Alzheimer's disease (AD), the leading cause of
dementia, is characterized by irreversible cognitive decline. AD
currently affects ~55 million people globally, a number projected
to nearly triple by 2050 (1).
Despite its prevalence, no effective treatments or preventive
measures exist, and a comprehensive understanding of its
pathogenesis remains elusive (1-3).
The hallmarks of AD include extracellular amyloid β-protein (Aβ)
plaques and neurofibrillary tangles (NFTs) composed of
hyperphosphorylated tau (p-tau) (4). Aβ plaques disrupt inter-neuronal
communication (5) and trigger
neuroinflammation, while NFTs impair neuronal function by affecting
axoplasmic transport and disrupting the cytoskeleton (6). Therapeutic strategies targeting the
amyloid cascade or p-tau have yielded unsatisfactory clinical
outcomes, indicating the limitations of single-target therapies
(7). AD pathology also involves
other critical alterations, including neuronal loss, synaptic
dysfunction, oxidative stress and aberrant energy metabolism
(8-10), which interact synergistically to
drive disease progression. While the exact pathogenesis of AD is
not fully understood, emerging evidence implicates impaired
autophagic clearance as a key cellular mechanism in AD pathogenesis
(11) and as a significant
influence on its progression (12,13).
Autophagy, an essential intracellular degradation
pathway, plays a critical role in clearing damaged organelles and
protein aggregates (14-16). This process is significantly
impaired in AD (17),
manifesting as defects in autophagosome formation and maturation,
dysregulated lysosomal function and inefficient
autophagosome-lysosome fusion (18). These disruptions compromise the
clearance of Aβ and tau proteins, leading to the accumulation of
toxic species both inside and outside neurons, which in turn
amplifies neuronal injury and cell death (19,20). Autophagy dysfunction also
contributes to mitochondrial deficits, oxidative stress and chronic
neuroinflammation; it ultimately blocks energy production, disrupts
energy utilization and perturbs metabolic homeostasis, thereby
disturbing neuronal energy metabolism, impairing synaptic function
and promoting apoptosis (21).
Thus, strategies aimed at restoring autophagic activity have become
a major focus of AD therapeutic research (22).
Over the past few decades, mounting epidemiological
and clinical evidence has unequivocally established that regular
physical activity confers substantial benefits for the prevention
and management of various chronic diseases, particularly
cardiovascular diseases, type 2 diabetes mellitus and certain
malignancies (for example, breast, colon and liver cancer)
(23-25). This evidence provides a framework
for developing non-pharmacological interventions in chronic disease
management. Beyond non-modifiable factors such as genetic
susceptibility (1), physical
inactivity is recognized as a significant modifiable risk factor
for AD, accounting for ~13% of cases worldwide (26,27). Clinical and preclinical evidence
shows that regular exercise not only reduces the risk of AD
incidence (28) but also
improves cognitive outcomes in patients with AD (29-32). Mechanistically, dysregulated
autophagy serves as a key pathogenic driver in AD, and exercise
modulates neurodegeneration by regulating autophagic pathways.
Reports have confirmed that physical activity significantly
promotes autophagic activation, alleviates cerebral
neuroinflammation and mitigates AD-related pathological damage in
human and animal models, such as transgenic mouse models of AD
(33,34). Thus, clarifying the molecular
signaling pathways underlying exercise-regulated autophagy in AD is
critical for elucidating the core mechanisms of exercise-based
interventions and developing targeted therapeutic strategies.
Although existing reviews have independently addressed the role of
autophagy in AD or the neuroprotective effects of exercise in AD, a
systematic overview of the mechanisms by which exercise modulates
autophagic signaling pathways to affect AD is lacking.
Clarification of this network pathway is required to translate
exercise-based interventions from basic research to clinical
practice in AD. The current review systematically explores the
recent research progress on how exercise influences the autophagy
signaling pathway in AD, emphasizing its core molecular mechanisms.
The review also assesses the potential synergy between exercise and
natural products in AD intervention, offering a theoretical
foundation and research direction for the future development and
clinical application of exercise-based AD-targeted
interventions.
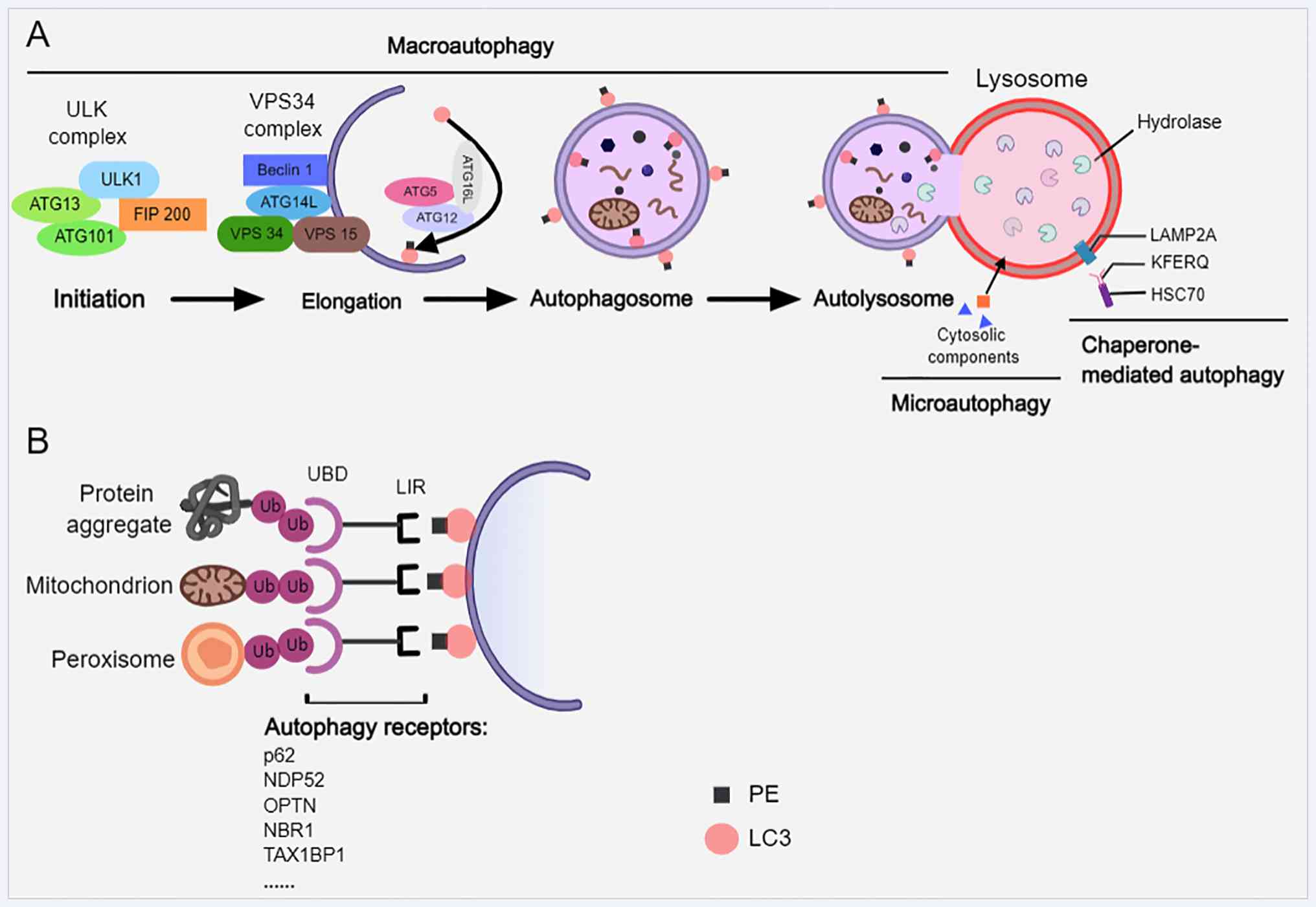
Neuronal autophagy is an essential intracellular
degradation system that mediates the delivery and breakdown of
cytoplasmic components within lysosomes (35,36). Autophagy is classified into three
forms based on the mechanisms of cargo delivery to lysosomes:
Macroautophagy, microautophagy and chaperone-mediated autophagy
(CMA) (19) (Fig. 1A). The initial step in
macroautophagy (commonly termed autophagy) is the formation of a
double-membrane vesicle known as the autophagosome (37). This process is regulated by a set
of highly conserved autophagy-associated proteins (ATGs) (36). The initial step involves the
activation of the UNC 51-like kinase (ULK) complex, consisting of
ULK1/2, ATG13, FAK family kinase-interacting protein of 200 kDa and
ATG101 (38,39). Subsequent phagophore nucleation
requires the class III PI3K complex, which includes vesicular
protein sorting 34 (VPS34), VPS15, beclin-1 and ATG14L (38). Phagophore expansion and
autophagosome maturation depend on two ubiquitin-like conjugation
systems: One results in the ATG5-ATG12-ATG16L complex that
facilitates membrane elongation, and the other conjugates
phosphatidylethanolamine to microtubule-associated protein 1 light
chain 3 (LC3), generating the lipidated form LC3-II that associates
with autophagosomal membranes (40). These steps are critical for
autophagosome biogenesis, cargo recognition and eventual fusion
with lysosomes to form autolysosomes (38).
By contrast, microautophagy involves the direct
engulfment of cytoplasmic material through lysosomal membrane
invagination (41). CMA, a
selective process, recognizes substrate proteins via the chaperone
heat shock cognate 70-kDa protein bearing a Kex2-Fer1-Endo U1-Qc-2
motif and translocates them into lysosomes through
lysosomal-associated membrane protein 2A (LAMP2A) (19,37,38). CMA deficiency has been linked to
exacerbated AD pathology (42).
Nutrient deprivation typically induces non-selective autophagy,
whereas selective autophagy targets damaged organelles such as
mitochondria and peroxisomes, as well as protein aggregates
(37,43) (Fig. 1B). Selective autophagy is
mediated by receptors such as p62, nuclear dot protein 52,
optineurin (OPTN), next to BRCA1 gene 1 and tax1 (human T-cell
leukemia virus type I) binding protein 1, which contain a
ubiquitin-binding domain and LC3 interaction region that connect
ubiquitinated cargo to the autophagic machinery (37,44,45).
Autophagic flux and the expression of
lysosome-associated proteins are significantly reduced in both
patients with AD and AD animal models (46). This dysregulation is hypothesized
to contribute to AD pathogenesis by impairing multiple steps in the
autophagy-lysosome pathway, from autophagosome formation to
lysosomal degradation (47). In
brains affected by AD, neuronal autophagy is disrupted at the
initiation phase, partly due to mammalian target of rapamycin
(mTOR) hyperactivation, which suppresses the ULK1 complex and
impedes autophagosome biogenesis (48,49). Moreover, mutations and the
downregulation of autophagy-related genes have been linked to AD.
For example, beclin-1 expression is markedly decreased in
AD-affected brains (50,51), and beclin-1-deficient microglia
in AD models have been reported to exhibit impaired phagocytosis
and reduced Aβ clearance (52).
LC3, a key autophagosomal membrane protein, shows altered
processing in AD, with reduced conversion of LC3-I to LC3-II,
thereby inhibiting autophagy initiation (53). Emerging evidence also implicates
non-canonical autophagic processes [e.g., LC3-associated
phagocytosis (LAP) and LC3-associated endocytosis (LANDO)] in AD
pathology, and this pathway has been discovered in microglia of the
brain and the central nervous system. LANDO deficiency exacerbates
neurodegeneration and cognitive deficits in AD mice (54,55). The mechanisms of LAP and LANDO
are distinct from those of autophagy (54). However, no current evidence
exists on how exercise affects these pathways. Therefore, the
present review will not elaborate further on these processes.
Lysosomal dysfunction further aggravates autophagic
impairment in AD. Decreased autolysosomal acidification occurs
prior to extracellular Aβ deposition (17), leading to defective proteolysis,
increased lysosomal membrane permeability (56,57) and impaired cathepsins, which are
lysosomal proteases essential in Aβ and tau clearance (58,59). Notably, Aβ accumulation both
results from and exacerbates lysosomal dysfunction, creating a
vicious cycle that promotes neurodegeneration (60). Restoring lysosomal function
enhances the clearance of Aβ and p-tau (57). The autophagy receptor
sequestosome 1 (SQSTM1/p62), which facilitates cargo delivery to
LC3, exerts protective effects in AD models by improving memory and
modulating autophagic activity (61,62).
Transcription factor EB (TFEB), a master regulator
of autophagy-lysosomal biogenesis, plays a critical role in AD
(63-65). The activity of TFEB is inhibited
by mTOR complex 1 (mTORC1); however, TFEB-mediated endocytosis is
essential for mTORC1 activation and autophagy function (57,66). Oxidative stress also contributes
to AD progression by promoting the amyloidogenic processing of
amyloid precursor protein (APP) and tau hyperphosphorylation
(67). Reactive oxygen species
(ROS) accumulation damages mitochondria, leading to dysfunction
that triggers mitophagy, a selective autophagic process essential
for maintaining mitochondrial quality (45,68). Collectively, these disruptions in
the autophagy-lysosome pathway significantly exacerbate AD
pathology (63,69,70).
Considering the pivotal role that autophagy
dysfunction plays in the development of AD, restoring compromised
autophagic flux has become a promising approach for therapy. Among
the different interventions available, exercise is as a
non-pharmacological treatment that is safe, cost-effective and easy
to implement. Recent studies have reported the significant
potential of exercise to regulate autophagy and ameliorate AD
pathology (71,72). The subsequent sections focus on
the ways in which exercise serves as a potential modulator to
restore autophagic homeostasis in AD.
Exercise provides a multi-targeted physiological
intervention strategy to address the complex autophagy dysfunction
aforementioned. Preclinical studies have shown that exercise can
improve memory, promote neurogenesis, and enhance hippocampal
structure and synaptic plasticity in AD transgenic mouse models
[e.g., APP/presenilin 1 (PS1) and proline-301-serine (P301S)]
(73,74). Both human and animal studies have
shown that a wide range of physical activities, ranging from
low-intensity walking to high-intensity training, support cognitive
health (75,76). For example, adolescents
performing treadmill exercises demonstrated significant cognitive
improvements, particularly at moderate to high intensity exercise
(28).
In one study, 16 weeks of voluntary running
significantly improved autophagy dysfunction in mice with five
familial Alzheimer's disease mutations (5xFAD transgenic mice) and
exerted neuroprotective effects (77). Notably, more recent studies
demonstrated that moderate-intensity exercise was more effective in
modulating brain autophagy in AD models than low or high-intensity
exercise, whereas low-intensity exercise was associated with higher
AD risk, and very high intensity increased oxidative stress
(78-82). Aerobic exercise modalities, such
as treadmill running, voluntary wheel exercise and swimming, were
shown to specifically facilitate the autophagy-mediated clearance
of pathogenic proteins (82,83).
Exercise enhances autophagic activity through
multiple pathways. In APP/PS1 mice, 12 weeks of aerobic treadmill
exercise restored autophagy-lysosomal flux, increased autophagosome
numbers in the hippocampus and cortex, promoted Aβ clearance, and
improved cognitive performance (71,84,85). Similarly, extended treadmill
exercise reduced tau pathology in P301S mice, and autophagy was
shown to play a crucial role in tau degradation through a
ubiquitin-mediated mechanism (84,86). Exercise also upregulates the
expression of core autophagy-related proteins such as LC3-II and
beclin-1, as observed following both high intensity interval
training (HIIT) and moderate intensity continuous training (MICT)
(78). However, a separate
animal study reported no significant changes in LC3, p62 or
beclin-1 in whole hippocampal lysates after HIIT, suggesting that
HIIT has specific effects on brain regions or motor programs
(81).
In summary, preclinical studies consistently
confirmed that moderate-intensity exercise enhances autophagic
flux, particularly in the hippocampus of AD model mice, in a manner
associated with spatial memory improvement. These preclinical
findings underscore a bidirectional, functionally significant
relationship between exercise and autophagy in AD.
Exercise exerts neuroprotective effects against AD
primarily by restoring impaired autophagy flux, which serves as a
pivotal link between exercise-induced signaling activation and the
amelioration of AD-specific pathological phenotypes. A
well-orchestrated causal chain underpins this process. Exercise
triggers the activation of key signaling pathways [(mTOR, sirtuin 1
(SIRT1) and adiponectin (APN) receptor 1 (AdipoR1)] and the
modulation of microRNAs (miRNAs/miRs)/irisin, which in turn
corrects defects in autophagosome biogenesis, lysosomal function
and autophagosome-lysosome fusion. This restoration of
physiological autophagic flux directly enhances the clearance of
neurotoxic Aβ aggregates and p-tau, mitigates chronic
neuroinflammation, preserves mitochondrial integrity by improving
mitophagy and rescues synaptic dysfunction (87). In summary, these effects
alleviate neuronal damage and apoptosis, and ultimately improve
cognitive function in AD models and patients. Notably, the
regulatory effect of exercise on autophagy is not mediated by a
single mechanism but is achieved through complex crosstalk between
signaling pathways and a cooperative regulatory network. These
interactions have not been fully elucidated and analyzing them can
be helpful in understanding the overall neuroprotective effect of
exercise.
mTOR, a serine/threonine kinase within the
PI3K-related kinase family, plays a central role in regulating
multiple cellular processes. mTOR functions through two distinct
complexes: mTORC1 and mTORC2 (88), of which mTORC1 acts as a key
negative regulator of autophagy (89,90). Under nutrient-sufficient
conditions, active mTORC1 phosphorylates and inhibits the ULK1
complex, thereby suppressing the initiation of autophagy. mTORC1
activity is suppressed during nutrient deprivation or cellular
stress, leading to the ULK1-mediated induction of autophagy
(89).
In AD studies, analyses of animal and post-mortem
human brain tissue have shown that mTORC1 is often hyperactivated
(48,91). Preclinical research has
demonstrated that regulating mTOR expression can salvage autophagy
deficiency and enhance cognitive function in AD models [e.g.,
triple-transgenic (PS1m146v/APPswe/TauP301L) (3xTg)-AD mice]
(92). The post-exercise
activation of mTORC1 in the brains of experimental animals has also
been associated with improved learning and memory (93). Moreover, treadmill exercise in
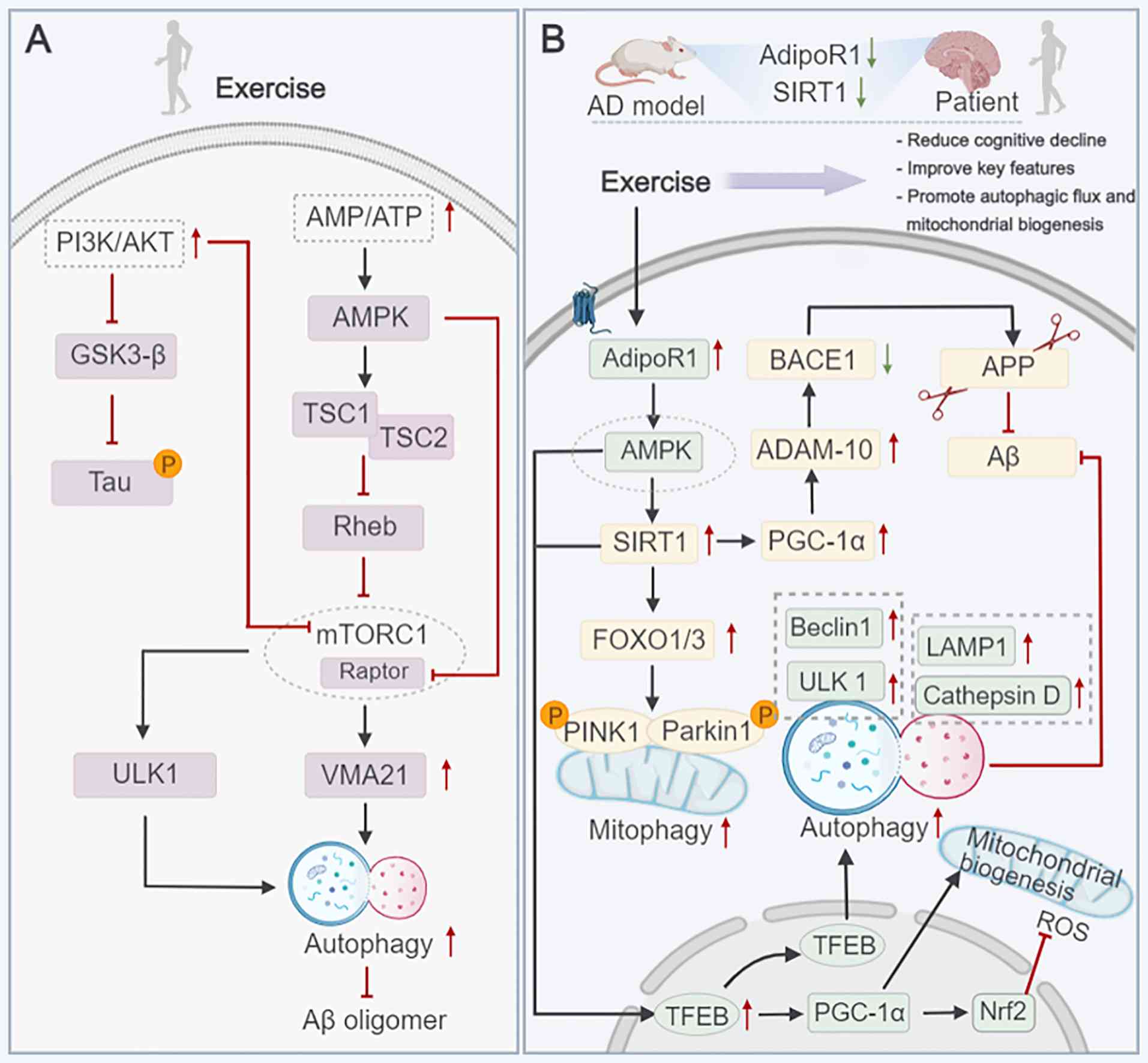
neuron-specific enolase/human tau23 (NSE/htau23) transgenic mice
reduced glycogen synthase kinase-3β (GSK-3β) activity and tau
hyperphosphorylation by enhancing PI3K/AKT signaling, an effect
potentially mediated by AMPK activation and downstream mTORC1
inhibition (94) (Fig. 2A). Notably, this inhibition of
mTORC1 by exercise directly reversed the pathological suppression
of the ULK1 complex, restoring autophagosome formation and the
subsequent clearance of p-tau aggregates. Additionally, both HIIT
and MICT were reported to inhibit overactive PI3K/AKT/mTOR
signaling in the hippocampus, with HIIT displaying a more prominent
inhibitory effect. This signaling suppression, in turn, facilitated
autophagy flux and mitigated Aβ deposition, which were mediated by
augmented lysosomal acidification and enhanced cathepsin activity
(78).
Exercise increases neuronal energy demand by
increasing the AMP/ATP ratio and activating AMPK. This
energy-sensitive kinase promotes autophagy by phosphorylating both
tuberous sclerosis complex (TSC)2 and TSC1, thereby enhancing the
inhibition of ras homolog enriched in brain, mTORC1 and raptor (a
core component of mTORC1), further inhibiting their activity
(95). Elevated AMPK
phosphorylation during exercise has been observed in the brains of
AD model animals (96).
Moreover, exercise-induced mTOR increased VMA21 expression in
APP/SP1 mice, contributed to the recovery of autolysosomal
function, reduced Aβ accumulation and improved cognitive impairment
(71) (Fig. 2A). Restoring autolysosomal
activity breaks the vicious cycle of Aβ accumulation and lysosomal
dysfunction in AD, ultimately reducing neuronal toxicity and
improving synaptic transmission.
SIRTs, a conserved family of nicotinamide adenine
dinucleotide-dependent deacetylases known as 'longevity proteins',
comprise seven isoforms (SIRT1-SIRT7) (97). SIRT1, one of the most extensively
studied members, is highly expressed in the hippocampus and
prefrontal cortex, and its levels are significantly reduced in AD
(98). Previous studies have
shown that serum SIRT1 levels are reduced in patients with mild
cognitive impairment and AD, highlighting its potential as a
therapeutic target (98-101). Aerobic exercise has been shown
to mitigate cognitive decline, improve synaptic function, promote
neuronal survival and reduce Aβ accumulation in AD models by
activating the SIRT1 pathway (65,102). Similarly, resistance training
was reported to increase hippocampal and prefrontal SIRT1 activity
and alleviate tau pathology in AD mice (82). Recent studies demonstrated that
high-intensity training over 6 weeks increased PPARγ coactivator-1α
(PGC-1α) and SIRT1 activity in human skeletal muscle (101). Further animal experiments
indicated that 12 weeks of moderate-intensity treadmill training
inhibited the production of Aβ in NSE/APPsw transgenic mice by
upregulating SIRT1, enhancing PGC-1α expression, increasing a
disintegrin and metalloprotease 10 levels, and reducing β-secretase
1 (BACE1) (a rate-limiting enzyme in APP processing and Aβ
production) activity (103)
(Fig. 2B). This evidence
supports the critical role of SIRT1 in AD progression.
Exercise also counteracts impaired mitophagy in AD.
The PTEN-induced kinase 1-Parkin pathway, essential for
mitochondrial quality control, is potentiated by treadmill training
via the SIRT1-FOXO1/3 axis, enhancing mitophagy and mitochondrial
integrity (102,104-106). This SIRT1-mediated mitophagy
not only reduces oxidative stress by eliminating dysfunctional
mitochondria but also decreases Aβ aggregation by reducing
ROS-induced APP amyloidogenic processing, while preserving synaptic
function by maintaining mitochondrial energy supply. In a study
using SIRT1-inhibited model mice, researchers found that acute
wheel running for 1 h increased TFEB levels in the nuclei of the
cerebral cortex within 2-4 h. This increase was driven by an
upregulation of the AMPK-SIRT1 signaling pathway. Moreover, after 8
weeks of wheel running, TFEB levels were significantly increased in
the cortex, hippocampus and striatum. Exercise effectively
activated the autophagy-lysosomal pathway, enhancing lysosomal
function and biogenesis in the mouse brain (107). Further research indicated that
inhibiting SIRT1 did not prevent TFEB nuclear translocation,
suggesting that other mechanisms might contribute to
exercise-induced TFEB increases (Fig. 2B). Similar to SIRT1, SIRT3 has
also been implicated in mitophagy regulation (108). Single-nucleus RNA sequencing in
a clinical study showed the decreased expression of neuronal SIRT5
in patients with AD, suggesting its role in autophagy flux
(46). However, the effects of
exercise intensity on SIRT activation and the specific roles of
SIRT3 and SIRT5 in exercise-induced neuroprotection require further
investigation.
APN, a protein hormone secreted by adipose tissue,
acts through receptors, including AdipoR1, AdipoR2 and T-cadherin.
AdipoR1 is highly expressed in the brain, where it regulates energy
homeostasis, hippocampal neurogenesis and synaptic plasticity
(109-111). Maintaining the APN metabolic
balance is increasingly recognized to be important in AD, with
reduced APN levels in patients with AD correlating with elevated
Aβ42 and p-tau in the cerebrospinal fluid and impaired
hippocampal function (111).
APN promotes hippocampal progenitor cell and Neuro2A
cell proliferation via AdipoR1 signaling, which suppresses GSK-3β
through p38-mitogen-activated protein kinase activation and
phosphorylation at Ser-389, thereby supporting neurogenesis
(109). Impaired AdipoR1
expression has been reported in both patients with AD and AD animal
models (111,112). The activation of autophagy
through the AdipoR1/AMPK pathway exerts neuroprotective effects in
AD progression (113). Recent
studies have found that APN enhances autophagy through the
AdipoR1/AMPK/SIRT1 pathway, thereby promoting the clearance of Aβ
in APP/SP1 mice (114).
Notably, Aβ deposition and associated pathology were significantly
reduced in APP/PS1 mice after 12 weeks of moderate-intensity
aerobic intervention. These changes were related to improvements in
lysosomal function and the recovery of autophagy flux through the
AdipoR1/AMPK/TFEB signaling pathway (115). Specifically, AdipoR1 activation
by exercise-induced APN upregulation phosphorylates AMPK, which in
turn activates TFEB. TFEB nuclear translocation upregulates genes
involved in autophagosome formation (e.g., ULK1 and beclin-1) and
lysosomal function (e.g., LAMP1 and cathepsin D), thereby enhancing
the clearance of Aβ and p-tau, reducing neuroinflammation by
activating microglial autophagy and preserving synaptic plasticity
by maintaining neuronal energy metabolism (115). Thus, the translocation of the
TFEB nucleus is not only regulated by SIRT1, but also by AMPK.
Additionally, Aβ aggregation increases ROS levels, triggers
oxidative stress and disrupts mitochondrial autophagy. Exercise was
reported to promote the nuclear translocation of TFEB, increase the
expression of PGC-1α and nuclear factor e2-related factor 2,
improve mitochondrial autophagy, and thereby reduce mitochondrial
damage and neuronal death (65).
However, the mechanism by which exercise regulates AdipoR1 and the
complex signaling pathways of AdipoR1 require further preclinical
research.
miRNAs are small endogenous non-coding RNAs that
post-transcriptionally regulate gene expression (116). The majority are highly
expressed in the nervous system and play important roles in
neuronal development, synaptic plasticity and the pathogenesis of
neurodegenerative diseases (117). In a previous study, clinical
data showed that miR-4763-3p was upregulated in the early stage of
AD compared with that in the healthy control group (118). A preclinical study has shown
that inhibiting miR-4763-3p can improve learning and memory
disorders in 3xTg-AD mice, accompanied by reduced
neuroinflammation, neuronal death and synaptic alterations
(118). miRNAs modulate
transcript stability and translation by binding to the 3'-UTR of
target mRNAs; their dysregulation can disrupt protein homeostasis,
leading to imbalances in Aβ production and clearance (119). Growing evidence indicates that
miRNA dysfunction contributes to impaired autophagy in AD, with
miRNAs indirectly influencing disease progression by regulating
autophagy (73,120).
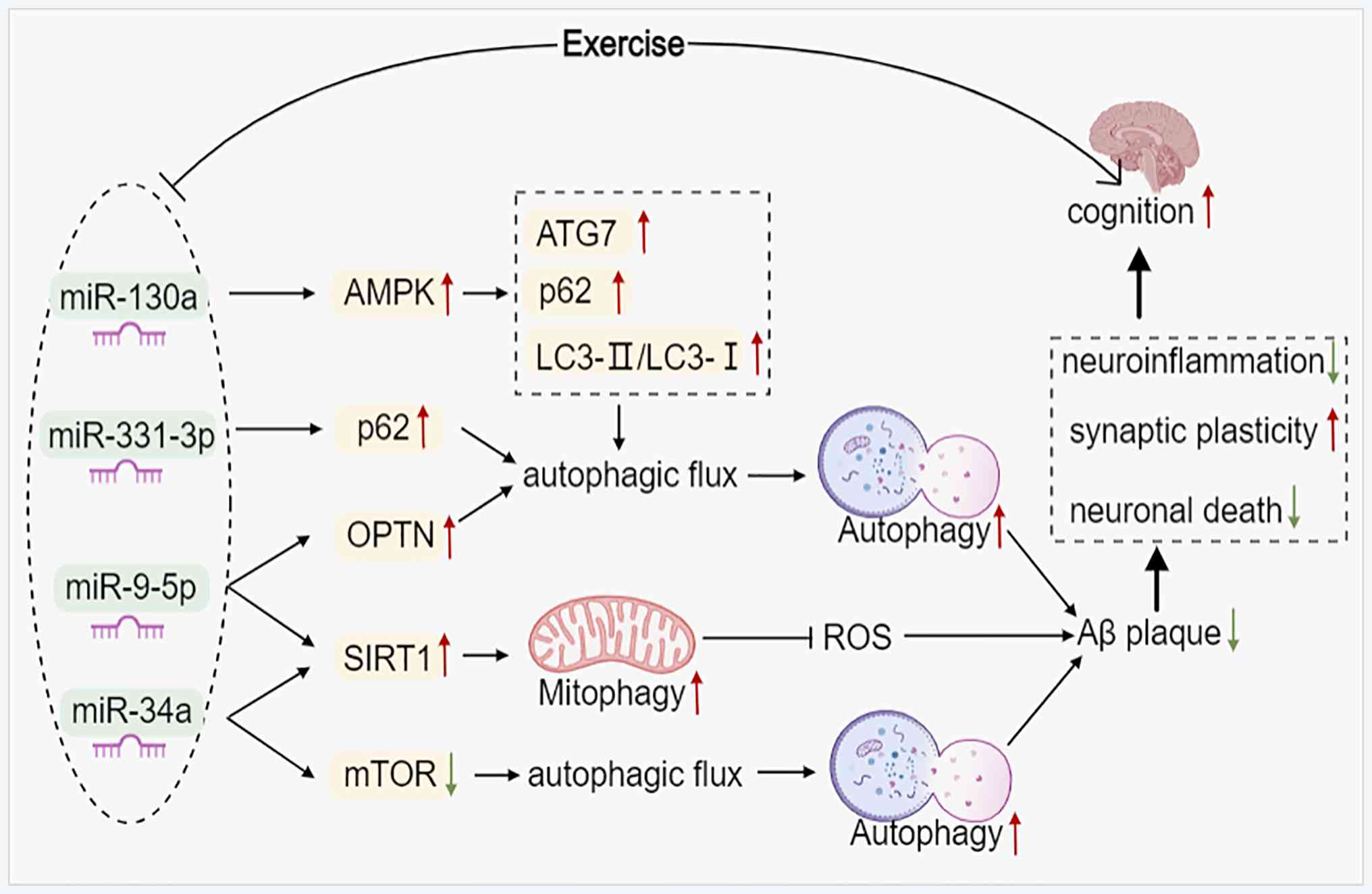
Studies have begun to reveal the role of miRNAs in
exercise-induced autophagy in AD. For instance, miR-34a and miR-130
have been implicated in mediating the effects of exercise on
autophagic processes (121)
(Fig. 3). Swimming training was
reported to improve cognitive function and delay aging in rats by
counteracting miR-34a-mediated autophagic impairment and abnormal
mitochondrial dynamics (122).
Another study has shown that miR-34a-mediated autophagy may be
related to the activation of the SIRT1 signaling pathway and the
inhibition of mTOR signaling (123). Furthermore, cell experiments
using SH-SY5Y cells have identified miR-331-3p and miR-9-5p as
regulators of autophagy receptors, targeting SQSTM1/p62 and OPTN,
respectively. Overexpressing these miRNAs in SH-SY5Y cells impaired
autophagy flux and promoted Aβ plaque formation (124) (Fig. 3). Ginsenoside Rg1 treatment of
APP/PS1 mice activated miR-9-5p/SIRT1-mediated mitophagy,
alleviated mitochondrial dysfunction and effectively improved
cognitive function in AD mice (125). However, all these studies lack
a connection with exercise. A preclinical study showed that the
expression of miR-130a in aging rats (21 months) was significantly
downregulated compared with that in young rats (3 months). However,
voluntary wheel running (8 weeks) significantly increased the
expression of miR-130a, whereas the expression of autophagy-related
protein p62 decreased. The increase in the LC3II/LC3I ratio and the
expression of ATG7 reduced apoptosis in senescent rats. The
expression of p-AMPK increased, while SIRT1 remained unchanged,
suggesting that voluntary wheel running activates the AMPK
signaling pathway but not the SIRT1 pathway in aging rats (96). Additionally, D-gal-induced
SH-SY5Y cells showed results consistent with the animal experiments
(96). miRNA can activate
multiple signaling pathways, which may be related to the targeted
activation of upstream signaling molecules in various signaling
pathways. Therefore, more research on using miRNA as a potential
target for AD treatment, in which its comprehensive regulatory
network in exercise-regulated AD autophagy is analyzed, is
required.
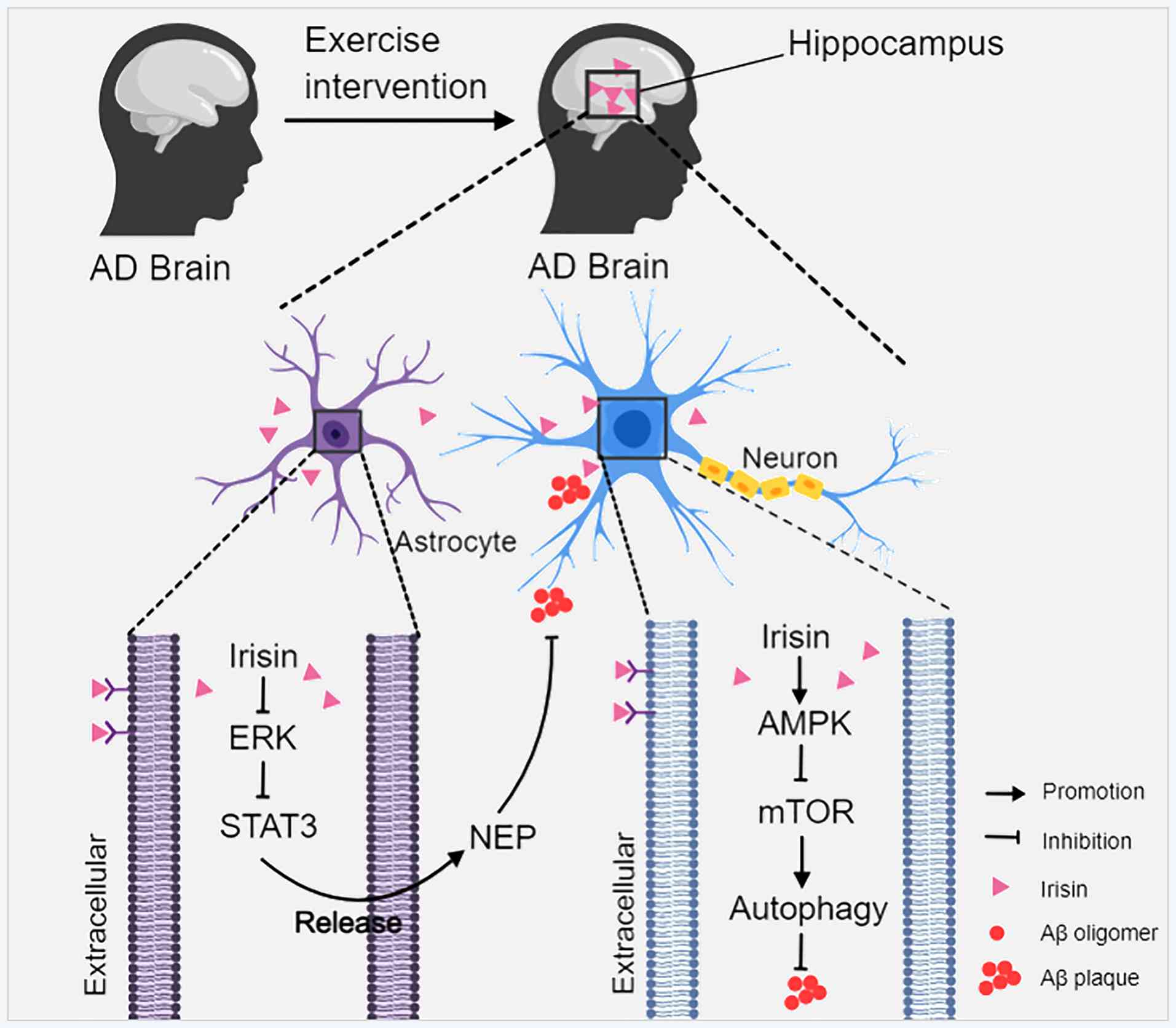
Irisin is a PGC-1α-dependent myokine that was
originally identified in skeletal muscle. Irisin is produced by the
proteolytic cleavage of the membrane protein fibronectin type III
structural domain-containing 5 protein (FNDC5) (126,127), is widely expressed in multiple
tissues and exhibits neuroprotective properties (128-130). Studies have reported decreased
levels of FNDC5/irisin in the hippocampus and cerebrospinal fluid
of patients with AD. Exercise-induced increases in irisin have been
shown to exert beneficial effects in AD models (127,131,132). A recent study demonstrated that
irisin suppressed Aβ aggregation by promoting the release of
enkephalin from astrocytes by inhibiting the ERK-STAT3 signaling
pathway (133). Research have
reported that exercise-induced irisin could regulate the AMPK
pathway by activating it and inhibiting mTOR, thereby increasing
autophagy and clearing amyloid proteins and toxic aggregates in the
brain (134-136) (Fig. 4). However, no experimental
evidence has verified that exercise regulates autophagy
dysregulation in AD by upregulating irisin. In addition, the miRNA
regulation of irisin may play an important role in the clinical
prevention and treatment of AD (128); however, relevant research data
are lacking. Therefore, this gap in the research field of
exercise-induced irisin autophagy in AD remains to be explored.
Given the complexity of AD pathology, single-target
therapies often fail in clinical trials. AD drug development
projects are targeting multiple mechanisms (1). Natural products derived from herbal
or dietary sources (e.g., curcumin, catechins and quercetin) have
shown favorable safety profiles and multi-target efficacy in
clinical studies focused on AD (137). For instance, quercetin and
curcumin have entered phase II clinical trials and have
significantly demonstrated improvements in cases of mild cognitive
impairment (1). These studies
highlight the essential role of natural products in treating AD.
Additionally, a number of studies have shown the effects of natural
products in regulating autophagy in AD (138-140). For instance, Esculentoside A is
a kind of triterpene saponin isolated from Phytolacca
esculenta that activates the autophagy pathway in an
AMPK-dependent manner, thereby promoting the clearance of p-tau and
alleviating cognitive decline in 3xTg-AD mice (138). Ginsenoside Rg2, a steroidal
glycoside derived from Panax ginseng, activates autophagy
via AMPK-dependent but mTOR-independent signaling pathways. This
activation facilitates the clearance of Aβ aggregates and enhances
cognitive function in 5xFAD transgenic mice (139). Notably, only a few studies have
focused on the synergistic effect of exercise and natural products
in the autophagy regulation of AD. Consequently, the following
review section will focus on representative natural products in AD
research. Their synergistic effects and underlying mechanisms in
regulating autophagy and alleviating AD pathology upon combination
with exercise will be systematically summarized.
Resveratrol, a natural polyphenolic compound
extracted from various plants (e.g., grapes and peanuts) (141), demonstrates antioxidant and
neuroprotective effects in vitro (142). Studies in AD models have shown
that resveratrol decreases Aβ aggregation and hippocampal toxicity,
promotes neurogenesis and mitigates hippocampal degeneration
(143-145). Clinical trials showed that
resveratrol sustainably decreased Aβ40 levels in the
cerebrospinal fluid and plasma of patients with AD, and that on
oral administration of high-dose resveratrol (500-1,000 mg/day) was
safe and well-tolerated (117,146). The therapeutic potential of
resveratrol in AD involves modulating AMPK-SIRT1-mediated autophagy
and miRNA-dependent pathways, as well as the direct activation of
autophagy via the mTOR signaling pathway, through which it
regulates tau hyperphosphorylation, neuroinflammation, BACE1
activity and Aβ accumulation (117). After 5 months of moderate to
vigorous exercise combined with resveratrol treatment (557
mg/kg/day, diet), the expression of autophagy-related markers
(LC3-I), lysosomal proteins (cathepsin B/D and LAMP2) and
ubiquitination markers (Ub1) was significantly reduced in 3xTg-AD
mice. The expression levels of p62 and SIRT1 proteins were
significantly increased, while the expression of AMPK was elevated
but not significantly different. These findings suggest that the
combination of exercise and resveratrol may synergistically enhance
autophagy via the AMPK-SIRT1 pathway, reduce Aβ oligomer levels,
decrease apoptosis and ultimately improve cognitive function
(147). However, differences in
the degree of AMPK-SIRT1 activation or that of other pathways,
cognitive improvements from different doses of resveratrol combined
with different exercise intensities, the impact of resveratrol
supplementation timing (such as before, after or simultaneously
with exercise) on bioavailability and synergistic effects, as well
as clinical trials, should all be explored in the future.
Luteolin, a natural flavonoid in a variety of
fruits, vegetables and herbs [e.g., honeysuckle (Lonicera
japonica) and Perilla (Perilla frutescens)] (148), confers significant
neuroprotection against glutamate-induced hippocampal neuronal
death (149). A clinical trial
of patients with AD showed that no dose-limiting toxicity occurred
when luteolin was administered at a dose of 100 mg/day (150). Preclinical studies have shown
that luteolin can improve cognitive function and protect AD neurons
by alleviating Aβ-induced oxidative stress, reducing mitochondrial
damage and alleviating neuroinflammation (148,151). In Aβ1-42
oligomer-induced AD mice, a combination of luteolin (100 mg/kg/day,
gavage) and 4 weeks of voluntary wheel exercise was more effective
than either treatment alone. This dual approach significantly
improved cognitive function, reduced Aβ accumulation and inhibited
microglial activation, thereby alleviating neuroinflammation. The
approach also enhanced autophagy activity in the hippocampus and
cortical regions, as indicated by a notable decrease in p-ULK1
protein expression and an increase in LC3II/LC3I expression in the
cerebral cortex. Notably, luteolin, exercise and their combination
each reduced p62 protein expression to varying degrees, although no
significant differences were observed among them (152). A recent study further confirmed
that luteolin combined with exercise could alleviate AD-related
cognitive impairment. Untargeted metabolomics was also used to
reveal the key role of autophagy in this combined mechanism
(153). In conclusion, further
research is warranted to elucidate how varying doses of luteolin
modulate its bioavailability and exert synergistic effects when
combined with distinct exercise intensities and supplementation
timing (before, after or during exercise). Additionally, more
robust clinical trials are necessary to investigate these
topics.
The combination of natural products and exercise
demonstrates synergistic potential in treating AD. Both resveratrol
and luteolin, two distinct types of natural products, have been
confirmed to possess neuroprotective effects and can modulate
autophagy pathways relevant to AD. Treatment of AD model mice with
either of the two natural products and exercise exerted effects
superior to individual interventions. This combined strategy more
significantly improves cognitive function, alleviates
neuroinflammation and reduces abnormal Aβ deposition.
In summary, the combined application of exercise and
natural products coordinated the regulation of autophagy processes
and pathological protein metabolism through multiple targets and
multiple pathways, and provides a promising intervention strategy
for preventing and treating AD. This combined protocol enhances the
autophagy-mediated neuroprotective cascade response, simultaneously
targeting multiple nodes of AD pathology, from the activation of
upstream signaling pathways to the clearance of downstream
pathological substrates, thereby avoiding the inherent limitations
of single-target interventions and achieving greater systemic
intervention efficacy.
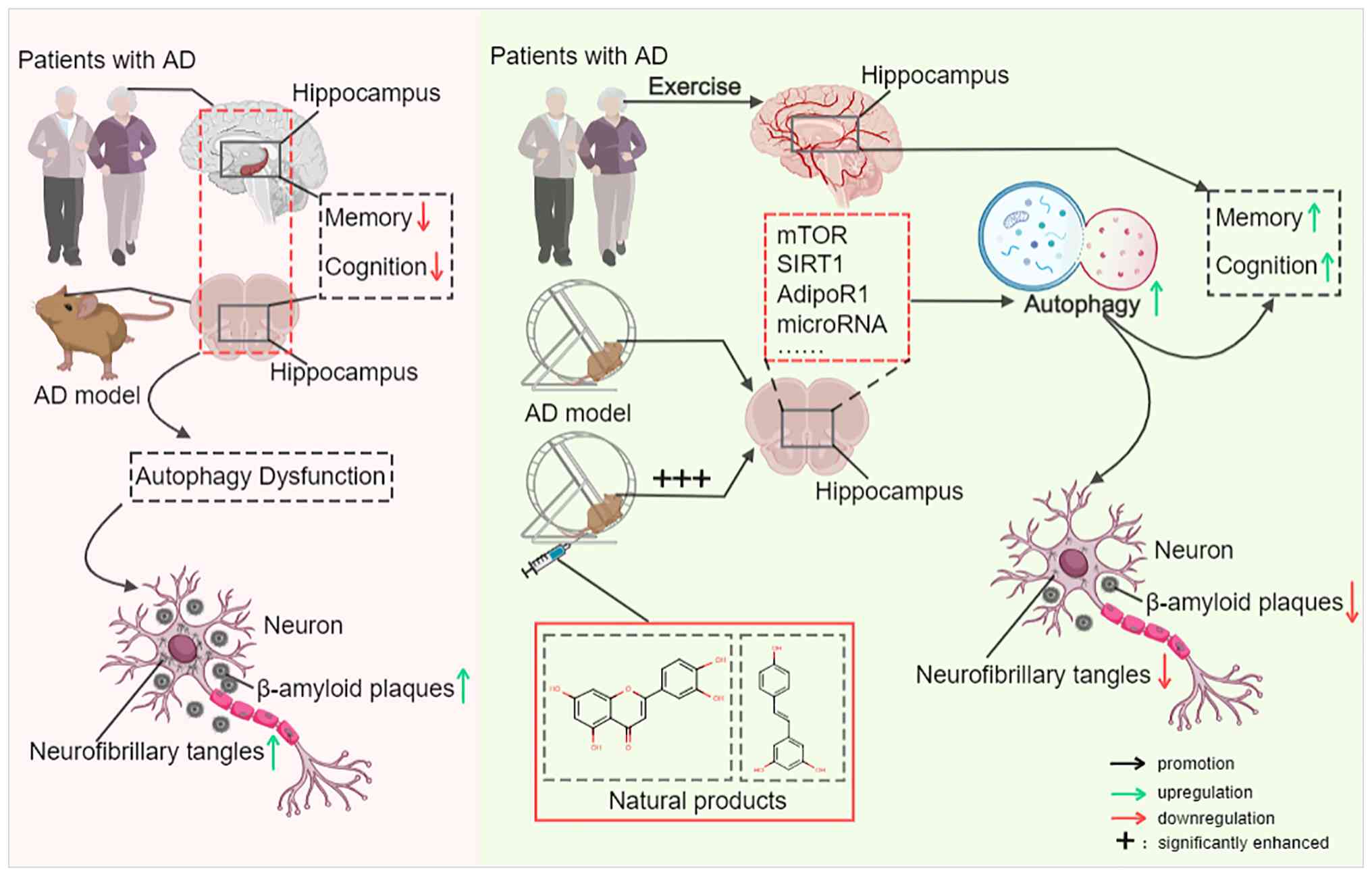
AD is a devastating neurodegenerative disorder with
no curative treatments. Autophagy dysfunction is a key pathogenic
driver underlying the accumulation of neurotoxic Aβ aggregates,
p-tau, neuronal loss and synaptic impairment. The present review
systematically highlights current evidence demonstrating that
mild-to-moderate intensity physical exercise represents a safe,
cost-effective and multi-targeted non-pharmacological intervention
for AD (Table I; Fig. 5). Specifically, exercise restores
impaired autophagic flux in AD by modulating core signaling
pathways (mTOR, SIRT1 and AdipoR1), regulating miRNAs such as
miR-34a and miR-130a, and upregulating irisin. These regulatory
effects collectively enhance autophagosome biogenesis, improve
lysosomal function and promote autophagosome-lysosome fusion,
thereby facilitating the clearance of Aβ and p-tau, alleviating
neuroinflammation and mitochondrial dysfunction, and ultimately
ameliorating cognitive deficits in both preclinical AD models and
clinical settings. Additionally, combining exercise with natural
products (e.g., resveratrol and luteolin) exerts synergistic
neuroprotective effects by enhancing autophagy-mediated clearance
of pathological proteins, highlighting a promising multi-modal
intervention strategy for AD.
Despite this valuable insight, this review has
several specific limitations that warrant acknowledgment. First,
while the roles of key signaling pathways (mTOR, SIRT1 and AdipoR1)
in exercise-mediated autophagy regulation are summarized, molecular
crosstalk among these pathways (e.g., the AMPK-mTOR-SIRT1
regulatory axis) remains incompletely elucidated, and how exercise
coordinates these interconnected networks to fine-tune autophagic
flux demands the further integration of multi-omics data. Second,
this review did not address the potential regulation of
non-canonical autophagic pathways (e.g., LAP and LANDO) through
exercise due to limited existing experimental evidence. These
pathways have been implicated in AD pathology but remain a critical
knowledge gap in the field. Third, the miRNA regulatory network
underlying exercise-induced autophagy restoration remains
incompletely understood, with only a handful of miRNAs (e.g.,
miR-34a and miR-130a) characterized, and interactions between
miRNAs and irisin/FNDC5 in AD remain largely unvalidated. Fourth,
preclinical studies on combinations of exercise and natural
products are scope-limited, and data on the optimization of key
parameters are lacking, including natural product dosage, exercise
intensity and intervention timing (e.g., pre- or post-exercise
supplementation), which hinders the translation of these
synergistic effects into clinical practice. Finally, the current
evidence is predominantly derived from rodent models, and the
generalizability of these mechanisms to human AD populations,
particularly across different disease stages and genetic
backgrounds, remains to be fully established.
Future research should focus on novel, practical and
translationally relevant directions to address these gaps and
advance the field. Leveraging single-cell RNA sequencing and
spatial metabolomics to dissect cell-type-specific mechanisms of
exercise-regulated autophagy (e.g., in neurons, microglia and
astrocytes) will yield unprecedented insight into cell-cell
communication networks underlying neuroprotection. High-throughput
CRISPR screening can identify uncharacterized miRNAs or long
non-coding RNAs that link exercise and autophagy regulation,
facilitating the development of targeted nucleic acid-based
therapeutics (e.g., miRNA mimics or inhibitors) for combination
with exercise interventions. Large-scale stratified clinical trials
are urgently needed to establish personalized exercise
prescriptions, using peripheral biomarkers (e.g., serum irisin and
TFEB) to tailor exercise intensity, duration and modality for
individual patients with AD or high-risk populations. Integrating
network pharmacology and in vitro high-throughput assays to
identify shared molecular targets of natural products and exercise
(e.g., the AMPK-SIRT1-TFEB axis) will expedite the development of
combination preclinical strategies with optimized synergistic
efficacy. In addition, genetically engineered AD mouse models will
clarify the role of non-canonical autophagy (e.g., LAP and LANDO)
in exercise-mediated neuroprotection, filling a critical knowledge
gap in the current literature. Finally, exploring the long-term
sustainability of exercise-induced autophagy restoration and its
impact on AD progression (e.g., delaying conversion from mild
cognitive impairment to clinical AD) will provide critical evidence
to support the integration of exercise into routine clinical
care.
In summary, exercise is a potent intervention to
counteract autophagy dysfunction in AD, supported by preclinical
and clinical evidence of its neuroprotective effects. Addressing
the aforementioned limitations and pursuing innovative,
translationally focused research directions will further unravel
the mechanistic complexity of exercise-autophagy crosstalk and
enable the development of more effective, personalized strategies
for AD prevention and management. Given the current challenges in
AD drug development, integrating exercise alone or its combination
with natural products into clinical practice holds considerable
theoretical significance and potential for improving patient
outcomes.
WL and WW conducted the literature search, drafted
the manuscript and prepared the figures. YS, XL and YL drafted,
edited and revised the manuscript. XW and TT contributed to figure
design and edited the manuscript. XH and LZ commented on and edited
the manuscript, and provided substantial improvements. All authors
have read and approved the final manuscript. Data authentication is
not applicable.
Not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present review was supported by the Chongqing Natural
Science Foundation (grant no. 2023NSCQ-MSX3083), the Outstanding
Youth Project of Department of Education of Hunan Province (grant
no. 24B1077) and the Chongqing Science and Health Joint Project
(grant no. 2026ZYYB027).
|
1
|
Scheltens P, De Strooper B, Kivipelto M,
Holstege H, Chételat G, Teunissen CE, Cummings J and van der Flier
WM: Alzheimer's disease. Lancet. 397:1577–1590. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reitz C, Pericak-Vance MA, Foroud T and
Mayeux R: A global view of the genetic basis of Alzheimer disease.
Nat Rev Neurol. 19:261–277. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Di Meco A, Curtis ME, Lauretti E and
Praticò D: Autophagy dysfunction in Alzheimer's Disease:
Mechanistic insights and new therapeutic opportunities. Biol
Psychiatry. 87:797–807. 2020. View Article : Google Scholar
|
|
4
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer's disease: Progress and problems on the
road to therapeutics. Science. 297:353–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De Strooper B and Karran E: The cellular
phase of Alzheimer's disease. Cell. 164:603–615. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iqbal K, Liu F and Gong CX: Tau and
neurodegenerative disease: The story so far. Nat Rev Neurol.
12:15–27. 2016. View Article : Google Scholar
|
|
7
|
Tong BC, Wu AJ, Huang AS, Dong R,
Malampati S, Iyaswamy A, Krishnamoorthi S, Sreenivasmurthy SG, Zhu
Z, Su C, et al: Lysosomal TPCN (two pore segment channel)
inhibition ameliorates beta-amyloid pathology and mitigates memory
impairment in Alzheimer disease. Autophagy. 18:624–642. 2022.
View Article : Google Scholar :
|
|
8
|
Nixon RA: Autophagy-lysosomal-associated
neuronal death in neurodegenerative disease. Acta Neuropathol.
148:422024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tzioras M, McGeachan RI, Durrant CS and
Spires-Jones TL: Synaptic degeneration in Alzheimer disease. Nat
Rev Neurol. 19:19–38. 2023. View Article : Google Scholar
|
|
10
|
Butterfield DA and Halliwell B: Oxidative
stress, dysfunctional glucose metabolism and Alzheimer disease. Nat
Rev Neurosci. 20:148–160. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deng Z, Dong Y, Zhou X, Lu JH and Yue Z:
Pharmacological modulation of autophagy for Alzheimer's disease
therapy: Opportunities and obstacles. Acta Pharm Sin B.
12:1688–1706. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee JH and Nixon RA: Autolysosomal
acidification failure as a primary driver of Alzheimer disease
pathogenesis. Autophagy. 18:2763–2764. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rubinsztein DC, Codogno P and Levine B:
Autophagy modulation as a potential therapeutic target for diverse
diseases. Nat Rev Drug Discov. 11:709–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Levine B and Kroemer G: Biological
functions of autophagy genes: A disease perspective. Cell.
176:11–42. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu L, Chen Y and Tooze SA: Autophagy
pathway: Cellular and molecular mechanisms. Autophagy. 14:207–215.
2018. View Article : Google Scholar :
|
|
16
|
Kaushik S, Tasset I, Arias E, Pampliega O,
Wong E, Martinez-Vicente M and Cuervo AM: Autophagy and the
hallmarks of aging. Ageing Res Rev. 72:1014682021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee JH, Yang DS, Goulbourne CN, Im E,
Stavrides P, Pensalfini A, Chan H, Bouchet-Marquis C, Bleiwas C,
Berg MJ, et al: Faulty autolysosome acidification in Alzheimer's
disease mouse models induces autophagic build-up of Aβ in neurons,
yielding senile plaques. Nat Neurosci. 25:688–701. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Uddin MS, Stachowiak A, Mamun AA, Tzvetkov
NT, Takeda S, Atanasov AG, Bergantin LB, Abdel-Daim MM and
Stankiewicz AM: Autophagy and Alzheimer's Disease: From molecular
mechanisms to therapeutic implications. Front Aging Neurosci.
10:042018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nixon RA: The role of autophagy in
neurodegenerative disease. Nat Med. 19:983–997. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Palmer JE, Wilson N, Son SM, Obrocki P,
Wrobel L, Rob M, Takla M, Korolchuk VI and Rubinsztein DC:
Autophagy, aging, and age-related neurodegeneration. Neuron.
113:29–48. 2025. View Article : Google Scholar
|
|
21
|
Fang EF, Hou Y, Palikaras K, Adriaanse BA,
Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, et
al: Mitophagy inhibits amyloid-β and tau pathology and reverses
cognitive deficits in models of Alzheimer's disease. Nat Neurosci.
22:401–412. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Menzies FM, Fleming A, Caricasole A, Bento
CF, Andrews SP, Ashkenazi A, Füllgrabe J, Jackson A, Jimenez
Sanchez M, Karabiyik C, et al: Autophagy and neurodegeneration:
Pathogenic mechanisms and therapeutic opportunities. Neuron.
93:1015–1034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sharif K, Watad A, Bragazzi NL, Lichtbroun
M, Amital H and Shoenfeld Y: Physical activity and autoimmune
diseases: Get moving and manage the disease. Autoimmun Rev.
17:53–72. 2018. View Article : Google Scholar
|
|
24
|
Zhu C, Ma H, He A, Li Y, He C and Xia Y:
Exercise in cancer prevention and anticancer therapy: Efficacy,
molecular mechanisms and clinical information. Cancer Lett.
544:2158142022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Andersen LL: Health promotion and chronic
disease prevention at the workplace. Annu Rev Public Health.
45:337–357. 2024. View Article : Google Scholar
|
|
26
|
Halon-Golabek M, Borkowska A,
Herman-Antosiewicz A and Antosiewicz J: Iron Metabolism of the
skeletal muscle and neurodegeneration. Front Neurosci. 13:1652019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
López-Ortiz S, Pinto-Fraga J, Valenzuela
PL, Martín-Hernández J, Seisdedos MM, García-López O, Toschi N, Di
Giuliano F, Garaci F, Mercuri NB, et al: Physical exercise and
Alzheimer's disease: Effects on pathophysiological molecular
pathways of the disease. Int J Mol Sci. 22:28972021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mahalakshmi B, Maurya N, Lee SD and
Bharath Kumar V: Possible neuroprotective mechanisms of physical
exercise in neurodegeneration. Int J Mol Sci. 21:58952020.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sujkowski A, Hong L, Wessells RJ and Todi
SV: The protective role of exercise against age-related
neurodegeneration. Ageing Res Rev. 74:1015432022. View Article : Google Scholar
|
|
30
|
De la Rosa A, Olaso-Gonzalez G,
Arc-Chagnaud C, Millan F, Salvador-Pascual A, García-Lucerga C,
Blasco-Lafarga C, Garcia-Dominguez E, Carretero A, Correas AG, et
al: Physical exercise in the prevention and treatment of
Alzheimer's disease. J Sport Health Sci. 9:394–404. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fox FAU, Diers K, Lee H, Mayr A, Reuter M,
Breteler MMB and Aziz NA: Association between accelerometer-derived
physical activity measurements and brain structure: A
Population-based cohort study. Neurology. 99:e1202–e1215. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yau WW, Kirn DR, Rabin JS, Properzi MJ,
Schultz AP, Shirzadi Z, Palmgren K, Matos P, Maa C, Pruzin JJ, et
al: Physical activity as a modifiable risk factor in preclinical
Alzheimer's disease. Nat Med. 31:4075–4083. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cotman CW, Berchtold NC and Christie LA:
Exercise builds brain health: Key roles of growth factor cascades
and inflammation. Trends Neurosci. 30:464–472. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kang J, Liu M, Yang Q, Dang X, Li Q, Wang
T, Qiu B, Zhang Y, Guo X, Li X, et al: Exercise training exerts
beneficial effects on Alzheimer's disease through multiple
signaling pathways. Front Aging Neurosci. 17:15580782025.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nixon RA and Rubinsztein DC: Mechanisms of
autophagy-lysosome dysfunction in neurodegenerative diseases. Nat
Rev Mol Cell Biol. 25:926–946. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kaur J and Debnath J: Autophagy at the
crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol.
16:461–472. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakatogawa H: Mechanisms governing
autophagosome biogenesis. Nat Rev Mol Cell Biol. 21:439–458. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nazio F and Cecconi F: Autophagy up and
down by outsmarting the incredible ULK. Autophagy. 13:967–968.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gui X, Yang H, Li T, Tan X, Shi P, Li M,
Du F and Chen ZJ: Autophagy induction via STING trafficking is a
primordial function of the cGAS pathway. Nature. 567:262–266. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li WW, Li J and Bao JK: Microautophagy:
Lesser-known self-eating. Cell Mol Life Sci. 69:1125–1136. 2012.
View Article : Google Scholar
|
|
42
|
Bourdenx M, Martín-Segura A, Scrivo A,
Rodriguez-Navarro JA, Kaushik S, Tasset I, Diaz A, Storm NJ, Xin Q,
Juste YR, et al: Chaperone-mediated autophagy prevents collapse of
the neuronal metastable proteome. Cell. 184:2696–2714.e25. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kerr JS, Adriaanse BA, Greig NH, Mattson
MP, Cader MZ, Bohr VA and Fang EF: Mitophagy and Alzheimer's
disease: Cellular and molecular mechanisms. Trends Neurosci.
40:151–166. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sulkshane P, Ram J, Thakur A, Reis N,
Kleifeld O and Glickman MH: Ubiquitination and receptor-mediated
mitophagy converge to eliminate oxidation-damaged mitochondria
during hypoxia. Redox Biol. 45:1020472021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang Z, Yang X, Song YQ and Tu J:
Autophagy in Alzheimer's disease pathogenesis: Therapeutic
potential and future perspectives. Ageing Res Rev. 72:1014642021.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Deng P, Fan T, Gao P, Peng Y, Li M, Li J,
Qin M, Hao R, Wang L, Li M, et al: SIRT5-mediated desuccinylation
of RAB7A protects against Cadmium-induced Alzheimer's disease-like
pathology by restoring autophagic flux. Adv Sci (Weinh).
11:e24020302024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xie ZS, Zhao JP, Wu LM, Chu S, Cui ZH, Sun
YR, Wang H, Ma HF, Ma DR, Wang P, et al: Hederagenin improves
Alzheimer's disease through PPARα/TFEB-mediated autophagy.
Phytomedicine. 112:1547112023. View Article : Google Scholar
|
|
48
|
Shafei MA, Harris M and Conway ME:
Divergent metabolic regulation of autophagy and mTORC1-early events
in Alzheimer's disease? Front Aging Neurosci. 9:1732017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang X and Jia J: Magnolol improves
Alzheimer's disease-like pathologies and cognitive decline by
promoting autophagy through activation of the AMPK/mTOR/ULK1
pathway. Biomed Pharmacother. 161:1144732023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang XW, Zhu XX, Tang DS and Lu JH:
Targeting autophagy in Alzheimer's disease: Animal models and
mechanisms. Zool Res. 44:1132–1145. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Salminen A, Kaarniranta K, Kauppinen A,
Ojala J, Haapasalo A, Soininen H and Hiltunen M: Impaired autophagy
and APP processing in Alzheimer's disease: The potential role of
Beclin 1 interactome. Prog Neurobiol. 106-107:33–54. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
O'Brien CE and Wyss-Coray T: Sorting
through the roles of beclin 1 in microglia and neurodegeneration. J
Neuroimmune Pharmacol. 9:285–292. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Longobardi A, Catania M, Geviti A, Salvi
E, Vecchi ER, Bellini S, Saraceno C, Nicsanu R, Squitti R, Binetti
G, et al: Autophagy markers are altered in Alzheimer's disease,
dementia with lewy bodies and frontotemporal dementia. Int J Mol
Sci. 25:11252024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Peña-Martinez C, Rickman AD and Heckmann
BL: Beyond autophagy: LC3-associated phagocytosis and endocytosis.
Sci Adv. 8:eabn17022022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Heckmann BL, Teubner BJW, Tummers B,
Boada-Romero E, Harris L, Yang M, Guy CS, Zakharenko SS and Green
DR: LC3-Associated Endocytosis facilitates β-Amyloid clearance and
mitigates neurodegeneration in murine Alzheimer's disease. Cell.
178:536–551.e14. 2019. View Article : Google Scholar
|
|
56
|
Udayar V, Chen Y, Sidransky E and Jagasia
R: Lysosomal dysfunction in neurodegeneration: Emerging concepts
and methods. Trends Neurosci. 45:184–199. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chae CW, Yoon JH, Lim JR, Park JY, Cho JH,
Jung YH, Choi GE, Lee HJ and Han HJ: TRIM16-mediated lysophagy
suppresses high-glucose-accumulated neuronal Aβ. Autophagy.
19:2752–2768. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Qian XH, Ding GY, Chen SY, Liu XL, Zhang M
and Tang HD: Blood cathepsins on the risk of Alzheimer's disease
and related pathological biomarkers: Results from observational
cohort and mendelian randomization study. J Prev Alzheimers Dis.
11:1834–1842. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Almeida MF, Bahr BA and Kinsey ST:
Endosomal-lysosomal dysfunction in metabolic diseases and
Alzheimer's disease. Int Rev Neurobiol. 154:303–324. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mançano ASF, Pina JG, Froes BR and Sciani
JM: Autophagy-lysosomal pathway impairment and cathepsin
dysregulation in Alzheimer's disease. Front Mol Biosci.
11:14902752024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Cecarini V, Bonfili L, Gogoi O, Lawrence
S, Venanzi FM, Azevedo V, Mancha-Agresti P, Drumond MM, Rossi G,
Berardi S, et al: Neuroprotective effects of p62(SQSTM1)-engineered
lactic acid bacteria in Alzheimer's disease: A pre-clinical study.
Aging. 12:15995–16020. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Thal DR, Gawor K and Moonen S: Regulated
cell death and its role in Alzheimer's disease and amyotrophic
lateral sclerosis. Acta Neuropathol. 147:692024. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zheng X, Lin W, Jiang Y, Lu K, Wei W, Huo
Q, Cui S, Yang X, Li M, Xu N, et al: Electroacupuncture ameliorates
beta-amyloid pathology and cognitive impairment in Alzheimer
disease via a novel mechanism involving activation of TFEB
(transcription factor EB). Autophagy. 17:3833–3847. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Settembre C, Fraldi A, Medina DL and
Ballabio A: Signals from the lysosome: A control centre for
cellular clearance and energy metabolism. Nat Rev Mol Cell Biol.
14:283–296. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Morais GP, de Sousa Neto IV, Marafon BB,
Ropelle ER, Cintra DE, Pauli JR and Silva A: The dual and emerging
role of physical exercise-induced TFEB activation in the protection
against Alzheimer's disease. J Cell Physiol. 238:954–965. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Nnah IC, Wang B, Saqcena C, Weber GF,
Bonder EM, Bagley D, De Cegli R, Napolitano G, Medina DL, Ballabio
A, et al: TFEB-driven endocytosis coordinates MTORC1 signaling and
autophagy. Autophagy. 15:151–164. 2019. View Article : Google Scholar :
|
|
67
|
Zhang J, Zhang Y, Wang J, Xia Y, Zhang J
and Chen L: Recent advances in Alzheimer's disease: Mechanisms,
clinical trials and new drug development strategies. Signal
Transduct Target Ther. 9:2112024. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Dewanjee S, Chakraborty P, Bhattacharya H,
Chacko L, Singh B, Chaudhary A, Javvaji K, Pradhan SR, Vallamkondu
J, Dey A, et al: Altered glucose metabolism in Alzheimer's disease:
Role of mitochondrial dysfunction and oxidative stress. Free Radic
Biol Med. 193:134–157. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chen WT, Lu A, Craessaerts K, Pavie B,
Sala Frigerio C, Corthout N, Qian X, Laláková J, Kühnemund M,
Voytyuk I, et al: Spatial transcriptomics and in situ sequencing to
study Alzheimer's disease. Cell. 182:976–991.e19. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Martini-Stoica H, Xu Y, Ballabio A and
Zheng H: The Autophagy-lysosomal pathway in neurodegeneration: A
TFEB perspective. Trends Neurosci. 39:221–234. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wu JJ, Yu H, Bi SG, Wang ZX, Gong J, Mao
YM, Wang FZ, Zhang YQ, Nie YJ and Chai GS: Aerobic exercise
attenuates autophagy-lysosomal flux deficits by ADRB2/β2-adrenergic
receptor-mediated V-ATPase assembly factor VMA21 signaling in
APP-PSEN1/PS1 mice. Autophagy. 20:1015–1031. 2024. View Article : Google Scholar :
|
|
72
|
Bi SG, Yu H, Gao TL, Wu JJ, Mao YM, Gong
J, Wang FZ, Yang L, Chen J, Lan ZC, et al: Aerobic exercise
attenuates Autophagy-lysosomal flux deficits via β2-AR-Mediated
ESCRT-III Subunit CHMP4B in mice with human MAPT P301L. Aging Cell.
24:e701842025. View Article : Google Scholar
|
|
73
|
Kou X, Chen D and Chen N: Physical
activity alleviates cognitive dysfunction of Alzheimer's disease
through regulating the mTOR signaling pathway. Int J Mol Sci.
20:15912019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Li HY, Rong SS, Hong X, Guo R, Yang FZ,
Liang YY, Li A and So KF: Exercise and retinal health. Restor
Neurol Neurosci. 37:571–581. 2019.PubMed/NCBI
|
|
75
|
Ungvari Z, Fazekas-Pongor V, Csiszar A and
Kunutsor SK: The multifaceted benefits of walking for healthy
aging: From Blue Zones to molecular mechanisms. Geroscience.
45:3211–3239. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Cassilhas RC, Tufik S and de Mello MT:
Physical exercise, neuroplasticity, spatial learning and memory.
Cell Mol Life Sci. 73:975–983. 2016. View Article : Google Scholar
|
|
77
|
Rocchi A, Yamamoto S, Ting T, Fan Y,
Sadleir K, Wang Y, Zhang W, Huang S, Levine B, Vassar R and He C: A
Becn1 mutation mediates hyperactive autophagic sequestration of
amyloid oligomers and improved cognition in Alzheimer's disease.
PLoS Genet. 13:e10069622017. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Li X, He Q, Zhao N, Chen X, Li T and Cheng
B: High intensity interval training ameliorates cognitive
impairment in T2DM mice possibly by improving PI3K/Akt/mTOR
Signaling-regulated autophagy in the hippocampus. Brain Res.
1773:1477032021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chen J, Zhu T, Yang X, Yang Z, Shen M, Gu
B, Wang D, Zhang Y, Zhang M, Sun S, et al: Treadmill exercise
alleviates STING-mediated microglia pyroptosis and polarization via
activating mitophagy post-TBI. Free Radic Biol Med. 239:155–176.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sun L, Wu L, Xu Z, Zeng W and Wang Y:
Running exercise alleviates depressive-like behaviors through the
activation of PINK1-Parkin mediated mitophagy in mice exposed to
chronic social defeat stress. Psychiatry Res. 352:1167142025.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhang Y, Liao B, Hu S, Pan SY, Wang GP,
Wang YL, Qin ZH and Luo L: High intensity interval training induces
dysregulation of mitochondrial respiratory complex and mitophagy in
the hippocampus of middle-aged mice. Behav Brain Res.
412:1133842021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Tan ZX, Dong F, Wu LY, Feng YS and Zhang
F: The beneficial role of exercise on treating Alzheimer's disease
by Inhibiting β-amyloid peptide. Mol Neurobiol. 58:5890–5906. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Gratuze M, Julien J, Morin F, Marette A
and Planel E: Differential effects of voluntary treadmill exercise
and caloric restriction on tau pathogenesis in a mouse model of
Alzheimer's disease-like tau pathology fed with Western diet. Prog
Neuropsychopharmacol Biol Psychiatry. 79:452–461. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Xu L, Li M, Wei A, Yang M, Li C, Liu R,
Zheng Y, Chen Y, Wang Z, Wang K, et al: Treadmill exercise promotes
E3 ubiquitin ligase to remove amyloid β and P-tau and improve
cognitive ability in APP/PS1 transgenic mice. J Neuroinflammation.
19:2432022. View Article : Google Scholar
|
|
85
|
Morais GP, de Sousa Neto IV, Veras ASC,
Teixeira GR, Paroschi LO, Pinto AP, Dos Santos JR, Alberici LC,
Cintra DEC, Pauli JR, et al: Chronic exercise protects against
cognitive deficits in an Alzheimer's disease model by enhancing
autophagy and reducing mitochondrial abnormalities. Mol Neurobiol.
62:12791–12810. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Ohia-Nwoko O, Montazari S, Lau YS and
Eriksen JL: Long-term treadmill exercise attenuates tau pathology
in P301S tau transgenic mice. Mol Neurodegener. 9:542014.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hussain MS, Agrawal N, Ilma B, M MR, Nayak
PP, Kaur M, Khachi A, Goyal K, Rekha A, Gupta S, et al: Autophagy
and cellular senescence in Alzheimer's disease: Key drivers of
neurodegeneration. CNS Neurosci Ther. 31:e705032025. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 168:960–976. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Norwitz NG and Querfurth H: mTOR
Mysteries: Nuances and questions about the mechanistic target of
rapamycin in neurodegeneration. Front Neurosci. 14:7752020.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Gourmaud S, Stewart DA, Irwin DJ, Roberts
N, Barbour AJ, Eberwine G, O'Brien WT, Vassar R, Talos DM and
Jensen FE: The role of mTORC1 activation in seizure-induced
exacerbation of Alzheimer's disease. Brain. 145:324–339. 2022.
View Article : Google Scholar :
|
|
92
|
Babygirija R, Sonsalla MM, Mill J, James
I, Han JH, Green CL, Calubag MF, Wade G, Tobon A, Michael J, et al:
Protein restriction slows the development and progression of
pathology in a mouse model of Alzheimer's disease. Nat Commun.
15:52172024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Watson K and Baar K: mTOR and the health
benefits of exercise. Semin Cell Dev Biol. 36:130–139. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kang EB and Cho JY: Effect of treadmill
exercise on PI3K/AKT/mTOR, autophagy, and Tau hyperphosphorylation
in the cerebral cortex of NSE/htau23 transgenic mice. J Exerc
Nutrition Biochem. 19:199–209. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Deneubourg C, Ramm M, Smith LJ, Baron O,
Singh K, Byrne SC, Duchen MR, Gautel M, Eskelinen EL, Fanto M, et
al: The spectrum of neurodevelopmental, neuromuscular and
neurodegenerative disorders due to defective autophagy. Autophagy.
18:496–517. 2022. View Article : Google Scholar :
|
|
96
|
Shen K, Liu X, Chen D, Chang J, Zhang Y
and Kou X: Voluntary wheel-running exercise attenuates brain aging
of rats through activating miR-130a-mediated autophagy. Brain Res
Bull. 172:203–211. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Wątroba M and Szukiewicz D: The role of
sirtuins in aging and age-related diseases. Adv Med Sci. 61:52–62.
2016. View Article : Google Scholar
|
|
98
|
Wang J, Zhou F, Xiong CE, Wang GP, Chen
LW, Zhang YT, Qi SG, Wang ZH, Mei C, Xu YJ, et al: Serum sirtuin1:
A potential blood biomarker for early diagnosis of Alzheimer's
disease. Aging (Albany NY). 15:9464–9478. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Liu L, Dai WZ, Zhu XC and Ma T: A review
of autophagy mechanism of statins in the potential therapy of
Alzheimer's disease. J Integr Neurosci. 21:462022. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Surya K, Manickam N, Jayachandran KS,
Kandasamy M and Anusuyadevi M: Resveratrol mediated regulation of
hippocampal neuroregenerative plasticity via SIRT1 pathway in
synergy with wnt signaling: Neurotherapeutic implications to
mitigate memory loss in Alzheimer's disease. J Alzheimers Dis.
94(Suppl): S125–S140. 2023. View Article : Google Scholar :
|
|
101
|
Mehramiz M, Porter T, O'Brien EK,
Rainey-Smith SR and Laws SM: A potential role for Sirtuin-1 in
Alzheimer's disease: Reviewing the biological and environmental
evidence. J Alzheimers Dis Rep. 7:823–843. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Zhao N, Xia J and Xu B: Physical exercise
may exert its therapeutic influence on Alzheimer's disease through
the reversal of mitochondrial dysfunction via
SIRT1-FOXO1/3-PINK1-Parkin-mediated mitophagy. J Sport Health Sci.
10:1–3. 2021. View Article : Google Scholar :
|
|
103
|
Koo JH, Kang EB, Oh YS, Yang DS and Cho
JY: Treadmill exercise decreases amyloid-β burden possibly via
activation of SIRT-1 signaling in a mouse model of Alzheimer's
disease. Exp Neurol. 288:142–152. 2017. View Article : Google Scholar
|
|
104
|
Han R, Liu Y, Li S, Li XJ and Yang W:
PINK1-PRKN mediated mitophagy: Differences between in vitro and in
vivo models. Autophagy. 19:1396–1405. 2023. View Article : Google Scholar :
|
|
105
|
Pallanck LJ: Culling sick mitochondria
from the herd. J Cell Biol. 191:1225–1227. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhao N, Zhang X, Li B, Wang J, Zhang C and
Xu B: Treadmill exercise improves PINK1/Parkin-Mediated mitophagy
activity against Alzheimer's disease pathologies by upregulated
SIRT1-FOXO1/3 Axis in APP/PS1 mice. Mol Neurobiol. 60:277–291.
2023. View Article : Google Scholar
|
|
107
|
Huang J, Wang X, Zhu Y, Li Z, Zhu YT, Wu
JC, Qin ZH, Xiang M and Lin F: Exercise activates lysosomal
function in the brain through AMPK-SIRT1-TFEB pathway. CNS Neurosci
Ther. 25:796–807. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Tyagi A and Pugazhenthi S: A Promising
strategy to treat neurodegenerative diseases by SIRT3 Activation.
Int J Mol Sci. 24:16152023. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Khoramipour K, Chamari K, Hekmatikar AA,
Ziyaiyan A, Taherkhani S, Elguindy NM and Bragazzi NL: Adiponectin:
Structure, physiological functions, role in diseases, and effects
of nutrition. Nutrients. 13:11802021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Bloemer J, Pinky PD, Govindarajulu M, Hong
H, Judd R, Amin RH, Moore T, Dhanasekaran M, Reed MN and
Suppiramaniam V: Role of adiponectin in central nervous system
disorders. Neural Plast. 2018:45935302018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Rehman IU, Park JS, Choe K, Park HY, Park
TJ and Kim MO: Overview of a novel osmotin abolishes abnormal
metabolic-associated adiponectin mechanism in Alzheimer's disease:
Peripheral and CNS insights. Ageing Res Rev. 100:1024472024.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ali T, Rehman SU, Khan A, Badshah H, Abid
NB, Kim MW, Jo MH, Chung SS, Lee HG, Rutten BPF and Kim MO:
Adiponectin-mimetic novel nonapeptide rescues aberrant neuronal
metabolic-associated memory deficits in Alzheimer's disease. Mol
Neurodegener. 16:232021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Liu B, Liu J, Wang JG, Liu CL and Yan HJ:
AdipoRon improves cognitive dysfunction of Alzheimer's disease and
rescues impaired neural stem cell proliferation through
AdipoR1/AMPK pathway. Exp Neurol. 327:1132492020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Sun F, Wang J, Meng L, Zhou Z, Xu Y, Yang
M, Li Y, Jiang T, Liu B and Yan H: AdipoRon promotes amyloid-β
clearance through enhancing autophagy via nuclear GAPDH-induced
sirtuin 1 activation in Alzheimer's disease. Br J Pharmacol.
181:3039–3063. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Jian Y, Yuan S, Yang J, Lei Y, Li X and
Liu W: Aerobic exercise alleviates abnormal autophagy in brain
cells of APP/PS1 mice by upregulating AdipoR1 levels. Int J Mol
Sci. 23:99212022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Lu TX and Rothenberg ME: MicroRNA. J
Allergy Clin Immunol. 141:1202–1207. 2018. View Article : Google Scholar
|
|
117
|
Kou X and Chen N: Resveratrol as a natural
autophagy regulator for prevention and treatment of Alzheimer's
disease. Nutrients. 9:9272017. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Qi W, Ying Y, Wu P, Dong N, Fu W, Liu Q,
Ward N, Dong X, Zhao RC and Wang J: Inhibition of miR-4763-3p
expression activates the PI3K/mTOR/Bcl2 autophagy signaling pathway
to ameliorate cognitive decline. Int J Biol Sci. 20:5999–6017.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Madadi S, Schwarzenbach H, Saidijam M,
Mahjub R and Soleimani M: Potential microRNA-related targets in
clearance pathways of amyloid-β: Novel therapeutic approach for the
treatment of Alzheimer's disease. Cell Biosci. 9:912019. View Article : Google Scholar
|
|
120
|
Kou X, Chen D and Chen N: The regulation
of microRNAs in Alzheimer's sisease. Front Neurol. 11:2882020.
View Article : Google Scholar
|
|
121
|
Zhang H, Liang J and Chen N: The potential
role of miRNA-Regulated autophagy in Alzheimer's disease. Int J Mol
Sci. 23:77892022. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Kou X, Li J, Liu X, Chang J, Zhao Q, Jia
S, Fan J and Chen N: Swimming attenuates d-galactose-induced brain
aging via suppressing miR-34a-mediated autophagy impairment and
abnormal mitochondrial dynamics. J Appl Physiol (1985).
122:1462–1469. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Chen P, Chen F, Lei J, Li Q and Zhou B:
Activation of the miR-34a-Mediated SIRT1/mTOR signaling pathway by
urolithin a attenuates D-Galactose-induced brain aging in mice.
Neurotherapeutics. 16:1269–1282. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Chen ML, Hong CG, Yue T, Li HM, Duan R, Hu
WB, Cao J, Wang ZX, Chen CY, Hu XK, et al: Inhibition of miR-331-3p
and miR-9-5p ameliorates Alzheimer's disease by enhancing
autophagy. Theranostics. 11:2395–2409. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wang Y, Sun X, He B and Yu S: Ginsenoside
Rg1 downregulates miR-9-5p expression to modulate SIRT1-Mediated
mitochondrial dysfunction and Ameliorate Alzheimer's disease. Mol
Neurobiol. 62:13044–13059. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Boström P, Wu J, Jedrychowski MP, Korde A,
Ye L, Lo JC, Rasbach KA, Boström EA, Choi JH, Long JZ, et al: A
PGC1-α-dependent myokine that drives brown-fat-like development of
white fat and thermogenesis. Nature. 481:463–468. 2012. View Article : Google Scholar
|
|
127
|
Lourenco MV, Frozza RL, de Freitas GB,
Zhang H, Kincheski GC, Ribeiro FC, Gonçalves RA, Clarke JR, Beckman
D, Staniszewski A, et al: Exercise-linked FNDC5/irisin rescues
synaptic plasticity and memory defects in Alzheimer's models. Nat
Med. 25:165–175. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Zhang H, Wu X, Liang J, Kirberger M and
Chen N: Irisin, an exercise-induced bioactive peptide beneficial
for health promotion during aging process. Ageing Res Rev.
80:1016802022. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
de Freitas GB, Lourenco MV and De Felice
FG: Protective actions of exercise-related FNDC5/Irisin in memory
and Alzheimer's disease. J Neurochem. 155:602–611. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Ratne N, Jari S, Tadas M, Katariya R, Kale
M, Kotagale N, Madia D, Umekar M and Taksande B: Neurobiological
role and therapeutic potential of exercise-induced irisin in
Alzheimer's disease management. Ageing Res Rev. 105:1026872025.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Islam MR, Valaris S, Young MF, Haley EB,
Luo R, Bond SF, Mazuera S, Kitchen RR, Caldarone BJ, Bettio LEB, et
al: Exercise hormone irisin is a critical regulator of cognitive
function. Nat Metab. 3:1058–1070. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Lourenco MV: Irisin limits amyloid-β
buildup in Alzheimer's disease. Trends Endocrinol Metab. 35:94–96.
2024. View Article : Google Scholar
|
|
133
|
Kim E, Kim H, Jedrychowski MP, Bakiasi G,
Park J, Kruskop J, Choi Y, Kwak SS, Quinti L, Kim DY, et al: Irisin
reduces amyloid-β by inducing the release of neprilysin from
astrocytes following downregulation of ERK-STAT3 signaling. Neuron.
111:3619–3633.e8. 2023. View Article : Google Scholar
|
|
134
|
Bellettini-Santos T, Batista-Silva H,
Marcolongo-Pereira C, Quintela-Castro FCA, Barcelos RM, Chiepe K,
Rossoni JV Jr, Passamani-Ambrosio R, da Silva BS, Chiarelli-Neto O,
et al: Move your body toward healthy aging: Potential
neuroprotective mechanisms of irisin in Alzheimer's disease. Int J
Mol Sci. 24:124402023. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Bujak AL, Crane JD, Lally JS, Ford RJ,
Kang SJ, Rebalka IA, Green AE, Kemp BE, Hawke TJ, Schertzer JD and
Steinberg GR: AMPK activation of muscle autophagy prevents
fasting-induced hypoglycemia and myopathy during aging. Cell Metab.
21:883–890. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Sánchez B, Muñoz-Pinto MF and Cano M:
Irisin enhances longevity by boosting SIRT1, AMPK, autophagy and
telomerase. Expert Rev Mol Med. 25:e42022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Rahman MM, Islam MR and Emran TB:
Clinically important natural products for Alzheimer's disease. Int
J Surg. 104:1068072022. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
He Z, Zhang H, Li X, Tu S, Wang Z, Han S,
Du X, Shen L, Li N and Liu Q: The protective effects of
Esculentoside A through AMPK in the triple transgenic mouse model
of Alzheimer's disease. Phytomedicine. 109:1545552023. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Fan Y, Wang N, Rocchi A, Zhang W, Vassar
R, Zhou Y and He C: Identification of natural products with
neuronal and metabolic benefits through autophagy induction.
Autophagy. 13:41–56. 2017. View Article : Google Scholar :
|
|
140
|
Thakral S, Yadav A, Singh V, Kumar M,
Kumar P, Narang R, Sudhakar K, Verma A, Khalilullah H, Jaremko M
and Emwas AH: Alzheimer's disease: Molecular aspects and treatment
opportunities using herbal drugs. Ageing Res Rev. 88:1019602023.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Agah S, Akbari A, Sadeghi E, Morvaridzadeh
M, Basharat Z, Palmowski A and Heshmati J: Resveratrol
supplementation and acute pancreatitis: A comprehensive review.
Biomed Pharmacother. 137:1112682021. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Li C, Wang N, Zheng G and Yang L: Oral
administration of Resveratrol-Selenium-Peptide nanocomposites
alleviates Alzheimer's disease-like pathogenesis by inhibiting Aβ
aggregation and regulating gut microbiota. ACS Appl Mater
Interfaces. 13:46406–46420. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Islam F, Nafady MH, Islam MR, Saha S,
Rashid S, Akter A, Or-Rashid MH, Akhtar MF, Perveen A, Md Ashraf G,
et al: Resveratrol and neuroprotection: An insight into prospective
therapeutic approaches against Alzheimer's disease from bench to
bedside. Mol Neurobiol. 59:4384–4404. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Chen JY, Zhu Q, Zhang S, OuYang D and Lu
JH: Resveratrol in experimental Alzheimer's disease models: A
systematic review of preclinical studies. Pharmacol Res.
150:1044762019. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Rahman MH, Akter R, Bhattacharya T,
Abdel-Daim MM, Alkahtani S, Arafah MW, Al-Johani NS, Alhoshani NM,
Alkeraishan N, Alhenaky A, et al: Resveratrol and neuroprotection:
Impact and its therapeutic potential in Alzheimer's disease. Front
Pharmacol. 11:6190242020. View Article : Google Scholar
|
|
146
|
Turner RS, Thomas RG, Craft S, van Dyck
CH, Mintzer J, Reynolds BA, Brewer JB, Rissman RA, Raman R and
Aisen PS: A randomized, double-blind, placebo-controlled trial of
resveratrol for Alzheimer disease. Neurology. 85:1383–1391. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Broderick TL, Rasool S, Li R, Zhang Y,
Anderson M, Al-Nakkash L, Plochocki JH, Geetha T and Babu JR:
Neuroprotective effects of chronic resveratrol treatment and
exercise training in the 3xTg-AD mouse model of Alzheimer's
disease. Int J Mol Sci. 21:73372020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Kou JJ, Shi JZ, He YY, Hao JJ, Zhang HY,
Luo DM, Song JK, Yan Y, Xie XM, Du GH and Pang XB: Luteolin
alleviates cognitive impairment in Alzheimer's disease mouse model
via inhibiting endoplasmic reticulum stress-dependent
neuroinflammation. Acta Pharmacol Sin. 43:840–849. 2022. View Article : Google Scholar
|
|
149
|
Vongthip W, Nilkhet S, Boonruang K,
Sukprasansap M, Tencomnao T and Baek SJ: Neuroprotective mechanisms
of luteolin in glutamate-induced oxidative stress and
autophagy-mediated neuronal cell death. Sci Rep. 14:77072024.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Ali F and Siddique YH: Bioavailability and
Pharmaco-therapeutic potential of luteolin in overcoming
Alzheimer's disease. CNS Neurol Disord Drug Targets. 18:352–365.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
He Z, Li X, Wang Z, Cao Y, Han S, Li N,
Cai J, Cheng S and Liu Q: Protective effects of luteolin against
amyloid beta-induced oxidative stress and mitochondrial impairments
through peroxisome proliferator-activated receptor γ-dependent
mechanism in Alzheimer's disease. Redox Biol. 66:1028482023.
View Article : Google Scholar
|
|
152
|
Tao X, Zhang R, Wang L, Li X and Gong W:
Luteolin and exercise combination therapy ameliorates Amyloid-β1-42
Oligomers-Induced cognitive impairment in AD Mice by mediating
neuroinflammation and autophagy. J Alzheimers Dis. 92:195–208.
2023. View Article : Google Scholar
|
|
153
|
Tao X, Wang L and Gong W: Untargeted
metabolomics reveals the mechanisms of luteolin and exercise
combination treatment against cognitive impairments in AD mice
through modulating autophagy. J Nutr Biochem. 145:1100112025.
View Article : Google Scholar : PubMed/NCBI
|