Sepsis is a life-threatening syndrome resulting from
a dysregulated host response to infection, which can progress to
multiple organ dysfunction syndrome with a mortality rate
approaching 40% (1,2). Immune homeostasis plays a pivotal
role in the complex pathophysiology of sepsis and is directly
linked to clinical outcomes. Sepsis disrupts immune balance,
leading to a state of immunosuppression. The mechanisms underlying
this immune dysregulation are multifactorial, including excessive
anti-inflammatory cytokine release, abnormal apoptosis of immune
effector cells, over-proliferation of immunosuppressive cells and
upregulation of immune checkpoint molecules (3). Sepsis inflicts extensive damage to
multiple organs, including the kidneys, liver, lungs, circulatory
system, gastrointestinal tract, blood and central nervous system.
Among these, the kidneys are often the first organs affected during
sepsis (4). Acute kidney injury
(AKI), one of the most common and severe complications of sepsis,
occurs in >50% of sepsis cases and is associated with
significantly increased mortality (5). In sepsis, the kidneys undergo a
series of alterations, including hemodynamic changes,
microcirculatory dysfunction, endothelial injury, inflammatory
responses, oxidative stress and direct tubular damage (6). Furthermore, immune responses driven
by various immune cells, such as macrophages, neutrophils,
dendritic cells (DCs), natural killer (NK) cells, natural killer T
(NKT) cells, B cells and T cells, play a pivotal role in the
development and progression of sepsis-associated AKI (SA-AKI)
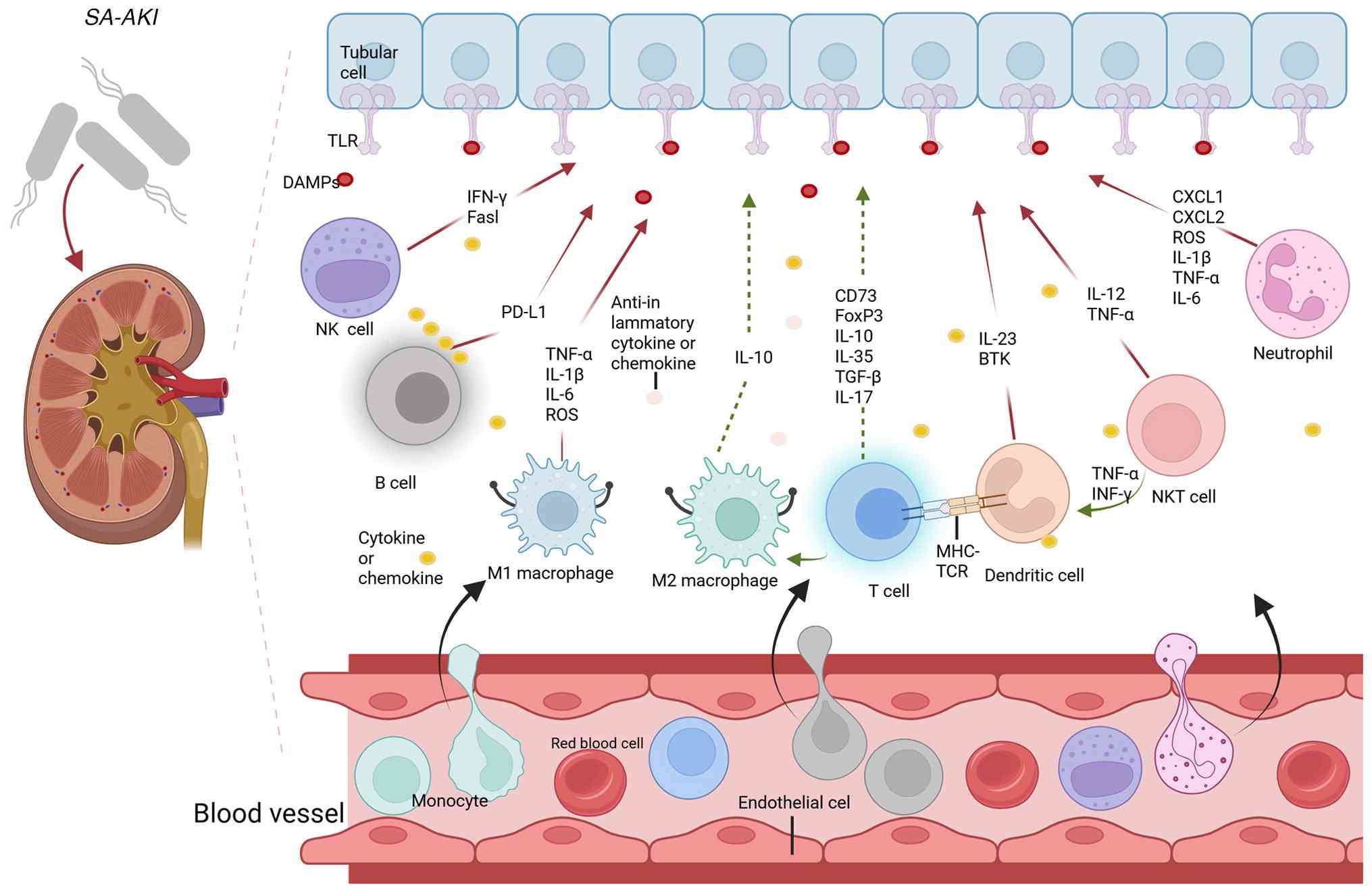
(Fig. 1). Damage-associated
molecular patterns (DAMPs) are recognized by Toll-like receptors
(TLRs), activating immune cells and initiating an inflammatory
cascade (7). Upon sensing damage
signals, innate immune cells not only help control tissue injury
but also initiate adaptive immune responses. These immune cells
synergistically contribute to the onset, progression and
persistence of disease. In sepsis, immune cell roles differ between
the early hyperinflammatory phase and the later immunosuppressive
phase. The early phase is characterized by a pro-inflammatory
response marked by a cytokine storm. Following this excessive
inflammation, patients either gradually recover or transition into
a state of persistent immunosuppression, characterized by immune
cell exhaustion (8-16); key features of this progression
are summarized in Table I.
However, the roles of mast cells and innate lymphoid cells (ILCs)
in SA-AKI remain insufficiently studied, and their exact mechanisms
are still unclear (Fig. 1).
Therefore, precise targeting of immune cells may offer a promising
strategy for preventing and treating SA-AKI.
Under physiological conditions, the kidneys harbor a
notable population of resident macrophages from birth. These
macrophages primarily include those derived from yolk sac erythroid
progenitors, fetal liver erythroid progenitors and hematopoietic
stem cells (17). During early
embryogenesis, yolk sac-derived macrophages play a predominant role
(18). In adulthood, macrophage
populations are sustained through two distinct mechanisms: The
self-renewal of fetal liver erythroid progenitor-derived and
hematopoietic stem cell-derived macrophages and the replacement of
macrophages by monocyte-derived cells (17). Kidney resident macrophages (KRMs)
and recruited macrophages perform distinct functions within the
kidney. In the context of SA-AKI, macrophages play a pivotal role
in driving both injury and repair through a complex interplay of
polarization, metabolic reprogramming and signaling pathways, as
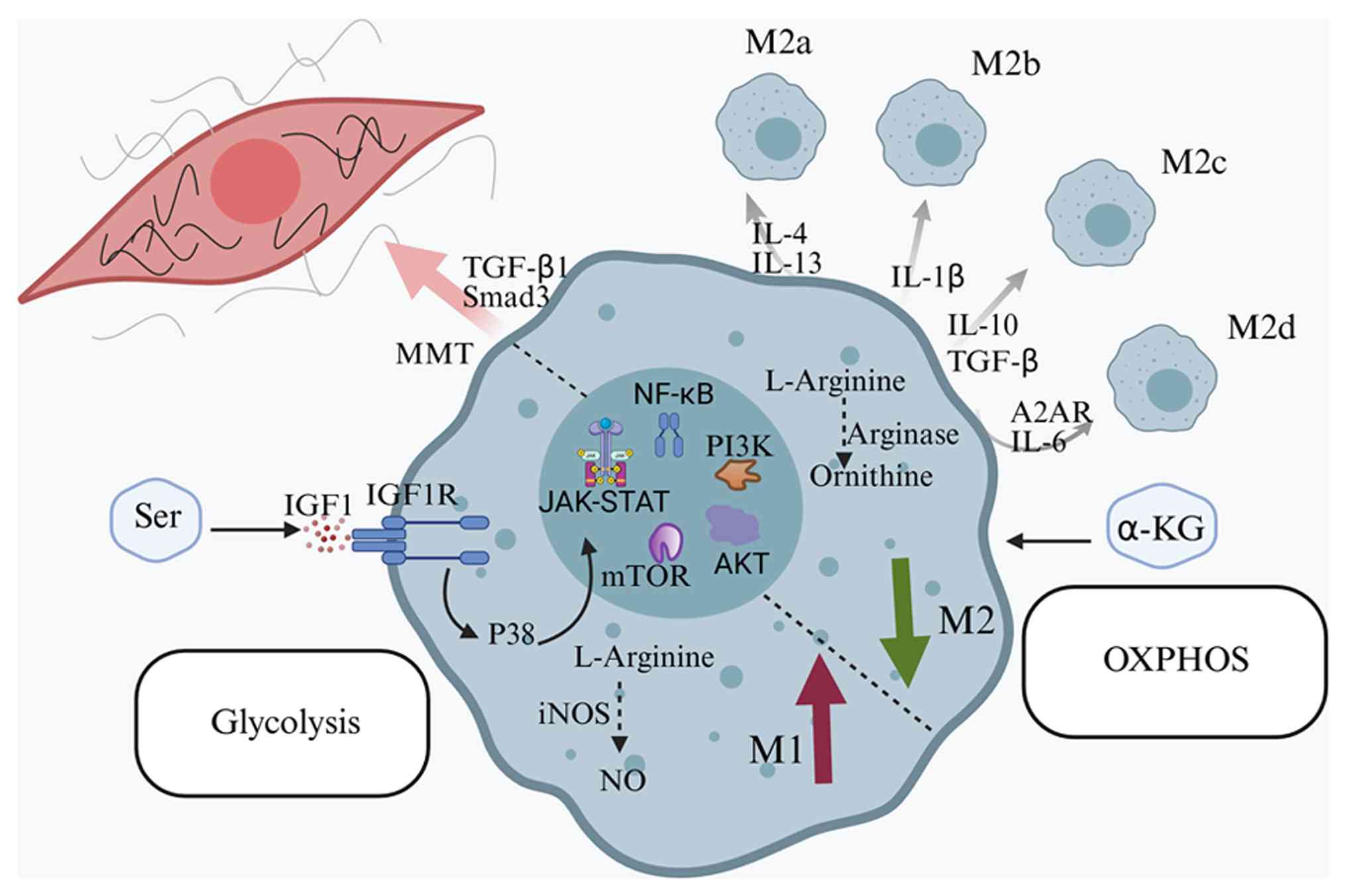
summarized in Fig. 2.
As tissue-resident immune cells, KRMs are essential
for maintaining normal kidney function. These macrophages are
complex, highly adaptable renal resident mononuclear phagocytes
with diverse functions, which originate from the yolk sac during
early embryogenesis and persist in the kidney throughout
development (19,20). KRMs help maintain systemic
homeostasis through various mechanisms, such as clearing cellular
debris, modulating local inflammation and promoting tissue repair
(21). In adult tissues, KRMs
possess self-renewal capabilities with minimal input from
peripheral blood, helping sustain renal homeostasis, monitor the
immune microenvironment, promote angiogenesis and mitigate AKI.
Following kidney injury, KRMs aid in tissue repair and regeneration
(22). Additionally, KRMs can
reduce renal injury by selectively inhibiting interleukin (IL)-6
production from endothelial cells through IL-1 receptor antagonist
expression (23). Furthermore,
KRMs regulate renal sympathetic nerve activity, influencing salt
and water balance and further promoting renal function recovery
(24).
In SA-AKI, monocyte recruitment is a critical
pathophysiological event. This process involves multiple mechanisms
that synergistically contribute to uncontrolled inflammation and
exacerbated renal injury. These mechanisms include the release of
chemoattractants, expression of adhesion molecules, activation of
inflammatory cascades and microcirculatory dysfunction (25). During sepsis, renal tubular
epithelial cells upregulate the expression of C-C motif ligand
chemokine 2 (CCL2) at both the mRNA and protein levels through the
combined actions of chemokine receptors CCR2 and C-X3-C motif
chemokine receptor 1 (CX3CR1); this upregulation drives monocyte
chemotaxis (26). Moreover, the
endothelial glycocalyx, produced by vascular endothelial cells,
undergoes degradation during sepsis due to inflammatory responses
and oxidative stress. The glycocalyx, a critical component of the
vascular endothelial barrier, when damaged, increases vascular
permeability, facilitating the contact and adhesion of monocytes to
endothelial cells (27).
Monocytes infiltrating the kidney can differentiate into M1 or M2
macrophages, exerting pro-inflammatory or anti-inflammatory
effects, respectively. Notably, a specific monocyte subset, such as
Ly6Chigh monocytes, strongly adheres to the renal vascular wall in
a CX3CR1-dependent manner during the early stages of sepsis. This
subset plays a protective role via CX3CR1-dependent adhesion
mechanisms (28).
Sepsis is primarily induced by lipopolysaccharide
(LPS) released from Gram-negative bacteria. LPS binds to TLRs on
renal cells, triggering the release of DAMPs (29). These molecules act as endogenous
'danger signals', recognized by receptors on macrophages, leading
to their activation. This activation results in the extensive
release of inflammatory cytokines, such as tumor necrosis factor-α
(TNF-α), IL-1β, IL-6 and the chemoattractant CCL2, initiating a
cytokine storm and exacerbating renal inflammatory injury (30,31). Macrophages exhibit high
plasticity and can polarize into distinct phenotypes, typically
classified as M1 and M2. M1 macrophages primarily contribute to
inflammation by releasing pro-inflammatory cytokines (such as
TNF-α, IL-1β and IL-6) and reactive oxygen species (ROS), driving
inflammation and tissue damage. By contrast, M2 macrophages are
involved in tissue repair and inflammation resolution (32). Thus, modulation of macrophage
polarization represents a potential strategy for alleviating renal
injury in SA-AKI. Macrophage polarization is a dynamic process and
excessive suppression of M1 polarization may impair pathogen
clearance, hindering tissue healing. Conversely, overactivation of
M2 macrophages can lead to immunosuppression, increasing infection
risk (33,34). Therefore, precise regulation of
the M1/M2 balance is critical to avoid adverse outcomes.
Macrophage metabolic reprogramming plays a pivotal
role in SA-AKI. During sepsis, macrophages undergo significant
metabolic changes to support the inflammatory response and cellular
survival (35). This
reprogramming involves a shift from fatty acid oxidation (FAO) to a
dual reliance on glycolysis and FAO. M1 macrophages predominantly
utilize aerobic glycolysis to rapidly initiate immune responses,
while M2 macrophages rely on oxidative phosphorylation (OXPHOS) to
exert anti-inflammatory effects and prevent tissue damage (36). In the early stages of sepsis,
macrophages activate the AKT/mammalian target of rapamycin
(mTOR)/hypoxia-inducible factor-1α signaling pathway, which
enhances glycolytic enzyme activity and suppresses the
tricarboxylic acid (TCA) cycle, initiating glycolysis to generate
ATP, supporting cell survival and pro-inflammatory responses
(35). However, excessive
glycolysis can induce immunosuppression, impairing host defense and
immune function. This immunosuppressive state is characterized by
pro-inflammatory cytokines (such as TNF-α, IL-6 and CCL2) and
T-cell-recruiting chemoattractants, as well as anti-inflammatory
cytokines (such as IL-4, IL-10) (37). Nevertheless, this metabolic shift
can impair mitochondrial function and reduce ATP production.
Limiting glycolysis can mitigate the inflammatory response
(38,39). While the transition from OXPHOS
to aerobic glycolysis is essential for renal survival in early
sepsis, failure to restore OXPHOS in the later stages may result in
persistent inflammation and renal fibrosis (35). During the late inflammatory
phase, cells activate peroxisome proliferator-activated receptor-γ
coactivator 1α and carnitine palmitoyltransferase 1 through
mediators such as sirtuin (Sirt)6, signal transducer and activator
of transcription 3 (STAT3) and sirtuin 1, promoting a metabolic
shift back to OXPHOS. Restoration of OXPHOS stimulates
mitochondrial biogenesis, generates ATP and exerts
anti-inflammatory effects, promoting renal function recovery
(35). Additionally, lactate, a
key metabolite in macrophage metabolism, influences the macrophage
phenotype through lactylation, further impacting disease
progression (40).
Amino acid metabolism, which regulates macrophage
activation, is one of the earliest metabolic alterations during
macrophage polarization. Arginine, glutamine and serine metabolism
play pivotal roles in this process (41). Arginine catabolism regulates
macrophage activation primarily through inducible nitric oxide
synthase (iNOS) and arginase 1 (Arg1). iNOS, predominantly
expressed in M1 macrophages, catalyzes the synthesis of nitric
oxide and L-citrulline from arginine. Nitric oxide, a ROS, helps M1
macrophages combat pathogen invasion (40-42). By contrast, Arg1, highly
expressed in M2 macrophages, catalyzes the hydrolysis of arginine
into urea and L-ornithine. The metabolites of L-ornithine,
including polyamines and proline, influence macrophage
proliferation and collagen synthesis (42-44). A distinctive feature of M2
macrophages is their increased glutamine metabolism, with one-third
of the carbon in TCA cycle metabolites of M2 macrophages derived
from glutamine. Glutaminolysis generates α-ketoglutarate (α-KG),
which enhances M2 macrophage activation and governs metabolic
reprogramming through Jmjd3-dependent regulation. Additionally,
α-KG, via a prolyl hydroxylase-dependent mechanism, inhibits the
NF-κB pathway, thereby limiting M1 macrophage activation and
modulating IKKβ activity (45).
Serine metabolism also plays a pivotal role in macrophage
functional polarization. It has been shown that serine metabolism
suppresses insulin-like growth factor-1 (IGF1) transcription by
increasing the promoter abundance of H3K27me3 through
S-adenosylmethionine. Deficiency in serine metabolism alters M1
macrophage polarization and modulates Janus kinase (JAK)/STAT1
signaling via IGF1-dependent p38 activation (46).
In conclusion, during SA-AKI, macrophage metabolic
reprogramming may be a critical component of the host defense
response. However, specific regulatory strategies require further
investigation to precisely modulate macrophage metabolism for
therapeutic purposes.
NF-κB is a dimeric transcription factor composed of
five protein family members and is present in nearly all human
cells (47); it plays a critical
role in inflammation and immunity by regulating the expression of a
broad range of chemoattractants, cytokines, transcription factors
and regulatory proteins (47).
The binding of LPS to its receptor, TLR4, on macrophages activates
the myeloid differentiation primary response 88 (MyD88)-dependent
signaling pathway. This pathway activates the IKK complex, which
phosphorylates IκB proteins, leading to their ubiquitination and
proteasomal degradation. Degradation of IκB releases NF-κB dimers,
such as p50/RelA (p65), allowing their translocation to the nucleus
(48). Within the nucleus, NF-κB
binds to specific κB sites on DNA via its Rel homology domain,
promoting the transcription of inflammatory genes, including TNF-α
and IL-1β, thereby amplifying the inflammatory response (48). The NF-κB pathway interacts
extensively with other signaling pathways. For instance, the
PI3K/AKT/NF-κB signaling pathway plays a pivotal role in regulating
macrophage polarization (49).
Moreover, complex regulatory interactions exist between NF-κB and
pathways such as the MAPK and STAT pathways.
The PI3K/AKT/mTOR signaling pathway is a critical
cascade that governs various cellular processes, including growth,
proliferation, survival, metabolism and migration (50). A study has shown that LPS-induced
activation of TLR4 in macrophages triggers the PI3K/AKT/mTOR
pathway, leading to the production of muscle-type pyruvate kinase
isozyme, which then facilitates the acetylation of high mobility
group box 1 (HMGB1). Acetylated HMGB1 promotes LPS uptake by
macrophages and inhibits macrophage apoptosis (51). This pathway is also essential for
regulating macrophage polarization towards M1 or M2 phenotypes.
Androulidaki et al (52)
demonstrated that AKT1 modulates macrophage polarization upon LPS
stimulation. AKT1 deficiency promotes M1 polarization, while AKT2
deficiency favors M2 polarization. For instance, Aquaporin 1 has
been shown to mitigate SA-AKI by promoting M2 polarization via the
PI3K/AKT pathway (53,54).
The JAK/STAT pathway is a key mechanism in cell
communication, enabling cells to respond to external stimuli
(55); it plays a critical role
in various physiological and pathological processes, including cell
proliferation, metabolism, immune response and inflammation
(55). During sepsis, the
release of large quantities of pro-inflammatory cytokines (such as
TNF-α, IL-6 and IL-1β) activates the JAK/STAT signaling pathway.
Activated STAT proteins translocate to the nucleus, driving the
transcription of additional inflammatory mediators, thus amplifying
the inflammatory response and contributing to kidney damage
(56,57). STAT inhibitors can effectively
reduce inflammation by restoring the CD11blowF4/80high macrophage
population, which exhibits potential anti-inflammatory properties,
thus protecting the kidneys from injury (58). For example, curcumin has been
demonstrated to alleviate inflammation and apoptosis by modulating
the JAK2/STAT3 pathway, mitigating SA-AKI (59).
The JAK/STAT pathway also regulates immune cell
function. In sepsis, this pathway influences macrophage activation
and differentiation, enabling macrophages to produce inflammatory
cytokines through receptor-interacting serine/threonine-protein
kinase and the NLRP3 inflammasome, thus impacting the intensity and
duration of the immune response (51,56). For instance, mesenchymal stem
cells (MSCs) with upregulated heme oxygenase-1 expression can
ameliorate SA-AKI by activating the JAK/STAT3 pathway (60). Moreover, blocking TLR4/TNF1 can
promote a phenotypic shift of M1 macrophages towards the M2
phenotype by inhibiting STAT1/STAT3 expression and increasing
Suppressor of cytokine signaling 3 expression (61). Acyl-CoA thioesterase 11, an
IL-1β-associated gene involved in fatty acid metabolism, promotes
the accumulation of fatty acids, such as eicosatetraenoic acid (EA)
and stearic acid, within macrophages (62). This accumulation inhibits
JAK/STAT signaling activation via palmitoylation of interferon-γ
(IFN-γ) receptor 2 at the C261 site (62). Eicosatetraenoic acid has also
been shown to alleviate sepsis-associated organ damage via the same
pathway (62). Additionally,
macrophages expressing the chemokine CCL5 activate several pathways
related to immune and inflammatory responses, including
IL-6/JAK/STAT3 signaling, TGF-β signaling and inflammatory
responses, recruiting neutrophils and exacerbating SA-AKI (63). In summary, the JAK/STAT signaling
pathway plays a complex role in the pathophysiology of SA-AKI. By
regulating inflammatory responses, immune modulation and apoptosis,
this pathway contributes to the initiation and progression of renal
injury. Thus, targeting the JAK/STAT pathway may offer novel
therapeutic strategies for SA-AKI.
Accumulative evidence suggests that the M1/M2
dichotomy does not fully encompass the complex behaviors and
functions of macrophages in vivo (26,64,65). Macrophages may exhibit multiple
phenotypes and even co-express both M1 and M2 markers
simultaneously. For example, macrophages can display mixed M1/M2
phenotypes, which are essential for regulating the assembly and
structure of the extracellular matrix (66). Additionally, M2 macrophages
represent not a single phenotype but several subtypes, including
M2a, M2b, M2c and M2d, each characterized by distinct surface
markers, cytokine secretion profiles and immune effector functions
(67,68). M2a and M2b macrophages are
particularly prevalent in renal tissue. M2a macrophages, typically
induced by IL-4 or IL-13, express high levels of Arg1 and the
mannose receptor and are primarily involved in wound healing,
fibrosis and allergic responses (67). M2b macrophages, stimulated by
immune complexes or IL-1β, exhibit immunomodulatory and
anti-inflammatory effects. These cells produce anti-inflammatory
cytokines such as IL-10, while also secreting pro-inflammatory
cytokines such as IL-1β and TNF-α, thereby participating in immune
complex-mediated diseases (69).
In the later stages of sepsis, promoting the M2b macrophage
phenotype may help maintain immune homeostasis and facilitate
tissue healing. However, the activity of M2b macrophages must be
precisely regulated to prevent exacerbation of inflammation or
suppression of immune responses. M2c macrophages, induced by IL-10
or TGF-β, are primarily involved in immunosuppression and tissue
remodeling. They express surface markers such as CD163 and signal
regulatory protein α (SIRPα) (67,68). M2d macrophages, induced by
adenosine A2A receptor agonists or IL-6, play a major role in tumor
angiogenesis and immunosuppression (67). Future research should focus on
investigating M2 macrophage subtypes and their specific functions
in SA-AKI, providing a theoretical foundation for more targeted
therapies.
Fibrosis, a common pathological feature of chronic
disorders, is characterized by excessive activation of
myofibroblasts and abnormal deposition of extracellular matrix in
tissues (70). Macrophages can
directly transdifferentiate into myofibroblasts via MMT, promoting
renal fibrosis (71,72). Recently, MMT has been recognized
as a novel source of myofibroblasts and plays a critical role in
fibrotic processes across multiple organs. The regulatory
mechanisms underlying MMT remain incompletely understood, but the
TGF-β1/Smad3 signaling cascade is the most extensively studied
pathway (73,74). Although no direct evidence
currently links MMT to SA-AKI, a study has confirmed increased
expression of TGF-β R2 and phosphorylated Smad3 in the kidneys of
septic mice (75). Given the
critical role of the TGF-β1/Smad3 pathway in MMT, it is
hypothesized that MMT may contribute to the development and
progression of renal fibrosis following SA-AKI. Therapeutic
strategies targeting MMT hold promise for improving the long-term
prognosis of SA-AKI, although further experimental validation is
needed.
Catalytic nanoparticles offer a promising
alternative to conventional anti-infective therapies by modulating
innate immune responses and engineering macrophages for
immunotherapy, thus providing innovative treatment options for
infectious diseases such as sepsis. For example,
ultrasound-responsive piezoelectric catalytic nanoparticles enhance
macrophage-mediated antibacterial phagocytosis and bactericidal
activity through intracellular piezoelectric catalysis (76). Bone marrow-derived macrophages
co-cultured with these piezoelectric nanoparticles form
piezoelectric macrophages, which, upon activation via ultrasound
irradiation, can be utilized in adoptive cell therapy. This
approach has proven effective against immunosuppressive bacterial
infections, including sepsis (76). However, challenges remain in
targeting these piezoelectric nanomaterials to macrophages and
kidneys, as they are typically non-degradable and considered
biologically inert, necessitating further investigation into their
long-term effects and biosafety. Another novel nanoparticle
modulates metabolic reprogramming in macrophages to mitigate the
septic cytokine storm (77).
Additionally, engineered apoptotic extracellular vesicles derived
from macrophages can alleviate symptoms in septic patients by
clearing toxins and regulating inflammatory responses (78). Nicotinamide adenine dinucleotide
(NAD+), an immunomodulator, shows promise for sepsis
treatment. Co-incubation of LPS-activated macrophages with
NAD+-loaded lipid nanoparticles not only reduces the
production of pro-inflammatory cytokines and ROS by macrophages but
also enhances their viability (79). Moreover, nanoparticles formed by
coordination between Ce3+ and astragalin (CeAst
nanoparticles), encapsulated with macrophage membranes and modified
with a kidney-targeting peptide, form the CeAst@MK system. This
nanoplatform enables precise kidney targeting and promotes M2
macrophage polarization (80).
The CeAst@MK system exhibits excellent biocompatibility and in
vivo safety, indicating notable potential for clinical
translation. A novel system of mannose-conjugated, PEGylated
graphene oxide nanoparticles loaded with a sesquiterpene lactone
(GMO@ GO@PEG@MAN) not only targets the kidneys but also binds to
macrophages and promotes the transition from M1 to M2 macrophages
(81). Given the promising
outcomes of these macrophage-based therapies in sepsis treatment,
they hold notable potential for managing SA-AKI. Ongoing
optimization of therapeutic strategies, deeper exploration of
underlying mechanisms and enhanced clinical translational research
are expected to improve the prognosis of patients with SA-AKI. As
an emerging therapeutic approach, macrophage-based therapy shows
broad potential in SA-AKI management. Through continuous refinement
of treatment strategies and further investigation into its
mechanisms, new avenues for improving patient outcomes in SA-AKI
are anticipated.
Neutrophils are a critical component of the first
line of defense against invading microorganisms, playing a key role
in infection control and the mediation of inflammatory responses
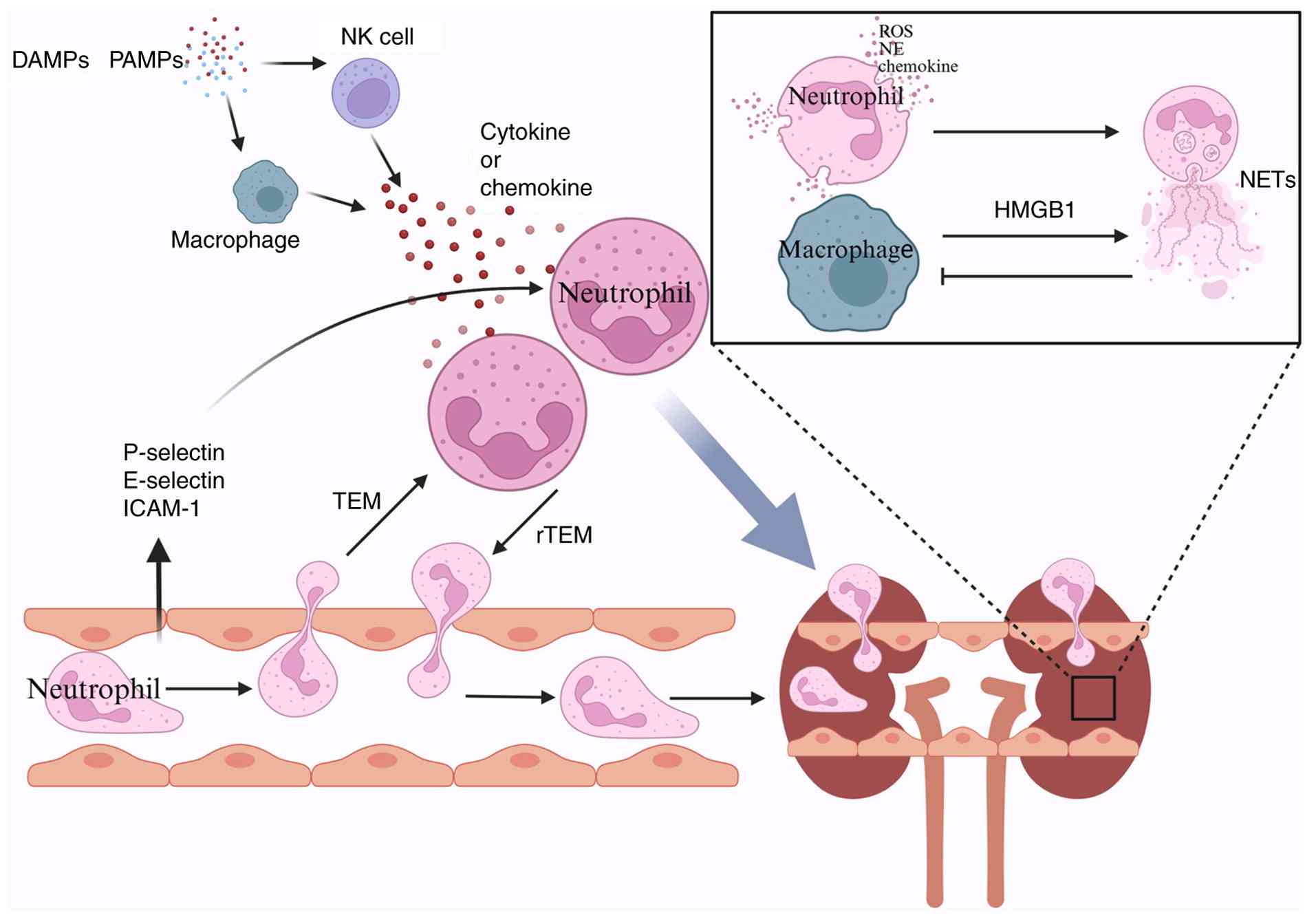
(Fig. 3). Neutrophils constitute
~70% of the total white blood cell count in circulation, making
them the most abundant type of leukocyte. However, under normal
physiological conditions, the number of neutrophils in the kidney
is relatively low. Neutrophils primarily function in immune
surveillance, clearing small quantities of pathogens or cellular
debris that enter the kidney (82); they are typically maintained in a
non-activated state to prevent unnecessary damage to renal tissues
(82). In sepsis, excessive
neutrophil activation can lead to an uncontrolled inflammatory
response. Analysis of the large MIMIC-IV database has shown that an
elevated neutrophil-to-lymphocyte-to-platelet ratio is strongly
associated with an increased risk and severity of SA-AKI (83), indicating a notable role for
neutrophils in the pathophysiology of SA-AKI. Similar to
macrophages, neutrophils display phenotypic heterogeneity, with N1
neutrophils being pro-inflammatory and N2 neutrophils being
anti-inflammatory (84).
Neutrophils isolated from the kidneys of cecal ligation and
puncture (CLP) mice exhibit a CD11bhigh, CD54high and CD95high
profile, indicating an N1 phenotype (85,86). Upon LPS stimulation, CD54
expression on neutrophils increases, which correlates with enhanced
phagocytosis and ROS production (87).
During SA-AKI, significant neutrophil recruitment
and infiltration occur in the kidneys. Sepsis triggers a systemic
inflammatory response, leading to endothelial cell activation and
upregulation of various adhesion molecules, such as P-selectin,
E-selectin and intercellular adhesion molecule-1 (88). These adhesion molecules interact
with ligands on the neutrophil surface, promoting neutrophil
adhesion from the circulation to the renal vascular endothelium and
their subsequent migration into renal tissue (88). Additionally, one study indicated
that the release of DAMPs and pathogen-associated molecular
patterns activates monocytes/macrophages and DCs, leading to the
secretion of pro-inflammatory cytokines and chemokines, which
initiate neutrophil recruitment into renal tissue (89-91). Other research has highlighted a
sharp increase in classical pro-inflammatory chemokines and
cytokines, such as C-X-C motif chemokine ligand (CXCL)1 and CXCL2,
during SA-AKI (88,89). Neutrophils follow the CXCL1
and/or CXCL2 gradient to the sites of infection and uncontrolled
levels of these chemokines may enhance tissue damage by
over-activating neutrophils (92,93). Once recruited to the kidneys,
neutrophils primarily accumulate around microvessels, with
subsequent infiltration into the renal tubulointerstitium (94). Upon tissue infiltration,
neutrophils are further activated, releasing inflammatory mediators
such as ROS, proteases (such as neutrophil elastase) and cytokines
(95). Excessive ROS production
plays a key role in sepsis and SA-AKI, causing direct damage to
renal tubular epithelial cells through lipid peroxidation, protein
denaturation and DNA damage (96). Neutrophil elastase degrades the
extracellular matrix, disrupting the structural integrity of renal
tissue (97). In contrast to
most studies that rely on reducing systemic neutrophil counts or
inflammatory cytokines, one study demonstrated that renal injury
during systemic inflammation was not significantly alleviated after
specifically targeting neutrophil recruitment into the kidney. This
finding suggests that the uncontrolled systemic inflammatory
response, rather than local neutrophil recruitment into the kidney,
dominates the pathogenesis of kidney injury in SA-AKI. However, the
authors of this study also acknowledged the limitation that the
specific mechanisms by which individual circulating proinflammatory
mediators contribute to renal injury remain to be elucidated
(98). This study likely focuses
on the role of circulating inflammatory mediators in the early
stages of the disease, whereas other studies emphasize the local
effects of neutrophils in the kidneys during disease progression.
Moreover, neutrophils recruited to sites of inflammation often
undergo apoptosis. Insufficient clearance of apoptotic neutrophils
can result in the persistence and worsening of inflammation
(99). In patients with SA-AKI,
peripheral blood mononuclear cells express high levels of the
anti-phagocytic signal, involving leukocyte surface antigen CD47
and SIRPα. This signal facilitates the clearance of apoptotic
neutrophils, promoting the resolution of inflammation and repair of
tissue injury (100).
Neutrophil metabolic reprogramming refers to the
adjustment of metabolic pathways in response to physiological or
pathological conditions. Typically, neutrophils utilize multiple
metabolic pathways, including glycolysis (101), the pentose phosphate pathway
(PPP) and FAO, to support immune functions such as chemotaxis, ROS
production, neutrophil extracellular trap (NET) formation and
degranulation (102). Under
normal conditions, glycolysis serves as the primary energy source.
However, in inflammatory or tumor microenvironments, neutrophils
may shift to alternative metabolic pathways, such as FAO, to
sustain effector functions. A study has shown that during sepsis,
levels of glycolysis, PPP and fatty acid amide hydrolase are
elevated in neutrophils (103).
2-deoxy-D-glucose alleviates excessive inflammatory responses by
enhancing neutrophil recruitment, highlighting the link between
metabolic regulation and infection/inflammation (104). Furthermore, during the acute
phase of sepsis, the long non-coding RNA GSEC (containing a
G-quadruplex sequence) is upregulated in sepsis-induced
neutrophils, enhancing the transcription and translation of
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 mRNA, which
promotes neutrophil glycolysis and the production of inflammatory
cytokines such as TNF-α, IL-1β and IL-6 (35). Thus, neutrophil metabolic
reprogramming in SA-AKI likely contributes to the increased release
of ROS, NETs and other factors, exacerbating disease progression.
This process represents an interesting area for future
investigation in SA-AKI.
Additionally, NETs play a notable role in the
development of SA-AKI. NETs are web-like structures composed of
DNA, histones and antimicrobial proteins, released by activated
neutrophils (105). The
formation of NETs is regulated by the pore-forming protein
gasdermin D and depends on NADPH oxidase and neutrophil elastase
activity (106). NETs can
suppress macrophage phagocytosis, leading to persistent
inflammation and tissue damage. Inhibiting NET formation may
alleviate SA-AKI by restoring the infiltration and survival of
GAS6+ macrophages, promoting the clearance of damaged
cells and the resolution of inflammation (107). However, while NETs provide a
fibrous scaffold to entangle bacteria or pathogens, excessive
activation of NETs can worsen inflammation and tissue injury
(108). Furthermore, prolonged
disturbances in the pro-/anti-inflammatory balance lead to
pathogenic NET formation, which is a hallmark of systemic
inflammatory response syndrome (107). NETs can exacerbate kidney
damage by releasing cytokines and histones. Histones within NETs
directly damage renal tubular epithelial cells (109), while the DNA component can
activate the complement system (110), thereby intensifying the
inflammatory response. Notably, defective NET formation has been
shown to be protective in SA-AKI models (111).
Neutrophils do not remain fixed at the site of
inflammation; they can exit via a process known as rTEM (112,113), in which neutrophils move back
across the endothelium from the interstitial space into the
bloodstream. This re-migration occurs through interactions between
neutrophil surface molecules, such as β2 integrins, and
corresponding ligands on vascular endothelial cells, with
regulation by various cytokines and chemoattractants. rTEM plays a
dual role: On the one hand, it may help reduce local inflammatory
damage by decreasing neutrophil numbers in the affected tissue
(114), while on the other
hand, activated neutrophils re-entering the circulation can carry
inflammatory mediators, releasing them into the bloodstream and
potentially causing remote organ damage (115). For example, one study showed
that extracellular vesicles derived from endothelial cells
exacerbate remote lung injury by promoting rTEM (116). It is plausible that rTEM also
contributes significantly to SA-AKI, warranting further
investigation.
Additionally, neutrophil gelatinase-associated
lipocalin (NGAL) is crucial in the pathogenesis and progression of
SA-AKI. NGAL functions as a notable renal growth factor,
facilitating the differentiation of renal progenitor cells into
renal tubular epithelial cells (117). During the initial stages of
AKI, NGAL is upregulated in a compensatory manner to promote
tubular repair and regeneration (117). NGAL, primarily secreted by
neutrophils, is also produced by endothelial cells upon stimulation
by TNF-α, LPS and IL-1β (118).
After the kidney sustains ischemic, septic or nephrotoxic injury,
it releases large amounts of NGAL into both urine and serum. Thus,
NGAL is considered a promising early biomarker for SA-AKI (119-122). Beyond serving as an early
diagnostic biomarker, serum and urine NGAL can predict the
progression of AKI to CKD in patients with SA-AKI (123).
Numerous innovative nanotechnology-based therapies
targeting neutrophils have been developed. In diseases associated
with neutrophil infiltration, neutrophils serve not only as
therapeutic targets but also as ideal carriers for targeted drug
delivery (124). For example, a
targeted nanodrug delivery platform, Ac-PGP (a peptide sequence
that targets the CXCR2 receptor on neutrophils)-tetrahedral
framework nucleic acid (tFNA), utilizes tFNA and a neutrophil
hitchhiking mechanism (125).
DNA-based nanorobots have also been employed to target, load and
modulate neutrophils for precise drug delivery and
anti-inflammatory effects (125,126). Ly6G nanoparticles loaded with a
selective phosphodiesterase 4 inhibitor can effectively suppress
the release of cytokines and chemoattractants from activated
neutrophils, preventing organ damage caused by excessive neutrophil
accumulation and migration (127). Additionally, nanomaterials
loaded with astragaloside IV, a natural compound with
anti-inflammatory and antioxidant properties, can precisely target
neutrophils and inhibit NET formation, mitigating sepsis-induced
inflammatory responses and organ damage (128). Similarly, nanomaterials loaded
with Coenzyme Q10, an antioxidant, significantly enhance kidney
delivery efficiency, offering a new approach to alleviating kidney
damage (129). Nanozymes,
nanomaterials with enzyme-like activities, also play a vital role
in SA-AKI treatment by catalyzing specific biochemical reactions
and protecting the kidneys through mechanisms such as scavenging
excess ROS and modulating inflammatory signaling pathways (130). Neutrophil-targeted nanodrug
delivery systems provide novel strategies for SA-AKI treatment by
enabling precise intervention in neutrophil-mediated kidney injury,
optimizing nanocarrier design and facilitating clinical
translation.
Although studies targeting neutrophil-mediated
injury pathways have shown promising results, neutropenia remains a
high-risk condition in critically ill septic patients (131). Compared with non-neutropenic
patients, severe sepsis triggers a distinct inflammatory response
in neutropenic patients (131).
Levels of IL-6, IL-8 and granulocyte colony-stimulating factor
(CSF) are significantly elevated in neutropenic septic patients and
are independently associated with an increased risk of AKI
(131). Therefore, the
development of therapeutic strategies targeting neutrophils must be
carefully balanced to avoid excessive suppression of their
functions, which could impair renal repair.
DCs, the most abundant resident leukocytes in the
kidney, play a critical role in renal immunity by forming a
sophisticated immune sentinel system that continuously monitors the
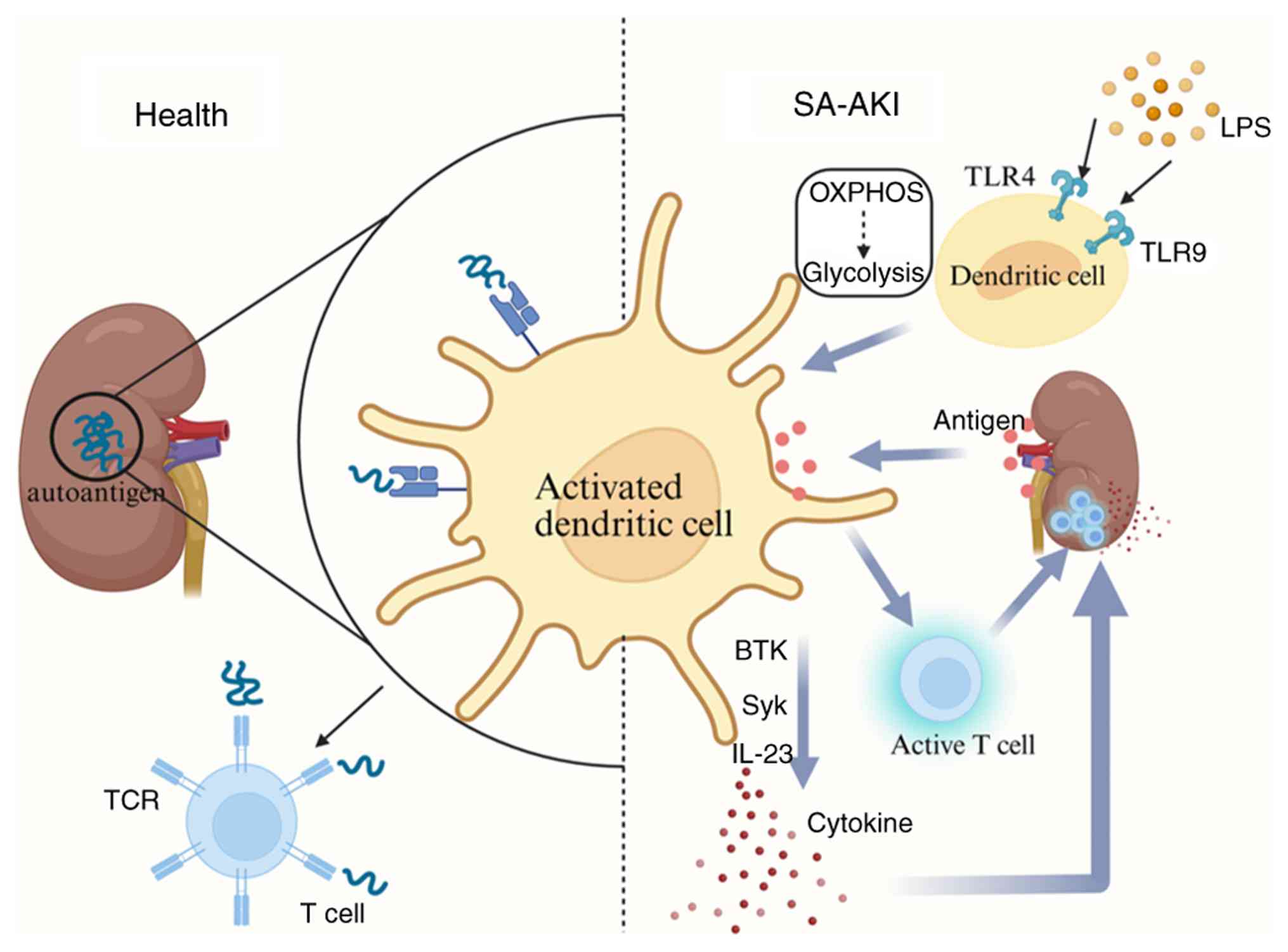
tubulointerstitial compartment (132) (Fig. 4). Under normal physiological
conditions, DCs primarily maintain immune tolerance and prevent
autoimmune responses (133);
they constantly sample and present autoantigens and
low-molecular-weight antigens from the glomeruli and tubules to T
lymphocytes, thereby promoting T cell tolerance and preventing
attacks on renal tissues (133,134). Additionally, DCs have the
capacity to recognize and clear apoptotic cells and debris,
preventing excessive activation of inflammatory responses (133). However, under pathological
conditions, DCs undergo notable changes. Upon recognition of DAMPs,
DCs undergo metabolic reprogramming, shifting from OXPHOS to
glycolysis to generate an immunogenic response (135). This metabolic shift is
accompanied by phenotypic maturation, enabling DCs to specialize in
T cell stimulation and activate innate immune cells (136). Mature DCs then migrate to
inflamed tissues and lymph nodes, enhancing their
antigen-presenting capacity by upregulating major
histocompatibility complex (MHC) molecules and T cell
co-stimulatory proteins on their cell membranes, thus facilitating
T cell activation and initiating an immune response (137,138).
DCs play an even more critical role in sepsis, where
they modulate the adaptive immune response to infection by
producing pro-inflammatory cytokines and chemokines that guide
lymphocyte activation and lineage commitment (139). DCs may also mediate T cell
infiltration into the kidney, initiating inflammatory responses
that contribute to the onset and progression of SA-AKI, potentially
through antigen uptake from peritubular capillaries (139). DCs mediate the link between
TLRs and sepsis. TLR4 and TLR9, key members of the TLR family, are
closely associated with sepsis-induced inflammatory responses. TLR4
serves as the cellular receptor for LPS on the DC surface; the
binding of LPS to TLR4 activates downstream signaling pathways,
leading to the activation of NF-κB, a transcription factor that
regulates gene expression during LPS-induced inflammatory responses
in both kidney damage and sepsis (140). Triggered by endogenously
released mitochondrial DNA during sepsis, TLR9 activates DCs. The
activated DCs then produce IL-23, which induces γδ T cells to
produce IL-17A, thereby promoting the development of SA-AKI
(141). Based on these
mechanisms, developing specific inhibitors targeting TLR4 or TLR9
could be an effective strategy to mitigate the progression of the
inflammatory cascade in SA-AKI (140). Furthermore, LPS activates TLR4
signaling through two distinct pathways: Via the plasma membrane
through the Toll/IL-1 receptor domain-containing adapter protein
(TIRAP)-MyD88 adapter complex and via endosomes through the
TRIF-related adaptor molecule-Toll/IL-1 receptor domain-containing
adapter inducing IFN-β (TRAM-TRIF) adapter complex. In DCs, the
p110δ subunit of PI3K acts as a key mediator, promoting the
transition of TLR4 signaling from the pro-inflammatory
TIRAP-MyD88-dependent phase to the anti-inflammatory
TRAM-TRIF-dependent phase (142). Therefore, using DCs as vehicles
through genetic modification or drug delivery approaches to enhance
the anti-inflammatory effects of the p110δ subunit presents a
promising therapeutic strategy for SA-AKI.
In addition to TLR4 and TLR9-related mechanisms, DCs
influence the progression of SA-AKI through the release of
inflammatory cytokines, a process regulated by the spleen tyrosine
kinase (Syk) signaling pathway (143). Inhibition of Syk signaling has
been shown to limit the inflammatory cascade in SA-AKI (144). Concurrently, research has
identified that activation of Bruton's tyrosine kinase (BTK) in DCs
leads to a significant increase in biochemical markers associated
with subacute kidney injury. During the onset of SA-AKI, BTK is
activated in DCs, neutrophils and B cells, and inhibiting BTK
signaling improves renal function following AKI (145). Therefore, inhibiting both the
Syk and BTK signaling pathways in innate immune cells may serve as
an effective strategy to attenuate the progression of the
inflammatory cascade in SA-AKI.
Mast cells are granule-rich immune cells distributed
throughout the body, particularly in areas commonly exposed to
microbes, such as mucosal tissues, skin and connective tissues
(150). These cells interact
with pathogens through surface and intracellular receptors,
including pattern recognition receptors, bacterial toxin receptors,
antimicrobial peptides, complement proteins and Fc receptors
(151). While mast cells are
traditionally associated with allergic reactions, atopic asthma and
other IgE-mediated allergic diseases (152), studies have highlighted their
broader immunomodulatory roles. These include enhancing host
resistance in certain bacterial or parasitic infection models and
even providing defense against specific animal venoms (152). This functional diversity is
especially evident in sepsis. In the early stages of infection,
mast cells activate neutrophils by releasing IL-6 and suppressing
TNF, thereby enhancing their bactericidal activity (150,153-156). However, in severe sepsis,
excessive mast cell activation exacerbates the systemic
inflammatory response and increases mortality. Despite this, in
models of sepsis and ischemia-reperfusion injury, mast cells can
modulate TNF levels through the release of anti-inflammatory
mediators, such as protease 4 (a homolog of mouse mast cell
protease), which suppresses neutrophil overactivation, reduces
inflammation and mitigates renal impairment (152,157). In the context of kidney
disease, mast cells display a dual role. In anti-glomerular
basement membrane antibody-induced glomerulonephritis, mast cells
reduce glomerular injury by initiating repair mechanisms (158). By contrast, in
cisplatin-induced AKI, mast cells exacerbate the condition through
a TNF-dependent pathway (159).
While the specific role of mast cells in SA-AKI remains unclear,
existing evidence suggests their potential importance. Mast cells
may combat infection via rapid inflammatory responses and influence
disease progression through immunomodulation. Current understanding
of this mechanism largely relies on non-sepsis models, such as
cisplatin-induced and ischemia-reperfusion injury models. However,
the unique immune microenvironment in sepsis may alter the
manifestation of these mechanisms, and their direct role in SA-AKI
requires further validation.
Given the complex and diverse roles of mast cells,
future multicenter, large-scale clinical studies are urgently
needed. These studies should focus on the specific association
between mast cells and SA-AKI and further elucidate their
immunoregulatory functions, repair mechanisms and interactions with
the renal microenvironment. Such investigations are expected to
provide novel insights and therapeutic strategies for the treatment
of SA-AKI.
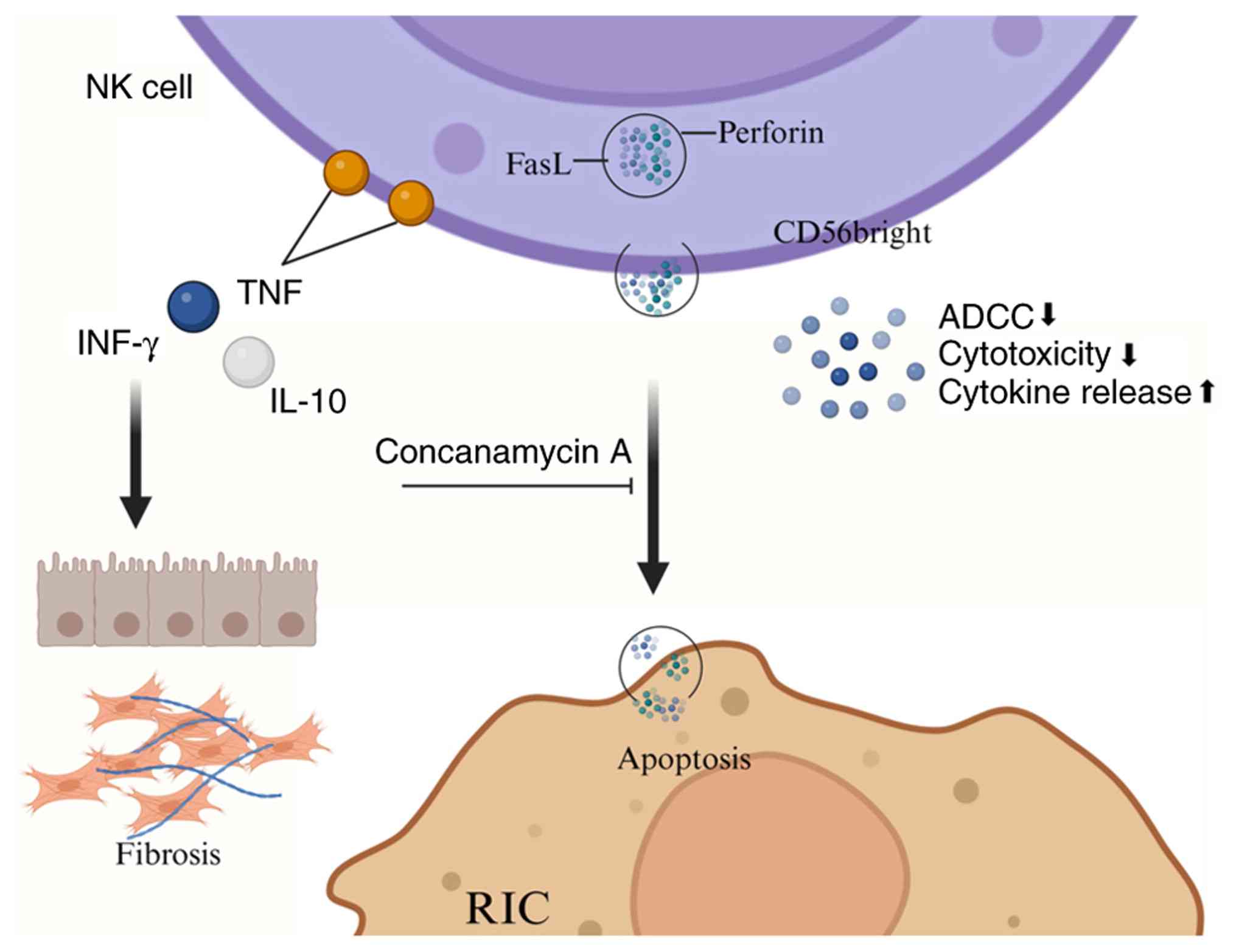
NK cells are large granular lymphocytes derived from
the bone marrow, constituting ~15% of all circulating lymphocytes.
Fig. 5 provides a schematic
overview of the roles and mechanisms of NK cells in the
pathogenesis of SA-AKI. As vital components of the innate immune
system, NK cells are capable of directly lysing tumor cells and
other target cells (160,161); however, their function is
dualistic. While NK cells are critical for immune defense,
excessive activation may result in attacks on normal cells, leading
to shock and multiple organ failure (MOF) (162). The mechanisms of NK cell action
manifest in two primary ways: i) Direct cytolysis via azurophilic
granules within the cytoplasm; and ii) the secretion of
pro-inflammatory cytokines, such as IFNs and TNF, to modulate the
activities of other immune cells (160,163). The activity of NK cells is
regulated by a balance of activating and inhibitory receptors on
their surface. Most healthy cells express MHC class I molecules,
which deliver inhibitory signals that suppress NK cell activity
(164). In humans, NK cells are
typically divided into two subsets: i) CD56bright NK cells,
predominantly found in lymphoid organs; and ii) CD56dim NK cells,
which are mainly located in peripheral blood. CD56bright NK cells
primarily exert immunoregulatory functions through cytokine
secretion, while CD56dim NK cells exhibit potent cytotoxic
capabilities (165,166). Notably, CD56bright NK cells
often express tissue-resident markers in the healthy kidney and may
play a key role in renal defense against infections (167). However, in patients with renal
fibrosis and chronic kidney disease (CKD), a substantial number of
CD56bright NK cells accumulate in the renal tubulointerstitium,
where they produce IFN-γ and contribute to the progression of renal
fibrosis (168). In the context
of SA-AKI, targeted treatment strategies could be developed based
on the distinct characteristics of these NK cell subsets. For
example, the cytotoxic function of CD56dim NK cells could be
harnessed to clear damaged cells, while the activity of CD56bright
NK cells could be modulated to prevent excessive inflammatory
responses. Furthermore, NK cells produce a wide range of cytotoxic
effector molecules, including Fas ligand (FasL) and perforin.
Notably, stimulation with bacterial components such as LPS leads to
a significant increase in the proportion of perforin-positive cells
(169,170). Stimulated human
CD56+ NK cell subsets can damage intrinsic renal cells,
including renal tubular epithelial cells and glomerular endothelial
cells. Inhibition of the perforin-mediated pathway with
concanamycin A can reduce the cytotoxicity of CD56+ NK
cells against glomerular endothelial cells (169,171).
In sepsis-related research, a decrease in human NK
cell counts has been observed within 1 day of sepsis onset, which
increases the risk of hospital-acquired infections and
sepsis-related complications (168). Additionally, NK cell function
is impaired in critically ill septic patients, accompanied by
reduced cytokine secretion (172). A further study has indicated
that lower levels of NK cells and other immune cells elevate
mortality risk in septic patients, whereas those with higher NK
cell levels tend to have longer survival times (173). A case report described a
patient with pulmonary squamous cell carcinoma who developed sepsis
and showed a poor response to conventional therapy. Following NK
cell infusion therapy, the patient's infection markers initially
decreased but later increased. While the long-term efficacy of NK
cell therapy requires further investigation, optimizing treatment
regimens holds promise for improving NK cell survival and
therapeutic outcomes (174).
The aforementioned case report suggests that NK cell therapy can
enhance patient physical function, increase cytokine levels with
antitumor activity and reduce pro-inflammatory cytokines; it also
provides insights for SA-AKI treatment: Monitoring NK cell count
and function during therapy and selecting the appropriate timing
for intervention. For instance, administering appropriate
stimulation or supplementation before NK cell numbers severely
decline may enhance treatment efficacy, whereas restoring function
is necessary if NK cell activity is already impaired. Future
research should focus on optimizing NK cell treatment strategies,
including selecting suitable cell sources and determining the
optimal infusion doses and frequencies, to improve their survival
and efficacy in SA-AKI treatment.
In addition to NK cells, NKT cells represent a
unique subset of immune cells that exhibit characteristics of both
T cells and NK cells. As innate lymphocytes bridging innate and
adaptive immune responses, NKT cells are present in extremely low
numbers in the blood, accounting for <0.1% of human peripheral
blood mononuclear cells (175).
Based on the variability of the T cell receptor (TCR) chain, NKT
cells can be classified into two types: Type I (iNKT cells) and
type II (vNKT cells) (174,176). Among these, iNKT cells are the
predominant subtype in humans, expressing the Va-24 and Ja-18
chains; they differentiate into mature iNKT cells upon recognition
of glycolipid antigens presented by CD1d molecules (177). Furthermore, various cytokines
secreted by iNKT cells play a potent regulatory role in the
maturation of DCs, positioning iNKT cells as a critical link
between innate and adaptive immunity (177). A reciprocal regulatory
relationship exists between iNKT and vNKT cells, with vNKT cells
suppressing iNKT cell-mediated inflammatory responses. This
interaction may influence disease progression during the onset and
development of SA-AKI. Therefore, in treating SA-AKI, it is
essential to consider not only the overall status of NKT cells but
also the interactions between different subsets (178,179). Like NK cells, NKT cells express
activating receptors, inhibitory receptors and cytokine receptors
(such as IL-12 and TNF-α receptors) (167); they also possess the NK1.1
antigen and an intermediate TCR (171). NKT cells recognize lipids and
glycolipids presented by CD1d molecules (180) and can be activated through TCR
engagement, exhibiting potent cytotoxic functions and directly
participating in the clearance of infectious agents (167). This dual functionality provides
a more comprehensive perspective for understanding the
pathophysiology of SA-AKI and offers potential therapeutic
targets.
The role of iNKT cells in immune functions is
multifaceted. On the one hand, a study has demonstrated that iNKT
cells can mediate macrophage inflammatory responses to pathogenic
microorganisms by regulating macrophage quantity and phenotype,
optimizing immune factor release, enhancing host defense capacity
and improving macrophage killing efficiency (181). On the other hand, research on
sepsis has revealed that iNKT cells exacerbate the inflammatory
response during the early stages of sepsis, worsening its severity.
iNKT cell-deficient mice exhibit a certain level of protection
against sepsis (182), whereas
CD1-deficient mice, lacking both iNKT and vNKT cells, do not show
this protective effect (183).
During the development of AKI, NKT cells are known to highly
express inflammatory regulators, modulating the production of
cytokines involved in injury and complement activation, indicating
their involvement in AKI pathogenesis through inflammatory
mechanisms (184). SA-AKI can
lead to MOF, including AKI (167,185). Upon stimulation by cytokines or
bacterial components, NKT cells are activated. Similar to NK cells,
they can damage renal tubular epithelial cells via the TNF-α/FasL
system and injure vascular endothelial cells through a
perforin-mediated pathway. These independent mechanisms contribute
to the induction of SA-AKI (171). Relevant studies support this
view: In patients with SA-AKI undergoing continuous renal
replacement therapy (CRRT), the expression of perforin in
CD56+ NK cells and CD56+ T cells (that is,
NKT cells) is elevated, with FasL expression significantly
upregulated in these CD56+ T cells (167,169). This suggests that both the
TNF-α/FasL system and perforin play critical roles in the
progression of SA-AKI. Future strategies for SA-AKI treatment could
focus on regulating NKT cell activation. Additionally, this
phenomenon suggests that CRRT may be linked to NKT cell activation.
Subsequent therapeutic approaches could consider combining CRRT
with treatments targeting NKT cells to enhance CRRT efficacy and
improve the prognosis of patients with SA-AKI by modulating NKT
cell activation.
ILCs are critical components of the innate immune
system, playing essential roles in immune responses, inflammation
and tissue healing (186).
Although ILCs lack lineage-specific surface markers or antigen
receptors typical of other immune cells, they are critical for
maintaining protective immunity, homeostasis and inflammatory
responses (187). The role of
ILCs in the kidney is becoming increasingly recognized, with
evidence suggesting their involvement in immune surveillance,
homeostasis maintenance and disease progression within the kidney
(188). Despite their lack of
antigen-specific reactivity, ILCs share key transcriptional
regulators with T lymphocytes and possess the ability to produce
cytokines (189). ILCs are
classified into three main subsets: Group 1 ILCs (ILC1s), group 2
ILCs (ILC2s) and group 3 ILCs (ILC3s), each functionally associated
with Th1, Th2 and Th17 cells, respectively (190). Among these, ILC2-derived
cytokines, such as IL-4 and IL-13, play a key role in driving
macrophage polarization towards the M2 phenotype (191). As aforementioned, appropriate
M2 macrophage activation is essential for ameliorating SA-AKI.
Kidney-resident ILC2s express receptors for IL-25
(IL-17RB) and IL-33 (T1/ST2), which, when activated by their
respective cytokines, promote ILC2 expansion in vivo
(192). While ILC2s exert
protective effects in the kidney, their impact depends on renal
IL-33 levels. During kidney injury and disease, IL-33, released
from vascular endothelial cells and/or renal tubular epithelial
cells, activates kidney-resident ILC2s (193). Studies on ILC2 function have
shown varying results. For instance, Akcay et al (194) found that recombinant IL-33
exacerbated AKI in a cisplatin-induced model by promoting
CD4+ T cell-mediated CXCL1 production. However, soluble
ST2, which neutralizes IL-33, reduced the severity of AKI. This
suggests that in cisplatin-induced AKI, IL-33 aggravates kidney
damage by activating the ST2 signaling pathway and promoting
inflammation. Conversely, Cao et al (195) observed that in an
ischemia-reperfusion-induced AKI model, IL-33 pretreatment improved
renal injury by activating ILC2s, promoting M2 macrophage
polarization and aiding renal function recovery. These studies
indicate that the role of IL-33 in AKI is context-dependent,
influenced by factors such as the type of injury, inflammatory
environment and immune cell composition. The role of ILC2s in
SA-AKI remains unclear. Unlike non-infection-induced AKI, SA-AKI
pathogenesis is more complex and it is uncertain whether IL-33
activates ILC2s through similar mechanisms. However, a study has
shown that during sepsis, plasma IL-33 levels are significantly
elevated in young mice, promoting the egress of ILC2s from the bone
marrow. This alteration may affect the accumulation and function of
ILC2s in the kidney (196).
Therefore, when studying the functions of ILC2s in SA-AKI, factors
such as the experimental model and conditions must be carefully
considered. Further research into the specific signaling pathways
activated by IL-33 in different AKI models, along with the roles of
ILC2s in these pathways, could provide a theoretical foundation for
developing more effective therapeutic strategies for SA-AKI.
Research on the role of ILCs in SA-AKI is still
limited and their precise mechanisms of action are not fully
understood. Given the preliminary evidence supporting the
involvement of ILCs in other types of AKI, further investigation
into the functions and mechanisms of ILC subsets in SA-AKI is
crucial for advancing our understanding and treatment of this
condition.
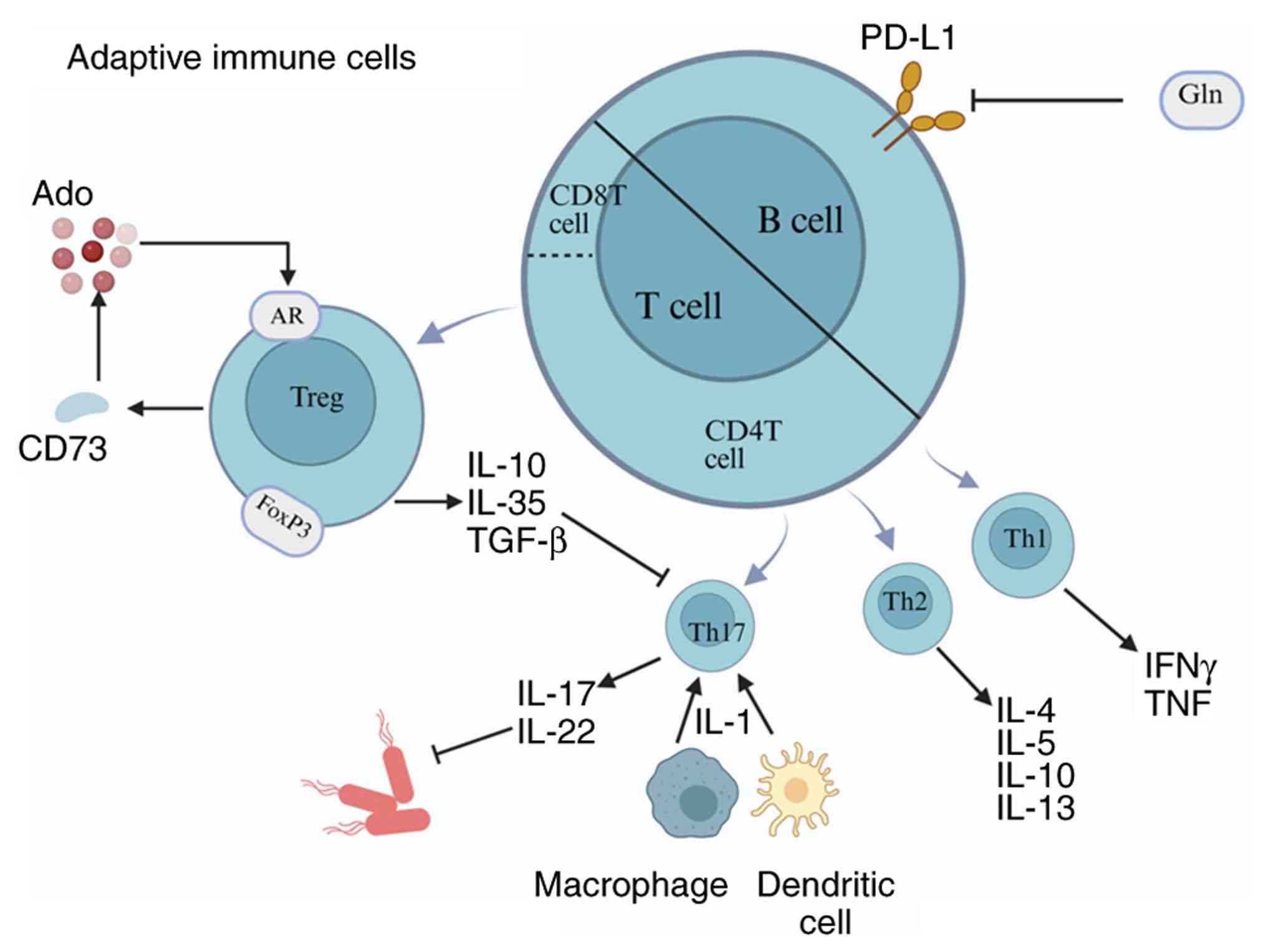
B cells, a key subset of lymphocytes, play an
essential role in the immune response. As summarized in Fig. 6, B cells produce antibodies,
presenting antigens and secreting cytokines, thereby mediating both
adaptive and innate immune responses, maintaining immune balance
and renal homeostasis (197).
However, due to their pathogenic role in organ transplantation and
autoimmune diseases, B cells are considered to have detrimental
effects in AKI (198). In a
cisplatin-induced AKI model, Inaba et al (199) observed that renal B cells were
a significant source of CCL7, promoting the recruitment of
neutrophils and monocytes to the injured kidney and exacerbating
AKI severity. Similarly, in a study of unilateral ureteral
obstruction-induced AKI, Han et al (200), found that early accumulation of
B cells in the kidney accelerated the mobilization and infiltration
of monocytes/macrophages, aggravating fibrosis induced by AKI.
Clinical studies on SA-AKI have shown an association between the
plasma lymphocyte ratio and renal function, with 1 study revealing
that septic patients who recovered renal function had fewer B
lymphocytes at hospital admission (201). Sepsis increases the expression
of programmed death-ligand 1 (PD-L1) on B lymphocytes. Programmed
cell death protein 1 (PD-1) impairs immunity by inducing apoptosis,
inhibiting T-cell activation, increasing IL-10 production and
rendering T cells unresponsive while reducing their cytokine
secretion, leading to T-cell exhaustion. Glutamine administration
has been shown to reduce the expression of PD-1/PD-L1 on B cells,
alleviating kidney damage (202).
Under normal physiological conditions, T cells play
a pivotal role in maintaining renal immune homeostasis (203) (Fig. 6); they regulate immune responses,
preventing autoimmune reactions and excessive inflammation, thereby
protecting the kidneys from damage. T cells continuously monitor
renal tissues, identifying and eliminating potential pathogens or
abnormal cells to preserve kidney health (204). Following the onset of SA-AKI, T
cells, as key immune cells combating pathogen infections, play a
decisive role in the pathogenesis and progression of the
disease.
T cell subsets exert distinct immunomodulatory
functions in the kidneys. T lymphocytes can be classified into two
major subsets: CD4+ Th cells and CD8+ T
cells. Th cells are further divided into Th1, Th2 and Th17 subsets.
Th1 cells primarily produce IFN-γ and TNF-α, exerting
pro-inflammatory effects. Th2 cells secrete IL-4, IL-5, IL-10 and
IL-13 (205), predominantly
mediating anti-inflammatory effects. Th17 cells mainly produce
IL-17 and IL-22 (206). After
kidney injury, IL-1 secreted by renal DCs and macrophages promotes
the differentiation and activation of Th17 cells (207). Th17 cells infiltrate the renal
parenchyma during murine AKI, defend against extracellular
pathogens through the production of their signature cytokine IL-17
and play a pivotal role in promoting autoimmune diseases and tissue
inflammation (208).
In sepsis, lymphocyte dysfunction occurs not only
in peripheral blood cells but also in target organs (219). T cell priming is regulated by a
balance of positive and negative co-stimulatory molecules, with
imbalances in these molecules (also known as immune checkpoint
molecules) being closely linked to immune dysfunction (220). A common feature of
sepsis-associated immunosuppression is impaired lymphocyte function
and increased expression of inhibitory checkpoint molecules such as
PD-1. Activation of the PD-1/PD-L1 pathway by lactate can induce
immunosuppression by promoting lymphocyte apoptosis in SA-AKI.
Blocking the lactate receptor or PD-1/PD-L1 can restore lymphocyte
function and alleviate kidney damage (221). Additionally, MSCs can restore
the balance between Th17 and Treg cells via the galectin-9/T-cell
immunoglobulin and mucin domain-containing molecule-3 pathway,
reduce inflammatory cell infiltration and reestablish the
equilibrium between pro-inflammatory and anti-inflammatory
responses, thus improving SA-AKI (222). Dysregulation of T cells can
also lead to the abnormal release of immunomodulatory molecules,
such as excessive inflammatory cytokines, exacerbating tissue
inflammatory injury. In this context, glutamine has been shown to
alleviate SA-AKI by balancing T cell polarization and reducing T
cell apoptosis (223). Given
the imbalance in T cell subsets in patients with SA-AKI, future
therapeutic strategies should aim to selectively expand protective
T lymphocyte subsets or suppress damaging T cell subsets.
Several potential therapeutic targets related to T
cells in SA-AKI have been identified. In addition to α7nAChR
agonists, MSCs and anti-PD-L1 antibodies, recombinant human soluble
thrombomodulin (rTM), a single-transmembrane, multi-domain
glycoprotein receptor for thrombin, has emerged as a promising
candidate (224). rTM not only
reduces the upregulation of pro-inflammatory cytokines and
chemoattractants induced by LPS, thereby decreasing leukocyte
infiltration into the kidneys, but also mitigates kidney damage by
reducing the accumulation of CD4+ T cells,
CD11c+ cells and F4/80+ cells in SA-AKI. This
effect is mediated through enhanced phosphorylation of c-Jun, which
diminishes cytokine production and apoptosis signaling (224). Additionally, CD28 plays a role
in regulating TNF-α and IL-10 homeostasis, influencing the
expression of chemoattractants. Through the CD28 pathway, T cells
modulate renal function during sepsis (225).
In addition to therapeutic targets, certain T
cell-related biomarkers may serve as predictors for SA-AKI. Ma
et al (226)
demonstrated that T cell knock-out mice exhibit reduced
inflammatory cytokine secretion in renal tissue after LPS injection
and that T cell suppression alleviates sepsis-induced inflammation
and kidney damage. Therefore, T cell hyperactivation or
upregulation could serve as a biomarker for worsening SA-AKI, and
therapies targeting T cells or blocking receptors for inflammatory
cytokine secretion may offer benefits in treating SA-AKI (226). Elevated plasma levels of IL-10
and soluble CD25 (a marker of Treg cells) in patients with SA-AKI
may also serve as novel biomarkers (227). Moreover, ATP content in
CD4+ T cells (ATP_CD4) has been shown to correlate with
survival in sepsis patients. Low ATP_CD4 levels at 48 h post-onset
may indicate a higher likelihood of complete renal recovery, making
it a potential prognostic indicator (228).
Future research should focus on elucidating the
mechanisms of T cells in SA-AKI, including interactions among
different subsets and signaling pathways. Integrating the
regulation of immune checkpoint molecules, MSCs application,
exploration of potential therapeutic targets and the development of
biomarkers will contribute to the formulation of comprehensive
treatment strategies to improve the prognosis of patients with
SA-AKI.
Although treatment for SA-AKI significantly
alleviates the complex inflammatory response within the kidney, the
potential for persistent adverse effects following treatment
warrants attention. Even after the acute phase of AKI resolves,
renal injury may result in permanent nephron loss or maladaptive
repair, ultimately leading to CKD or accelerating the progression
of pre-existing CKD (229).
Furthermore, sepsis-induced immunosuppression represents a
prolonged and complex state of immune dysfunction. Even after the
acute inflammatory response is controlled, the immune system may
remain dysregulated, increasing the risk of subsequent infections
and abnormal responses during tissue healing (16). Additionally, sepsis often
involves multiple organs, leading to intricate organ-organ
interactions (230). AKI can
modulate the function of other vital organs through inter-organ
crosstalk. TNF-α and IL-1, key mediators of renal injury, also play
central roles in the pathophysiology of cardiac dysfunction during
sepsis (231). During SA-AKI,
renal injury not only triggers immune responses in lung tissue,
including monocyte, neutrophil and CD8+ T cell
infiltration, but also increases inflammatory cytokine production
and activates immune cells that impair intestinal barrier function
and increase permeability. Intestinal hyperpermeability is a common
factor exacerbating renal failure (232). These complex interactions
suggest that dysfunction in organs beyond the kidney may persist or
worsen, adversely impacting renal recovery. Therefore, the
therapeutic strategy for SA-AKI should focus not only on mitigating
renal inflammation but also on addressing the potential for
long-term adverse effects following treatment.
The present review systematically summarized the
mechanisms of various immune cells in SA-AKI and discussed
potential therapeutic strategies, encompassing both innate and
adaptive immunity and introducing innovative approaches such as
nanotechnology and cell therapy. However, several limitations
remain. First, some mechanistic studies cited in the present review
are based on non-septic AKI models (such as cisplatin-induced or
ischemia-reperfusion injury), which, while simulating certain
pathological features of kidney injury, differ significantly from
the immune microenvironment of patients with sepsis.
Sepsis-specific phenomena, such as immunoparalysis and endotoxin
tolerance, are difficult to replicate fully in these models,
limiting the direct applicability of their findings to SA-AKI.
Second, the roles of certain immune cells (such as mast cells and
ILCs) in SA-AKI are still discussed primarily through inference or
indirect evidence, particularly concerning their specific functions
in sepsis. Further experimental validation using sepsis-specific
models is necessary to confirm these roles. Finally, while the
present review has introduced various novel therapeutic strategies
based on nanotechnology and cell therapy, the application of these
strategies in treating SA-AKI remains challenging. Most
nanomaterials lack clinical evaluations for long-term safety and
biocompatibility. Although some animal studies have shown positive
results, the human immune system's response to nanomaterials may
differ. Additionally, targeted therapies for the kidneys require
further research. The stability of the preparation process and
quality control in the clinical translation of nanomaterials are
also key factors affecting long-term safety. Therefore, long-term
safety testing and large-scale clinical studies are needed to
ensure the sustainability and reliability of these treatments. In
conclusion, future research should focus on the development and
application of sepsis-specific animal models, enhance
spatio-temporal analysis of the dynamic functions of immune cells
and promote the translation of basic research into clinical
applications, ultimately providing a more reliable foundation for
precision immunotherapy in SA-AKI.
Not applicable.
LX, WJ, JY and RZ wrote the main manuscript text.
LS and JW modified the details of the manuscript. Data
authentication is not applicable. All of the authors read and
approved the final version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
We sincerely thank Dr Keran Shi (Department of
Critical Care Medicine, The Yangzhou Clinical College of Xuzhou
Medical University, Yangzhou, Jiangsu 225001, P.R. China), Dr
Yuanjin Pan (Department of Critical Care Medicine, Northern Jiangsu
People's Hospital Affiliated to Yangzhou University, Yangzhou,
Jiangsu 225001, P.R. China), Dr. Yuchen Wang (Department of
Critical Care Medicine, The Yangzhou Clinical College of Xuzhou
Medical University, Yangzhou, Jiangsu 225001, P.R. China) and Dr
Luanluan Li (Department of Critical Care Medicine, Northern Jiangsu
People's Hospital Affiliated to Yangzhou University, Yangzhou,
Jiangsu 225001, P.R. China) for their continuous encouragement and
valuable insights during the preparation of this review. Their
constructive suggestions and enthusiastic support not only
alleviated the challenges encountered during the writing process
but also inspired us to refine the content with greater depth and
precision.
This work was supported by grants from Health and Wellness
Development and Promotion Project (grant no. QS-XFGCJWZZ-0062);
National Key Clinical Specialty, Financial Appropriations of
National [grant no. 176.(2022)]; Flagship Institution of Chinese
and Western Medicine Coordination, Financial Appropriations of
National (grant no. Jiangsu 60(2023)]; Jiangsu Provincial Medical
Key Discipline Cultivation Unit (grant no. JSDW20221); Yangzhou
Social Development Project (grant no. YZ2023105); Special
Scientific Research Fund of Yangzhou Health Commission (grant no.
2023-1-02) and Management Project of Northern Jiangsu People's
Hospital (grant no. YYGL202306).
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seymour CW, Kennedy JN, Wang S, Chang CH,
Elliott CF, Xu Z, Berry S, Clermont G, Cooper G, Gomez H, et al:
Derivation, validation, and potential treatment implications of
novel clinical phenotypes for sepsis. JAMA. 321:2003–2017. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu D, Huang SY, Sun JH, Zhang HC, Cai QL,
Gao C, Li L, Cao J, Xu F, Zhou Y, et al: Sepsis-induced
immunosuppression: Mechanisms, diagnosis and current treatment
options. Mil Med Res. 9:562022.PubMed/NCBI
|
|
4
|
Wang D, Sun T and Liu Z: Sepsis-associated
acute kidney injury. Intensive Care Res. 3:251–258. 2023.
View Article : Google Scholar
|
|
5
|
Uchino S, Kellum JA, Bellomo R, Doig GS,
Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, et al:
Acute renal failure in critically ill patients: A multinational,
multicenter study. JAMA. 294:813–818. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Balkrishna A, Sinha S, Kumar A, Arya V,
Gautam AK, Valis M, Kuca K, Kumar D and Amarowicz R:
Sepsis-mediated renal dysfunction: Pathophysiology, biomarkers and
role of phytoconstituents in its management. Biomed Pharmacother.
165:1151832023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee K, Jang HR and Rabb H: Lymphocytes and
innate immune cells in acute kidney injury and repair. Nat Rev
Nephrol. 20:789–805. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao X, Cai S, Li X and Wu G:
Sepsis-induced immunosuppression: Mechanisms, biomarkers and
immunotherapy. Front Immunol. 16:15771052025. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Z and Wang Z: The role of macrophages
polarization in sepsis-induced acute lung injury. Front Immunol.
14:12094382023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kwok AJ, Allcock A, Ferreira RC,
Cano-Gamez E, Smee M, Burnham KL, Zurke YX; Emergency Medicine
Research Oxford (EMROx); McKechnie S, Mentzer AJ, et al:
Neutrophils and emergency granulopoiesis drive immune suppression
and an extreme response endotype during sepsis. Nat Immunol.
24:767–779. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qi X, Yu Y, Sun R, Huang J, Liu L, Yang Y,
Rui T and Sun B: Identification and characterization of neutrophil
heterogeneity in sepsis. Crit Care. 25:502021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao F, Xiao C, Evans KS, Theivanthiran T,
DeVito N, Holtzhausen A, Liu J, Liu X, Boczkowski D, Nair S, et al:
Paracrine Wnt5a-β-catenin signaling triggers a metabolic program
that drives dendritic cell tolerization. Immunity. 48:147–160.e7.
2018. View Article : Google Scholar
|
|
13
|
Flohé SB, Agrawal H, Schmitz D, Gertz M,
Flohé S and Schade FU: Dendritic cells during polymicrobial sepsis
rapidly mature but fail to initiate a protective Th1-type immune
response. J Leukoc Biol. 79:473–481. 2006. View Article : Google Scholar
|
|
14
|
Tang J, Shang C, Chang Y, Jiang W, Xu J,
Zhang L, Lu L, Chen L, Liu X, Zeng Q, et al: Peripheral
PD-1+NK cells could predict the 28-day mortality in
sepsis patients. Front Immunol. 15:14260642024. View Article : Google Scholar
|
|
15
|
Nascimento DC, Viacava PR, Ferreira RG,
Damaceno MA, Piñeros AR, Melo PH, Donate PB, Toller-Kawahisa JE,
Zoppi D, Veras FP, et al: Sepsis expands a CD39+
plasmablast population that promotes immunosuppression via
adenosine-mediated inhibition of macrophage antimicrobial activity.
Immunity. 54:2024–2041.e8. 2021. View Article : Google Scholar
|
|
16
|
Kox M, Bauer M, Bos LDJ, Bouma H, Calandra
T, Calfee CS, Chousterman BG, Derde LPG, Giamarellos-Bourboulis EJ,
Gómez H, et al: The immunology of sepsis: Translating new insights
into clinical practice. Nat Rev Nephrol. 22:30–49. 2026. View Article : Google Scholar
|
|
17
|
Dick SA, Wong A, Hamidzada H, Nejat S,
Nechanitzky R, Vohra S, Mueller B, Zaman R, Kantores C, Aronoff L,
et al: Three tissue resident macrophage subsets coexist across
organs with conserved origins and life cycles. Sci Immunol.
7:eabf77772022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bell RMB and Conway BR: Macrophages in the
kidney in health, injury and repair. Int Rev Cell Mol Biol.
367:101–147. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zimmerman KA, Yang Z, Lever JM, Li Z,
Croyle MJ, Agarwal A, Yoder BK and George JF: Kidney resident
macrophages in the rat have minimal turnover and replacement by
blood monocytes. Am J Physiol Renal Physiol. 321:F162–F169. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cheung MD, Erman EN, Moore KH, Lever JM,
Li Z, LaFontaine JR, Ghajar-Rahimi G, Liu S, Yang Z, Karim R, et
al: Resident macrophage subpopulations occupy distinct
microenvironments in the kidney. JCI Insight. 7:e1610782022.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao J, Andreev I and Silva HM: Resident
tissue macrophages: Key coordinators of tissue homeostasis beyond
immunity. Sci Immunol. 9:eadd19672024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qu Z, Chu J, Jin S, Yang C, Zang J, Zhang
J, Xu D and Cheng M: Tissue-resident macrophages and renal
diseases: Landscapes and treatment directions. Front Immunol.
16:15480532025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Burns KD and Douvris A: Protecting the
kidney in sepsis: Resident macrophages to the rescue. Kidney Int.
103:461–463. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu Q, Xiao L, Cheng G, He J, Yin C, Wang
L, Wang Q, Li L, Wei B, Weng Y, et al: Self-maintaining macrophages
within the kidney contribute to salt and water balance by
modulating kidney sympathetic nerve activity. Kidney Int.
104:324–333. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi C and Pamer EG: Monocyte recruitment
during infection and inflammation. Nat Rev Immunol. 11:762–774.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gong N, Wang W, Fu Y, Zheng X, Guo X, Chen
Y, Chen Y, Zheng S and Cai G: The crucial role of metabolic
reprogramming in driving macrophage conversion in kidney disease.
Cell Mol Biol Lett. 30:722025. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Zhang Z, Qu X and Zhou G: Role of
the endothelial cell glycocalyx in sepsis-induced acute kidney
injury. Front Med (Lausanne). 12:15356732025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chousterman BG, Boissonnas A, Poupel L,
Baudesson de Chanville C, Adam J, Tabibzadeh N, Licata F,
Lukaszewicz AC, Lombès A, Deterre P, et al: Ly6Chigh monocytes
protect against kidney damage during sepsis via a CX3CR1-dependent
adhesion mechanism. J Am Soc Nephrol. 27:792–803. 2016. View Article : Google Scholar :
|
|
29
|
Gao Z, Lu L and Chen X: Release of HMGB1
in podocytes exacerbates lipopolysaccharide-induced acute kidney
injury. Mediators Inflamm. 2021:52202262021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kounatidis D, Tzivaki I, Daskalopoulou S,
Daskou A, Adamou A, Rigatou A, Sdogkos E, Karampela I, Dalamaga M
and Vallianou NG: Sepsis-associated acute kidney injury: What's new
regarding its diagnostics and therapeutics? Diagnostics (Basel).
14:28452024. View Article : Google Scholar
|
|
31
|
Jia P, Xu S, Wang X, Wu X, Ren T, Zou Z,
Zeng Q, Shen B and Ding X: Chemokine CCL2 from proximal tubular
epithelial cells contributes to sepsis-induced acute kidney injury.
Am J Physiol Renal Physiol. 323:F107–F119. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu W, Hou H, Yang W, Tang W and Sun L:
Immunologic role of macrophages in sepsis-induced acute liver
injury. Int Immunopharmacol. 143:1134922024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nie J, Zhou L, Tian W, Liu X, Yang L, Yang
X, Zhang Y, Wei S, Wang DW and Wei J: Deep insight into cytokine
storm: From pathogenesis to treatment. Signal Transduct Target
Ther. 10:1122025. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ovali MA and Percin S: Sepsis-associated
immunosuppression: Mechanistic Insights, Biomarkers, and
therapeutic perspectives. Mol Biol Rep. 53:1482025. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu C, Wei W, Huang Y, Fu P, Zhang L and
Zhao Y: Metabolic reprogramming in septic acute kidney injury:
Pathogenesis and therapeutic implications. Metabolism.
158:1559742024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Freemerman AJ, Johnson AR, Sacks GN,
Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P,
Rathmell JC and Makowski L: Metabolic reprogramming of macrophages:
glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a
proinflammatory phenotype. J Biol Chem. 289:7884–7896. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Morris M and Li L: Molecular mechanisms
and pathological consequences of endotoxin tolerance and priming.
Arch Immunol Ther Exp (Warsz). 60:13–18. 2012. View Article : Google Scholar
|
|
38
|
Gauthier T and Chen W: Modulation of
macrophage immunometabolism: A new approach to fight infections.
Front Immunol. 13:7808392022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pålsson-McDermott EM and O'Neill LAJ:
Targeting immunometabolism as an anti-inflammatory strategy. Cell
Res. 30:300–314. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu B, Liu Y, Li N and Geng Q: Lactate and
lactylation in macrophage metabolic reprogramming: Current progress
and outstanding issues. Front Immunol. 15:13957862024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kieler M, Hofmann M and Schabbauer G: More
than just protein building blocks: How amino acids and related
metabolic pathways fuel macrophage polarization. FEBS J.
288:3694–3714. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Munder M, Eichmann K and Modolell M:
Alternative metabolic states in murine macrophages reflected by the
nitric oxide synthase/arginase balance: Competitive regulation by
CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol.
160:5347–5354. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rath M, Müller I, Kropf P, Closs EI and
Munder M: Metabolism via arginase or nitric oxide synthase: Two
competing arginine pathways in macrophages. Front Immunol.
5:5322014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bronte V and Zanovello P: Regulation of
immune responses by L-arginine metabolism. Nat Rev Immunol.
5:641–654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu PS, Wang H, Li X, Chao T, Teav T,
Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M, et al:
α-ketoglutarate orchestrates macrophage activation through
metabolic and epigenetic reprogramming. Nat Immunol. 18:985–994.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shan X, Hu P, Ni L, Shen L, Zhang Y, Ji Z,
Cui Y, Guo M, Wang H, Ran L, et al: Serine metabolism orchestrates
macrophage polarization by regulating the IGF1-p38 axis. Cell Mol
Immunol. 19:1263–1278. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mussbacher M, Derler M, Basílio J and
Schmid JA: NF-κB in monocytes and macrophages-an inflammatory
master regulator in multitalented immune cells. Front Immunol.
14:11346612023. View Article : Google Scholar
|
|
48
|
Ekihiro S and David AB: NF-κB. Signaling
Pathways in Liver Diseases. 2015.
|
|
49
|
Wang J, Zhao X, Wang Q, Zheng X, Simayi D,
Zhao J, Yang P, Mao Q and Xia H: FAM76B regulates
PI3K/Akt/NF-κB-mediated M1 macrophage polarization by influencing
the stability of PIK3CD mRNA. Cell Mol Life Sci. 81:1072024.
View Article : Google Scholar
|
|
50
|
Geng H, Zhang H, Cheng L and Dong S:
Sivelestat ameliorates sepsis-induced myocardial dysfunction by
activating the PI3K/AKT/mTOR signaling pathway. Int
Immunopharmacol. 128:1114662024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
He J, Zhao S and Duan M: The Response of
macrophages in sepsis-induced acute kidney injury. J Clin Med.
12:11012023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Androulidaki A, Iliopoulos D, Arranz A,
Doxaki C, Schworer S, Zacharioudaki V, Margioris AN, Tsichlis PN
and Tsatsanis C: The kinase Akt1 controls macrophage response to
lipopolysaccharide by regulating microRNAs. Immunity. 31:220–231.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zeng Y, Yuan W, Feng C, Peng L, Xie X,
Peng F, Li T, Lin M, Zhang H and Dai H: Trametinib alleviates
lipopolysaccharide-induced acute kidney injury by inhibiting
macrophage polarization through the PI3K/Akt pathway. Transpl
Immunol. 89:1021832025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Liu C, Li B, Tang K, Dong X, Xue L, Su G
and Jin Y: Aquaporin 1 alleviates acute kidney injury via
PI3K-mediated macrophage M2 polarization. Inflamm Res. 69:509–521.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xue C, Yao Q, Gu X, Shi Q, Yuan X, Chu Q,
Bao Z, Lu J and Li L: Evolving cognition of the JAK-STAT signaling
pathway: Autoimmune disorders and cancer. Signal Transduct Target
Ther. 8:2042023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cai B, Cai JP, Luo YL, Chen C and Zhang S:
The specific roles of JAK/STAT signaling pathway in sepsis.
Inflammation. 38:1599–1608. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou
X, Ma H, Wei D and Sun S: The role of JAK/STAT signaling pathway
and its inhibitors in diseases. Int Immunopharmacol. 80:1062102020.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lee SH, Kim KH, Lee SM, Park SJ, Lee S,
Cha RH, Lee JW, Kim DK, Kim YS, Ye SK and Yang SH: STAT3 blockade
ameliorates LPS-induced kidney injury through macrophage-driven
inflammation. Cell Commun Signal. 22:4762024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhu H, Wang X, Wang X, Liu B, Yuan Y and
Zuo X: Curcumin attenuates inflammation and cell apoptosis through
regulating NF-κB and JAK2/STAT3 signaling pathway against acute
kidney injury. Cell Cycle. 19:1941–1951. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yan X, Cheng X, He X, Zheng W, Yuan X and
Chen H: HO-1 overexpressed mesenchymal stem cells ameliorate
sepsis-associated acute kidney injury by activating JAK/stat3
pathway. Cell Mol Bioeng. 11:509–518. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sawoo R and Bishayi B: TLR4/TNFR1 blockade
suppresses STAT1/STAT3 expression and increases SOCS3 expression in
modulation of LPS-induced macrophage responses. Immunobiology.
229:1528402024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang M, Liu S, Wu Z, Wang C, Zhang H,
Yang J, Guo C, Dai M, Wang X and Ren W: Porcine GWAS identifies
ACOT11 as regulator for macrophage IL-1β maturation via IFNGR2
palmitoylation. Sci China Life Sci. 68:3037–3050. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Fan W, Wang C, Xu K, Liang H and Chi Q:
Ccl5+ Macrophages drive pro-inflammatory responses and
neutrophil recruitment in sepsis-associated acute kidney injury.
Int Immunopharmacol. 143:1133392024. View Article : Google Scholar
|
|
64
|
Shi Y, Zhang Q, Bi H, Lu M, Tan Y, Zou D,
Ge L, Chen Z, Liu C, Ci W and Ma L: Decoding the multicellular
ecosystem of vena caval tumor thrombus in clear cell renal cell
carcinoma by single-cell RNA sequencing. Genome Biol. 23:872022.
View Article : Google Scholar
|
|
65
|
Nalio Ramos R, Missolo-Koussou Y,
Gerber-Ferder Y, Bromley CP, Bugatti M, Núñez NG, Tosello Boari J,
Richer W, Menger L, Denizeau J, et al: Tissue-resident
FOLR2+ macrophages associate with CD8+ T cell
infiltration in human breast cancer. Cell. 185:1189–1207.e25. 2022.
View Article : Google Scholar
|
|
66
|
Mitsi E, Kamng'ona R, Rylance J, Solórzano
C, Jesus Reiné J, Mwandumba HC, Ferreira DM and Jambo KC: Human
alveolar macrophages predominately express combined classical M1
and M2 surface markers in steady state. Respir Res. 19:662018.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sezginer O and Unver N: Dissection of
pro-tumoral macrophage subtypes and immunosuppressive cells
participating in M2 polarization. Inflamm Res. 73:1411–1423. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Fuchs AL, Costello SM, Schiller SM, Tripet
BP and Copié V: Primary human M2 macrophage subtypes are
distinguishable by aqueous metabolite profiles. Int J Mol Sci.
25:24072024. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang LX, Zhang SX, Wu HJ, Rong XL and Guo
J: M2b macrophage polarization and its roles in diseases. J Leukoc
Biol. 106:345–358. 2019. View Article : Google Scholar
|
|
70
|
Di X, Li Y, Wei J, Li T and Liao B:
Targeting fibrosis: From molecular mechanisms to advanced
therapies. Adv Sci (Weinh). 12:e24104162025. View Article : Google Scholar
|
|
71
|
Meng XM, Wang S, Huang XR, Yang C, Xiao J,
Zhang Y, To KF, Nikolic-Paterson DJ and Lan HY: Inflammatory
macrophages can transdifferentiate into myofibroblasts during renal
fibrosis. Cell Death Dis. 7:e24952016. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kuppe C, Ibrahim MM, Kranz J, Zhang X,
Ziegler S, Perales-Patón J, Jansen J, Reimer KC, Smith JR, Dobie R,
et al: Decoding myofibroblast origins in human kidney fibrosis.
Nature. 589:281–286. 2021. View Article : Google Scholar :
|
|
73
|
Liu H, Guan Q, Zhao P and Li J:
TGF-β-induced CCR8 promoted macrophage transdifferentiation into
myofibroblast-like cells. Exp Lung Res. 1–14. 2022.Epub ahead of
print. View Article : Google Scholar
|
|
74
|
Tang PC, Chung JY, Xue VW, Xiao J, Meng
XM, Huang XR, Zhou S, Chan AS, Tsang AC, Cheng AS, et al: Smad3
promotes cancer-associated fibroblasts generation via
macrophage-myofibroblast transition. Adv Sci (Weinh).
9:e21012352022. View Article : Google Scholar
|
|
75
|
Wang L, Tang W, Jiang L, Zhang D, Wang Z,
Guo R, Wang J and Xiao D: Inhibition of USP11 attenuates
sepsis-associated acute kidney injury by downregulating
TGFBR2/Smad3 signaling. Front Mol Biosci. 12:15715932025.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Liu X, Xu W, Feng J, Wang Y, Li K, Chen Y,
Wang W, Zhao W, Ge S and Li J: Adoptive cell transfer of
piezo-activated macrophage rescues immunosuppressed rodents from
life-threating bacterial infections. Nat Commun. 16:13632025.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhuang J, Hai Y, Lu X, Sun B, Fan R, Zhang
B, Wang W, Han B, Luo L, Yang L, et al: A self-assembled metabolic
regulator reprograms macrophages to combat cytokine storm and boost
sepsis immunotherapy. Research (Wash DC). 8:06632025.
|
|
78
|
Li Y, Qu G, Dou G, Ren L, Dang M, Kuang H,
Bao L, Ding F, Xu G, Zhang Z, et al: Engineered extracellular
vesicles driven by erythrocytes ameliorate bacterial sepsis by iron
recycling, toxin clearing and inflammation regulation. Adv Sci
(Weinh). 11:e23068842024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ye M, Zhao Y, Wang Y, Xie R, Tong Y, Sauer
JD and Gong S: NAD(H)-loaded nanoparticles for efficient sepsis
therapy via modulating immune and vascular homeostasis. Nat
Nanotechnol. 17:880–890. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhang S, Xie Y, Zhang L, Qi Y, Liao Q, Xu
C, Yang S, Zhou H, Tan Q and Qi S: A pH-responsive biomimetic
antioxidant nanoplatform with dual renal targeting for synergistic
therapy of acute kidney injury. Adv Sci (Weinh). 13:e156642026.
View Article : Google Scholar
|
|
81
|
Wang Y, Wang S, Luo Q, Zhou Y, Li X, Shang
Y, Wang H, Plotnikov EY, Liao X, Feng W, et al: Engineered
macrophage-targeted germacrone-bearing nanosheet achieves
synergistic treatment of acute kidney injury through macrophage
polarization modulation and oxidative stress alleviation. Chem Eng
J. 520:1656022025. View Article : Google Scholar
|
|
82
|
Ganesh K and Joshi MB: Neutrophil
sub-types in maintaining immune homeostasis during steady state,
infections and sterile inflammation. Inflamm Res. 72:1175–1192.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Xiao W, Lu Z, Liu Y, Hua T, Zhang J, Hu J,
Li H, Xu Y and Yang M: Influence of the initial neutrophils to
lymphocytes and platelets ratio on the incidence and severity of
sepsis-associated acute kidney injury: A double robust estimation
based on a large public database. Front Immunol. 13:9254942022.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL,
Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY and Lindsey
ML: Temporal neutrophil polarization following myocardial
infarction. Cardiovasc Res. 110:51–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ohms M, Möller S and Laskay T: An attempt
to polarize human neutrophils toward N1 and N2 phenotypes in vitro.
Front Immunol. 11:5322020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Garley M and Jabłońska E: Heterogeneity
among neutrophils. Arch Immunol Ther Exp (Warsz). 66:21–30. 2018.
View Article : Google Scholar
|
|
87
|
Woodfin A, Beyrau M, Voisin MB, Ma B,
Whiteford JR, Hordijk PL, Hogg N and Nourshargh S:
ICAM-1-expressing neutrophils exhibit enhanced effector functions
in murine models of endotoxemia. Blood. 127:898–907. 2016.
View Article : Google Scholar :
|
|
88
|
Herter JM, Rossaint J, Spieker T and
Zarbock A: Adhesion molecules involved in neutrophil recruitment
during sepsis-induced acute kidney injury. J Innate Immun.
6:597–606. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wu L, Gokden N and Mayeux PR: Evidence for
the role of reactive nitrogen species in polymicrobial
sepsis-induced renal peritubular capillary dysfunction and tubular
injury. J Am Soc Nephrol. 18:1807–1815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Bianchi ME and Manfredi AA: High-mobility
group box 1 (HMGB1) protein at the crossroads between innate and
adaptive immunity. Immunol Rev. 220:35–46. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Leliefeld PH, Wessels CM, Leenen LP,
Koenderman L and Pillay J: The role of neutrophils in immune
dysfunction during severe inflammation. Crit Care. 20:732016.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
De Filippo K, Dudeck A, Hasenberg M, Nye
E, van Rooijen N, Hartmann K, Gunzer M, Roers A and Hogg N: Mast
cell and macrophage chemokines CXCL1/CXCL2 control the early stage
of neutrophil recruitment during tissue inflammation. Blood.
121:4930–4937. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Sawant KV, Poluri KM, Dutta AK, Sepuru KM,
Troshkina A, Garofalo RP and Rajarathnam K: Chemokine CXCL1
mediated neutrophil recruitment: Role of glycosaminoglycan
interactions. Sci Rep. 6:331232016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Molema G, Zijlstra JG, van Meurs M and
Kamps JAAM: Renal microvascular endothelial cell responses in
sepsis-induced acute kidney injury. Nat Rev Nephrol. 18:95–112.
2022. View Article : Google Scholar
|
|
95
|
Aguilar MG, AlHussen HA, Gandhi PD, Kaur
P, Pothacamuri MA, Talikoti MAH, Avula N, Shekhawat P, Silva AB,
Kaur A and Rai M: Sepsis-associated acute kidney injury:
Pathophysiology and treatment modalities. Cureus.
16:e759922024.
|
|
96
|
Tang D, Kang R, Berghe TV, Vandenabeele P
and Kroemer G: The molecular machinery of regulated cell death.
Cell Res. 29:347–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Okeke EB, Louttit C, Fry C, Najafabadi AH,
Han K, Nemzek J and Moon JJ: Inhibition of neutrophil elastase
prevents neutrophil extracellular trap formation and rescues mice
from endotoxic shock. Biomaterials. 238:1198362020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Li Z, Ludwig N, Thomas K, Mersmann S,
Lehmann M, Vestweber D, Pittet JF, Gomez H, Kellum JA, Rossaint J
and Zarbock A: The pathogenesis of ischemia-reperfusion induced
acute kidney injury depends on renal neutrophil recruitment whereas
sepsis-induced AKI does not. Front Immunol. 13:8437822022.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Singhal A and Kumar S: Neutrophil and
remnant clearance in immunity and inflammation. Immunology.
165:22–43. 2022. View Article : Google Scholar
|
|
100
|
Jia Y, Li JH, Hu BC, Huang X, Yang X, Liu
YY, Cai JJ, Yang X, Lai JM, Shen Y, et al: Targeting SLC22A5
fosters mitophagy inhibition-mediated macrophage immunity against
septic acute kidney injury upon CD47-SIRPα axis blockade. Heliyon.
10:e267912024. View Article : Google Scholar
|
|
101
|
Borregaard N and Herlin T: Energy
metabolism of human neutrophils during phagocytosis. J Clin Invest.
70:550–557. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Jeon JH, Hong CW, Kim EY and Lee JM:
Current understanding on the metabolism of neutrophils. Immune
Netw. 20:e462020. View Article : Google Scholar
|
|
103
|
Stojkov D, Gigon L, Peng S, Lukowski R,
Ruth P, Karaulov A, Rizvanov A, Barlev NA, Yousefi S and Simon HU:
Physiological and pathophysiological roles of metabolic pathways
for NET formation and other neutrophil functions. Front Immunol.
13:8265152022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Tan C, Gu J, Chen H, Li T, Deng H, Liu K,
Liu M, Tan S, Xiao Z, Zhang H and Xiao X: Inhibition of Aerobic
glycolysis promotes neutrophil to influx to the infectious site via
CXCR2 in sepsis. Shock. 53:114–123. 2020. View Article : Google Scholar
|
|
105
|
Masucci MT, Minopoli M, Del Vecchio S and
Carriero MV: The emerging role of neutrophil extracellular traps
(NETs) in tumor progression and metastasis. Front Immunol.
11:17492020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kovacs SB, Oh C, Maltez VI, McGlaughon BD,
Verma A, Miao EA and Aachoui Y: Neutrophil caspase-11 is essential
to defend against a cytosol-invasive bacterium. Cell Rep.
32:1079672020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ni Y, Hu BC, Wu GH, Shao ZQ, Zheng Y,
Zhang R, Jin J, Hong J, Yang XH, Sun RH, et al: Interruption of
neutrophil extracellular traps formation dictates host defense and
tubular HOXA5 stability to augment efficacy of anti-Fn14 therapy
against septic AKI. Theranostics. 11:9431–9451. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Kaplan MJ and Radic M: Neutrophil
extracellular traps: Double-edged swords of innate immunity. J
Immunol. 189:2689–2695. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Wei Z, Wang J, Wang Y, Wang C, Liu X, Han
Z, Fu Y and Yang Z: Effects of neutrophil extracellular traps on
bovine mammary epithelial cells in vitro. Front Immunol.
10:10032019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wang H, Wang C, Zhao MH and Chen M:
Neutrophil extracellular traps can activate alternative complement
pathways. Clin Exp Immunol. 181:518–527. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Biron BM, Chung CS, Chen Y, Wilson Z,
Fallon EA, Reichner JS and Ayala A: PAD4 deficiency leads to
decreased organ dysfunction and improved survival in a dual insult
model of hemorrhagic shock and sepsis. J Immunol. 200:1817–1828.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Woodfin A, Voisin MB, Beyrau M, Colom B,
Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE,
et al: The junctional adhesion molecule JAM-C regulates polarized
transendothelial migration of neutrophils in vivo. Nat Immunol.
12:761–769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Garner H and de Visser KE: Neutrophils
take a round-trip. Science. 358:42–43. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
de Oliveira S, Rosowski EE and
Huttenlocher A: Neutrophil migration in infection and wound repair:
Going forward in reverse. Nat Rev Immunol. 16:378–391. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Bordon Y: Innate immunity: Neutrophil
U-turn fans the flames. Nat Rev Immunol. 11:4982011. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zi SF, Wu XJ, Tang Y, Liang YP, Liu X,
Wang L, Li SL, Wu CD, Xu JY, Liu T, et al: Endothelial cell-derived
extracellular vesicles promote aberrant neutrophil trafficking and
subsequent remote lung injury. Adv Sci (Weinh). 11:e24006472024.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Ya-Fen Z, Jing C, Yue-Fei Z and Chang-Ping
D: Reduction in NGAL at 48 h predicts the progression to CKD in
patients with septic associated AKI: A single-center clinical
study. Int Urol Nephrol. 56:607–613. 2024. View Article : Google Scholar
|
|
118
|
Strieter RM, Kunkel SL, Showell HJ, Remick
DG, Phan SH, Ward PA and Marks RM: Endothelial cell gene expression
of a neutrophil chemotactic factor by TNF-alpha, LPS, and IL-1
beta. Science. 243:1467–1469. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Zhang A, Cai Y, Wang PF, Qu JN, Luo ZC,
Chen XD, Huang B, Liu Y, Huang WQ, Wu J and Yin YH: Diagnosis and
prognosis of neutrophil gelatinase-associated lipocalin for acute
kidney injury with sepsis: A systematic review and meta-analysis.
Crit Care. 20:412016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Tavris BS, Morath C, Rupp C, Szudarek R,
Uhle F, Sweeney TE, Liesenfeld O, Fiedler-Kalenka MO, Dubler S,
Zeier M, et al: Complementary role of transcriptomic endotyping and
protein-based biomarkers for risk stratification in
sepsis-associated acute kidney injury. Crit Care. 29:1362025.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Bagshaw SM, Bennett M, Haase M,
Haase-Fielitz A, Egi M, Morimatsu H, D'amico G, Goldsmith D,
Devarajan P and Bellomo R: Plasma and urine neutrophil
gelatinase-associated lipocalin in septic versus non-septic acute
kidney injury in critical illness. Intensive Care Med. 36:452–461.
2010. View Article : Google Scholar
|
|
122
|
Mårtensson J, Bell M, Oldner A, Xu S,
Venge P and Martling CR: Neutrophil gelatinase-associated lipocalin
in adult septic patients with and without acute kidney injury.
Intensive Care Med. 36:1333–1340. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Tuan PNH, Quyen DBQ, Van Khoa H, Loc ND,
Van My P, Dung NH, Toan ND, Quyet D and Thang LV: Serum and urine
neutrophil gelatinase-associated lipocalin levels measured at
admission predict progression to chronic kidney disease in
sepsis-associated acute kidney injury patients. Dis Markers.
2020:88834042020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Mathur R, Elsafy S, Press AT, Brück J,
Hornef M, Martin L, Schürholz T, Marx G, Bartneck M, Kiessling F,
et al: Neutrophil hitchhiking enhances liposomal dexamethasone
therapy of sepsis. ACS Nano. 18:28866–28880. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Zhou M, Tang Y, Lu Y, Zhang T, Zhang S,
Cai X and Lin Y: Framework nucleic acid-based and neutrophil-based
nanoplatform loading baicalin with targeted drug delivery for
anti-inflammation treatment. ACS Nano. 19:3455–3469. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Zhou M, Lu Y, Tang Y, Zhang T, Xiao D,
Zhang M, Zhang S, Li J, Cai X and Lin Y: A DNA-based nanorobot for
targeting, hitchhiking, and regulating neutrophils to enhance
sepsis therapy. Biomaterials. 318:1231832025. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Chang YT, Lin CY, Chen CJ, Hwang E,
Alshetaili A, Yu HP and Fang JY: Neutrophil-targeted combinatorial
nanosystems for suppressing bacteremia-associated hyperinflammation
and MRSA infection to improve survival rates. Acta Biomater.
174:331–344. 2024. View Article : Google Scholar
|
|
128
|
Wu S, Zhou M, Zhou H, Han L and Liu H:
Astragaloside IV-loaded biomimetic nanoparticles target IκBα to
regulate neutrophil extracellular trap formation for sepsis
therapy. J Nanobiotechnology. 23:1552025. View Article : Google Scholar
|
|
129
|
Liu Z, Liu X, Yang Q, Yu L, Chang Y and Qu
M: Neutrophil membrane-enveloped nanoparticles for the amelioration
of renal ischemia-reperfusion injury in mice. Acta Biomater.
104:158–166. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Yang Y, Du J, Gan J, Song X, Shu J, An C,
Lu L, Wei H, Che J and Zhao X: Neutrophil-mediated nanozyme
delivery system for acute kidney injury therapy. Adv Healthc Mater.
13:e24011982024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Reilly JP, Anderson BJ, Hudock KM, Dunn
TG, Kazi A, Tommasini A, Charles D, Shashaty MG, Mikkelsen ME,
Christie JD and Meyer NJ: Neutropenic sepsis is associated with
distinct clinical and biological characteristics: A cohort study of
severe sepsis. Crit Care. 20:2222016. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Kurts C: Dendritic cells: Not just another
cell type in the kidney, but a complex immune sentinel network.
Kidney Int. 70:412–414. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Kurts C, Ginhoux F and Panzer U: Kidney
dendritic cells: Fundamental biology and functional roles in health
and disease. Nat Rev Nephrol. 16:391–407. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Weisheit CK, Engel DR and Kurts C:
Dendritic cells and macrophages: Sentinels in the kidney. Clin J Am
Soc Nephrol. 10:1841–1851. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Sun Y, Oravecz-Wilson K, Bridges S,
McEachin R, Wu J, Kim SH, Taylor A, Zajac C, Fujiwara H, Peltier
DC, et al: miR-142 controls metabolic reprogramming that regulates
dendritic cell activation. J Clin Invest. 129:2029–2042. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Trombetta ES, Ebersold M, Garrett W,
Pypaert M and Mellman I: Activation of lysosomal function during
dendritic cell maturation. Science. 299:1400–1403. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Efron P and Moldawer LL: Sepsis and the
dendritic cell. Shock. 20:386–401. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Worbs T, Hammerschmidt SI and Förster R:
Dendritic cell migration in health and disease. Nat Rev Immunol.
17:30–48. 2017. View Article : Google Scholar
|
|
139
|
Carson WF, Cavassani KA, Dou Y and Kunkel
SL: Epigenetic regulation of immune cell functions during
post-septic immunosuppression. Epigenetics. 6:273–283. 2011.
View Article : Google Scholar :
|
|
140
|
Fan HY, Qi D, Yu C, Zhao F, Liu T, Zhang
ZK, Yang MY, Zhang LM, Chen DQ and Du Y: Paeonol protects
endotoxin-induced acute kidney injury: Potential mechanism of
inhibiting TLR4-NF-κB signal pathway. Oncotarget. 7:39497–39510.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Naito Y, Tsuji T, Nagata S, Tsuji N,
Fujikura T, Ohashi N, Kato A, Miyajima H and Yasuda H: IL-17A
activated by Toll-like receptor 9 contributes to the development of
septic acute kidney injury. Am J Physiol Renal Physiol.
318:F238–F247. 2020. View Article : Google Scholar
|
|
142
|
Aksoy E, Taboubi S, Torres D, Delbauve S,
Hachani A, Whitehead MA, Pearce WP, Berenjeno IM, Nock G, Filloux
A, et al: The p110δ isoform of the kinase PI(3)K controls the
subcellular compartmentalization of TLR4 signaling and protects
from endotoxic shock. Nat Immunol. 13:1045–1054. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Mócsai A, Ruland J and Tybulewicz VLJ: The
SYK tyrosine kinase: A crucial player in diverse biological
functions. Nat Rev Immunol. 10:387–402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Al-Harbi NO, Nadeem A, Ahmad SF, Alanazi
MM, Aldossari AA and Alasmari F: Amelioration of sepsis-induced
acute kidney injury through inhibition of inflammatory cytokines
and oxidative stress in dendritic cells and neutrophils
respectively in mice: Role of spleen tyrosine kinase signaling.
Biochimie. 158:102–110. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Nadeem A, Ahmad SF, Al-Harbi NO, Ibrahim
KE, Alqahtani F, Alanazi WA, Mahmood HM, Alsanea S and Attia SM:
Bruton's tyrosine kinase inhibition attenuates oxidative stress in
systemic immune cells and renal compartment during sepsis-induced
acute kidney injury in mice. Int Immunopharmacol. 90:1071232021.
View Article : Google Scholar
|
|
146
|
Qi C, Liu C, Gong J, Liu D, Wang X, Zhang
P, Qin Y, Ge S, Zhang M, Peng Z, et al: Claudin18.2-specific CAR T
cells in gastrointestinal cancers: Phase 1 trial final results. Nat
Med. 30:2224–2234. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Bol KF, Schreibelt G, Bloemendal M, van
Willigen WW, Hins-de Bree S, de Goede AL, de Boer AJ, Bos KJH,
Duiveman-de Boer T, Olde Nordkamp MAM, et al: Adjuvant dendritic
cell therapy in stage IIIB/C melanoma: The MIND-DC randomized phase
III trial. Nat Commun. 15:16322024. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Benjamim CF, Lundy SK, Lukacs NW, Hogaboam
CM and Kunkel SL: Reversal of long-term sepsis-induced
immunosuppression by dendritic cells. Blood. 105:3588–3595. 2005.
View Article : Google Scholar
|
|
149
|
Wang HW, Yang W, Gao L, Kang JR, Qin JJ,
Liu YP and Lu JY: Adoptive transfer of bone marrow-derived
dendritic cells decreases inhibitory and regulatory T-cell
differentiation and improves survival in murine polymicrobial
sepsis. Immunology. 145:50–59. 2015. View Article : Google Scholar :
|
|
150
|
Piliponsky AM, Acharya M and Shubin NJ:
Mast cells in viral, bacterial, and fungal infection immunity. Int
J Mol Sci. 20:28512019. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Pundir P, Liu R, Vasavda C, Serhan N,
Limjunyawong N, Yee R, Zhan Y, Dong X, Wu X, Zhang Y, et al: A
connective tissue mast-cell-specific receptor detects bacterial
quorum-sensing molecules and mediates antibacterial immunity. Cell
Host Microbe. 26:114–122.e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Piliponsky AM, Chen CC, Rios EJ, Treuting
PM, Lahiri A, Abrink M, Pejler G, Tsai M and Galli SJ: The chymase
mouse mast cell protease 4 degrades TNF, limits inflammation, and
promotes survival in a model of sepsis. Am J Pathol. 181:875–886.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Sutherland RE, Olsen JS, McKinstry A,
Villalta SA and Wolters PJ: Mast cell IL-6 improves survival from
Klebsiella pneumonia and sepsis by enhancing neutrophil killing. J
Immunol. 181:5598–5605. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Ramos L, Peña G, Cai B, Deitch EA and
Ulloa L: Mast cell stabilization improves survival by preventing
apoptosis in sepsis. J Immunol. 185:709–716. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Urb M and Sheppard DC: The role of mast
cells in the defence against pathogens. PLoS Pathog.
8:e10026192012. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Amponnawarat A, Torres MDT, Krishnan R, de
la Fuente-Nunez C and Ali H: Synthetic peptides targeting a mast
cell-specific G protein-coupled receptor MRGPRB2 display
anti-infective potential. Cell Biomaterials. 1:1000882025.
View Article : Google Scholar
|
|
157
|
Madjene LC, Danelli L, Dahdah A, Vibhushan
S, Bex-Coudrat J, Pacreau E, Vaugier C, Claver J, Rolas L, Pons M,
et al: Mast cell chymase protects against acute ischemic kidney
injury by limiting neutrophil hyperactivation and recruitment.
Kidney Int. 97:516–527. 2020. View Article : Google Scholar
|
|
158
|
Blank U, Essig M, Scandiuzzi L, Benhamou M
and Kanamaru Y: Mast cells and inflammatory kidney disease. Immunol
Rev. 217:79–95. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Summers SA, Chan J, Gan PY, Dewage L,
Nozaki Y, Steinmetz OM, Nikolic-Paterson DJ, Kitching AR and
Holdsworth SR: Mast cells mediate acute kidney injury through the
production of TNF. J Am Soc Nephrol. 22:2226–2236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Wang F, Cui Y, He D, Gong L and Liang H:
Natural killer cells in sepsis: Friends or foes? Front Immunol.
14:11019182023. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Cooper MA, Fehniger TA and Caligiuri MA:
The biology of human natural killer-cell subsets. Trends Immunol.
22:633–640. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Seki S, Nakashima H, Nakashima M and
Kinoshita M: Antitumor immunity produced by the liver Kupffer
cells, NK cells, NKT cells, and CD8 CD122 T cells. Clin Dev
Immunol. 2011:8683452011. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Choi YH, Lim EJ, Kim SW, Moon YW, Park KS
and An HJ: IL-27 enhances IL-15/IL-18-mediated activation of human
natural killer cells. J Immunother Cancer. 7:1682019. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Maskalenko NA, Zhigarev D and Campbell KS:
Harnessing natural killer cells for cancer immunotherapy:
Dispatching the first responders. Nat Rev Drug Discov. 21:559–577.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Fehniger TA, Cooper MA, Nuovo GJ, Cella M,
Facchetti F, Colonna M and Caligiuri MA: CD56bright natural killer
cells are present in human lymph nodes and are activated by T
cell-derived IL-2: A potential new link between adaptive and innate
immunity. Blood. 101:3052–3057. 2003. View Article : Google Scholar
|
|
166
|
Brodin P, Kärre K and Höglund P: NK cell
education: Not an on-off switch but a tunable rheostat. Trends
Immunol. 30:143–149. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Uchida T, Seki S and Oda T: Infections,
reactions of natural killer T cells and natural killer cells, and
kidney injury. Int J Mol Sci. 23:4792022. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Schuster IS, Sng XYX, Lau CM, Powell DR,
Weizman OE, Fleming P, Neate GEG, Voigt V, Sheppard S, Maraskovsky
AI, et al: Infection induces tissue-resident memory NK cells that
safeguard tissue health. Immunity. 56:2173–2174. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Uchida T, Nakashima H, Ito S, Ishikiriyama
T, Nakashima M, Seki S, Kumagai H and Oshima N: Activated natural
killer T cells in mice induce acute kidney injury with hematuria
through possibly common mechanisms shared by human CD56+
T cells. Am J Physiol Renal Physiol. 315:F618–F627. 2018.
View Article : Google Scholar
|
|
170
|
Sato A, Nakashima H, Kinoshita M,
Nakashima M, Ogawa Y, Shono S, Ikarashi M and Seki S: The effect of
synthetic C-reactive protein on the in vitro immune response of
human PBMCs stimulated with bacterial reagents. Inflammation.
36:781–792. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Uchida T, Ito S, Kumagai H, Oda T,
Nakashima H and Seki S: Roles of natural killer T cells and natural
killer cells in kidney injury. Int J Mol Sci. 20:24872019.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Forel JM, Chiche L, Thomas G, Mancini J,
Farnarier C, Cognet C, Guervilly C, Daumas A, Vély F, Xéridat F, et
al: Phenotype and functions of natural killer cells in
critically-ill septic patients. PLoS One. 7:e504462012. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Giamarellos-Bourboulis EJ, Tsaganos T,
Spyridaki E, Mouktaroudi M, Plachouras D, Vaki I, Karagianni V,
Antonopoulou A, Veloni V and Giamarellou H: Early changes of
CD4-positive lymphocytes and NK cells in patients with severe
Gram-negative sepsis. Crit Care. 10:R1662006. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Tang J, Xie L, Liu H, Wu L, Li X, Du H,
Wang X, Li X and Yang Y: The effect of NK cell therapy on sepsis
secondary to lung cancer: A case report. Open Life Sci.
18:202207022023. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Zhou X, Wang Y, Dou Z, Delfanti G,
Tsahouridis O, Pellegry CM, Zingarelli M, Atassi G, Woodcock MG,
Casorati G, et al: CAR-redirected natural killer T cells
demonstrate superior antitumor activity to CAR-T cells through
multimodal CD1d-dependent mechanisms. Nat Cancer. 5:1607–1621.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Godfrey DI, MacDonald HR, Kronenberg M,
Smyth MJ and Van Kaer L: NKT cells: What's in a name? Nat Rev
Immunol. 4:231–237. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Liu Y, Wang G, Chai D, Dang Y, Zheng J and
Li H: iNKT: A new avenue for CAR-based cancer immunotherapy. Transl
Oncol. 17:1013422022. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Ambrosino E, Terabe M, Halder RC, Peng J,
Takaku S, Miyake S, Yamamura T, Kumar V and Berzofsky JA:
Cross-regulation between type I and type II NKT cells in regulating
tumor immunity: A new immunoregulatory axis. J Immunol.
179:5126–5136. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Halder RC, Aguilera C, Maricic I and Kumar
V: Type II NKT cell-mediated anergy induction in type I NKT cells
prevents inflammatory liver disease. J Clin Invest. 117:2302–2312.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Kezić A, Stajic N and Thaiss F: Innate
immune response in kidney ischemia/reperfusion injury: Potential
target for therapy. J Immunol Res. 2017:63054392017. View Article : Google Scholar
|
|
181
|
Heffernan DS, Chun TT, Monaghan SF, Chung
CS and Ayala A: Invariant natural killer T cells modulate the
peritoneal macrophage response to polymicrobial sepsis. J Surg Res.
300:211–220. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Hu CK, Venet F, Heffernan DS, Wang YL,
Horner B, Huang X, Chung CS, Gregory SH and Ayala A: The role of
hepatic invariant NKT cells in systemic/local inflammation and
mortality during polymicrobial septic shock. J Immunol.
182:2467–2475. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Etogo AO, Nunez J, Lin CY, Toliver-Kinsky
TE and Sherwood ER: NK but not CD1-restricted NKT cells facilitate
systemic inflammation during polymicrobial intra-abdominal sepsis.
J Immunol. 180:6334–6345. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Tang R, Jin P, Shen C, Lin W, Yu L, Hu X,
Meng T, Zhang L, Peng L, Xiao X, et al: Single-cell RNA sequencing
reveals the transcriptomic landscape of kidneys in patients with
ischemic acute kidney injury. Chin Med J (Engl). 136:1177–1187.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Zhang J, Han C, Dai H, Hou J, Dong Y, Cui
X, Xu L, Zhang M and Xia Q: Hypoxia-Inducible factor-2α limits
natural killer T cell cytotoxicity in renal ischemia/reperfusion
injury. J Am Soc Nephrol. 27:92–106. 2016. View Article : Google Scholar
|
|
186
|
Mohammadi H, Sharafkandi N, Hemmatzadeh M,
Azizi G, Karimi M, Jadidi-Niaragh F, Baradaran B and Babaloo Z: The
role of innate lymphoid cells in health and disease. J Cell
Physiol. 233:4512–4529. 2018. View Article : Google Scholar
|
|
187
|
Wang L, Tang J, Yang X, Zanvit P, Cui K,
Ku WL, Jin W, Zhang D, Goldberg N, Cain A, et al: TGF-β induces ST2
and programs ILC2 development. Nat Commun. 11:352020. View Article : Google Scholar
|
|
188
|
Wang Y, Luo P and Wuren T: Narrative
review of mesenchymal stem cell therapy in renal diseases:
Mechanisms, clinical applications, and future directions. Stem
Cells Int. 2024:86582462024. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Ryu S, Lee EY, Kim DK, Kim YS, Chung DH,
Kim JH, Lee H and Kim HY: Reduction of circulating innate lymphoid
cell progenitors results in impaired cytokine production by innate
lymphoid cells in patients with lupus nephritis. Arthritis Res
Ther. 22:632020. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Jacquelot N, Luong K and Seillet C:
Physiological regulation of innate lymphoid cells. Front Immunol.
10:4052019. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Gieseck RL III, Wilson MS and Wynn TA:
Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol.
18:62–76. 2018. View Article : Google Scholar
|
|
192
|
Becker M, Gnirck AC and Turner JE: Innate
lymphoid cells in renal inflammation. Front Immunol. 11:722020.
View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Nagashima R and Iyoda M: The roles of
kidney-resident ILC2 in renal inflammation and fibrosis. Front
Immunol. 12:6886472021. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Akcay A, Nguyen Q, He Z, Turkmen K, Won
Lee D, Hernando AA, Altmann C, Toker A, Pacic A, Ljubanovic DG, et
al: IL-33 exacerbates acute kidney injury. J Am Soc Nephrol.
22:2057–2067. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Cao Q, Wang Y, Niu Z, Wang C, Wang R,
Zhang Z, Chen T, Wang XM, Li Q, Lee VWS, et al: Potentiating
tissue-resident type 2 innate lymphoid cells by IL-33 to prevent
renal ischemia-reperfusion injury. J Am Soc Nephrol. 29:961–976.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Lai D, Chen W, Zhang K, Scott MJ, Li Y,
Billiar TR, Wilson MA and Fan J: GRK2 regulates group 2 innate
lymphoid cell mobilization in sepsis. Mol Med. 28:322022.
View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Dong X, Tu H, Qin S, Bai X, Yang F and Li
Z: Insights into the roles of B cells in patients with sepsis. J
Immunol Res. 2023:74089672023. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Sun S, Chen R, Dou X, Dai M, Long J, Wu Y
and Lin Y: Immunoregulatory mechanism of acute kidney injury in
sepsis: A narrative review. Biomed Pharmacother. 159:1142022023.
View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Inaba A, Tuong ZK, Riding AM, Mathews RJ,
Martin JL, Saeb-Parsy K and Clatworthy MR: B lymphocyte-derived
CCL7 augments neutrophil and monocyte recruitment, exacerbating
acute kidney injury. J Immunol. 205:1376–1384. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Han H, Zhu J, Wang Y, Zhu Z, Chen Y, Lu L,
Jin W, Yan X and Zhang R: Renal recruitment of B lymphocytes
exacerbates tubulointerstitial fibrosis by promoting monocyte
mobilization and infiltration after unilateral ureteral
obstruction. J Pathol. 241:80–90. 2017. View Article : Google Scholar
|
|
201
|
Coelho S, Cabral MG, Salvador R, Andrade
C, Martins A, Correia B, Freitas P, Cruzado JM and Jacinto A:
Urinary immune cell phenotype of severe AKI in critically ill
patients. Int Urol Nephrol. 54:2047–2055. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Hu YM, Hsiung YC, Pai MH and Yeh SL:
Glutamine administration in early or late septic phase
downregulates lymphocyte PD-1/PD-L1 expression and the inflammatory
response in mice with polymicrobial sepsis. JPEN J Parenter Enteral
Nutr. 42:538–549. 2018. View Article : Google Scholar
|
|
203
|
Liu Z, Dai B, Bao J and Pan Y: T cell
metabolism in kidney immune homeostasis. Front Immunol.
15:14988082024. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Waterhölter A, Krebs CF and Panzer U: γδ T
cells in immune-mediated kidney disease. Eur J Immunol.
54:e24510692024. View Article : Google Scholar
|
|
205
|
Abbas AK, Murphy KM and Sher A: Functional
diversity of helper T lymphocytes. Nature. 383:787–793. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Chung Y, Chang SH, Martinez GJ, Yang XO,
Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q and Dong
C: Critical regulation of early Th17 cell differentiation by
interleukin-1 signaling. Immunity. 30:576–587. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Dong X, Bachman LA, Miller MN, Nath KA and
Griffin MD: Dendritic cells facilitate accumulation of IL-17 T
cells in the kidney following acute renal obstruction. Kidney Int.
74:1294–1309. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Yang XY, Song J, Hou SK, Fan HJ, Lv Q, Liu
ZQ, Ding H, Zhang YZ, Liu JY, Dong WL and Wang X: Ulinastatin
ameliorates acute kidney injury induced by crush syndrome
inflammation by modulating Th17/Treg cells. Int Immunopharmacol.
81:1062652020. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
D'Alessio FR, Kurzhagen JT and Rabb H:
Reparative T lymphocytes in organ injury. J Clin Invest.
129:2608–2618. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Wardell CM, Boardman DA and Levings MK:
Harnessing the biology of regulatory T cells to treat disease. Nat
Rev Drug Discov. 24:93–111. 2025. View Article : Google Scholar
|
|
211
|
Kinsey GR, Huang L, Jaworska K,
Khutsishvili K, Becker DA, Ye H, Lobo PI and Okusa MD: Autocrine
adenosine signaling promotes regulatory T cell-mediated renal
protection. J Am Soc Nephrol. 23:1528–1537. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Wang Y and Tao Y: Research progress on
regulatory T cells in acute kidney injury. J Immunol Res.
2015:1741642015. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Sakaguchi S, Yamaguchi T, Nomura T and Ono
M: Regulatory T cells and immune tolerance. Cell. 133:775–787.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Kinsey GR, Sharma R, Huang L, Li L, Vergis
AL, Ye H, Ju ST and Okusa MD: Regulatory T cells suppress innate
immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol.
20:1744–1753. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Zhou X, Yao J, Lin J, Liu J, Dong L and
Duan M: Th17/regulatory T-cell imbalance and acute kidney injury in
patients with sepsis. J Clin Med. 11:40272022. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Shi X, Li J, Han Y, Wang J, Li Q, Zheng Y
and Li W: The α7 nicotinic acetylcholine receptor agonist
PNU-282987 ameliorates sepsis-induced acute kidney injury through
CD4+CD25+ regulatory T cells in rats. Bosn J Basic Med Sci.
22:882–893. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Li G, Zhang Y, He GL, Hao W and Hu W:
Impaired T lymphocyte subsets early predict acute kidney injury in
sepsis patients. Authorea. 2024.
|
|
218
|
Shih JM, Shih YM, Pai MH, Hou YC, Yeh CL
and Yeh SL: Fish oil-based fat emulsion reduces acute kidney injury
and inflammatory response in antibiotic-treated polymicrobial
septic mice. Nutrients. 8:1652016. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
de Pablo R, Monserrat J, Prieto A and
Alvarez-Mon M: Role of circulating lymphocytes in patients with
sepsis. Biomed Res Int. 2014:6710872014. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Guo XL, Lu CX, Luo Y, Wang PP, Su WS, Yang
SJ and Zhan LH: Circulating T-lymphocyte subsets as promising
biomarkers for the identification of sepsis-induced acute kidney
injury. J Chin Med Assoc. 87:1068–1077. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Xu J, Ma X, Yu K, Wang R, Wang S, Liu R,
Liu H, Gao H, Yu K and Wang C: Lactate up-regulates the expression
of PD-L1 in kidney and causes immunosuppression in septic acute
renal injury. J Microbiol Immunol Infect. 54:404–410. 2021.
View Article : Google Scholar
|
|
222
|
Luo C, Luo F, Man X, Liu X, Zhao L, Che L,
Zhang W, Guo J, Cai S, Wang D and Xu Y: Mesenchymal stem cells
attenuate sepsis-associated acute kidney injury by changing the
balance of Th17 cells/Tregs via Gal-9/Tim-3. Curr Stem Cell Res
Ther. 18:540–550. 2023. View Article : Google Scholar
|
|
223
|
Hou YC, Wu JM, Chen KY, Chen PD, Lei CS,
Yeh SL and Lin MT: Effects of prophylactic administration of
glutamine on CD4+ T cell polarisation and kidney injury
in mice with polymicrobial sepsis. Br J Nutr. 122:657–665. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Nozaki Y, Ri J, Sakai K, Niki K, Funauchi
M and Matsumura I: Protective effects of recombinant human soluble
thrombomodulin on lipopolysaccharide-induced acute kidney injury.
Int J Mol Sci. 21:25192020. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Singbartl K, Bockhorn SG, Zarbock A,
Schmolke M and Van Aken H: T cells modulate neutrophil-dependent
acute renal failure during endotoxemia: Critical role for CD28. J
Am Soc Nephrol. 16:720–728. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Ma K, Luo L, Yang M and Meng Y: The
suppression of sepsis-induced kidney injury via the knockout of T
lymphocytes. Heliyon. 10:e233112023. View Article : Google Scholar
|
|
227
|
Cho E, Lee JH, Lim HJ, Oh SW, Jo SK, Cho
WY, Kim HK and Lee SY: Soluble CD25 is increased in patients with
sepsis-induced acute kidney injury. Nephrology (Carlton).
19:318–324. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Patschan D, Heeg M, Brier M, Brandhorst G,
Schneider S, Müller GA and Koziolek MJ: CD4+ lymphocyte adenosine
triphosphate-a new marker in sepsis with acute kidney injury? BMC
Nephrol. 15:2032014. View Article : Google Scholar
|
|
229
|
Ostermann M, Forni LG, Joannidis M,
Kane-Gill SL, Legrand M, Lumlertgul N, McNicholas B, Meersch M,
Monard C, Pickkers P, et al: State of the art: Renal recovery after
AKI-from basic science to clinical practice. Intensive Care Med.
51:1490–1507. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Borges A and Bento L: Organ crosstalk and
dysfunction in sepsis. Ann Intensive Care. 14:1472024. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Virzì G, Day S, de Cal M, Vescovo G and
Ronco C: Heart-kidney crosstalk and role of humoral signaling in
critical illness. Crit Care. 18:2012014. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Li X, Yuan F and Zhou L: Organ crosstalk
in acute kidney injury: Evidence and mechanisms. J Clin Med.
11:66372022. View Article : Google Scholar : PubMed/NCBI
|