Introduction
Multiple sclerosis (MS)
MS is a complex neurological disorder. Extensive
research on its global epidemiology and societal impact has
highlighted its significant effects on both individuals and
communities. Data from 2015 ranked MS as the 10th most common
neurological condition, with an estimated 2.01 million cases at
that time (1). According to
updated data, an estimated 2.9 million individuals are living with
MS globally, with the prevalence having increased in most countries
reporting updated data (2). This
data represents a significant increase compared to earlier
estimates, and the rising prevalence of the disease underscores the
importance of ongoing research efforts. However, MS incidence
varies considerably by region. Countries with higher socioeconomic
status often report a greater frequency of MS, possibly influenced
by environmental factors, genetic predispositions and lifestyle
choices. For instance, MS is more prevalent in regions like North
America and Northern Europe, while it is less common in parts of
Asia and Africa. MS typically manifests in young adults,
particularly affecting women aged 20 to 40 years, with women being
affected at roughly three times the rate of men (3). This gender disparity may be
attributed to hormonal, immune and genetic factors.
The management and treatment of MS require
significant medical resources, placing a heavy burden on both
individuals and society (4).
Because MS requires long-term multidisciplinary management,
including medication, rehabilitation and psychological support,
patients often face substantially higher healthcare costs than
individuals without the condition (5). Furthermore, MS often leads to
reduced work capacity, contributing to higher unemployment and
absenteeism rates. Studies show that individuals with MS experience
diminished work abilities throughout their careers, affecting both
their financial independence and quality of life (6). Mental health challenges, such as
anxiety and depression, are also common among those with MS,
further deteriorating their overall health and amplifying the
demand for healthcare services (7).
Inadequate social support networks can exacerbate
these issues (8). Additionally,
societal attitudes and understanding of MS vary widely across
cultures and communities (9).
Numerous patients face social stigma and misunderstanding, which
negatively affects their psychological well-being and social
interactions. Therefore, raising public awareness of MS is
essential to improving social integration and support for affected
individuals (10). Global
epidemiological data highlight the rising prevalence of MS as a
significant public health concern, with profound implications for
both individuals and society (11). Given its rising incidence and
impact, advancing research and public health initiatives is crucial
to improving the lives of those affected and alleviating the strain
on society (12).
Definition and classification of
autophagy
Adapter proteins are crucial in selective autophagy,
identifying distinct autophagic substrates and attaching them to
autophagosomes. Through this method, cells can effectively mark and
separate substances requiring degradation (13). This mechanism plays a crucial
role in maintaining autophagy selectivity, guaranteeing the
breakdown of only certain substances without impacting other cell
parts.
Multistep process
Selective autophagy involves initially identifying
and isolating the autophagic substrate, then creating the
autophagosome, merging it with the lysosome and finally breaking it
down. The procedure entails controlling various intracellular
signaling routes to guarantee autophagy's efficiency and
specificity (14).
Selective autophagy varieties
Depending on the specific substrate and its role,
there are multiple subtypes of selective autophagy, including
mitochondrial autophagy (mitophagy), which specifically focuses on
impaired or dysfunctional mitochondria. Through mitochondrial
autophagy, cells are able to eliminate impaired mitochondria,
thereby averting oxidative stress and cellular demise. Plasmalemmal
autophagy is a type of autophagy that targets certain areas of the
cell membrane, eliminating detrimental elements or pathogens to
preserve the cell's integrity and functionality (15). Endoplasmic reticulum (ER)
autophagy concentrates on eliminating irregular or improperly
folded proteins in the ER to preserve its functionality and health
(16).
The biological roles of autophagy
Autophagy plays a crucial role in MS pathogenesis by
regulating the balance of cellular stress and inflammation
(17). In MS, autophagy is
particularly involved in controlling oxidative stress and
mitigating damage to oligodendrocytes, which are critical for
myelin regeneration (18). Three
recent studies consistently emphasize that autophagy is not
intrinsically beneficial or harmful, but a context-dependent
regulatory process whose impact in diseases such as MS depends on
cell type, timing and magnitude of activation (19-21). According to the conceptual
framework proposed by the three recently studies, autophagy is a
highly dynamic and tightly regulated cellular process rather than a
uniformly protective pathway. Their critical evaluation of
autophagy in MS explicitly challenges the notion of autophagy as an
inherently beneficial process, emphasizing that its effects vary
across immune and central nervous system (CNS) cell types, disease
stages and inflammatory environments. Autophagy is also essential
for maintaining cellular quality (22). It selectively eliminates
dysfunctional organelles, such as damaged mitochondria, and removes
harmful proteins, preventing the accumulation of toxic substances
within the cell (23). This
process plays a key role in protecting cells from stress and
damage. As an adaptive mechanism, autophagy helps cells adjust to
environmental changes, particularly during starvation or stress
(24). By breaking down internal
cellular components, autophagy provides essential nutrients,
ensuring ongoing cell survival and functionality. Additionally,
autophagy is integral to immune responses (25). It is involved in both
anti-infective and immune reactions by eliminating intracellular
pathogens and promoting the activation and growth of immune cells
through antigen processing (26). As a result, modulating autophagy
may offer a novel therapeutic strategy for treating MS (27). In summary, autophagy serves
multiple biological roles, from energy distribution and cell
quality control to adaptive responses and immune function.
Understanding the mechanisms underlying autophagy and its impact on
diseases can inspire innovative approaches for studying and
treating conditions like MS. Regulating autophagy holds promise for
improving disease outcomes, enhancing neuroprotection and slowing
disease progression. Seminal work by Mizushima et al
(28-30) has established autophagy as a
fundamental cellular process essential for maintaining
intracellular and tissue homeostasis. Through a series of
influential studies and comprehensive reviews, autophagy has been
conceptualized not merely as a degradation pathway but as a dynamic
system governing cellular quality control, metabolic adaptation and
tissue renovation. Extending from the cellular to the tissue level,
autophagy has been described as a 'renovation' mechanism that
supports long-term tissue maintenance and functional stability,
particularly in organs with limited regenerative capacity such as
the CNS. This conceptual framework provides a critical foundation
for understanding how autophagy may exert both protective and
detrimental effects in CNS immune-mediated disorders, including MS,
depending on cell type, disease stage and microenvironmental
cues.
Control systems governing autophagy
Pathway of mammalian target of
rapamycin (mTOR)
The mTOR is a central nutrient- and energy-sensing
kinase that plays a major, but context-dependent, role in autophagy
regulation (31). Rather than
acting as a simple inhibitory switch, mTOR integrates multiple
environmental cues to dynamically modulate autophagy. In
particular, mTOR complex 1 (mTORC1) suppresses autophagy under
nutrient-rich conditions by phosphorylating components of the
unc-51-like autophagy-activated kinase 1 (ULK1) initiation complex,
thereby preventing autophagosome formation (32,33). Growth factor signaling, such as
insulin-like growth factor (IGF) binding to IGF1 receptor (IGF1R),
activates the PI3K-Akt pathway, leading to inhibition of the
tuberous sclerosis 1 (TSC1)/TSC2 complex and subsequent activation
of mTORC1, which suppresses autophagy (34-36). Conversely, amino acid deprivation
and energy stress suppress mTORC1 activity, enabling autophagy
initiation. mTORC1 activity is also regulated spatially through its
association with lysosomal membranes, highlighting the importance
of subcellular localization in autophagy control (37,38).
Pathway of adenosine
monophosphate-activated protein kinase (AMPK)
AMPK functions as a key metabolic sensor that
regulates autophagy in response to cellular energy stress (39). Upon glucose deprivation, AMPK
activation can promote autophagy by directly phosphorylating ULK1.
However, this effect is modulated by mTORC1 activity, as
mTORC1-dependent phosphorylation of ULK1 may counteract
AMPK-mediated activation (40).
In addition, AMPK indirectly influences autophagy through the
TSC1/2-mTOR axis, highlighting its role as an upstream regulator
within a broader signaling network rather than a unidirectional
activator (41).
Pathway of protein kinase A (PKA)
PKA senses glucose availability and has been
reported to negatively regulate autophagy under nutrient-rich
conditions, primarily through phosphorylation of autophagy-related
proteins such as autophagy-related protein 1 (Atg1) and Atg13
(42). Furthermore, PKA may
indirectly suppress autophagy by modulating mTORC1 and AMPK
signaling, suggesting an indirect and context-dependent regulatory
role (43).
Pathway of Jnk-1
The JNK-1 pathway has been implicated in autophagy
induction, particularly under stress conditions such as nutrient
deprivation and ER stress (44).
Activation of JNK-1 can lead to phosphorylation of Bcl-2, thereby
disrupting its interaction with Beclin 1 (BECN1) and facilitating
autophagy initiation (45).
However, JNK-mediated autophagy is highly dependent on stress
intensity, duration and cell type, underscoring its role as a
stress-responsive modulator rather than a universal autophagy
activator.
Pathway of P53
Nuclear p53 can transcriptionally activate
autophagy-related genes through the TSC1-mTOR pathway, whereas
cytoplasmic p53 has been shown to suppress basal autophagy.
Consequently, pharmacological inhibition of p53 may induce
autophagy in certain cellular contexts but suppress it in others,
highlighting the complexity of p53-mediated autophagy regulation
(40).
Free fatty acids
Lipid metabolism is closely connected to autophagy
regulation (46). Free fatty
acids can activate EIF2S1/eIF2α and MAPK8 through the
EIF2AK2/PKR-dependent pathway or inhibit mTORC1, thereby promoting
autophagy (47). Lipid droplets,
which are dietary fats stored in cells, trigger autophagy, forming
autophagosomes that envelop and transport them to lysosomes for
degradation. Degradation products, such as free fatty acids,
regulate autophagy and prevent lipotoxicity (25).
Pathway of inositol
1,4,5-trisphosphate (IP3)
Inositol monophosphatase (IMPase) reduces the levels
of unbound inositol and IP3, which in turn promotes calcium ion
release from the ER. This process occurs via the binding of IP3 to
its receptors on the reticulum's surface, activating a cascade of
calcium-dependent proteases and inhibiting autophagy by cleaving
and activating stimulatory alpha subunit of heterotrimeric G
proteins (Gsα), resulting in significant cyclic (c)AMP production
(48). Additional mechanisms may
contribute to this pathway, such as the reduction in IP3 levels,
which limits calcium entry from the ER into mitochondria,
potentially impairing the mitochondrial respiratory chain and
subsequently enhancing autophagy via the AMPK pathway. Furthermore,
the IP3 receptor interacts with BECN1, suppressing autophagy
(16). Neuropsychotropic drugs
such as sodium valproate and carbamazepine are known to stimulate
autophagy through this pathway (49).
Molecular impacts of autophagy
The primary mechanism driving autophagy involves the
activity of ATG proteins. This process is further regulated by AMPK
and mTOR, which modulate autophagy through the phosphorylation of
key proteins like ULK1, ULK2 and ATG13. Phosphorylated ULK1
activates BECN1, a homolog of yeast Vps30/Atg6, initiating the
formation of autophagosomes through the ULK1-BECN1 complex. As part
of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex,
BECN1 generates PtdIns3P on the membranes of phagocytic vesicles,
serving as a binding site for additional autophagy-related
proteins. A pair of ubiquitin-like conjugation systems, comprising
ATG12-ATG5-ATG16L1 and the Atg8 family proteins such as
microtubule-associated protein 1 light chain 3 (MAP1LC3, commonly
referred to as LC3) and γ-aminobutyric acid receptor-associated
protein, are essential for the growth and expansion of phagocytic
vesicles and the attachment of specific autophagic types (16,48,50) (Fig. 1).
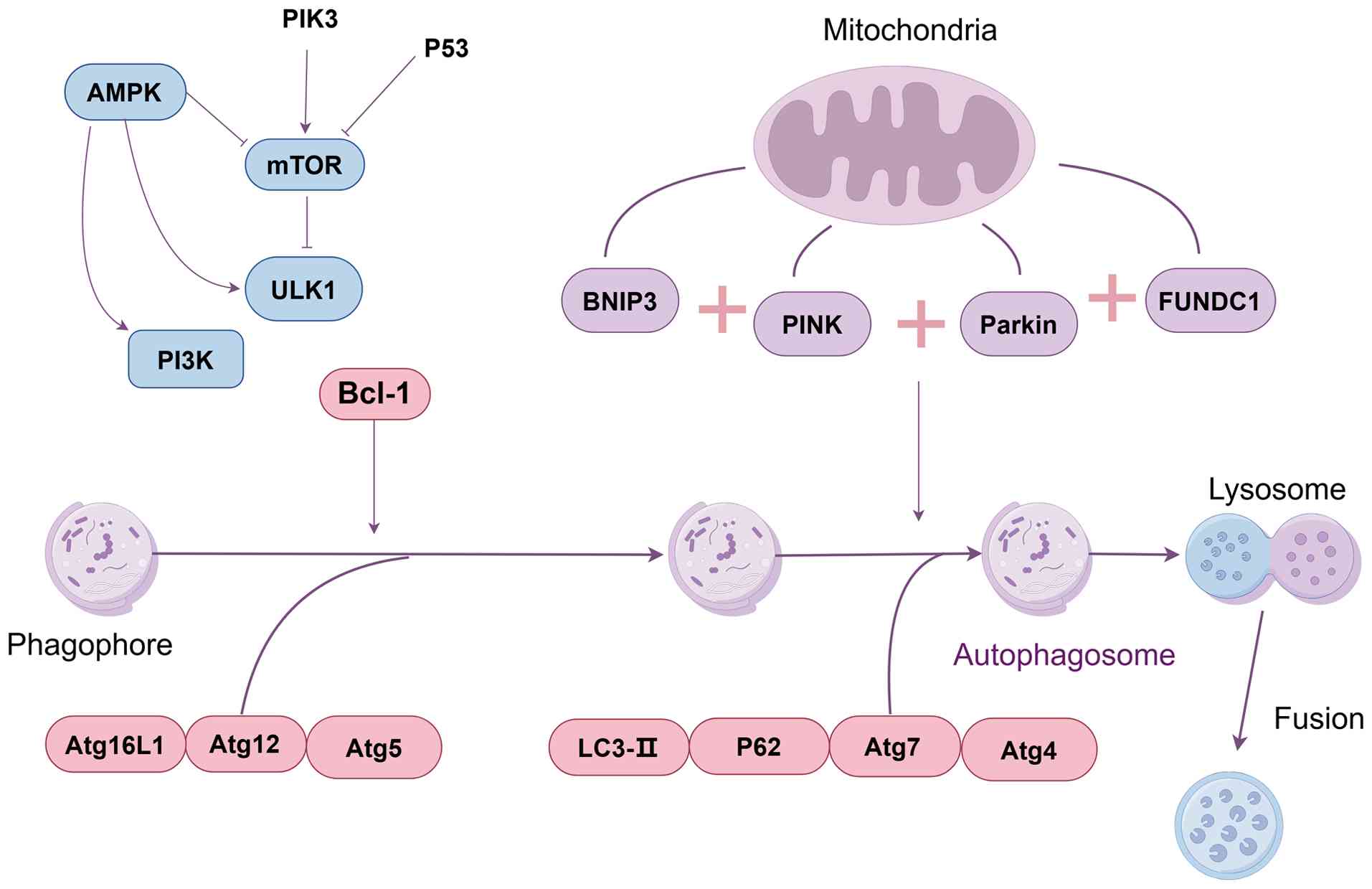 | Figure 1Major signaling pathways regulating
autophagy. Autophagy is regulated by multiple signaling pathways,
including mTOR, AMPK, PKA, JNK-1, p53, free fatty acid-related
pathways and IP3 signaling. The mTOR pathway functions as a central
negative regulator of autophagy in response to nutrient
availability, whereas AMPK activates autophagy under energy stress.
PKA generally suppresses autophagy under high-glucose conditions.
JNK-1 promotes autophagy by modulating the Beclin 1-Bcl-2
interaction during nutrient deprivation. p53 can either induce or
inhibit autophagy depending on its cellular localization and
regulatory context. Free fatty acids and lipid droplets modulate
autophagy through metabolic stress-related signaling, while IP3
signaling inhibits autophagy via calcium-dependent mechanisms and
BECN1binding. IP3, inositol 1,4,5-trisphosphate; mTOR, mammalian
target of rapamycin; AMPK, adenosine monophosphate-activated
protein kinase; PKA, protein kinase A; ULK1, unc-51-like
autophagy-activated kinase 1; Atg12, autophagy-related protein 12;
BECN1, Beclin 1. |
Collectively, these pathways illustrate that
autophagy is governed by an intricate and highly interconnected
regulatory network rather than by linear signaling cascades. The
functional outcome of autophagy signaling depends on nutrient
availability, cellular context, stress intensity and disease
state.
This review was based on a comprehensive literature
search of peer-reviewed articles related to autophagy and MS. The
primary databases searched included PubMed (https://pubmed.ncbi.nlm.nih.gov/), Web of Science
(https://www.webofscience.com/) and
Scopus (https://www.scopus.com/). Relevant
studies were identified using combinations of key words such as
'multiple sclerosis', 'autophagy', 'mitophagy', 'oxidative stress',
'immune regulation' and 'neurodegeneration'. Articles were selected
based on their relevance to the role of autophagy in MS
pathogenesis, progression and potential therapeutic implications.
Priority was given to original research articles, high-quality
reviews and recent studies that provided mechanistic insights or
experimental evidence. Studies focusing on non-MS neurodegenerative
diseases were included when they offered conceptual or mechanistic
relevance to MS. Non-English publications and studies lacking
sufficient experimental or clinical relevance were excluded.
Connection between autophagy and
neurological conditions
Neuronal structures are highly susceptible to
disruptions due to the interaction between endosomes/autophagosomes
and lysosomes, particularly in the aging brain. Genetic aging and
alterations in autophagic processes are linked to the onset of
various neurodegenerative diseases, such as Alzheimer's disease
(AD), familial Parkinson's disease (PD) and amyotrophic lateral
sclerosis (ALS), which arise from impaired lysosomal clearance.
Autophagic dysfunction is associated with the accumulation of
cytoplasmic aggregates, protein buildup in neurons and
mitochondrial dysfunction. These diseases share similarities with
MS, characterized by suppressed autophagy. Dysfunction in autophagy
leads to cellular dysfunction, further promoting the progression of
these diseases. Unlike other cell types, neurons cannot eliminate
damaged organelles or misfolded proteins through cell division. The
presence of synapses, dendrites and axons, along with their
significant distance from the cytosol, further complicates
autophagy and intercellular communication (28,37,51,52).
PD
PD, a neurodegenerative disorder, results from the
selective degeneration of dopaminergic neurons in the substantia
nigra. Neuropathologically, PD is characterized by the deposition
of Lewy bodies, which consist of cellular accumulations of
ubiquitin and α-synuclein (SNCA). Autophagy plays a critical role
in preventing PD onset by degrading SNCA through both
macroautophagy and chaperone-mediated autophagy (CMA). Inhibition
of these processes leads to the accumulation of both mutant and
normal forms of SNCA (53).
BECN1 promotes autophagy, thereby enhancing SNCA degradation. The
role of BECN1 in promoting autophagy is equally important in MS.
The accumulation of SNCA disrupts macroautophagy, as high SNCA
levels lead to irregular distribution of ATG9, inhibiting omegasome
formation, a precursor to autophagic vesicles, and suppressing CMA,
which contributes to progressive neuronal damage (40). The autophagy process in neurons
is more complex than in other cell types, further complicating its
role in neurodegenerative diseases.
Of note, autophagy may paradoxically contribute to
neuronal degeneration under certain conditions. Overactivation of
autophagy can exacerbate dopaminergic neuron death, with heightened
autophagic activity in PD potentially triggered by oxidative stress
that suppresses genes involved in mTOR activation. Supporting this
view, autophagy stimulants have been shown to worsen neuronal death
in the context of oxidative stress, suggesting that autophagy may
act as a double-edged sword in PD (54). Additionally, parkin RBR E3
ubiquitin ligase (PRKN) plays a key role in mitochondrial autophagy
by removing damaged mitochondria. Defective PRKN in PD leads to
mitochondrial dysfunction and neurodegeneration. Of note,
mitochondrial autophagy is dysfunctional in MS, not merely
suppressed (55).
A comparative analysis between 20 patients with PD
and 20 individuals with idiopathic tremor revealed significantly
lower plasma concentrations of the lysosomal hydrolase CTSD
(histone D) and BAG chaperone 2, a regulator of the PTEN-induced
kinase 1 (PINK1)-PRKN pathway, in those with PD. These findings
suggest a reduction in autophagic activity in PD. Similarly,
peripheral blood mononuclear cells from patients with PD showed
diminished levels of heat shock protein family A member 8/heat
shock cognate protein of 71 kDa, a critical component of CMA
(56). As a result, autophagy is
notably impaired in PD, though it can be either suppressed or
upregulated under different circumstances.
Conversely, inhibiting mTOR may offer therapeutic
potential in AD by promoting autophagy (57). In AD, autophagy, a vital process
for microglia to clear amyloid-β (Aβ), becomes dysfunctional.
Experimental studies have shown that microglia in AD mice exhibit
elevated levels of microRNAs (miRNAs), which suppress the
expression of autophagy-related proteins. Of note, Aβ accumulation
around microglia precedes autophagy impairment in AD (58). Extended exposure to Aβ also
triggers miRNA expression in peripheral neurons (59).
Impaired autophagic processes, particularly the
dysfunctional formation and transport of autophagosomes, lead to
the progressive accumulation of autophagic vesicles around neurons
in AD. This accumulation, exacerbated by Aβ, promotes
neurodegeneration (60). In
addition, a reduction in BECN1 levels, both in AD and aging,
results in diminished autophagic activity. This decrease in BECN1
triggers neurodegeneration through enhanced Aβ accumulation and
disruptions in autophagy and APP metabolism (17). These findings suggest that
excessive autophagy may contribute to the neuropathology of AD,
although autophagy malfunctions could also be secondary to disease
progression.
Furthermore, the autophagy and BECN1regulatory
factor 1 (AMBRA1) protein, which regulates autophagy in the CNS,
interacts with BECN1 to strengthen its association with the lipid
kinase phosphatidylinositol 3-kinase catalytic subunit type 3
(PIK3C3/VPS34). Together with PIK3C3/VPS34 and BECN1, AMBRA1 is
considered a key component of the autophagy core complex, which is
critical for regulating this process in AD. The autophagy core
complex itself plays an essential role in modulating autophagic
activity (61).
Sclerosis of the amyotrophic lateral
system
ALS, a progressive neurodegenerative disorder, is
characterized by the gradual degeneration of motor neurons. Of the
two ALS subtypes, 90% of cases are sporadic, while the familial
form accounts for the remaining 10% (62). Autophagy plays a critical role in
ALS pathogenesis. Excessive macroautophagy accelerates spinal motor
neuron degeneration in ALS mice with the mutant superoxide
dismutase 1 (SOD1), which is implicated in ~20% of familial ALS
cases (63). Notably, the motor
neuron degeneration observed in these mice is closely linked to
autophagic dysfunction and autophagosome formation (64). Chen et al (65) highlighted the association between
impaired autophagic regulation and ALS-associated proteins, such as
mutant SOD1, along with other gene products that contribute to
abnormal autophagic activity.
Furthermore, optic nerve phosphatase (OPTN), a
highly conserved protein, is essential for regulating various
cellular processes, including vesicular transport, inflammation and
autophagy (66). Genetic
alterations in OPTN are linked to inflammation and glaucoma in
patients with ALS, while the loss of OPTN is associated with
cerebral atrophy (67).
In summary, the various stages of autophagy
influence a range of neurodegenerative disorders in distinct ways.
For instance, in AD and ALS, there is an increase in autophagy
initiation, while the fusion and maturation stages are disrupted in
both conditions. In PD, the nucleation phase is notably diminished.
These stage-specific autophagic dysfunctions may be linked to the
unique pathophysiology of each neurodegenerative disease.
Investigating autophagy's function in
MS
Autophagy plays a dual role in the pathogenesis of
MS, similar to its involvement in other diseases. Recent studies
have highlighted that autophagy-dependent ferroptosis contributes
to the onset of MS (68).
Microglia-derived extracellular vesicles (EVs) carry bioactive
cargo that modulates neuroinflammation and glia-neuronal
interactions in MS, and dysregulation of microglial autophagy has
been linked to impaired myelin-debris handling and poorer recovery
in demyelinating models. MS-focused studies indicate that altered
microglial EV signaling and autophagic dysfunction may contribute
to neurodegeneration and disease progression in specific
pathological contexts (69).
The link between autophagy and MS was first
established in animal models, where altered autophagy markers were
observed in hippocampal neurons. One study identified significant
increases in the expression of Beclin-1, LC3 and p62, alongside
disturbances in the Akt/mTOR signaling pathway, upon examining key
autophagy proteins. When compared to control groups, mice with MS
exhibited notably reduced Beclin-1/LC3II and p62 levels. Molecular
studies further revealed abnormalities in Akt/mTOR pathways,
indicating that autophagy activation occurs in hippocampal neurons
within this experimental model (70). Additionally, autophagy and
mitochondrial autophagy-related markers were found to be
upregulated in the bodily fluids of patients with MS (71).
These findings suggest a robust association between
autophagy and MS pathology. Of note, ALS-associated proteins,
including transactive response DNA-binding protein of 43 kDa and
SOD1, have also been implicated in autophagic dysregulation.
Several genetic mutations in ALS interfere with autophagy in motor
neurons, reinforcing the notion that autophagy dysregulation plays
a central role in ALS pathogenesis (72). Furthermore, ATG16L2, an
autophagy-related gene, has been proposed as a potential serum
biomarker for MS, identified using microbead-based proteomics
technology (73).
In addition, bioinformatics combined with machine
learning approaches has been used to explore the role of
autophagy-related genes in MS. The 9 autophagy genes most strongly
associated with MS were identified, with bioinformatics and
experimental data indicating their significant involvement in
autophagy regulation and MS development (74). The 9 autophagy genes with the
strongest correlations were as follows: Becn1, fibroblast growth
factor receptor 2 (Fgfr2), integrin alpha 3 (Itga3), Napsin A
aspartic peptidase (Napsa), neurotrophic tyrosine kinase, receptor,
type 2 (Ntrk2), ataxia telangiectasia mutated (Atm), solute carrier
family 36 member 4 (Slc36a4), member RAS oncogene family (Rab10)
and Rous sarcoma oncogene (Src). BECN1 BECN1is one of the core
genes involved in autophagy; in MS, reduced expression of BECN1is
associated with impaired myelin regeneration. Changes in Fgfr2
expression levels in patients with MS may be associated with
neuroinflammation and myelin damage. In MS, Itga3 expression is
upregulated, potentially affecting immune cell migration and nerve
damage. Napsa may influence immune responses within the nervous
system in MS. Reduced expression of Ntrk2 correlates with
neurodegeneration and impaired myelin repair. Atm gene defects are
associated with enhanced neuroinflammation and immune responses.
Slc36a4 may influence neuroinflammation by regulating immune
responses. Downregulation of Rab10 may affect autophagosome
maturation and the process of neural repair. Upregulation of Src
expression may exacerbate immune responses and neuroinflammation in
MS. Gene expression profiles of autophagy-related genes in MS also
reflected similar patterns, further emphasizing the connection
between autophagy and MS (75).
These findings collectively highlight the complex relationship
between autophagy and MS. The varying stages of autophagy and the
progression of MS warrant further investigation to understand how
these processes are interlinked and how they can be leveraged for
potential therapeutic strategies.
The process of autophagy fosters the
progression of MS
Numerous studies have emphasized the protective role
of autophagy in preventing the onset and progression of MS
(76). Autophagy helps mitigate
the development of MS by neutralizing reactive oxygen species (ROS)
(77). Compared to healthy
individuals, patients with MS show elevated levels of oxidized
phospholipids, DNA and oxidative stress markers (78). A reduction in autophagic activity
correlates with increased oxidative stress and inflammation due to
impaired ROS clearance, leading to higher IL1B/IL-1β production
(79). Research has extensively
explored the interplay between hypoxia-inducible factor 1, ROS and
autophagy in MS (80). The
mechanisms through which ER stress and autophagy contribute to MS
pathogenesis are well documented.
Similarly, fenofibrate, a peroxisome
proliferator-activated receptor-α (PPAR-α) agonist, has been shown
to reduce inflammation in MS by modulating autophagy and inhibiting
helper T cell 17 (Th17) cell differentiation, along with decreasing
pro-inflammatory signals (81).
Studies also indicate that miRNA-223 (miR-223) impedes autophagy
and exacerbates inflammation in MS by targeting ATG16L1 (82). Furthermore, blocking autophagy
has been found to trigger NOD-, LRR-, and pyrin domain-containing
protein 3 (NLRP3) inflammasome activation in microglia via
phosphodiesterase 10A-cyclic AMP signaling, which leads to
increased IL1B/IL-1β levels and enhanced macrophage migration
inhibitory factor (MIF) production. Inhibition of NLRP3
inflammasomes with the MCC950 inhibitor reduced MIF expression,
alleviated neuroinflammation and reversed neuronal damage in the
substantia nigra (83).
In addition to ROS, reactive nitrogen species (RNS),
such as nitric oxide and peroxynitrite, are involved in
neurodegeneration in MS (84).
These molecules can nitrate or nitrosylate the dynamin-related
protein 1 (Drp1)/Parkin/PINK1 pathway, promoting excessive
mitochondrial autophagy and amplifying neuronal damage. Targeting
the RNS-driven excessive autophagy/mitochondrial autophagy pathway
holds potential for developing novel anti-MS therapies (85).
Notably, the mTOR pathway, which regulates
autophagy, also influences microglial-driven inflammation in MS
(86). Targeting specific
inhibitors within the PI3K-mTOR signaling axis could offer a
promising therapeutic strategy for MS treatment.
Extensive research also supports the inhibitory role
of autophagy in MS, potentially through prolonged activation of
mTORC1, which may depend on the progression of the lesions. The
present findings deepen the understanding of autophagy's role in
various stages of MS pathology, suggesting that the mTORC1 pathway
could serve as a key regulator, influencing CNS homeostasis and
neuroinflammation in MS (87).
In this context, rapamycin, an mTOR inhibitor, may stimulate
autophagy and promote myelin regeneration by reducing inflammation.
Additionally, curcumin has been shown to affect MS metabolism by
interacting with AMPK, PPARG coactivator 1 α/PPARγ and the
PI3K/Akt/mTOR signaling pathways (88). Several reviews have discussed the
mechanisms through which the mTOR complex pathway influences MS
(89).
Furthermore, the increase in autophagy during MS
could represent an adaptive response to the heightened unfolded
protein response (UPR) triggered by ER stress. The UPR serves a
cytoprotective function by regulating ER activity, supporting
oligodendrocyte function and enhancing myelin production in MS.
Thus, stimulating the UPR may reduce MS neuropathology by promoting
autophagy.
Echoing previous research, numerous studies
emphasize the importance of protein homeostasis in disease
development. Impairment of the autophagy-lysosomal pathway, coupled
with the detrimental effects of C9orf72 repeat RNA and dipeptide
repeat proteins, contributes to disease progression (90). Additionally, optineurin has been
shown to initiate autophagy by clearing protein aggregates via a
ubiquitin-independent mechanism, thus facilitating the progression
of MS (91).
During the active phase of MS, significant increases
in autophagy and mitochondrial autophagy markers have been observed
in the biological fluids of patients, indicating the activation of
these pathways. In line with this, both in vitro and in
vivo MS models (induced by pro-inflammatory cytokines, lysyl
ovalbumin and copper ketones) exhibit mitochondrial dysfunction,
triggering lactate metabolism, enhancing autophagic flux and
increasing mitochondrial autophagy. Various autophagy inhibitors,
differing in structure and mechanism, have been found to promote
myelin synthesis and normalize axonal myelination, while the
antipsychotics haloperidol and clozapine significantly reduced
dyskinesia induced by copper ketones (92).
Previous studies have also highlighted the role of
autophagy in macrophages for MS repair (93). Lipophagy in macrophages has been
shown to accelerate MS recovery, with microglia containing lipid
droplets playing a pivotal role in MS pathology. Notably,
miR-223-mediated suppression of cathepsin B in microglia enhances
lipophagy following lysolecithin-induced demyelination in mice,
indicating that activation of selective autophagy pathways
contributes to myelin lipid degradation during demyelinating injury
(94).
A recent study introduced a novel theory involving
partial neuronal cell death and an autophagy-positive feedback
loop, highlighting the role of the neuronal-astrocyte
glutamate-glutamine cycle in ALS. Under optimal conditions,
disrupting these cycles could slow ALS progression. Brain vascular
damage, such as recurrent embolisms and strokes, may initiate
neuronal cell death followed by autophagy. Furthermore, ALS impedes
the fusion of autophagosomes and lysosomes, exacerbating cell
death. Treatment with alginate has been shown to restore the
defective fusion phase and significantly delay disease onset,
suggesting its potential as a preventative treatment for ALS
(95). This study aligns with
our long-held belief that autophagy may indirectly influence the
development of MS.
Additionally, inhibiting autophagy leads to
mitochondrial dysfunction and oxidative stress, which may increase
the risk of MS development. There is a notable imbalance in
neuronal autophagy in proteopathies, indicating a link between
autophagic dysfunction and the onset of neurodegenerative diseases.
Patients with MS exhibit a reduction in CD46 receptors, which
facilitate autophagy, further supporting the involvement of
autophagy in MS pathology. However, the precise role of
mitochondrial autophagy in MS remains unclear. Mitochondrial
autophagy, a specialized form of autophagy, is crucial for
maintaining mitochondrial function and controlling oxidative stress
by eliminating damaged mitochondria. This process often involves
the activation of PARKIN and PINK1 on the mitochondrial outer
membrane, mediated by calcium-binding and coiled-coil
domain-containing protein 2 (CALCOCO2)/nuclear dot protein 52 and
OPTN receptors. These receptors enhance mitochondrial autophagy by
promoting the interaction between phagocytic vesicle membranes and
organellar proteins, aiding mitochondrial sequestration within the
autophagosome. Alterations in the CALCOCO2 gene are associated with
MS progression, contributing to the secretion of pro-inflammatory
cytokines like TNF-α. Supporting evidence shows that CALCOCO2 is
primarily expressed in B cells of peripheral blood mononuclear
cells, where its stimulation through mitochondrial autophagy
reduces pro-inflammatory cytokine production. This highlights
CALCOCO2's protective function in B cells and its potential
involvement in autoimmune diseases like MS (96).
Together, these findings suggest that autophagy
plays a protective role in MS by reducing oxidative stress and
inflammatory responses, thereby hindering disease onset and
progression. Furthermore, the aforementioned molecular mechanisms
are summarized in Fig. 2.
Although numerous studies have indicated that mitochondrial
autophagy in MS is closely associated with neurological damage,
these investigations often focus on describing the fundamental
processes of autophagy while lacking in-depth analysis of its
molecular mechanisms. For instance, while PINK1 and Parkin play
crucial roles in clearing damaged mitochondria, the changes in
their expression levels among patients with MS remain unclear.
Furthermore, research generally lacks experimental data to
elucidate how they regulate autophagy activity.
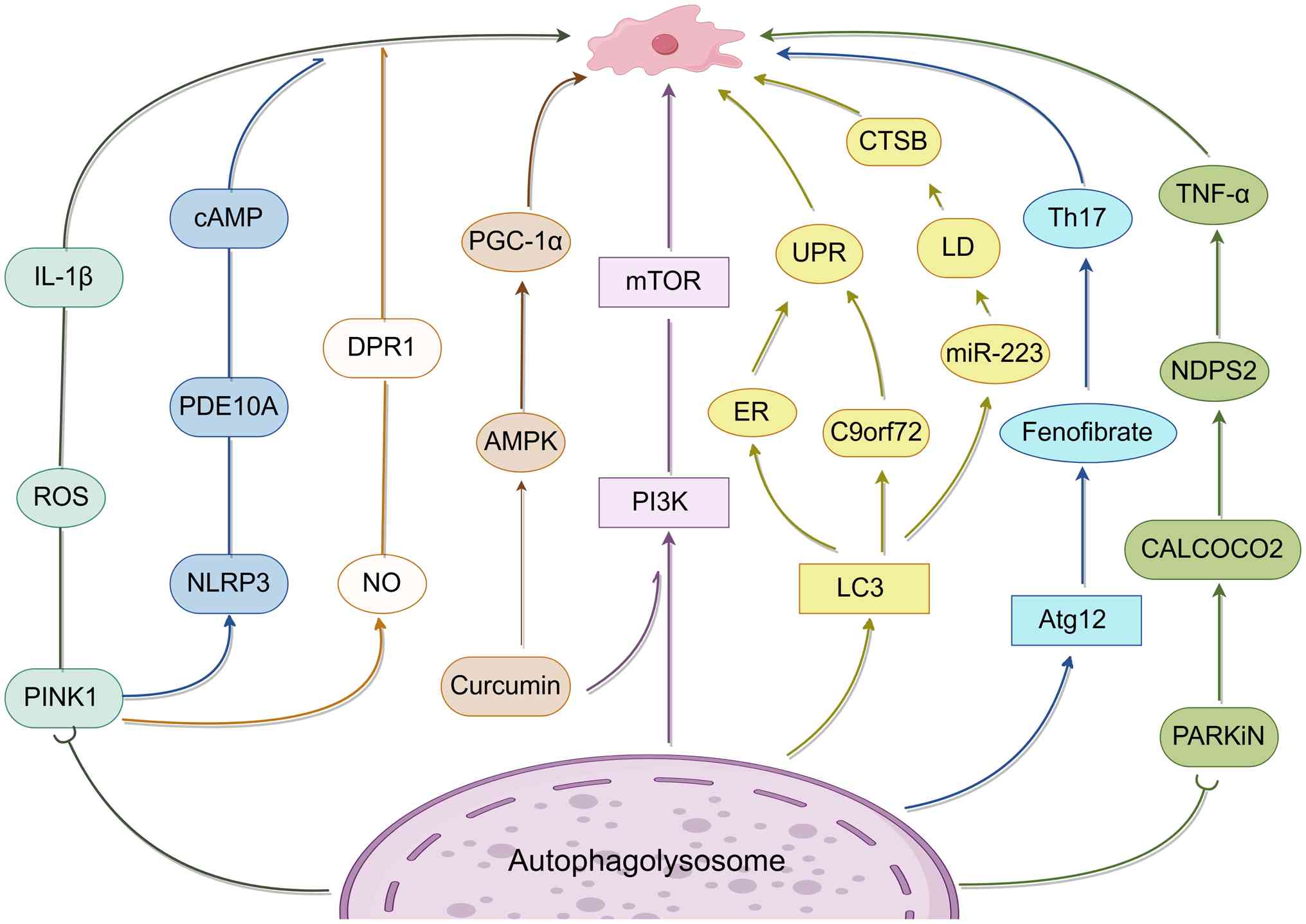 | Figure 2Autophagy-associated pathways
involved in MS. Multiple autophagy-related pathways are implicated
in the pathogenesis of MS. Dysregulation of autophagy influences
inflammatory signaling, mitochondrial quality control, endoplasmic
reticulum stress responses and lipid metabolism in neural and glial
cells. Key regulators include miR-223-ATG16L1 signaling, NLRP3
inflammasome activation, mitophagy-related pathways and selective
autophagy receptors. MS, multiple sclerosis; IL-1β, interleukin-1β;
TNF-α, tumor necrosis factor α; Th17, T helper 17 cells; NLRP3, NLR
family pyrin domain containing 3; LC3, microtubule-associated
protein 1 light chain 3; Atg12, autophagy-related protein 12;
PINK1, PTEN-induced kinase 1; PARKIN, Parkinson protein 2, E3
ubiquitin-protein ligase; CALCOCO2, calcium-binding and coiled-coil
domain-containing protein 2; CTSB, cathepsin B; ER, endoplasmic
reticulum; UPR, unfolded protein response; LD lipid droplet; AMPK,
adenosine monophosphate-activated protein kinase; mTOR, mammalian
target of rapamycin; PI3K, phosphatidylinositol 3-kinase; PGC-1α,
peroxisome proliferator-activated receptor-γ coactivator-1α; cAMP,
cyclic adenosine monophosphate; PDE10A, phosphodiesterase 10A; ROS,
reactive oxygen species; NO, nitric oxide. |
The process of autophagy acts as an
inhibitor to the progression of MS
Contrary to the previously discussed findings,
several studies-both preclinical and clinical-suggest that
autophagy may have a harmful effect on the progression of MS
(97). Elevated expression of
immune-related GTPase M 1 and ATG5 has been observed in T cells
from individuals with relapsing-remitting MS (98). Notably, administration of the
mTOR inhibitor rapamycin accelerated the progression of
relapsing-remitting MS (99). In
parallel, MS lesions displayed increased oxidative stress and
inflammation, along with a reduced LC3-II:LC3-I ratio, suggesting
altered or impaired autophagic flux in demyelinated regions
(100). Autophagy has been
implicated in both removal of myelin debris and regulation of
remyelination, but defective or dysregulated autophagy may delay
efficient clearance of myelin fragments and contribute to sustained
neuroinflammation and neuronal injury in MS pathology (18). Additionally, stimulating
autophagy by activating STAT1 in microglia led to significant white
matter damage in mice. A marked increase in T-cell autophagy
correlates with the severity and progression of MS.
Consequently, ATG16L2, a regulator of T-cell
autophagy, has potential as a biomarker for predicting relapse in
patients with MS (101).
Clinical trial data reveal a decrease in ATG16L2 mRNA levels in T
cells of individuals with MS, suggesting that targeting autophagy
could be a viable therapeutic approach in MS (102).
Notably, there is an upregulation of
autophagy-related genes and markers in T lymphocytes and tissues in
MS (103). For instance, Atg5
mRNA levels are associated with neurological impairments in
patients with MS, with autophagy exacerbating the severity of
relapsing-remitting MS (104).
Furthermore, the heightened levels of mitochondrial autophagy and
general autophagic activity in the biological fluids of patients
with active MS point to their potential use as indicators of
disease severity (105).
Previous studies have demonstrated a significant
upregulation of autophagy in T cells from individuals with MS, as
evidenced by elevated ATG5 expression, suggesting a close
association between ATG5-driven autophagy and enhanced inflammatory
capacity in autoreactive lymphocytes (106,107). In this context, autophagy not
only supports T-cell survival under inflammatory stress but also
influences the functional balance between pathogenic Th17 cells and
immunosuppressive regulatory T cells (Tregs), a key determinant of
MS disease activity. Dysregulated autophagy has been proposed to
favor Th17 polarization while impairing Treg stability, thereby
exacerbating immune imbalance and neuroinflammation (108). Therapeutically, rapamycin has
been shown to modulate autophagy through mTOR inhibition and to
partially restore immune homeostasis by promoting Treg expansion
and dampening neuroinflammation (109). Although early clinical studies
indicate that rapamycin is generally well tolerated in MS, its cell
type-specific effects on T cells, microglia and oligodendrocytes
remain insufficiently characterized. Finally, accumulating evidence
links MS pathogenesis to both T- and B-cell dysfunction, with
Epstein-Barr virus-mediated B-cell activation further amplifying
autoimmune responses (110).
How autophagy integrates these peripheral and CNS-specific
mechanisms into a unified pathogenic framework warrants further
investigation.
Mitochondrial dysfunction in MS is accompanied by
activation of mitochondrial autophagy, and experimental inhibition
of this process has been shown to enhance myelin synthesis in
oligodendrocytes and promote axonal remyelination. Consistently,
liraglutide attenuates CNS inflammation and demyelination through
modulation of the AMPK and pyroptosis-related NLRP3 pathway,
indicating a functional link between mitochondrial stress
responses, autophagy and demyelinating pathology (111). Furthermore, antipsychotic
medications like clozapine and haloperidol alleviate locomotor
impairments in mice caused by copper ketone by inhibiting
mitochondrial autophagy. FK506-binding protein 5 (FKBP5) has been
identified as a regulator of PPAR-γ, playing a role in mitigating
mitochondrial autophagy to create a favorable environment for
myelin regeneration. By contrast, a lack of myelin triggers
PINK1/Parkin-mediated mitochondrial autophagy, where FKBP5 acts as
a critical modulator through PPAR-γ. This discovery underscores
FKBP5's role in controlling mitochondrial autophagy during the
recovery process in demyelinating conditions, offering a potential
target for treating demyelinating disorders (112) (Fig. 3).
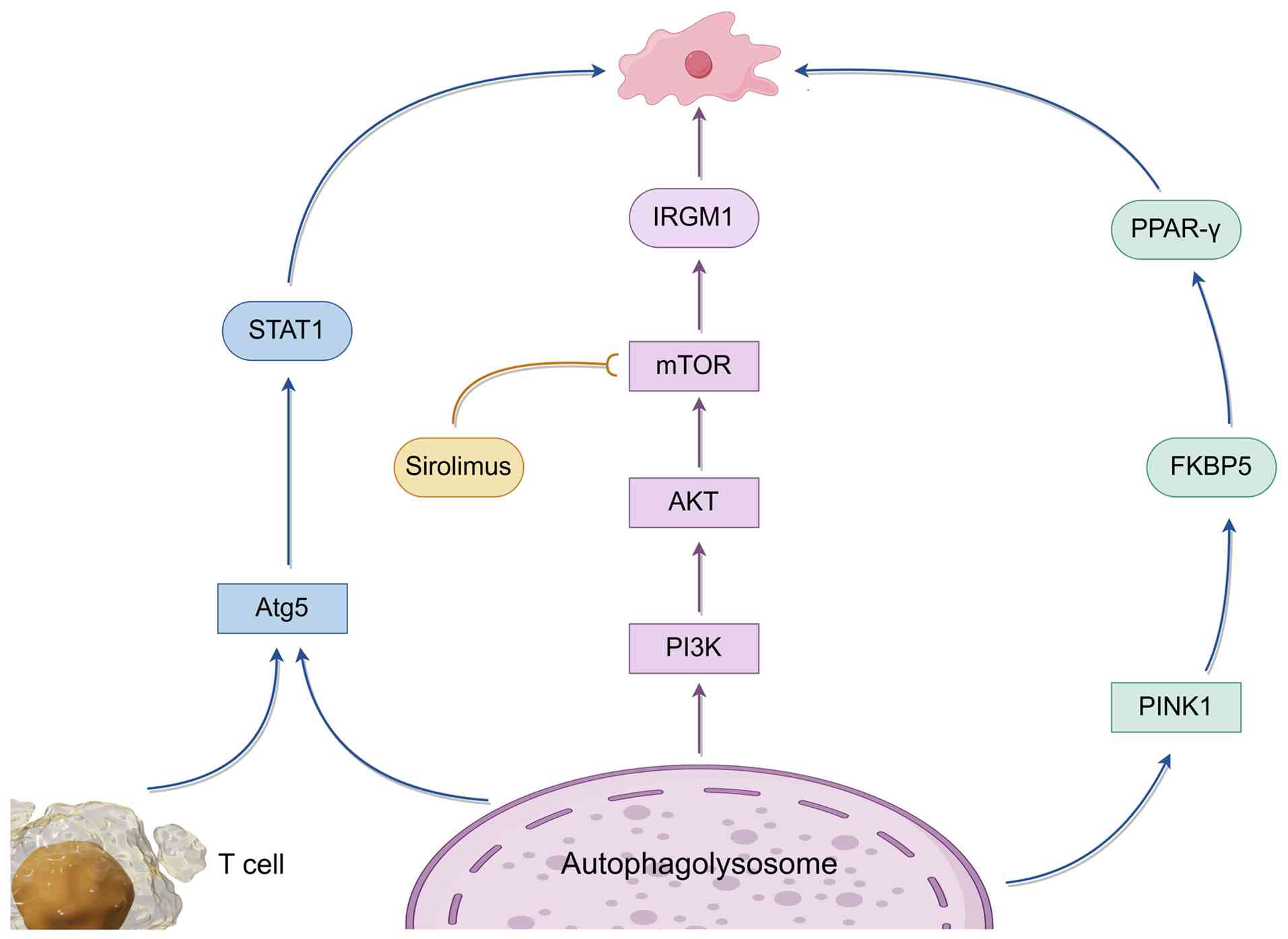 | Figure 3Context-dependent roles of autophagy
and mitophagy in MS. Autophagy and mitophagy can exert both
detrimental and protective effects during MS progression.
Dysregulation of autophagy-related genes in T cells, mitochondrial
dysfunction and excessive mitophagy contribute to inflammation and
impaired remyelination. Pharmacological modulation of
autophagy-related pathways influences neuroinflammation,
demyelination and myelin regeneration. MS, multiple sclerosis;
Atg5, autophagy-related protein 5; LC3, microtubule-associated
protein 1 light chain 3; PINK1, PTEN-induced kinase 1; IRGM1,
immunity-related GTPase family M member 1; PI3K,
phosphatidylinositol 3-kinase; AKT, protein kinase B; mTOR,
mammalian target of rapamycin; AMPK, adenosine
monophosphate-activated protein kinase; STAT1, signal transducer
and activator of transcription 1; NLRP3, NLR family pyrin domain
containing 3; PPAR-γ, peroxisome proliferator-activated receptor γ;
FKBP5, FK506-binding protein 5. |
Overall, these findings indicate that excessive
autophagy activation may drive MS neuropathology, suggesting that
autophagy inhibitors could be a promising strategy for slowing the
progression of MS. In MS research, although autophagy is recognized
as playing a crucial role in disease onset and progression, current
studies still face significant limitations in understanding its
mechanisms.
Collectively, the evidence summarized above suggests
that autophagy in MS does not exert a uniform biological effect but
instead operates within a context-dependent spectrum ranging from
neuroprotection to neurotoxicity. We propose an overarching
conceptual framework in which the functional outcome of autophagy
in MS is determined by four interrelated factors: Disease stage,
cell type, autophagy subtype and inflammatory-metabolic
context.
In the early or compensatory phases of MS, moderate
activation of autophagy, particularly selective forms such as
mitophagy and lipophagy, appears to be predominantly protective. In
this context, autophagy promotes mitochondrial quality control,
limits oxidative and nitrosative stress, facilitates lipid
clearance in microglia and macrophages, and supports
oligodendrocyte survival and myelin regeneration. These effects
collectively contribute to the maintenance of CNS homeostasis and
the attenuation of neuroinflammation.
By contrast, during active or chronic stages of MS,
sustained inflammatory signaling, immune cell hyperactivation and
metabolic dysregulation may shift autophagy toward a maladaptive
state. Excessive or dysregulated autophagy, particularly within T
lymphocytes, microglia and neurons, can amplify immune-mediated
damage, promote demyelination and exacerbate axonal injury. In this
setting, heightened autophagic flux or prolonged mitophagy may lead
to energy depletion, impaired cellular repair and reinforcement of
pro-inflammatory circuits.
Importantly, this framework reconciles seemingly
contradictory findings in the literature by emphasizing that
autophagy intensity alone is insufficient to predict biological
outcome. Rather, the balance between selective and non-selective
autophagy, the cellular compartment involved, and the temporal
dynamics of MS pathology jointly determine whether autophagy
functions as a protective adaptive response or a driver of
neurodegeneration. This integrative model underscores the need for
stage-specific and cell type-targeted modulation of autophagy,
rather than global activation or inhibition, as a rational
therapeutic strategy in MS.
Discussion
MS is a chronic immune-mediated disorder of the CNS
characterized by demyelination, axonal injury and progressive
neurodegeneration. While inflammatory immune responses have long
been regarded as the primary drivers of MS pathology, increasing
evidence suggests that cell-intrinsic stress responses, including
autophagy, play a critical modulatory role in disease progression
rather than acting as mere downstream consequences of inflammation.
Accumulating experimental and clinical studies indicate that
autophagy is dysregulated in MS across multiple cellular
compartments. Altered expression of canonical autophagy markers,
such as Beclin-1, has been observed in animal models and
patient-derived samples, alongside perturbations in key regulatory
pathways, including Akt/mTOR signaling. These findings collectively
suggest that autophagic flux is disturbed in MS. However, most
available evidence remains correlative, relying heavily on static
measurements of autophagy-related markers, which limits the ability
to distinguish between increased autophagy induction and impaired
autophagic degradation. This methodological limitation represents a
major barrier to establishing causality between autophagy
dysfunction and MS pathogenesis. Notably, emerging data
increasingly implicate mitochondrial dysfunction and mitochondrial
autophagy (mitophagy) in MS. Elevated levels of mitophagy-related
markers in cerebrospinal fluid and serum, together with
associations between mitochondrial injury and neuroaxonal
degeneration, support the notion that mitochondrial quality control
is particularly vulnerable in MS. Nevertheless, it remains elusive
whether mitophagy activation represents a compensatory
neuroprotective response or a maladaptive process contributing to
cellular energy failure and neurodegeneration. Importantly, the
relative contribution of selective mitophagy vs. non-selective
autophagy has not been systematically addressed, highlighting a
critical knowledge gap in the field.
Comparative insights from other neurodegenerative
diseases further emphasize the complexity of autophagy regulation
in neurodegeneration. Although MS and ALS differ substantially in
etiology, both diseases exhibit convergent features, including
mitochondrial impairment, oxidative stress and defective autophagic
clearance. These parallels suggest that shared autophagy-related
mechanisms may underlie neurodegenerative vulnerability, while
disease-specific regulatory contexts likely determine whether
autophagy exerts protective or detrimental effects.
Despite substantial progress, several unresolved
questions warrant further investigation. First, the cell-type
specificity of autophagy dysregulation in MS remains poorly
defined. Most studies focus on neurons or oligodendrocytes in
isolation, neglecting the dynamic interactions among immune cells,
glial populations and neurons within the inflammatory
microenvironment. Second, advances in high-resolution imaging,
single-cell transcriptomics and artificial intelligence-based data
integration offer unprecedented opportunities to dissect autophagy
dynamics at spatial and temporal resolutions previously
unattainable. The emerging association between autophagy-related
pathways and MRI outcomes further underscores the potential
clinical relevance of these approaches (113,114). In patients with
relapsing-remitting MS, circulating and cerebrospinal fluid markers
of autophagy and mitophagy were found to be significantly elevated
during active disease phases and showed quantitative correlations
with MRI activity (115).
Specifically, levels of Parkin and BNIP3 were increased by ~1.5-2.5
fold in patients with gadolinium-enhancing lesions compared with
radiologically inactive patients, and these markers correlated
positively with the number of contrast-enhancing lesions
(r≈0.40-0.55, P<0.01). Furthermore, autophagy- and
mitophagy-related markers displayed moderate associations with T2
lesion load (r≈0.35-0.48), suggesting a link between dysregulated
autophagy and MRI-defined inflammatory burden in MS. While MRI
metrics reflecting long-term neurodegeneration, such as brain
atrophy, were not directly assessed in these studies (113-115), their established relationship
with axonal injury markers (e.g., neurofilament light chain)
indicates that autophagy-related alterations may be indirectly
connected to MRI measures of disease progression.
Finally, although autophagy represents an attractive
therapeutic target, its dual role in MS poses significant
translational challenges. Both insufficient and excessive autophagy
may exacerbate pathology, suggesting that context-dependent and
stage-specific modulation, rather than global activation or
inhibition, will be required. Beyond mitophagy, relatively
underexplored selective autophagy pathways, such as ER autophagy
and lipophagy, may also contribute to MS pathology and warrant
systematic investigation. Bridging the gap between mechanistic
insights and clinical application will require integrative
strategies combining molecular biology, systems-level analysis and
longitudinal patient studies.
In conclusion, autophagy emerges as a central but
complex regulator in MS, positioned at the intersection of immune
dysregulation, mitochondrial dysfunction and neurodegeneration.
Clarifying its precise mechanistic roles and therapeutic potential
remains a critical priority for future research.
Availability of data and materials
Not applicable.
Authors' contributions
DW, MW and HF wrote and revised the review. HS, QF,
YZ, GL, YB, YY prepared figures. All authors reviewed the
manuscript and have read and approved the final manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by the Joint Fund of Henan Provincial
Science and Technology R&D Project (grant no. 242103810034),
Henan Academy of Innovations in Medical Science 'Three Hundreds'
Plan (grant no. HNCMS202437), Henan Provincial Young and
Middle-aged Health Science and Technology Innovation Leading Talent
Training Program (grant no. LJRC2024019) and Heluo Young Talents
Support Program (grant no. 2024HLTJ08).
References
|
1
|
Oh J, Vidal-Jordana A and Montalban X:
Multiple sclerosis: Clinical aspects. Curr Opin Neurol. 31:752–759.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
MS International Federation: New
prevalence and incidence data now available in the Atlas of MS. MS
International Federation; 2023
|
|
3
|
Doshi A and Chataway J: Multiple
sclerosis, a treatable disease. Clin Med (Lond). 16(Suppl 6):
s53–s59. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mohseni SO, Au KM, Issa W, Ruan L, Stuve O
and Wang AZ: Multiple sclerosis treatments a review of current
biomedical engineering approaches. Biomaterials. 313:1228072025.
View Article : Google Scholar
|
|
5
|
Kornberg MD and Calabresi PA: Multiple
sclerosis and other acquired demyelinating diseases of the central
nervous system. Cold Spring Harb Perspect Biol. 17:a0413742025.
View Article : Google Scholar
|
|
6
|
Kooistra SM and Schirmer L: Multiple
sclerosis: Glial cell diversity in time and space. Glia.
73:574–590. 2025. View Article : Google Scholar :
|
|
7
|
Marcus R: What is multiple sclerosis?
JAMA. 328:20782022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Katz Sand I: Classification, diagnosis,
and differential diagnosis of multiple sclerosis. Curr Opin Neurol.
28:193–205. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eriksson AM, Emini N, Harbo HF and Berge
T: Is DEXI a multiple sclerosis susceptibility gene? Int J Mol Sci.
26:11752025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Quan M, Zhang H, Deng X, Liu H, Xu Y and
Song X: Neutrophils, NETs and multiple sclerosis: A mini review.
Front Immunol. 16:14878142025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Correale J, Gaitán MI, Ysrraelit MC and
Fiol MP: Progressive multiple sclerosis: From pathogenic mechanisms
to treatment. Brain. 140:527–546. 2017.
|
|
12
|
De Keersmaecker E, Guida S, Denissen S,
Dewolf L, Nagels G, Jansen B, Beckwée D and Swinnen E: Virtual
reality for multiple sclerosis rehabilitation. Cochrane Database
Syst Rev. 1:CD0138342025.PubMed/NCBI
|
|
13
|
Berglund R, Guerreiro-Cacais AO, Adzemovic
MZ, Zeitelhofer M, Lund H, Ewing E, Ruhrmann S, Nutma E, Parsa R,
Thessen-Hedreul M, et al: Microglial autophagy-associated
phagocytosis is essential for recovery from neuroinflammation. Sci
Immunol. 5:eabb50772020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
D'Arcy MS: Cell death: A review of the
major forms of apoptosis, necrosis and autophagy. Cell Biol Int.
43:582–592. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumariya S, Ubba V, Jha RK and Gayen JR:
Autophagy in ovary and polycystic ovary syndrome: Role, dispute and
future perspective. Autophagy. 17:2706–2733. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen T, Tu S, Ding L, Jin M, Chen H and
Zhou H: The role of autophagy in viral infections. J Biomed Sci.
30:52023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Palmer JE, Wilson N, Son SM, Obrocki P,
Wrobel L, Rob M, Takla M, Korolchuk VI and Rubinsztein DC:
Autophagy, aging, and age-related neurodegeneration. Neuron.
113:29–48. 2025. View Article : Google Scholar
|
|
18
|
Misrielal C, Mauthe M, Reggiori F and
Eggen BJL: Autophagy in multiple sclerosis: Two sides of the same
coin. Front Cell Neurosci. 14:6037102020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Al-Kuraishy HM, Jabir MS, Al-Gareeb AI,
Saad HM, Batiha GES and Klionsky DJ: The beneficial role of
autophagy in multiple sclerosis: Yes or no? Autophagy. 20:259–274.
2024. View Article : Google Scholar :
|
|
20
|
Klionsky DJ, Petroni G, Amaravadi RK,
Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cadwell K,
Cecconi F, Choi AMK, et al: Autophagy in major human diseases. EMBO
J. 40:e1088632021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Parzych KR and Klionsky DJ: An overview of
autophagy: Morphology, mechanism, and regulation. Antioxid Redox
Signal. 20:460–473. 2014. View Article : Google Scholar
|
|
22
|
Ito-Silva VI, Smith BJ and
Martins-de-Souza D: The autophagy proteome in the brain. J
Neurochem. 169:e162042025. View Article : Google Scholar
|
|
23
|
Dafsari HS, Martinelli D, Saffari A,
Ebrahimi-Fakhari D, Fanto M, Dionisi-Vici C and Jungbluth H: An
update on autophagy disorders. J Inherit Metab Dis. 48:e127982025.
View Article : Google Scholar
|
|
24
|
Righes G, Semenzato L, Koutsikos K, Zanato
V, Pinton P, Giorgi C and Patergnani S: The role of autophagy in
the pathogenesis and treatment of multiple sclerosis. Autophagy
Rep. 4:25291962025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li W, He P, Huang Y, Li YF, Lu J, Li M,
Kurihara H, Luo Z, Meng T, Onishi M, et al: Selective autophagy of
intracellular organelles: Recent research advances. Theranostics.
11:222–256. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moreno-Blas D, Adell T and
González-Estévez C: Autophagy in tissue repair and regeneration.
Cells. 14:2822025. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang XR, Ye L, An N, Wu CY, Wu HL, Li HY,
Huang YH, Ye QR, Liu MD, Yang LW, et al: Macrophage autophagy
protects against acute kidney injury by inhibiting renal
inflammation through the degradation of TARM1. Autophagy.
21:120–140. 2025. View Article : Google Scholar :
|
|
28
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mizushima N and Levine B: Autophagy in
human diseases. N Engl J Med. 383:1564–1576. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu Y, Wang H and Xu H: Autophagy-lysosome
pathway in insulin & glucagon homeostasis. Front Endocrinol
(Lausanne). 16:15417942025. View Article : Google Scholar
|
|
32
|
Liu S, Yao S, Yang H, Liu S and Wang Y:
Autophagy: Regulator of cell death. Cell Death Dis. 14:6482023.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma L and Cao Z: Periodontopathogen-related
cell autophagy-a double-edged sword. Inflammation. 48:1–14. 2025.
View Article : Google Scholar
|
|
34
|
Zhang J, Zhang J and Yang C: Autophagy in
brain tumors: Molecular mechanisms, challenges, and therapeutic
opportunities. J Transl Med. 23:522025. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gupta S, Cassel SL, Sutterwala FS and
Dagvadorj J: Regulation of the NLRP3 inflammasome by autophagy and
mitophagy. Immunol Rev. 329:e134102025. View Article : Google Scholar :
|
|
36
|
Liao H, Liu S, Ma Q, Huang H, Goel A,
Torabian P, Mohan CD and Duan C: Endoplasmic reticulum stress
induced autophagy in cancer and its potential interactions with
apoptosis and ferroptosis. Biochim Biophys Acta Mol Cell Res.
1872:1198692025. View Article : Google Scholar
|
|
37
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ma Y, Lv W, Guo Y, Yin T, Bai Y, Liu Z,
Chen C, Yang W, Feng J, Qian W, et al: Histone demethylases in
autophagy and inflammation. Cell Commun Signal. 23:242025.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jin S, Li Y, Xia T, Liu Y, Zhang S, Hu H,
Chang Q and Yan M: Mechanisms and therapeutic implications of
selective autophagy in nonalcoholic fatty liver disease. J Adv Res.
67:317–329. 2025. View Article : Google Scholar :
|
|
40
|
Levine B and Kroemer G: Biological
functions of autophagy genes: A disease perspective. Cell.
176:11–42. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Luo PY, Zou JR, Chen T, Zou J, Li W, Chen
Q, Cheng L, Zheng LY and Qian B: Autophagy in erectile dysfunction:
focusing on apoptosis and fibrosis. Asian J Androl. 27:166–176.
2025. View Article : Google Scholar :
|
|
42
|
Yang S, Li M, Lian G, Wu Y, Cui J and Wang
L: ABHD8 antagonizes inflammation by facilitating
chaperone-mediated autophagy-mediated degradation of NLRP3.
Autophagy. 21:338–351. 2025. View Article : Google Scholar :
|
|
43
|
Miller DR and Thorburn A: Autophagy and
organelle homeostasis in cancer. Dev Cell. 56:906–918. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu CZ, Li GZ, Lyu HF, Lu YY, Li Y and
Zhang XN: Modulation of autophagy by melatonin and its receptors:
Implications in brain disorders. Acta Pharmacol Sin. 46:525–538.
2025. View Article : Google Scholar
|
|
45
|
Stanigut AM, Tuta L, Pana C, Alexandrescu
L, Suceveanu A, Blebea NM and Vacaroiu IA: Autophagy and mitophagy
in diabetic kidney disease-a literature review. Int J Mol Sci.
26:8062025. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen Y, Wang Z, Ma Q and Sun C: The role
of autophagy in fibrosis: Mechanisms, progression and therapeutic
potential (Review). Int J Mol Med. 55:612025. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhou R, Zhang Z, Li X, Duan Q, Miao Y,
Zhang T, Wang M, Li J, Zhang W, Wang L, et al: Autophagy in
high-fat diet and streptozotocin-induced metabolic cardiomyopathy:
Mechanisms and therapeutic implications. Int J Mol Sci.
26:16682025. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim KH and Lee MS: Autophagy-a key player
in cellular and body metabolism. Nat Rev Endocrinol. 10:322–337.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nutma E, Marzin MC, Cillessen SA and Amor
S: Autophagy in white matter disorders of the CNS: Mechanisms and
therapeutic opportunities. J Pathol. 253:133–147. 2021. View Article : Google Scholar :
|
|
50
|
Marsh T and Debnath J: Autophagy
suppresses breast cancer metastasis by degrading NBR1. Autophagy.
16:1164–1165. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhu Y, Runwal G, Obrocki P and Rubinsztein
DC: Autophagy in childhood neurological disorders. Dev Med Child
Neurol. 61:639–645. 2019. View Article : Google Scholar
|
|
52
|
Li G, Sherchan P, Tang Z and Tang J:
Autophagy & phagocytosis in neurological disorders and their
possible cross-talk. Curr Neuropharmacol. 19:1912–1924. 2021.
View Article : Google Scholar
|
|
53
|
Zhang L, Dai L and Li D: Mitophagy in
neurological disorders. J Neuroinflammation. 18:2972021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Vargas JNS, Hamasaki M, Kawabata T, Youle
RJ and Yoshimori T: The mechanisms and roles of selective autophagy
in mammals. Nat Rev Mol Cell Biol. 24:167–185. 2023. View Article : Google Scholar
|
|
55
|
de Barcelos IP, Troxell RM and Graves JS:
Mitochondrial dysfunction and multiple sclerosis. Biology (Basel).
8:372019.
|
|
56
|
Ghosh I, Sankhe R, Mudgal J, Arora D and
Nampoothiri M: Spermidine, an autophagy inducer, as a therapeutic
strategy in neurological disorders. Neuropeptides. 83:1020832020.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fang C, Gu L, Smerin D, Mao S and Xiong X:
The interrelation between reactive oxygen species and autophagy in
neurological disorders. Oxid Med Cell Longev. 2017:84951602017.
View Article : Google Scholar
|
|
58
|
Chu CT: Autophagy in neurological
diseases: An update. Neurobiol Dis. 122:1–2. 2019. View Article : Google Scholar
|
|
59
|
Samarasinghe H and Xu J: Hybrids and
hybridization in the cryptococcus neoformans and cryptococcus
gattii species complexes. Infect Genet Evol. 66:245–255. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Evans CS and Holzbaur ELF: Autophagy and
mitophagy in ALS. Neurobiol Dis. 122:35–40. 2019. View Article : Google Scholar
|
|
61
|
Yamamoto H and Matsui T: Molecular
Mechanisms of macroautophagy, microautophagy, and
chaperone-mediated autophagy. J Nippon Med Sch. 91:2–9. 2024.
View Article : Google Scholar
|
|
62
|
Wu L, Lin Y, Song J, Li L, Rao X, Wan W,
Wei G, Hua F and Ying J: TMEM175: A lysosomal ion channel
associated with neurological diseases. Neurobiol Dis.
185:1062442023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hermans G and Van den Berghe G: Clinical
review: Intensive care unit acquired weakness. Crit Care.
19:2742015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Choi AMK, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chen Y, Liu H, Guan Y, Wang Q, Zhou F, Jie
L, Ju J, Pu L, Du H and Wang X: The altered autophagy mediated by
TFEB in animal and cell models of amyotrophic lateral sclerosis. Am
J Transl Res. 7:1574–1587. 2015.PubMed/NCBI
|
|
66
|
Galluzzi L, Bravo-San Pedro JM, Levine B,
Green DR and Kroemer G: Pharmacological modulation of autophagy:
Therapeutic potential and persisting obstacles. Nat Rev Drug
Discov. 16:487–511. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ojha CR, Lapierre J, Rodriguez M, Dever
SM, Zadeh MA, DeMarino C, Pleet ML, Kashanchi F and El-Hage N:
Interplay between autophagy, exosomes and HIV-1 associated
neurological disorders: New insights for diagnosis and therapeutic
applications. Viruses. 9:1762017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Tang D, Kang R and Klionsky DJ:
Autophagy-dependent ferroptosis mediates multiple sclerosis.
Autophagy. 21:257–259. 2025. View Article : Google Scholar :
|
|
69
|
Dolcetti E, Bruno A, Guadalupi L, Rizzo
FR, Musella A, Gentile A, De Vito F, Caioli S, Bullitta S, Fresegna
D, et al: Emerging role of extracellular vesicles in the
pathophysiology of multiple sclerosis. Int J Mol Sci. 21:73362020.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ceccariglia S, Sibilia D, Parolini O,
Michetti F and Di Sante G: Altered expression of autophagy
biomarkers in hippocampal neurons in a multiple sclerosis animal
model. Int J Mol Sci. 24:132252023. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Patergnani S, Castellazzi M, Bonora M,
Marchi S, Casetta I, Pugliatti M, Giorgi C, Granieri E and Pinton
P: Autophagy and mitophagy elements are increased in body fluids of
multiple sclerosis-affected individuals. J Neurol Neurosurg
Psychiatry. 89:439–441. 2018. View Article : Google Scholar
|
|
72
|
Chen S, Zhang X, Song L and Le W:
Autophagy dysregulation in amyotrophic lateral sclerosis. Brain
Pathol. 22:110–116. 2012. View Article : Google Scholar
|
|
73
|
Yin L, Liu J, Dong H, Xu E, Qiao Y, Wang
L, Zhang L, Jia J, Li L and Geng X: Autophagy-related gene16L2, a
potential serum biomarker of multiple sclerosis evaluated by
bead-based proteomic technology. Neurosci Lett. 562:34–38. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wang JQ, Li Q, He JY, Zhou F, Huang ZH,
Wang LB, Zhang Y and Li X: Autophagy in multiple sclerosis:
Phagocytosis and autophagy of oligodendrocyte precursor cells. Mol
Neurobiol. 61:6920–6933. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Igci M, Baysan M, Yigiter R, Ulasli M,
Geyik S, Bayraktar R, Bozgeyik İ, Bozgeyik E, Bayram A and Cakmak
EA: Gene expression profiles of autophagy-related genes in multiple
sclerosis. Gene. 588:38–46. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Cheng Z, Gan W, Xiang Q, Zhao K, Gao H,
Chen Y, Shi P, Zhang A, Li G, Song Y, et al: Impaired degradation
of PLCG1 by chaperone-mediated autophagy promotes cellular
senescence and intervertebral disc degeneration. Autophagy.
21:352–373. 2025. View Article : Google Scholar :
|
|
77
|
Kausar MA, Anwar S, Khan YS, Saleh AA,
Ahmed MAA, Kaur S, Iqbal N, Siddiqui WA and Najm MZ: Autophagy and
cancer: Insights into molecular mechanisms and therapeutic
approaches for chronic myeloid leukemia. Biomolecules. 15:2152025.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bao Y, Ma Y, Huang W, Bai Y, Gao S, Xiu L,
Xie Y, Wan X, Shan S, Chen C and Qu L: Regulation of autophagy and
cellular signaling through non-histone protein methylation. Int J
Biol Macromol. 291:1390572025. View Article : Google Scholar
|
|
79
|
Lei Y and Klionsky DJ: Autophagy as a way
to remove DNA lesions. Autophagy. 21:497–499. 2025. View Article : Google Scholar :
|
|
80
|
Asgari R, Yarani R, Mohammadi P and Emami
Aleagha MS: HIF-1α in the crosstalk between reactive oxygen species
and autophagy process: A review in multiple sclerosis. Cell Mol
Neurobiol. 42:2121–2129. 2022. View Article : Google Scholar
|
|
81
|
Abulaban AA, Al-Kuraishy HM, Al-Gareeb AI,
Elekhnawy E, Alanazi A, Alexiou A, Papadakis M and Batiha GE: Role
of fenofibrate in multiple sclerosis. Eur J Med Res. 29:1132024.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Li Y, Zhou D, Ren Y, Zhang Z, Guo X, Ma M,
Xue Z, Lv J, Liu H, Xi Q, et al: Mir223 restrains autophagy and
promotes CNS inflammation by targeting ATG16L1. Autophagy.
15:478–492. 2019. View Article : Google Scholar :
|
|
83
|
Cheng J, Liao Y, Dong Y, Hu H, Yang N,
Kong X, Li S, Li X, Guo J, Qin L, et al: Microglial autophagy
defect causes parkinson disease-like symptoms by accelerating
inflammasome activation in mice. Autophagy. 16:2193–2205. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Liu X, Tuerxun H and Zhao Y, Li Y, Wen S,
Li X and Zhao Y: Crosstalk between ferroptosis and autophagy:
Broaden horizons of cancer therapy. J Transl Med. 23:182025.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Li W, Wu M, Li Y and Shen J: Reactive
nitrogen species as therapeutic targets for autophagy/mitophagy
modulation to relieve neurodegeneration in multiple sclerosis:
Potential application for drug discovery. Free Radic Biol Med.
208:37–51. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Li S, Wang Y, Liang X and Li Y: Autophagy
intersection: Unraveling the role of the SNARE complex in lysosomal
fusion in Alzheimer's disease. J Alzheimers Dis. 103:979–993. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Misrielal C, Alsema AM, Wijering MHC,
Miedema A, Mauthe M, Reggiori F and Eggen BJL: Transcriptomic
changes in autophagy-related genes are inversely correlated with
inflammation and are associated with multiple sclerosis lesion
pathology. Brain Behav Immun Health. 25:1005102022. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yazdani Y, Zamani ARN, Majidi Z,
Sharafkandi N, Alizadeh S, Mofrad AME, Valizadeh A, Idari G, Radvar
AD, Safaie N and Faridvand Y: Curcumin and targeting of molecular
and metabolic pathways in multiple sclerosis. Cell Biochem Funct.
41:779–787. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Vakrakou AG, Alexaki A, Brinia ME,
Anagnostouli M, Stefanis L and Stathopoulos P: The mTOR signaling
pathway in multiple sclerosis; from animal models to human data.
Int J Mol Sci. 23:80772022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Beckers J, Tharkeshwar AK and Van Damme P:
C9orf72 ALS-FTD: Recent evidence for dysregulation of the
autophagy-lysosome pathway at multiple levels. Autophagy.
17:3306–3322. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ying H and Yue BYJT: Optineurin: The
autophagy connection. Exp Eye Res. 144:73–80. 2016. View Article : Google Scholar
|
|
92
|
Patergnani S, Bonora M, Ingusci S,
Previati M, Marchi S, Zucchini S, Perrone M, Wieckowski MR,
Castellazzi M, Pugliatti M, et al: Antipsychotic drugs counteract
autophagy and mitophagy in multiple sclerosis. Proc Natl Acad Sci
USA. 118:e20200781182021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Haidar M, Loix M, Vanherle S, Dierckx T,
Vangansewinkel T, Gervois P, Wolfs E, Lambrichts I, Bogie JFJ and
Hendriks JJA: Targeting lipophagy in macrophages improves repair in
multiple sclerosis. Autophagy. 18:2697–2710. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ma H, Ou ZL, Alaeiilkhchi N, Cheng YQ,
Chen K, Chen JY, Guo RQ, He MY, Tang SY, Zhang X, et al: MiR-223
enhances lipophagy by suppressing CTSB in microglia following
lysolecithin-induced demyelination in mice. Lipids Health Dis.
23:1942024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Yuan S, Zhang ZW and Li ZL: Cell
death-autophagy loop and glutamate-glutamine cycle in amyotrophic
lateral sclerosis. Front Mol Neurosci. 10:2312017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Di Rita A and Strappazzon F: A protective
variant of the autophagy receptor CALCOCO2/NDP52 in multiple
sclerosis (MS). Autophagy. 17:1565–1567. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Xu TT, Deng YY, Yu XY, Li M and Fu YY:
Natural autophagy modulators in non-communicable diseases: From
autophagy mechanisms to therapeutic potential. Acta Pharmacol Sin.
46:8–32. 2025. View Article : Google Scholar
|
|
98
|
Lorentzen KC, Prescott AR and Ganley IG:
Artificial targeting of autophagy components to mitochondria
reveals both conventional and unconventional mitophagy pathways.
Autophagy. 21:315–337. 2025. View Article : Google Scholar :
|
|
99
|
Lu Z, Yan H and Wang H: Autophagy
modulates male fertility in Arabidopsis. Autophagy. 21:686–688.
2025. View Article : Google Scholar :
|
|
100
|
Liang P and Le W: Role of autophagy in the
pathogenesis of multiple sclerosis. Neurosci Bull. 31:435–444.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Jin M and Klionsky DJ: Nuclear proteasomes
as a backup for autophagy: Interconnected proteostasis pathways.
Autophagy. 21:1–2. 2025. View Article : Google Scholar :
|
|
102
|
Cooper KF: Cargo hitchhiking autophagy-a
hybrid autophagy pathway utilized in yeast. Autophagy. 21:500–512.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Kim J, Lee Y, Jeon T, Ju S, Kim JS, Kim MS
and Kang C: Autophagy-dependent splicing control directs
translation toward inflammation during senescence. Dev Cell.
60:364–378.e7. 2025. View Article : Google Scholar
|
|
104
|
White J, Choi YB, Zhang J, Vo MT, He C,
Shaikh K and Harhaj EW: Phosphorylation of the selective autophagy
receptor TAX1BP1 by TBK1 and IKBKE/IKKi promotes ATG8-family
protein-dependent clearance of MAVS aggregates. Autophagy.
21:160–177. 2025. View Article : Google Scholar :
|
|
105
|
Jia N, Ganesan D, Guan H, Jeong YY, Han S,
Rajapaksha G, Nissenbaum M, Kusnecov AW and Cai Q: Mitochondrial
bioenergetics stimulates autophagy for pathological MAPT/Tau
clearance in tauopathy neurons. Autophagy. 21:54–79. 2025.
View Article : Google Scholar :
|
|
106
|
Tan N, Luo H, Li W, Ling G, Wei Y, Wang W
and Wang Y: The dual function of autophagy in doxorubicin-induced
cardiotoxicity: Mechanism and natural products. Semin Cancer Biol.
109:83–90. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Paunovic V, Petrovic IV, Milenkovic M,
Janjetovic K, Pravica V, Dujmovic I, Milosevic E, Martinovic V,
Mesaros S, Drulovic J and Trajkovic V: Autophagy-independent
increase of ATG5 expression in T cells of multiple sclerosis
patients. J Neuroimmunol. 319:100–105. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Hu H, Li H, Li R, Liu P and Liu H:
Re-establishing immune tolerance in multiple sclerosis: Focusing on
novel mechanisms of mesenchymal stem cell regulation of Th17/Treg
balance. J Transl Med. 22:6632024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Mandrioli J, D'Amico R, Zucchi E, De Biasi
S, Banchelli F, Martinelli I, Simonini C, Lo Tartaro D, Vicini R,
Fini N, et al: Randomized, double-blind, placebo-controlled trial
of rapamycin in amyotrophic lateral sclerosis. Nat Commun.
14:49702023. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Morandi E, Jagessar SA, 't Hart BA and
Gran B: EBV Infection Empowers Human B cells for autoimmunity: Role
of autophagy and relevance to multiple sclerosis. J Immunol.
199:435–448. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Song S, Guo R, Mehmood A, Zhang L, Yin B,
Yuan C, Zhang H, Guo L and Li B: Liraglutide attenuate central
nervous inflammation and demyelination through AMPK and
pyroptosis-related NLRP3 pathway. CNS Neurosci Ther. 28:422–434.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Sun X, Qian M, Li H, Wang L, Zhao Y, Yin
M, Dai L and Bao H: FKBP5 activates mitophagy by ablating PPAR-γ to
shape a benign remyelination environment. Cell Death Dis.
14:7362023. View Article : Google Scholar
|
|
113
|
Genovese AV, Hagemeier J, Bergsland N,
Jakimovski D, Dwyer MG, Ramasamy DP, Lizarraga AA, Hojnacki D, Kolb
C, Weinstock-Guttman B and Zivadinov R: Atrophied Brain T2 lesion
volume at MRI is associated with disability progression and
conversion to secondary progressive multiple sclerosis. Radiology.
293:424–433. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Gill AJ, Schorr EM, Gadani SP and
Calabresi PA: Emerging imaging and liquid biomarkers in multiple
sclerosis. Eur J Immunol. 53:e22502282023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Castellazzi M, Patergnani S, Donadio M,
Giorgi C, Bonora M, Fainardi E, Casetta I, Granieri E, Pugliatti M
and Pinton P: Correlation between auto/mitophagic processes and
magnetic resonance imaging activity in multiple sclerosis patients.
J Neuroinflammation. 16:1312019. View Article : Google Scholar : PubMed/NCBI
|














