Introduction
Autoimmune diseases, once considered rare, now
affect a substantial proportion of the global population and arise
from a breakdown of immune tolerance, whereby adaptive and innate
responses fail to discriminate self from non-self (1). While inherited risk loci explain
part of disease susceptibility, mounting evidence implicates
epigenetic mechanisms, heritable yet reversible changes in gene
regulation, including DNA methylation, histone modifications and
non-coding RNAs, as key determinants of pathogenic immune programs
(2,3). These mechanisms are metabolically
gated: The one-carbon (folate) network supplies methyl groups for
DNA and histone methylation, thereby coupling nutrient status to
chromatin state and immune function (4). This metabolic-epigenetic axis
carries out a central role in integrating environmental factors,
nutrition and cellular stress responses, thereby shaping
immune-system development across the lifespan.
Within this network, methylenetetrahydrofolate
reductase (MTHFR) reduces 5,10-methylene-THF to 5-methyl-THF,
enabling remethylation of homocysteine to methionine and sustaining
S-adenosylmethionine (SAM), the universal methyl donor. Common
MTHFR variants 677C>T and 1298A>C lower enzymatic flux
to varying degrees and, particularly under low folate conditions,
are associated with hyperhomocysteinemia and reduced methylation
capacity (5-8). Beyond genetics, emerging research
indicates that MTHFR itself is subject to epigenetic
regulation (promoter methylation, chromatin context, microRNAs and
lncRNAs) (9-12), positioning the enzyme both as a
modulator and as a target within the metabolism-epigenome
interface. This dual role underscores why MTHFR alterations
can propagate broadly across the immune, vascular and neurological
systems, especially in environments of chronic inflammation or
increased methylation demand.
The research landscape shows convergent epigenetic
phenotypes across immune-mediated conditions, for example, synovial
and T-cell hypomethylation in rheumatoid arthritis (RA),
interferon-driven signatures in systemic lupus erythematosus (SLE),
mucosal (and saliva-detectable) alterations in celiac disease (CeD)
and therapy-responsive methylomes in multiple sclerosis (MS)
(2,3,13,14). These findings are consistent with
constraints on methyl-group availability and inflammation-linked
suppression of the methylation machinery (2,3).
Yet key controversies remain. First, associations between
MTHFR variants and autoimmunity are heterogeneous across
ancestries and clinical phenotypes and appear conditional on
environmental factors (dietary folate/B-vitamin status) and
medications (such as methotrexate; MTX) (5-8).
Second, the direction of causality is debated: Epigenetic
abnormalities may be primary drivers, secondary consequences of
inflammation and treatment or both and locus-resolved evidence in
primary immune cells for MTHFR regulation is still limited.
Moreover, the majority of existing studies examine isolated
components of the pathway (2,15-17), highlighting the need for
integrative analyses that jointly evaluate genetic variants,
methylation capacity, inflammatory signaling and nutrient
availability.
The present review synthesizes current literature on
the folate-MTHFR-SAM axis as a modulator of epigenetic
stability in autoimmune disease. Specifically, the present review
i) summarizes epigenetic regulation of MTHFR (DNA
methylation, histone marks and non-coding RNAs); ii) outlines
one-carbon biochemistry and gene-nutrient interactions that tune
methylation capacity; iii) integrates disease-specific epigenomic
findings across RA, SLE, MS, CeD and fibromyalgia (FM); and iv)
discusses translational implications for biomarkers and
nutritionally informed or epigenetic adjuncts to immunotherapy. The
present review highlights areas of agreement and active debate and
delineates priorities for cell type-resolved and mechanism-anchored
studies. By doing so, the present review aims to bridge molecular
insights relevant to autoimmune diseases, emphasizing how
folate-dependent epigenetic regulation can contribute both to
disease-risk stratification and to the development of personalized
therapeutic strategies.
Literature search
For the present review, a literature search was
performed in the PubMed (https://pubmed.ncbi.nlm.nih.gov/) and PubMed Central
(PMC) (https://pmc.ncbi.nlm.nih.gov/)
databases using topics and subtopics associated with the role of
one-carbon metabolism (such as MTHFR, folate and homocysteine),
epigenetic mechanisms (such as DNA methylation and histone
modification) and their impact on autoimmune diseases such as RA,
SLE, MS, cEd and FM. The reviewed publications included mechanistic
studies, clinical trials and systematic reviews involving human
subjects that report relevant findings on direct epigenetic
evidence (such as DNA methylation levels), biochemical markers or
clinical outcomes associated with disease activity and therapeutic
response.
Folates
History and discovery
In 1931, studies investigating anemia during
pregnancy identified nutritional deficiency as a key etiological
factor. Experimental work by Wills and Stewart (18) demonstrated that supplementation
with yeast extract (Marmite®) or animal protein could
reverse severe anemia in animal models. These findings led to the
identification of an unknown nutritional factor distinct from
vitamin B2, called the 'Wills factor', which was later
recognized as folate (18-21).
In 1941, Mitchell et al (21) published the first study
describing the concentration of folic acid, a compound named after
the Latin word folium (leaf). Their study revealed that folic acid
was capable of promoting the proliferation of Lactobacillus
casei, Lactobacillus delbreuckii and Streptococcus
lactis. The isolation of the pure, crystalline form of folic
acid (pteroglutamic acid) was first achieved by Robert Stokstad and
Lederle Laboratories in 1943. This achievement was notable because
it enabled researchers to study the characteristics and properties
of the compound. The utilization of folic acid-fermenting
microorganisms was instrumental in achieving this goal (22).
In 1945, Angier et al (23) determined that the chemical
synthesis of pteroglutamic acid from liver samples (21). This development represented a
pivotal shift in the management of megaloblastic anemias due to the
ability to produce folic acid on a large scale for clinical use
(23).
Although early research on folic acid focused on
treating a specific form of anemia, Kumar (24), was the first to identify key
elements of folic acid metabolism associated with acute
lymphoblastic leukemia in children. This work and its broader
implications have been discussed by Kumar et al (24). Despite recognition of the
essential roles of folate in DNA synthesis and neural tube defect
prevention, its metabolic pathway remained poorly characterized. In
1971, Kutzbach and Stokstad (25) isolated and described the enzyme,
MTHFR, for the first time. The enzyme was found to be subject to
allosteric regulation by SAM, associating it with methionine
metabolism, DNA synthesis, cardiovascular disease and neural tube
defects.
In 1973, Tamura and Stokstad (26) evaluated the bioavailability of
naturally occurring folates compared with five synthetic
derivatives. They observed increased bioavailability for synthetic
folates compared with food sources such as liver, yeast, banana,
orange juice and lettuce. Based on advancements in folate
metabolism research, in 1980, scientists described the 'folate
trap', a phenomenon in which the enzyme methionine synthase (MTR),
in the absence of its cofactor (vitamin B12), is unable
to convert homocysteine into methionine. This traps folate in its
5-methyltetrahydrofolate form and inhibits purine and thymidine
synthesis for DNA, even when folate levels are sufficient (27).
In 1990, Frosst et al (28) identified a C677T polymorphism in
the MTHFR gene that reduced enzyme activity and altered
folate concentrations. Epidemiological evidence accumulated since
the discovery of folate, its sources and bioavailability has served
as a foundation for translating knowledge into preventive and
therapeutic public health strategies. Efforts to fortify food
products with synthetic folic acid have been implemented in several
countries, including the United States, Canada, Argentina, Chile
and others across Europe and Latin America (29). These initiatives have led to
substantial reductions in the prevalence of neural tube defects in
newborns, with decreases ranging from ~25 to >60%, depending on
the country and the specifics of program implementation (13,29). Despite these successes, some
populations may exceed recommended folate intake, underscoring the
importance of careful monitoring. Conversely, countries such as
Mexico and Colombia lack comprehensive surveillance and monitoring
systems to evaluate the impact of mandatory folic acid
fortification on population health (29-31).
Chemical structure
During its discovery, folic acid was assigned
multiple names. One of the most widely recognized designations is
folic acid, although it is more commonly referred to as vitamin
B9 or folate (32-34). The term folate refers to a family
of molecules with similar chemical structures that exert beneficial
effects in various health conditions, ranging from anemia to
cardiovascular diseases, cancer and inflammatory processes
(29,35). The chemical structure of folic
acid (C19H19N7O6) serves as the foundation for the diverse chemical
forms of folates. The structural components of folates can be
categorized as follows: i) A heterocyclic pterin structure in
oxidized or reduced form, consisting of a pyrimidine ring, a
pyrazine ring and a methyl-group at carbon 6 that serves as a
bridge for acid linkage; ii) p-aminobenzoic acid; and iii) a mono
or polyglutamate chain of variable length (Fig. 1A).
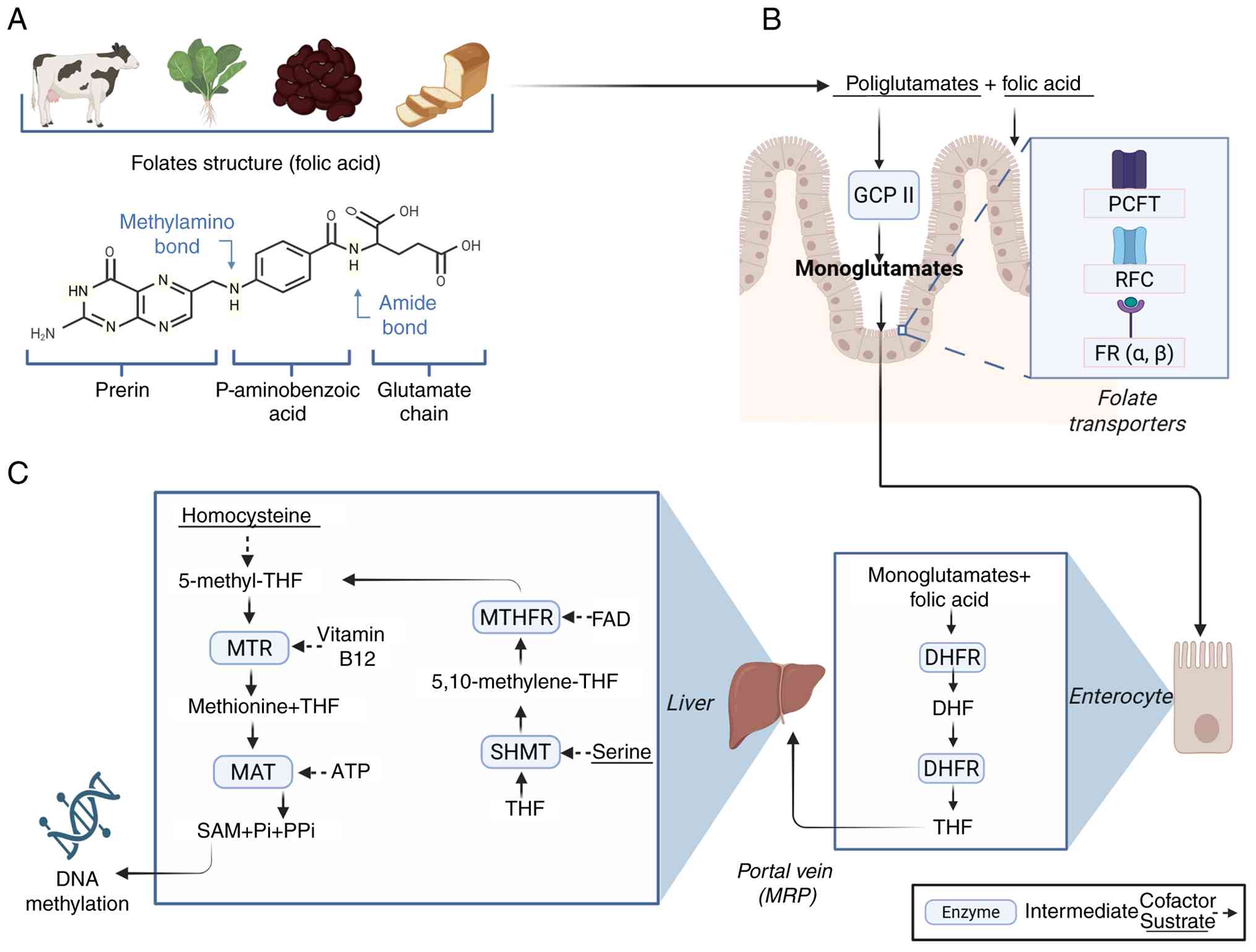 | Figure 1(A) Dietary sources of folate and
general structure. Dietary folates, comprising three main
components (pterin, p-aminobenzoic acid and a glutamate
chain), are ingested as polyglutamates and hydrolyzed to
monoglutamates by the enzyme GCPII in the intestinal brush border.
(B) Intestinal absorption of folate. Folate uptake occurs through
transporters located in enterocytes, primarily the PCFT and the
RFC, as well as through FRα/FRβ present in other cell types. (C) In
folate metabolism, within intestinal cells, folate is sequentially
reduced by DHFR to DHF and ultimately to its active form, THF. THF
is transported to 5,10-methylenetetrahydrofolate (5,10-MTHF) by
serine hydroxymethyltransferase (SHMT) and subsequently reduced by
the FAD-dependent enzyme MTHFR to 5-methylenetetrahydrofolate
(5-MTHF). The latter donates its methyl group to homocysteine to
regenerate methionine, in a reaction catalyzed by MTR that requires
vitamin B12. Methionine is then converted by MAT into
SAM, the universal methyl donor, in an ATP-dependent process. This
sequence associates folate metabolism with methylation reactions
that are key for nucleotide synthesis and epigenetic regulation.
GCPII, glutamate carboxypeptidase II; PCFT, proton-coupled folate
transporter; RFC, reduced folate carrier; FR, folate receptor;
DHFR, dihydrofolate reductase; THF, tetrahydrofolate; 5,10-MTFH,
5,10-methylenetetrahydrofolate; SHMT, serine
hydroxymethyltransferase; MTR, methionine synthase; MAT, methionine
adenosyltransferase; SAM, S-adenosylmethionine. |
A carbon unit may be associated with either the
pterin or the p-aminobenzoic ring, or with both. The classification
of folates depends on the oxidation state of the pterin and carbon
unit, as well as the polyglutamylation state. Consequently, folate
derivatives such as dihydro, tetrahydro, methyl and formyl forms
are expected, invariably conjugated with p-aminobenzoyl-glutamate
as mono-, di-, tri- or polyglutamates (36-38).
This group of compounds, associated with
water-soluble-B complex vitamins, cannot be synthesized by
mammalian cells; therefore, dietary intake is essential, either for
natural sources (tetrahydrofolate, THF) or synthetic
supplementation (folic acid) (31). In total, ~150 biochemical
derivatives with metabolic activity have been described, among
which tetrahydrofolates are the most relevant due to their roles in
DNA and RNA synthesis, cell division, methylation reactions and as
cofactors in multiple metabolic pathways (30,36).
Folic acid, the most oxidized and stable form of
folate, is reduced by dihydrofolate reductase (DHFR) at nitrogen 8
to produce dihydrofolate (DHF). Further reduction at nitrogen 5
yields THF, the active form of the vitamin that functions as a
coenzyme (Fig. 1). THF accepts
carbon atoms at nitrogen positions 5 and 10, generating cofactor
derivatives with specific physiological functions: 5-methyl-THF,
5,10-methyl-THF, 10-formyl-THF and 5-formyl-THF (37).
THF is the principal dietary form of folate in the
body, acting as a carrier in one-carbon cycle biosynthesis. Its
derivative, 5-methyl-THF, is the predominant active form of folate
in blood and supports the conversion of methionine to SAM for
methylation processes. Folic acid, a synthetic and fully oxidized
compound used in supplementation and food fortification, requires
hepatic DHFR for biological activity. By contrast, folinic acid can
yield 5,10-methyl-THF or 5-methyl-THF without requiring DHFR,
making it particularly useful for counteracting the effects of
chemotherapeutic agents (38,39).
The metabolic reactions of folate and its
derivatives are closely associated with the one-carbon cycle and
subject to feedback regulation, emphasizing the generation of
methyl groups that influence epigenetic modifications (36). In addition, folate intermediates
participate in the synthesis of purines, pyrimidines and
methionine, in the interconversion of serine to glycine and in the
catabolism of the latter (40).
Dietary sources
In mammals, the main source of folate comes from
dietary intake. Folate is a component of various food groups,
including vegetables, cereals, fruits and foods of animal origin
(Table I). It can also be
produced as a metabolite by the intestinal microbiota from
different phyla such as bacteroidetes, fusobacteria, proteobacteria
and actinobacteria (41-44).
 | Table INatural foods rich in folate and
industrialized foods fortified with folic acid. |
Table I
Natural foods rich in folate and
industrialized foods fortified with folic acid.
| Food | Folate
(μg/100 g) | Scientific
source |
|---|
| Cooked beef
liver | 290 | USDA |
| Raw spinach | 194 | USDA |
| Cooked lentils | 181 | USDA |
| Cooked white
beans | 172 | BEDCA |
| Cooked
broccoli | 108 | USDA |
| Cooked turnip
greens | 106 | FAO/INFOODS |
| Avocado | 81 | USDA |
| Cooked pork
kidney | 77 | BEDCA |
| Roquefort
cheese | 50 | BEDCA |
| Cooked egg | 25 | FAO/INFOODS |
| Fortified breakfast
cereal | 150-400 | USDA |
| Enriched wheat
pasta | 100-150 | BEDCA |
| Enriched white
bread | 120 | USDA |
| Fortified corn
flour | 80-120 | FAO/INFOODS |
| Enriched white
rice | 90-110 | USDA |
Bioavailability and intestinal
absorption
In mammals, the main source of folate is dietary
intake. Folate is abundant in various food groups, including
vegetables, cereals, fruits and foods of animal origin, and it may
also be produced as a metabolite by the intestinal microbiota. Once
ingested, the efficiency with which folate becomes available for
metabolic reactions depends not only on its chemical form but also
on how it is processed and absorbed in the body. Dietary folates
are typically present as polyglutamates, which are chemically
labile and can be lost during food processing and cooking.
Depending on the food type and preparation, losses can range from
20-60% in vegetables and 30-70% in cereals (45). The bioavailability of folate
refers to the proportion that is absorbed and available for
metabolic reactions or storage. Thus, foods may be rich in folate
yet exhibit low bioavailability (46,47).
The bioavailability of dietary folates can be
compromised by several factors: i) Incomplete release of the
molecule from the original food matrix; ii) degradation of the
molecule within the gastrointestinal tract; and iii) incomplete
hydrolysis of the molecule due to the presence of other dietary
components, such as fatty acids. Additionally, individual
characteristics (such as sex and genetic variations), folate stores
and the availability of other nutrients (vitamin C, vitamin
B12, vitamin B6, niacin, riboflavin or
choline) influence the bioavailability of both dietary and
synthetic folate (46).
Folate absorption primarily occurs in the proximal
segments of the small intestine, the duodenum and jejunum, where an
acidic environment facilitates folate transport (48-50). Dietary polyglutamates undergo
hydrolysis at the intestinal brush border through the action of
glutamate carboxypeptidase II (GCPII), converting them into
monoglutamates that can be absorbed in a manner similar to
synthetic folic acid (48,49). Due to the charge and hydrophilic
nature of the molecule, passive diffusion across cell membranes is
inefficient (50). Reduced
folate carriers (RFCs) serve as the main transporters mediating
systemic folate metabolism (50,51). In addition, dietary folates are
absorbed primarily via proton-coupled folate transporters (PCFTs),
which are located mainly in the upper gastrointestinal tract and in
certain tumors (Fig. 1B)
(48,50,52). Folate receptors (FRα and FRβ)
located in the cell membrane possess a glycosylphosphoinositol
anchor and mediate endocytosis of folate at neutral or slightly
acidic pH (50,51). Across the basolateral membrane,
folates are subsequently transported into the vascular system via
ABCC proteins, particularly ABCC3 (50).
Folate metabolism
The transformation of dietary folate into its active
monoglutamate form requires the action of the enzyme GCPII, located
on the intestinal brush border. GCPII catalyzes the hydrolysis of
folate, generating monoglutamate, which is then internalized by
PCFT within enterocytes. After absorption, DHFR sequentially
reduces monoglutamate to DHF and then to 5-methyl-THF. The
resulting metabolite is exported via the portal vein through
interacting with multidrug resistance proteins until it enters the
bloodstream and reaches tissues, where it is taken up by the RFC
system or by folate receptors. Once inside cells, folate is
converted to polyglutamate forms within tissues, while its active
form is primarily metabolized in the liver (53,54).
5-methyl-THF enters cells via transporters or
receptors. Inside the cell, it participates in the remethylation of
homocysteine to methionine through the activity of MTR and its
cofactor vitamin B12. Methionine is subsequently
converted by methionine adenosyltransferase (MAT) into SAM, the
universal methyl donor for DNA methylation (Fig. 1C). Following methyl group
donation, SAM is converted to S-adenosylhomocysteine (SAH), which
is then hydrolyzed by SAH hydrolase into homocysteine and adenosine
(55). To maintain balanced
production of bioactive folate metabolites, interconversion occurs
at both the mitochondrial and cytosolic levels. This process
involves the cytosolic enzyme serine hydroxymethyltransferase
(SHMT1), which catalyzes the conversion of serine to glycine and
transfers a one-carbon unit to THF to form
5,10-methylenetetrahydrofolate. The mitochondrial isoform (SHMT2)
catalyzes the reverse reaction, synthesizing serine from glycine.
The newly formed serine serves as a feedback substrate in the
metabolic pathways for thymidylate and methionine synthesis and
supports DNA methylation (56).
These reactions underscore the close interconnection
between folate metabolism, the one-carbon cycle and methionine
metabolism. The capacity of folate to accept and donate methyl
groups is fundamental to epigenetic regulation, influencing gene
expression and protein synthesis through methylation processes
(54). MTX, the primary
treatment for inflammatory autoimmune diseases such as RA and MS,
acts as a folate antagonist. It directly inhibits folate metabolism
and indirectly disrupts associated pathways, including purine and
pyrimidine synthesis. Within these pathways, folate-derived
metabolites such as 5,10-methylenetetrahydrofolate (5,10-THF) and
10-formyl-THF carry out essential roles. MTX inhibits key enzymes
of folate metabolism, including DHFR, thymidylate synthase, MTHFR
and SHMT (57,58).
In addition to these pharmacological effects, MTX
interacts with genetic variants in the MTHFR gene (C677T and
A1298C), which independently reduce biologically active folate
levels and elevate homocysteine concentrations. These alterations
contribute to gastrointestinal and hematologic toxicity,
inflammation, oxidative stress and increased cardiovascular risk.
Therefore, maintaining adequate levels of biologically active
folate is essential to preserve cellular homeostasis, support
methylation-dependent processes and minimize systemic dysfunction
across multiple organ systems (59).
Normal values and clinical evaluation of
folate status
Alterations in folate concentration have been
associated with various health complications due to the essential
role of folate in DNA replication, cell proliferation and growth.
This has led to widespread implementation of supplementation and
fortification programs in staple foods across several countries
(60). The prevailing scientific
consensus indicates that high folate intake does not adversely
affect healthy individuals. However, in individuals with
preexisting neoplastic conditions, excessive intake, particularly
of synthetic folic acid, may increase cancer risk, although this
relationship remains to be fully elucidated (61). The erythrocyte folate
concentrations (RBC folate) are considered a stable and reliable
long-term indicator of folate status, as it reflects intracellular
stores, whereas plasma folate concentrations are more transient and
influenced by recent dietary intake. Similarly, analysis of plasma
homocysteine concentrations also facilitates the identification of
disturbances in the methylation cycle (Table II) (62-64).
| Table IIReference concentrations of folate in
serum and erythrocytes (62). |
Table II
Reference concentrations of folate in
serum and erythrocytes (62).
A, Serum or plasma
folate concentration
|
|---|
| Concentration,
ng/ml (nmol/l) | Interpretation |
|---|
| >20 (45.3) | Elevated |
| 6-20
(13.5-45.3) | Normal range |
| 3-5.9
(6.8-13.4) | Possible
deficiency |
| <3
(<6.8) | Deficiency |
|
| B, Erythrocyte
folate concentrations |
|
| Concentration ng/ml
(nmol/l) | Interpretation |
|
| >140
(>317.5) | Elevated |
| 100-140
(226.5-317.5) | Normal range |
| <100
(<226.5) | Possible
deficiency |
Recommended daily intake (RDI)
Although the Food and Drug Administration (FDA)
established the mandatory fortification of all enriched cereal
grain products with folic acid in 1996, full implementation,
providing 140 μg per 100 g of product, was achieved in 1998
(65). Subsequently, in 2016,
the FDA issued a voluntary recommendation for the fortification of
cornmeal. In the United States, the primary dietary sources of
folate include enriched grain products, fortified cornmeal,
ready-to-eat cereals containing 100-400 μg per serving and
adult supplements providing 400-800 μg of folic acid
(66).
Folate requirements depend on age and
physiological status, particularly in women and adolescents of
reproductive age
The RDI is defined as the amount of nutrients
necessary to meet the nutritional needs of 97-98% of the healthy
population. For folate, the average recommended intake is expressed
as micrograms (μg) of dietary folate equivalents (DFE). For
adults, the recommended amount is 400 μg per day (Table III) (67,68).
| Table IIIRecommended daily intake of folate by
age group and physiological condition. |
Table III
Recommended daily intake of folate by
age group and physiological condition.
| Age
group/condition | Daily
recommendation (μg DFE per day)a |
|---|
| Infants 0-6
months | 65 |
| Infants 7-12
months | 80 |
| Children 1-3
years | 150 |
| Children 4-8
years | 200 |
| Children 9-13
years | 300 |
| Adolescents 14-18
years (men) | 400 |
| Adolescents 14-18
years (women) | 400 |
| Adults (≥19
years) | 400 |
| Pregnancy | 600 |
| Lactation | 500 |
The MTHFR: Genetics and
regulation
Genomic location and structure
MTHFR is located on chromosome 1p36.22 and
spans ~20.3 kb, comprising 12 exons in the human genome. Its
promoter is GC-rich and TATA-less, containing Sp1, AP-1, AP-2 and
CAAT elements consistent with housekeeping-type regulation.
Multiple transcription start sites and alternative splicing events
generate two protein isoforms (70 and 77 kDa) and heterogeneous
mRNA 5' and 3'untranslated regions (UTRs), reflecting complex
transcriptional control. Predicted and observed protein lengths
across transcripts range from 656 to 700 amino acids (69-71). The gene encodes MTHFR, a
cytosolic flavoprotein that catalyzes the reduction of
5,10-methylene-THF to 5-methyl-THF, thereby associating the folate
and methionine cycles. Human MTHFR contains an N-terminal catalytic
domain that binds FAD and the folate substrate, and a C-terminal
regulatory domain that binds SAM to mediate allosteric inhibition.
Phosphorylation of N-terminal residues further sensitizes the
enzyme to SAM-dependent feedback (72-74). At the genetic level, two common
functional polymorphisms, C677T (rs1801133) and A1298C (rs1801131),
are widely studied for their effects on enzyme thermolability and
folate/homocysteine status. By contrast, rare truncating or severe
missense variants across catalytic or regulatory domains underlie
classical MTHFR deficiency (75-77).
Epigenetic regulation of MTHFR (DNA
methylation, histone marks and non-coding RNAs)
Expression of the MTHFR gene is finely
controlled by multiple epigenetic mechanisms, including DNA
methylation, histone modifications and non-coding RNAs.
Functionally, MTHFR bridges one-carbon metabolism with the
epigenome by generating 5-methyl-THF for methionine remethylation
and SAM synthesis, thereby determining the methyl-group supply
available for DNA and histone methyltransferases. Evidence shows
that MTHFR acts both as a modulator and a target of
epigenetic regulation: Promoter DNA methylation, chromatin state
and non-coding RNAs can alter its expression, while reduced MTHFR
activity, whether genetic or epigenetic, reduces SAM levels,
limiting methyltransferase capacity and favoring hypomethylation at
methylation-sensitive loci. The result is a metabolic-epigenetic
feedback loop in which folate flux and chromatin control
co-regulate each other (78,79).
The first regulatory layer involves DNA methylation,
the addition of a methyl group to cytosine bases (typically in CpG
promoter regions), which generally represses gene transcription. In
MTHFR, promoter methylation modulates expression in a tissue
and context-dependent manner. For example, in sperm DNA from men
with idiopathic infertility, a case-control study reported
MTHFR promoter hypermethylation in 45% (41/94) of cases vs.
15% (8/54) of fertile controls, with higher methylation levels in
the oligozoospermic subgroup (80). These findings illustrate that
absolute percentages vary widely by CpG site, tissue and
methodology, and that direct data from autoimmune cohorts remain
scarce (81-83).
A second regulatory layer involves histone
post-translational modifications, such as acetylation (for example
H3K9ac) or methylation (for example H3K9me3 and H3K27me3), which
remodel chromatin to activate or repress transcription. This
process is highly sensitive to the metabolic state. During
2-acetylaminofluorene-induced hepatocarcinogenesis in rats,
MTHFR expression is downregulated early; concomitant
promoter-level increases in repressive marks (H3K27me3 gain and
H3K18ac loss) were also observed in the associated methylation gene
Mat1a, while MTHFR repression was mechanistically
associated with miR-22 and miR-29b (84). Models of folate stress likewise
show global reductions in H3K27 and H3K9 methylation, consistent
with SAM limitation (85). In
acute myeloid leukemia cells, reduced MTHFR function, whether due
to polymorphisms or pharmacologic inhibition, decreases
intracellular SAM, leading to loss of the repressive histone marks
H3K27me3 and H3K9me3 and de-repression of transcription factors
such as SPI1 (85). In
neuronal (SH-SY5Y) cell models, MTHFR acts as a metabolic buffer:
Excessive folate disturbs histone-modifying enzyme expression and
shifts the H3K4me3/H3K9me2 balance (86,87). These effects are markedly
amplified when MTHFR is deficient, emphasizing its role in
safeguarding the epigenome against nutrient fluctuations (86-87).
A third regulatory layer involves non-coding RNAs.
MicroRNAs (miRNAs) bind mRNA to inhibit translation and can
modulate MTHFR expression. For instance, miR-22-3p and
miR-149-5p bind the 3'UTR of MTHFR mRNA, and under folate
deficiency, their upregulation suppresses MTHFR protein, further
restricting one-carbon flux. Long non-coding RNAs (lncRNAs) can
also guide chromatin modifiers: The lncRNA HOTAIR, for example,
recruits protein complexes to the MTHFR promoter in
esophageal cancer cells, depositing repressive H3K27me3 marks and
silencing transcription (88-91).
While the majority of direct evidence of
MTHFR epigenetic regulation derives from reproductive and
cancer models, autoimmune contexts offer a compelling rationale for
analogous studies in immune cells. Both RA and SLE exhibit
pronounced DNA-methylation defects in T cells (such as
hypomethylation of type-I-interferon-stimulated genes such as
IFI44L) and show responsiveness to SAM-linked chromatin
mechanisms. These parallels make MTHFR-centered regulation a
plausible contributor in autoimmune pathogenesis and a promising
target for future cell-specific investigations (92-94).
The biochemical axis: One-carbon
metabolism
One-carbon metabolism, SAM and
homocysteine
The one-carbon network integrates the folate and
methionine cycles to supply methyl groups for biosynthesis and
epigenetic regulation. Folate coenzymes carry and transform
one-carbon units primarily derived from serine and glycine. MTHFR
catalyzes the reduction of 5,10-methylene-THF to 5-methyl-THF,
which donates a methyl group to homocysteine via MTR (a vitamin
B12-dependent enzyme) to regenerate methionine.
Methionine is subsequently adenylated by MAT to form SAM, the
universal methyl donor utilized by DNA, RNA and histone
methyltransferases.
After methyl transfer, SAH is formed and hydrolyzed
by adenosylhomocysteinase (AHCY) into homocysteine and adenosine,
thereby removing a potent inhibitor of methyltransferases.
Consequently, the SAM:SAH ratio serves as a proximate index of
cellular methylation capacity. In the liver and kidney, an
alternative remethylation pathway involves betaine-homocysteine
methyltransferase (BHMT), while homocysteine can also leave the
cycle through transsulfuration, catalyzed by cystathionine
β-synthase and cystathionine γ-lyase, to generate cysteine and
glutathione. Collectively, these reactions couple nutrient status
to epigenetic regulation via SAM production and SAH clearance
(Fig. 2) (95-99).
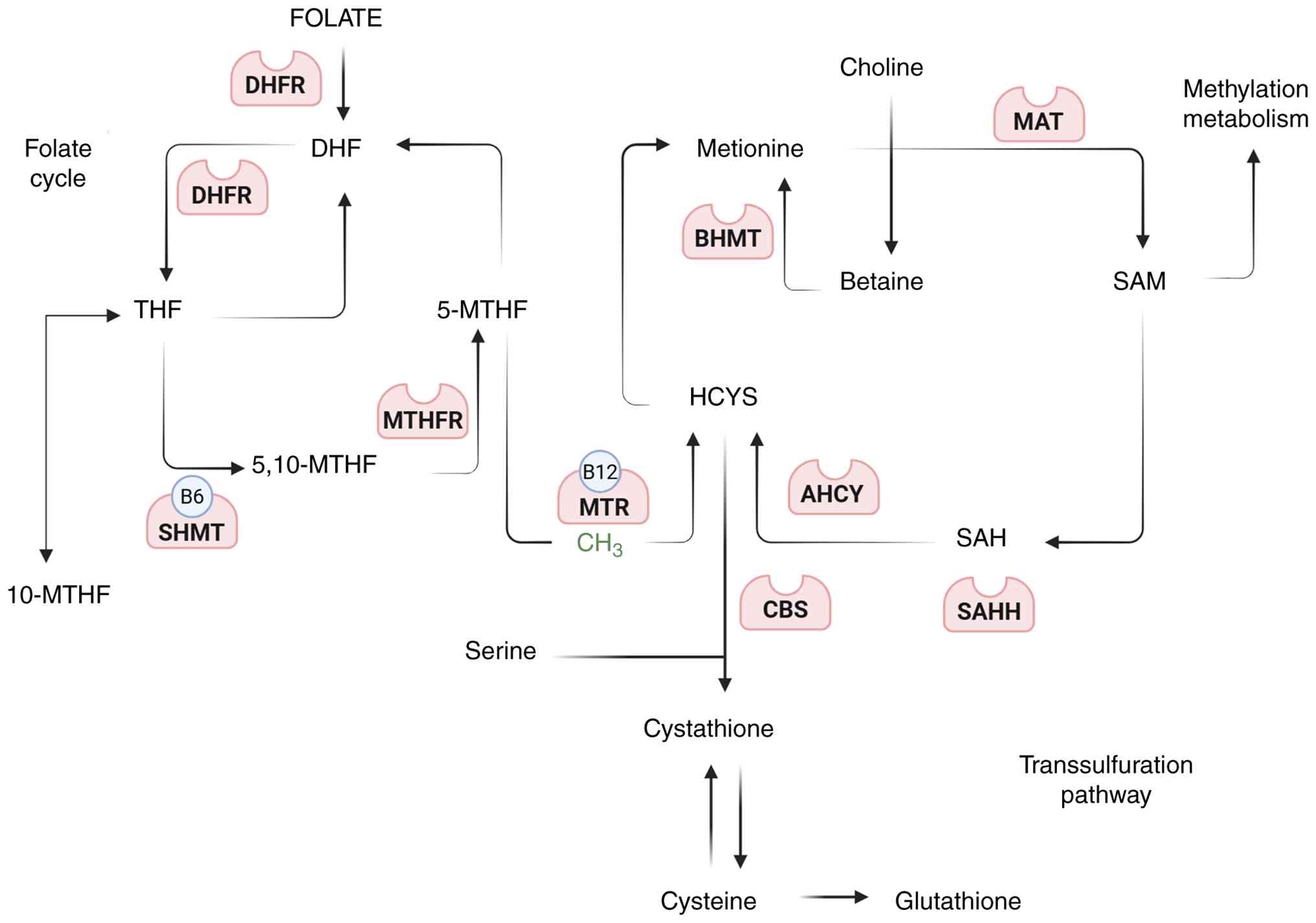 | Figure 2Schematic representation of the
folate and methionine cycles illustrating their interconnection
with one-carbon metabolism, methylation reactions and
transsulfuration. Dietary folate is initially reduced to DHF and
THF by DHFR. THF is converted to 5,10-MTHF by SHMT (a vitamin
B6-dependent enzyme) and subsequently reduced to 5-MTHF
by MTHFR. 5-MTHF donates a methyl group to Hcy via MTR (a vitamin
B12-dependent enzyme), thereby regenerating methionine.
Methionine is then converted to SAM by MAT. SAM serves as the
universal methyl donor for DNA, RNA, protein and lipid methylation.
Following methyl donation, SAM is converted to SAH, which is
hydrolyzed by AHCY to Hcy. Homocysteine can be remethylated by BHMT
using betaine as a methyl donor or diverted into the
transsulfuration pathway via CBS to generate cystathionine,
cysteine and ultimately glutathione. This pathway highlights the
biochemical integration of folate status, methylation potential and
redox homeostasis (97-99). DHR, dihydrofolate; THF,
tetrahydrofolate; DHFR, dihydrofolate reductase; 5,10-MTHF;
5,10-methylene-THF, SHMT, serine hydroxymethyltransferase; 5-MTHF,
5-methyl-THF; 5-MTHF, 5-methyl-THF; MTHFR,
methylenetetrahydrofolate reductase; Hcy, homocysteine; MTR,
methionine synthase; SAM, S-adenosylmethionine; MAT, methionine
adenosyltransferase; SAH; S-adenosylhomocysteine; AHCY,
adenosylhomocysteinase; BHMT, betaine-homocysteine
methyltransferase; CBS, cystathionine β-synthase; SAHH,
S-adenosylhomocysteine hydrolase. |
A fundamental biochemical principle is that SAH
inhibits the majority of methyltransferases with low-micromolar
inhibition constants (Ki). Accumulation of SAH, or a reduction in
SAM, thus constrains DNA and histone methylation even when
substrate (cytosine or lysine) is available. Manipulating AHCY
activity or methionine flux can therefore alter global methylation
states. This inhibitory 'SAH brake' explains why the SAM:SAH ratio,
rather than SAM alone, more accurately reflects methylation
capacity in cells and tissues (98,100,101).
In humans, the MTHFR C677T variant decreases
enzymatic activity and interacts with folate status to influence
genomic DNA methylation and homocysteine levels, with the lowest
methylation observed in TT homozygotes under low-folate conditions.
In mice, MTHFR deficiency decreases SAM, increases SAH and reduces
global DNA methylation, directly associating impaired MTHFR flux to
a hypomethylated genome (102,103). Dietary interventions further
demonstrate causal control of leukocyte methylation by one-carbon
nutrients. Randomized and longitudinal studies have shown that
folic acid and vitamin B12 supplementation modify DNA
methylation profiles, both globally and at specific loci,
consistent with enhanced methyl-group availability for
DNMT-mediated reactions (104-106). Epigenome-wide analyses of
habitual folate and B12 intake corroborate these
associations. Although the magnitude and direction of methylation
change are locus-specific, the collective evidence supports
nutrient-sensitive modulation of the blood methylome through the
folate-MTHFR-SAM axis (104-106).
Finally, plasma homocysteine serves as a clinically
accessible marker of one-carbon imbalance. Elevated concentrations
often indicate insufficient folate or vitamin B12
intake, or reduced MTHFR or MTR activity, and are typically
accompanied by a low SAM:SAH ratio and reduced methylation
potential. In hepatic and renal tissues, BHMT provides a
compensatory remethylation pathway that becomes particularly
relevant when folate-dependent remethylation is limited,
underscoring tissue-specific buffering within the one-carbon
network. These biochemical relationships demonstrate that folate
availability and MTHFR activity define the upper limit for DNA and
histone methylation, providing a mechanistic bridge to the
epigenetic phenotypes described in autoimmune diseases (95-97,99).
Crosstalk with inflammatory and
immunological pathways
Epigenetic regulation intersects immune function
through the one-carbon network that maintains cellular methylation
potential in leukocytes. DNA methylation at promoters and
enhancers, together with SAM-dependent histone methylation,
regulates cytokine programs and lineage stability. Because both
processes depend on the folate-MTHFR-SAM axis and are inhibited by
SAH, immune activation is tightly associated with one-carbon flux
(107,108).
In RA, inflammatory signaling actively suppresses
the methylation machinery. IL-1 rapidly downregulates DNMT1 and
DNMT3A in synovial fibroblasts at picogram concentrations, a change
associated with DNA hypomethylation and sustained inflammatory gene
expression. In SLE, oxidative stress inhibits ERK signaling in
CD4+ T cells, decreases DNMT1 expression and induces
promoter demethylation with aberrant overexpression of normally
silenced genes, thus mechanistically associating inflammatory
stress to erosion of the T-cell methylome (109-111).
Beyond T and stromal compartments, innate immune
cells can also rely on one-carbon-supported SAM to mount
proinflammatory responses. Upon LPS stimulation, macrophages
upregulate serine synthesis and one-carbon metabolism, fueling
epigenetic reprogramming that licenses IL-1β expression.
Inhibition of serine metabolism, in turn, blunts IL-1β production
both in vitro and in vivo. Complementing these
disease-specific examples, epigenome-wide studies in type 1
diabetes (T1D) have demonstrated methylation abnormalities in
immune effector cells (CD4+ T cells, B cells and
monocytes), underscoring that immune epigenomes are broadly
sensitive to both inflammatory context and metabolic state
(112,113).
Converging evidence associates folate status and
homocysteine to inflammatory tone. Folate deficiency increases
oxidative and nitrosative stress, activating NF-κB, while
homocysteine triggers NF-κB activation and IL-6/IL-1β production in
vascular and myeloid cells, biochemical routes through which
impaired remethylation amplifies inflammation. In
macrophage-lineage cells, experimental folate restriction enhances
proinflammatory responses, consistent with a model in which limited
methyl-donor availability constrains methyltransferase activity and
shifts signaling toward activation (114,115).
Collectively, these findings support a bidirectional
feedback loop: Inflammatory cues (such as IL-1 and oxidative
stress) suppress DNMT expression and activity, eroding genomic
methylation, while low folate or MTHFR-limited flux and the
resulting homocysteine/SAM:SAH imbalance promote oxidative and
NF-κB signaling. Together, these mechanisms stabilize a
proinflammatory, hypomethylated epigenetic state within immune
tissues. This mechanistic crosstalk provides the biochemical bridge
associating nutrient status and MTHFR activity to the
immune-epigenetic phenotypes observed across RA, SLE, T1D and other
immune-mediated diseases (107,108,111,116-118).
MTHFR-mediated epigenetic
dysregulation in autoimmune diseases
RA
RA is a chronic, systemic autoimmune disorder
characterized by persistent synovitis, pannus formation and
extra-articular manifestations. Superimposed on this pathogenetic
framework is a robust epigenetic component. Drug-naïve patients
already exhibit disease-associated DNA-methylation differences in
circulating T-cell subsets and synovial tissue, including global
hypomethylation in early disease and cell-type-specific changes
across naïve and memory CD4+ lineages (119,120). In synovial fibroblasts,
proinflammatory cues directly impair methylation capacity: IL-1
rapidly downregulates DNMT1 and DNMT3A/3B expression and activity,
promoting demethylation and stable activation of inflammatory gene
programs (116).
At the gene level, methylation alterations converge
on RA-relevant loci. Hypomethylation at the TNF locus
associates with increased expression and, importantly, predicts
response to biological therapies in clinical cohorts (121). An additional chromatin study in
synovial fibroblasts reveal histone-methylation/STAT3 crosstalk
controlling IL-6-driven effector genes, underscoring how
methyl-donor availability and chromatin state co-regulate cytokine
programs (122).
Regulatory-T-cell instability also arises through epigenetic
mechanisms: Patients with RA exhibit reduced FOXP3
expression and insufficient demethylation of the FOXP3 TSDR,
while MTX therapy can restore Treg function by demethylating a
FOXP3 enhancer, associating pharmacologic intervention to
methylome repair (123,124).
Biochemically, these patterns align with one-carbon
control. The folate-MTHFR-SAM axis determines the methyl-group
supply for DNMTs. Common MTHFR variants (C677T and A1298C)
and low folate levels reduce SAM and increase SAH, limiting
methyltransferase capacity. Consistently, baseline leukocyte DNA
methylation (including global indices) predicts MTX non-response in
early RA, while multiple cohorts reveal methylation signatures
associated with MTX response, pointing to a pharmaco-epigenetic
interface between folate metabolism and RA therapy (125-127). Preliminary evidence further
suggests MTHFR variants may modulate anti-TNF responses in
an allele-dose manner, although results remain population-specific
(128). Overall, folate status,
MTHFR genotype and DNMT activity appear to co-determine the
epigenetic tone of RA tissues and the likelihood of therapeutic
control (129).
SLE
SLE is a chronic autoimmune disease that causes
widespread inflammation and tissue damage affecting the skin,
joints, kidneys, brain, lungs and heart. A defining molecular
hallmark is global DNA hypomethylation in lymphocytes, especially
CD4+ T cells, which associates with disease activity and
stabilizes a type-I-interferon-driven program. Among
interferon-stimulated genes, IFI44L promoter hypomethylation
is exceptionally consistent and has emerged as a diagnostic
biomarker across tissues (130). Mechanistically, SLE T cells
exhibit reduced DNMT1 (maintenance methyltransferase) and
dysregulated DNMT3A/3B, partly due to oxidative-stress mediated
inhibition of ERK signaling, directly associating inflammatory
stress to methylome erosion (131,132).
This enzymatic deficit is compounded by a
one-carbon bottleneck. MTHFR activity is essential for generating
SAM, the universal methyl donor for all DNMTs. Polymorphic
depression of MTHFR activity (such as C677T) and
folate/B12 insufficiency favor hyperhomocysteinemia and
a low SAM:SAH ratio, both of which inhibit methyltransferases.
Meta-analyses confirm associations between MTHFR C677T and
SLE susceptibility; in SLE cohorts, the 677TT genotype and elevated
homocysteine levels associate with subclinical atherosclerosis,
emphasizing the clinical consequences of impaired remethylation
(133,134).
Functionally, SLE exhibits promoter hypomethylation
and overexpression of proinflammatory cytokines (such as IL-6,
particularly in T cells and affected tissues) alongside IL-17 axis
activation. Conversely, tolerance-maintaining pathways become
epigenetically repressed, such as IL-2 transcriptional silencing
and FOXP3 locus instability, where insufficient TSDR
demethylation undermines Treg stability (135,136). Altogether, this evidence
positions the MTHFR-SAM-DNMT axis at the core of SLE
immunoepigenetics: Metabolic constraint yields methylation defects
that hardwire the interferon-skewed, proinflammatory state. The
reversible nature of DNA methylation underscores its diagnostic and
therapeutic potential, motivating interventions that restore
one-carbon flux or target epigenetic writers and erasers (130).
MS
MS is a chronic, immune-mediated neurodegenerative
disorder of the central nervous system, characterized by
demyelination, axonal injury and progressive neurological
impairment (137). Epigenetic
deregulation contributes to its pathogenesis by promoting a
proinflammatory phenotype in peripheral immune cells. Cell-sorted
studies have revealed widespread methylation shifts in T cells and
monocytes, including reproducible changes in CD8+ T
cells and distinct profiles relative to CD4+ subsets,
indicative of compartment-specific immune methylome remodeling
(138-141). These patterns are clinically
dynamic: Both global and locus-specific methylation signals
associate with disability scores and can be modified by
disease-modifying therapies, highlighting the plasticity of the MS
methylome (138,139). In line with immune
polarization, proinflammatory pathways (Th1/Th17) and regulatory
circuits (Treg networks) represent methylation-sensitive axes in MS
(140,141). Central nervous system tissue
analyses further reveal lesion-associated methylation changes
affecting myelin biology and glial programs, associating epigenetic
remodeling to demyelination and repair potential (137).
These epigenetic alterations are biochemically
associated with one-carbon metabolism. The folate-MTHFR-SAM axis
supplies the methyl donor required by DNMTs. Across cohorts, plasma
homocysteine levels are elevated in MS (with folate and
B12 largely unchanged), consistent with impaired
methyl-group homeostasis and a reduced SAM:SAH ratio. Associations
between MTHFR C677T and MS vary across populations, some
case-control studies identify risk signals, while others do not,
supporting a 'substrate-limitation' model where genetic
predisposition interacts with B-vitamin status to constrain DNMT
activity and stabilize a proinflammatory methylation landscape
(7,142,143).
Several agents modulate immune cell methylomes.
Dimethyl fumarate induces coordinated DNA methylation changes in
circulating leukocytes (including CD4+ T cells and
monocytes), while interferon-β produces targeted,
cell-type-specific methylation shifts, demonstrating that
MS-relevant epigenetic programs are reversible and may be corrected
in tandem with the restoration of one-carbon flux (138,144-146).
CeD
CeD is an autoimmune enteropathy triggered by
dietary gluten in genetically susceptible individuals carrying
HLA-DQ2 or HLA-DQ8 haplotypes (147). Beyond its genetic
predisposition, CeD exhibits distinctive DNA methylation
alterations in intestinal mucosa and saliva, revealing the
importance of gene-environment interactions mediated by epigenetic
mechanisms (147-149). Genome-wide studies demonstrate
differential methylation within the HLA region, partly independent
of genotype, and tissue-specific analyses confirm mucosal
remodeling (148). Remarkably,
saliva methylation profiles associate with intestinal patterns,
supporting their potential as non-invasive biomarkers (150).
A compelling mechanistic explanation arises from
the interplay between intestinal pathology and one-carbon
metabolism. Untreated CeD leads to villous atrophy, causing
malabsorption of key nutrients, including folate, thereby
disrupting the MTHFR-dependent remethylation cycle. Common
MTHFR variants may further impair this pathway. Consistent
with this model, hyperhomocysteinemia is frequently observed at
diagnosis and typically improves with a gluten-free diet; however,
in patients with MTHFR variants, elevated homocysteine may
persist despite supplementation. At the tissue level, global
hypomethylation signals, such as LINE-1 hypomethylation, have been
identified in CeD-associated intestinal mucosa, consistent with
substrate limitation of SAM-dependent methylation (151).
This convergence of factors constrains SAM
synthesis, limiting DNMT activity and thereby compromising
maintenance of the methylome. It provides a biochemical explanation
for the epigenetic alterations observed in CeD (152-154). The resulting model establishes
a mechanistic cascade associating dietary triggers (gluten),
intestinal injury (malabsorption), metabolic disruption
(folate/MTHFR deficiency) and epigenetic dysregulation (SAM/DNMTs
imbalance). Clinically, residual folate insufficiency has been
documented even in treated cohorts, emphasizing the need to ensure
methyl-donor adequacy. Methylation profiles, including saliva-based
assays, may assist in diagnosis, monitoring and assessment of
dietary adherence (148,153,154).
FM
Although FM is not a classical autoimmune disease,
it consistently presents with epigenetic abnormalities intersecting
immune and neuroendocrine pathways. Genome-wide and
candidate-region studies reveal global DNA hypomethylation and
locus-specific changes in peripheral blood, enriched for
stress-response, immune-inflammatory and central-sensitization
pathways. These alterations, replicated across independent cohorts,
involve disease-relevant loci such as COMT and BDNF
(155-160).
Mechanistically, these epigenetic findings align
with one-carbon metabolism. A 'substrate-limitation' model in FM is
supported by elevated homocysteine levels observed in FM/chronic
fatigue syndrome cohorts, by associations between symptoms and the
MTHFR C677T (rs1801133) variant, and by gene-environment
interactions showing that rs1801133 modifies the effect of physical
activity on fatigue. Together, these findings indicate that genetic
variation and B-vitamin status can limit SAM availability and
consequently DNMT activity (161-163).
The translational implications are direct. Blood
methylation panels already demonstrate diagnostic and prognostic
potential. Clinical trials further provide proof-of-concept:
Vitamin B12 supplementation markedly improves symptom
severity and anxiety in patients with FM, consistent with
restoration of one-carbon flux and normalization of
methylation-dependent pathways. Similarly, folate and
B12 supplementation have been shown to enhance leukocyte
and mucosal DNA methylation in colorectal adenoma cohorts,
confirming that methyl-donor interventions can modulate the human
methylome. Despite heterogeneity and modest sample sizes, these
findings establish a functional association between nutrient
availability, MTHFR-dependent SAM synthesis and epigenetic
control, underscoring the rationale for larger, mechanistically
informed intervention studies (106,164,165).
These disease-specific epigenetic and metabolic
patterns, encompassing RA, SLE, MS, CeD and FM, are summarized in a
comparative framework (Table
IV), providing an integrated overview of autoimmune diseases
and related disorders.
| Table IVEpigenetic and metabolic comparison
of autoimmune and associated disorders, a summary of converging
evidence associating one-carbon metabolism dysregulation to
epigenetic alterations, particularly DNA hypomethylation, across
autoimmune and associated disorders. |
Table IV
Epigenetic and metabolic comparison
of autoimmune and associated disorders, a summary of converging
evidence associating one-carbon metabolism dysregulation to
epigenetic alterations, particularly DNA hypomethylation, across
autoimmune and associated disorders.
| Feature | RA | SLE | MS | CeD | FM |
|---|
| Primary
pathophysiology | Chronic systemic
autoimmune disorder characterized by persistent joint inflammation
(synovitis). | Systemic autoimmune
disease-causing widespread inflammation and multi-organ
damage. | Immune-mediated
neurodegenerative disorder of the CNS causing demyelination. | Autoimmune
enteropathy triggered by gluten, resulting in villous intestinal
malabsorption. | Neuro-sensory
disorder with chronic pain and fatigue; not a classical autoimmune
disease but involves immune dysregulation. |
| Key epigenetic
signature | Global
hypomethylation in early diseases and cell-type specific
changes. | Global DNA
hypomethylation in lymphocytes (especially CD4+ T cells)
as a defining molecular hallmark. | Widespread, dynamic
methylation shifts in T cells and monocytes. | Altered DNA
methylation in intestinal mucosa and saliva; hypomethylation in the
HLA region. | Global DNA
hypomethylation in peripheral blood, involving stress and
pain-regulation pathways. |
| Affected
cells/tissues | CD4+ T
cells, synovial fibroblasts, circulating leukocytes. | CD4+ T
cells, lymphocytes. |
CD4+/CD8+ T cells,
monocytes, CNS tissue. | Intestinal
epithelium/mucosa, saliva. | Peripheral blood
leukocytes. |
| Role of one-carbon
metabolism | MTHFR
variants and low folate reduce SAM levels, impairing methylation.
Methylation status predicts response to MTX. | MTHFR
variants (C677T) increase susceptibility. Low folate/B12
leads to a reduced SAM:SAH ratio, inhibiting methylation. | Elevated
homocysteine indicates impaired methyl-group metabolism. A
'substrate-limitation' model has been proposed. | Gluten-induced
malabsorption causes folate deficiency, leading to
hyperhomocysteinemia and limited SAM availability for
methylation. | Elevated
homocysteine and symptom association with the MTHFR C677T
support a 'substrate-limitation' model. |
| Key genes and
pathways | TNF,
IL-6, FOXP3 (Treg stability). | IFI44L
(biomarker), IL-6, IL-17, IL-2, FOXP3
(Treg stability). | Th1/Th17
proinflammatory programs, Treg networks, myelin-associated
genes. | HLA region,
RNF5. | Stress-response
genes (COMT, BDNF), immune-inflammatory
pathways. |
| Clinical
implications | TNF
methylation predicts biological response; methylation profiles
predict MTX response. | IFI44L
hypomethylation serves as a diagnostic biomarker; restoration of
one-carbon flux is a potential therapy. | Methylation
patterns associate with disability and are modified by treatments
(for example dimethyl fumarate), suggesting therapeutic
targets. | Saliva methylation
profiles may serve as non-invasive biomarkers; diet and folate
status are key for management. | Blood methylation
panels show diagnostic potential; B-vitamin supplementation has
demonstrated clinical benefits. |
Clinical implications and therapeutic
potential
Dietary interventions and folate
supplementation
A systematic review and meta-analysis evaluated the
dose-response relationship between folate intake and changes in
blood biomarkers. The review included 120 clinical trials in
healthy participants with study durations ranging from 3 to 144
weeks. Doses of 375-570 μg/day produced a 1.7-fold increase
in erythrocyte folate concentrations relative to baseline (95% CI,
1.66-1.93), reaching normalization by week 36. The analysis
reported moderate heterogeneity among studies (posterior predictive
interval=1.37-2.34) (166).
Individuals carrying the MTHFR C677T TT
genotype (homozygous mutant) exhibit lower folate and higher
homocysteine concentrations, often presenting with fatigue,
irritability and megaloblastic anemia. By contrast, those with the
MTHFR A1298C CC genotype (homozygous mutant) tend to have
higher folate concentrations without overt clinical symptoms,
although vitamin B12 deficiency may be masked (63).
In RA, methotrexate, a folate antagonist with
gastrointestinal toxicity, is a cornerstone therapy. Meta-analytic
data indicate that folic or folinic acid supplementation <5
mg/day reduces gastrointestinal adverse effects by 79% (odds
ratio=0.21; 95% CI, 0.10-0.44) but does not notably modify disease
activity as measured by tender-joint count (167).
Conversely, in SLE, methyl-donor-rich diets or
folic acid supplementation have been proposed to modulate
epigenetic mechanisms underlying proinflammatory gene expression.
Experimental evidence has demonstrated that folic acid can
epigenetically silence the IRF5 gene, a key driver of TNF-α
synthesis and suppress type I and III interferon pathways, both
commonly overexpressed in SLE (168).
In MS, a recent systematic review and meta-analysis
assessed folate metabolism and epigenetic implications. No notable
differences were found in folate levels between patients and
controls (weighted mean difference [WMD]=0.00 μg/l; 95%
CI,−0.01 to 0.01; I2=0%). However, homocysteine levels
were notably increased in MS (WMD=2.47 μmol/; 95% CI, 0.40
to 4.55; I2=92%), suggesting a potential association
between altered one-carbon metabolism, inflammation and disease
activity (169).
Patients with CeD frequently exhibit low folate
concentrations due to persistent enteropathy, inadequate adherence
to a gluten-free diet or consumption of non-fortified gluten-free
products. Consequently, management strategies should emphasize a
well-planned gluten-free diet, folic acid supplementation,
nutritional education and the fortification of gluten-free foods
(170).
In FM, low dietary folate intake has been inversely
associated with disease severity. A study using the Fibromyalgia
Impact Questionnaire-Revised reported a negative association
between dietary folate intake and symptom burden (r=-0.250;
P=0.017) (171). This
relationship likely reflects the role of folate in neurotransmitter
synthesis, epigenetic regulation, DNA methylation and the
modulation of inflammation and oxidative stress (172).
Therapeutic strategies based on
epigenetic modulation
A meta-analysis of intervention studies (folic acid
supplementation, fortified foods and natural folate sources) in
individuals stratified by MTHFR C677T genotype included six
randomized controlled trials and four quasi-experimental studies,
each lasting ≥4 weeks and using doses of 400-1,670 μg DFE.
Homozygous TT carriers displayed higher baseline homocysteine and
lower folate concentrations compared with CT and CC genotypes.
Following supplementation, homocysteine levels decreased across all
genotypes; however, serum folate increases were smaller among TT
carriers. These data highlight a persistent biochemical
vulnerability in individuals with reduced MTHFR activity.
Populations of Asian and Latin American ancestry carrying the TT
genotype may require higher daily folate intakes, direct
supplementation with 5-methyltetrahydrofolate (5-MTHF), prolonged
interventions and/or combined B-vitamin therapy (173).
Genotype-specific differences in folate and
homocysteine metabolism have implications for methylation-dependent
regulatory pathways. Given the central role of folate in one-carbon
metabolism, optimizing folate status in TT carriers could enhance
epigenetic stability, particularly in tissues sensitive to
methylation imbalance (174).
Limitations and future perspectives
The main strength of the present review is its
comprehensive integration of the biological mechanisms associating
one-carbon metabolism and epigenetic regulation in autoimmune
diseases, which contributes to the literature, as, to the best of
our knowledge, few studies have synthesized these complex
interactions. However, it is important to note that the analysis of
the present review focused primarily on the folate-dependent
pathway, which constitutes a limitation because it excludes other
nutrients involved in epigenetic regulation, such as choline,
serine and vitamins B12 and B6, all of which
participate in SAM availability and overall methylation capacity
(175,176).
Current evidence also poses challenges due to the
heterogeneity of study designs and methodologies. Several studies
rely on small cohorts that do not stratify participants by folate
level or MTHFR genotype and employ disparate methods to
quantify DNA methylation (for example, global, LINE-1 or
locus-specific promoter assays) (177-182). These methodological differences
hinder the ability to compare findings and to draw firm conclusions
regarding the mechanisms associating methylation changes to
autoimmune diseases. In addition, the predominance of
cross-sectional designs limits the capacity to determine causality
between folate deficiency and disease progression.
Future research must move beyond associations
toward establishing causality. This requires longitudinal,
multi-omics studies that integrate genomics, epigenomics and
metabolomics in disease-specific cohorts. Such approaches will
enable the identification of more precise molecular markers of
one-carbon metabolism dysfunction and its relationship to immune
regulation. Once improved definition of these mechanisms are
established, the next step should involve targeted interventions.
Clinical trials are needed to determine whether restoring
methylation capacity, through folate supplementation or strategies
that increase SAM availability, actually reduces inflammatory
phenotypes. Ultimately, the goal is to validate sensitive
biomarkers that support the development of personalized prevention
and treatment strategies tailored to the genotype of each
patient.
Conclusions
The MTHFR-folate axis represents a pivotal
intersection between metabolism, immune homeostasis and epigenetic
regulation in autoimmune diseases. Genetic and environmental
perturbations affecting this axis have been consistently associated
with epigenetic alterations underlying the pathophysiology of RA
and SLE. By contrast, evidence regarding MS, CeD and FM remains
heterogeneous and warrants further clarification. Importantly, this
axis not only influences DNA and histone methylation but also
modulates inflammatory signaling and cellular stress responses,
highlighting its potential as both a biomarker and a therapeutic
target.
Future research should prioritize clinical trials
that integrate genetic background, nutritional status, inflammatory
load and emerging biomarkers, such as homocysteine levels and the
SAM:SAH ratio, while evaluating the effects of targeted nutritional
interventions involving folic acid and vitamin B12.
Complementary studies using primary immune cells and
tissue-specific models are essential to elucidate how
MTHFR-related methylation changes translate into functional
dysregulation of the immune system. This integrative approach will
facilitate the characterization of disease-specific epigenetic and
metabolomic profiles, supporting the development of personalized,
mechanism-based therapeutic strategies in autoimmune
conditions.
Availability of data and materials
Not applicable.
Authors' contributions
PMNR, RFBS, FJTH and HRCG contribute to
conceptualization; PMNR and RFBS contributed to investigation; JFMV
contributed to project administration; FJTH, HRCG, JHB and JFV
contributed to supervision; FJTH, HRCG, JHB, COHR, SRDLS and JFMV
contributed to validation; PPMNR, RFBS and FJTH contributed to
visualization; PMNR and RFBS contributed to writing of the original
draft; FJTH, HRCG, JHB, COHR, SRDLS and JFMV contributed to review
and editing. All authors read and approved the final manuscript.
Data authentication not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
No funding was received.
References
|
1
|
Song Y, Li J and Wu Y: Evolving
understanding of autoimmune mechanisms and new therapeutic
strategies of autoimmune disorders. Signal Transduct Target Ther.
9:2632024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Danieli MG, Casciaro M, Paladini A,
Bartolucci M, Sordoni M, Shoenfeld Y and Gangemi S: Exposome:
Epigenetics and autoimmune diseases. Autoimmun Rev. 23:1035842024.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gurugubelli KR and Ballambattu VB:
Perspectives on folate with special reference to epigenetics and
neural tube defects. Reprod Toxicol. 125:1085762024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Souza LL, da Mota JCNL, Carvalho LM,
Ribeiro AA, Caponi CA, Pinhel MAS, Costa-Fraga N, Diaz-Lagares A,
Izquierdo AG, Nonino CB, et al: Genome-wide impact of folic acid on
DNA methylation and gene expression in lupus adipocytes: An in
vitro study on obesity. Nutrients. 17:10862025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu M, Peng K, Song L, Luo L, Liang P and
Liang Y: Association between Genetic polymorphisms in
Methylenetetrahydrofolate reductase and risk of autoimmune
diseases: A systematic review and meta-analysis. Dis Markers.
2022:45681452022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsai TY, Lee TH, Wang HH, Yang TH, Chang
IJ and Huang YC: Serum Homocysteine, Folate, and vitamin
B12 levels in patients with systemic lupus
Erythematosus: A meta-analysis and meta-regression. J Am Coll Nutr.
40:443–453. 2021. View Article : Google Scholar
|
|
7
|
Dardiotis E, Arseniou S, Sokratous M,
Tsouris Z, Siokas V, Mentis AA, Michalopoulou A, Andravizou A,
Dastamani M, Paterakis K, et al: Vitamin B12, folate, and
homocysteine levels and multiple sclerosis: A meta-analysis. Mult
Scler Relat Disord. 17:190–197. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nomair AM, Abdelati A, Dwedar FI, Elnemr
R, Kamel YN and Nomeir HM: The impact of folate pathway variants on
the outcome of methotrexate therapy in rheumatoid arthritis
patients. Clin Rheumatol. 43:971–983. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Crider KS, Yang TP, Berry RJ and Bailey
LB: Folate and DNA methylation: A review of molecular mechanisms
and the evidence for Folate's role. Adv Nutr. 3:21–38. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Coppedè F, Denaro M, Tannorella P and
Migliore L: Increased MTHFR promoter methylation in mothers of Down
syndrome individuals. Mutat Res. 787:1–6. 2016.PubMed/NCBI
|
|
11
|
Sun H, Song K, Zhou Y, Ding JF, Tu B, Yang
JJ, Sha JM, Zhao JY, Zhang Y and Tao H: MTHFR epigenetic
derepression protects against diabetes cardiac fibrosis. Free Radic
Biol Med. 193:330–341. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Prasad S, Adivikolanu H, Banerjee A,
Mittal M, Lemos JRN, Mittal R and Hirani K: The role of microRNAs
and long non-coding RNAs in epigenetic regulation of T cells:
Implications for autoimmunity. Front Immunol. 16:16958942025.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nuermaimaiti K, Li T, Li N, Shi T, Liu W,
Abulaiti P, Abulaihaiti K and Gao F: Vitamin and trace elements
imbalance are very common in adult patients with newly diagnosed
Celiac disease. Sci Rep. 15:283152025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fusco R, Siracusa R, D'Amico R, Peritore
AF, Cordaro M, Gugliandolo E, Crupi R, Impellizzeri D, Cuzzocrea S
and Di Paola R: Melatonin plus folic acid treatment ameliorates
reserpine-induced fibromyalgia: An evaluation of pain, oxidative
stress, and inflammation. Antioxidants (Basel). 8:6282019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nielsen HM and Tost J: Epigenetic Changes
in Inflammatory and Autoimmune Diseases. Epigenetics: Development
and Disease. Kundu TK: 61. Springer; Netherlands, Dordrecht: pp.
455–478. 2013
|
|
16
|
Surace AEA and Hedrich CM: The role of
epigenetics in autoimmune/inflammatory disease. Front Immunol.
10:15252019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Funes SC, Fernández-Fierro A,
Rebolledo-Zelada D, Mackern-Oberti JP and Kalergis AM: Contribution
of Dysregulated DNA methylation to autoimmunity. Int J Mol Sci.
22:118922021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wills L and Stewart A: Experimental anæmia
in monkeys, with special reference to macrocytic nutritional
anæmia. Br J Exp Pathol. 16:444–453. 1935.
|
|
19
|
Bastian H: Lucy Wills (1888-1964): The
life and research of an adventurous independent woman. J R Coll
Phys Edinb. 38:89–91. 2008. View Article : Google Scholar
|
|
20
|
Viswanathan M, Urrutia RP, Hudson KN,
Middleton JC and Kahwati LC: Folic acid supplementation to prevent
neural tube defects: Updated evidence report and systematic review
for the US Preventive Services Task Force. JAMA. 330:460–466. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mitchell HK, Snell EE and Williams RJ:
Journal of the American Chemical Society, Vol. 63, 1941: The
concentration of 'folic acid' by Herschel K. Mitchell, Esmond E.
Snell, and Roger J. Williams. Nutr Rev. 46:324–325. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rosenberg IH: A history of the isolation
and identification of folic acid (folate). Ann Nutr Metab.
61:231–235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Angier RB, Boothe JH, Hutchings BL, Mowat
JH, Semb J, Stokstad EL, Subbarow Y, Waller CW, Cosulich DB,
Fahrenbach MJ, et al: Synthesis of a compound identical with the L.
casei factor isolated from liver. Science. 102:227–228. 1945.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kumar Upadhyay A, Prakash A, Kumar A, Jena
S, Sinha N and Sharma S: Dr. Sidney Farber (1903-1973): Founder of
pediatric pathology and the father of modern chemotherapy. Cureus.
16:e682862024.PubMed/NCBI
|
|
25
|
Kutzbach C and Stokstad ELR: Mammalian
methylenetetrahydrofolate reductase. Partial purification,
properties, and inhibition by S-adenosylmethionine. Biochim Biophys
Acta. 250:459–477. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tamura T and Stokstad ELR: The
availability of food folate in man. Br J Haematol. 25:513–532.
1973. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Castillo LF, Pelletier CM, Heyden KE and
Field MS: New insights into folate-vitamin B12 interactions. Annu
Rev Nutr. 45:23–39. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Frosst P, Blom HJ, Milos R, Goyette P,
Sheppard CA, Matthews RG, Boers GJ, den Heijer M, Kluijtmans LA,
van den Heuvel LP, et al: A candidate genetic risk factor for
vascular disease: A common mutation in methylenetetrahydrofolate
reductase. Nat Genet. 10:111–113. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Loperfido F, Sottotetti F, Bianco I, El
Masri D, Maccarini B, Ferrara C, Limitone A, Cena H and De Giuseppe
R: Folic acid supplementation in European women of reproductive age
and during pregnancy with excessive weight: A systematic review.
Reprod Health. 22:132025. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Arynchyna-Smith A, Arynchyn AN, Kancherla
V, Anselmi K, Aban I, Hoogeveen RC, Steffen LM, Becker DJ,
Kulczycki A, Carlo WA and Blount JP: Improvement of serum folate
status in the US women of reproductive age with fortified iodised
salt with folic acid (FISFA study). Public Health Nutr.
27:e2182024. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Quinn M, Halsey J, Sherliker P, Pan H,
Chen Z, Bennett DA and Clarke R: Global heterogeneity in folic acid
fortification policies and implications for prevention of neural
tube defects and stroke: A systematic review. EClinicalMedicine.
67:1023662023. View Article : Google Scholar
|
|
32
|
He Q and Li J: The evolution of folate
supplementation-from one size for all to personalized, precision,
poly-paths. J Transl Int Med. 11:128–137. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Scaglione F and Panzavolta G: Folate,
folic acid and 5-methyltetrahydrofolate are not the same thing.
Xenobiotica. 44:480–488. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hoffbrand AV and Weir DG: The history of
folic acid. Br J Haematol. 113:579–589. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mishra VK, Rodriguez-Lecompte JC and Ahmed
M: Nanoparticles mediated folic acid enrichment. Food Chem.
456:1399642024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wusigale and Liang L: Folates: Stability
and interaction with biological molecules. J Agric Food Res.
2:1000392020.
|
|
37
|
Siatka T, Mát'uš M, Moravcová M, Harčárová
P, Lomozová Z, Matoušová K, Suwanvecho C, Kujovská Krčmová L and
Mladěnka P: Biological, dietetic and pharmacological properties of
vitamin B9. NPJ Sci Food. 9:302025. View Article : Google Scholar
|
|
38
|
Erşan S, Chen Y and Park JO: Comprehensive
profiling of folates across polyglutamylation and one-carbon
states. Metabolomics. 21:712025. View Article : Google Scholar
|
|
39
|
Yang M, Wang D, Wang X, Mei J and Gong Q:
Role of folate in liver diseases. Nutrients. 16:18722024.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Revuelta JL, Serrano-Amatriain C,
Ledesma-Amaro R and Jiménez A: Formation of folates by
microorganisms: Towards the biotechnological production of this
vitamin. Appl Microbiol Biotechnol. 102:8613–8620. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shulpekova Y, Nechaev V, Kardasheva S,
Sedova A, Kurbatova A, Bueverova E, Kopylov A, Malsagova K, Dlamini
JC and Ivashkin V: The concept of folic acid in health and disease.
Molecules. 26:37312021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
U.S. Department of Agriculture, National
Agricultural Library: Nutrient Lists from Standard Reference
Legacy. 2018, https://www.nal.usda.gov/human-nutrition-and-food-safety/nutrient-lists-standard-reference-legacy-2018.
Accessed Dec 17, 2025
|
|
43
|
Spanish Agency for Food Safety and
Nutrition: Composition of Foods. 2025, https://www.aesan.gob.es/en/AECOSAN/web/seguridad_alimentaria/subseccion/composicion_alimentos_BD.htm.
Accessed Dec 17, 2025
|
|
44
|
Food and Agriculture Organization of the
United Nations: FAO/INFOODS Food Composition Database. https://www.fao.org/infoods/infoods/tables-and-databases/faoinfoods-databases/en/.
Accessed Dec 17, 2025
|
|
45
|
Li J, Duan H, Ramaswamy H and Wang C: A
comprehensive review of fortification, bioavailability, and health
benefits of folate. Int J Mol Sci. 26:77032025. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Beltramo B, Urlings M, Padilla-Díaz CM,
Bast A, Diliën H and De Boer A: Bioavailability of vitamins C, B2
and B9 (Folate) in nutrition and health claims: A critical
appraisal. Food Prod Process Nutr. 7:552025. View Article : Google Scholar
|
|
47
|
Liu F, Kariluoto S, Edelmann M and
Piironen V: Bioaccessibility of folate in faba bean, oat, rye and
wheat matrices. Food Chem. 350:1292592021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Visentin M, Diop-Bove N, Zhao R and
Goldman ID: The intestinal absorption of folates. Annu Rev Physiol.
76:251–274. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pietrzik K, Bailey L and Shane B: Folic
acid and L-5-methyltetrahydrofolate: Comparison of clinical
pharmacokinetics and pharmacodynamics. Clin Pharmacokinet.
49:535–548. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhao R, Matherly LH and Goldman ID:
Membrane transporters and folate homeostasis: Intestinal absorption
and transport into systemic compartments and tissues. Expert Rev
Mol Med. 11:e42009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
O'Connor C, Wallace-Povirk A, Ning C,
Frühauf J, Tong N, Gangjee A, Matherly LH and Hou Z: Folate
transporter dynamics and therapy with classic and tumor-targeted
antifolates. Sci Rep. 11:63892021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Matherly LH, Schneider M, Gangjee A and
Hou Z: Biology and therapeutic applications of the proton-coupled
folate transporter. Expert Opin Drug Metab Toxicol. 18:695–706.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Alpers DH: Absorption and blood/cellular
transport of folate and cobalamin: Pharmacokinetic and
physiological considerations. Biochimie. 126:52–56. 2016.
View Article : Google Scholar :
|
|
54
|
Ebara S: Nutritional role of folate.
Congenit Anom (Kyoto). 57:138–141. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Froese DS, Fowler B and Baumgartner MR:
Vitamin B12, folate, and the methionine remethylation
cycle-biochemistry, pathways, and regulation. J Inherit Metab Dis.
42:673–685. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zarou MM, Vazquez A and Vignir Helgason G:
Folate metabolism: A re-emerging therapeutic target in
haematological cancers. Leukemia. 35:1539–1551. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bedoui Y, Guillot X, Sélambarom J, Guiraud
P, Giry C, Jaffar-Bandjee MC, Ralandison S and Gasque P:
Methotrexate an old drug with new tricks. Int J Mol Sci.
20:50232019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Cronstein BN and Aune TM: Methotrexate and
its mechanisms of action in inflammatory arthritis. Nat Rev
Rheumatol. 16:145–154. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhao X, Wu P, Yang Z and Miao RR:
Relationship between the efficacy and adverse effects of
methotrexate and gene polymorphism. Egypt J Med Hum Genet.
25:892024. View Article : Google Scholar
|
|
60
|
Rogers LM, Cordero AM, Pfeiffer CM,
Hausman DB, Tsang BL, De-Regil LM, Rosenthal J, Razzaghi H, Wong
EC, Weakland AP and Bailey LB: Global folate status in women of
reproductive age: A systematic review with emphasis on
methodological issues. Ann N Y Acad Sci. 1431:35–57. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Colapinto CK, O'Connor DL, Sampson M,
Williams B and Tremblay MS: Systematic review of adverse health
outcomes associated with high serum or red blood cell folate
concentrations. J Public Health (Oxf). 38:e84–e97. 2016. View Article : Google Scholar
|
|
62
|
Cordero AM, Crider KS, Rogers LM, Cannon
MJ and Berry RJ: Optimal serum and red blood cell folate
concentrations in women of reproductive age for prevention of
neural tube defects: World Health Organization guidelines. MMWR
Morb Mortal Wkly Rep. 64:421–423. 2015.PubMed/NCBI
|
|
63
|
Novaković R, Geelen A, Ristić-Medić D,
Nikolić M, Souverein OW, McNulty H, Duffy M, Hoey L, Dullemeijer C,
Renkema JMS, et al: Systematic review of observational studies with
Dose-response meta-analysis between folate intake and status
biomarkers in adults and the elderly. Ann Nutr Metab. 73:30–43.
2018. View Article : Google Scholar
|
|
64
|
Zhou Y, Sinnathamby V, Yu Y, Sikora L,
Johnson CY, Mossey P and Little J: Folate intake, markers of folate
status and oral clefts: An updated set of systematic reviews and
meta-analyses. Birth Defects Res. 112:1699–1719. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Choumenkovitch SF, Selhub J, Wilson PW,
Rader JI, Rosenberg IH and Jacques PF: Folic acid intake from
fortification in United States exceeds predictions. J Nutr.
132:2792–2798. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhou Y, Wang A, Yeung LF, Qi YP, Pfeiffer
CM and Crider KS: Folate and vitamin B12 usual intake and biomarker
status by intake source in United States adults aged ≥19 y: NHANES
2007-2018. Am J Clin Nutr. 118:241–254. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
National Institutes of Health: Office of
Dietary Supplements: Folate-Consumer. 2022.
|
|
68
|
National Academies Press: Dietary
Reference Intakes for Calcium and Vitamin D. Washington, DC:
2011
|
|
69
|
Zarembska E, Ślusarczyk K and Wrzosek M:
The implication of a polymorphism in the methylenetetrahydrofolate
reductase gene in homocysteine metabolism and related civilisation
diseases. Int J Mol Sci. 25:1932023. View Article : Google Scholar
|
|
70
|
Tran P, Leclerc D, Chan M, Pai A, Hiou-Tim
F, Wu Q, Goyette P, Artigas C, Milos R and Rozen R: Multiple
transcription start sites and alternative splicing in the
methylenetetrahydrofolate reductase gene result in two enzyme
isoforms. Mamm Genome. 13:483–492. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Homberger A, Linnebank M, Winter C,
Willenbring H, Marquardt T, Harms E and Koch HG: Genomic structure
and transcript variants of the human methylenetetrahydrofolate
reductase gene. Eur J Hum Genet. 8:725–729. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Araszkiewicz AF, Jańczak K, Wójcik P,
Białecki B, Kubiak S, Szczechowski M and Januszkiewicz-Lewandowska
D: MTHFR gene polymorphisms: A single gene with wide-ranging
clinical implications-a review. Genes (Basel). 16:4412025.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wrzosek M and Ślusarczyk K:
Methylenetetrahydrofolate reductase C677T gene variant in relation
to body mass index and folate concentration in a polish population.
Biomedicines. 10:31402022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Froese DS, Kopec J, Rembeza E, Bezerra GA,
Oberholzer AE, Suormala T, Lutz S, Chalk R, Borkowska O,
Baumgartner MR and Yue WW: Structural basis for the regulation of
human 5,10-methylenetetrahydrofolate reductase by phosphorylation
and S-adenosylmethionine inhibition. Nat Commun. 9:22612018.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Sibani S, Christensen B, O'Ferrall E,
Saadi I, Hiou-Tim F, Rosenblatt DS and Rozen R: Characterization of
six novel mutations in the methylenetetrahydrofolate reductase
(MTHFR) gene in patients with homocystinuria. Hum Mutat.
15:280–287. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Weisberg I, Tran P, Christensen B, Sibani
S and Rozen R: A second genetic polymorphism in
methylenetetrahydrofolate reductase (MTHFR) associated with
decreased enzyme activity. Mol Genet Metab. 64:169–172. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Liew SC and Gupta ED:
Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism:
Epidemiology, metabolism and the associated diseases. Eur J Med
Genet. 58:1–10. 2015. View Article : Google Scholar
|
|
78
|
Bhatia M, Thakur J, Suyal S, Oniel R,
Chakraborty R, Pradhan S, Sharma M, Sengupta S, Laxman S,
Masakapalli SK and Bachhawat AK: Allosteric inhibition of MTHFR
prevents futile SAM cycling and maintains nucleotide pools in
one-carbon metabolism. J Biol Chem. 295:16037–16057. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wang G and Han JJ: Connections between
metabolism and epigenetic modifications in cancer. Med Rev (Berl).
1:199–221. 2022. View Article : Google Scholar
|
|
80
|
Wu W, Shen O, Qin Y, Niu X, Lu C, Xia Y,
Song L, Wang S and Wang X: Idiopathic male infertility is strongly
associated with aberrant promoter methylation of
methylenetetrahydrofolate reductase (MTHFR). PLoS One.
5:e138842010. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Kulac T, Hekim N, Kocamanoglu F, Beyaz C,
Gunes S and Asci R: Methylation patterns of
methylenetetrahydrofolate reductase gene promoter in infertile
males. Andrologia. 53:e139422021. View Article : Google Scholar
|
|
82
|
Shaker MM, Shalabi TA and Amr KS:
Correlation of methylation status in MTHFR promoter region with
recurrent pregnancy loss. J Genet Eng Biotechnol. 19:442021.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Saraswathy KN, Kaur L, Talwar S, Mishra J,
Huidrom S, Sachdeva MP and Puri M: Methylenetetrahydrofolate
reductase gene-specific methylation and recurrent miscarriages: A
case-control study from North India. J Hum Reprod Sci. 11:142–147.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Koturbash I, Melnyk S, James SJ, Beland FA
and Pogribny IP: Role of epigenetic and miR-22 and miR-29b
alterations in the downregulation of Mat1a and Mthfr genes in early
preneoplastic livers in rats induced by 2-acetylaminofluorene. Mol
Carcinog. 52:318–327. 2013. View Article : Google Scholar
|
|
85
|
Su A, Ling F, Vaganay C, Sodaro G,
Benaksas C, Dal Bello R, Forget A, Pardieu B, Lin KH, Rutter JC, et
al: The folate cycle enzyme MTHFR is a critical regulator of cell
response to MYC-targeting therapies. Cancer Discov. 10:1894–1911.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Clark DF, Schmelz R, Rogers N, Smith NE
and Shorter KR: Acute high folic acid treatment in SH-SY5Y cells
with and without MTHFR function leads to gene expression changes in
epigenetic modifying enzymes changes in epigenetic marks and
changes in dendritic spine densities. PLoS One. 16:e02450052021.
View Article : Google Scholar
|
|
87
|
Al Sayed R, Smith W, Rogers N, Smith N,
Clark D, Castillo G, McLeod H, Glenister S and Shorter KR: A 2x
folic acid treatment affects epigenetics and dendritic spine
densities in SHSY5Y cells. Biochem Biophys Rep.
20:1006812019.PubMed/NCBI
|
|
88
|
Wu C, Gong Y, Sun A, Zhang Y, Zhang C,
Zhang W, Zhao G, Zou Y and Ge J: The human MTHFR rs4846049
polymorphism increases coronary heart disease risk through
modifying miRNA binding. Nutr Metab Cardiovasc Dis. 23:693–698.
2013. View Article : Google Scholar
|
|
89
|
Li C, Ni J, Liu YX, Wang H, Liang ZQ and
Wang X: Response of MiRNA-22-3p and MiRNA-149-5p to folate
deficiency and the differential regulation of MTHFR expression in
normal and cancerous human hepatocytes. PLoS One. 12:e01680492017.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zhang S, Zheng F, Zhang L, Huang Z, Huang
X, Pan Z, Chen S, Xu C, Jiang Y, Gu S, et al: LncRNA
HOTAIR-mediated MTHFR methylation inhibits 5-fluorouracil
sensitivity in esophageal cancer cells. J Exp Clin Cancer Res.
39:1312020. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Li C, Li X, Wang H, Guo X, Xue J, Wang X
and Ni J: MicroRNA-22-3p and MicroRNA-149-5p inhibit human
hepatocellular carcinoma cell growth and metastasis properties by
regulating methylenetetrahydrofolate reductase. Curr Issues Mol
Biol. 44:952–962. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Glossop JR, Emes RD, Nixon NB, Haworth KE,
Packham JC, Dawes PT, Fryer AA, Mattey DL and Farrell WE:
Genome-wide DNA methylation profiling in rheumatoid arthritis
identifies disease-associated methylation changes that are distinct
to individual T- and B-lymphocyte populations. Epigenetics.
9:1228–1237. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhao M, Zhou Y, Zhu B, Wan M, Jiang T, Tan
Q, Liu Y, Jiang J, Luo S, Tan Y, et al: IFI44L promoter methylation
as a blood biomarker for systemic lupus erythematosus. Ann Rheum
Dis. 75:1998–2006. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang J, Dang X, Wu X, Xiang Z, Li Y, Fu Y
and Shen T: DNA methylation of IFI44L as a potential blood
biomarker for childhood-onset systemic lupus erythematosus. Pediatr
Res. 96:494–501. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Li F, Feng Q, Lee C, Wang S, Pelleymounter
LL, Moon I, Eckloff BW, Wieben ED, Schaid DJ, Yee V and
Weinshilboum RM: Human betaine-homocysteine methyltransferase
(BHMT) and BHMT2: Common gene sequence variation and functional
characterization. Mol Genet Metab. 94:326–335. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Delgado-Reyes CV, Wallig MA and Garrow TA:
Immunohistochemical detection of betaine-homocysteine
S-methyltransferase in human, pig, and rat liver and kidney. Arch
Biochem Biophys. 393:184–186. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Sunden SLF, Renduchintala MS, Park EI,
Miklasz SD and Garrow TA: Betaine-homocysteine methyltransferase
expression in porcine and human tissues and chromosomal
localization of the human gene. Arch Biochem Biophys. 345:171–174.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Tehlivets O, Malanovic N, Visram M,
Pavkov-Keller T and Keller W: S-adenosyl-L-homocysteine hydrolase
and methylation disorders: Yeast as a model system. Biochim Biophys
Acta. 1832:204–215. 2013. View Article : Google Scholar :
|
|
99
|
Stipanuk MH: Sulfur amino acid metabolism:
Pathways for production and removal of homocysteine and cysteine.
Annu Rev Nutr. 24:539–577. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Chiang PK: Biological effects of
inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol Ther.
77:115–134. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Mentch SJ, Mehrmohamadi M, Huang L, Liu X,
Gupta D, Mattocks D, Gómez Padilla P, Ables G, Bamman MM,
Thalacker-Mercer AE, et al: Histone methylation dynamics and gene
regulation occur through the sensing of one-carbon metabolism. Cell
Metab. 22:861–873. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Chen Z, Karaplis AC, Ackerman SL, Pogribny
IP, Melnyk S, Lussier-Cacan S, Chen MF, Pai A, John SW, Smith RS,
et al: Mice deficient in methylenetetrahydrofolate reductase
exhibit hyperhomocysteinemia and decreased methylation capacity,
with neuropathology and aortic lipid deposition. Hum Mol Genet.
10:433–443. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Friso S, Choi SW, Girelli D, Mason JB,
Dolnikowski GG, Bagley PJ, Olivieri O, Jacques PF, Rosenberg IH,
Corrocher R, et al: A common mutation in the
5,10-methylenetetrahydrofolate reductase gene affects genomic DNA
methylation through an interaction with folate status. Proc Natl
Acad Sci USA. 99:5606–5611. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Mandaviya PR, Joehanes R, Brody J,
Castillo-Fernandez JE, Dekkers KF, Do AN, Graff M, Hänninen IK,
Tanaka T, de Jonge EAL, et al: Association of dietary folate and
vitamin B-12 intake with genome-wide DNA methylation in blood: A
large-scale epigenome-wide association analysis in 5841
individuals. Am J Clin Nutr. 110:437–450. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Kok DEG, Dhonukshe-Rutten RAM, Lute C,
Heil SG, Uitterlinden AG, van der Velde N, van Meurs JB, van Schoor
NM, Hooiveld GJ, de Groot LC, et al: The effects of long-term daily
folic acid and vitamin B12 supplementation on genome-wide DNA
methylation in elderly subjects. Clin Epigenetics. 7:1212015.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Pufulete M, Al-Ghnaniem R, Khushal A,
Appleby P, Harris N, Gout S, Emery PW and Sanders TA: Effect of
folic acid supplementation on genomic DNA methylation in patients
with colorectal adenoma. Gut. 54:648–653. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Lingappan K: NF-κB in oxidative stress.
Curr Opin Toxicol. 7:81–86. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yu W, Wang Z, Zhang K, Chi Z, Xu T, Jiang
D, Chen S, Li W, Yang X, Zhang X, et al: One-carbon metabolism
supports S-Adenosylmethionine and histone methylation to drive
inflammatory macrophages. Mol Cell. 75:1147–1160.e5. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Strickland FM, Li Y, Johnson K, Sun Z and
Richardson BC: CD4(+) T cells epigenetically modified by oxidative
stress cause lupus-like autoimmunity in mice. J Autoimmun.
62:75–80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Gorelik GJ, Yarlagadda S, Patel DR and
Richardson BC: Protein kinase Cδ oxidation contributes to ERK
inactivation in lupus T cells. Arthritis Rheum. 64:2964–2974. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Li Y, Gorelik G, Strickland FM and
Richardson BC: Oxidative stress, T cell DNA methylation, and lupus.
Arthritis Rheumatol. 66:1574–1582. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Paul DS, Teschendorff AE, Dang MAN, Lowe
R, Hawa MI, Ecker S, Beyan H, Cunningham S, Fouts AR, Ramelius A,
et al: Increased DNA methylation variability in type 1 diabetes
across three immune effector cell types. Nat Commun. 7:135552016.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Cerna M: Epigenetic regulation in etiology
of type 1 diabetes mellitus. Int J Mol Sci. 21:362019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Cheng M, Yang L, Dong Z, Wang M, Sun Y,
Liu H, Wang X, Sai N, Huang G and Zhang X: Folic acid deficiency
enhanced microglial immune response via the Notch1/nuclear factor
kappa B p65 pathway in hippocampus following rat brain I/R injury
and BV2 cells. J Cell Mol Med. 23:4795–4807. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Kolb AF and Petrie L: Folate deficiency
enhances the inflammatory response of macrophages. Mol Immunol.
54:164–172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Nakano K, Boyle DL and Firestein GS:
Regulation of DNA methylation in rheumatoid arthritis synoviocytes.
J Immunol. 190:1297–1303. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wang G, Siow YL and O K: Homocysteine
stimulates nuclear factor kappaB activity and monocyte
chemoattractant protein-1 expression in vascular smooth-muscle
cells: A possible role for protein kinase C. Biochem J.
352:817–826. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Au-Yeung KKW, Woo CWH, Sung FL, Yip JCW,
Siow YL and O K: Hyperhomocysteinemia activates nuclear
factor-kappaB in endothelial cells via oxidative stress. Circ Res.
94:28–36. 2004. View Article : Google Scholar
|
|
119
|
Webster AP, Plant D, Ecker S, Zufferey F,
Bell JT, Feber A, Paul DS, Beck S, Barton A, Williams FMK, et al:
Increased DNA methylation variability in rheumatoid
arthritis-discordant monozygotic twins. Genome Med. 10:642018.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Guderud K, Sunde LH, Flåm ST, Mæhlen MT,
Mjaavatten MD, Lillegraven S, Aga AB, Evenrød IM, Norli ES,
Andreassen BK, et al: Rheumatoid arthritis patients, both newly
diagnosed and methotrexate treated, show more DNA methylation
differences in CD4+ memory than in CD4+ Naïve
T cells. Front Immunol. 11:1942020. View Article : Google Scholar
|
|
121
|
Pitaksalee R, Parmar R, Hodgett R, Emery P
and Ponchel F: DNA Hypomethylation in the TNF-alpha gene predicts
rheumatoid arthritis classification in patients with early
inflammatory symptoms. Cells. 12:23762023. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Zafari P, Yari K, Mostafaei S, Iranshahi
N, Assar S, Fekri A and Taghadosi M: Analysis of Helios gene
expression and Foxp3 TSDR methylation in the newly diagnosed
Rheumatoid Arthritis patients. Immunol Invest. 47:632–642. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Cribbs AP, Kennedy A, Penn H, Amjadi P,
Green P, Read JE, Brennan F, Gregory B and Williams RO:
Methotrexate restores regulatory T cell function through
demethylation of the FoxP3 upstream enhancer in patients with
rheumatoid arthritis. Arthritis Rheumatol. 67:1182–1192. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Adams C, Nair N, Plant D, Verstappen SMM,
Quach HL, Quach DL, Carvidi A, Nititham J, Nakamura M, Graf J, et
al: Identification of cell-specific differential DNA methylation
associated with methotrexate treatment response in rheumatoid
arthritis. Arthritis Rheumatol. 75:1088–1097. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Nair N, Plant D, Verstappen SM, Isaacs JD,
Morgan AW, Hyrich KL, Barton A and Wilson AG; MATURA investigators:
Differential DNA methylation correlates with response to
methotrexate in rheumatoid arthritis. Rheumatology (Oxford).
59:1364–1371. 2020. View Article : Google Scholar
|
|
126
|
Gosselt HR, van Zelst BD, de Rotte MCFJ,
Hazes JMW, de Jonge R and Heil SG: Higher baseline global leukocyte
DNA methylation is associated with MTX non-response in early RA
patients. Arthritis Res Ther. 21:1572019. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Ravaei A, Pulsatelli L, Assirelli E,
Ciaffi J, Meliconi R, Salvarani C, Govoni M and Rubini M: MTHFR
c.665C>T and c.1298A>C polymorphisms in tailoring
personalized Anti-TNF-α therapy for rheumatoid arthritis. Int J Mol
Sci. 24:41102023. View Article : Google Scholar
|
|
128
|
Nemtsova MV, Zaletaev DV, Bure IV,
Mikhaylenko DS, Kuznetsova EB, Alekseeva EA, Beloukhova MI,
Deviatkin AA, Lukashev AN and Zamyatnin AA Jr: Epigenetic changes
in the pathogenesis of rheumatoid arthritis. Front Genet.
10:5702019. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Yang C, Li D, Teng D, Zhou Y, Zhang L,
Zhong Z and Yang GJ: Epigenetic regulation in the pathogenesis of
rheumatoid arthritis. Front Immunol. 13:8594002022. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Jeffries MA, Dozmorov M, Tang Y, Merrill
JT, Wren JD and Sawalha AH: Genome-wide DNA methylation patterns in
CD4+ T cells from patients with systemic lupus erythematosus.
Epigenetics. 6:593–601. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Hanaei S, Sanati G, Zoghi S, Gharibzadeh
S, Ziaee V and Rezaei N: The status of FOXP3 gene methylation in
pediatric systemic lupus erythematosus. Allergol Immunopathol
(Madr). 48:332–338. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Ribeiro AA, Carvalho LM, Da Mota JCNL,
Nonino CB, Gualano B, Nunes JAV, Martinez JA and Nicoletti CF:
Diet, DNA methylation and systemic lupus erythematosus: Evidence
and perspectives focused on personalized nutrition. Lifestyle
Genomics. 17:31–40. 2024.
|
|
133
|
Liu X, Zhou S, Huang M, Zhao M, Zhang W,
Liu Q, Song K, Wang X, Liu J, OuYang Q, et al: DNA methylation and
whole-genome transcription analysis in CD4+ T cells from
systemic lupus erythematosus patients with or without renal damage.
Clin Epigenetics. 16:982024. View Article : Google Scholar
|
|
134
|
da Mota JCNL, Carvalho LM, Ribeiro AA,
Souza LL, Borba EF, Roschel H, Gualano B and Nicoletti CF:
Methyl-donor supplementation in women with systemic lupus
erythematosus with different nutritional status: The protocol for a
randomised, double-blind, placebo-controlled trial. Lupus Sci Med.
11:e0012792024. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Ferreira RC, Simons HZ, Thompson WS,
Rainbow DB, Yang X, Cutler AJ, Oliveira J, Castro Dopico X, Smyth
DJ, Savinykh N, et al: Cells with Treg-specific FOXP3 demethylation
but low CD25 are prevalent in autoimmunity. J Autoimmun. 84:75–86.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Coit P, Jeffries M, Altorok N, Dozmorov
MG, Koelsch KA, Wren JD, Merrill JT, McCune WJ and Sawalha AH:
Genome-wide DNA methylation study suggests epigenetic accessibility
and transcriptional poising of interferon-regulated genes in naïve
CD4+ T cells from lupus patients. J Autoimmun. 43:78–84. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Tiane A, Schepers M, Reijnders RA, van
Veggel L, Chenine S, Rombaut B, Dempster E, Verfaillie C, Wasner K,
Grünewald A, et al: From methylation to myelination: Epigenomic and
transcriptomic profiling of chronic inactive demyelinated multiple
sclerosis lesions. Acta Neuropathol. 146:283–299. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Carlström KE, Ewing E, Granqvist M,
Gyllenberg A, Aeinehband S, Enoksson SL, Checa A, Badam TVS, Huang
J, Gomez-Cabrero D, et al: Therapeutic efficacy of dimethyl
fumarate in relapsing-remitting multiple sclerosis associates with
ROS pathway in monocytes. Nat Commun. 10:30812019. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Pinto-Medel MJ, Oliver-Martos B,
Urbaneja-Romero P, Hurtado-Guerrero I, Ortega-Pinazo J,
Serrano-Castro P, Fernández Ó and Leyva L: Global methylation
correlates with clinical status in multiple sclerosis patients in
the first year of IFNbeta treatment. Sci Rep. 7:87272017.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Maltby VE, Graves MC, Lea RA, Benton MC,
Sanders KA, Tajouri L, Scott RJ and Lechner-Scott J: Genome-wide
DNA methylation profiling of CD8+ T cells shows a distinct
epigenetic signature to CD4+ T cells in multiple sclerosis
patients. Clin Epigenetics. 7:1182015. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Bos SD, Page CM, Andreassen BK,
Elboudwarej E, Gustavsen MW, Briggs F, Quach H, Leikfoss IS,
Bjølgerud A, Berge T, et al: Genome-wide DNA methylation profiles
indicate CD8+ T cell hypermethylation in multiple sclerosis. PLoS
One. 10:e01174032015. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Azizi S, Shamshirian A, Alizadeh-Navaei R,
Jafarpour H, Asemi Z, Tamtaji OR, Vaziri MS, Homayounfar R, Rezaei
Shahmirzadi A and Alipoor R: A genetic association study of MTHFR
C677T polymorphism with risk of metabolic syndrome: A systematic
review and meta-analysis. Galen Med J. 8:e14722019. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Cevik B, Yigit S, Karakus N, Aksoy D, Kurt
S and Ates O: Association of methylenetetrahydrofolate reductase
gene C677T polymorphism with multiple sclerosis in Turkish
patients. J Investig Med. 62:980–984. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Xavier A, Campagna MP, Maltby VE,
Kilpatrick T, Taylor BV, Butzkueven H, Ponsonby AL, Scott RJ,
Jokubaitis VG, Lea RA, et al: Interferon beta treatment is a potent
and targeted epigenetic modifier in multiple sclerosis. Front
Immunol. 14:11627962023. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Holm Hansen R, Højsgaard Chow H,
Christensen JR, Sellebjerg F and von Essen MR: Dimethyl fumarate
therapy reduces memory T cells and the CNS migration potential in
patients with multiple sclerosis. Mult Scler Relat Disord.
37:1014512020. View Article : Google Scholar
|
|
146
|
Maltby VE, Lea RA, Ribbons KA, Sanders KA,
Kennedy D, Min M, Scott RJ and Lechner-Scott J: DNA methylation
changes in CD4+ T cells isolated from multiple sclerosis
patients on dimethyl fumarate. Mult Scler J Exp Transl Clin.
4:20552173187878262018.
|
|
147
|
Cielo D, Galatola M, Fernandez-Jimenez N,
De Leo L, Garcia-Etxebarria K, Loganes C, Tommasini A, Not T,
Auricchio R, Greco L, et al: Combined analysis of methylation and
gene expression profiles in separate compartments of small bowel
mucosa identified celiac disease patients' signatures. Sci Rep.
9:100202019. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Fernandez-Jimenez N, Garcia-Etxebarria K,
Plaza-Izurieta L, Romero-Garmendia I, Jauregi-Miguel A, Legarda M,
Ecsedi S, Castellanos-Rubio A, Cahais V, Cuenin C, et al: The
methylome of the celiac intestinal epithelium harbours
genotype-independent alterations in the HLA region. Sci Rep.
9:12982019. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Fernandez-Jimenez N, Castellanos-Rubio A,
Plaza-Izurieta L, Irastorza I, Elcoroaristizabal X, Jauregi-Miguel
A, Lopez-Euba T, Tutau C, de Pancorbo MM, Vitoria JC, et al:
Coregulation and modulation of NFκB-related genes in celiac
disease: Uncovered aspects of gut mucosal inflammation. Hum Mol
Genet. 23:1298–1310. 2014. View Article : Google Scholar
|
|
150
|
Hearn NL, Coleman AS, Ho V, Chiu CL and
Lind JM: Comparing DNA methylation profiles in saliva and
intestinal mucosa. BMC Genomics. 20:1632019. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Libera L, Vanoli A, Sahnane N, Adnan M,
Guerini C, Arpa G, Bianchi PI, Lenti MV, Corazza GR, La Rosa S, et
al: LINE-1 hypomethylation characterizes the inflammatory response
in coeliac disease associated-intestinal mucosa and small bowel
adenocarcinomas. J Pathol. 265:99–109. 2025. View Article : Google Scholar
|
|
152
|
Cardo A, Churruca I, Lasa A, Navarro V,
Vázquez-Polo M, Perez-Junkera G and Larretxi I: Nutritional
imbalances in adult celiac patients following a gluten-free diet.
Nutrients. 13:28772021. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Valente FX, Campos TN, Moraes LFDS,
Hermsdorff HH, Cardoso LM, Pinheiro-Sant'Ana HM, Gilberti FA and
Peluzio MC: B vitamins related to homocysteine metabolism in adults
celiac disease patients: A cross-sectional study. Nutr J.
14:1102015. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Wierdsma NJ, van Bokhorst-de van der
Schueren MA, Berkenpas M, Mulder CJ and van Bodegraven AA: Vitamin
and mineral deficiencies are highly prevalent in newly diagnosed
celiac disease patients. Nutrients. 5:3975–3992. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Polli A, Ghosh M, Bakusic J, Ickmans K,
Monteyne D, Velkeniers B, Bekaert B, Godderis L and Nijs J: DNA
methylation and brain-derived neurotrophic factor expression
account for symptoms and widespread hyperalgesia in patients with
chronic fatigue syndrome and comorbid fibromyalgia. Arthritis
Rheumatol. 72:1936–1944. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Lee YH, Kim JH and Song GG: Association
between the COMT Val158Met polymorphism and fibromyalgia
susceptibility and fibromyalgia impact questionnaire score: A
meta-analysis. Rheumatol Int. 35:159–166. 2015. View Article : Google Scholar
|
|
157
|
Ovrom EA, Mostert KA, Khakhkhar S, McKee
DP, Yang P and Her YF: A comprehensive review of the genetic and
epigenetic contributions to the development of fibromyalgia.
Biomedicines. 11:11192023. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Gerra MC, Carnevali D, Ossola P,
González-Villar A, Pedersen IS, Triñanes Y, Donnini C, Manfredini
M, Arendt-Nielsen L and Carrillo-de-la-Peña MT: DNA methylation
changes in fibromyalgia suggest the role of the immune-inflammatory
response and central sensitization. J Clin Med. 10:49922021.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Przybylowicz PK, Sokolowska KE, Rola H and
Wojdacz TK: DNA methylation changes in blood cells of fibromyalgia
and chronic fatigue syndrome patients. J Pain Res. 16:4025–4036.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Ciampi de Andrade D, Maschietto M,
Galhardoni R, Gouveia G, Chile T, Victorino Krepischi AC, Dale CS,
Brunoni AR, Parravano DC, Cueva Moscoso AS, et al: Epigenetics
insights into chronic pain: DNA hypomethylation in fibromyalgia-a
controlled pilot-study. Pain. 158:1473–1480. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Estévez-López F, Salazar-Tortosa DF,
Camiletti-Moirón D, Gavilán-Carrera B, Aparicio VA, Acosta-Manzano
P, Segura-Jiménez V, Álvarez-Gallardo IC, Carbonell-Baeza A,
Munguía-Izquierdo D, et al: Fatigue in women with fibromyalgia: A
gene-physical activity interaction study. J Clin Med. 10:19022021.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Inanir A, Yigit S, Tekcan A, Pinarli FA,
Inanir S and Karakus N: Angiotensin converting enzyme and
methylenetetrahydrofolate reductase gene variations in fibromyalgia
syndrome. Gene. 564:188–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Regland B, Forsmark S, Halaouate L,
Matousek M, Peilot B, Zachrisson O and Gottfries CG: Response to
vitamin B12 and folic acid in myalgic encephalomyelitis and
fibromyalgia. PLoS One. 10:e01246482015. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Gharibpoor F, Ghavidel-Parsa B, Sattari N,
Bidari A, Nejatifar F and Montazeri A: Effect of vitamin B12 on the
symptom severity and psychological profile of fibromyalgia
patients; a prospective pre-post study. BMC Rheumatol. 6:512022.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Huang C, Zhang N, Wei M, Pan Q, Cheng C,
Lu KE, Mo J and Chen Y: Methylation factors as biomarkers of
fibromyalgia. Ann Transl Med. 11:169. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Crider KS, Devine O, Qi YP, Yeung LF,
Sekkarie A, Zaganjor I, Wong E, Rose CE and Berry RJ: Systematic
review and Bayesian Meta-analysis of the Dose-response relationship
between folic acid intake and changes in blood folate
concentrations. Nutrients. 11:712019. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Rennie KL, Hughes J, Lang R and Jebb SA:
Nutritional management of rheumatoid arthritis: A review of the
evidence. J Hum Nutr Diet. 16:97–109. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Nikolova-Ganeva K and Tchorbanov A: Folic
acid in systemic lupus erythematosus-a new aspect. Clin Rheumatol.
42:1729–1730. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Kirsty CW, Mary H and Sumner J: The
relationship of cobalamin and/or folate to the patient-centred
outcomes in multiple sclerosis: A systematic review and
meta-analysis. Nutr Health. 28:527–542. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Lamjadli S, Oujamaa I, Souli I, Eddehbi
FE, Lakhouaja N, M'raouni B, Salami A, Guennouni M, Belghali MY,
Hazime R and Admou B: Micronutrient deficiencies in patients with
celiac disease: A systematic review and meta-analysis. Int J
Immunopathol Pharmacol. 39:39463202413134262025. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Bennett RM, Friend R, Jones KD, Ward R,
Han BK and Ross RL: The revised fibromyalgia impact questionnaire
(FIQR): Validation and psychometric properties. Arthritis Res Ther.
11:R1202009. View
Article : Google Scholar : PubMed/NCBI
|
|
172
|
Correa-Rodríguez M, Rueda-Medina B,
Casas-Barragán A, Tapia-Haro RM, Molina F and Aguilar-Ferrándiz ME:
Dietary intake assessment, severity of symptoms, and pain in women
with fibromyalgia. Clin Nurs Res. 30:1164–1173. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Colson NJ, Naug HL, Nikbakht E, Zhang P
and McCormack J: The impact of MTHFR 677 C/T genotypes on folate
status markers: A meta-analysis of folic acid intervention studies.
Eur J Nutr. 56:247–260. 2017. View Article : Google Scholar
|
|
174
|
Kumar A, Palfrey HA, Pathak R, Kadowitz
PJ, Gettys TW and Murthy SN: The metabolism and significance of
homocysteine in nutrition and health. Nutr Metab (Lond). 14:782017.
View Article : Google Scholar
|
|
175
|
Łoboś P and Regulska-Ilow B: Link between
methyl nutrients and the DNA methylation process in the course of
selected diseases in adults. Rocz Panstw Zakl Hig. 72:123–136.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
van Vliet MM, Schoenmakers S, Gribnau J
and Steegers-Theunissen RPM: The one-carbon metabolism as an
underlying pathway for placental DNA methylation-a systematic
review. Epigenetics. 19:23185162024. View Article : Google Scholar
|
|
177
|
Wernimont SM, Clark AG, Stover PJ, Wells
MT, Litonjua AA, Weiss ST, Gaziano JM, Tucker KL, Baccarelli A,
Schwartz J, et al: Folate network genetic variation, plasma
homocysteine, and global genomic methylation content: A genetic
association study. BMC Med Genet. 12:1502011. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Bakulski KM, Dou JF, Feinberg JI, Brieger
KK, Croen LA, Hertz-Picciotto I, Newschaffer CJ, Schmidt RJ and
Fallin MD: Prenatal multivitamin use and MTHFR genotype are
associated with newborn cord blood DNA methylation. Int J Environ
Res Public Health. 17:91902020. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
de Oliveira NFP, Persuhn DC and Dos Santos
MCLG: Can global DNA methylation be influenced by polymorphisms in
genes involved in epigenetic mechanisms? A review. Genes (Basel).
15:15042024. View Article : Google Scholar
|
|
180
|
Dević Pavlić S, Šverko R, Barišić A,
Mladenić T, Vraneković J, Stanković A, Peterlin A, Peterlin B,
Ostojić S and Pereza N: MTHFR gene polymorphisms and DNA
methylation in idiopathic spontaneous preterm birth. Medicina
(Kaunas). 60:20282024. View Article : Google Scholar
|
|
181
|
Majstorović D, Stoccoro A, Barišić A,
Buretić Tomljanović A, Giangreco M, Nicolì V, Coppedè F and
Vraneković J: Increased methylation levels of the MTHFR gene
promoter in Down syndrome. Epigenomics. 17:1141–1151. 2025.
View Article : Google Scholar
|
|
182
|
Tsymbalova EA, Chernyavskaya EA, Bisaga
GN, Polushin AY, Lopatina EI, Abdurasulova IN and Lioudyno VI:
LINE-1 methylation status in multiple sclerosis patients is
associated with changes in folate metabolism. Acta Nat. 17:94–103.
2025. View Article : Google Scholar
|














