
171
|
Lai Q, Li W, Hu D, Huang Z, Wu M, Feng S
and Wan Y: Hepatic stellate cell-targeted chemo-gene therapy for
liver fibrosis using fluorinated peptide-lipid hybrid
nanoparticles. J Control Release. 376:601–617. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Lee SW, Kim SM, Hur W, Kang BY, Lee HL,
Nam H, Yoo SH, Sung PS, Kwon JH, Jang JW, et al: Tenofovir
disoproxil fumarate directly ameliorates liver fibrosis by inducing
hepatic stellate cell apoptosis via downregulation of PI3K/Akt/mTOR
signaling pathway. PLoS One. 16:e2610672021. View Article : Google Scholar
|
|
173
|
Fondevila MF, Novoa E, Gonzalez-Rellan MJ,
Fernandez U, Heras V, Porteiro B, Parracho T, Dorta V, Riobello C,
da Silva Lima N, et al: p63 controls metabolic activation of
hepatic stellate cells and fibrosis via an HER2-ACC1 pathway. Cell
Rep Med. 5:1014012024. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Trinh VQ, Lee TF, Lemoinne S, Ray KC,
Ybanez MD, Tsuchida T, Carter JK, Agudo J, Brown BD, Akat KM, et
al: Hepatic stellate cells maintain liver homeostasis through
paracrine neurotrophin-3 signaling that induces hepatocyte
proliferation. Sci Signal. 16:f66962023. View Article : Google Scholar
|
|
175
|
Yang F, Li H, Li Y, Hao Y, Wang C, Jia P,
Chen X, Ma S and Xiao Z: Crosstalk between hepatic stellate cells
and surrounding cells in hepatic fibrosis. Int Immunopharmacol.
99:1080512021. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Vyas K and Patel MM: Insights on drug and
gene delivery systems in liver fibrosis. Asian J Pharm Sci.
18:1007792023.PubMed/NCBI
|
|
177
|
Lawitz EJ, Shevell DE, Tirucherai GS, Du
S, Chen W, Kavita U, Coste A, Poordad F, Karsdal M, Nielsen M, et
al: BMS-986263 in patients with advanced hepatic fibrosis: 36-week
results from a randomized, placebo-controlled phase 2 trial.
Hepatology. 75:912–923. 2022. View Article : Google Scholar
|
|
178
|
Matsumoto Y, Itami S, Kuroda M, Yoshizato
K, Kawada N and Murakami Y: MiR-29a assists in preventing the
activation of human stellate cells and promotes recovery from liver
fibrosis in mice. Mol Ther. 24:1848–1859. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Zhang C, Teng Y, Li F, Ho W, Bai X, Xu X
and Zhang XQ: Nanoparticle-Mediated RNA therapy attenuates
nonalcoholic steatohepatitis and related fibrosis by targeting
activated hepatic stellate cells. ACS Nano. 17:14852–14870. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Weber F, Casalini T, Valentino G,
Brülisauer L, Andreas N, Koeberle A, Kamradt T, Contini A and
Luciani P: Targeting transdifferentiated hepatic stellate cells and
monitoring the hepatic fibrogenic process by means of
IGF2R-specific peptides designed in silico. J Mater Chem B.
9:2092–2106. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Puche JE, Lee YA, Jiao J, Aloman C, Fiel
MI, Muñoz U, Kraus T, Lee T, Yee HF Jr and Friedman SL: A novel
murine model to deplete hepatic stellate cells uncovers their role
in amplifying liver damage in mice. Hepatology. 57:339–350. 2013.
View Article : Google Scholar
|
|
182
|
Cortes E, Lachowski D, Rice A, Thorpe SD,
Robinson B, Yeldag G, Lee DA, Ghemtio L, Rombouts K and Del Río
Hernández AE: Tamoxifen mechanically deactivates hepatic stellate
cells via the G protein-coupled estrogen receptor. Oncogene.
38:2910–2922. 2019. View Article : Google Scholar :
|
|
183
|
Harrison SA, Abdelmalek MF, Caldwell S,
Shiffman ML, Diehl AM, Ghalib R, Lawitz EJ, Rockey DC, Schall RA,
Jia C, et al: Simtuzumab is ineffective for patients with bridging
fibrosis or compensated cirrhosis caused by nonalcoholic
steatohepatitis. Gastroenterology. 155:1140–1153. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Masuda A, Nakamura T, Abe M, Iwamoto H,
Sakaue T, Tanaka T, Suzuki H, Koga H and Torimura T: Promotion of
liver regeneration and anti-fibrotic effects of the TGF-β receptor
kinase inhibitor galunisertib in CCl4-treated mice. Int J Mol Med.
46:427–438. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Huang J, Huang H, Wang Y, Xu B, Lin M, Han
S, Yuan Y, Wang Y and Shuai X: Retinol-binding protein-hijacking
nanopolyplex delivering siRNA to cytoplasm of hepatic stellate cell
for liver fibrosis alleviation. Biomaterials. 299:1221342023.
View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Ramani K, Mavila N, Abeynayake A, Tomasi
ML, Wang J, Matsuda M and Seki E: Targeting A-kinase anchoring
protein 12 phosphorylation in hepatic stellate cells regulates
liver injury and fibrosis in mouse models. Elife. 11:e784302022.
View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Hazra S, Xiong S, Wang J, Rippe RA,
Krishna V, Chatterjee K and Tsukamoto H: Peroxisome
proliferator-activated receptor gamma induces a phenotypic switch
from activated to quiescent hepatic stellate cells. J Biol Chem.
279:11392–11401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Nakano Y, Kamiya A, Sumiyoshi H, Tsuruya
K, Kagawa T and Inagaki Y: A deactivation factor of fibrogenic
hepatic stellate cells induces regression of liver fibrosis in
mice. Hepatology. 71:1437–1452. 2020. View Article : Google Scholar
|
|
189
|
Luo J, Li L, Chang B, Zhu Z, Deng F, Hu M,
Yu Y, Lu X, Chen Z, Zuo D and Zhou J: Mannan-binding lectin via
interaction with cell surface calreticulin promotes senescence of
activated hepatic stellate cells to limit liver fibrosis
progression. Cell Mol Gastroenterol Hepatol. 14:75–99. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Guo Q, Chen M, Chen Q, Xiao G, Chen Z,
Wang X and Huang Y: Silencing p53 inhibits interleukin 10-induced
activated hepatic stellate cell senescence and fibrotic degradation
in vivo. Exp Biol Med (Maywood). 246:447–458. 2021. View Article : Google Scholar
|
|
191
|
Huang YH, Chen MH, Guo QL, Chen ZX, Chen
QD and Wang XZ: Interleukin-10 induces senescence of activated
hepatic stellate cells via STAT3-p53 pathway to attenuate liver
fibrosis. Cell Signal. 66:1094452020. View Article : Google Scholar
|
|
192
|
Barcena-Varela M, Caruso S, Llerena S,
Álvarez-Sola G, Uriarte I, Latasa MU, Urtasun R, Rebouissou S,
Alvarez L, Jimenez M, et al: Dual targeting of histone
methyltransferase G9a and DNA-Methyltransferase 1 for the treatment
of experimental hepatocellular carcinoma. Hepatology. 69:587–603.
2019. View Article : Google Scholar
|
|
193
|
Barcena-Varela M, Paish H, Alvarez L,
Uriarte I, Latasa MU, Santamaria E, Recalde M, Garate M, Claveria
A, Colyn L, et al: Epigenetic mechanisms and metabolic
reprogramming in fibrogenesis: Dual targeting of G9a and DNMT1 for
the inhibition of liver fibrosis. Gut. 70:388–400. 2021. View Article : Google Scholar
|
|
194
|
Lu P, Yan M, He L, Li J, Ji Y and Ji J:
Crosstalk between epigenetic modulations in valproic acid
deactivated hepatic stellate cells: An integrated protein and miRNA
profiling study. Int J Biol Sci. 15:93–104. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Yang J, Lu Y, Yang P, Chen Q, Wang Y, Ding
Q, Xu T, Li X, Li C, Huang C, et al: MicroRNA-145 induces the
senescence of activated hepatic stellate cells through the
activation of p53 pathway by ZEB2. J Cell Physiol. 234:7587–7599.
2019. View Article : Google Scholar
|
|
196
|
Song G, Pacher M, Balakrishnan A, Yuan Q,
Tsay HC, Yang D, Reetz J, Brandes S, Dai Z, Pützer BM, et al:
Direct reprogramming of hepatic myofibroblasts into hepatocytes in
vivo attenuates liver fibrosis. Cell Stem Cell. 18:797–808. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Rezvani M, Espanol-Suner R, Malato Y,
Dumont L, Grimm AA, Kienle E, Bindman JG, Wiedtke E, Hsu BY, Naqvi
SJ, et al: In vivo hepatic reprogramming of myofibroblasts with AAV
Vectors as a therapeutic strategy for liver fibrosis. Cell Stem
Cell. 18:809–816. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Heslop JA and Duncan SA: FoxA factors: The
chromatin key and doorstop essential for liver development and
function. Genes Dev. 34:1003–1004. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Battle MA, Konopka G, Parviz F, Gaggl AL,
Yang C, Sladek FM and Duncan SA: Hepatocyte nuclear factor 4alpha
orchestrates expression of cell adhesion proteins during the
epithelial transformation of the developing liver. Proc Natl Acad
Sci USA. 103:8419–8424. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Mannaerts I, Leite SB, Verhulst S,
Claerhout S, Eysackers N, Thoen LF, Hoorens A, Reynaert H, Halder G
and van Grunsven LA: The Hippo pathway effector YAP controls mouse
hepatic stellate cell activation. J Hepatol. 63:679–688. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Du K, Hyun J, Premont RT, Choi SS,
Michelotti GA, Swiderska-Syn M, Dalton GD, Thelen E, Rizi BS, Jung
Y and Diehl AM: Hedgehog-YAP signaling pathway regulates
glutaminolysis to control activation of hepatic stellate cells.
Gastroenterology. 154:1465–1479.e13. 2018. View Article : Google Scholar : PubMed/NCBI
|