Introduction
Cardiovascular disease (CVD) persists as a foremost
contributor to global morbidity and mortality, posing a pervasive
disease burden that critically undermines population health metrics
(1). Atherosclerosis (AS),
serving as the pathological foundation and primary etiology of CVD,
manifests as a chronic vascular disorder characterized by
lipid-laden arterial wall deposition coupled with persistent
oxidative stress and inflammatory responses (2,3).
The pathogenesis of AS involves multiple processes: endothelial
dysfunction, lipoprotein retention, macrophage activation,
inflammatory cytokine release, oxidized low density lipoprotein
(ox-LDL)-driven foam cell formation and vascular smooth muscle cell
migration. Oxidative stress plays a central role by impairing
endothelial function, promoting ox-LDL accumulation, and enhancing
foam cell formation, collectively accelerating plaque development
(4). These molecular
interactions synergistically drive plaque instability through
matrix metalloproteinase activation and fibrous cap attenuation.
Preserving redox balance therefore represents a critical
therapeutic strategy for decelerating atherogenesis and sustaining
vascular homeostasis.
Under pathophysiological stress, the excessive
production of reactive oxygen species (ROS) that surpasses the
endogenous antioxidant capacity perturbs redox homeostasis, thereby
inducing oxidative modifications to cellular nucleic acids,
proteins, and lipids, which culminate in multi-level tissue damage
(5). Substantial evidence
identifies vascular endothelial dysfunction as the seminal event in
atherosclerotic pathogenesis (6). Under physiological conditions, the
vascular endothelium sustains homeostatic balance via orchestrated
regulation of vasomotor tone, thrombotic homeostasis, and selective
permeability. During atherosclerotic initiation, ROS-driven
oxidative stress disrupts this equilibrium, manifesting as: i)
Structural barrier compromise, ii) loss of thromboresistant
capacity, and iii) transition to pro-inflammatory phenotype
(7). This pathognomonic triad
perpetuates a vicious cycle of endothelial activation,
subendothelial lipoprotein retention, and vascular remodeling that
drives atherosclerotic plaque progression.
Nuclear factor erythroid 2-related factor 2 (Nrf2),
the key transcriptional regulator of cellular redox balance,
coordinates intrinsic antioxidant mechanisms to neutralize
ROS-driven oxidative damage (8).
Vascular endothelium employs tonic Nrf2 signaling to mediate
oxidation-sensitive activation, orchestrating cytoprotective
transcriptional programs through redox-responsive gene networks.
Nrf2 deficiency dysregulates adaptive redox homeostasis, amplifying
free radical flux and compromising reparative angiogenesis via
blunted endothelial tip cell specification and dysfunctional
angiocrine signaling (9). Under
redox homeostasis, Keap1 orchestrates Nrf2 cytoplasmic retention by
directing constitutive ubiquitination-proteasome turnover, thereby
enforcing Nrf2 transcriptional dormancy. Electrophilic stress
initiates redox-gated conformational switches within the Keap1-Nrf2
axis via cysteine sensor modification, uncoupling ubiquitin ligase
activity while liberating Nrf2 for nuclear import. Nrf2 forms
heterodimers with small Maf proteins in the nucleus, activating the
expression of heme oxygenase-1 (HO-1), NAD(P)H quinone
oxidoreductase 1 (NQO1) and glutamate-cysteine ligase modifier
subunit (GCLM) through binding to antioxidant response elements
(ARE) (10,11). This regulated genetic response
orchestrates a reduction in ROS production, a bolstering of free
radical scavenging capacity, and a stabilization of the
intracellular redox state, thereby attenuating oxidative tissue
damage.
The Nod-like receptor protein 3 (NLRP3) inflammasome
serves as a molecular signaling nexus that integrates
stress-activated pathways with inflammatory cascades, functionally
linking canonical pyroptosis to atherosclerotic pathogenesis
through regulated secretion of pro-inflammatory cytokines (12). Excessive ROS act as potent
activators of the NLRP3 inflammasome, triggering its
oligomerization through protein-protein interactions to form the
NLRP3-ASC-pro-cysteinyl aspartate specific proteinase (Caspase-1)
supramolecular complex (13).
Activated Caspase-1 subsequently cleaves pro-Interleukin-1β (IL-1β)
and pro-Interleukin-18 (IL-18) into their biologically active
forms, driving the secretion of these inflammatory mediators while
simultaneously inducing cellular osmotic imbalance through
gasdermin-D-mediated membrane pore formation (14). This dual mechanism mediates the
execution of distinctive pyroptotic events-cellular swelling,
plasma membrane rupture, and release of cytoplasmic contents-the
dissemination of which, as damage-associated molecular patterns,
thereby potentiates inflammatory responses. The cyclical interplay
between ROS overproduction, NLRP3 inflammasome activation, and
persistent inflammation establishes a self-perpetuating vicious
cycle that exacerbates vascular endothelial dysfunction and plaque
instability in AS (15).
Therapeutic strategies targeting ROS-dependent NLRP3 inflammasome
activation demonstrate significant potential to disrupt this
deleterious cycle, thereby attenuating inflammatory burden and
ameliorating atherosclerotic progression.
Ginkgo biloba L. (Ginkgoaceae; MPNS-verified)
a unique tree species native to China, is valued for its
neuroprotective properties in addressing cardio-cerebrovascular
diseases and neurological disorders (16). Contemporary pharmacological
research has elucidated the multifaceted pharmacodynamics of foliar
extracts, demonstrating their capacity to confer dose-dependent
cardioprotection, modulate neurovascular homeostasis, and suppress
neoplastic proliferation (17).
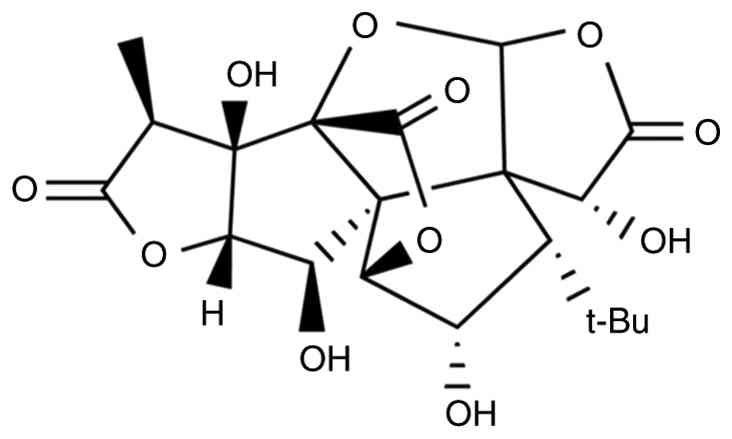
Of particular scientific interest is ginkgolide C (GC) (Fig. 1), a unique terpenoid trilactone
isolated from Ginkgo biloba leaves characterized by its
distinctive pentacyclic structure. Previous studies have
characterized GC as a pleiotropic therapeutic agent, demonstrating
potent anti-inflammatory efficacy and the capacity to confer
multi-organ protection in experimental models of cardiac and
cerebral ischemia-reperfusion injury (18,19). Atherosclerotic progression is
increasingly recognized as a critical pathogenic link between
ischemic events and cerebral/cardiovascular comorbidities, driven
by interconnected pathways of endothelial dysfunction and amplified
oxidative stress (20). However,
the potential of GC to mitigate atherosclerotic lesions through
suppression of ROS-induced NLRP3 inflammasome activation and
subsequent attenuation of inflammatory cascades remains
experimentally unestablished.
The present study aimed to elucidate the
anti-atherosclerotic mechanisms of GC through a combined
experimental approach both in vivo and in vitro,
focusing specifically on its dual-pathway regulatory activity. The
experimental findings demonstrated that GC ameliorates
atherosclerotic pathology through endothelial Nrf2 pathway
activation, which coordinates upregulation of antioxidant defense
proteins, suppression of ROS generation and sequential inhibition
of NLRP3 inflammasome assembly, ultimately attenuating
pro-inflammatory cytokine production and inflammatory cascade
propagation.
Materials and methods
Materials and reagents
GC (PubChem CID: 161120) and Atorvastatin (AVT;
PubChem CID: 60823) were commercially acquired from
MilliporeSigma-Aldrich. Anti-NLRP3 (cat. no. 15101S) was obtained
from Cell Signaling Technology, Inc. Anti-Nrf2 (cat. no.
16396-1-AP) was obtained from Proteintech Group, Inc. Anti-HO-1
(cat. no. PAA584Hu01), anti-NOQ1 (cat. no. PAL969Hu01), anti-GCLM
(cat. no. PAB038Hu01), anti-Caspase-1 (cat. no. PAB592Hu01),
anti-β-actin (cat. no. PAB340Mi01) and anti-histone (cat. no.
PAS091Ge01) antibodies were obtained from Cloud-Clone Corp. The
following analytical-grade materials were commercially sourced from
Beyotime Institute of Biotechnology: BCA protein quantification
kits (cat. no. P0010S), PVDF western blot membranes (cat. no.
FFP28), SDS-PAGE electrophoresis systems (cat. no. PG012) and ECL
Plus chemiluminescence substrates (cat. no. P0018M).
Randomization and blinding
procedures
Randomization was applied to animal/cell grouping,
and blinding was strictly maintained during data collection and
analysis to minimize potential bias and ensure experimental
objectivity.
Animal experiments
A total of 6 4-6-week-old male C57BL/6 mice weighing
14-16 g and 30 Apolipoprotein E-deficient (ApoE−/−) male
mice weighing 14-16 g were obtained from Huazhong Agricultural
University and Guangdong Yaokang Biotechnology Co., Ltd.,
respectively. All animals were maintained under standardized
laboratory conditions. Ambient temperature was regulated at
20-25°C, relative humidity was stabilized at 40-60%, and a 12-h
photocycle was enforced. A certified rodent diet and water were
provided ad libitum. All experimental procedures strictly
adhered to the Ethics Committee of the Shandong Provincial Hospital
Affiliated to Shandong First Medical University (approval no.
2022-661; Jinan, China).
Mice were randomized into 6 groups (n=6/group):
control group, AS model group, 2.5 mg/kg AVT group, and 12, 24, 48
mg/kg GC groups. ApoE−/− mice used for AS modeling, AVT
and GC treatment underwent disease induction through three
sequential interventions: Initial preconditioning via a single
cholecalciferol injection (300,000 IU/kg in 0.2 ml saline),
followed by 16 weeks of high-fat/high-cholesterol diet (40% of
calories from fat), and culminating in a final intraperitoneal
administration of vitamin D3 (100,000 IU/kg). Control
mice were maintained on standard chow and purified water under
controlled environmental conditions (22±1°C, 55±5% humidity, 12-h
light cycle) throughout the study. After confirming successful AS
development, daily tail vein injections of AVT or GC were
administered continuously for 7 days to the AVT or GC groups. At
the end of the experiment, euthanasia was performed by an
intraperitoneal injection of an overdose of sodium pentobarbital
(150 mg/kg). Death was confirmed by the cessation of heartbeat and
respiration, assessed by visual inspection and palpation.
Plaque assessment
After 24-h fixation in 4% paraformaldehyde at 4°C,
aortic root specimens were dehydrated in 30% sucrose and embedded
in OCT compound in the coronal orientation. Serial
10-μm-thick sections were obtained along the aortic valve
axis using a cryostat. For analysis, 10 representative sections
were stained with Oil Red O (Sigma-Aldrich) for lipid visualization
or with hematoxylin and eosin (H&E) for structural assessment.
Plaque areas were quantified in ImageJ software (version 1.8.0;
National Institutes of Health) applying standardized threshold
parameters.
Transmission electron microscope (TEM)
observation
Aortic root specimens were primarily fixed with 3%
glutaraldehyde in phosphate buffer (Ph 7.2) for 48-72 h, followed
by thorough PBS rinsing and subsequent graded ethanol dehydration.
Following embedding and polymerization in Epon 812 resin, tissue
blocks were subjected to ultrathin sectioning. Sections of 90 nm
thickness, prepared with an ultramicrotome equipped with a diamond
knife, were imaged using a Hitachi H-800 TEM at an acceleration
voltage of 80 kV to assess ultrastructural details.
Immunofluorescence (IF) staining
IF analysis cellular localization of Nrf2 and NLRP3
was assessed through IF. Fixed cells (pre-cooled methanol, −20°C,
10 min) underwent triple PBS washing (5 min each, pH 7.4) followed
by non-specific binding blockade with 3% BSA (MilliporeSigma)/0.1%
Tween-20 (1 h, RT). Primary antibodies included anti-Nrf2 (1:200;
cat. no. ab62352; Abcam) and anti-NLRP3 (1:500; cat. no. ab263899;
Abcam). Corresponding Secondary antibodies (1:500; cat. nos.
ab205718 and ab97051 Abcam) were purchased from Abcam. Nuclear
visualization employed DAPI staining (0.1 μg/ml PBS; 5 min;
cat. no. ab285390; Abcam). Confocal microscopy (Leica TCS SP8, 40x
oil lens, NA 1.30) captured images using LAS X 3.7.4 with
standardized 358/461 nm excitation/emission and z-axis imaging
(1-μm intervals).
Serum lipid quantification
Blood samples were centrifuged at 2,000 × g for 20
min at 4°C, followed by a 1:1 dilution of the resulting supernatant
with phosphate-buffered saline (PBS). Total cholesterol (TC),
triglycerides (TG), low-density lipoprotein cholesterol (LDL-C) and
high-density lipoprotein cholesterol (HDL-C) were tested using a
Beckman DXC700au system (NIST SRM 1951c-calibrated) with endpoint
analysis at 500 nm under standardized conditions (37±0.2°C, <3%
intra-assay CV).
Cell experiments
Primary mouse aortic endothelial cells (MAECs) were
obtained through enzymatic digestion of thoracic aortas isolated
aseptically from 8-12-week-old mice. Following the removal of
adventitial tissue and longitudinal incision, vessels were
enzymatically digested with collagenase I (1 mg/ml) at 37°C for
15-20 min. Digested cells were filtered through 40-μm
strainers and plated onto gelatin-coated dishes in
endothelial-specific medium (10% FBS; 30 μg/ml ECGS; 100
μg/ml heparin; all from Gibco; Thermo Fisher Scientific,
Inc.). Cellular purity was enhanced through differential adhesion
(three sequential 30-min platings at 37°C, 5% CO2).
Cells were maintained in DMEM (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% heat-inactivated fetal bovine serum and
1% penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.).
Cultures were kept at 37°C under a 5% CO2 atmosphere
with 95% humidity in a tri-gas incubator (Thermo Fisher Scientific,
Inc.). To simulate atherosclerotic inflammatory responses,
confluent monolayers were sequentially challenged with ox-LDL (100
μg/ml, 12 h).
The cells were randomized into 5 groups (n=3/group):
control group, cells in DMEM without ox-LDL stimulation; Model
group, cells treated with ox-LDL stimulation; GC group, cells
pretreated with GC (1, 10, 100 μM) for 24 h prior to ox-LDL
co-stimulation.
Cell viability
The assessment of cell viability was performed using
the MTT reduction method. Interventions were performed upon the
cells reaching a density of 5,000 cells per well. Following
experimental interventions, the culture medium was replaced with 5
mg/ml MTT solution and incubated at 37°C for 4 h. Subsequently, the
supernatant was carefully removed, and the formazan crystals were
solubilized in DMSO under 300 rpm orbital agitation for 15 min. The
optical density at 490 nm was recorded with a microplate reader and
normalized relative to the control group.
ROS assay
ROS levels were measured via
2',7'-dichlorofluorescein diacetate (DCFDA; Abcam). Cells
(1×105 cells/well) in 6-well plates were incubated with
10 μM DCFDA at 37°C for 20 min. Fluorescence images were
captured with a fluorescence microscope (BD Biosciences), and ROS
fluorescence intensity was analyzed using Image-Pro Plus software
(version 6.0; Media Cybernetics Inc.).
Detections of inflammatory factors and
oxidative stress factors
IL-1β, IL-18 and tumor necrosis factor (TNF)-α
concentrations in serum and cell culture supernatants were
determined by ELISA kits (IL-1β, cat. no. 88-7013A-88; IL-18, cat.
no. KMC0181; TNF-α, cat. no. BMS607-3; Thermo Fisher Scientific,
Inc.) following the manufacturer's instructions. The levels of
superoxide dismutase (SOD; cat. no. A001-3), malondialdehyde (MDA;
cat. no. A003-1), lactate dehydrogenase (LDH; cat. no. A020-2),
creatine kinase (CK; cat. no. A032-1-1), catalase (CAT; cat. no.
A007-1-1), glutathione (GSH; cat. no. A006-2-1) and NADPH (cat. no.
A115-1-1) in serum and cell culture supernatants were detected by
chemical kits of Nanjing Jiancheng Engineering Institute.
TUNEL staining
DNA fragmentation in individual cells was detected
by TUNEL staining with a fluorescence kit (Roche Diagnostics).
Following fixation with 4% paraformaldehyde (20 min) and
permeabilization with 0.1% Triton X-100 (10 min) at room
temperature, MAECs on coverslips were subjected to TUNEL assay by
incubation with the reagent at 37°C for 60 min. Fluorescent signals
were visualized and recorded with an Olympus FV3000 confocal
microscopy system.
Western blot analysis for Nrf2, NLRP3,
HO-1, NQO1, GCLM and Caspase-1 expression levels in MAECs
Total protein quantification was performed using BCA
assay. Subcellular fractionation was conducted with a
Nuclear-Cytoplasmic Protein Isolation Kit (cat. no. P1250; Applygen
Technologies, Inc.) following established protocols. Protein
samples (50 μg) underwent SDS-PAGE (10% gel) electrophoresis
and subsequent PVDF membrane transfer. Membranes were blocked with
5% non-fat milk at room temperature for 1 h, then incubation with
primary antibodies (1:800 dilution) targeting Nrf2, NLRP3, HO-1,
NQO1, GCLM and total Caspase-1 at 37°C for 4 h. Membranes were
subsequently probed with HRP-conjugated secondary antibodies
diluted at 1:1,000. Specific signals were visualized with ECL Plus
reagent and quantified via densitometry using a Bio-Rad Gel Imaging
System and Quantity One software (version 5.0; Bio-Rad
Laboratories, Inc.).
Total protein quantification was determined via BCA
assay. Nuclear-cytoplasmic fractionation employed the
nuclear-cytoplasmic protein isolation kit (cat. no. P1250; Applygen
Technologies, Inc.) according to the manufacturer's instructions. A
total of 50 μg protein samples were separated through
SDS-PAGE and electrotransferred onto PVDF membranes. After 5% skim
milk blocking, membranes were probed with primary antibodies
(Nrf2/NLRP3/HO-1/NQO1/GCLM/Caspase-1; 1:800) at 37°C (4 h),
followed by HRP-secondary antibodies (1:1,000). Chemiluminescent
detection was performed with ECL Plus reagent, followed by
densitometric analysis of the bands using the Quantity One software
(version 5.0; Bio-Rad Laboratories, Inc.) imaging system.
Statistical analysis
Triplicate experimental data are expressed as the
mean ± SD. Intergroup comparisons were assessed through one-way
ANOVA with Bonferroni post hoc adjustment (GraphPad Prism version
5.0; Dotmatics). P<0.05 was considered to indicate a
statistically significant significance.
Results
GC alleviates atherosclerotic
pathological damage in AS model mice
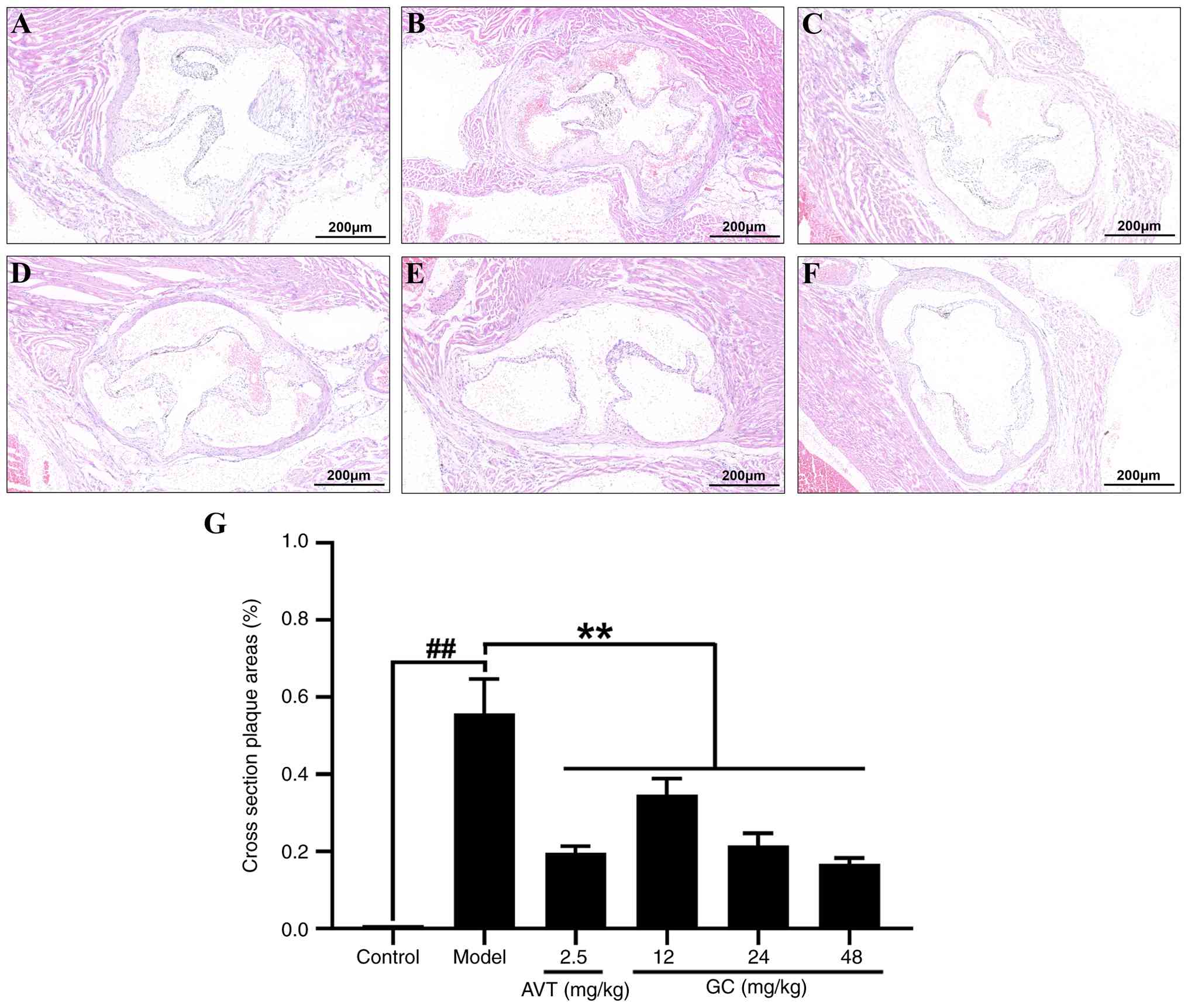
To observe the histopathological changes in the
aortic root following atherosclerotic injury, H&E staining was
performed. As depicted in Fig.
2A, aortic sections from the control group demonstrated a
well-defined trilaminar architecture: The tunica intima was lined
by a continuous endothelial monolayer; the tunica media showed
regularly arranged, wavy elastic fibers interspersed with smooth
muscle cells; and the tunica adventitia was composed of loose
connective tissue. No pathological alterations such as lipid
deposition, foam cell formation, inflammatory infiltration, luminal
disruption, or vascular wall thickening were observed. In the model
group (Fig. 2B), marked intimal
thickening was observed, featuring foam cells (vacuolated
cytoplasm), cholesterol clefts, inflammatory infiltration, and
fibrous caps overlying lipid cores. The tunica media showed
fragmented elastic fibers with reduced smooth muscle cells, while
the adventitia presented neovascularization and chronic
inflammation, accompanied by a narrowed and irregular lumen. In 2.5
mg/kg AVT group (Fig. 2C) and
12, 24, 48 mg/kg GC groups (Fig.
2D-F), the aortic intima displayed a significant decrease in
pathological thickening with parallel reductions in atherosclerotic
plaque deposition (Fig. 2G).
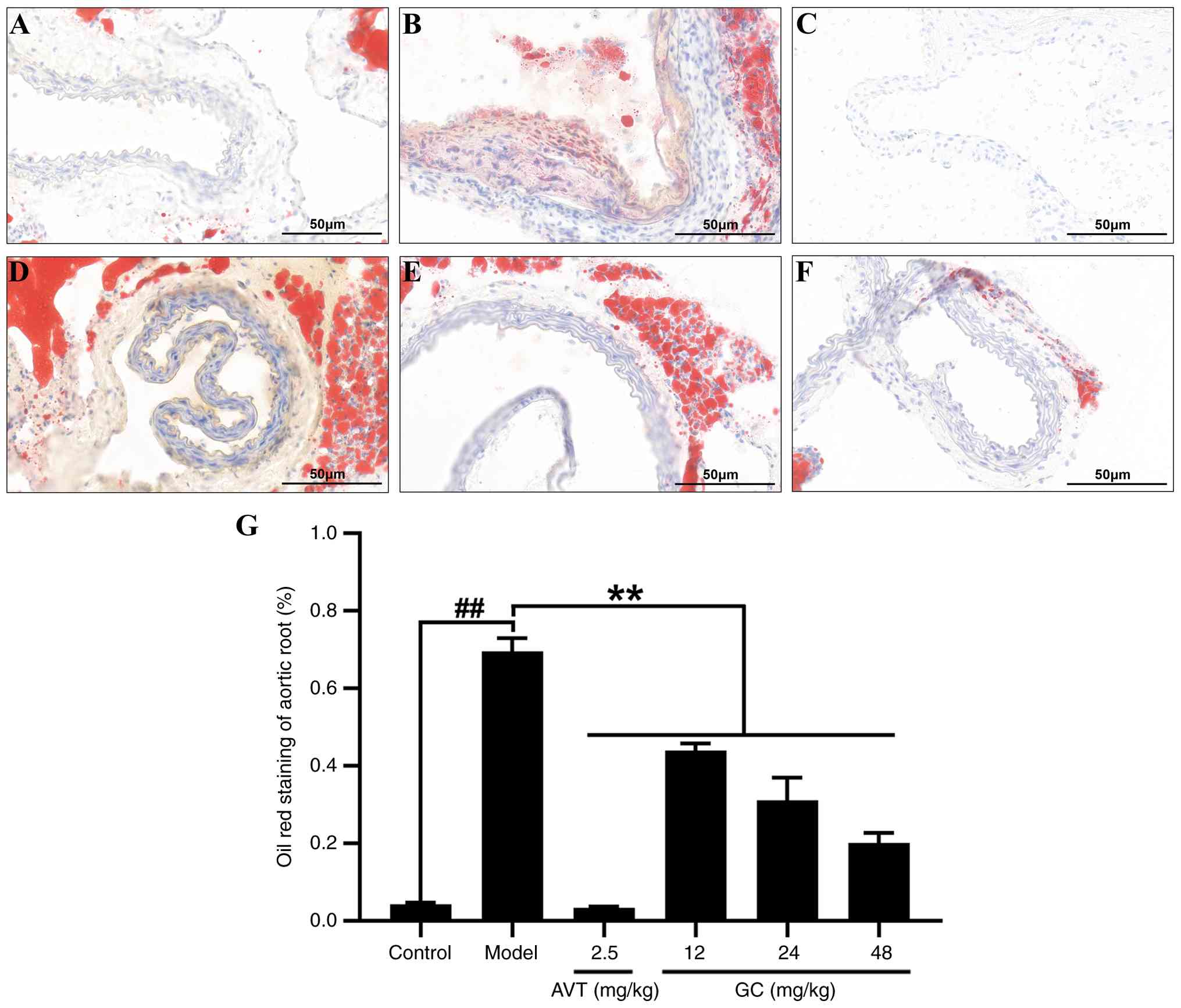
GC reduces atherosclerotic lipid
deposition in AS model mice
To investigate the effect of GC on lipid deposition
and examine its impact on plaque characteristics following
atherosclerotic injury, Oil Red O staining was performed. As shown
in Fig. 3A, the control group
exhibited no Oil Red O-positive red signals in any aortic wall
layers (intima, media, adventitia), with only physiological lipid
staining observed in periadventitial adipose tissue. The luminal
surface appeared smooth without lipid droplet adhesion, indicating
no pathological lipid deposition. By contrast, the model group
displayed bright red mass-like lipid deposits (plaque lipid cores)
in the aortic sinus and subintimal regions, accompanied by diffuse
punctate red infiltration (early lipid infiltration) at plaque
margins. Strongly positive lipid halos surrounded cholesterol
clefts in some areas, suggesting lipid metabolic disturbances and
extensive foam cell aggregation (Fig. 3B). However, Oil Red O staining
demonstrated a reduction in lipid core size, significant
attenuation of red-positive deposition area, and faded lipid halos
surrounding cholesterol clefts, indicative of ameliorated lipid
metabolic dysregulation and enhanced plaque stability after
treatment of 2.5 mg/kg AVT (Fig.
3C) and 12, 24, 48 mg/kg GC (Fig. 3D-F). Quantitative analysis of
atherosclerotic plaque area based on oil red O staining is
demonstrated in Fig. 3G.
GC reduces blood lipid levels in AS model
mice
The result in Table
I showed that the blood lipid levels in model group were
markedly changed (TG: 5.58±0.26 mM; TC: 41.59±5.49 mM; LDL-C:
16.14±2.41 mM and HDL-C: 1.46±0.24 mM, all P<0.01 vs. control
group). 2.5 mg/kg AVT and 12, 24, 48 mg/kg GC significantly
improved the blood lipid levels (all P<0.01 vs. model group).
The findings revealed that GC exhibited hypolipidemic effects and
attenuated atherosclerotic development.
 | Table IEffect of GC on serum lipid levels in
atherosclerosis model mice. |
Table I
Effect of GC on serum lipid levels in
atherosclerosis model mice.
| Group | Dose (mg/kg) | TG (mM) | TC (mM) | LDL-C (mM) | HDL-C (mM) |
|---|
| Control | | 1.39±0.26 | 3.63±0.17 | 1.50±0.33 | 4.97±0.64 |
| Model | | 5.58±0.26b | 41.59±5.49b | 16.14±2.41b | 1.46±0.24b |
| AVT | 2.5 | 1.53±0.36a | 16.51±4.14a | 1.67±0.62a | 3.96±0.60a |
| GC | 12 | 3.47±0.25a | 26.45±2.18a | 5.52±1.48a | 2.72±0.84a |
| 24 | 2.71±0.39a | 17.83±2.00a | 4.30±1.51a | 3.26±0.34a |
| 48 | 1.67±0.54a | 11.75±1.23a | 2.37±1.03a | 4.44±0.54a |
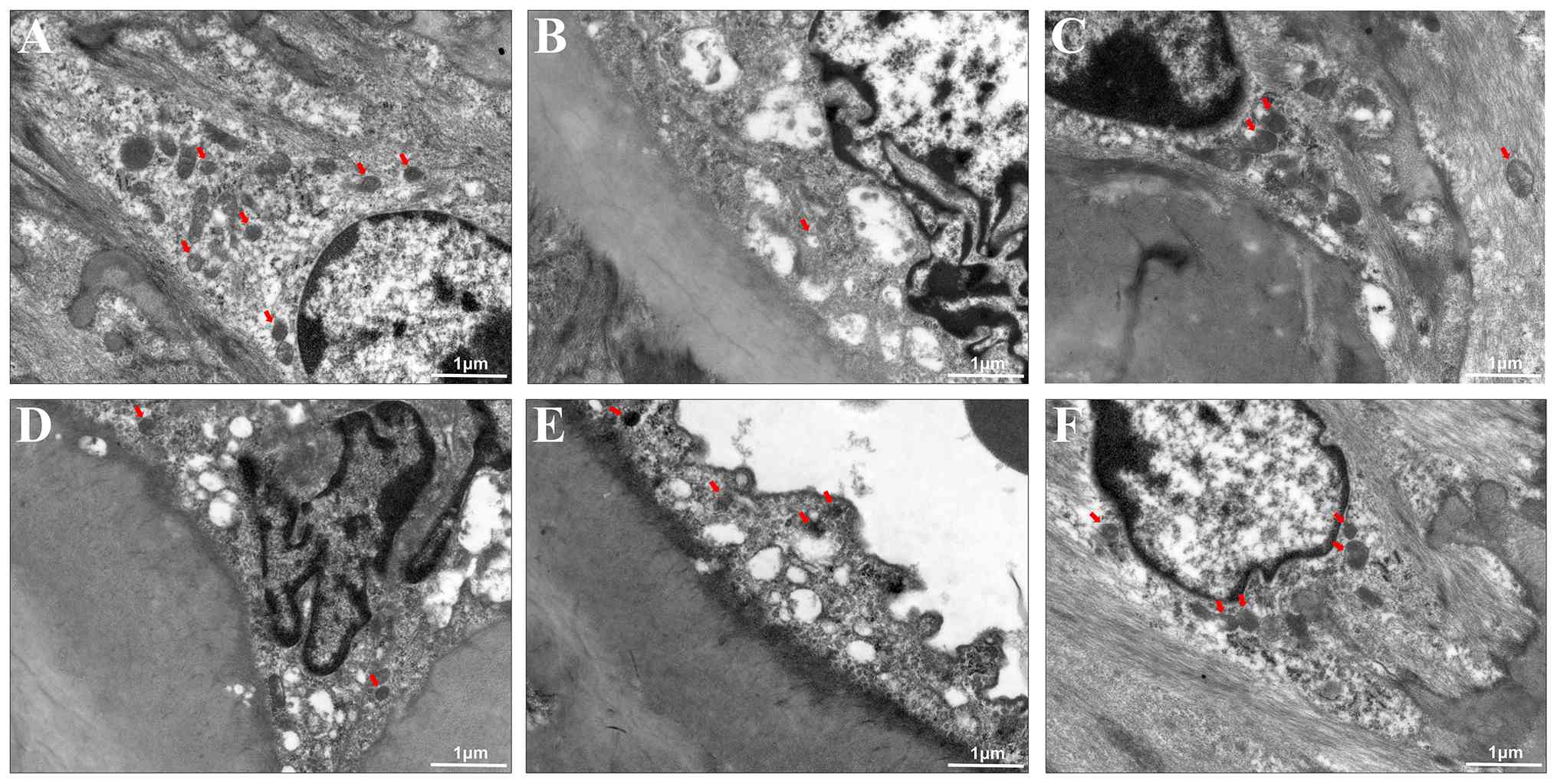
GC improves atherosclerotic
ultrastructural changes in AS model mice
Additionally, ultrastructural changes induced by GC
were analyzed by TEM. In control group, the endothelial cells
formed a continuous monolayer with intact tight junctions; the
medial smooth muscle cells were densely packed with contractile
myofilaments and surrounded by orderly arranged elastic fibers; the
extracellular matrix showed no lipid droplet deposition,
inflammatory infiltration, or structural disruption (Fig. 4A). However, in the model group,
endothelial disruption with lipid deposition, smooth muscle foam
cell transformation (marked by contractile myofilament loss and
lipid droplet-induced organelle compression), and elastic fiber
fragmentation were prominently observed. Concurrently,
extracellular collagen disorganization and cholesterol crystal
deposition synergistically drove plaque instability (Fig. 4B). The results in Fig. 4C-F revealed that 2.5 mg/kg AVT
and 12, 24, 48 mg/kg GC significantly improved the endothelial
continuity restoration, smooth muscle contractile phenotype
reconstruction (marked by myofilament regeneration and lipid
droplet clearance), and elastic-collagen reorganization in the
extracellular matrix. Plaque cholesterol crystal dissolution with
attenuated inflammatory infiltration indicates GC effectively
reversed core pathological cascades of AS.
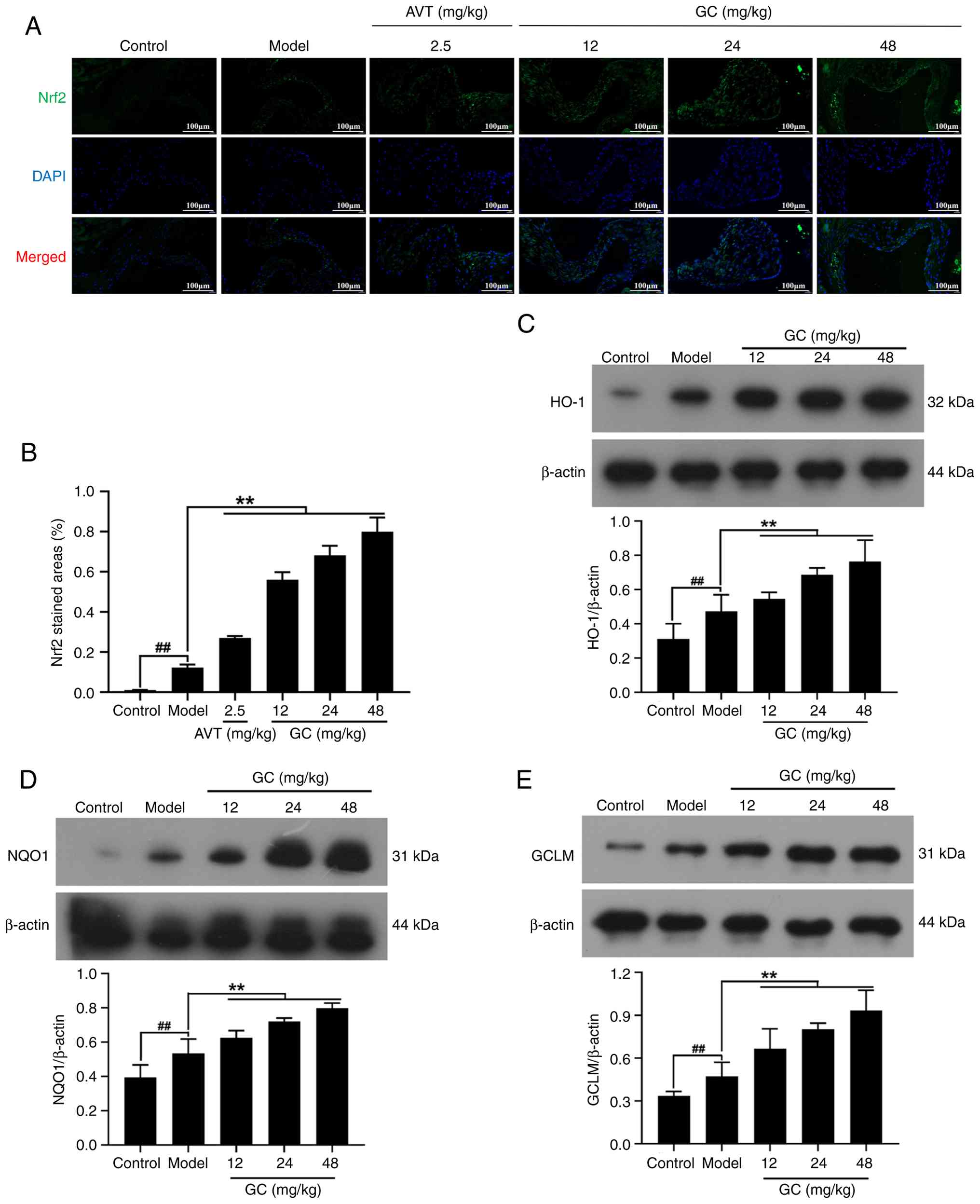
GC upregulates the expression levels of
Nrf2 and its downstream antioxidant proteins HO-1, NQO1 and GCLM in
AS model mice
As shown in Fig. 5A
and B through IF analysis, Nrf2 expression demonstrated
relatively low baseline levels in the control group. The model
group exhibited marginal elevation in Nrf2 expression compared with
the control group. However, GC administration at doses of 12, 24
and 48 mg/kg induced significant upregulation of Nrf2 expression
levels (all P<0.01 vs. model group). Moreover, 2.5 mg/kg AVT
slightly elevated the expression of Nrf2 (P<0.01 vs. model
group). Furthermore, baseline expression levels of HO-1, NQO1 and
GCLM were markedly diminished in the control group. While the model
group demonstrated modest elevation of these antioxidant markers
relative to controls, 12, 24, 48 mg/kg GC produced statistically
significant inductions in HO-1, NQO1 and GCLM expression (Fig. 5C-E; all P<0.01 vs. model
group).
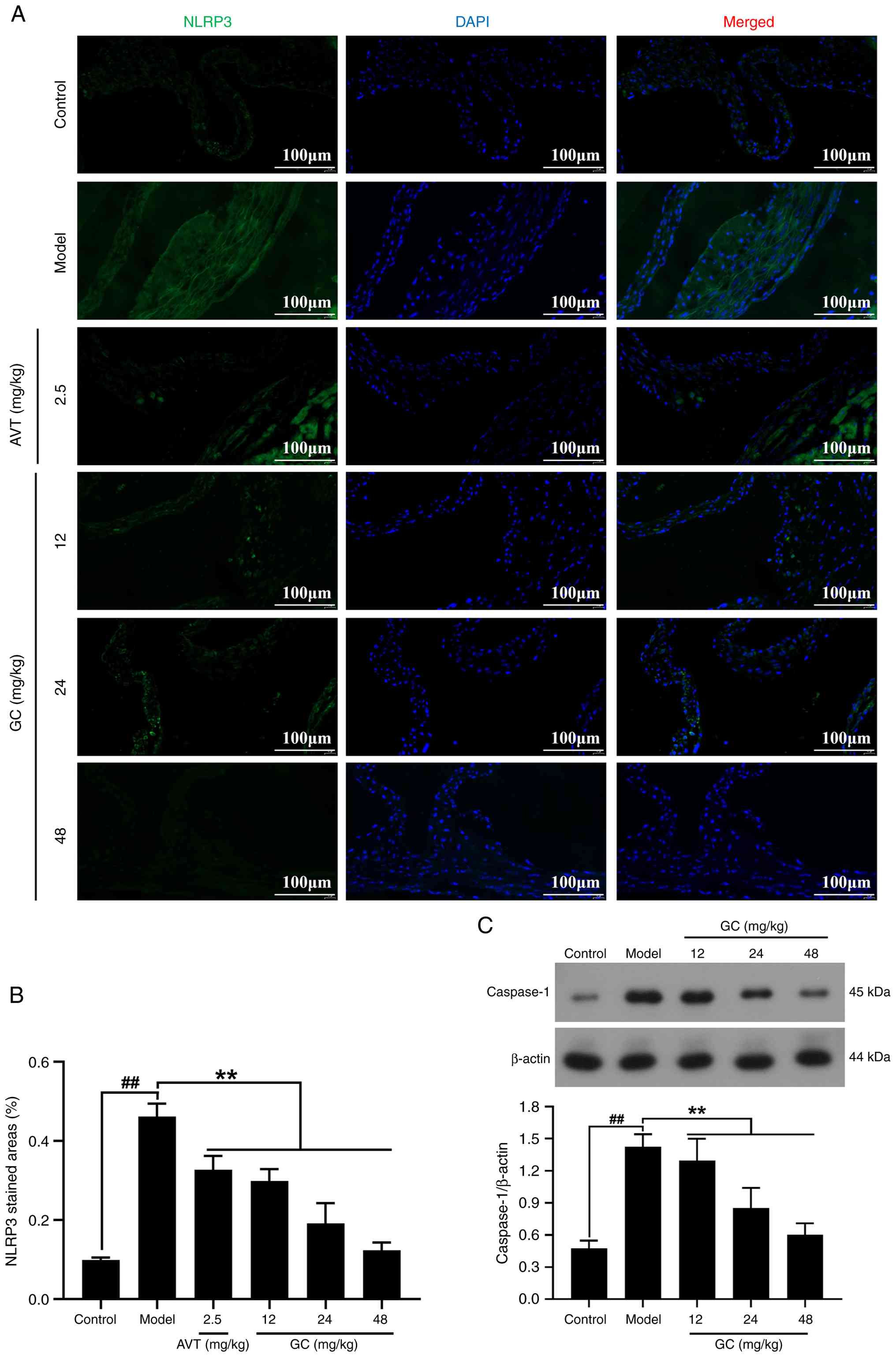
GC downregulates the expression levels of
NLRP3 and its downstream Caspase-1
As demonstrated in Fig. 6A-C, pathological activation of
NLRP3 inflammasome components was significantly elevated in the
model group, showing significant upregulation of both NLRP3 and
Caspase-1. Conversely, GC treatment at ascending doses (12, 24, 48
mg/kg) exhibited progressive suppressions of NLRP3 and Caspase-1
(all P<0.01 vs. model group).
GC inhibits oxidative stress and
inflammatory damage in AS model mice
Next, the effect of GC on the expression of
inflammatory cytokines was further investigated in the serum of AS
model mice. As revealed in Table
II, the levels of IL-1β, IL-18 and TNF-α in the control group
were low. After modeling, the levels of IL-1β, IL-18 and TNF-α
increased to 361.85±62.80 pg/ml, 96.41±8.10 pg/ml, 97.90±6.64
pg/ml, respectively (all P<0.01 vs. control group). However, 2.5
mg/kg AVT and 12, 24, 48 mg/kg GC could dose-dependently reduced
the expression levels of IL-1β, IL-18 and TNF-α (all P<0.01 vs.
model group).
| Table IIEffect of GC on serum inflammatory
cytokines in atherosclerosis model mice. |
Table II
Effect of GC on serum inflammatory
cytokines in atherosclerosis model mice.
| Group | Dose (mg/kg) | IL-1β (pg/ml) | IL-18 (pg/ml) | TNF-α (pg/ml) |
|---|
| Control | | 29.28±3.14 | 16.24±1.05 | 6.30±1.50 |
| Model | |
361.85±62.80b | 96.41±8.10b | 97.90±6.64b |
| AVT | 2.5 |
104.85±10.95a | 59.46±6.90a | 33.04±6.82a |
| GC | 12 |
133.27±18.50a | 57.47±2.07a | 40.35±3.55a |
| 24 | 84.12±12.52a | 28.25±8.88a | 35.27±3.80a |
| 48 | 38.01±4.01a | 17.52±2.27a | 24.39±6.21a |
As demonstrated in Table III, the levels of SOD, MDA,
LDH, CK, CAT, GSH and NADPH in the control group were 156.49±13.59
U/ml, 1.39±0.18 nM, 3513.01±663.44 U/l, 0.34±0.10 U/ml, 9.78±1.21
U/ml, 267.07±22.48 μM and 4.07±0.20 μM, respectively.
After modeling, the cohort exhibited significant reductions in
antioxidant defense markers (SOD, CAT and GSH) concomitant with
significant elevations in oxidative stress indicators (MDA, LDH and
CK) and NADPH generation (all P<0.01 vs. control group). 2.5
mg/kg AVT and 12, 24, 48 mg/kg GC significantly upregulated SOD,
CAT and GSH while downregulating MDA, LDH, CK and NADPH, with all
changes reaching statistical significance (P<0.05 vs. model
group).
| Table IIIEffect of GC on antioxidant enzymes
in atherosclerosis model mice. |
Table III
Effect of GC on antioxidant enzymes
in atherosclerosis model mice.
| Group | Control | Model | AVT | GC |
|---|
| Dose (mg/kg) | | | 2.5 | 12 | 24 | 48 |
| SOD (U/ml) | 156.49±13.59 | 47.66±9.25b | 82.01±4.51a | 93.69±9.46a | 101.95±6.02a | 133.82±5.59a |
| MDA (nM) | 1.39±0.18 | 8.10±0.60b | 4.48±0.34a | 4.20±0.67a | 3.29±0.63a | 2.37±0.26a |
| LDH (U/l)
3,513.01±663.44 |
9,953.69±510.21b |
4,724.05±641.13a |
6,460.10±728.44a |
5,125.62±239.46a |
4,242.19±692.69a |
| CK (U/ml) | 0.34±0.10 | 1.37±0.22b | 0.55±0.06a | 0.89±0.14a | 0.60±0.10a | 0.48±0.05a |
| CAT (U/ml) | 9.78±1.21 | 2.64±1.16b | 6.10±0.73b | 5.54±0.78a | 6.50±1.23a | 7.77±1.28a |
| GSH
(μM) | 267.07±22.48 | 79.55±15.10b | 129.25±6.20b |
133.67±21.23a |
181.02±19.21a |
219.30±15.46a |
| NADPH
(μM) | 4.07±0.20 | 8.20±0.25b | 5.48±0.39b | 5.90±0.39a | 4.45±0.40a | 4.10±0.73a |
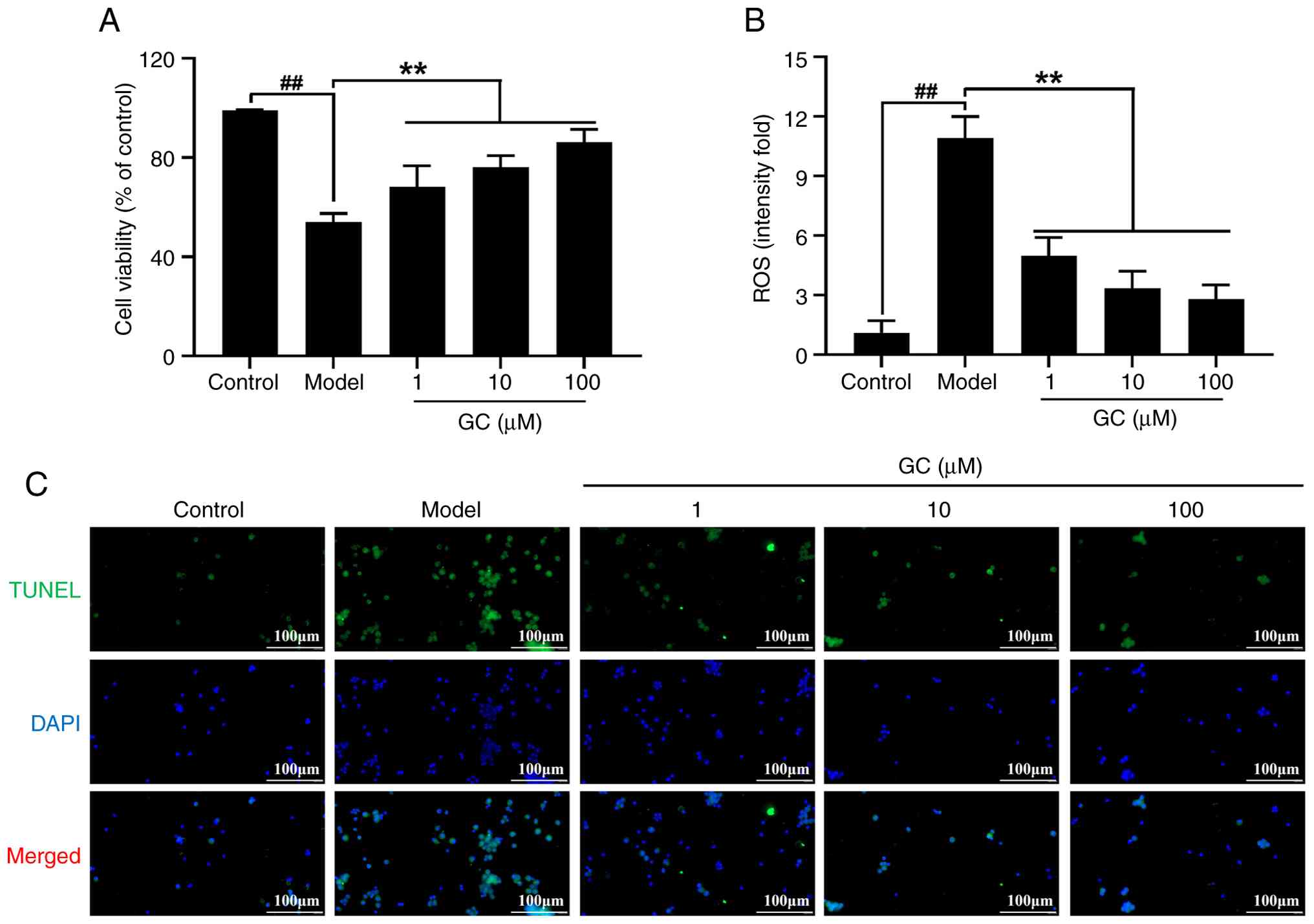
GC promotes cell survival, suppresses ROS
generation, and inhibits apoptosis in ox-LDL-stimulated MAECs
As shown in Fig.
7A, ox-LDL-treated MAECs exhibited a significant reduction in
cell viability (54.08±3.36%; P<0.01 vs. control group) as
determined by MTT assay. Treatment of GC (1, 10, 100 μM)
demonstrated dose-responsive enhancement of MAECs viabilities
(68.11±8.67%, 76.05±4.71%, 86.19±5.11%, P<0.01 vs. model
group).
Then, DCFDA was used for determining the level of
ROS in MAECs (Fig. 7B). In
contrast to the marked increase observed in ox-LDL-treated groups,
ROS levels in the control group were substantially lower. In
contrast to the model group, GC treatment at 1, 10 and 100
μM significantly reduced ROS levels (all P<0.01).
TUNEL staining result in Fig. 7C indicated that minimal apoptosis
in control group, whereas the model group exhibited significant
apoptotic cells. Treatment with 1, 10 and 100 μM GC markedly
reduced apoptosis.
GC enhances Nrf2 nuclear translocation
with upregulation of HO-1/NQO1/GCLM, while suppressing
NLRP3/Caspase-1 expression
As revealed in Fig.
8A and B, Nrf2 was primarily localized in the cytoplasm with
barely detectable nuclear levels in control cells. By contrast,
ox-LDL stimulation promoted its nuclear translocation. However, GC
(1, 10, 100 μM) significantly enhanced the
cytoplasmic-to-nuclear translocation of Nrf2. Therefore, GC
significantly activated the Nrf2 signaling pathway, with increased
cytoplasmic-to-nuclear translocation of Nrf2 leading to
upregulation of HO-1, NQO1 and GCLM expression levels (Fig. 8C-E). The results in Fig. 8F and G showed that ox-LDL
stimulation notably upregulated the expression of NLRP3 and
Caspase-1. Whereas treatment of 1, 10 and 100 μM GC
demonstrated dose-dependent suppression of NLRP3/Caspase-1 cascade
activation.
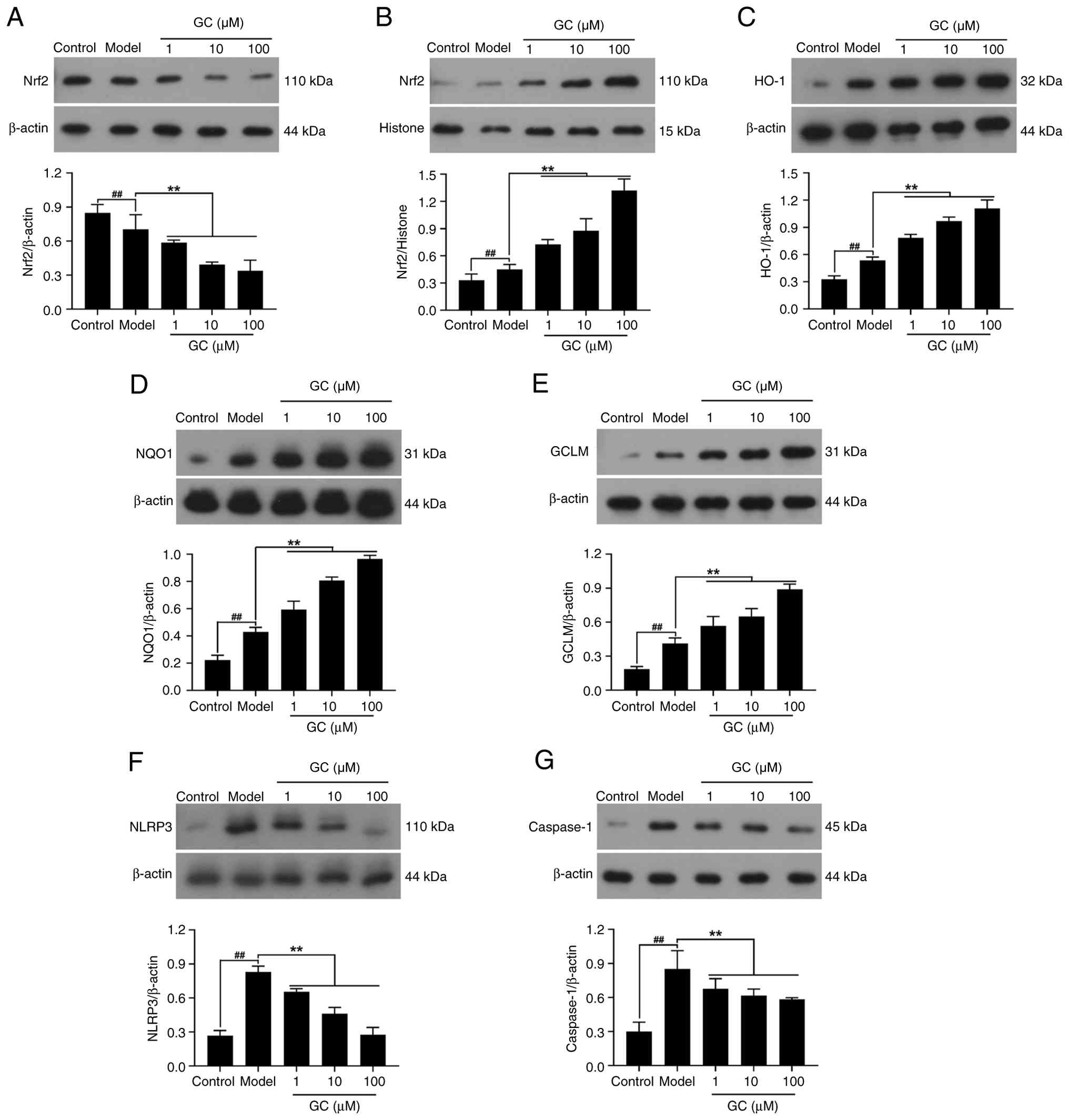 | Figure 8Effect of GC on expression levels of
Nrf2, HO-1, NQO1, GCLM, NLRP3 and Caspase-1 in oxidized-low density
lipoprotein stimulated mouse aortic endothelial cells. (A-G)
Western blot was applied to quantify the expression levels of (A)
cytoplasmic Nrf2, (B) nuclear Nrf2, (C) HO-1, (D) NQO1, (E) GCLM,
(F) NLRP3 and (G) Caspase-1. Results were expressed as
protein/reference protein ratio. Data are expressed as the mean ±
SD. ##P<0.01 vs. control group;
**P<0.01 vs. model group. GC, ginkgolide C; Nrf2,
nuclear factor erythroid 2-related factor 2; HO-1, heme
oxygenase-1; NQO1, NAD(P)H quinone oxidoreductase 1; GCLM,
glutamate-cysteine ligase modifier subunit; NLRP3, Nod-like
receptor protein 3. |
GC also improves antioxidant capacity and
suppresses inflammatory reaction in ox-LDL-stimulated MAECs
Results were consistent with in vivo
experiments (Tables IV and
V). ox-LDL exposure triggered
concurrent elevation of pro-inflammatory cytokines (IL-1β, IL-18
and TNF-α) and oxidative markers (MDA, LDH, CK and NADPH),
paralleled by significant depletion of antioxidant defenses (SOD,
CAT and GSH) (all P<0.01 vs. control). However, 1, 10, and 100
μM GC could significantly improve the inflammatory response
and oxidative stress damage induced by ox-LDL.
| Table IVEffect of GC on the supernatant
inflammatory cytokines of oxidized-low density lipoprotein
stimulated mouse aortic endothelial cells. |
Table IV
Effect of GC on the supernatant
inflammatory cytokines of oxidized-low density lipoprotein
stimulated mouse aortic endothelial cells.
| Group | Dose
(μM) | IL-1β (pg/ml) | IL-18 (pg/ml) | TNF-α (pg/ml) |
|---|
| Control | | 1.80±0.53 | 11.73±2.90 | 6.94±1.47 |
| Model | |
1,085.96±135.24b |
121.00±19.23b |
588.98±111.82b |
| GC | 1 |
509.94±113.03a | 34.43±5.09a |
245.31±20.99a |
| 10 |
252.86±39.04a | 21.12±6.34a |
169.81±33.94a |
| 100 |
105.53±13.05a | 12.27±1.34a | 65.90±9.20a |
| Table VEffect of GC on antioxidant enzymes
of oxidized-low density lipoprotein stimulated mouse aortic
endothelial cells. |
Table V
Effect of GC on antioxidant enzymes
of oxidized-low density lipoprotein stimulated mouse aortic
endothelial cells.
| Group | Control | Model | GC |
|---|
| Dose
(μM) | | | 1 | 10 | 100 |
| SOD (U/mg pro) | 296.04±24.49 |
129.08±23.70b | 206.48±7.53a | 241.78±8.70a |
273.96±18.58a |
| MDA (nM/mg
pro) | 6.18±0.24 | 21.87±0.71b | 15.91±1.66a | 11.53±3.93a | 9.39±1.77a |
| LDH (U/l) | 702.27±38.14 |
1,257.27±212.96b |
1,022.00±83.18a |
891.48±38.90a |
805.04±66.59a |
| CK (U/mg pro) | 0.40±0.35 | 1.45±0.28b | 1.01±0.41a | 0.95±0.38a | 0.83±0.21a |
| CAT (U/ml) | 7.80±2.06 | 0.97±0.05b | 2.38±0.58a | 4.08±0.71a | 6.17±0.38a |
| GSH (mg/g pro) | 3.76±1.05 | 1.57±0.42b | 2.45±0.41a | 2.74±0.30a | 3.22±0.19a |
| NADPH
(μM) | 9.72±2.52 | 34.60±4.18b | 23.62±3.17a | 16.51±0.54a | 12.19±3.33a |
The data demonstrate a significant dual benefit of
GC: It conferred a dose-dependent enhancement in oxidative stress
tolerance concurrent with a suppression of inflammation. This
effect, achievable through the potential activation of endogenous
pathways such as Nrf2, highlights GC's role in simultaneously
addressing two key pathological processes.
Discussion
CVDs remain the globally predominant cause of
mortality, with AS serving as a central pathological driver
(21). AS manifests as a
multifactorial vascular pathology originating from disrupted
endothelial homeostasis, clinically characterized by impaired
vasomotor regulation, dyslipidemia, oxidative stress and chronic
inflammatory cytokine activation (22,23). Exacerbated oxidative stress
induces pathological ROS overproduction, elevating oxidative
derivatives and provoking systemic oxidative damage. Subsequent
pathological ROS accumulation directly activates the NLRP3
inflammasome, thereby potentiating proinflammatory cascades that
exacerbate atherosclerotic lesion progression (24). Crucially, Nrf2, serving as a
master regulator of redox homeostasis, negatively modulates NLRP3
inflammasome-mediated inflammation through redox-sensitive
suppression of ROS-mediated signaling pathways (25). This mechanistic framework
supports the author's hypothesis that GC attenuates atherosclerotic
injury via Nrf2 activation-mediated ROS inhibition, consequently
attenuating NLRP3 inflammasome activation and reducing
atherosclerotic plaque progression.
Ginkgo biloba, a monotypic genus in the
family Ginkgoaceae and the sole extant species of this lineage, is
recognized as a 'living fossil' that occupies a pivotal node in
plant evolutionary history (26,27). Modern pharmacological research
demonstrates that the biological activity of Ginkgo biloba
leaf extracts predominantly originates from its characteristic
secondary metabolites, specifically terpene lactones (6%) and
flavonoid glycosides (24%) as principal active components. The
latter category encompasses specific compounds such as ginkgolides
A/B/C/J and bilobalide, whose unique diterpene trilactone molecular
structure confers multi-target pharmacological activities (28). Guided by cumulative preclinical
evidence, GC was designated as the lead therapeutic candidate.
Experimental investigations employing in vitro and in
vivo models have established that both isolated constituents
manifest concentration-dependent cytoprotective efficacy against
ischemia-reperfusion pathophysiology in both myocardial and
cerebral compartments (18,19). The pathophysiological continuum
of AS, constituting the common etiological substrate for
cardio-cerebrovascular disorders, is mechanistically underpinned by
dysregulated endothelial homeostasis, pathogenic leukocyte
recruitment, and atheroma destabilization, pathobiological
processes intrinsically linked to redox imbalance and dysregulated
proinflammatory signaling pathways (29,30). Leveraging GC's multifaceted
anti-inflammatory pharmacodynamics and multiphasic vascular
protective actions, the present investigation systematically
interrogates the compound's interactome within AS pathobiology,
with dual objectives: i) Characterization of bioactive effector
moieties governing its therapeutic efficacy, and ii) comprehensive
mapping of pleiotropic therapeutic modalities encompassing atheroma
stabilization, endothelial homeostatic restoration, and
immunomodulatory microenvironment remodeling.
Of particular note is the atherogenic diet-fed
ApoE−/− mouse model employed in the present study, which
allowed for the evaluation of GC interventions under controlled,
dose-escalating conditions. The experimental approach integrated
serum lipid profiling, histological examination of plaques, and
ultrastructural visualization via electron microscopy.
Pathophysiologically, atherogenesis is widely understood to be
initiated by dyslipidemia, wherein LDL-C undergoes oxidative
modification to ox-LDL, triggering endothelial activation,
inflammatory recruitment, and foam cell formation-central events in
plaque development (31,32). Epidemiological observations
consistently link hypercholesterolemia with increased
cardiovascular risk, whereas HDL-C is recognized for its protective
role via reverse cholesterol transport (33). In light of these established
mechanisms, the lipid-modulating effects observed following GC
intervention provide a substantive basis for discussing its
potential therapeutic mechanism. The results suggest that GC may
exert a dual regulatory influence, not only reducing atherogenic
lipids but also promoting functional HDL activity, thereby
contributing to the restoration of lipid homeostasis. This
positions GC as a candidate for further investigation into its role
in modulating key pathways involved in AS progression. Notably, the
pleiotropic modulation of both pro-atherogenic lipoprotein
suppression and reverse cholesterol transport enhancement suggests
GC's unique position in contemporary anti-atherosclerotic
therapeutic strategies, potentially bridging current gaps in
targeting complex lipid flux dysregulation. In the present study,
GC was administered for only 7 days in a murine model of
atherosclerotic lesions, yet AS is a chronic, long-term disease
process. Whether GC can be sustainably used or possesses the
potential to reverse atherosclerotic lesions remains unclear.
Consequently, future studies are needed to systematically evaluate
extended GC treatment regimens (for example, 6-12 weeks) to
characterize its pharmacodynamic effects on plaque stabilization
and regression.
The multifaceted antiatherogenic profile of GC
warrants particular attention in the context of current therapeutic
challenges. Rather than focusing solely on lipid reduction, GC
appears to concurrently address plaque stabilization through
collagenous cap preservation and endothelial restoration, as
reflected in histopathological and ultrastructural observations.
This suggests a synergistic mechanism that targets multiple
pathological pathways-lipid retention, inflammatory signaling, and
extracellular matrix degradation-which collectively contribute to
atherosclerotic progression (34). Such a multiphasic therapeutic
approach may offer distinct advantages over conventional
monotherapeutic strategies, particularly in its ability to
intervene at both early and advanced stages of the disease
(35). By reinforcing plaque
microstructure and reducing lipid core vulnerability, GC holds
potential clinical utility in managing high-risk plaques,
especially in cases where statin therapy alone fails to mitigate
persistent inflammatory risks. The convergence of these mechanisms
supports GC's promise as a multimodal agent, meriting further
investigation into its translational application in complex
atherosclerotic disease.
The antioxidant properties of GC and their
implications for AS management merit further discussion within the
broader context of redox imbalance in CVD. Oxidative stress,
characterized by disrupted redox homeostasis, plays a
well-established role in driving endothelial dysfunction and plaque
instability, largely through the persistent generation of ROS and
subsequent lipid peroxidation (36). In this regard, GC appears to
modulate key components of the oxidative cascade, influencing both
the initiation and propagation of oxidative damage. Its potential
to enhance endogenous antioxidant defenses while simultaneously
mitigating peroxidative processes suggests a biphasic mechanism
that may effectively disrupt the ROS-lipid oxidation-inflammatory
axis, a critical pathway in AS progression. Notably, such a
multitarget antioxidant strategy differs from conventional
approaches by addressing oxidative stress at multiple levels, not
only scavenging free radicals but also promoting enzymatic
homeostasis (37). This may
offer a more sustainable and physiologically integrated means of
reducing oxidative injury in vascular tissues. Furthermore, the
potential involvement of the Nrf2 pathway, as inferred from
indirect evidence, hints at epigenetically mediated enhancement of
cellular antioxidant capacity, positioning GC as a modulator of
redox signaling beyond mere scavenging activity. Such pleiotropic
regulation could be particularly relevant in clinical settings
involving high residual oxidative stress, such as statin-resistant
or advanced atherosclerotic cases, where monotherapeutic
antioxidant strategies have shown limited success (38). These mechanistic insights
encourage further investigation into GC's translational potential
for preventing oxidative stress-driven cardiovascular events.
The central role of the Nrf2-ARE pathway in
maintaining cellular redox homeostasis provides a compelling
framework for discussing antioxidant-based interventions in AS. As
a key transcriptional regulator, Nrf2 responds to oxidative stress
by translocating to the nucleus and binding to ARE, thereby
orchestrating the expression of cytoprotective genes (39,40). This mechanism enables a
coordinated upregulation of endogenous defenses, which has
attracted interest as a therapeutic strategy in vascular diseases
driven by oxidative injury (41). Within this context, GC appears to
engage the Nrf2-ARE axis, suggesting a capacity to modulate
oxidative signaling beyond simple antioxidant scavenging. By
potentially enhancing multiple defense mechanisms, such as
HO-1-mediated detoxification, NQO1-regulated redox cycling and
glutathione biosynthesis, GC may reinforce antioxidant capacity
across cellular compartments, offering a more integrated approach
to managing redox imbalance in AS.
Such multi-level regulation could be particularly
advantageous in resolving the spatial and compositional
heterogeneity of oxidative stress within atherosclerotic plaques
(42,43). Unlike conventional monofunctional
antioxidants, the potential epigenetic influence of GC on Nrf2
signaling may enable sustained modulation of redox homeostasis,
positioning it as a candidate for addressing complex clinical
scenarios, including statin-resistant cases where persistent
oxidative stress contributes to residual cardiovascular risk. These
considerations underscore the relevance of further exploring GC's
role in targeting the interplay between oxidative stress, lipid
metabolism and inflammation, key drivers of plaque progression and
vulnerability.
The therapeutic strategy of concurrently targeting
the NLRP3 inflammasome and potentiating the Nrf2 pathway represents
a promising direction in AS research, particularly for disrupting
the self-perpetuating cycle of oxidative stress, inflammation and
lipid dysregulation (29,44).
GC's putative dual modulation of these pathways suggests a capacity
to intervene at multiple nodal points within the atherogenic
cascade, not only by potentially mitigating oxidative triggers via
Nrf2-mediated mechanisms but also by suppressing downstream
inflammasome activation and cytokine release. Such an integrated
approach may effectively uncouple the ROS-inflammation-lipid
deposition triad, which is central to plaque progression and
destabilization.
By simultaneously addressing oxidative insult and
inflammatory amplification, GC could offer a therapeutic advantage
over agents that target only single components of this pathogenic
network. This may be especially relevant in advanced
atherosclerotic lesions, where metabolic dysfunction and chronic
inflammation coexist. The ability to stabilize plaque phenotype by
modulating endothelial activation and attenuating leukocyte
recruitment further highlights its potential vascular-protective
role (45). From a clinical
perspective, this bifunctional anti-inflammatory and antioxidant
profile may hold particular value in managing complex patient
subsets, such as those with pyroptosis-prone plaques or inadequate
responses to conventional lipid-lowering therapy, underscoring the
need for further investigation into its translational applicability
(46,47).
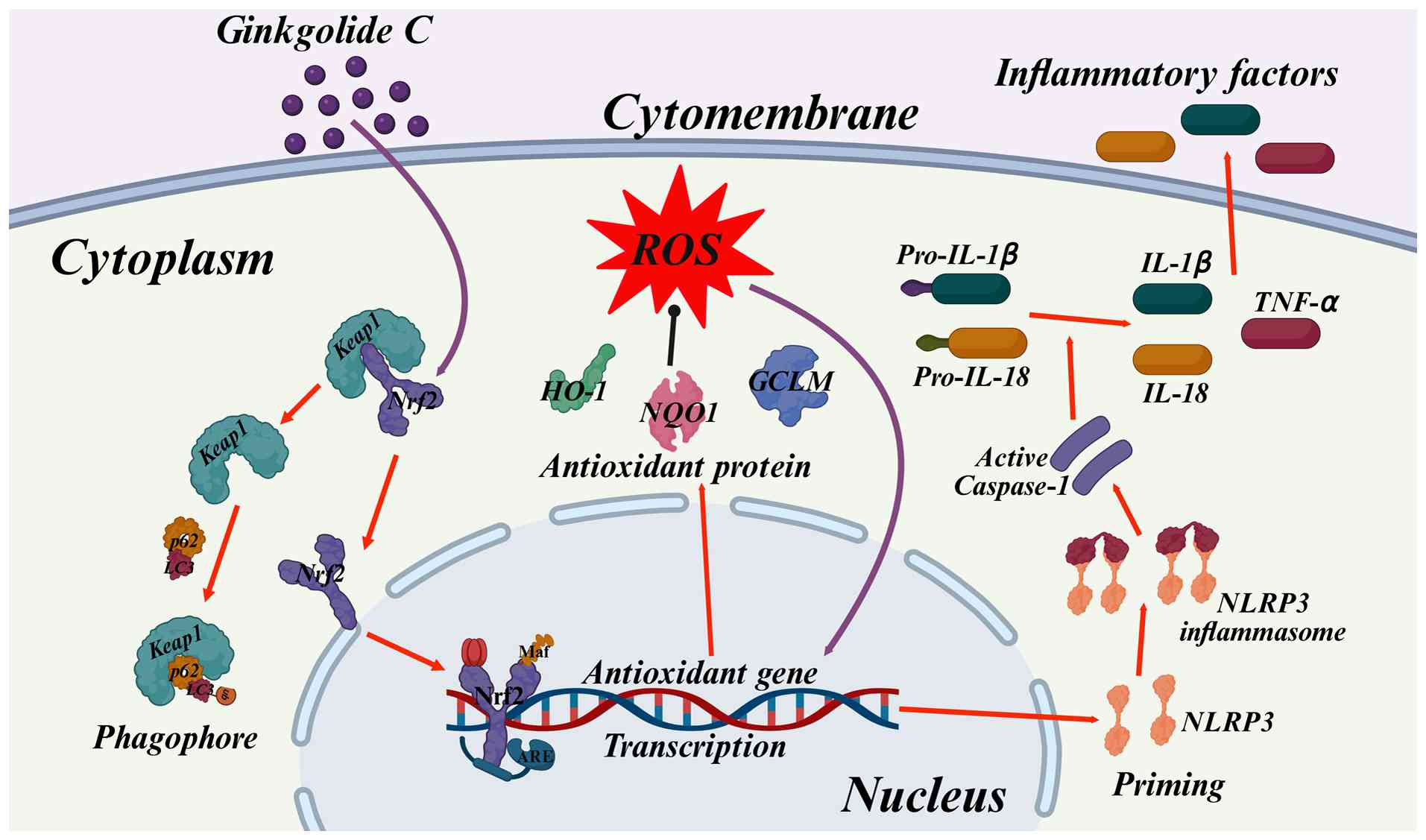
Collectively, this investigation mechanistically
demonstrates GC-mediated cytoprotection against AS through
Nrf2-dependent redox homeostasis modulation, concomitantly
attenuating NLRP3 inflammasome activation and downstream
pro-inflammatory effector signaling (Fig. 9). These findings establish a
preclinical rationale for advancing Ginkgo biloba bioactive
constituents as multi-mechanistic therapeutics in atherosclerotic
CVD management.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
RZ conducted investigation, formal analysis,
conceptualization, developed methodology, acquired funding, curated
and visualized data, wrote, reviewed and edited the manuscript. The
author read and approved the final version of the manuscript. RZ
confirms the authenticity of all the raw data.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Shandong Provincial Hospital Affiliated to
Shandong First Medical University (approval no. 2022-661; Jinan,
China).
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
No funding was received.
References
|
1
|
Boffa MB and Koschinsky ML: Lipoprotein(a)
and cardiovascular disease. Biochem J. 481:1277–1296. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hassan HA, Ahmed HS and Hassan DF: Free
radicals and oxidative stress: Mechanisms and therapeutic targets.
Hum Antibodies. 32:151–167. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aleksandrowicz M, Konop M, Rybka M,
Mazurek Ł, Stradczuk-Mazurek M, Kciuk M, Bądzyńska B, Dobrowolski L
and Kuczeriszka M: Dysfunction of microcirculation in
atherosclerosis: Implications of nitric oxide, oxidative stress,
and inflammation. Int J Mol Sci. 26:64672025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Batty M, Bennett MR and Yu E: The role of
oxidative stress in atherosclerosis. Cells. 11:38432022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jakubowski H and Witucki Ł: Homocysteine
metabolites, endothelial dysfunction, and cardiovascular disease.
Int J Mol Sci. 26:7462025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu L, Liao Y and Jiang B: Role of ROS and
autophagy in the pathological process of atherosclerosis. J Physiol
Biochem. 80:743–756. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Howden R: Nrf2 and cardiovascular defense.
Oxid Med Cell Longev. 2013:1043082013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jomova K, Raptova R, Alomar SY, Alwasel
SH, Nepovimova E, Kuca K and Valko M: Reactive oxygen species,
toxicity, oxidative stress, and antioxidants: Chronic diseases and
aging. Arch Toxicol. 97:2499–2574. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mimura J and Itoh K: Role of Nrf2 in the
pathogenesis of atherosclerosis. Free radical biology &
medicine. 88:221–232. 2015. View Article : Google Scholar
|
|
11
|
O'Rourke SA, Shanley LC and Dunne A: The
Nrf2-HO-1 system and inflammaging. Front Immunol. 15:14570102024.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grebe A, Hoss F and Latz E: NLRP3
Inflammasome and the IL-1 pathway in atherosclerosis. Circ Res.
122:1722–1740. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang K, Liu H, Sun W, Guo J, Jiang Z, Xu S
and Miao Z: Eucalyptol alleviates avermectin exposure-induced
apoptosis and necroptosis of grass carp hepatocytes by regulating
ROS/NLRP3 axis. Aquat Toxicol. 264:1067392023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fu J and Wu H: Structural mechanisms of
NLRP3 inflammasome assembly and activation. Annu Rev Immunol.
41:301–316. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tall AR and Bornfeldt KE: Inflammasomes
and atherosclerosis: A mixed picture. Circ Res. 132:1505–1520.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hort J, Duning T and Hoerr R: Ginkgo
biloba Extract EGb 761 in the treatment of patients with mild
neurocognitive impairment: A systematic review. Neuropsychiatr Dis
Treat. 19:647–660. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Boateng ID: Ginkgols and bilobols in
Ginkgo biloba L. A review of their extraction and bioactivities.
Phytother Res. 37:3211–3223. 2023. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang R, Han D, Li Z, Shen C, Zhang Y, Li
J, Yan G, Li S, Hu B, Li J and Liu P: Corrigendum: Ginkgolide C
alleviates myocardial ischemia/reperfusion-induced inflammatory
injury via inhibition of CD40-NF-κB pathway. Front Pharmacol.
15:14925202024. View Article : Google Scholar
|
|
19
|
Li B, Zhang B, Li Z, Li S, Li J, Wang A,
Hou J, Xu J and Zhang R: Ginkgolide C attenuates cerebral
ischemia/reperfusion-induced inflammatory impairments by
suppressing CD40/NF-κB pathway. J Ethnopharmacol. 312:1165372023.
View Article : Google Scholar
|
|
20
|
Emanueli C, Schmidt AM and Golledge J:
Unveiling intriguing links between limb ischemia, systemic
inflammation, and progressive atherosclerosis: A tangled and
interconnected web. Arterioscler Thromb Vasc Biol. 43:907–909.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tasouli-Drakou V, Ogurek I, Shaikh T,
Ringor M, DiCaro MV and Lei K: Atherosclerosis: A comprehensive
review of molecular factors and mechanisms. Int J Mol Sci.
26:13642025. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang X, Shen Y, Shang M, Liu X and Munn
LL: Endothelial mechanobiology in atherosclerosis. Cardiovasc Res.
119:1656–1675. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang DR, Wang MY, Zhang CL and Wang Y:
Endothelial dysfunction in vascular complications of diabetes: A
comprehensive review of mechanisms and implications. Front
Endocrinol (Lausanne). 15:13592552024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He B, Nie Q, Wang F, Wang X, Zhou Y, Wang
C, Guo J, Fan X, Ye Z, Liu P and Wen J: Hyperuricemia promotes the
progression of atherosclerosis by activating endothelial cell
pyroptosis via the ROS/NLRP3 pathway. J Cell Physiol.
238:1808–1822. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang B, Yu J, Bao L, Feng D, Qin Y, Fan
D, Hong X and Chen Y: Cynarin inhibits microglia-induced pyroptosis
and neuroinflammation via Nrf2/ROS/NLRP3 axis after spinal cord
injury. Inflamm Res. 73:1981–1994. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin J, Sun X and Yang L: Effects and
safety of Ginkgo biloba on depression: A systematic review and
meta-analysis. Front Pharmacol. 15:13640302024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fang WH, Bonavida V, Agrawal DK and
Thankam FG: Hyperlipidemia in tendon injury: Chronicles of
low-density lipoproteins. Cell Tissue Res. 392:431–442. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Silva H and Martins FG: Cardiovascular
activity of Ginkgo biloba-An insight from healthy subjects. Biology
(Basel). 12:152022.
|
|
29
|
Kong P, Cui ZY, Huang XF, Zhang DD, Guo RJ
and Han M: Inflammation and atherosclerosis: Signaling pathways and
therapeutic intervention. Signal Transduct Target Ther. 7:1312022.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Weber C, Habenicht AJR and von
Hundelshausen P: Novel mechanisms and therapeutic targets in
atherosclerosis: Inflammation and beyond. Eur Heart J.
44:2672–2681. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang S, Hong F, Ma C and Yang S: Hepatic
lipid metabolism disorder and atherosclerosis. Endocr Metab Immune
Disord Drug Targets. 22:590–600. 2022. View Article : Google Scholar
|
|
32
|
Lubrano V, Ndreu R and Balzan S: Classes
of lipid mediators and their effects on vascular inflammation in
atherosclerosis. Int J Mol Sci. 24:16372023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bu LL, Yuan HH, Xie LL, Guo MH, Liao DF
and Zheng XL: New dawn for atherosclerosis: Vascular endothelial
cell senescence and death. Int J Mol Sci. 24:151602023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pintó X, Fanlo M, Esteve V and Millán J:
Remnant cholesterol, vascular risk, and prevention of
atherosclerosis. Clin Investig Arterioscler. 35:206–217.
2023.PubMed/NCBI
|
|
35
|
Gaggini M, Gorini F and Vassalle C: Lipids
in atherosclerosis: Pathophysiology and the role of calculated
lipid indices in assessing cardiovascular risk in patients with
hyperlipidemia. Int J Mol Sci. 24:752022. View Article : Google Scholar
|
|
36
|
Dabravolski SA, Churov AV, Beloyartsev DF,
Kovyanova TI, Lyapina IN, Sukhorukov VN and Orekhov AN: The role of
NRF2 function and regulation in atherosclerosis: An update. Mol
Cell Biochem. 480:3935–3949. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jomova K, Alomar SY, Valko R, Liska J,
Nepovimova E, Kuca K and Valko M: Flavonoids and their role in
oxidative stress, inflammation, and human diseases. Chem Biol
Interact. 413:1114892025. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yan Q, Liu S, Sun Y, Chen C, Yang S, Lin
M, Long J, Yao J, Lin Y, Yi F, et al: Targeting oxidative stress as
a preventive and therapeutic approach for cardiovascular disease. J
Transl Med. 21:5192023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Z, Dai Y, Xiao Y and Liu Q:
Protective effects of catalpol on cardio-cerebrovascular diseases:
A comprehensive review. J Pharm Anal. 13:1089–1101. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Han H, Zhang G, Zhang X and Zhao Q:
Nrf2-mediated ferroptosis inhibition: A novel approach for managing
inflammatory diseases. Inflammopharmacology. 32:2961–2986. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Meng T, Li X, Li C, Liu J, Chang H, Jiang
N, Li J, Zhou Y and Liu Z: Natural products of traditional Chinese
medicine treat atherosclerosis by regulating inflammatory and
oxidative stress pathways. Front Pharmacol. 13:9975982022.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang Z, Wu M, Zeng L and Wang D: The
beneficial role of Nrf2 in the endothelial dysfunction of
atherosclerosis. Cardiol Res Pract. 2022:42877112022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pfefferlé M and Vallelian F: Transcription
factor NRF2 in shaping myeloid cell differentiation and function.
Adv Exp Med Biol. 1459:159–195. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang XN, Yu ZL, Chen JY, Li XY, Wang ZP,
Wu M and Liu LT: The crosstalk between NLRP3 inflammasome and gut
microbiome in atherosclerosis. Pharmacol Res. 181:1062892022.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tanase DM, Valasciuc E, Gosav EM, Ouatu A,
Buliga-Finis ON, Floria M, Maranduca MA and Serban IL: Portrayal of
NLRP3 inflammasome in atherosclerosis: Current knowledge and
therapeutic targets. Int J Mol Sci. 24:81622023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang H and Dhalla NS: The role of
Pro-inflammatory cytokines in the pathogenesis of cardiovascular
disease. Int J Mol Sci. 25:10822024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Asemi R, Omidi Najafabadi E, Mahmoudian Z,
Reiter RJ, Mansournia MA and Asemi Z: Melatonin as a treatment for
atherosclerosis: Focus on programmed cell death, inflammation and
oxidative stress. J Cardiothorac Surg. 20:1942025. View Article : Google Scholar : PubMed/NCBI
|