Introduction
Vasoplegia is a key driver of poor prognosis in
sepsis, a severe complication characterized by persistent vasomotor
dysfunction and catecholamine-resistant hypotension (1). Sepsis-induced vasoplegia
contributes directly to multiple organ dysfunction syndrome, and
associated mortality through circulatory failure and impaired
oxygen utilization (1,2). According to the Global Burden of
Diseases, Injuries, and Risk Factors Study, it was estimated that
in 2021 there were 166 million (95% uncertainty interval: 135-201
million) sepsis cases worldwide, with 21.4 million (20.3-22.5
million) all-cause sepsis-related deaths, accounting for 31.5% of
total global mortality (3).
Consequently, elucidating the pathogenesis and regulatory pathways
of vasoplegia in sepsis, and developing effective management and
targeted therapeutic strategies specifically addressing this
complication, are imperative for improving sepsis management and
patient survival.
Conventional paradigms in sepsis research focus
predominantly on endothelial dysfunction, encompassing barrier
disruption, glycocalyx degradation and dysregulated nitric oxide
(NO)/cyclic guanosine monophosphate (cGMP) signaling, as the
primary mediator of vasoplegia (4-6).
However, a growing body of evidence from translational
investigations challenges this endothelial-centric model,
highlighting vascular smooth muscle cells (VSMCs), the ultimate
effectors of vascular tone regulation, as equally important
contributors. In rodent models of cecal ligation and puncture
(CLP), the contractile impairment of VSMCs exhibits biphasic
kinetics (7,8): Early impairment is influenced by
endothelial-derived factors associated with sepsis, whereas
late-phase dysfunction results from direct injury to VSMCs
(7). Recent metabolomic
profiling has further identified sepsis-associated lactic acidosis
as a key driver of the phenotypic plasticity of VSMCs, facilitating
transitions towards senescent, osteogenic and proinflammatory
phenotypes that accelerate vascular stiffening (8). Therefore, understanding the role of
VSMCs in vasoplegia during sepsis is crucial for elucidating the
pathogenesis of sepsis-induced circulatory insufficiency and
identifying novel targets for therapeutic intervention.
Notably, despite increasing recognition of the
pivotal role of VSMCs, previous reviews in this field remain
limited in several crucial aspects. Most reviews have concentrated
on isolated mechanisms of VSMC dysfunction without sufficiently
integrating upstream triggers, including endothelial injury and the
cytokine storm, or examining intercellular crosstalk (7-9).
Others have retained an endothelial-centric viewpoint, relegating
VSMCs to a subordinate 'effector' role rather than recognizing them
as central regulatory elements (4-6).
Furthermore, few reviews successfully summarize preclinical data
with clinical translational challenges, such as the disparity
between promising VSMC-targeted therapies in animal models and
their lack of efficacy in human trials, or engage with unresolved
controversies, for example, the reversibility of septic VSMC
dedifferentiation, which is critical for informing therapeutic
strategies (1,6). These shortcomings have hindered the
development of a comprehensive, translation-oriented understanding
of VSMC-driven vasoplegia.
The present review advances the field by addressing
these crucial limitations and building upon the existing
literature. Specifically, it moves beyond fragmented,
mechanism-focused accounts to develop an integrated framework
linking upstream sepsis-related stimuli, such as inflammatory
mediators, metabolic disturbances and endothelial crosstalk, to
downstream VSMC dysfunction. Furthermore, it explicitly engages
with fundamental scientific controversies and bridges preclinical
insights with clinical translational challenges, a perspective
rarely emphasized in prior reviews. By thoroughly examining the
complex role of VSMCs in the pathogenesis of sepsis-induced
vasoplegia, including their signaling pathways, inflammatory
responses and regulatory interactions, the current review aims to
provide a comprehensive, controversy-conscious and translationally
oriented understanding of vascular dysfunction in this context.
Ultimately, it not only consolidates current knowledge but also
delineates critical gaps, thereby guiding future preclinical and
clinical investigations, and addressing the limitations of the
narrow or fragmented perspectives of previous literature.
Anatomical localization and functional
properties of VSMCs
Within the pathological context of sepsis, VSMCs
serve as crucial hubs for signal integration, processing cues from
inflammatory mediators, alterations in mechanical stress, patterns
of injury and fragments of extracellular matrix (ECM) degradation
(10).
Cellular localization of VSMCs:
Anatomical distribution and structural context
Anatomically, the vascular wall (excluding
capillaries and lymphatic capillaries) is organized into three
concentric layers from the lumen outwards: The intima, media and
adventitia (11). Functionally,
the media acts as the central hub for vascular tension regulation,
housing circumferentially arranged VSMCs intertwined with
elastic/collagen fiber networks. This arrangement endows vessels
with both active contractility and passive elastic recoil (12). Ultrastructurally, VSMCs exhibit
an elongated spindle-shaped morphology, containing cytoplasmic
contractile units, force-transducing dense bodies and
calcium-regulating sarcoplasmic reticulum (13). However, in sepsis, this
structural organization is disrupted: VSMC detachment from the ECM
and degradation of elastic fibers impair vascular compliance,
exacerbating hypotension. The changes in VSMCs from physiological
conditions to septic conditions are shown in Fig. 1.
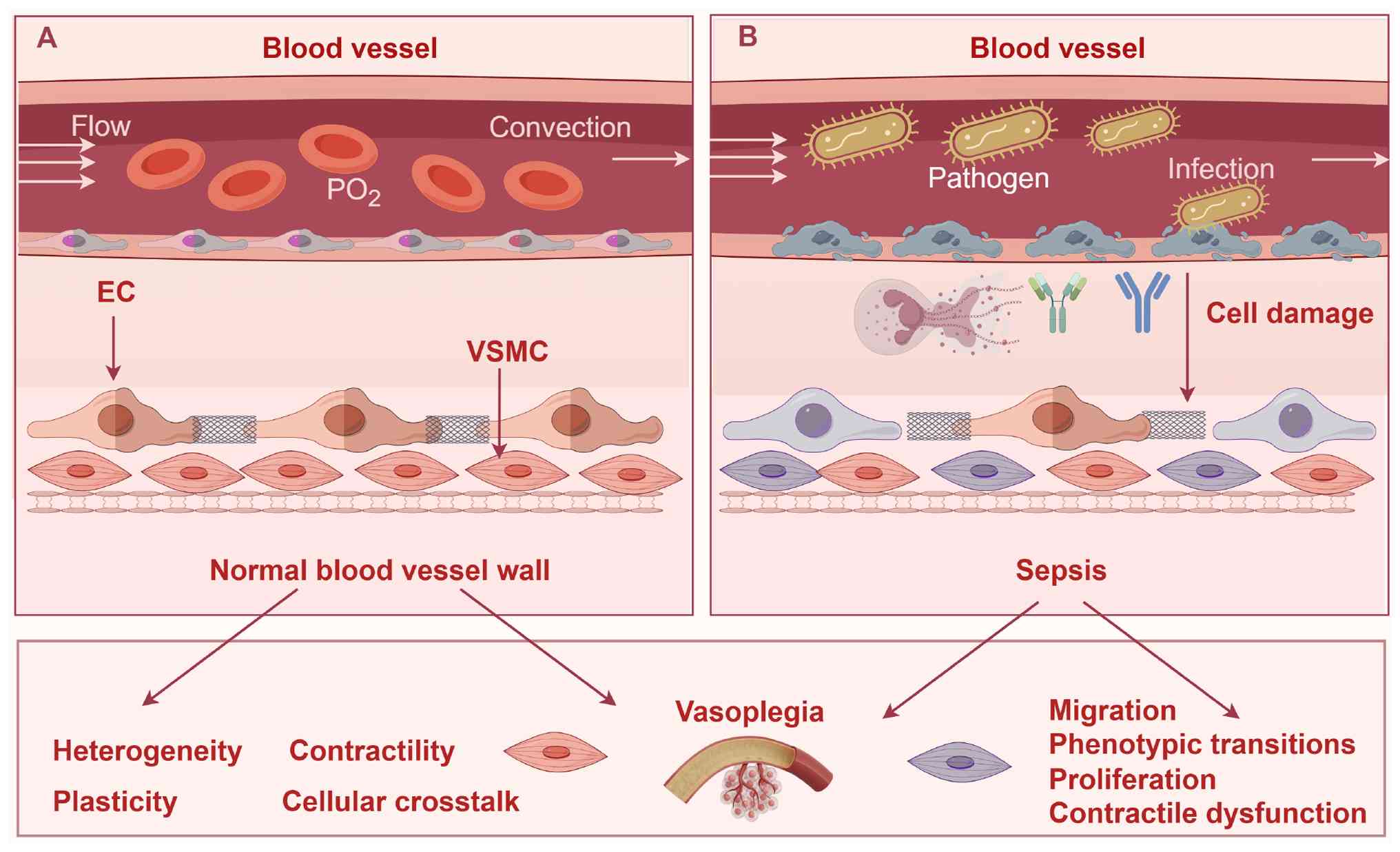 | Figure 1VSMCs in physiological and septic
conditions. (A) VSMCs exhibit a quiescent, contractile phenotype
characterized by organized cytoskeletal architecture and intact
regulatory pathways. Key features include: i) Stable anatomical
distribution within the vascular media, maintaining vascular wall
integrity; ii) homogeneous lineage-specific phenotype with minimal
heterogeneity; iii) robust contractility driven by intact calcium
homeostasis and myofilament calcium sensitivity; iv) limited
phenotypic plasticity, with minimal switching from contractile to
synthetic states; v) balanced cellular crosstalk: Intricate
bidirectional interactions with neighboring cells, primarily ECs.
EC-VSMC communication relies on core pathways to maintain vascular
homeostasis, enabling adaptation to mechanical injury, shear stress
and chemical stimuli. (B) Sepsis induces profound structural and
functional perturbations in VSMCs, characterized by five core
changes: i) Altered cellular localization: Disrupted anatomical
distribution within the vascular media due to sepsis-induced cell
injury, compromising vascular wall stability; ii) enhanced cellular
heterogeneity: Expanded phenotypic diversity with an increased
proportion of synthetic VSMCs (arising from lineage switching or
progenitor cell recruitment), contributing to vascular dysfunction;
iii) impaired cellular contractility: Diminished contractile
capacity driven by dysregulated calcium homeostasis and
downregulated contractile proteins; iv) exaggerated cellular
plasticity: Prominent phenotypic switching from contractile to
synthetic states, associated with increased proliferation,
migration and secretion of proinflammatory mediators; v)
dysregulated cellular crosstalk: Collapse of bidirectional EC-VSMC
communication, cytokine secretion becomes proinflammatory
(increased release of proinflammatory cytokines), preventing
adaptation to stressors. The figure was constructed using Figdraw
2.0 tool (https://www.figdraw.com/#/), with
official authorization obtained by the authors (authorization no.:
PTSIYbe897).VSMCs, vascular smooth muscle cells; ECs, endothelial
cells; PO2, partial pressure of oxygen. |
Cellular heterogeneity of VSMCs: Lineage
origins and phenotypic diversity
Studies employing single-cell transcriptomics and
lineage tracing techniques have revealed the distinct origins of
VSMCs: hindlimb/inguinal-axillary venous VSMCs predominantly stem
from the lateral plate mesenchyme (14); SMCs in the proximal thoracic
aorta are derived from the second heart field and cardiac neural
crest (15); and coronary artery
SMCs partly originate from epicardial cells that migrate into the
heart during development (16).
These differences in embryonic origins, combined with varied
responses to local microenvironmental signals, confer upon VSMCs
tissue-specific molecular profiles and functional
characteristics.
Cellular contractility of VSMCs:
Mechanisms and regulatory pathways
Under physiological conditions, VSMCs maintain a
robust contractile capacity, which is crucial for the dynamic
regulation of blood flow and pressure (17). This contractile ability is
regulated by the phosphorylation of myosin light chain (MLC)
through two primary pathways: The calcium-dependent MLC kinase
(MLCK)/MLC signaling pathway and the calcium-sensitized
Rho/Rho-associated protein kinase (ROCK) signaling pathway.
Regarding the MLCK/MLC signaling pathway, an
increase in the intracellular calcium ion concentration
([Ca2+]i) occurs via the opening of voltage-gated or
receptor-gated calcium channels, or release from intracellular
stores (18). Free calcium ions
bind to calmodulin, activating MLCK; MLCK catalyzes the
phosphorylation of the 19th serine residue on regulatory MLC,
activating the Mg2+-ATPase activity of myosin heads.
This induces a conformational change in myosin, enhancing its
binding to actin (19). MLC
phosphatase (MLCP)-mediated dephosphorylation of MLC reduces
myosin-actin binding, inhibiting contraction. Regarding the
Rho/ROCK signaling pathway (20), activation of RhoA leads to ROCK
activation, which inhibits MLCP activity, thereby maintaining or
increasing MLC phosphorylation levels by preventing
dephosphorylation (21). ROCK
can also directly phosphorylate MLC to promote myosin contraction
(22). Additionally, the
calcium-sensitized pathway is associated with actin cytoskeleton
remodeling, such as via cofilin phosphorylation, which inhibits
actin filament depolymerization, further enhancing contraction
(23). In summary, these two
signaling pathways work together to regulate VSMC contractility:
The calcium-dependent pathway facilitates acute responses to
fluctuations in [Ca2+]i, whereas the calcium-sensitized
pathway prolongs contraction.
Cellular plasticity of VSMCs: Phenotypic
switching and functional adaptation
Upon encountering injury or stimulation by growth
factors, VSMCs undergo a response characterized by enhanced
proliferation, migration and synthesis of extracellular components,
a phenomenon known as phenotypic switching (24). The main subtypes of VSMCs are as
follows: i) Contractile VSMCs are characterized by a high
concentration of myofilaments, a slender spindle-shaped morphology
and robust contractility. ii) Synthetic VSMCs are characterized by
an abundance of rough endoplasmic reticulum and Golgi apparatus,
downregulation of contractile proteins, and upregulation of ECM
components and inflammatory factors. Other identified phenotypes
include osteogenic, macrophage-like, mesenchymal-like and
fibroblast-like VSMCs (24).
Under physiological conditions, VSMCs typically exhibit a quiescent
state characterized by low proliferative and migratory activity.
However, in response to pathological stimuli, such as vascular
injury, diabetes or hypertension, VSMCs undergo phenotypic
plasticity, transitioning from a contractile state to
secretory/inflammatory phenotypes with increased proliferative,
migratory and matrix-synthetic functions (25).
Cellular crosstalk involving VSMCs:
Interactions with neighboring cells
Effective communication between endothelial cells
(ECs) and VSMCs is a complex physiological process essential for
maintaining vascular structure and function, allowing cells to
adapt to mechanical injury, shear stress and chemical stimuli
through pathways such as Notch signaling, cytokine secretion and
exosomal trafficking (12,24). The conserved Notch signaling
pathway facilitates interactions between adjacent cells: ECs
express Jagged1 ligands that bind to Notch3 receptors on VSMCs,
inhibiting their transition from contractile to synthetic
phenotypes (24). Additionally,
ECs regulate VSMC metabolism, influencing phenotypic plasticity. In
coculture models, hypoxic ECs increase lactate production, whereas
the knockout of lactate dehydrogenase A (LDHA) reduces osteogenic
marker expression and VSMC apoptosis (26). Cytokine secretion serves as
another crucial regulatory mechanism. Factors such as prostanoids,
arachidonic acid, acid metabolites and NO, which are released by
ECs, have distinct effects on VSMC function (12). Furthermore, exosomes serve a
notable role in vascular remodeling; in atherosclerosis models,
endothelial autophagy facilitates the transfer of microRNA
(miRNA/miR)-204-5p via exosomes to VSMCs, inhibiting endothelial
apoptosis and VSMC calcification (27). This multidimensional crosstalk
highlights the interconnection of ECs and VSMCs in preserving
vascular health or contributing to pathological processes.
Multifactorial impairment of VSMCs in septic
vasoplegia
Metabolic disturbances in VSMCs during
sepsis-induced vasoplegia
Sepsis induces marked metabolic reprogramming in
VSMCs, disrupting their function and contributing to disease
pathogenesis. Systemic circulatory failure and microcirculatory
dysfunction collectively impair cellular oxygen uptake and
utilization, triggering metabolic remodeling (5). Under septic conditions, the energy
metabolism of VSMCs transitions from oxidative phosphorylation
(OXPHOS) to aerobic glycolysis, a metabolic reprogramming observed
across various cell types. This glycolytic switch, characterized by
increased glucose uptake and upregulation of hexokinase 2, supports
the migratory activity of VSMCs (28).
Upregulated glycolysis results in excess lactate
production, leading to lactic acidosis. This metabolic disturbance
inhibits mitochondrial respiration (OXPHOS) and glutamine
catabolism, decreasing cellular ATP levels and altering the
NAD+/NADH ratio (8).
These sequential effects contribute to the phenotypic transition
and functional decline of VSMCs, exacerbating sepsis-induced
vascular dysfunction. Notably, glycolysis and lactate directly
promote VSMC migration and proliferation. In sepsis-induced
systemic cellular injury, elevated plasma LDH levels are indicative
of tissue injury, a common feature observed in severe sepsis
(29,30). At the mechanistic level, lysine 5
crotonylation enhances the tetramer formation of LDHA, increasing
lactate production to drive the migration and proliferation of
VSMCs (31). These combined
metabolic disturbances impair VSMCs contractility and provoke
phenotypic alterations, worsening the vascular pathophysiology
induced by sepsis.
Shear stress and VSMCs dysfunction in
sepsis-induced vasoplegia
Shear stress is the fluid dynamic force exerted by
blood flow on the vascular wall, and is influenced by factors such
as vessel diameter, flow pulsatility, blood viscosity and velocity
(32). Physiological shear
stress is crucial for vascular homeostasis, whereas abnormal shear
stress serves as a notable mechanical trigger for vascular
pathology. Abnormal shear stress in sepsis arises from two mutually
reinforcing mechanisms: Hemodynamic disturbances and vascular
structural damage.
First, sepsis-induced hyperdynamic circulation
creates a paradoxical hemodynamic state: Inflammatory vasodilation
and hypovolemia drive hypotension, whereas compensatory cardiac
responses attempt to maintain tissue perfusion (33). This imbalance is exacerbated by
microcirculatory dysfunction, characterized by vasoregulatory
failure, capillary shunting and microthrombosis, which further
disrupts blood flow patterns and amplifies tissue hypoperfusion
(34). Collectively, these
changes alter flow velocity, pulsatility and distribution, laying
the foundation for abnormal shear stress.
Second, sepsis-mediated vascular structural damage
compromises the ability of the vascular wall to buffer shear
stress. Pathological alterations include glycocalyx degradation,
reduced red blood cell deformability, redistribution of membrane
phospholipids, and endothelial injury driven by platelet and
neutrophil extracellular trap formation (35,36). When the endothelial barrier is
disrupted, VSMCs are directly exposed to pulsatile shear stress, an
insult that is normally attenuated by the endothelial layer,
directly triggering functional dysregulation of VSMCs (37). This structural breakdown forms
the anatomical basis for abnormal shear stress in sepsis.
Notably, invasive therapies for sepsis can further
perturb hemodynamics by altering flow velocity and patterns,
thereby modifying intravascular shear stress (38-40). These treatment-related changes
may either mitigate or exacerbate the phenotypic plasticity of
VSMCs, highlighting the need for tailored clinical management to
minimize iatrogenic vascular damage.
Cellular crosstalk involving VSMCs in
sepsis-associated vasoplegia
Sepsis-induced vascular dysfunction arises from a
marked disruption of the EC-VSMC interaction network. In a
physiological state, the close crosstalk between ECs and VSMCs
serves a crucial role in maintaining vascular tone, structure and
functional equilibrium (41).
The presence of physiological laminar shear stress acting on ECs
helps preserve the contractile phenotype of VSMCs, characterized by
high α-smooth muscle actin (α-SMA) expression (42). Key paracrine pathways, such as
endothelial NO synthase (eNOS)/NO, have a role in regulating
platelet-derived growth factor signaling to facilitate
flow-dependent vasodilation and adaptive remodeling processes
(43,44). The ECM, which offers structural
support, influences cellular signaling through its compositional
and mechanical attributes (12,45).
Sepsis disrupts this balance through several
mechanisms: i) Mechanical force-sensing dysregulation: Hemodynamic
disturbances (such as hypotension and microcirculatory failure)
alter physiological shear stress, compromising its role in
preserving VSMCs contractility (46). ii) Aberrations in signaling
pathways: Sepsis-induced inflammation, oxidative stress and
endotoxemia disrupt communication between ECs and VSMCs. In acute
kidney injury induced by lipopolysaccharide (LPS), endothelial
calpain activation leads to p38 phosphorylation and upregulation of
inducible NO synthase (iNOS), resulting in excessive production of
NO and reactive oxygen species (ROS) that leads to EC apoptosis
(47). Multiple signaling
factors involved in EC-VSMC crosstalk exhibit altered expression or
activity in response to sepsis. iii) ECM remodeling dysfunction:
Inflammatory mediators trigger excessive ECM deposition (48), imbalanced degradation (for
example, altered metalloproteinase activity) and abnormal
cross-linking, disrupting vascular mechanics and intercellular
chemical/mechanical signaling. iv) Altered vesicle-mediated
communication: Extracellular vesicles (EVs) serve a crucial role in
EC-VSMC communication (49).
Sepsis alters the release and contents (proteins, mRNAs and miRNAs)
of EVs: EVs derived from ECs containing miR-539 promote VSMC
proliferation (49), potentially
contributing to vascular repair or pathological remodeling. Other
sepsis-regulated miRNAs similarly modulate EC-VSMC interactions
through EVs.
These collective disruptions force VSMCs to switch
from a normal contractile phenotype to a pathological,
proinflammatory, proliferative, migratory or synthetic state. This
phenotypic transition serves as the fundamental cellular mechanism
contributing to sepsis-associated vasoplegia, increased
permeability, abnormal remodeling and organ hypoperfusion.
Calcium homeostasis dysregulation in
VSMCs in sepsis-associated vasoplegia
Alterations in [Ca2+]i are crucial events
for initiating vascular contraction, and calcium levels and the
sensitivity of contractile proteins collectively determine the
strength of contraction. Bacterial LPS, a prominent
pathogen-associated molecular pattern (PAMP), serves a central role
in sepsis and septic shock. In a rat model of endotoxemia, the
administration of LPS has been shown to result in systolic
hypotension, tachycardia, peritoneal neutrophil migration and
elevated alanine aminotransferase levels at 24 h. Concurrently,
VSMCs displayed disturbances in calcium homeostasis, such as
reduced calcium influx, depletion of sarcoplasmic reticulum
calcium, inactivation of Orai1 channels, and ultimately,
sepsis-associated vasoplegia (50).
The precise mechanisms through which LPS mediates
intracellular calcium dysregulation have not been fully elucidated.
In microglia, LPS-induced mitochondrial fragmentation hinders the
capacity for and rate of calcium uptake, leading to disruption of
calcium homeostasis (51). In
cardiomyocytes, the interaction between pyruvate kinase M2 (PKM2)
and sarcoplasmic/endoplasmic reticulum calcium ATPase 2a is crucial
for maintaining calcium balance; PKM2 deficiency exacerbates the
LPS-induced disruption of calcium homeostasis and cardiac
contractile dysfunction (52).
Another crucial mechanism is the reduced sensitivity
of contractile proteins to calcium: Intravenous LPS administration
in adult rabbits has been reported to result in a depression of the
force-calcium relationship in cardiac tissue despite an increase in
[Ca2+]i, indicating endotoxemia-induced ineffective
calcium cycling and desensitization of myofibrils (53). Additionally, sepsis-associated
vasoplegia is associated with a decrease in the expression of
contractile proteins: LPS inhibits TGF-β control elements on the
α-SMA promoter, leading to reduced α-SMA transcription and protein
levels in human aortic and coronary VSMCs, and in rat aortic VSMCs
(54).
The potential regulatory mechanisms of calcium
homeostasis in VSMCs are illustrated in Fig. 2. Physiologically, Ca2+
enters VSMCs via voltage-dependent calcium channel,
receptor-operated calcium channel, transient receptor potential
channel and store-operated calcium channel, and intracellular
calcium handling is regulated by plasmalemmal calcium ATPase,
sodium-calcium exchanger and organelles (mitochondria and
endoplasmic reticulum). GPCR activation triggers a signaling
cascade via phospholipase Cβ/γ, which hydrolyze
phosphatidylinositol 4,5-bisphosphate into inositol
1,4,5-trisphosphate (IP3); IP3 binds to endoplasmic reticulum
receptors to release Ca2+ and elevate cytoplasmic
Ca2+ levels. In sepsis and septic shock, LPS disrupts
this finely balanced regulatory network by impairing calcium
homeostasis through interference with calcium channels,
intracellular calcium stores and calcium transporters. It also
reduces myofibrillar calcium sensitivity and suppresses α-actinin
expression (54). This decrease
in α-actinin further undermines the structural integrity of the
actin cytoskeleton, synergistically weakening myosin-actin
interactions and VSMC contractility.
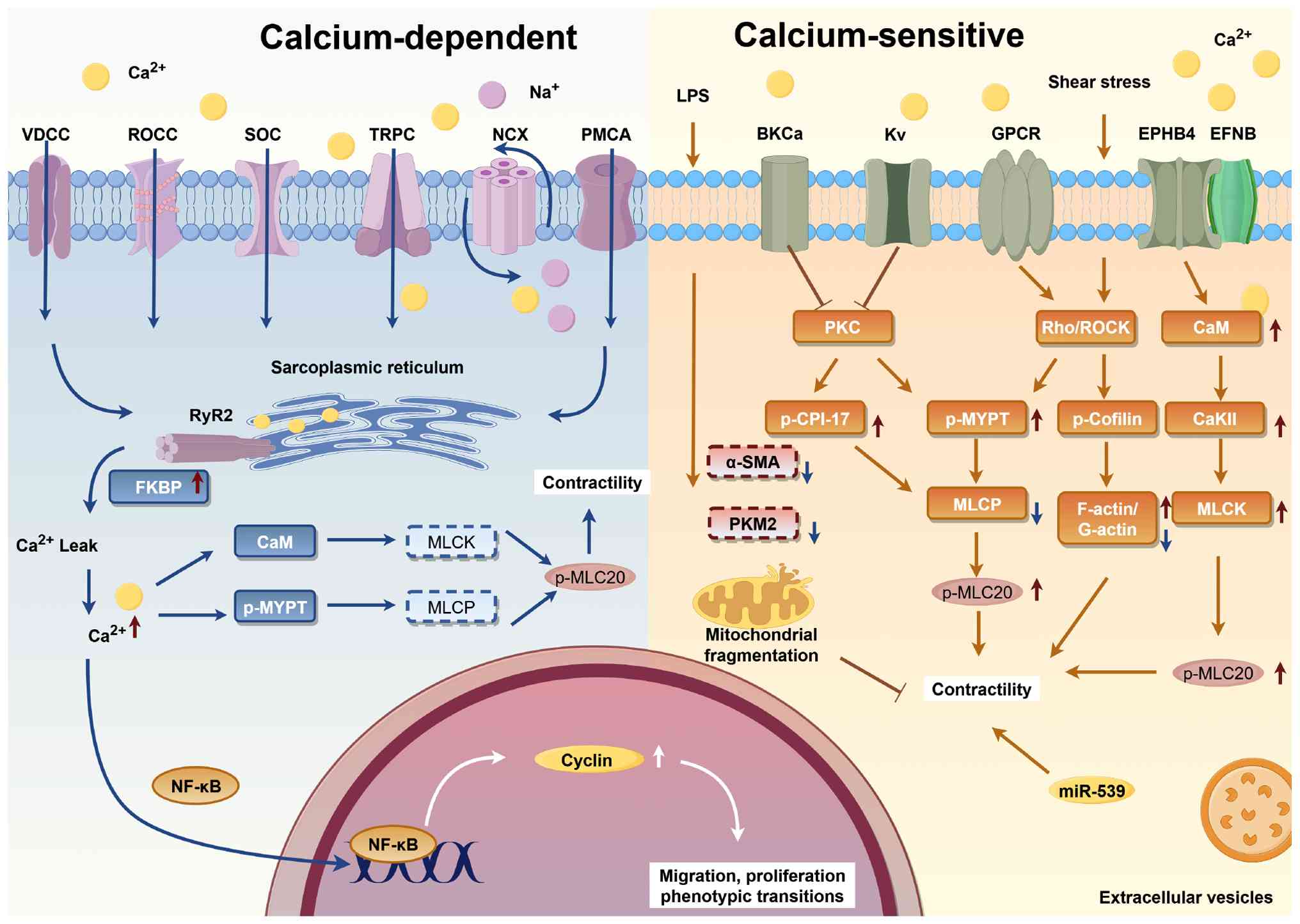 | Figure 2Regulation of Ca2+ and
signal transduction in VSMCs. Ca2+ enters VSMCs via
multiple pathways, including VDCC, ROCC, TRPC and SOC.
Intracellular calcium handling relies on the PMCA and NCX at the
cell membrane, as well as organelle-mediated transport in
mitochondria and the endoplasmic reticulum. VSMC contraction is
regulated by two primary mechanisms: Calcium-dependent and
calcium-sensitive pathways. In the calcium-dependent pathway,
increased intracellular Ca2+ forms a complex with CaM,
activating MLCK. This activation enhances Mg2+-ATPase
activity, driving myosin-actin interaction and subsequent cell
contraction. The calcium-sensitive pathway, mainly the Rho/ROCK
pathway, is activated by sepsis-associated stimuli, including
angiotensin II, leptin and mechanical stretch. PKC also modulates
calcium influx by regulating the activity of calcium and potassium
ion channels at the cell membrane. Additionally, miRNAs fine-tune
these regulatory processes, adding another layer of complexity to
calcium homeostasis and smooth muscle function. The figure was
constructed using Figdraw 2.0 tool (https://www.figdraw.com/#/), with official
authorization obtained by the authors (authorization no.:
PPTAA80b0b).α-SMA, α-smooth muscle actin; BKCa, big-conductance
calcium-activated potassium channel; Ca2+, calcium ions;
CaKII, calcium/calmodulin-dependent protein kinase II; CaM,
calmodulin; EPHB4, Eph receptor B4; EFNB, ephrin B; FKBP, FK506
binding proteins; GPCR, G-protein coupled receptors; Kv,
voltage-gated potassium channel; LPS, lipopolysaccharide; MLCK,
myosin light chain kinase; MLCP, MLC phosphatase; miRNA/miR,
microRNA; NCX, sodium-calcium exchanger; p-CPI-17, phosphorylated
protein kinase C-potentiated inhibitor protein-17; p-MLC20,
phosphorylated myosin light chain 20; p-MYPT, phosphorylated myosin
phosphatase target subunit; PKC, protein kinase C; PKM2, pyruvate
kinase M2; PLC, phospholipase C; PMCA, plasmalemmal calcium ATPase;
ROCC, receptor-operated calcium channel; ROCK, Rho-associated
protein kinase; RyR2, ryanodine receptor 2; SOC, store-operated
calcium channel; TRPC, transient receptor potential channel; VDCC,
voltage-dependent calcium channel. |
Mechanisms underlying VSMC injury in
sepsis
The pathophysiology of VSMC injury encompasses a
complex interplay of direct and indirect injury. These mechanisms
intersect to disrupt vascular structure and function, leading to
implications for sepsis-induced vasoplegia. Fig. 3 delineates the mechanisms by
which pathogens, endothelial injury, cytokine storms and calcium/NO
imbalances collectively contribute to VSMC dysfunction.
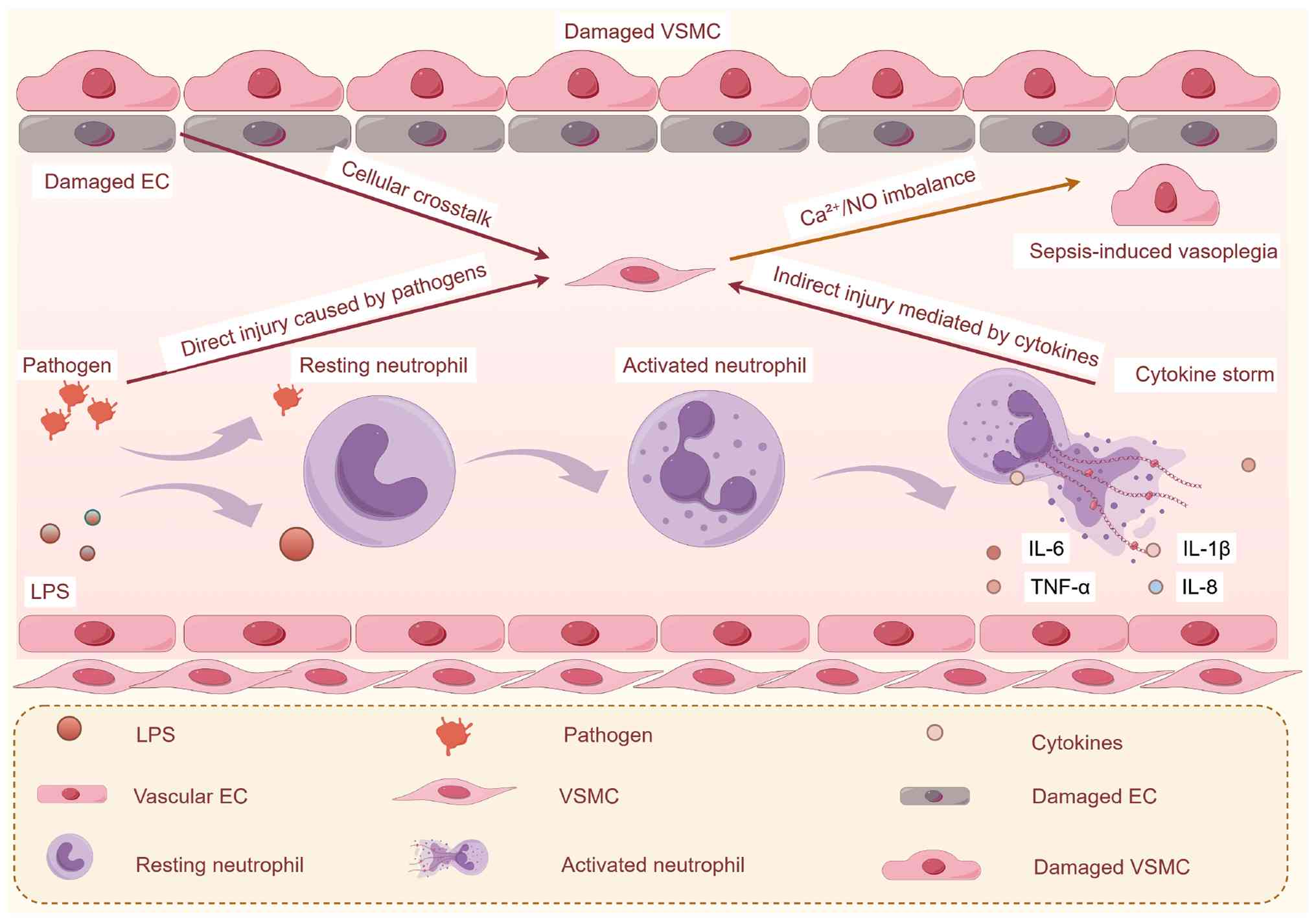 | Figure 3Conceptual overview of the
convergence of endothelial injury, cytokine storm and
Ca2+/NO imbalance in sepsis-induced VSMC dysfunction.
Sepsis (LPS or polymicrobial infection) initiates three
interconnected pathological processes: Endothelial injury, cytokine
storm and Ca2+/NO imbalance, which synergistically drive
VSMC dysfunction and subsequent sepsis-induced vasoplegia.
Collectively, these events result in VSMC hypocontractility,
phenotypic switching and apoptosis, ultimately causing vascular
hyporeactivity and catecholamine resistance in septic vasoplegia.
The figure was constructed using Figdraw 2.0 tool (https://www.figdraw.com/#/), with official
authorization obtained by the authors (authorization no.:
PWSOU68e36).Ca2+, calcium ions; EC, endothelial cell;
eNOS, endothelial NO synthase; iNOS, inducible NO synthase; LPS,
lipopolysaccharide; NO, nitric oxide; ROS, reactive oxygen species;
VSMC, vascular smooth muscle cell. |
Direct VSMC injury caused by
pathogens
The injury to VSMCs induced by sepsis results from a
complex interaction of factors, including direct invasion by
pathogens and injury caused by virulence factors. Pathogens disrupt
cellular structures through their metabolic activities or through
the action of secreted virulence factors, such as pore-forming
toxins, exfoliative toxins and superantigens, leading to
inflammatory processes that ultimately result in tissue necrosis
(6). For example, bacterial
sepsis, the predominant form of sepsis (55), has been shown to induce injury to
VSMCs and impair their contractile function in a rat model of CLP
(56). Different pathogens have
unique effects: Chlamydia pneumoniae infection triggers an
increase in mitochondrial ROS (mtROS) via Toll-like receptor
(TLR)2, activating the JunB-Fra-1/matrix metalloproteinase-2
pathway to facilitate VSMC migration (57). By contrast, human cytomegalovirus
enhances the expression of a disintegrin and metalloproteinase
domain 9, promoting VSMC proliferation, migration and transition
from a contractile to a synthetic phenotype (58). Notably, COVID-19 infection also
induces a shift in the phenotype of VSMCs, worsening vascular
dysfunction (59). These
mechanisms underscore the direct involvement of pathogens and their
virulence factors in inducing injury to VSMCs.
Moreover, pathogens indirectly damage VSMCs through
the release of virulence factors, such as endotoxins and exotoxins.
These molecules, which are essential for pathogen-induced tissue
injury or immune response evasion, include LPS, exotoxins (such as
cytotoxins, neurotoxins and enterotoxins), secretory systems and
catalases (6). For example, LPS,
a crucial component of gram-negative bacteria, serves a pivotal
role in the pathogenesis of sepsis. Research has demonstrated that
LPS enhances the expression of regulator of G-protein signaling 1,
activates the JNK-p38 signaling pathway, and induces a shift in
VSMCs from a contractile to a synthetic phenotype, thereby
promoting proliferation, and contributing to the development and
rupture of infectious intracranial aneurysms (60). In vitro studies have
revealed that lipoteichoic acid from Staphylococcus aureus
or Streptococcal Streptolysin O stimulates the expression of
iNOS in rat VSMCs, leading to excessive NO production and
subsequent vascular dysfunction, mechanisms closely associated with
vasoplegia in gram-positive septic shock (61,62). Additionally, staphylococcal
α-toxin directly causes coronary vasoconstriction and impairs
myocardial contractility, a process mediated by the production of
thromboxane A2 (63). In
conclusion, sepsis-induced VSMC injury involves a variety of
mechanisms, including direct invasion by pathogens, the release of
virulence factors and the activation of inflammatory cascades.
These insults disrupt the contractility of VSMCs and promote
phenotypic transition, thereby contributing to the development of
vasoplegia. Table I presents a
summary of the pathogenic effects and molecular mediators of common
pathogens on VSMCs (57-67).
 | Table IEffects of common pathogens on VSMCs
and their mechanisms of action. |
Table I
Effects of common pathogens on VSMCs
and their mechanisms of action.
A, Virulence
factors
|
|---|
| First author,
year | Pathogenic
mechanisms | Model | Effector
molecule | Molecular effects
on cells | (Refs.) |
|---|
| Hu, 2024 | Gram-negative
bacteria LPS | HASMCs | RGS-1
upregulation | Phenotypic
switching | (60) |
| Hattori, 1998 | Staphylococcus
aureus LTA | Rat VSMCs | iNOS
upregulation | Vasodilation | (61) |
| Sibelius, 2000 |
Staphylococcus α-toxin | Rat CA | Thromboxane
upregulation |
Vasoconstriction | (63) |
| Seki, 2025 |
Streptococcus SLO | Rat ASMCs | iNOS
upregulation | Vasodilation | (62) |
| Kook, 1999 | Vibrio
vulnificus hemolysin | Rat ASMCs | cGMP
upregulation | Vasodilation | (64) |
| DelVechio,
2023 | Candida
CAWS | Rat ASMCs | COX2
upregulation | Vascular
injury | (65) |
|
| B, Direct
injury |
|
| Zhao, 2022 |
Chlamydia | Rat VSMCs | JunB-Fra-1
upregulation | Migration | (57) |
| Zhang, 2013 | Porphyromonas
gingivalis | HASMCs | Notch and TGF-β
upregulation | Promotes
proliferation | (66) |
| He, 2023 | HCMV | Human VSMCs | ADAM9
upregulation | Phenotypic
switching | (58) |
| Oseghale, 2022 | Influenza A
virus | Pregnant mice
ASMCs | CD69
upregulation | Vasodilation | (67) |
| Richards, 2024 | COVID-19 | hPSC-derived
SMCs | IFN-α/IFN-γ
upregulation | Phenotypic
switching | (59) |
VSMC injury mediated by cytokines in
sepsis
PAMPs released by invading microorganisms are
recognized by the host immune system, initiating innate immunity
and the subsequent cytokine storm, a characteristic feature of
sepsis (6,68). Cytokine release syndrome involves
the extensive secretion of ILs, interferons (IFNs), TNF and
colony-stimulating factors, driving systemic inflammation (69,70). These cytokines serve crucial
roles not only in regulating immunity and tissue repair, but also
in influencing the physiology of VSMCs and the progression of
diseases. Numerous studies have shown that inflammatory factors
modulate the phenotype and function of VSMCs through distinct
mechanisms. For example, the constitutive expression of the IL-1α
precursor stimulates proliferation in human saphenous vein VSMCs
(71), whereas IL-1β enhances
the migration and invasion of human aortic SMCs through the p38
MAPK/angiopoietin-2 signaling pathway (72). TNF-α rapidly alters the
expression of contractile and synthetic markers in vitro,
promoting VSMC proliferation and migration at low concentrations
(73). In sepsis, immune cells,
including macrophages, neutrophils, T cells and natural killer
cells, secrete substantial amounts of pro-inflammatory cytokines,
with IFN-γ serving as a central mediator in regulating VSMC
function (69). A previous study
in a non-septic setting has demonstrated that IFN-γ secreted by
decidual natural killer cells induces long non-coding RNA MEG3,
thereby modulating VSMC migration and apoptosis, suggesting a
conserved role for IFN-γ-regulated pathways in VSMC function
(74). Furthermore, various
other cytokines also participate in this regulatory network.
Table II provides a summary of
representative inflammatory factors, their regulatory effects on
VSMCs and the underlying signaling pathways (71-101). These mechanisms collectively
contribute to sepsis-induced vasoplegia.
| Table IIPathogen-induced direct injury and
virulence factors on VSMCs. |
Table II
Pathogen-induced direct injury and
virulence factors on VSMCs.
A, ILs
|
|---|
| First author,
year | Cytokine | Model | Key
molecular/signaling pathway | Molecular effects
on cells | (Refs.) |
|---|
| Beasley, 1999 | IL-1α | HSVSMCs | Not given | Promotes
proliferation | (71) |
| Xu, 2022 | IL-1β | HASMCs | p38
MAPK/Angpt-2 | Migration and
invasion | (72) |
| Arumugam, 2019 | IL-2 | Human iliac
artery | PI3K/Akt/mTOR | Migration and
proliferation | (75) |
| Brizzi, 2001 | IL-3 | Human umbilical
cord | ERK1/2 | Migration and
proliferation | (76) |
| Hofbauer, 2006 | IL-4 | HCASMCs |
Osteoprotegerin | Calcification | (77) |
| Cimmino, 2021 | IL-6 | Rat ASMCs | Bcl-xL and p53 | Reduces
apoptosis | (78) |
| Yue, 1994 | IL-8 | Rat ASMCs | MAPK | Promotes
proliferation | (79) |
| Mazighi, 2004 | IL-10 | Rat ASMCs | NF-κB/IκB | Inhibits
activation | (80) |
| Zimmerman,
2002 | IL-11 | Human VSMCs | NF-κB | Inhibits
proliferation | (81) |
| Cho, 2013 | IL-13 | HPASMCs | IL-13Rα2-Arg2 | Phenotypic
switching | (82) |
| Iwasaki, 2007 | IL-15 | Rat ductus
arteriosus | PDGF | Inhibits
proliferation | (83) |
| Park, 2015 | IL-16 | Rat ASMCs | p38
MAPK/Sp-1/MMP-9 | Enhanced migration
and invasion | (84) |
| Duncan, 2020 | IL-17 | Rat ASMCs | MAPK/βENaC | Inhibits
proliferation | (85) |
| Zhang, 2017 | IL-18 | HASMCs | TRPM7 channel | Calcification | (86) |
| Cuneo, 2010 | IL-19 | Human VSMCs | HuR | Reduces
activation | (87) |
| Dale, 2019 | IL-21 | Mice ASMCs | Angiotensin II | Inhibits
proliferation | (88) |
| Rattik, 2015 | IL-22 | Mice ASMCs | α-actin and
caldesmon | Phenotypic
switching | (89) |
| Conway, 2018 | IL-23 | Human temporal
artery | Not given | Promotes
proliferation | (90) |
| Lee, 2012 | IL-24 | Rat ASMCs | Wnt/β-catenin | Inhibits
calcification | (91) |
| Hao, 2023 | IL-29 | Rat ASMCs |
JAK2/STAT3/BMP2 | Calcification | (92) |
| Son, 2017 | IL-32 | Mice ASMCs | MicroRNA-205 | Reduces
activation | (93) |
| DeVallance,
2014 | IL-33 | Rat ASMCs | ERK1/2 | Increases vascular
tone | (94) |
| Skowron, 2015 | IL-35 | HASMCs | ICAM-1 | Immune
homeostasis | (95) |
| Ding, 2022 | IL-37 | HASMCs | RIPK3 | Phenotypic
switching | (96) |
|
| B, IFNs |
|
| Niessner, 2007 | IFN-α | Human carotid
artery | STAT | Enhances
apoptosis | (97) |
| Sano, 2015 | IFN-β | HCASMCs | Caspase-3 | Enhances
apoptosis | (98) |
| Liu, 2017 | IFN-γ | VSMCs | Long non-coding RNA
MEG3 | Enhances both
migration and apoptosis | (74) |
|
| C, TNF
superfamily |
|
| Sun, 2024 | TNF-α | Calf ASMCs | Not given | Phenotypic
switching | (73) |
|
| D, CSFs |
|
| Vasudevan,
2003 | M-CSF | VSMCs | ICAM-1 | Enhances
apoptosis | (99) |
| Plenz, 1999 | GM-CSF | HASMCs | Collagen VIII | Modulates
collagen | (100) |
| Rinaldi, 2016 | G-CSF | Rat CCA | Foxo3a mRNA | Promotes
differentiation | (101) |
VSMC injury-related signaling pathways in
sepsis
The immunopathogenesis of sepsis is characterized by
dysregulated immune responses, involving both hyperinflammation and
immunosuppression (102). In
sepsis, PAMPs and damage-associated molecular patterns (DAMPs)
activate the host immune system, leading to cytokine storms and the
aberrant activation of signaling pathways; events that compromise
the function of VSMCs. These dysregulated signaling cascades are
closely associated with VSMC inflammation, phenotypic switching and
functional impairment. In addition to classical inflammatory
pathways, emerging mechanisms, including NLRP3 inflammasome
activation, mtROS release and the dysregulation of mechanosensitive
ion channels, have recently been identified as critical regulators
of VSMCs pathobiology in sepsis.
Notch signaling: A regulator of VSMC
phenotypic stability
The Notch signaling pathway is a highly conserved
intercellular communication system that includes receptors, ligands
and effector molecules. In mammals, this pathway comprises four
Notch receptors (Notch1-4) and five ligands (Jagged1, Jagged2,
Delta1, Delta3 and Delta4) (103). Under normal physiological
conditions, Notch signaling serves a role in regulating VSMC
differentiation and phenotype maintenance (24). However, under septic conditions,
such as in a mouse model of CLP, the activity of the Notch pathway,
particularly that of Notch3, is markedly reduced. This reduction is
associated with the downregulation of the expression of Notch3 and
its ligands Jagged1 and Delta4, resulting in impaired VSMC
contractility (104). Moreover,
in a mouse model of LPS-induced sepsis, Notch3 expression has been
reported to be notably decreased in lung tissue, highlighting the
essential role of Notch signaling in modulating VSMC function
during sepsis (105).
AMPK/FOXO axis: Metabolic stress sensors
in VSMC senescence
The AMPK/FOXO pathway serves a critical role in
cellular energy balance and the response to oxidative stress. AMPK,
a key energy sensor, is activated under metabolic stress
conditions, whereas FOXO transcription factors act as important
downstream effectors of stress signaling. Upon activation, AMPK
phosphorylates FOXO proteins, augmenting their transcriptional
activity and consequently upregulating the expression of
antioxidant and cytoprotective genes, such as SOD1 and SOD2,
which facilitate peroxide degradation and mitigate the accumulation
of ROS (106). In sepsis,
tissue damage results in the release of extracellular histones,
which serve as DAMPs. These external histones induce an
inflammatory response and senescence in VSMCs in a dose-dependent
manner through activation of AMPK/FOXO4 signaling (107). This signaling cascade connects
the sensing of metabolic stress to the regulation of VSMC
dysfunction during septic conditions, highlighting its role in
integrating energy metabolism with inflammatory signaling
networks.
NF-κB: The central inflammatory hub in
VSMCs
The NF-κB pathway serves as a central
transcriptional regulatory hub that controls cell proliferation,
apoptosis, inflammatory responses and immune homeostasis (108). In sepsis, stimuli from
pathogens and proinflammatory cytokines activate NF-κB, promoting
its translocation to the nucleus and subsequently inducing
downstream inflammatory genes, including HIF-1α (6). Preclinical studies have shown that
the pharmacological inhibition of NF-κB signaling effectively
reduces VSMC proliferation and activation, leading to the
mitigation of pathological vascular remodeling (80,81). These findings underscore the
crucial role of NF-κB in linking septic inflammation to VSMC
dysfunction, positioning it as a potential therapeutic target for
vascular complications associated with sepsis.
p38 MAPK: Mediating inflammatory VSMC
migration
p38 MAPK is a crucial member of the MAPK family,
which regulates cellular responses to environmental stressors and
inflammatory stimuli (109).
Consisting of four isoforms (α, β, γ and δ), p38 MAPK acts as a
central transducer of signals from cell surface receptors to
nuclear effectors, and is activated by various stressors and
proinflammatory cytokines (110,111). In the context of sepsis,
inflammatory factors such as IL-1β and IL-16 stimulate p38 MAPK
activation, facilitating VSMC migration and invasion, processes
that are fundamental to pathological vascular remodeling and
dysfunction (72,84). This pathway links inflammatory
signaling to alterations in VSMCs behavior, emphasizing its role in
mediating sepsis-induced vascular pathology.
PI3K/Akt/mTOR: Regulating VSMC
proliferation in sepsis
The PI3K/Akt signaling pathway serves as a central
regulator of cell survival, proliferation and metabolic homeostasis
(112). In sepsis, this pathway
modulates inflammatory responses and vascular cell behavior.
Research has indicated that the IL-2/IL-2 receptor system promotes
VSMC proliferation and migration via the PI3K/Akt/mTOR axis,
exacerbating vascular injury (75). Table III summarizes the key signaling
pathways involved in VSMC injury during sepsis (24,72,75,80,81, 84,104-112).
| Table IIISummary of the principal signaling
pathways implicated in VSMC injury in sepsis. |
Table III
Summary of the principal signaling
pathways implicated in VSMC injury in sepsis.
| Pathway | Primary function in
VSMCs | Key dysregulation
in sepsis | Functional outcome
in vasoplegia |
|---|
| Notch3 | Maintains
contractile phenotype (24). | Downregulation of
Notch3/Jagged 1/Delta 4 (104,105). | Reduced
vasoconstriction, phenotypic switching (104,105). |
| AMPK/FOXO4 | Energy sensing,
stress response (106). | Activation by
extracellular histones (107). | Senescence, SASP
secretion (107). |
| NF-κB | Inflammatory gene
transcription (108). | IKK-mediated
nuclear translocation (80,81). | Cytokine
production, vascular leak (80,81). |
| p38 MAPK | Inflammatory
signaling (109-111). | Activation by
IL-1β/IL-16 (72,84). | Enhanced migration,
amplified NF-κB activity (72,84). |
| PI3K/Akt/mTOR | Proliferation,
survival (112). | Hyperactivation via
IL-2/IL-2R (75). | Neointimal
thickening, paracrine NO elevation (75). |
Emerging mechanisms of VSMC regulation in
sepsis-induced vasoplegia
Sepsis-induced vasoplegia is closely associated with
dysregulated VSMC function, with emerging evidence highlighting
three nonclassical regulatory mechanisms: NLRP3 inflammasome
activation, mtROS overproduction and mechanosensitive ion channel
dysfunction, which complement classical inflammatory pathways.
The NLRP3 inflammasome, which comprises the sensor
NLRP3, adaptor ASC and effector pro-caspase-1, is activated through
a two-stage pathway in sepsis: NF-κB-driven upregulation of
NLRP3/pro-IL-1β (initiation) and assembly triggered by PAMPs or
DAMPs (activation) (113,114). Direct evidence for its role in
septic VSMC dysfunction remains scarce, with most data derived from
indirect observations. For example, histones released as DAMPs by
severely damaged cells in sepsis promote ASC-NLRP3 interactions in
VSMCs, mediating VSMC inflammation and senescence (107), processes implicated in impaired
VSMC contractility. Although characterized in the context of
chronic kidney disease-related vascular calcification,
Prevotella copri-derived LPS (a PAMP) has been shown to
induce VSMC osteogenic differentiation in vitro via
activation of the TLR4-NF-κB-NLRP3 inflammasome axis (115); notably, NF-κB blockade can
abolish both inflammasome activation and the resulting VSMC
phenotypic shift in a rat model of chronic kidney disease.
Extrapolating from this mechanistic framework, these findings
suggest that PAMP-mediated VSMC phenotypic perturbation in sepsis
may also occur via inflammasome signaling pathways. Notably, NLRP3
is transcriptionally regulated by Runx2, which coordinates vascular
matrix stiffness and VSMC inflammatory phenotypes (116). Although direct evidence in
sepsis is lacking, matrix stiffness-induced VSMC dysfunction is a
recognized contributor to decreased vascular compliance, a feature
that overlaps with sepsis-induced vasoplegia (25,116). Given the critical role of NLRP3
inflammasome signaling in IL-1β production in VSMCs (117), crosstalk with the classical
NF-κB pathway may be a possibility; for example, IL-1β released by
activated VSMCs may further activate NF-κB, resulting in the
formation of a feed-forward loop that amplifies VSMC inflammation
and functional impairment.
Oxidative stress serves a pivotal role in the
pathogenesis of sepsis. When the body is subjected to severe
external insults, such as burns, shock or serious infections,
cellular structural alterations lead to mitochondrial injury and a
sudden surge in ROS and reactive nitrogen species (RNS) levels. An
imbalance between oxidant and antioxidant systems allows oxidative
stress products, including ROS and RNS, to inflict mitochondrial
damage and compromise vital cellular components, such as lipids,
proteins and nucleic acids (118). Sepsis-induced inflammatory
stress triggers the production of excessive amounts of mtROS
(119), a critical upstream
regulator of VSMC dysfunction and NLRP3 inflammasome activation
(120). Mechanistically, mtROS
upregulate NLRP3 at the translational level rather than directly
activating the assembled inflammasome (121), although the specific molecular
events involved in sepsis remain unclear. Beyond NLRP3 modulation,
mtROS enhance MAPK activity and promote DNA synthesis, stimulating
VSMC proliferation (122,123). This may perturb vascular wall
integrity and exacerbate sepsis-induced decreases in vascular
compliance, but direct evidence linking mtROS-driven proliferation
to vasoplegia is lacking.
Mechanosensitive ion channels, which transduce
mechanical stimuli into electrochemical signals, are potential
regulators of septic VSMC dysfunction. Hemodynamic instability and
inflammatory mediators induced by sepsis have been implicated in
disrupting the function of key mechanosensitive channels in VSMCs,
such as Piezo1, transient receptor potential vanilloid (TRPV)
channels and two-pore domain potassium channels (124-126). Existing data primarily come
from non-VSMC tissues or systemic models: Piezo1 has been shown to
be upregulated in the intestinal tissues of CLP-induced septic mice
(125), and its established
role in VSMC Ca2+ influx suggests potential perturbation
of Ca2+ homeostasis (127). In addition, TRPV1 has been
reported to be upregulated in a rat model of endotoxemia (126), which may promote
Ca2+-dependent NO production and vasodilation in VSMCs.
CLP-induced mice exhibit downregulation of TASK-1, TASK-2 and
TREK-1 (124), with TREK 1
downregulation potentially altering VSMC membrane potential and
inhibiting voltage-gated Ca2+ channel activation.
Targeting VSMC dysfunction in sepsis-induced
vasoplegia
As aforementioned, exposure to stimuli such as
metabolic disturbances, shear stress, cellular crosstalk and
disruption of calcium homeostasis in sepsis triggers the transition
of VSMCs from a differentiated contractile phenotype to a synthetic
dedifferentiated phenotype (128). The core scientific controversy
surrounding this process lies in whether VSMC dedifferentiation is
irreversible. On one side, cellular dedifferentiation is
traditionally regarded as irreversible due to stable epigenetic
reprogramming and persistent alterations in gene expression
profiles that lock VSMCs in a synthetic state. This perspective
implicitly underpins early assumptions about septic VSMC
dysfunction, where long-term phenotypic shifts were thought to
preclude functional recovery without targeted intervention
(25). On the other side,
accumulating evidence challenges this irreversibility and supports
the potential for redifferentiation. For example, VSMCs subjected
to serum deprivation (a model of reduced pro-dedifferentiation
stimuli) fully regain a spindle-like morphology, increased
contractile filament density and restored expression of
VSMC-specific contractile proteins (such as α-SMA, calponin),
demonstrating functional redifferentiation (129). Preclinical studies have further
identified agents that reverse septic VSMC dedifferentiation:
Dehydrocorydaline sustains the contractile phenotype via Spta1
upregulation (130), and
atorvastatin fully reverses morphological and functional
abnormalities (including proliferation, medial layer rearrangement
and impaired vasoreactivity) in a rat model of LPS-induced carotid
artery inflammation (131),
directly supporting the reversibility of VSMC dysfunction in
inflammatory contexts relevant to sepsis. Notably, clinical
observations of sepsis survivors have also documented partial
recovery of vascular reactivity over time, indirectly suggesting
that VSMC dedifferentiation may not be permanent (132). The current review focuses on
pharmacological strategies targeting VSMC dysfunction in
sepsis-induced vasoplegia (conventional drugs, calcium homeostasis
modulators and NO-reducing therapies) precisely because of this
unresolved controversy: If dedifferentiation were strictly
irreversible, therapeutic efforts would focus solely on symptom
relief, whereas evidence for potential reversibility justifies
exploring mechanism-based interventions to restore intrinsic VSMC
contractile function.
Conventional clinical drugs modulating
VSMC function
Sepsis-induced vasoplegia is characterized by VSMC
hypocontractility, impaired vasomotor tone and resistance to
catecholamines, prompting clinical investigations into conventional
agents targeting VSMC function. The current review presents a
comprehensive analysis of four principal drug classes, vasopressin
analogues, phosphodiesterase (PDE) inhibitors, antioxidants and
calcium sensitizers, integrating preclinical mechanisms, clinical
trial data and key translational challenges, with an emphasis on a
critical and balanced appraisal of their therapeutic potential and
limitations. Table IV
summarizes the preclinical and clinical evidence, underlying
mechanisms and translational challenges of conventional
VSMC-targeted therapies for sepsis-induced vasoplegia (133-156).
| Table IVSubclass-specific comparison of
preclinical and clinical evidence, mechanisms and translational
barriers for VSMC-targeted conventional drugs in sepsis-induced
vasoplegia. |
Table IV
Subclass-specific comparison of
preclinical and clinical evidence, mechanisms and translational
barriers for VSMC-targeted conventional drugs in sepsis-induced
vasoplegia.
| Drug class | Representative
agents | Mechanisms of
action | Preclinical
evidencea | Clinical
evidenceb | Core
limitationsc |
|---|
| Vasopressin and its
analogues | Arginine
vasopressin, terlipressin, selepressin | V1a receptor
activation: Triggers PLC/IP3-mediated Ca2+mobilization
and MLC phosphorylation to restore VSMC contractility. Inhibits
NF-κB nuclear translocation (133-134). | LPS/CLP models:
Restored aortic vasoreactivity and reduced plasma NO metabolites
(135). | VASST trial: No
28-day mortality reduction; post hoc analysis showed reduced AKI
progression (136,137). TERLIVAP trial: Reduced
catecholamine requirements and rebound hypotension (138). SEPSIS-ACT trial: Selepressin
failed to reduce ventilator/vasopressor-free days (139). | Ischemic off-target
effects (digital gangrene, mesenteric ischemia). |
| PDE inhibitors | PDE3: Milrinone,
enoximone. PDE4: Roflumilast; PDE5: Tadalafil, sildenafil | Inhibits cAMP/cGMP
hydrolysis: Modulates VSMC Ca2+ sensitivity, suppresses
inflammation and stabilizes microvascular barrier (140). | Roflumilast:
Improved renal perfusion but worsened MAP/cardiac output in CLP
rats (141,142). Tadalafil: Enhanced renal blood
flow but not survival in CLP models (143). Milrinone: Restored mesenteric
villus perfusion but exacerbated systemic hypo-tension in
endotoxemia (144,145). | Scarce and
inconclusive. | Context-dependent
efficacy (beneficial in mild microvascular dysfunction, harmful in
severe vasoplegia). |
| Antioxidants | Vitamin C, vitamin
E, MitoQ | Vitamin C:
Scavenges cytosolic ROS (146).
Vitamin E: Preserves thiol-disulfide homeostasis to protect VSMC
contractile proteins (147).
MitoQ: TPP+-mediated mitochondrial targeting; scavenges
mtROS + activates Keap1/Nrf2 pathway (148). | Vitamin C:
Mitigated cardiomyopathy and restored VSMC contractility in
LPS-induced rats (146).
Vitamin E: Reduced oxidative/inflammatory injury and preserved
vasoreactivity in LPS-induced mice (147). MitoQ: Inhibited VSMC phenotypic
switching and apoptosis (149). | Vitamin C:
Meta-analysis (12 RCTs) indicated reduced vasopressor duration/SOFA
scores (150); 1 RCT reported
higher 28-day mortality (151).
Vitamin E: Reduced 28-day mortality in a retrospective cohort study
(152). MitoQ: No clinical data
in sepsis-induced vasoplegia. | Poor
bioavailability/tissue penetration in sepsis. Conflicting
clinicalevidence. Non-selective antioxidant effects mayblunt
anti-infective immunity. |
| Calcium
sensitizers | Levosimendan | Calcium
sensitization and mitochondrial protection (153,154). | Relaxed thoracic
aorta via PKC inhibition and Kir channel activation (153). | Meta-analysis:
Improved hemodynamics and lactate clearance, and reduced
in-hospital mortality in sepsis-induced cardiomyopathy (155). Prospective RCT: Increased
sublingual micro-circulatory flow index (156). Dose-dependent hypotension
reported in patients with septic shock (153). | Vasodilatory
effects exacerbate hypotension in severe vasoplegia. |
Vasopressin and its analogues
As the most extensively studied agents for
sepsis-induced vasoplegia, vasopressin analogues [including
arginine vasopressin (AVP), terlipressin and selepressin]
constitute a cornerstone in the clinical management of VSMC
dysfunction. However, their therapeutic application is constrained
by a delicate equilibrium between hemodynamic efficacy, off-target
toxicity and patient heterogeneity; these are critical barriers
that have limited translational success beyond symptomatic relief.
Unlike catecholamines, which rely on intact adrenergic signaling
(often compromised in sepsis due to receptor desensitization),
vasopressin analogues directly restore vascular contractility via
V1a receptors expressed on VSMCs (133). Activation of V1a receptors
triggers downstream signaling of phospholipases A, C and D,
promoting inositol triphosphate-mediated intracellular
Ca2+ mobilization from the sarcoplasmic reticulum and
increasing MLC phosphorylation (134). Moreover, in LPS-challenged
rats, terlipressin suppresses aortic iNOS expression and activity
by inhibiting NF-κB nuclear translocation, thereby disrupting the
pathogenic calcium-NO feedback loop and ameliorating aortic
vasoplegia in response to vasoconstrictors (135). This dual mechanism provides
vasopressin analogues with a distinct advantage in targeting the
fundamental pathophysiology of sepsis-induced vasoplegia rather
than merely alleviating hemodynamic instability.
Clinically, evidence for vasopressin analogues
remains mixed, reflecting the complexity of balancing efficacy and
safety. A multicenter, randomized, double-blind trial (n=778)
comparing low-dose AVP with norepinephrine in
catecholamine-dependent septic shock showed no significant
reduction in 28-day mortality (35.4% vs. 39.3%, P=0.26) (136), indicating that restoring
vascular tone alone is insufficient to reverse sepsis-related
multiorgan dysfunction. Nonetheless, post hoc analysis of the VASST
trial revealed a promising renal protective effect of AVP: In
high-risk patients (a ≥1.5-fold increase in serum creatinine from
baseline or >25% glomerular filtration rate (GFR) reduction
within 7 days, sustained for >24 h), AVP was associated with a
significantly lower incidence of AKI progression to 'Failure'
[serum creatinine >3-fold baseline; ≥4.0 mg/dl (353.6
μmol/l) with an acute ≥0.5 mg/dl rise; or >75% GFR
reduction] or 'Loss' (permanent renal failure requiring dialysis
for >4 weeks) than norepinephrine (20.8% vs. 39.6%, P=0.03)
(137). This benefit may stem
from reduced renal vasodilation and preserved glomerular filtration
pressure (137). The phase II
TERLIVAP trial (n=45) demonstrated that, compared with continuous
infusion of 0.03 U/h vasopressin or 15 μg/min
norepinephrine, administration of 1.3 μg/kg/h terlipressin,
a longer-acting prodrug of vasopressin, significantly reduced
catecholamine requirements needed to achieve hemodynamic stability
within 48 h (0.8±1.3 vs. 1.2±1.4 vs. 0.2±0.4 μg/kg/min; each
P≤0.05) (138). Moreover,
terlipressin treatment was associated with a lower incidence of
rebound hypotension compared with the two comparator groups
(P<0.05) (138).
Selepressin, a selective V1a agonist designed to mitigate
off-target toxicity, failed to meet the primary endpoint of
ventilator- and vasopressor-free day reduction in the SEPSIS-ACT
trial (n=868) (139).
PDE inhibitors
PDE inhibitors modulate VSMC function by inhibiting
the hydrolysis of cyclic nucleotides (cyclin adenosine
monophosphate/cGMP), representing a mechanistically distinct
approach to targeting sepsis-induced vasoplegia (140). However, their translational
potential is constrained by context-dependent efficacy, off-target
effects and a paucity of sepsis-specific clinical data, reflecting
the gap between preclinical mechanistic insights and clinical
reality. Mammalian PDEs are classified into 11 families, with PDE3,
PDE4 and PDE5 being the most relevant to septic VSMC dysfunction
because of their tissue distribution and cyclic nucleotide
selectivity (157). Commonly
used PDE inhibitors in clinical practice are classified based on
their target PDE isoforms, as follows: PDE3 inhibitors, which
target PDE3 (predominantly expressed in the heart and circulatory
system), with milrinone as a prototypical agent; PDE4 inhibitors,
which target PDE4 (primarily localized in the respiratory system,
particularly the bronchial tissues), such as roflumilast; and PDE5
inhibitors, which target PDE5 (mainly expressed in the lungs and
penile tissues), with sildenafil as a representative drug.
Preclinical evidence for the role of PDE inhibitors
in sepsis is conflicting, highlighting their model-dependent
efficacy. In CLP-induced septic rats, the PDE4 inhibitor
roflumilast reduced VSMC-derived inflammatory cytokine release
(TNF-α and IL-6) and apoptosis, while stabilizing the microvascular
barrier and improving renal perfusion (141,142). However, roflumilast worsened
hemodynamic parameters [mean arterial pressure (MAP) and cardiac
output] and failed to improve survival, likely because of its
nonselective vasodilatory effects in the context of systemic
inflammation (142). In the
colon ascendens stent peritonitis model (a more clinically relevant
polymicrobial sepsis model), PDE4 inhibition has been shown to
stabilize the microvascular barrier and improve microcirculatory
flow, supporting its potential in targeting sepsis-induced
microvascular dysfunction (158). Similarly, the PDE5 inhibitor
tadalafil improved basal renal blood flow in CLP-induced rats by
increasing VSMC calcium sensitivity, but did not increase survival,
suggesting that the benefits of isolated organ perfusion do not
translate to systemic hemodynamic stability (143). Preclinical data on PDE3
inhibitors (for example, milrinone) present inherent
contradictions, reflecting their context-dependent pleiotropic
effects: Although milrinone restored mesenteric intestinal villus
perfusion in rat models of endotoxemia, it concurrently aggravated
systemic hypotension. Notably, despite this systemic hemodynamic
deterioration, milrinone ameliorated intestinal mucosal
hypoperfusion, underscoring a dissociation between systemic
vascular tone and regional microcirculatory function (144,145).
Clinical evidence for the use of PDE inhibitors in
sepsis-induced vasoplegia remains limited and inconclusive. A
multicenter cohort study of 229 patients with septic shock revealed
that compared with standard care, PDE3 inhibitors (milrinone and
enoximone) did not improve lactate clearance, organ failure, length
of Intensive Care Unit (ICU) or hospital stay, or mortality
(159). Notably, the effects of
PDE3 inhibitors on cardiogenic shock have also been evaluated, with
a systematic review showing no differences in outcomes (early
death, cardiac arrest, renal replacement therapy initiation) when
PDE3 inhibitors were combined with other inotropes, findings that
may be extrapolated to sepsis-related cardiomyopathy but not
specifically to VSMC-mediated vasoplegia (160). To date, no large-scale
randomized controlled trials (RCTs) have evaluated PDE4 or PDE5
inhibitors for sepsis-induced vasoplegia.
Antioxidants
Antioxidants exist in various forms, each
characterized by distinct mechanisms of action and clinical
indications. They can broadly be categorized into those already in
clinical use and others still under experimental investigation. As
a water-soluble antioxidant, ascorbic acid (vitamin C) participates
in numerous enzymatic and nonenzymatic reactions, and serves as a
cofactor in multiple biological processes (161). The therapeutic potential of
vitamin C in sepsis and critical illness has been studied for
several decades; however, its clinical utility remains unclear.
In vitro studies have demonstrated that vitamin C, when
administered orally or added directly to cell cultures, promotes EC
growth while inhibiting SMC proliferation (162,163). Preclinically, in LPS-induced
septic rats, vitamin C has been shown to mitigate endotoxin-induced
cardiomyopathy by inhibiting oxidative stress-related cytokine
expression, thereby protecting myocardial tissue from damage
(146). Clinical evidence
remains contradictory: A meta-analysis of 12 RCTs revealed that
intravenous vitamin C supplementation markedly reduced the duration
of vasopressor therapy and improved Sequential Organ Failure
Assessment scores in patients with septic shock, findings
indirectly attributed to restored VSMC contractility, reducing
catecholamine dependence (150). However, another randomized
placebo-controlled trial reported that compared with placebo
recipients, adults with sepsis receiving vasopressor therapy who
were treated with intravenous vitamin C exhibited a greater risk of
death at 28 days (risk ratio 1.17; 95% CI 0.98-1.40) (151). This contradiction is partly
explained by sepsis-specific pharmacokinetic alterations:
Critically ill patients have an increased volume of distribution
and reduced renal clearance of vitamin C, leading to subtherapeutic
concentrations in VSMCs despite high plasma levels.
Natural vitamin E (α-tocopherol), a lipid-soluble
antioxidant, is selectively depleted in septic VSMCs because of
increased oxidative consumption, with deficiency independently
associated with severe septic shock (adjusted OR 6.75; 95% CI
2.45-18.60; P<0.001) (164).
In an LPS-induced sepsis mouse model, vitamin E notably protected
against sepsis-induced oxidative and inflammatory damage by
preserving thiol-disulfide homeostasis and attenuating cytokine
production (147). However,
clinical evidence remains limited to retrospective analyses: a
cohort study of 523 patients with sepsis in the ICU revealed that
vitamin E supplementation was associated with reduced 28-day
mortality (HR 0.75; 95% CI 0.59-0.95; P=0.019) (152). Given the current scarcity of
robust clinical evidence, which is limited primarily to the results
of retrospective cohort studies indicating possible benefits in
vitamin E-deficient subpopulations, and persistent translational
challenges, the therapeutic efficacy of vitamin E in patients with
sepsis-induced vasoplegia requires substantiation through
rigorously designed, prospective, multicenter RCT.
Mitoquinone mesylate (MitoQ) is a
mitochondrion-targeted antioxidant characterized by a triphenyl
phosphonium cation conjugated to ubiquinone, this structural design
enables selective accumulation in mitochondria via membrane
potential-dependent uptake, allowing it to specifically scavenge
mtROS and protect cells from oxidative stress-induced mitochondrial
dysfunction (148). MitoQ, a
key regulator of VSMC homeostasis, has been validated in multiple
preclinical models to mitigate vascular pathologies related to
sepsis-induced VSMC dysfunction. In human aortic VSMCs, MitoQ has
been reported to attenuate PM2.5-induced vascular fibrosis by
modulating mitochondrial dynamics; specifically, it can suppress
the transition of VSMCs from a contractile to a synthetic
phenotype, and alleviate mitochondrial fragmentation and mitophagy
(149). This
phenotype-stabilizing effect is highly relevant to sepsis, in which
VSMC phenotypic dedifferentiation is a key driver of vasoplegia. In
an adenine-induced rat model of aortic calcification, MitoQ
inhibited VSMC oxidative stress and apoptosis via activation of the
Keap1/Nrf2 signaling pathway, downregulating the activity of
oxidative factors and upregulating the activity of antioxidant
enzymes, thereby attenuating vascular calcification and preserving
VSMC contractile potential (165). Notably, in endotoxin-induced
cardiac dysfunction models, Supinski et al (166) demonstrated that MitoQ may
protect against cardiac mitochondrial damage by inhibiting mtROS
overproduction, and suppressing the activation of caspase-9 and
caspase-3. These preclinical findings underscore the multifaceted
role of MitoQ in regulating VSMCs phenotype, mitochondrial
function, and survival, supporting its potential as a targeted
therapy for sepsis-induced vasoplegia.
Calcium sensitizers (levosimendan)
Calcium sensitizers are a novel class of potential
therapeutic agents for sepsis-induced vasoplegia that increase VSMC
contractility without increasing intracellular calcium levels,
thereby avoiding the risk of calcium overload and arrhythmia linked
to catecholamines and vasopressin analogues (153-156). Levosimendan, a prototypical
agent initially designed as an inotrope, has shown promise for
treating septic vasoplegia because of its combined effects on
cardiac cells and VSMCs, although its clinical use is limited by
context-dependent vasodilation (156).
At the molecular level, levosimendan exerts
VSMC-specific effects via two core mechanisms: i) Calcium
sensitization: It binds to the N-terminal domain of troponin C
(TnC) in VSMCs with high affinity, increasing the sensitivity of
TnC to [Ca2+]i and promoting actin-myosin cross-bridge
formation, even at physiological [Ca2+]i levels, thereby
restoring contractile function compromised by NO-mediated calcium
desensitization in sepsis (164). ii) Mitochondrial protection: It
activates ATP-sensitive potassium (K-ATP) channels in VSMC
mitochondria, reducing mtROS production and inhibiting
caspase-9/-3-dependent apoptosis, thus preserving VSMC viability
and the contractile phenotype in the context of septic oxidative
stress (165). Unlike
catecholamines, which rely on intact adrenergic signaling (often
desensitized in sepsis), the direct modulation of contractile
proteins by levosimendan is effective in catecholamine-resistant
states.
Clinically, the efficacy of levosimendan in
sepsis-related cardiovascular dysfunction has been supported by
accumulating evidence, although data specific to sepsis-induced
vasoplegia remain nuanced, reflecting a dual effect with
context-dependent benefits. A systematic review and meta-analysis
of 12 RCTs comparing levosimendan with dobutamine in patients with
sepsis-induced cardiomyopathy revealed that levosimendan markedly
improved hemodynamic parameters, tissue perfusion and biomarkers of
myocardial injury, while reducing in-hospital mortality and ICU
length of stay (155). With
respect to the treatment of septic shock-related vasoplegia, a
prospective, double-blind RCT demonstrated that compared with a
placebo, levosimendan (0.2 μg/kg/min) improved sublingual
microcirculatory blood flow, reflected by a 32% increase in the
microcirculatory flow indices of small and medium vessels, which
suggested that enhanced regional tissue perfusion was mediated by
VSMC function restoration (156). Despite these benefits, the
vasodilatory properties of levosimendan pose a notable constraint
to its use in sepsis-induced vasoplegia. The activation of Kir
channels and inhibition of protein kinase C by the drug in systemic
VSMCs can induce dose-dependent vasodilation, leading to transient
hypotension, an adverse effect that may exacerbate hemodynamic
instability in patients with severe vasoplegia requiring high-dose
vasopressors (153). This
paradox highlights the context-dependent nature of the effects of
levosimendan; while its effects on calcium sensitization and
mitochondrial protection are beneficial for VSMC dysfunction, its
vasodilatory actions may be detrimental in the context of notable
hypotension.
Modulating calcium homeostasis
Calcium homeostasis is a pivotal regulator of VSMC
contractile function, and its disruption represents a central
mechanism underlying vascular hyporeactivity in sepsis-induced
vasoplegia. Pharmacological agents targeting calcium-handling
mechanisms have diverse modes of action, including receptor
modulation and ion channel blockade, and interact within a complex
regulatory network. Table V
summarizes the emerging therapeutic agents targeting VSMCs in
sepsis-induced vasoplegia, focusing on modulating calcium
homeostasis and reducing NO production (154,167-174).
| Table VPrincipal medications targeting VSMC
for improved vascular hyporeactivity in sepsis-induced
vasoplegia. |
Table V
Principal medications targeting VSMC
for improved vascular hyporeactivity in sepsis-induced
vasoplegia.
A, Calcium
modulators
|
|---|
| First author,
year | Agent name | Core mechanism | Experimental
model | Key efficacy
outcomes | (Refs.) |
|---|
| Lei, 2020 | Calhex-231 | Inhibited
mitochondrial fragmentation | Rat model of
traumatic hemorrhagic shock | Sustained calcium
sequestration and release in VSMC | (154) |
| Schmid, 2011 | Glibenclamide | Reduction of
calcium influx into VSMCs | In ex vivo
endotoxemia model | Prevented calcium
overload and desensitization of the contractile machinery | (167) |
| Zhou, 2010 | IB-MECA | Blocked
overactivation of ryanodine receptor-mediated calcium release | Rat model of
hemorrhagic shock | Antagonized
vascular hyporeactivity caused by aberrant calcium release | (168) |
| Li, 2008 | Ang-II | Mediated calcium
sensitivity | Rat model of
hemorrhagic shock | Improved Vascular
hyporeactivity | (169) |
|
| B, iNOS
inhibitors |
|
| Gibraeil, 2000 | 4-ABH4 | Inhibition of iNOS
activity | Pig
pulmonary/coronary vasculature model | Reduced NO-mediated
vasodilation | (170) |
| Squadrito,
2000 | CsA | Inhibition of iNOS
activity | Rat model of
splanchnic artery occlusion shock | Decreased vascular
NO levels | (171) |
| Luo, 2020 | Tubeimoside I | Inhibition of iNOS
activity | Septic mouse
model | Suppressed
NO-driven vasodilation and preserved vascular reactivity | (172) |
| Liu, 2013 |
Andrographolide | Downregulation of
iNOS expression | LPS-induced rat
endotoxemia model | Reversed
LPS-induced upregulation of PVAT iNOS | (173) |
| Altavilla,
1999 | U-74389G | Inhibition of iNOS
activity | Septic models | Reduced NO-mediated
vascular hyporeactivity | (174) |
One critical node in calcium homeostasis regulation
is the calcium-sensing receptor (CaSR), a key mediator of
extracellular calcium sensing that is pathologically overactivated
in sepsis. In a rat model of traumatic hemorrhagic shock,
Calhex-231 inhibited mitochondrial fragmentation and preserved
mitochondrial morphology (154). Mitochondria serve a dual role
in calcium buffering and energy production, and their structural
integrity is vital for maintaining calcium sequestration and
release in VSMCs. Calhex-231 has the potential to improve vascular
reactivity in sepsis-induced vasoplegia, but its systemic
administration may trigger adverse off-target reactions, primarily
because of nonselective binding to CaSR in diverse tissues and
interference with tissue-specific calcium signaling. As
demonstrated in a study on pregnant human myometrium, Calhex-231
partially inhibits oxytocin-induced uterine contractions (175).
A contrasting approach to restoring
calcium-dependent VSMC function involves the targeting of ion
channels, with K-ATP channels emerging as key therapeutic nodes.
Glibenclamide, a nonselective K-ATP channel blocker, has been shown
to restore vascular tone in animal models of septic shock by
preventing potassium ion efflux-induced VSMC hyperpolarization
(176). In an ex vivo
model of hypoxic human endotoxemia, its anti-inflammatory effect
was attributed to reduced depolarization of hypoxic monocytes,
which in turn decreased calcium influx (167). However, its nonselectivity
poses critical off-target risks: K-ATP channels are highly
expressed in pancreatic β cells and immune cells, and systemic
glibenclamide administration may cause hypoglycemia and suppress
monocyte phagocytic activity (139), both of which worsen outcomes in
patients with sepsis. Additionally, a randomized, double-blind,
placebo-controlled crossover pilot study (177) revealed a critical translational
gap: The lack of efficacy of glibenclamide in reducing the dosage
of norepinephrine compared with that of a placebo in patients with
septic shock may be attributed to individual patient variations,
such as the severity of the condition and the etiology, leading to
diverse responses to and efficacy of the drug.
Whereas glibenclamide targets calcium influx via
voltage-gated calcium channel, another agent,
chloro-N6-(3-iodobenzyl) adenosine-5'-N-methyluronamide (IB-MECA),
targets intracellular calcium release, highlighting the diversity
of calcium handling defects in sepsis (168). IB-MECA blocks the
overactivation of ryanodine receptor (RyR)-mediated Ca2+
release, a pathological feature of hypoxic VSMCs and hemorrhagic
shock-associated vascular hyporeactivity (168). Compared with control cells,
hypoxic VSMCs exhibit a marked increase in [Ca2+]i in
response to RyR activation by caffeine, which is associated with
the loss of vascular responsiveness to norepinephrine. The
stimulation of A3 adenosine receptor (A3AR, a G
protein-coupled receptor subtype regulating calcium signaling)
activity by IB-MECA directly counteracts this excessive RyR
activity, thereby normalizing intracellular calcium dynamics.
However, A3AR is also expressed by immune cells
(178), and the nonselective
activation by IB-MECA may modulate cytokine production while
impairing T-cell proliferation, a trade-off that could exacerbate
immunosuppression in late-stage sepsis.
Complementing agents that target calcium flux or
release are therapies that increase calcium sensitivity, a
mechanism that is particularly critical in advanced sepsis where
calcium-handling machinery is severely disrupted. AVP and
angiotensin II (Ang-II), which have been evaluated in rat models of
hemorrhagic shock, increase calcium sensitivity in VSMCs (169,179). Unlike agents targeting calcium
flux, they increase the affinity of myofilaments for calcium, a
mechanism particularly relevant in sepsis, where calcium
sensitivity is often impaired. These findings position them as
potentially more effective in advanced sepsis, where
calcium-handling machinery is severely disrupted. However, their
clinical use is limited by nonselective vasoconstriction in
noncritical vascular beds (such as renal and splanchnic
circulation) and potential off-target immunomodulatory effects. For
example, in early experimental hypotensive hyperdynamic sepsis, the
intravenous infusion of Ang-II has been shown to decrease renal
blood flow (180).
Reducing NO production
Excessive NO production by iNOS in VSMCs and other
vascular cells is a major driver of vasodilation and vascular
hyporeactivity in sepsis. This pathological process is mediated by
NO-cGMP-protein kinase G signaling, which reduces VSMC calcium
sensitivity and enhances intracellular calcium leakage. To
counteract this, four key pharmacological agents have been
developed, each of which target iNOS through distinct mechanisms
and exhibit varying degrees of clinical translation potential. The
present review has provided a summary for each of these agents,
with a focus on how their mechanisms align with the pathophysiology
of sepsis and where gaps persist in translating preclinical success
to patient care.
Among iNOS-targeted therapies, selectivity for VSMC
iNOS over eNOS is a critical criterion to avoid compromising
endothelial barrier function, a common pitfall of nonspecific NOS
inhibitors. Notably, 4-amino analogue of tetrahydrobiopterin
(4-ABH4) addresses this need as a potent and selective inhibitor of
SMC iNOS (170). Its mechanism
relies on competing with BH4, a cofactor essential for iNOS
catalytic activity, for binding to VSMC iNOS while sparing eNOS
(due to the markedly greater affinity of eNOS for BH4). In an in
vitro model of endotoxemia, this selectivity translated to
tangible benefits: 4-ABH4 specifically inhibited VSMC iNOS
activity, preventing excessive NO production without disrupting
eNOS-mediated endothelial protection (170). As a result, it effectively
blocked endotoxin-induced vascular hyporeactivity, making it a
promising candidate for sepsis subtypes in which EC-VSMC crosstalk
is preserved.
While 4-ABH4 directly targets iNOS catalytic
activity, another agent, cyclosporin-A (CsA), takes an upstream
approach by suppressing iNOS activation, highlighting the diversity
of strategies for inhibiting iNOS. CsA reduces iNOS activation,
decreases plasma nitrite/nitrate levels, increases blood pressure
and markedly improves survival in rats with splanchnic artery
occlusion shock (171). Despite
these preclinical benefits, its clinical translation is hindered by
two interconnected challenges: Nephrotoxicity, a condition that
poses a marked risk in patients with sepsis and pre-existing renal
impairment, and off-target immunosuppression. CsA induces distal
renal tubular acidosis by inhibiting H pumps in the distal nephron
(181). Furthermore, the
primary mechanism through which CsA inhibits calcineurin-NFAT
signaling is not restricted to VSMCs; it also suppresses T-cell
proliferation and cytokine production, exacerbating the
immunosuppressive phase of sepsis and increasing susceptibility to
opportunistic infections (182).
Beyond direct or upstream iNOS inhibition,
targeting the inflammatory signaling cascades that induce iNOS
represents another viable strategy, one that aligns with the
polymicrobial nature of clinical sepsis. Tubeimoside I (TBM), a
triterpenoid saponin, reduces iNOS expression by inhibiting the
TLR4-MyD88-NF-κB-iNOS signaling pathway (172). In a CLP-induced sepsis model,
an intraperitoneal injection of 4 mg/kg TBM 1 h before surgery
improved survival, ameliorated the MAP, and enhanced vascular
responsiveness to norepinephrine and KCl in wild-type septic mice
(172). Notably, iNOS gene
knockout completely abrogated the protective effects of TBM,
confirming that its therapeutic efficacy is mediated by reducing
excessive NO production. Nevertheless, the translational
applicability of TBM is hindered by nonspecific NF-κB inhibition.
Off-target NF-κB inhibition in immune cells could attenuate the
synthesis of proinflammatory cytokines during the hyperinflammatory
stage, yet it might also impede macrophage phagocytosis.
Complementing single-mechanism iNOS inhibitors are
agents that combine iNOS inhibition with adjunctive effects to
address coexisting sepsis pathologies, such as oxidative stress,
which amplifies VSMC dysfunction by modifying calcium-handling
proteins (for example, RyRs). Andrographolide, evaluated in
endotoxemia rats, restores vascular reactivity by downregulating
iNOS in perivascular adipose tissue (173). U-74389G, which has been tested
in septic models, combines iNOS inhibition with vascular protection
(174); it reduces NO-mediated
hyporeactivity via iNOS inhibition, reverses vascular failure and
protects against endotoxin shock, addressing the broader
pathological cascade of sepsis. However, andrographolide has poor
bioavailability, and neither agent has validated biomarkers for
iNOS activity or oxidative stress to enable precise patient
stratification, hindering their clinical translation. Moreover, the
nonselective tissue distribution of U-74389G increases the risk of
off-target effects in organs with high metabolic activity, where it
may disrupt normal NO signaling and exacerbate organ
dysfunction.
Outstanding questions in VSMC-targeted
therapy for sepsis-induced vasoplegia
The preceding assessment of calcium-modulating and
iNOS-targeted strategies emphasizes the identification of promising
VSMC-directed pathways for mitigating sepsis-induced vasoplegia in
preclinical studies. However, the attainment of translational
efficacy is consistently hindered by sepsis-specific obstacles in
drug delivery and off-target effects. The following is a
consolidation of the core challenges and actionable future
directions to address them (25,34,70,150,178): i) Disrupted tissue distribution
and bioavailability: Sepsis-induced increases in vascular
permeability and organ dysfunction disrupt drug distribution,
leading to reduced accumulation at VSMC sites and increased
systemic exposure. ii) Lack of VSMC-specific targeting: Currently,
agents demonstrate limited selectivity for VSMCs, resulting in
off-target effects on immune cells and vital organs. iii)
Formulation and dosing limitations: Challenges such as poor aqueous
solubility, short half-lives and the need for parenteral
administration present logistical barriers in critically ill
patients with sepsis. Additionally, the lack of validated
biomarkers hinders precise patient stratification, resulting in
variable clinical responses. iv) Immunosuppressive paradox: A
number of VSMC-targeted pathways serve vital roles in immune cell
function. Nonspecific blockade of these pathways risks exacerbating
sepsis-induced immunosuppression and increasing susceptibility to
secondary infections, a notable unmet challenge in translating
preclinical efficacy into clinical benefit.
Conclusion and prospects
The present review identifies VSMCs as a core
driver of sepsis-induced vasoplegia, with dysfunction stemming from
two linked cascades: Dysregulated calcium homeostasis and excessive
iNOS-derived NO. These factors impair VSMC contractility and
vascular tone, highlighting VSMCs as a key therapeutic target. In
conclusion, VSMCs are critical for the management of sepsis-induced
vasoplegia, and aligning mechanistic insights with translational
approaches may drive progress in VSMC-focused therapies to improve
outcomes in treating catecholamine-resistant sepsis.
Availability of data and materials
Not applicable.
Authors' contributions
HR was responsible for literature collection and
the initial draft of the manuscript. XYS and SYL contributed to the
analysis and integration of data and table data extracted from
published literatures collected in this review, focusing on
vascular smooth muscle cell function in sepsis, and participated in
the writing of this review; they were additionally responsible for
image rendering and critically revised the manuscript for content
accuracy. SSL contributed to revising the manuscript critically for
intellectual content and approved the final version for
publication. Data authentication is not applicable. All authors
reviewed the manuscript critically for intellectual content, and
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
ASMCs
|
aortic smooth muscle cells
|
|
AVP
|
arginine vasopressin
|
|
CLP
|
cecal ligation and puncture
|
|
cGMP
|
cyclic guanosine monophosphate
|
|
ECs
|
endothelial cells
|
|
eNOS
|
endothelial nitric oxide synthase
|
|
ECM
|
extracellular matrix
|
|
HASMCs
|
human ASMCs
|
|
HCASMCs
|
human coronary ASMCs
|
|
HCMV
|
human cytomegalovirus
|
|
iNOS
|
inducible nitric oxide synthase
|
|
LDHA
|
lactate dehydrogenase A
|
|
LPS
|
lipopolysaccharide
|
|
LTA
|
lipoteichoic acid
|
|
M-CSF
|
macrophage colony-stimulating
factor
|
|
PVAT
|
perivascular adipose tissue
|
|
RCT
|
randomized controlled trial
|
|
RNS
|
reactive nitrogen species
|
|
ROS
|
reactive oxygen species
|
|
TRPV
|
transient receptor potential
vanilloid
|
|
VSMCs
|
vascular smooth muscle cells
|
Acknowledgements
Not applicable.
Funding
This study was supported by the China Postdoctoral Science
Foundation (grant no. 413059).
References
|
1
|
Lambden S, Creagh-Brown BC, Hunt J,
Summers C and Forni LG: Definitions and pathophysiology of
vasoplegic shock. Critical Care. 22:1742018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu Y, Bao D, She H, Zhang Z, Shao S, Wu
Z, Wu Y, Li Q, Wang L, Li T and Liu L: Role of Hippo/ACSL4 axis in
ferroptosis-induced pericyte loss and vascular dysfunction in
sepsis. Redox Biol. 78:1033532024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
GBD 2021 Global Sepsis Collaborators:
Global, regional, and national sepsis incidence and mortality,
1990-2021: A systematic analysis. Lancet Global Health.
13:e2013–e2026. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ran X, Zhang Q and Li S, Yu Z, Wan L, Wu
B, Wu R and Li S: Tissue Kallikrein exacerbating sepsis-induced
endothelial hyperpermeability is highly predictive of severity and
mortality in sepsis. J Inflamm Res. 14:3321–3333. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ruan H, Li YZ, Zhang Q, Wang BR, Wu R, Li
SS and Ran X: Identification and clinical validation of
hypoxia-inducible factor 1α protein as the potential biomarker in
patients with sepsis. Shock. 59:855–863. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ruan H, Zhang Q, Zhang YP, Li SS and Ran
X: Unraveling the role of HIF-1α in sepsis: From pathophysiology to
potential therapeutics-a narrative review. Crit Care. 28:1002024.
View Article : Google Scholar
|
|
7
|
Wurster SH, Wang P, Dean RE and Chaudry
IH: Vascular smooth muscle contractile function is impaired during
early and late stages of sepsis. J Surg Res. 56:556–561. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Terpe P, Ruhs S, Dubourg V, Bucher M and
Gekle M: The synergism of cytosolic acidosis and reduced NAD+/NADH
ratio is responsible for lactic acidosis-induced vascular smooth
muscle cell impairment in sepsis. J Biomed Sci. 31:32024.
View Article : Google Scholar
|
|
9
|
Liu R, Jia L, Yu L, Lai D, Li Q, Zhang B,
Guo E, Xu K and Luo Q: Interaction between post-tumor inflammation
and vascular smooth muscle cell dysfunction in sepsis-induced
cardiomyopathy. Front Immunol. 16:15607172025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Donadon M and Santoro MM: The origin and
mechanisms of smooth muscle cell development in vertebrates.
Development. 148:dev1973842021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tinajero MG and Gotlieb AI: Recent
developments in vascular adventitial pathobiology: The dynamic
adventitia as a complex regulator of vascular disease. Am J Pathol.
190:520–534. 2020. View Article : Google Scholar
|
|
12
|
Méndez-Barbero N, Gutiérrez-Muñoz C and
Blanco-Colio LM: Cellular crosstalk between endothelial and smooth
muscle cells in vascular wall remodeling. Int J Mol Sci.
22:72842021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tang HY, Chen AQ, Zhang H, Gao XF, Kong XQ
and Zhang JJ: Vascular smooth muscle cells phenotypic switching in
cardiovascular diseases. Cells. 11:40602022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Arroyo-Ataz G, Yagüe AC, Breda JC,
Mazzilli SA and Jones D: Single-cell transcriptomics and lineage
tracing unveil parallels in lymphatic muscle and venous smooth
muscle development, identity, and function. Arterioscler Thromb
Vasc Biol. 45:1207–1225. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sawada H, Rateri DL, Moorleghen JJ,
Majesky MW and Daugherty A: Smooth muscle cells derived from second
heart field and cardiac neural crest reside in spatially distinct
domains in the media of the ascending aorta-brief report.
Arterioscler Thromb Vasc Biol. 37:1722–1726. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mikawa T and Gourdie RG: Pericardial
mesoderm generates a population of coronary smooth muscle cells
migrating into the heart along with ingrowth of the epicardial
organ. Dev Biol. 174:221–232. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Khachigian LM, Black BL, Ferdinandy P, De
Caterina R, Madonna R and Geng YJ: Transcriptional regulation of
vascular smooth muscle cell proliferation, differentiation and
senescence: Novel targets for therapy. Vascular Pharmacol.
146:1070912022. View Article : Google Scholar
|
|
18
|
Martinsen A, Dessy C and Morel N:
Regulation of calcium channels in smooth muscle: New insights into
the role of myosin light chain kinase. Channels (Austin).
8:402–413. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ito M, Okamoto R, Ito H, Zhe Y and Dohi K:
Regulation of myosin light-chain phosphorylation and its roles in
cardiovascular physiology and pathophysiology. Hypertens Res.
45:40–52. 2022. View Article : Google Scholar
|
|
20
|
Phelps C, Chess-Williams R and Moro C: The
role of intracellular calcium and Rho kinase pathways in G
protein-coupled receptor-mediated contractions of urinary bladder
urothelium and lamina propria. Am J Physiol Cell Physiol.
324:C787–C797. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ran Q, Li A, Tan Y, Zhang Y, Zhang Y and
Chen H: Action and therapeutic targets of myosin light chain
kinase, an important cardiovascular signaling mechanism. Pharmacol
Res. 206:1072762024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rezaei M, Mehta JL, Zadeh GM, Khedri A and
Rezaei HB: Myosin light chain phosphatase is a downstream target of
Rho-kinase in endothelin-1-induced transactivation of the TGF-β
receptor. Cell Biochem Biophys. 82:1109–1120. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weng W, Gu X, Yang Y, Zhang Q, Deng Q,
Zhou J, Cheng J, Zhu MX, Feng J, Huang O and Li Y: N-terminal
α-amino SUMOylation of cofilin-1 is critical for its regulation of
actin depolymerization. Nat Commun. 14:56882023. View Article : Google Scholar
|
|
24
|
Cao G, Xuan X, Hu J, Zhang R, Jin H and
Dong H: How vascular smooth muscle cell phenotype switching
contributes to vascular disease. Cell Commun Signal. 20:1802022.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sorokin V, Vickneson K, Kofidis T, Woo CC,
Lin XY, Foo R and Shanahan CM: Role of vascular smooth muscle cell
plasticity and interactions in vessel wall inflammation. Front
Immunol. 11:5994152020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu Y, Zhang JL, Yan XJ, Ji Y and Wang FF:
Exploring a new mechanism between lactate and VSMC calcification:
PARP1/POLG/UCP2 signaling pathway and imbalance of mitochondrial
homeostasis. Cell Death Dis. 14:5982023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tian Z, Ning H, Wang X, Wang Y, Han T and
Sun C: Endothelial autophagy promotes atheroprotective
communication between endothelial and smooth muscle cells via
Exosome-Mediated delivery of miR-204-5p. Arterioscler Thromb Vasc
Biol. 44:1813–1832. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Heiss EH, Schachner D, Donati M, Grojer CS
and Dirsch VM: Increased aerobic glycolysis is important for the
motility of activated VSMC and inhibited by indirubin-3'-monoxime.
Vascul Pharmacol. 83:47–56. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zein JG, Lee GL, Tawk M, Dabaja M and
Kinasewitz GT: Prognostic significance of elevated serum lactate
dehydrogenase (LDH) in patients with severe sepsis. Chest.
126(Suppl 1): 873S2004. View Article : Google Scholar
|
|
30
|
Yu H, Yang Q, Qian Y, Luo S, Kong T, Yang
G, Chen W, Xie F, Chen J, Xiong X, et al: A positive correlation
between serum lactate dehydrogenase level and in-hospital mortality
in ICU sepsis patients: Evidence from two large databases. Eur J
Med Res. 29:5252024. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen ZH, Cao SH, Ren ZY, Zhang T, Jiang
HM, Hu ZK and Dong LH: Lactate dehydrogenase A crotonylation and
Mono-Ubiquitination maintains vascular smooth muscle cell growth
and migration and promotes neointima hyperplasia. J Am Heart Assoc.
14:e0363772025. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Meng F, Cheng H, Qian J, Dai X, Huang Y
and Fan Y: In vitro fluidic systems: Applying shear stress on
endothelial cells. Med Novel Technol Devices. 15:1001432022.
View Article : Google Scholar
|
|
33
|
Di Giantomasso D, May CN and Bellomo R:
Vital organ blood flow during hyperdynamic sepsis. Chest.
124:1053–1059. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
McMullan RR, McAuley DF, O'Kane CM and
Silversides JA: Vascular leak in sepsis: Physiological basis and
potential therapeutic advances. Crit Care. 28:972024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bateman RM, Sharpe MD, Singer M and Ellis
CG: The effect of sepsis on the erythrocyte. Int J Mol Sci.
18:19322017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang Y, Ouyang Y, Liu B, Ma X and Ding R:
Platelet activation and antiplatelet therapy in sepsis: A narrative
review. Thromb Res. 166:28–36. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Haga JH, Li YSJ and Chien S: Molecular
basis of the effects of mechanical stretch on vascular smooth
muscle cells. J Biomech. 40:947–960. 2007. View Article : Google Scholar
|
|
38
|
Sun W, Wang S, Chen Z, Zhang J, Li T,
Arias K, Griffith BP and Wu ZJ: Impact of high mechanical shear
stress and oxygenator membrane surface on blood damage relevant to
thrombosis and bleeding in a pediatric ECMO circuit. Artif Organs.
44:717–726. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin C, Zheng X, Lin S, Zhang Y, Wu J and
Li Y: Mechanotransduction regulates the interplays between alveolar
epithelial and vascular endothelial cells in lung. Front Physiol.
13:8183942022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shirazian S, Rios-Rojas L, Drakakis J,
Dikkala S, Dutka P, Duey M, Cho DJ and Fishbane S: The effect of
hemodialysis ultrafiltration on changes in whole blood viscosity.
Hemodial Int. 16:342–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xie M, Li X, Chen L, Zhang Y, Chen L, Hua
H and Qi J: The crosstalks between vascular endothelial cells,
vascular smooth muscle cells, and adventitial fibroblasts in
vascular remodeling. Life Sci. 361:1233192025. View Article : Google Scholar
|
|
42
|
Hiroshima Y, Oyama Y, Sawasaki K, Nakamura
M, Kimura N, Kawahito K, Fujie H and Sakamoto N: A compressed
collagen construct for studying Endothelial-smooth muscle cell
interaction under high shear stress. Ann Biomed Eng. 50:951–963.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sawasaki K, Nakamura M, Kimura N, Kawahito
K, Yamazaki M, Fujie H and Sakamoto N: Endothelial-derived nitric
oxide impacts vascular smooth muscle cell phenotypes under high
wall shear stress condition. Biochem Biophys Res Commun.
740:1510052024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yu J, Zhang Y, Zhang X, Rudic RD, Bauer
PM, Altieri DC and Sessa WC: Endothelium derived nitric oxide
synthase negatively regulates the PDGF-survivin pathway during
flow-dependent vascular remodeling. PLoS One. 7:e314952012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sack KD, Teran M and Nugent MA:
Extracellular matrix stiffness controls VEGF signaling and
processing in endothelial cells. J Cell Physiol. 231:2026–2039.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ploppa A, Schmidt V, Hientz A, Reutershan
J, Haeberle HA and Nohé B: Mechanisms of leukocyte distribution
during sepsis: An experimental study on the interdependence of cell
activation, shear stress and endothelial injury. Crit Care.
14:R2012010. View
Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu Z, Ji J, Zheng D, Su L, Peng T and
Tang J: Protective role of endothelial calpain knockout in
lipopolysaccharide-induced acute kidney injury via attenuation of
the p38-iNOS pathway and NO/ROS production. Exp Mol Med.
52:702–712. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fan Y, Moser J, van Meurs M, Kiers D, Sand
JMB, Leeming DJ, Pickkers P, Burgess JK, Kox M and Pillay J:
Neo-epitope detection identifies extracellular matrix turnover in
systemic inflammation and sepsis: An exploratory study. Crit Care.
28:1202024. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fontaine M, Herkenne S, Ek O, Paquot A,
Boeckx A, Paques C, Nivelles O, Thiry M and Struman I:
Extracellular vesicles mediate communication between endothelial
and vascular smooth muscle cells. Int J Mol Sci. 23:3312021.
View Article : Google Scholar
|
|
50
|
Nonato AO, Olivon VC, Dela Justina V,
Zanotto CZ, Webb RC, Tostes RC, Lima VV and Giachini FR: Impaired
Ca(2+) homeostasis and decreased orai1 expression modulates
arterial hyporeactivity to vasoconstrictors during endotoxemia.
Inflammation. 39:1188–1197. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pereira OR, Ramos VM, Cabral-Costa JV and
Kowaltowski AJ: Changes in mitochondrial morphology modulate
LPS-induced loss of calcium homeostasis in BV-2 microglial cells. J
Bioenerg Biomembr. 53:109–118. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ni L, Lin B, Shen M, Li C, Hu L, Fu F,
Chen L, Yang J and Shi D: PKM2 deficiency exacerbates gram-negative
sepsis-induced cardiomyopathy via disrupting cardiac calcium
homeostasis. Cell Death Discov. 8:4962022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Takeuchi K, del Nido PJ, Ibrahim AE,
Poutias DN, Glynn P, Cao-Danh H, Cowan DB and McGowan FX: Increased
myocardial calcium cycling and reduced myofilament calcium
sensitivity in early endotoxemia. Surgery. 126:231–238. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sandbo N, Taurin S, Yau DM, Kregel S,
Mitchell R and Dulin NO: Downregulation of smooth muscle
alpha-actin expression by bacterial lipopolysaccharide. Cardiovasc
Res. 74:262–269. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dyck B, Unterberg M, Adamzik M and Koos B:
The impact of pathogens on sepsis prevalence and outcome.
Pathogens. 13:892024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang C, Mansard A, Giummelly P and
Atkinson J: Decreased aortic smooth muscle contraction in a rat
model of multibacterial sepsis. Fundam Clin Pharmacol. 18:679–683.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhao X, Miao G and Zhang L, Zhang Y, Zhao
H, Xu Z, Wang B and Zhang L: Chlamydia pneumoniae infection induces
vascular smooth muscle cell migration and atherosclerosis through
mitochondrial reactive oxygen Species-mediated JunB-Fra-1
activation. Front Cell Dev Biol. 10:8790232022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
He H, Tan Y, Tang Z, Wang L, Liu S and Wu
G: ADAM9: A regulator between HCMV infection and function of smooth
muscle cells. J Med Virol. 95:e283522023. View Article : Google Scholar
|
|
59
|
Richards A, Khalil AS, Friesen M,
Whitfield TW, Gao X, Lungjangwa T, Kamm RD, Wan Z, Gehrke L, Mooney
D and Jaenisch R: SARS-CoV-2 infection of human pluripotent stem
cell-derived vascular cells reveals smooth muscle cells as key
mediators of vascular pathology during infection. Nat Commun.
15:107542024. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hu X, He X, Zhang W, Jin C, Deng C, Ma Y,
Chen P, Ma S, Zhao R and Shi B: LPS induces RGS-1 to promote
infectious intracranial aneurysm formation and rupture by
accelerating smooth muscle cell phenotypic switching. Int
Immunopharmacol. 142:1132032024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hattori Y, Kasai K, Nakanishi N, Gross SS
and Thiemermann C: Induction of nitric oxide and
tetrahydrobiopterin synthesis by lipoteichoic acid from
Staphylococcus aureus in vascular smooth muscle cells. J Vasc Res.
35:104–108. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sibelius U, Grandel U, Buerke M, Mueller
D, Kiss L, Kraemer HJ, Braun-Dullaeus R, Haberbosch W, Seeger W and
Grimminger F: Staphylococcal alpha-toxin provokes coronary
vasoconstriction and loss in myocardial contractility in perfused
rat hearts: Role of thromboxane generation. Circulation. 101:78–85.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Seki M, Mukohda M, Tajima H, Morikita N,
Imai R, Itaya K, Mizuno R and Ozaki H: Long-term treatment with the
streptococcal exotoxin streptolysin O inhibits vascular smooth
muscle contraction by inducing iNOS expression in endothelial
cells. J Pharmacol Exp Ther. 392:1000112025. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kook H, Rhee JH, Lee SE, Kang SY, Chung
SS, Cho KW and Baik YH: Activation of particulate guanylyl cyclase
by Vibrio vulnificus hemolysin. Eur J Pharmacol. 365:267–272. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
DelVechio M, Alves JV, Saiyid AZ, Singh S,
Galley J, Awata WMC, Costa RM, Bruder-Nascimento A and
Bruder-Nascimento T: Progression of vascular function and blood
pressure in a mouse model of Kawasaki disease. Shock. 59:74–81.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang B, Elmabsout AA, Khalaf H, Basic VT,
Jayaprakash K, Kruse R, Bengtsson T and Sirsjö A: The periodontal
pathogen Porphyromonas gingivalis changes the gene expression in
vascular smooth muscle cells involving the TGFbeta/Notch signalling
pathway and increased cell proliferation. BMC Genomics. 14:7702013.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Oseghale O, Liong S, Coward-Smith M, To
EE, Erlich JR, Luong R, Liong F, Miles M, Norouzi S, Martin C, et
al: Influenza A virus elicits peri-vascular adipose tissue
inflammation and vascular dysfunction of the aorta in pregnant
mice. PLoS Pathog. 18:e10107032022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liu MW, Duan SX, Zhao XY, Wang QF, Yang
SL, Ma N and Li X: Research status and advances in dexmedetomidine
for sepsis-induced multiple organ dysfunction syndrome (review).
Int J Mol Med. 55:942025. View Article : Google Scholar
|
|
69
|
Jarczak D and Nierhaus A: Cytokine
Storm-definition, causes, and implications. Int J Mol Sci.
23:117402022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Reddy H, Javvaji CK, Malali S, Kumar S,
Acharya S and Toshniwal S: Navigating the cytokine storm: A
comprehensive review of chemokines and cytokines in sepsis. Cureus.
16:e542752024.PubMed/NCBI
|
|
71
|
Beasley D and Cooper AL: Constitutive
expression of interleukin-1alpha precursor promotes human vascular
smooth muscle cell proliferation. Am J Physiol. 276:H901–H912.
1999.PubMed/NCBI
|
|
72
|
Xu A, Pei J, Yang Y, Hua B and Wang J:
IL-1β promotes A7r5 and HASMC migration and invasion via the
p38-MAPK/Angpt-2 pathway. Eur J Med Res. 27:1532022. View Article : Google Scholar
|
|
73
|
Sun XH, Jiang HJ, Liu Q, Xiao C, Xu JY, Wu
Y, Mei JY, Wu ST and Lin ZY: Low concentrations of TNF-α in vitro
transform the phenotype of vascular smooth muscle cells and enhance
their survival in a three-dimensional culture system. Artif Organs.
48:839–848. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Liu W, Liu X, Luo M, Liu X, Luo Q, Tao H,
Wu D, Lu S, Jin J, Zhao Y and Zou L: dNK derived IFN-γ mediates
VSMC migration and apoptosis via the induction of LncRNA MEG3: A
role in uterovascular transformation. Placenta. 50:32–39. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Arumugam P, Carroll KL, Berceli SA,
Barnhill S and Wrenshall LE: Expression of a Functional IL-2
receptor in vascular smooth muscle cells. J Immunol. 202:694–703.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Brizzi MF, Formato L, Dentelli P, Rosso A,
Pavan M, Garbarino G, Pegoraro M, Camussi G and Pegoraro L:
Interleukin-3 stimulates migration and proliferation of vascular
smooth muscle cells: A potential role in atherogenesis.
Circulation. 103:549–554. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Hofbauer LC, Schrader J, Niebergall U,
Viereck V, Burchert A, Hörsch D, Preissner KT and Schoppet M:
Interleukin-4 differentially regulates osteoprotegerin expression
and induces calcification in vascular smooth muscle cells. Thromb
Haemost. 95:708–714. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Cimmino I, Prisco F, Orso S, Agognon AL,
Liguoro P, De Biase D, Doti N, Ruvo M, Paciello O, Beguinot F, et
al: Interleukin 6 reduces vascular smooth muscle cell apoptosis via
Prep1 and is associated with aging. FASEB J. 35:e219892021.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yue TL, Wang X, Sung CP, Olson B, McKenna
PJ, Gu JL and Feuerstein GZ: Interleukin-8. A mitogen and
chemoattractant for vascular smooth muscle cells. Circ Res. 75:1–7.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Mazighi M, Pellé A, Gonzalez W, Mtairag el
M, Philippe M, Hénin D, Michel JB and Feldman LJ: IL-10 inhibits
vascular smooth muscle cell activation in vitro and in vivo. Am J
Physiol Heart Circ Physiol. 287:H866–H871. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zimmerman MA, Selzman CH, Reznikov LL,
Raeburn CD, Barsness K, McIntyre RC Jr, Hamiel CR and Harken AH:
Interleukin-11 attenuates human vascular smooth muscle cell
proliferation. Am J Physiol Heart Circ Physiol. 283:H175–H180.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Cho WK, Lee CM, Kang MJ, Huang Y, Giordano
FJ, Lee PJ, Trow TK, Homer RJ, Sessa WC, Elias JA and Lee CG: IL-13
receptor α2-arginase 2 pathway mediates IL-13-induced pulmonary
hypertension. Am J Physiol Lung Cell Mol Physiol. 304:L112–L124.
2013. View Article : Google Scholar
|
|
83
|
Iwasaki S, Minamisawa S, Yokoyama U,
Akaike T, Quan H, Nagashima Y, Nishimaki S, Ishikawa Y and Yokota
S: Interleukin-15 inhibits smooth muscle cell proliferation and
hyaluronan production in rat ductus arteriosus. Pediatr Res.
62:392–398. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Park SL, Hwang B, Lee SY, Kim WT, Choi YH,
Chang YC, Kim WJ and Moon SK: p21WAF1 is required for
Interleukin-16-Induced migration and invasion of vascular smooth
muscle cells via the p38MAPK/Sp-1/MMP-9 pathway. PLoS One.
10:e01421532015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Duncan JW, Granger JP, Ryan MJ and
Drummond HA: Interleukin-17 reduces βENaC via MAPK signaling in
vascular smooth muscle cells. Int J Mol Sci. 21:29532020.
View Article : Google Scholar
|
|
86
|
Zhang K, Zhang Y, Feng W, Chen R, Chen J,
Touyz RM, Wang J and Huang H: Interleukin-18 enhances vascular
calcification and osteogenic differentiation of vascular smooth
muscle cells through TRPM7 activation. Arterioscler Thromb Vasc
Biol. 37:1933–1943. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Cuneo AA, Herrick D and Autieri MV: Il-19
reduces VSMC activation by regulation of mRNA regulatory factor HuR
and reduction of mRNA stability. J Mol Cell Cardiol. 49:647–654.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Dale BL, Pandey AK, Chen Y, Smart CD,
Laroumanie F, Ao M, Xiao L, Dikalova AE, Dikalov SI, Elijovich F,
et al: Critical role of Interleukin 21 and T follicular helper
cells in hypertension and vascular dysfunction. JCI Insight.
5:e1292782019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Rattik S, Hultman K, Rauch U, Söderberg I,
Sundius L, Ljungcrantz I, Hultgårdh-Nilsson A, Wigren M, Björkbacka
H, Fredrikson GN and Nilsson J: IL-22 affects smooth muscle cell
phenotype and plaque formation in apolipoprotein E knockout mice.
Atherosclerosis. 242:506–514. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Conway R, O'Neill L, McCarthy GM, Murphy
CC, Fabre A, Kennedy S, Veale DJ, Wade SM, Fearon U and Molloy ES:
Interleukin 12 and interleukin 23 play key pathogenic roles in
inflammatory and proliferative pathways in giant cell arteritis.
Ann Rheum Dis. 77:1815–1824. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lee KM, Kang HA, Park M, Lee HY, Choi HR,
Yun CH, Oh JW and Kang HS: Interleukin-24 attenuates
β-glycerophosphate-induced calcification of vascular smooth muscle
cells by inhibiting apoptosis, the expression of calcification and
osteoblastic markers, and the Wnt/β-catenin pathway. Biochem
Biophys Res Commun. 428:50–55. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Hao N, Zhou Z, Zhang F, Li Y, Hu R, Zou J,
Zheng R, Wang L, Xu L, Tan W, et al: Interleukin-29 accelerates
vascular calcification via JAK2/STAT3/BMP2 signaling. J Am Heart
Assoc. 12:e0272222023. View Article : Google Scholar :
|
|
93
|
Son DJ, Jung YY, Seo YS, Park H, Lee DH,
Kim S, Roh YS, Han SB, Yoon DY and Hong JT: Interleukin-32α
inhibits endothelial inflammation, vascular smooth muscle cell
activation, and atherosclerosis by upregulating Timp3 and reck
through suppressing microRNA-205 biogenesis. Theranostics.
7:2186–2203. 2017. View Article : Google Scholar
|
|
94
|
DeVallance E, Bowdridge E, Garner K,
Griffith J, Seman M, Batchelor T, Velayutham M, Goldsmith WT,
Hussain S, Kelley EE and Nurkiewicz TR: The alarmin,
interleukin-33, increases vascular tone via extracellular signal
regulated kinase-mediated Ca2+ sensitization and endothelial nitric
oxide synthase inhibition. J Physiol. 602:6087–6107. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Skowron W, Zemanek K, Wojdan K, Gorzelak
P, Borowiec M, Broncel M and Chalubinski M: The effect of
interleukin-35 on the integrity, ICAM-1 expression and apoptosis of
human aortic smooth muscle cells. Pharmacol Rep. 67:376–381. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Ding Y, Wang Y, Cai Y, Pan C, Yang C, Wang
M, Qi X, Ye J, Ji Q, Yu J, et al: IL-37 expression in patients with
abdominal aortic aneurysm and its role in the necroptosis of
vascular smooth muscle cells. Oxid Med Cell Longev.
2022:18065132022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Niessner A, Shin MS, Pryshchep O, Goronzy
JJ, Chaikof EL and Weyand CM: Synergistic proinflammatory effects
of the antiviral cytokine interferon-alpha and Toll-like receptor 4
ligands in the atherosclerotic plaque. Circulation. 116:2043–2052.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Sano E, Tashiro S, Tsumoto K and Ueda T:
Differential effects of IFN-β on the survival and growth of human
vascular smooth muscle and endothelial cells. Biores Open Access.
4:1–15. 2015. View Article : Google Scholar
|
|
99
|
Vasudevan SS, Lopes NH, Seshiah PN, Wang
T, Marsh CB, Kereiakes DJ, Dong C and Goldschmidt-Clermont PJ:
Mac-1 and Fas activities are concurrently required for execution of
smooth muscle cell death by M-CSF-stimulated macrophages.
Cardiovasc Res. 59:723–733. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Plenz G, Reichenberg S, Koenig C,
Rauterberg J, Deng MC, Baba HA and Robenek H:
Granulocyte-macrophage colony-stimulating factor (GM-CSF) modulates
the expression of type VIII collagen mRNA in vascular smooth muscle
cells and both are codistributed during atherogenesis. Arterioscler
Thromb Vasc Biol. 19:1658–1668. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Rinaldi B, Finicelli M, Donniacuo M, Di
Bernardo G, Gritti G, Gaudio SD, Forte A, Peluso G, Cipollaro M,
Rossi F and Galderisi U: G-CSF contributes at the healing of tunica
media of arteriotomy-injured rat carotids by promoting
differentiation of vascular smooth muscle cells. J Cell Physiol.
231:215–223. 2016. View Article : Google Scholar
|
|
102
|
Li Y, Ren S and Zhou S: Advances in sepsis
research: Insights into signaling pathways, organ failure, and
emerging intervention strategies. Exp Mol Pathol. 142:1049632025.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Chen C, Du Y, Nie R, Wang S, Wang H and Li
P: Notch signaling in cancers: Mechanism and potential therapy.
Front Cell Dev Biol. 13:15429672025. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Singh V, Akash R, Chaudhary G, Singh R,
Choudhury S, Shukla A, Prabhu SN, Gangwar N and Garg SK: Sepsis
downregulates aortic Notch signaling to produce vascular
hyporeactivity in mice. Sci Rep. 12:29412022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zeng H, He X, Tuo QH, Liao DF, Zhang GQ
and Chen JX: LPS causes pericyte loss and microvascular dysfunction
via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2α/Notch3
pathways. Sci Rep. 6:209312016. View Article : Google Scholar
|
|
106
|
Guan G, Chen Y and Dong Y: Unraveling the
AMPK-SIRT1-FOXO pathway: The In-depth analysis and breakthrough
prospects of oxidative Stress-induced diseases. Antioxidants
(Basel). 14:702025. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Yang H, Luo YY, Zhang LT, He KR and Lin
XJ: Extracellular histones induce inflammation and senescence of
vascular smooth muscle cells by activating the AMPK/FOXO4 signaling
pathway. Inflamm Res. 71:1055–1066. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Millar MW, Fazal F and Rahman A:
Therapeutic targeting of NF-κB in acute lung injury: A Double-edged
sword. Cells. 11:33172022. View Article : Google Scholar
|
|
109
|
Canovas B and Nebreda AR: Diversity and
versatility of p38 kinase signalling in health and disease. Nat Rev
Mol Cell Biol. 22:346–366. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Liu Z, Ting Y, Li M, Li Y, Tan Y and Long
Y: From immune dysregulation to organ dysfunction: Understanding
the enigma of Sepsis. Front Microbiol. 15:14152742024. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Miao S, Zhang R, Guo G, Wang X, Zhang B,
Li L, Wang J, Weng Y, Shi X, Ma S and Lu C: p38 protein as a
therapeutic target for sepsis-induced organ dysfunction. Eur J
Pharmacol. 1002:1778332025. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Chan CH, Jo U, Kohrman A, Rezaeian AH,
Chou PC, Logothetis C and Lin HK: Posttranslational regulation of
Akt in human cancer. Cell Biosci. 4:592014. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Liu Y, Meng J, Liu J, Zhang F, An C and
Yang J: NLRP3 inflammasome in acute lung injury: Cumulative
evidence for traditional Chinese medicine and natural products.
Chin Herbal Med. 17:703–719. 2025. View Article : Google Scholar
|
|
114
|
Zhan X, Li Q, Xu G, Xiao X and Bai Z: The
mechanism of NLRP3 inflammasome activation and its pharmacological
inhibitors. Front Immunol. 13:11099382022. View Article : Google Scholar
|
|
115
|
Hao QY, Yan J, Wei JT, Zeng YH, Feng LY,
Que DD, Li SC, Guo JB, Fan Y, Ding YF, et al: Prevotella copri
promotes vascular calcification via lipopolysaccharide through
activation of NF-κB signaling pathway. Gut Microbes.
16:23515322024. View Article : Google Scholar
|
|
116
|
Li Z, Wu H, Yao F, Li Y, Li Y, Xie SA, Yu
F, Fu Y, Wang L, Zhou J and Kong W: Runx2-NLRP3 axis orchestrates
matrix stiffness-evoked vascular smooth muscle cell inflammation.
Am J Physiol Cell Physiol. 328:C467–C482. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Kim EJ, Park SY, Baek SE, Jang MA, Lee WS,
Bae SS, Kim K and Kim CD: HMGB1 Increases IL-1β production in
vascular smooth muscle cells via NLRP3 inflammasome. Front Physiol.
9:3132018. View Article : Google Scholar
|
|
118
|
James AM and Murphy MP: How mitochondrial
damage affects cell function. J Biomed Sci. 9:475–487. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Tsuji N, Tsuji T, Yamashita T, Hayase N,
Hu X, Yuen PS and Star RA: BAM15 treats mouse sepsis and kidney
injury, linking mortality, mitochondrial DNA, tubule damage, and
neutrophils. J Clin Invest. 133:e1524012023. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Hu D, Sheeja Prabhakaran H, Zhang YY, Luo
G, He W and Liou YC: Mitochondrial dysfunction in sepsis:
Mechanisms and therapeutic perspectives. Critical Care. 28:2922024.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Bauernfeind F, Bartok E, Rieger A, Franchi
L, Núñez G and Hornung V: Cutting edge: Reactive oxygen species
inhibitors block priming, but not activation, of the NLRP3
inflammasome. J Immunol. 187:613–617. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Li PF, Dietz R and von Harsdorf R:
Differential effect of hydrogen peroxide and superoxide anion on
apoptosis and proliferation of vascular smooth muscle cells.
Circulation. 96:3602–3609. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Brown MR, Miller FJ Jr, Li WG, Ellingson
AN, Mozena JD, Chatterjee P, Engelhardt JF, Zwacka RM, Oberley LW,
Fang X, et al: Overexpression of human catalase inhibits
proliferation and promotes apoptosis in vascular smooth muscle
cells. Circ Res. 85:524–533. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Bieling F, Uhle F, Weissmüller K, Weigand
MA and Henrich M: Sepsis varies arterial two-pore-domain potassium
channel messenger RNA in mice. J Surg Res. 193:816–824. 2015.
View Article : Google Scholar
|
|
125
|
Yan Z, Niu L, Wang S, Gao C and Pan S:
Intestinal Piezo1 aggravates intestinal barrier dysfunction during
sepsis by mediating Ca2+ influx. J Transl Med. 22:3322024.
View Article : Google Scholar
|
|
126
|
Orliac ML, Peroni RN, Abramoff T, Neuman
I, Podesta EJ and Adler-Graschinsky E: Increases in vanilloid TRPV1
receptor protein and CGRP content during endotoxemia in rats. Eur J
Pharmacol. 566:145–152. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zhang FR, Tang J, Lai Y, Mo SQ, Lin ZM,
Lei QQ, Han CC, Zhou AD, Lv XF, Wang C, et al: Smooth muscle cell
Piezo1 is essential for phenotypic switch and neointimal
hyperplasia. Br J Pharmacol. 182:2031–2048. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Zhao C, Shen J, Lu Y, Ni H, Xiang M and
Xie Y: Dedifferentiation of vascular smooth muscle cells upon
vessel injury. Int Immunopharmacol. 144:1136912025. View Article : Google Scholar
|
|
129
|
Han M, Wen JK, Zheng B, Cheng Y and Zhang
C: Serum deprivation results in redifferentiation of human
umbilical vascular smooth muscle cells. Am J Physiol Cell Physiol.
291:C50–C58. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Dang YY, Chen C, Wei QF, Gao LF, Zhang SC,
Li YX and Dai XY: Dehydrocorydaline maintains the vascular smooth
muscle cell contractile phenotype by upregulating Spta1. Acta
Pharmacol Sin. 46:1303–1316. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zhang HS, Hong H, Zeng DY, Xie LN, Cheng
Q, Pang XF and Guan QG: Atorvastatin suppresses vascular
hypersensitivity and remodeling induced by transient adventitial
administration of lipopolysaccharide in rats. Ann Transl Med.
7:3862019. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Kazune S, Piebalga A, Strike E and Vanags
I: Impaired vascular reactivity in sepsis-a systematic review with
meta-analysis. Arch Med Sci Atheroscler Dis. 4:e151–e161. 2019.
View Article : Google Scholar
|
|
133
|
Glavaš M, Gitlin-Domagalska A, Dębowski D,
Ptaszyńska N, Łęgowska A and Rolka K: Vasopressin and its
analogues: From natural hormones to multitasking peptides. Int J
Mol Sci. 23:30682022. View Article : Google Scholar
|
|
134
|
Yoshimura M, Conway-Campbell B and Ueta Y:
Arginine vasopressin: Direct and indirect action on metabolism.
Peptides. 142:1705552021. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Moreau R, Barrière E, Tazi KA, Lardeux B,
Dargère D, Urbanowicz W, Poirel O, Chauvelot-Moachon L, Guimont MC,
Bernuau D and Lebrec D: Terlipressin inhibits in vivo aortic iNOS
expression induced by lipopolysaccharide in rats with biliary
cirrhosis. Hepatology. 36:1070–1078. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Russell JA, Walley KR, Singer J, Gordon
AC, Hébert PC, Cooper DJ, Holmes CL, Mehta S, Granton JT, Storms
MM, et al: Vasopressin versus norepinephrine infusion in patients
with septic shock. N Engl J Med. 358:877–887. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Gordon AC, Russell JA, Walley KR, Singer
J, Ayers D, Storms MM, Holmes CL, Hébert PC, Cooper DJ, Mehta S, et
al: The effects of vasopressin on acute kidney injury in septic
shock. Intensive Care Med. 36:83–91. 2010. View Article : Google Scholar
|
|
138
|
Morelli A, Ertmer C, Rehberg S, Lange M,
Orecchioni A, Cecchini V, Bachetoni A, D'Alessandro M, Van Aken H,
Pietropaoli P and Westphal M: Continuous terlipressin versus
vasopressin infusion in septic shock (TERLIVAP): A randomized,
controlled pilot study. Crit Care. 13:R1302009. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Laterre PF, Berry SM, Blemings A, Carlsen
JE, François B, Graves T, Jacobsen K, Lewis RJ, Opal SM, Perner A,
et al: Effect of selepressin vs placebo on Ventilator- and
Vasopressor-free days in patients with septic shock: The SEPSIS-ACT
randomized clinical trial. JAMA. 322:1476–1485. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Chaudhary A, Bansal S, Saha S, Arora S,
Akram W and Kumar S: Phosphodiesterase inhibitors for diabetes:
From mechanistic insights to therapeutic innovations. N Emirates
Med J. 5:2024.
|
|
141
|
Zhang Z, Liang M and Wan X: Roflumilast, a
type of phosphodiesterase 4 inhibitor, can reduce intestinal injury
caused by sepsis. Exp Ther Med. 22:13982021. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Ferreira Alves G, Oliveira JG, Nakashima
MA, Delfrate G, Sordi R, Assreuy J, da Silva-Santos JE, Collino M
and Fernandes D: Cardiovascular effects of Roflumilast during
sepsis: Risks or benefits? Eur J Pharmacol. 983:1770152024.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Nakashima MA, Delfrate G, Albino LB, Alves
GF, Oliveira JG and Fernandes D: Impact of tadalafil on
cardiovascular and organ dysfunction induced by experimental
sepsis. Acute Crit Care. 40:46–58. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Schmidt W, Tinelli M, Secchi A, Gebhard
MM, Martin E and Schmidt H: Milrinone improves intestinal villus
blood flow during endotoxemia. Can J Anaesth. 47:673–679. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
de Miranda ML, Pereira SJ, Santos AO,
Villela NR, Kraemer-Aguiar LG and Bouskela E: Milrinone attenuates
arteriolar vasoconstriction and capillary perfusion deficits on
endotoxemic hamsters. PLoS One. 10:e01170042015. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Shati AA, Zaki MSA, Alqahtani YA,
Al-Qahtani SM, Haidara MA, Dawood AF, AlMohanna AM, El-Bidawy MH,
Alaa Eldeen M and Eid RA: Antioxidant activity of Vitamin C against
LPS-Induced septic cardiomyopathy by Down-regulation of oxidative
stress and inflammation. Curr Issues Mol Biol. 44:2387–2400. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Pehlivan VF, Pehlivan B, Duran E, Taskın
A, Koyuncu I and Çakmak Y: Effects of Vitamin E on Redox balance in
regulating thiol/disulfide homeostasis in sepsis: An antioxidant
therapy perspective. PLoS One. 20:e03363342025. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Mao H, Zhang Y, Xiong Y, Zhu Z, Wang L and
Liu X: Mitochondria-targeted antioxidant mitoquinone maintains
mitochondrial homeostasis through the Sirt3-Dependent pathway to
mitigate oxidative damage caused by renal Ischemia/Reperfusion.
Oxid Med Cell Longev. 2022:22135032022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Ning R, Li Y, Du Z, Li T, Sun Q, Lin L, Xu
Q, Duan J and Sun Z: The mitochondria-targeted antioxidant MitoQ
attenuated PM2.5-induced vascular fibrosis via regulating
mitophagy. Redox Biol. 46:1021132021. View Article : Google Scholar
|
|
150
|
Liang B, Su J, Shao H, Chen H and Xie B:
The outcome of IV vitamin C therapy in patients with sepsis or
septic shock: A meta-analysis of randomized controlled trials. Crit
Care. 27:1092023. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Lamontagne F, Masse MH, Menard J, Sprague
S, Pinto R, Heyland DK, Cook DJ, Battista MC, Day AG, Guyatt GH, et
al: Intravenous vitamin C in adults with sepsis in the intensive
care unit. N Engl J Med. 386:2387–2398. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
He Y, Li Y, Lao Q, Qin T, Xie X and Jiang
W: Association between vitamin E and 28-day mortality in adult
sepsis patients: A retrospective cohort analysis. Am J Med Sci.
370:552–558. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Yang CH, Qiu HQ, Wang C, Tang YT, Zhang
CR, Fan YY and Jiao XY: Levosimendan relaxes thoracic aortic smooth
muscle in mice by inhibiting PKC and activating inwardly rectifying
potassium channels. J Cardiovasc Pharmacol. 83:474–481. 2024.
View Article : Google Scholar
|
|
154
|
Lei Y, Peng X, Hu Y, Xue M, Li T, Liu L
and Yang G: The calcilytic drug calhex-231 ameliorates vascular
hyporesponsiveness in traumatic hemorrhagic shock by inhibiting
oxidative stress and miR-208a-Mediated mitochondrial fission. Oxid
Med Cell Longev. 2020:41327852020. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Rodríguez EE, Jaramillo GAD, Cuellar LMR,
Herran SEP, Lima DRR and Herpain A: Levosimendan vs. Dobutamine in
patients with septic shock: A systematic review and Meta-analysis
with trial sequential analysis. J Clin Med. 14:54962025. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Morelli A, Donati A, Ertmer C, Rehberg S,
Lange M, Orecchioni A, Cecchini V, Landoni G, Pelaia P, Pietropaoli
P, et al: Levosimendan for resuscitating the microcirculation in
patients with septic shock: A randomized controlled study. Crit
Care. 14:R2322010. View
Article : Google Scholar : PubMed/NCBI
|
|
157
|
Nielsen J, Langgård M, Tengberg JF and
Kehler J: The molecular mechanism of PDE1 Regulation. Cells.
14:17222025. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Flemming S, Schlegel N, Wunder C, Meir M,
Baar W, Wollborn J, Roewer N, Germer CT and Schick MA:
Phosphodiesterase 4 inhibition dose dependently stabilizes
microvascular barrier functions and microcirculation in a rodent
model of polymicrobial sepsis. Shock. 41:537–545. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Tai-Passmann S, Slegers CAD, Hemelaar P,
Waalders N, Koopmans M, van den Bogaard B, van Lookeren Campagne M,
Goedegebuur J, Kuindersma M, Schroten N, et al: Phosphodiesterase 3
inhibitors do not influence lactate kinetics and clinical outcomes
in patients with septic shock: A multicentre cohort study. J Crit
Care. 83:1548272024. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Yamamoto M, Hosoya Y, Hanada H, Osawa T,
Arai M, Sakamoto K, Okazaki Y, Katasako-Yabumoto A, Ishizu T, Kondo
T, et al: Clinical importance of Phosphodiesterase 3 inhibitors on
outcomes in patients with cardiogenic shock-A systematic review.
Circ Rep. 7:1021–1028. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Sahoo DK, Wong D, Patani A, Paital B,
Yadav VK, Patel A and Jergens AE: Exploring the role of
antioxidants in sepsis-associated oxidative stress: A comprehensive
review. Front Cell Infect Microbiol. 14:13487132024. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Kakade S and Mani G: A comparative study
of the effects of vitamin C, sirolimus, and paclitaxel on the
growth of endothelial and smooth muscle cells for cardiovascular
medical device applications. Drug Des Devel Ther. 7:529–544.
2013.PubMed/NCBI
|
|
163
|
Ulrich-Merzenich G, Zeitler H, Panek D,
Bokemeyer D and Vetter H: Vitamin C promotes human endothelial cell
growth via the ERK-signaling pathway. Eur J Nutr. 46:87–94. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Dang H, Li J, Liu C and Xu F: The
association between Vitamin E deficiency and critically Ill
children with sepsis and septic shock. Front Nutr. 8:6484422021.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Cui L, Zhou Q, Zheng X, Sun B and Zhao S:
Mitoquinone attenuates vascular calcification by suppressing
oxidative stress and reducing apoptosis of vascular smooth muscle
cells via the Keap1/Nrf2 pathway. Free Radic Biol Med. 161:23–31.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Supinski GS, Murphy MP and Callahan LA:
MitoQ administration prevents endotoxin-induced cardiac
dysfunction. Am J Physiol Regul Integr Comp Physiol.
297:R1095–R1102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Schmid D, Svoboda M, Sorgner A, Moravcevic
I, Thalhammer T, Chiba P and Möslinger T: Glibenclamide reduces
proinflammatory cytokines in an ex vivo model of human
endotoxinaemia under hypoxaemic conditions. Life Sci. 89:725–734.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Zhou R, Chen F, Li Q, Hu DY and Liu LM:
Stimulation of the adenosine A3 receptor reverses vascular
hyporeactivity after hemorrhagic shock in rats. Acta Pharmacol Sin.
31:413–420. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Li T, Liu L, Liu J, Ming J, Xu J, Yang G
and Zhang Y: Mechanisms of Rho kinase regulation of vascular
reactivity following hemorrhagic shock in rats. Shock. 29:65–70.
2008. View Article : Google Scholar
|
|
170
|
Gibraeil HD, Dittrich P, Saleh S and Mayer
B: Inhibition of endotoxin-induced vascular hyporeactivity by
4-amino-tetrahydrobiopterin. Br J Pharmacol. 131:1757–1765. 2000.
View Article : Google Scholar
|
|
171
|
Squadrito F, Altavilla D, Squadrito G,
Ferlito M, Deodato B, Arlotta M, Minutoli L, Campo GM, Bova A,
Quartarone C, et al: Protective effects of Cyclosporin-A in
splanchnic artery occlusion shock. Br J Pharmacol. 130:339–344.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Luo MH, Luo SX, Cheng Z, Yang X, Lv D, Li
X, Guo Y, Li C and Yan J: Tubeimoside I improves survival of mice
in sepsis by inhibiting inducible nitric oxide synthase expression.
Biomed Pharmacother. 126:1100832020. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Hai-Mei L, Song-Yin H, Run-Mei L,
Xiao-Huang X, Le-Quan Z, Xiao-Ping L and Jin-Wen X: Andrographolide
protects against Lipopolysaccharide-induced vascular hyporeactivity
by suppressing the expression of inducible nitric oxide in
periaortic adipose. J Cardiovasc Pharmacol. 62:154–159. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Altavilla D, Squadrito F, Campo GM,
Squadrito G, Arlotta M, Urna G, Sardella A, Quartarone C, Saitta A
and Caputi AP: The lazaroid, U-74389G, inhibits inducible nitric
oxide synthase activity, reverses vascular failure and protects
against endotoxin shock. Eur J Pharmacol. 369:49–55. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Crankshaw DJ, Pistilli MJ, O'Brien YM,
Sweeney EM, Dockery P, Holloway AC and Morrison JJ: The effects of
extracellular calcium-sensing receptor ligands on the contractility
of pregnant human myometrium in vitro. Reprod Sci. 20:882–890.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Winter A, Nepper P, Hermann M, Bayer F,
Riess S, Salem R, Hlavicka J, Prinzing A, Hecker F, Holubec T, et
al: Glibenclamide serves as a potent vasopressor to treat
vasoplegia after cardiopulmonary bypass and reperfusion in a
porcine model. Int J Mol Sci. 26:40402025. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Warrillow S, Egi M and Bellomo R:
Randomized, double-blind, placebo-controlled crossover pilot study
of a potassium channel blocker in patients with septic shock. Crit
Care Med. 34:980–985. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Corriden R, Self T, Akong-Moore K, Nizet
V, Kellam B, Briddon SJ and Hill SJ: Adenosine-A3 receptors in
neutrophil microdomains promote the formation of bacteria-tethering
cytonemes. EMBO Rep. 14:726–732. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Yang G, Xu J, Li T, Ming J, Chen W and Liu
L: Role of V1a receptor in AVP-induced restoration of vascular
hyporeactivity and its relationship to MLCP-MLC20 phosphorylation
pathway. J Surg Res. 161:312–320. 2010. View Article : Google Scholar
|
|
180
|
Wan L, Langenberg C, Bellomo R and May CN:
Angiotensin II in experimental hyperdynamic sepsis. Crit Care.
13:R1902009. View
Article : Google Scholar : PubMed/NCBI
|
|
181
|
Tsuruoka S, Schwartz GJ, Wakaumi M,
Nishiki K, Yamamoto H, Purkerson JM and Fujimura A: Nitric oxide
production modulates cyclosporin A-Induced distal renal tubular
acidosis in the rat. J Pharmacol Exp Ther. 305:840–845. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Gui Q, Jiang Z and Zhang L: Insights into
the modulatory role of cyclosporine A and its research advances in
acute inflammation. Int Immunopharmacol. 93:1074202021. View Article : Google Scholar : PubMed/NCBI
|














