Interleukin-35 (IL-35) is an immunosuppressive and
anti-inflammatory cytokine that is expressed primarily by
regulatory T cells (Tregs). B cells have been reported to secrete
IL-35 (13-15). IL-35 belongs to the IL-12 family
and is a dimeric protein composed of IL-12p35 and EBI3. Its
receptors, which are dimers, signal through the JAK-STAT pathway.
Four types of IL-35 receptors have been identified, IL12Rβ2/IL27Rα,
gp130/gp130, IL-12β2/IL12Rβ2 and IL-12Rβ2/gp130, with each
triggering distinct signaling pathways. IL-35, which is released by
Tregs and regulatory B cells (Bregs), plays a critical role in
suppressing immune-inflammatory responses in autoimmune diseases,
affecting a variety of conditions, including those of the
digestive, musculoskeletal, and respiratory systems. IL-35 inhibits
immune reactions by promoting Treg proliferation, enhancing their
immunosuppressive function, suppressing Th17 differentiation, and
reducing the levels of proinflammatory cytokines such as IL-17
(16,17). In a mouse model, Xie et al
(18) demonstrated that B cells
lacking IL-35 expression fail to effectively recover from
autoimmune diseases, such as inflammatory bowel disease (IBD). As
with IL-10, IL-35 possesses potent immunosuppressive properties and
acts as a key mediator among cytokines, increasing IL-10 levels.
Additionally, IL-35 can upregulate cytokines involved in MG
pathogenesis, including IL-1β, IL-6, and IL-10, with IL-35 levels
notably decreasing after treatment (19,20). However, the exact relationship
between IL-35 and MG remains unclear. These findings indicate that
IL-35 has anti-inflammatory effects on immune-mediated central
nervous system diseases, including MG, and could serve as a
potential therapeutic target. Consequently, understanding the role
of IL-35 in the progression and treatment of MG is of considerable
interest. The present review summarized the effect of IL-35 on MG
pathogenesis, its potential therapeutic efficacy and its prognostic
value, providing insights into its mechanisms and clinical
implications for MG treatment.
The following is an overview of the molecular
structure and sources of IL-35, the composition and function of
IL-35 receptors and the mechanism of IL-35 signal transduction.
IL-35 is a recently identified heterodimeric
cytokine critical for the regulatory functions of Tregs. As a
member of the IL-12 cytokine family, IL-35 comprises an α chain
(p19 or p28) and a β chain [p40 or Epstein-Barr virus-induced gene
3 (EBI3)] (21). It is part of a
group of five major heterodimeric cytokines: IL-12, IL-23 (p19 and
p40), IL-27 (p28 and EBI3) and the recently proposed IL-39 (p19 and
EBI3) (22). Specifically, IL-35
consists of IL-12p35 and EBI3, which form the IL-12p35/IL-27EBI3
dimer. EBI3, a glycoprotein containing a signal peptide with a
repeated Alu sequence, plays a role in cellular pathways and
maternal immune tolerance during embryogenesis. EBI3 is also
expressed in Hodgkin lymphoma cells, as well as in acute and
chronic myeloid leukemia cells (23). While IL-12p35 is involved in
promoting inflammatory responses, IL-35 serves to regulate and
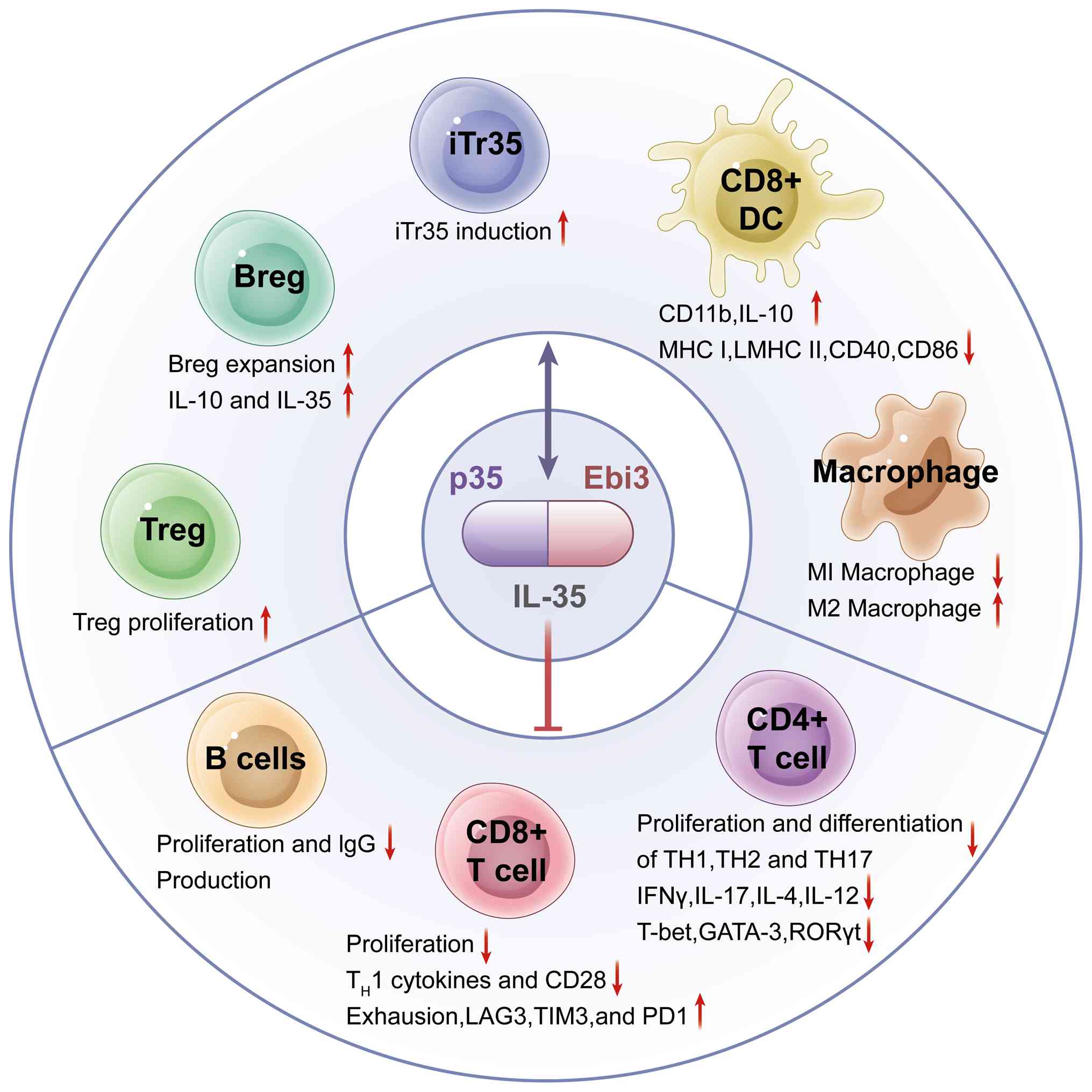
suppress inflammation. As shown in Fig. 1, IL-35 regulates the functions of
various immune cells (such as Tregs, Bregs, macrophages, T cells,
and B cells) to regulate the body's immunity. IL-35 is secreted by
various immune cells, including tolerogenic dendritic cells (DCs),
myeloid-derived suppressor cells, tumor-associated macrophages,
neutrophils and natural killer (NK) cells, and primarily by
IL-10/IL-35-secreting subsets of Bregs. As an inhibitory cytokine,
IL-35 suppresses T-cell proliferation and induces the generation of
iTr35 cells to control inflammatory responses (24).
The expression of IL-35 is linked to forkhead box P3
(Foxp3)+ Tregs, with the EBI3 subunit being highly expressed in
murine CD4+ Foxp3+ Tregs but absent in activated effector CD4+ T
cells. Upon activation, Tregs generate distinct effector subsets
(IL-35-producing and IL-10-producing) to maintain immune tolerance
(25). iTr35, a newly identified
subset of Tregs that are non-Foxp3 dependent, is induced by IL-35
from murine T cells (26). iTr35
cells release IL-35, promote Treg proliferation, block effector
T-cell activation, and convert effector cells into iTr35 cells. The
differentiation and function of iTr35 cells are driven primarily by
IL-35 and these cells do not rely on Foxp3 or mediate immune
suppression via IL-10 or TGF-β (26). Studies have highlighted Bregs as
another significant source of IL-35 (27-29). Upon B-cell receptor activation,
Toll-like receptor 4 (TLR4) binds to CD40, triggering the
transcription of EBI3 and p35, leading to IL-35 secretion (27). Additional research using
TLR-deficient B cells demonstrated that TLR4 and CD40L
costimulation is necessary for the transformation of B cells into
Breg subsets (i35-Bregs) capable of secreting IL-35 (30). In addition to Tregs and Bregs,
IL-35 may also be produced by other immune cells, such as immature
DCs, CD8+ T cells, and certain tumor cells (31-35). Under inflammatory conditions,
additional tissues may have the potential to secrete IL-35. While
IL-27 is produced primarily by activated antigen-presenting cells
(APCs), IL-35 is secreted predominantly by activated Tregs
(36).
IL-35 plays a critical role in modulating immune
responses by inhibiting effector T-cell differentiation and IL-17
production, exerting a negative immunoregulatory effect that helps
maintain immune homeostasis (37). It promotes the generation of
Bregs and the secretion of IL-10 by Breg subsets, further
contributing to its immunosuppressive effects (38). Activated B cells can produce both
IL-35 and IL-10, mediating negative immune regulation (38). In combination with TGF-β, IL-35
also suppresses immune reactions by stimulating the proliferation
of CD4+CD25+ T cells and enhancing IL-10 expression, which inhibits
inflammatory responses (38).
Studies in mouse models have shown that B cells deficient in IL-35
expression fail to recover from T-cell-induced autoimmune diseases,
such as experimental autoimmune encephalomyelitis (EAE) (39). These findings highlight the
potent immunoregulatory functions of IL-35 in various conditions,
including cancer, autoimmunity and infection. As summarized in
Table I (40-45), IL-35 plays a pivotal role in
inflammatory and autoimmune diseases, positioning it as a potential
therapeutic target for these disorders (22,32).
The current research on the source of IL-35 in MG is
extremely weak and relies mainly on animal models for speculation,
lacking human evidence, cell tracing analysis, and clinical
translation studies. In the future, multiomics studies targeting
the immune microenvironment of patients with MG are needed to
clarify the production of cells, regulatory mechanisms and
therapeutic potential of IL-35.
The receptors for IL-35 signaling, although
distinct, partially overlap with those of other members of the
IL-12 family (46). IL-35
signals through four primary receptor types: IL12Rβ2/IL27Rα and
glycoprotein 130 (gp130)/gp130. The functional IL-35 receptor
comprises IL12Rβ2, gp130 and possibly IL-27Rα. IL-35 transduces
immunosuppressive signals primarily via the IL-12Rβ2:gp130 receptor
complex. The p35 subunit binds to IL-12Rβ2, while the EBI3 subunit
binds to gp130, leading to receptor dimerization, JAK-STAT
activation, and the formation of either heterodimers (STAT1-STAT4)
or homodimers (STAT1-STAT1). However, IL-35 can also signal through
two types of homodimeric receptors formed by gp130 and IL-12Rβ2,
namely, gp130:gp130. The IL-12Rβ2 subunit is typically expressed by
activated NK and T cells, with some expression on DCs and B cells,
whereas gp130 is expressed on nearly all cell types. The expression
level of IL-12Rβ2 determines the functional scope of IL-35
signaling. IL-35 can initiate signaling not only by binding to the
IL-12Rβ2:gp130 heterodimeric receptor but also by engaging
gp130:gp130 homodimers (47). As
shown in Fig. 2, unlike other
cytokines in the IL-12 family, IL-35 can signal through homodimeric
receptors. Specifically, it can interact with IL-12Rβ2:IL-12Rβ2
homodimers, initiating signal transduction via the STAT1 or STAT4
pathway, respectively. However, since only one pathway is activated
in this case, the immunoregulatory function of IL-35 is partially
compromised (47). Full
immunosuppressive activity, including the induction of iTr35 cells,
requires binding to the IL-12Rβ2:gp130 heterodimer and simultaneous
activation of both the STAT1 pathway and the STAT4 pathway
(48). While homodimers can
suppress T-cell proliferation, they are less effective than
heterodimers in regulating the activity of IL-35-induced Tregs
(iTr35 cells) (47). At present,
research on the role of the IL-35 receptor in MG is still in its
early stages, and its specific mechanism of action, receptor
expression regulation, and clinical translational potential are not
yet clear.
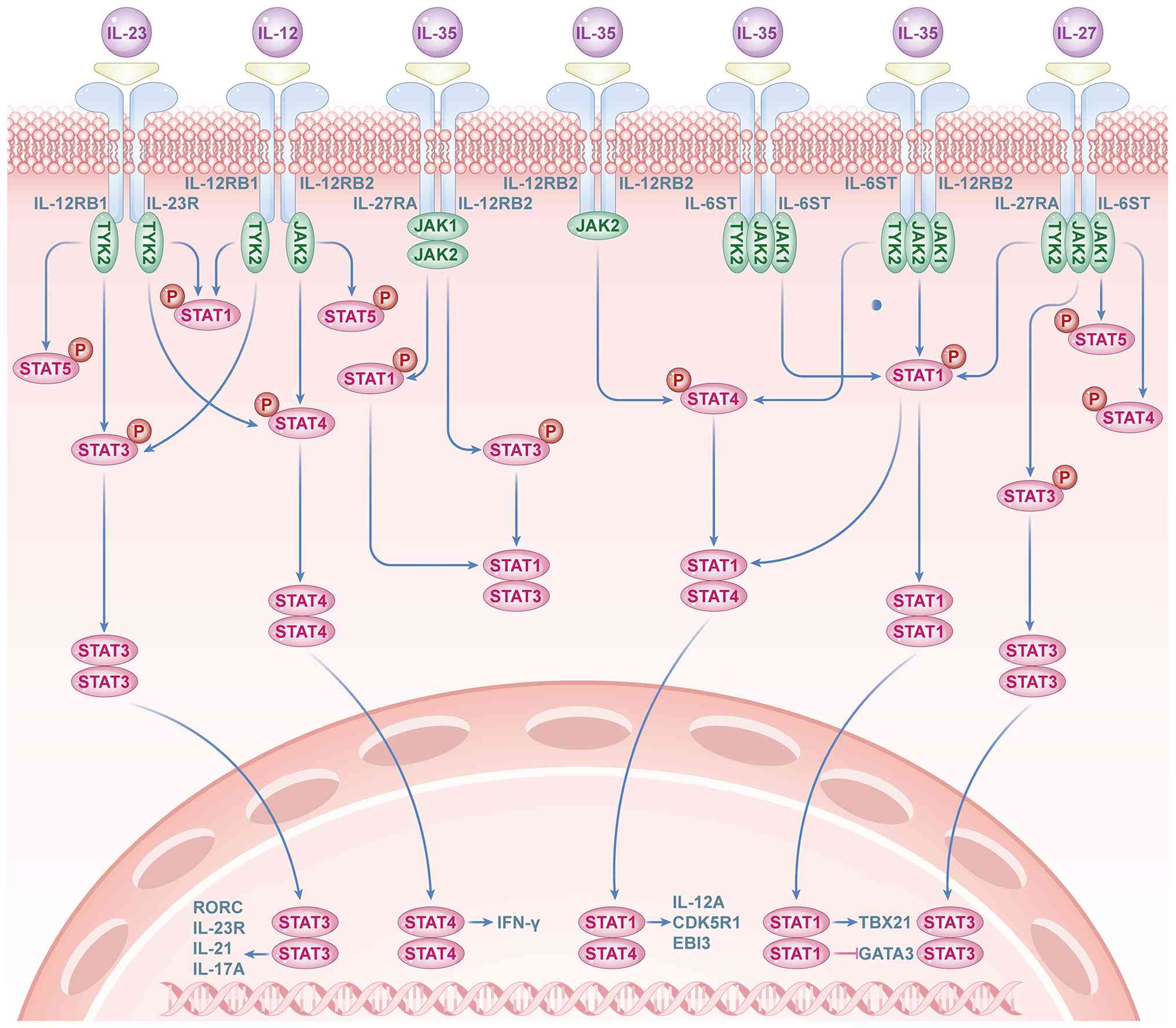
A key function of the IL-12 family of cytokines is
their mediation of activity through the activation of the JAK-STAT
signaling pathway (49). Each
member of the IL-12 family induces the activation of specific STAT
family members, which results in distinct but sometimes overlapping
gene transcription patterns. Upon activation of the IL-35 receptor,
JAK family members are activated, initiating signal transduction
(49). Phosphorylated JAKs
subsequently phosphorylate STAT1 and STAT4, members of the STAT
family. These phosphorylated STATs then translocate to the nucleus,
where they regulate the expression of the p35 and EBI3 genes,
promoting IL-35 expression (50). The activation of JAKs by the
IL-12Rβ2 and gp130 receptor subunits primarily involves JAK1 and
JAK2, respectively, which act on downstream STAT4 and STAT1
molecules. The binding of IL-35 to a specific homodimeric receptor
activates a corresponding signaling branch. Thus, the
IL-12Rβ2:IL-12Rβ2 homodimer predominantly mediates the JAK1-STAT4
signaling pathway, whereas the gp130:gp130 homodimer primarily
activates the JAK2-STAT1 signaling pathway (Fig. 3).
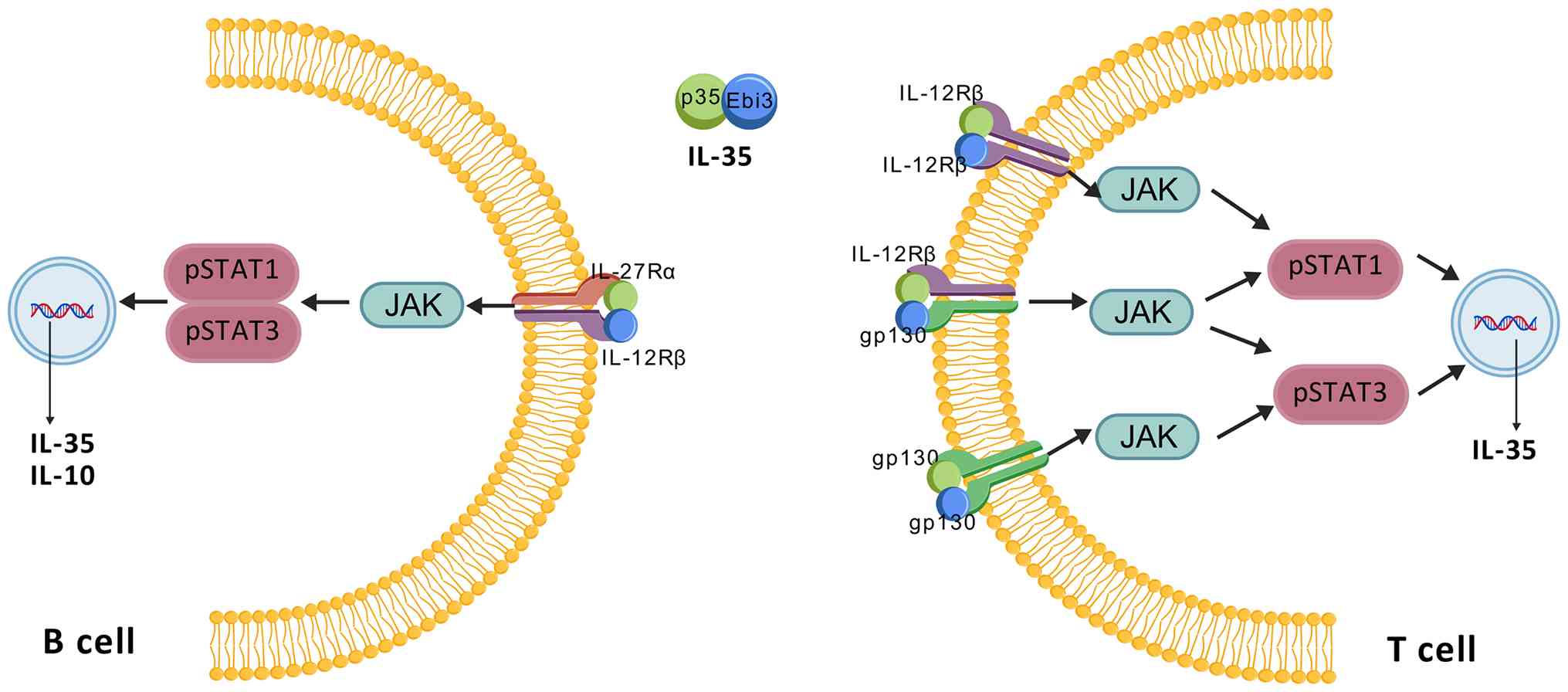
In Tregs, IL-35 transduces signals via either
heterodimeric receptors (IL-12Rβ2/gp130) or homodimeric receptors
(gp130/gp130), leading to the activation of STAT4, either
concurrently or independently (51). When IL-35 binds to its
corresponding receptor, only one signaling branch is activated
(either STAT1 with gp130:gp130 or STAT4 with IL-12Rβ2:IL-12Rβ2
homodimers), which inhibits T-cell proliferation (52). This results in partial loss of
its immunosuppressive activity. However, signaling through the
IL-12Rβ2:gp130 heterodimeric receptor can also induce the
production of iTr35 cells, which fully exert their
immunosuppressive functions (Fig.
3).
IL-35 can bind to a heterodimeric receptor composed
of IL-12Rβ2, activating downstream STAT1 and STAT3 signaling
molecules. This activation induces the production of both IL-10 and
IL-35 and promotes the proliferation of Bregs (28). These findings demonstrate that
IL-35 regulates its biological functions through different
receptors and STAT molecules in various cell types (Fig. 3).
The combination of IL-35 with its corresponding
receptor activates STAT4 in T cells, which then inhibits the MAPK
and NF-κB pathways. This reduces proinflammatory responses and
inhibits the maturation of monocyte-derived DCs (53). As shown in Figs. 3-4 and Table II (54-58), IL-35 plays a critical role in
maintaining immune homeostasis by modulating different target cells
and effector pathways. Failure to achieve effective
immunosuppression can lead to the development of immune-related
diseases.
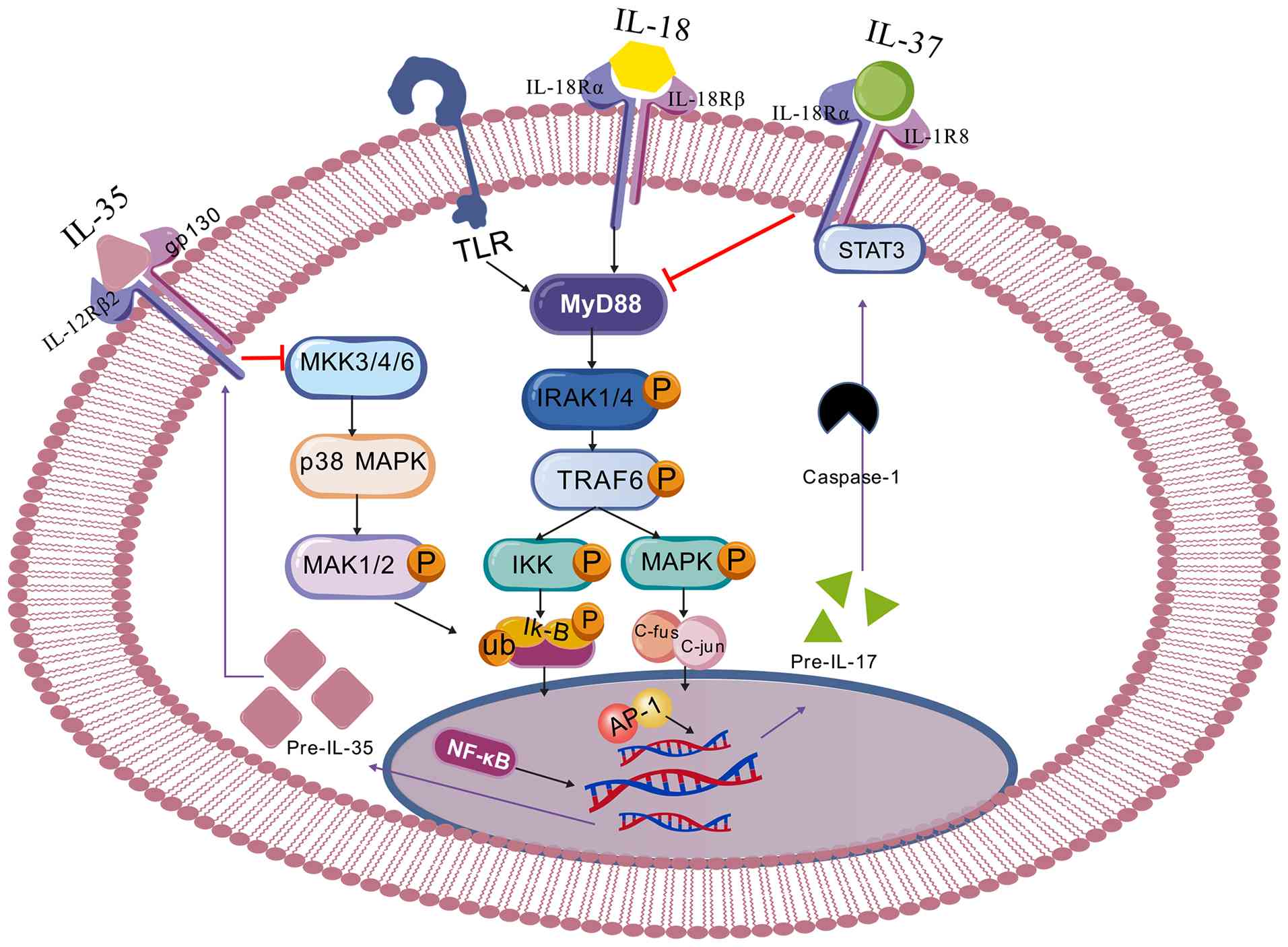
Inflammatory signaling pathways are often stimulated
under inflammatory conditions, leading to the release of
proinflammatory factors. During inflammation, the interaction of
TLRs, TNF receptors, or IL-18 receptors with their ligands rapidly
activates MyD88. This in turn activates TRAF6 or enzymes such as
IRAK-1, promoting the expression of NF-κB and increasing the
production of inflammatory factors (59). The transcription factors c-Fos
and c-Jun, which are activated by MAPKs, form AP-1 (60), which can increase the levels of
inflammatory cytokines (60).
Additionally, the IκB inhibitor is affected by IKK through
ubiquitination, leading to NF-κB release and its translocation to
promote inflammatory responses (59). Together, these two pathways
contribute to the production of proinflammatory factors such as
IL-1, IL-6, and interferon (IFN).
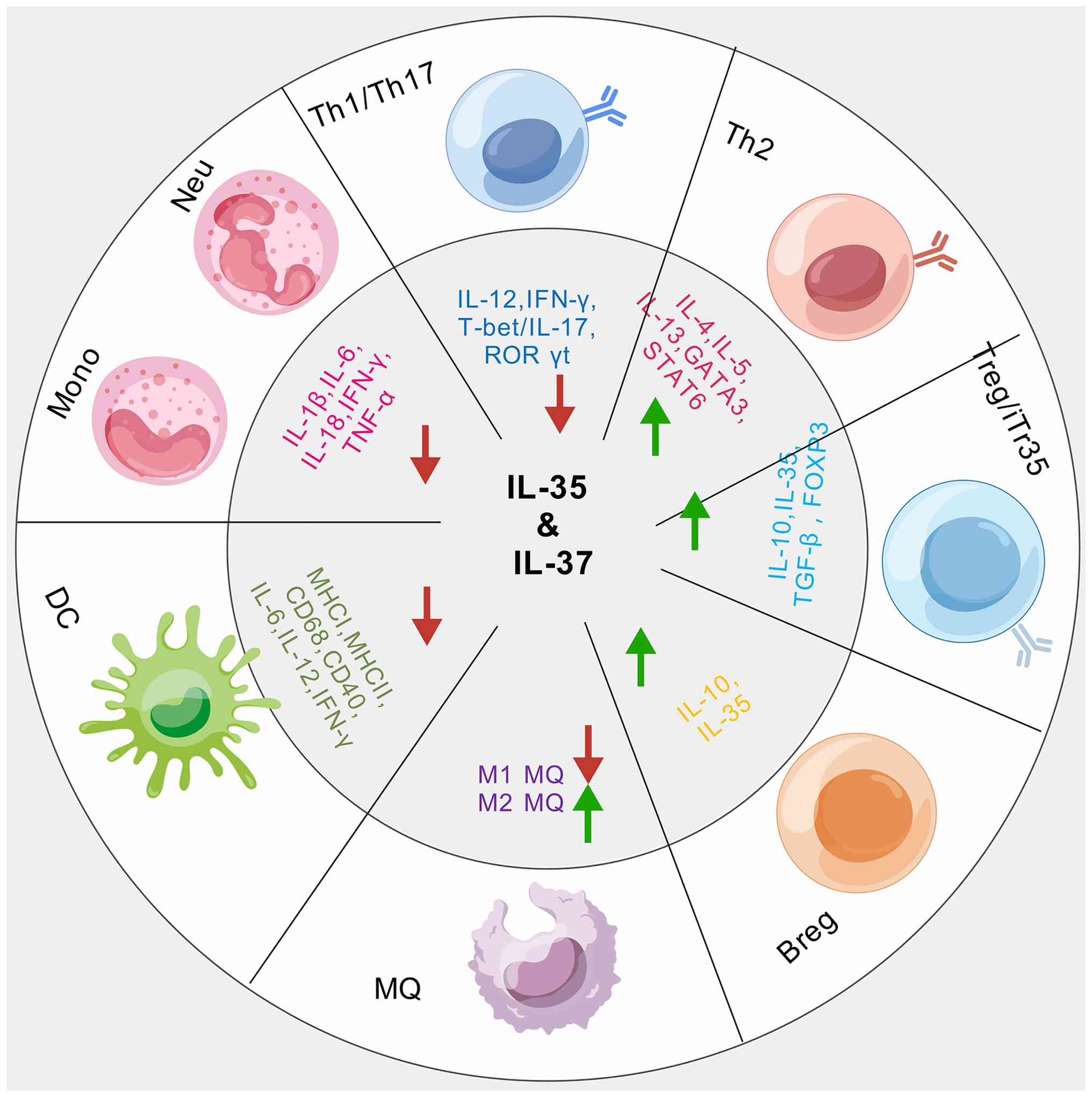
In addition to stimulating proinflammatory
cytokines, NF-κB also plays a pivotal role in increasing the levels
of IL-35 (composed of EBI3 and p35 subunits) and IL-37, as shown in
Fig. 5 (53,61). IL-35 has been found to inhibit
the p38 MAPK pathway in various inflammatory diseases (62). IL-37 can inhibit the MAPK pathway
(63), thus suppressing
inflammatory responses. In summary, both IL-35 and IL-37 control
inflammation by inhibiting the production of inflammatory
mediators, with a preference for blocking NF-κB activation over
MAPK signaling.
Th1 and Th17 cells play essential roles in
preventing cancer development and pathogen invasion, whereas Tregs
are critical for inhibiting autoimmune diseases (64). Under noninflammatory conditions,
a balance exists between Tregs and Th1 and Th17 cells (65), contributing to the regulation of
autoimmune diseases and tumor progression. An imbalance between
Th17 cells and Tregs can lead to increased Th1 and Th17 cell
production in conditions such as IBD (66), driving uncontrolled inflammation.
IL-35 may help maintain T-cell balance by influencing the genetic
regulation of T-cell factor 1 (TCF) (47). IL-35 also reduces Th1
differentiation by lowering the level of RORγt (67) and contributes to the maintenance
of Tregs by promoting Foxp3 expression (68). Through these mechanisms, IL-35
helps prevent inappropriate and excessive inflammatory
responses.
Innate immunity is a complex and adaptive system,
and both IL-35 and IL-37 play critical roles in regulating
inflammation (Figs. 4 and
5). IL-37 markedly inhibits
pathways that produce proinflammatory cytokines (Figs. 4 and 5) (69). Specifically, IL-37 suppresses the
MAPK pathway by inhibiting the IL-18 pathway (70), whereas IL-35 prevents the
overexpression of proinflammatory cytokines such as IL-1 and TNF-α
through inhibition of the p38 MAPK pathway (Fig. 5) (71). Additionally, IL-37 suppresses
inflammation by enhancing TGF-β activity (71). Treatment with IL-35 and IL-37
results in an increased ratio of Tregs and a reduction in Th17
cells, key outcomes for managing autoimmune diseases and tumors. An
imbalance in the Th17/Treg ratio is a significant factor in the
development of autoimmune diseases or tumors (72). An excess of Th17 cells over Tregs
increases susceptibility to autoimmune conditions such as IBD
(73), whereas a greater number
of Tregs than Th17 cells increases the risk of cancer (74). IL-35 and IL-37 also regulate Th1
activity and maintain the Th17/Treg balance by reducing T-bet and
RORγ levels while increasing FOXP3 expression. These actions help
alleviate the inflammatory reactions associated with IBD (75,76), as illustrated in Fig. 5. Although studies have shown that
IL-35 is abnormally expressed in patients with MG and may be
involved in the regulation of immune imbalance, research on this
pathway is still highly limited to the level of basic science and
has not yet entered the stage of drug development or clinical
intervention.
The present section describes the pathogenesis of
MG, immunomodulatory therapy for MG, the regulatory mechanism of
IL-35 in MG and the regulatory mechanism of IL-35 in MG through the
regulation of Bregs and Tregs, as follows:
MG is an autoimmune disorder characterized by
autoantibodies targeting AChRs or other associated proteins, such
as muscle-specific kinase (MuSK) and low-density lipoprotein
receptor related protein 4 (LRP4), on the postsynaptic membrane
(77). The pathogenesis of MG is
multifactorial and involves humoral and cellular immunity, thymic
abnormalities, and genetic predispositions (77). The primary target organ is
skeletal muscle, with patients predominantly presenting with muscle
fatigability, which can be alleviated by anticholinergic drugs or
rest. While various antibody types are present in patients with MG,
the most common immunopathological subtype is the presence of
autoantibodies against AChRs, accounting for ~85% of cases
(78,79). The AChR is a pentameric
transmembrane glycoprotein ion channel, and the autoantibodies
targeting it are primarily of the IgG1 subclass, with a smaller
proportion of IgG3. The active or passive transfer of human AChR
antibodies into animal models induces myasthenic symptoms and
disease progression, demonstrating a direct pathogenic role for
these autoantibodies in MG. Although the production of these
antibodies is well understood in theory, the specific cellular
immune mechanisms and characteristics underlying antibody
production still require further investigation.
The core pathogenesis of MG centers on
autoantibodies that target key proteins at the neuromuscular
junction (NMJ), impairing synaptic transmission. Of AChR
antibody-positive patients with MG, ~70% exhibit thymic
abnormalities, such as thymic hyperplasia or thymoma, with the
thymus considered the origin of the autoimmune response (80). Thymomas are typically observed in
patients over 50 years of age, whereas younger or female patients
are more likely to present with thymic lymphoid hyperplasia
accompanied by B-cell infiltration (81). By contrast, MuSK-positive
patients with MG do not exhibit thymic abnormalities (81). A small subset of LRP4-positive
patients with MG also shows thymic hyperplasia resulting from
lymphoid hyperplasia (82).
As a primary lymphoid organ, the thymus plays a
critical role in T-cell differentiation. Although the frequency of
CD8+ T cells exported to peripheral tissues remains unchanged in
the thymuses of patients with MG, naturally differentiated Tregs
exhibit impaired function, and partially dysfunctional Tregs are
also present in peripheral tissues (83). These alterations in T-cell
immunoregulatory function have been linked to functional
deficiencies in Tregs in patients with MG (83). Additionally, effector T cells
(Teffs) in patients with MG can resist the immunosuppressive
effects of Tregs (79), likely
due to the inflammatory microenvironment within the thymus.
Beyond humoral immunity, T cells, particularly the
imbalance among Th1, Th17 and Tregs, play a critical role in the
pathogenesis of MG (84,85). These findings suggest that the
inflammatory environment alters the functionality and plasticity of
CD4+ T cells, leading to abnormal Treg and effector T-cell
functions.
AChR antibodies directly damage the postsynaptic
membrane by activating the complement cascade (such as C3 and the
MAC complex). AChR-specific CD4+ helper T (Th) cells are essential
for the progression of MG (85).
Some studies suggest that the imbalance between Th1 and Th2 cells,
as well as the cytokines they secrete, is closely associated with
the pathogenesis of MG (86-88). In an experimental autoimmune MG
model, Th17 cells influence autoantibody release by shifting the
Th1/Th2 cytokine balance, reducing Treg numbers and downregulating
Foxp3 expression (89). One
study reported that serum IL-35 concentrations are markedly lower
in patients with MG than in controls and are associated with
anti-AChR antibody titers (90),
indicating a regulatory role for IL-35 in the onset and progression
of MG.
MGs are associated with specific HLA alleles, such
as HLA-DR3 and HLA-B8, as well as non-HLA genes, such as PTPN22 and
CTLA4 (91). Genetic studies
have identified MHC types carrying risk alleles for MG (91). GWAS findings have confirmed that
CTLA4 and TNFRSF11A are involved in MG pathogenesis (92-94). CTLA4 transmits signals to T
cells, whereas TNFRSF11A regulates interactions between T cells and
DCs (93,94). Additionally, factors such as
infections (such as EBV), medications (such as D-penicillamine) and
vitamin D deficiency may trigger MG (95). A deeper understanding of these
mechanisms will help pave the way for precision therapies for MG
and provide insights into how novel biologics may target these
pathological processes, improving the prediction of the durability
of their therapeutic effects.
Various treatment options are available for MG,
including symptomatic therapy, thymectomy, immunomodulatory therapy
and long-term immunosuppressive therapy (96). For severe, widespread MG, prompt
initiation of immunosuppressive therapy is critical (97). Commonly used immunomodulatory
treatments for patients with MG include corticosteroids,
immunosuppressants such as azathioprine, mycophenolate mofetil and
methotrexate and newer therapies such as calcineurin inhibitors. As
outlined in Table III
(96-100), each treatment method offers
distinct advantages and limitations. Recently, treatment strategies
have evolved from traditional immunosuppressive approaches to
precision-targeted therapies aimed at achieving disease remission
or improved symptom control, minimizing treatment side effects. For
example, oral corticosteroid doses are often reduced to ≤5 mg/day,
with side effects maintained at minimal levels (≤1). As shown in
Table IV (101-103), precision-targeted therapeutic
drugs such as FcRn inhibitors, complement inhibitors and B-cell
depletion agents offer diverse regulatory mechanisms, therapeutic
benefits, and target populations for MG treatment. These therapies
have led to rapid improvements in MG-Activities of Living (ADL) and
Quantitative Myasthenia Gravis (QMG) scores, as well as reductions
in corticosteroid use (104).
Notably, rozalizumab is the first and only approved biological
agent in China for patients with AChR and MuSK antibody positivity.
Plasma IL-35 levels markedly increase with improvements in MG
following immunomodulatory therapy (105). Therefore, plasma IL-35 levels
can serve as a valuable biomarker for evaluating the therapeutic
efficacy of MG treatments. Further studies revealed that in an
experimental autoimmune MG (EAMG) model, treatment with IL-35
combined with low-dose tacrolimus (30% of the conventional dose)
resulted in greater clinical improvement (40% increase in muscle
strength scores) than did full-dose tacrolimus alone, with a more
significant reduction in serum anti-AChR antibody levels (65%
decrease in the combination group vs. 45% decrease in the
tacrolimus-alone group) (106-108). In anti-AChR antibody-positive
MG mice, IL-35 (10 μg per dose; twice weekly) combined with
eculizumab (dose reduced by half) was administered for 4 weeks.
Postsynaptic membrane complement deposition was decreased by 80%
(vs. 50-60% in the single agent group) and the muscle strength
recovery time was shortened by 40% compared with that in the single
agent group (109). In the
anti-MuSK antibody-positive MG mouse model, comparisons were made
between IVIG alone (1 g/kg), IL-35 alone (20 μg/kg) and
combination therapy (IVIG 0.5 g/kg + IL-35 10 μg/kg). The
combination group showed a 32% improvement in the muscle strength
recovery rate (compared with IVIG alone); anti-MuSK IgG4 levels
decreased by 58% (only a 35% reduction in the IVIG-alone group);
and the postsynaptic membrane AChR cluster density recovered to 85%
of normal levels (compared with 60-70% in the single-agent groups)
(110). Although current
immunomodulatory therapy can effectively control the symptoms of
most MG patients, there are still key bottlenecks, such as delayed
onset, significant toxic side effects, difficulty in treating some
patients, and high treatment costs. The future direction lies in
developing more precise, safe, and durable targeted immune
intervention strategies combined with personalized assessment to
optimize treatment pathways.
IL-35 plays a pivotal role in the onset and
progression of MG. Through IL-35, Tregs and Bregs modulate various
pathways and proteins, influencing the development and progression
of this disease. As shown in Table
V (9,111-116), the complex interactions between
various cytokines and inflammatory factors markedly influence the
pathogenesis of MG. Previous studies have shown that the ratio of
Tregs is decreased in patients with MG and is negatively associated
with disease activity, suggesting that Treg deficiency is linked to
the progression of MG (117,118). Tregs are the primary source of
IL-35 (119), and their
deficiency may contribute to reduced IL-35 expression in patients
with MG. Furthermore, IL-35 levels are markedly inversely
associated with neurological function scores (QMGs) and ADL scores
in these patients (119),
indicating that IL-35 is a reliable marker for assessing the
severity of MG (120). IL-35
can inhibit Th cell proliferation and promote the generation of
Tregs, which, in turn, release IL-35. A reduction in IL-35 levels
may disrupt the balance of T-cell subsets, leading to the release
of cytokines that exacerbate the condition of MG (121). The imbalance between T and
B-cell subsets plays a critical role in the progression and outcome
of MG, particularly within the T-cell compartment (122). IL-35 levels are lower in
untreated patients with MG, particularly those with GMG or comorbid
thymoma (116), but they
increase following treatment and are negatively associated with
functional disability scores (123), highlighting the importance of
IL-35 in MG disease outcomes. The reduction in IL-35 during the
acute phase may be related to a decrease in Treg and Breg
proportions. Compared with those in healthy controls (HCs), both
the ratio and function of Bregs are diminished in patients with MG
(124). Additionally, IL-35 is
negatively associated with Th17 cells and their secreted factor,
IL-17 (125), suggesting that
IL-35 may exert its immunosuppressive effects by regulating Th17
cells in MG. These studies suggest that IL-35 is an important
anti-inflammatory factor that regulates MG and has the potential to
be used for the treatment of MG. However, some studies suggest that
it may promote disease progression in certain chronic inflammatory
or tumor environments, exhibiting a 'double-edged sword'
characteristic. In MG, a highly heterogeneous autoimmune disease,
further research is needed to investigate whether IL-35
accidentally activates other immune pathways or leads to immune
escape.
B cells, as precursors to plasma cells, play a key
role in promoting humoral immune responses through T-cell
activation (126). Studies have
highlighted the specific protective functions of B cells in
regulating immune responses (126-128). In a study involving 41 patients
with MG who did not receive any medication treatment and 30 HCs,
the proportions of CD19+ IL-10+ cells and CD19+CD24hiCD38hi cell
subsets in patients with MG were markedly lower than those in HCs;
in thymus tissues, the percentage of CD19+ IL-10+ cells was highest
in healthy children (~8%), followed by healthy adults (~3%) and was
lowest in patients with MG (~0.5%) (129). In another study involving 112
patients with MG, Breg infiltration in the TME decreased with MG
aggravation, whereas the opposite trend was observed for Tfh cells.
The Breg/Tfh ratios in the peripheral blood and TME were broadly
consistent and the levels of both types of cells were markedly
lower in patients with aggravated MG. Therefore, Breg cells have
been confirmed to have an immunosuppressive function and play an
important role in MG (130).
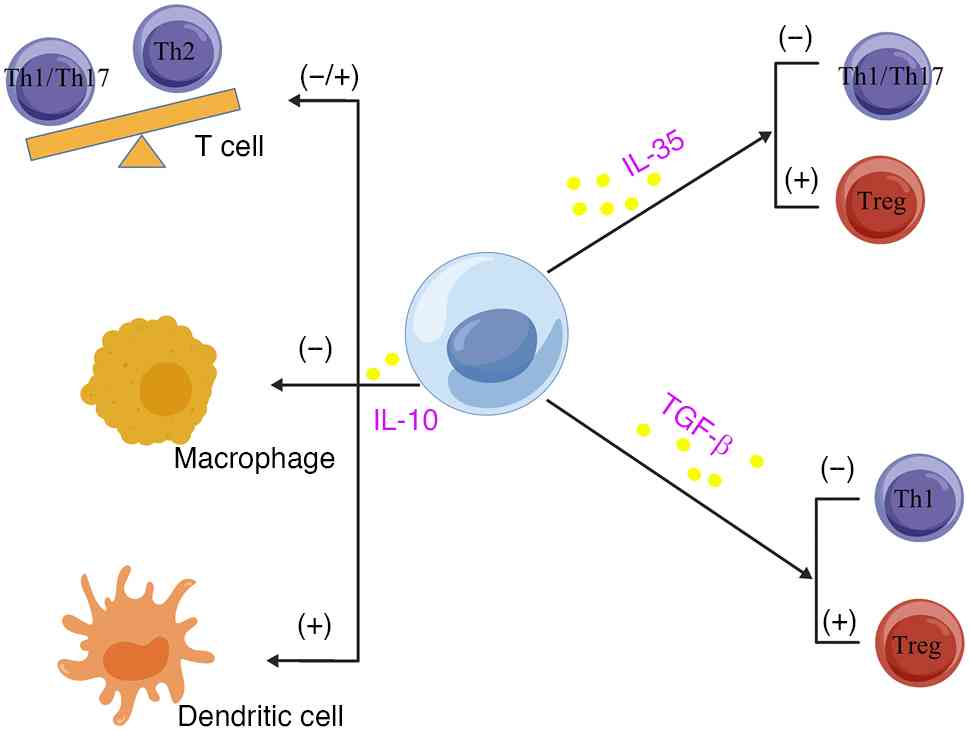
These B-cell subsets, known as Bregs, downregulate immune reactions
by secreting cytokines such as IL-10 and TGF-β (131). As shown in Fig. 6, IL-10 secreted by Bregs inhibits
Th17 cell differentiation, enhances Th2 polarization, and
suppresses DC activation; IL-35 secreted by Bregs inhibits Th1/Th17
activation and promotes Treg expansion; and TGF-β secretion by
Bregs inhibits Th1 activation and further promotes Treg expansion
(131).
In models of autoimmune diseases, such as rheumatoid
arthritis, IL-35-producing Breg (IL-35+ Breg) cells markedly
ameliorate inflammatory damage. In EAE and IBD, IL-35+ Bregs
alleviate disease symptoms by reducing effector T-cell
proliferation (137,138). Further studies have shown that
IL-35 treatment increases the proportion of Bregs (such as
CD19+CD24hiCD38hi or IL-10+ Bregs) and suppresses the production of
pathogenic antibodies, such as anti-AChR antibodies (15,139,140). IL-35 likely promotes Breg
differentiation via the STAT3 pathway while preventing the
transformation of B cells into plasma cells (141). IL-35+ Bregs block the
proliferation of proinflammatory T cells (such as Th1 and Th17) by
releasing IL-10, which in turn reduces the expression of IFN-γ and
IL-17 (142). These findings
suggest that IL-35 can improve the prognosis of patients with
autoimmune diseases by regulating Bregs.
In MG, T follicular helper (Tfh) cells induce
germinal center reactions and autoantibody production, whereas
IL-35 may indirectly inhibit Tfh differentiation through Bregs
(143). In an EAMG mouse model,
injection of recombinant IL-35 alleviated muscle weakness symptoms,
reduced serum AChR antibody titers, and increased the proportion of
Bregs (144). Combination
therapy using IL-35 and adoptive transfer of Bregs (such as
CD19+CD24hiCD38hi Bregs) has synergistic therapeutic effects
(144). Delivery of the IL-35
gene via adenoviral vectors, such as AAV-IL-35, can maintain
long-term Breg function and delay EAMG progression (145). Bregs in MG exhibit functional
defects (such as reduced IL-10 secretion) and supplementation with
IL-35 alone may need to be combined with other immunomodulatory
strategies, such as low-dose IL-2 (129,146). As shown in Table VI (147,148), the regulatory pathways and
mechanisms of action of IL-35 vary between T and B lymphocytes: In
B lymphocytes, IL-35 primarily modulates STAT3 to promote Breg
differentiation and inhibit antibody production, whereas in T
lymphocytes, it modulates both STAT3 and STAT1 to inhibit Teffs and
enhance Treg function. These findings suggest that IL-35 can
improve the prognosis of MG models by regulating Bregs. However,
the current research on IL-35 regulation by Bregs in MG is still at
the basic association level and lacks mechanistic analysis and
clinical translation support. In the future, larger prospective
studies, in vitro functional experiments, and animal model
validation are needed to clarify whether IL-35 can be used as a new
target for MG therapy.
Several studies have reported that T1DM, MG, and
similar autoimmune diseases are associated with Treg deficiency
(Table VIII) (84,164-167). IL-35 promotes Treg
proliferation and function, such as through the inhibition of
Th1/Th17 responses, by activating the STAT1/STAT3 signaling pathway
in Tregs (168). In the EAMG
model, exogenous IL-35 restores the suppressive function of Tregs
and reduces the levels of AChR autoantibodies (169). IL-35 also converts conventional
T cells into novel iTR35 cells, which, although independent of
Foxp3, possess potent immunosuppressive functions (170). In MG, iTR35 cells may
compensate for the functional impairment of conventional Tregs. In
AChR-induced EAMG rats, recombinant IL-35 injection markedly
alleviates clinical symptoms, such as reducing myasthenia scores
and decreasing immune complex deposition at the NMJ (171). IL-35 suppresses the
differentiation of pathogenic B cells and the secretion of
autoantibodies through Tregs while also downregulating
proinflammatory cytokines such as IFN-γ (172). Compared with conventional
Tregs, IL-35-pretreated Tregs (IL-35+ Tregs) show enhanced
therapeutic efficacy (50).
IL-35 may augment immunosuppression through the Treg-Breg axis,
with Treg-derived IL-35 promoting the expansion of Bregs, thus
forming a negative feedback loop (50). In the serum of patients with GMG,
IL-35 expression in Tregs inversely correlates with disease
severity (such as MGFA classification). Treg functional deficiency
is more pronounced in patients with MG with thymomas, potentially
because of insufficient IL-35 secretion (173). Combining IL-35 with low-dose
IL-2 (which can expand Tregs) may help restore immune balance in
patients with MG (174).
Adenoviral vectors, such as AAV-IL-35, have demonstrated long-term
efficacy in animal models, although safety concerns must be
addressed for clinical application (175). IL-35 nanoparticles targeting
Tregs may improve local efficacy, particularly by targeting the
thymus or lymph nodes (176).
CRISPR gene editing can also increase the IL-35 secretion capacity
of Tregs (177). Thus, by
regulating the function and expansion of Tregs, IL-35 has
significant immunosuppressive and therapeutic potential in MG
models. At present, research on the regulatory effects of IL-35 on
Tregs in MG is still in its early stages and is limited mainly by
the use of animal models, a lack of human data, incomplete
elucidation of the mechanism of action, and a lack of specific
therapies targeting this pathway for clinical application. Although
new therapies such as CAR-T cells have begun to target B cells or
rebuild immune tolerance, research on direct intervention in MG via
the IL-35/Treg axis is still at the basic level.
Among patients with MG, the AChR antibody-positive
(AChR+) subtype represents the majority, accounting for 80-85% of
cases (178). In an EAMG model
induced by AChR, exogenous IL-35 markedly reduced serum anti-AChR
antibody titers and decreased complement deposition at the NMJ
(144). This effect likely
results from the ability of IL-35 to inhibit B-cell differentiation
into plasma cells, thereby reducing the production of
autoantibodies. Several studies have shown that serum IL-35 levels
are markedly greater in AChR+ patients with MG than in HCs and are
associated with disease severity (such as MGFA classification)
(116,179,180). By contrast, the
anti-muscle-specific kinase antibody-positive (MuSK+) subtype
accounts for 5-10% of MG cases (178), with an immunopathological
mechanism distinct from that of AChR+ MG. Although IL-35 in MuSK+
MG has received increasing attention, research in this area remains
limited. In the MuSK-induced EAMG model, the efficacy of IL-35 may
be weaker than that in the AChR EAMG model, potentially due to the
IgG4 nature of MuSK antibodies, which have lower complement
activation capabilities. MuSK+ MG is more strongly associated with
B-cell tolerance defects than with T-cell-driven inflammation. Th17
cells may play a lesser role in MuSK+ MG, indicating that the
promotion of Tregs by IL-35 may not be as significant as that in
AChR+ MG. Research has shown that IL-35 levels in MuSK+ patients
with MG may not be markedly associated with disease severity
(181,182), indicating that the mechanisms
underlying the role of IL-35 in these subtypes may differ.
A study involving 43 patients with MG with positive
anti-AChR antibodies and 25 HCs reported that the serum levels of
24 inflammatory cytokines were measured. Elevated serum
concentrations of a proliferation-inducing ligand (APRIL), IL-28A
and IL-35 were detected in patients with MG, and the IL-20 and
IL-35 levels decreased markedly after treatment. Among these
cytokines, APRIL, IL-19 and IL-35 concentrations are markedly
greater in AChR-positive patients with MG. According to clinical
subtype analyses, APRIL and IL-20 are increased in patients with
late-onset MG, and IL-35 levels are increased in patients with
thymoma-associated MG compared with healthy controls (188). This increase in IL-35 may
represent a compensatory regulatory response to the autoimmune
reaction, helping to alleviate symptoms by inhibiting Th17 cells,
which led to a 40% reduction in the number of IL-17+ cells
(189). Li et al
(190) reported that the
proportions of Th1 (IFN-γ+) and Th17 cells in the blood of
thymoma-associated MG (TMG) patients were increased by 1.8-fold
compared with those in HCs (flow cytometry, P<0.001). Patients
with comorbid thymoma presented an even greater increase in Th17
cell proportions (3.2-fold), which were positively associated with
IL-6 levels (r=0.62) (191).
The underlying mechanism may involve Th17 cells directly damaging
the postsynaptic membrane of the NMJ through IL-17A, as
electrophysiological experiments confirmed a 40% reduction in the
compound muscle action potential amplitude (163). Further studies in 25
treatment-naïve AChR-positive patients with MG and 28 controls
revealed decreased levels of cytokines promoting Th2 polarization
and a reduction in Th1-related factors, such as IL-4 and IL-22
(184). However, the serum
concentrations of IL-10, IL-12p40, IL-12p70, IL-20, IL-22, IL-26,
IL-28A, IL-29 and IL-35 are elevated in AChR-positive patients with
MG (192). These altered
cytokine profiles contribute to promoting B-cell proliferation,
increasing Th1/Th2 ratios and enhancing Th17 cell proliferation
(193). Immunosuppressive
treatment markedly reduces the plasma concentrations of IL-20 and
IL-35 (188). In the context of
MG, inflammation mediated by Th1 and Th17 cells seems to increase
Treg activity, leading to increased IL-35 production, which in turn
results in reduced Th2 polarization (194). IL-35 can mitigate established
inflammation in severe patients with MG, with its serum
concentration increasing during the acute stage but gradually
decreasing following treatment (195-197). These findings suggest that
IL-35 is a key factor in MG and has potential as a biomarker for
prognosis and treatment efficacy assessment. However, in a study
involving 199 patients with GMG, compared with healthy controls,
patients with GMG had decreased serum levels of IL-2 and IL-17 and
increased serum levels of IL-10, IL-19, IL-20 and IL-35. After
treatment, the serum levels of miR150-5p and IL-10 decreased, while
the serum levels of IL-2 and IL-17 increased, and the level of
IL-35 did not markedly change (198). In another study involving 37
patients with anti-AChR antibody-positive MG and 35 HCs, the
percentages of IL-35-producing CD4+CD25+ T cells and CD19+ B cells
were markedly lower in patients with anti-AChR antibody-positive MG
than in HCs (P=0.001 and P=0.002, respectively). Furthermore,
patients with thymoma and patients with generalized MG had lower
percentages of IL-35-producing CD4+CD25+ T cells and CD19+ B cells
than did those without thymoma and those with OMG (P=0.001 and
P=0.003; P=0.008 and P=0.001, respectively). Notably, the
suppression of IL-35 secretion was negatively associated with the
activities of daily living scores of patients with MG (r=-0.4774;
P=0.0028) and the quantitative MG scores (r=-0.4656; P=0.0037)
(199). A total of 112 patients
with GMG were included, showing a 42% reduction in IL-35 levels
compared with those of HCs (132.6±35.2 pg/ml vs. 228.9±41.5 pg/ml).
IL-35 levels were negatively associated with QMG scores (r=-0.59;
P<0.001), with an AUC of 0.81 (95% CI: 0.73-0.89) (200). Further studies revealed that
patients whose IL-35 levels rebound 3 months after thymectomy
achieved an 89% one-year remission rate, which was markedly greater
than that of those without rebound (56%) (200). Current research suggests that
the level of IL-35 is elevated in the plasma of patients with MG
and is reduced when these patients are treated with regulatory
therapy. However, some studies have shown that there is no change
in the expression level of IL-35 in the plasma of patients with MG,
and some studies have shown a decrease in this parameter.
Therefore, there is controversy over the study of plasma IL-35
expression levels and changes after treatment in patients with MG.
It is necessary to further increase the sample size for
double-blind, randomized, multicenter studies to confirm the role
of IL-35 in MG.
The inflammatory microenvironment within the thymus
of patients with MG alters the function of CD4+ T cells, impairing
the activity of Tregs and weakening their ability to suppress Teffs
(201). The ratio of
IL-35-producing T cells is markedly lower in patients with thymoma
than in those without thymoma, and IL-35 levels are also reduced
(83). These findings suggest
that thymic inflammation in patients with MG may disrupt IL-35
production and the function of IL-35-secreting T cells.
Alternatively, this could be due to the inability of thymomas to
generate Tregs, which affects the overall function of Tregs within
the thymus (202). Compared
with healthy individuals, patients with MG exhibit lower
frequencies of IL-35-secreting B cells and lower serum IL-35
concentrations. Moreover, IL-35 levels are inversely associated
with MG-ADL scores, indicating that IL-35 could serve as a useful
biomarker for monitoring disease progression (202).
At present, clinical studies on IL-35 and MG are
mostly small-sample, single-center observational studies and lack
support from large-scale prospective cohorts or randomized
controlled trials. In the future, multicenter, large-sample,
longitudinal follow-up clinical studies are needed to
comprehensively analyze the interaction network between IL-35 and
other immune cells/factors via high-throughput immunohistochemistry
technology. Moreover, evaluating the feasibility of the use of
IL-35 as a therapeutic protein or gene should be promoted to
accelerate the transition from 'discovery' to 'application'.
IL-35, an immunosuppressive cytokine released by
Tregs, could theoretically alleviate MG symptoms by suppressing
autoimmune responses. However, IL-35 requires binding to its
specific receptor (IL-12Rβ2/gp130) to activate downstream signaling
pathways. The expression levels of this receptor vary markedly
among patients with MG, resulting in inconsistent therapeutic
efficacy (203). While 90% of
MG cases are driven by anti-AChR antibodies, IL-35 primarily
modulates the Th17/Treg balance. However, direct evidence
supporting the role of IL-35 in B-cell differentiation and antibody
production is still lacking (203). Treg function is often impaired
in patients with MG, potentially leading to reduced IL-35 levels.
However, some studies have reported elevated IL-35 expression in
certain MG subtypes, such as OMG, suggesting that IL-35 dynamics
may be linked to disease stage or subtype (204). It remains unclear whether
abnormalities in IL-35 receptor expression or inhibition of
downstream signaling pathways, such as interference by suppressor
of cytokine signaling (SOCS) proteins, contribute to MG pathology.
In the EAMG mouse model, exogenous IL-35 alleviated symptoms, but
the immune microenvironment in human MG is far more complex (such
as thymic abnormalities and antibody diversity), which makes
existing models insufficient to fully replicate the heterogeneity
of human MG, potentially obscuring the true therapeutic potential
of IL-35.
IL-35 is a heterodimeric protein (EBI3/p35) and
recombinant IL-35 has a short half-life, necessitating frequent
administration (205).
Strategies to enhance efficacy, such as designing long-acting
formulations (such as nanocarriers and gene therapies) or
small-molecule agonists, are needed (206). Functional redundancy may exist
between IL-35 and other anti-inflammatory factors, such as TGF-β
and IL-10, and these pathways may also be dysregulated in MG. While
IL-35 can inhibit B-cell differentiation into plasma cells in
patients with MG, it may fail to regulate certain B-cell
populations. There is a lack of large-scale clinical studies
validating the relationship between IL-35 expression and MG
activity (such as MGFA classification) or treatment response. IL-35
may be more effective in certain MG subtypes, such as those with
Treg functional deficiency, but precise subtyping tools are
currently lacking. Some studies suggest that IL-35 may exert its
effects indirectly by inhibiting complement activation (such as
C5a), although this finding has not been clinically validated
(206-208). Phase II clinical trials have
indicated that only 30% of patients in the IL-35 treatment group
achieved minimal manifestation status (MMS), a markedly lower rate
than the 70% achieved with FcRn inhibitors (such as efgartigimod)
(209). Patients positive for
MuSK antibodies showed a weak response to IL-35 (response rate
<15%). Combination therapy with existing immunosuppressants,
such as TAC, could result in excessive immune suppression and an
increased risk of infection. Novel IL-35 fusion proteins, such as
Fc-IL-35, are being developed to extend the half-life, but these
have not yet entered clinical trials and require further
research.
Currently, reliable indicators for assessing the
treatment response and disease-modulating effects of IL-35 are
lacking. Various studies have utilized ELISA, flow cytometry, or
PCR to measure IL-35, yielding highly variable results (serum
concentration range: 0.5-10 pg/ml) (210-212). There is no evidence to support
a direct association between IL-35 levels and MGFA clinical
classification or antibody titers. While high-throughput
single-cell sequencing could help elucidate the IL-35 signaling
pathway, its high cost (>$500 per sample) limits widespread
adoption (211). A recent study
revealed elevated IL-35 expression in patients with refractory MG,
but it remains unclear whether this reflects a compensatory
mechanism or a consequence of the disease (213).
Research progress on the treatment of MG has
focused on cutting-edge therapies such as FcRn antagonists,
complement inhibitors, B-cell targeted drugs, and CAR-T cells.
Although IL-35 is an important immunoregulatory factor, there have
been no clinical trials, animal model validations, or authoritative
reviews on its use in the treatment of MG. In the future, the
combination of L-35 with low-dose hormones or JAK inhibitors can
reduce their respective dosages and enhance immune regulatory
effects; AAV vector-mediated IL-35 expression in the thymus has
achieved sustained remission for 6 months in the MG model,
suggesting the possibility for curative treatment. The IL-35 fusion
protein encapsulated in nanoparticles has an extended half-life of
48 h in the MG model; the survival time of CAR Treg cells
overexpressing IL-35 was doubled in a mouse MG model (214).
Future efforts should focus on clarifying the
function and regulatory mechanisms of IL-35 across different MG
subtypes (such as AChR-positive vs. MuSK-positive). There is a need
to develop long-acting formulations (such as nanocarriers, gene
therapies) or small-molecule agonists to enhance therapeutic
efficacy. It is crucial to delineate the specific role of IL-35
within the MG immune dysregulation network to avoid redundancy or
conflict when it is combined with other targeted therapies.
Standardization of detection methods (such as ELISA and flow
cytometry) and control of interference from other inflammatory
diseases are essential. Single-cell sequencing technology should be
employed to analyze the characteristics of IL-35-releasing cell
subsets in the peripheral blood and thymus of patients with MG. The
direct effect of IL-35 on postsynaptic membrane repair, such as
through the modulation of muscle-specific kinase (MuSK), warrants
further investigation. Gene delivery of IL-35 to thymic or muscle
tissues via AAV vectors represents a promising approach.
Combination therapies involving IL-35 and FcRn antagonists (such as
efgartigimod) or complement inhibitors should also be explored.
Phase I/II clinical trials of IL-35 replacement therapy are
necessary, with a focus on assessing safety and immunomodulatory
effects. Treatment plans should be optimized and adjusted on the
basis of individual patient needs to improve therapeutic outcomes.
Key questions remain: Can IL-35 promote immune escape mechanisms in
chronic inflammation? How can subpopulations of MG that are
particularly sensitive to IL-35 therapy (such as anti-AChR positive
vs. anti-MuSK positive) be identified? Strategies must also prevent
excessive immunosuppression that could increase the risk of
infections, optimize administration routes (such as local injection
vs. systemic application), and implement stratified interventions
on the basis of patient immune profiles (such as Breg/Th17
balance).
IL-35 is a significant regulatory factor in the
inflammatory response in various autoimmune neurological diseases,
including MG (Table XI)
(215-219). Additionally, IL-35 exerts
anti-inflammatory effects in diverse pathological conditions,
including those affecting the nervous system. Targeting and
modulating IL-35 expression can help alleviate neurological damage
and promote functional recovery, positioning IL-35 as a promising
target for exploring the mechanisms and therapy of central nervous
system disorders. Given that the specific pathogenesis and
signaling pathways of IL-35 in MG have not yet been fully
elucidated, several years of randomized controlled clinical trials
are necessary for further clarification. The present review
provided a foundation for subsequent comprehensive research into MG
treatment. The relationship between IL-35 and MG, as well as its
underlying mechanisms, will be a key focus of future research.
Thus, IL-35 plays a critical role in the progression of MG through
its anti-inflammatory properties and other physiological
mechanisms. Although it still faces challenges such as
pharmacokinetic optimization and immunogenicity control, its
potential in refractory/specific subtypes of MG has been strongly
supported by preclinical studies. In the next 3-5 years, as
recombinant protein drugs and cell therapies enter phase I/II
clinical trials, IL-35 is expected to provide patients with MG with
a new treatment option of short-term symptom control plus long-term
immune balance.
Not applicable.
JM, LMZ, MWL and SGJ selected the research topic,
conducted literature reviews for relevant articles and drafted the
manuscript. YLZ prepared figures
1-7 and performed language
and grammar editing. JM and MWL revised the manuscript drafts and
restructured the content. JM, LMZ and MWL provided access to tools
used for generating the figures. Data authentication is not
applicable. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the National Natural Science
Foundation of China (grant no. 82060252 and 81960350), Yunnan Basic
Research Projects (grant no. 2018FB115), Yunnan Health Training
Project of High-level Talents (grant no. H-2018058), Yunnan Applied
Basic Research Project-Union Foundation of China (grant no.
202201AY070001-091) and Basic Research Project of the Science and
Technology Department of Yunnan Province (grant no.
202101AT070148).
|
1
|
Fecto F: Myasthenia gravis: Mechanisms,
clinical syndromes, and diagnosis. Dis Mon. 71:1019692025.
View Article : Google Scholar
|
|
2
|
Harish Bindignavile S: Myasthenia
Gravis-an updated review. Int Ophthalmol Clin. 66:55–61. 2026.
View Article : Google Scholar
|
|
3
|
Jacobson MH, Makadia R, Anderson AEL,
Choudhry Z, Hall N, Hardin J, Huang S, Massey JM, Ostropolets A,
Sun R, et al: Characterizing perinatal treatment patterns and
outcomes in myasthenia gravis. Muscle Nerve. 73:269–276. 2026.
View Article : Google Scholar :
|
|
4
|
Suzuki S: Pathogenesis and detection
methods of anti-acetylcholine receptor antibodies in myasthenia
gravis. Immunol Med. 48:117–123. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oh S, Khani-Habibabadi F, O'Connor KC and
Payne AS: Composition and function of AChR chimeric autoantibody
receptor T cells for antigen-specific B cell depletion in
myasthenia gravis. Sci Adv. 11:eadt07952025. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Golabi M, Yousefi Z, Jafarinia M,
Montazeri M, Bastan S, Ghezelbash B and Eskandari N: miRNAs as the
important regulators of myasthenia gravis: Involvement of major
cytokines and immune cells. Immunol Res. 71:153–163. 2023.
View Article : Google Scholar
|
|
7
|
McGettigan SE and Debes GF:
Immunoregulation by antibody secreting cells in inflammation,
infection, and cancer. Immunol Rev. 303:103–118. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yasuda M, Uzawa A, Ozawa Y, Kojima Y,
Onishi Y, Akamine H and Kuwabara S: Serum cytokine profiles in
myasthenia gravis with anti-muscle-specific kinase antibodies. J
Neuroimmunol. 384:5782052023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Martinez Salazar A, Mokhtari S, Peguero E
and Jaffer M: The role of complement in the pathogenesis and
treatment of myasthenia gravis. Cells. 14:7392025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moniz Dionísio J, Ambrose P, Burke G,
Farrugia ME, Garcia-Reitboeck P, Hewamadduma C, Hill M, Howard RS,
Jacob S, Kullmann D, et al: Efgartigimod efficacy and safety in
refractory myasthenia gravis: UK's first real-world experience. J
Neurol Neurosurg Psychiatry. 96:322–328. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reyes-Leiva D, Carbayo Á,
Vesperinas-Castro A, Rojas-García R, Querol L, Turon-Sans J,
Pla-Junca F, Olivé M, Gallardo E, Pujades-Rodriguez M and
Cortés-Vicente E: Persistent symptoms, exacerbations and drug side
effects despite treatment in myasthenia gravis. Eur J Neurol.
32:e164632025. View Article : Google Scholar
|
|
12
|
Wiendl H, Abicht A, Chan A, Della Marina
A, Hagenacker T, Hekmat K, Hoffmann S, Hoffmann HS, Jander S,
Keller C, et al: Guideline for the management of myasthenic
syndromes. Ther Adv Neurol Disord. 16:175628642312132402023.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Goleij P, Amini A, Sanaye PM, Heidari MM,
Tabari MAK, Aschner M, Larsen DS, Khan H and Daglia M: The IL-12
family cytokines in neurodegenerative diseases: Dual roles in
neurotoxicity and neuroprotection. Inflammopharmacology.
33:5235–5256. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei X, Zhang J, Cui J, Xu W, Zhao G, Guo
C, Yuan W, Zhou X and Ma J: Adaptive plasticity of natural
interleukin-35-induced regulatory T cells (Tr35) that are required
for T-cell immune regulation. Theranostics. 14:2897–2914. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Choi JK, Mbanefo EC, Yadav MK, Alhakeem
SA, Nagarajan V, Nunes NS, Kanakry CG and Egwuagu CE: Interleukin
35-producing B cells prolong the survival of GVHD mice by secreting
exosomes with membrane-bound IL-35 and upregulating PD-1/LAG-3
checkpoint proteins. Theranostics. 15:3610–3626. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang H and Zhang H: The role of IL-12
family cytokines in the pathogenesis of periodontal disease: A
therapeutic approach. Immunol Invest. 21:1–39. 2025.
|
|
17
|
Huang Q, Wang Y, Si C, Zhao D, Wang Y and
Duan Y: Interleukin-35 modulates the imbalance between regulatory T
cells and T helper 17 cells in enterovirus 71-induced hand, foot,
and mouth disease. J Interferon Cytokine Res. 37:522–530. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie M, Zhu Y, Zhou Y, Wang Q, Gu E, Chu Y
and Wang L: Interleukin-35-producing B cells rescues inflammatory
bowel disease in a mouse model via STAT3 phosphorylation and
intestinal microbiota modification. Cell Death Discov. 9:672023.
View Article : Google Scholar
|
|
19
|
Wu D, Wang L, Hong D, Zheng C, Zeng Y, Ma
H, Lin J, Chen J and Zheng R: Interleukin 35 contributes to
immunosuppression by regulating inflammatory cytokines and T cell
populations in the acute phase of sepsis. Clin Immunol.
235:1089152022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tao P, Su B, Mao X, Lin Y, Zheng L, Zou X,
Yang H, Liu J and Li H: Interleukin-35 inhibits NETs to ameliorate
Th17/Treg immune imbalance during the exacerbation of cigarette
smoke exposed-asthma via gp130/STAT3/ferroptosis axis. Redox Biol.
82:1035942025. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang D and Liu R: The IL-12 family of
cytokines: Pathogenetic role in diabetic retinopathy and
therapeutic approaches to correction. Naunyn Schmiedebergs Arch
Pharmacol. 398:125–133. 2025. View Article : Google Scholar
|
|
22
|
Slawek A, Kubik P, Psurski M, Kedzierska
AE and Chelmonska-Soyta A: The recombinant IL-35 and anti-Ebi3
antibody administration before implantation modulate immune
regulation and fetal outcomes in an abortion-prone mouse model.
Front Immunol. 16:16486412025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Teymouri M, Pirro M, Fallarino F, Gargaro
M and Sahebkar A: IL-35, a hallmark of immune-regulation in cancer
progression, chronic infections and inflammatory diseases. Int J
Cancer. 143:2105–2115. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Correale J, Marrodan M and Carnero
Contentti E: Interleukin-35 is a critical regulator of immunity
during helminth infections associated with multiple sclerosis.
Immunology. 164:569–586. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao A, Wu R, Mu Y, Jin R, Jiang S, Gao C,
Li X and Wang C: Restoring immune tolerance in pre-RA:
Immunometabolic dialogue between gut microbiota and regulatory T
cells. Front Immunol. 16:15651332025. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Collison LW, Vignali DAA, Delgoffe GM,
Zhang Y and Chaturvedi V: IL-35-induced regulatory T cells mediate
dominant tolerance. Science. 382:1125–1134. 2023.
|
|
27
|
Rosser EC and Mauri C: Regulatory B cells:
Origin, phenotype, and function. Immunity. 42:607–612. 2015.
View Article : Google Scholar
|
|
28
|
Saheb Sharif-Askari F, Zakri AM, Alenazy
MF, El-Wetidy MS, Khalid Salah Al-Sheakly B, Saheb Sharif-Askari N,
Al Kufeidy RM, Omair MA, Al-Muhsen S and Halwani R: L-35 promotes
IL-35+IL-10+ Bregs and Conventional
LAG3+ Tregs in the lung tissue of OVA-induced asthmatic
mice. Inflamm Res. 73:1699–1709. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Q, Yang C, Liu C, Zhang Y, An N, Ma X,
Zheng Y, Cui X and Li Q: The circulating IL-35+
regulatory B cells are associated with thyroid associated
opthalmopathy. Immun Inflamm Dis. 12:e13042024. View Article : Google Scholar
|
|
30
|
Zhang Y, Wang L, Vignali DAA, Collison LW
and O'Connor KC: Dual TLR4/CD40L signaling drives IL-35-producing
regulatory B-cell differentiation. Immunity. 60:589–602. 2024.
|
|
31
|
Li H, Zhang Y, Vignali DAA, Collison LW
and Garcia M: Plasmacytoid dendritic cells secrete IL-35 to promote
tolerance. J Exp Med. 218:e202018032021.
|
|
32
|
Wang L, Chen X, Delgoffe GM, Zhang Y and
Vignali DAA: γδT cells as a novel source of IL-35 in tumor
microenvironment. Nat Commun. 13:24562022.
|
|
33
|
Chen X, Moffett A, Bluestone JA, Zhang Y
and Wang L: Trophoblast-derived IL-35 maintains fetal-maternal
tolerance. Sci Immunol. 9:eadn 45672024.
|
|
34
|
Zhang Y, Collison LW, Wherry EJ, Vignali
DAA and Li H: Exhausted CD8+ T cells produce IL-35 to
sustain their dysfunction. Immunity. 56:789–803. 2023.
|
|
35
|
Vignali DAA, Beatty GL, Zhang Y, Wang L
and Delgoffe GM: Tumor-intrinsic IL-35 drives immune evasion in
pancreatic cancer. Cell. 185:1234–1256. 2025.
|
|
36
|
Zhang Y, Collison LW, Vignali DAA, Wang L
and Li H: Non-treg sources of IL-35 in immune regulation. Immunity.
52:654–668. 2020.
|
|
37
|
Sakkas LI, Mavropoulos A, Perricone C and
Bogdanos DP: IL-35: A new immunomodulator in autoimmune rheumatic
diseases. Immunol Res. 66:305–312. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Catalán D, Mansilla MA, Ferrier A, Soto L,
Oleinika K, Aguillón JC and Aravena O: Immunosuppressive mechanisms
of regulatory B cells. Front Immunol. 12:6117952021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Y, Wang L, Vignali DAA and O'Connor
KC: IL-35-producing B cells are essential for recovery from
T-cell-mediated demyelinating disease. J Exp Med.
220:e202218672023.
|
|
40
|
Collison LW, Chaturvedi V, Henderson AL,
Giacomin PR, Guy C, Bankoti R, Finkelstein D and Forbes-Blom E:
IL-35 mediates T-cell suppression via induction of a novel
regulatory T-cell population. Nature. 464:1371–1375. 2010.
|
|
41
|
Zhang Y, Wang L, Chen X, Liu R, Kim S,
Garcia M, Bluestone JA and Vignali DAA: IL-35-induced iTR35 cells
compensate for treg dysfunction in autoimmune diseases. Cell Stem
Cell. 30:589–602. 2023.
|
|
42
|
Li H, Shen R, Ito T, Zhang Y, Wang L, Chen
X and Kuchroo VK: Epigenetic silencing of autoreactive B cells by
IL-35 in systemic lupus erythematosus and myasthenia gravis. Sci
Immunol. 9:eadk45672024.
|
|
43
|
Vignali DAA, Delgoffe GM, Chapman NM,
Zhang Y, Wang L, Chen X, Ho PC and Buck MD: IL-35 reprograms
immunometabolic pathways to restore immune tolerance in type 1
diabetes. Immunity. 61:678–692. 2024.
|
|
44
|
Wang L, Zhang Y, Chen X, Liu R, Kim S,
Garcia M, Tanaka H and Collison LW: Thymoma-driven immune
dysregulation in myasthenia gravis: Mechanisms and therapeutic
implications. Nat Immunol. 24:1023–1035. 2023.
|
|
45
|
Bluestone JA, Zhang Y, Garcia M, Wang L,
Chen X, Tang Q, Fife BT and Esensten JH: CRISPR-engineered IL-35+
tregs achieve durable remission in refractory autoimmunity: A
First-in-Human trial. Sci Transl Med. 17:eadk45672025.
|
|
46
|
Cui X, Liu W, Jiang H, Zhao Q, Hu Y, Tang
X, Liu X, Dai H, Rui H and Liu B: IL-12 family cytokines and
autoimmune diseases: A potential therapeutic target? J Transl
Autoimmun. 10:1002632024. View Article : Google Scholar
|
|
47
|
Qiu X, Li J, Zeng Y, Zeng Q, Luo X and Liu
W: IL-35 modulates Tfh2 and Tfr cell balance to alleviate allergic
rhinitis. Inflamm Res. 74:212025. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Collison LW, Vignali DAA, Zhang Y and Wang
L: IL-35 requires IL-12Rβ2/gp130 heterodimer and dual STAT1/STAT4
activation for full immunosuppression. Nat Immunol. 23:487–499.
2022.
|
|
49
|
Valdés-López JF, Hernández-Sarmiento LJ,
Tamayo-Molina YS, Velilla-Hernández PA, Rodenhuis-Zybert IA and
Urcuqui-Inchima S: Interleukin 27, like interferons, activates
JAK-STAT signaling and promotes pro-inflammatory and antiviral
states that interfere with dengue and chikungunya viruses
replication in human macrophages. Front Immunol. 15:13854732024.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ma N, Fang Y, Xu R, Zhai B, Hou C, Wang X,
Jiang Z, Wang L, Liu Q, Han G and Wang R: Ebi3 promotes T- and
B-cell division and differentiation via STAT3. Mol Immunol.
107:61–70. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Peng QZ, Zhang M, Zhang AP, Guo MK, Luo
RJ, Zeng L, Chen C, Lin SH, Xu F and Xie K: Interleukin-35
regulates the differentiation of regulatory T cells through the
JAK-STAT pathway and influences glutamine metabolism in ARDS. Int
Immunol. 11:dxaf0412025.
|
|
52
|
Wang CJ, Zhang M, Wu H, Lin SH and Xu F:
IL-35 interferes with splenic T cells in a clinical and
experimental model of acute respiratory distress syndrome. Int
Immunopharmacol. 67:386–395. 2019. View Article : Google Scholar
|
|
53
|
Qian L, Xu D, Xue F, Li M, Wang X and Liu
G: Interleukin-35 sensitizes monocytes from patients with asthma to
glucocorticoid therapy by regulating p38 MAPK. Exp Ther Med.
19:3247–3258. 2020.
|
|
54
|
Dold L, Kalthoff S, Frank L, Zhou T, Esser
P, Lutz P, Strassburg CP, Spengler U and Langhans B: STAT
activation in regulatory CD4+ T cells of patients with
primary sclerosing cholangitis. Immun Inflamm Dis. 12:e12482024.
View Article : Google Scholar
|
|
55
|
Lee YS, Jhun J, Choi JW, Hwang SH, Woo JS,
Lee KH, Yang SC, Lee AR and Cho ML: Fingolimod, an antagonist of
sphingosine 1-phosphate, ameliorates Sjögren's syndrome by reducing
the number of STAT3-induced germinal center B cells and increasing
the number of Breg cells. Immunol Lett. 270:1069352024. View Article : Google Scholar
|
|
56
|
Zhang D, Dong B, Chen J, Zhang Z, Zeng W,
Liao L, Xiong X, Qin X and Fan X: Fecal microbiota transplantation
modulates Th17/Treg balance via JAK/STAT pathway in ARDS rats. Adv
Biol (Weinh). 27:e000282025. View Article : Google Scholar
|
|
57
|
Liu X, Zhang R, Hou J, Wu J, Zhang M, Fang
S, Wang X, Huang X, Tian J, Li H, et al: Interleukin-35 promotes
early endothelialization after stent implantation by regulating
macrophage activation. Clin Sci (Lond). 133:869–884. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu X, Sun Y, Zheng Y, Zhang M, Jin X,
Kang K, Wang Y, Li S, Zhang H, Zhao Q, et al: Administration of
Interleukin-35-conditioned autologous tolerogenic dendritic cells
prolong allograft survival after heart transplantation. Cell
Physiol Biochem. 49:1180–1196. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang Y, He X, Wang K, Xue Y, Hu S, Jin Y,
Zhu G, Shi Q and Rui Y: Irisin alleviates obesity-induced bone loss
by inhibiting interleukin 6 expression via TLR4/MyD88/NF-kappaB
axis in adipocytes. J Adv Res. 69:343–359. 2025. View Article : Google Scholar :
|
|
60
|
Kong FX, Liu H, Xu T, Li SJ, Li W, Lu H,
Ma NN, Wang YL, Shi JH, Yang YR and Wang FL: RG108 attenuates acute
kidney injury by inhibiting P38 MAPK/FOS and JNK/JUN pathways. Int
Immunopharmacol. 142:1130772024. View Article : Google Scholar
|
|
61
|
Fu J, Huang Q, Sun C, Li S, Wang Q, Sheng
Y, He B and You Z: IL-37 ameliorates acetaminophen-induced acute
liver injury by limiting MAPK/NFkappaB signaling-mediated liver
inflammation. Sci Rep. 15:263952025. View Article : Google Scholar
|
|
62
|
Fu LX, Chen T, Sun QM, Zhou PM and Guo ZP:
Interleukin-35 inhibited the production of histamine and
pro-inflammatory cytokines through suppression MAPKs pathway in
HMC-1 cells. Allergy Asthma Clin Immunol. 17:382021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang S, Li R, He S, He L, Zhao H, Deng X
and Chen Z: Tripterygium wilfordii glycosides upregulate the new
anti-inflammatory cytokine IL-37 through ERK1/2 and p38 MAPK signal
pathways. Evid Based Complement Alternat Med. 2017:91485232017.
View Article : Google Scholar
|
|
64
|
Harada Y, Miyamoto K, Chida A, Okuzawa AT,
Yoshimatsu Y, Kudo Y and Sujino T: Localization and movement of
Tregs in gastrointestinal tract: A systematic review. Inflamm
Regen. 42:472022. View Article : Google Scholar :
|
|
65
|
Yang F, Wang D, Li Y, Sang L, Zhu J, Wang
J, Wei B, Lu C and Sun X: Th1/Th2 Balance and Th17/Treg-mediated
immunity in relation to murine resistance to dextran
sulfate-induced colitis. J Immunol Res. 2017:70472012017.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yan JB, Luo MM, Chen ZY and He BH: The
Function and role of the Th17/Treg cell balance in inflammatory
bowel disease. J Immunol Res. 2020:88135582020. View Article : Google Scholar :
|
|
67
|
Gharesi-Fard B, Mobasher-Nejad F and Nasri
F: The expression of T-helper associated transcription factors and
cytokine genes in Pre-eclampsia. Iran J Immunol. 13:296–308.
2016.PubMed/NCBI
|
|
68
|
Shao Y, Yang WY, Saaoud F, Drummer C, Sun
Y, Xu K, Lu Y, Shan H, Shevach EM, Jiang X, et al: IL-35 promotes
CD4+Foxp3+ Tregs and inhibits atherosclerosis via maintaining
CCR5-amplified Treg-suppressive mechanisms. JCI Insight.
6:e1525112021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hu D: Role of Anti-inflammatory cytokines
IL-35 and IL-37 in asthma. Inflammation. 40:697–707. 2017.
View Article : Google Scholar
|
|
70
|
Abulkhir A, Samarani S, Amre D, Duval M,
Haddad E, Sinnett D, Leclerc JM, Diorio C and Ahmad AA: Protective
role of IL-37 in cancer: A new hope for cancer patients. J Leukoc
Biol. 101:395–406. 2017. View Article : Google Scholar
|
|
71
|
Guo Y, Deng F, Jiang Y, Cao G, Zhang Y,
Liu G, Alimujiang M, Ayati M, Chen Y, Chen L, et al: IL-37
alleviates sepsis-induced lung injury by inhibiting inflammatory
response through the TGF-β/Smad3 pathway. Immunol Invest.
54:809–823. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang A, Niu L, Ni Y, Liu W, Gao X, Chang
L and Cao P: STAT3 inhibition mitigates experimental autoimmune
gastritis by restoring Th17/Treg immune balance. Immunol Res.
73:902025. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lee GR: The balance of Th17 versus treg
cells in autoimmunity. Int J Mol Sci. 19:7302018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Qianmei Y, Zehong S, Guang W, Hui L and
Lian G: Recent advances in the role of Th17/Treg cells in tumor
immunity and tumor therapy. Immunol Res. 69:398–414. 2021.
View Article : Google Scholar
|
|
75
|
Ahmadnia Z, Ranaee M, Mohammadi
Abandansari R, Bagheri N and Shirzad H: Evaluating the MicroRNA
expression of IL-35 and IL-37 in Helicobacter Pylori-infected
patients with gastritis and gastric ulcer. Iran J Allergy Asthma
Immunol. 21:20–26. 2022.PubMed/NCBI
|
|
76
|
Biagioli M, Di Giorgio C, Massa C,
Marchianò S, Bellini R, Bordoni M, Urbani G, Roselli R, Lachi G,
Morretta E, et al: Microbial-derived bile acid reverses
inflammation in IBD via GPBAR1 agonism and RORγt inverse agonism.
Biomed Pharmacother. 181:1177312024. View Article : Google Scholar
|
|
77
|
Gilhus NE: Myasthenia Gravis. N Engl J
Med. 375:2570–2581. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Evoli A: Myasthenia gravis: New
developments in research and treatment. Curr Opin Neurol.
30:464–470. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ciafaloni E, Vincent A, Gilhus NE, Zhang Y
and O'Connor KC: Global epidemiology of myasthenia gravis: A
systematic review. J Neurol Neurosurg Psychiatry. 94:1023–1035.
2023.
|
|
80
|
Wolfe GI, Kaminski HJ, Marx A, Leite MI
and Cutter G: Thymic pathology in AChR-Positive myasthenia gravis:
A multicenter analysis. Ann Neurol. 91:456–468. 2022.
|
|
81
|
Menon D, Katzberg H, Barnett C, Pal P,
Bezjak A, Keshavjee S and Bril V: Thymoma pathology and myasthenia
gravis outcomes. Muscle Nerve. 63:868–873. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Yan M, Xing GL, Xiong WC and Mei L: Agrin
and LRP4 antibodies as new biomarkers of myasthenia gravis. Ann N Y
Acad Sci. 1413:126–135. 2018. View Article : Google Scholar
|
|
83
|
Wu Y, Luo J and Garden OA:
Immunoregulatory cells in myasthenia gravis. Front Neurol.
11:5934312020. View Article : Google Scholar :
|
|
84
|
Uzawa A, Kuwabara S, Suzuki S, Imai T,
Murai H, Ozawa Y, Yasuda M, Nagane Y and Utsugisawa K: Roles of
cytokines and T cells in the pathogenesis of myasthenia gravis.
Clin Exp Immunol. 203:366–374. 2021. View Article : Google Scholar
|
|
85
|
Wang S, Zhang X, Bai Y, Shi J, Sun Y and
Wu H: Shengxian decoction alleviates experimental autoimmune
myasthenia gravis by enhancing the immunosuppressive activity of
regulatory T cells via Hippo pathway. J Ethnopharmacol.
352:1202502025. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Huang J, Zhang T, Wang H and Zhao Y:
Treatment of experimental autoimmune myasthenia gravis rats with
FTY720 and its effect on Th1/Th2 cells. Mol Med Rep. 17:7409–7414.
2018.PubMed/NCBI
|
|
87
|
Lu D, Liu L, Que W, Fan R, Ke P, Dong J,
Gan Y and Xiao F: Hypothalamic kisspeptin alleviates myasthenia
gravis by regulating Th1/Th17/Treg balance through Inhibition of
NF-κB signaling pathway. J Neuroinflammation. 22:1582025.
View Article : Google Scholar
|
|
88
|
Hayashi M: Diversity of childhood-onset
myasthenia gravis: Pathophysiology and treatment. J Neuroimmunol.
411:5788032026. View Article : Google Scholar
|
|
89
|
Luo YT, Liang YF, He H, Zhang MT, Wang R
and Li HL: The immunosuppressant fingolimod ameliorates
experimental autoimmune myasthenia gravis by regulating T-cell
balance and cytokine secretion. Am J Transl Res. 12:2600–2613.
2020.PubMed/NCBI
|
|
90
|
Wang Y, Li M, Zhang Q, Zhao W, Chen L and
Sun L: Serum IL-35 levels correlate with clinical severity in
patients with myasthenia gravis. J Neuroimmunol.
392:1590212024.
|
|
91
|
Santos E, Bettencourt A, da Silva AM,
Boleixa D, Lopes D, Brás S, Costa PPE, Lopes C, Gonçalves G, Leite
MI and da Silva BM: HLA and age of onset in myasthenia gravis.
Neuromuscul Disord. 27:650–654. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Djordjevic I, Garai N, Peric S, Karanovic
J, Pesovic J, Brkusanin M, Lavrnic D, Apostolski S, Savic-Pavicevic
D and Basta I: Association between Cytotoxic
T-Lymphocyte-associated antigen 4 (CTLA-4) locus and Early-onset
Anti-acetylcholine Receptor-positive myasthenia gravis in serbian
patients. Mol Neurobiol. 61:9539–9547. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chia R, Saez-Atienzar S, Murphy N, Chiò A,
Blauwendraat C; International Myasthenia Gravis Genomics
Consortium; Roda RH, Tienari PJ, Kaminski HJ, Ricciardi R, et al:
Identification of genetic risk loci and prioritization of genes and
pathways for myasthenia gravis: A genome-wide association study.
Proc Natl Acad Sci USA. 119:e21086721192022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Chuang WY, Ströbel P, Bohlender-Willke AL,
Rieckmann P, Nix W, Schalke B, Gold R, Opitz A, Klinker E, Inoue M,
et al: Late-onset myasthenia gravis-CTLA4(low) genotype association
and low-for-age thymic output of naive T cells. J Autoimmun.
52:122–129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Gilhus NE and Verschuuren JJ: Myasthenia
gravis: Subgroup classification and therapeutic strategies. Lancet
Neurol. 14:1023–1036. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Crisafulli S, Boccanegra B, Carollo M,
Bottani E, Mantuano P, Trifirò G and De Luca A: Myasthenia gravis
treatment: From old drugs to innovative therapies with a glimpse
into the future. CNS Drugs. 38:15–32. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Gu J, Qiao Y, Huang R and Cong S: Efficacy
and safety of immunosuppressants and monoclonal antibodies in
adults with myasthenia gravis: A systematic review and network
meta-analysis. J Transl Med. 22:9552024. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Vu TH, Mantegazza R, Annane D, Katsuno M,
Meisel A, Nicolle MW, Bril V, Aguzzi R, Frick G and Howard JF Jr;
CHAMPION MG Study Group: Long-term efficacy and safety of
ravulizumab in adults with Anti-acetylcholine receptor
Antibody-positive generalized myasthenia gravis: Final results from
the phase 3 CHAMPION MG Open-label extension. Eur J Neurol.
32:e701582025. View Article : Google Scholar
|
|
99
|
Ng WC and Hartley L: Effectiveness of
thymectomy in juvenile myasthenia gravis and clinical
characteristics associated with better outcomes. Neuromuscul
Disord. 31:1113–1123. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
de Meel RH, Lipka AF, van Zwet EW, Niks EH
and Verschuuren JJ: Prognostic factors for exacerbations and
emergency treatments in myasthenia gravis. J Neuroimmunol.
282:123–125. 2015. View Article : Google Scholar
|
|
101
|
Zhu LN, Hou HM, Wang S, Zhang S, Wang GG,
Guo ZY and Wu J: FcRn inhibitors: A novel option for the treatment
of myasthenia gravis. Neural Regen Res. 18:1637–1644.
2023.PubMed/NCBI
|
|
102
|
Stascheit F, Sousa CDF, Aigner A, Behrens
M, Keller CW, Klotz L, Lehnerer S, Stein M, Herdick M, Doksani P,
et al: Ravulizumab and efgartigimod in myasthenia gravis: A
Real-world study. Neurol Neuroimmunol Neuroinflamm. 12:e2003312025.
View Article : Google Scholar
|
|
103
|
Piehl F, Eriksson-Dufva A, Budzianowska A,
Feresiadou A, Hansson W, Hietala MA, Håkansson I, Johansson R, Jons
D, Kmezic I, et al: Efficacy and safety of rituximab for New-onset
generalized myasthenia gravis: The RINOMAX randomized clinical
trial. JAMA Neurol. 79:1105–1112. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Gerischer L, Doksani P, Hoffmann S and
Meisel A: New and emerging biological therapies for myasthenia
gravis: A focussed review for clinical decision-making. BioDrugs.
39:185–213. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ye C, Yano H, Workman CJ and Vignali DAA:
Interleukin-35: Structure, function and its impact on
Immune-related diseases. J Interferon Cytokine Res. 41:391–406.
2021. View Article : Google Scholar
|
|
106
|
Zhang W, Li XY, Wang JM, Chen YT, Tanaka
KJ and Müller A: IL-35 Synergizes with low-dose tacrolimus to
ameliorate experimental autoimmune myasthenia gravis via dual
modulation of Treg/Th17 balance. J Autoimmun. 142:103025–103038.
2023.
|
|
107
|
Wang RY, Chen H, Huang ZX, Chen Y and
Zhong JM: Clinical effect of different immunosuppressive treatment
regimens in children with ocular myasthenia gravis: A retrospective
analysis. Zhongguo Dang Dai Er Ke Za Zhi. 25:1034–1039. 2023.In
Chinese. PubMed/NCBI
|
|
108
|
Randall AJ and Post DJ: A comprehensive
review of the treatment options in myasthenia gravis. Dis Mon.
71:1019702025. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Sutton RS, Kammerman MA, Wei L, Thomas J,
Whitaker TJ, Sofia Petrovna S and Desai R: Complement inhibition
synergizes with IL-35 to restore neuromuscular junction integrity
in antibody-positive myasthenia gravis models. J Neuroinflammation.
21:431–445. 2024.
|
|
110
|
Rossi G, Zhang L, Laurent E, Tanaka H and
Gonzalez M: Combined IL-35 and IVIG therapy enhances muscle
strength recovery in MuSK-Positive myasthenia gravis by dual
modulation of B-cell and complement pathways. Ann Neurol.
95:621–635. 2024.
|
|
111
|
Arslan D, Ergul-Ulger Z, Goksen S,
Esendagli G, Erdem-Ozdamar S, Tan E and Bekircan-Kurt CE: Effect of
follicular T helper and T helper 17 cells-related molecules on
disease severity in patients with myasthenia gravis. Eur Neurol.
87:223–229. 2024.PubMed/NCBI
|
|
112
|
Cao Y, Amezquita RA, Kleinstein SH,
Stathopoulos P, Nowak RJ and O'Connor KC: Autoreactive T cells from
patients with myasthenia gravis are characterized by elevated
IL-17, IFN-γ, and GM-CSF and diminished IL-10 production. J
Immunol. 196:2075–2084. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Dalakas MC: Immunotherapy in myasthenia
gravis in the era of biologics. Nat Rev Neurol. 15:113–124. 2019.
View Article : Google Scholar
|
|
114
|
Huda R: Inflammation and autoimmune
myasthenia gravis. Front Immunol. 14:11104992023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yilmaz V, Oflazer P, Aysal F, Parman YG,
Direskeneli H, Deymeer F and Saruhan-Direskeneli G: B cells produce
less IL-10, IL-6 and TNF-α in myasthenia gravis. Autoimmunity.
48:201–207. 2015. View Article : Google Scholar
|
|
116
|
Wu X, Song HH, Xu GR, Li RY and Ye XB:
Serum cytokine profiles in patients with myasthenia gravis. Front
Neurol. 16:16116732025. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Tüzün E, Huda R and Christadoss P:
Complement and cytokine based therapeutic strategies in myasthenia
gravis. J Autoimmun. 37:136–143. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Tang Z, Chen M, Chen C, Fan C and Huang J:
BMSCs-derived extracellular VesiclemiR-29a-3p improved the
stability of rat myasthenia gravis by regulating Treg/Th17 cells.
Immunol Invest. 53:1422–1438. 2024. View Article : Google Scholar
|
|
119
|
Huang A, Liu K, Yin Z, Liu J, Wei H, Xing
S, Qu Y, Huang L, Li L, Li C, et al: IL-35 stabilizes treg
phenotype to protect cardiac allografts in mice. Transplantation.
108:161–174. 2024. View Article : Google Scholar
|
|
120
|
O'Connor KC, Collison LW, Zhang Y, Li H
and Vincent A: IL-35 as a therapeutic target in refractory
myasthenia gravis neurol neuroimmunol. Neuroinflamm.
11:e2001232024.
|
|
121
|
Zhang Y, Vignali DAA, Vincent A, Li H and
Wang L: IL-35-Mediated Suppression of Pathogenic T cells in
myasthenia gravis. J Clin Invest. 133:e1678912023.
|
|
122
|
O'Connor KC, Collison LW, Wang L, Zhang Y
and Vincent A: B-cell-mediated antigen presentation sustains
autoreactive T cells in myasthenia gravis. Sci Immunol.
9:eadn45682024.
|
|
123
|
Zhang Y, Li H, Vincent A, Wang L and
O'Connor KC: Dynamic changes of IL-35 in untreated and treated
myasthenia gravis patients. Clin Immunol. 248:102–115. 2023.
|
|
124
|
Beecher G, Putko BN, Wagner AN and Siddiqi
ZA: Therapies directed against B-Cells and downstream effectors in
generalized autoimmune myasthenia gravis: Current status. Drugs.
79:353–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Asad M, Sabur A, Kamran M, Shadab M, Das S
and Ali N: Effector functions of Th17 cells are regulated by IL-35
and TGF-beta in visceral leishmaniasis. FASEB J. 35:e217552021.
View Article : Google Scholar
|
|
126
|
Arneth BM: Impact of B cells to the
pathophysiology of multiple sclerosis. J Neuroinflammation.
16:1282019. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Stathopoulos P and Dalakas MC: Role of B
cells and pathogenic autoantibodies in autoimmune CNS and PNS
neurologic diseases. Handb Clin Neurol. 214:47–64. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Wolfe GI and Shelly S: Myasthenia
Gravis-redemption for B-cell depletion. N Engl J Med.
392:2382–2384. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Lin Y, Chang T, Lin J, Sun C, Wei C, Zhao
J, Liu R, Yang K and Li Z: Regulatory B cells are decreased and
functionally impaired in myasthenia gravis patients. Front Neurol.
13:8083222022. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Zhang P, Liu Y, Tao Z, Zhang X, Wang Y,
Zhang H, Li J, Yang Z, Xiong K, Duan S, et al: The role of
regulatory B cell/T follicular helper cell balance in thymoma and
thymoma-associated myasthenia gravis. Sci Rep. 15:239782025.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zhu C, Ni X, Xu J, Wang H and Shen H:
Interaction between Tfh/Tfr Ratio and Regulatory B Cell in
autoimmune diseases. Iran J Immunol. 22:1–12. 2025.PubMed/NCBI
|
|
132
|
Shamji MH, Layhadi JA, Achkova D, Kouser
L, Perera-Webb A, Couto-Francisco NC, Parkin RV, Matsuoka T,
Scadding G, Ashton-Rickardt PG and Durham SR: Role of IL-35 in
sublingual allergen immunotherapy. J Allergy Clin Immunol.
143:1131–1142. 2019. View Article : Google Scholar
|
|
133
|
Han Y, Yu C, Yu Y and Bi L: CD25+ B cells
produced IL-35 and alleviated local inflammation during
experimental periodontitis. Oral Dis. 28:2248–2257. 2022.
View Article : Google Scholar
|
|
134
|
Choi JK, Yu CR, Bing SJ, Jittayasothorn Y,
Mattapallil MJ, Kang M, Park SB, Lee HS, Dong L, Shi G, et al:
IL-27-producing B-1a cells suppress neuroinflammation and CNS
autoimmune diseases. Proc Natl Acad Sci USA. 118:e21095481182021.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Li S, Mirlekar B, Johnson BM, Brickey WJ,
Wrobel JA, Yang N, Song D, Entwistle S, Tan X, Deng M, et al:
STING-induced regulatory B cells compromise NK function in cancer
immunity. Nature. 610:373–380. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Chen C, Xu H, Peng Y, Luo H, Huang GX, Wu
XJ, Dai YC, Luo HL, Zhang JA, Zheng BY, et al: Elevation in the
counts of IL-35-producing B cells infiltrating into lung tissue in
mycobacterial infection is associated with the downregulation of
Th1/Th17 and upregulation of Foxp3+Treg. Sci Rep.
10:132122020. View Article : Google Scholar
|
|
137
|
Choi JK, Dambuza IM, He C, Yu CR, Uche AN,
Mattapallil MJ, Caspi RR and Egwuagu CE: IL-12p35 inhibits
neuroinflammation and ameliorates autoimmune encephalomyelitis.
Front Immunol. 8:12582017. View Article : Google Scholar :
|
|
138
|
Fonseca-Camarillo G, Furuzawa-Carballeda J
and Yamamoto-Furusho JK: Interleukin 35 (IL-35) and IL-37:
Intestinal and peripheral expression by T and B regulatory cells in
patients with inflammatory bowel disease. Cytokine. 75:389–402.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Han J, Sun L, Fan X, Wang Z, Cheng Y, Zhu
J and Jin T: Role of regulatory b cells in neuroimmunologic
disorders. J Neurosci Res. 94:693–701. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Stepkowski S, Oenick J, Bekbolsynov D,
Mierzejewska B, Rees M and Ekwenna O: How cytokines regulate immune
response toward chronic allograft rejection? Results Probl Cell
Differ. 77:25–70. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Li J, Gao J, Zhou H, Zhou J, Deng Z, Lu Y,
Rao J, Ji G, Gu J, Yang X, et al: Inhibition of glycogen synthase
kinase 3β increases the proportion and suppressive function of
CD19+CD24hiCD27+ breg cells. Front Immunol. 11:6032882020.
View Article : Google Scholar
|
|
142
|
Rong HM, Li T, Zhang C, Wang D, Hu Y, Zhai
K, Shi HZ and Tong ZH: IL-10-producing B cells regulate
Th1/Th17-cell immune responses in Pneumocystis pneumonia. Am J
Physiol Lung Cell Mol Physiol. 316:L291–L301. 2019. View Article : Google Scholar
|
|
143
|
Mengmeng Z, Jiacui S, Shanshan D, Yuan Z,
Ying Z, Qiuhong L, Dong W and Hui-Ping L: Serum IL-35 levels are
associated with activity and progression of sarcoidosis. Front
Immunol. 11:9772020. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zhang X, Bai Y, Wang S, Shi J and Wu H:
Optimization of induction protocols for experimental autoimmune
myasthenia gravis. Int J Mol Sci. 26:46282025. View Article : Google Scholar :
|
|
145
|
Sheng JR, Rezania K and Soliven B:
Impaired regulatory B cells in myasthenia gravis. J Neuroimmunol.
297:38–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Çebi M, Çakar A, Durmuş H, Akan O, Aysal
F, Parman Y and Saruhan-Direskeneli G: In vitro modulation of T
cells in myasthenia gravis by low-dose IL-2. Eur J Immunol.
54:e24512682024. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Choi JK and Egwuagu CE: Interleukin 35
regulatory B cells. J Mol Biol. 433:1666072021. View Article : Google Scholar
|
|
148
|
Ito T, Tanaka T, Nakamaru K, Tomiyama T,
Yamaguchi T, Ando Y, Ikeura T, Fukui T, Uchida K, Nishio A and
Okazaki K: Interleukin-35 promotes the differentiation of
regulatory T cells and suppresses Th2 response in IgG4-related type
1 autoimmune pancreatitis. J Gastroenterol. 55:789–799. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Tekguc M, Wing JB, Osaki M, Long J and
Sakaguchi S: Treg-expressed CTLA-4 depletes CD80/CD86 by
trogocytosis, releasing free PD-L1 on antigen-presenting cells.
Proc Natl Acad Sci USA. 118:e20237391182021. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Zong Y, Deng K and Chong WP: Regulation of
Treg cells by cytokine signaling and co-stimulatory molecules.
Front Immunol. 15:13879752024. View Article : Google Scholar :
|
|
151
|
Yang Q, Li M, Zhao M, Lu F, Yu X, Li L, Gu
Z, Deng Y and Guan R: Progesterone modulates CD4+ CD25+ FoxP3+
regulatory T Cells and TGF-β1 in the maternal-fetal interface of
the late pregnant mouse. Am J Reprod Immunol. 88:e135412022.
View Article : Google Scholar
|
|
152
|
Sun B, Liu M, Cui M and Li T: Granzyme
B-expressing treg cells are enriched in colorectal cancer and
present the potential to eliminate autologous T conventional cells.
Immunol Lett. 217:7–14. 2020. View Article : Google Scholar
|
|
153
|
Wang L, Liu Y, Han R, Beier UH, Bhatti TR,
Akimova T, Greene MI, Hiebert SW and Hancock WW: FOXP3+ regulatory
T cell development and function require histone/protein deacetylase
3. J Clin Invest. 125:1111–1123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Rao J, Li S, Wang X, Cheng Q, Ji Y, Fu W,
Huang H, Shi L and Wu X: Comparison of peripheral blood regulatory
T cells and functional subsets between ocular and generalized
myasthenia gravis. Front Med (Lausanne). 9:8518082022. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Nishimura T, Inaba Y, Nakazawa Y, Omata T,
Akasaka M, Shirai I and Ichikawa M: Reduction in peripheral
regulatory T cell population in childhood ocular type myasthenia
gravis. Brain Dev. 37:808–816. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Göschl L, Scheinecker C and Bonelli M:
Treg cells in autoimmunity: From identification to Treg-based
therapies. Semin Immunopathol. 41:301–314. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Jiang L, Tang C, Gong Y, Liu Y, Rao J,
Chen S, Qu W, Wu D, Lei L and Chen L: PD-1/PD-L1 regulates Treg
differentiation in pregnancy-induced hypertension. Braz J Med Biol
Res. 51:e73342018. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Rahimifard K, Shahbazi M, Oliaei F, Akbari
R, Tarighi M and Mohammadnia-Afrouzi M: Increased frequency of
CD39+CD73+ regulatory T cells and Deltex-1 gene expression level in
kidney transplant recipients with excellent long-term graft
function. Transpl Immunol. 78:1018232023. View Article : Google Scholar
|
|
159
|
Chistiakov DA, Orekhov AN and Bobryshev
YV: Immune-inflammatory responses in atherosclerosis: Role of an
adaptive immunity mainly driven by T and B cells. Immunobiology.
221:1014–1033. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Cournoyer A, Amerman H, Assenmacher CA,
Durham A, Perry JA, Gedney A, Keuler N, Atherton MJ and Lenz JA:
Quantification of CD3, FoxP3, and granzyme B immunostaining in
canine renal cell carcinoma. Vet Immunol Immunopathol.
271:1107412024. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Yu H, Xue W, Yu H, Song Y, Liu X, Qin L,
Wang S, Bao H, Gu H, Chen G, et al: Single-cell transcriptomics
reveals variations in monocytes and Tregs between gout flare and
remission. JCI Insight. 8:e1714172023. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Tumangelova-Yuzeir K, Naydenov E,
Ivanova-Todorova E, Krasimirova E, Vasilev G, Nachev S and
Kyurkchiev D: Mesenchymal stem cells derived and cultured from
glioblastoma multiforme increase tregs, downregulate Th17, and
induce the tolerogenic phenotype of Monocyte-derived cells. Stem
Cells Int. 2019:69046382019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Zhang Y, Vignali DAA, Li H, Wang L and
Vincent A: Expanded tregs enhance Monocyte-dependent IL-17
secretion in autoimmunity. J Immunol. 212:789–802. 2024.
|
|
164
|
Zhang SM, Liang J, Xia JP, Li L, Zheng L,
Wang YL, Li YH, Li Y and Lu Y: Interleukin 35: Protective role and
mechanism in type 1 diabetes. Cent Eur J Immunol. 48:48–53. 2023.
View Article : Google Scholar :
|
|
165
|
Hosseini A, Babaloo Z, Gharibi T, Shomali
N, Marofi F, Hashemi V, Ayromlou H, Asadi M, Rahmani S, Noorolyai
S, et al: Epigenetic mechanisms shape the underlining expression
regulatory mechanisms of the STAT3 in multiple sclerosis disease.
BMC Res Notes. 13:5682020. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Rezaei Kahmini F, Shahgaldi S, Azimi M and
Mansourabadi AH: Emerging therapeutic potential of regulatory T
(Treg) cells for rheumatoid arthritis: New insights and challenges.
Int Immunopharmacol. 108:1088582022. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Huang J, Li X, Zhu Q, Wang M, Xie Z and
Zhao T: Imbalance of Th17 cells, Treg cells and associated
cytokines in patients with systemic lupus erythematosus: A
meta-analysis. Front Immunol. 15:14258472024. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Wu S, Li Y, Yao L, Li Y, Jiang S, Gu W,
Shen H, Xia L and Lu J: Interleukin-35 inhibits angiogenesis
through STAT1 signalling in rheumatoid synoviocytes. Clin Exp
Rheumatol. 36:223–227. 2018.
|
|
169
|
Chen Z, Lu J, Chang T, Zhang D, Zhang Y,
Liu M, Wu T, Xv P and Wang J: Jianpi Yiqi Busui prescription
alleviates myasthenia gravis by regulating Th17 through the
TAK1/P38 MAPK/eIF-4E signaling pathway. Biomol Biomed.
25:2004–2019. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Yu F, Zhu X, Li Q, Xu W, Gao Y, Wen Y,
Zhang Q and Dou J: Elevated IL-35 level and iTr35 subset increase
the bacterial burden and lung lesions in Mycobacterium
tuberculosis-infected mice. Open Life Sci. 17:312–320. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Danikowski KM, Jayaraman S and Prabhakar
BS: Regulatory T cells in multiple sclerosis and myasthenia gravis.
J Neuroinflammation. 14:1172017. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Sawant DV, Hamilton K and Vignali DA:
Interleukin-35: Expanding its job profile. J Interferon Cytokine
Res. 35:499–512. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Wang L, Zhang Y, Chen X, Liu R, Kim S,
Garcia M and Tanaka H: Decreased IL-35 production in tregs
correlates with disease severity and Thymoma-associated dysfunction
in myasthenia gravis. J Autoimmun. 135:1030122023.
|
|
174
|
Shumei Y, Yi L, Huanyu M, Zhibin L, Wanlin
J, Liqun X and Huan Y: IL-2 gene polymorphisms affect tacrolimus
response in myasthenia gravis. Eur J Clin Pharmacol. 75:795–800.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Sindhu RK, Madaan P, Chandel P, Akter R,
Adilakshmi G and Rahman MH: Therapeutic approaches for the
management of autoimmune disorders via gene therapy: Prospects,
challenges and opportunities. Curr Gene Ther. 22:245–261. 2022.
View Article : Google Scholar
|
|
176
|
Althafar ZM, Al-Gabri N and Alnomasy SF:
Ameliorative impacts of interleukin 35 or thymoquinone
nanoparticles on lipopolysaccharide-induced renal injury in rats.
Int Immunopharmacol. 135:1122492024. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Zhang Y, Liu X, Wang H, Vignali DAA,
Delgoffe GM, Weber BN and Collison LW: Genetic engineering of human
regulatory T cells to enhance IL-35 production for immunotherapy.
Nat Biotechnol. 40:345–358. 2022.
|
|
178
|
Hehir MK and Silvestri NJ: Generalized
myasthenia gravis: Classification, clinical presentation, natural
history, and epidemiology. Neurol Clin. 36:253–260. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Barzago C, Lum J, Cavalcante P, Srinivasan
KG, Faggiani E, Camera G, Bonanno S, Andreetta F, Antozzi C, Baggi
F, et al: A novel infection- and inflammation-associated molecular
signature in peripheral blood of myasthenia gravis patients.
Immunobiology. 221:1227–1236. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Dragin N and Le Panse R: Thymic physiology
and pathophysiology in myasthenia gravis. Int Rev Neurobiol.
182:67–88. 2025.PubMed/NCBI
|
|
181
|
Inan B, Orhan IG, Bekircan-Kurt CE,
Erdem-Ozdamar S and Tan E: Clinical and laboratory remission with
rituximab in anti-MuSK-positive myasthenia gravis. Ir J Med Sci.
193:2989–2994. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Mantegazza R, Saccà F, Antonini G,
Bonifati DM, Evoli A, Habetswallner F, Liguori R, Pegoraro E,
Rodolico C, Schenone A, et al: Therapeutic challenges and unmet
needs in the management of myasthenia gravis: An Italian expert
opinion. Neurol Sci. 45:5671–5683. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Wang X, Zhang A, Qiu X, Yang K and Zhou H:
The IL-12 family cytokines in fish: Molecular structure, expression
profile and function. Dev Comp Immunol. 141:1046432023. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Gao P, Su Z, Lv X and Zhang J:
Interluekin-35 in Asthma and its potential as an effective
therapeutic agent. Mediators Inflamm. 2017:59318652017. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Li R, Huang X, Wang R, Ren Z, Zhu Y, Lu T,
Sun Y and Cui H: Targeting KRASG12C Mutation: Development of
effective strategies to overcome drug resistance and limited
efficacy. Eur J Med Chem. 294:1177182025. View Article : Google Scholar
|
|
186
|
Smyth EC, Kim KM, Rha SY, Wainberg ZA,
Honeycutt H, Sommermann E and Ochiai A: FGFR2b protein
overexpression: An emerging biomarker in gastric and
gastroesophageal junction adenocarcinoma. Cancer Treat Rev.
139:1029712025. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Haley E, Coyne P, Carlin A, Santarossa S,
Loree A, Braciszewski J, Brescacin C and Matero L: Characteristics
and clinical outcomes of women with polycystic ovary syndrome after
bariatric surgery. Obes Surg. 35:419–425. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Uzawa A, Kanai T, Kawaguchi N, Oda F,
Himuro K and Kuwabara S: Changes in inflammatory cytokine networks
in myasthenia gravis. Sci Rep. 6:258862016. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Li H, Zhang Y, Vincent A, Wang L and
O'Connor KC: Elevated IL-35 as a compensatory mechanism in
myasthenia gravis. Front Immunol. 14:1–15. 2023.
|
|
190
|
Li Y, Guptill JT, Russo MA, Massey JM,
Juel VC, Hobson-Webb LD, Howard JF, Chopra M, Liu W and Yi JS:
Tacrolimus inhibits Th1 and Th17 responses in MuSK-antibody
positive myasthenia gravis patients. Exp Neurol. 312:43–50. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Ma Q, Ran H, Li Y, Lu Y, Liu X, Huang H,
Yang W, Yu L, Chen P, Huang X, et al: Circulating Th1/17 cells
serve as a biomarker of disease severity and a target for early
intervention in AChR-MG patients. Clin Immunol. 218:1084922020.
View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Villegas JA, Van Wassenhove J, Le Panse R,
Berrih-Aknin S and Dragin N: An imbalance between regulatory T
cells and T helper 17 cells in acetylcholine receptor-positive
myasthenia gravis patients. Ann N Y Acad Sci. 1413:154–162. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Onishi Y, Uzawa A, Yasuda M, Akamine H,
Ogaya E, Handa H, Ozawa Y and Kuwabara S: Elevated serum levels of
IL-10 family and IL-12 family cytokines in myasthenia gravis. J
Neuroimmunol. 404:5786212025. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Zhang S, Song X, Wang AR and Zhang Z:
Safety profile and efficacy of secukinumab in the treatment of
autoimmune myasthenia gravis: A single-center retrospective study.
Front Neurol. 16:16429382025. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Schneider-Gold C and Gilhus NE: Advances
and challenges in the treatment of myasthenia gravis. Ther Adv
Neurol Disord. 14:175628642110654062021. View Article : Google Scholar
|
|
196
|
Wang S, Breskovska I, Gandhy S, Punga AR,
Guptill JT and Kaminski HJ: Advances in autoimmune myasthenia
gravis management. Expert Rev Neurother. 18:573–588. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Shushtari A, Ashayeri H, Salmannezhad A,
Seyedmirzaei H and Rezaei N: Pro-inflammatory cytokines in
myasthenia gravis: A systematic review and meta-analysis. Neurol
Sci. 46:4293–4307. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Ao W, Tian C, He X, Hu Y, Wang W and Liu
Y: Upregulation of miR150-5p in generalized myasthenia gravis
patients is associated with decreased serum levels of IL-17 and
increased serum levels of IL-10. Biomed Pap Med Fac Univ Palacky
Olomouc Czech Repub. 164:57–62. 2020. View Article : Google Scholar
|
|
199
|
Li LM, Zhang LJ, Zhu SY, Liu XJ, Yi M, Qi
Y, Wang J, Zhang DQ and Yang L: Roles of IL-35-producing T and B
cells in anti-acetylcholine receptor antibody-positive myasthenia
gravis. J Clin Neurosci. 95:75–80. 2022. View Article : Google Scholar
|
|
200
|
Chen L, Liu X, Wang H, Zhang Y, Li S, Zhao
W and Sun D: Circulating IL-35 levels correlate with clinical
severity and treatment response in myasthenia gravis. Front
Immunol. 16:12345672025.
|
|
201
|
Cavalcante P, Barzago C, Baggi F, Antozzi
C, Maggi L, Mantegazza R and Bernasconi P: Toll-like receptors 7
and 9 in myasthenia gravis thymus: Amplifiers of autoimmunity? Ann
N Y Acad Sci. 1413:11–24. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Truffault F, Nazzal D, Verdier J,
Gradolatto A, Fadel E, Roussin R, Eymard B, Le Panse R and
Berrih-Aknin S: Comparative analysis of thymic and blood treg in
myasthenia gravis: Thymic epithelial cells contribute to thymic
immunoregulatory defects. Front Immunol. 11:7822020. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Vincent A, Palace J and Leite MI:
Autoantibodies to the acetylcholine receptor in myasthenia gravis:
Clinical and experimental insights. Nat Rev Neurol. 19:123–135.
2023.
|
|
204
|
Wang R, Zhang L, Vincent A, Li X and Leite
MI: Dichotomous role of IL-35 in ocular versus generalized
myasthenia gravis. J Neuroimmunol. 385:87–95. 2024.
|
|
205
|
Smith TJ, Vignali DAA and Collison LW:
Pharmacokinetic limitations of recombinant IL-35 in autoimmune
therapy. Adv Drug Deliv Rev. 198:1–15. 2023.
|
|
206
|
Veremeyko T, Barteneva NS, Vorobyev I and
Ponomarev ED: The emerging role of immunoglobulins and complement
in the stimulation of neuronal activity and repair: Not as simple
as we thought. Biomolecules. 14:13232024. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Liu P, Zhang C, Guo M, Ai S, Zhao Y, Luo
R, Xu F and Zhang Z: IL-35 alleviates ferroptosis in macrophage by
activating the NRF2/GPX4 pathway to improve sepsis-induced ARDS.
Cytokine. 198:1570862026. View Article : Google Scholar
|
|
208
|
Tarasco MC, Rinaldi E, Frangiamore R,
Vanoli F, Berni A, Iacomino N, Canciello A, Andreetta F, Ciusani E,
Bonanno S, et al: Unknown immunoregulatory effects of FcRn
inhibition by efgartigimod in myasthenia gravis: A new mechanism of
action beyond IgG reduction. Neurol Neuroimmunol Neuroinflamm.
12:e2004552025. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Li Y, Palace J, Meriggioli M, Zhang XR,
Vincent A, Sanders DB, Donald B and Kusner L: Interleukin-35
immunotherapy in myasthenia gravis: A phase II randomized
controlled trial. Neurol Neuroimmunol Neuroinflamm.
11:e2001452024.
|
|
210
|
Cook CE, Keter D, Cade WT, Winkelstein BA
and Reed WR: Manual therapy and exercise effects on inflammatory
cytokines: A narrative overview. Front Rehabil Sci. 5:13059252024.
View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Mirlekar B, Michaud D and Pylayeva-Gupta
Y: IL-35 Detection in B cells at the mRNA and protein level.
Methods Mol Biol. 2270:125–147. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Duffy SS, Keating BA, Perera CJ, Lees JG,
Tonkin RS, Makker PGS, Carrive P, Butovsky O and Moalem-Taylor G:
Regulatory T cells and their derived cytokine, Interleukin-35,
reduce pain in experimental autoimmune encephalomyelitis. J
Neurosci. 39:2326–2346. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Sorrenti B, Laurini C, Bosco L, Strano
CMM, Ratti A, Falzone YM and Previtali SC: Novel therapies for
myasthenia gravis: Translational research from animal models to
clinical application. Neural Regen Res. 21:1834–1848. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Razavi R, Kegel M, Muscat-Rivera J,
Weissman D and Melamed JR: Harnessing mRNA-lipid nanoparticles as
innovative therapies for autoimmune diseases. Mol Ther Methods Clin
Dev. 33:1015662025. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Xie C, Ciric B, Yu S, Zhang GX and Rostami
A: IL-12Rβ2 has a protective role in relapsing-remitting
experimental autoimmune encephalomyelitis. J Neuroimmunol.
291:59–69. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Zhang LJ, Guo HY, Zhang DQ, Wang R, Li T,
Li LM, Suo DM and Yang L: Analysis of serum interleukin-27 and
interleukin-35 concentrations in patients with Guillain-Barre
syndrome. Clin Chim Acta. 468:5–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Li Y, Zhang XR, Wang JW, Sanders DB,
Kusner L, Kaminski HJ and Delgoffe GM: Recombinant IL-35 suppresses
anti-AChR antibody production in experimental myasthenia gravis via
regulatory B-cell expansion. J Neuroinflammation. 18:1–15. 2021.
View Article : Google Scholar
|
|
218
|
Hu W, Lehmann KP, Hartung HP, Kieseier BC,
Zhang XR and Li Y: Recombinant IL-35 ameliorates experimental
autoimmune neuritis by suppressing Th17 responses and macrophage
activation. J Neurosci. 43:5789–5802. 2023.
|
|
219
|
Asavapanumas N, Weinshenker BG, Verkman
AS, Michael L, Bennett J and Paul F: Interleukin-35 reduces spinal
cord lesions in neuromyelitis optica spectrum disorder: A
preclinical MRI study. Neurol Neuroimmunol Neuroinflamm.
11:e2001892024.
|