Introduction
Abdominal aortic aneurysm (AAA) is a lethal
degenerative vascular disease that mainly affects older adults and
has a high mortality rate (>80%) upon rupture (1). The features of AAA include dilation
of the aortic diameter to >30 mm or 50% (2), and repair by either open or
minimally invasive surgery is performed for large, asymptomatic AAA
or symptomatic or ruptured AAA of any size (3). Aortic rupture is not only
associated with increasing aneurysm diameter but also results from
characteristic changes that involve progressive expansion and
weakening of the three layers of the aorta: The intima, media, and
adventitia (4). Multiple
pathological processes, including extracellular matrix (ECM)
breakdown, inflammation, phenotype switching of vascular smooth
muscle cells (VSMCs), oxidative stress, and neovascularization,
contribute to the occurrence and development of AAA (5).
VSMCs are major components of the vessel wall and
perform critical roles in maintaining vascular structure and
homeostasis (6). Various changes
involving phenotype switching, apoptosis and migration of VSMCs are
important causes of AAA formation (7). In normal vasculature, VSMCs reside
in the tunica media and were quiescent and contractile (8). Once vascular injury and repair
occur, VSMCs dedifferentiate in response to pathophysiological
stimuli (9). The homeostasis of
VSMCs during AAA is disturbed, and VSMCs phenotypic
switching-mediated vascular pathology contributed to AAA formation
(10). The proteolytic enzymes
degrade the ECM, facilitating the detachment of VSMCs from the ECM
and promoting VSMCs migration (11). Therefore, identifying key
molecules involved in phenotypic switching and migration of VSMCs
may provide potential targets for AAA diagnosis and treatment.
MicroRNAs (miRNAs or miRs) are ~20-nucleotide,
single-stranded RNA molecules that target mRNA through partial
complementarity, thereby inhibiting translation or inducing mRNA
degradation (12). miRNAs belong
to a conserved class of endogenous, small, non-coding,
single-stranded RNAs (13), and
are involved in diverse cellular functions, including
proliferation, differentiation, apoptosis, migration, invasion and
angiogenesis (14,15). Furthermore, numerous miRNAs play
important roles in the occurrence and development of AAA (16), including miRNA-29b (17), miRNA-33b (18), miRNA-21 (19), miRNA-24 (20), miRNA-194 (21) and miRNA-195 (22).
Previous studies revealed that miRNA-378a-5p
(miR-378-5p) exerts broad biological effects. miR-378a-5p inhibits
colorectal cancer (CRC) cell proliferation by targeting cell
cycle-dependent protein kinase 1 (CDK1) (23), miRNA-378a-5p is also a key
mediator in regulating VSMCs proliferation and migration by
targeting the CDK1/cyclin-dependent kinase inhibitor 1A (p21)
signaling pathway (24).
However, whether miR-378a-5p is involved in the development of AAA
remains unclear.
Actin-binding LIM protein 1 (ABLIM1), which contains
four LIM domains, a coiled-coil domain, and an HP domain, belongs
to the large LIM domain protein family (25). ABLIM1 modulates actin
polymerization, which is essential for cell proliferation and
migration (26). Additionally,
ABLIM1 interacts with F-actin and colocalizes with F-actin in the
retina, suggesting that ABLIM1 may regulate the actin cytoskeleton
in the retina (27). A genetic
study involving humans revealed abnormal splicing of ABLIM1 in the
skeletal muscles of patients with myotonic dystrophy type 1 (DM1)
characterized by muscle weakness and heart defects (28). However, its role of ABLIM1 in AAA
development remains unknown.
VSMCs respond to vascular injury by regulating their
phenotypes, from quiescent cells expressing high levels of genes
encoding contractions and cytoskeletal proteins to proliferating
cells expressing high levels of genes encoding cytokines, growth
factors and the ECM. Most VSMCs marker genes, including α-smooth
muscle actin (α-SMA), calponin 1 (CNN1) and smooth muscle 22α
(SM22-α), contain multiple CArG [CC(AT-rich)6GG] elements in the
promoter-enhancer regions, and expression of these genes was
controlled by the ubiquitously expressed trans binding factor,
serum response factor (SRF) and its coactivators (29). Megakaryoblastic leukemia 1 (MKL1,
also referred to as MRTF-A) is a member of the myocardin-related
transcription factor family (30) and induces the transcription of
multiple CArG-containing smooth muscle cells (SMCs) marker genes,
including α-SMA, CNN1 and SM22-α (31). Acts as a transcriptional
co-activator of SRF, once released for nuclear translocation, MKL1
can bind and activate SRF (32).
During MKL1−/− lactation/early lactation,
differentiation defects in mammary myoepithelial cells were
observed, manifested by severe reduction or loss of gene expression
encoding SMCs restricted contractile proteins including actin,
myosin heavy chain, calmodulin 1 and tropomyosin 2 (33).
In the present study, it was aimed to clarify the
role and mechanism of miR-378a-5p in the AAA development in
vivo and in vitro. It was found that miR-378a-5p exerted
a critical protective effect against AAA and inhibited the
phenotypic switching and migration of VSMCs, by directly targeting
the ABLIM1-MKL1 pathway. The results of the present study may
assist in the early diagnosis, prevention and treatment of AAA.
Materials and methods
Human specimen collection
Human serum and aortic tissue samples were collected
at the General Hospital of Northern Theater Command (Shenyang,
China) between January 20, 2021, and January 19, 2023. Serum
samples were obtained from 40 participants, including 20 healthy
individuals (aged 40±10 years) and 20 patients diagnosed with AAA
(aged 50±10 years). During the same period, aortic tissue samples
were collected from patients diagnosed with aortic dissection (AD)
who underwent open surgical repair at the same hospital. Patients
with valvular heart disease, chronic kidney disease, autoimmune
disease, or other cardiopulmonary organic diseases were excluded.
All participants were men, and there were no significant
differences in age, diabetes mellitus, hypertension, or smoking
between the control and AAA or AD groups. Blood samples were
obtained from all participants; serum was obtained and stored at
−80°C. The present study was conducted in accordance with the World
Medical Association Code of Ethics (Declaration of Helsinki)
[approval no. Y(2021)002] and was approved by the Ethics Committee
of General Hospital of Northern Theater Command. Written informed
consent was obtained from all individual participants included in
the study. All participants consented to the use of their
surgically resected tissue samples and anonymized clinical data for
scientific research.
Animals
A total of 198 male 8-weeks-old
ApoE−/− mice, with body weights ranging from 20-
25 g, were purchased from GemPharmatech Co., Ltd. The experiment
started after 1 week of acclimatization. All mice were housed under
temperature-controlled (22±1-2°C) and specific pathogen-free
conditions on a 12/12-h day/night cycle with free access to food
and water.
All experiments were approved by the Subcommittee on
Animal Medical Research Ethics, General Hospital of Northern
Theater Command (approval no. 2022-20; Shenyang, China) and
conducted in accordance with the existing guidelines on the care
and use of laboratory animals. All animal care and experimental
protocols complied with the National Institute of Health Guide for
the Care and Use of Laboratory Animals.
Animal experiments in the present study were divided
into four parts. In the first part of the in vivo
experiments, a total of 30 ApoE−/− mice were
randomly divided into four groups: i) Angomir-negative control (NC)
+ saline (n=5); ii) Angomir-NC + Ang II (n=10); iii)
Angomir-378a-5p + saline (n=5); iv) Angomir-378a-5p + Ang II
(n=10). In the second part of the in vivo experiments, a
total of 40 ApoE−/− mice were randomly divided
into four groups: i) Antagomir-NC + saline (n=5); ii) Antagomir-NC
+ Ang II (n=15); iii) Antagomir-378a-5p + saline (n=5); iv)
Antagomir-378a-5p + Ang II (n=15). In the third part of the in
vivo experiments, a total of 60 ApoE−/− mice
were randomly divided into four groups: i) AAV-SM22-shNC + saline
(n=5); ii) AAV-SM22-short hairpin (sh)NC + Ang II (n=25); iii)
AAV-SM22-sh Ablim1 + saline (n=5); iv) AAV-SM22-shAblim1
+ Ang II (n=25). In the fourth part of the in vivo
experiments, a total of 68 ApoE−/− mice were
randomly divided into four groups: i) AAV-SM22-NC + saline (n=9);
ii) AAV-SM22-NC + Ang II (n=25); iii) AAV-SM22 +
Ablim1-saline (n=9); iv) AAV-SM22-Ablim1 + Ang II
(n=25).
To minimize animal suffering, the following
predefined humane endpoints were strictly observed, and any animal
meeting one or more criteria was euthanized immediately: i)
sustained weight loss exceeding 20% of baseline body weight within
72 h; ii) Impaired mobility or inability to access food or water
autonomously; iii) Signs of severe distress or pain unrelieved by
analgesia; iv) Ulceration, necrosis, or exceeding a tumor volume of
1.5 cm3 in any dimension in tumor-bearing models and v)
Clinical signs indicating severe systemic illness. Animals were
monitored daily, and any mouse reaching a predefined humane
endpoint was euthanized humanely by CO2 inhalation. No
animals in the present study reached the predefined humane
endpoints prior to the scheduled experimental endpoint. All animals
were euthanized at the planned conclusion of the study for tissue
collection, in accordance with the approved protocol.
Angiotensin II (Ang II)-induced AAA mice
model
ApoE−/− mice (8-week-old) were
infused with saline (0.9% sodium chloride) or Ang II (1,000
ng/kg/min) using an osmotic pump (Alzet model 1004; AlzaCorp;
https://alzet.com/products/alzet_pumps/) for 28 days.
Ang II (A1042) was purchased from APeXBIO Technology LLC. Briefly,
mice were anesthetized with 2-3% isoflurane (RWD Life Science) in
oxygen. During surgery, anesthesia was maintained with 1.5-2%
isoflurane. Minipumps were implanted into the subcutaneous space of
the mice at the back of the neck. All surgeries were performed
under aseptic conditions. Animals were monitored daily for signs of
distress.
At the experimental endpoint (Day 28), mice were
euthanized by CO2 inhalation at a flow rate displacing
30-70% of the chamber volume per min, followed by secondary
cervical dislocation. Death was confirmed by the absence of
heartbeat (via palpation), cessation of breathing, and fixed,
dilated pupils. Aortic tissues were then harvested for subsequent
analysis. The aortic diameters were measured using small animal
ultrasound (Visual Sonics) at 28 days after Ang II infusion. Aortic
tissues were harvested for RNA, protein, morphological and
histological analyses. AAA incidence was defined as an increase in
the external aortic diameter by 50% or greater than that in the
aortas from saline-infused mice.
Blood pressure measurement
Blood pressure was measured in conscious mice using
a non-invasive tail-sleeve system (CODA-6; Kent Scientific). The
mice were placed in a holding tube within a heating chamber set at
37°C (Model LE5510; Panlab). All animals were acclimatized to the
instrument for at least 1 week before baseline measurements and
osmotic pump implantation. To avoid changes in blood pressure due
to circadian cycles, all measurements were taken between 8 a.m. and
10 a.m. Each mouse received 10 initial pressure assessments so that
they can adapt to the procedure, and then 10 additional readings
were recorded to obtain average systolic and diastolic blood
pressure. The acceptable standard was to consider at least 10 of
the 20 acquired measurements and a standard deviation (SD) of
<30 mmHg for each session (34).
miRNA microarray analysis
As previously described (35), miRNA expression in mouse aortas
was analyzed by Bio-Miao Biological Technology Co., Ltd. using the
Agilent Mouse miRNA Microarray Kit, Release 21.0,8×60 K (Design ID:
070,155; Agilent Technologies, Inc.), which contained 1,902 probes
for mature miRNA (35). Tukey's
bi-weight average (log2) intensity was analyzed using the analysis
of variance (ANOVA). Differentially expressed miRNAs were defined
as those exhibiting an absolute average log2 fold change of ≥2.0
and an adjusted P<0.05.
miRNA isolation from serum and reverse
transcription-quantitative PCR (RT-qPCR)
Conditions of amplification reactions were as
follows: 95°C for 15 min, followed by 40 cycles of 95°C for 30 sec,
55°C for 1 min, and 72°C for 30 sec. Peripheral venous blood was
collected from patients into EDTA-containing tubes and centrifuged
at 3,000 × g for 10 min at 4°C, and serum was stored at −80°C.
Total RNA was extracted from 200 μl serum samples using the
miRNeasy Serum/Plasma kit (Qiagen GmbH) and cDNA was synthesized
using a reverse transcription kit (Guangzhou RiboBio Co., Ltd.)
according to the manufacturer's instructions. After equal volume
dilution of cDNA with DNase/RNase-free deionized water, the
expression levels of miRNAs were evaluated by RT-qPCR using
specific primers and the miRNA RT-qPCR kit (Guangzhou RiboBio Co.,
Ltd.) following the manufacturer's protocol. Reactions were
performed on a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad
Laboratories, Inc.). Each reaction was performed in triplicate, and
the relative expression level of miRNAs were calculated based on
cycle threshold (Ct) values using the following formula:
2-∆∆Cq (36). The
expression levels of miRNAs in the tissues and cells were
normalized to the expression levels of U6 snRNA, and those in the
serum were normalized to the expression levels of the external
reference cel-miR-39-3p.
Besides, total RNA from aortic tissues and VSMCs was
extracted using TRIzol reagent (Qiagen). The isolated RNA was used
to synthesize cDNA using PrimeScript RT with a gDNA Eraser kit
(Takara), RT-qPCR was performed in triplicate using SYBR Premix Ex
Taq II (Takara Bio, Inc.). The primer sequences are shown in
Table SI.
RNA fluorescent in-situ hybridization
(FISH)
The probe 5'-CUCCUGACUCCAGGUCCUGUGU'-3 was labeled
by FAM and Cy3 and were synthesized from the sequence of
hsa-miR-378a-5p. In situ hybridization was conducted
according to the instructions of the FISH Detection Kit (Qiagen).
In brief, 5-μm of human aorta tissue was digested with
proteinase K and incubated in a blocking buffer for 30 min (37°C).
The FAM and Cy3-labeled hsa-miR-378a-5p fluorescent probe working
solutions were prepared at a volume ratio of 1:1. The aortic tissue
slices were incubated for 14 h (37°C) using an in-situ
hybridization instrument. The sections were then washed with
deionized formamide at 43°C to denature the unhybridized probes.
The sections were washed three times with sodium citrate buffer
(60°C). FISH images were then captured by confocal microscopy.
In vivo administration of angomir-378a-5p
or antagomir-378a-5p
Angomir-378a-5p and angomiR-NC were purchased from
MedChemExpress. 8-week-old ApoE−/− mice were
injected with angomir-378a-5p or angomiR-NC via the tail vein 3
times per week for 4 weeks (20 nmol each time). Additionally,
antagomir-378a-5p and antagomir-NC were purchased from Guangzhou
RiboBio Co., Ltd. 6-week-old ApoE−/− mice were
injected with antagomir-378a-5p and antagomir-NC via the tail vein
3 times per week for 4 weeks (50 nmol each time).
AAV2/9 virus injection
Adeno-associated virus serotype 2/9 (AAV2/9)
carrying the Ablim1 coding sequence (CDS) with a
Sm22α promoter (AAV-SM22-Ablim1) or the control virus
(AAV-SM22-NC) were constructed by OBiO Technology Corp., Ltd.
6-week-old ApoE−/− mice were injected with
AAV-SM22-Ablim1 or the control virus (AAV-SM22-NC) via the
tail vein at a dosage of 5×1011 vg per mouse. A total of
3 weeks later, aortas were collected to access overexpression
performance.
To achieve VSMCs-specific knockdown of
Ablim1, adeno-associated virus serotype 2/9 (AAV2/9)
carrying mouse shAblim1 sequence with a Sm22α
promoter (AAV-SM22-shAblim1) or the control virus
(AAV-SM22-shNC) were constructed by OBiO Technology Corp., Ltd.
6-week-old ApoE−/− mice were injected with
AAV-SM22-shAblim1 or the control virus (AAV-SM22-NC) via the
tail vein at a dosage of 5×1011 vg per mouse. After 3
weeks of injection, aortas were harvested for evaluation of
Ablim1 knockdown efficiency.
Small animal ultrasonography for AAA
mice
To check the incidence of AAA, three representative
parameters including the diameters of the aorta, superior renal
artery and inferior renal artery were examined using a small animal
ultrasound system (Vevo 2100 apparatus; Visual Sonics) equipped
with a 30-MHz probe. Each mouse was anesthetized with 2% isoflurane
throughout the ultrasonographic procedure. Arterial diameters were
assessed by a blinded researcher.
Histomorphology analysis
Mice were euthanized and the whole aortas were
perfused with saline and fixed with 4% paraformaldehyde for 24 h at
room temperature. The aortas were isolated from the ascending aorta
to the entrances of both iliac arteries for macroscopic analysis.
The aortas were then segmented to obtain suprarenal abdominal
aortas. The aortic samples were harvested, fixed for 24 h, and
embedded in paraffin. Histology was examined in cross sections
(5-μm) that were taken from these aortic samples.
Paraffin-embedded sections were used for staining. Hematoxylin and
eosin (H&E) staining was used for morphological evaluation.
Sirius Red staining (Beijing Solarbio Science & Technology Co.,
Ltd.) was used to evaluate collagen deposition, and Verhoeff Van
Gieson Elastic staining (MilliporeSigma) was used to evaluate
elastin. Elastin degradation was scored as follows: 1, no
degradation and well-organized lamina; 2, mild degradation with
some interruptions or breaks in the lamina; 3, moderate degradation
with multiple interruptions or breaks in the lamina; and 4, severe
fragmentation, loss, or aortic rupture.
Immunofluorescence staining
Paraffin-embedded sections (5-μm) were
deparaffinized in xylene and rehydrated through a graded ethanol
series (100, 95, 70%) to distilled water and blocked with 5% goat
serum (cat. no. X0907; Fuzhou Maixin Biotechnology Co., Ltd.) for 1
h at room temperature. The sections were incubated with anti-ABLIM1
(1:200; cat. no. PA5-70451; Thermo Fisher Scientific, Inc.),
anti-α-SMA (1:200; cat. no. Ab7817; Abcam) primary antibodies
overnight at 4°C. After washing with phosphate-buffered saline
(PBS) 3 times, Alexa Fluor 488/594-conjugated secondary antibodies
(1:200; cat. nos. A-11012 and A-11008; Thermo Fisher Scientific,
Inc.) were applied for 1 h at 37°C in the dark. Sections were
mounted using ProLong Gold anti-fade reagent with
4',6-diamidino-2-phenylindole (DAPI; cat. no. P36931; 1
μg/ml; Thermo Fisher Scientific, Inc.) for fluorescence
microscopy (Carl Zeiss AG).
Cell culture and treatment
Primary mouse VSMCs were isolated from the freshly
dissected aortas. Briefly, the tunica media were separated by
peeling off the tunica intima from the aortic tissue in PBS. The
tunica media were cut into ~2-mm pieces and digested with 0.15%
type II collagenase (MilliporeSigma). VSMCs were seeded in 6-well
plates and cultured in fresh Dulbecco's modified Eagle's medium
(DMEM) containing 20% fetal bovine serum (FBS; Biochrom Ltd.).
Positive immunofluorescence staining of α-SMA and SM22-α were used
to confirm VSMCs. VSMCs were stimulated with different
concentrations of tumor necrosis factor α (TNFα; MilliporeSigma)
for 24 h.
RNA interference and cell
transfection
miR-378a-5p mimics, miR-378a-5p inhibitor, and
matched controls were purchased from Guangzhou RiboBio Co., Ltd.
The concentration of miR-378a-5p mimics was 50 nM and miR-378a-5p
inhibitor was 100 nM. Ablim1 and Mkl1 small interfering RNA (siRNA)
and matched controls were purchased from Shanghai GenePharma Co.,
Ltd. The concentration of siRNA was 50 nM. Mouse Ablim1
overexpression plasmid (pcDNA3.1-Flag-Ablim1), cellular
Myelocytomatosis oncogene (c-Myc) overexpression plasmid
(pcDNA3.1-Flag-c-Myc), and Mkl1 overexpression plasmid
(pcDNA3.1-His-Mkl1) were designed and constructed by the OBiO
Technology Corp., Ltd. The quality of plasmids was 1 μg/ml.
Lipofectamine 2000 (Thermo Fisher Scientific, Inc.) was mixed with
250 μl of serum- and antibiotic-free medium, and incubated
for 5 min. The siRNA or plasmids and lipofectamine 2000 solutions
were mixed and incubated at room temperature for 20 min. The
transfection mixture was added to the cells in serum-free culture
and incubated for 8 h and then replaced with normal serum and
antibiotic-containing growth medium. The cells were incubated for
48 h before collection for testing. Sequences of miRNA mimic, miRNA
inhibitor, and small interfering RNA are included in Table SII. It was first demonstrated
that VSMCs could be successfully transfected with both siRNAs and
overexpression plasmids, which resulted in effective gene silencing
and overexpression of the targets (Fig. S1).
Western blotting
Cells and tissues were homogenized in ice-cold
suspension buffer (RIPA Lysis Buffer) supplemented with a
proteinase inhibitor cocktail (MilliporeSigma). Briefly, protein
concentrations were determined using a BCA protein assay kit
(Thermo Fisher Scientific, Inc.). Equal amounts (20 μg) of
protein were fractionated on SDS polyacrylamide gels and
transferred to a polyvinylidene fluoride (PVDF) membrane. The
concentration of the acrylamide gel was chosen based on the size of
the target proteins. Usually, proteins with a size range from 10 to
30, 30 to 100, and >100 kD are separated on 12, 10 and 8% gels,
respectively. After blocking with 5 % non-fat milk at room
temperature for 1 h, the membrane was incubated with primary
antibodies at 4°C overnight. The membranes followed by
immunoblotting with the primary antibodies: anti-α-SMA (1:1,000;
cat. no. Ab7817; Abcam), anti-SM22-α (1:1,000; cat. no. Ab14016;
Abcam), anti-CNN1 (1:1,000; cat. no. Ab46794; Abcam), anti-MMP2
(1:1,000; cat. no. Ab92536; Abcam), anti-ABLIM1 (1:1,000; cat. no.
PA5-70451; Thermo Fisher Scientific, Inc.), anti-MKL1 (1:1,000;
cat. no. Ab219981; Abcam), anti-Flag (1:1,000; cat. no. sc-166355;
Santa Cruz Biotechnology, Inc.), anti-His (1:1,000; cat. no.
66005-1-Ig; Proteintech Group, Inc.), anti-c-MYC (1:1,000;. cat.
no. 10828-1-AP; Proteintech Group, Inc.), anti-MYOD1 (1:1,000; cat.
no. 18943-1-AP; Abcam) and anti-β-actin (1:100; cat. no. sc-517582;
Santa Cruz Biotechnology, Inc.). The membranes were then incubated
with mouse or rabbit appropriate peroxidase-conjugated secondary
antibody (1:1,000; cat. no. 32460; Thermo Fisher Scientific, Inc.)
for 1 h at room temperature. Specific protein bands were detected
using ECL detection reagent (Beijing New Create Life Science
Biotechnology Co., Ltd.) and specific protein bands were visualized
by enhanced chemiluminescence using Amersham Imager 680
(Cytiva).
Transwell assay
The cells in each group were digested using trypsin,
and were resuspended using DMEM without serum to adjust the cell
density to 1×105 cells/ml, and 300 μl of cell
suspension was added to the upper compartment of the 0.8-μm
Transwell chamber (Corning, Inc.); meanwhile, 700 μl of DMEM
containing 10% FBS was added to the bottom compartment and the
cells were cultured at 37°C, 5% CO2 for 24 h. After the
chamber was removed, the cells on the bottom of the membrane were
fixed with 4% paraformaldehyde for 15 min at room temperature,
stained with 0.1% crystal violet solution for 15 min, and the
remaining crystal violet solution was washed off using PBS; the
cells in the upper chamber were cleaned using a cotton swab. A
total of five fields of the membrane were used to count the number
of cells using an optical microscope, and the average was
calculated to indicate the migration ability of the VSMCs.
Scratch wound healing assay
VSMCs were cultured in six-well plates. A scratch
was made using 10-μl pipette tips. After washing to remove
cell debris, the medium was replaced with FBS-free DMEM and
incubated for 24 h at 37°C. Migratory cells were visualized using a
phase-contrast microscope (Olympus Corporation).
Luciferase assay
The 3' untranslated region (3'UTR) of mouse
Ablim1, Ddx5 (Dead-box helicase 5),
Slc7a1(Cationic amino acid transporter 1) gene was amplified
and cloned into the pMIR-REPORT Luciferase (OBiO Technology
Company) to construct pMIR-REPORT Luciferase 3'UTR wild-type (WT)
plasmids. A mutation was introduced at the seed sequence of
miR-378a-5p to create pMIR-REPORT Luciferase-3'UTR mutated (MUT)
plasmids. 293T cells were transfected with miR-378a-5p mimic or
control (50 nM), together with pMIR-REPORT Luciferase-3'UTR (WT)
(1,000 ng) or pMIR-REPORT Luciferase-3'UTR (MUT) (1,000 ng) and pRL
(Renilla)-CMV (500 ng) for 48 h. Transfection was performed
with Lipofectamine 2000. At 48 h after transfection, luciferase
activity was measured using a dual-luciferase analysis system kit
(Promega Corporation). The ratio of luciferase
activity/Renilla Luciferase activity was normalized to the
control and presented as the relative transcriptional activity.
Bioinformatic analysis
JASPAR (https://jaspar.elixir.no/) and TRANSMIR (http://cmbi.bjmu.edu.cn/transmir) were used to
predict the upstream transcription factors of miR-378a-5p.
TargetScan7.0 (http://www.targetscan.org/) and miRDB (http://www.mirdb.org) databases were used to predict
the target genes of miR-378a-5p. Finally, intersecting genes were
obtained from the two databases for subsequent analysis.
A total of 3 datasets of AAA and control aortic
samples were obtained from the Gene Expression Omnibus (GEO)
database (https://www.ncbi.nlm.nih.gov/geo/). GSE183464 is an
RNA sequencing analysis of abdominal aorta tissues from 14
participants, including 7 patients with AAA and 7 control
individuals. GSE237229 is an RNA sequencing dataset of human aortic
SMCs isolated from 5 patients with AAA and 3 non-AAA donors. The
human thoracic aortic aneurysm (TAA) single-cell RNA sequence
(scRNA-seq) dataset (GSE155468) was also downloaded from the GEO
database.
Co-immunoprecipitation (Co-IP)
Co-IP was performed to validate the interaction
between ABLIM1 and MKL1. Briefly, 293T cells were transfected with
pcDNA3.1-Flag-Ablim1 and pcDNA3.1-His-Mkl1 for 48 h
and lysed in 1 ml IP lysis containing protease and phosphatase
inhibitors. The cell lysate was collected and centrifuged at 12,000
× g for 15 min at 4°C, followed by incubation with 50 μl
magnetic beads (MBL International Co.) as suggested at 4°C for 1 h.
The tube was placed on a magnetic rack for a few sec, and the
supernatant was removed. A total of 1 ml of cold wash buffer was
added, and the magnetic beads were resuspended 3 times. The
magnetic beads were resuspended in the loading buffer and heated
for 5 min, and the tube was placed on a magnetic rack for a few
sec. Total or separate cell contents and immunoprecipitants were
separated using SDS-PAGE gels. Consistent with the aforementioned
description, the concentration of the acrylamide gel was chosen
based on the size of the target proteins. The proteins were
transferred onto PVDF membranes and incubated with the
corresponding primary antibodies at 4°C overnight. The primary
antibodies were as follows: anti-FLAG (1:1,000; cat. no. sc-166355;
Santa Cruz Biotechnology, Inc.), anti-HIS (1:1,000; cat. no.
66005-1-Ig; Proteintech Group, Inc.), anti-ADM (1:1,000; cat. no.
Ab190819; Abcam), anti-LMOD1 (1:1,000; cat. no. 15117-1-AP;
Proteintech Group, Inc.) and anti-PRKCD (1:100; cat. no. sc-365969;
Santa Cruz Biotechnology, Inc.). HRP-conjugated secondary
antibodies were added, and the proteins were examined using a
chemiluminescence imaging system.
LC-MS/MS analysis
ABLIM1 antibody was added to 293T lysates for IP.
Rabbit IgG was used as a negative control. Then, the LC-MS/MS
analysis was carried out by PTM Bio Co., Ltd. Finally, the
substrate proteins that could bind to ABLIM1 were screened out
according to the score and the mass of detected proteins.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. P<0.05 was considered to indicate a statistically
significant difference. Statistical analyses were performed using
GraphPad Prism 9.3 (GraphPad Software Inc.; Dotmatics). For
statistical comparisons, it was first evaluated whether the data
were normally distributed using the Shapiro-Wilk normality test.
Non-parametric tests were used when data were not normally
distributed. Normality tests were performed using Shapiro-Wilk
statistics. Differences between the two groups were compared using
the unpaired Student's t-test. Differences between three or more
groups were compared using one-way ANOVA, followed by Tukey's
post-hoc test for multiple pairwise comparisons. The Kaplan-Meier
survival curve was used to analyze the survival percentage of
saline- or Ang II-infused mice.
Results
miR-378a-5p expression is reduced during
the formation of AAA
To establish a mouse AAA model, 8-week-old
ApoE−/− mice were infused with Ang II via an osmotic
pump for 28 days. Mice infused with saline served as the control
group. To identify miRNAs associated with AAA, the miRNA array was
performed to determine the differentially expressed miRNAs in the
aortas of AAA and control mice. Compared with the control group, 64
differentially expressed miRNAs were found in the aortas of AAA
group mice, of which 27 miRNAs were upregulated, and 37 miRNAs were
downregulated. The heatmap illustrated the top 10 miRNAs that
exhibited the most significant decrease, with those highlighted in
red indicating a high degree of homology between human and mice
(miR-200b-3p, miR-200c-3p, miR-342-5p, miR-149-5p, miR-150-3p and
miR-378a-5p) (Figs. 1A and B and
S2A). The results of RT-qPCR
demonstrated that miR-200b-3p, miR-200c-3p, miR-342-5p, miR-150-3p,
and miR-378a-5p were significantly decreased in the aortas of AAA
mice compared with control mice (Fig. 1C). To further clarify whether
these miRNAs played a role in AAA, serum samples were collected
from 20 patients with AAA and 20 healthy individuals. Compared with
normal group, the expression of miR-378a-5p in the serum levels of
patients with AAA were reduced; however, the expression levels of
miR-200b-3p, miR-200c-3p, miR-342-5p and miR-150-3p were unchanged
between the two groups (Fig.
1D). Furthermore, compared with the saline group, circulating
miR-378a-5p level was significantly reduced in the serum of AAA
mice (Fig. 1E).
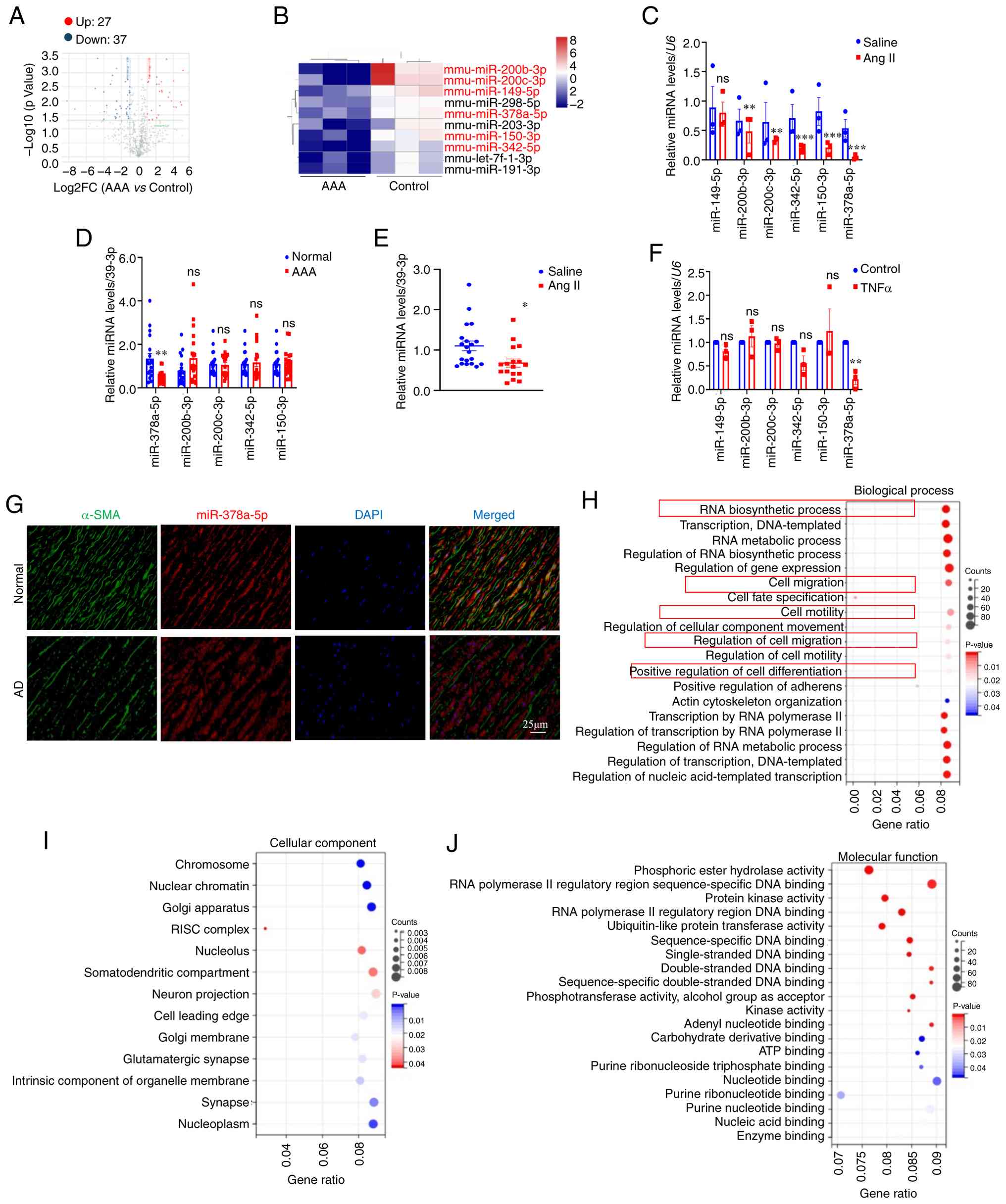 | Figure 1miR-378a-5p is downregulated in mice
with Ang II-induced AAA and TNFα-induced VSMCs. (A) Volcano plot of
64 differential miRNA expression in the aortas of Ang II and
saline-treated mice. (B) Heatmap of differentially expressed miRNAs
in mice treated with Ang II compared with the saline group,
|log2(Fold change)|≥2, P<0.05. Blue indicated low relative
expression, while red indicated high relative expression. (C)
RT-qPCR analysis of miRNAs in the aortas of Ang II and
saline-treated mice (n=3 per group). (D) RT-qPCR analysis of miRNAs
in the serum of patients with AAA and normal individuals (n=20 per
group). (E) RT-qPCR analysis of miRNAs in the serum of Ang II and
saline-treated mice (n=20 per group). (F) RT-qPCR analysis of
miRNAs in the TNFα-treated VSMCs (n=3 per group). (G) Fluorescence
in situ hybridization was performed to detect the
miR-378a-5p expression in human aorta tissues. (H) The
GO-Biological Process signaling pathways of the target genes of
miR-378a-5p were analyzed using DIANA TOOLS-miRPath algorithm. (I)
The GO-Cellular Component signaling pathways of the target genes of
miR-378a-5p were analyzed using DIANA TOOLS-miRPath algorithm. (J)
The GO-Molecular Function signaling pathways of the target genes of
miR-378a-5p were analyzed using DIANA TOOLS-miRPath algorithm. Data
are presented as the mean±SEM. P-values were calculated by
Student's t test. *P<0.05, **P<0.01 and
***P<0.001 vs. saline, Normal or Control. miR or
miRNA, microRNA; Ang II, angiotensin-II; AAA, abdominal aortic
aneurysm; VSMCs, vascular smooth muscle cells; RT-qPCR, reverse
transcription-quantitative PCR; GO, Gene Ontology; ns, not
significant. |
VSMCs were isolated and identified by
immunofluorescence staining using SM22-α and α-SMA (Fig. S2B). TNFα is a well-established
direct suppressor of VSMCs contractile phenotype, rapidly
downregulating contractile genes and impairing function (37). To simulate the pathological
process of AAA in vitro, TNFα was used to stimulate VSMCs.
As shown in Fig. S2C and D,
TNFα inhibited the protein expression levels of VSMCs
differentiation markers (α-SMA, CNN1 and SM22-α) and increased the
protein level of metal matrix degrading enzyme 2 (MMP2) in a dose
dependent manner. TNFα also significantly decreased miR-378a-5p
levels in VSMCs. However, the levels of miR-149-5P, miR-200b-3p,
miR-200c-3p, miR-342-5p and miR-150-3p were unchanged in
TNFα-treated VSMCs compared with control group (Fig. 1F). Besides, miR-378a-5p
fluorescence was also decreased in human AD aortas by FISH analysis
(Fig. 1G).
To identify the biological functions of miR-378a-5p
and its target genes in AAA pathogenesis, miRNA-associated
signaling pathways were analyzed using the DIANA TOOLS-miRPath
algorithm (http://www.microrna.gr/miRPathv4) (38). For the target genes of
miR-378-5p, most biological processes enriched in cell migration,
differentiation, RNA synthesis and degradation (Fig. 1H-J). These findings suggested
that miR-378a-5p may play a critical role in AAA pathogenesis.
miR-378a-5p is regulated by c-MYC in
AAA
To clarify why miR-378a-5p was downregulated in AAA
formation, two transcription factor prediction websites, JASPAR and
TRANSMIR, were used to predict the transcription factors of
miR-378a-5p. Two possible transcription factors were identified:
c-MYC and myogenic differentiation 1 (MYOD1) (Fig. S2E). Next, upregulation of c-MYC
protein expression was detected in TNFα-treated VSMCs, but the
expression levels of MYOD1 had no significant changes (Fig. S2F). Besides, the increase of
c-MYC was also detected in human AD tissues (Fig. S2G). Luciferase assays revealed
that c-MYC-dependent miR-378a-5p suppression was maintained upon
transfection with a luciferase vector containing the miR-378a-5p
promoter, and this inhibitory effect was reversed by mutations in
the miR-378a-5p promoter region (Fig. S2H). C-MYC knockdown resulted in
a significant increase in miR-378a-5p levels in VSMCs, whereas
c-MYC overexpression decreased miR-378a-5p levels in VSMCs
(Fig. S2I and J). These results
indicated that c-MYC directly bound to the promoter of miR-378a-5p
and negatively regulated miR-378a-5p expression in VSMCs.
Overexpression of miR-378a-5p prevents
Ang II-induced AAA formation
ApoE−/− mice were injected with
angomir-NC or angomir-378a-5p for 4 weeks through tail vein before
AAA modeling (Fig. S3A). No
difference in body weight was observed between all the groups
(Fig. S3B). Blood pressure
increased similarly upon Ang II infusion in both angomir-NC and
angomir-378a-5p-injected mice (Fig.
S3C and D). Compared with the angomir-NC group, the expression
of miR-378a-5p was significantly increased in the aortas of the
angomir-378a-5p group (Fig.
S3E).
In the presence of Ang II, mice in the angomir-NC
and angomir-378a-5p groups developed aortic dilations and
aneurysms, which were mitigated in the angomir-378a-5p group
(Fig. 2A). miR-378a-5p
overexpression blunted the AAA incidence induced by Ang II
(Fig. 2B); however, there was no
significant difference in the survival rates among all groups
(Fig. S3F). All images of the
abdominal aortic specimens were displayed (Fig. S3G). Compared with the angomir-NC
group, miR-378a-5p overexpression inhibited the aortic enlargement
at 28 days post-Ang II infusion (Fig. 2C and D). H&E staining
indicated the alleviated aortic dilatation in angomir-378a-5p mice
in response to Ang II. Concomitantly, the collagen deposition and
media degeneration were significantly reduced in angomir-378a-5p
mice in response to Ang II (Fig.
2E-G). The results of western blotting demonstrated that Ang II
reduced the protein expression levels of contractile markers
(α-SMA, CNN1 and SM22-α) and elevated protein expression of MMP2 in
the aortas, indicating that VSMCs dedifferentiation occurred in AAA
formation, and miR-378a-5p overexpression could inhibit the
dedifferentiation of VSMCs (Fig. 2H
and I). These results indicated that miR-378a-5p overexpression
effectively alleviates Ang II-induced AAA formation.
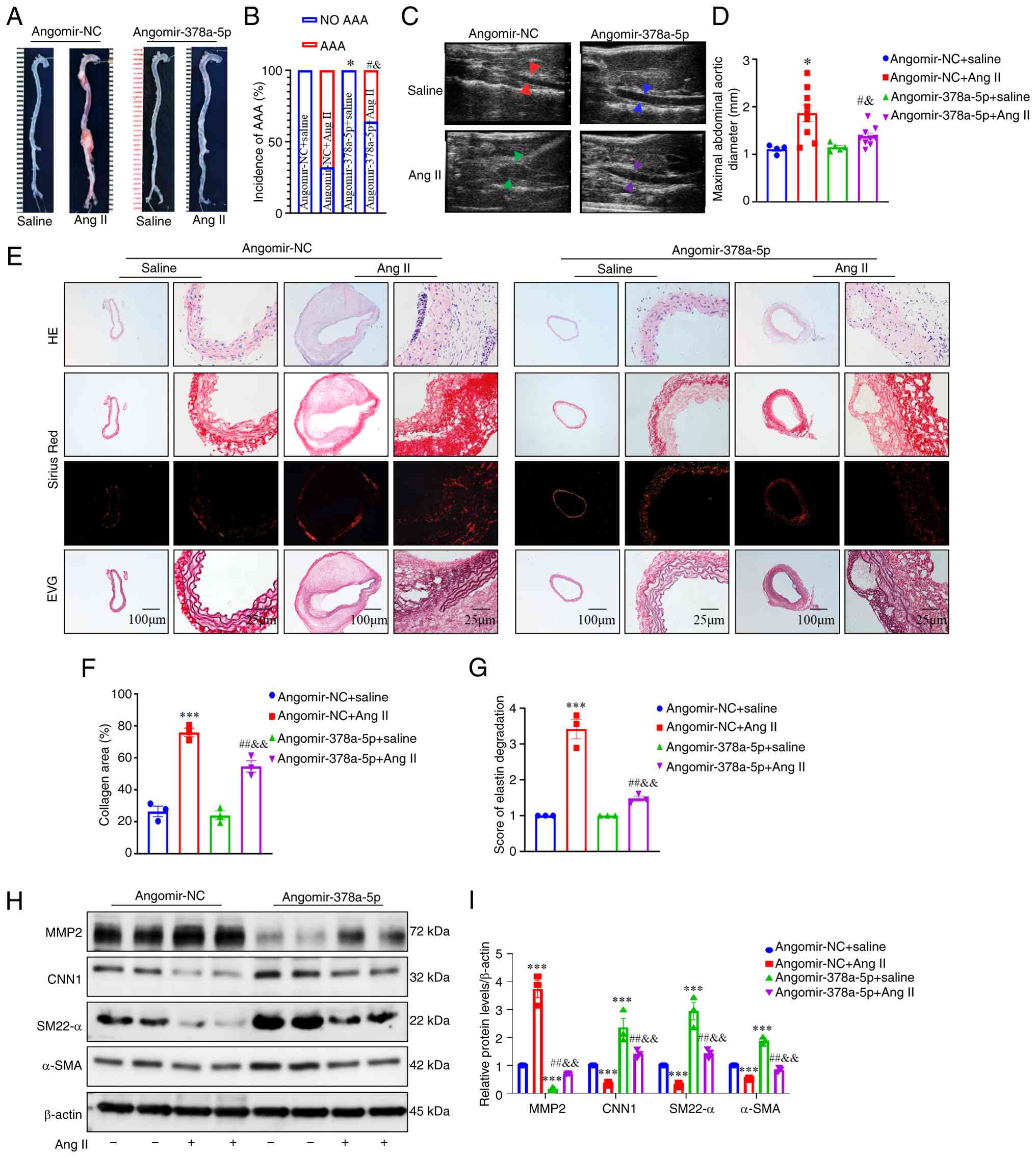 | Figure 2Overexpression of miR-378a-5p
prevents Ang II-induced AAA formation in apolipoprotein E-deficient
mice. (A) Gross specimen image of aortas from AAA models treated
with angomir-NC or angomir-378a-5p, followed by Ang II infusion.
(B) The incidence of AAA in Ang II-infused mice treated with
angomir-NC or angomir-378a-5p (n=10 in Ang II groups and n=5 in
saline groups). (C and D) Ultrasound images and inner diameter
quantification of the suprarenal abdominal aorta (n=10 in Ang II
groups and n=5 in saline groups). (E) Representative H&E,
Sirius red and EVG staining of the abdominal aorta in different
groups. (F) Quantification of fibrosis in aorta tissues (n=3 per
group). (G) Quantification of the degree of elastic fiber
degradation levels in the abdominal aortic wall (n=3 per group). (H
and I) Representative western blots and quantification of the
protein levels of MMP2, CNN1, α-SMA and SM22-α in aortas from AAA
models treated with angomir-NC or angomir-378a-5p (n=3 per group).
Data are presented as the mean ± SEM. P-values were calculated by
Student's t test (for C), two-way ANOVA with Holm-Sidak multiple
comparisons test (for F, G and I), AAA incidence was analyzed with
the Fisher exact test (for B). *P<0.05 and
***P<0.001 vs. angomir-NC + saline;
#P<0.05 and ##P<0.01 vs. angomir-NC +
Ang II; &P<0.05 and
&&P<0.01 vs. angomir-378a-5p. miR or miRNA,
microRNA; Ang II, angiotensin-II; AAA, abdominal aortic aneurysm;
NC, negative control; EVG, Verhoeff-Van Gieson; α-SMA, α-smooth
muscle actin; SM22-α, smooth muscle 22α; MMP2, matrix
metalloproteinase 2; CNN1, calponin 1. |
Knockdown of miR-378a-5p aggravates Ang
II-induced AAA formation
Given the aforementioned data, male
ApoE−/− mice were injected with a locked nucleic
acid-modified antagomir-378a-5p or a scrambled mir control
(antagomir-NC). At 4 weeks after the injection, the mice were
treated with Ang II or saline for an additional 4 weeks (Fig. S4A). Body weight remained
consistent across all groups (Fig.
S4B). Ang II treatment group had a significant increase in
blood pressure, but there was no significant difference between the
antagomir-378a-5p + Ang II and antagomir-NC + Ang II groups
(Fig. S3C and D).
Antagomir-378a-5p significantly reduced the expression of
miR-378a-5p in the aortas (Fig.
S4E). The incidence of AAA in the antagomir-NC + Ang II group
was 60%, and the incidence of AAA increased by 20% in
antagomir-378a-5p + Ang II group (Fig. S4F). There was no significant
difference in the survival rate or incidence of AAA between
antagomir-NC + Ang II and antagomir-378a-5p + Ang II groups
(Fig. S4G).
All images of the abdominal aortic specimens were
displayed in Fig. S4H. Mice
with antagomir-378a-5p treatment were susceptible to aortic
dilation and exhibited severe aneurysms after Ang II treatment
(Fig. 3A). Ultrasound images
revealed that the maximum abdominal aortic diameter was increased
by antagomir-378a-5p injection (Fig.
3B and C). Compared with the antagomir-NC + Ang II group,
increased collagen disruption and elastin degradation were observed
in the abdominal aortas of the antagomir-378a-5p + Ang II group
(Fig. 3D-F).
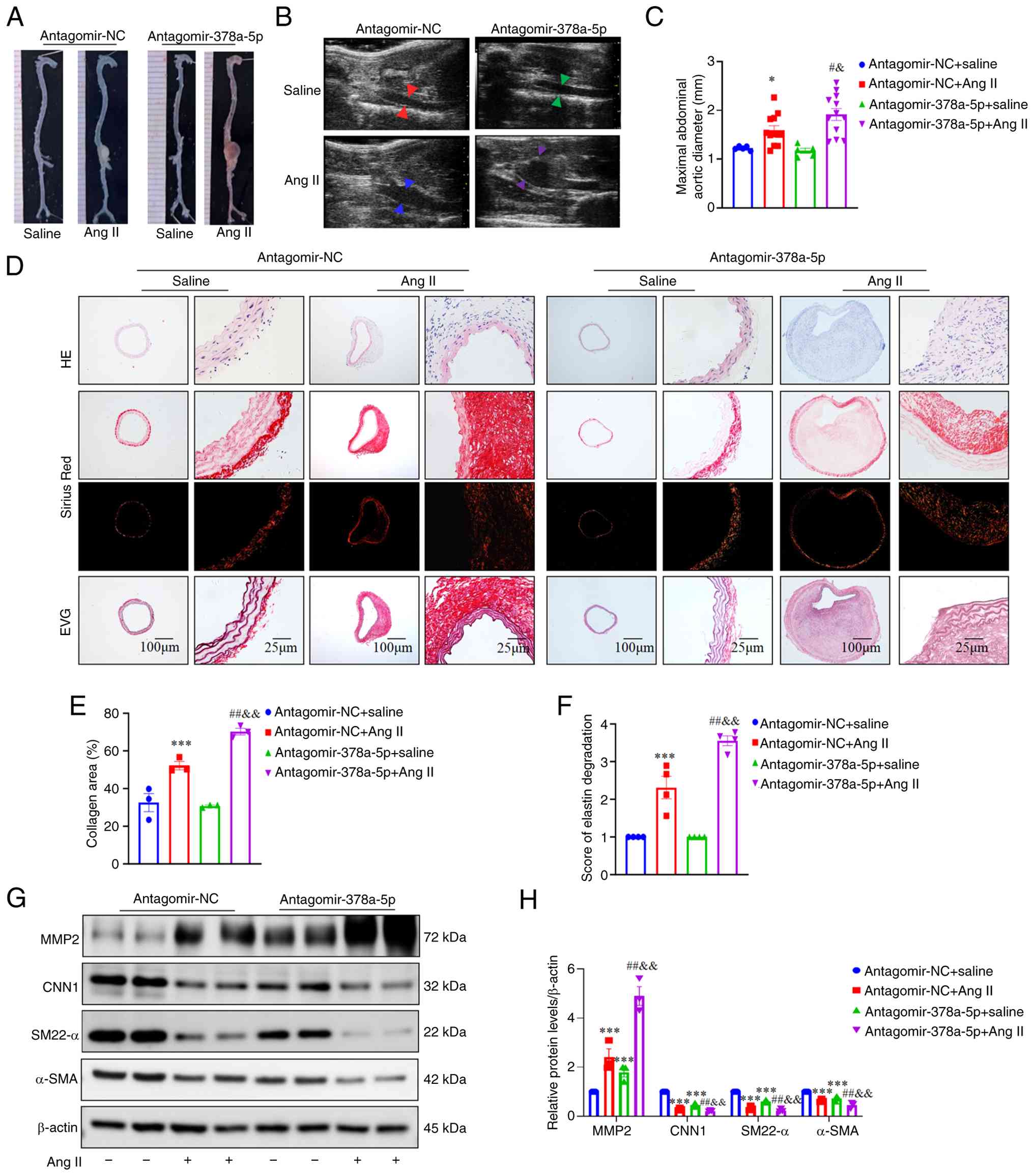 | Figure 3Knockdown of miR-378a-5p promotes AAA
formation in Ang II-infused apolipoprotein E-deficient mice. (A)
Gross specimen image of aortas from AAA models treated with
antagomir-NC or antagomir-378a-5p. (B and C) Ultrasound images and
inner diameter quantification of the suprarenal abdominal aorta
(n=15 in Ang II groups and n=5 in saline groups). (D)
Representative H&E, Sirius red and EVG staining of the
abdominal aorta in different groups. (E) Quantification of fibrosis
in aorta tissues (n=3 per group). (F) Quantification of the degree
of elastic fiber degradation levels in the abdominal aortic wall
(n=4 per group). (G and H) Representative western blots and
quantification of the protein expression levels of MMP2, CNN1,
α-SMA and SM22-α in aortas from AAA models treated with
antagomir-NC or antagomir-378a-5p (n=3 per group). Data are
presented as the mean ± SEM. P-values were calculated by Student's
t test (for C), two-way ANOVA with Holm-Sidak multiple comparisons
test (for E, F and H), *P<0.05 and
***P<0.001 vs. antagomir-NC + saline;
#P<0.05 and ##P<0.01 vs. antagomir-NC +
Ang II; &P<0.05 and
&&P<0.01 vs. antagomir-378a-5p. miR or miRNA,
microRNA; Ang II, angiotensin-II; AAA, abdominal aortic aneurysm;
NC, negative control; EVG, Verhoeff-Van Gieson; α-SMA, α-smooth
muscle actin; SM22-α, smooth muscle 22α; MMP2, matrix
metalloproteinase 2; CNN1, calponin 1. |
Western blot demonstrated that the protein
expression levels of α-SMA, CNN1 and SM22-α were significnalty
downregulated in the antagomir-378a-5p + Ang II group compared with
the antagomir-NC + Ang II group, while the protein expression of
MMP2 was significantly higher in the antagomir-378a-5p + Ang II
group (Fig. 3G and H). These
results revealed that the inhibition of miR-378a-5p aggravated Ang
II-induced AAA.
Overexpression of miR-378a-5p preserves
the VSMCs contractile phenotype in vitro
The loss of VSMCs in the medial layer of the aortic
wall is an early hallmark of AAA development (39). To further evaluate the cellular
effects of miR-378a-5p on VSMCs function, miR-378a-5p mimics were
transfected into mouse primary VSMCs. miR-378a-5p mimics
significantly increased the expression of miR-378a-5p in VSMCs
(Fig. S5A).
TNFα decreased the protein expression of the
contractile genes including α-SMA, SM22-α and CNN1 in VSMCs, and
increased the protein expression of MMP2. With or without TNFα
stimulation, miR-378a-5p overexpression significantly increased the
protein expression of VSMCs contractile markers, and decreased MMP2
protein expression (Fig. 4A).
The results of RT-qPCR were consistent with those of western
blotting (Fig. 4B).
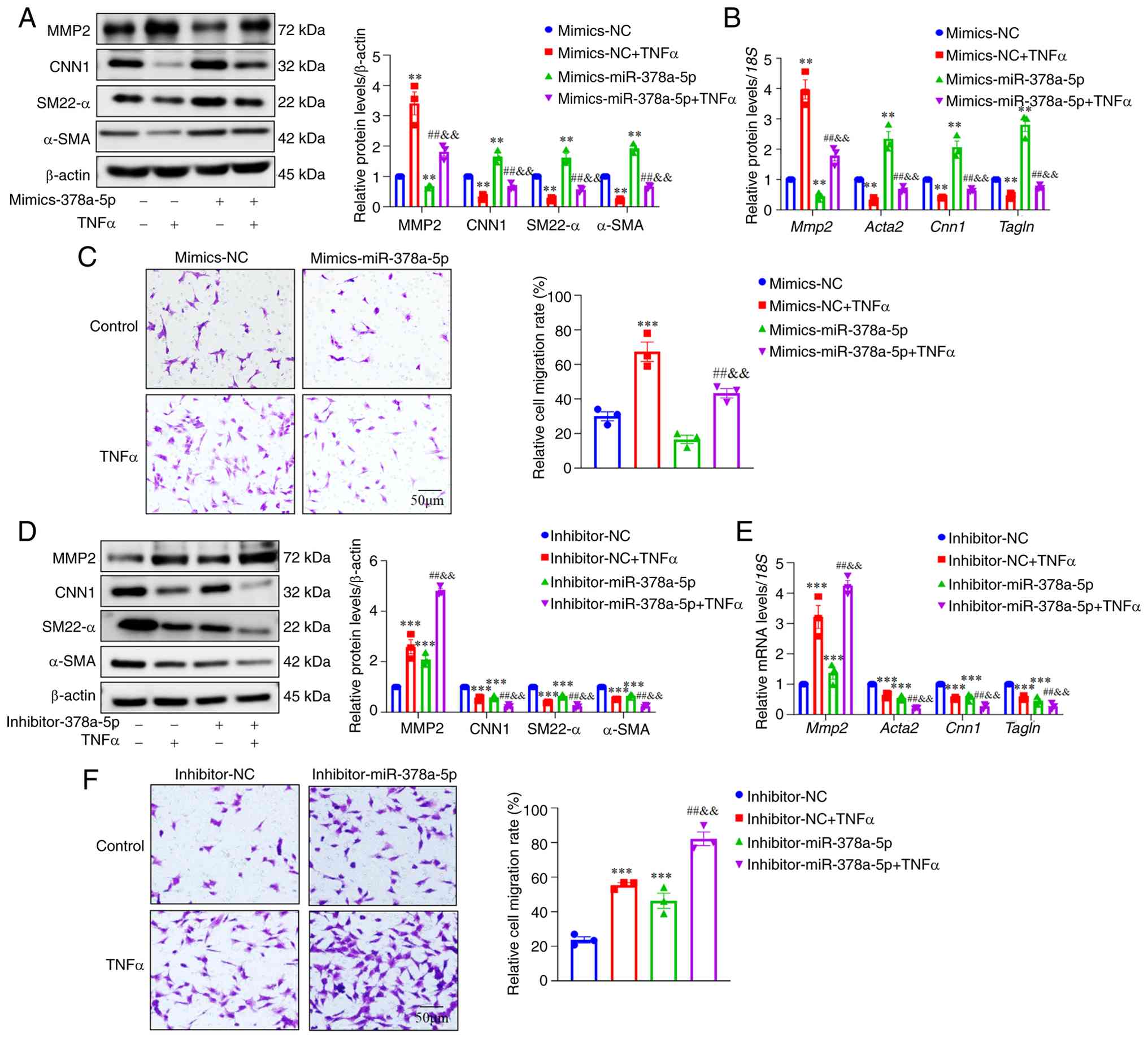 | Figure 4miR-378a-5p inhibitor promotes VSMCs'
phenotypic transformation and migration, while miR-378a-5p mimics
inhibits VSMCs' phenotypic transformation and migration. (A) VSMCs
were transfected with mimics-miR-378a-5p or mimics-NC for 24 h,
followed by TNFα treatment (10 ng/ml) for additional 24 h.
Representative western blots and quantification of MMP2, CNN1,
α-SMA and SM22-α were shown. (B) The mRNA expression of
Mmp2, Cnn1, Acta2 and Tagln was
determined by RT-qPCR. (C) Cell migration was assessed using a
Transwell assay in mimics-miR-378a-5p or mimics-NC
transfected-VSMCs. (D) VSMCs were transfected with
inhibitor-miR-378a-5p or inhibitor-NC for 24 h, followed by TNFα
treatment (10 ng/ml) for additional 24 h. Representative western
blots and quantification of MMP2, CNN1, α-SMA and SM22-α were
shown. (E) The mRNA expression of Mmp2, Cnn1,
Acta2 and Tagln was determined by RT-qPCR. (F) Cell
migration was assessed using a Transwell assay in
inhibitor-miR-378a-5p and inhibitor-NC transfected-VSMCs. Data are
presented as the mean ± SEM (n=3 per group). P-values were
calculated by two-way ANOVA with Holm-Sidak multiple comparisons
test. **P<0.01 and ***P<0.001 vs.
mimics-NC or inhibitor-NC; ##P<0.01 vs. mimics-NC +
TNFα or inhibitor-NC + TNFα; &&P<0.01 vs.
mimics-miR-378a-5p or inhibitor-miR-378a-5p. miR or miRNA,
microRNA; VSMCs, vascular smooth muscle cells; α-SMA, α-smooth
muscle actin; SM22-α, smooth muscle 22α; MMP2, matrix
metalloproteinase 2; CNN1, calponin 1; RT-qPCR, reverse
transcription-quantitative PCR; NC, negative control. |
The balance between contractile and synthetic VSMCs
shifts toward synthetic VSMCs, facilitating the detachment of VSMCs
from the ECM and promoting VSMCs' migration (40). Therefore, it was also examined
whether miR-378a-5p influenced VSMCs' migration. miR-378a-5p
overexpression could inhibit migration of VSMCs induced by TNFα
stimulation (Figs. 4C and
S5B).
miR-378a-5p inhibitor destroys the VSMCs'
contractile phenotype in vitro
To further evaluate the effects of miR-378a-5p
inhibitor on VSMCs function, VSMCs were transfected with
miR-378a-5p inhibitors. miR-378a-5p inhibitor significantly reduced
the expression of miR-378a-5p in VSMCs (Fig. S5A).
With or without TNFα stimulation, the miR-378a-5p s
inhibitor significantly reduced the mRNA and protein expression of
VSMCs' contractile markers (α-SMA, SM22-α and CNN1) and increased
the mRNA and protein expression of MMP2 (Fig. 4D and E). Transwell and wound
healing assays confirmed that miR-378a-5p inhibitor promoted the
migration of VSMCs induced by TNFα (Figs. 4F and S5C).
ABLIM1 is predicted as a target gene of
miR-378a-5p
To elucidate the molecular mechanism by which
miR-378a-5p regulated VSMCs' phenotypic transformation, the
predicted target candidates of miR-378a-5p were screened in
silico. The target genes of miR-378a-5p were predicted using
miRDB and starBase. Moreover, two datasets of AAA and control
aortic samples were obtained from the GEO database (GSE183464 and
GSE237229). GEO2R was used to analyze differentially expressed
genes (DEGs) in datasets, and 3,001 DEGs were revealed with the
cut-off criterion of adjusted P≤0.05 and |log2 (fold change)≥1,
containing 1,473 upregulated and 1,528 downregulated genes in
GSE183464. There were 1,009 DEGs in GSE237229, which included 536
upregulated and 473 downregulated genes. A volcano plot was used to
depict the expression patterns of DEGs in the dataset (Fig. 5A). The intersection of DEGs and
predicted target genes are presented in a Venn diagram, which
showed three intersecting genes: ABLIM1, DDX5 and
SLC7A1 (Fig. 5A).
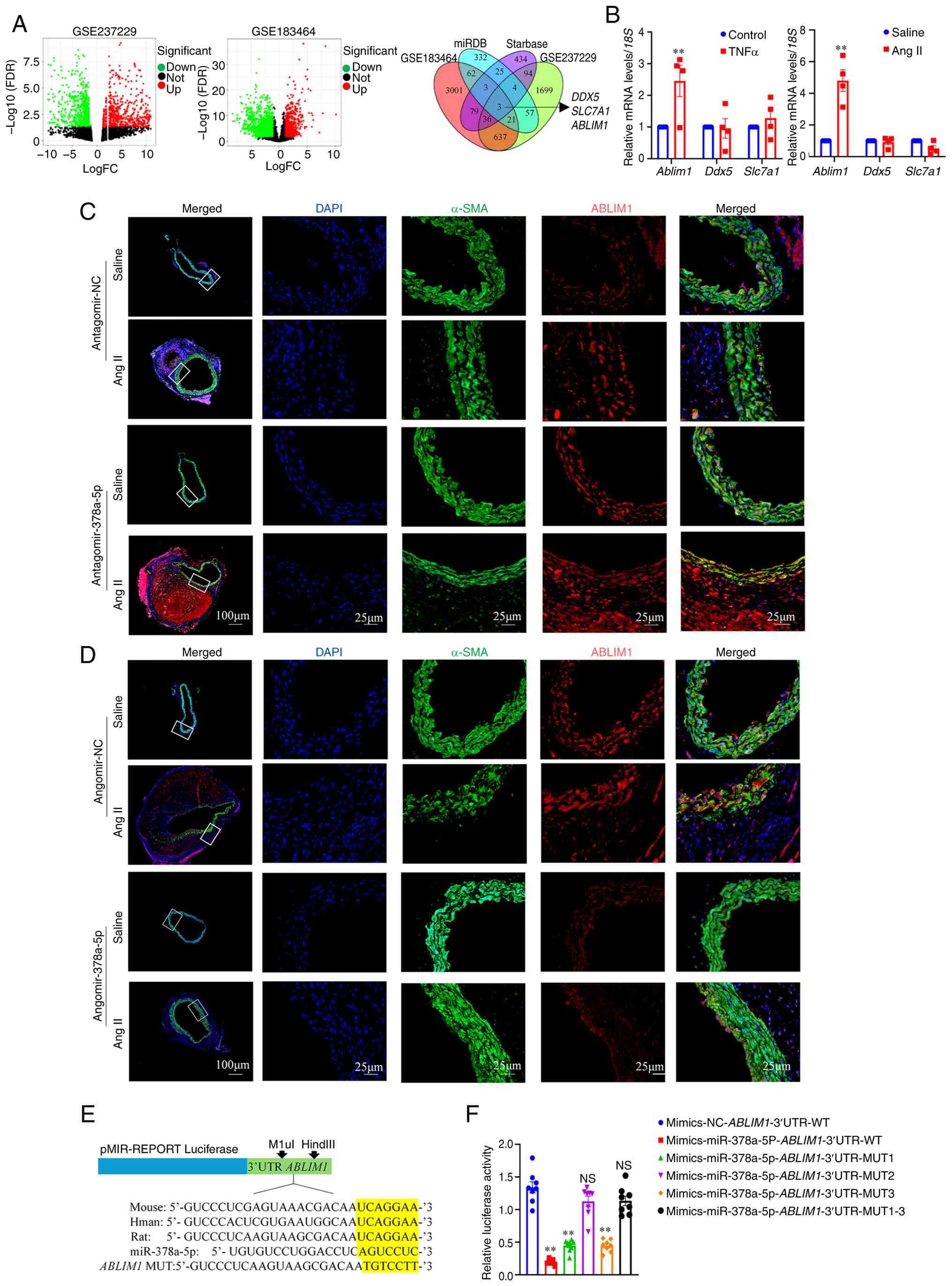 | Figure 5ABLIM1 is a downstream target of
miR-378a-5p and involves in abdominal aortic aneurysm development.
(A) Volcano plot of differently expressed genes in GSE183464 and
GSE237229 database. Venn diagram showed intersection of
differentially expressed genes and predicted target genes including
ABLIM1, DDX5 and SLC7A1. (B) RT-qPCR analysis
of target genes Ablim1, Ddx5 and Slc7a1 in the
TNFα-treated vascular smooth muscle cells (n=3 per group). RT-qPCR
analysis of Ablim1, Ddx5 and Slc7a1 in the
aortas of Ang II-treated mice (n=3 per group). (C) ABLIM1
expression in the aortas treated-with antagomir-NC or
antagomir-378a-5p was identified using by immunofluorescence
staining. ABLIM1 (red), α-SMA (green) and DAPI (blue). (D)
Representative images of immunofluorescence staining identified
ABLIM1 expression in aortas with angomir-NC or angomir-378a-5p.
ABLIM1 (red), α-SMA (green) and DAPI (blue). (E) Conservatism
analysis of the binding site for miR-378a-5p and ABLIM1 in in
humans, mice and rat. (F) Luciferase activity in 293T cells
transfected with mimics-miR-378a-5p together with
ABLIM1-3'UTR-wild type or mutant plasmid. Data are presented
as the mean ± SEM. P-values were calculated by Student's t test
(for B). **P<0.01 vs. Control or saline or
mimics-NC-ABLIM1-3'UTR-WT. ABLIM1, actin-binding LIM protein
1; miR or miRNA, microRNA; RT-qPCR, reverse
transcription-quantitative PCR; α-SMA, α-smooth muscle actin; NC,
negative control; UTR, untranslated region; WT, wild-type; MUT,
mutated. |
Furthermore, the mRNA levels of three intersecting
genes were detected in TNFα-treated VSMCs. The results identified
that the transcription level of Ablim1 was significantly
increased in VSMCs after TNFα stimulation, and no change in the
transcription levels of Ddx5 and Slc7a1 occurred in
TNFα-treated VSMCs (Fig. 5B).
Subsequently, the transcript levels of Ablim1, Ddx5
and Slc7a1 were measured in the aortic tissues of Ang
II-treated and control mice. The results also revealed a
significant increase in the transcript level of Ablim1 in
the aortic tissues of Ang II-treated, however, no changes occurred
in the transcript levels of Ddx5 and Slc7a1 (Fig. 5B). These results revealed that
ABLIM1 may be a direct downstream of miR-378a-5p.
ABLIM1 expression is negatively regulated
by miR-378a-5p
To test whether ABLIM1 was involved in the
occurrence and development of AAA, the expression of ABLIM1 was
first detected in the aortic tissues of patients with AD and normal
individuals using immunofluorescence staining and western blotting.
The expression of ABLIM1 were significantly increased in the aortic
tissues of patients with AD (Fig.
S6A and B). It was also found that the expression levels of
Ablim1 were increased in the aortic tissue of AAA mice (Fig. S6C and D) and in TNFα-treated
VSMCs (Fig. S6E and F).
To further validate the relationship between ABLIM1
and miR-378a-5p, the expression of Ablim1 was detected in the
aortas of antagomir-378a-5p + Ang II and angomir-378a-5p + Ang II
mice. Compared with the corresponding control group, Ablim1
expression was increased in the aortic tissue of the
antagomir-378a-5p + Ang II group (Figs. 5C and S6G) and decreased in the aortic tissue
of the angomir-378a-5p + Ang II group (Figs. 5D and S6H). It was also found that
miR-378a-5p mimics significantly decreased Ablim1 expression levels
(Fig. S6I and J), whereas the
miR-378a-5p inhibitor increased Ablim1 expression levels (Fig. S6K and L).
Bioinformatic analysis suggested that the binding
sites for miR-378a-5p and ABLIM1 were highly conserved in humans,
mice and rats (Fig. 5E). The
three predicted binding sites are shown in Fig. S7A. To determine whether
miR-378a-5p could directly bind to the 3'UTR of ABLIM1, the
WT ABLIM1 3'UTR and the MUT ABLIM1 3'UTR were
reconstituted into the pMIR-REPORT Luciferase vector (Fig. S7B). Compared with the
mimics-NC-ABLIM1-3'UTR-WT group, the luciferase activity in
the mimics-miR-378a-5p-ABLIM1-3'UTR-WT group,
mimics-miR-378a-5p-ABLIM11-3'UTR-MUT1 group, and
mimics-miR-378a-5p-ABLIM1-3'UTR-MUT3 group was significantly
decreased, but there was no difference in the
mimics-miR-378a-5p-ABLIM1-3'UTR-MUT2 and mimics-m
iR-378a-5p-ABLIM1-3'UTR-MUT1-3 groups (Fig. 5F). These results indicated the
position 1403-1409 of ABLIM1-3' UTR was the binding site of
miR-378a-5p.
miR-378a-5p regulates the contractile
phenotype of VSMCs by targeting ABLIM1
To verify whether miR-378a-5p regulated the VSMCs
biological effects by directly regulating the expression of ABLIM1,
the role of ABLIM1 in the occurrence and development of AAA was
firstly explored by re-analyzing the GSE155468 dataset, which
contained high-quality single-cell transcriptome data from 8
patients with TAA and 3 healthy donor thoracic aortas. A total of
42,611 cells with 20,551 genes remained after the unqualified cells
and genes were filtered. The unsupervised clustering algorithm
clustered the 42,611 cells into 26 cell populations (Fig. S8A and B). These cells were
divided into seven groups (Fig.
S8C), and each group was identified and named according to the
expression of biomarker genes, including B cells (CD69 and CCR7),
blood cells (MZB1), endothelial cells (VWF and IFI27), macrophages
(CD14, CD68 and S100A9), mast cells (CPA3 and HPGD), SMCs (MYL9,
ACTA2, MYH11 and TAGLN) and T cells (NKG7 and ZGMA) (Fig. S8D). These cells exhibited
consistently high biomarker expression, thereby validating the
robustness of the categorization.
Given that VSMCs lesions were the main mechanism of
AAA, VSMCs were categorized into eight subpopulations (Fig. S8E and F). Based on the
expression of ABLIM1, VSMCs were divided into two subsets:
ABLIM1 high expression (ABLIM1+) and low
expression (ABLIM1-) (Fig. S8G-I). Gene Ontology and Kyoto
Encyclopedia of Gens and Genomes analyses of DEGs in the
ABLIM1+ and ABLIM1-groups
showed that most of the DEGs were related to VSMCs' contraction
(Fig. S8J and K). Subsequently,
it was found that the expression of contractile marker ACTA2,
CNN1 and TAGLN in ABLIM1+ subsets was
reduced and MMP2 expression was increased (Fig. 6A), which supported the
deleterious role for ABLIM1 in AAA pathophysiology.
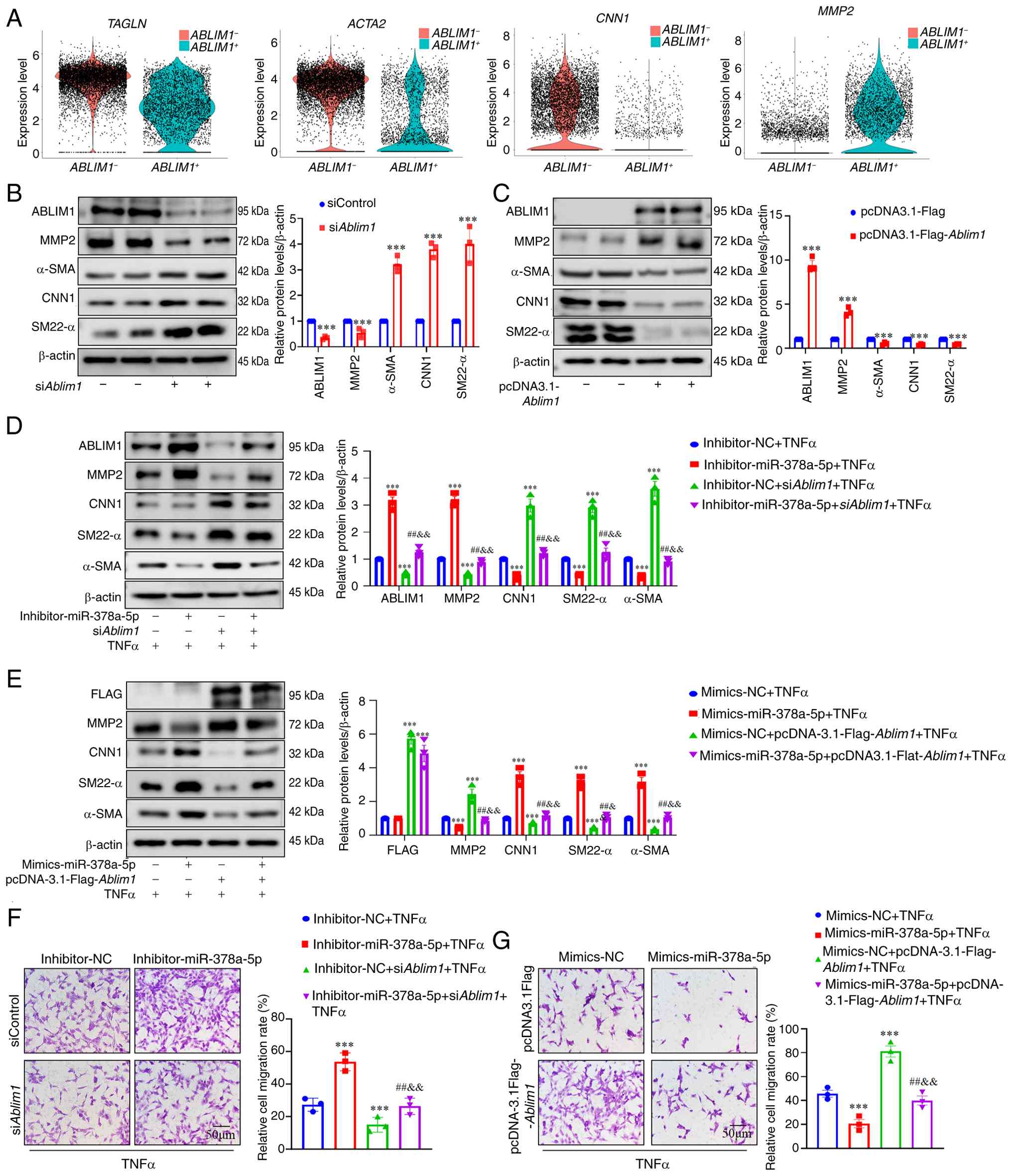 | Figure 6miR-378a-5p regulates phenotypic
transformation and migration of VSMCs by targeting ABLIM1. (A)
Normalized expression of ACTA2, TAGLN, CNN1 and
MMP2 transcripts in aortic VSMCs expressing ABLIM1
(blue) or not (red). (B) Western blots and quantification to
evaluate the expression of MMP2, CNN1, α-SMA, SM22-α in the VSMCs
transfected with siAblim1 or siControl (n=3 per group). (C)
Western blots to evaluate the expression of MMP2, CNN1, α-SMA,
SM22-α in the VSMCs transfected with pcDNA3.1-Flag-Ablim1 or
pcDNA3.1-Flag. (D) VSMCs were transfected with
inhibitor-miR-378a-5p or inhibitor-NC and followed by
siAblim1 transfection. Representative western blots and
quantification of MMP2, CNN1, α-SMA and SM22-α are shown. (E) VSMCs
were transfected with mimics-miR-378a-5p or mimics-NC, followed by
pcDNA3.1-Flag-Ablim1 transfection. Representative western
blots and quantification of MMP2, CNN1, α-SMA and SM22-α are
presented. (F) VSMCs migration was assessed using a Transwell assay
in VSMCs transfected with inhibitor-miR-378a-5p or inhibitor-NC
together with siAblim1 transfection. (G) VSMCs migration was
assessed using a Transwell assay in VSMCs transfected with
mimics-miR-378a-5p or mimics-NC together with
pcDNA3.1-Flag-Ablim1 transfection. Data are presented as the
the mean±SEM (n=3 per group). P-values were calculated by two-way
ANOVA with Holm-Sidak multiple comparisons test.
***P<0.001 vs. siControl or pcDNA3.1-Flag,
mimics-NC+TNFα or inhibitor-NC + TNFα; ##P<0.01 vs
mimics-NC + pcDNA3.1-Flag1-Ablim1 + TNFα or
inhibitor-NC+siAblim1 + TNFα; &&P<0.01
vs. mimics-miR-378a-5p + TNFα or inhibitor-miR-378a-5p + TNFα. miR
or miRNA, microRNA; VSMCs, vascular smooth muscle cells; α-SMA,
α-smooth muscle actin; SM22α, smooth muscle 22α; MMP2, matrix
metalloproteinase 2; CNN1, calponin 1; si-, small interfering; NC,
negative control. |
The results showed that Ablim1 knockdown led
to suppress the protein expression of MMP2 and increased
contractile markers (α-SMA, SM22-α and CNN1) (Fig. 6B), and overexpression of
Ablim1 significantly increased the protein expression of
MMP2 and suppressed expression of contractile markers (α-SMA,
SM22-α and CNN1) in VSMCs (Fig.
6C). Under TNFα stimulation, the effects of miR-378a-5p
inhibitor on the expression of VSMCs' contractile markers and MMP2
as well as migration were reversed by ABLIM1 knockdown. In
addition, the protective role of the miR-378a-5p overexpression
against VSMCs' contractile markers and migration was abolished by
ABLIM1 overexpression (Fig.
6D-G). These aforementioned results indicated that miR-378a-5p
regulates the phenotypic switching and secretion of MMP2 by
attenuating the expression of ABLIM1 in VSMCs.
Knockdown of ABLIM1 prevents Ang
II-induced AAA formation
The role of ABLIM1 in the development of AAA was
investigated in vivo. ApoE−/− mice were
injected with AAV-SM22-shNC or AAV-SM22-shAblim1 for 3 weeks
through the tail vein before AAA modeling (Fig. S9A). It was found that
Ablim1 was specifically knocked down in the aortas (Fig. S9B-D). There was no difference in
body weight between the groups (Fig. S10A). After infusion of Ang II
into AAV-SM22-shNC or AAV-SM22-shAblim1 mice, blood pressure
was increased (Fig. S10B).
There was no significant difference in survival rate between
AAV-SM22-shNC + Ang II group and AAV-SM22-shAblim1 + Ang II
group (Fig. S10C).
Compared with AAV-SM22-shNC + Ang II group, ABLIM1
knockdown reduced the incidence of AAA (40 vs. 74%, Fig. 7A and B). In the presence of Ang
II, mice in the AAV-SM22-shNC and AAV-SM22-shAblim1 groups
showed aortic dilation and aneurysms, whereas the symptoms in the
AAV-SM22-shAblim1 treatment group were relieved (Fig. 7C and D). Moreover, histological
analysis revealed that aortic dilatation, collagen deposition and
medium degeneration were alleviated in AAV-SM22-shAblim1
mice in response to Ang II (Figs.
7E and S10D and E). All
images of the abdominal aortic specimens were displayed in Fig. S11. These results indicated that
ABLIM1 knockdown alleviated Ang II-induced AAA formation.
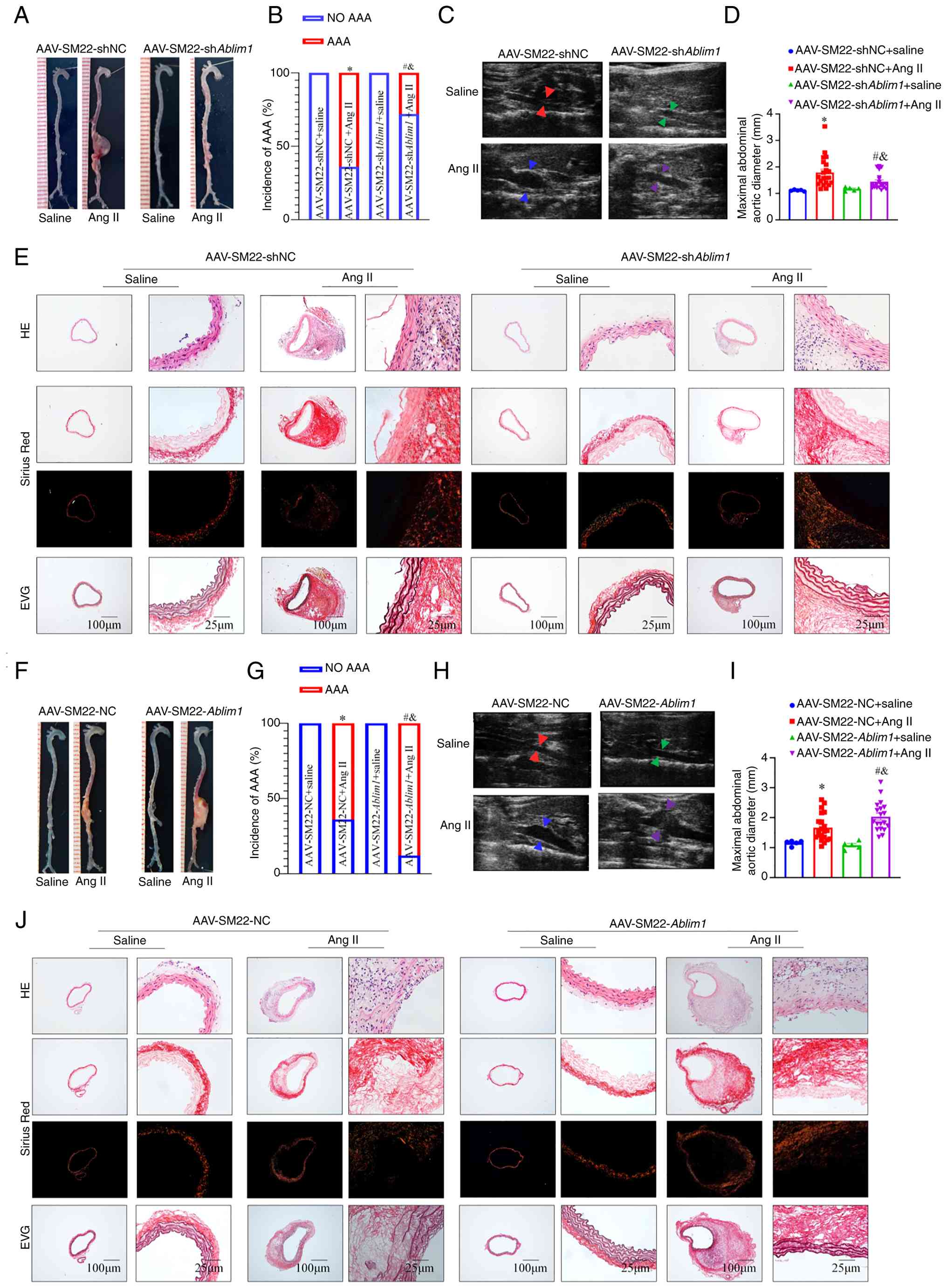 | Figure 7Knockdown of ABLIM1 prevents
Ang II-induced AAA formation, and overexpression of A BLIM1
aggravates Ang II-induced AAA formation. (A) Gross specimen image
of aortas from AAA models treated with AAV-SM22-shAblim1 or
AAV-SM22-shNC. (B) The incidence of AAA in Ang II-infused mice
treated with AAV-SM22-shAblim1 or AAV-SM22-shNC (n=25 in Ang
II groups and n=5 in saline groups). (C and D) Ultrasound images
and inner diameter quantification of the suprarenal abdominal aorta
(n=25 in Ang II groups and n=5 in saline groups). (E)
Representative H&E, Sirius red and EVG staining of the
abdominal aorta in different groups. (F) Gross specimen image of
aortas from AAA models treated with AAV-SM22-Ablim1 or
AAV-SM22-NC. (G) The incidence of AAA in Ang II-infused mice
treated with AAV-SM22-Ablim1 or AAV-SM22-NC (n=25 in Ang II
groups and n=9 in saline groups). (H and I) Ultrasound images and
inner diameter quantification of the suprarenal abdominal aorta
(n=25 in Ang II groups and n=5 in saline groups). (J)
Representative H&E, Sirius red and EVG staining of the
abdominal aorta in different groups. Data are presented as the mean
± SEM. P-values were calculated by two-way ANOVA with Holm-Sidak
multiple comparisons test (for B, D, G and I).
*P<0.05 vs. AAV-SM22-NC + saline or AAV-SM22-shNC +
saline; #P<0.05 vs. AAV-SM22-Ablim1 + saline
or AAV-SM22-shAblim1 + saline; &P<0.05 vs.
AAV-SM22-NC + Ang II or AAV-SM22-shNC + Ang II. ABLIM1,
actin-binding LIM protein 1; Ang II, angiotensin-II; AAA, abdominal
aortic aneurysm; sh-, short hairpin; NC, negative control; EVG,
Verhoeff-Van Gieson. |
Overexpression of ABLIM1 aggravates Ang
II-induced AAA formation
ApoE−/− mice were injected with
AAV-SM22-NC or AAV-SM22-Ablim1 for 3 weeks through the tail
vein before AAA modeling (Fig.
S12A). There was no difference in body weight between all the
groups (Fig. S12B). Blood
pressure was similarly increased following Ang II infusion in both
AAV-SM22-NC and AAV-SM22-Ablim1 mice (Fig. S12C). There was no significant
difference in survival rate between AAV-SM22-NC + Ang II and
AAV-SM22-Ablim1 + Ang II group (Fig. S12D). As expected, the incidence
of aortic aneurysms was significantly higher in Ang II-infused
AAV-SM22-Ablim1 + Ang II mice compared with AAV-SM22-NC +
Ang II mice (Fig. 7F and G).
In vivo ultrasound showed that overexpression of ABLIM1 mice
exhibited larger maximal internal diameters than control mice in
response to Ang II (Fig. 7H and
I). Furthermore, histological analysis revealed that
exacerbated aortic dilatation, collagen deposition, and disruption
of the medial architecture were aggravated in AAV-SM22-Ablim1
+ Ang II mice compared with AAV-SM22-NC + Ang II mice (Figs. 7J and S12E and F). All images of the
abdominal aortic specimens were displayed in Fig. S13. These results indicated that
ABLIM1 overexpression aggravated Ang II-induced AAA formation.
ABLIM1 aggravates the VSMCs contractile
phenotype through MKL1
To further investigate the potential mechanisms
underlying ABLIM1-related VSMCs phenotypic switching, a Co-IP assay
and mass spectrometry were performed to identify potential
downstream effectors (Fig. 8A).
A total of four proteins associated with VSMCs' contraction were
validated: Mitogen-activated protein kinase 1 (MAPK1),
adrenomedullin (ADM), protein kinase C delta (PRKCD) and MKL1
(Fig. 8B). It was found that
ABLIM1 could interact with MKL1 (Fig. 8C and D). In addition, molecular
docking predictions suggested that ABLIM1 bound to MKL1 (Fig. 8E). A total of 3 other proteins
(ADM, PRKCD and MAPK1) did not interact with ABLIM1 (Fig. S14).
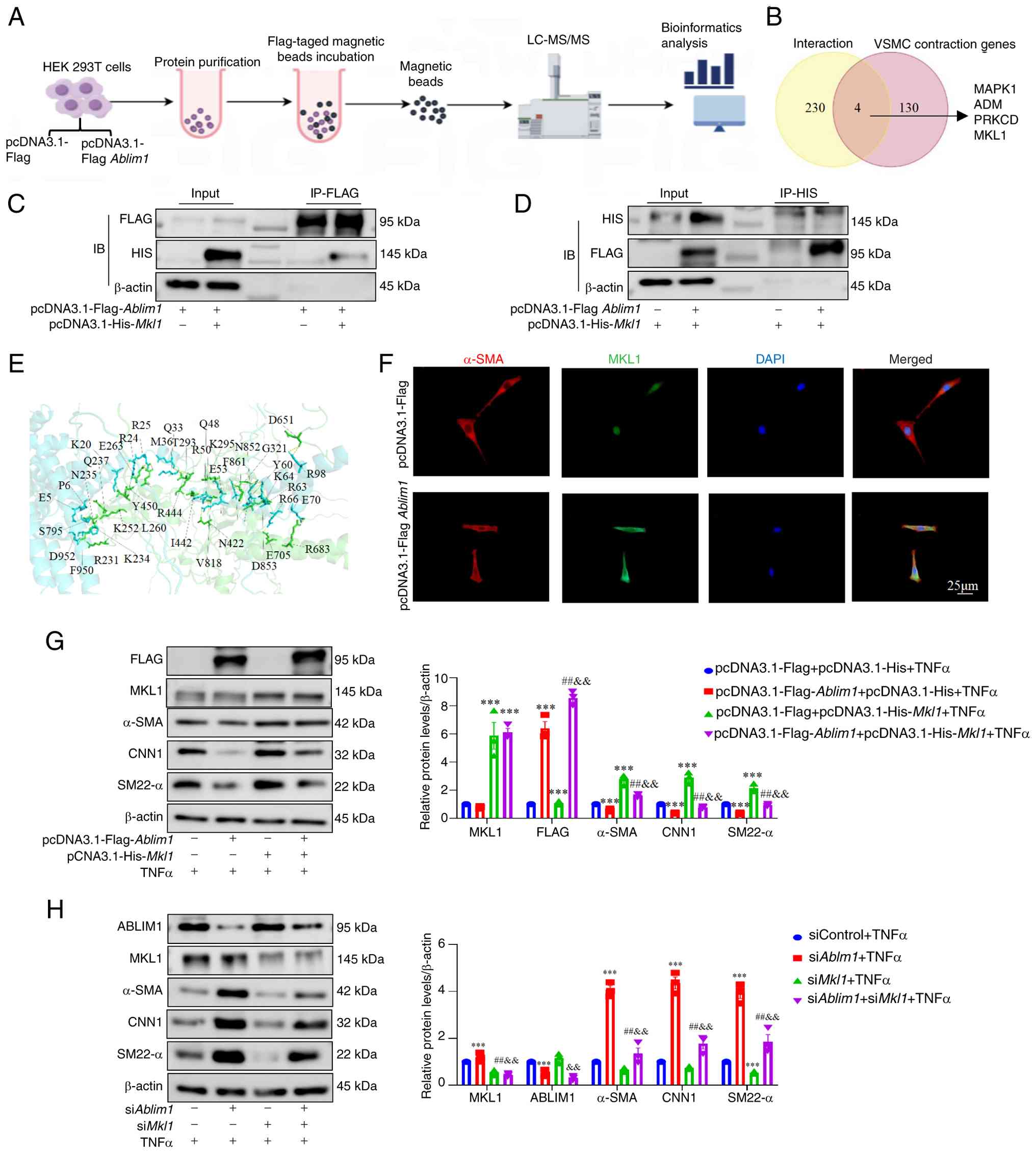 | Figure 8ABLIM1 regulates VSMCs' contractile
phenotype through interacting with MKL1. (A) Experimental flowchart
of Co-immunoprecipitation assay combined with mass spectrometry.
(B) Venn diagram showed the intersection of four proteins. (C) 293T
cells were transfected with pcDNA3.1-Flag-Ablim1 and
pcDNA3.1-His-Mkl1. Lysates were immunoprecipitated with
anti-Flag magnetic beads and blotted with anti-Flag and anti-His
antibodies. (D) 293T cells were transfected with
pcDNA3.1-Flag-Ablim1 and pcDNA3.1-His-Mkl1. Lysates
were immunoprecipitated with anti-His magnetic beads and blotted
with anti-Flag and anti-His antibodies. (E) Molecular docking mode
of ABLIM1 and MKL1. (F) Immunofluorescence images stained with
anti-α-SMA, anti-MKL1 antibody in the VSMCs transfected with
pcDNA3.1-Flag-Ablim1 and pcDNA3.1-Flag. (G) VSMCs were
transfected with pcDNA3.1-Flag-Ablim1 and
pcDNA3.1-His-Mkl1, followed by TNFα treatment.
Representative western blots and quantification of Flag, MKL1,
MMP2, CNN1, α-SMA and SM22-α were shown. (H) VSMCs were transfected
with siAblim1 and siMkl1, followed by TNFα treatment.
Representative western blots and quantification of ABLIM1, MKL1,
MMP2, CNN1, α-SMA and SM22-α were shown. Data are presented as the
mean ± SEM (n=3 per group). P-values were calculated by two-way
ANOVA with Holm-Sidak multiple comparisons test.
***P<0.001 vs. pcDNA3.1-Flag + pcDNA3.1-His + TNFα or
siControl + TNFα; ##P<0.01 vs. pcDNA3.
1-Flag-Ablim1 + pcDNA3.1-His + TNFα or siAblim1 +
TNFα, &&P<0.01 vs. pcDNA3.1-Flag +
pcDNA3.1-His-Mkl1 + TNFα or siMkl1 + TNFα. ABLIM1,
actin-binding LIM protein 1; VSMCs, vascular smooth muscle cells;
VSMCs, vascular smooth muscle cells; MKL1, megakaryoblastic
leukemia 1; α-SMA, α-smooth muscle actin; SM22α, smooth muscle 22α;
MMP2, matrix metalloproteinase 2; CNN1, calponin 1; si-, small
interfering. |
MKL1 (also known as MRTF-A) was a member of the
myocardin-related transcription family. MKL1 transduce
extracellular signals through the cytoskeleton that promote SMC
differentiation and modulate SMC phenotype (31). MKL1 functions as a transcription
cofactor through the nuclear cytoplasmic shuttle. It was found that
ABLIM1 overexpression reduced nuclear MKL1 expression and increased
cytoplasmic MKL1 expression (Fig.
8F). To further verify that whether the adverse effect of
ABLIM1 on VSMCs phenotypic switching was dependent on MKL1, MKL1
overexpression was forced and the consequences of ABLIM1
overexpression on phenotypic switching of VSMCs were assessed. The
results showed that MKL1 overexpression reversed the ABLIM1
overexpression-mediated phenotypic switching of VSMCs (Fig. 8G). Furthermore, MKL1 knockdown
weakened the role of ABLIM1 knockdown-mediated phenotypic switching
of VSMCs (Fig. 8H). These
aforementioned results indicated that ABLIM1 regulates the VSMCs
contractile phenotype by interacting with MKL1 and inhibiting the
expression of MKL1.
Discussion
In the present study, a novel role for miR-378a-5p
was identified in the pathogenesis of AAA. miR-378a-5p expression
was reduced in the serum and aortas of patients with AAA and mice.
Overexpression of miR-378a-5p prevented Ang II-induced AAA
formation, while knockdown of miR-378a-5p aggravated Ang II-induced
AAA formation. Moreover, miR-378a-5p overexpression promoted VSMCs'
differentiation and inhibited the migration of VSMCs, miR-378a-5p
knockdown played the opposite roles. Mechanistically, it was found
that miR-378a-5p played a protective role in AAA development by
regulating ABLIM1-MKL1 pathway. The mechanism diagram is shown in
Fig. S15.
VSMCs are plastic and undergo reversible phenotypic
changes in response to pathological stimulations. VSMCs'
dedifferentiation is observed during the early onset of various
vascular diseases including Marfan syndrome, AAA and AD (7,40-42). Weakness of the aortic wall is the
most important factor in AAA pathogenesis. The loss of VSMCs in the
aorta promotes AAA development. After converting from a contractile
to a secretory phenotype, VSMCs secrete large amounts of MMPs,
chemokines and pro-inflammatory cytokines, causing VSMCs apoptosis,
ECM degradation and the recruitment and activation of white blood
cells, eventually leading to aneurysm dilation and rupture.
Furthermore, the apoptosis and migration of VSMCs in the aortic
media further exacerbate the weakness of the aortic wall (6,9,43,44). Therefore, dysfunction of VSMCs is
a critical for AAA development (45). In the current cytological
experiments, TNF-α was utilized to simulate the pathological state
of AAA. TNF-α is a pivotal cytokine upregulated in both human AAA
and the Ang II-induced mouse model of the present study, where it
drives critical pathological events such as VSMC apoptosis,
phenotypic switching and matrix degradation.
While numerous miRNAs have been linked to AAA
through effects on inflammation or matrix remodeling (16,19,22,46-48), the present results reveal a
unique role for miR-378a-5p in preserving VSMCs' contractility,
adding a new mechanistic dimension to miRNA-mediated regulation in
AAA. miR-378a-5p mediates a wide range of biological processes
involved in cancer and angiogenesis (49). miR-378a-5p is a suppressor of
various cancers and serves as a serum biomarker for cancer
(23,24,50,51). miR-378a-5p has anti-apoptotic
functions and regulated SMCs' migration and invasion in breast
cancer (52-54). Moreover, miR-378a-5p is a
critical mediator of the regulation of VSMCs proliferation by
targeting the CDK1/p21 signaling pathway (51-55). The present results showed that
the expression of miR-378a-5p is reduced in the serum and aortas of
patients with AAA and Ang II-induced AAA mice, and TNFα-induced
VSMCs. Overexpression of miR-378a-5p prevented Ang II-induced AAA
formation, while knockdown of miR-378a-5p aggravated Ang II-induced
AAA formation. Moreover, overexpression of miR-378a-5p promoted
VSMCs differentiation and inhibited the migration of VSMCs;
knockdown of miR-378a-5p inhibited VSMCs differentiation and
increased the migration of VSMCs. These aforementioned results
showed that miR-378a-5p played a protective role in AAA development
by regulating the differentiation and migration of VSMCs.
miRNAs play an important role in AAA development by
directly regulating target genes (16). Bioinformatics prediction
indicated that ABLIM1 may be a target gene of miR-378a-5p. ABLIM1
is a cytoskeletal actin-binding protein implicated in interactions
between actin filaments and cytoplasmic targets (56). ABLIM1 splicing is abnormal in the
heart of patients with DM1 (57). Another study indicates that
abnormal splicing of ABLIM1 exon 11 occurred in the skeletal
muscles of patients with DM1 (28). ABLIM1 negatively controls
osteoclast differentiation by regulating cell migration and fusion
mediated by actin formation (55). ABLIM1 is also a novel E3 ligase
of IKBα (NF-kappa-B inhibitor alpha), and its abnormally high
expression activated NF-ĸB (nuclear factor kappa B) signaling,
thereby promoting CRC growth and metastasis in vitro and
in vivo (25). ABLIM1 is
also an F-actin crosslinking protein that ensures the formation of
a dense cortical actin meshwork for cells to resist mechanical
tension-induced blebbing (58)
and can crosslink and bundle F-actin to induce dense F-actin
network formation (59).
However, the role of ABLIM1 in AAA and VSMCs' function has not been
reported. In the experiments of the present study, it was found
that overexpression of ABLIM1 inhibited differentiation of VSMCs,
whereas interference with Ablim1 promoted differentiation
in vitro. In addition, miR-378a-5p overexpression inhibited
the phenotypic transformation and migration of VSMCs by inhibiting
the expression of ABLIM1, whereas miR-378a-5p knockdown promoted
the phenotypic transformation and migration of VSMCs by increasing
the expression of ABLIM1. Moreover, the current results revealed
that ABLIM1 knockdown mitigates AAA progression in vivo.
Interaction between SRF and coactivators was a
critical determinant of VSMCs' development. MKL1 is a
transcriptional co-activator of SRF which involved in a wide range
of pathophysiological processes in the cardiovascular system
(60,61). MKL1 induces transcription of
multiple SMCs marker genes containing CArG, including α-SMA, CNN1
and SM22-α (60). Reduction of
MKL1 in the nucleus suppressed the transcription of contraction
genes in VSMCs activated by SRF. The current results indicated that
ABLIM1 interacts with MKL1 and inhibits its nuclear translocation.
MKL1 overexpression reversed the ABLIM1 overexpression-mediated
phenotypic switching of VSMCs, and MKL1 knockdown weakened the role
of ABLIM1 knockdown-mediated phenotypic switching of VSMCs. These
aforementioned results indicated that miR-378a-5p plays a
protective role in AAA development by regulating the ABLIM1-MKL1
pathway.
The present study has several limitations. In
cellular assays, TNFα was only employed to model the inflammatory
stress driving VSMC phenotypic switching; however, the direct
effect of miR-378a-5p on inflammatory pathways was not examined. In
future study, the potential effect of miR-378a-5p on VSMC
inflammation should be carried out. Besides, VSMCs exhibit high
phenotypic plasticity, and the present study focused only on the
contractile phenotype. Other phenotypes of VSMCs and their roles in
AAA remain unclear, which need to be further clarified.
In conclusion, it is noteworthy that the present
study elucidates a previously unrecognized function of miR-378a-5p,
delineating its dedicated role in preserving VSMCs contractility-a
crucial yet underexplored mechanism in the pathogenesis of AAA.
Mechanistically, miR-378a-5p governs AAA progression by targeting
the ABLIM1-MKL1 axis, thereby regulating VSMCs' differentiation and
migration. Therefore, targeting the miR-378a-5p/ABLIM1-MKL1 axis
could inform new approaches for the prevention, early diagnosis and
treatment of AAA.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
found in the Gene Expression Omnibus under accession number
GSE280216 or at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE280216
and in the ProteomeXchange under accession number PXD073033 or at
the following URL: https://proteomecentral.proteomexchange.org/?view=datasets&search=PXD073033.
The data generated in the present study may be requested from the
corresponding author.
Authors' contributions
YH, DL, JW and YZ contributed to experiments
design, data analysis, and manuscript writing. YW, ZY, XS and DL
contributed to reviewing the bioinformatics analysis. CY and KX
contributed to experimental design and review of the manuscript. HS
and CY contributed to data analysis, and review and revise of the
manuscript. All authors read and approved the final version of the
manuscript. YH and DL confirm the authenticity of all raw data.
Ethics approval and consent to
participate
Human studies were conducted in accordance with the
World Medical Association Code of Ethics (Declaration of Helsinki)
[approval no. Y(2021)002] and was approved by the Ethics Committee
of General Hospital of Northern Theater Command (Shenyang, China).
All animal experiments were approved (approval no. 2022-20) by the
Ethics Committee of the General Hospital of Northern Theater
Command (Shenyang, China) and conducted in accordance with the
existing guidelines on the care and use of laboratory animals. All
animal care and experimental protocols complied with the National
Institute of Health Guide for the Care and Use of Laboratory
Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
AAA
|
abdominal aortic aneurysm
|
|
Ang II
|
angiotensin-II
|
|
ApoE-/-
|
apolipoprotein E-deficient
|
|
MMP
|
matrix metalloproteinase
|
|
VSMCs
|
vascular smooth muscle cells
|
|
Tagln
|
transgelin
|
|
TNF-α
|
tumor necrosis factor-α
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
miRNA or miR
|
microRNA
|
|
SRF
|
serum response factor
|
|
MKL1
|
megakaryoblastic leukemia 1
|
|
ABLIM1
|
actin-binding LIM protein 1
|
|
ECM
|
extracellular matrix
|
|
PBS
|
phosphate-buffered saline
|
|
α-SMA
|
α-smooth muscle actin
|
|
CNN1
|
calponin1
|
|
SM22-α
|
smooth muscle 22α
|
|
DMEM
|
Dulbecco's modified Eagle's
medium
|
|
FBS
|
fetal bovine serum
|
|
AAV
|
adeno-associated virus
|
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 82270449 and 82200535), the China
National Key R&D Project (grant no. 2022YFC2503403) and the
Natural Science Foundation of Liaoning (grant no. 2025-MS-348).
References
|
1
|
Pinard A, Jones GT and Milewicz DM:
Genetics of thoracic and abdominal aortic diseases. Circ Res.
124:588–606. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wanhainen A, Van Herzeele I, Bastos
Goncalves F, Bellmunt Montoya S, Berard X, Boyle JR, D'Oria M,
Prendes CF, Karkos CD, Kazimierczak A, et al: Editor's
choice-European society for vascular surgery (ESVS) 2024 clinical
practice guidelines on the management of abdominal aortoiliac
artery aneurysms. Eur J Vasc Endovasc Surg. 67:192–331. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Golledge J, Thanigaimani S, Powell JT and
Tsao PS: Pathogenesis and management of abdominal aortic aneurysm.
Eur Heart J. 44:2682–2697. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Golledge J: Abdominal aortic aneurysm:
Update on pathogenesis and medical treatments. Nat Rev Cardiol.
16:225–242. 2019. View Article : Google Scholar
|
|
5
|
Gao J, Cao H, Hu G, Wu Y, Xu Y, Cui H, Lu
HS and Zheng L: The mechanism and therapy of aortic aneurysms.
Signal Transduct Target Ther. 8:552023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rombouts KB, van Merrienboer TAR, Ket JCF,
Bogunovic N, van der Velden J and Yeung KK: The role of vascular
smooth muscle cells in the development of aortic aneurysms and
dissections. Eur J Clin Invest. 52:e136972022. View Article : Google Scholar :
|
|
7
|
Zhuge Y, Zhang J, Qian F, Wen Z, Niu C, Xu
K, Ji H, Rong X, Chu M and Jia C: Role of smooth muscle cells in
cardiovascular disease. Int J Biol Sci. 16:2741–2751. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Owens GK, Kumar MS and Wamhoff BR:
Molecular regulation of vascular smooth muscle cell differentiation
in development and disease. Physiol Rev. 84:767–801. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Frismantiene A, Philippova M, Erne P and
Resink TJ: Smooth muscle cell-driven vascular diseases and
molecular mechanisms of VSMC plasticity. Cell Signal. 52:48–64.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang HY, Chen AQ, Zhang H, Gao XF, Kong XQ
and Zhang JJ: Vascular smooth muscle cells phenotypic switching in
cardiovascular diseases. Cells. 11:40602022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang S, Liu D, Zhang X and Tian X:
Regulation of matrix metalloproteinase-2 and matrix
metalloproteinase-9 in abdominal aortic aneurysm. Cardiol Discov.
3:212–220. 2023.
|
|
12
|
Chen H: microRNA-based cancer diagnosis
and therapy. Int J Mol Sci. 25:2302023. View Article : Google Scholar
|
|
13
|
Bhaskaran M and Mohan M: MicroRNAs:
History, biogenesis, and their evolving role in animal development
and disease. Vet Pathol. 51:759–774. 2024. View Article : Google Scholar
|
|
14
|
Mohr AM and Mott JL: Overview of microRNA
biology. Semin Liver Dis. 35:3–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang G, Luo Y, Gao X, Liang Y, Yang F, Wu
J, Fang D and Luo M: MicroRNA regulation of phenotypic
transformations in vascular smooth muscle: Relevance to vascular
remodeling. Cell Mol Life Sci. 80:1442023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nguyen DND, Chilian WM, Zain SM, Daud MF
and Pung YF: MicroRNA regulation of vascular smooth muscle cells
and its significance in cardiovascular diseases. Can J Physiol
Pharmacol. 99:827–838. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maegdefessel L, Azuma J, Toh R, Merk DR,
Deng A, Chin JT, Raaz U, Schoelmerich AM, Raiesdana A, Leeper NJ,
et al: Inhibition of microRNA-29b reduces murine abdominal aortic
aneurysm development. J Clin Invest. 122:497–506. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yamasaki T, Horie T, Koyama S, Nakao T,
Baba O, Kimura M, Sowa N, Sakamoto K, Yamazaki K, Obika S, et al:
Inhibition of microRNA-33b specifically ameliorates abdominal
aortic aneurysm formation via suppression of inflammatory pathways.
Sci Rep. 12:119842022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maegdefessel L, Azuma J, Toh R, Deng A,
Merk DR, Raiesdana A, Leeper NJ, Raaz U, Schoelmerich AM, McConnell
MV, et al: MicroRNA-21 blocks abdominal aortic aneurysm development
and nicotine-augmented expansion. Sci Transl Med. 4:122ra222012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maegdefessel L, Spin JM, Raaz U, Eken SM,
Toh R, Azuma J, Adam M, Nakagami F, Heymann HM, Chernogubova E, et
al: miR-24 limits aortic vascular inflammation and murine abdominal
aneurysm development. Nat Commun. 5:52142014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang H, Wang Y, Bian X and Yin H:
MicroRNA-194 acts as a suppressor during abdominal aortic aneurysm
via inhibition of KDM3A-mediated BNIP3. Life Sci. 277:1193092021.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma X, Yao H, Yang Y, Jin L, Wang Y, Wu L,
Yang S and Cheng K: miR-195 suppresses abdominal aortic aneurysm
through the TNF-α/NF-κB and VEGF/PI3K/Akt pathway. Int J Mol Med.
41:2350–2358. 2018.PubMed/NCBI
|
|
23
|
Li K, Zhang J, Zhang M, Wu Y, Lu X and Zhu
Y: miR-378a-5p inhibits the proliferation of colorectal cancer
cells by downregulating CDK1. World J Surg Oncol. 19:542021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nadeem U, Ye G, Salem M and Peng C:
MicroRNA-378a-5p targets cyclin G2 to inhibit fusion and
differentiation in BeWo cells. Biol Reprod. 91:762014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He Y, Shi Q, Ling Y, Guo H, Fei Y, Wu R,
Tang C, Zhang X and Yao L: ABLIM1, a novel ubiquitin E3 ligase,
promotes growth and metastasis of colorectal cancer through
targeting IĸBα ubiquitination and activating NF-ĸB signaling. Cell
Death Differ. 31:203–216. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu D, Wang X, Liu Y, Li C, Zhang Z and Lv
P: Actin-Binding LIM 1 (ABLIM1) inhibits glioblastoma progression
and serves as a novel prognostic biomarker. Dis Markers.
2022:95168082022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roof DJ, Hayes A, Adamian M, Chishti AH
and Li T: Molecular characterization of abLIM, a novel
actin-binding and double zinc finger protein. J Cell Biol.
138:575–588. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ohsawa N, Koebis M, Mitsuhashi H, Nishino
I and Ishiura S: ABLIM1 splicing is abnormal in skeletal muscle of
patients with DM1 and regulated by MBNL, CELF and PTBP1. Genes
Cells. 20:121–134. 2015. View Article : Google Scholar
|
|
29
|
McDonald OG and Owens GK: Programming
smooth muscle plasticity with chromatin dynamics. Circ Res.
100:1428–1441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miralles F, Posern G, Zaromytidou AI and
Treisman R: Actin dynamics control SRF activity by regulation of
its coactivator MAL. Cell. 113:329–342. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cen B, Selvaraj A, Burgess RC, Hitzler JK,
Ma Z, Morris SW and Prywes R: Megakaryoblastic leukemia 1, a potent
transcriptional coactivator for serum response factor (SRF), is
required for serum induction of SRF target genes. Mol Cell Biol.
23:6597–6608. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Friedman RM, Truong HD, Aronson MR, Brown
EA, Angelozzi M, Chen JF, Zur KB, Lefebvre V and Gottardi R:
Inhibition of the MRTF-A/SRF signaling axis alleviates vocal fold
scarring. Matrix Biol. 137:1–11. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun Y, Boyd K, Xu W, Ma J, Jackson CW, Fu
A, Shillingford JM, Robinson GW, Hennighausen L, Hitzler JK, et al:
Acute myeloid leukemia-associated Mkl1 (Mrtf-a) is a key regulator
of mammary gland function. Mol Cell Biol. 26:5809–5826. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu H, Wang J, Bu Y, Li J, Li Y, Jing Q,
Wang X, Yan C, Liu D and Han Y: Pentamethylquercetin attenuates
angiotensin II-induced abdominal aortic aneurysm formation by
blocking nuclear translocation of C/EBPβ at Lys253. Biochim Biophys
Acta Mol Basis Dis. 1870:1672242024. View Article : Google Scholar
|
|
35
|
Yang L, Sui HG, Wang MM, Li JY, He XF, Li
JY and Wang XZ: MiR-30c-1-3p targets matrix metalloproteinase 9
involved in the rupture of abdominal aortic aneurysms. J Mol Med
(Berl). 100:1209–1221. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
37
|
Wen Y, Liu Y, Li Q, Tan J, Fu X, Liang Y,
Tuo Y, Liu L, Zhou X, LiuFu D, et al: Spatiotemporal ATF3
expression determines VSMC fate in abdominal aortic aneurysm. Circ
Res. 134:1495–1511. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tastsoglou S, Skoufos G, Miliotis M,
Karagkouni D, Koutsoukos I, Karavangeli A, Kardaras FS and
Hatzigeorgiou AG: DIANA-miRPath v4.0: Expanding target-based miRNA
functional analysis in cell-type and tissue contexts. Nucleic Acids
Res. 51:W154–W159. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hu Y, Cai Z and He B: Smooth muscle
heterogeneity and plasticity in health and aortic aneurysmal
disease. Int J Mol Sci. 24:117012023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao G, Zhao Y, Lu H, Chang Z, Liu H, Wang
H, Liang W, Liu Y, Zhu T, Rom O, et al: BAF60c prevents abdominal
aortic aneurysm formation through epigenetic control of vascular
smooth muscle cell homeostasis. J Clin Invest. 132:e1583092022.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen M, Yang D, Zhou Y, Yang C, Lin W, Li
J, Liu J, Ye J, Huang W, Ma W, et al: Colchicine blocks abdominal
aortic aneurysm development by maintaining vascular smooth muscle
cell homeostasis. Int J Biol Sci. 20:2092–2110. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nappi F: Current opinion in acute aortic
dissection. Cardiol Discov. Jan 3–2025.Epub ahead of print.
|
|
43
|
Lu H, Du W, Ren L, Hamblin MH, Becker RC,
Chen YE and Fan Y: Vascular smooth muscle cells in aortic aneurysm:
From genetics to mechanisms. J Am Heart Assoc. 10:e0236012021.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Petsophonsakul P, Furmanik M, Forsythe R,
Dweck M, Schurink GW, Natour E, Reutelingsperger C, Jacobs M, Mees
B and Schurgers L: Role of vascular smooth muscle cell phenotypic
switching and calcification in aortic aneurysm formation.
Arterioscler Thromb Vasc Biol. 39:1351–1368. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ailawadi G, Moehle CW, Pei H, Walton SP,
Yang Z, Kron IL, Lau CL and Owens GK: Smooth muscle phenotypic
modulation is an early event in aortic aneurysms. J Thorac
Cardiovasc Surg. 138:1392–1399. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li X, Yang Y, Wang Z, Jiang S, Meng Y,
Song X, Zhao L, Zou L, Li M and Yu T: Targeting non-coding RNAs in
unstable atherosclerotic plaques: Mechanism, regulation,
possibilities, and limitations. Int J Biol Sci. 17:3413–3427. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhou Y, Wang M, Zhang J, Xu P and Wang H:
MicroRNA-29a-3p regulates abdominal aortic aneurysm development and
progression via direct interaction with PTEN. J Cell Physiol.
235:9414–9423. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shi X, Ma W, Li Y, Wang H, Pan S, Tian Y,
Xu C and Li L: MiR-144-5p limits experimental abdominal aortic
aneurysm formation by mitigating M1 macrophage-associated
inflammation: Suppression of TLR2 and OLR1. J Mol Cell Cardiol.
143:1–14. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Machado IF, Teodoro JS, Palmeira CM and
Rolo AP: miR-378a: A new emerging microRNA in metabolism. Cell Mol
Life Sci. 77:1947–1958. 2020. View Article : Google Scholar
|
|
50
|
Pan X, Zhao L, Quan J, Liu K, Lai Y, Li Z,
Zhang Z, Xu J, Xu W, Guan X, et al: MiR-378a-5p acts as a tumor
suppressor in renal cell carcinoma and is associated with the good
prognosis of patients. Am J Transl Res. 11:2207–2218.
2019.PubMed/NCBI
|
|
51
|
Zhang Y, Zhang P and Deng C: miR-378a-5p
regulates CAMKK2/AMPK pathway to contribute to cerebral
ischemia/reperfusion injury-induced neuronal apoptosis. Folia
Histochem Cytobiol. 59:57–65. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sha T, Li J, Sun S, Li J, Zhao X, Li Z and
Cui Z: YEATS domain-containing 2 (YEATS2), targeted by microRNA
miR-378a-5p, regulates growth and metastasis in head and neck
squamous cell carcinoma. Bioengineered. 12:7286–7296. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bhandari R, Shaikh II, Bhandari R and
Chapagain S: LINC01023 promotes the hepatoblastoma tumorigenesis
via miR-378a-5p/WNT3 axis. Mol Cell Biochem. 478:1867–1885. 2023.
View Article : Google Scholar
|
|
54
|
Yan C and Jin Y: Silencing of long
noncoding RNA MIAT inhibits the viability and proliferation of
breast cancer cells by promoting miR-378a-5p expression. Open Med
(Wars). 18:202306762023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu S, Yang Y, Jiang S, Xu H, Tang N, Lobo
A, Zhang R, Liu S, Yu T and Xin H: MiR-378a-5p regulates
proliferation and migration in vascular smooth muscle cell by
targeting CDK1. Front Genet. 10:222019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Narahara H, Sakai E, Yamaguchi Y, Narahara
S, Iwatake M, Okamoto K, Yoshida N and Tsukuba T: Actin binding LIM
1 (abLIM1) negatively controls osteoclastogenesis by regulating
cell migration and fusion. J Cell Physiol. 234:486–499. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Koshelev M, Sarma S, Price RE, Wehrens XH
and Cooper TA: Heart-specific overexpression of CUGBP1 reproduces
functional and molecular abnormalities of myotonic dystrophy type
1. Hum Mol Genet. 19:1066–1075. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang S, Liu C, Guo Y, Li G, Li D, Yan X
and Zhu X: Self-construction of actin networks through phase
separation-induced abLIM1 condensates. Proc Natl Acad Sci USA.
119:e21224201192022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Li G, Huang S, Yang S, Wang J, Cao J,
Czajkowsky DM, Shao Z and Zhu X: abLIM1 constructs non-erythroid
cortical actin networks to prevent mechanical tension-induced
blebbing. Cell Discov. 4:422018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhou J, Zhang M, Fang H, El-Mounayri O,
Rodenberg JM, Imbalzano AN and Herring BP: The SWI/SNF chromatin
remodeling complex regulates myocardin-induced smooth
muscle-specific gene expression. Arterioscler Thromb Vasc Biol.
29:921–928. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang DZ, Li S, Hockemeyer D, Sutherland L,
Wang Z, Schratt G, Richardson JA, Nordheim A and Olson EN:
Potentiation of serum response factor activity by a family of
myocardin-related transcription factors. Proc Natl Acad Sci USA.
99:14855–14860. 2002. View Article : Google Scholar : PubMed/NCBI
|














