B-cells develop from hematopoietic stem cells (HSCs)
in the bone marrow (BM) of adult mammals or in the fetal liver
during embryogenesis (1). The
sequential and coordinated expression of lineage-specifying
transcription factors, including E2A, PU.1, Ikaros and EBF1, drives
HSCs into early lymphoid progenitors (ELPs), which activate
lymphoid-restricted gene programs indicative of B-cell potential
(2). ELPs, characterized by
interleukin (IL)-7 receptor (IL-7R, CD127) negativity, subsequently
differentiate into common lymphoid progenitors (CLPs) in the BM.
CLPs serve as immediate precursors not only for B-cells, but also
for T-cells, natural killer cells and dendritic cells (3). B-cell maturation proceeds through
discrete stages, including pro-B, pre-B, immature B and mature
B-cells, under the coordinated control of transcription factors
(including E2A, Pax5, PU.1, Ikaros, EBF1, Sox4, IRF4 and IRF8) and
cytokines such as IL-7 (1).
During these stages, developing B-cells undergo tightly ordered
V(D)J recombination events to assemble a functional,
non-autoreactive heterodimeric B-cell receptor (BCR), thereby
equipping them for antigen recognition and adaptive immune
responses. Once they migrate to secondary lymphoid organs, mature
B-cells can be activated via T-cell-dependent (TD) or
T-cell-independent (TI) pathways and differentiate into
antibody-secreting plasma cells (PCs) and long-lived memory B-cells
(MBCs) (4).
In contrast to genetic mechanisms, epigenetic
mechanisms regulate gene expression without altering the DNA
sequence, thereby modulating cellular metabolism and function
(5). Emerging evidence indicates
that epigenetic regulation plays a crucial role in establishing
B-cell lineage identity and effector functions. The dysregulation
of DNA methylation, histone modifications and the altered
expression of long non-coding RNAs (lncRNAs) and microRNAs
(miRNAs/miRs) can impair B-cell tolerance and contribute to the
pathogenesis of systemic lupus erythematosus (SLE), rheumatoid
arthritis (RA), diffuse large B-cell lymphoma (DLBCL), follicular
lymphoma (FL) and mantle cell lymphoma (MCL) (6-8).
Dissecting the specific epigenetic perturbations that drive these
pathologies may reveal novel biomarkers and therapeutic targets.
Indeed, clinical and preclinical studies of autoimmune disorders
and B-cell malignancies have shown that agents capable of reversing
aberrant epigenetic states, such as histone deacetylase and DNA
methyltransferase inhibitors, hold significant therapeutic
potential (7,9). A comprehensive understanding of
epigenetic dysregulation in disease is crucial for elucidating
pathogenic mechanisms and designing targeted epigenetic therapies.
The present review aimed to provide an overview of current insights
into the epigenetic regulation of B-cell development and the
consequences of its dysregulation in autoimmune diseases and B-cell
lymphomas.
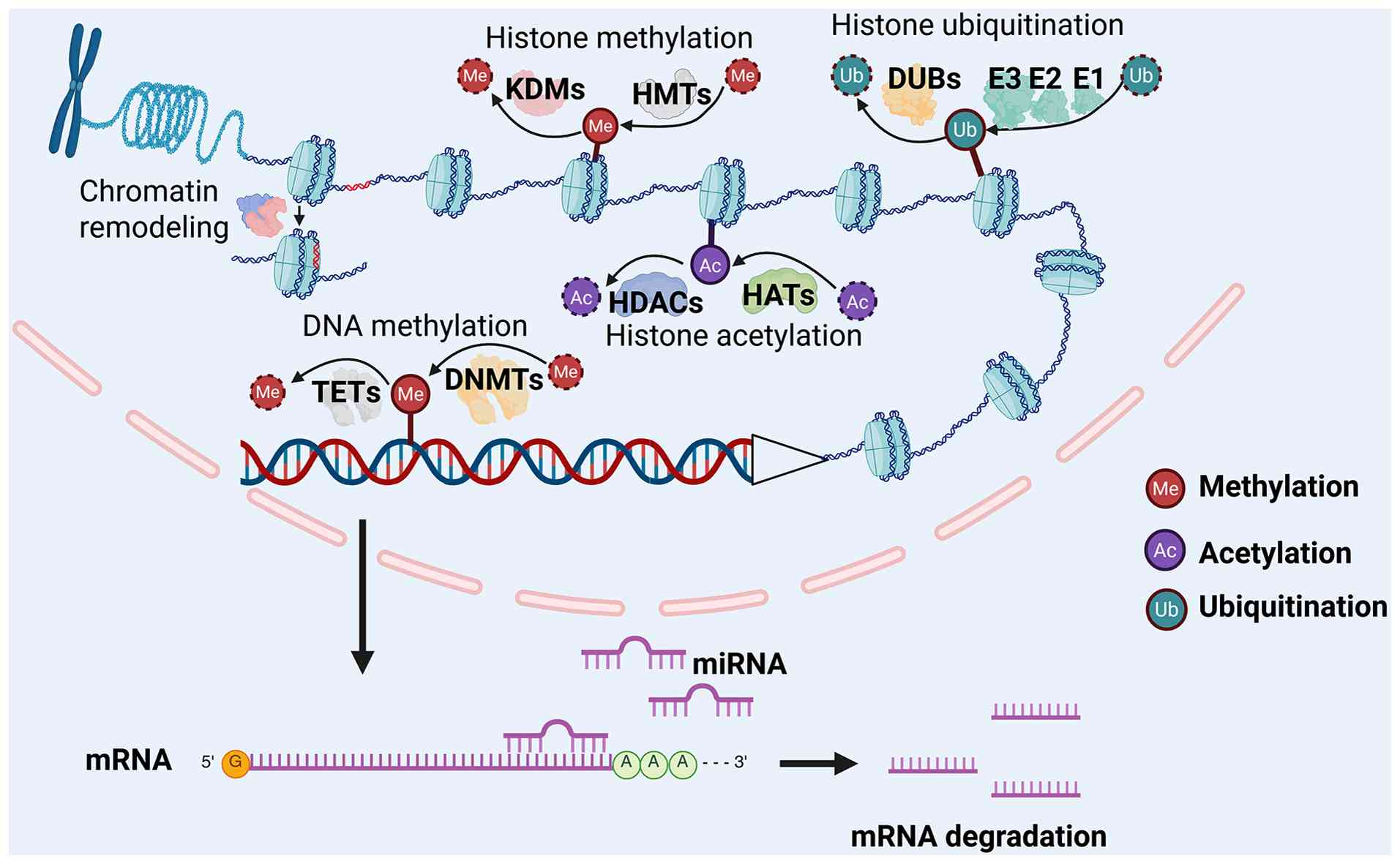
Eukaryotic DNA is organized into chromatin, with the
nucleosome as the fundamental repeating unit, consisting of ~147
base pairs of DNA wrapped around an octamer of core histones
(10). Nucleosomes are further
compacted and folded to form higher-order chromatin structures.
Dynamic changes in chromatin structure play a crucial role in gene
transcription. Generally, the chromatin in the transcriptional
silencing region is highly packaged, while the chromatin structure
is loosed in the active transcriptional region, which facilitates
the binding of transcription regulators (11,12). Increasing evidence indicates that
epigenetic modifications, including chromatin remodeling, DNA
methylation, histone modifications, lncRNAs and miRNA activity, are
key drivers of chromatin structural dynamics and gene regulation
(13,14). The processes of chromatin
remodeling (with nucleosome sliding as an illustrative example),
DNA methylation, major histone modifications, and miRNA-mediated
regulation are illustrated in Fig.
1.
Chromatin remodeling is an ATP-dependent process
that modulates nucleosome architecture to regulate both
transcription initiation and elongation. Of note, four principal
families of remodelers have been defined based on their
Swi2/Snf2-like ATPase domains: SWI/SNF (such as BAF, PBAF and WINAC
complexes), ISWI (such as ACF and NURF complexes), CHD (such as
NURD complexes) and INO80/SWR1 (such as SRCAP and Tip60/EP400
complexes) (15,16). These complexes target specific
genomic loci through interactions with sequence-specific
transcription factors or histone marks, where they function as
coactivators or corepressors. Mechanistically, chromatin remodelers
promote gene activation or repression by sliding or evicting
nucleosomes, exchanging core histones for histone variants, or
altering nucleosome spacing to fine-tune DNA accessibility for the
transcriptional machinery (15,17). Beyond transcriptional control,
remodelers also coordinate with histone-modifying enzymes and the
RNA polymerase II complex to ensure proper promoter clearance,
pause release and elongation processivity, and they play critical
roles in development, cell differentiation and disease pathogenesis
(18).
As a prototypical epigenetic mark, DNA methylation
persists throughout the lifespan of the cell. During the
methylation process, a methyl group is covalently added to a
specific base in the DNA sequence, catalyzed by DNA
methyltransferases (DNMTs). DNMT3A, DNMT3B, and the catalytically
inactive DNMT3L establish de novo methylation patterns in
mammals, while DNMT1 maintains these patterns during DNA
replication (19). DNA
methylation is reversible; it can be actively removed by ten-eleven
translocation (TET) enzymes or passively lost through
replication-dependent dilution (20). In eukaryotes, methylation occurs
predominantly at cytosine residues in CpG dinucleotides
(5'-CpG-3'), although adenine methylation has also been observed.
DNA methylation exerts a profound impact on gene expression by
interfering with transcription factor binding, influencing
chromatin compaction, and recruiting methyl-CpG binding domain
proteins (21).
Four core histones and their variant isoforms
assemble into the fundamental repeating unit of chromatin, the
nucleosome. These nucleosomal histones undergo a variety of
covalent post-translational modifications, such as methylation,
acetylation, phosphorylation, ADP-ribosylation, ubiquitination, and
SUMOylation, which collectively constitute a complex 'histone code'
(22). A cohort of specialized
enzymes catalyzes the 'writing', 'erasing' and 'reading' of these
marks. For instance, histone acetylation is catalyzed by histone
acetyltransferases (HATs) such as PCAF, GCN5 and the CREBBP/p300
complex, whereas histone deacetylases (HDACs), including HDAC1-3
and the sirtuin family (SIRT1-7), remove acetyl groups to
facilitate chromatin compaction and transcriptional repression
(23,24). Histone methylation is installed
by histone methyltransferases (HMTs), including the KMT2 family,
PRC2 complex, SUV39H1/2, and PRMT1, which deposit mono-, di- and
tri-methyl marks on lysine (K) or arginine (R) residues (25). Conversely, histone demethylases
(KDMs), such as LSD1/KDM1A and members of the jumonji domain
family, 'erase' methyl modifications in a residue- and
methylation-state-specific manner (25). Histone ubiquitination involves a
cascade of reactions that covalently attach ubiquitin to lysine
side chains, mediated by the E1 activating enzyme, E2 conjugating
enzyme, and E3 ligases (such as RNF20/40 and RING1A/B). This
modification is reversed by deubiquitinases, such as USP16, USP22
and BAP1 (26,27). Functionally, these histone
modifications modulate chromatin biophysics, govern the recruitment
or exclusion of transcriptional regulators, and delineate distinct
chromatin domains, such as euchromatin and heterochromatin, thereby
providing a pivotal epigenetic layer of gene expression control
(28).
miRNAs are a class of non-coding RNAs, ~21 to 25
nucleotides in length, that primarily regulate gene expression at
the post-transcriptional level (29). Intergenic miRNAs possess
independent promoters and are transcribed as discrete
transcriptional units, whereas intragenic miRNAs share promoters
with their host genes; additionally, a number of miRNAs are
arranged in polycistronic clusters and co-transcribed as a single
primary transcript (primary microRNAs, pri-miRNAs) (30). Within the nucleus, the
microprocessor complex (Drosha-DGCR8) cleaves pri-miRNAs to release
~70-nucleotide stem-loop precursor miRNAs (pre-miRNAs) (31). Pre-miRNAs are exported from the
nucleus by Exportin-5 in a Ran-GTP-dependent manner. In the
cytoplasm, the RNase III enzyme Dicer and TRBP cleave pre-miRNA to
produce a miRNA duplex (32,33). This duplex is then loaded into an
Argonaute protein in an ATP-dependent manner, leading to passenger
strand ejection and mature single-stranded miRNA formation
(34). In addition to this
canonical pathway, certain miRNAs arise via non-canonical
(Drosha-independent and/or Dicer-independent) pathways (35). Accumulating evidence indicates
that miRNAs orchestrate diverse biological processes, such as cell
differentiation, apoptosis and immune responses, and that aberrant
miRNA expression contributes to various pathologies, highlighting
their potential as therapeutic targets (30,36).
lncRNAs are transcripts typically >200
nucleotides with minimal protein-coding potential (37,38). The majority of lncRNAs are
transcribed by RNA polymerase II and undergo 5' capping, splicing
and 3' polyadenylation. Despite low sequence conservation and
generally weaker expression than protein-coding genes, lncRNAs
exhibit highly tissue-, cell type- and developmental stage-specific
expression. Advances in RNA sequencing, single-cell
transcriptomics, multi-omics and RNA-protein interaction studies
have established that lncRNAs play critical regulatory roles in
embryonic development, immune modulation, and tumorigenesis, rather
than representing mere transcriptional noise (38,39).
lncRNAs regulate gene expression through diverse
mechanisms, including epigenetic, transcriptional,
post-transcriptional and macromolecular complex-mediated processes
(40,41). Some recruit chromatin modifiers,
such as PRC2, to induce H3K27me3 and other repressive histone
marks, thereby modulating chromatin accessibility and gene
silencing (42).
Enhancer-associated lncRNAs regulate nearby gene transcription by
altering enhancer-promoter interactions and local chromatin
architecture (43). Certain
lncRNAs function as competing endogenous RNAs, sequestering miRNAs
and relieving repression of target mRNAs (41,44). lncRNAs can also interact with
mRNAs, miRNAs, or RNA-binding proteins to influence splicing,
stability, localization and translation (41).
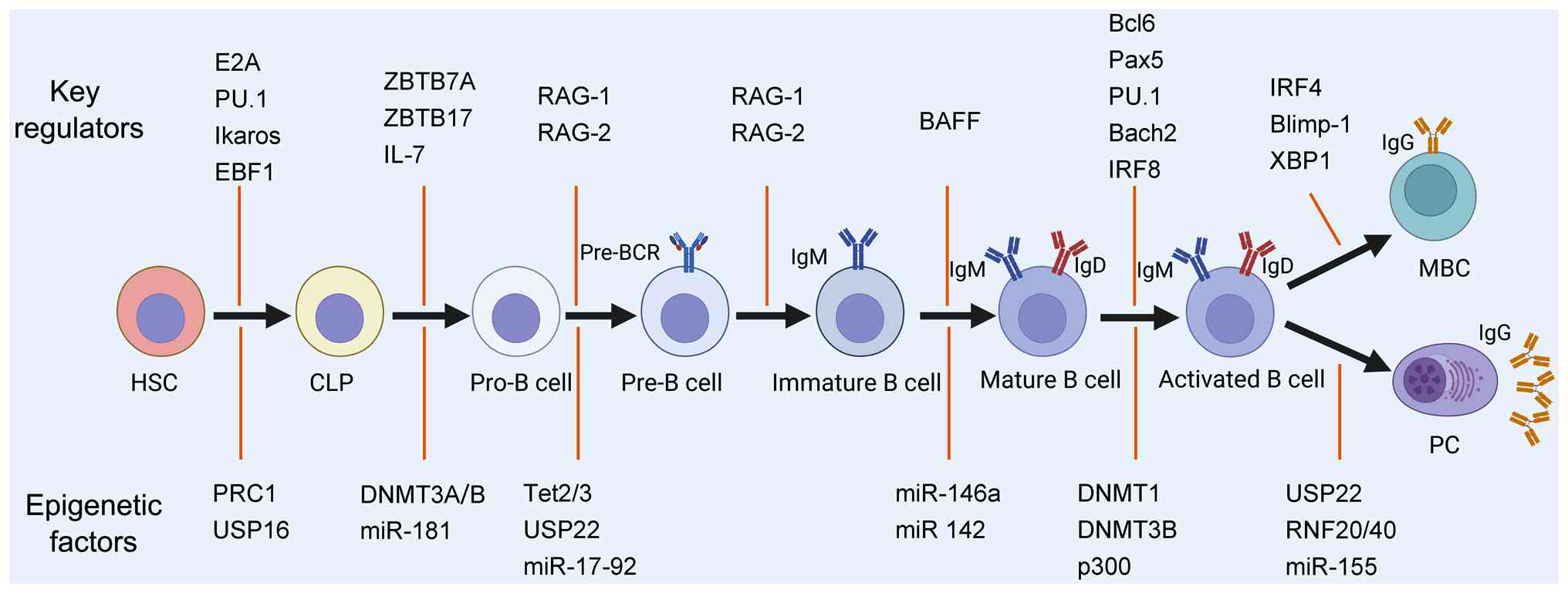
Early B-cell development in the BM is orchestrated
by chemokines (such as C-X-C motif chemokine ligand 12 and C-X-C
motif chemokine ligand 13) and cytokines (such as IL-7 and stem
cell factor) produced by stromal cells (45-47). The transcription factors, ZBTB7A
and ZBTB17, are indispensable for the transition of CLPs to
pre-pro-B-cells through modulation of Notch signaling, JAK-STAT5
pathway and IL-7R signaling (48,49). Rearrangement of the
immunoglobulin heavy-chain gene segments is the hallmark of
pro-B-cells and requires expression of RAG-1 and RAG-2 (50). In addition, pro-B-cells express
terminal deoxynucleotidyl transferase, which increases
antigen-receptor diversity by adding non-templated nucleotides at
V(D)J junctions. Pro-B-cells also express the surrogate light chain
(VpreB/λ5), which pairs with μ heavy chains to form the
pre-B-cell receptor (pre-BCR). Pre-BCR signaling then drives
progression to the large and subsequently small pre-B-cell stages.
Subsequently, light-chain gene rearrangement occurs under strict
allelic and isotypic exclusion, ensuring each B-cell expresses
either κ or λ light chains exclusively. B-cells enter
IgM-expressing immature B-cells after completing both heavy and
light chains rearrangement.
In peripheral lymphoid tissues, B-cell activator of
the TNF-α family (BAFF) signals provided by the follicle are
important for B-cell survival (51). The mature B-cells differentiate
into MBCs and PCs when activated by antigens in the peripheral
lymphoid tissues. In addition to antigenic signals, TD or TI
co-stimulation signals are also required for the B-cell activation.
TI antigens typically drive differentiation into short-lived PCs,
whereas TD responses initiate germinal center (GC) response
(52). Within GCs,
immunoglobulin genes undergo class-switch recombination (CSR) and
somatic hypermutation (SHM) (53). GC responses are governed by a
transcriptional network in which Bcl6, Pax5, PU.1, Bach2 and IRF8
enforce the GC B-cell program, whereas Blimp-1 (encoded by
Prdm1), IRF4 and XBP1 drive differentiation into MBCs and
PCs (54). Bcl6 is essential for
GC B-cell identity by upregulating Bach2 and repressing
Prdm1, thereby preventing premature Blimp-1-mediated
differentiation (55). Blimp-1
represses Bcl6 and other B-cell fate factors, while inducing
Irf4 and Xbp1. Deletion of Prdm1 in mice
abolishes PC formation (56).
IRF4 further reinforces PC differentiation by activating
Prdm1 and repressing Bcl6 (54).
In addition to the intricate networks of
transcription factors, an increasing body of evidence suggests that
epigenetic regulation plays a pivotal role in B-cell development
and differentiation. The involvement of epigenetic regulators in
B-cell lymphopoiesis is illustrated in Fig. 2.
In the immune system, B-cells exhibit a highly
dynamic DNA methylation landscape, particularly during
differentiation. Approximately one-third of CpG sites undergo
methylation changes throughout B-cell development (57). The transition from naïve B-cells
to GC B-cells is characterized by extensive methylome remodeling,
whereas subsequent shifts from GC B-cells to MBCs and PCs involve
comparatively modest alterations (58). Notably, CTCF-binding sites
exhibit dynamic methylation changes during B-cell maturation,
indicating subset-specific epigenetic regulation within the immune
system (59). Furthermore, DNA
methylation also influences SHM by targeting V(D)J gene segments
and promoting an open chromatin state in coordination with histone
modifications (60,61).
B-cells respond to a wide array of immunological
stimuli, such as pathogens, commensal microbes, tumors and other
environmental cues (1,62). During immune responses,
activation-induced cytidine deaminase (AID) drives SHM and CSR by
deaminating deoxycytidine to deoxyuracil, thereby generating U:G
mismatches that are processed to effect antibody diversification
(63). The precise control of
AID expression is essential to maintain genomic integrity. In naïve
B-cells, Aicda (the gene encoding AID) is transcriptionally
silenced by promoter hypermethylation (64), whereas upon activation, it
undergoes demethylation-dependent induction. Moreover, the
differentiation of MBC is dependent on DNA methylation, where high
levels of DNMTs are observed (58).
Histone post-translational modifications are a core
epigenetic mechanism that regulate gene expression by remodeling
chromatin and recruiting effector proteins (22,69). In general, histone acetylation is
associated with transcriptional activation, whereas specific
methylation marks are frequently associated with gene repression
(70). Histone phosphorylation,
mediated by protein kinases, modulates DNA-histone interactions and
facilitates the assembly of DNA repair and transcriptional
complexes. In addition, the ubiquitination of histones H2A and H2B
contribute to both gene regulation and DNA damage repair (71).
B-cell development occurs in two sequential phases:
An antigen-independent stage in the BM and an antigen-dependent
stage in peripheral lymphoid tissues (1). During the latter, naive B-cells
undergo CSR and SHM upon activation, eventually differentiating
into MBCs and PCs (4). These
differentiation steps coincide with marked changes in histone
modification patterns. In resting naïve B-cells, chromatin at the
immunoglobulin heavy-chain (IgH) locus is enriched in
repressive marks and depleted of activating modifications,
resulting in a compact configuration and low transcriptional
activity (72). Upon activation,
extensive histone remodeling leads to the induction of genes
essential for B-cell effector function. For example, stimulation
with lipopolysaccharide and IL-4 has been shown to result in a
marked increase in histone H3 acetylation at regulatory elements of
the Aicda locus (73).
These elements also become enriched in H3K9ac, H3K14ac, and H3K4me3
and undergo DNA demethylation, thereby enabling robust AID
expression (73). Moreover,
activated B-cells exhibit increased expression of the histone
acetyltransferase p300, resulting in global histone
hyperacetylation. p300-mediated histone lysine acetylation at the
Bruton's tyrosine kinase (Btk) promoter enhances Btk
transcription, thereby potentiating BCR signaling and B-cell
activation (74).
Histone ubiquitination also critically influences
B-cell development. The major H2A ubiquitin ligase, polycomb
repressive complex 1 (PRC1), along with the deubiquitinase USP16,
controls H2A ubiquitination. PRC1 is essential for HSC self-renewal
(75), and Usp16 deletion
significantly reduces CLPs (76), highlighting a role for H2A
ubiquitination in early lymphopoiesis. Similarly, the
ubiquitination of H2BK120ub, catalyzed by the RNF20/RNF40 complex
and removed by the SAGA-associated deubiquitinase USP22, is
essential for CSR by facilitating DNA double-strand break repair
during activation (77,78). Rnf20 or Rnf40
knockdown diminishes H2Bub levels and impairs CSR in CH12 cells
(78), and the conditional
deletion of Usp22 in pre-B-cells disrupts classical
non-homologous end joining and antigen-specific IgG1 production
without affecting early B-cell development in the BM and periphery
(77). In summary, these
findings demonstrate that histone ubiquitination is particularly
critical for DNA repair processes during antigen-driven B-cell
differentiation.
Previous studies have delineated stage-specific
miRNA networks governing B-cell biology. For example, miR-181 and
the miR-17-92 cluster promote early B-cell development (79,80). By contrast, the constitutive
expression of miR-150 and miR-34a blocks pro-B-cell to pre-B-cell
transition. Mechanistically, miR-150 and miR-34a interfere with
early B-cell development by targeting the transcription factors,
C-Myb and Foxp1, respectively (81,82). Moreover, studies have
demonstrated that the miR-212/132 cluster regulates immunoglobulin
gene rearrangement by directly targeting Sox4 mRNA (83).
During peripheral maturation, transitional B-cells
differentiate into splenic follicular or marginal zone (MZ) B-cell
subsets. miR-146a deficiency in murine models causes selective
depletion of MZ B-cells via Numb-mediated inhibition of Notch2
signaling (84). A complex
network of cytokines (such as IL-6, IL-21 and BAFF) and
transcription factors (such as Bcl6, Blimp-1 and IRF4) regulates GC
responses and antibody affinity maturation (54,55). miR-155 regulates GC reactions and
IgG1+ B-cell differentiation by modulating cytokine
production and targeting multiple genes, including the
transcription factor PU.1 (85,86). Another critical regulator is
miR-142, which plays a pivotal role in B-cell homeostasis.
miR-142-deficient mice develop immunoproliferative disorders
characterized by an expansion of MZ B-cells and a reduction in B1
B-cells, partly due to the derepression of BAFF (87). Collectively, these findings
underscore the intricate miRNA-mediated networks that ensure proper
B-cell lineage specification, function and immune competence.
In B-cell development and differentiation, lncRNAs
have emerged as key regulatory nodes (88). Numerous lncRNAs display
stage-specific expression from hematopoietic stem cells to mature
B-cells, plasma cells and memory B-cells (88,89). Expression profiling across
developmental stages reveals thousands of lncRNAs associated with
proliferation, V(D)J recombination, maturation and immune memory
formation (90,91). Early B-cells exhibit IgH
locus-associated lncRNAs enriched at topologically associating
domain anchors and enhancers, suggesting roles in 3D genome
organization and antibody repertoire diversification (92,93). Moreover, lncRNA-CSRIgA
promotes the recruitment of regulatory proteins to a proximal
CTCF-binding site, thereby reshaping chromosomal interactions
within the topologically associated domain
(TADlncCSRIgA) and long-range contacts with the 3' RR
super-enhancer, ultimately facilitating CSR to IgA (93). LncHSC-1 and LncHSC-2 are required
for HSC self-renewal and differentiation; their knockdown in stem
and progenitor cells (Sca-1+) impairs commitment to the
B-cell lineage (94).
Furthermore, XIST is necessary for the sustained silencing of a set
of X-linked immune-related genes (such as TLR7) in B-cells; XIST
depletion reactivates these loci and drives differentiation of
CD11c+ atypical memory B-cells implicated in the
pathogenesis of SLE (95).
The emergence of autoreactive B-cells and their
secreted autoantibodies during B-cell development and
differentiation drives the onset of autoimmune diseases, primarily
including SLE, RA, primary Sjögren's syndrome (pSS), multiple
sclerosis (MS) and type 1 diabetes mellitus (T1DM). Emerging
evidence suggests that epigenetic dysregulation in B-cells
contributes to aberrant B-cell development and differentiation,
thereby promoting autoimmune pathogenesis (summarized in Table SI).
SLE is a chronic, relapsing-remitting systemic
autoimmune disease characterized by immune dysregulation (96). B-cells, as the main source of
pathogenic autoantibodies, play a pivotal role in the pathogenesis
of SLE (97). The low
concordance rate of SLE among monozygotic twins indicates that
epigenetic regulation is critically involved in the development of
SLE (98). In recurrent SLE twin
pairs, interferon-stimulated genes (ISGs) in B-cells exhibit
hypomethylation, whereas key upstream regulators such as TNF
and EP300 are hypermethylated (98). Similarly, the persistent
hypomethylation of CpG sites within ISGs has been observed in SLE
patient B-cells (99). Fali
et al (100) reported
that DNA hypomethylation in SLE B-cells leads to the overexpression
of HRES1/p28, being associated with disease activity. Furthermore,
compared with healthy children and adult patients with SLE, B-cells
from pediatric patients with SLE display a distinctive chromatin
accessibility landscape, with increased accessibility at non-coding
genomic regions that regulate inflammatory activation, indicating
that epigenetic dysregulation-induced aberrant expression of
regulatory elements controlling B-cell activation plays a critical
role in the pathogenesis of pediatric SLE (101). In a murine model, the adoptive
transfer of B-cells treated with a DNMT1 inhibitor into syngeneic
mice resulted in increased antinuclear antibody production
(102). Conversely, the
deficiency of the demethylases Tet2 and Tet3 leads to
hyperactivation of lymphocytes and spontaneous SLE-like
autoimmunity (103).
Mechanistically, Tet2 and Tet3 remove methylation marks and
cooperate with HDAC1/2-mediated deacetylation to suppress
Cd86 expression upon self-antigen engagement, thereby
preventing excessive autoreactive B-cell activation (103). Collectively, these studies
demonstrate that alterations in B-cell DNA methylation are closely
linked to the pathogenesis of SLE.
In addition to DNA methylation, the dysregulation of
histone methylation and acetylation critically contributes to the
pathogenesis of SLE. During B-cell activation, T-helper
cell-derived co-stimulatory signals upregulate the histone
demethylases KDM4A and KDM4C, coincident with global reductions in
H3K9me2 and H3K9me3 levels (104). These demethylases cooperate
with NF-κB p65 to target and activate WDR5, which mediates H3K4
methylation at cyclin-dependent kinase inhibitor (CDKN) gene
loci, thereby restraining excessive B-cell proliferation (104). In B-cells from patients with
SLE, the downregulation of KDM4A/C and WDR5,
accompanied by reduced CDKN expression, is associated with
heightened B-cell activation and proliferation (104). Moreover, in a previous study,
the overexpression of the HMT EZH2 was observed in peripheral blood
B-cells and leukocytes from patients with SLE (105). The inhibition of EZH2 with
3-deazaneplanocin A in MRL/lpr mice reduced splenocyte H3K27me3
levels, attenuated anti-dsDNA antibody production and ameliorated
lupus-like pathology (105).
Compared with healthy donors, B-cells from patients with SLE
exhibit the hypoacetylation of histones H3 and H4 (106). The expression and enzymatic
activity of the histone deacetylases HDAC6 and HDAC9 are markedly
upregulated in B-cells from MRL/lpr mice relative to control mice
(107). Moreover, the loss of
histone acetyltransferase activity from a single Ep300
allele in mouse B-cells leads to the development of an autoimmune
disease that closely resembles the pathological features of SLE
(108). In the area of
chromatin remodeling, the literature search performed for the
present review identified no available studies to date that have
reported alterations in chromatin-remodeling complexes in B-cells
and their associations with SLE and other autoimmune diseases.
Further investigations in this area are thus warranted (7).
The dysregulated expression of miRNAs in B-cells
also contributes to autoimmune pathogenesis. In patients with SLE,
B-cells exhibit a decreased expression of Lyn, a critical negative
regulator of B-cell activation (109). miR-30a specifically targets Lyn
mRNA through the binding to the 3' untranslated region (3'-UTR),
and the observed upregulation of miR-30a in SLE B-cells is strongly
associated with a decreased expression of Lyn and subsequent B-cell
hyperactivation (109). EBF1
promotes B-cell activation and proliferation via the activation of
the AKT signaling pathway. However, miR-1246 counteracts this
process by interacting with the 3'-UTR of EBF1 mRNA to mediate its
degradation (110). In SLE
B-cells, the abnormal activation of AKT-p53 signaling downregulates
miR-1246, thereby enhancing EBF1 expression and exacerbating B-cell
activation (110). In MRL/lpr
mice, miR-7 is upregulated and targets PTEN to down-modulate the
PTEN/AKT pathway, thereby driving spontaneous GC formation and the
generation of autoreactive antibody-secreting PCs (111). B6.Sle123 mice, a spontaneous
genetic model of SLE, exhibit an increased expression of miR-21 in
both B- and T-cells, which regulates cellular proliferation and
apoptosis (112). Notably, the
in vivo silencing of miR-21 using tiny seed-targeting LNA
significantly ameliorates splenomegaly, a hallmark autoimmune
manifestation in these mice (112). Research on B/W lupus mice has
revealed elevated miR-15a levels in B-10 cells, being associated
with autoantibody titers (113). Moreover, mice with the
lymphocyte-specific overexpression of the miR-17-92 cluster develop
spontaneous autoimmune manifestations. Mechanistically, the
miR-17-92 cluster drives lymphocyte proliferation and breaks
self-tolerance by directly repressing the pro-apoptotic protein Bim
and the tumor suppressor, PTEN (114).
RA is a chronic, systemic autoimmune disorder
characterized by progressive synovial inflammation, cartilage
degradation and bone erosion, primarily affecting diarthrodial
joints (115,116). As one of the most prevalent
inflammatory arthropathies, RA affects ~0.5-1% of the global
population (116). Synovial
tissue samples from patients with RA demonstrate prominent
infiltration of B-cells and PCs. Notably, approximately 80% of
patients with well-established RA exhibit characteristic
autoantibodies, such as anti-citrullinated protein antibodies and
rheumatoid factor, highlighting the pivotal role of B-cell-mediated
immunity in the pathogenesis of RA (117).
In an epigenome-wide association study (EWAS) on
patients with RA, 64 differentially methylated CpG sites and six
dysregulated biological pathways were consistently identified in
B-cells across three different replication cohorts (118). Among these, the key B-cell
activation gene CD1C exhibited significant hypermethylation
in B-cells from RA patients (118). B-cells from patients with
early-stage RA also exhibit a decreased expression of DNMT1 and
DNMT3A, concomitant with a global decrease in DNA methylation
(119). Methotrexate (MTX)
treatment partially reversed these epigenetic alterations by
restoring DNMT1 and DNMT3A expression in B-cells (119). In the proteoglycan-induced
arthritis (PGIA) murine model, the promoter hypomethylation-driven
Zbtb38 overexpression in B-cells promotes the progression of
arthritis by suppressing IL-1 receptor 2 expression and inhibiting
key anti-inflammatory pathways (120). Conversely, the administration
of the DNA methyltransferase inhibitor 5-azacytidine (AzaC) has
been shown to ameliorate autoimmune arthritis in PGIA mice by
downregulating AID in B-cells, impairing CSR and GC responses, and
reducing IgG1 production (121). As demonstrated in a previous
study, compared with healthy controls, the expression of class I
HDACs was significantly decreased in peripheral blood mononuclear
cells from patients with RA, whereas nuclear HAT activity was
markedly increased (122). This
imbalance resulted in increased acetylation of histone H3 and
upregulation of genes encoding pro-inflammatory cytokines, thereby
contributing to the pathogenesis of RA (122).
The dysregulated expression of miRNAs in B-cells
further contributes to the development of RA. In a previous study,
small RNA sequencing revealed the differential expression of 27
miRNAs in B-cells from patients with RA in remission following
treatment with MTX, compared to the healthy controls. The predicted
targets of these miRNAs were enriched in pathways governing B-cell
activation, differentiation, and BCR signaling (123). Compared with healthy controls,
miR-155 (an essential regulator of GC and PC differentiation) is
markedly upregulated in peripheral blood B-cells from patients with
RA (124). In synovial tissue,
miR-155 expression inversely correlates with the transcription
factor PU.1. The inhibition of endogenous miR-155 in RA B-cells
restores PU.1 levels and reduces autoantibody production (124). A high expression of miR-155 has
also been observed in CD14+ synovial cells, where it
downregulates the anti-inflammatory phosphatase SHIP-1 and promotes
the release of pro-inflammatory cytokines (125). Conversely, miR-155 inhibition
in RA CD14+ synovial cells has been shown to decrease
TNF-α secretion (125).
Moreover, the genetic deletion of miR-155 prevents collagen-induced
arthritis in mice, identifying miR-155 as a potential therapeutic
target in RA (125).
pSS is a chronic systemic autoimmune disease
characterized by the lymphocytic infiltration of exocrine glands,
resulting in inflammation and tissue destruction of the salivary
and lacrimal glands, and clinically manifesting as xerostomia (dry
mouth) and keratoconjunctivitis sicca (dry eyes) (126). Beyond classic exocrine
involvement, pSS often presents with systemic complications,
including interstitial lung disease, renal tubular acidosis,
peripheral neuropathy, as well as hematological abnormalities, such
as cytopenias and B-cell lymphoproliferative disorders, reflecting
its complex immunopathogenesis (127,128). Emerging evidence implicates
epigenetic dysregulation and B-cell hyperactivity with autoantibody
production as key drivers of pSS. For example, anti-SSA/Ro
antibodies are detected in ~33-74% of cases, and anti-SSB/La
antibodies are detected in 23-52% of cases (128).
Epigenome-wide analyses have demonstrated that DNA
methylation changes in peripheral B-cells of patients with pSS far
exceed those observed in T-cells, with genes involved in B-cell
signaling, inflammation and autoimmunity exhibiting significant
methylation alterations (129).
Specifically, ISGs, including PARP9, IFI44L and
MX1 are markedly hypomethylated in B-cells of patients with
pSS, being associated with their transcriptional upregulation and
B-cell expansion (129,130). In a genome-wide DNA methylation
study of labial salivary gland biopsies, 7,820 differentially
methylated sites associated with disease status were identified,
5,699 hypomethylated and 2,121 hypermethylated, and enrichment
analysis revealed that genes crucial for B-cell differentiation and
function (such as SPI1, CD19, CD79B,
PTPRCAP and TNFRSF13B) were predominantly
hypomethylated, highlighting the pivotal role of B-cell DNA
methylation alterations in pSS (131). Furthermore, in another study,
active histone marks, including H3K4me2, H3K4me3, H3K36me3, H3K9ac
and H3K27ac, were shown to be significantly enriched at promoters
and enhancers harboring Sjögren's syndrome risk variants in B-cells
from patients with pSS, suggesting that epigenetic mechanisms
contribute to the regulation of these promoters and enhancers
(132).
The transcriptome sequencing of B-cell subsets from
patients with pSS has revealed the significant upregulation of the
lncRNA LINC00487 in CD38+IgD+
(immature B-cells), CD38−IgD+ (Bm1 cells),
CD38highIgD+ (pre-GC B-cells) and
CD38±IgD− (MBCs) subsets, with
LINC00487 expression levels being positively associated with
the clinical disease activity score (133). Furthermore, in a previous
study, comparative miRNA expression profiling revealed significant
disparities between B- and T-cells in patients with pSS. Peripheral
B-cells from patients with pSS exhibited the significant
dysregulation of 24 miRNAs, with 11 upregulated and 13
downregulated miRNAs, compared to the healthy controls (134). In both discovery and
independent replication cohorts, miR-30b-5p, miR-222-3p,
miR-19b-3p, miR-26a-5p, and miR-378a-3p were consistently
dysregulated (134). Notably,
miR-30b-5p targets the 3'-UTR of BAFF, and its downregulation in
pSS B-cells is associated with BAFF overexpression (134). Elevated BAFF levels have also
been observed in salivary gland-infiltrating B-cells (135). Collectively, these studies
highlight the critical contribution of non-coding RNA dysregulation
in the B-cell-mediated pathogenesis of pSS.
MS is a chronic immune-mediated demyelinating
disorder of the central nervous system (CNS), characterized by
inflammatory lesions, axonal damage and progressive neurological
disability (136,137). Accumulating evidence over the
past decade has demonstrated that B-cells play a pivotal role in
the pathogenesis of MS by producing autoantibodies, presenting
antigens, and secreting pro-inflammatory cytokines, thereby
exacerbating damage to the CNS (138,139). An abnormal cytokine profile has
been observed in B-cells from patients with MS, with activated
B-cells exhibiting the excessive production of pro-inflammatory
cytokines, including TNF-α, lymphotoxin and granulocyte-macrophage
colony-stimulating factor (140,141). Moreover, clinical trials of
B-cell-depleting therapies in relapsing-remitting MS (RRMS) have
yielded strikingly positive outcomes, further underscoring the
contribution of B-cells to disease activity (142).
The genome-wide DNA methylation profiling of
T-cells, monocytes and B-cells from patients with RRMS, secondary
progressive MS and healthy controls has revealed a preponderance of
differentially methylated positions (DMPs) in B-cells (143). These DMPs are predominantly
located within genes associated with lymphocyte signaling pathways
(143). In a previous study on
patients with RRMS in remission under various treatments, the
analysis of CD19+ B-cell methylomes identified a large
hypermethylated region encompassing the transcriptional start site
of the lymphotoxin-α (LTA) locus (144). Additionally, four MS-associated
loci, including SLC44A2, LTBR, CARD11 and
CXCR5, exhibited significant methylation changes (144), suggesting that B-cell-specific
DNA methylation, particularly at the LTA locus, may
contribute to the pathogenesis of MS. Moreover, in another study,
an integrative analysis of MS immune-cell GWAS data and
histone-modification profiles revealed that MS genetic risk loci in
B-cells are enriched at active enhancers and promoters marked by
H3K4me1, H3K4me3 and H3K27ac, indicating that MS genetic risk
associations preferentially localize to active enhancer and
promoter regions (145).
The dysregulated expression of miRNAs in B-cells
also influences the progression of MS. In a previous study, the
transcriptomic analyses of peripheral B-cells from untreated
patients with MS revealed the marked downregulation of IRF1
and CXCL10; the suppression of the IRF1/CXCL10 axis may
foster a pro-survival phenotype in these cells (146). Mechanistically, the
upregulation of miR-424 in B-cells of patients mediates the
downregulation of IRF1 and CXCL10 (146). In RRMS lymphocyte subsets,
purified CD19+ B-cells exhibit an increased expression
of miR-497 alongside decreased levels of miR-92, miR-153, miR-135b,
miR-422a and miR-189 relative to healthy volunteers (147). A comprehensive expression
analysis of 1,059 miRNAs in B-cells from healthy volunteers,
treatment-naïve patients with RRMS and natalizumab-treated patients
with RRMS identified 49 differentially expressed miRNAs in
untreated patients compared to healthy volunteers, including
miR-25, miR-19b, miR-106b, miR-93 and miR-181a (148). Target prediction and pathway
analyses indicated that these miRNAs predominantly regulate B-cell
receptor signaling pathway, PI3K pathway, and PTEN pathway
(148). Notably, natalizumab
treatment upregulated 10 miRNAs, including miR-106b, miR-19b and
miR-191 (148). Furthermore,
hyperactivated B-cells in MS secrete pro-inflammatory cytokines,
including TNF-α and lymphotoxin. The elevated expression of miR-132
in these cells enhances TNF-α and lymphotoxin production by
inhibiting SIRT1 activity (149).
T1DM is a chronic autoimmune disorder characterized
by the immune-mediated destruction of insulin-producing pancreatic
β-cells, culminating in absolute insulin deficiency (150). Although T-cells have
traditionally been considered the primary mediators of pancreatic
tissue injury, accumulating evidence indicates that B-cells also
contribute significantly to the pathogenesis of T1DM (151). To investigate the influence of
B-cell epigenetic factors on the progression of T1DM, a genome-wide
DNA methylation study was performed on purified B-cells from three
pairs of monozygotic twins discordant for T1DM and six pairs of
unaffected monozygotic twins. This analysis identified 88 CpG sites
with altered methylation in B-cells from the T1DM-affected twins
(152). Functional enrichment
analysis revealed that genes harboring these differentially
methylated sites are predominantly involved in immune-response
pathways (152). In a separate
investigation involving 52 pairs of monozygotic twins discordant
for T1DM, methylation profiling at 406,365 CpG sites in B-cells,
monocytes, and CD4+ T-cells uncovered multiple
cell-type-specific differentially variable positions (DVPs) related
to cell-cycle control and metabolic processes in the T1DM-affected
twins compared with their healthy co-twins and unrelated healthy
controls (153). Notably,
significant DVPs were observed at loci encoding the B-cell
transcriptional regulators NRF1 and FOXP1, highlighting a postnatal
role for DNA methylation in the pathogenesis of T1DM (153). Compared with healthy controls,
peripheral blood lymphocytes from individuals with T1D have been
shown to exhibit a significant increase in H3K9me2 at genes
involved in autoimmune and inflammatory pathways (154). These findings indicate that
altered histone methylation at key genomic loci in lymphocytes is
associated with T1DM (154).
Complementary studies in murine models have demonstrated that the
diabetes-protective Idd9.3 locus encodes miRNA-34a, which
impairs B-cell development and maturation by targeting the core
regulatory factor Foxp1, thereby conferring resistance to the onset
of T1DM (155).
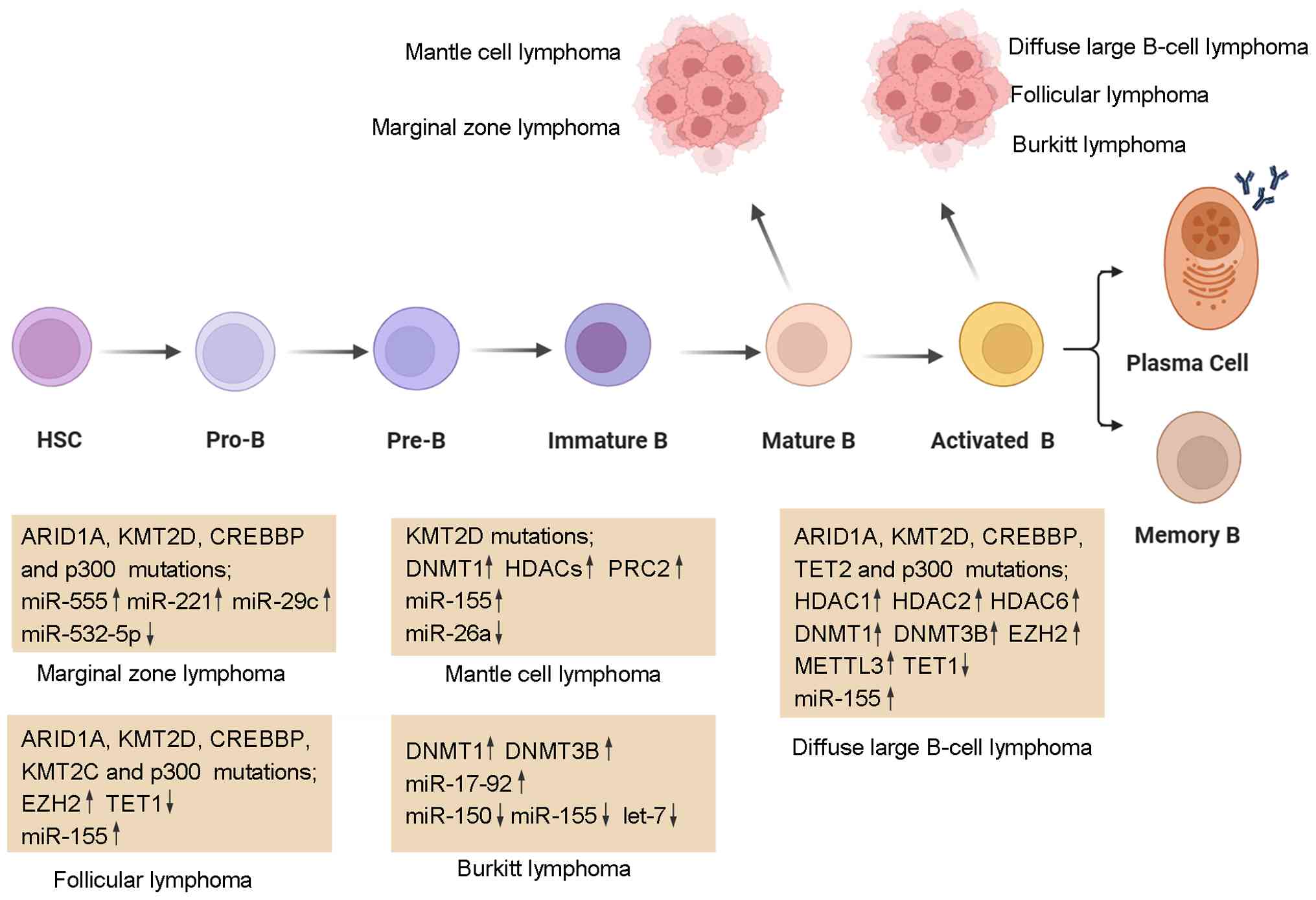
B-cell non-Hodgkin lymphoma (B-NHL) is a highly
heterogeneous malignancy originating from B-cells and accounts for
~85-90% of all lymphoma cases (6). Based on the cell of origin,
molecular characteristics and clinical behavior, B-NHL can be
classified into several distinct subtypes, including DLBCL, FL,
MCL, Burkitt lymphoma (BL) and marginal zone lymphoma (MZL). The
present review focuses on the role of dysregulated epigenetic
mechanisms in the pathogenesis and progression of B-NHL
(illustrated in Fig. 3). The
roles of dysregulated epigenetic enzymes in B-NHL are summarized in
Table SII.
DLBCL is the most common and aggressive subtype of
B-NHL, representing ~30-40% of all B-NHL cases (156,157). Gene expression profiling
further stratifies DLBCL into GC B-cell-like (GCB) DLBCL, activated
B-cell-like (ABC) DLBCL and unclassified groups (158). Loss-of-function mutations in
the SET domain of histone lysine N-methyltransferase 2D (KMT2D)
represent the most frequent epigenetic abnormality in DLBCL,
occurring in ~30-40% of cases (159,160). Mechanistically, KMT2D
deficiency impairs H3K4 methylation and dysregulates the expression
of specific gene sets, including those involved in JAK-STAT,
Toll-like receptor, CD40 and BCR signaling pathways, as well as
tumor suppressors, such as SOCS3, TNFRSF14 and
TNFAIP3 (161).
Consequently, KMT2D mutations promote malignant B-cell
proliferation by disrupting the coordinated activation of B-cell
signaling cascades and the expression of anti-oncogenic regulators
(161). Furthermore,
loss-of-function mutations in the histone acetyltransferases CREBBP
and p300 are found in ~40% of DLBCL cases (159,162). These mutations reduce the
acetylation of BCL6 and p53, thereby perturbing BCL6 inactivation
and p53-mediated tumor suppression and enhancing tumor cell
resistance to DNA damage (163). The overexpression of the HDACs,
HDAC1, HDAC2 and HDAC6, together with global H4 hypoacetylation,
also contributes to transcriptional repression (164). In particular, HDAC6 plays a
critical role in coping with proteotoxic stress by regulating the
acetylation-dependent stability of the HSP90 and mediating the
binding of misfolded proteins to the dynein motor for transport to
aggresomes (165-167).
Dysregulated DNA methylation further contributes to
the pathogenesis of DLBCL. DNMT1 and DNMT3B are overexpressed in
DLBCL, and are associated with an advanced clinical stage,
therapeutic resistance, and, specifically for DNMT3B, significantly
shorter overall survival and progression-free survival (168). Research has demonstrated that
DNMT1 modulates cell-cycle progression and DNA replication, as
DNMT1 knockdown in GCB-DLBCL cell lines markedly suppresses the
expression of CDK1, CCNA2, E2F2, PCNA,
RFC5 and POLD3 (169). By contrast, the DNA demethylase
TET2 functions as a tumor suppressor and harbors loss-of-function
mutations in DLBCL (170).
Murine models demonstrate that the knockout of Tet1 and
Tet2 promotes the development of DLBCL (171,172). Moreover, transcriptional
downregulation of TET1 has been observed in patients with
DLBCL and FL (173). EZH2, the
catalytic subunit of PRC2, is overexpressed in >70% of
aggressive B-cell lymphomas and is associated with high
proliferation rates (174).
Furthermore, gain-of-function mutations of EZH2 (most frequently at
Y641 and A677) are predominate in GC-type DLBCL and FL (175). Moreover, a mutation of EZH2
promotes the growth of GC-type DLBCL by recruiting PRC2 to silence
PRDM1 (176).
Recent evidence has also highlighted dysregulated
RNA methylation and miRNAs in DLBCL. The
N6-Methyladenosine (m6A) 'writer'
METTL3 and global m6A levels are upregulated in DLBCL
tissues and cell lines, and METTL3 knockdown inhibits proliferation
by reducing m6A modification of pigment
epithelium-derived factor mRNA, thereby impairing DLBCL progression
(177). The miR-17-92 cluster
is overexpressed in DLBCL and FL. Mechanistically, overexpressed
miR-17-92 cooperates with c-MYC to promote B-cell lymphoma growth
by enhancing lymphoma cell proliferation and suppressing apoptosis
(178,179). In a previous study, the
analysis of miRNA profiles from 58 DLBCL cases revealed that,
compared to normal tissues, miR-155, miR-210, miR-106a, and
miR-17-5p were significantly upregulated, whereas miR-95, miR-150,
miR-139, miR-145, miR-149, miR-328, miR-10a, miR-99a, miR-320,
miR-151 and let-7e were downregulated (180). Notably, miR-155 plays a crucial
role in the pathogenesis of DLBCL by inhibiting apoptosis and
promoting cell proliferation through the activation of the PI3K/AKT
signaling pathway (181).
FL, the second most common subtype of B-NHL,
accounts for ~25% of all B-NHL cases. FL originates from GC B-cells
and is characterized by the t(14;18)(q32;q21) translocation, which leads
to the overexpression of the anti-apoptotic gene BCL2
(182). Among the KMT2 family
members, KMT2D and KMT2C have been implicated in the pathogenesis
of FL (183,184). Of note, ~72% of patients with
FL harbor KMT2D mutations, the majority of which are nonsense or
frameshift alterations resulting in reduced KMT2D protein
expression (183). In addition,
gain-of-function mutations in EZH2 are observed in ~7.2% of FL
cases (185). Mechanistically,
mutant EZH2 drives FL initiation by epigenetically reprogramming
B-cells and remodeling the immune microenvironment: It promotes the
follicular dendritic cell (FDC)-mediated replacement of T-cell
help, thereby facilitating the gradual expansion of GC B-cells
(186). CREBBP is also
recurrently mutated in over 50% of FL cases (163). In a Bcl2-overexpressing
mouse model, GC-specific Crebbp deletion accelerates FL
development (187). Mutations
in the SWI/SNF complex subunit ARID1A reduce RUNX3
expression, resulting in decreased RUNX3/ETS1-driven FAS
transcription (188). The
consequent downregulation of FAS renders FL cells resistant to FAS
ligand-induced apoptosis (188). The comparative profiling of
miRNA expression in FL and DLBCL vs. normal lymph nodes has
revealed the overexpression of miR-155, miR-106a, miR-149, miR-210
and miR-139 in both malignancies. Notably, miR-20a/b and miR-194
are selectively upregulated in FL; these miRNAs enhance tumor cell
proliferation and survival by targeting CDKN1A and
SOCS2, respectively (189).
Epigenetic dysregulation also plays a pivotal role
in the pathogenesis of MCL. Genome-wide DNA methylation profiling
has revealed highly heterogeneous methylation patterns in MCL tumor
tissues compared with normal lymphoid counterparts. Specifically,
WNT pathway inhibitors and several tumor suppressor genes show
significant hypermethylation (195). Moreover, DNMT1 is upregulated
in MCL, and treatment with arsenic trioxide inhibits WNT/β-catenin
signaling and reduces DNMT1 levels, thereby inducing apoptosis and
attenuating proliferation in MCL cell lines (196). Hypomethylation within the
downstream region of the SOX11 oncogene has been identified
in MCL tumors, suggesting that epigenetic mechanisms may regulate
SOX11 expression (197).
Histone modifications further contribute to MCL
biology. HDACs are overexpressed in MCL, and pharmacologic HDAC
inhibition induces cell-cycle arrest and apoptosis in these cells
(198,199). KMT2D is among the most
frequently mutated genes in both DLBCL and MCL (200). Loss-of-function KMT2D mutations
reduce H3K4 methylation and drive neoplastic proliferation in MCL
(201). Furthermore, the PRC2
complex is overexpressed in MCL cell lines, promoting tumor-cell
proliferation and survival by epigenetically silencing the
cyclin-dependent kinase inhibitor gene CDKN2B (202).
miRNAs provide an additional layer of epigenetic
regulation in MCL. A study of the miRNA expression profiles in 30
patients with MCL revealed that, compared to normal B-cells, 18
miRNAs were significantly downregulated and 21 miRNAs were
significantly upregulated in MCL tissues (203). Notably, miR-142-3p/5p,
miR-29a/b/c and miR-150 were significantly downregulated, whereas
miR-155 and miR-124a were markedly upregulated (203). A separate study reported that
miR-26a, miR-31, miR-27b and miR-148a were significantly
downregulated, whereas miR-370, miR-617 and miR-654 were
upregulated in MCL tissues compared to reactive lymphoid tissues.
Notably, MAP3K2 (a predicted target of miR-26a) was upregulated in
MCL tissues and plays a crucial role in the activation of the
alternative NF-κB pathway (204).
BL is a GC-derived B-NHL, accounting for ~1-5% of
all NHL cases. BL is characterized by MYC dysregulation secondary
to chromosomal translocations such as t(8;14) (q24;q32). Overexpression of DNA
methyltransferases DNMT1 and DNMT3B has been observed in primary BL
specimens, and treatment of BL cell lines with the DNMT inhibitor
5-aza-2'-deoxycytidine significantly inhibits cellular
proliferation by reducing the protein levels of DNMT1 and DNMT3B
(205). miR-155 expression is
significantly downregulated in BL, where it binds to the 3'-UTR of
AID mRNA, promoting its degradation and thereby suppressing
AID-mediated MYC-IGH translocations (206). miR-150 is also downregulated in
BL and directly targets c-Myb and Survivin (207). The re-expression of miR-150 in
the Raji cell line has been shown to diminish cell proliferation,
suggesting that miR-150 may serve as a potential therapeutic target
of BL (207). Compared with
other types of NHL, BL exhibits the upregulation of the miR-17-92
cluster and the downregulation of let-7 family miRNAs, miR-146a,
miR-155 and the miR-29 family (208). In addition, reduced levels of
let-7 family, miR-132, miR-125b-1 and miR-154 contribute to
increased expression of MYC and other oncogenes in BL (209).
MZL accounts for ~5-15% of all B-NHL. It can be
subdivided into three distinct subtypes: Extranodal marginal zone
lymphoma (EMZL), splenic marginal zone lymphoma (SMZL) and nodal
marginal zone lymphoma (NMZL) (210). EMZL represents >60% of MZL
cases and is associated with chronic inflammation triggered by
autoimmune disorders or infectious agents. This subtype is
characterized by recurrent genetic translocations, including
t(11;18)(p21;q21) in API2-MALT1, t(14;18)(p32;q21) in IGH-MALT1 and
t(1;14) (p22;q32) in BCL10-IGH, which
collectively drive transcriptional dysregulation of BCL10,
MALT1 and FOXP1 (211-213). By contrast, these
translocations are not detected in NMZL and SMZL (210). However, NMZL and SMZL share
recurrent somatic mutations in KMT2D, NOTCH2, PTPRD, TNFAPI3
and KLF2 (214).
Epigenetic alterations play a critical role in the
pathogenesis of MZL. In SMZL, frequent mutations occur in chromatin
regulators, such as KMT2D, ARID1A, p300 and CREBBP (215). The genome-wide methylation
profiling of SMZL has revealed hypermethylation and the
transcriptional silencing of multiple tumor suppressors
(KLF4, DAPK1, CDKN1C, CDKN2D and
CDH1/2) alongside hypomethylation and overexpression of
oncogenes in the NF-κB, AKT/PI3K, BCR and IL-2 signaling pathways
(216). Compared with reactive
lymphoid hyperplasia, NMZL exhibits the upregulation of miR-555,
miR-221 and miR-29c, and the downregulation of miR-532-5p.
Predicted targets of miR-555 and miR-221 include CD10 and LMO2,
respectively, whereas miR-532-5p targets BCR-related kinases, such
as SYK and LYN, the overexpression of which is associated with
enhanced tumor cell proliferation (217).
Epigenetic modifications are dynamic and
reversible, and serve as valuable biomarkers in B-cell-mediated
autoimmune diseases and lymphomas (summarized in Table SIII). Alterations in DNA
methylation, histone modification and non-coding RNAs contribute to
disease classification, prognosis and to the prediction of the
therapeutic response (218). In
SLE, B-cells exhibit global hypomethylation, particularly at
interferon-related genes such as IFI44L, PARP9 and MX1, which are
strongly associated with disease activity (219). Additional regulators involved
in inflammatory and chromatin networks also display aberrant
epigenetic states (220). In
patients with SLE, defective DNA methylation in B-cells leads to
the overexpression of the endogenous retrovirus HRES1/p28, with the
dysregulation of the Erk/DNMT1 signaling pathway and autocrine IL-6
signaling playing key roles (100). Notably, intervention with
anti-IL-6 receptor antibodies can restore DNA methylation and
effectively suppress HRES1/p28 expression, providing a novel
therapeutic strategy for the disease. At the miRNA level, miR-30a
upregulation promotes B-cell activation by suppressing Lyn
(109), whereas reduced
miR-1246 increases EBF1 expression and enhances AKT signaling,
promoting B-cell activation and proliferation (110). In pSS, B-cells display
widespread methylome and transcriptome dysregulation affecting
inflammatory and B-cell signaling pathways, including ISGs; the
hypomethylation of PARP9, IFI44L and MX1 is associated with B-cell
expansion (221,222). The downregulation of miR-30b-5p
contributes to the excess expression of BAFF.
In RA, EWAS analyses have identified multiple
differentially methylated loci with biomarker potential (223). Early-stage RA is characterized
by decreased DNMT1/3A expression and global hypomethylation, while
MTX may partially restore methylation patterns and predict
treatment response (119). The
upregulation of miR-155 enhances autoantibody production by
suppressing PU.1, contributing to pathogenic B-cell responses
(124). In MS, B-cell
methylation alterations are prominent. Patients with RRMS exhibit
differential methylation, including the hypermethylation of the
IL2RA promoter and changes at SLC44A2, LTBR, CARD11, and CXCR5,
implicating epigenetic regulation in disease susceptibility and
progression (224). Upregulated
miR-424 suppresses the IRF1-CXCL10 pathway and promotes a survival
phenotype, whereas miR-132 enhances TNF-α and lymphotoxin
expression by targeting SIRT1, strengthening inflammatory B-cell
responses (146,149).
Recent studies have identified two prognostic
molecular signatures in DLBCL: A six-gene m6A regulator
signature (ALKBH5, FMR1, HNRNPC, RBM15B, YTHDC2 and YTHDF1) and an
eight-gene mitochondria-related signature (PCK2, PUSL1, ACP6, PDK4,
ALDH4A1, C15orf61, THNSL1 and COX7A1) (225,226). Both signatures demonstrated
potent prognostic performance, stratifying patients into high- and
low-risk groups with distinct survival patterns, immune
infiltration characteristics, and predicted responses to
immunotherapy. These findings underscore the prognostic relevance
of m6A regulation and mitochondrial metabolism in DLBCL,
and provide promising biomarkers for precision risk assessment and
therapeutic guidance. The overexpression of DNMT3B predicts a poor
survival, whereas the loss of TET2 disrupts AID-mediated DNA
demethylation in GC B-cells, resulting in aberrant DNA
hypermethylation that alters gene regulation and promotes
lymphomagenesis (168,227).
In FL, gain-of-function EZH2 mutations (such as
Y641) reprogram GC B-cells by reducing their dependence on
T-follicular helper cells, promoting centrocyte survival and slow
expansion within FDC networks, thereby establishing an aberrant
immunological niche that underlies indolent tumor growth (186). Concurrently, the epigenetic
dysregulation of GC B-cells through mutations in KMT2D and CREBBP
perturbs transcriptional programs controlling proliferation,
differentiation and microenvironment interactions, contributing to
malignant transformation and immune niche remodeling (228). Collectively, these epigenetic
alterations highlight critical mechanisms by which GC B-cells
evolve into FL and provide potential targets for
microenvironment-focused therapeutic strategies. In MCL,
integrative DNA methylome analyses reveal widespread epigenetic
heterogeneity with hypermethylation of B-cell-unrelated regions and
recurrent hypomethylated differentially methylated regions
associated with SOX11, suggesting that epigenetic
reprogramming of distal regulatory elements contributes to
tumorigenesis and clinical behavior (197).
Epigenetic regulatory pathways provide multiple
druggable targets for B-cell-associated autoimmune diseases and
B-cell lymphomas, and several inhibitors have already entered
clinical use or advanced stages of clinical development (summarized
in Table SIII). Mutations in
EZH2 are observed in ~22% of GC-derived lymphomas, including FL and
DLBCL (175). The FDA-approved
EZH2 inhibitor, tazemetostat, has demonstrated clinical activity in
patients with EZH2 mutant relapsed or refractory FL
(229,230). An open-label, single-arm, phase
II clinical trial reported that oral tazemetostat administered at
800 mg twice daily achieved an objective response rate of 69% in
the EZH2 mutant cohort vs. 35% in the EZH2 wild-type
cohort (230). Moreover, in
GC-DLBCL cases that are insensitive to EZH2 inhibition, combination
therapeutic approaches targeting compensatory pathways, for
example, the cholesterol biosynthesis pathway, have shown promise
in preclinical models (231).
KMT2D loss-of-function mutations represent another major alteration
in GC-derived lymphomas (159).
Haploinsufficiency of KMT2D disrupts H3K4 methylation at
enhancers, resulting in global impairment of enhancer activity and
consequent dysregulation of genes involved in differentiation and
immune regulation (161).
Preclinical studies have shown that pharmacological inhibition of
members of the KDM5 family of H3K4 demethylases, which functionally
antagonize KMT2D, can restore H3K4 methylation levels and suppress
tumor growth in KMT2D-mutant models (232). However, as KDM5 family members
exhibit significant functional redundancy, therapeutic efficacy may
require the simultaneous inhibition of multiple paralogs (232). CREBBP alterations impair
histone acetylation at enhancers and promoters that regulate genes
involved in immune responses and cellular differentiation (233). Mechanistic analyses have
indicated that CREBBP loss creates a dependency on class I histone
deacetylase activity, notably HDAC3 (233). The selective inhibition of
HDAC3 reverses the epigenetic reprogramming induced by CREBBP
mutations, restores the expression of tumor-suppressor and
antigen-presentation genes, and demonstrates anti-B-cell lymphoma
activity in preclinical models (233,234). In addition, combination
regimens pairing the pan-HDAC inhibitor vorinostat with rituximab
(a monoclonal antibody targeting CD20) have improved outcomes in
patients with B-cell lymphoma (235). Moreover, in vitro
research has demonstrated that the PRC2 inhibitor, pyrroloquinoline
quinone, exerts potent inhibitory effects on the proliferation of
B-cell lymphoma cells (236).
The aberrant expression of HDACs plays a crucial
role in the development of autoimmune diseases, and inhibitors
targeting HDACs have shown immense promise in the treatment of
these disorders (237,238). Compared with age-matched
control mice, MRL/lpr lupus model mice exhibit an elevated
expression and activity of HDAC6 and HDAC9 during both the early
and late stages of disease (107). Preclinical studies have
demonstrated that the HDAC6-selective inhibitor, ACY-738, suppress
multiple B-cell activation pathways, reduces PC formation and
attenuates GC responses in lupus-prone mice (238,239). Treatment with the DNMT
inhibitor, 5'-azacytidine, an FDA-approved anticancer agent,
induces hypomethylation of the Ahr gene, downregulates
Aicda expression, attenuates GC responses and diminishes
IgG1 production, ultimately leading to marked amelioration of
RA-like disease in mice (121).
Taken together, these findings establish a strong rationale for
developing novel epigenetic therapies in autoimmune diseases and
lymphomas.
B-cells allow response against a large number of
antigens from various pathogens and confer the important feature of
immune memory. Extensive research has shown that DNA methylation,
post-translational histone modifications, and miRNAs together
constitute the epigenetic landscape governing B-cell
differentiation and function. Technological advances, particularly
in single-cell transcriptomics, whole-genome bisulfite sequencing
and next-generation sequencing, have greatly deepened our
understanding of these regulatory mechanisms during B-cell
development and differentiation. Specifically, distinct B-cell
subsets exhibit dynamic epigenetic profiles, while immature B-cells
display genome-wide DNA hypermethylation and histone deacetylation,
whereas activated B-cells exhibit elevated histone lysine
methylation. Dysregulation of epigenetic modifications frequently
contributes to the pathogenesis of autoimmune diseases and
lymphomas.
Over the past decades, high-throughput sequencing
approaches have provided extensive insights into the mechanisms
through which epigenomic dysregulation contributes to
B-cell-mediated autoimmunity and lymphomagenesis. Aberrant DNA
methylation patterns, altered histone-modifying enzyme activities,
and imbalances in miRNA and lncRNA expression act in concert to
promote autoreactive B-cell expansion, pathogenic autoantibody
production and pro-inflammatory cytokine release, thereby
facilitating malignant transformation. Unlike irreversible genetic
mutations, epigenetic modifications are reversible and thus
represent attractive therapeutic targets. Preclinical and clinical
evidence demonstrates that selective inhibitors targeting these
epigenetic enzymes, such as EZH2 inhibitors, KDM5 inhibitors,
HDAC3/HDAC6 inhibitors and DNMT inhibitors, can restore normal
epigenetic landscapes, suppress malignant B-cell proliferation,
attenuate autoimmune responses, and improve disease outcomes
(121,229,232,234). Moreover, combination strategies
that integrate epigenetic modulators with chemotherapeutic agents
or therapies targeting compensatory pathways further enhance
therapeutic efficacy and overcome resistance mechanisms (231,240). These findings collectively
provide a strong rationale for the continued development and
clinical translation of epigenetic therapies, highlighting their
promise as precision interventions for both B-cell malignancies and
autoimmune disorders.
The pathogenesis of B-cell-derived autoimmune
diseases and lymphomas involves complex interactions among genetic,
epigenetic and microenvironmental factors. Although significant
progress has been made in elucidating epigenetic contributions,
further studies are required to define the precise molecular events
that drive disease initiation and progression. Future efforts
integrating multi-omics analyses, single-cell sequencing and
spatial epigenomics will be critical for elucidating these
mechanisms. Moreover, the identification of reliable biomarkers is
essential for early diagnosis and the development of targeted
therapies. The comprehensive characterization of B-cell epigenetic
landscapes using high-throughput technologies will accelerate
biomarker discovery and facilitate personalized treatment
strategies in B-cell autoimmune diseases and lymphomas.
Not applicable.
YL, YG and JZ wrote the manuscript. RR provided the
overall concept and framework of the manuscript and revised it. All
authors have read and agreed to the published version of the
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the Gansu Province University
Teacher Innovation Fund Project (grant no. 2025A-052), the Youth
Science Fund of Lanzhou Jiaotong University (grant no. 1200061323),
Peking University Medicine Sailing Program for Young Scholars'
Scientific and Technological Innovation (grant no.
BMU2025YFJHPY035), and the Fundamental Research Funds for Central
Universities.
|
1
|
Pieper K, Grimbacher B and Eibel H: B-cell
biology and development. J Allergy Clin Immunol. 131:959–971. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hardy RR, Kincade PW and Dorshkind K: The
protean nature of cells in the B lymphocyte lineage. Immunity.
26:703–714. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jensen CT, Strid T and Sigvardsson M:
Exploring the multifaceted nature of the common lymphoid progenitor
compartment. Curr Opin Immunol. 39:121–126. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Suan D, Sundling C and Brink R: Plasma
cell and memory B cell differentiation from the germinal center.
Curr Opin Immunol. 45:97–102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaliman P: Epigenetics and meditation.
Curr Opin Psychol. 28:76–80. 2019. View Article : Google Scholar
|
|
6
|
Silkenstedt E, Salles G, Campo E and
Dreyling M: B-cell non-Hodgkin lymphomas. Lancet. 403:1791–1807.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mu S, Wang W, Liu Q, Ke N, Li H, Sun F,
Zhang J and Zhu Z: Autoimmune disease: A view of epigenetics and
therapeutic targeting. Front Immunol. 15:14827282024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ghafouri-Fard S, Khoshbakht T, Hussen BM,
Taheri M and Jamali E: The emerging role non-coding RNAs in B
cell-related disorders. Cancer Cell Int. 22:912022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sermer D, Pasqualucci L, Wendel HG,
Melnick A and Younes A: Emerging epigenetic-modulating therapies in
lymphoma. Nat Rev Clin Oncol. 16:494–507. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gamarra N and Narlikar GJ: Collaboration
through chromatin: Motors of transcription and chromatin structure.
J Mol Biol. 433:1668762021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng H and Xie W: The role of 3D genome
organization in development and cell differentiation. Nat Rev Mol
Cell Biol. 20:535–550. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zaret KS: Pioneer Transcription factors
initiating gene network changes. Annu Rev Genet. 54:367–385. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Klemm SL, Shipony Z and Greenleaf WJ:
Chromatin accessibility and the regulatory epigenome. Nat Rev
Genet. 20:207–220. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chiarella AM, Lu D and Hathaway NA:
Epigenetic control of a local chromatin landscape. Int J Mol Sci.
21:9432020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nodelman IM and Bowman GD: Biophysics of
chromatin remodeling. Annu Rev Biophys. 50:73–93. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tan T, Shi P, Abbas MN, Wang Y, Xu J, Chen
Y and Cui H: Epigenetic modification regulates tumor progression
and metastasis through EMT (review). Int J Oncol. 60:702022.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lorch Y and Kornberg RD:
Chromatin-remodeling for transcription. Q Rev Biophys. 50:e52017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tanny JC: Chromatin modification by the
RNA polymerase II elongation complex. Transcription. 5:e9880932014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moore LD, Le T and Fan G: DNA methylation
and its basic function. Neuropsychopharmacology. 38:23–38. 2013.
View Article : Google Scholar
|
|
20
|
López-Moyado IF, Ko M, Hogan PG and Rao A:
TET enzymes in the immune system: From DNA demethylation to
immunotherapy, inflammation, and cancer. Annu Rev Immunol.
42:455–488. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gigek CO, Chen ES and Smith MA:
Methyl-CpG-binding protein (MBD) family: Epigenomic read-outs
functions and roles in tumorigenesis and psychiatric diseases. J
Cell Biochem. 117:29–38. 2016. View Article : Google Scholar
|
|
22
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shvedunova M and Akhtar A: Modulation of
cellular processes by histone and non-histone protein acetylation.
Nat Rev Mol Cell Biol. 23:329–349. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sabari BR, Zhang D, Allis CD and Zhao Y:
Metabolic regulation of gene expression through histone acylations.
Nat Rev Mol Cell Biol. 18:90–101. 2017. View Article : Google Scholar :
|
|
25
|
Jambhekar A, Dhall A and Shi Y: Roles and
regulation of histone methylation in animal development. Nat Rev
Mol Cell Biol. 20:625–641. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mattiroli F and Penengo L: Histone
ubiquitination: An integrative signaling platform in genome
stability. Trends Genet. 37:566–581. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oss-Ronen L, Sarusi T and Cohen I: Histone
mono-ubiquitination in transcriptional regulation and its mark on
life: Emerging roles in tissue development and disease. Cells.
11:24042022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lim PS, Li J, Holloway AF and Rao S:
Epigenetic regulation of inducible gene expression in the immune
system. Immunology. 139:285–293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Su N, Yu X, Duan M and Shi N: Recent
advances in methylation modifications of microRNA. Genes Dis.
12:1012012023. View Article : Google Scholar
|
|
30
|
Saliminejad K, Khorram Khorshid HR,
Soleymani Fard S and Ghaffari SH: An overview of microRNAs:
Biology, functions, therapeutics, and analysis methods. J Cell
Physiol. 234:5451–5465. 2019. View Article : Google Scholar
|
|
31
|
Nguyen TA, Park J, Dang TL, Choi YG and
Kim VN: Microprocessor depends on hemin to recognize the apical
loop of primary microRNA. Nucleic Acids Res. 46:5726–5736. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wilson RC, Tambe A, Kidwell MA, Noland CL,
Schneider CP and Doudna JA: Dicer-TRBP complex formation ensures
accurate mammalian MicroRNA biogenesis. Mol Cell. 57:397–407. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fareh M, Yeom KH, Haagsma AC, Chauhan S,
Heo I and Joo C: TRBP ensures efficient Dicer processing of
precursor microRNA in RNA-crowded environments. Nat Commun.
7:136942016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bartel DP: Metazoan MicroRNAs. Cell.
173:20–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Abdelfattah AM, Park C and Choi MY: Update
on non-canonical microRNAs. Biomol Concepts. 5:275–287. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fabian MR, Sonenberg N and Filipowicz W:
Regulation of mRNA translation and stability by microRNAs. Annu Rev
Biochem. 79:351–379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Coyle MC and King N: The evolutionary
foundations of transcriptional regulation in animals. Nat Rev
Genet. 26:812–827. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen LL and Kim VN: Small and long
non-coding RNAs: Past, present, and future. Cell. 187:6451–6485.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Winkle M, Kluiver JL, Diepstra A and van
den Berg A: Emerging roles for long noncoding RNAs in B-cell
development and malignancy. Crit Rev Oncol Hematol. 120:77–85.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mattick JS, Amaral PP, Carninci P,
Carpenter S, Chang HY, Chen LL, Chen R, Dean C, Dinger ME,
Fitzgerald KA, et al: Long non-coding RNAs: Definitions, functions,
challenges and recommendations. Nat Rev Mol Cell Biol. 24:430–447.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Statello L, Guo CJ, Chen LL and Huarte M:
Gene regulation by long non-coding RNAs and its biological
functions. Nat Rev Mol Cell Biol. 22:96–118. 2021. View Article : Google Scholar
|
|
42
|
Gail EH, Healy E, Flanigan SF, Jones N, Ng
XH, Uckelmann M, Levina V, Zhang Q and Davidovich C: Inseparable
RNA binding and chromatin modification activities of a
nucleosome-interacting surface in EZH2. Nat Genet. 56:1193–1202.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tan JY and Marques AC: The activity of
human enhancers is modulated by the splicing of their associated
lncRNAs. PLoS Comput Biol. 18:e10097222022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Y, Liu H, Niu M, Wang Y, Xu R, Guo Y
and Zhang C: Roles of long noncoding RNAs in human inflammatory
diseases. Cell Death Discov. 10:2352024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Corfe SA and Paige CJ: The many roles of
IL-7 in B cell development; mediator of survival, proliferation and
differentiation. Semin Immunol. 24:198–208. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Egawa T, Kawabata K, Kawamoto H, Amada K,
Okamoto R, Fujii N, Kishimoto T, Katsura Y and Nagasawa T: The
earliest stages of B cell development require a chemokine stromal
cell-derived factor/pre-B cell growth-stimulating factor. Immunity.
15:323–334. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhiming W, Luman W, Tingting Q and Yiwei
C: Chemokines and receptors in intestinal B lymphocytes. J Leukoc
Biol. 103:807–819. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Maeda T, Merghoub T, Hobbs RM, Dong L,
Maeda M, Zakrzewski J, van den Brink MR, Zelent A, Shigematsu H,
Akashi K, et al: Regulation of B versus T lymphoid lineage fate
decision by the proto-oncogene LRF. Science. 316:860–866. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kosan C, Saba I, Godmann M, Herold S,
Herkert B, Eilers M and Möröy T: Transcription factor miz-1 is
required to regulate interleukin-7 receptor signaling at early
commitment stages of B cell differentiation. Immunity. 33:917–928.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bosticardo M, Pala F and Notarangelo LD:
RAG deficiencies: Recent advances in disease pathogenesis and novel
therapeutic approaches. Eur J Immunol. 51:1028–1038. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rolink AG, Tschopp J, Schneider P and
Melchers F: BAFF is a survival and maturation factor for mouse B
cells. Eur J Immunol. 32:2004–2010. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
De Silva NS and Klein U: Dynamics of B
cells in germinal centres. Nat Rev Immunol. 15:137–148. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Stavnezer J, Guikema JE and Schrader CE:
Mechanism and regulation of class switch recombination. Annu Rev
Immunol. 26:261–292. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nera KP, Kyläniemi MK and Lassila O:
Regulation of B cell to plasma cell transition within the
follicular B cell response. Scand J Immunol. 82:225–234. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Victora GD and Nussenzweig MC: Germinal
centers. Annu Rev Immunol. 30:429–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shapiro-Shelef M, Lin KI,
McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG and Calame K:
Blimp-1 is required for the formation of immunoglobulin secreting
plasma cells and pre-plasma memory B cells. Immunity. 19:607–620.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Martin-Subero JI and Oakes CC: Charting
the dynamic epigenome during B-cell development. Semin Cancer Biol.
51:139–148. 2018. View Article : Google Scholar
|
|
58
|
Lai AY, Mav D, Shah R, Grimm SA, Phadke D,
Hatzi K, Melnick A, Geigerman C, Sobol SE, Jaye DL and Wade PA: DNA
methylation profiling in human B cells reveals immune regulatory
elements and epigenetic plasticity at Alu elements during B-cell
activation. Genome Res. 23:2030–2041. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Schuyler RP, Merkel A, Raineri E, Altucci
L, Vellenga E, Martens JHA, Pourfarzad F, Kuijpers TW, Burden F,
Farrow S, et al: Distinct trends of DNA methylation patterning in
the innate and adaptive immune systems. Cell Rep. 17:2101–2111.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Odegard VH, Kim ST, Anderson SM, Shlomchik
MJ and Schatz DG: Histone modifications associated with somatic
hypermutation. Immunity. 23:101–110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Begum NA, Stanlie A, Nakata M, Akiyama H
and Honjo T: The histone chaperone Spt6 is required for
activation-induced cytidine deaminase target determination through
H3K4me3 regulation. J Biol Chem. 287:32415–32429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cyster JG and Allen CDC: B cell responses:
Cell interaction dynamics and decisions. Cell. 177:524–540. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Victora GD and Nussenzweig MC: Germinal
centers. Annu Rev Immunol. 40:413–442. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Fujimura S, Matsui T, Kuwahara K, Maeda K
and Sakaguchi N: Germinal center B-cell-associated DNA
hypomethylation at transcriptional regions of the AID gene. Mol
Immunol. 45:1712–1719. 2008. View Article : Google Scholar
|
|
65
|
Barwick BG, Scharer CD, Martinez RJ, Price
MJ, Wein AN, Haines RR, Bally APR, Kohlmeier JE and Boss JM: B cell
activation and plasma cell differentiation are inhibited by de novo
DNA methylation. Nat Commun. 9:19002018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Manoharan A, Du Roure C, Rolink AG and
Matthias P: De novo DNA Methyltransferases Dnmt3a and Dnmt3b
regulate the onset of Igκ light chain rearrangement during early
B-cell development. Eur J Immunol. 45:2343–2355. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Shaknovich R, Cerchietti L, Tsikitas L,
Kormaksson M, De S, Figueroa ME, Ballon G, Yang SN, Weinhold N,
Reimers M, et al: DNA methyltransferase 1 and DNA methylation
patterning contribute to germinal center B-cell differentiation.
Blood. 118:3559–3569. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Orlanski S, Labi V, Reizel Y, Spiro A,
Lichtenstein M, Levin-Klein R, Koralov SB, Skversky Y, Rajewsky K,
Cedar H and Bergman Y: Tissue-specific DNA demethylation is
required for proper B-cell differentiation and function. Proc Natl
Acad Sci USA. 113:5018–5023. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Millán-Zambrano G, Burton A, Bannister AJ
and Schneider R: Histone post-translational modifications-cause and
consequence of genome function. Nat Rev Genet. 23:563–580. 2022.
View Article : Google Scholar
|
|
70
|
Konuma T and Zhou MM: Distinct histone H3
lysine 27 modifications dictate different outcomes of gene
transcription. J Mol Biol. 436:1683762024. View Article : Google Scholar
|
|
71
|
Weake VM and Workman JL: Histone
ubiquitination: Triggering gene activity. Mol Cell. 29:653–663.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chowdhury M, Forouhi O, Dayal S, McCloskey
N, Gould HJ, Felsenfeld G and Fear DJ: Analysis of intergenic
transcription and histone modification across the human
immunoglobulin heavy-chain locus. Proc Natl Acad Sci USA.
105:15872–15877. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Crouch EE, Li Z, Takizawa M,
Fichtner-Feigl S, Gourzi P, Montaño C, Feigenbaum L, Wilson P, Janz
S, Papavasiliou FN and Casellas R: Regulation of AID expression in
the immune response. J Exp Med. 204:1145–1156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Liu Z, Mai A and Sun J: Lysine acetylation
regulates Bruton's tyrosine kinase in B cell activation. J Immunol.
184:244–254. 2010. View Article : Google Scholar
|
|
75
|
Iwama A, Oguro H, Negishi M, Kato Y and
Nakauchia H: Epigenetic regulation of hematopoietic stem cell
self-renewal by polycomb group genes. Int J Hematol. 81:294–300.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Gu Y, Jones AE, Yang W, Liu S, Dai Q, Liu
Y, Swindle CS, Zhou D, Zhang Z, Ryan TM, et al: The histone H2A
deubiquitinase Usp16 regulates hematopoiesis and hematopoietic stem
cell function. Proc Natl Acad Sci USA. 113:E51–E60. 2016.
View Article : Google Scholar :
|
|
77
|
Li C, Irrazabal T, So CC, Berru M, Du L,
Lam E, Ling AK, Gommerman JL, Pan-Hammarström Q and Martin A: The
H2B deubiquitinase Usp22 promotes antibody class switch
recombination by facilitating non-homologous end joining. Nat
Commun. 9:10062018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
So CC, Ramachandran S and Martin A: E3
ubiquitin ligases RNF20 and RNF40 are required for double-stranded
break (DSB) repair: Evidence for monoubiquitination of histone H2B
lysine 120 as a novel axis of DSB signaling and repair. Mol Cell
Biol. 39:e00488–18. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chen CZ, Li L, Lodish HF and Bartel DP:
MicroRNAs modulate hematopoietic lineage differentiation. Science.
303:83–86. 2004. View Article : Google Scholar
|
|
80
|
Ventura A, Young AG, Winslow MM, Lintault
L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone
JR, et al: Targeted deletion reveals essential and overlapping
functions of the miR-17 through 92 family of miRNA clusters. Cell.
132:875–886. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Rao DS, O'Connell RM, Chaudhuri AA,
Garcia-Flores Y, Geiger TL and Baltimore D: MicroRNA-34a perturbs B
lymphocyte development by repressing the forkhead box transcription
factor Foxp1. Immunity. 33:48–59. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Xiao C, Calado DP, Galler G, Thai TH,
Patterson HC, Wang J, Rajewsky N, Bender TP and Rajewsky K: MiR-150
controls B cell differentiation by targeting the transcription
factor c-Myb. Cell. 131:146–159. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Mehta A, Mann M, Zhao JL, Marinov GK,
Majumdar D, Garcia-Flores Y, Du X, Erikci E, Chowdhury K and
Baltimore D: The microRNA-212/132 cluster regulates B cell
development by targeting Sox4. J Exp Med. 212:1679–1692. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
King JK, Ung NM, Paing MH, Contreras JR,
Alberti MO, Fernando TR, Zhang K, Pellegrini M and Rao DS:
Regulation of marginal zone B-cell differentiation by
MicroRNA-146a. Front Immunol. 7:6702017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Thai TH, Calado DP, Casola S, Ansel KM,
Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, et
al: Regulation of the germinal center response by microRNA-155.
Science. 316:604–608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Vigorito E, Perks KL, Abreu-Goodger C,
Bunting S, Xiang Z, Kohlhaas S, Das PP, Miska EA, Rodriguez A,
Bradley A, et al: microRNA-155 regulates the generation of
immunoglobulin class-switched plasma cells. Immunity. 27:847–859.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kramer NJ, Wang WL, Reyes EY, Kumar B,
Chen CC, Ramakrishna C, Cantin EM, Vonderfecht SL, Taganov KD, Chau
N and Boldin MP: Altered lymphopoiesis and immunodeficiency in
miR-142 null mice. Blood. 125:3720–3730. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wigton EJ and Ansel KM: Noncoding RNAs in
B cell responses. RNA Biol. 18:633–639. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Brazão TF, Johnson JS, Müller J, Heger A,
Ponting CP and Tybulewicz VL: Long noncoding RNAs in B-cell
development and activation. Blood. 128:e10–e19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tschumper RC, Hoelzinger DB, Walters DK,
Davila JI, Osborne CA and Jelinek DF: Stage-specific non-coding RNA
expression patterns during in vitro human B cell differentiation
into antibody secreting plasma cells. Noncoding RNA.
8:152022.PubMed/NCBI
|
|
91
|
Petri A, Dybkær K, Bøgsted M, Thrue CA,
Hagedorn PH, Schmitz A, Bødker JS, Johnsen HE and Kauppinen S: Long
noncoding RNA expression during human B-cell development. PLoS One.
10:e01382362015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Mielczarek O, Rogers CH, Zhan Y, Matheson
LS, Stubbington MJT, Schoenfelder S, Bolland DJ, Javierre BM,
Wingett SW, Várnai C, et al: Intra- and interchromosomal contact
mapping reveals the Igh locus has extensive conformational
heterogeneity and interacts with B-lineage genes. Cell Rep.
42:1130742023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Rothschild G, Zhang W, Lim J, Giri PK,
Laffleur B, Chen Y, Fang M, Chen Y, Nair L, Liu ZP, et al:
Noncoding RNA transcription alters chromosomal topology to promote
isotype-specific class switch recombination. Sci Immunol.
5:eaay58642020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Luo M, Jeong M, Sun D, Park HJ, Rodriguez
BA, Xia Z, Yang L, Zhang X, Sheng K, Darlington GJ, et al: Long
non-coding RNAs control hematopoietic stem cell function. Cell Stem
Cell. 16:426–438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Yu B, Qi Y, Li R, Shi Q, Satpathy AT and
Chang HY: B cell-specific XIST complex enforces X-inactivation and
restrains atypical B cells. Cell. 184:1790–1803.e17. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kiriakidou M and Ching CL: Systemic lupus
erythematosus. Ann Intern Med. 172:ITC81–ITC96. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Dai X, Fan Y and Zhao X: Systemic lupus
erythematosus: Updated insights on the pathogenesis, diagnosis,
prevention and therapeutics. Signal Transduct Target Ther.
10:1022025. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Ulff-Møller CJ, Asmar F, Liu Y, Svendsen
AJ, Busato F, Grønbaek K, Tost J and Jacobsen S: Twin DNA
methylation profiling reveals flare-dependent interferon signature
and B cell promoter hypermethylation in systemic lupus
erythematosus. Arthritis Rheumatol. 70:878–890. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Absher DM, Li X, Waite LL, Gibson A,
Roberts K, Edberg J, Chatham WW and Kimberly RP: Genome-wide DNA
methylation analysis of systemic lupus erythematosus reveals
persistent hypomethylation of interferon genes and compositional
changes to CD4+ T-cell populations. PLoS Genet. 9:e10036782013.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Fali T, Le Dantec C, Thabet Y, Jousse S,
Hanrotel C, Youinou P, Brooks WH, Perl A and Renaudineau Y: DNA
methylation modulates HRES1/p28 expression in B cells from patients
with lupus. Autoimmunity. 47:265–271. 2014. View Article : Google Scholar :
|
|
101
|
Hui-Yuen J, Jiang K, Malkiel S, Eberhard
BA, Walters H, Diamond B and Jarvis J: B lymphocytes in
treatment-naive paediatric patients with lupus are epigenetically
distinct from healthy children. Lupus Sci Med. 10:e0009212023.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Mazari L, Ouarzane M and Zouali M:
Subversion of B lymphocyte tolerance by hydralazine, a potential
mechanism for drug-induced lupus. Proc Natl Acad Sci USA.
104:6317–6322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Tanaka S, Ise W, Inoue T, Ito A, Ono C,
Shima Y, Sakakibara S, Nakayama M, Fujii K, Miura I, et al: Tet2
and Tet3 in B cells are required to repress CD86 and prevent
autoimmunity. Nat Immunol. 21:950–961. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hung KH, Woo YH, Lin IY, Liu CH, Wang LC,
Chen HY, Chiang BL and Lin KI: The KDM4A/KDM4C/NF-κB and WDR5
epigenetic cascade regulates the activation of B cells. Nucleic
Acids Res. 46:5547–5560. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Rohraff DM, He Y, Farkash EA, Schonfeld M,
Tsou PS and Sawalha AH: Inhibition of EZH2 ameliorates lupus-like
disease in MRL/lpr mice. Arthritis Rheumatol. 71:1681–1690. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Gautam P, Sharma A and Bhatnagar A: Global
histone modification analysis reveals hypoacetylated H3 and H4
histones in B Cells from systemic lupus erythematosus patients.
Immunol Lett. 240:41–45. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Regna NL, Vieson MD, Gojmerac AM, Luo XM,
Caudell DL and Reilly CM: HDAC expression and activity is
upregulated in diseased lupus-prone mice. Int Immunopharmacol.
29:494–503. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Forster N, Gallinat S, Jablonska J, Weiss
S, Elsässer HP and Lutz W: p300 protein acetyltransferase activity
suppresses systemic lupus erythematosus-like autoimmune disease in
mice. J Immunol. 178:6941–6948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Liu Y, Dong J, Mu R, Gao Y, Tan X, Li Y,
Li Z and Yang G: MicroRNA-30a promotes B cell hyperactivity in
patients with systemic lupus erythematosus by direct interaction
with Lyn. Arthritis Rheum. 65:1603–1611. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Luo S, Liu Y, Liang G, Zhao M, Wu H, Liang
Y, Qiu X, Tan Y, Dai Y, Yung S, et al: The role of microRNA-1246 in
the regulation of B cell activation and the pathogenesis of
systemic lupus erythematosus. Clin Epigenetics. 7:242015.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Wang M, Chen H, Qiu J, Yang HX, Zhang CY,
Fei YY, Zhao LD, Zhou JX, Wang L, Wu QJ, et al: Antagonizing miR-7
suppresses B cell hyperresponsiveness and inhibits lupus
development. J Autoimmun. 109:1024402020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Garchow BG, Bartulos Encinas O, Leung YT,
Tsao PY, Eisenberg RA, Caricchio R, Obad S, Petri A, Kauppinen S
and Kiriakidou M: Silencing of microRNA-21 in vivo ameliorates
autoimmune splenomegaly in lupus mice. EMBO Mol Med. 3:605–615.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Yuan Y, Kasar S, Underbayev C,
Vollenweider D, Salerno E, Kotenko SV and Raveche E: Role of
microRNA-15a in autoantibody production in interferon-augmented
murine model of lupus. Mol Immunol. 52:61–70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Xiao C, Srinivasan L, Calado DP, Patterson
HC, Zhang B, Wang J, Henderson JM, Kutok JL and Rajewsky K:
Lymphoproliferative disease and autoimmunity in mice with increased
miR-17-92 expression in lymphocytes. Nat Immunol. 9:405–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Jang S, Kwon EJ and Lee JJ: Rheumatoid
arthritis: Pathogenic roles of diverse immune cells. Int J Mol Sci.
23:9052022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Smolen JS, Aletaha D and McInnes IB:
Rheumatoid arthritis. Lancet. 388:2023–2038. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Scott DL, Wolfe F and Huizinga TW:
Rheumatoid arthritis. Lancet. 376:1094–1108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Julià A, Absher D, López-Lasanta M, Palau
N, Pluma A, Waite Jones L, Glossop JR, Farrell WE, Myers RM and
Marsal S: Epigenome-wide association study of rheumatoid arthritis
identifies differentially methylated loci in B cells. Hum Mol
Genet. 26:2803–2811. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
de Andres MC, Perez-Pampin E, Calaza M,
Santaclara FJ, Ortea I, Gomez-Reino JJ and Gonzalez A: Assessment
of global DNA methylation in peripheral blood cell subpopulations
of early rheumatoid arthritis before and after methotrexate.
Arthritis Res Ther. 17:2332015. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Ocskó T, Tóth DM, Hoffmann G, Tubak V,
Glant TT and Rauch TA: Transcription factor Zbtb38 downregulates
the expression of anti-inflammatory IL1r2 in mouse model of
rheumatoid arthritis. Biochim Biophys Acta Gene Regul Mech.
1861:1040–1047. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Tóth DM, Ocskó T, Balog A, Markovics A,
Mikecz K, Kovács L, Jolly M, Bukiej AA, Ruthberg AD, Vida A, et al:
Amelioration of autoimmune arthritis in mice treated with the DNA
methyltransferase inhibitor 5'-azacytidine. Arthritis Rheumatol.
71:1265–1275. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Li Y, Zhou M, Lv X, Song L, Zhang D, He Y,
Wang M, Zhao X, Yuan X, Shi G and Wang D: Reduced activity of HDAC3
and increased acetylation of histones H3 in peripheral blood
mononuclear cells of patients with rheumatoid arthritis. J Immunol
Res. 2018:73135152018. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Heinicke F, Zhong X, Flåm ST, Breidenbach
J, Leithaug M, Mæhlen MT, Lillegraven S, Aga AB, Norli ES,
Mjaavatten MD, et al: MicroRNA expression differences in
blood-derived CD19+ B cells of methotrexate treated rheumatoid
arthritis patients. Front Immunol. 12:6637362021. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Alivernini S, Kurowska-Stolarska M,
Tolusso B, Benvenuto R, Elmesmari A, Canestri S, Petricca L,
Mangoni A, Fedele AL, Di Mario C, et al: MicroRNA-155 influences
B-cell function through PU.1 in rheumatoid arthritis. Nat Commun.
7:129702016. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Kurowska-Stolarska M, Alivernini S,
Ballantine LE, Asquith DL, Millar NL, Gilchrist DS, Reilly J, Ierna
M, Fraser AR, Stolarski B, et al: MicroRNA-155 as a proinflammatory
regulator in clinical and experimental arthritis. Proc Natl Acad
Sci USA. 108:11193–11198. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Baldini C, Fulvio G, La Rocca G and Ferro
F: Update on the pathophysiology and treatment of primary Sjögren
syndrome. Nat Rev Rheumatol. 20:473–491. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Luppi F, Sebastiani M, Sverzellati N,
Cavazza A, Salvarani C and Manfredi A: Lung complications of
Sjogren syndrome. Eur Respir Rev. 29:2000212020. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Rischmueller M, Tieu J and Lester S:
Primary Sjogren's syndrome. Best Pract Res Clin Rheumatol.
30:189–220. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Miceli-Richard C, Wang-Renault SF,
Boudaoud S, Busato F, Lallemand C, Bethune K, Belkhir R, Nocturne
G, Mariette X and Tost J: Overlap between differentially methylated
DNA regions in blood B lymphocytes and genetic at-risk loci in
primary Sjögren's syndrome. Ann Rheum Dis. 75:933–940. 2016.
View Article : Google Scholar
|
|
130
|
Imgenberg-Kreuz J, Sandling JK, Almlöf JC,
Nordlund J, Signér L, Norheim KB, Omdal R, Rönnblom L, Eloranta ML,
Syvänen AC and Nordmark G: Genome-wide DNA methylation analysis in
multiple tissues in primary Sjögren's syndrome reveals regulatory
effects at interferon-induced genes. Ann Rheum Dis. 75:2029–2036.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Cole MB, Quach H, Quach D, Baker A, Taylor
KE, Barcellos LF and Criswell LA: Epigenetic signatures of salivary
gland inflammation in Sjogren's syndrome. Arthritis Rheumatol.
68:2936–2944. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Konsta OD, Le Dantec C, Charras A, Brooks
WH, Arleevskaya MI, Bordron A and Renaudineau Y: An in silico
approach reveals associations between genetic and epigenetic
factors within regulatory elements in B cells from primary
Sjögren's syndrome patients. Front Immunol. 6:4372015. View Article : Google Scholar
|
|
133
|
Inamo J, Suzuki K, Takeshita M, Kassai Y,
Takiguchi M, Kurisu R, Okuzono Y, Tasaki S, Yoshimura A and
Takeuchi T: Identification of novel genes associated with
dysregulation of B cells in patients with primary Sjögren's
syndrome. Arthritis Res Ther. 22:1532020. View Article : Google Scholar
|
|
134
|
Wang-Renault SF, Boudaoud S, Nocturne G,
Roche E, Sigrist N, Daviaud C, Bugge Tinggaard A, Renault V,
Deleuze JF, Mariette X and Tost J: Deregulation of microRNA
expression in purified T and B lymphocytes from patients with
primary Sjögren's syndrome. Ann Rheum Dis. 77:133–140. 2018.
View Article : Google Scholar
|
|
135
|
Daridon C, Devauchelle V, Hutin P, Le
Berre R, Martins-Carvalho C, Bendaoud B, Dueymes M, Saraux A,
Youinou P and Pers JO: Aberrant expression of BAFF by B lymphocytes
infiltrating the salivary glands of patients with primary Sjögren's
syndrome. Arthritis Rheum. 56:1134–1144. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Oh J, Vidal-Jordana A and Montalban X:
Multiple sclerosis: Clinical aspects. Curr Opin Neurol. 31:752–759.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Wang Y, Wang J and Feng J: Multiple
sclerosis and pregnancy: Pathogenesis, influencing factors, and
treatment options. Autoimmun Rev. 22:1034492023. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Comi G, Bar-Or A, Lassmann H, Uccelli A,
Hartung HP, Montalban X, Sørensen PS, Hohlfeld R and Hauser SL:
Expert Panel of the 27th Annual Meeting of the European Charcot
Foundation: Role of B cells in multiple sclerosis and related
disorders. Ann Neurol. 89:13–23. 2021. View Article : Google Scholar
|
|
139
|
Cencioni MT, Mattoscio M, Magliozzi R,
Bar-Or A and Muraro PA: B cells in multiple sclerosis-from targeted
depletion to immune reconstitution therapies. Nat Rev Neurol.
17:399–414. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Bar-Or A, Fawaz L, Fan B, Darlington PJ,
Rieger A, Ghorayeb C, Calabresi PA, Waubant E, Hauser SL, Zhang J
and Smith CH: Abnormal B-cell cytokine responses a trigger of
T-cell-mediated disease in MS? Ann Neurol. 67:452–461. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Li R, Rezk A, Miyazaki Y, Hilgenberg E,
Touil H, Shen P, Moore CS, Michel L, Althekair F, Rajasekharan S,
et al: Proinflammatory GM-CSF-producing B cells in multiple
sclerosis and B cell depletion therapy. Sci Transl Med.
7:310ra1662015. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Kappos L, Li D, Calabresi PA, O'Connor P,
Bar-Or A, Barkhof F, Yin M, Leppert D, Glanzman R, Tinbergen J and
Hauser SL: Ocrelizumab in relapsing-remitting multiple sclerosis: A
phase 2, randomised, placebo-controlled, multicentre trial. Lancet.
378:1779–1787. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Ewing E, Kular L, Fernandes SJ,
Karathanasis N, Lagani V, Ruhrmann S, Tsamardinos I, Tegner J,
Piehl F, Gomez-Cabrero D and Jagodic M: Combining evidence from
four immune cell types identifies DNA methylation patterns that
implicate functionally distinct pathways during multiple sclerosis
progression. EBioMedicine. 43:411–423. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Maltby VE, Lea RA, Graves MC, Sanders KA,
Benton MC, Tajouri L, Scott RJ and Lechner-Scott J: Genome-wide DNA
methylation changes in CD19+ B cells from
relapsing-remitting multiple sclerosis patients. Sci Rep.
8:174182018. View Article : Google Scholar
|
|
145
|
Ma Q, Shams H, Didonna A, Baranzini SE,
Cree BAC, Hauser SL, Henry RG and Oksenberg JR: Integration of
epigenetic and genetic profiles identifies multiple sclerosis
disease-critical cell types and genes. Commun Biol. 6:3422023.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Annibali V, Umeton R, Palermo A, Severa M,
Etna MP, Giglio S, Romano S, Ferraldeschi M, Buscarinu MC,
Vecchione A, et al: Analysis of coding and non-coding transcriptome
of peripheral B cells reveals an altered interferon response factor
(IRF)-1 pathway in multiple sclerosis patients. J Neuroimmunol.
324:165–171. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Lindberg RLP, Hoffmann F, Mehling M, Kuhle
J and Kappos L: Altered expression of miR-17-5p in CD4+ lymphocytes
of relapsing-remitting multiple sclerosis patients. Eur J Immunol.
40:888–898. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Sievers C, Meira M, Hoffmann F, Fontoura
P, Kappos L and Lindberg RLP: Altered microRNA expression in B
lymphocytes in multiple sclerosis: Towards a better understanding
of treatment effects. Clin Immunol. 144:70–79. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Miyazaki Y, Li R, Rezk A, Misirliyan H,
Moore C, Farooqi N, Solis M, Goiry LG, de Faria O Junior, Dang VD,
et al: A novel microRNA-132-sirtuin-1 axis underlies aberrant
B-cell cytokine regulation in patients with relapsing-remitting
multiple sclerosis [corrected]. PLoS One. 9:e1054212014. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Syed FZ: Type 1 diabetes mellitus. Ann
Intern Med. 175:ITC33–ITC48. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Wang YN, Li R, Huang Y, Chen H, Nie H, Liu
L, Zou X, Zhong J, Zheng B and Gong Q: The role of B cells in the
pathogenesis of type 1 diabetes. Front Immunol. 15:14503662024.
View Article : Google Scholar
|
|
152
|
Stefan M, Zhang W, Concepcion E, Yi Z and
Tomer Y: DNA methylation profiles in type 1 diabetes twins point to
strong epigenetic effects on etiology. J Autoimmun. 50:33–37. 2014.
View Article : Google Scholar :
|
|
153
|
Paul DS, Teschendorff AE, Dang MAN, Lowe
R, Hawa MI, Ecker S, Beyan H, Cunningham S, Fouts AR, Ramelius A,
et al: Increased DNA methylation variability in type 1 diabetes
across three immune effector cell types. Nat Commun. 7:135552016.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Miao F, Smith DD, Zhang L, Min A, Feng W
and Natarajan R: Lymphocytes from patients with type 1 diabetes
display a distinct profile of chromatin histone H3 lysine 9
dimethylation: An epigenetic study in diabetes. Diabetes.
57:3189–3198. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Berry GJ, Budgeon LR, Cooper TK,
Christensen ND and Waldner H: The type 1 diabetes resistance locus
B10 Idd9.3 mediates impaired B-cell lymphopoiesis and implicates
microRNA-34a in diabetes protection. Eur J Immunol. 44:1716–1727.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Zou D, Feng S, Hu B, Guo M, Lv Y, Ma R, Du
Y and Feng J: Bromodomain proteins as potential therapeutic targets
for B-cell non-Hodgkin lymphoma. Cell Biosci. 14:1432024.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Martelli M, Ferreri AJM, Agostinelli C, Di
Rocco A, Pfreundschuh M and Pileri SA: Diffuse large B-cell
lymphoma. Crit Rev Oncol Hematol. 87:146–171. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Melnik BC, Stadler R, Weiskirchen R,
Leitzmann C and Schmitz G: Potential pathogenic impact of Cow's
milk consumption and bovine milk-derived exosomal MicroRNAs in
diffuse large B-cell lymphoma. Int J Mol Sci. 24:61022023.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Morin RD, Mendez-Lago M, Mungall AJ, Goya
R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field
M, et al: Frequent mutation of histone-modifying genes in
non-Hodgkin lymphoma. Nature. 476:298–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
You H, Xu-Monette ZY, Wei L, Nunns H, Nagy
ML, Bhagat G, Fang X, Zhu F, Visco C, Tzankov A, et al: Genomic
complexity is associated with epigenetic regulator mutations and
poor prognosis in diffuse large B-cell lymphoma. Oncoimmunology.
10:19283652021. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Ortega-Molina A, Boss IW, Canela A, Pan H,
Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al: The
histone lysine methyltransferase KMT2D sustains a gene expression
program that represses B cell lymphoma development. Nat Med.
21:1199–1208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Pasqualucci L, Trifonov V, Fabbri G, Ma J,
Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, et
al: Analysis of the coding genome of diffuse large B-cell lymphoma.
Nat Genet. 43:830–837. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
163
|
Pasqualucci L, Dominguez-Sola D, Chiarenza
A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma
J, et al: Inactivating mutations of acetyltransferase genes in
B-cell lymphoma. Nature. 471:189–195. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Marquard L, Poulsen CB, Gjerdrum LM, de
Nully Brown P, Christensen IJ, Jensen PB, Sehested M, Johansen P
and Ralfkiaer E: Histone deacetylase 1, 2, 6 and acetylated histone
H4 in B- and T-cell lymphomas. Histopathology. 54:688–698. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Cosenza M and Pozzi S: The therapeutic
strategy of HDAC6 inhibitors in lymphoproliferative disease. Int J
Mol Sci. 19:23372018. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance
JM, Ito A and Yao TP: The deacetylase HDAC6 regulates aggresome
formation and cell viability in response to misfolded protein
stress. Cell. 115:727–738. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Kovacs JJ, Murphy PJM, Gaillard S, Zhao X,
Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB and Yao TP: HDAC6
regulates Hsp90 acetylation and chaperone-dependent activation of
glucocorticoid receptor. Mol Cell. 18:601–607. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Amara K, Ziadi S, Hachana M, Soltani N,
Korbi S and Trimeche M: DNA methyltransferase DNMT3b protein
overexpression as a prognostic factor in patients with diffuse
large B-cell lymphomas. Cancer Sci. 101:1722–1730. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Loo SK, Ab Hamid SS, Musa M and Wong KK:
DNMT1 is associated with cell cycle and DNA replication gene sets
in diffuse large B-cell lymphoma. Pathol Res Pract. 214:134–143.
2018. View Article : Google Scholar
|
|
170
|
Asmar F, Punj V, Christensen J, Pedersen
MT, Pedersen A, Nielsen AB, Hother C, Ralfkiaer U, Brown P,
Ralfkiaer E, et al: Genome-wide profiling identifies a DNA
methylation signature that associates with TET2 mutations in
diffuse large B-cell lymphoma. Haematologica. 98:1912–1920. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Mouly E, Ghamlouch H, Della-Valle V,
Scourzic L, Quivoron C, Roos-Weil D, Pawlikowska P, Saada V, Diop
MK, Lopez CK, et al: B-cell tumor development in Tet2-deficient
mice. Blood Adv. 2:703–714. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Dominguez PM, Ghamlouch H, Rosikiewicz W,
Kumar P, Béguelin W, Fontán L, Rivas MA, Pawlikowska P, Armand M,
Mouly E, et al: TET2 deficiency causes germinal center hyperplasia,
impairs plasma cell differentiation, and promotes B-cell
lymphomagenesis. Cancer Discov. 8:1632–1653. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap
YS, Bakogianni S, Yu Y, Bhattacharyya S, Shaknovich R, Geng H,
Lobry C, et al: TET1 is a tumor suppressor of hematopoietic
malignancy. Nat Immunol. 16:653–662. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Tian X, Pelton A, Shahsafaei A and Dorfman
DM: Differential expression of enhancer of zeste homolog 2 (EZH2)
protein in small cell and aggressive B-cell non-Hodgkin lymphomas
and differential regulation of EZH2 expression by p-ERK1/2 and MYC
in aggressive B-cell lymphomas. Mod Pathol. 29:1050–1057. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
175
|
McCabe MT, Ott HM, Ganji G, Korenchuk S,
Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A III,
Diaz E, et al: EZH2 inhibition as a therapeutic strategy for
lymphoma with EZH2-activating mutations. Nature. 492:108–112. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Caganova M, Carrisi C, Varano G, Mainoldi
F, Zanardi F, Germain PL, George L, Alberghini F, Ferrarini L,
Talukder AK, et al: Germinal center dysregulation by histone
methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest.
123:5009–5022. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Cheng Y, Fu Y, Wang Y and Wang J: The m6A
methyltransferase METTL3 is functionally implicated in DLBCL
development by regulating m6A modification in PEDF. Front Genet.
11:9552020. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
He L, Thomson JM, Hemann MT,
Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe
SW, Hannon GJ and Hammond SM: A microRNA polycistron as a potential
human oncogene. Nature. 435:828–833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Tagawa H, Karube K, Tsuzuki S, Ohshima K
and Seto M: Synergistic action of the microRNA-17 polycistron and
Myc in aggressive cancer development. Cancer Sci. 98:1482–1490.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Roehle A, Hoefig KP, Repsilber D, Thorns
C, Ziepert M, Wesche KO, Thiere M, Loeffler M, Klapper W,
Pfreundschuh M, et al: MicroRNA signatures characterize diffuse
large B-cell lymphomas and follicular lymphomas. Br J Haematol.
142:732–744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Lawrie CH, Chi J, Taylor S, Tramonti D,
Ballabio E, Palazzo S, Saunders NJ, Pezzella F, Boultwood J,
Wainscoat JS and Hatton CS: Expression of microRNAs in diffuse
large B cell lymphoma is associated with immunophenotype, survival
and transformation from follicular lymphoma. J Cell Mol Med.
13:1248–1260. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Carbone A, Roulland S, Gloghini A, Younes
A, von Keudell G, López-Guillermo A and Fitzgibbon J: Follicular
lymphoma. Nat Rev Dis Primers. 5:832019. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Green MR: Chromatin modifying gene
mutations in follicular lymphoma. Blood. 131:595–604. 2018.
View Article : Google Scholar :
|
|
184
|
Green MR, Kihira S, Liu CL, Nair RV,
Salari R, Gentles AJ, Irish J, Stehr H, Vicente-Dueñas C,
Romero-Camarero I, et al: Mutations in early follicular lymphoma
progenitors are associated with suppressed antigen presentation.
Proc Natl Acad Sci USA. 112:E1116–E1125. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Morin RD, Johnson NA, Severson TM, Mungall
AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et
al: Somatic mutations altering EZH2 (Tyr641) in follicular and
diffuse large B-cell lymphomas of germinal-center origin. Nat
Genet. 42:181–185. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
186
|
Béguelin W, Teater M, Meydan C, Hoehn KB,
Phillip JM, Soshnev AA, Venturutti L, Rivas MA, Calvo-Fernández MT,
Gutierrez J, et al: Mutant EZH2 induces a pre-malignant lymphoma
niche by reprogramming the immune response. Cancer Cell.
37:655–673.e11. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Zhang J, Vlasevska S, Wells VA, Nataraj S,
Holmes AB, Duval R, Meyer SN, Mo T, Basso K, Brindle PK, et al: The
CREBBP acetyltransferase is a haploinsufficient tumor suppressor in
B-cell lymphoma. Cancer Discov. 7:322–337. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Antoniolli M, Solovey M, Hildebrand JA,
Freyholdt T, Strobl CD, Bararia D, Keay WD, Adolph L, Heide M,
Passerini V, et al: ARID1A mutations protect follicular lymphoma
from FAS-dependent immune surveillance by reducing
RUNX3/ETS1-driven FAS-expression. Cell Death Differ. 32:899–910.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Wang W, Corrigan-Cummins M, Hudson J,
Maric I, Simakova O, Neelapu SS, Kwak LW, Janik JE, Gause B, Jaffe
ES and Calvo KR: MicroRNA profiling of follicular lymphoma
identifies microRNAs related to cell proliferation and tumor
response. Haematologica. 97:586–594. 2012. View Article : Google Scholar :
|
|
190
|
Ryan CE, Armand P and LaCasce AS:
Frontline management of mantle cell lymphoma. Blood. 145:663–672.
2025. View Article : Google Scholar
|
|
191
|
López C, Silkenstedt E, Dreyling M and Beà
S: Biological and clinical determinants shaping heterogeneity in
mantle cell lymphoma. Blood Adv. 8:3652–3664. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Hill HA, Qi X, Jain P, Nomie K, Wang Y,
Zhou S and Wang ML: Genetic mutations and features of mantle cell
lymphoma: A systematic review and meta-analysis. Blood Adv.
4:2927–2938. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Greiner TC, Dasgupta C, Ho VV,
Weisenburger DD, Smith LM, Lynch JC, Vose JM, Fu K, Armitage JO,
Braziel RM, et al: Mutation and genomic deletion status of ataxia
telangiectasia mutated (ATM) and p53 confer specific gene
expression profiles in mantle cell lymphoma. Proc Natl Acad Sci
USA. 103:2352–2357. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Kridel R, Meissner B, Rogic S, Boyle M,
Telenius A, Woolcock B, Gunawardana J, Jenkins CE, Cochrane C,
Ben-Neriah S, et al: Whole transcriptome sequencing reveals
recurrent NOTCH1 mutations in mantle cell lymphoma. Blood.
119:1963–1971. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Enjuanes A, Albero R, Clot G, Navarro A,
Beà S, Pinyol M, Martín-Subero JI, Klapper W, Staudt LM, Jaffe ES,
et al: Genome-wide methylation analyses identify a subset of mantle
cell lymphoma with a high number of methylated CpGs and aggressive
clinicopathological features. Int J Cancer. 133:2852–2863. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Li XY, Li Y, Zhang L, Liu X, Feng L and
Wang X: The antitumor effects of arsenic trioxide in mantle cell
lymphoma via targeting Wnt/β-catenin pathway and DNA
methyltransferase-1. Oncol Rep. 38:3114–3120. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Queirós Ana C, Beekman R, Vilarrasa-Blasi
R, Duran-Ferrer M, Clot G, Merkel A, Raineri E, Russiñol N,
Castellano G, Beà S, et al: Decoding the DNA methylome of mantle
cell lymphoma in the light of the entire B cell lineage. Cancer
Cell. 30:806–821. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Xargay-Torrent S, López-Guerra M,
Saborit-Villarroya I, Rosich L, Campo E, Roué G and Colomer D:
Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by
acetylation of proapoptotic BH3-only gene promoters. Clin Cancer
Res. 17:3956–3968. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Zhang J, Jima D, Moffitt AB, Liu Q, Czader
M, Hsi ED, Fedoriw Y, Dunphy CH, Richards KL, Gill JI, et al: The
genomic landscape of mantle cell lymphoma is related to the
epigenetically determined chromatin state of normal B cells. Blood.
123:2988–2996. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Zhang J, Dominguez-Sola D, Hussein S, Lee
JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al:
Disruption of KMT2D perturbs germinal center B cell development and
promotes lymphomagenesis. Nat Med. 21:1190–1198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Demosthenous C, Gupta SK, Sun J, Wang Y,
Troska TP and Gupta M: Deregulation of polycomb repressive
complex-2 in mantle cell lymphoma confers growth advantage by
epigenetic suppression of cdkn2b. Front Oncol. 10:12262020.
View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Zhao JJ, Lin J, Lwin T, Yang H, Guo J,
Kong W, Dessureault S, Moscinski LC, Rezania D, Dalton WS, et al:
microRNA expression profile and identification of miR-29 as a
prognostic marker and pathogenetic factor by targeting CDK6 in
mantle cell lymphoma. Blood. 115:2630–2639. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Di Lisio L, Gómez-López G, Sánchez-Beato
M, Gómez-Abad C, Rodríguez ME, Villuendas R, Ferreira BI, Carro A,
Rico D, Mollejo M, et al: Mantle cell lymphoma: Transcriptional
regulation by microRNAs. Leukemia. 24:1335–1342. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Robaina MC, Mazzoccoli L, Arruda VO, Reis
FR, Apa AG, de Rezende LM and Klumb CE: Deregulation of DNMT1,
DNMT3B and miR-29s in Burkitt lymphoma suggests novel contribution
for disease pathogenesis. Exp Mol Pathol. 98:200–207. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Dorsett Y, McBride KM, Jankovic M,
Gazumyan A, Thai TH, Robbiani DF, Di Virgilio M, Reina San-Martin
B, Heidkamp G, Schwickert TA, et al: MicroRNA-155 suppresses
activation-induced cytidine deaminase-mediated Myc-Igh
translocation. Immunity. 28:630–638. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Wang M, Yang W, Li M and Li Y: Low
expression of miR-150 in pediatric intestinal Burkitt lymphoma. Exp
Mol Pathol. 96:261–266. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Di Lisio L, Sánchez-Beato M, Gómez-López
G, Rodríguez ME, Montes-Moreno S, Mollejo M, Menárguez J, Martínez
MA, Alves FJ, Pisano DG, et al: MicroRNA signatures in B-cell
lymphomas. Blood Cancer J. 2:e572012. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Bueno MJ, Gómez de Cedrón M, Gómez-López
G, Pérez de Castro I, Di Lisio L, Montes-Moreno S, Martínez N,
Guerrero M, Sánchez-Martínez R, Santos J, et al: Combinatorial
effects of microRNAs to suppress the Myc oncogenic pathway. Blood.
117:6255–6266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Zucca E, Arcaini L, Buske C, Johnson PW,
Ponzoni M, Raderer M, Ricardi U, Salar A, Stamatopoulos K,
Thieblemont C, et al: Marginal zone lymphomas: ESMO clinical
practice guidelines for diagnosis, treatment and follow-up. Ann
Oncol. 31:17–29. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Dierlamm J, Baens M, Wlodarska I,
Stefanova-Ouzounova M, Hernandez JM, Hossfeld DK, De Wolf-Peeters
C, Hagemeijer A, Van den Berghe H and Marynen P: The apoptosis
inhibitor gene API2 and a novel 18q gene, MLT, are recurrently
rearranged in the t(11;18)(q21;q21) associated with
mucosa-associated lymphoid tissue lymphomas. Blood. 93:3601–3609.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Willis TG, Jadayel DM, Du MQ, Peng H,
Perry AR, Abdul-Rauf M, Price H, Karran L, Majekodunmi O, Wlodarska
I, et al: Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell
lymphoma and mutated in multiple tumor types. Cell. 96:35–45. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Streubel B, Lamprecht A, Dierlamm J,
Cerroni L, Stolte M, Ott G, Raderer M and Chott A:
T(14;18)(q32;q21) involving IGH and MALT1 is a frequent chromosomal
aberration in MALT lymphoma. Blood. 101:2335–2339. 2003. View Article : Google Scholar
|
|
214
|
Bühler MM, Martin-Subero JI,
Pan-Hammarström Q, Campo E and Rosenquist R: Towards precision
medicine in lymphoid malignancies. J Intern Med. 292:221–242. 2022.
View Article : Google Scholar
|
|
215
|
Mirandari A, Parker H, Ashton-Key M,
Stevens B, Walewska R, Stamatopoulos K, Bryant D, Oscier DG, Gibson
J and Strefford JC: The genomic and molecular landscape of splenic
marginal zone lymphoma, biological and clinical implications.
Explor Target Antitumor Ther. 5:877–901. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Arribas AJ, Rinaldi A, Mensah AA, Kwee I,
Cascione L, Robles EF, Martinez-Climent JA, Oscier D, Arcaini L,
Baldini L, et al: DNA methylation profiling identifies two splenic
marginal zone lymphoma subgroups with different clinical and
genetic features. Blood. 125:1922–1931. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Arribas AJ, Campos-Martín Y, Gómez-Abad C,
Algara P, Sánchez-Beato M, Rodriguez-Pinilla MS, Montes-Moreno S,
Martinez N, Alves-Ferreira J, Piris MA and Mollejo M: Nodal
marginal zone lymphoma: Gene expression and miRNA profiling
identify diagnostic markers and potential therapeutic targets.
Blood. 119:e9–e21. 2012. View Article : Google Scholar
|
|
218
|
Xiao F, Rui K, Shi X, Wu H, Cai X, Lui KO,
Lu Q, Ballestar E, Tian J, Zou H and Lu L: Epigenetic regulation of
B cells and its role in autoimmune pathogenesis. Cell Mol Immunol.
19:1215–1234. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Lanata CM, Paranjpe I, Nititham J, Taylor
KE, Gianfrancesco M, Paranjpe M, Andrews S, Chung SA, Rhead B,
Barcellos LF, et al: A phenotypic and genomics approach in a
multi-ethnic cohort to subtype systemic lupus erythematosus. Nat
Commun. 10:39022019. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Imgenberg-Kreuz J, Almlöf JC, Leonard D,
Sjöwall C, Syvänen AC, Rönnblom L, Sandling JK and Nordmark G:
Shared and unique patterns of DNA methylation in systemic lupus
erythematosus and primary Sjögren's syndrome. Front Immunol.
10:16862019. View Article : Google Scholar
|
|
221
|
Imgenberg-Kreuz J, Rasmussen A, Sivils K
and Nordmark G: Genetics and epigenetics in primary Sjögren's
syndrome. Rheumatology (Oxford). 60:2085–2098. 2021. View Article : Google Scholar
|
|
222
|
Zhang K, Luo Z, Yao X, Lu D, Hong T, Zhu
X, Chen M and Wang X: Identification of epigenetic alteration of
the IFI44L gene in B cells of sjogren's syndrome as a clinical
biomarker and molecular significance. J Inflamm Res. 18:2499–2512.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Nair N, Plant D, Verstappen SM, Isaacs JD,
Morgan AW, Hyrich KL, Barton A and Wilson AG; MATURA investigators:
Differential DNA methylation correlates with response to
methotrexate in rheumatoid arthritis. Rheumatology (Oxford).
59:1364–1371. 2020. View Article : Google Scholar
|
|
224
|
Kiselev IS, Kulakova OG, Boyko AN and
Favorova OO: DNA methylation as an epigenetic mechanism in the
development of multiple sclerosis. Acta Naturae. 13:45–57. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Wang F, Gao Y, Chen Y, Li P, Zeng Y, Chen
Y, Yin Y, Jia Y and Wang Y: Development of a mitochondria-related
gene signature for prognostic assessment in diffuse large B cell
lymphoma. Front Oncol. 15:15428292025. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Xie Z, Li M, Hong H, Xu Q, He Z and Peng
Z: Expression of N6-methyladenosine (m6A)
regulators correlates with immune microenvironment characteristics
and predicts prognosis in diffuse large cell lymphoma (DLBCL).
Bioengineered. 12:6115–6133. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Rosikiewicz W, Chen X, Dominguez PM,
Ghamlouch H, Aoufouchi S, Bernard OA, Melnick A and Li S: TET2
deficiency reprograms the germinal center B cell epigenome and
silences genes linked to lymphomagenesis. Sci Adv. 6:eaay58722020.
View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Amin R and Braza MS: The follicular
lymphoma epigenome regulates its microenvironment. J Exp Clin
Cancer Res. 41:212022. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Straining R and Eighmy W: Tazemetostat:
EZH2 inhibitor. J Adv Pract Oncol. 13:158–163. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Morschhauser F, Tilly H, Chaidos A, McKay
P, Phillips T, Assouline S, Batlevi CL, Campbell P, Ribrag V, Damaj
GL, et al: Tazemetostat for patients with relapsed or refractory
follicular lymphoma: An open-label, single-arm, multicentre, phase
2 trial. Lancet Oncol. 21:1433–1442. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Niccolai R, Göbel C, Ndoj K, Kreft M,
Kuiken HJ, Lieftink C, Morris B, Yska SD, Hendrix S, van den Broek
B, et al: Cholesterol biosynthesis as a drug-induced vulnerability
in diffuse large B cell lymphoma insensitive to EZH2 inhibition.
Neoplasia. 70:1012432025. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Heward J, Konali L, D'Avola A, Close K,
Yeomans A, Philpott M, Dunford J, Rahim T, Al Seraihi AF, Wang J,
et al: KDM5 inhibition offers a novel therapeutic strategy for the
treatment of KMT2D mutant lymphomas. Blood. 138:370–381. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Jiang Y, Ortega-Molina A, Geng H, Ying HY,
Hatzi K, Parsa S, McNally D, Wang L, Doane AS, Agirre X, et al:
CREBBP inactivation promotes the development of HDAC3-dependent
lymphomas. Cancer Discov. 7:38–53. 2017. View Article : Google Scholar
|
|
234
|
Mondello P, Tadros S, Teater M, Fontan L,
Chang AY, Jain N, Yang H, Singh S, Ying HY, Chu CS, et al:
Selective inhibition of HDAC3 targets synthetic vulnerabilities and
activates immune surveillance in lymphoma. Cancer Discov.
10:440–459. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Straus DJ, Hamlin PA, Matasar MJ, Lia
Palomba M, Drullinsky PR, Zelenetz AD, Gerecitano JF, Noy A,
Hamilton AM, Elstrom R, et al: Phase I/II trial of vorinostat with
rituximab, cyclophosphamide, etoposide and prednisone as palliative
treatment for elderly patients with relapsed or refractory diffuse
large B-cell lymphoma not eligible for autologous stem cell
transplantation. Br J Haematol. 168:663–670. 2015. View Article : Google Scholar
|
|
236
|
Jiang M, Huang F, Hong X, Xu C, Zhang B,
Hu S, Wang G, Hu D, Sun W, Lu Q, et al: PQQ inhibits PRC2
methyltransferase activity and suppresses the proliferation of
B-cell lymphoma in vitro. Chem Biodivers. 22:e2025001982025.
View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Vijaykrishnaraj M, Patil P, Ghate SD,
Bhandary AK, Haridas VM and Shetty P: Efficacy of HDAC inhibitors
and epigenetic modulation in the amelioration of synovial
inflammation, cellular invasion, and bone erosion in rheumatoid
arthritis pathogenesis. Int Immunopharmacol. 122:1106442023.
View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Regna NL, Vieson MD, Luo XM, Chafin CB,
Puthiyaveetil AG, Hammond SE, Caudell DL, Jarpe MB and Reilly CM:
Specific HDAC6 inhibition by ACY-738 reduces SLE pathogenesis in
NZB/W mice. Clin Immunol. 162:58–73. 2016. View Article : Google Scholar
|
|
239
|
Ren J, Catalina MD, Eden K, Liao X, Read
KA, Luo X, McMillan RP, Hulver MW, Jarpe M, Bachali P, et al:
Selective histone deacetylase 6 inhibition normalizes B cell
activation and germinal center formation in a model of systemic
lupus erythematosus. Front Immunol. 10:25122019. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Rosenthal AC, Munoz JL and Villasboas JC:
Clinical advances in epigenetic therapies for lymphoma. Clin
Epigenetics. 15:392023. View Article : Google Scholar : PubMed/NCBI
|