Introduction
The human epidermal growth factor receptor 2
(HER2)-positive subtype accounts for 15-20% of breast cancer cases
and is characterized by aggressive nature and poor clinical
outcomes (1). Over the past two
decades, the implementation of HER2-targeted therapeutics,
including monoclonal antibodies, small molecule tyrosine kinase
inhibitors and antibody-drug conjugates such as trastuzumab
emtansine and trastuzumab deruxtecan, has markedly improved patient
outcomes in patients with HER2-positive breast cancer (2). In the metastatic setting, these
therapeutic advances are associated with a median overall survival
exceeding 50 months, as demonstrated by clinical trials and
real-world studies conducted in North America and Europe (3-5).
Despite advances in HER2-targeted therapies, approximately 15-24%
of patients with HER2-positive breast cancer develop metastatic
disease, highlighting a persistent risk of disease progression
(6). Therapeutic resistance
remains a major limitation, underscoring the need for more
effective and durable therapeutic strategies (2,7).
In breast cancer, cancer stem cells (CSCs)
constitute a distinct subpopulation characterized by self-renewal
capacity, multilineage differentiation potential and resistance to
standard chemotherapy. These cells are commonly defined by specific
phenotypic markers, most notably a
CD44high/CD24low surface profile and elevated
aldehyde dehydrogenase 1 (ALDH1) activity, both of which are
associated with tumor initiation, progression, metastasis and poor
clinical outcomes (8-10). HER2-positive breast cancer
exhibits pronounced tumor heterogeneity and biological complexity,
typically harboring increased CSC-like populations, which actively
contribute to resistance mechanisms (11,12). In these resistant tumors, CSCs
exhibit increased activation of survival pathways, including
PI3K/AKT and Notch signaling, and CSC characteristics are further
promoted by HER2/HER3-mediated signaling, creating a
self-reinforcing cycle of resistance (13). Consequently, simultaneously
targeting HER2/HER3 signaling and CSC pathways may offer an
effective strategy to overcome trastuzumab resistance.
Recently, drug repurposing, which involves
identifying novel anticancer indications for existing
non-oncological agents, has emerged as a rational and
cost-effective strategy in therapeutic development (14). Disulfiram, an ALDH inhibitor that
targets CSC-like properties, has also been shown to inhibit HER2
signaling through proteasome inhibition and copper-dependent
oxidative stress (15,16), and is being evaluated in a Phase
II trial with copper in metastatic breast cancer (trial no.
NCT03323346). Metformin, a widely used antidiabetic agent, improves
clinical and pathological responses when combined with neoadjuvant
doxorubicin and cyclophosphamide followed by a taxane, in breast
cancer (trial no. NCT04170465) (17) and shows potential survival
benefits in the HER2-positive subgroup in the large-scale MA.32
Phase III trial (trial no. NCT01101438) (18). Together, findings suggest that
disulfiram and metformin are promising drug repurposing candidates
for breast cancer therapy.
Ebastine is a second-generation antihistamine with
favorable pharmacokinetic and safety profiles, including high oral
bioavailability, minimal central nervous system (CNS) penetration
and low systemic toxicity (19,20). Although primarily used for
allergic conditions, recent in vitro and in vivo
preclinical studies have shown its antitumor potential in various
types of cancer through diverse mechanisms such as EZH2 (enhancer
of zeste homolog 2) inhibition, autophagy induction and suppression
of angiogenesis (21-23). Our previous study demonstrated
that ebastine suppresses metastatic progression in triple-negative
breast cancer by targeting focal adhesion kinase, leading to
inhibition of STAT3/ERK signaling and decreased CSC-like properties
(22). These findings highlight
the potential of ebastine as a multi-targeted anticancer, agent
although its mechanism of action in the context of HER2-positive
breast cancer remains to be fully elucidated. The present study
investigated the potential of ebastine to overcome trastuzumab
resistance by targeting HER2/HER3 heterodimerization and CSC-like
properties in vitro and evaluated its antitumor efficacy
in vivo using a trastuzumab-resistant xenograft model.
Materials and methods
Reagents and antibodies
Ebastine was purchased from Selleck Chemicals.
Propidium iodide (PI), Triton X-100 and DMSO were obtained from
Sigma-Aldrich (Merck KGaA). Phosphatase and protease inhibitor
cocktail tablets were obtained from Roche Applied Sciences. RNase A
was purchased from Invitrogen (Thermo Fisher Scientific, Inc.).
Primary antibodies were as follows: Ki-67 (cat. no. ab16667), CD31
(cat. no. ab28364), ALDH1A1 (Abcam; cat. no. ab52492), Bcl-2
(Abcam; cat. no. ab692) and CD44 (all Abcam; cat. no. ab254530);
HER2 (Cell Signaling Technology, Inc.; cat. no. 2165), HER3 (Cell
Signaling Technology, Inc.; cat. no. 12708), phosphorylated
(p-)HER2 (Y1221/1222; Cell Signaling Technology, Inc.; cat. no.
2243), p-HER3 (Y1289; Cell Signaling Technology, Inc.; cat. no.
2842), Akt (Cell Signaling Technology, Inc.; cat. no. 9272), p-Akt
(S473; Cell Signaling Technology, Inc.; cat. no. 4060), PARP (Cell
Signaling Technology, Inc.; cat. no. 9542), cleaved PARP (Cell
Signaling Technology, Inc.; cat. no. 5625), caspase-3 (Cell
Signaling Technology, Inc.; cat. no. 7148), -7 (Cell Signaling
Technology, Inc.; cat. no. 12827) and -8 (Cell Signaling
Technology, Inc.; cat. no. 4790), cleaved caspase-3 (Cell Signaling
Technology, Inc.; cat. no. 9664), -7 (Cell Signaling Technology,
Inc.; cat. no. 8438) and -8 (Cell Signaling Technology, Inc.; cat.
no. 9496), Bax (Cell Signaling Technology, Inc.; cat. no. 2772) and
vimentin (Cell Signaling Technology, Inc.; cat. no. 5741);
anti-intracellular domain (ICD) HER2 clone 4B5 (Ventana Medical
Systems; cat. no. 790-4493) and GAPDH (Invitrogen; Thermo Fisher
Scientific, Inc.; cat. no. MA5-15738). Secondary antibodies
included HRP-conjugated anti-mouse (Bio-Rad Laboratories, Inc.;
cat. no. 1721011) and anti-rabbit IgG (Bio-Rad Laboratories, Inc.;
cat. no. 1706515), as well as Alexa Fluor 594-conjugated goat
anti-rabbit IgG (Invitrogen; Thermo Fisher Scientific, Inc.; cat.
no. A-11037), Alexa Fluor 488-conjugated goat anti-rabbit IgG
(Invitrogen; Thermo Fisher Scientific, Inc.; cat. no. A-11008),
Alexa Fluor 594-conjugated goat anti-mouse IgG (Invitrogen; Thermo
Fisher Scientific, Inc.; cat. no. A-11032) and Alexa Fluor
488-conjugated goat anti-mouse IgG (Invitrogen; Thermo Fisher
Scientific, Inc.; cat. no. A-11001).
Breast cancer cell lines
The human breast cancer cell lines SKBR3, BT474,
MDA-MB-453 (American Type Culture Collection) and JIMT-1 (Leibnitz
Institute DSMZ-German Collection of Microorganisms and Cell
Cultures GmbH) were cultured in DMEM, MEM or RPMI-1640 (all
Sigma-Aldrich; Merck KGaA) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.) and 100 U/ml penicillin-streptomycin at
37°C in a humidified atmosphere of 5% CO2. All cell
lines were passaged for <6 months and were authenticated by
short tandem repeat profiling performed by Macrogen, Inc. The study
design is summarized in Fig.
S1.
Cell viability assay
Cell viability was measured using the CellTiter
96® Aqueous One Solution Cell Proliferation Assay (MTS)
according to the manufacturer's instructions (Promega Corporation).
The quantity of formazan product was determined by measuring
absorbance at 490 nm using a SpectraMax® 190 microplate
reader (Molecular Devices LLC).
Sub-G1 analysis and Annexin V/PI
assay
For cell cycle analysis, JIMT-1, MDA-MB-453, SKBR3
and BT474 cells were fixed with pre-chilled 95% ethanol containing
0.5% Tween-20 at 4°C for 24 h, then incubated with PI and RNase A
(both 50 μg/ml) at room temperature for 30 min. Apoptotic
cell death was determined as the sum of early apoptotic (Annexin
V+/PI−) and late apoptotic (Annexin
V+/PI+) populations, using the FITC Annexin V
Apoptosis Detection kit (BD Biosciences), according to the
manufacturer's instructions. Stained cells were analyzed by flow
cytometry using a BD LSRFortessa™ X-20 Cell Analyzer (BD
Biosciences), and BD FACSDiva™ software (version 8.0.1; BD
Biosciences).
Aldefluor-positivity assay and
CD44high/CD24low staining
An Aldefluor™ assay kit (Stemcell Technologies,
Inc.) was used to assess ALDH1 activity, according to the
manufacturer's instructions. BT474 and SKBR3 cells were incubated
for 45 min at 37°C in Aldefluor assay buffer containing the ALDH
substrate BODIPY-aminoacetaldehyde (1 μM/0.5x106
cells). The ALDH1-specific inhibitor diethylamino-benzaldehyde (50
mM) was used to define the baseline Aldefluor fluorescence. For
CD44/CD24 analysis, cells (1x106) were immunostained at
4°C for 30 min with FITC-conjugated anti-CD24 (cat. no. 555427),
PE-conjugated anti-CD44 (BD Biosciences; cat. no. 555479),
FITC-(cat. no. 553456) or PE-conjugated anti-mouse IgG (all 1:50;
all BD Biosciences; cat. no. 555749), followed by flow cytometric
analysis using a BD LSRFortessa X-20 flow cytometer (BD
Biosciences). Data acquisition and analysis were performed using BD
FACSDiva software (version 8.0.1; BD Biosciences).
Immunoblot analysis
JIMT-1, SKBR3 and BT474 cells were lysed in cold
lysis buffer [0.5% Triton X-100, 30 mM NaCl, 50 mM Tris-HCl (pH
7.4)] containing protease and phosphatase inhibitor cocktail
tablets. The supernatant was collected following centrifugation
(14,000 × g at 4°C for 20 min), and protein concentrations were
quantified using a Quick Start™ Bradford Protein Assay (Bio-Rad
Laboratories, Inc.). Equal amounts of protein (25 μg/lane)
were separated by SDS-PAGE using 8-15% gradient gels and
transferred to PVDF membranes. Membranes were blocked with 5%
skimmed milk for 30 min at room temperature and incubated overnight
at 4°C with primary antibodies diluted in 5% BSA (Sigma-Aldrich;
Merck KGaA; cat. no. A7906-100G) as follows: HER2 (1:2,000), p-HER2
(1:1,000), HER3, p-HER3, Akt, p-Akt, PARP, cleaved PARP, caspase-3,
-7 and -8, cleaved caspase-3, -7 and -8, Bcl-2, Bax (all 1:2,000)
or GAPDH (1:3,000). Following washing with 1X PBST, membranes were
incubated with HRP-conjugated anti-mouse (1:3,000-1:10,000; cat.
no. 1721011) or anti-rabbit secondary antibodies (1:3,000-1:5,000;
both Bio-Rad Laboratories, cat. no. 1706515) for 1 h at room
temperature. Signal intensity was detected using a
Chemiluminescence kit (Thermo Fisher Scientific, Inc.) on X-ray
film (AGFA HealthCare) and quantified using AlphaEaseFC software
(version 4.0.0; Alpha Innotech).
Immunoprecipitation assay
A Dynabeads™ Protein G Immunoprecipitation kit
(Thermo Fisher Scientific Inc.) was used to evaluate
protein-protein interactions according to the manufacturer's
instructions. Cells were lysed in Pierce® IP lysis
buffer (Thermo Fisher Scientific, Inc.) supplemented with
phosphatase and protease inhibitor cocktails. The supernatant was
collected by centrifugation (14,000 × g at 4°C for 20 min) and
equal amounts (1,000 μg) were incubated with 10 μg
anti-HER2 antibody conjugated to Dynabeads Protein G at 4°C
overnight. The protein complexes were eluted by boiling the beads
in a mixture of SDS-PAGE sample buffer and elution buffer (1:1),
followed by SDS-PAGE and immunoblotting as described above.
Cellular thermal shift assay (CETSA)
CETSA was performed to evaluate the direct
engagement of ebastine with endogenous HER2. 293T (American Type
Culture Collection) cells were cultured overnight at 37°C in a
humidified atmosphere with 5% CO2, and treated with
either DMSO (vehicle) or 30 μM ebastine for 1 h at 37°C.
Cells were harvested, resuspended in PBS containing protease and
phosphatase inhibitor cocktails (Roche Applied Science) and
aliquoted into PCR tubes. Samples were heated at 45-63°C for 3 min
using a thermal cycler, followed by cooling at room temperature for
3 min. The heated cells were lysed by three freeze-thaw cycles
consisting of freezing in liquid nitrogen (-196 °C) for 3 min
followed by thawing at room temperature for 1 min, and soluble
fractions were obtained by centrifugation (14,000 × g at 4°C for 20
min). Supernatants were collected and analyzed by immunoblotting
using anti-HER2 antibody, as aforementioned.
Mammosphere formation assay
BT474 (5x104/ml) and JIMT-1 cells
(1.5x104/ml) were plated in ultra-low attachment dishes
and cultured under serum-free suspension conditions in HuMEC basal
serum-free medium (Gibco; Thermo Fisher Scientific, Inc.) for 5
days (BT474) or 8 days (JIMT-1) supplemented with B27 (1:50,
Invitrogen; Thermo Fisher Scientific, Inc.), 20 ng/ml human EGF and
basic fibroblast growth factor (both Sigma-Aldrich; Merck KGaA), 1%
antibiotic-antimycotic (Gibco; Thermo Fisher Scientific, Inc.; cat.
no. 15240-062), 4 μg/ml heparin and 15 μg/ml
gentamycin at 37°C in a 5% CO2 atmosphere. The number
and volume of mammospheres were assessed using an inverted light
microscope, CKX53 (Olympus Corporation), and calculated using the
formula: Volume=(4/3) × π × r3, where r is the
radius.
Animals and in vivo xenograft
experiment
All animal procedures were conducted in accordance
with the Guide for the Care and Use of Laboratory Animals and
approved by the Institutional Animal Care and Use Committee
(approval no. KOREA-2021-0070-C1) of Korea University College of
Medicine, Seoul, Republic of Korea. Female BALB/c nude mice (age, 5
weeks; n=10; initial body weight, 16-17 g) were purchased from NARA
Biotech, housed under standard specific pathogen-free conditions
(temperature, 22±2°C; humidity, 50±10%; 12-h light/dark cycle) and
acclimated for 1 week with free access to food and water. JIMT-1
cells (3.5x106) were inoculated into the fourth mammary
fat pads of the mice. When the mean tumor volume reached 100
mm3, mice were randomized into two groups (n=5/group)
and administered either a solvent control (DMSO/corn oil, 1:9) or
ebastine (20 mg/kg body weight/day) via intraperitoneal injection
every other day for 46 days. Tumor volume and body weight were
measured twice/week. Tumor volume was calculated using the formula:
Volume=(length × width2)/2. At study termination, the
largest tumor measured 14.42 mm in diameter with a volume of
1,873.55 mm3, which was within the tumor-size limits
permitted by the approved IACUC protocol. Mice were euthanized
using a gradual-fill CO2 method, starting at an initial
concentration of 20% CO2 and gradually increasing to 70%
CO2, followed by cervical dislocation to ensure complete
euthanasia, in accordance with the IACUC-approved protocol. Death
was confirmed by the absence of respiration and heartbeat.
Molecular modeling and docking
analysis
Molecular docking studies were performed using
publicly available platforms for protein-ligand virtual screening,
including GalaxySagittarius (galaxy.seoklab.org/), DockThor (dockthor.lncc.br/) and CB-Dock2 (cadd.labshare.cn/cb-dock2/). Following completion
of the docking simulations, 2D and 3D visualization of
protein-ligand interactions, along with predicted binding
affinities and binding energies, was performed using UCSF Chimera
1.16 (cgl.ucsf.edu/chimera/) and BIOVIA
Discovery Studio 2021 (discover.3ds.com/discovery-studio-visualizer-download/).
Serum biochemistry for liver and renal
injury biomarkers
At the time of sacrifice, blood samples (0.5-1.0 ml)
were collected from each animal, and serum was obtained by
centrifugation at 1,100 × g for 20 min at 4°C. Serum biochemical
markers of liver and renal function, including aspartate
aminotransferase (AST), alanine aminotransferase (ALT), total
bilirubin (TBL), blood urea nitrogen (BUN) and creatinine, were
evaluated using a serum biochemistry profiling service by DKKorea
Inc.
Immunohistochemistry with apoptosis
in-situ localization (TUNEL)
Tumors were fixed in 4% paraformaldehyde at room
temperature for 24 h and embedded in paraffin. Tissue sections (5
μm thickness) were mounted on positively charged glass
slides, deparaffinized with xylene and rehydrated through a graded
ethanol series to water. For antigen retrieval, sections were
boiled in citric acid buffer (pH 6.0) at ~95-100°C for 20 min. The
sections were incubated overnight at 4°C with primary antibodies
diluted in antibody diluent [Ki-67, CD31, HER2, 4B5, HER3, ALDH1A1,
CD44 (all 1:100) and vimentin (1:300)], followed by incubation with
Alexa Fluor® 488- or 594-conjugated secondary antibodies
at room temperature for 2 h. Slides were mounted with ProLong™ Gold
Antifade Reagent with DAPI at room temperature for 1 h. In
situ TUNEL assays were performed on tissue sections using a
TUNEL kit (Roche Applied Sciences; cat. no. 11684795910), according
to the manufacturer's instructions. Briefly, sections were
incubated with the TUNEL reaction mixture at 37°C for 1 h, and
fluorescence signals were evaluated in at least five randomly
selected fields of view per section. All images were acquired using
a confocal microscope, and fluorescence intensity was analyzed
using the histogram tool in ZEN Blue software (version 3.2; Carl
Zeiss Microscopy GmbH).
Hematoxylin and eosin (H&E)
staining
Organs (kidney, liver and lung) were excised, fixed
in 10% neutral-buffered formalin at room temperature for 24-48 h,
embedded in paraffin, and sectioned at 4 μm thickness.
Sections were deparaffinized in xylene, rehydrated through graded
ethanol to water, and stained with hematoxylin for 3 min followed
by eosin for 1 min at room temperature, followed by dehydration,
clearing and mounting. H&E-stained sections were examined under
a light microscope to assess histopathological abnormality.
Whole-slide images were acquired using a slide scanner (Axio Scan,
Z1; Carl Zeiss Microscopy GmbH), and digital images were used for
histopathological evaluation and quantitative analysis.
Public datasets and bioinformatics
analysis
For survival analyses, mRNA expression data and
clinical information of patients with breast cancer were obtained
from the UCSC Xena TCGA-BRCA cohort (tcga.xenahubs.net; dataset IDs:
TCGA.BRCA.sampleMap/HiSeqV2 and TCGA.BRCA.
sampleMap/BRCA_clinicalMatrix) (24) and the GENT2 database (gent2.appex.kr/gent2/; GEO-derived; GPL570 and GPL96)
(25). Patients were stratified
into high- and low-expression groups based on the median expression
of each gene (TCGA: CD44=13.023, ALDH1A1=8.5464, ERBB2=12.707; GEN
T2: CD4 4 =9.894818, A LDH1A1=7.870365, ERBB2=9.210671). Survival
regression curves were generated using GraphPad Prism 9.0 software
(Dotmatics) and overall survival was analyzed for up to 200 months
using the log-rank test.
Statistical analysis
All data were analyzed using GraphPad Prism 9.0
statistical software (Dotmatics). All data are presented as the
mean ± SD from at least three independent experiments. Statistical
comparisons were performed using unpaired Student's t-test or one-
or two-way ANOVA. For multiple group comparisons, the Bonferroni
post hoc test was applied. Spearman's rank correlation coefficient
was calculated to determine the association between ALDH1A1 and
CD44 expression in patients with HER2-positive breast cancer.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Ebastine induces apoptosis in
trastuzumab-resistant HER2-positive breast cancer cells
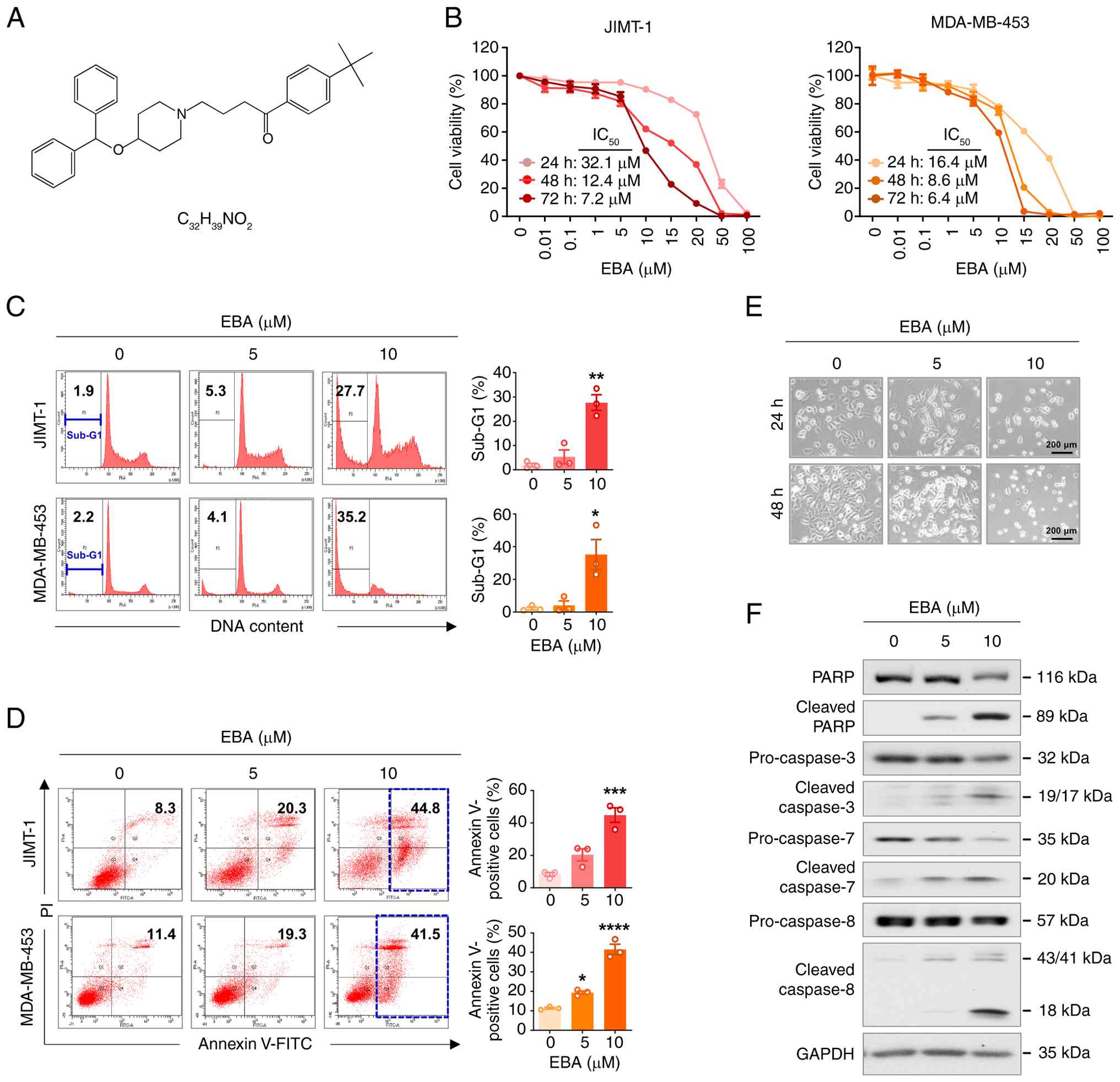
Ebastine is a piperidine derivative containing a
diphenylmethoxy group, connected via a butan-1-one chain to a
4-tert-butylphenyl group. This structural configuration enhances
membrane permeability and metabolic stability (Fig. 1A) (19,26). To evaluate its therapeutic
potential in overcoming trastuzumab resistance, the present study
examined whether ebastine inhibited the viability of HER2-positive
breast cancer cells with established trastuzumab resistance. The
JIMT-1 cell line, derived from a patient with intrinsic resistance
to trastuzumab, and the MDA-MB-453 cell line, established from the
pleural effusion of a patient with metastatic breast cancer and
known to be insensitive to trastuzumab, were used as representative
models (27-29). Compared with
trastuzumab-sensitive BT474 and SKBR3 cells, these resistant lines
showed notably lower basal HER2/HER3 expression and
phosphorylation, consistent with their decreased responsiveness to
trastuzumab (Fig. S2).
Ebastine (0-100 μM, 24-72 h) decreased
viability in JIMT-1 and MDA-MB-453 cells in a time- and
dose-dependent manner (Fig. 1B).
To determine whether this inhibition was associated with apoptosis,
the present study performed DNA content analysis to quantify the
sub-G1 population. A significant accumulation of HER2-positive
breast cancer cells in the sub-G1 phase was occurred following
ebastine treatment (10 μM, 48 h, Fig. 1C). Ebastine treatment led to an
increase in early and late apoptotic JIMT-1 and MDA-MB-453 cells
(Fig. 1D). Consistent with these
findings, ebastine-treated JIMT-1 cells exhibited characteristic
morphological features of apoptosis, including nuclear
condensation, cytoplasmic shrinkage and detachment from the culture
substrate (Fig. 1E).
Mechanistically, ebastine-induced apoptosis involved caspase
activation, evidenced by the cleavage of caspase-8 and activation
of executioner caspases-3, -7, and -8 (Fig. 1F). Notable PARP cleavage
indicated that cell death occurred through a caspase-dependent
pathway.
The present study evaluated the effect of ebastine
on trastuzumab-sensitive HER2-positive breast cancer BT474 and
SKBR3 cells. Ebastine (0-100 μM, up to 72 h) decreased cell
viability in a dose- and time-dependent manner (Fig. S3A and B). The apoptotic features
included notable morphological changes (Fig. S3C), a marked increase in the
sub-G1 population (Fig. S3D),
and an elevated proportion of Annexin V-positive cells (Fig. S3E) following ebastine treatment
(5-10 μM, 48 h). These effects coincided with activation of
caspases-3 and -7, increased PARP cleavage (Fig. S3F and G) and increased p18Bax
levels without altering Bcl-2 expression (Fig. S3H and I).
Ebastine downregulates HER2/HER3 and
truncated-p95HER2
Trastuzumab resistance is primarily driven by HER2,
HER3 and the truncated isoform p95HER2, which maintain pro-survival
PI3K/AKT signaling through persistent HER2/HER3 heterodimerization
(30). Ebastine (5-10 μM,
48 h) notably decreased total and p-HER2 (Y1221/1222), as well as
HER3 and p-HER3 (Y1289) in JIMT-1 (Fig. 2A and B) cells. p95HER2, which
lacks the extracellular domain required for trastuzumab binding,
retains HER2 kinase activity and contributes to therapeutic
resistance (31,32). Ebastine also downregulated
cleaved p95HER2 and suppressed the expression of AKT and p-AKT
(Fig. 2A and B). Since AKT
functions downstream of the HER2/HER3 signaling pathway, the
present study examined its phosphorylation status as an indicator
of downstream signaling (33,34). Immunoprecipitation analysis using
anti-HER2 revealed that ebastine substantially impaired the
heterodimerization of HER2 with both HER3 and EGFR (Fig. 2C). Similarly, in
trastuzumab-sensitive BT474 and SKBR3 cells, ebastine decreased the
phosphorylation of HER2, HER3, and p95HER2, leading to decreased
AKT activation (Fig. S4). The
present study performed molecular docking simulation to determine
whether this was due to the binding of ebastine to HER2 (Fig. 2D-F). Docking studies using the
crystal structure of the HER2 kinase domain retrieved from the
Protein Data Bank revealed that ebastine fit into the ATP-binding
pocket near the hinge region between the N- and C-lobe of the
tyrosine kinase domain (Fig. 2D and
F). This interaction was stabilized by three hydrogen bonds,
several π-interactions and hydrophobic contacts. Ebastine formed
hydrogen bonds with key residues within the ATP-binding site of
HER2-KD (HER2 kinase domain), including Thr862, Asp863 and Lys753
and a π-cation interaction occurred between the active site residue
Lys753 and a phenyl ring of ebastine (Fig. 2E). To validate this predicted
interaction, CETSA was performed using 293T cells. Ebastine (30
μM, 1 h) markedly enhanced the thermal stability of
endogenous HER2 compared with DMSO-treated controls. In
DMSO-treated samples, the soluble fraction of HER2 was largely lost
at temperatures >60°C, whereas ebastine-treated cells
demonstrated detectable HER2 up to ~63°C (Fig. 2G). These results indicated that
ebastine stabilized HER2 in cells, supporting the hypothesis of a
physical interaction between the proteins. Collectively, molecular
docking and CETSA data suggested that ebastine may interact with
the HER2 kinase domain and was associated with decreased HER2
phosphorylation, which may overcome trastuzumab resistance in
HER2-positive breast cancer.
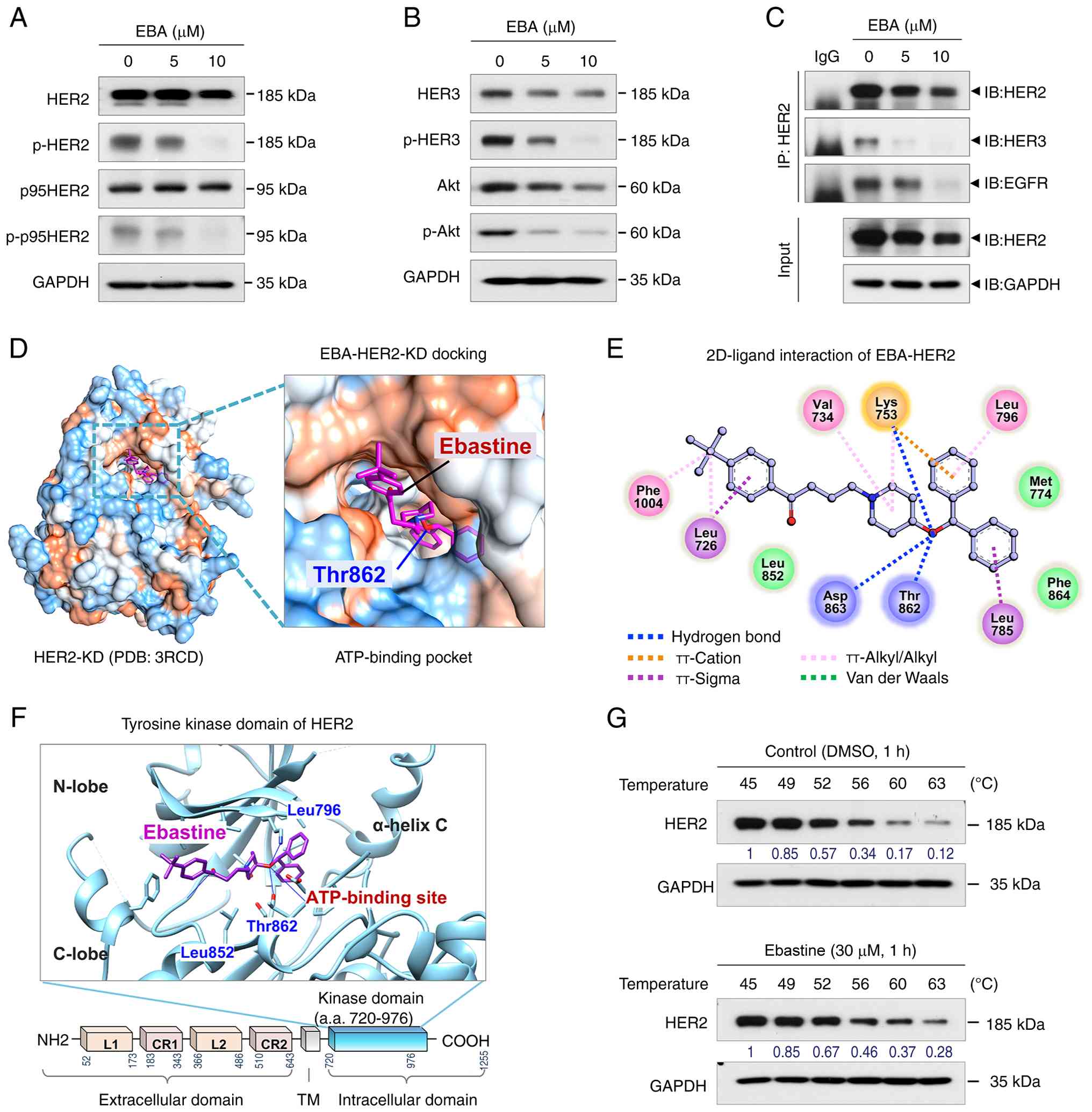 | Figure 2EBA downregulates HER2, p95HER2, HER3
and AKT expression. (A) Immunoblot analysis of HER2, p95HER2 and
p-HER2 (Y1221/1222) in JIMT-1 cells treated with EBA for 48 h. (B)
Immunoblot analysis of HER3, p-HER3 (Y1289), AKT and p-AKT
following treatment with EBA (48 h) in JIMT-1 cells. (C) Immunoblot
analysis of HER2, HER3 and EGFR following IP with anti-HER2
antibody in JIMT-1 cells treated with EBA. In silico
molecular docking of EBA with the crystal structure of HER2-KD. (D)
Surface map of lipophilic and hydrophilic properties at the
ATP-binding site of HER2-KD (red, hydrophobic; blue, hydrophilic).
(E) 2D interaction diagram showing intermolecular interactions
between EBA and HER2-KD. Key amino acid residues within the binding
pocket are shown. (F) Predicted binding pose of EBA (purple stick
model) within the tyrosine kinase domain of HER2 (blue ribbon). (G)
293T cells were treated with DMSO or EBA for 1 h at 37°C, followed
by heating for 3 min. Soluble fractions were collected following
centrifugation and analyzed by immunoblotting using an anti-HER2
antibody. EBA, ebastine; p-, phosphorylated; IP,
immunoprecipitation; IB, immunoblotting; KD, kinase domain PCB,
protein complex binding; TM, transmembrane; a.a., amino acid. |
Ebastine targets breast CSC (BCSC)-like
properties in HER2-positive breast cancer
BCSCs are defined by their self-renewal and
tumor-initiating capacity and characterized by distinct molecular
markers and functional attributes (35). ALDH1, a detoxifying enzyme for
intracellular aldehydes, contributes to maintaining the stem-like
state in tumor cells with activated cancer hallmarks (10). HER2-positive breast cancer
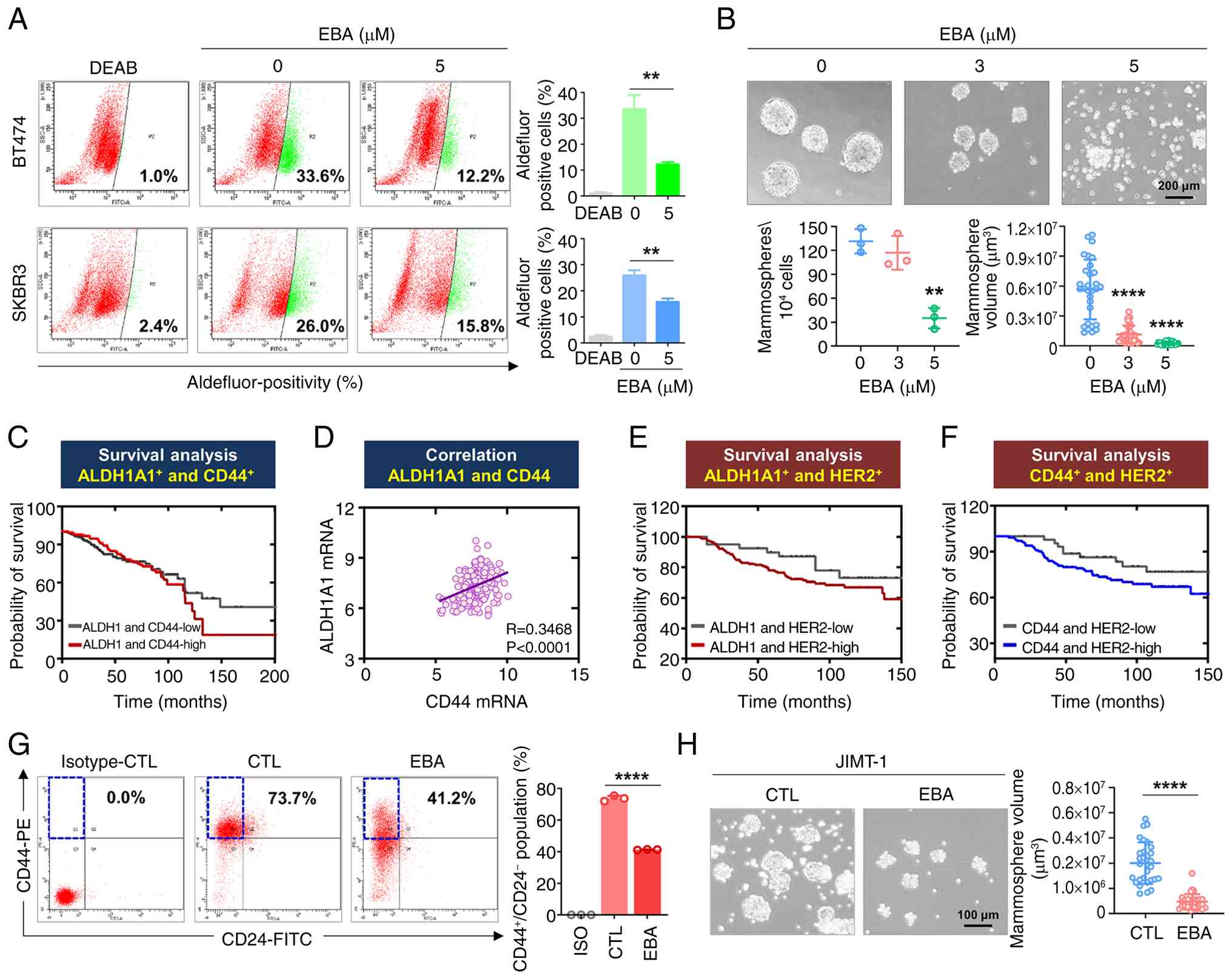
exhibits the highest ALDH1 activity among all subtypes (15). Ebastine significantly decreased
ALDH1 activity in BT474 and SKBR3 cells (Fig. 3A). To evaluate self-renewal, the
present study performed 3D mammosphere assay, which functionally
assess the tumor-initiating potential of BCSCs (36). Ebastine significantly decreased
mammosphere number and volume in BT474 cells (P<0.01, Fig. 3B), indicating impaired
self-renewal.
The CD44high/CD24low phenotype
marks a CSC-like population in breast cancer and is associated with
increased invasiveness, a key feature of early metastasis (10,37). Tumorigenesis can be initiated by
a limited number of cells, requiring ~500 Aldefluor-positive cells
or 20 ALDH+/CD44+/CD24− cells
(10). Kaplan-Meier analysis
showed that patients with breast cancer with high expression of
ALDH1A1 and CD44 had a shorter overall survival (Fig. 3C). Gene expression analysis of
GENT2 dataset showed a significant positive correlation between
CD44 and ALDH1A1 in patients with HER2-high breast cancer (Fig. 3D). High expression of either
ALDH1A1 (Fig. 3E) or CD44
(Fig. 3F) was associated with
poor prognosis in this HER2-overexpressing patient group. These
findings indicated that CSC markers were clinically associated with
poor outcomes in patients with HER2 overexpression, supporting the
mechanistic relevance of ebastine CSC-targeting effects observed
in vitro. CD44-associated gene expression is positively
associated with trastuzumab response, suggesting CD44 as a
potential biomarker (38).
Ebastine treatment significantly decreased the
CD44high/CD24low stem-like population
(Fig. 3G) and impaired
mammosphere formation in trastuzumab-resistant JIMT-1 cells
(Fig. 3H), demonstrating that
ebastine effectively targeted CSC-like properties in
trastuzumab-resistant contexts.
Ebastine exerts anti-tumor activity in
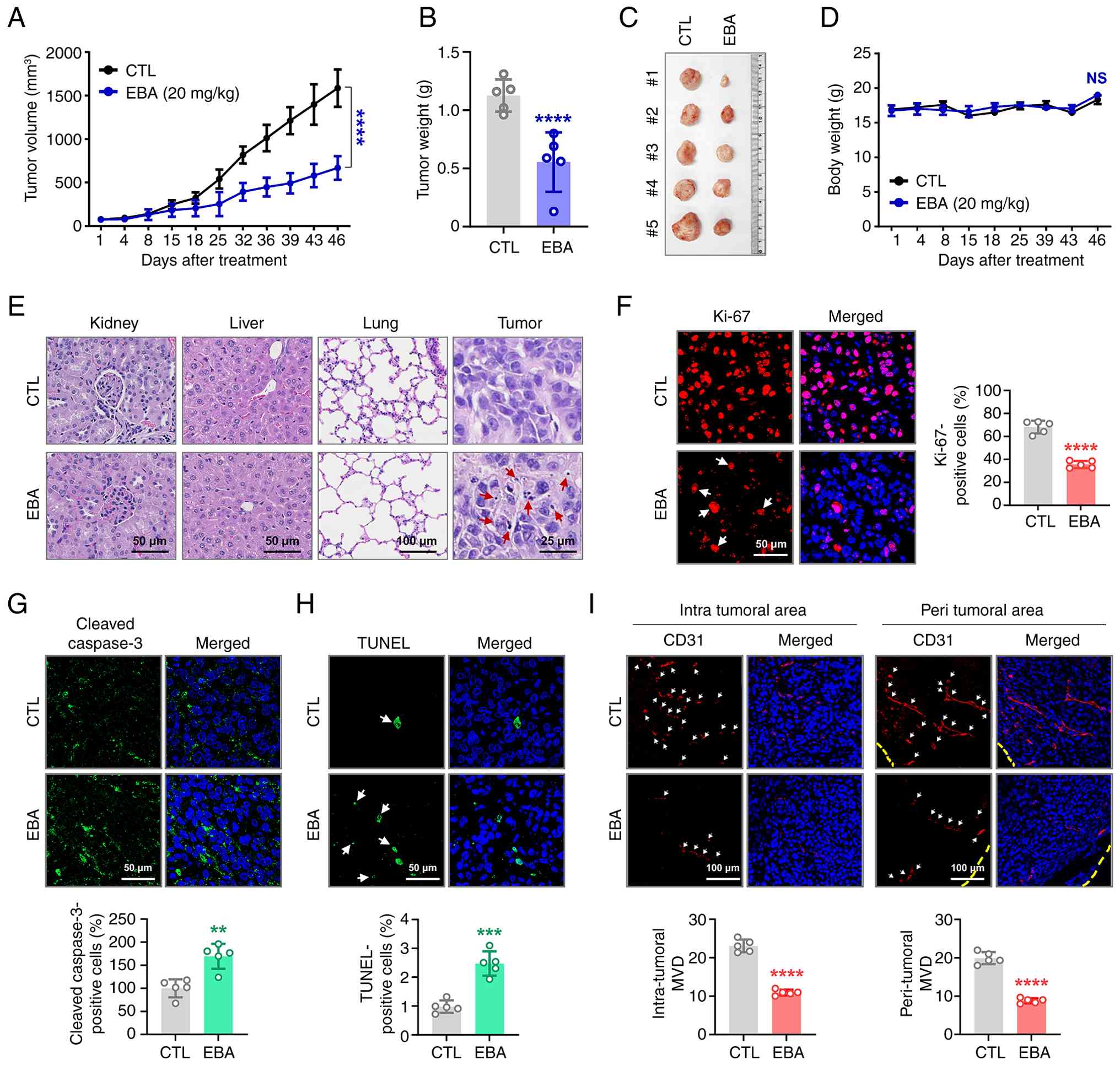
trastuzumab-resistant HER2-positive breast cancer in vivo
To assess the physiological relevance of the in
vitro findings, the present study established a
trastuzumab-resistant JIMT-1 xenograft model to evaluate the
antitumor effect of ebastine in vivo (Fig. 4A). When tumor volumes reached ~50
mm3, mice were administered ebastine (20 mg/kg, every
other day) or vehicle control. At day 46, ebastine led to a
significant suppression of tumor volume (Fig. 4A) and tumor weight (Fig. 4B and C), without affecting body
weight (Fig. 4D).
Histopathological examination using hematoxylin and eosin (H&E)
staining revealed no observable abnormality in the kidney, liver or
lung tissue of ebastine-treated mice compared with controls. Tumor
tissue from the ebastine-treated group exhibited a notable number
of cells with nuclear condensation and fragmentation, indicative of
treatment-induced apoptosis (Fig.
4E). Immunohistochemical analysis revealed a significant
reduction in the Ki-67 proliferation index following ebastine
treatment (Fig. 4F), supporting
its antiproliferative activity. Apoptosis induction was evidenced
by a significant increase in DNA fragmentation, as determined by
the in situ TUNEL assay (Fig.
4H), accompanied by enhanced caspase-3 cleavage (Fig. 4G). The antiangiogenic potential
of ebastine was assessed by quantifying microvessel density using
the endothelial marker CD31. A significant decrease in the number
of CD31-positive microvessels was observed in both intratumoral and
peritumoral (Fig. 4I) regions
following ebastine treatment.
Ebastine downregulates HER2/HER3
expression and CSC-associated markers in vivo
To validate the in vitro observations, the
present study examined the expression of full-length HER2, ICD-HER2
and HER3 in trastuzumab-resistant xenograft tumors using
immunohistochemistry. The present study assessed ICD-HER2
expression with 4B5, an FDA-approved antibody that specifically
recognizes an epitope within the HER2 intracellular domain. Tumors
from ebastine-treated mice exhibited significantly decreased
expression of full-length HER2 (Fig.
5A), ICD-HER2 (Fig. 5B) and
HER3 (Fig. 5C) relative to
control tumors. Consistent with the in vitro abrogation of
the CSC-like phenotype, ebastine administration significantly
decreased the expression of ALDH1A1 (Fig. 5D) and CD44 (Fig. 5E) in tumor tissues. Expression of
the mesenchymal marker vimentin was also significantly decreased in
the ebastine-treated group (Fig.
5F).
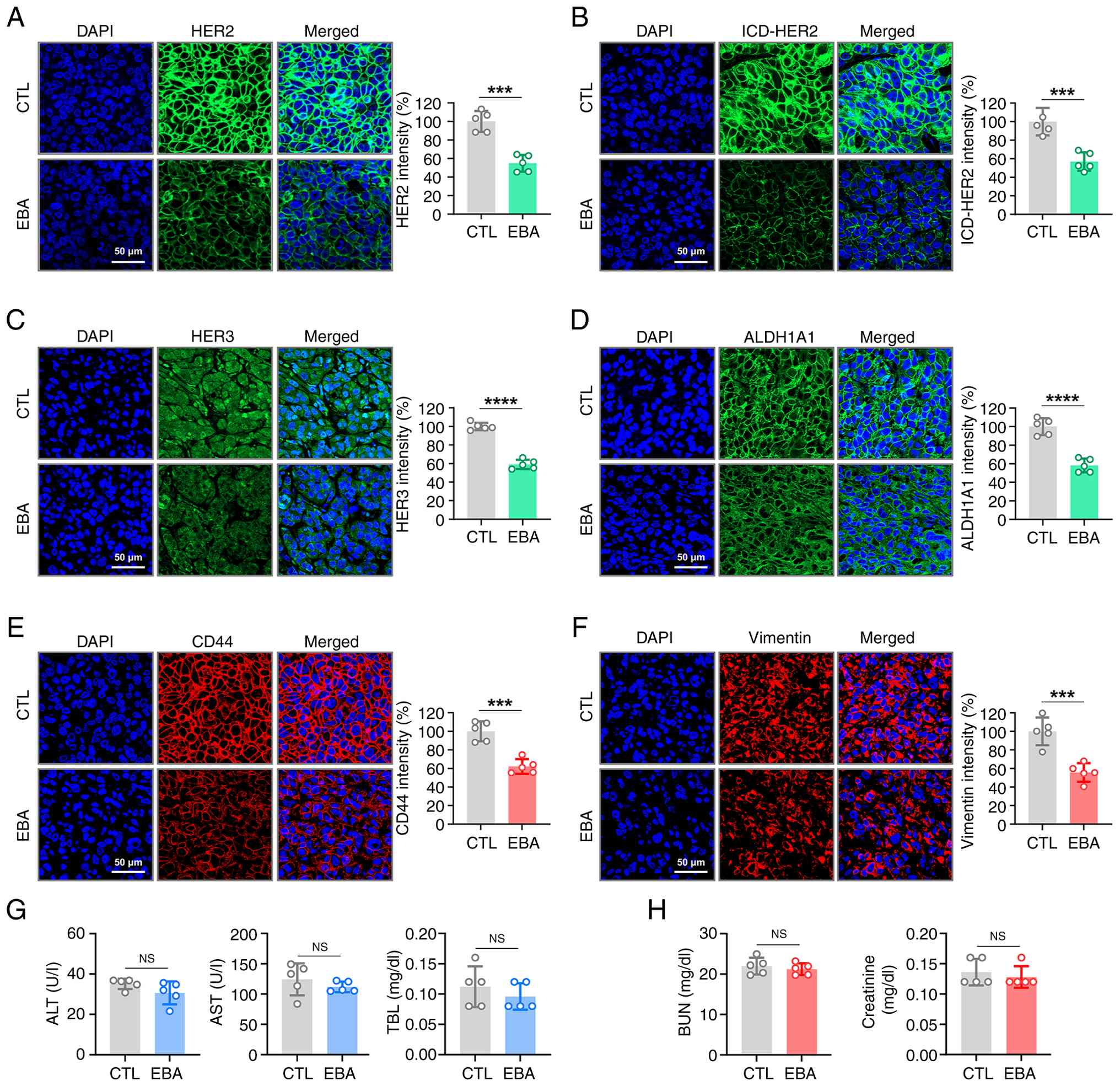 | Figure 5EBA downregulates HER2, ICD-HER2,
HER3, ALDH1A1, CD44 and vimentin in JIMT-1 xenograft tumors.
Immunofluorescence staining of JIMT-1 xenograft tumor tissue for
(A) full-length HER2 (green), (B) ICD-HER2 (green) and (C) HER3.
Immunohistochemical analysis of (D) ALDH1A1 (green) and (E) CD44
(red) in tumor tissue. (F) Tumor sections were immunostained for
vimentin (red). Magnification, x500. Fluorescence intensities were
quantified. Serum biochemical analysis for (G) liver and (H) kidney
function in EBA-treated or CTL mice (n=5). Serum levels of ALT,
AST, TBL, BUN and creatinine were assessed.
***P<0.001, ****P<0.0001. ALT, alanine
aminotransferase; AST, aspartate aminotransferase; BUN, blood urea
nitrogen; EBA, ebastine; ICD, intracellular domain; ALDH, aldehyde
dehydrogenase; CTL, control; NS, not significant. |
Ebastine exhibits no significant
hepatotoxicity or nephrotoxicity in vivo
To evaluate the potential hepatotoxicity of
ebastine, serum samples from treated mice were analyzed for AST,
ALT and TBL. No significant alterations were detected in these
hepatic toxicity markers (Fig.
5G). BUN and creatinine levels, key indicators of renal
function, remained within the normal range (Fig. 5H). These biochemical results
indicated that ebastine did not cause hepatic or renal dysfunction,
even at doses that produce anticancer efficacy.
Discussion
Repurposing existing drugs for new therapeutic
indications has attracted attention in oncology as a cost-effective
and time-efficient alternative to traditional drug development
(14,39,40). As these agents already possess
established safety and pharmacokinetic profiles, they can be more
rapidly transitioned into clinical use, offering a practical
strategy to address issues such as treatment resistance and tumor
heterogeneity (14,40). The present study investigated the
anticancer potential of ebastine, an FDA-approved second-generation
antihistamine, in the context of trastuzumab-resistant
HER2-positive breast cancer. First-generation antihistamines, such
as diphenhydramine, often cause CNS side effects due to their
ability to cross the blood-brain barrier (41). By contrast, ebastine has minimal
CNS penetration, high oral bioavailability and low toxicity, making
it more suitable for long-term use (19,42). These advantages highlight its
potential as a safe and effective anticancer agent for drug
repurposing in oncology.
In ebastine, the diphenylmethoxy-substituted
piperidine and 4-tert-butylphenyl group enhance flexibility and
hydrophobic interactions at protein binding sites. Docking
simulations showed that ebastine fit into the ATP-binding pocket of
the HER2 kinase domain. Its aromatic rings form π-π and π-cation
interactions, while the piperidine group forms hydrogen bonds with
key residues such as Thr862, Asp863 and Lys753. This interaction
likely facilitates the stable binding of ebastine to HER2, notably
downregulating p-HER2 and HER3, as well as trastuzumab
resistance-associated p95HER2. This results in the disruption of
HER2/HER3 and HER2/EGFR heterodimerization, a key mechanism
underlying persistent downstream signaling in HER2-positive tumors
refractory to standard therapy. Trastuzumab and HER2-targeted
tyrosine kinase inhibitors (TKIs; such as lapatinib, neratinib and
tucatinib) differ mechanistically, with trastuzumab targeting the
extracellular HER2 domain and TKIs inhibiting intracellular kinase
activity, however, these agents typically share common mechanisms
of therapeutic resistance (43).
A notable mechanism involves HER2/HER3 heterodimerization, where
HER3, despite lacking intrinsic kinase function, serves as a potent
co-activator by recruiting PI3K to its docking sites. This
interaction activates downstream PI3K/AKT signaling, enabling tumor
cells to bypass HER2-targeted inhibition (2,43). Additionally, genetic alterations
such as activating mutations in PIK3CA or loss of PTEN limit the
efficacy of these targeted agents (44).
To evaluate the in vivo relevance of the
present findings, a trastuzumab-resistant xenograft model was
established using JIMT-1 cells, which were originally derived from
a patient exhibiting intrinsic resistance to trastuzumab (27). Notably, JIMT-1 cells display
resistance not only to trastuzumab but also to numerous
HER2-targeted TKIs (45),
rendering them a representative preclinical model for studying
refractory HER2-positive breast cancer. In the xenograft model,
ebastine treatment resulted in a significant decrease in the
expression of full-length HER2, ICD-HER2 and HER3 within tumor
tissues, further substantiating its ability to interfere with key
therapeutic resistance pathways.
As a transmembrane receptor for hyaluronic acid,
CD44 binds to its polymerized form, contributing to the formation
of a physical barrier that obscures HER2 and diminishes the
efficacy of HER2-targeted antibody therapy (46). CD44 is notably overexpressed in
HER2-positive breast cancer cells exhibiting trastuzumab resistance
compared with sensitive cell lines (47). Consistently, the
trastuzumab-resistant cell line JIMT-1 exhibited a high proportion
(~50%) of CD44high/CD24low cells, compared
with the low frequencies observed in sensitive cell lines such as
SKBR3 (0.03%) and BT474 (0.36%) in our previous studies (15,47). Suppression of CD44 enhances
trastuzumab sensitivity and decreases the invasive behavior and
anchorage-independent proliferation of resistant cells (38). Furthermore, CD44 activates Rho
GTPases, PI3K/AKT and MAPK/ERK signaling pathways, which
collectively promote cell survival, proliferation and invasion,
thereby contributing to the aggressive phenotype of tumor cells
(48,49). In this context, ebastine
treatment significantly decreased the
CD44+/CD24− subpopulation in vitro and
decreased CD44 expression in trastuzumab-resistant JIMT-1 xenograft
tumors in vivo, highlighting its potential to overcome
resistance by targeting CD44. In parallel, ebastine also
effectively attenuated stem-like phenotypes in HER2-positive breast
cancer by decreasing ALDH1 enzymatic activity and impairing the
mammosphere-forming capacity. Elevated expression of ALDH1A1 and
CD44 is associated with shorter overall survival and poor prognosis
in HER2-overexpressing breast cancer, highlighting their potential
as biomarkers for trastuzumab resistance. These findings indicate
that ebastine may improve therapeutic outcomes by targeting
CD44+/ALDH1+ stem-like tumor populations.
Vimentin, a key indicator of epithelial-mesenchymal
transition, promotes tumor progression in breast cancer by
enhancing cell plasticity and motility through cytoskeletal
remodeling (50,51). Vimentin is frequently upregulated
in aggressive breast cancer subtypes, such as triple-negative
breast cancer, and is highly expressed in HER2-positive breast
cancer exhibiting resistance to trastuzumab (52,53). Its expression is associated with
the acquisition of stem-like characteristics, including the
CD44+/CD24− phenotype (54,55). In addition to its role in
promoting stemness, vimentin enhances endothelial cell motility and
structural organization and promotes tumor angiogenesis by
regulating VEGF-dependent signaling pathways (56,57). Although HER2 is typically
associated with an epithelial phenotype, the trastuzumab-resistant
JIMT-1 cell line exhibits mesenchymal features marked by elevated
vimentin expression during tumor progression. Here, ebastine
administration markedly decreased vimentin levels, which may
contribute to suppression of tumor angiogenesis and growth in
vivo.
Although HER2-targeted TKIs effectively inhibit
HER2 signaling, several studies have reported that TKI exposure can
enrich CSC-like populations (58-60). For example, lapatinib has been
shown to increase mammosphere formation in HER2-overexpressing
breast cancer cells, suggesting CSC enrichment (58,61). By contrast, the present data
showed that ebastine decreased ALDH activity and the
CD44high/CD24low fraction and impaired
mammosphere formation, indicating suppression of CSC-like traits.
Several drug-repurposing candidates, including disulfiram,
metformin and niclosamide, have also been explored for their
potential to modulate CSC characteristics by targeting pathways
involved in self-renewal, metabolism and therapeutic resistance
(14,61,62). Disulfiram exerts its anticancer
and CSC-suppressive effects primarily via ALDH inhibition and
proteasome-mediated cytotoxic mechanisms (15,16,63). However, disulfiram has limited
clinical success because its anticancer activity is highly
copper-dependent, resulting in unstable and inconsistent
therapeutic efficacy in vivo (64,65). By contrast, ebastine offers
advantages for drug repurposing due to its metal-independent
mechanisms, stable pharmacokinetic profile and predictable
formation of the active metabolite carebastine (19,66). Metformin is a promising drug
repurposing candidate because it has a long-established safety
profile and low cost, while exerting anticancer effects partly via
AMPK activation, mitochondrial complex I inhibition and suppression
of CSCs (67,68). Although metformin downregulates
HER2 expression through inhibition of the mTOR effector p70S6K1
(69), evidence for its direct
targeting of HER2 signaling remains limited, whereas ebastine
decreases both full-length HER2 and p95HER2 expression and
modulates key signaling nodes associated with trastuzumab
resistance. Niclosamide suppresses key oncogenic pathways,
including Wnt/β-catenin and STAT3 signaling, demonstrating broad
antitumor potential. However, its poor oral bioavailability
necessitates the use of salt forms, prodrugs or nano-based delivery
systems to achieve adequate systemic exposure (61,70). By contrast, ebastine shows high
oral bioavailability, predictable pharmacokinetics and reliable
in vivo exposure, making it a more practical and clinically
feasible option for drug repurposing (19,26,66).
Ebastine may offer strategic therapeutic advantages
by concurrently attenuating the HER2/HER3 signaling pathway, one of
the key drivers of trastuzumab resistance, and suppressing
CSC-associated phenotypes, thereby exhibiting multifaceted
anticancer activity. Taken together, the present findings
demonstrate that ebastine inhibited HER2 kinase activation and
HER2/HER3 heterodimerization, induced caspase-mediated apoptosis
and downregulated CSC-associated markers such as CD44, ALDH1 and
vimentin, supporting its potential as a promising drug-repurposing
candidate for trastuzumab-resistant HER2-positive breast
cancer.
While the present study provided evidence that
ebastine inhibited HER2 signaling and overcomes trastuzumab
resistance, it has several limitations. Although the molecular
docking and CETSA results support a cellular interaction between
ebastine and HER2, these findings do not constitute definitive
biochemical evidence of direct HER2 inhibition. Additional
biochemical and biophysical analyses, such as in vitro
kinase assay, surface plasmon resonance and isothermal titration
calorimetry, are required to confirm direct binding and quantify
binding affinity. Furthermore, site-directed mutagenesis of key
residues (Lys753, Thr862 and Asp863) will be necessary to validate
the predicted binding sites. Future studies should also focus on
evaluating the pharmacokinetic and pharmacodynamic profiles of
ebastine in tumor-bearing models, exploring its combinatorial
efficacy with trastuzumab or HER2-targeted tyrosine kinase
inhibitors and conducting early-phase clinical investigations to
assess its translational potential. Collectively, these approaches
are essential to elucidate the molecular mechanism of the
ebastine-HER2 interaction and facilitate its clinical development
as a repurposed anticancer agent.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
EJ, YJK, JYK and JHS conceived and designed the
study. EJ, DK, JS, SL, DL, SK, KL, YJK and JYK performed the
experiments. EJ, DK, JS, MP, SK, SP, YKK, KDN, YJK and JYK analyzed
the data. EJ, YJK and JYK wrote the manuscript. JYK and JHS confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were performed in strict
accordance with institutional guidelines and were approved by the
Institutional Animal Care and Use Committee of Korea University
College of Medicine, Seoul, Republic of Korea (approval no.
KOREA-2021-0070-C1; approved on May 23, 2022). All procedures were
conducted in compliance with the Animal Research: Reporting of
In Vivo Experiments guidelines and relevant national
regulations for the care and use of laboratory animals. The study
did not involve human participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Korea Health Technology
R&D Project through the Korea Health Industry Development
Institute, funded by the Ministry of Health & Welfare, Republic
of Korea (grant no. HR20C0021), the National Research Foundation
funded by the Korean government (grant nos. 2021R1A2C2009723,
2023R1A2C3004010, RS-2024-00342677, RS-2025-02634306 and
RS-2025-00558356), a Korea University Guro Hospital Grant (grant
no. O2411391) and the Brain Korea 21 Plus Program (grant no.
T2024656).
References
|
1
|
Slamon DJ, Clark GM, Wong SG, Levin WJ,
Ullrich A and McGuire WL: Human breast cancer: Correlation of
relapse and survival with amplification of the HER-2/neu oncogene.
Science. 235:177–182. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Swain SM, Shastry M and Hamilton E:
Targeting HER2-positive breast cancer: Advances and future
directions. Nat Rev Drug Discov. 22:101–126. 2023. View Article : Google Scholar
|
|
3
|
Swain SM, Baselga J, Kim SB, Ro J,
Semiglazov V, Campone M, Ciruelos E, Ferrero JM, Schneeweiss A,
Heeson S, et al: Pertuzumab, trastuzumab, and docetaxel in
HER2-positive metastatic breast cancer. N Engl J Med. 372:724–734.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu L, Xie Y, Gou Q, Cai R, Bao R, Huang Y
and Tang R: HER2-targeted therapies for HER2-positive early-stage
breast cancer: present and future. Front Pharmacol. 15:14464142024.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu K, Yang X, Tai H, Zhong X, Luo T and
Zheng H: HER2-targeted therapies in cancer: A systematic review.
Biomark Res. 12:162024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sánchez-Lorenzo L, Bachiller A, Gea C and
Espinós J: Current management and future perspectives in metastatic
HER2-positive breast cancer. Semin Oncol Nurs. 40:1515542024.
View Article : Google Scholar
|
|
7
|
Arteaga CL and Engelman JA: ERBB
receptors: From oncogene discovery to basic science to
mechanism-based cancer therapeutics. Cancer Cell. 25:282–303. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chu X, Tian W, Ning J, Xiao G, Zhou Y,
Wang Z, Zhai Z, Tanzhu G, Yang J and Zhou R: Cancer stem cells:
Advances in knowledge and implications for cancer therapy. Signal
Transduct Target Ther. 9:1702024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pattabiraman DR and Weinberg RA: Tackling
the cancer stem cells-what challenges do they pose? Nat Rev Drug
Discov. 13:497–512. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ginestier C, Hur MH, Charafe-Jauffret E,
Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG,
Liu S, et al: ALDH1 is a marker of normal and malignant human
mammary stem cells and a predictor of poor clinical outcome. Cell
Stem Cell. 1:555–567. 2007. View Article : Google Scholar
|
|
11
|
Shah D and Osipo C: Cancer stem cells and
HER2 positive breast cancer: The story so far. Genes Dis.
3:114–123. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Duru N, Fan M, Candas D, Menaa C, Liu HC,
Nantajit D, Wen Y, Xiao K, Eldridge A, Chromy BA, et al:
HER2-associated radioresistance of breast cancer stem cells
isolated from HER2-negative breast cancer cells. Clin Cancer Res.
18:6634–6647. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Korkaya H and Wicha MS: HER2 and breast
cancer stem cells: More than meets the eye. Cancer Res.
73:3489–3493. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xia Y, Sun M, Huang H and Jin WL: Drug
repurposing for cancer therapy. Signal Transduct Target Ther.
9:922024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim JY, Cho Y, Oh E, Lee N, An H, Sung D,
Cho TM and Seo JH: Disulfiram targets cancer stem-like properties
and the HER2/Akt signaling pathway in HER2-positive breast cancer.
Cancer Lett. 379:39–48. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen D, Cui QC, Yang H and Dou QP:
Disulfiram, a clinically used anti-alcoholism drug and
copper-binding agent, induces apoptotic cell death in breast cancer
cultures and xenografts via inhibition of the proteasome activity.
Cancer Res. 66:10425–10433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Serageldin MA, El-Bassiouny NA, El-Kerm Y,
Aly RG, Helmy MW, El-Mas MM and Kassem AB: A randomized controlled
study of neoadjuvant metformin with chemotherapy in nondiabetic
breast cancer women: The METNEO study. Br J Clin Pharmacol.
90:3160–3175. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goodwin PJ, Chen BE, Gelmon KA, Whelan TJ,
Ennis M, Lemieux J, Ligibel JA, Hershman DL, Mayer IA, Hobday TJ,
et al: Effect of metformin vs placebo on invasive disease-free
survival in patients with breast cancer: The MA.32 randomized
clinical trial. JAMA. 327:1963–1973. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roberts DJ: A preclinical overview of
ebastine. Studies on the pharmacological properties of a novel
histamine H1 receptor antagonist. Drugs. 52(Suppl 1): S8–S14. 1996.
View Article : Google Scholar
|
|
20
|
Noveck RJ, Preston RA and Swan SK:
Pharmacokinetics and safety of ebastine in healthy subjects and
patients with renal impairment. Clin Pharmacokinet. 46:525–534.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Q, Liu KY, Liu Q, Wang G, Jiang W, Meng
Q, Yi Y, Yang Y, Wang R, Zhu S, et al: Antihistamine drug ebastine
inhibits cancer growth by targeting polycomb group protein EZH2.
Mol Cancer Ther. 19:2023–2033. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seo J, Park M, Ko D, Kim S, Park JM, Park
S, Nam KD, Farrand L, Yang J, Seok C, et al: Ebastine impairs
metastatic spread in triple-negative breast cancer by targeting
focal adhesion kinase. Cell Mol Life Sci. 80:1322023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pan Z, Li SJ, Guo H, Li ZH, Fei X, Chang
SM, Yang QC and Cheng DD: Ebastine exerts antitumor activity and
induces autophagy by activating AMPK/ULK1 signaling in an
IPMK-dependent manner in osteosarcoma. Int J Biol Sci. 19:537–551.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cancer Genome Atlas Network: Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Park SJ, Yoon BH, Kim SK and Kim SY:
GENT2: An updated gene expression database for normal and tumor
tissues. BMC Med Genomics. 12(Suppl 5): S1012019. View Article : Google Scholar
|
|
26
|
Wiseman LR and Faulds D: Ebastine. a
review of its pharmacological properties and clinical efficacy in
the treatment of allergic disorders. Drugs. 51:260–277. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tanner M, Kapanen AI, Junttila T, Raheem
O, Grenman S, Elo J, Elenius K and Isola J: Characterization of a
novel cell line established from a patient with Herceptin-resistant
breast cancer. Mol Cancer Ther. 3:1585–1592. 2004. View Article : Google Scholar
|
|
28
|
Vranic S, Gatalica Z and Wang ZY: Update
on the molecular profile of the MDA-MB-453 cell line as a model for
apocrine breast carcinoma studies. Oncol Lett. 2:1131–1137. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park M, Jung E, Park JM, Park S, Ko D, Seo
J, Kim S, Nam KD, Kang YK, Farrand L, et al: The HSP90 inhibitor
HVH-2930 exhibits potent efficacy against trastuzumab-resistant
HER2-positive breast cancer. Theranostics. 14:2442–2463. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang ZH, Zheng ZQ, Jia SC, Liu SN, Xiao
XF, Chen GY, Liang WQ and Lu XF: Trastuzumab resistance in
HER2-positive breast cancer: Mechanisms, emerging biomarkers and
targeting agents. Front Oncol. 12:10064292022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chandarlapaty S, Scaltriti M, Angelini P,
Ye Q, Guzman M, Hudis CA, Norton L, Solit DB, Arribas J, Baselga J
and Rosen N: Inhibitors of HSP90 block p95-HER2 signaling in
trastuzumab-resistant tumors and suppress their growth. Oncogene.
29:325–334. 2010. View Article : Google Scholar
|
|
32
|
Yang M, Li Y, Kong L, Huang S, He L, Liu
P, Mo S, Lu X, Lin X, Xiao Y, et al: Inhibition of DPAGT1
suppresses HER2 shedding and trastuzumab resistance in human breast
cancer. J Clin Invest. 133:e1644282023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Baselga J and Swain SM: Novel anticancer
targets: Revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer.
9:463–475. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gajria D and Chandarlapaty S:
HER2-amplified breast cancer: Mechanisms of trastuzumab resistance
and novel targeted therapies. Expert Rev Anticancer Ther.
11:263–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Korkaya H, Paulson A, Iovino F and Wicha
MS: HER2 regulates the mammary stem/progenitor cell population
driving tumorigenesis and invasion. Oncogene. 27:6120–6130. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Oh E, Kim YJ, An H, Sung D, Cho TM,
Farrand L, Jang S, Seo JH and Kim JY: Flubendazole elicits
anti-metastatic effects in triple-negative breast cancer via STAT3
inhibition. Int J Cancer. 143:1978–1993. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Idowu MO, Kmieciak M, Dumur C, Burton RS,
Grimes MM, Powers CN and Manjili MH: CD44(+)/CD24(-/low) cancer
stem/progenitor cells are more abundant in triple-negative invasive
breast carcinoma phenotype and are associated with poor outcome.
Hum Pathol. 43:364–373. 2012. View Article : Google Scholar
|
|
38
|
Boulbes DR, Chauhan GB, Jin Q,
Bartholomeusz C and Esteva FJ: CD44 expression contributes to
trastuzumab resistance in HER2-positive breast cancer cells. Breast
Cancer Res Treat. 151:501–513. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pushpakom S, Iorio F, Eyers PA, Escott KJ,
Hopper S, Wells A, Doig A, Guilliams T, Latimer J, McNamee C, et
al: Drug repurposing: Progress, challenges and recommendations. Nat
Rev Drug Discov. 18:41–58. 2019. View Article : Google Scholar
|
|
40
|
Armando RG, Mengual Gómez DL and Gomez DE:
New drugs are not enough-drug repositioning in oncology: An update.
Int J Oncol. 56:651–684. 2020.PubMed/NCBI
|
|
41
|
Simons FER and Simons KJ: H1
antihistamines: Current status and future directions. World Allergy
Organ J. 1:145–155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hurst M and Spencer CM: Ebastine: An
update of its use in allergic disorders. Drugs. 59:981–1006. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Schlam I, Tarantino P and Tolaney SM:
Overcoming resistance to HER2-directed therapies in breast cancer.
Cancers (Basel). 14:39962022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Berns K, Horlings HM, Hennessy BT,
Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM,
Stemke-Hale K, Hauptmann M, et al: A functional genetic approach
identifies the PI3K pathway as a major determinant of trastuzumab
resistance in breast cancer. Cancer Cell. 12:395–402. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pourjamal N, Yazdi N, Halme A, Joncour VL,
Laakkonen P, Saharinen P, Joensuu H and Barok M: Comparison of
trastuzumab emtansine, trastuzumab deruxtecan, and disitamab
vedotin in a multiresistant HER2-positive breast cancer lung
metastasis model. Clin Exp Metastasis. 41:91–102. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pályi-Krekk Z, Barok M, Isola J, Tammi M,
Szöllosi J and Nagy P: Hyaluronan-induced masking of ErbB2 and
CD44-enhanced trastuzumab internalisation in trastuzumab resistant
breast cancer. Eur J Cancer. 43:2423–2433. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim YJ, Sung D, Oh E, Cho Y, Cho TM,
Farrand L, Seo JH and Kim JY: Flubendazole overcomes trastuzumab
resistance by targeting cancer stem-like properties and HER2
signaling in HER2-positive breast cancer. Cancer Lett. 412:118–130.
2018. View Article : Google Scholar
|
|
48
|
Chen C, Zhao S, Karnad A and Freeman JW:
The biology and role of CD44 in cancer progression: Therapeutic
implications. J Hematol Oncol. 11:642018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ouhtit A, Rizeq B, Saleh HA, Rahman MM and
Zayed H: Novel CD44-downstream signaling pathways mediating breast
tumor invasion. Int J Biol Sci. 14:1782–1790. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Berr AL, Wiese K, Dos Santos G, Koch CM,
Anekalla KR, Kidd M, Davis JM, Cheng Y, Hu YS and Ridge KM:
Vimentin is required for tumor progression and metastasis in a
mouse model of non-small cell lung cancer. Oncogene. 42:2074–2087.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Grasset EM, Dunworth M, Sharma G, Loth M,
Tandurella J, Cimino-Mathews A, Gentz M, Bracht S, Haynes M, Fertig
EJ and Ewald AJ: Triple-negative breast cancer metastasis involves
complex epithelial-mesenchymal transition dynamics and requires
vimentin. Sci Transl Med. 14:eabn75712022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Winter M, Meignan S, Völkel P, Angrand PO,
Chopin V, Bidan N, Toillon RA, Adriaenssens E, Lagadec C and Le
Bourhis X: Vimentin promotes the aggressiveness of triple negative
breast cancer cells surviving chemotherapeutic treatment. Cells.
10:15042021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li F, Zhang L, Feng F, Zheng K, Li Y, Wang
T and Ren G: Livin participates in resistance to trastuzumab
therapy for breast cancer through ERK1/2 and AKT pathways and
promotes EMT-like phenotype. RSC Adv. 8:28588–28601. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu
Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, et al:
Breast cancer stem cells transition between epithelial and
mesenchymal states reflective of their normal counterparts. Stem
Cell Reports. 2:78–91. 2013. View Article : Google Scholar
|
|
55
|
Usman S, Waseem NH, Nguyen TKN, Mohsin S,
Jamal A, Teh MT and Waseem A: Vimentin Is at the heart of
epithelial mesenchymal transition (EMT) mediated metastasis.
Cancers (Basel). 13:49852021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
van Beijnum JR, Huijbers EJM, van Loon K,
Blanas A, Akbari P, Roos A, Wong TJ, Denisov SS, Hackeng TM,
Jimenez CR, et al: Extracellular vimentin mimics VEGF and is a
target for anti-angiogenic immunotherapy. Nat Commun. 13:28422022.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Dave JM and Bayless KJ: Vimentin as an
integral regulator of cell adhesion and endothelial sprouting.
Microcirculation. 21:333–344. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shah D, Wyatt D, Baker AT, Simms P,
Peiffer DS, Fernandez M, Rakha E, Green A, Filipovic A, Miele L and
Osipo C: Inhibition of HER2 increases JAGGED1-dependent breast
cancer stem cells: Role for membrane JAGGED1. Clin Cancer Res.
24:4566–4578. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hangauer MJ, Viswanathan VS, Ryan MJ, Bole
D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL,
et al: Drug-tolerant persister cancer cells are vulnerable to GPX4
inhibition. Nature. 551:247–250. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Huang WC, Hung CM, Wei CT, Chen TM, Chien
PH, Pan HL, Lin YM and Chen YJ: Interleukin-6 expression
contributes to lapatinib resistance through maintenance of stemness
property in HER2-positive breast cancer cells. Oncotarget.
7:62352–62363. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kim JH, Park S, Jung E, Shin J, Kim YJ,
Kim JY, Sessler JL, Seo JH and Kim JS: A dual-action
niclosamide-based prodrug that targets cancer stem cells and
inhibits TNBC metastasis. Proc Natl Acad Sci USA.
120:e23040811202023. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ajmeera D and Ajumeera R: Drug
repurposing: A novel strategy to target cancer stem cells and
therapeutic resistance. Genes Dis. 11:148–175. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kim JY, Lee N, Kim YJ, Cho Y, An H, Oh E,
Cho TM, Sung D and Seo JH: Disulfiram induces anoikis and
suppresses lung colonization in triple-negative breast cancer via
calpain activation. Cancer Lett. 386:151–160. 2017. View Article : Google Scholar
|
|
64
|
Kannappan V, Ali M, Small B, Rajendran G,
Elzhenni S, Taj H, Wang W and Dou QP: Recent advances in
repurposing disulfiram and disulfiram derivatives as
copper-dependent anticancer agents. Front Mol Biosci. 8:7413162021.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cvek B: The promiscuity of disulfiram in
medicinal research. ACS Med Chem Lett. 14:1610–1614. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Vincent J, Liminana R, Meredith PA and
Reid JL: The pharmacokinetics, antihistamine and
concentration-effect relationship of ebastine in healthy subjects.
Br J Clin Pharmacol. 26:497–502. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Rocha GZ, Dias MM, Ropelle ER,
Osório-Costa F, Rossato FA, Vercesi AE, Saad MJ and Carvalheira JB:
Metformin amplifies chemotherapy-induced AMPK activation and
antitumoral growth. Clin Cancer Res. 17:3993–4005. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Saini N and Yang X: Metformin as an
anti-cancer agent: Actions and mechanisms targeting cancer stem
cells. Acta Biochim Biophys Sin (Shanghai). 50:133–143. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Vazquez-Martin A, Oliveras-Ferraros C and
Menendez JA: The antidiabetic drug metformin suppresses HER2
(erbB-2) oncoprotein overexpression via inhibition of the mTOR
effector p70S6K1 in human breast carcinoma cells. Cell Cycle.
8:88–96. 2009. View Article : Google Scholar
|
|
70
|
Tan M, Ye W, Liu Y, Chen X, Huttad L, Chua
MS and So S: Niclosamide prodrug enhances oral bioavailability and
efficacy against hepatocellular carcinoma by targeting vasorin-TGFβ
signalling. Br J Pharmacol. 182:5517–5535. 2025. View Article : Google Scholar : PubMed/NCBI
|