Sepsis represents a form of systemic inflammatory
response syndrome triggered by severe infections characterized by
systemic dissemination disease and capable of causing multi-organ
impairment. It accounts for a substantial global burden of
morbidity and mortality (1),
contributing to 20-40% of in-hospital mortalities (2,3).
The Global Burden of Diseases, Injuries and Risk Factors Study 2021
showed that there were ~166 million sepsis cases in the world in
2021, resulting in 21.4 million sepsis related mortalities. This
notable figure accounts for 31.5% of the total mortalities in the
world, indicating that nearly one third of human mortality is due
to sepsis (1,4). The epidemiological situation is
also not optimistic. Although the number of sepsis related
mortalities due to infection decreased before 2019, a sharp
increase was observed between 2020 and 2021. This is mainly caused
by the coronavirus disease 2019 pandemic (5). In addition, sepsis is increasingly
considered as a fatal complication of non-communicable diseases. In
2021, stroke, chronic obstructive pulmonary disease and cirrhosis
led to 5.81 million sepsis related mortalities (4). This demographic change, especially
since 1990, has seen an increase of 230% in adult morbidity.
Therefore, the innate immune response of the host to the inducement
of infection and chronic metabolic dysfunction must also be taken
into account (4). A hallmark of
sepsis is profound immune dysregulation, leading to tissue damage,
organ failure and ultimately mortality, distinguishing it from
uncomplicated infections (6,7).
Excessive cytokine release, commonly termed a 'cytokine storm', can
exacerbate tissue damage and promote systemic inflammatory response
syndrome (SIRS) (8). Notably,
sepsis involves the concurrent occurrence of hyperinflammation and
immune suppression (2), which
can lead to mortality either during the acute inflammatory phase,
frequently associated with multiple organ dysfunction syndrome
(MODS), or through progression to protracted inflammation, immune
paralysis and organ failure (9).
Macrophages are central to immune homeostasis and
inflammation regulation, notably influencing the onset and
progression of sepsis (10).
They exhibit remarkable heterogeneity and plasticity, polarizing
into classically activated (M1, pro-inflammatory) or alternatively
activated (M2, anti-inflammatory) states in response to
environmental cues (11). During
sepsis, macrophage metabolism, primarily involving glucose, lipid
and amino acid pathways, deviates markedly from its physiological
state. This metabolic reprogramming critically regulates immune
function, supplying cells with the nutrients and energy required to
adapt to environmental stresses and immune challenges (12).
The present review aims to reframe the understanding
of macrophage metabolism in sepsis. We hypothesize that metabolic
reprogramming is not merely a passive consequence of activation but
an active driver of macrophage function. While previous studies
have extensively cataloged individual metabolic pathways, such as
the Warburg effect or the kynurenine pathway in isolation, the
present review distinguishes them by proposing an integrated model
(13-15). How glucose, lipid, and amino acid
fluxes are hierarchically synchronized by specific metabolic
checkpoints is delineated. Furthermore, the present review uniquely
bridges basic signaling hubs with clinical biomarkers, providing a
stage-specific therapeutic roadmap that addresses the dynamic
nature of sepsis. It should be noted that although the M1/M2
classification provides a basic conceptual baseline, it is
increasingly recognized that macrophage activation in sepsis is a
multidimensional and dynamic process, rather than two discrete
states. The present review, while using these terms for
clarification, also acknowledges that the sepsis microenvironment
determines a complex activation environment. Targeting
immunometabolic crosstalk in macrophages is key to developing more
effective and personalized sepsis therapies.
In order to ensure a comprehensive and up-to-date
analysis of macrophage metabolic reprogramming in sepsis, a
systematic survey of relevant literature was performed. Articles
published from its establishment to February 2026 were searched on
websites such as PubMed (https://pubmed.ncbi.nlm.nih.gov/) and Web of Science
(https://www.webofscience.com/), with a
particular focus on high-impact research from the past 5 to 10
years. The search strategy employed Boolean logic with specific
term combinations, such as ('sepsis' OR 'septic shock') AND
('macrophage') AND ('glycolysis' OR 'fatty acid oxidation' OR
'immunometabolism' OR 'metabolic reprogramming').
The selection of papers was based on the following
inclusion criteria: i) Peer-reviewed English original research
articles or comprehensive reviews; ii) studies of the molecular
mechanism of metabolic flux of macrophages in sepsis; or iii)
clinical trials or observational studies evaluating metabolic
biomarkers and therapeutic interventions for sepsis. Exclusion
criteria included: i) Conference abstracts or preprints that are
not peer-reviewed; ii) research focusing only on non-infectious
inflammatory conditions; or iii) reports with insufficient
experimental details or unreliable methods.
In the early stage of sepsis, which is the
hyper-acute phase, the encounter between pathogen associated
molecular patterns (PAMPs) and toll-like receptors (TLRs), such as
TLR4, triggers a robust pro-inflammatory cascade. This 'cytokine
storm' is characterized by the dominance of cells exhibiting
M1-like features, driven by the nuclear factor κ-B (NF-κB) pathway
and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB or
Akt)/mammalian target of rapamycin (mTOR) axis. While this phase is
essential for initial pathogen sequestration, persistent
hyper-inflammation often precipitates SIRS and acute organ
dysfunction (3,8,10,16).
As sepsis progresses, the host environment will
transition to the stage of immune suppression or regression. Under
the influence of anti-inflammatory mediators such as interleukin-10
(IL-10) and IL-4, macrophages undergo a compensatory shift toward
M2-like states via the Janus Kinase/signal transducer and activator
of transcription 6 (STAT6) and peroxisome proliferator activated
receptor γ (PPARγ) pathways. While this transition is nominally
reparative, the sustained presence of these cells often leads to
immune paralysis, characterized by defective antigen presentation
and impaired phagocytic capacity, thereby increasing susceptibility
to secondary opportunistic infections (10,17,18).
However, the plasticity of macrophage function
during sepsis represented by M1/M2 is overly simplified. A previous
study has found that the existence of an atypical pro-inflammatory
M2 (M2INF) phenotype, indicating that glycolysis
presents an M2INF pro-inflammatory phenotype, while
inhibition of glycolysis weakens the M2INF phenotype
(19). The coexistence of
inflammatory and inhibitory markers within the same cell population
is also a characteristic of clinical progression in sepsis. For
example, the mTOR-hypoxia inducible factor-1α (HIF-1α) axis is not
only a hub for M1 phenotype transfer, but also a core driver of
enhanced glycolysis (Warburg effect). At the same time, the
adenosine 5'-monophosphate (AMP)-activated protein kinase (AMPK)
pathway can regulate fatty acid oxidation (FAO) by inhibiting mTOR
activity, thereby supporting the transition to the M2 phenotype.
The conversion of pathways affects the phenotypic transfer of
macrophages in sepsis, directly guiding their immune function and
metabolic remodeling and determining the host's defense strategies
against infection, clinical manifestations and prognosis (13,20,21).
In recent years, the advent of single-cell RNA
sequencing (scRNA-seq) has fundamentally reshaped the understanding
of macrophage individual development and activation, with
macrophage activation in sepsis now viewed as a multidimensional
continuum rather than a discrete state. A prominent example is the
discovery of lipid associated macrophages (LAMs), characterized by
triggering receptor expressed on myeloid cells 2 (TREM2)-dependent
transcriptional features. In sepsis, these LAMs coordinate lipid
uptake and energy homeostasis. However, their excessive activation
can damage FAO through the SHP1/Bruton's tyrosine kinase (BTK)
axis, exacerbating the transition to immune paralysis (22-25). Network analysis also suggests
that individual macrophages can simultaneously express markers of
pro-inflammatory and anti-inflammatory programs, a phenomenon known
as the 'mixed' or 'intermediate' phenotype (26,27). With the development of scRNA-seq,
more subtypes of macrophages will be discovered in sepsis, allowing
for targeted regulation of specific macrophage populations based on
the real-time immune status of the patient (28-30).
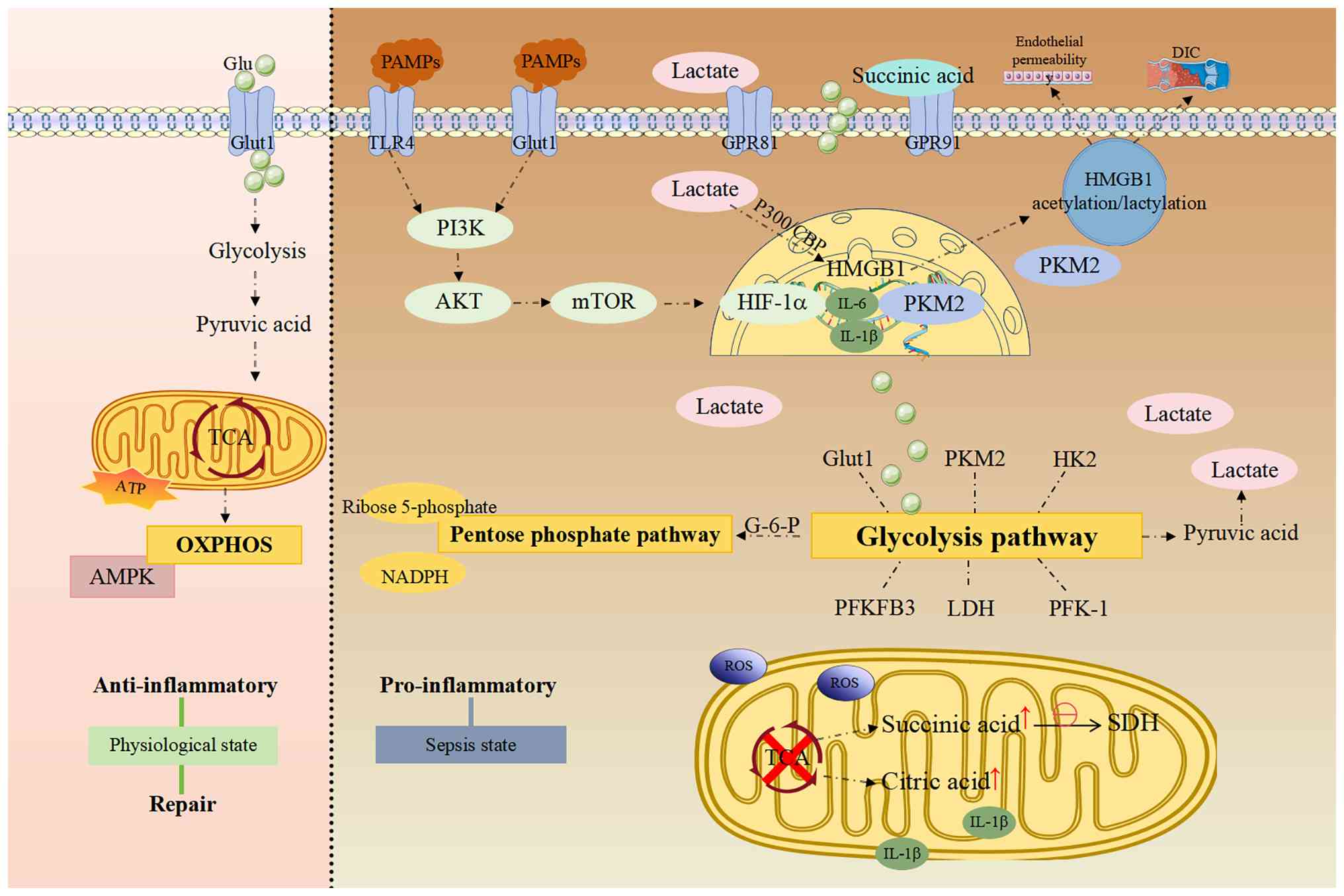
Glucose metabolism is the foremost and most
extensively studied metabolic pathway disrupted in septic
macrophages. Under physiological conditions, macrophages utilize
oxidative phosphorylation (OXPHOS), supported by a complete
tricarboxylic acid (TCA) cycle and electron transport chain (ETC),
to provide stable energy for M2 polarization and tissue repair
(14,31). During early sepsis, PAMPs
activate macrophages, inducing the Warburg effect where
inflammation drives a shift from mitochondrial respiration to
aerobic glycolysis (32-34). In the present review, alterations
in glycolysis, the TCA cycle and the pentose phosphate pathway
(PPP) are integrated within the context of sepsis progression
(Fig. 1).
In early sepsis, M1 macrophages primarily rely on
glycolysis, exhibiting enhanced glycolytic flux and diminished
oxygen consumption (31,35), which disrupts the TCA cycle. This
reprogramming enables rapid generation of energy and metabolic
intermediates necessary for macrophage activity and function during
infection (36). Upregulation of
glycolytic enzymes, including pyruvate kinase M2 (PKM2), glucose
transporter 1 (GLUT1), hexokinase (HK), phosphofructokinase-1 and
6-phosphofructose-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3),
along with increased lactate production, further amplifies
glycolytic activity and promotes M1 polarization (37,38).
Mechanistically, the metabolic-transcriptional
interface in septic macrophages is orchestrated by the functional
transformation of PKM2 and its synergy with HIF-1a (39,40). Under inflammatory cues, PKM2
undergoes post-translational modifications, specifically
phosphorylation and acetylation, triggering its transition to a
dimeric form that translocates to the nucleus (39,41). Elevated serum PKM2 levels have
been shown to be strongly associated with disease severity and
organ damage (42). Once in the
nucleus, PKM2 acts as a co-activator for HIF-1a to directly promote
the transcription of pro-inflammatory genes and essential
glycolytic enzymes (16,43). Notably, this axis is further
stabilized by the accumulation of succinate resulting from TCA
cycle disruption at the succinate dehydrogenase (SDH) site
(15,44). Intracellular succinate prevents
HIF-1α degradation by inhibiting prolyl hydroxylases, thereby
driving IL-1β production (45,46). Furthermore, extracellular
succinate functions as a signaling molecule via G protein-coupled
receptor 91 (GPR91) to sustain the pro-inflammatory M1 phenotype
(47). Together, the
PKM2-HIF-1α-succinate axis represents a metabolic checkpoint that
bridges mitochondrial dysfunction with persistent glycolytic
reprogramming.
HIF-1α upregulates glycolytic enzyme genes [such as
HK2, PFKFB3 and lactate dehydrogenase (LDH)] and promotes GLUT1
synthesis. Moreover, it inhibits pyruvate entry into mitochondria
and enhances its conversion to lactate by increasing LDH
expression, thereby fueling glycolysis (48-50). Glycolysis, in turn, enhances
HIF-1α translation and stability while promoting GLUT1 expression,
creating a self-reinforcing loop that sustains glycolytic
reprogramming (16,43). In the cytoplasm, dimeric PKM2
interacts with molecules such as high mobility group box-1 (HMGB1),
enhancing transcription of glycolytic enzymes (GLUT1, LDH and HK)
and skewing immune cells toward glycolysis, thereby amplifying the
inflammatory response of M1 macrophages (40,41,51). These changes promote the release
of late-phase pro-inflammatory mediators such as HMGB1 from
macrophages (52). Extracellular
lactate is taken up primarily via monocarboxylate transporters,
facilitating HMGB1 lactylation through a p300 and CREB-binding
protein-dependent mechanism.
Lactate can also be recruited to the nucleus via
GPR81 to stimulate HMGB1 acetylation. Lactylated/acetylated HMGB1
is released via exosomes, increasing endothelial permeability and
accelerating the progression of polymicrobial sepsis (53). HMGB1, a nuclear protein released
extracellularly, exacerbates immune responses through TLR
stimulation, direct cytotoxicity and platelet activation,
contributing to disseminated intravascular coagulation and
functioning as a damage-associated molecular pattern (54). Early clinical studies
demonstrated elevated HMGB1 levels in sepsis, positively
associating with disease severity and mortality (55,56). A previous study of 218 critically
ill patients (145 with sepsis, 73 without) also reported a positive
correlation between blood HMGB1 and lactate levels (r=0.144;
P=0.035), supporting the interplay between HMGB1 and lactate during
sepsis (57). The clinical
correlation observed between PKM2, lactate and HMGB1 levels forms a
metabolic inflammatory feedback loop in the pathogenesis of sepsis.
The elevation of these markers is not only a result of tissue
damage, but also an indicator of the enhanced glycolytic state
before organ dysfunction. This indicates that the therapeutic
window for glycolytic inhibitors or PKM2 modulators must be
strictly aligned with the initial surge of these circulating
biomarkers, as their peak likely represents a point of no return
for HMGB1-driven vascular failure.
Beyond signaling pathways, macrophages in
sepsis-induced acute lung injury (ALI) often overexpress the
Sprouty RTK signaling antagonist 4 (Spry4) gene. Notably, Spry4
deficiency has been shown to alleviate lung injury by activating
the calcium/calmodulin dependent protein kinase kinase 2 pathway,
contrasting with reports that Spry4 can aggravate sepsis
progression (58). Another key
metabolic regulator in sepsis is the PI3K/Akt/mTOR pathway, which
also contributes to ALI pathogenesis (59). PI3K activation leads to
downstream Akt activation, enhancing GLUT1 expression and glucose
uptake. Activated Akt further increases HIF-1α expression via mTOR,
explaining the preference for glycolysis even under aerobic
conditions (60). In addition,
the PI3K/Akt/mTOR axis overactivation not only prioritizes
glycolytic flux, but also governs the intracellular availability of
essential amino acids such as glutamine (Gln) and arginine,
effectively linking glucose consumption to the protein synthesis
machinery required for cytokine production. Taken together, these
findings underscore the glycolysis-driven metabolic reprogramming
in macrophages mediated by AMPK inhibition, PI3K/Akt/mTOR
activation and PKM2 functional transformation, which promotes M1
polarization and inflammatory mediator release, ultimately driving
immunometabolic imbalance and multi-organ damage in sepsis.
Under physiological conditions, the TCA cycle
maintains a balance between isocitrate dehydrogenase
(IDH)-catalyzed conversion of isocitrate to α-ketoglutarate (α-KG)
and SDH-catalyzed conversion of succinate to fumarate (14,31). In sepsis, the TCA cycle undergoes
notable remodeling, characterized by two metabolic changes at the
IDH and SDH sites (15,61).
In M1 macrophages, the activity of key TCA enzymes
such as IDH and SDH is reduced, disrupting the mitochondrial
oxidative respiration chain. This leads to partial TCA cycle
blockade, enhanced glycolysis and accumulation of intermediates
such as succinate and citrate (61). Cytosolic ATP-citrate lyase
cleaves accumulated citrate to provide acetyl-CoA for fatty acid
synthesis, supporting production of inflammatory mediators such as
prostaglandins and nitric oxide (NO) (62,63). Furthermore, excess
citrate-derived acetyl-CoA affects epigenetic regulation by serving
as a core donor for histone acetylation, which upregulates genes
involved in inflammatory responses, such as IL-6 and IL-1β
(64,65). Epigenetically, mitochondrial
sirtuin 3 deficiency leads to hyperacetylation of TCA enzymes,
increasing lactate and nicotinamide adenine dinucleotide production
and contributing to sepsis-induced myocardial dysfunction (66).
Enhanced glycolysis and TCA disruption jointly
increase lactate production. Lactate can serve as a precursor for
histone lactylation [such as histone H3 lysine 18 lactylation
(H3K18la], upregulating M2-related genes including arginase 1
(Arg1). This mechanism may serve a role in late-stage sepsis
immunosuppression (67). M2
macrophages maintain an intact TCA cycle. Clinical data show
elevated H3K18la levels in peripheral blood mononuclear cells from
critically ill patients with sepsis, positively associating with
Arg1 mRNA expression and disease severity (68). This suggests that early
inflammatory responses driven by TCA interruption and its
metabolites may lay the groundwork for later epigenetic
reprogramming and phenotypic shifts. Other studies demonstrated
that alveolar macrophages (AMs) in early ALI exhibit weak
glycolytic capacity, predominantly displaying an M2 phenotype that
relies on OXPHOS for cytokine production during lipopolysaccharide
(LPS) activation (69). However,
under extreme hypoxia, such as when ALI progresses to acute
respiratory distress syndrome, HIF-1α activation promotes a shift
in AMs to an M1 phenotype, transitioning from OXPHOS to glycolysis
(70,71).
The PPP branches from glycolysis (at
glucose-6-phosphate and fructose-6-phosphate) and serves two key
roles in septic macrophages (13,72): Synthesis of ribose-5-phosphate
and generation of nicotinamide adenine dinucleotide phosphate
hydrogen (NADPH).
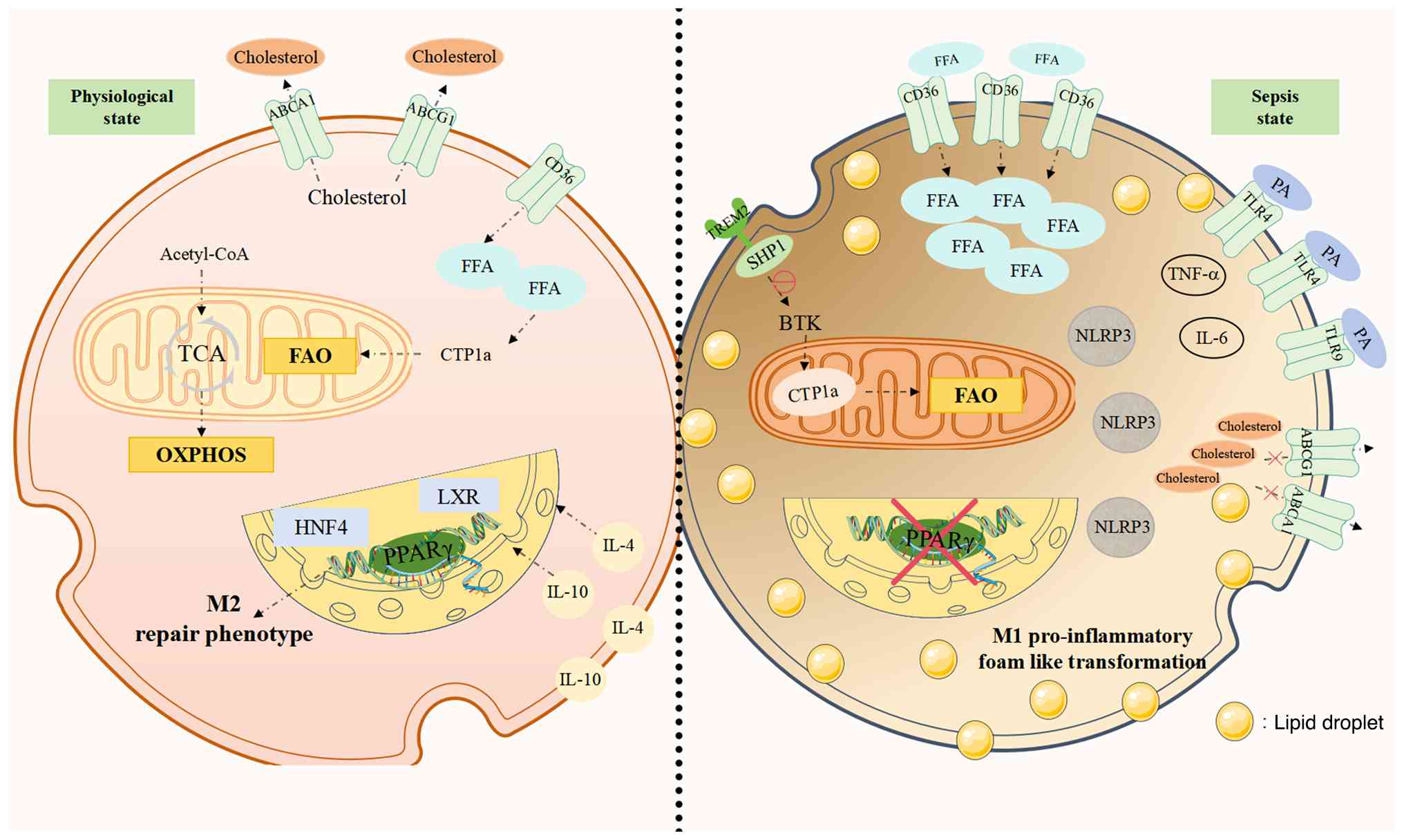
Under physiological conditions, macrophages maintain
lipid homeostasis through balanced FAO for energy and cholesterol
efflux for membrane integrity (14,63,74,75). In sepsis, this balance is
disrupted: Increased lipid uptake, impaired FAO and disturbed
cholesterol metabolism collectively promote inflammation,
lipotoxicity and organ damage (Fig.
2).
During sepsis, macrophage lipid uptake capacity is
enhanced, increasing fatty acid metabolic activity (76). This uptake depends largely on the
expression of fatty acid transporters such as cluster of
differentiation 36 and carnitine palmitoyltransferase 1a (CPT1a).
Upregulation of these transporters facilitates efficient fatty acid
acquisition (63,77). Additionally, mitochondrial STAT3
further promotes FAO by stabilizing CPT1a via ubiquitin specific
peptidase 50-mediated deubiquitination (78), enhancing fatty acid entry into
mitochondria. Lipid metabolites can also act as immune signaling
molecules; for example, saturated fatty acids such as palmitic acid
promote pro-inflammatory responses in macrophages via TLR
signaling, increasing tumor necrosis factor-α (TNF-α) and IL-6
production (79,80). These pro-inflammatory cytokines,
in turn, further enhance fatty acid uptake and activate metabolic
regulators such as AMPK and mTOR (81,82). Although AMPK activation can
promote autophagy and FAO to counteract inflammation (77,83), its activity is typically
suppressed in sepsis.
In late-stage sepsis, macrophages rely mainly on
mitochondrial OXPHOS and FAO for tissue repair (15,84). However, reliance on FAO often
leads to imbalances. First, anti-inflammatory cytokines such as
IL-4 and IL-10 may contribute to immune suppression (10). Second, rapid glycogen depletion
causes transient hyperglycemia and increased triglyceride
lipolysis, elevating free fatty acid (FFA) and glycerol levels and
manifesting as impaired FAO in sepsis. Dysfunction of PPARα and the
glucocorticoid receptor exacerbates metabolic imbalance, leading to
ketone body and glucose deficiency and promoting inflammation
(85-87).
Excessive lipid accumulation disrupts intracellular
homeostasis and aggravates inflammation. However, not all lipid
metabolites are detrimental. For instance, ketone bodies directly
protect cells and negatively regulate inflammation by activating
the β-hydroxybutyrate receptor GPR109A and inhibiting the NOD-like
receptor domain-containing protein 3 inflammasome (88,89). Prostaglandin E2 (PGE2) inhibits
TNF-α and IL-6 production, while lipoxin A4, derived from
arachidonic acid, inhibits PGE2 signaling and promotes M2
macrophage polarization (90).
Moreover, other fatty acid metabolites also suppress inflammatory
responses. Unsaturated fatty acids promote tissue repair by
activating anti-inflammatory pathways such as PPARγ (91). Omega-3 fatty acids attenuate
sepsis-induced inflammation and oxidative stress by increasing
notch receptor 3 expression via downregulation of micro-RNA
(miR)-1-3p and blocking the Smad pathway, thereby mitigating
intestinal epithelial injury (92). Nevertheless, persistent lipid
metabolic dysregulation can sustain M1 polarization, perpetuating
chronic inflammation and tissue damage (3,63,93,94).
Clinically, lipid metabolism is linked to sepsis
prognosis. Elevated serum FFA levels are positively associated with
disease severity and can exacerbate sepsis by activating specific
inflammatory pathways, further contributing to MODS (95). Alterations in essential fatty
acid metabolism may disrupt the balance between pro- and
anti-inflammatory mediators (such as eicosanoids and cytokines),
leading to immune dysregulation in sepsis (96). Additionally, elevated heart-type
fatty acid-binding protein serves as a biomarker for early
diagnosis and prognosis of sepsis-induced cardiomyopathy (97). These findings suggest that
targeting lipid metabolic pathways may improve macrophage function
and attenuate sepsis-induced inflammation. Therapeutically, mTOR
inhibitors such as rapamycin promote FAO and autophagy to limit
tissue damage and prevent excessive immune responses (98). Preclinical studies show that
PPARα agonists improve survival in septic mice by restoring FAO and
reducing lipotoxicity (87,99). Although this strategy awaits
validation in human trials, it highlights the potential of PPAR
activation in alleviating sepsis-related metabolic disorders
(100).
Recent single-cell landscape studies have identified
a specific subpopulation called LAMs, which differs from the
classical M1/M2 classification system (22-24). The characteristic of LAM is the
TREM2 dependent transcriptional program, which serves as a
metabolic sensor to coordinate lipid uptake, lysosomal function and
energy homeostasis. In sepsis, TREM2 serves a double-edged sword
role in this subgroup. On the one hand, TREM2 signaling in LAMs
helps prevent systemic lipotoxicity, promote the clearance of
apoptotic cells and improve sepsis outcomes in liver and heart
injury models (23,24). On the other hand, overactivation
of TREM2+ LAMs can lead to systemic hypercholesterolemia
and increase susceptibility to sepsis by over-activating the
SHP1/BTK axis, which in turn impairs FAO (22). Studies have found that knocking
out TREM2 in macrophages reduces inflammation, organ damage,
triglyceride accumulation and enhances FAO, improving survival in
septic mouse models (25). This
indicates that LAMs represent the metabolic adaptation of
macrophages to the lipid-rich sepsis microenvironment, and its
functional direction depends on the severity and stage of
infection.
Cholesterol, a sterol lipid, is a precursor for
steroid hormones, bile acids and oxysterols (101,102), and also regulates various
cellular functions while forming structural components of cell
membranes along with cholesteryl esters (103). Both cholesterol and its
lipoprotein carriers possess immunomodulatory properties, binding
and neutralizing endotoxins to prevent PAMPs activation of TLRs
(104).
During sepsis, serum levels of total cholesterol,
high-density lipoprotein cholesterol (HDL-C) and low-density
lipoprotein cholesterol (LDL-C) are markedly reduced (105,106), associating with increased
mortality risk. Early clinical data indicate that LDL-C levels are
lowest at diagnosis, while HDL-C levels typically reach their nadir
around the third day in hospital (107). However, previous studies
suggest that while low levels of HDL-C may be a key contributor to
mortality risk, reduced LDL-C concentrations may not be causative
(106,108). A cohort study investigating the
non-HDL-C/HDL-C ratio in patients with sepsis revealed a U-shaped
relationship with 28-day mortality, both excessively high and low
ratios were associated with increased mortality (109), highlighting the importance of
monitoring this ratio. The U-shaped relationship between the
comprehensive cholesterol ratio and mortality rate indicates that
lipid homeostasis can serve as a buffering system. In addition to
serving as energy precursors, lipoproteins also act as innate
scavengers of pathogenic endotoxins. The precipitous drop in HDL-C
observed in non-survivors reflects a critical failure in this
protective capacity, positioning cholesterol profiles as functional
indicators of the host's residual innate defense reserves rather
than a simple metabolite. Animal studies demonstrate
hypocholesterolemia during sepsis, with serum cholesterol levels
inversely associated with inflammatory markers (110-112). Although well-documented, the
mechanisms underlying hypocholesterolemia in sepsis remain
unclear.
Cholesterol levels intricately influence macrophage
signaling, particularly through lipid raft domains that modulate
TLR4 and TLR9 signaling (113).
Impaired cholesterol transport (such as due to ATP-binding cassette
protein A1/ATP-binding cassette protein G1 defects) hinders efflux,
leading to cellular cholesterol accumulation and lipid raft
enlargement. This heightens macrophage sensitivity to TLR4
signaling and LPS, exacerbating inflammatory responses (113-117). Cholesterol metabolites also
activate nuclear receptors such as hepatocyte nuclear factor 4a and
liver X receptors. In infected macrophages, altered lipid
metabolism can activate these receptors, modulating inflammatory
mediator expression (118).
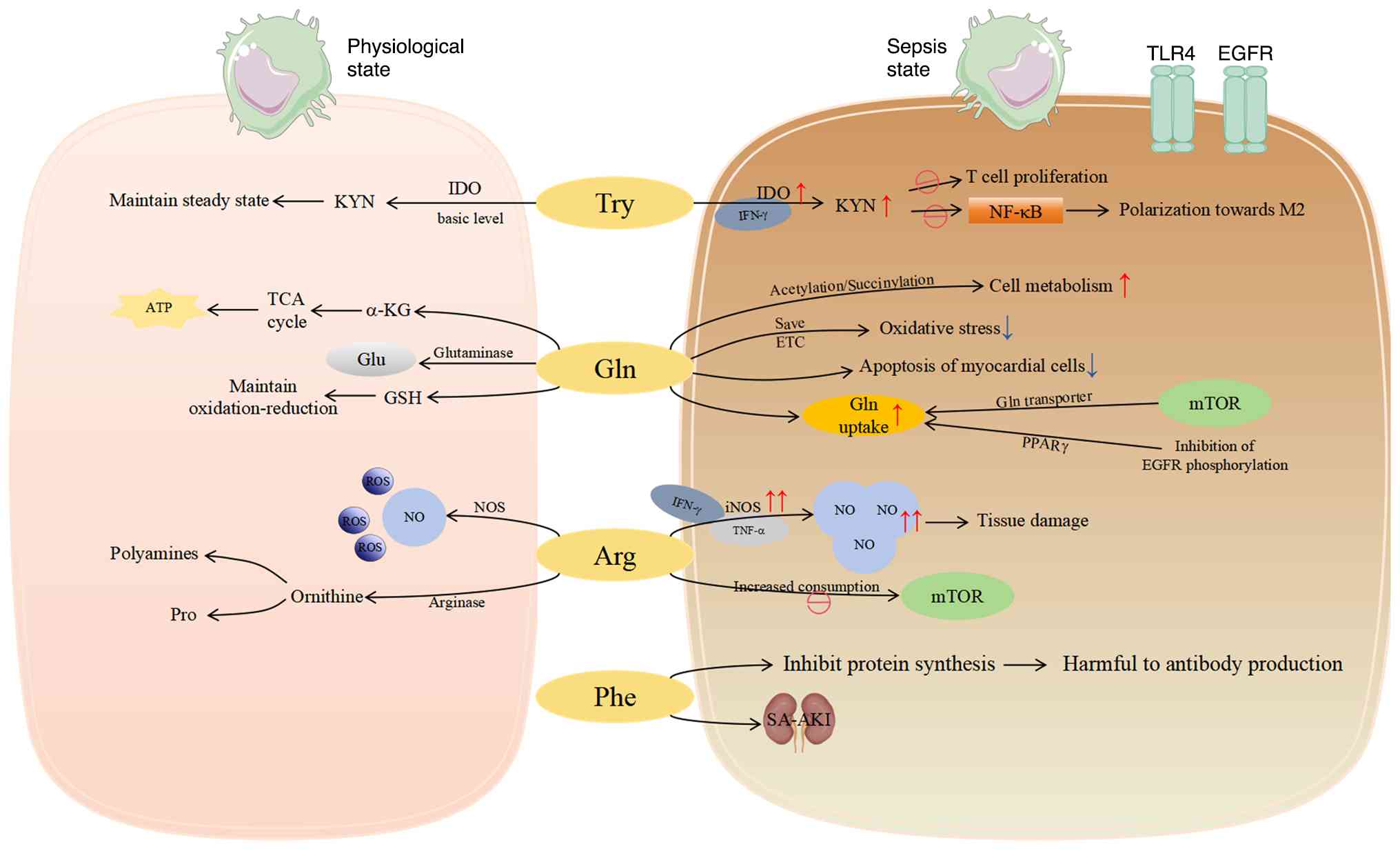
Amino acids serve not only as fundamental building
blocks of cellular metabolism but also as key signaling molecules
and regulators of immune cell functions, including those of
macrophages (119). Sepsis
primarily reprograms three key amino acid pathways in macrophages,
aromatic amino acid, Gln and arginine metabolism, each prominent at
different disease stages (inflammatory response and immune
suppression) (Fig. 3).
Aromatic amino acids tyrosine, tryptophan and
phenylalanine, are among the most markedly altered metabolites in
sepsis. A previous study noted marked increases in intermediates
such as phenylpyruvate and L-phenylalanine, highlighting prominent
dysregulation of aromatic amino acid metabolism in sepsis (120).
Tryptophan metabolism is particularly relevant to
immune escape mechanisms in sepsis (121). Inflammatory cytokines such as
interferon-γ induce indoleamine 2,3-dioxygenase gene transcription
in macrophages, enhancing tryptophan degradation via the kynurenine
pathway. Kynurenine and its derivatives suppress T-cell
proliferation and modulate macrophage activity, promoting
immunosuppression (121-123).
Notably, kynurenic acid, a kynurenine pathway metabolite,
facilitates the transition from M1 to M2 macrophages by inhibiting
NF-κB signaling and alleviating septic colon injury (124).
The phenylalanine/tyrosine ratio also reflects
immune activation status, and studies have identified phenylalanine
and histidine metabolism as among the most markedly altered in
sepsis (120,125). Excessive phenylalanine can
inhibit protein synthesis and exert toxic effects on antibodies
(120). A study of 63 patients
with sepsis also found strong associations between phenylalanine
metabolism and sepsis-associated acute kidney injury.
In macrophages, Gln is converted to glutamate by
glutaminase and further metabolized to a-KG, which enters the TCA
cycle for energy production (75). Gln metabolism-driven glutathione
(GSH) synthesis provides energy and intermediates while helping
maintain redox balance (13,14). Studies show a close association
between Gln and M2 polarization; α-KG restores pyruvate
dehydrogenase activity and supports M2 differentiation (13,14). Additionally, inflammatory
mediator-stimulated metabolic reprogramming in macrophages depends
on Gln. Macrophages can activate mTOR and other pathways to enhance
Gln transporter expression, forming a positive feedback loop that
amplifies immune responses (126,127). In sepsis-induced muscle
atrophy, Gln therapy activates the mTOR pathway to alleviate muscle
degradation (128). These
cellular mechanisms indicate the potential of Gln in restoring
metabolic balance and reducing organ damage in sepsis.
Substantial evidence suggests that Gln regulates
cellular metabolism and function through post-translational
modifications such as acetylation and succinylation, particularly
in burn-induced sepsis (129,130). Gln also reduces oxidative
stress by rescuing dysfunctional mitochondrial ETC complexes,
protecting hepatocytes from inflammation-induced injury, a
protective mechanism in burn sepsis (131). Moreover, Gln supplementation
reduces sepsis-induced cardiomyocyte apoptosis in rat models
(132,133). However, translating these
beneficial mechanisms to clinical practice has yielded complex
results. Some previous studies question the benefits of Gln in
critically ill intensive care unit (ICU) patients and even
associate its supplementation with development of chronic critical
illness (134,135).
In the ICU, the benefit of Gln supplementation is
highly dependent on timing, dose and host baseline status.
Regarding timing, a study of 1,223 critically ill patients found
that patients receiving Gln had a trend toward higher 28-day
mortality compared with non-recipients (32.4 vs. 27.2%; adjusted
odds ratio 1.28; 95% CI 1.00-1.64; P=0.05). In-hospital and 6-month
mortality were also significantly higher in the Gln group (both
P=0.02) (136). Dose-dependence
was also significant, as evidenced by the significantly higher
frequency of high urea levels in the glutamine group (13.4 vs.
4.0%; P<0.001). A large multicenter randomized trial indicated
that high-dose parenteral Gln (>0.5 g/kg/day) should be avoided
early in critical illness. This caution is mainly due to the
metabolic substrate overload that occurs during the hyperacute
phase of sepsis. In patients with multiple organ dysfunction,
especially kidney and liver damage, the body's ability to handle
nitrogen load is markedly reduced. High doses of exogenous Gln can
lead to excessive accumulation of metabolic byproducts such as
ammonia and urea, which may exacerbate uremic toxicity or hepatic
encephalopathy.
In addition, in the early stages of critical
illness, the host may trigger large-scale catabolic reactions to
mobilize endogenous amino acids. At this point, adding high doses
of exogenous Gln may interfere with beneficial autophagy processes
or inhibit the host's adaptive metabolic stress response, leading
to increased mortality. The different results of large-scale Gln
trials combine a fundamental clinical paradox, namely that the
therapeutic benefits of amino acid supplementation depend on the
metabolic stage of the patient rather than absolute dosage. The
increase in mortality rate during the hyperacute phase indicates
that exogenous nitrogen load may cause severe liver damage, while
the same intervention during the recovery phase helps with redox
balance and tissue repair. Therefore, a more cautious dosage of
0.3-0.5 g/kg/day should only be considered after the acute phase
has subsided and organ function has stabilized (137). Regarding host status, patients
with high metabolic demands (such as burns or trauma) may benefit
from supplementation due to increased Gln consumption (elevating
GSH and reducing oxidative stress). It may also benefit
experimental neonatal endotoxemia and very low birth weight preterm
infants. However, intracellular heat shock protein 70 deficiency
due to Gln deprivation may increase post-sepsis mortality (138).
Ultimately, the clinical benefit of Gln is not
absolute. Future research should move beyond the simplistic
question of whether Gln is beneficial or harmful. Efforts should
clarify these regulatory factors to identify sepsis subgroups most
likely to benefit from Gln supplementation and develop
individualized treatment strategies targeting related metabolic
pathways, thereby improving sepsis outcomes.
In macrophages, arginine is metabolized via two
primary pathways: The nitric oxide synthase (NOS) pathway producing
NO, and the arginase pathway producing ornithine. Regarding
phenotype, NO can prevent M1 macrophages from repolarizing to M2,
while inducible NOS (iNOS) inhibition facilitates M1-to-M2
transition (139). By contrast,
M2 macrophages metabolize arginine via Arg1, producing ornithine
and urea.
Arginase activity is also altered during sepsis. Arg
exists in two isoforms: Arg1 (cytosolic, liver-specific) and Arg2
(mitochondrial). Arg1 participates in the urea cycle, typically
utilizing extracellular arginine to regulate its availability to
neighboring cells (149). In M2
macrophages, Arg1 expression is promoted via the STAT6 pathway
(150) and can be
synergistically induced by IL-10 and LPS (151). In M2 macrophages, Arg1
expression is promoted via the STAT6 pathway and can be
synergistically induced by IL-10 and LPS (152). Arg1 hydrolyzes arginine to
ornithine and urea (153,154). Ornithine serves as a precursor
for polyamine and proline synthesis; polyamines (such as putrescine
and spermidine) participate in cell proliferation, while proline is
crucial for collagen synthesis (155). Similar to Arg1, Arg2 expression
is upregulated in human and mouse macrophages after LPS treatment
(151). Arg2 downregulates
succinate levels by promoting SDH (complex II) activity, further
downregulating HIF-1α and IL-1 expression, promoting the M2
anti-inflammatory phenotype and enhancing mitochondrial respiration
(156,157). However, in chronic inflammation
models (such as atherosclerosis), Arg2 can also produce
proinflammatory effects by increasing mitochondrial ROS. This
suggests that it may have a similar dual role in sepsis, but
further confirmation is needed (158). In summary, the balance between
the NO and arginase pathways is disrupted; overactivation of iNOS
consumes large amounts of arginine for NO production, affecting the
arginase pathway and impairing normal macrophage functions such as
phagocytosis and antigen presentation during the sepsis (159).
Current sepsis management remains largely
supportive, focusing on three main strategies: i) Early infection
control; ii) fluid resuscitation and vasopressors; and iii)
mechanical ventilation (160,161). Antimicrobial therapy is
essential early in sepsis, typically involving broad-spectrum
antibiotics to control infection and prevent progression (162). However, this approach carries a
growing risk of antimicrobial resistance, estimated to contribute
to 215,000 neonatal sepsis mortalities annually (163). Other source control measures
(such as abscess drainage and debridement) and fluid resuscitation
are also critical for correcting tissue hypoperfusion in septic
patients (162). Nonetheless,
these measures are often insufficient to improve prognosis for
numerous patients (160).
Notable macrophage metabolic reprogramming occurs
in sepsis, exacerbating inflammation while impairing immune
regulation and tissue repair (164). Table I summarizes promising
drugs/treatments, their mechanisms and key considerations based on
this reprogramming (76,78,165-188). Although targeting macrophage
metabolic pathways represents a strategic move toward precision
immunomodulation, these interventions require a critical evaluation
of their systemic trade-offs. For instance, while blocking the
glycolysis-HIF-1α axis via 2-deoxy-d-glucose (2-DG) or PKM2
inhibitors effectively curbs the initial cytokine storm, such
systemic inhibition often risks blunting the energy-intensive
responses necessary for initial pathogen sequestration and
clearance. Furthermore, the temporal application of mitochondrial
restorers, such as itaconate or PPAR agonists, remains an
evaluative challenge; promoting OXPHOS too early may lead to
premature immunosuppression, whereas late administration may fail
to rescue bioenergetically exhausted cells. Evaluating the
metabolic changes and functional effects of macrophages must also
consider disease tolerance, with the goal of maintaining organ
function under pathogen load. The success of the aforementioned
metabolic targeting strategies mainly depends on overcoming the
current lack of macrophage specific delivery systems and inaccurate
monitoring of real-time metabolic flux in patients.
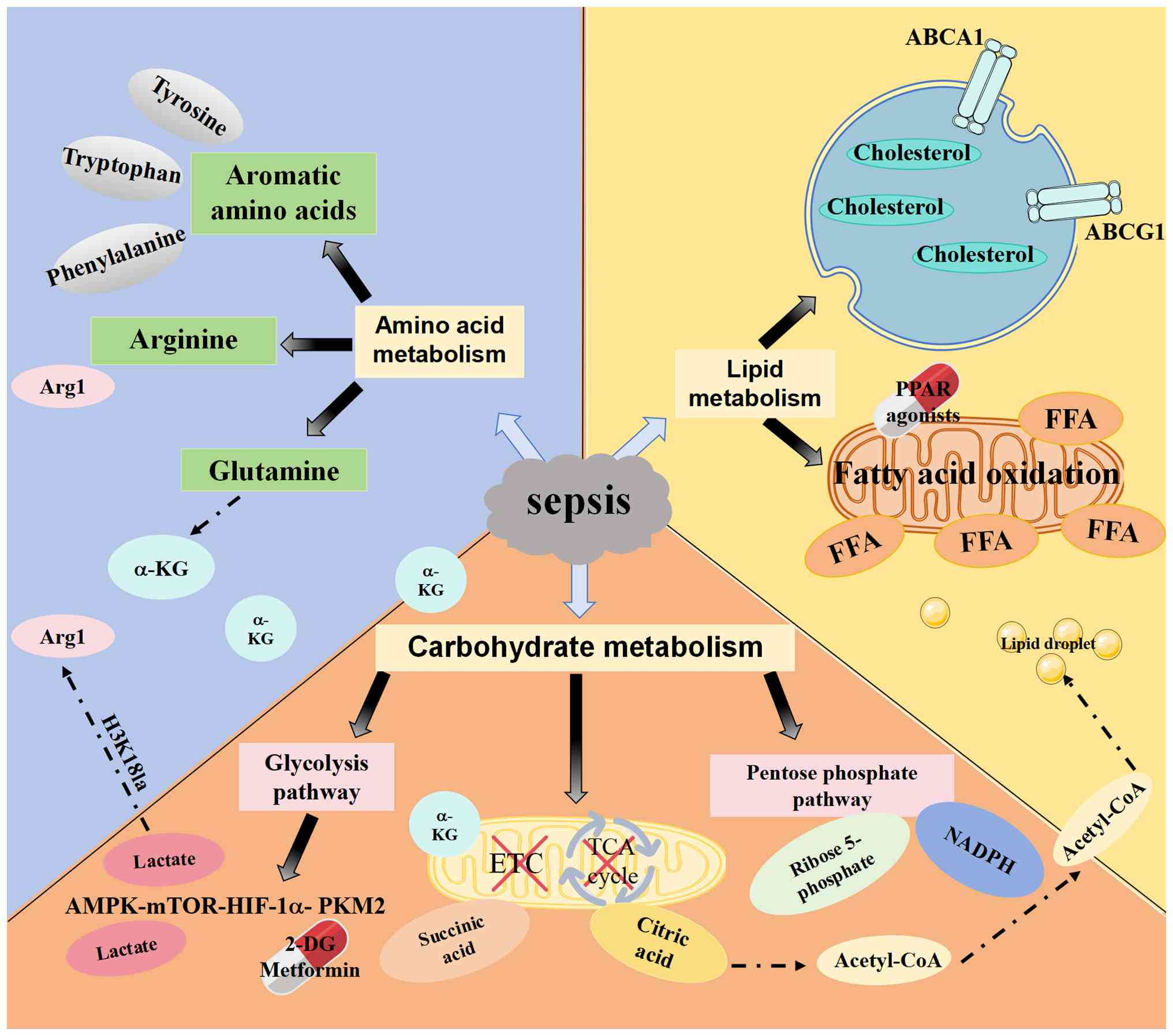
A critical paradigm shift in immunometabolism is
the recognition that metabolic reprogramming is not merely a
collateral consequence of sepsis but a primary determinant of
macrophage fate. We hypothesize that there can be a framework
concept based on metabolic control in sepsis. In this framework,
some nodes control the transition of sepsis stages. Central to the
early hyper-inflammatory phase is the mTOR-HIF-1a-PKM2 axis, which
facilitates the rapid glycolytic flux necessary for cytokine
production. This pathway, while essential for initial pathogen
clearance, concurrently primes the cell for late-stage exhaustion
by generating specific metabolic signals. Figs. 1-3 depict the main metabolic changes in
sepsis; however, these pathways do not operate in parallel and
isolation, but rather function as an interdependent regulatory
network. In order to understand the non-linear characteristics of
immune metabolism, the complex interactions between glucose, lipids
and amino acids are depicted in Fig.
4. A single substrate is not linear, but is internally coupled
through metabolic centers. For instance, the citrate accumulated
from the truncated TCA cycle (Fig.
1) serves as the indispensable part of the de novo
lipogenesis and lipid droplet formation observed in septic
macrophages (Fig. 2).
Simultaneously, the Gln-derived a-KG (Fig. 3) acts as a key energy buffer to
sustain mitochondrial output when glycolytic flux is redirected
toward lactate production. This metabolic interconnectedness
implies that macrophage fate is determined by the global
integration of these fluxes rather than isolated enzymatic
changes.
We hypothesize that the lactate-H3K18la epigenetic
axis, the TREM2-SHP1-BTK signaling pathway and itaconate-mediated
mitochondrial modulation regulatory nodes in sepsis can function as
synchronous elements rather than isolated metabolic changes
(25,68,189,190). Specifically, the
PKM2-HIF-1α-succinic acid circuit persists during the highly
inflammatory phase, and accumulated lactate triggers the lactate
H3K18la epigenetic axis. This histone modification controls
macrophages in an immunosuppressive state by upregulating
anti-inflammatory genes such as Arg1. Concurrently, the
TREM2-SHP1-BTK axis serves as a critical regulatory node in
response to systemic lipid dysregulation. Its overactivation
inhibits mitochondrial FAO, leading to the bioenergetic failure
observed in immunosuppressed macrophages (25). Additionally, the diversion of
metabolic flux toward itaconate production regulates the TCA cycle
by inhibiting SDH. In addition, the activation of nuclear factor
erythroid 2-related factor 2 mediated antioxidant response limits
oxidative damage and promotes the resolution of inflammation. These
regulatory nodes combine glycolysis, TCA cycle (via itaconate) and
lipid sensors to form a theoretical framework for metabolic
interventions targeting specific stages of sepsis. A unique
contribution of the present review is linking the classification of
therapeutic targets with stage specificity. As shown in Table I, drugs such as 2-DG or metformin
have an early stabilizing effect by limiting the peak of
pro-inflammatory glycolysis. And PPARα agonists or itaconic acid
derivatives serve a late stage repair role to maintain
mitochondrial homeostasis. Compared with the traditional methods,
such as fluid resuscitation, broad-spectrum antibiotics and the
static immunomodulatory strategies, this dynamic immune metabolism
intervention may be beneficial for septic patient therapy.
Second, intervention timing is crucial and must
align with the dynamic metabolic phenotype of macrophages.
Metabolic patterns shift markedly from early to late sepsis. Early
sepsis is characterized by a glycolytic, pro-inflammatory M1
phenotype (Fig. 1), suggesting a
therapeutic window for glycolytic inhibitors [such as 2-DG
(171,172)] or PKM2 inhibitors. By contrast,
advanced sepsis typically features immune suppression. At this
stage, enhancing mitochondrial function [such as via PPARa agonists
(87,99)] or carefully dosing IL-10
(182) may be more pertinent.
Using glycolysis inhibitors during immunosuppression could further
impair macrophage rescue capacity, underscoring the double-edged
nature of targeting core metabolic pathways.
Third, the organ-specific context of macrophage
niches necessitates tailored approaches. Metabolic reprogramming of
alveolar macrophages in the lungs (Fig. 1) likely differs from that of
Kupffer cells in the liver, potentially leading to divergent
clinical manifestations even with the same pathogen. Systemically
administered drugs may have varying effects on macrophages in
different organs, offsetting overall therapeutic benefits. For
instance, for biomimetic nanomodulators, such as macrophage
membrane-coated antioxidant/anti-inflammatory nanoparticles (mAOI
NPs), alleviating sepsis-associated encephalopathy may not
necessarily ameliorate ALI or AKI (188).
Interventions targeting macrophage metabolism often
aim to regulate organ function or improve long-term outcomes rather
than prevent early mortality. Focusing on organ-specific endpoints,
resolution of immunosuppression or metabolic biomarkers [such as
circulating succinate, HMGB1 or lactate levels (42,44,57)] could provide more sensitive
measures of drug efficacy in specific sepsis phases or patient
subpopulations.
Despite the identification of key metabolic nodes,
several critical limitations in current immunometabolism research
must be acknowledged. Firstly, the majority of mechanistic insights
are derived from LPS-induced or cecal ligation and puncture (CLP)
rodent models. While CLP is considered the gold standard for
polymicrobial sepsis, both models exhibit notable genomic and
physiological discrepancies when compared with the clinical
progression of human sepsis, particularly regarding the temporal
dynamics of the inflammatory response (160). Therefore, direct clinical
application requires careful verification. Secondly, the present
review frequently generalizes 'macrophages' without fully
distinguishing between tissue-resident macrophages (TRMs) and
recruited monocyte-derived macrophages (MDMs). TRMs, such as
alveolar macrophages and Kupffer cells, possess distinct ontogeny
and metabolic baselines dictated by their organ-specific niches,
whereas MDMs exhibit rapid, high-flux metabolic reprogramming upon
recruitment to the infection site. The failure to account for this
heterogeneity may lead to the misidentification of metabolic
targets. Furthermore, macrophage metabolism does not function in
isolation; it is markedly shaped by the inflammatory milieu
provided by other cell types. Endothelial cells, neutrophils and
lymphocytes influence macrophage metabolic states through the
secretion of paracrine factors and competition for extracellular
substrates such as glucose and Gln. Finally, metabolic centers that
have been reported and studied were prioritized, as well as
emerging therapeutic targets, which may naturally overlook
less-characterized or niche signaling pathways that could also
contribute to sepsis pathogenesis.
Macrophage metabolic reprogramming is a cornerstone
of sepsis pathogenesis. It is an active driving process that shapes
the immune response from initial inflammatory outburst to eventual
immune paralysis. The present review redefines sepsis as an
interconnected pathological network in which glucose, lipid and
amino acid metabolism disorders are interrelated, ultimately
locking the host in a pathological state. Clinical biomarker data
(such as serum PKM2 and HMGB1) was combined with basic signaling
nodes such as the TREM2-SHP1 axis to propose a treatment framework
arranged in chronological order, combining metabolic interventions
with the dynamic stages of sepsis.
The therapeutic potential of targeting macrophage
metabolism is immense yet complex. Multi-omics techniques
(metabolomics and transcriptomics) should be used to define sepsis
processes based on immunometabolic profiles. Tools for real-time
patient metabolic monitoring need development. Finally, reliable
metabolic biomarkers need to be identified to test targeted
therapies in precisely defined patient populations at optimal
timepoints during the disease course. Understanding and
manipulating immunometabolism is not merely an adjunct to sepsis
research but a fundamental framework for deciphering the complexity
of the disease.
Not applicable.
TZ wrote, reviewed and edited the original draft.
WZ wrote and reviewed the original draft. ZR reviewed the original
draft. XF conceptualized and supervised the study and was involved
with investigation and reviewing and editing the manuscript. Data
authentication is not applicable. All authors read and approved the
final version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present work was supported by the National Natural Science
Foundation of China (grant no. 82073911 to XF), the Taishan
Scholars Program (grant no. Tsqn202211220 to XF), Shandong Province
Natural Science Foundation (grant no. ZR2025MS1431 to XF) and the
Joint Innovation Team for Clinical and Basic Research (grant no.
202409).
|
1
|
Rudd KE, Johnson SC, Agesa KM, Shackelford
KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer
S, et al: Global, regional, and national sepsis incidence and
mortality, 1990-2017: Analysis for the Global Burden of Disease
Study. Lancet. 395:200–211. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Deutschman CS and Tracey KJ: Sepsis:
Current dogma and new perspectives. Immunity. 40:463–475. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Willmann K and Moita LF: Physiologic
disruption and metabolic reprogramming in infection and sepsis.
Cell Metab. 36:927–946. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
GBD 2021 Global Sepsis Collaborators:
Global, regional, and national sepsis incidence and mortality,
1990-2021: A systematic analysis. Lancet Glob Health.
13:e2013–e2026. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alhazzani W, Møller MH, Arabi YM, Loeb M,
Gong MN, Fan E, Oczkowski S, Levy MM, Derde L, Dzierba A, et al:
Surviving sepsis campaign: Guidelines on the management of
critically ill adults with coronavirus disease 2019 (COVID-19).
Intensive Care Med. 46:854–887. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Crunkhorn S: New route to sepsis therapy.
Nat Rev Drug Discov. 18:251. 2019.
|
|
7
|
Weis S, Carlos AR, Moita MR, Singh S,
Blankenhaus B, Cardoso S, Larsen R, Rebelo S, Schäuble S, Del
Barrio L, et al: Metabolic adaptation establishes disease tolerance
to sepsis. Cell. 169:1263–1275.e14. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wiersinga WJ, Leopold SJ, Cranendonk DR
and van der Poll T: Host innate immune responses to sepsis.
Virulence. 5:36–44. 2014. View Article : Google Scholar :
|
|
9
|
Darden DB, Kelly LS, Fenner BP, Moldawer
LL, Mohr AM and Efron PA: Dysregulated Immunity and Immunotherapy
after Sepsis. J Clin Med. 10:17422021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen XS, Liu YC, Gao YL, Shou ST and Chai
YF: The roles of macrophage polarization in the host immune
response to sepsis. Int Immunopharmacol. 96:1077912021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chinetti-Gbaguidi G and Staels B:
Macrophage polarization in metabolic disorders: Functions and
regulation. Curr Opin Lipidol. 22:365–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dominski A, Krawczyk M, Konieczny T,
Kasprów M, Foryś A, Pastuch-Gawołek G and Kurcok P: Biodegradable
pH-responsive micelles loaded with 8-hydroxyquinoline
glycoconjugates for Warburg effect based tumor targeting. Eur J
Pharm Biopharm. 154:317–329. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Neill LA, Kishton RJ and Rathmell J: A
guide to immunometabolism for immunologists. Nat Rev Immunol.
16:553–565. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jha AK, Huang SC, Sergushichev A,
Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart
KM, Ashall J, Everts B, et al: Network integration of parallel
metabolic and transcriptional data reveals metabolic modules that
regulate macrophage polarization. Immunity. 42:419–430. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arts RJ, Gresnigt MS, Joosten LA and Netea
MG: Cellular metabolism of myeloid cells in sepsis. J Leukoc Biol.
101:151–164. 2017. View Article : Google Scholar
|
|
16
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 169:361–371. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hotchkiss RS, Monneret G and Payen D:
Sepsis-induced immunosuppression: From cellular dysfunctions to
immunotherapy. Nat Rev Immunol. 13:862–874. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
King EG, Bauzá GJ, Mella JR and Remick DG:
Pathophysiologic mechanisms in septic shock. Lab Invest. 94:4–12.
2014. View Article : Google Scholar
|
|
19
|
Dang B, Gao Q, Zhang L, Zhang J, Cai H,
Zhu Y, Zhong Q, Liu J, Niu Y, Mao K, et al: The glycolysis/HIF-1α
axis defines the inflammatory role of IL-4-primed macrophages. Cell
Rep. 42:1124712023. View Article : Google Scholar
|
|
20
|
Giamarellos-Bourboulis EJ, Kotsaki A,
Kotsamidi I, Efthymiou A, Koutsoukou V, Ehler J, Paridou A,
Frantzeskaki F, Müller MCA, Pickkers P, et al: Precision
immunotherapy to improve sepsis outcomes: The ImmunoSep randomized
clinical trial. JAMA. 335:775–786. 2026. View Article : Google Scholar
|
|
21
|
Chen XS, Wang SH, Liu CY, Gao YL, Meng XL,
Wei W, Shou ST, Liu YC and Chai YF: Losartan attenuates
sepsis-induced cardiomyopathy by regulating macrophage polarization
via TLR4-mediated NF-κB and MAPK signaling. Pharmacol Res.
185:1064732022. View Article : Google Scholar
|
|
22
|
Jaitin DA, Adlung L, Thaiss CA, Weiner A,
Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et
al: Lipid-associated macrophages control metabolic homeostasis in a
Trem2-dependent manner. Cell. 178:686–698.e14. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hou J, Zhang J, Cui P, Zhou Y, Liu C, Wu
X, Ji Y, Wang S, Cheng B, Ye H, et al: TREM2 sustains
macrophage-hepatocyte metabolic coordination in nonalcoholic fatty
liver disease and sepsis. J Clin Invest. 131:e1351972021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang K, Wang Y, Chen S, Mao J, Jin Y, Ye
H, Zhang Y, Liu X, Gong C, Cheng X, et al: TREM2hi resident
macrophages protect the septic heart by maintaining cardiomyocyte
homeostasis. Nat Metab. 5:129–146. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ming S, Li X, Xiao Q, Qu S, Wang Q, Fang
Q, Liang P, Xu Y, Yang J, Yang Y, et al: TREM2 aggravates sepsis by
inhibiting fatty acid oxidation via the SHP1/BTK axis. J Clin
Invest. 135:e1594002024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu X, Zhang J, Zeigler AC, Nelson AR,
Lindsey ML and Saucerman JJ: Network analysis reveals a distinct
axis of macrophage activation in response to conflicting
inflammatory cues. J Immunol. 206:883–891. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Murray PJ, Allen JE, Biswas SK, Fisher EA,
Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence
T, et al: Macrophage activation and polarization: Nomenclature and
experimental guidelines. Immunity. 41:14–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ye Q, Lai X, Liu Y, Zhang Z, Fu Y, Luo J,
Liu C, Duan J, Ding H, Liu Y, et al: Single-cell multi-omic
landscape reveals anatomical-specific immune features in adult and
pediatric sepsis. Nat Immunol. 27:150–165. 2026. View Article : Google Scholar
|
|
29
|
Murao A, Jha A, Aziz M and Wang P:
Transcriptomic profiling of immune cells in murine polymicrobial
sepsis. Front Immunol. 15:13474532024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mo Q, Mo Q and Mo F: Single-cell RNA
sequencing and transcriptomic analysis reveal key genes and
regulatory mechanisms in sepsis. Biotechnol Genet Eng Rev.
40:1636–1658. 2024. View Article : Google Scholar
|
|
31
|
Viola A, Munari F, Sánchez-Rodríguez R,
Scolaro T and Castegna A: The metabolic signature of macrophage
responses. Front Immunol. 10:14622019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Alves-Filho JC and Pålsson-McDermott EM:
Pyruvate Kinase M2: A potential target for regulating inflammation.
Front Immunol. 7:1452016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Van Wyngene L, Vandewalle J and Libert C:
Reprogramming of basic metabolic pathways in microbial sepsis:
Therapeutic targets at last? EMBO Mol Med. 10:e87122018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu D, Huang W, Sheng M, Zhang S, Pan H,
Ren F, Luo L, Zhou J, Huang D and Tang L: Angiotensin-(1-7)
modulates the Warburg effect to alleviate inflammation in
LPS-induced macrophages and septic mice. J Inflamm Res. 17:469–485.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hard GC: Some biochemical aspects of the
immune macrophage. Br J Exp Pathol. 51:97–105. 1970.PubMed/NCBI
|
|
36
|
Ma G, Wu X, Qi C, Yu X and Zhang F:
Development of macrophage-associated genes prognostic signature
predicts clinical outcome and immune infiltration for sepsis. Sci
Rep. 14:20262024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pan L, Hu L, Zhang L, Xu H, Chen Y, Bian
Q, Zhu A and Wu H: Deoxyelephantopin decreases the release of
inflammatory cytokines in macrophage associated with attenuation of
aerobic glycolysis via modulation of PKM2. Int Immunopharmacol.
79:1060482020. View Article : Google Scholar
|
|
38
|
Chen Y, Zhang P, Han F, Zhou Y, Wei J,
Wang C, Song M, Lin S, Xu Y and Chen X: MiR-106a-5p targets PFKFB3
and improves sepsis through regulating macrophage pyroptosis and
inflammatory response. J Biol Chem. 300:1073342024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang F, Wang K, Xu W, Zhao S, Ye D, Wang
Y, Xu Y, Zhou L, Chu Y, Zhang C, et al: SIRT5 desuccinylates and
activates pyruvate kinase M2 to block macrophage IL-1β production
and to prevent DSS-induced colitis in mice. Cell Rep. 19:2331–2344.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li T, Han J, Jia L, Hu X, Chen L and Wang
Y: PKM2 coordinates glycolysis with mitochondrial fusion and
oxidative phosphorylation. Protein Cell. 10:583–594. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W,
Kang R, Lotze MT, Billiar TR, Wang H, et al: PKM2 regulates the
Warburg effect and promotes HMGB1 release in sepsis. Nat Commun.
5:44362014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang L, Tang D and Zhang P: Changes of
serum pyruvate kinase M2 level in patients with sepsis and its
clinical value. Infect Drug Resist. 16:6437–6449. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Palmer CS, Ostrowski M, Balderson B,
Christian N and Crowe SM: Glucose metabolism regulates T cell
activation, differentiation, and functions. Front Immunol. 6:12015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tannahill GM, Curtis AM, Adamik J,
Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ,
Kelly B, Foley NH, et al: Succinate is an inflammatory signal that
induces IL-1β through HIF-1α. Nature. 496:238–242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Palsson-McDermott EM, Curtis AM, Goel G,
Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR,
Domingo-Fernandez R, Johnston DG, et al: Pyruvate kinase M2
regulates Hif-1α activity and IL-1β induction and is a critical
determinant of the warburg effect in LPS-activated macrophages.
Cell Metab. 21:65–80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Van den Bossche J, O'Neill LA and Menon D:
Macrophage immunometabolism: Where are we (Going)? Trends Immunol.
38:395–406. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Littlewood-Evans A, Sarret S, Apfel V,
Loesle P, Dawson J, Zhang J, Muller A, Tigani B, Kneuer R, Patel S,
et al: GPR91 senses extracellular succinate released from
inflammatory macrophages and exacerbates rheumatoid arthritis. J
Exp Med. 213:1655–1662. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cheng SC, Scicluna BP, Arts RJ, Gresnigt
MS, Lachmandas E, Giamarellos-Bourboulis EJ, Kox M, Manjeri GR,
Wagenaars JA, Cremer OL, et al: Broad defects in the energy
metabolism of leukocytes underlie immunoparalysis in sepsis. Nat
Immunol. 17:406–413. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Düvel K, Yecies JL, Menon S, Raman P,
Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S,
et al: Activation of a metabolic gene regulatory network downstream
of mTOR complex 1. Mol Cell. 39:171–183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ganeshan K and Chawla A: Metabolic
regulation of immune responses. Annu Rev Immunol. 32:609–634. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Damasceno LEA, Prado DS, Veras FP, Fonseca
MM, Toller-Kawahisa JE, Rosa MH, Públio GA, Martins TV, Ramalho FS,
Waisman A, et al: PKM2 promotes Th17 cell differentiation and
autoimmune inflammation by fine-tuning STAT3 activation. J Exp Med.
217:e201906132020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Huang J, Liu K, Zhu S, Xie M, Kang R, Cao
L and Tang D: AMPK regulates immunometabolism in sepsis. Brain
Behav Immun. 72:89–100. 2018. View Article : Google Scholar
|
|
53
|
Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F,
Gill PS, Ha T, Liu L, Williams DL and Li C: Lactate promotes
macrophage HMGB1 lactylation, acetylation, and exosomal release in
polymicrobial sepsis. Cell Death Differ. 29:133–146. 2022.
View Article : Google Scholar :
|
|
54
|
Yang H, Ochani M, Li J, Qiang X, Tanovic
M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, et al:
Reversing established sepsis with antagonists of endogenous
high-mobility group box 1. Proc Natl Acad Sci USA. 101:296–301.
2004. View Article : Google Scholar :
|
|
55
|
Angus DC, Yang L, Kong L, Kellum JA,
Delude RL, Tracey KJ and Weissfeld L; GenIMS Investigators:
Circulating high-mobility group box 1 (HMGB1) concentrations are
elevated in both uncomplicated pneumonia and pneumonia with severe
sepsis. Crit Care Med. 35:1061–1067. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Karlsson S, Pettilä V, Tenhunen J,
Laru-Sompa R, Hynninen M and Ruokonen E: HMGB1 as a predictor of
organ dysfunction and outcome in patients with severe sepsis.
Intensive Care Med. 34:1046–1053. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yagmur E, Buendgens L, Herbers U, Beeretz
A, Weiskirchen R, Koek GH, Trautwein C, Tacke F and Koch A: High
mobility group box 1 as a biomarker in critically ill patients. J
Clin Lab Anal. 32:e225842018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen R, Cao C, Liu H, Jiang W, Pan R, He
H, Ding K and Meng Q: Macrophage Sprouty4 deficiency diminishes
sepsis-induced acute lung injury in mice. Redox Biol.
58:1025132022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xie T, Xu Q, Wan H, Xing S, Shang C, Gao Y
and He Z: Lipopolysaccharide promotes lung fibroblast proliferation
through autophagy inhibition via activation of the PI3K-Akt-mTOR
pathway. Lab Invest. 99:625–633. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Li R, Ren T and Zeng J: Mitochondrial
coenzyme Q protects sepsis-induced acute lung injury by activating
PI3K/Akt/GSK-3β/mTOR pathway in rats. Biomed Res Int.
2019:52408982019. View Article : Google Scholar
|
|
61
|
Mills EL and O'Neill LA: Reprogramming
mitochondrial metabolism in macrophages as an anti-inflammatory
signal. Eur J Immunol. 46:13–21. 2016. View Article : Google Scholar
|
|
62
|
Mills E and O'Neill LA: Succinate: A
metabolic signal in inflammation. Trends Cell Biol. 24:313–320.
2014. View Article : Google Scholar
|
|
63
|
Huang SC, Everts B, Ivanova Y, O'Sullivan
D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY,
O'Neill CM, et al: Cell-intrinsic lysosomal lipolysis is essential
for alternative activation of macrophages. Nat Immunol. 15:846–855.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Williams NC and O'Neill LAJ: A role for
the Krebs cycle intermediate citrate in metabolic reprogramming in
innate immunity and inflammation. Front Immunol. 9:1412018.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lauterbach MA, Hanke JE, Serefidou M,
Mangan MSJ, Kolbe CC, Hess T, Rothe M, Kaiser R, Hoss F, Gehlen J,
et al: Toll-like receptor signaling rewires macrophage metabolism
and promotes histone acetylation via ATP-citrate Lyase. Immunity.
51:997–1011.e7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Xu Y, Zhang S, Rong J, Lin Y, Du L, Wang Y
and Zhang Z: Sirt3 is a novel target to treat sepsis induced
myocardial dysfunction by acetylated modulation of critical enzymes
within cardiac tricarboxylic acid cycle. Pharmacol Res.
159:1048872020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ma W, Ao S, Zhou J, Li J, Liang X, Yang X,
Zhang H, Liu B, Tang W, Liu H, et al: Methylsulfonylmethane
protects against lethal dose MRSA-induced sepsis through promoting
M2 macrophage polarization. Mol Immunol. 146:69–77. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chu X, Di C, Chang P, Li L, Feng Z, Xiao
S, Yan X, Xu X, Li H, Qi R, et al: Lactylated histone H3K18 as a
potential biomarker for the diagnosis and predicting the severity
of septic shock. Front Immunol. 12:7866662021. View Article : Google Scholar
|
|
69
|
Pereverzeva L, van Linge CCA, Schuurman
AR, Klarenbeek AM, Ramirez Moral I, Otto NA, Peters-Sengers H,
Butler JM, Schomakers BV, van Weeghel M, et al: Human alveolar
macrophages do not rely on glucose metabolism upon activation by
lipopolysaccharide. Biochim Biophys Acta Mol Basis Dis.
1868:1664882022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Woods PS, Kimmig LM, Sun KA, Meliton AY,
Shamaa OR, Tian Y, Cetin-Atalay R, Sharp WW, Hamanaka RB and Mutlu
GM: HIF-1α induces glycolytic reprograming in tissue-resident
alveolar macrophages to promote cell survival during acute lung
injury. Elife. 11:e774572022. View Article : Google Scholar
|
|
71
|
Svedberg FR, Brown SL, Krauss MZ, Campbell
L, Sharpe C, Clausen M, Howell GJ, Clark H, Madsen J, Evans CM, et
al: The lung environment controls alveolar macrophage metabolism
and responsiveness in type 2 inflammation. Nat Immunol. 20:571–580.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Russell DG, Huang L and VanderVen BC:
Immunometabolism at the interface between macrophages and
pathogens. Nat Rev Immunol. 19:291–304. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Haschemi A, Kosma P, Gille L, Evans CR,
Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, et al:
The sedoheptulose kinase CARKL directs macrophage polarization
through control of glucose metabolism. Cell Metab. 15:813–826.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
de Boer JF, Kuipers F and Groen AK:
Cholesterol transport revisited: A new turbo mechanism to drive
cholesterol excretion. Trends Endocrinol Metab. 29:123–133. 2018.
View Article : Google Scholar
|
|
75
|
Chauhan P and Saha B: Metabolic regulation
of infection and inflammation. Cytokine. 112:1–11. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Colaço HG, Barros A, Neves-Costa A, Seixas
E, Pedroso D, Velho T, Willmann KL, Faisca P, Grabmann G, Yi HS, et
al: Tetracycline antibiotics induce host-dependent disease
tolerance to infection. Immunity. 54:53–67.e57. 2021. View Article : Google Scholar :
|
|
77
|
Huen SC: Metabolism as disease tolerance:
Implications for sepsis-associated acute kidney injury. Nephron.
146:291–294. 2022. View Article : Google Scholar
|
|
78
|
Li R, Li X, Zhao J, Meng F, Yao C, Bao E,
Sun N, Chen X, Cheng W, Hua H, et al: Mitochondrial STAT3
exacerbates LPS-induced sepsis by driving CPT1a-mediated fatty acid
oxidation. Theranostics. 12:976–998. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Korbecki J and Bajdak-Rusinek K: The
effect of palmitic acid on inflammatory response in macrophages: An
overview of molecular mechanisms. Inflamm Res. 68:915–932. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Tian H, Liu C, Zou X, Wu W, Zhang C and
Yuan D: MiRNA-194 regulates Palmitic acid-induced toll-like
receptor 4 inflammatory responses in THP-1 cells. Nutrients.
7:3483–3496. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wen Y, Gu J, Chakrabarti SK, Aylor K,
Marshall J, Takahashi Y, Yoshimoto T and Nadler JL: The role of
12/15-lipoxygenase in the expression of interleukin-6 and tumor
necrosis factor-alpha in macrophages. Endocrinology. 148:1313–1322.
2007. View Article : Google Scholar
|
|
82
|
Yang Y, Zhong ZT, Xiao YG and Chen HB: The
activation of AMPK/NRF2 pathway in lung epithelial cells is
involved in the protective effects of kinsenoside on
lipopolysaccharide-induced acute lung injury. Oxid Med Cell Longev.
2022:35892772022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Ma L, Li W, Zhang Y, Qi L, Zhao Q, Li N,
Lu Y, Zhang L, Zhou F, Wu Y, et al: FLT4/VEGFR3 activates AMPK to
coordinate glycometabolic reprogramming with autophagy and
inflammasome activation for bacterial elimination. Autophagy.
18:1385–1400. 2022. View Article : Google Scholar :
|
|
84
|
Russo S, Kwiatkowski M, Govorukhina N,
Bischoff R and Melgert BN: Meta-inflammation and metabolic
reprogramming of macrophages in diabetes and obesity: The
importance of metabolites. Front Immunol. 12:7461512021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Vandewalle J and Libert C: Sepsis: A
failing starvation response. Trends Endocrinol Metab. 33:292–304.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Vandewalle J, Timmermans S, Paakinaho V,
Vancraeynest L, Dewyse L, Vanderhaeghen T, Wallaeys C, Van Wyngene
L, Van Looveren K, Nuyttens L, et al: Combined glucocorticoid
resistance and hyperlactatemia contributes to lethal shock in
sepsis. Cell Metab. 33:1763–1776.e5. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Van Wyngene L, Vanderhaeghen T, Timmermans
S, Vandewalle J, Van Looveren K, Souffriau J, Wallaeys C, Eggermont
M, Ernst S, Van Hamme E, et al: Hepatic PPARα function and lipid
metabolic pathways are dysregulated in polymicrobial sepsis. EMBO
Mol Med. 12:e113192020. View Article : Google Scholar
|
|
88
|
Singh N, Gurav A, Sivaprakasam S, Brady E,
Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, et
al: Activation of Gpr109a, receptor for niacin and the commensal
metabolite butyrate, suppresses colonic inflammation and
carcinogenesis. Immunity. 40:128–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Youm YH, Nguyen KY, Grant RW, Goldberg EL,
Bodogai M, Kim D, D'Agostino D, Planavsky N, Lupfer C, Kanneganti
TD, et al: The ketone metabolite β-hydroxybutyrate blocks NLRP3
inflammasome-mediated inflammatory disease. Nat Med. 21:263–269.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Das UN: Essential fatty acids and their
metabolites in the pathobiology of inflammation and its resolution.
Biomolecules. 11:18732021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Vats D, Mukundan L, Odegaard JI, Zhang L,
Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ and Chawla A:
Oxidative metabolism and PGC-1beta attenuate macrophage-mediated
inflammation. Cell Metab. 4:13–24. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Chen YL, Xie YJ, Liu ZM, Chen WB, Zhang R,
Ye HX, Wang W, Liu XY and Chen HS: Omega-3 fatty acids impair
miR-1-3p-dependent Notch3 down-regulation and alleviate
sepsis-induced intestinal injury. Mol Med. 28:92022. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Fu Y, Gong T, Loughran PA, Li Y, Billiar
TR, Liu Y, Wen Z and Fan J: Roles of TLR4 in macrophage immunity
and macrophage-pulmonary vascular/lymphatic endothelial cell
interactions in sepsis. Commun Biol. 8:4692025. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Liu L, Lu Y, Martinez J, Bi Y, Lian G,
Wang T, Milasta S, Wang J, Yang M, Liu G, et al: Proinflammatory
signal suppresses proliferation and shifts macrophage metabolism
from Myc-dependent to HIF1α-dependent. Proc Natl Acad Sci USA.
113:1564–1569. 2016. View Article : Google Scholar
|
|
95
|
Wellhoener P, Vietheer A, Sayk F, Schaaf
B, Lehnert H and Dodt C: Metabolic alterations in adipose tissue
during the early phase of experimental endotoxemia in humans. Horm
Metab Res. 43:754–759. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Das UN: The dysregulation of essential
fatty acid (EFA) metabolism may be a factor in the pathogenesis of
sepsis. Medicina (Kaunas). 60:9342024. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Chang X, Guo Y, Wang J, Liu J, Ma Y, Lu Q
and Han Y: Heart-type fatty acid binding protein (H-FABP) as an
early biomarker in sepsis-induced cardiomyopathy: A prospective
observational study. Lipids Health Dis. 23:2832024. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wang Z, Li Y, Yang X, Zhang L, Shen H, Xu
W and Yuan C: Protective effects of rapamycin induced autophagy on
CLP septic mice. Comp Immunol Microbiol Infect Dis. 64:47–52. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Chawla A: Control of macrophage activation
and function by PPARs. Circ Res. 106:1559–1569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Crossland H, Constantin-Teodosiu D and
Greenhaff PL: The regulatory roles of PPARs in skeletal muscle fuel
metabolism and inflammation: Impact of PPAR Agonism on muscle in
chronic disease, contraction and sepsis. Int J Mol Sci.
22:97752021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Marin JJ, Macias RI, Briz O, Banales JM
and Monte MJ: Bile acids in physiology, pathology and pharmacology.
Curr Drug Metab. 17:4–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Mutemberezi V, Guillemot-Legris O and
Muccioli GG: Oxysterols: From cholesterol metabolites to key
mediators. Prog Lipid Res. 64:152–169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Duan Y, Gong K, Xu S, Zhang F, Meng X and
Han J: Regulation of cholesterol homeostasis in health and
diseases: From mechanisms to targeted therapeutics. Signal
Transduct Target Ther. 7:2652022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Varshney P, Yadav V and Saini N: Lipid
rafts in immune signalling: Current progress and future
perspective. Immunology. 149:13–24. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Golucci A, Marson FAL, Ribeiro AF and
Nogueira RJN: Lipid profile associated with the systemic
inflammatory response syndrome and sepsis in critically ill
patients. Nutrition. 55-56:7–14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Walley KR, Boyd JH, Kong HJ and Russell
JA: Low low-density lipoprotein levels are associated with, but do
not causally contribute to, increased mortality in sepsis. Crit
Care Med. 47:463–466. 2019. View Article : Google Scholar
|
|
107
|
van Leeuwen HJ, Heezius EC, Dallinga GM,
van Strijp JA, Verhoef J and van Kessel KP: Lipoprotein metabolism
in patients with severe sepsis. Crit Care Med. 31:1359–1366. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Trinder M, Genga KR, Kong HJ, Blauw LL, Lo
C, Li X, Cirstea M, Wang Y, Rensen PCN, Russell JA, et al:
Cholesteryl ester transfer protein influences high-density
lipoprotein levels and survival in sepsis. Am J Respir Crit Care
Med. 199:854–862. 2019. View Article : Google Scholar
|
|
109
|
Chang L, Chen X and Lian C: The
association between the non-HDL-cholesterol to HDL-cholesterol
ratio and 28-day mortality in sepsis patients: A cohort study. Sci
Rep. 12:34762022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Hardy JP, Streeter EM and DeCook RR:
Retrospective evaluation of plasma cholesterol concentration in
septic dogs and its association with morbidity and mortality: 51
cases (2005-2015). J Vet Emerg Crit Care (San Antonio). 28:149–156.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Morel J, Hargreaves I, Brealey D,
Neergheen V, Backman JT, Lindig S, Bläss M, Bauer M, McAuley DF and
Singer M: Simvastatin pre-treatment improves survival and
mitochondrial function in a 3-day fluid-resuscitated rat model of
sepsis. Clin Sci (Lond). 131:747–758. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Vavrova L, Rychlikova J, Mrackova M,
Novakova O, Zak A and Novak F: Increased inflammatory markers with
altered antioxidant status persist after clinical recovery from
severe sepsis: A correlation with low HDL cholesterol and albumin.
Clin Exp Med. 16:557–569. 2016. View Article : Google Scholar
|
|
113
|
Fessler MB and Parks JS: Intracellular
lipid flux and membrane microdomains as organizing principles in
inflammatory cell signaling. J Immunol. 187:1529–1535. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Luo J, Yang H and Song BL: Mechanisms and
regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol.
21:225–245. 2020. View Article : Google Scholar
|
|
115
|
Tabas I and Lichtman AH:
Monocyte-macrophages and T cells in atherosclerosis. Immunity.
47:621–634. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Pownall HJ, Rosales C, Gillard BK and
Gotto AM Jr: High-density lipoproteins, reverse cholesterol
transport and atherogenesis. Nat Rev Cardiol. 18:712–723. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Hofmaenner DA, Kleyman A, Press A, Bauer M
and Singer M: The many roles of cholesterol in sepsis: A review. Am
J Respir Crit Care Med. 205:388–396. 2022. View Article : Google Scholar :
|
|
118
|
Van Dender C, Timmermans S, Paakinaho V,
Vanderhaeghen T, Vandewalle J, Claes M, Garcia B, Roman B, De Waele
J, Croubels S, et al: A critical role for HNF4α in polymicrobial
sepsis-associated metabolic reprogramming and death. EMBO Mol Med.
16:2485–2515. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Yang L, Chu Z, Liu M, Zou Q, Li J, Liu Q,
Wang Y, Wang T, Xiang J and Wang B: Amino acid metabolism in immune
cells: Essential regulators of the effector functions, and
promising opportunities to enhance cancer immunotherapy. J Hematol
Oncol. 16:592023. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Chen Q, Liang X, Wu T, Jiang J, Jiang Y,
Zhang S, Ruan Y, Zhang H, Zhang C, Chen P, et al: Integrative
analysis of metabolomics and proteomics reveals amino acid
metabolism disorder in sepsis. J Transl Med. 20:1232022. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Hezaveh K, Shinde RS, Klötgen A, Halaby
MJ, Lamorte S, Ciudad MT, Quevedo R, Neufeld L, Liu ZQ, Jin R, et
al: Tryptophan-derived microbial metabolites activate the aryl
hydrocarbon receptor in tumor-associated macrophages to suppress
anti-tumor immunity. Immunity. 55:324–340.e328. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Roager HM and Licht TR: Microbial
tryptophan catabolites in health and disease. Nat Commun.
9:32942018. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Mulder K, Patel AA, Kong WT, Piot C,
Halitzki E, Dunsmore G, Khalilnezhad S, Irac SE, Dubuisson A,
Chevrier M, et al: Cross-tissue single-cell landscape of human
monocytes and macrophages in health and disease. Immunity.
54:1883–1900.e5. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Wang F, Zhang M, Yin L, Zhou Z, Peng Z, Li
W, Chen H, Yu G and Tang J: The tryptophan metabolite kynurenic
acid ameliorates septic colonic injury through activation of the
PPARγ signaling pathway. Int Immunopharmacol. 147:1136512025.
View Article : Google Scholar
|
|
125
|
Zangerle R, Kurz K, Neurauter G, Kitchen
M, Sarcletti M and Fuchs D: Increased blood phenylalanine to
tyrosine ratio in HIV-1 infection and correction following
effective antiretroviral therapy. Brain Behav Immun. 24:403–408.
2010. View Article : Google Scholar
|
|
126
|
Shang M, Cappellesso F, Amorim R, Serneels
J, Virga F, Eelen G, Carobbio S, Rincon MY, Maechler P, De Bock K,
et al: Macrophage-derived glutamine boosts satellite cells and
muscle regeneration. Nature. 587:626–631. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Wang B, Pei J, Xu S, Liu J and Yu J: A
glutamine tug-of-war between cancer and immune cells: Recent
advances in unraveling the ongoing battle. J Exp Clin Cancer Res.
43:742024. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Hou YC, Wu JM, Chen KY, Wu MH, Yang PJ,
Lee PC, Chen PD, Yeh SL and Lin MT: Glutamine and leucine
administration attenuates muscle atrophy in sepsis. Life Sci.
314:1213272023. View Article : Google Scholar
|
|
129
|
Wu D, Su S, Zha X, Wei Y, Yang G, Huang Q,
Yang Y, Xia L, Fan S and Peng X: Glutamine promotes O-GlcNAcylation
of G6PD and inhibits AGR2 S-glutathionylation to maintain the
intestinal mucus barrier in burned septic mice. Redox Biol.
59:1025812023. View Article : Google Scholar
|
|
130
|
Chen K, Rao Z, Dong S, Chen Y, Wang X, Luo
Y, Gong F and Li X: Roles of the fibroblast growth factor signal
transduction system in tissue injury repair. Burns Trauma.
10:tkac0052022. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Yang Y, Chen Q, Fan S, Lu Y, Huang Q, Liu
X and Peng X: Glutamine sustains energy metabolism and alleviates
liver injury in burn sepsis by promoting the assembly of
mitochondrial HSP60-HSP10 complex via SIRT4 dependent protein
deacetylation. Redox Rep. 29:23123202024. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Li W, Tao S, Wu Q, Wu T, Tao R and Fan J:
Glutamine reduces myocardial cell apoptosis in a rat model of
sepsis by promoting expression of heat shock protein 90. J Surg
Res. 220:247–254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Briassouli E, Tzanoudaki M, Goukos D,
Vardas K, Briassoulis P, Ilia S, Kanariou M, Routsi C, Nanas S,
Daikos GL and Briassoulis G: Ex vivo evaluation of glutamine
treatment in sepsis and trauma in a human peripheral blood
mononuclear cells model. Nutrients. 15:2522023. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Jennaro TS, Viglianti EM, Ingraham NE,
Jones AE, Stringer KA and Puskarich MA: Serum levels of
acylcarnitines and amino acids are associated with liberation from
organ support in patients with septic shock. J Clin Med.
11:6272022. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
van Zanten AR, Dhaliwal R, Garrel D and
Heyland DK: Enteral glutamine supplementation in critically ill
patients: A systematic review and meta-analysis. Crit Care.
19:2942015. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Heyland D, Muscedere J, Wischmeyer PE,
Cook D, Jones G, Albert M, Elke G, Berger MM and Day AG; Canadian
Critical Care Trials Group: A randomized trial of glutamine and
antioxidants in critically ill patients. N Engl J Med.
368:1489–1497. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Mulherin DW and Sacks GS: Uncertainty
about the safety of supplemental glutamine: An editorial on 'A
randomized trial of glutamine and antioxidants in critically ill
patients'. Hepatobiliary Surg Nutr. 4:76–79. 2015.PubMed/NCBI
|
|
138
|
Briassouli E and Briassoulis G: Glutamine
randomized studies in early life: The unsolved riddle of
experimental and clinical studies. Clin Dev Immunol.
2012:7491892012. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Van den Bossche J, Baardman J, Otto NA,
van der Velden S, Neele AE, van den Berg SM, Luque-Martin R, Chen
HJ, Boshuizen MC, Ahmed M, et al: Mitochondrial dysfunction
prevents repolarization of inflammatory macrophages. Cell Rep.
17:684–696. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Schairer DO, Chouake JS, Nosanchuk JD and
Friedman AJ: The potential of nitric oxide releasing therapies as
antimicrobial agents. Virulence. 3:271–279. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Bogdan C: Nitric oxide and the immune
response. Nat Immunol. 2:907–916. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Mills CD: Anatomy of a discovery: M1 and
m2 macrophages. Front Immunol. 6:2122015. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wijnands KA, Castermans TM, Hommen MP,
Meesters DM and Poeze M: Arginine and citrulline and the immune
response in sepsis. Nutrients. 7:1426–1463. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Chantranupong L, Scaria SM, Saxton RA,
Gygi MP, Shen K, Wyant GA, Wang T, Harper JW, Gygi SP, Sabatini DM,
et al: The CASTOR proteins are arginine sensors for the mTORC1
pathway. Cell. 165:153–164. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Wijnands KA, Vink H, Briedé JJ, van
Faassen EE, Lamers WH, Buurman WA and Poeze M: Citrulline a more
suitable substrate than arginine to restore NO production and the
microcirculation during endotoxemia. PLoS One. 7:e374392012.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Yeh CL, Pai MH, Shih YM, Shih JM and Yeh
SL: Intravenous arginine administration promotes proangiogenic
cells mobilization and attenuates lung injury in mice with
polymicrobial sepsis. Nutrients. 9:5072017. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Yeh CL, Tanuseputero SA, Wu JM, Tseng YR,
Yang PJ, Lee PC, Yeh SL and Lin MT: Intravenous arginine
administration benefits CD4+ T-cell homeostasis and attenuates
liver inflammation in mice with polymicrobial sepsis. Nutrients.
12:10472020. View Article : Google Scholar
|
|
148
|
Martí ILAA and Reith W: Arginine-dependent
immune responses. Cell Mol Life Sci. 78:5303–5324. 2021. View Article : Google Scholar
|
|
149
|
Karadima E, Chavakis T and Alexaki VI:
Arginine metabolism in myeloid cells in health and disease. Semin
Immunopathol. 47:112025. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Qualls JE, Neale G, Smith AM, Koo MS,
DeFreitas AA, Zhang H, Kaplan G, Watowich SS and Murray PJ:
Arginine usage in mycobacteria-infected macrophages depends on
autocrine-paracrine cytokine signaling. Sci Signal. 3:ra622010.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Dowling JK, Afzal R, Gearing LJ,
Cervantes-Silva MP, Annett S, Davis GM, De Santi C, Assmann N,
Dettmer K, Gough DJ, et al: Mitochondrial arginase-2 is essential
for IL-10 metabolic reprogramming of inflammatory macrophages. Nat
Commun. 12:14602021. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Hannemann N, Cao S, Eriksson D, Schnelzer
A, Jordan J, Eberhardt M, Schleicher U, Rech J, Ramming A, Uebe S,
et al: Transcription factor Fra-1 targets arginase-1 to enhance
macrophage-mediated inflammation in arthritis. J Clin Invest.
129:2669–2684. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Yang Z and Ming XF: Functions of arginase
isoforms in macrophage inflammatory responses: Impact on
cardiovascular diseases and metabolic disorders. Front Immunol.
5:5332014. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Zhang Y, Higgins CB, Tica S, Adams JA, Sun
J, Kelly SC, Zong X, Dietzen DJ, Pietka T, Ballentine SJ, et al:
Hierarchical tricarboxylic acid cycle regulation by hepatocyte
arginase 2 links the urea cycle to oxidative metabolism. Cell
Metab. 36:2069–2085.e8. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Canè S, Geiger R and Bronte V: The roles
of arginases and arginine in immunity. Nat Rev Immunol. 25:266–284.
2025. View Article : Google Scholar
|
|
156
|
De Santi C, Nally FK, Afzal R, Duffy CP,
Fitzsimons S, Annett SL, Robson T, Dowling JK, Cryan SA and McCoy
CE: Enhancing arginase 2 expression using target site blockers as a
strategy to modulate macrophage phenotype. Mol Ther Nucleic Acids.
29:643–655. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Fitzsimons S, Muñoz-San Martín M, Nally F,
Dillon E, Fashina IA, Strowitzki MJ, Ramió-Torrentà L, Dowling JK,
De Santi C, McCoy CE, et al: Inhibition of pro-inflammatory
signaling in human primary macrophages by enhancing arginase-2 via
target site blockers. Mol Ther Nucleic Acids. 33:941–959. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Ming XF, Rajapakse AG, Yepuri G, Xiong Y,
Carvas JM, Ruffieux J, Scerri I, Wu Z, Popp K, Li J, et al:
Arginase II promotes macrophage inflammatory responses through
mitochondrial reactive oxygen species, contributing to insulin
resistance and atherogenesis. J Am Heart Assoc. 1:e0009922012.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Li R, Li Y, Jiang K, Zhang L, Li T, Zhao
A, Zhang Z, Xia Y, Ge K, Chen Y, et al: Lighting up arginine
metabolism reveals its functional diversity in physiology and
pathology. Cell Metab. 37:291–304.e9. 2025. View Article : Google Scholar
|
|
160
|
Velho TR, Santos I, Póvoa P and Moita LF:
Sepsis: The need for tolerance not complacency. Swiss Med Wkly.
146:W142762016. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Cecconi M, Evans L, Levy M and Rhodes A:
Sepsis and septic shock. Lancet. 392:75–87. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Evans L, Rhodes A, Alhazzani W, Antonelli
M, Coopersmith CM, French C, Machado FR, Mcintyre L, Ostermann M,
Prescott HC, et al: Surviving sepsis campaign: International
guidelines for management of sepsis and septic shock 2021.
Intensive Care Med. 47:1181–1247. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Soni M, Handa M, Singh KK and Shukla R:
Recent nanoengineered diagnostic and therapeutic advancements in
management of Sepsis. J Control Release. 352:931–945. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Qiu P, Liu Y and Zhang J: Review: The role
and mechanisms of macrophage autophagy in sepsis. Inflammation.
42:6–19. 2019. View Article : Google Scholar
|
|
165
|
Zhong H, Tang R, Feng JH, Peng YW, Xu QY,
Zhou Y, He ZY, Mei SY and Xing SP: Metformin mitigates
sepsis-associated pulmonary fibrosis by promoting AMPK activation
and inhibiting HIF-1α-induced aerobic glycolysis. Shock.
61:283–293. 2024. View Article : Google Scholar
|
|
166
|
Liang H, Song H, Zhang X, Song G, Wang Y,
Ding X, Duan X, Li L, Sun T and Kan Q: Metformin attenuated
sepsis-related liver injury by modulating gut microbiota. Emerg
Microbes Infect. 11:815–828. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Saraiva IE, Hamahata N, Huang DT,
Kane-Gill SL, Rivosecchi RM, Shiva S, Nolin TD, Chen X, Minturn J,
Chang CH, et al: Metformin for sepsis-associated AKI: A protocol
for the randomized clinical trial of the safety and FeasibiLity of
metformin as a treatment for sepsis-associated AKI (LiMiT AKI). BMJ
Open. 14:e0811202024. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Fan SY, Zhao ZC, Liu XL, Peng YG, Zhu HM,
Yan SF, Liu YJ, Xie Q, Jiang Y and Zeng SZ: Metformin mitigates
sepsis-induced acute lung injury and inflam-mation in young mice by
suppressing the S100A8/A9-NLRP3-IL-1β signaling pathway. J Inflamm
Res. 17:3785–3799. 2024. View Article : Google Scholar
|
|
169
|
Quan H, Yin M, Kim J, Jang EA, Yang SH,
Bae HB and Jeong S: Resveratrol suppresses the reprogramming of
macrophages into an endotoxin-tolerant state through the activation
of AMP-activated protein kinase. Eur J Pharmacol. 899:1739932021.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Li J, Zeng X, Yang F, Wang L, Luo X, Liu
R, Zeng F, Lu S, Huang X, Lei Y and Lan Y: Resveratrol: Potential
application in sepsis. Front Pharmacol. 13:8213582022. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Pandey S, Anang V, Singh S, Seth S, Bhatt
AN, Kalra N, Manda K, Soni R, Roy BG, Natarajan K and Dwarakanath
BS: Dietary administration of the glycolytic inhibitor
2-deoxy-D-glucose reduces endotoxemia-induced inflammation and
oxidative stress: Implications in PAMP-associated acute and chronic
pathology. Front Pharmacol. 14:9401292023. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Tan C, Gu J, Li T, Chen H, Liu K, Liu M,
Zhang H and Xiao X: Inhibition of aerobic glycolysis alleviates
sepsis-induced acute kidney injury by promoting lactate/Sirtuin
3/AMPK-regulated autophagy. Int J Mol Med. 47:192021. View Article : Google Scholar
|
|
173
|
Orsini EM, Roychowdhury S, Gangadhariah M,
Cross E, Abraham S, Reinhardt A, Grund ME, Zhou JY, Stuehr O, Pant
B, et al: TRPV4 regulates the macrophage metabolic response to
limit Sepsis-induced lung injury. Am J Respir Cell Mol Biol.
70:457–467. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Shao C, Lin S, Liu S, Jin P, Lu W, Li N,
Zhang Y, Bo L and Bian J: HIF1α-Induced glycolysis in macrophage is
essential for the protective effect of ouabain during endotoxemia.
Oxid Med Cell Longev. 2019:71365852019. View Article : Google Scholar
|
|
175
|
Zhang Z, Deng W, Kang R, Xie M, Billiar T,
Wang H, Cao L and Tang D: Plumbagin protects mice from lethal
sepsis by modulating immunometabolism upstream of PKM2. Mol Med.
22:162–172. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Virga F, Cappellesso F, Stijlemans B,
Henze AT, Trotta R, Van Audenaerde J, Mirchandani AS,
Sanchez-Garcia MA and Vandewalle J: Macrophage miR-210 induction
and metabolic reprogramming in response to pathogen interaction
boost life-threatening inflammation. Sci Adv. 7:eabf04662021.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Xu J, Gao C, He Y, Fang X, Sun D, Peng Z,
Xiao H, Sun M, Zhang P, Zhou T, et al: NLRC3 expression in
macrophage impairs glycolysis and host immune defense by modulating
the NF-κB-NFAT5 complex during septic immunosuppression. Mol Ther.
31:154–173. 2023. View Article : Google Scholar
|
|
178
|
Wang D, Li Y, Yang H, Shen X, Shi X, Li C,
Zhang Y, Liu X, Jiang B, Zhu X, et al: Disruption of TIGAR-TAK1
alleviates immunopathology in a murine model of sepsis. Nat Commun.
15:43402024. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Sfogliarini C, Pepe G, Dolce A, Della
Torre S, Cesta MC, Allegretti M, Locati M and Vegeto E: Tamoxifen
twists again: On and off-targets in macrophages and infections.
Front Pharmacol. 13:8790202022. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Pepe G, Sfogliarini C, Rizzello L,
Battaglia G, Pinna C, Rovati G, Ciana P, Brunialti E, Mornata F,
Maggi A, et al: ERα-independent NRF2-mediated immunoregulatory
activity of tamoxifen. Biomed Pharmacother. 144:1122742021.
View Article : Google Scholar
|
|
181
|
Xu Y, An X, Liu L, Cao X, Wu Z, Jia W, Sun
J, Wang H, Huo J, Sun Z, et al: Self-cascade redox modulator
trilogically renovates intestinal microenvironment for mitigating
endotoxemia. ACS Nano. 18:2131–2148. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Yeung ST, Ovando LJ, Russo AJ, Rathinam VA
and Khanna KM: CD169+ macrophage intrinsic IL-10 production
regulates immune homeostasis during sepsis. Cell Rep.
42:1121712023. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Saxton RA, Tsutsumi N, Su LL, Abhiraman
GC, Mohan K, Henneberg LT, Aduri NG, Gati C and Garcia KC:
Structure-based decoupling of the pro- and anti-inflammatory
functions of interleukin-10. Science. 371:eabc84332021. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Wu Y, Wang Q, Li M, Lao J, Tang H, Ming S,
Wu M, Gong S, Li L, Liu L and Huang X: SLAMF7 regulates the
inflammatory response in macrophages during polymicrobial sepsis. J
Clin Invest. 133:1502242023. View Article : Google Scholar
|
|
185
|
Zhu S, Chen Y, Lao J, Wu C, Zhan X, Wu Y,
Shang Y, Zou Z, Zhou J, Ji X, et al: Signaling lymphocytic
activation molecule Family-7 alleviates corneal inflammation by
promoting M2 polarization. J Infect Dis. 223:854–865. 2021.
View Article : Google Scholar
|
|
186
|
Shi Y, Zhu ML, Wu Q, Huang Y, Xu XL and
Chen W: The potential of drug delivery nanosystems for sepsis
treatment. J Inflamm Res. 14:7065–7077. 2021. View Article : Google Scholar
|
|
187
|
Hou X, Zhang X, Zhao W, Zeng C, Deng B,
McComb DW, Du S, Zhang C, Li W and Dong Y: Vitamin lipid
nanoparticles enable adoptive macrophage transfer for the treatment
of multi-drug-resistant bacterial sepsis. Nat Nanotechnol.
15:41–46. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Qu H, Wu J, Pan Y, Abdulla A, Duan Z,
Cheng W, Wang N, Chen H, Wang C, Yang J, et al: Biomimetic
nanomodulator regulates oxidative and inflammatory stresses to
treat Sepsis-associated encephalopathy. ACS Nano. 18:28228–28245.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Zhang D, Tang Z, Huang H, Zhou G, Cui C,
Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al: Metabolic
regulation of gene expression by histone lactylation. Nature.
574:575–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Mills EL, Ryan DG, Prag HA, Dikovskaya D,
Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E,
et al: Itaconate is an anti-inflammatory metabolite that activates
Nrf2 via alkylation of KEAP1. Nature. 556:113–117. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Lindell RB and Meyer NJ: Charting a course
for precision therapy trials in sepsis. Lancet Respir Med.
12:265–267. 2024. View Article : Google Scholar : PubMed/NCBI
|