Introduction
Hepatic ischemia-reperfusion injury (HIRI)
represents a critical clinical challenge in liver transplantation
and major hepatectomy, serving as a key determinant of
postoperative liver dysfunction, graft rejection and patient
mortality (1,2). HIRI pathogenesis mainly involves
hypoxic damage during the ischemic phase, which is subsequently
dominated by a robust sterile inflammatory response triggered upon
reperfusion. This response manifests as oxidative stress,
activation of immune cells and programmed cell death (3). During this process, neutrophils, as
the earliest immune cells recruited to the injury site, react
swiftly and play a complex, multi-layered role in the progression
of HIRI. Traditionally, neutrophils have been considered primary
executors of tissue damage, exacerbating HIRI through the
generation of reactive oxygen species (ROS), release of granular
proteases and formation of neutrophil extracellular traps (NETs)
(4,5). Notably, NETs not only possess
antibacterial functions but also can entrap host cells and release
damage-associated molecular patterns (DAMPs), thereby further
amplifying inflammatory responses and thrombosis, which intensifies
tissue damage and microcirculatory dysfunction.
However, with the continuous development of
immunomodulatory strategies and advances in cell tracking
technologies (6), the
understanding of neutrophil roles in HIRI has gradually shifted.
Studies have revealed that neutrophils are not merely terminal
effector cells participating in damage but also act as dynamic
regulators in the processes of injury and repair (7-10). The functional fate of
neutrophils, whether they polarize towards a pro-inflammatory (N1)
or an anti-inflammatory (N2) phenotype, plays a pivotal role in the
course of HIRI. This phenotypic shift determines whether HIRI is
exacerbated or alleviated (11,12). Although some progress has been
made regarding neutrophil functions and their mechanisms in HIRI,
significant research gaps remain in this field (13,14). Currently, the most critical
questions are: What are the key molecules regulating the transition
from the N1 to the N2 phenotype? More importantly, how can this
mechanism be translated into therapeutic strategies that precisely
modulate neutrophil function, moving beyond simple immune cell
depletion? The present review aimed to address these conceptual
gaps by proposing and elaborating an 'injury-repair balance'
theoretical framework to reinterpret the multifaceted roles of
neutrophils in HIRI. Furthermore, the present review explored the
potential of emerging therapeutic strategies, such as targeting
neutrophil phenotypic regulation, intervening in NETs formation and
utilizing neutrophil-derived vesicles. These investigations not
only provide a novel perspective for understanding HIRI but also
establish a theoretical foundation for precise immunomodulatory
interventions, potentially paving new avenues for future
therapeutic strategies.
Pathological mechanism of HIRI
HIRI refers to the interruption of blood flow to the
liver during the ischemic phase, causing liver cells to lose oxygen
and nutrients. Subsequently, upon the restoration of blood flow
(reperfusion phase), the liver undergoes sterile inflammatory
damage caused by excessive oxidative stress and inflammatory
responses (15). In liver
transplantation, HIRI not only leads to functional impairment of
the donor liver but also exacerbates the issue of donor shortage
(16). Healthy livers and
marginal donor livers are both susceptible to IRI. Reducing the
occurrence of HIRI can help improve the success rate of
transplantation and expand the available donor pool. Although the
success rate of liver transplantation has increased in recent
years, the persistent growth in the number of patients awaiting a
liver means that HIRI remains a pressing clinical challenge
(17).
The liver is composed of parenchymal cells (such as
hepatocytes) and non-parenchymal cells [such as Kupffer cells,
liver sinusoidal endothelial cells (LSECs) and stellate cells].
HIRI can be divided into warm IRI and cold IRI (1,4,18,19). Warm IRI primarily occurs in
situations such as liver surgery, liver transplantation,
hypovolemic shock, certain toxic liver injuries, venous embolic
diseases and Budd-Chiari syndrome. On the other hand, cold IRI
occurs mainly during the preservation process of donor livers
before transplantation. Although both types of HIRI share similar
pathological mechanisms, their specific mechanisms differ. The
present review focused on the pathogenesis of warm IRI (Fig. 1).
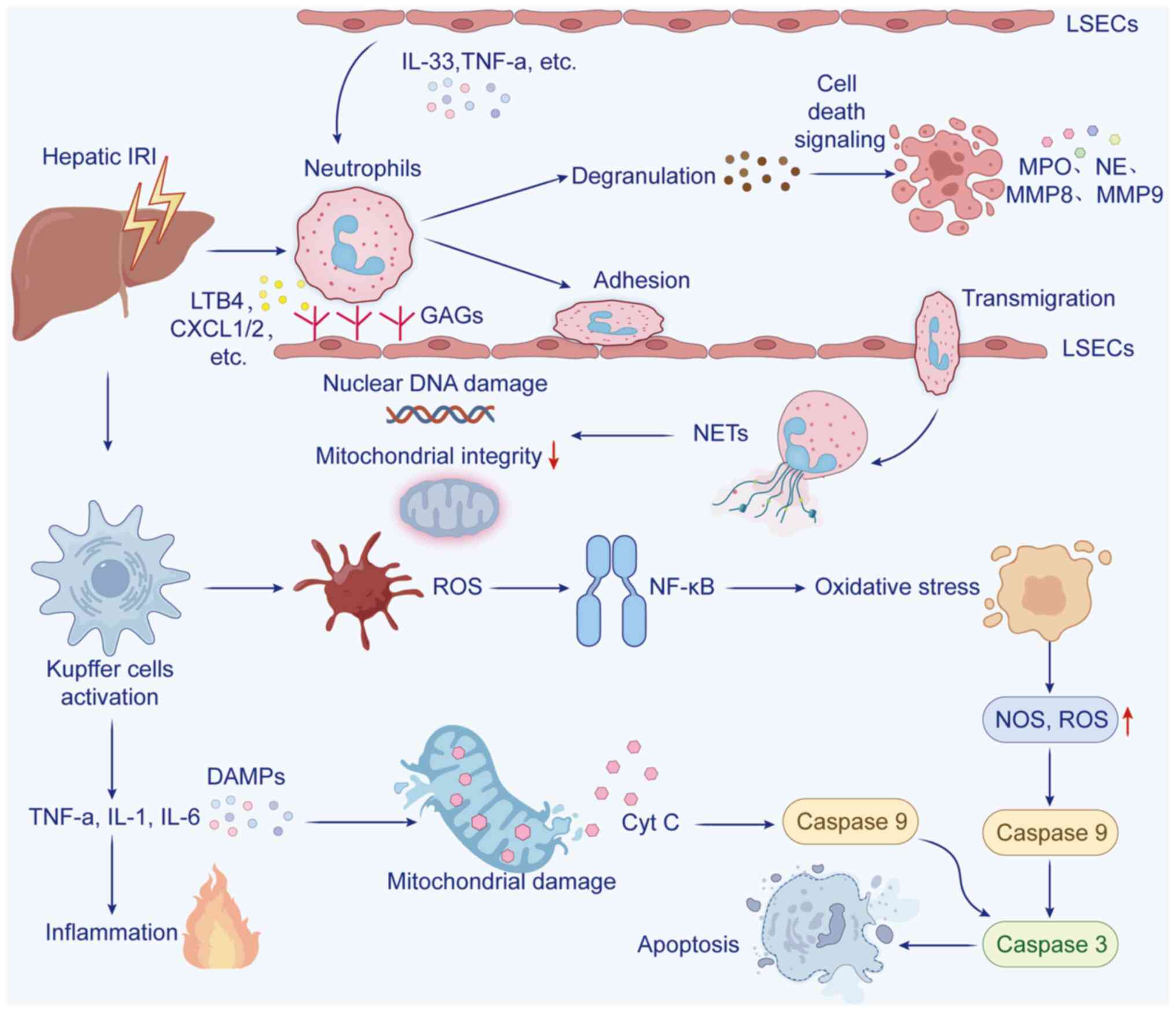 | Figure 1The pathological mechanism of HIRI
and the role of neutrophils in HIRI. HIRI, hepatic
ischemia-reperfusion injury; IRI, ischemia-reperfusion injury;
LSECs, liver sinusoidal endothelial cells; GAGs,
glycosaminoglycans; DAMP, damage associated molecular pattern;
NETs, neutrophil extracellular traps; ROS, reactive oxygen species;
NF-κB, nuclear factor-kappa B; TNF-α, tumor necrosis factor alpha;
LTB4, leukotriene B4; MPO, myeloperoxidase; NE, neutrophil
elastase; MMP8, matrix metallopeptidase 8; MMP9, matrix
metallopeptidase 9; NOS, nitric oxide synthase; Cyt C, Cytochrome
c. |
In the pathogenesis of HIRI, hepatocyte necrosis is
the primary manifestation of cellular damage. During the ischemic
phase, hepatocytes suffer from a lack of oxygen and nutrients,
leading to suppressed metabolism, inhibited ATP synthesis and a
gradual accumulation of an acidic intracellular environment,
ultimately causing structural and functional damage to the liver
cells (20-22). However, although the restoration
of blood flow is essential during reperfusion, it is accompanied by
a dramatic increase in oxidative stress and produce excessive ROS
that damage cell membranes, mitochondria and DNA (23-25). This oxidative damage-induced cell
death affects hepatocytes and involves LSECs and cholangiocytes.
ROS generation can directly damage the parenchymal liver cells and
activate immune cells and endothelial cells, leading to the release
of various inflammatory factors such as tumor necrosis factor alpha
(TNF-α), interferon beta and gamma, interleukins (IL-1β, IL-6) and
vascular endothelial growth factor (VEGF). This further exacerbates
tissue injury. It has been reported that the alternating effects of
ischemia and reperfusion can induce oxidative damage to cell
membranes, lipid peroxidation, endothelial dysfunction,
intracellular calcium overload and cell cycle arrest, all of which
contribute to liver injury (26-28).
During the HIRI process, immune responses are one of
the key factors in the pathological progression. The immune cascade
involves the activation of Kupffer cells in the liver and the
infiltration of circulating lymphocytes, neutrophils and monocytes.
Macrophages are strategically distributed throughout the body,
phagocytosing dead cells and debris. Liver contains a large number
of macrophages, which play a crucial role in the mechanism of HIRI.
They sense the initial damage-associated signals, triggering
inflammation and recruiting host immune cells and promote
inflammation resolution and tissue repair for maintaining
homeostasis. Neutrophils, a major immune cell in steady-state blood
circulation, serve as the first line of defense against invading
pathogens. They are recruited to the injury site where they enhance
hepatocyte damage and act as a biomarker for the severity of HIRI
(29) as 'non-specialized'
innate effector cells. In ischemic tissue, neutrophils produce
hydrogen peroxide and myeloperoxidase (MPO) (30). Hydrogen peroxide induces
intracellular oxidative stress within hepatocytes by directly
diffusing into the cells. MPO utilizes hydrogen peroxide to produce
hypochlorous acid, which enters target cells and causes damage.
Activated neutrophils also release neutrophil elastase (NE), which
inhibits the production of prostacyclin I2 (PGI2). PGI2 is a
vasodilator released from endothelial cells that plays a protective
role by maintaining normal hepatic circulation during liver
ischemia-reperfusion. When activated neutrophils release NE, the
protective effect of PGI2 is suppressed and liver damage ensues
(31,32).
Additionally, T lymphocytes and natural killer T
(NKT) cells play important roles in the immune response during HIRI
(33-35). Deficiency of T cells or
disruption of their infiltration markedly reduces HIRI injury,
while NKT cells exacerbate inflammation by secreting IFN-γ. The
activation of T cells, particularly CD4+ Th1 and
CD8+ T cells, relies on the classical 'two-signal'
model: T cell receptor recognition of the peptide-MHC complex
presented by antigen-presenting cells (APCs) (first signal), along
with co-stimulatory signals (such as B7-CD28 interactions) provided
by APCs. In HIRI, hepatic APCs (e.g., Kupffer cells, dendritic
cells) upregulate co-stimulatory molecules upon activation, thereby
driving T cell clonal expansion and effector function
differentiation. Activated T cells aggravate hepatocyte damage
directly or indirectly by releasing cytokines such as IFN-γ and
TNF-α, as well as cytotoxic mediators such as granzyme/perforin
(36). Studies have shown that
sulfonylurea-mediated activation of type II NKT cells can reduce
the secretion of INF-γ by type I NKT cells, thus alleviating HIRI
damage (37,38).
Following ischemia, the hypoxic local environment in
the liver leads to the release of large amounts of DAMPs into the
bloodstream. These DAMPs activate the innate immune response via
pattern recognition receptors (PRRs). The activation of receptors
such as Toll-like receptors (TLRs) and NOD-like receptors (39) triggers intracellular signaling
pathways, which in turn activate inflammatory transcription factors
such as nuclear factor-kappa B (NF-κB) and promote the production
of various inflammatory cytokines (40,41). Moreover, among various PRRs, the
recently characterized cyclic GMP-AMP synthase-stimulator of
interferon genes (cGAS-STING) signaling pathway has emerged as a
key mediator of inflammation and innate immune responses in HIRI.
cGAS-STING activation occurs in response to cytoplasmic DNA, which
becomes abundant during ischemia-reperfusion due to mitochondrial
dysfunction and cellular damage. Once activated, STING initiates
downstream signaling pathways such as TANK-binding kinase-1
(TBK1)-interferon regulatory factor-3 (IRF3) and NF-κB, not only
enhancing the generation of ROS but also amplifying inflammatory
responses by promoting the adhesion and migration of immune cells
(42). Furthermore, the
interplay between the cGAS-STING pathway and the NOD-like receptor
family pyrin domain-containing 3 (NLRP3) inflammasome is
increasingly recognized as a critical axis under various conditions
(43,44). Activation of the NLRP3
inflammasome represents a key step in IL-1β production during HIRI,
further promoting neutrophil recruitment and amplification of the
inflammatory response (45). The
STING-NLRP3 axis links innate immune sensing to pyroptosis, a form
of programmed cell death, which exacerbates hepatocyte injury and
enhances the recruitment of immune cells to the liver. Activation
of this axis has been shown to contribute markedly to endothelial
dysfunction, hepatocyte necrosis and impaired liver regeneration in
HIRI. Therefore, targeting the inflammasome and the production of
inflammatory factors represents a potential therapeutic
strategy.
Microcirculatory dysfunction is also a significant
manifestation in the pathological process of HIRI (46). During ischemia, LSECs undergo
vacuolization and swelling due to energy depletion and ion
transport dysfunction, leading to the narrowing of the sinusoidal
lumen and obstructing smooth blood flow (47). Although the restoration of blood
flow during reperfusion helps provide oxygen, it may exacerbate the
microcirculatory disorder, further aggravating liver damage.
Preliminary studies suggested that the adhesion and accumulation of
neutrophils may be a major cause of microcirculatory dysfunction.
However, subsequent research indicated that despite accumulating in
the microcirculation, neutrophils do not directly cause severe
obstruction of sinusoidal perfusion (48). The isolated rat liver model has
also demonstrated that the accumulation of neutrophils within the
microcirculation does not significantly affect hepatic sinusoidal
perfusion (49). Subsequently,
local vasoconstrictors such as endothelin have been identified as
the primary cause of post-ischemic microcirculatory dysfunction.
Agonists of the adenosine or nitric oxide pathways, endothelin
antagonists and prostaglandins can exert protective effects by
regulating the sinusoidal diameter, preventing neutrophil adhesion,
inhibiting platelet aggregation and scavenging ROS (50).
Furthermore, the renin-angiotensin-aldosterone
system (RAAS) plays a key role in the pathophysiology of HIRI.
During reperfusion, blood recirculates into the ischemic liver
tissue, triggering a surge in ROS and pro-inflammatory mediators,
thereby activating the RAAS. Angiotensin II (Ang II) is a key
effector of the RAAS, exerting pro-inflammatory, pro-fibrotic and
vasoconstrictive effects through its interaction with the
angiotensin II type 1 receptor (AT1R). Upon binding to AT1R, Ang II
primarily activates Gq proteins, leading to the activation of
phospholipase C and the generation of second messengers IP3 and
DAG. This results in intracellular calcium release and protein
kinase C (PKC) activation. PKC further activates NADPH oxidase,
producing large amounts of ROS and activating the NF-κB and AP-1
pathways, thereby forming a positive feedback loop of ROS and
inflammation (51). Ang II
promotes the recruitment of inflammatory cells, increases oxidative
stress, stimulates the production of cytokines such as TNF-α and
IL-1β and further exacerbates hepatocyte damage. Aldosterone,
another key component of the RAAS, amplifies infection and fibrosis
formation, worsening liver injury volume. Abouzed et al
(52) demonstrated that using
angiotensin receptor blockers, such as valsartan, to inhibit the
RAAS and LCZ696 to inhibit neprilysin reduced liver IRI damage in a
mouse model.
In summary, the initiation and progression of HIRI
are orchestrated by a complex network of cytokines, primarily
released by early-activated Kupffer cells and later-infiltrating
neutrophils (Fig. 2).
Mechanistically, TNF-α acts as a central mediator by binding to its
receptor tumor necrosis factor receptor 1, subsequently triggering
the activation of NF-κB, mitogen-activated protein kinases (MAPKs)
and c-Jun N-terminal kinase (JNK) pathways (53). Furthermore, TNF-α enhances the
expression of adhesion molecules on sinusoidal endothelial cells,
particularly intercellular adhesion molecule-1 (ICAM-1) and
vascular cell adhesion molecule 1(VCAM-1), promoting firm adhesion
and aggregation of neutrophils and aggravating microvascular
obstruction and tissue damage (54). In addition to TNF-α, a series of
interleukins contribute to the immunopathology of HIRI. IL-6,
IL-12, IL-23, VEGF and hepatocyte growth factor (HGF) play critical
yet distinct roles (55).
Pro-inflammatory interleukins such as IL-12 and IL-23 stimulate
CD4+ T cells to secrete IL-17, a potent chemoattractant
for neutrophils, creating a positive feedback loop that exacerbates
liver injury (51). Moreover,
IL-12 and IL-23 increase TNF-α production via NF-κB activation,
while IL-23 further activates the IFN-γ/IRF-1 axis, promoting the
propagation of inflammation. Conversely, the post-inflammatory
phase is mediated by counter-regulatory cytokines. IL-6, VEGF and
HGF exhibit protective and regenerative properties. IL-6 promotes
hepatocyte proliferation and alleviates oxidative stress by
enhancing glutathione synthesis and activating the STAT3 signaling
pathway (56). VEGF demonstrates
a dual role in IRI; while endogenous VEGF may promote injury
initiation, exogenous administration has been shown to facilitate
angiogenesis and regeneration in post-ischemic liver (57). Similarly, HGF exerts potent
protective effects by inhibiting oxidative stress, stimulating
hepatocyte proliferation via ERK1/2 activation, upregulating
glutathione and downregulating ICAM-1 expression, thereby
suppressing neutrophil adhesion and migration (58). In conclusion, the pathogenesis of
HIRI is a complex, multi-factorial and multi-stage process
involving interactions among oxidative stress, immune cell
activation, cytokine release, microcirculatory dysfunction and
damage-repair mechanisms. Although our understanding of its
pathological mechanisms has advanced, effectively intervening in
and treating HIRI, particularly in clinical settings such as liver
transplantation, remains a key focus of current research.
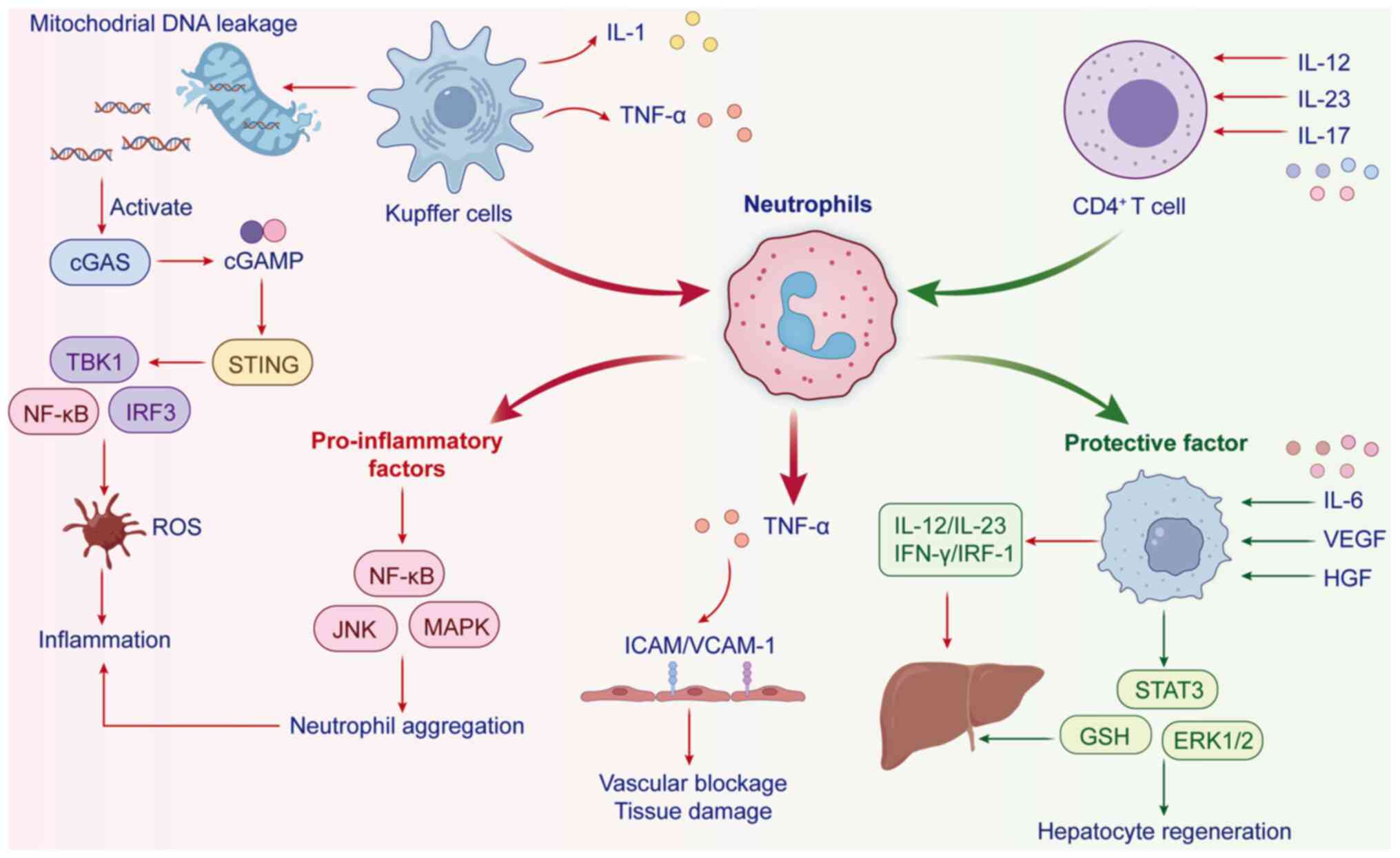 | Figure 2Integrated signaling network in HIRI:
Inflammation amplification via cGAS-STING and cytokine crosstalk.
mtDNA release activates cGAS-STING, driving TBK1-IRF3/NF-κB to
enhance ROS and immune cell recruitment. Kupffer
cell/neutrophil-derived IL-1/TNF-α and IL-12/IL-23/IL-17 axes
promote inflammation, while IL-6, VEGF and HGF support regeneration
via STAT3/ERK1/2, antioxidant effects and reduced adhesion. HIRI,
hepatic ischemia-reperfusion injury; cGAS-STING, cyclic GMP-AMP
synthase-stimulator of interferon genes; mtDNA, mitochondrial DNA;
ROS, reactive oxygen species; NF-κB, nuclear factor-kappa B; IRF3,
interferon regulatory factor 3; JNK, c-Jun N-terminal kinase; MAPK,
mitogen-activated protein kinase; ICAM-1, intercellular adhesion
molecule-1; VCAM-1, vascular cell adhesion molecule-1; VEGF,
vascular endothelial growth factor; HGF, hepatocyte growth factor ;
STAT3, signal transducer and activator of transcription 3; GSH,
glutathione. |
The key role of neutrophils in HIRI
In HIRI, neutrophils play a central and dynamic role
as effector cells. Their function is not fixed but evolves in an
orderly manner along with the injury process. During the early
phase, neutrophils are rapidly recruited to the ischemic liver,
where they directly mediate hepatocyte damage by releasing ROS and
proteases and form NETs, thereby amplifying local inflammation and
microcirculatory dysfunction (4,5,59). As the pathological process
progresses, neutrophils exhibit remarkable functional plasticity
(60,61). A subset of these cells can
undergo phenotypic switching, enhancing phagocytic activity to
clear necrotic debris and secreting anti-inflammatory cytokines,
thereby actively contributing to inflammation resolution and tissue
repair (11). Ultimately,
neutrophils are cleared from the inflammatory site through
controlled processes such as programmed apoptosis, NETosis, or
reverse transendothelial migration (rTEM) (62,63). Therefore, neutrophils are not
merely terminal effectors but are deeply integrated into a
coordinated immune response program. The dynamic balance between
their pro-damage and pro-repair functions directly determines
whether the tissue outcome shifts toward sustained injury or
initiates orderly repair. This core mechanism establishes
neutrophils as a key therapeutic target in HIRI. Future
intervention strategies should focus on precisely modulating the
nodes of their functional transition rather than applying
non-specific suppression.
Neutrophil activation and
recruitment
In the early phase of HIRI, DAMPs activate resident
immune cells in the liver, particularly Kupffer cells and LSECs,
which subsequently induce the production of various inflammatory
mediators such as chemokines (CXCL1, CXCL2), cytokines (TNF-α,
IL-1β) and ROS (64). This
cascade of reactions collectively promotes the directed recruitment
of neutrophils to the liver and initiates a systemic inflammatory
response. Notably, the extent of neutrophil recruitment is markedly
associated with the duration of ischemia, the longer the ischemic
period, the more intense the local and systemic inflammatory
responses, ultimately leading to aggravated liver injury.
During the HIRI process, leukotriene B4 (LTB4) is
one of the earliest neutrophil chemokines to be released and its
strong chemotactic effect plays a crucial role in the early
inflammatory response (65). In
addition, FMIT released from mitochondria binds to the formyl
peptide receptor 1 (FPR1) receptor on the surface of neutrophils
during cell necrosis, promoting their migration to the necrotic
area. Neutrophils play a role in HIRI in the initial immune
response and by activating the complement system, which further
enhances the inflammatory response. C5a in the complement system is
one of the strongest chemotactic factors (65) and promotes neutrophil recruitment
and infiltration to the damage site by binding to the C5aR1
receptor on the neutrophil surface (66). Zhang et al (67) reduced neutrophil recruitment and
ROS production by using single-stranded oligonucleotides with high
affinity for C5a, coupled to nanoparticles, thus alleviating
inflammation and damage associated with HIRI.
LSECs upregulate the expression of CXCL1 during
HIRI, which not only promotes neutrophil NETs formation but also
exacerbates the formation of liver sinusoids and the occurrence of
portal hypertension (68).
However, chemokines alone are insufficient for efficient neutrophil
recruitment. For optimal chemotactic effects, the concentration of
chemokines must be maintained within a precise range, a process
depends on the expression of glycosaminoglycans (GAGs) on LSECs
(69). GAGs can bind chemokines
via their sulfated domains, anchoring them to the endothelial
surface and thereby establishing and maintaining a local
high-concentration chemokine gradient, which is essential for the
specific recruitment of neutrophils (70,71). Research shows that blocking the
expression of heparan sulfate significantly inhibits
chemokine-mediated neutrophil migration (72); similarly, disrupting the
interaction between GAGs and chemokines effectively suppresses
neutrophil extravasation in gout models (73). In summary, chemokines, cytokines,
the complement system and GAGs form a synergistic regulatory
network that precisely controls the activation and recruitment of
neutrophils in the early stage of HIRI.
Neutrophil adhesion in the sinusoids
After reperfusion, neutrophils are activated and
adhere to LSECs to enter the injury area. The unique structure of
LSECs determines the specificity of neutrophil migration. Unlike
capillaries in other organs, LSECs possess a porous structure, have
relatively little subendothelial basement membrane material and
lack intercellular junctions. These structural characteristics
allow large molecules such as lipoproteins to pass through, but
prevent the passage of chylomicrons (74). Additionally, LSECs in the liver
do not express Weibel-Palade bodies and are devoid of E- and
P-selectins (75). As a result,
selectin-mediated neutrophil rolling is not observed on LSECs
(76). This feature indicates
that the accumulation of neutrophils in the hepatic sinusoids is
less dependent on these adhesion molecules. In vivo
microscopic observations show that ~80% of neutrophils adhere to
LSECs, while 20% adhere to post-sinusoidal venules, highlighting
the critical role of LSECs in adhesion (77). Among these, CD44-mediated
interactions between hyaluronic acid (HA) receptors play an
important role in neutrophil adhesion to LSECs. The binding of CD44
to the extracellular matrix (ECM) of LSECs enhances neutrophil
contact with the liver microenvironment (78,79). In models of endotoxin-induced
liver injury, blocking the CD44-HA interaction markedly inhibits
neutrophil adhesion in the sinusoids but does not markedly affect
adhesion in post-sinusoidal venules. This indicates that the
CD44-HA pathway has a specific role in HIRI. CD44 activates the p38
MAPK and PI3K/Akt pathways, mediates cytoskeletal rearrangement and
strengthens firm adhesion, while upregulating integrin expression
to stabilize neutrophil-endothelial interactions (78).
In HIRI models, neutrophil interactions with LSECs
involve the binding of the integrin molecule Mac-1 (CD11b/CD18)
with ICAM-1 on LSECs. After ischemic injury, LSECs can sense DAMPs
through Toll-like receptor 9, thereby directly inducing the release
of IL-1β and IL-18. The upregulation of these cytokines further
enhances ICAM-1 expression, promoting neutrophil migration to the
liver sinusoidal spaces (80).
Neutrophil transendothelial migration
(TEM) and rTEM in the liver sinusoids
Neutrophils migrate into the liver parenchyma via
TEM, a process that occurs mainly through paracellular or
transcellular routes, with the paracellular pathway dominating in
peripheral circulation (81). A
key step in TEM is the binding of β2-integrin (CD11b/CD18) to
VCAM-1 (82). Under acute
inflammatory conditions, LSECs rapidly activate the endothelial
Notch signaling pathway (83),
increasing the expression of TNF-α and IL-33, which further
exacerbates liver damage (84).
Additionally, after neutrophils cross the vascular
endothelium, their migration within the ECM depends on the dynamic
balance of matrix metalloproteinases (MMPs) and tissue inhibitors
of metalloproteinases (TIMPs). MMPs are responsible for hydrolyzing
ECM components, while TIMPs regulate MMP activity. Among them,
gelatinases (MMP2-MMP9) play an important role in HIRI (85). Fibronectin is a key component of
the ECM glycoprotein family and activates the integrin α4β1
receptor on neutrophils in HIRI mice, leading to the production of
MMP9. Inhibitors of MMP9 markedly reduce neutrophil infiltration
and alleviate liver damage. Alterations in the MMP-TIMP balance are
associated with pathological processes involving ECM degradation,
such as tumor invasion, angiogenesis and tissue repair. Duarte
et al (86) verified that
animals lacking TIMP1 suffered more severe liver dysfunction and
tissue damage. In this model, the mortality rate of TIMP1
double-knockout mice following reperfusion was 60%, while all
wild-type mice survived (86).
Additionally, the lack of TIMP1 was associated with increased MMP9
activity, which facilitated neutrophil migration across the
vascular barrier during liver IRI. Indeed, TIMP double knockout
mice exhibited marked neutrophil infiltration in the liver,
accompanied by upregulation of proinflammatory mediators such as
TNF-α, IFN-γ and inducible nitric oxide synthase. Therefore, MMPs
and TIMPs play crucial roles in maintaining liver homeostasis and
can serve as therapeutic targets for improving liver injury.
Beyond TEM, increasing evidence with the advance of
imaging techniques suggests that neutrophils can also undergo rTEM
(63). Neutrophils can migrate
within tissues and subsequently clear the infection site through
this mechanism. However, dysregulation of this process may trigger
systemic inflammatory responses. The mechanisms of rTEM are still
under active investigation. Some studies have identified potential
regulatory factors that play key roles in neutrophil rTEM. For
example, increased levels of chemokines ultimately lead to
neutrophil migration in the opposite direction along the
microvenular wall (87),
suggesting that the concentration of chemotactic factors is one of
the main determinants of accelerating neutrophil rTEM. Furthermore,
dynamic regulation of chemokine receptor expression on the
neutrophil surface or its sensitivity to corresponding ligands is
critical for neutrophil rTEM (88). In addition to these chemokine-
and receptor-mediated events, junctional adhesion molecule-C
(JAM-C) is the most widely studied molecule regulating neutrophil
rTEM and has been widely expressed on endothelial cells (EC). JAM-C
knockout mice exhibited increased rTEM neutrophils, with more than
50% of total transendothelial migration events, compared with ~10%
observed in wild-type animals (80). This group also described the
JAM-C-mediated leukocyte rTEM regulation mechanism mediated by the
LTB4 and NE axis (89).
It is worth noting that, since neutrophil rTEM
functions by clearing excess neutrophils from local tissues,
regulating T and B cell proliferation, NET formation and inducing
systemic inflammation and immune system interactions (81,90-92), targeting neutrophil rTEM may
represent a new therapeutic strategy to address inflammation.
Future research strategies may focus on modulating chemokine
gradients, stabilizing endothelial junctions, or targeting the
MMP-TIMP axis to achieve this balance, offering novel avenues for
treating HIRI and other inflammatory diseases.
NETs
Another important function of neutrophils in HIRI is
the formation of NETs. NETs consist of an extracellular scaffold of
nuclear DNA loaded with histones and granular proteins such as NE
and MPO. The mechanism of NET formation involves NADPH oxidase- and
peptidylarginine deiminase 4 (PAD4)-mediated citrullination of
histones, translocation of neutrophil NE and MPO to the nucleus,
chromatin decondensation and extracellular DNA release promoted by
proteolysis of histones.
Neutrophils produce and release NETs through a
process called NETosis. While NETs serve as a beneficial defense
strategy against pathogens by trapping and combating bacteria,
fungi and protozoa (92), in
sterile inflammation such as HIRI, 'suicidal NETosis' (that is,
neutrophil programmed death associated with NET release)
predominantly occurs. By exposing intracellular proteins to the
extracellular space, NETosis promotes the presentation of
autoantigens and the release of DAMPs, thereby amplifying the
ongoing inflammatory response. Research (93) shows that in liver
transplantation-associated HIRI, plasma levels of NET markers were
markedly elevated in LT recipients, peaking at the end of
transplantation and correlating with coagulation activation.
Compared with healthy controls, levels of the more specific NET
marker MPO-DNA complexes were already markedly increased at the
beginning of transplantation (94). Besides plasma, NETs were detected
in liver tissue 30 min after reperfusion. Excessive NET formation
exacerbates tissue damage.
The dual role of neutrophils in HIRI:
From inflammatory effector to repair and regulation
In recent years, the dual role of neutrophils in
sterile inflammation, both driving tissue injury and promoting
inflammation resolution and tissue repair, has garnered widespread
attention. Traditionally viewed as 'first responders' that promote
inflammation and tissue destruction, growing evidence indicates
that neutrophils possess high functional plasticity. Under specific
microenvironmental regulation, they can shift toward
anti-inflammatory, pro-repair phenotypes and participate in
steering HIRI from a 'damage-dominant' to a 'repair-dominant'
transition. In liver-related injury models, the reparative
functions of neutrophils have been preliminarily revealed. For
instance, Wang et al (7)
found that in a fully repaired sterile thermal liver injury model,
neutrophils also penetrated the injury site, performing critical
tasks such as dismantling damaged vessels and creating channels for
new vascular regrowth. These neutrophils performing key repair
functions neither died nor were phagocytosed. Instead, they
returned to the circulation via rTEM, likely became inactivated or
reprogrammed and then migrated selectively to the bone marrow via
CXCR4 to end their life cycle. Deng et al (8) further elucidated how the
post-injury liver signals the bone marrow to release neutrophils
and how reparative neutrophils signal hepatocytes to re-enter the
cell cycle. Using liver-specific leukemia inhibitory factor
receptor (LIFR) knockout mice, the authors demonstrated that
hepatocyte LIFR promotes the secretion of CXCL1 and cholesterol in
a STAT3-dependent manner, recruiting and activating neutrophils.
These neutrophils, in turn, secrete the hepatocyte mitogen HGF,
accelerating liver injury repair and regeneration. Lin et al
(9) discovered that during
allogeneic tissue transplantation (mouse heart, kidney and human
cardiac tissue), neutrophils are indispensable for creating new
vascular networks, further confirming their pivotal role in tissue
repair. Marques et al (10) found that compared with Kupffer
cell depletion, neutrophil depletion using anti-Ly-6G alleviated
acetaminophen-induced liver injury. In this liver injury model, a
small number of neutrophils contributed to the clearance of
cellular debris, whereas a large number enhanced liver injury and
inflammation. Thus, the role of neutrophils in this process is
determined by the quantity of toxic substances in the liver and the
number of infiltrating neutrophils. Consequently, how to regulate
neutrophil function to promote the shift from injury to repair has
become a focus of our attention.
Neutrophil polarization
Similar to macrophages, the diverse biological
functions of neutrophils are influenced by timing and specific
tissue environments. Analogous to the ability of macrophages to
polarize toward M1/M2 phenotypes under different stimuli (95), studies indicate that specific
microenvironments can also drive neutrophils toward distinct
phenotypes such as N1/N2 (60,61,96,97). Currently, N1 and N2 neutrophil
populations are primarily defined by their functional phenotypes,
based on their capacities for degranulation, cytokine release and
migration. The same phenotypes have been identified as
pro-inflammatory (N1) and anti-inflammatory (N2) neutrophils.
Mihaila et al (98)
characterized phenotypic and functional differences between N1 and
N2 neutrophils in vitro based on transcriptomics and
functional assays. The authors found that compared with N2
neutrophils, N1 neutrophils exhibited higher levels of ROS and
oxidative burst, greater MPO and MMP-9 activity and stronger
chemotactic responses. N1 neutrophils also showed elevated
expression of NADPH oxidase subunits and activation of signaling
molecules ERK and the p65 subunit of NF-κB. Furthermore, they
identified for the first time that the alarmin S100A8/9, abundantly
secreted by activated neutrophils, serves as a critical promoter of
the aggressive pro-inflammatory N1 phenotype through an autocrine
mechanism.
Although no direct studies have explicitly
identified N2-type neutrophils or systematically resolved their
polarization trajectory in HIRI models, multiple cross-disease
model studies provide strong support for this hypothesis (11,12,99). In the early phase of HIRI,
initially infiltrating neutrophils mostly exhibit an 'N1-like'
phenotype with high activity, ROS production and strong chemotactic
capacity, releasing mediators such as MPO, NE and NETs that
exacerbate oxidative stress and microcirculatory dysfunction. As
reperfusion time extends, the microenvironment gradually shifts
from pro-inflammatory to pro-repair and some neutrophils begin to
display anti-inflammatory, pro-angiogenic and immunomodulatory
characteristics, suggesting the occurrence of functional
polarization. This functional duality makes neutrophil polarization
a central node in the 'injury-resolution balance,' directly
influencing the prognosis of HIRI. This theory has been
corroborated in ischemia models of the heart and brain. In a
myocardial infarction model, the predominant neutrophil subset in
the infarct area was N1-type neutrophils rich in pro-inflammatory
factors, peaking on day 1; subsequently increased N2-type
neutrophils corresponded to the anti-inflammatory phenotype after
infarction (11). In a mouse
stroke model, peripheral blood neutrophil counts peaked at 12 h
after cerebral ischemia, with peak brain injury occurring 1-2 days
later. In stroke lesions, neutrophil clearance peaked 2 days
post-stroke and extracellular traps were mostly detected 2-3 days
after stroke. Among neutrophils infiltrating stroke lesions,
N2-phenotype neutrophils promoted macrophage-mediated neutrophil
clearance. These processes are highly similar to the pathological
progression of HIRI, that is, the dynamic transition from early
inflammatory storm to later repair and regeneration (12). Therefore, it is reasonable to
hypothesize that a similar neutrophil phenotypic switch exists in
HIRI, though it has not yet been systematically identified and
deeply explored.
Currently, research on neutrophil polarization and
repair primarily focuses on cancer models (100). However, the role of neutrophil
polarization in tissue repair is also gradually emerging in
non-neoplastic diseases. In a mouse brain inflammation model,
rosiglitazone treatment shifted the neutrophil population toward an
N2 phenotype, suppressing inflammatory responses, promoting
neutrophil clearance and reducing brain injury (101).
In summary, although direct evidence for N1/N2
polarization switching in HIRI is currently lacking, multisystem,
cross-disease studies consistently indicate that neutrophil
function is highly dynamic and context-dependent. Their shift from
'pro-inflammatory effectors' to 'repair coordinators' may be a
critical node determining the prognosis of HIRI. Therefore,
promoting neutrophil polarization toward an N2-like phenotype could
be a potential therapeutic strategy to alleviate HIRI and foster
liver regeneration. Future research should focus on the specific
mechanisms of N1/N2 polarization, including whether a polarization
process exists and whether it is driven by 'transcriptional
reprogramming' of existing neutrophils or by 'epigenetic
remodeling' of newly recruited cells due to environmental
changes.
Targeted neutrophil therapy strategies
The role of neutrophils in the initiation and
progression of various pathological conditions makes them an
attractive therapeutic target. However, the critical requirement of
neutrophils for antimicrobial host defense limits the utility of
therapies that broadly reduce neutrophil numbers or functional
responses. Current treatments targeting single inflammatory
mediators may have limited efficacy due to redundancy within the
innate immune system and the potential for eventual
immunosuppression. Arguably, the ideal therapeutic strategy would
be to prevent or reverse neutrophil-mediated tissue damage without
compromising their ability to control microbial invasion.
Furthermore, one innovative approach for developing treatments for
inflammatory disorders is to harness the biological properties of
neutrophils to enhance the resolution of inflammation. Based on the
functional plasticity of neutrophils, the present review proposed
the 'injury-repair balance' theoretical framework. This framework
leverages the evolving multifunctional roles of neutrophils in
HIRI, from a 'pro-inflammatory (N1)' to an
'anti-inflammatory/reparative (N2)' phenotype, to design
alternative therapies for pro-inflammatory diseases. By preventing
N1 polarization or inducing N2 polarization, this approach opens
new therapeutic avenues for achieving precise immunomodulation and
developing highly effective, low-toxicity organ-protective
strategies in the future. Building on this, the present review
further explore multi-dimensional intervention strategies targeting
neutrophils. These include inhibiting excessive recruitment and
activation, regulating migration to reduce local accumulation,
suppressing NET formation and promoting their clearance, as well as
combining antioxidant and anti-inflammatory treatments to
re-establish immune homeostasis. Additionally, EVs, owing to their
excellent targeted delivery and immunomodulatory capabilities, have
emerged as a potential tool for achieving precise regulation of
neutrophil function (Fig.
3).
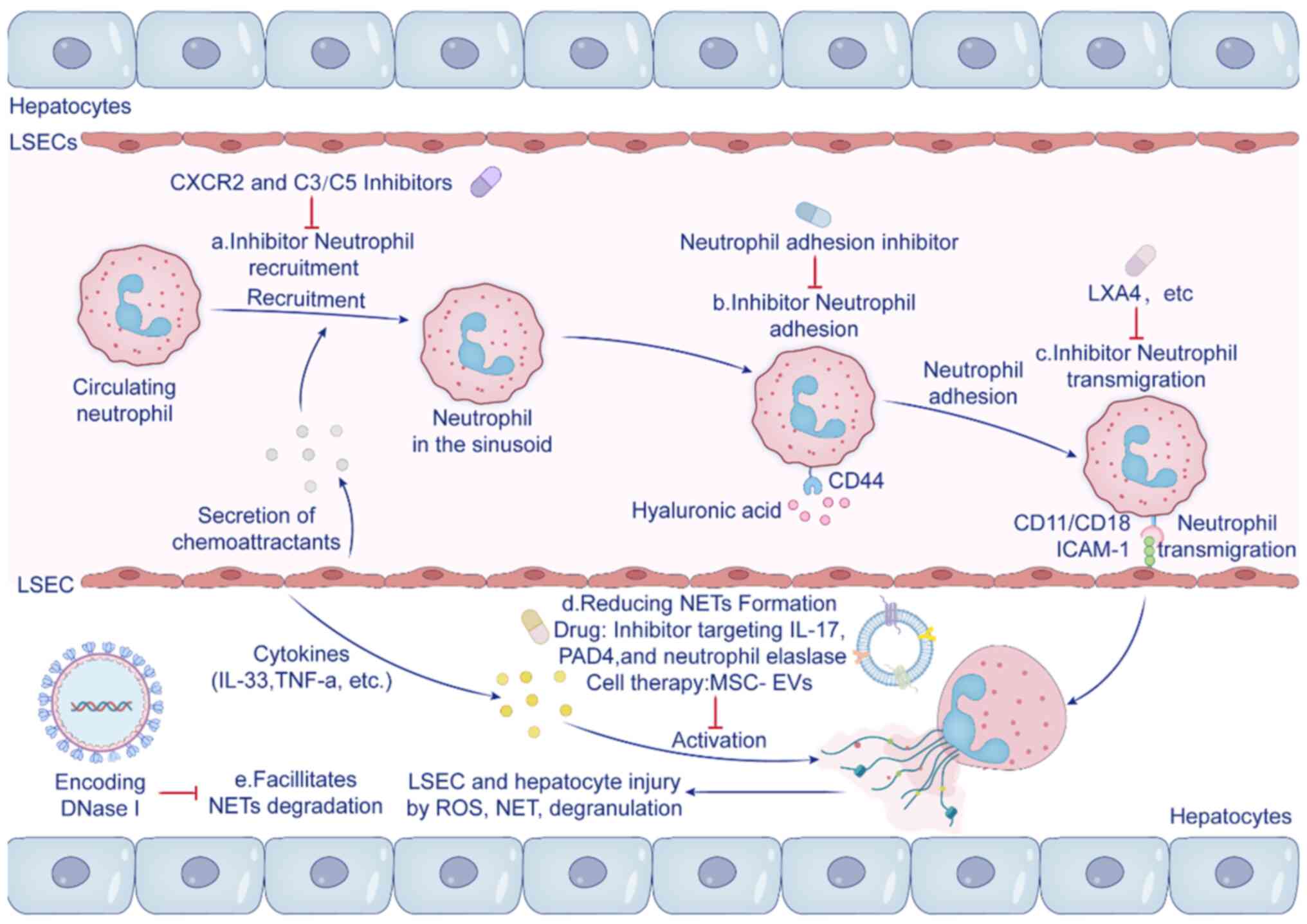 | Figure 3Neutrophil-targeted therapeutic
strategies for hepatic ischemia-reperfusion injury. Strategies
include targeting chemokine receptors and complement inhibitors to
regulate neutrophil recruitment, anti-adhesion and migration
therapies to prevent neutrophil accumulation, reducing NETs
formation and enhancing their degradation, antioxidant and
anti-inflammatory treatments to mitigate oxidative stress and the
application of extracellular vesicles as a novel approach for
targeted therapeutic delivery. LSECs, liver sinusoidal endothelial
cells; LXA4, lipoxin A4; ICAM-1, intercellular adhesion molecule-1;
NETs, neutrophil extracellular traps; PAD4, peptidylarginine
deiminase 4; MSCs-EVs, mesenchymal stem cells-extracellular
vesicles; TNF-α, tumor necrosis factor alpha; ROS, reactive oxygen
species; DNase I, Deoxyribonuclease I. |
Chemokine receptors and complement
inhibitors
Chemokine receptors, as core targets regulating
neutrophil recruitment, have become an important direction in HIRI
treatment. Chemokines such as CXCL1 and CXCL2 precisely guide
neutrophils toward inflammatory sites by binding to the CXCR2
receptor. Currently, drug development targeting CXCR2 has achieved
phased progress. In animal models, the dual-target inhibitor
Ladarixin (targeting CXCR1 and CXCR2) has been shown to effectively
block neutrophil infiltration across the vascular basement membrane
while preserving their rolling and adhesion functions, thereby
suppressing the inflammatory response (102). Furthermore, small-molecule
CXCR2 antagonists such as Repertaxin, Navarixin, Danirixin and
AZD5069 have also demonstrated potential in reducing neutrophil
recruitment in animal disease models (103), showing significant efficacy
particularly in sustained inflammatory conditions such as cystic
fibrosis, severe asthma and chronic obstructive pulmonary disease
(COPD). It is noteworthy that CXCR2-selective inhibitors have not
caused significant impairment to neutrophil host defense functions,
providing a key basis for their safety profile. Most of these drugs
are still in clinical development (such as, Phase II trial
NCT001006616 for COPD) (104),
indicating that preliminary human safety data are being
accumulated. However, no clinical trials targeting HIRI have yet
been reported. Clinical investigations of CXCR2-selective
inhibitors for other diseases are ongoing, such as studies on their
role in combination with oseltamivir for influenza treatment
(105). These preliminary
results have validated the feasibility of the strategy to inhibit
neutrophil recruitment by targeting chemokine receptors.
The complement system represents another key
regulatory network for neutrophil recruitment, demonstrating unique
advantages in mitigating HIRI when suppressed. C3a, C5a and the
membrane attack complex (MAC) play central roles in liver IRI by
activating the complement cascade and exacerbating tissue damage.
Preclinical studies have confirmed that multi-strategy
interventions targeting the complement system (such as blocking C3,
C5, or enhancing endogenous regulatory mechanisms) can markedly
improve liver injury outcomes. For example, the humanized
monoclonal antibody Eculizumab (targeting C5), an approved drug,
has established its safety profile in diseases such as paroxysmal
nocturnal hemoglobinuria and atypical hemolytic uremic syndrome
(106). However, its
application for human HIRI has not been reported. For mouse models,
anti-C5 monoclonal antibody (BB5.1) treatment not only reduced ALT
levels and inhibited the expression of inflammatory mediators such
as IL-6 and TNF-α, but also provided cellular protection by
reducing neutrophil infiltration and oxidative stress at both early
(2 h) and late (6 h) reperfusion stages (66). Furthermore, C3 inhibitors such as
the compstatin analog AMY-101, by blocking C3 convertase, reduced
kidney damage and systemic inflammation in hemorrhagic shock
models. The mechanism involves inhibiting the production of
anaphylatoxins, regulating Kupffer cell phagocytosis and blocking
the complement positive feedback loop (107,108). A promising strategy involves
using recombinant regulatory molecules to mimic endogenous
regulatory mechanisms [such as soluble CR1 (sCR1) and CR2-CD59
fusion proteins] (109,110), which can precisely modulate
complement activation without inducing immunosuppression. In liver
IRI models, these approaches have shown the potential to reduce MAC
deposition, protect microvascular integrity and preserve
C3a/C5a-mediated regenerative signaling. While clinical evidence
for complement inhibitors in cardiac surgery has accumulated
(110), targeted clinical
trials are still needed to validate efficacy in humans for hepatic
IRI. Nevertheless, existing data strongly support their potential
as breakthrough therapies for surgery- and transplant-associated
liver injury.
Anti-adhesion and migration molecule
therapy
Neutrophil adhesion and migration depend on
interactions with adhesion molecules on endothelial cells, such as
ICAM-1 and P-selectin. Therefore, anti-adhesion molecule therapy
has become an important strategy for targeting neutrophils. ICAM-1
blockers reduce inflammation in rat pancreatic transplant models
and improve microvascular perfusion (111). In rat models, NE inhibitor
sivelestat sodium inhibits neutrophil migration to the vessel wall,
reducing HIRI (112). In 2025,
Xia et al (113)
developed a macrophage membrane-disguised manganese-based
anti-aging nanzyme (MB@LM) for targeted delivery and oxidative
stress relief in acute kidney injury (AKI) treatment. MB@LM
selectively targets damaged kidney tissue overexpressing adhesion
molecules such as ICAM-1 and VCAM-1, promoting enhanced
accumulation at the injury site. In an AKI mouse model, MB@LM
treatment markedly reduced renal injury, restored microvascular
perfusion assessed by ultrasound imaging and alleviated
inflammation, demonstrating marked therapeutic efficacy. The
present review provides strong support for the treatment of liver
injury.
In addition to conventional systemic
immunosuppressive therapies, some studies have proposed local
immunosuppressive strategies that involve the topical
administration of immunosuppressants or regulatory factors to
reduce neutrophil activation. For example, the use of specific
pro-resolving mediators such as lipoxin A4 (LXA4) can inhibit
neutrophil activation and migration (114,115), thereby alleviating hepatic
inflammatory responses. LXA4 acts by activating the ALX/FPR2
receptor, suppressing ROS production and NETs formation by
neutrophils and thus reducing liver inflammation induced by
reperfusion injury. The LXA4/FPR2 signaling pathway mitigates
ferroptosis in alveolar epithelial cells during lung IRI via an
NRF2-pathway-dependent mechanism (116), which may offer a novel
therapeutic target for HIRI.
Reducing NETs formation and promoting
clearance
In addition to regulating neutrophil activation and
migration, targeting the inhibition of NETs formation and promoting
their degradation is also an important direction for treating HIRI.
The formation of NETs relies on neutrophil oxidative stress and the
synergistic regulation of multiple molecular signaling pathways,
making drug interventions targeting the NETosis process a potential
therapeutic target for alleviating HIRI. Superoxide induces NET
release through the TLR4/NOX pathway and the combination of
allopurinol (superoxide inhibitor) and diphenyl iodinium (NOX
inhibitor) can markedly suppress NET formation in mice and
alleviate liver injury (117).
Elevated serum IL-17 levels are directly associated with increased
neutrophil infiltration and NET formation in the liver, suggesting
that IL-17 is a potent NET inducer (118). Similarly, in the kidney IRI
mouse model, infiltrated neutrophils were the main source of IL-17
production, further promoting neutrophil migration (119). Moreover, research by Lin et
al (120) indicates that
IL-17-positive neutrophils, abundant in human psoriatic lesions,
can release IL-17 through the induction of NETs. Therefore,
targeting IL-17 with neutralizing antibodies can reduce NET
formation and mitigate IR-induced damage. Histone citrullinase PAD4
is a key mediator of NET formation and its inhibitors (YW3-56 and
YW4-03) can markedly reduce liver injury in HIRI mice (40). Similar effects were observed in
mouse models when targeting NE or using specific MPO inhibitors.
Compared with wild-type animals, NE inhibitor-treated PAD4KO or
wild-type animals showed markedly smaller tumors, reduced
neutrophil infiltration and fewer NETs (121), suggesting that anti-PAD4
strategies may provide a new pathway for HIRI treatment. Yang et
al (122) found that Aldh2
deficiency induced NOX2-dependent NETosis through the endoplasmic
reticulum stress/GST2/LTC4 pathway and the LTC4 receptor antagonist
montelukast might be a potential therapeutic option. Other studies
have indicated that acrolein promotes NET formation by activating
the NOX2/P38MAPK signaling pathway, delaying postoperative liver
recovery (123). Targeting NOX2
and P38MAPK pathways can suppress NET generation in chronic liver
disease patients but improve postoperative liver function (123). These strategies effectively
block the source of NET formation and reduce liver tissue damage.
However, excessive NET accumulation can still trigger localized
inflammatory damage, making the promotion of NET degradation a key
aspect of treatment. Under physiological conditions, serum
deoxyribonuclease (DNase; e.g., DNase1) is the main nuclease
responsible for degrading NETs (124,125). Liu et al (126) innovatively constructed a
stroke-homing peptide (SHp) fused with DNase1 (SHp-DNase1), which
enhances DNase stability and targets NETs in thromboembolism
events. Combined with ceftriaxone sodium therapy, this approach
synergistically controls inflammation and resolves NET-induced
microcirculatory impairment. The validation of these
multidimensional intervention strategies in the HIRI model provides
an innovative direction for precisely regulating NET biology and
overcoming the therapeutic bottleneck in HIRI treatment.
Antioxidant and anti-inflammatory
therapy
Neutrophils contribute to liver reperfusion injury
through migration and release of inflammatory mediators and by
generating ROS, which exacerbate tissue damage. Excess ROS can
attack cell membrane lipids, mitochondrial structures and DNA,
triggering oxidative stress, leading to hepatocyte apoptosis or
necrosis, worsening inflammation and impairing liver function
(127). Hence, targeting
neutrophil-mediated oxidative stress has emerged as a key strategy
for preventing and treating HIRI, with antioxidant therapies
gaining increasing attention.
Antioxidants can effectively alleviate liver
reperfusion injury by scavenging free radicals and reducing
oxidative stress (127). In
animal models, various antioxidants have shown protective effects,
including N-acetylcysteine (NAC) (117), vitamin E, vitamin C (128). NAC, as a precursor for
glutathione (GSH) synthesis, replenishes intracellular cysteine,
promotes GSH production and enhances endogenous antioxidant
capacity, thereby reducing ROS-induced liver damage (117). Vitamin E, a lipid-soluble
antioxidant primarily localized in cell membranes, interrupts the
chain reaction of lipid peroxidation. Vitamin C, a water-soluble
antioxidant, directly neutralizes free radicals and regenerates
vitamin E, synergistically exerting protective effects (128).
In recent years, nano-selenium (nano-Se) has gained
popularity in antioxidant therapy due to its high efficiency and
low toxicity. Selenium is a necessary cofactor for various
antioxidant enzymes, such as glutathione peroxidase, superoxide
dismutase and catalase and it helps regulate cellular redox
balance. Nano-Se not only directly scavenges free radicals such as
DPPH and ABTS but also enhances the activity of the endogenous
antioxidant system (129).
Nano-Se particles modified with chitosan of different molecular
weights exhibit superior free radical scavenging capacity and
concentration-dependent ROS inhibition in skin and intestinal
cells, higher selenium concentrations yield stronger inhibitory
effects (130). Compared with
traditional selenium preparations, nano-Se has higher
bioavailability and targeting potential. It exerts hepatoprotective
effects through multiple mechanisms, including ROS clearance,
enzyme activity enhancement and apoptosis inhibition, with lower
toxicity, showing broad potential in preventing and treating HIRI
(131).
However, single antioxidant therapy is insufficient
to comprehensively control the complex pathological process of
HIRI. Regarding the interplay between oxidative stress and
inflammation, which forms a 'vicious cycle' (51), combined antioxidant and
anti-inflammatory therapy strategies are considered more
advantageous (132). On the one
hand, antioxidants can alleviate oxidative stress and inhibit
neutrophil activation and infiltration; on the other hand,
anti-inflammatory drugs, such as non-steroidal anti-inflammatory
drugs or corticosteroids, can effectively reduce local liver
inflammation (133,134). Their synergistic effects hold
promises for more efficiently mitigating liver tissue damage and
promoting functional recovery (132).
Notably, following HIRI, the expression of polo-like
kinase 2 was markedly upregulated, indicating its involvement in
regulating the injury process. Biocompatible Prussian blue (PB)
scavengers possess ROS-scavenging and anti-inflammatory properties
(135) and may be used for HIRI
treatment. PB scavengers are primarily distributed in the liver and
have good alleviating effects on cell apoptosis, tissue damage and
organ dysfunction following HIRI. In 2025, Shen et al
(136) developed a PB nanzyme
carrier system (met@PBN@Neu-CVs) based on neutrophil membrane
coating for the delivery of metformin. This biomimetic nanoplatform
utilizes the chemotactic properties of neutrophil membranes to
actively target the inflamed liver, markedly enhancing drug
accumulation in damaged tissues. This system can efficiently clear
ROS, suppress inflammation and promote macrophage polarization
toward the anti-inflammatory M2 phenotype. Consequently, it reduces
HIRI-induced liver injury in a multidimensional manner,
demonstrating strong therapeutic potential. The present review
provided conceptual support for liver injury therapy; however,
direct evidence regarding its role in HIRI still comes from animal
models.
In conclusion, targeting neutrophil-driven oxidative
stress and inflammation cascades, developing nano-antioxidant-based
therapies combined with anti-inflammatory interventions, offers new
approaches and effective methods for clinical prevention and
treatment of liver reperfusion injury.
Application of extracellular vesicles: a
new strategy for targeted treatment of IRI
Nano-scale targeted drug delivery systems have shown
broad application prospects in reducing IRI and promoting tissue
function recovery. Biomimetic nanoparticles inherit the surface
antigen profiles of their source cells and possess multiple
biological functions, including precise targeted delivery, high
accumulation in lesion sites and the ability to regulate the immune
microenvironment (137). As key
effector cells in inflammation, neutrophils play a central role in
recognizing and responding to tissue damage. Their surface
expresses various adhesion molecules, chemokine receptors and
damage-associated molecular patterns, allowing them to migrate
directionally and accumulate at inflammation sites (25). Based on this characteristic,
neutrophil membrane-coated liposome delivery systems can
effectively bind to local chemokines. For example, neutrophil
membrane-coated liposome drug delivery systems effectively target
and accumulate at rheumatoid arthritis lesion sites with
joint-specific precision (138)
and neutrophil membrane-wrapped therapeutic liposomes can target
acute lung injury treatment (139).
Furthermore, in vitro and in vivo
studies have shown that neutrophil-derived extracellular vesicles
not only efficiently load therapeutic drugs but also specifically
bind to damaged endothelial cells, markedly alleviating endothelial
dysfunction and improving sepsis-induced lung injury. Yuan et
al (140) developed a
ROS-responsive neutrophil-derived vesicle system (SOD2-Fer-1@CVs),
which could specifically accumulate in inflammation tissues with
high oxidative stress, thus enabling the controlled release of
superoxide dismutase 2 (SOD2) and ferroptosis inhibitor Fer-1 upon
ROS stimulation. SOD2-Fer-1@CVs intervene in the core pathological
processes of IRI from multiple dimensions: alleviating oxidative
stress, adsorbing and neutralizing pro-inflammatory cytokines,
inhibiting ferroptosis, restoring endothelial barrier integrity and
promoting macrophage polarization to the anti-inflammatory M2
phenotype. Ultimately, it markedly alleviates IRI following lung
transplantation, demonstrating strong multi-effect synergistic
therapeutic potential.
At the same time, stem cell therapy, as an important
direction in regenerative medicine, has shown great potential in
various disease fields. In liver IRI, mesenchymal stem cells (MSCs)
have garnered widespread attention due to their immune regulatory,
anti-inflammatory and tissue repair functions. MSCs can secrete
anti-inflammatory factors such as IL-10 and TGF-β, regulating
neutrophil activation and reducing their infiltration in the liver,
thereby inhibiting excessive inflammatory responses. Additionally,
MSCs can promote hepatocyte proliferation and regeneration,
alleviating liver failure caused by reperfusion. Notably, EVs
secreted by MSCs, particularly exosomes, are gradually being
regarded as a cell-free alternative to full-cell therapies and are
entering the clinical translation stage for liver disease
treatment. These nano-sized vesicles are rich in bioactive
components such as microRNA, proteins, lipids and functional
mitochondria and can modulate immune responses through
intercellular communication mechanisms. Research has demonstrated
that human umbilical cord-derived MSC-EVs (hUC-MSC-EVs) effectively
inhibit the formation of neutrophil extracellular traps (NETs) in
mouse models (141), thereby
ameliorating HIRI. The mechanism involves hUC-MSC-EVs transferring
functional mitochondria to liver neutrophils, triggering
mitochondrial fusion processes, repairing damaged mitochondrial
structure and function and subsequently inhibiting abnormal NET
release. This 'mitochondrial rescue' strategy reveals a novel
protective mechanism whereby MSC-EVs exert effects by modulating
the neutrophil metabolic-immune axis.
Hope and challenges
Although targeting neutrophils has shown therapeutic
potential in animal models of HIRI, clinical translation still
faces significant challenges. These challenges stem not only from
inherent interspecies differences in immunobiology but also from an
insufficient understanding of the functional complexity of
neutrophils. There is an urgent need to re-frame research paradigms
and intervention strategies under the guidance of the
'injury-repair balance' theory.
The primary challenge lies in the significant
heterogeneity between human and mouse neutrophil biology. Studies
indicate that human neutrophils constitute a much higher proportion
(40-70%) of circulating leukocytes compared with mice (10-20%) and
their phenotypic characteristics and functions differ markedly
(142). For instance, whether
human neutrophils express key inflammatory cytokines such as IL-4
and IL-6 remains debated, while the expression patterns of surface
molecules (such as TLRs, MHC-II) and chemokine receptors also
diverge substantially from those in mice (142). Such phylogenetic and regulatory
discrepancies often lead to therapeutic deviations or failures when
rodent-model-based interventions are applied to humans. Moreover,
HIRI is a dynamic process involving the coordinated participation
of multiple cell types, including intricate signaling crosstalk
among LSECs, Kupffer cells, platelets and parenchymal cells. The
current systematic understanding of this interactive network
remains incomplete. Therefore, establishing research models that
more closely mimic human pathophysiology, such as humanized mice,
organoid co-culture systems, or single-cell spatiotemporal atlas
platforms, is a critical step in bridging the 'translational gap'
between basic research and clinical application.
Secondly, existing therapeutic strategies generally
lack spatiotemporal specificity and functional selectivity. Most
conventional approaches rely on systemic immunosuppression or
broad-spectrum anti-inflammatory interventions, which, while
capable of mitigating tissue injury to some extent, inevitably
carry the risk of immune imbalance due to off-target effects. For
example, broadly blocking the complement pathway may compromise
host defense, increase postoperative infection rates and interfere
with liver regeneration. Notably, the complement cascade is rapidly
activated within minutes after reperfusion, requiring therapeutic
intervention to be precisely timed within a minute-level window,
posing a major challenge for clinical dosing regimen design.
Although emerging strategies such as C5aR antagonists or CR2-CD59
fusion proteins have demonstrated in animal models the ability to
achieve tissue-specific inhibition by targeting the C5a-C5aR axis,
thereby reducing inflammatory damage while preserving physiological
complement functions (143),
their clinical translation is still hampered by difficulties in
target validation, substantial inter-individual variability in
response and a lack of long-term safety data. Consequently,
developing targeted drugs with both high selectivity and efficacy
remains a critical bottleneck. Emerging strategies are now focusing
on targeted or pathway-specific inhibition, such as C5aR
antagonists or fusion proteins such as CR2-CD59, which enable
selective inhibition of tissue-damaging pathways while preserving
beneficial complement functions. These approaches also reduce
systemic side effects and offer more personalized treatment
options, particularly in patients with variable complement
activation due to pre-existing liver disease, steatosis, or
autoimmune backgrounds (144).
A deeper challenge arises from the multifaceted
functionality and phenotypic plasticity of neutrophils themselves.
Neutrophil polarization reflects a spatiotemporally coordinated,
microenvironment-driven process transitioning from a
damage-promoting state to a repair-promoting state. The
'injury-repair balance' model we propose for neutrophils in HIRI
argues against comprehensive neutrophil depletion and instead
advocates for stage-specific interventions targeting the
polarization process. Potential therapeutic strategies include
inhibiting early N1-driving factors, administering pro-resolving
mediators to promote an N2 shift and facilitating the conversion to
an N2 phenotype during the recovery phase. A study has shown that
through specific interventions, it is possible to promote the
transition of neutrophils from the N1 to the N2 phenotype. For
instance, combining IFN-γ and lipopolysaccharide (LPS) can
successfully induce polarization toward an N1 phenotype, whereas
induction with TGF-β or IL-4 can promote their conversion to an N2
phenotype (12). Although in
vitro models offer valuable insights, the dynamics of
neutrophils in vivo are far more complex. Cells in
vivo do not simply switch to one of two polarized phenotypes
but instead exhibit a spectrum of lineages with overlapping
functions. Some studies indicate that N2 polarization of
neutrophils may involve multiple signaling pathways. For example,
the miR-193a-5p/TLR4/JNK/p38 MAPK pathway has been found to skew
neutrophils toward an N2 anti-inflammatory phenotype (13). Another study demonstrated that
activating the STAT6/SOCS1 signaling pathway promotes
anti-inflammatory polarization of neutrophils and suppresses their
pro-inflammatory polarization (14). While these studies provide strong
clues for understanding neutrophil polarization, the interactions
and synergistic effects among different signaling pathways remain
an important area for future exploration. Therefore, further
research is needed to elucidate the precise mechanisms of these
pathways and how to effectively modulate neutrophil polarization to
achieve precise inflammation control and tissue repair.
Future breakthroughs will depend on achieving
'precision reprogramming' of neutrophil function rather than simple
depletion. This requires the development of high-dimensional
dynamic monitoring technologies, such as single-cell multi-omics,
spatial transcriptomics, live imaging, or multiparameter flow
analysis, to resolve in real time the distribution, interactions
and functional states of neutrophil subsets across different
disease stages. Furthermore, intervention strategies based on the
'injury-repair balance' framework should enable precise modulation
according to the features of each phase, thereby shifting from
'passive suppression' to 'active regulation'. Hence, developing
technologies that can dynamically monitor the distribution and
functional states of neutrophil subsets and designing
subset-specific regulatory strategies, such as promoting the
polarization of protective subsets or targeting the clearance of
harmful subsets, will be key to achieving precision therapy.
Additionally, integrating advances in targeted delivery systems and
smart responsive materials can enable spatiotemporally controlled
drug release within specific cell subsets or microenvironments,
thereby minimizing off-target effects.
In summary, the dual role of neutrophils in HIRI
makes them both a therapeutic target and a potential source of
therapeutic risk. Future therapeutic strategies should no longer
focus merely on whether to target neutrophils but should instead
concentrate on when to target, which state to target and how to
modulate their fate. Guided by the 'injury-repair balance' theory,
the ideal intervention would be a stage-specific,
phenotype-directed and microenvironment-responsive integration,
inhibiting N1 polarization and NET formation in the early phase,
regulating inflammation resolution in the middle phase and
promoting N2-mediated tissue regeneration in the late phase.
Achieving this goal demands a deeper understanding of neutrophil
fate-determining mechanisms in basic research, particularly at the
levels of metabolic reprogramming, intercellular communication and
epigenetic regulation; promoting the development of high-precision
monitoring tools and intelligent delivery systems in translational
research; and formulating personalized dosing regimens based on
dynamic biomarkers in clinical trial design. Only in this way can
we advance clinical management from 'attenuating injury' to
'actively promoting regeneration', ushering HIRI treatment into a
new era of precise immune modulation.
Availability of data and materials
Not applicable.
Authors' contributions
SL was responsible for conceptualization, formal
analysis, investigation and writing the original draft. JT was
responsible for conceptualization, funding acquisition, project
administration, resources, supervision, writing, reviewing and
editing. JJ was responsible for data curation, investigation,
visualization, writing, reviewing and editing. QZ was responsible
for data curation, investigation, visualization, writing, reviewing
and editing. YH was responsible for conceptualization, supervision
and writing the original draft. Data authentication is not
applicable. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
IRI
|
ischemia-reperfusion injury
|
|
HIRI
|
hepatic ischemia-reperfusion
injury
|
|
NETs
|
neutrophil extracellular traps
|
|
EVs
|
extracellular vesicles
|
|
DAMPs
|
damage-associated molecular
patterns
|
|
LSECs
|
liver sinusoidal endothelial
cells
|
|
ROS
|
reactive oxygen species
|
|
NF-κB
|
nuclear factor-kappa B
|
|
TNF-α
|
tumor necrosis factor alpha
|
|
IFN-β
|
interferon beta
|
|
IFN-γ
|
interferon gamma
|
|
VEGF
|
vascular endothelial growth
factor
|
|
MPO
|
myeloperoxidase
|
|
NE
|
neutrophil elastase
|
|
PGI2
|
prostacyclin I2
|
|
NKT
|
natural killer T cells
|
|
APCs
|
antigen-presenting cells
|
|
PRRs
|
pattern recognition receptors
|
|
TLRs
|
Toll-like receptors
|
|
cGAS
|
cyclic GMP-AMP synthase
|
|
STING
|
stimulator of interferon genes
|
|
DNase
|
deoxyribonuclease
|
Acknowledgements
Not applicable.
Funding
The present review was supported by the Natural Science
Foundation of Chongqing (grant no. CSTB2022NSCQ-MSX1509) and the
China Foundation for International Medical Exchange (grant no.
z-2016-23-2101-05).
References
|
1
|
Nakamura K, Kageyama S, Kaldas FM, Hirao
H, Ito T, Kadono K, Dery KJ, Kojima H, Gjertson DW, Sosa RA, et al:
Hepatic CEACAM1 expression indicates donor liver quality and
prevents early transplantation injury. J Clin Invest.
130:2689–2704. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hudcova J, Scopa C, Rashid J, Waqas A,
Ruthazer R and Schumann R: Effect of early allograft dysfunction on
outcomes following liver transplantation. Clin Transplant. Dec
22–2016.Epub ahead of print. PubMed/NCBI
|
|
3
|
Rampes S and Ma D: Hepatic
ischemia-reperfusion injury in liver transplant setting: Mechanisms
and protective strategies. J Biomed Res. 33:221–234. 2019.
View Article : Google Scholar :
|
|
4
|
Nakamura K, Kageyama S and
Kupiec-Weglinski JW: The evolving role of neutrophils in liver
transplant Ischemia-reperfusion injury. Curr Transplant Rep.
6:78–89. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oliveira THC, Marques PE, Proost P and
Teixeira MMM: Neutrophils: A cornerstone of liver ischemia and
reperfusion injury. Lab Invest. 98:51–62. 2018. View Article : Google Scholar
|
|
6
|
McDonald B, Pittman K, Menezes GB, Hirota
SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA and Kubes P:
Intravascular danger signals guide neutrophils to sites of sterile
inflammation. Science. 330:362–366. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang J, Hossain M, Thanabalasuriar A,
Gunzer M, Meininger C and Kubes P: Visualizing the function and
fate of neutrophils in sterile injury and repair. Science.
358:111–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deng Y, Zhao Z, Sheldon M, Zhao Y, Teng H,
Martinez C, Zhang J, Lin C, Sun Y, Yao F, et al: LIFR regulates
cholesterol-driven bidirectional hepatocyte-neutrophil cross-talk
to promote liver regeneration. Nat Metab. 6:1756–1774. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin RZ, Lee CN, Moreno-Luna R, Neumeyer J,
Piekarski B, Zhou P, Moses MA, Sachdev M, Pu WT, Emani S, et al:
Host non-inflammatory neutrophils mediate the engraftment of
bioengineered vascular networks. Nat Biomed Eng. 1:00812017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marques PE, Amaral SS, Pires DA, Nogueira
LL, Soriani FM, Lima BH, Lopes GA, Russo RC, Avila TV, Melgaço JG,
et al: Chemokines and mitochondrial products activate neutrophils
to amplify organ injury during mouse acute liver failure.
Hepatology. 56:1971–1982. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL,
Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY and Lindsey
ML: Temporal neutrophil polarization following myocardial
infarction. Cardiovasc Res. 110:51–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cai W, Liu S, Hu M, Huang F, Zhu Q, Qiu W,
Hu X, Colello J, Zheng SG and Lu Z: Functional dynamics of
neutrophils after ischemic stroke. Transl Stroke Res. 11:108–121.
2020. View Article : Google Scholar
|
|
13
|
Fan Y, Yang J, Xie Y, Yang X, Zhu H, Liu
Y, Xia Z, Ji S and Yang R: Inflammatory memory-activated biomimetic
nanovesicles regulate neutrophil plasticity and metabolic
reprogramming for rapid diabetic wound healing via targeting
miR-193a-5p/TLR4/JNK/P38 MAPK pathways. J Nanobiotechnology.
23:1152025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang Y, Wang N, Liu J, Ren H, Jiang W,
Lei Y, Fu X, Hao M, Lang X, Liu Y, et al: Intranasal delivery of
hMSC-derived supernatant for treatment of ischemic stroke by
inhibiting the pro-inflammatory polarization of neutrophils. Stem
Cell Res Ther. 16:432025. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhai Y, Petrowsky H, Hong JC, Busuttil RW
and Kupiec-Weglinski JW: Ischaemia-reperfusion injury in liver
transplantation-from bench to bedside. Nat Rev Gastroenterol
Hepatol. 10:79–89. 2013. View Article : Google Scholar :
|
|
17
|
Feng S, Roll GR, Rouhani FJ and Sanchez
Fueyo A: The future of liver transplantation. Hepatology.
80:674–697. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu X, Cao H, Li J, Wang B, Zhang P, Dong
Zhang X, Liu Z, Yuan H and Zhan Z: Autophagy induced by DAMPs
facilitates the inflammation response in lungs undergoing
ischemia-reperfusion injury through promoting TRAF6 ubiquitination.
Cell Death Differ. 24:683–693. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou J, Chen J, Wei Q, Saeb-Parsy K and Xu
X: The Role of Ischemia/reperfusion injury in early hepatic
allograft dysfunction. Liver Transpl. 26:1034–1048. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Teoh NC and Farrell GC: Hepatic ischemia
reperfusion injury: Pathogenic mechanisms and basis for
hepatoprotection. J Gastroenterol Hepatol. 18:891–902. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gores GJ, Nieminen AL, Wray BE, Herman B
and Lemasters JJ: Intracellular pH during 'chemical hypoxia' in
cultured rat hepatocytes. Protection by intracellular acidosis
against the onset of cell death. J Clin Invest. 83:386–396. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Semenza GL: Cellular and molecular
dissection of reperfusion injury: ROS within and without. Circ Res.
86:117–118. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fujii Y, Johnson ME and Gores GJ:
Mitochondrial dysfunction during anoxia/reoxygenation injury of
liver sinusoidal endothelial cells. Hepatology. 20:177–185.
1994.PubMed/NCBI
|
|
24
|
Nishimura Y, Romer LH and Lemasters JJ:
Mitochondrial dysfunction and cytoskeletal disruption during
chemical hypoxia to cultured rat hepatic sinusoidal endothelial
cells: The pH paradox and cytoprotection by glucose, acidotic pH,
and glycine. Hepatology. 27:1039–1049. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaltenmeier C, Yazdani HO, Handu S, Popp
B, Geller D and Tohme S: The role of neutrophils as a driver in
hepatic ischemia-reperfusion injury and cancer growth. Front
Immunol. 13:8875652022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev
Cell Mol Biol. 298:229–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Saleh H and El-Shorbagy HM: Chitosan
protects liver against ischemia-reperfusion injury via regulating
Bcl-2/Bax, TNF-α and TGF-β expression. Int J Biol Macromol.
164:1565–1574. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bektas S, Karakaya K, Can M, Bahadir B,
Guven B, Erdogan N and Ozdamar SO: The effects of tadalafil and
pentoxifylline on apoptosis and nitric oxide synthase in liver
ischemia/reperfusion injury. Kaohsiung J Med Sci. 32:339–347. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kageyama S, Nakamura K, Fujii T, Ke B,
Sosa RA, Reed EF, Datta N, Zarrinpar A, Busuttil RW and
Kupiec-Weglinski JW: Recombinant relaxin protects liver transplants
from ischemia damage by hepatocyte glucocorticoid receptor: From
bench-to-bedside. Hepatology. 68:258–273. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Adams DH, Ju C, Ramaiah SK, Uetrecht J and
Jaeschke H: Mechanisms of immune-mediated liver injury. Toxicol
Sci. 115:307–321. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Okajima K, Harada N, Uchiba M and Mori M:
Neutrophil elastase contributes to the development of
ischemia-reperfusion-induced liver injury by decreasing endothelial
production of prostacyclin in rats. Am J Physiol Gastrointest Liver
Physiol. 287:G1116–G1123. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kawai M, Harada N, Takeyama H and Okajima
K: Neutrophil elastase contributes to the development of
ischemia/reperfusion-induced liver injury by decreasing the
production of insulin-like growth factor-I in rats. Transl Res.
155:294–304. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang L, Li J, He S, Liu Y, Chen H, He S,
Yin M, Zou D, Chen S, Luo T, et al: Resolving the graft
ischemia-reperfusion injury during liver transplantation at the
single cell resolution. Cell Death Dis. 12:5892021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fahrner R, Trochsler M, Corazza N,
Graubardt N, Keogh A, Candinas D, Brunner T, Stroka D and Beldi G:
Tumor necrosis factor-related apoptosis-inducing ligand on NK cells
protects from hepatic ischemia-reperfusion injury. Transplantation.
97:1102–1109. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhai Y, Busuttil RW and Kupiec-Weglinski
JW: Liver ischemia and reperfusion injury: New insights into
mechanisms of innate-adaptive immune-mediated tissue inflammation.
Am J Transplant. 11:1563–1569. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Heo MJ, Suh JH, Poulsen KL, Ju C and Kim
KH: Updates on the immune cell basis of hepatic
ischemia-reperfusion injury. Mol Cells. 46:527–534. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Arrenberg P, Maricic I and Kumar V:
Sulfatide-mediated activation of type II natural killer T cells
prevents hepatic ischemic reperfusion injury in mice.
Gastroenterology. 140:646–655. 2011. View Article : Google Scholar
|
|
38
|
Yang SH, Lee JP, Jang HR, Cha RH, Han SS,
Jeon US, Kim DK, Song J, Lee DS and Kim YS: Sulfatide-reactive
natural killer T cells abrogate ischemia-reperfusion injury. J Am
Soc Nephrol. 22:1305–1314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang HF, Zeng Z, Wang KH, Zhang HY, Wang
S, Zhou WX, Wang ZB, Xu WG and Duan J: Heme oxygenase-1 protects
rat liver against warm ischemia/reperfusion injury via
TLR2/TLR4-triggered signaling pathways. World J Gastroenterol.
21:2937–2948. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang H, Tohme S, Al-Khafaji AB, Tai S,
Loughran P, Chen L, Wang S, Kim J, Billiar T, Wang Y, et al:
Damage-associated molecular pattern-activated neutrophil
extracellular trap exacerbates sterile inflammatory liver injury.
Hepatology. 62:600–614. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xiahou Z, Wang X, Shen J, Zhu X, Xu F, Hu
R, Guo D, Li H, Tian Y, Liu Y and Liang H: NMI and IFP35 serve as
proinflammatory DAMPs during cellular infection and injury. Nat
Commun. 8:9502017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen R, Du J, Zhu H and Ling Q: The role
of cGAS-STING signalling in liver diseases. JHEP Rep. 3:1003242021.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu J, Zhou J, Luan Y, Li X, Meng X, Liao
W, Tang J and Wang Z: cGAS-STING, inflammasomes and pyroptosis: An
overview of crosstalk mechanism of activation and regulation. Cell
Commun Signal. 22:222024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen R, Yang T, Jiang Z, Long Y, Qian B
and Fu W: Novel perspectives in hepatic ischemia-reperfusion
injury: The cGAS-STING pathway. J Inflamm Res. 18:16427–16448.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
de Carvalho Ribeiro M and Szabo G: Role of
the inflammasome in liver disease. Annu Rev Pathol. 17:345–365.
2022. View Article : Google Scholar
|
|
46
|
Nemeth N, Peto K, Magyar Z, Klarik Z,
Varga G, Oltean M, Mantas A, Czigany Z and Tolba RH:
Hemorheological and microcirculatory factors in liver
Ischemia-Reperfusion Injury-an update on pathophysiology, molecular
mechanisms and protective strategies. Int J Mol Sci. 22:18642021.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Vollmar B, Glasz J, Leiderer R, Post S and
Menger MD: Hepatic microcirculatory perfusion failure is a
determinant of liver dysfunction in warm ischemia-reperfusion. Am J
Pathol. 145:1421–1431. 1994.PubMed/NCBI
|
|
48
|
Vollmar B, Richter S and Menger MD:
Leukocyte stasis in hepatic sinusoids. Am J Physiol. 270:G798–G803.
1996.PubMed/NCBI
|
|
49
|
Zhang JX, Jones DV and Clemens MG: Effect
of activation on neutrophil-induced hepatic microvascular injury in
isolated rat liver. Shock. 1:273–278. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yamanaka K, Houben P, Bruns H, Schultze D,
Hatano E and Schemmer P: A systematic review of pharmacological
treatment options used to reduce ischemia reperfusion injury in rat
liver transplantation. PLoS One. 10:e01222142014. View Article : Google Scholar
|
|
51
|
Dery KJ, Chiu R, Kasargod A and
Kupiec-Weglinski JW: Feedback loops shape oxidative and immune
interactions in hepatic Ischemia-reperfusion injury. Antioxidants
(Basel). 14:9442025. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Elsayed Abouzed DE, Bafail DA, Refaie SM,
Aboelez MO, Elsayed AA, Mallasiy LO, Bayoumy NMK and Hagar H:
Protective effect of valsartan alone and in combination with
neprilysin inhibitor (valsartan + sacubitril) against hepatic
ischemia-reperfusion injury: Targeting angiotensin II
receptor-neprilysin and modulating SMAD-4/NF-κβ/JNK pathways in
rats. Naunyn Schmiedebergs Arch Pharmacol. 398:8797–8811. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Schwabe RF and Brenner DA: Mechanisms of
liver injury. I. TNF-alpha-induced liver injury: Role of IKK, JNK,
and ROS pathways. Am J Physiol Gastrointest Liver Physiol.
290:G583–G589. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Oe S, Hiros T, Fujii H, Yasuchika K,
Nishio T, Iimuro Y, Morimoto T, Nagao M and Yamaoka Y: Continuous
intravenous infusion of deleted form of hepatocyte growth factor
attenuates hepatic ischemia-reperfusion injury in rats. J Hepatol.
34:832–839. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Csak T, Ganz M, Pespisa J, Kodys K,
Dolganiuc A and Szabo G: Fatty acid and endotoxin activate
inflammasomes in mouse hepatocytes that release danger signals to
stimulate immune cells. Hepatology. 54:133–144. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Teoh N, Field J and Farrell G:
Interleukin-6 is a key mediator of the hepatoprotective and
pro-proliferative effects of ischaemic preconditioning in mice. J
Hepatol. 45:20–27. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Tsurui Y, Sho M, Kuzumoto Y, Hamada K,
Akashi S, Kashizuka H, Ikeda N, Nomi T, Mizuno T, Kanehiro H and
Nakajima Y: Dual role of vascular endothelial growth factor in
hepatic ischemia-reperfusion injury. Transplantation. 79:1110–1115.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Xiao X, Liu D, Chen S, Li X, Ge M and
Huang W: Sevoflurane preconditioning activates HGF/Met-mediated
autophagy to attenuate hepatic ischemia-reperfusion injury in mice.
Cell Signal. 82:1099662021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
McDonald B, Urrutia R, Yipp BG, Jenne CN
and Kubes P: Intravascular neutrophil extracellular traps capture
bacteria from the bloodstream during sepsis. Cell Host Microbe.
12:324–333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Silvestre-Roig C, Fridlender ZG, Glogauer
M and Scapini P: Neutrophil diversity in health and disease. Trends
Immunol. 40:565–583. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tsuda Y, Takahashi H, Kobayashi M,
Hanafusa T, Herndon DN and Suzuki F: Three different neutrophil
subsets exhibited in mice with different susceptibilities to
infection by methicillin-resistant Staphylococcus aureus. Immunity.
21:215–226. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
de Oliveira THC and Gonçalves GKN: Liver
ischemia reperfusion injury: Mechanisms, cellular pathways, and
therapeutic approaches. Int Immunopharmacol. 150:1142992025.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nourshargh S, Renshaw SA and Imhof BA:
Reverse migration of neutrophils: Where, When, How, and Why? Trends
Immunol. 37:273–286. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wilson GC, Kuboki S, Freeman CM, Nojima H,
Schuster RM, Edwards MJ and Lentsch AB: CXC chemokines function as
a rheostat for hepatocyte proliferation and liver regeneration.
PLoS One. 10:e01200922015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Petri B and Sanz MJ: Neutrophil
chemotaxis. Cell Tissue Res. 371:425–436. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kusakabe J, Hata K, Tamaki I, Tajima T,
Miyauchi H, Wang Y, Nigmet Y, Okamura Y, Kubota T, Tanaka H, et al:
Complement 5 inhibition ameliorates hepatic Ischemia/reperfusion
injury in mice, dominantly via the C5a-mediated cascade.
Transplantation. 104:2065–2077. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhang X, Hu J, Becker KV, Engle JW, Ni D,
Cai W, Wu D and Qu S: Antioxidant and C5a-blocking strategy for
hepatic ischemia-reperfusion injury repair. J Nanobiotechnology.
19:1072021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Hilscher MB, Sehrawat T, Arab JP, Zeng Z,
Gao J, Liu M, Kostallari E, Gao Y, Simonetto DA, Yaqoob U, et al:
Mechanical stretch increases expression of CXCL1 in liver
sinusoidal endothelial cells to recruit neutrophils, generate
sinusoidal microthombi, and promote portal hypertension.
Gastroenterology. 157:193–209.e9. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Verkaar F, van Offenbeek J, van der Lee
MMC, van Lith LHCJ, Watts AO, Rops ALWMM, Aguilar DC, Ziarek JJ,
van der Vlag J, Handel TM, et al: Chemokine cooperativity is caused
by competitive glycosaminoglycan binding. J Immunol. 192:3908–3914.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Rajarathnam K and Desai UR: Structural
insights into how proteoglycans determine chemokine-CXCR1/CXCR2
interactions: Progress and challenges. Front Immunol. 11:6602020.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sepuru KM and Rajarathnam K: Structural
basis of chemokine interactions with heparan sulfate, chondroitin
sulfate, and dermatan sulfate. J Biol Chem. 294:15650–15661. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bao X, Moseman EA, Saito H, Petryniak B,
Thiriot A, Hatakeyama S, Ito Y, Kawashima H, Yamaguchi Y, Lowe JB,
et al: Endothelial heparan sulfate controls chemokine presentation
in recruitment of lymphocytes and dendritic cells to lymph nodes.
Immunity. 33:817–829. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Vanheule V, Janssens R, Boff D, Kitic N,
Berghmans N, Ronsse I, Kungl AJ, Amaral FA, Teixeira MM, Van Damme
J, et al: The positively charged COOH-terminal
Glycosaminoglycan-binding CXCL9(74-103) peptide inhibits
CXCL8-induced neutrophil extravasation and monosodium Urate
Crystal-induced gout in mice. J Biol Chem. 290:21292–21304. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Song J, He Z, Yang M, Yu T, Wang X, Liu B
and Li J: HepaticIschemia/Reperfusion Injuryinvolves functional
tryptase/PAR-2 signaling in liver sinusoidal endothelial cell
population. Int Immunopharmacol. 100:1080522021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Rossaint J and Zarbock A: Tissue-specific
neutrophil recruitment into the lung, liver, and kidney. J Innate
Immun. 5:348–357. 2013. View Article : Google Scholar
|
|
76
|
Wang Y and Liu Y: Neutrophil-induced liver
injury and interactions between neutrophils and liver sinusoidal
endothelial cells. Inflammation. 44:1246–1262. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wong J, Johnston B, Lee SS, Bullard DC,
Smith CW, Beaudet AL and Kubes P: A minimal role for selectins in
the recruitment of leukocytes into the inflamed liver
microvasculature. J Clin Invest. 99:2782–2790. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Khan AI, Kerfoot SM, Heit B, Liu L,
Andonegui G, Ruffell B, Johnson P and Kubes P: Role of CD44 and
hyaluronan in neutrophil recruitment. J Immunol. 173:7594–7601.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
McDonald B, Jenne CN, Zhuo L, Kimata K and
Kubes P: Kupffer cells and activation of endothelial TLR4
coordinate neutrophil adhesion within liver sinusoids during
endotoxemia. Am J Physiol Gastrointest Liver Physiol.
305:G797–G806. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Benedicto A, Herrero A, Romayor I, Marquez
J, Smedsrød B, Olaso E and Arteta B: Liver sinusoidal endothelial
cell ICAM-1 mediated tumor/endothelial crosstalk drives the
development of liver metastasis by initiating inflammatory and
angiogenic responses. Sci Rep. 9:131112019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Woodfin A, Voisin MB, Beyrau M, Colom B,
Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE,
et al: The junctional adhesion molecule JAM-C regulates polarized
transendothelial migration of neutrophils in vivo. Nat Immunol.
12:761–769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ley K, Laudanna C, Cybulsky MI and
Nourshargh S: Getting to the site of inflammation: The leukocyte
adhesion cascade updated. Nat Rev Immunol. 7:678–689. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang P, Yue K, Liu X, Yan X, Yang Z, Duan
J, Xia C, Xu X, Zhang M, Liang L, et al: Endothelial Notch
activation promotes neutrophil transmigration via downregulating
endomucin to aggravate hepatic ischemia/reperfusion injury. Sci
China Life Sci. 63:375–387. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yazdani HO, Chen HW, Tohme S, Tai S, van
der Windt DJ, Loughran P, Rosborough BR, Sud V, Beer-Stolz D,
Turnquist HR, et al: IL-33 exacerbates liver sterile inflammation
by amplifying neutrophil extracellular trap formation. J Hepatol.
Sep 21–2017.Epub ahead of print. PubMed/NCBI
|
|
85
|
Kato H, Duarte S, Liu D, Busuttil RW and
Coito AJ: Matrix Metalloproteinase-2 (MMP-2) gene deletion enhances
MMP-9 activity, impairs PARP-1 degradation, and exacerbates hepatic
ischemia and reperfusion injury in mice. PLoS One. 10:e01376422015.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Duarte S, Matian P, Ma S, Busuttil RW and
Coito AJ: Adeno-associated Virus-mediated gene transfer of tissue
inhibitor of Metalloproteinases-1 impairs neutrophil extracellular
trap formation and ameliorates hepatic ischemia and reperfusion
injury. Am J Pathol. 188:1820–1832. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Tharp WG, Yadav R, Irimia D, Upadhyaya A,
Samadani A, Hurtado O, Liu SY, Munisamy S, Brainard DM, Mahon MJ,
et al: Neutrophil chemorepulsion in defined interleukin-8 gradients
in vitro and in vivo. J Leukoc Biol. 79:539–554. 2006. View Article : Google Scholar
|
|
88
|
Buckley CD, Ross EA, McGettrick HM,
Osborne CE, Haworth O, Schmutz C, Stone PC, Salmon M, Matharu NM,
Vohra RK, et al: Identification of a phenotypically and
functionally distinct population of long-lived neutrophils in a
model of reverse endothelial migration. J Leukoc Biol. 79:303–311.
2006. View Article : Google Scholar
|
|
89
|
Colom B, Bodkin JV, Beyrau M, Woodfin A,
Ody C, Rourke C, Chavakis T, Brohi K, Imhof BA and Nourshargh S:
Leukotriene B4-Neutrophil elastase axis drives neutrophil reverse
transendothelial cell migration in vivo. Immunity. 42:1075–1086.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Burn T and Alvarez JI: Reverse
transendothelial cell migration in inflammation: To help or to
hinder? Cell Mol Life Sci. 74:1871–1881. 2017. View Article : Google Scholar
|
|
91
|
de Oliveira S, Rosowski EE and
Huttenlocher A: Neutrophil migration in infection and wound repair:
Going forward in reverse. Nat Rev Immunol. 16:378–391. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Brinkmann V and Zychlinsky A: Neutrophil
extracellular traps: Is immunity the second function of chromatin?
J Cell Biol. 198:773–783. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
von Meijenfeldt FA, Stravitz RT, Zhang J,
Adelmeijer J, Zen Y, Durkalski V, Lee WM and Lisman TJ: Generation
of neutrophil extracellular traps in patients with acute liver
failure is associated with poor outcome. Hepatology. 75:623–633.
2022. View Article : Google Scholar :
|
|
94
|
Middleton EA, He XY, Denorme F, Campbell
RA, Ng D, Salvatore SP, Mostyka M, Baxter-Stoltzfus A, Borczuk AC,
Loda M, et al: Neutrophil extracellular traps contribute to
immunothrombosis in COVID-19 acute respiratory distress syndrome.
Blood. 136:1169–1179. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Shapouri-Moghaddam A, Mohammadian S,
Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi
A, Afshari JT and Sahebkar A: Macrophage plasticity, polarization,
and function in health and disease. J Cell Physiol. 233:6425–6440.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Shaul ME, Levy L, Sun J, Mishalian I,
Singhal S, Kapoor V, Horng W, Fridlender G, Albelda SM and
Fridlender ZG: Tumor-associated neutrophils display a distinct N1
profile following TGFβ modulation: A transcriptomics analysis of
pro-vs. antitumor TANs. Oncoimmunology. 5:e12322212016. View Article : Google Scholar
|
|
97
|
Zou JM, Qin J, Li YC, Wang Y, Li D, Shu Y,
Luo C, Wang SS, Chi G, Guo F, et al: IL-35 induces N2 phenotype of
neutrophils to promote tumor growth. Oncotarget. 8:33501–33514.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Mihaila AC, Ciortan L, Macarie RD, Vadana
M, Cecoltan S, Preda MB, Hudita A, Gan AM, Jakobsson G, Tucureanu
MM, et al: Transcriptional profiling and functional analysis of
N1/N2 neutrophils reveal an immunomodulatory effect of
S100A9-blockade on the pro-inflammatory N1 subpopulation. Front
Immunol. 12:7087702021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
García-Culebras A, Durán-Laforet V,
Peña-Martínez C, Moraga A, Ballesteros I, Cuartero MI, de la Parra
J, Palma-Tortosa S, Hidalgo A, Corbí AL, et al: Role of TLR4
(Toll-Like Receptor 4) in N1/N2 neutrophil programming after
stroke. Stroke. 50:2922–2932. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Giese MA, Hind LE and Huttenlocher A:
Neutrophil plasticity in the tumor microenvironment. Blood.
133:2159–2167. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Cuartero MI, Ballesteros I, Moraga A,
Nombela F, Vivancos J, Hamilton JA, Corbí ÁL, Lizasoain I and Moro
MA: N2 neutrophils, novel players in brain inflammation after
stroke: Modulation by the PPARγ agonist rosiglitazone. Stroke.
44:3498–3508. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Napoli M, Sitaru S, Budke A, Kammerl A,
Querner G, Immler R, Rohwedder I, Seifert S, Ferraro B, Weckbach L,
et al: The dual non-competitive CXCR1/2 inhibitor ladarixin impairs
neutrophil extravasation without altering intravascular adhesion.
Haematologica. Aug 28–2025.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Chapman RW, Phillips JE, Hipkin RW, Curran
AK, Lundell D and Fine JS: CXCR2 antagonists for the treatment of
pulmonary disease. Pharmacol Ther. 121:55–68. 2009. View Article : Google Scholar
|
|
104
|
Rennard SI, Dale DC, Donohue JF, Kanniess
F, Magnussen H, Sutherland ER, Watz H, Lu S, Stryszak P, Rosenberg
E and Staudinger H: CXCR2 antagonist MK-7123. A phase 2
Proof-of-Concept trial for chronic obstructive pulmonary disease.
Am J Respir Crit Care Med. 191:1001–1011. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Madan A, Chen S, Yates P, Washburn ML,
Roberts G, Peat AJ, Tao Y, Parry MF, Barnum O, McClain MT, et al:
Efficacy and safety of danirixin (GSK1325756) Co-administered with
Standard-of-Care antiviral (Oseltamivir): A Phase 2b, global,
randomized study of adults hospitalized with influenza. Open Forum
Infect Dis. 6:ofz1632019. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Howard JF Jr, Utsugisawa K, Benatar M,
Murai H, Barohn RJ, Illa I, Jacob S, Vissing J, Burns TM, Kissel
JT, et al: Safety and efficacy of eculizumab in anti-acetylcholine
receptor antibody-positive refractory generalised myasthenia gravis
(REGAIN): A phase 3, randomised, double-blind, placebo-controlled,
multi-centre study. Lancet Neurol. 16:976–986. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Mastellos DC, Ricklin D and Lambris JD:
Clinical promise of next-generation complement therapeutics. Nat
Rev Drug Discov. 18:707–729. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Melgaço JG, Veloso CE, Pacheco-Moreira LF,
Vitral CL and Pinto MA: Complement system as a target for therapies
to control liver Regeneration/damage in acute liver failure induced
by viral hepatitis. J Immunol Res. 2018:39170322018. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Di Bona D, Montalto G, Clemenza L, Bascone
F, Accardo P, Bellavia D, Craxì A and Brai M: Soluble complement
receptor type 1 (sCR1) in chronic liver diseases: Serum levels at
different stages of liver diseases. Clin Exp Immunol. 114:102–105.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Stahl GL, Shernan SK, Smith PK and Levy
JH: Complement activation and cardiac surgery: A novel target for
improving outcomes. Anesth Analg. 115:759–771. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Preissler G, Eichhorn M, Waldner H, Winter
H, Kleespies A and Massberg S: Intercellular adhesion molecule-1
blockade attenuates inflammatory response and improves
microvascular perfusion in rat pancreas grafts. Pancreas.
41:1112–1118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Sakai S, Tajima H, Miyashita T, Nakanuma
S, Makino I, Hayashi H, Nakagawara H, Kitagawa H, Fushida S,
Fujimura T, et al: Sivelestat sodium hydrate inhibits neutrophil
migration to the vessel wall and suppresses hepatic
ischemia-reperfusion injury. Dig Dis Sci. 59:787–794. 2014.
View Article : Google Scholar
|
|
113
|
Xia F, Xiang S, Qiu Y, Lou H, Wang S, Liu
Y, Yu F and Li S: Macrophage Membrane-camouflaged nanozymes for
combating the oxidative Stress-microvascular perfusion negative
feedback in Ischemia-reperfusion induced acute kidney injury. Adv
Healthc Mater. 14:e24050902025. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Luan H, Wang C, Sun J, Zhao L, Li L, Zhou
B, Shao S, Shen X and Xu Y: Resolvin D1 protects against
Ischemia/Reperfusion-Induced acute kidney injury by increasing treg
percentages via the ALX/FPR2 pathway. Front Physiol. 11:2852020.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Oda H, Tanaka S, Shinohara M, Morimura Y,
Yokoyama Y, Kayawake H, Yamada Y, Yutaka Y, Ohsumi A, Nakajima D,
et al: Specialized proresolving lipid meditators agonistic to
formyl peptide receptor type 2 attenuate Ischemia-reperfusion
injury in rat lung. Transplantation. 106:1159–1169. 2022.
View Article : Google Scholar
|
|
116
|
Kollareth DJM, Leroy V, Tu Z,
Woolet-Stockton MJ, Kamat M, Garrett TJ, Atkinson C, Cai G,
Upchurch GR Jr, Sharma AK, et al: Lipoxin A4/FPR2 signaling
mitigates ferroptosis of alveolar epithelial cells via
NRF2-Dependent pathway during lung Ischemia-reperfusion injury.
FASEB J. 39:e705452025. View Article : Google Scholar
|
|
117
|
Sener G, Tosun O, Sehirli AO, Kaçmaz A,
Arbak S, Ersoy Y and Ayanoğlu-Dülger G: Melatonin and
N-acetylcysteine have beneficial effects during hepatic ischemia
and reperfusion. Life Sci. 72:2707–2718. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Tohme S, Yazdani HO, Sud V, Loughran P,
Huang H, Zamora R, Simmons RL, Vodovotz Y and Tsung A:
Computational analysis supports IL-17A as a central driver of
neutrophil extracellular Trap-mediated injury in liver ischemia
reperfusion. J Immunol. 202:268–277. 2019. View Article : Google Scholar
|
|
119
|
Li L, Huang L, Vergis AL, Ye H, Bajwa A,
Narayan V, Strieter RM, Rosin DL and Okusa MD: IL-17 produced by
neutrophils regulates IFN-gamma-mediated neutrophil migration in
mouse kidney ischemia-reperfusion injury. J Clin Invest.
120:331–342. 2010. View Article : Google Scholar :
|
|
120
|
Lin AM, Rubin CJ, Khandpur R, Wang JY,
Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ and Bruce
AT: Mast cells and neutrophils release IL-17 through extracellular
trap formation in psoriasis. J Immunol. 187:490–500. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Yazdani HO, Roy E, Comerci AJ, van der
Windt DJ, Zhang H, Huang H, Loughran P, Shiva S, Geller DA,
Bartlett DL, et al: Neutrophil extracellular traps drive
mitochondrial homeostasis in tumors to augment growth. Cancer Res.
79:5626–5639. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Yang K, Gao R, Chen H, Hu J, Zhang P, Wei
X, Shi J, Chen Y, Zhang L, Chen J, et al: Myocardial reperfusion
injury exacerbation due to ALDH2 deficiency is mediated by
neutrophil extracellular traps and prevented by leukotriene C4
inhibition. Eur Heart J. 45:1662–1680. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Arumugam S, Girish Subbiah K, Kemparaju K
and Thirunavukkarasu C: Neutrophil extracellular traps in acrolein
promoted hepatic ischemia reperfusion injury: Therapeutic potential
of NOX2 and p38MAPK inhibitors. J Cell Physiol. 233:3244–3261.
2018. View Article : Google Scholar
|
|
124
|
Belvončíková P, Feješ A, Gromová B,
Janovičová Ľ, Farkašová A, Babál P and Gardlík R: Therapeutic
effects of DNase I on peripheral and local markers of liver injury
and neutrophil extracellular traps in a model of Alcohol-related
liver disease. Int J Mol Sci. 26:18932025. View Article : Google Scholar
|
|
125
|
Zeng FL, Zhang Y, Wang ZH, Zhang H, Meng
XT, Wu YQ, Qian ZZ, Ding YH, Li J, Ma TT and Huang C: Neutrophil
extracellular traps promote acetaminophen-induced acute liver
injury in mice via AIM2. Acta Pharmacol Sin. 45:1660–1672. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Liu T, Lv X, Xu Q, Qi X, Qiu S, Luan Y,
Shen N, Cheng J, Jin L, Tian T, et al: Stroke-homing Peptide-DNase1
alleviates intestinal ischemia reperfusion injury by selectively
degrading neutrophil extracellular traps. Cell Prolif.
58:e700102025. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
George J, Lu Y, Tsuchishima M and Tsutsumi
M: Cellular and molecular mechanisms of hepatic
ischemia-reperfusion injury: The role of oxidative stress and
therapeutic approaches. Redox Biol. 75:1032582024. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Rhee JE, Jung SE, Shin SD, Suh GJ, Noh DY,
Youn YK, Oh SK and Choe KJ: The effects of antioxidants and nitric
oxide modulators on hepatic ischemic-reperfusion injury in rats. J
Korean Med Sci. 17:502–506. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Loeschner K, Hadrup N, Hansen M, Pereira
SA, Gammelgaard B, Møller LH, Mortensen A, Lam HR and Larsen EH:
Absorption, distribution, metabolism and excretion of selenium
following oral administration of elemental selenium nanoparticles
or selenite in rats. Metallomics. 6:330–337. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Huang B, Zhang J, Hou J and Chen C: Free
radical scavenging efficiency of Nano-Se in vitro. Free Radic Biol
Med. 35:805–813. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Deng X, Ouyang P, Xu W, Yang E, Bao Z, Wu
Y, Gong J and Pan J: Research progress of nano selenium in the
treatment of oxidative stress injury during hepatic
ischemia-reperfusion injury. Front Pharmacol. 13:11034832022.
View Article : Google Scholar
|
|
132
|
Mohamed RA, Ghoneim ME, Tawfiq RA and El
Sayed NF: Amygdalin synergizes with the TNF-α monoclonal antibody
infliximab to modulate HSP90 and related
necro-inflammatory/oxidative stress pathways in a rat model of
hepatic I/R. Naunyn Schmiedebergs Arch Pharmacol. Dec 17–2025.Epub
ahead of print. View Article : Google Scholar
|
|
133
|
Chaabani R, Bejaoui M, Ben Jeddou I,
Zaouali MA, Haouas Z, Belgacem S, Peralta C and Ben Abdennebi H:
Effect of the Non-steroidal Anti-inflammatory drug diclofenac on
Ischemia-reperfusion injury in rat liver: A Nitric Oxide-dependent
mechanism. Inflammation. 46:1221–1235. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Montalvo-Javé EE, Ortega-Salgado JA,
Castell A, Carrasco-Daza D, Jay D, Gleason R, Muñoz E,
Montalvo-Arenas C, Hernández-Muñoz R and Piña E: Piroxicam and
meloxicam ameliorate hepatic oxidative stress and protein
carbonylation in Kupffer and sinusoidal endothelial cells promoted
by ischemia-reperfusion injury. Transpl Int. 24:489–500. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Huang Y, Xu Q, Zhang J, Yin Y, Pan Y,
Zheng Y, Cai X, Xia Q and He K: Prussian blue scavenger ameliorates
hepatic ischemia-reperfusion injury by inhibiting inflammation and
reducing oxidative stress. Front Immunol. 13:8913512022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Shen P, Huang K, Zhang X, Qin M, Wang X
and Fan Z: Neutrophil-Mimicking Prussian blue nanozymes for
synergistic targeted immuno-metabolic therapy of hepatic
ischemia-reperfusion injury. J Nanobiotechnology. 23:7362025.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Zhang Y, Wu D, Zhou C, Bai M, Wan Y, Zheng
Q, Fan Z, Wang X and Yang C: Engineered extracellular vesicles for
tissue repair and regeneration. Burns Trauma. 12:tkae0622024.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Thomas BL, Montero-Melendez T, Oggero S,
Kaneva MK, Chambers D, Pinto AL, Nerviani A, Lucchesi D,
Austin-Williams S, Hussain MT, et al: Molecular determinants of
neutrophil extracellular vesicles that drive cartilage regeneration
in inflammatory arthritis. Arthritis Rheumatol. 76:1705–1718. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Huang Z, Wang H, Long J, Lu Z, Chun C and
Li X: Neutrophil membrane-coated therapeutic liposomes for targeted
treatment in acute lung injury. Int J Pharm. 624:1219712022.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Yuan HX, Ye YY, Shen P, Zhang J, Meng QF,
Chen Y, Xu X, Zhang XL, Rao L, Fan ZJ, et al: Engineering
neutrophil vesicles for synergistic protection against
Ischemia/Reperfusion injury after lung transplant. Adv Sci (Weinh).
12:e061272025. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Lu T, Zhang J, Cai J, Xiao J, Sui X, Yuan
X, Li R, Li Y, Yao J, Lv G, et al: Extracellular vesicles derived
from mesenchymal stromal cells as nanotherapeutics for liver
ischaemia-reperfusion injury by transferring mitochondria to
modulate the formation of neutrophil extracellular traps.
Biomaterials. 284:1214862022. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Mantovani A, Cassatella MA, Costantini C
and Jaillon S: Neutrophils in the activation and regulation of
innate and adaptive immunity. Nat Rev Immunol. 11:519–531. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Conigliaro P, Triggianese P, Ballanti E,
Perricone C, Perricone R and Chimenti MS: Complement, infection,
and autoimmunity. Curr Opin Rheumatol. 31:532–541. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Geller A and Yan J: The role of membrane
bound complement regulatory proteins in tumor development and
cancer immunotherapy. Front Immunol. 10:10742019. View Article : Google Scholar : PubMed/NCBI
|














