Introduction
In the 21st century, cardiovascular disease (CVD)
has become one of the leading causes of mortality worldwide.
Statistics from the World Economic Forum show that CVD causes
>50% of non-communicable disease deaths and it is expected that
CVD will cause >22.2 million mortalities by 2030 (1). In China, the incidence and
lethality of CVD top the list, with cardiovascular-induced deaths
accounting for 46.74-44.26% of the total causes of death in rural
and urban areas, respectively, in 2019 (2). Since 1990, the health burden
imposed by CVD has been continuously escalating across most
countries worldwide, a trend closely linked to changes in the
exposure level to harmful risk factors, population growth and the
accelerated aging of the population. The Global Burden of Disease
Study 2023 provided updated insights into the epidemiological
profile of cardiovascular diseases worldwide (3). Data from this study indicate that
the global disability-adjusted life years (DALYs) of cardiovascular
diseases in 2023 reached 401-465 million, a 1.4-fold escalation
relative to the 1990 level (292-344 million). Among the various
cardiovascular conditions, ischemic heart disease, intracerebral
hemorrhage, ischemic stroke and hypertensive heart disease
accounted for the majority of the disease burden in terms of DALYs.
Regional disparities in the disease burden were evident: The
age-standardized DALY rate of cardiovascular diseases was the
highest in low and low-middle Socio-demographic Index (SDI) regions
and the lowest in high-SDI regions. In parallel with the growing
DALY burden, global mortality from cardiovascular diseases surged
from 12.2-14 million in 1990 to 17.4-20.4 million in 2023,
underscoring the persistent and worsening threat of cardiovascular
diseases to global public health (3,4).
The study projected that from 2025-2050, the global prevalence of
cardiovascular diseases would increase by 90.0%, the crude
mortality rate by 73.4% and the crude DALYs by 54.7%. Specifically,
the number of mortalities attributable to cardiovascular diseases
was estimated to reach 35.6 million in 2050, representing a marked
increase from the 20.5 million recorded in 2025 (5). CVD brings a heavy burden to society
and the study of the pathophysiological mechanism of its
development may help to improve the effectiveness of CVD diagnosis
and treatment. CVD includes atherosclerosis (AS), hypertension,
myocardial ischemia-reperfusion injury (MIRI), myocardial
remodeling and heart failure (HF) (6). Despite the significant improvement
in the treatment of CVD patients, there is still no effective drug
available for the cure of CVD. The pathogenesis of CVD is complex
and involves a variety of cytopathic processes such as cell
migration, proliferation, apoptosis, hypertrophy, regeneration,
endothelial cell dysfunction, cellular oxidative stress injury,
inflammatory response and myocardial fibrosis (7,8).
Members of the E26 transformation-specific (ETS)
transcription factor family share a highly conserved DNA-binding
domain and are involved in the regulation of a variety of
biological processes, including oncogenic transformation,
angiogenesis, differentiation and apoptosis (9,10). ETS-1/ETS-2 belongs to a subgroup
of ETS family members that can be expressed in different cell types
and tissues and play a variety of roles in both physiological and
pathological conditions play various roles (11). Currently, most of the ETS-1/ETS-2
studies are focused on tumor invasion and other aspects and
relatively few studies have been conducted in the field of the
cardiovascular system, but they have received more and more
attention from cardiovascular system researchers in recent years
because they are closely related to angiogenesis and cardiac
development and they can also promote cardiac remodeling and
vascular remodeling and other pathological processes. Therefore,
the present study hoped to elucidate the possible mechanisms of the
role of ETS-1/ETS-2 in related CVD, to provide new ideas for the
treatment of cardiovascular system diseases.
ETS family
ETS was originally discovered in the avian leukemia
retrovirus E26, which was transduced from a homologous gene in the
chicken genome and encodes a portion of a hybrid viral protein
(12). The ETS transcription
factor has long been thought to be associated with tumorigenesis
and its expression level correlates with tumor progression in
leukemia (13), thyroid cancer
(14), pancreatic cancer
(15), gastric cancer (16), hepatocellular carcinoma (17), prostate cancer (18), colon cancer (19), lung cancer (20) and breast cancer (21) and definitive evidence for its
role in human malignancies was initially discovered through the
discovery of recurrent translocations between the EWSR1 gene on
chr22 and the FLI1 gene on chr11 in Ewing's sarcoma (22). The ETS family is encoded by 28
genes and acts as transcriptional activators or repressors to
regulate target genes (23). The
ETS family proteins all contain a conserved sequence region known
as the ETS domain, which is a winged helix-turn-helix DNA-binding
domain composed of highly conserved 85 amino acids. This domain
forms three α-helices and a four-stranded β-sheet. It can bind to
the purine-rich GGAA/T core motifs present in the promoters and
enhancers of different target genes by interacting with other
transcription factors, cofactors and cis-elements close to the ETS
binding site (24-26). In Drosophila, the ETS
family of transcription factors consists of eight proteins:
Ecdysone-induced protein 74EF (EIP74EF), ETS-21c, ETS65A, ETS96B,
ETS97D, ETS98B, Pointed (PNT) and Anterior Open (Yan). Of these,
PNT and Yan are the most widely studied ETS factors (27). Among them, the PNT gene encodes
three isoforms, including PNT.P1, PNT.P2 and PNT.P3, all of which
share an ETS structural domain and exhibit similar DNA binding
activity (28). Human ETS
factors are divided into 11 subgroups: ETS (ETS-1/2), ERG, FLI1,
ETV (PEA3, ETV1/4/5), TEL (ETV6/7), ELG (GABPα), TCF (ELK1/3/4),
ELF (ELF1/2/4), SPI1 (SPI1/B/C), ERF (ERF, ETV3, ETV3L) and FEV
(29), all contain a conserved
PNT structural domain, which are involved in protein-protein
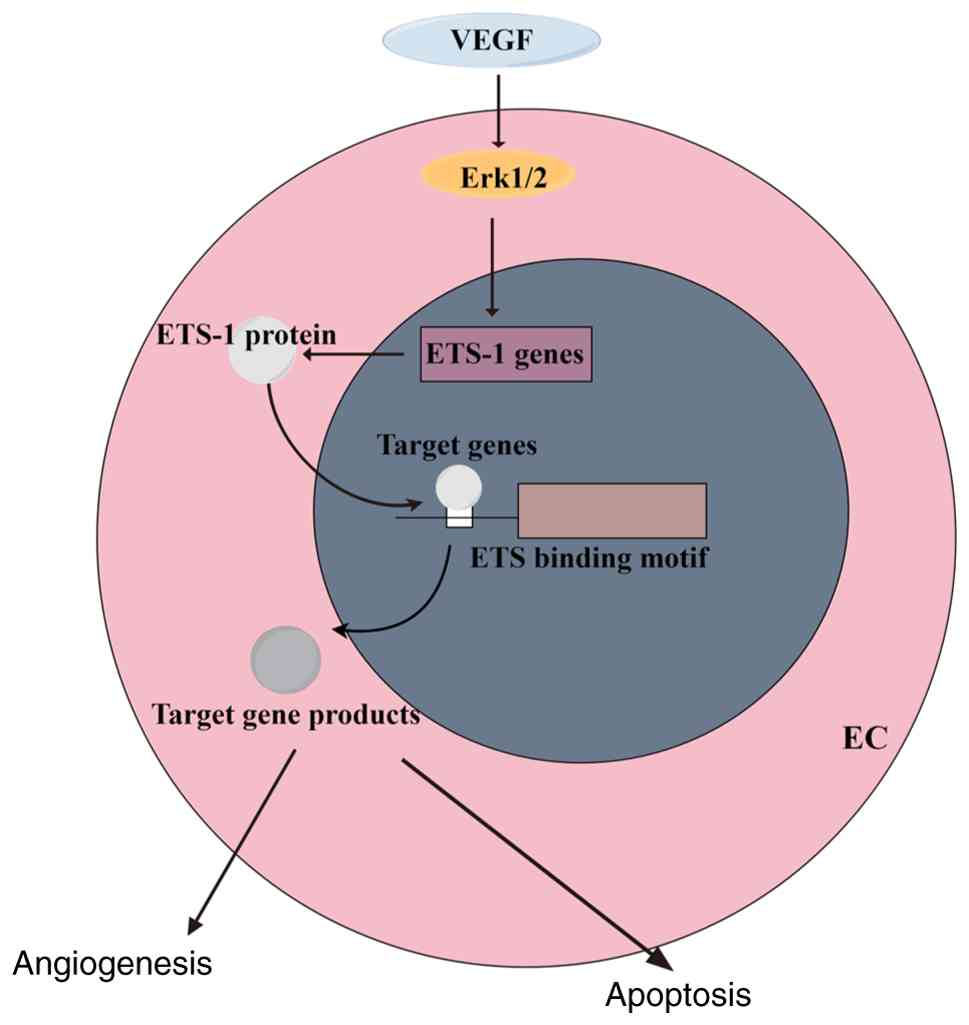
interactions (Fig. 1). During
early vertebrate embryogenesis, at least 13 ETS genes were found to
be expressed in hematopoietic or endothelial cells, including
ETS-1, ETS-2, Etv2, Etv6, Fli1, Erg, Fev, Gabpa, Elf1, Elf2, Spi1,
Spib and Elk4 (30,31). Moreover, ETS proteins regulate
various processes in embryonic development, such as mediating cell
proliferation, differentiation, transformation, apoptosis,
angiogenesis, or mediating the development of the hematopoietic or
cardiovascular system, by binding to the DNA-bound GGAA/T core
(32) (Fig. 2).
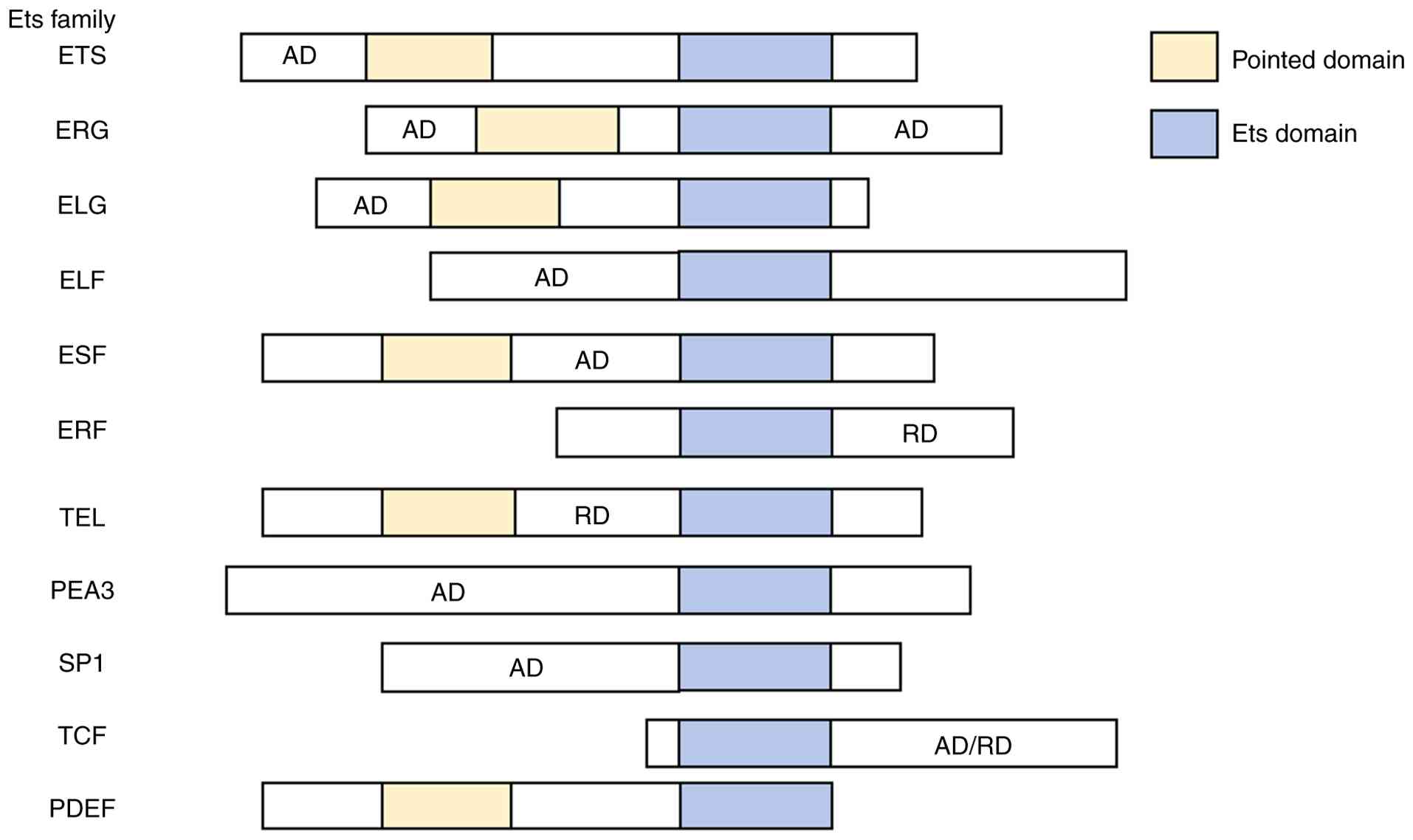 | Figure 1List of the members of the ETS family
and their domains. AD, activation domain; RD, repression domain;
ETS, E26 transformation-specific; ERG, ETS transcription factor;
ELG, ETS-like gene; ELF, eukaryotic translation release factor 3;
ESE, epithelial-specific ETS; ERF, ETS2 repressor factor; TEL,
translocation ETS leukemia; PEA3, polyomavirus enhancer activator
3; SP1, specificity protein 1; TCF, T-cell factor/lymphoid enhancer
factor; PDEF, prostate-derived ETS factor. |
Different signaling pathways and protein chaperones
can regulate the ETS factor, which is upregulated and activated by
various signaling pathways, including the Ras/mitogen-activated
protein kinase (MAPK) pathway and the phosphoinositide 3-kinase
(PI3K)/protein kinase B (AKT) pathway, in response to stimulation
by a variety of growth factors (33). ETS-1 and ETS-2 are representative
members of the ETS family of transcription factors, which are
identified on chromosomes 11 and 21, respectively and are
downstream effectors of the RAS/RAF/extracellular signal-regulated
kinase (ERK) pathway that enhance the expression of ETS-1 target
genes (34-37). Overexpression of
dominant-negative forms of ETS-1 or ETS-2 blocks Ras translation
(38), suggesting that members
of the ETS family play an essential role in this process.
ETS-1 and ETS-2
ETS-1 belongs to the ETS family of transcription
factors, also known as ETS proto-oncogene 1, and is the founding
member of the ETS family and was first identified as a fusion
protein expressed in conjunction with the E26 avian erythropoietic
virus GAG and MYB genes, which recognizes the conserved GGAA/T
motifs and binds to DNA through the winged helix-turn-helix motifs
known as ETS structural domains (39). In humans, the ETS-1 gene is
located on chromosome 11 (11q24.3). The human ETS-1 gene without
TATA contains eight exons, called exon A (the first exon) and exons
III-IX (the last seven exons) (40) and its product is the major
isoform of ETS-1 with 441 amino acids. The ETS-1 mRNA undergoes
selective splicing to produce three isoforms of the
protein-full-length protein (termed p51 or p54), a protein lacking
the sequence encoded in exon VII (termed p42) and a protein lacking
the sequence encoded in exons III-VI (termed p27) (41). A total of three different
targeting alleles of ETS-1 have been reported. The first was
reported by the BORIES laboratory, which produced an allele that
deleted the last two exons of the gene, which encode the
DNA-binding structural domain and are null alleles (42). Second, the allele reported by the
MUTHUSAMY laboratory is missing exon IV and part of the exon
(43). The last allele was
developed by HIGUCHI and targets the exon VII of the gene. This
targeting allele produces only the p42 isoform of ETS-1, which
lacks a self-repressor sequence and is therefore more active than
full-length ETS-1 (44). ETS-1
is a 54 kDa nuclear protein that primarily acts as a
transcriptional activator but also represses gene transcription
(45). The transcriptional
activity of the transcriptional activity of ETS can be regulated by
post-translational modifications (including phosphorylation,
ubiquitination, acetylation and sumoylation), transcription factors
and nuclear translocation (46).
ETS-1 is expressed in a variety of cells, including lymphocytes,
endothelial cells, vascular smooth muscle cells and epithelial
cells, but it is predominantly expressed in lymphocytes and, as
such, it plays a key role in immunity (47-49). In addition, it mediates
extracellular matrix degradation, cell migration, angiogenesis and
drug resistance (50).
ETS-2, like its closely related homolog ETS-1, is a
member of the ETS family of DNA-binding transcription factors
encoded in a 56 kDa nuclear protein containing 469 amino acids and
is also a downstream regulator of RAS-mediated transformation.ETS-2
is located on human chromosome 21q22.3 and has been reported to be
overexpressed in the brain and fibroblasts of subjects with Down
syndrome (DS) (51-53). The common core binding sequence
of ETS-2 is 5'-GGAA/T-3' and when combined with this core sequence,
ETS-2 supports a variety of cellular functions, such as cell
proliferation, adhesion, migration, survival and angiogenesis and
plays an important role in a variety of human diseases (54). Currently, previous studies have
demonstrated that the ETS-2 protein is associated with the
development of a variety of tumors and is an oncogene (55-57).
Relationship between ETS-1/ETS-2 and
CVD
Currently, CVD remains one of the leading causes of
mortality and morbidity in the global population. Despite the many
efforts made in the last few decades, the number of patients with
CVD is still high. Therefore, the need for the development of new
therapeutic strategies is crucial for a better understanding of the
biology of the cardiovascular system.
Traditionally, cancer and CVD have been viewed as
separate pathological entities. The only established association
between them was that cancer treatment could increase the risk of
cardiotoxicity and subsequent cardiovascular complications.
Nevertheless, mounting clinical evidence has demonstrated that
cancer patients are at a higher risk of developing CVD (58). Moreover, cancer and CVD share a
multitude of common risk factors and underlying pathogenic
mechanisms (59), including
diabetes mellitus, dyslipidemia, cachexia and impaired immune
function. Notably, the widely used 10-year risk score for
atherosclerotic cardiovascular disease is also predictive of cancer
incidence (60). Similarly,
cancer is a systemic disease that affects the cardiovascular system
through multiple mechanisms, including the release of small
molecules, modulation of immune cell activity and metastatic
lesions (61), thereby
exacerbating the progression of adverse outcomes and elevating the
risk of mortality in patients with CVD. A previous study based on
the Surveillance, Epidemiology and End Results (SEER) database,
which included more than 3.23 million U.S. cancer patients, clearly
demonstrated that CVD accounted for ~11.3% of all mortalities among
cancer patients (62). The
CVD-related mortality risk and all-cause mortality risk were both
markedly increased in this population. Such survival disadvantages
have been observed across multiple types of cancer, including
esophageal cancer, hepatocellular carcinoma, gastrointestinal
cancer, ovarian cancer, breast cancer, prostate cancer and lung
cancer (63-66). Wang et al (67) conducted a population-based cohort
study using data from the SEER program of the U.S. National Cancer
Institute. The authors reported that the incidence rate ratios
(IRR) of cardiovascular disease mortality among patients with lung
cancer were higher than those in the general population [IRR 1.74;
95% confidence interval (CI) 1.71-1.77]. Lung cancer in patients
aged 30-79 years was markedly associated with an increased risk of
cardiovascular disease mortality (IRR 2.61; 95% CI 2.55-2.66) and
this association was most pronounced in individuals aged 30-34
years (IRR 48.93; 95% CI 21.98-108.92). In 2025, Sabaté-Tormos
et al (68) performed a
retrospective cohort study enrolling 3,626 patients admitted to the
emergency department for suspected acute coronary syndrome between
2012 and 2013. Using univariate and multivariate Cox regression
models, the authors analyzed the associations of clinical variables
and a history of cancer with all-cause mortality over a 4-year
follow-up period. The results demonstrated that all-cause mortality
was markedly higher in cancer patients with myocardial infarction
(68.8%) than in cancer patients without myocardial infarction
(32.4%) and in non-cancer patients either with or without
myocardial infarction (11.3%). Multivariate analysis revealed that
a diagnosis of cancer was an independent predictor of mortality,
particularly in patients with concomitant myocardial infarction.
Although the link between cancer and cardiovascular-related
mortality has been well established, the core regulatory molecules
underlying cardiovascular system injury, imbalanced inflammatory
responses and abnormal tissue remodeling during the comorbidity of
cancer and cardiovascular disease remain to be elucidated.
Recently, ETS-1/ETS-2 has been recognized as a major
regulator of cardiovascular system development, playing an
important role in normal coronary artery and myocardial development
as well as in regulating vascular inflammation and remodeling
(69,70). For example, Wang et al
(71) demonstrated that
endothelial and/or endocardial ETS-1 is essential for the
regulation of ventricular cardiomyocyte proliferation and
homeostasis, as well as coronary vascular growth during cardiac
development. The data from this study showed that deletion of ETS-1
in endothelial cells resulted in ventricular noncompactification,
recapitulating the phenotype caused by the overall deletion of
ETS-1. Endothelial-specific deletion of ETS-1 reduces the levels of
six important angiogenesis-related genes, Alk1, Cldn5, Sox18,
Robo4, Esm1 and KDR, in endothelial cells, resulting in defective
development of the coronary vasculature system, which is associated
with reduced proliferation of cardiomyocytes in the dense zone.
Notably, defects in the coronary vasculature system cause the
development of congenital heart defects that can progress to early
heart failure. Therefore, further understanding of the important
mediating mechanisms of ETS-1/ETS-2 in related CVD is crucial and
provides promising therapeutic targets for the treatment of
CVD.
Est-1/ETS-2 and vascular
inflammation/vascular remodeling
Transient inflammation and its resolution after
vascular injury are necessary for wound healing and repair, whereas
prolonged excessive inflammation leads to pathological vascular
remodeling manifested as neointimal hyperplasia (72). Among other things, vascular
inflammation induces endothelial dysfunction and confers a
pro-adhesive and pro-thrombotic phenotype to endothelial cells,
which has a significant effect on the development and progression
of various CVDs, most notably AS, thrombosis and congestive HF
(73). Vascular remodeling is a
complex pathological process that is a fundamental pathological
process characterized by abnormal changes in vascular cell
morphology, structure and function in various diseases, such as
migration, proliferation, hypertrophy and apoptosis and is mostly
caused by vascular endothelial injury, medial smooth muscle
proliferation, extracellular matrix remodeling, oxidative stress
and vascular inflammation, which in turn leads to increased
vascular tone and thus the development of vascular disease
(74,75). Currently, studies have confirmed
that the role of ETS factors in regulating endothelial-specific
gene expression (76-78) and studies support a role for
several ETS family members in the regulation of vascular
inflammation and vascular remodeling, including endothelial
activation in response to inflammatory mediators, recruitment of
inflammatory cells to the vascular wall and proliferation and
migration of vascular smooth muscle cells (VSMCs) (79-81) (Fig. 3).
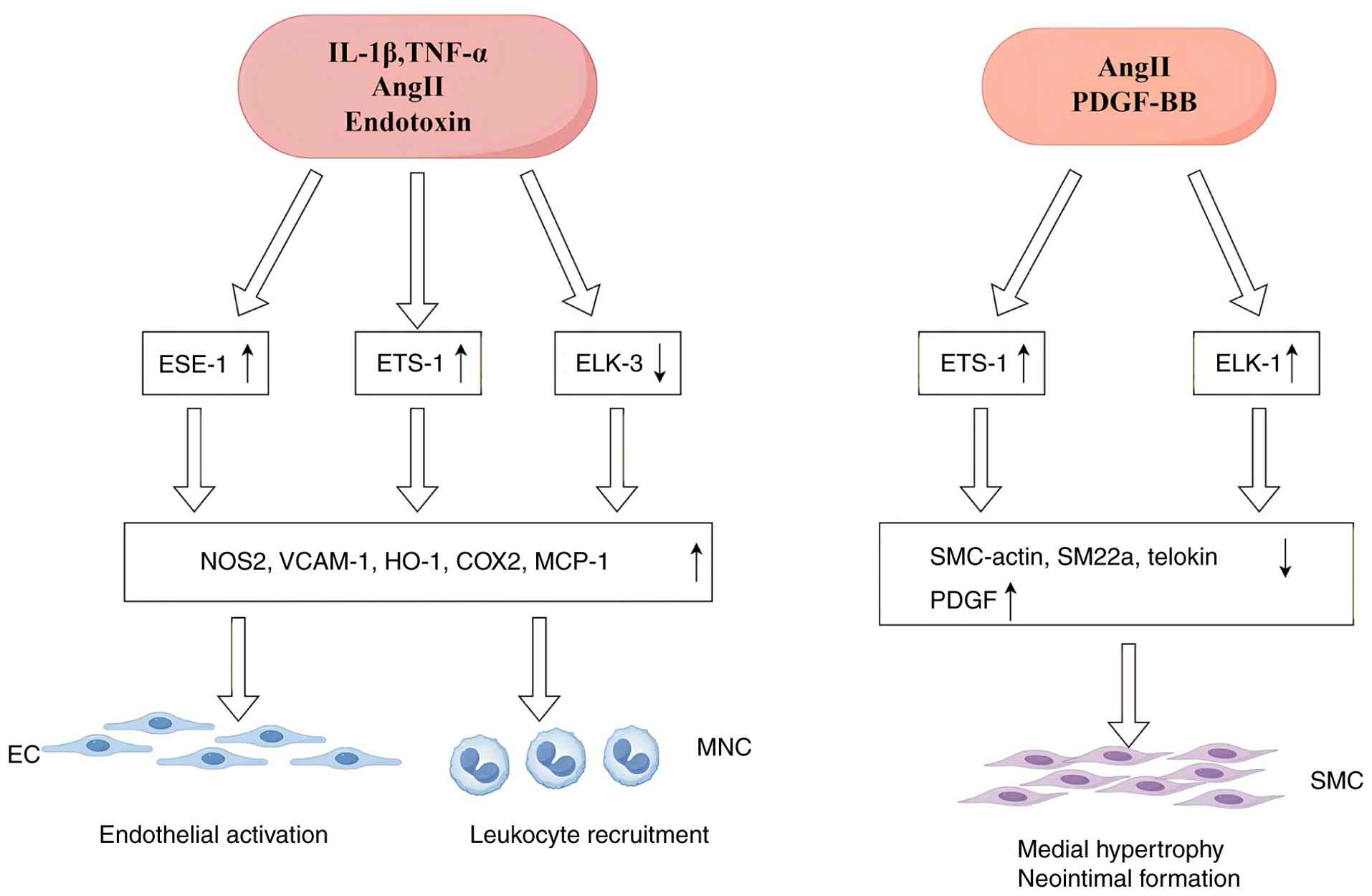 | Figure 3Role of ETS transcription factors in
the regulation of vascular inflammation: members of the ETS family
of transcription factors play a crucial role in the regulation of
vascular inflammation. Transcription factors such as ESE-1, ETS-1
and Elk-3 regulate the expression of related genes, thereby
influencing the activation of endothelial cells, the recruitment of
leukocytes and the proliferation and migration of vascular smooth
muscle cells (VSMCs). These processes are essential components of
the vascular inflammatory response (The diagram was drawn using
Figdraw 2.0; https://www.figdraw.com). ETS, E26
transformation-specific; IL-1β, interleukin-1β; TNF-α, tumor
necrosis factor-α; Ang II, angiotensin II; PDGF-BB,
platelet-derived growth factor-BB; EC, endothelial cell; SMC,
smooth muscle cell; ESE-1, epithelial-specific ETS 1; ELK-1,
ETS-like protein 1; NOS2, nitric oxide synthase 2; VCAM-1, vascular
cell adhesion molecule 1; HO-1, heme oxygenase 1; COX2,
cyclooxygenase 2; MCP-1, monocyte chemoattractant protein 1;
SMC-actin, smooth muscle cell actin; SM22α, smooth muscle 22α;
PDGF, platelet-derived growth factor. |
Angiotensin II (Ang II) is a central mediator of
vascular inflammation and remodeling and Zhan et al
(82) found in vascular smooth
muscle and endothelial cells from mouse thoracic aorta that the
transcription factor ETS-1 was rapidly induced in response to
systemic Ang II infusion and that compared with control mice,
ETS-1−/− mice had markedly reduced arterial wall
thickening, perivascular fibrosis and cardiac hypertrophy. Also,
cell cycle-dependent kinase inhibitor p21CIP (associated with cell
hypertrophy, cell cycle arrest, apoptosis and endothelial
dysfunction), plasminogen activator inhibitor-1 (PAI-1; which
promotes the development of perivascular fibrosis) and monocyte
chemotactic protein-1 (MCP-1; a central mediator of inflammatory
responses in hypertensive vascular disease) were identified in the
study as ETS-1 targets, all of which were reduced in expression in
ETS-1−/− mice in response to Ang II and resulted in a
significant reduction in T cell and macrophage recruitment to the
vessel wall compared with control mice. This suggests that ETS-1
plays a key role in Ang II-induced vascular inflammation and
remodeling. Vascular inflammation plays an important role in the
pathogenesis of hypertension, atherosclerosis, restenosis and other
forms of vascular disease. In particular, carotid balloon injury is
a model of vascular injury characterized by increased expression of
inflammatory mediators and marked leukocyte infiltration, which
leads to a process of vascular remodeling and neointimal formation
after luminal injury (83,84). Feng et al (85) found that the expression of ETS-1
was markedly increased after balloon injury, which mediated the
increased expression of pro-inflammatory molecules and adhesion
molecules in carotid balloon injury, including MCP-1, P-selectin
and E-selectin and that the blockade of ETS-1 before balloon injury
not only reduced leukocyte infiltration but also prevented the
development of neointima after balloon injury. Taken together,
ETS-1 plays a key role as a transcriptional mediator of
inflammation and neointima formation induced by intraluminal
vascular injury and has the potential to be a new strategy for the
treatment and prevention of diseases associated with vascular
injury.
Est-1/ETS-2 and angiogenesis
Angiogenesis is a complex event that requires
endothelial cell germination, lumen formation and tubulogenesis and
is regulated by the coordinated action of different transcription
factors. Angiogenesis is involved in a variety of biological
processes, including atherosclerosis and cancer. Many transcription
factors have been implicated in vascular development and
angiogenesis and their interactions lead to endothelial cell
differentiation and the acquisition of arterial, venous and
lymphatic properties (86). For
example, Di et al (87)
found that miR33a-5p/ETS-1/Dickkopf1 (DKK1) signaling was
pro-angiogenic in ox-LDL-induced HUVECs.
Endothelial cells (ECs) are located in the innermost
layer of blood vessels and form the vascular lining, which is
important for maintaining vascular homeostasis and normal
circulation (88). In response
to endothelial dysfunction caused by stimulation by pathological
(hyperbaric or hypoxic) and chemical (oxidized low-density
lipoprotein or infection) factors, ECs are activated, increasing
their permeability, releasing cytokines to promote inflammation and
thrombosis and increasing adhesion molecule recruitment and
adhesion to inflammatory factors (89). Endothelial dysfunction and
endothelial activation are thought to play a key role in the
initiation of the atherosclerotic process and the progression of
advanced AS (90). AS is a
chronic vascular inflammatory disease and is one of the leading
causes of cardiovascular morbidity and mortality (91). The development of AS is
associated with structural vascular lesions and the pathologic
process involves endothelial damage, lipid deposition, inflammatory
cell infiltration, foam cell formation and plaque formation,
leading to the formation of atherosclerotic plaques (92). These plaques narrow the lumen of
the coronary arteries, leading to episodic or persistent angina. If
the plaque ruptures, it can lead to thrombus formation, myocardial
infarction and even death (93).
AS involves complex interactions between various cell types,
including macrophages, ECs and smooth muscle cells (SMC) (94). AS is a complex disease involving
multiple factors. Currently, it has been demonstrated that
ETS-1/ETS-2 plays a role in the regulation of vascular inflammation
in AS, including endothelial activation in response to inflammatory
mediators, recruitment of inflammatory cells to the vascular wall
and proliferation and migration of VSMCs (79). ETS-1 is upregulated in
vitro upon exposure to agonists such as serum and is expressed
in damaged vessels and it is also expressed in SMCs in human
carotid atherosclerotic lesions (95). Similarly, it has been shown that
in ApoE−/− mice experiments using a vulnerable plaque
model, ETS-2 expression is increased under atherogenic conditions
and is specifically enhanced in vulnerable plaques compared to
stable plaques. ETS-2 levels are associated with microvessel
formation within the plaque and proinflammatory in human vulnerable
atherosclerotic plaques ETS-2 levels correlate with intraplaque
microvessel formation and proinflammatory cytokine levels in human
susceptible atherosclerotic plaques and overexpression of ETS-2
promotes lesion growth with neovascularization, hemorrhage and
plaque destabilization (96).
Currently, it has been shown that ETS-2 is critical
for a variety of inflammatory functions in human macrophages and
that macrophage activation, cytokine production, reactive oxygen
species (ROS) production, phagocytosis and migration are induced by
overexpression of ETS-2 in a dose-dependent manner and that
overexpression of ETS-2 increases the secretion of pro-inflammatory
cytokines. RNA sequencing of ETS-2 edited and unedited inflammatory
macrophages from multiple donors reveals that disruption of ETS-2
results in extensive transcriptional changes and reduced expression
of many inflammatory genes. This suggests that macrophage ETS-2
signaling may play a central role in a variety of inflammatory
diseases (97). Moreover, Jiang
et al (80) found that
ETS-1 promotes hyperglycemia-mediated endothelial inflammation by
upregulating protein tyrosine phosphatase 1B (PTP1B) expression,
which is involved in high glucose-induced elevation of endothelial
cell inflammatory factor levels). In addition, it was found that
hypoxia-inducible factor-1α (HIF-1α), vascular endothelial growth
factor (VEGF) and ETS-1 coexist in the deeper layers of plaques in
carotid atherosclerosis, with significant angiogenesis. Notably,
the major lesions of plaques exhibit markedly elevated levels of
HIF-1α, VEGF and ETS-1. This suggests that HIF-1α induces
angiogenesis through the expression of VEGF and ETS-1 (98). In addition, Katsume et al
(99) also found that the early
inflammatory response in atherosclerosis is mediated by
proline-rich tyrosine kinase/reactive oxygen species, which induces
the release of tumor necrosis factor α (TNF-α) and promotes the
expression of TNF-α-dependent pro-inflammatory molecules through
the p21Cip1/ETS-1/p300 transcriptional regulatory
system. Endothelial dysfunction represents the initial step of AS.
Endothelial dysfunction is the initial step of AS (100). 17β-estradiol (E2) can delay
atherosclerosis formation by improving lipid levels, accelerating
endothelial repair by promoting endothelial proliferation and
migration, inhibiting the proliferation and migration of vascular
smooth muscle cells and modulating the function of immune cells to
alleviate the vascular inflammatory response (101-103). Li et al (104) demonstrated the effect of E2 on
miR-126-3p expression (which is critical in the proliferation,
migration and tube formation) in a cellular model by utilizing
human venous umbilical cord endothelial cells. Using human venous
umbilical cord endothelial cells, Li et al (104) demonstrated the effect of E2 on
microRNA (miRNA/miR) 126-3p expression (which plays a critical role
in endothelial cell proliferation, migration and tube formation),
which prevents endothelial cell apoptosis and slows down
atherogenesis in a cellular model and identified the key role of
ETS-1, a transcription factor for miR-126-3p. It was found that E2
increased the expression level of miR-126 through ETS-1 in HUVEC
and then miR-126-3p bound to its targets Spred1 and vascular cell
adhesion molecule-1 (VCAM-1) mRNA, leading to their degradation and
silencing of protein expression and finally promoted endothelial
proliferation, atherogenesis and atherosclerosis. In addition, Nie
et al (105) showed that
the ETS-2 transcription factor is a potent regulator of
angiogenesis and mediates immune activation of the endothelium in
the late stages of AS. miR-203 promotes the proliferation of
vascular smooth muscle cells and was found to be involved in the
process of atherosclerotic plaque formation through the regulation
of ETS-2. Immediately after, Nie et al (105) found that shangou, a common
traditional aromatic herb, reduced atherosclerotic plaque area and
serum antioxidant low-density lipoprotein (LDL) autoantibodies in
endogenous high angiotensin II ApoE−/− mice by
increasing miR-203 expression and decreasing ETS-2 expression,
which was beneficial in preventing or treating the onset and
progression of AS. In addition, it has been demonstrated that
tyrosine kinase receptor B (TrkB) plays an important role in
protecting the integrity of the endothelial barrier during the
process of atherogenesis and Jiang et al (106) found that TrkB can promote
VE-calmodulin expression in endothelial cells by inducing and
activating the expression of ETS-1, which binds to two ETS binding
sites in the promoter of the VE-calmodulin gene in endothelial
cells. ETS-1 promotes VE- cadherin expression by binding to two ETS
binding sites in the VE- cadherin promoter in endothelial cells to
protect endothelial integrity during atherogenesis.
Inflammation is observed in all stages of
atherosclerosis, the initial phase of which is characterized by
leukocyte recruitment to activated ECs. Zhu et al (107) used bioinformatics analysis to
show that ETS-1 is a key endothelial transcription factor in
inflammation and tube formation and is a candidate target for the
miR-155 and miR-221/222 clusters and that miR-155 and miR-221 are
highly expressed in human umbilical vein endothelial cells (HUVECs)
and VSMCs, confirming that angiotensin II type 1 receptor (AT1R) is
a target of miR-155 in HUVECs. It was found that HUVEC with high
miR-155 expression may co-target AT1R and ETS-1, while miR-221/222
targets ETS-1, which indirectly regulates the expression of several
inflammatory molecules in ECs, thereby attenuating the adhesion of
Jurkat T-cells to activated HUVECs and decreasing the migration of
HUVECs and thus reducing the inflammation of endothelial cells and
ameliorating atherosclerosis.
ETS-1/ETS-2 and MIRI
Ischemic heart disease has become the leading cause
of morbidity and mortality worldwide (108). Myocardial cell death plays a
central role in the pathogenesis of ischemic heart disease;
therefore, timely restoration of coronary blood flow, that is,
reperfusion, is the best method to reduce myocardial cell loss
induced by ischemic injury (109). However, myocardial reperfusion
can lead to further damage to the heart, termed MIRI (110). MIRI leads to myocardial
dysfunction, cardiomyocyte loss and fibrosis. The process of
myocardial ischemia/reperfusion can lead to several types of
cardiomyocyte death, including necrosis, apoptosis, autophagy and
iron death (111). These new
forms of regulated cell death lead to cardiomyocyte loss and
exacerbate MIRI by influencing ROS production, calcium stress and
inflammatory cascade responses, which subsequently mediate adverse
remodeling, cardiac insufficiency and HF. Therefore, prevention of
cardiac apoptosis and fibrosis has been the goal of some therapies
that interfere with MIRI.
Lee et al (112) found that adipose-derived stem
cell conditioned medium (ADSC-CM) had a significant effect on
apoptosis and fibrosis in MIRI-treated hearts and
hypoxia/reoxygenation (H/R)-treated cardiomyocytes. ADSC-CM
attenuates cardiac apoptosis and fibrosis by downregulating the
expression of p53 upregulated modulator of apoptosis (PUMA) and
ETS-1 via the miR-221/222/p38/NF-κB pathway, thereby alleviating
MIRI. The approximate mechanisms are as follows:
ADSC-CM treatment markedly reduced the expression of
apoptosis-related proteins, such as PUMA, phosphorylated (p-)p53
and B-cell lymphoma 2 (Bcl2), as well as the fibrosis-related
proteins, ETS-1, fibronectin and collagen 3, and markedly
attenuated the cardiac injury and fibrosis in both MIRI-treated
mice and H/R-treated cardiomyocytes.
ADSC-CM is abundant in miR-221/222, which can target
and regulate the protein levels of PUMA or ETS-1. Knockout of PUMA
and ETS-1 reduced the induction of apoptosis and fibrosis,
respectively, and overexpression of miR-221/222 achieved similar
results. Furthermore, MIRI significantly increased apoptosis and
fibrosis in miR-221/222 knockout (KO) mice, whereas ADSC-CM
attenuated these effects.
Increased phosphorylation of p38 and NF-kB not only
mediates myocardial apoptosis through the PUMA/p53/Bcl2 pathway,
but also regulates fibrosis through the ETS-1/fibronectin/collagen
3 pathway. In addition, Lai et al (113) also found that both PUMA and
ETS-1 are target genes of miR-221/222. The addition of
H2O2 to cultured cardiomyocytes increases the
levels of PUMA and ETS-1 and treatment with adipose-derived stem
cell exosomes markedly inhibits the levels of PUMA and ETS-1 and
reduces apoptosis and hypertrophy through the
miR-221/miR-222/PUMA/ETS-1 pathway, thereby preventing cardiac I/R
injury.
Apoptosis, ROS and inflammation-induced myocardial
injury are the most critical factors in MIRI (114). It has been demonstrated that
the lysine (K)-specific demethylase 3A (KDM3A), plays a key role in
MIRI. Guo et al (115)
found that KDM3A knockdown exacerbated myocardial dysfunction and
cardiomyocyte injury in vivo and in vitro, while
worsening mitochondrial apoptosis, ROS and inflammation were
observed. By contrast, KDM3A overexpression was ameliorated in
MIRI. At the same time, it was found that KDM3A, which is known to
target ETS-1, directly regulates ETS-1 expression by binding to its
promoter region. KDM3A overexpression leads to higher ETS-1
transcription, resulting in reduced apoptosis, ROS and
inflammation, which plays a protective role during myocardial
injury. Thus, the data from this study suggest that the protective
effect of KDM3A on MIRI is partially attributable to epigenetic
transactivation of ETS-1.
Thus, it is clear that targeting ETS-1 plays a
critical role in preventing MIRI-induced apoptosis and hypertrophy.
However, there is no experimental data to analyze whether ETS-2 can
prevent MIRI through certain mechanisms.
ETS-1/ETS-2 and cardiac remodeling
Cardiac remodeling, defined as genomic, molecular,
cellular and interstitial alterations clinically manifested as
changes in cardiac size, shape and function in response to
cardiovascular injury and pathogenic risk factors, is at the core
of most CVD and persistent cardiac remodeling almost always ends in
HF and mortality (116).
Cardiac remodeling is categorized as physiological (in response to
growth, exercise and pregnancy) and pathological (in response to
inflammation, ischemia, MIRI, biomechanical stress, excessive
neurohormonal activation and pressure overload). Among these,
pathological cardiac remodeling is the process of structural and
functional changes in the left ventricle as a result of internal or
external cardiovascular injury or pathogenic risk factors (117,118). Pathological changes in
pathological cardiac remodeling include cardiomyocyte apoptosis,
cardiac hypertrophy and cardiac fibrosis and persistent adverse
remodeling can lead to HF (119,120). Therefore, it is crucial to
understand the mechanisms leading to cardiac remodeling and to
prevent adverse remodeling. Research has demonstrated the critical
role of ETS-1/ETS-2 in the development of cardiac remodeling.
The process of diabetic cardiomyopathy involves a
series of sequential and interrelated steps, including myocardial
apoptosis, hypertrophy and fibrosis. Cardiomyocyte apoptosis is key
to this process and to myocardial remodeling. Wang et al
(121) found that elevated
levels of high mobility group box 1 (HMGB1) may promote
cardiomyocyte apoptosis induced by high glucose (HG) or
hyperglycemia. HG can activate ETS-1; however, inhibition of HMGB1
can attenuate HG-induced ETS-1 activation through ERK1/2. However,
inhibition of HMGB1 attenuates HG-induced ETS-1 activation through
ERK1/2 signaling. In addition, inhibition of ETS-1 markedly reduces
HG-induced cardiomyocyte apoptosis. In conclusion, inhibition of
HMGB1 may prevent HG-induced cardiomyocyte apoptosis by
downregulating ERK-dependent activation of ETS-1.
The two major cellular signaling pathways that drive
cardiac hypertrophy are the calcineurin/nuclear factor of activated
T-cells (NFAT) pathway and the MAPK/ERK pathway and sustained
activation of either of these pathways causes myocardial
hypertrophy (122,123). Luo et al (124) found that ETS-2 is activated
during hypertrophic myocardial growth, and ETS-2 deficiency
attenuates cardiac hypertrophy induced by pressure overload. In
addition to this, ETS-2 was observed to be activated by Erk1/2 in
hypertrophic mouse hearts and in human dilated cardiomyopathy
tissues in this study. ETS-2 is also required for pressure overload
and calmodulin-induced cardiac hypertrophy and, as a transcription
factor, ETS-2 is able to bind to the promoters of hypertrophic
hallmark genes, such as atrial natriuretic peptide, brain
natriuretic peptide and Rcan1.4, which is an established downstream
target of NFAT. ETS-2 forms a complex with NFAT, stimulating
transcriptional activity by enhancing NFAT binding to the promoters
of at least two hypertrophy-stimulating genes: Rcan1.4 and miR-223
(which are downstream targets of ETS-2 and NFAT in cardiomyocytes).
Inhibiting miR-223 in cardiomyocytes suppresses
calcineurin-mediated cardiac hypertrophy, revealing that
microRNA-223 is a novel pro-hypertrophic target of the
calcineurin/NFAT and Erk1/2-ETS-2 pathways. In conclusion, ETS-2
plays a critical role in cardiac hypertrophy driven by the
calmodulin/NFAT pathway and reveals a molecular link between the
Erk1/2 activation of ETS-2 and the expression of NFAT/ETS-2 target
genes. Furthermore, Zhan et al (82) demonstrated that ETS-1 is a key
mediator of vascular remodeling and inflammation in response to Ang
II. The authors showed that vascular proliferation, perivascular
fibrosis and cardiac hypertrophy, were markedly reduced in
ETS-1−/− mice in response to systemic administration of
Ang II compared to control mice.
ETS-1, as a prototypical member of the ETS family of
transcription factors, has now been demonstrated in several studies
to play an important role in myocardial fibrosis. Hao et al
(125) identified a role for
ETS-1 in the pro-fibrotic effects of Ang II in cardiac fibroblasts
(CFs) and in the heart in vivo. In the left ventricle of Ang
II-infused rats, ETS-1, plasminogen activator inhibitor-1 (PAI-1)
and connective tissue growth factor (CTGF) protein levels were
concomitantly upregulated, while activation of ERK and c-Jun
NH2-terminal kinase was increased. Knockdown of ETS-1 by short
interfering RNA markedly inhibits the induction of cell
proliferation and CTGF and PAI-1 expression by Ang II. Thus, ETS-1
may mediate Ang II-induced cardiac fibrotic effects. These findings
point for the first time to the potential role of ETS-1 in cardiac
fibrosis in vitro and in vivo, contributing to a
fundamental understanding of the molecular mechanisms underlying
myocardial fibrosis as a pathological process and laying the
foundation for further studies. Subsequently, it was suggested that
endothelial-mesenchymal transition (EMT) promotes Ang II-mediated
cardiac fibrosis and that ETS-1 has a potential role in Ang
II-mediated myocardial fibrosis. Xu et al (126) found that endothelial
ETS-1-deficient mice exhibited markedly reduced cardiac fibrosis
and hypertrophy after Ang II infusion, accompanied by decreased
expression of fibrotic stromal genes, EMT (which is a possible
source of myofibroblasts and has been shown to cause myocardial
fibrosis) as well as its downstream transcription factors
(including Snail family transcriptional repressor 1, Snail family
transcriptional repressor 2, Twist-related protein 1 and Zinc
finger E-box binding homeobox 1), as well as reduced expression of
cardiac hypertrophy and hypertrophy-related genes. Moreover, in
vitro studies using cultured H5V cells further demonstrated
that ETS-1 knockdown inhibited TGF-β1-induced EMT. Thus, it is
evident that deletion of endothelial ETS-1 attenuates Ang
II-induced myocardial fibrosis through inhibition of EMT and that
inhibition of ETS-1 may be a potential therapeutic strategy for
delaying myocardial fibrosis.
Aldosterone plays an important role in the
pathophysiology of cardiac remodeling and recently, spironolactone
(an aldosterone receptor antagonist) has been found to improve
cardiac structure, function and prognosis and delay the progression
of myocardial fibrosis in patients with HF. Wang et al
(127) found that
spironolactone reduced the levels of inflammatory cytokines in the
myocardial tissues of autoimmune myocarditis (EAM) and had a
positive effect on experimental myocardial fibrosis in EAM mice.
This study found that ETS-1 was activated through the
TGF-β1/Smad-2/3 signaling pathway and is involved in EAM-induced
cardiac fibrosis in EAM mice and the use of spironolactone markedly
inhibited the activation of ETS-1 and the phosphorylation of
TGF-β1/Smad2/3 and suppressed ETS-1-mediated myocarditis-induced
myocardial fibrosis. In addition, inhibition of ETS-1 also reduced
the expression and activity of matrix metalloproteinase (MMP)-2 and
MMP-9 in EAM mice, which reduced myocardial fibrosis. The results
of this study suggest that the improvement of myocardial fibrosis
by spironolactone may be related to the TGF-β1/Smad-2/3/ETS-1
signaling pathway in EAM mice.
In addition, abnormal levels of chitinase-3-like
protein 1 (CH3L1) and long non-coding RNA (lnc)TUG1 are associated
with myocardial fibrosis. Sun et al (128) found that CH3L1 upregulated the
expression of lncTUG1 and lncTUG1 weakened the inhibitory effect on
ETS-1 through sponge adsorption of miR -495-3p, which promoted the
process of myocardial fibrosis. Similarly, Li et al
(129) isolated exosomes from
MSC and administered them in vitro to CFs and male infarcted
Sprague-Dawley rat hearts in vivo with vericiguat-treated
mesenchymal stem cell-derived exosomes (MSCVER-Exos) and found that
MSCVER-Exo was able to inhibit the proliferation of CFs. MSCVER-Exo
was found to inhibit CFs proliferation, migration and pro-fibrotic
gene expression. Meanwhile, miR-1180-3p was found to be enriched in
MSCVER-Exo and ETS-1 was a downstream target of miR-1180-3p.
miR-1180-3p could be delivered to CFs through Exo and attenuate
TGF-β1-induced fibrosis by inhibiting ETS-1 signaling. Thus,
miR-1180-3p targeting ETS-1 plays an important role in
anti-fibrosis.
ETS-1/ETS-2 and Jacobsen syndrome
(JS)
JS is a rare syndrome caused by partial chromosomal
deletions in the 11q23 region between sub-band 11q23.3 and
telomeres, ranging from approximately 7 Mb to 20 Mb (130). The condition was first
described in 1973 by Dr Jacobsen, in which multiple members of a
family inherited an unbalanced 11;21 translocation from parents who
carried a balanced translocation chromosome (131). Since Jacobsen's first report,
more than 200 cases have been noted (132). In cases of JS diagnosed after
birth in the neonatal period, the majority of children with JS have
prolonged hospitalizations, most commonly due to feeding
difficulties, heart problems, or bleeding problems. Of these
children, ~20% succumb within the first two years of life, most
commonly related to complications of congenital heart disease, most
of which require surgical intervention. Of patients with heart
defects, ~1/3 have membranous ventricular septal defects, another
third have left ventricular outflow tract defects with varying
degrees of hypoplasia or obstruction of the mitral valve, left
ventricle, aortic valve, or aorta and the final third present with
a variety of heart defects, including right ventricular double
outlet, transposition of the great arteries, atrioventricular
septal defect, ostium secundum atrial septal defect, dextrocardia,
anomalous right subclavian artery, patent ductus arteriosus,
persistent left superior vena cava, tricuspid atresia, interrupted
aortic arch type B, persistent truncus arteriosus and pulmonary
valve stenosis (133,134). It has been shown that ETS-1
transcription factors play an important role in mammalian heart
development, thus providing important insights into some of the
most common congenital heart diseases. For example, Ye et al
(135) identified an ~7
megabase (Mb) cardiac critical region distal to the long arm of
chromosome 11 that contained a gene for congenital heart disease,
ETS-1, which is a candidate for causing congenital heart disease in
at least some of the patients with 11q- JS. In addition, phenotypic
analysis of gene-targeted ETS-1 KO mice by Ye et al
(135) showed that deletion of
ETS-1 in a pure C57/B6 background resulted in a high prevalence of
ectopic, large membranous ventricular septal defects and frequently
abnormal ventricular morphology characterized by a bifid apex.
Thus, in summary, these results suggest a role for ETS-1 in
ventricular development and a gene in the critical region of
Jacobsen syndrome, the deletion of which leads to ventricular
septal defects and abnormal ventricular morphology in mice.
Ventricular septal defects, including double outlet
right ventricle, are one of the most common congenital heart
diseases (CHDs) in JS. One possible mechanism for CHDs and other
problems in Jacobsen syndrome is a defective function of neural
crest cells (cNCCs), which are required for normal mouse cardiac
development to accurately regulate the differentiation of cNCCs. Ye
et al (136) again found
that ETS-1 is strongly expressed in mouse cNCCs and that the
expression of Sox10, a key regulator of the gene regulatory network
of neural crest cells and an early marker of NCCs, helps to analyze
the function of cNCCs during embryonic development. However, this
study found that deletion of ETS-1 leads to a decrease in migrating
cells expressing Sox10 in E10.5 C57/B6 mouse embryos, which results
in impaired cardiac NCC function and causes congenital heart
defects.
ETS-1/ETS-2 and HF
HF has become a rapidly growing global health
problem affecting more than 37.7 million people worldwide. The
prevalence of HF has been increasing in this decade as the
population over the age of 65 increases (137). Although many different
approaches have been investigated to help manage HF, including
medical devices, medications and telemonitoring, the clinical
management of HF remains relatively poor (138). Therefore, there is an urgent
need for clinicians to investigate new therapies for the treatment
of HF.
Dysregulation of gene expression is a common
mechanism in HF and transcription factors (TFs) are thought to play
an important role in the regulation of gene expression. Li et
al (139) analyzed the
expression levels of miR-155, G protein-coupled receptor 18 (GRP18)
and ETS-2 in clinical HF samples through the use of bioinformatics
and experimental analysis and functional validation was performed
in H9c2 cells. A total of three miRNAs and eight TFs were
identified in the TF/miRNA target network. clinical validation
showed that the expression level of miR-155 was markedly reduced in
HF samples, whereas the expression levels of ETS-2 and GPR18 were
markedly increased in HF samples and GPR18 was found to be targeted
by both ETS-2 and miR-155. Thus, the data suggest that miR-155 and
ETS-2 may play a key role in the development of HF by targeting
GPR18 and this finding may help to provide new information for
understanding and treating HF. In addition to this, Tan et
al (140) found that ETS-2
could promote cardiomyocyte apoptosis and autophagy in HF by
regulating the lncRNA TUG1/miR-129-5p/ATG7 axis. In this
experiment, it was found that H2O2
stimulation increased the expression of ETS-2, TUG1 and ATG7 and
decreased the expression of miR-129-5p in AC16 cells, which
stimulated cardiomyocyte apoptosis and autophagy, whereas the
absence of ETS-2, the silencing of TUG1, or the upregulation of
miR-129-5p reversed this phenomenon.
DNA methyltransferase 1 (DNMT1) expression is
rapidly upregulated in cardiac tissues of mice with pressure
overload and adriamycin-induced HF, which promotes the development
of HF (141). In addition to
this, dysregulation of mitochondrial autophagy has been associated
with the progression of HF and aging (142). Deng et al (143) pointed out that upregulation of
miR-152-3p plays a protective role in cardiomyocytes and the STRING
database predicted that there is an interaction between ETS-1 and
Ras homologous gene family member H (RhoH) and that ETS-1/RhoH may
inhibit mitochondrial autophagy and exacerbate HF. Therefore,
targeting the ETS-1/RhoH axis may be a potential therapeutic
approach. Meanwhile, Deng et al (143) found that DNMT1 may inhibit
miR-152-3p expression by promoting the methylation of miR-152-3p
and enhancing the expression of ETS-1, which induces RHOH
transcriptional activation and inhibits mitochondrial autophagy and
ultimately promotes the development of HF.
Treatment
ETS-1 and ETS-2, key members of the ETS
transcription factor family, play a central role in regulating cell
proliferation, angiogenesis, inflammatory responses and fibrosis,
which renders them potential therapeutic targets for various
diseases, including cardiovascular diseases, cancer and autoimmune
disorders (144). However, the
translational potential of ETS-1/ETS-2 has been disproportionately
explored in the field of oncology, whereas research targeting
non-neoplastic diseases (e.g., cardiovascular lesions) has been
mainly confined to preclinical basic studies with limited
translational progress. To date, a large number of mechanistic
studies, preclinical models and even early exploratory trials have
elucidated the therapeutic value of modulating ETS-1/ETS-2 activity
for cancer intervention, covering diverse strategies such as
small-molecule inhibitors. By contrast, the therapeutic targeted
application of ETS-1/ETS-2 in cardiovascular and other
non-malignant diseases is still in its infancy. Given the
significant discrepancy in research maturity, this section will
focus specifically on the current status of ETS-1/ETS-2-targeted
therapeutic strategies in tumorigenesis, summarizing key advances,
challenges and directions for future clinical translation.
ETS-1/ETS-2 are members of the ETS family
characterized by broad tissue distribution and potent
transcriptional regulatory capacity. Their biological functions
span the development and differentiation of lymphocytes and they
can markedly regulate the proliferation, migration and invasion
phenotypes of multiple cell types. In the tumor pathological
process, the dysregulated expression of ETS-1/ETS-2 is closely
associated with the progression of various malignant tumors. By
participating in key steps of cancer invasion, including
angiogenesis, extracellular matrix degradation and tumor
metastasis, ETS-1/ETS-2 serve as important molecules mediating
tumor malignant phenotypes (145). Therefore, ETS-1/ETS-2 are
regarded as promising targets for cancer therapy and their
comprehensive investigation holds substantial scientific and
clinical value.
For ETS-1, targeted intervention strategies have
exhibited considerable potential across multiple tumor types: in
breast cancer, ETS-1 overexpression is found to enhance the
angiogenic capacity of tumor cells by modulating the interaction
between paracrine cells and endothelial cells and in vivo
experiments have demonstrated that ETS-1 inhibition can suppress
the angiogenic patterns of experimental breast tumors (146); in hepatocellular carcinoma
(HCC), ETS-1 has been identified as a mediator of sorafenib
resistance through regulating the mitochondrial ROS pathway via the
ETS-1-GPX-2 signaling axis and it effectively restores the
sensitivity of sorafenib-resistant HCC cells to sorafenib (147). As for ETS-2, targeted silencing
has shown remarkable anti-tumor efficacy in esophageal squamous
cell carcinoma: RNA interference-mediated ETS-2 depletion promotes
apoptosis, inhibits proliferation and invasion, induces
G0/G1 cell cycle arrest in vitro and
markedly suppresses tumor formation and metastasis in xenograft
mouse models by inactivating the mTOR/p70S6K signaling pathway
(148). Furthermore, ETS-2 in
tumor-associated macrophages (TAMs) drives breast cancer lung
metastasis by suppressing angiogenesis-inhibitory genes and
conditional depletion of ETS-2 in TAMs has been shown to reduce
tumor angiogenesis and metastatic burden in multiple murine breast
tumor models (149).
Overall, these preclinical studies have underscored
the feasibility of targeting ETS-1/ETS-2 for cancer therapy.
Moreover, with the in-depth understanding of the mechanisms
underlying the roles of ETS-1/ETS-2 in tumors and the development
of their inhibitors has emerged as a research priority. Currently,
the strategies for ETS-1 inhibition include small-molecule
compounds (150), antisense
oligonucleotides (151) and
CRISPR/Cas9-based gene editing tools (152) Although preliminary data have
demonstrated the potential therapeutic value of ETS-1 inhibitors,
they are still in the preclinical development stage. In animal
models, ETS-1 inhibitors have been shown to markedly suppress tumor
growth and metastasis while enhancing anti-tumor immune responses
(153). Nevertheless, the
clinical translation process still faces multiple challenges,
including key issues such as target specificity, therapeutic
efficacy, drug safety and optimal administration regimens. Future
clinical trials should focus on evaluating the human safety,
tolerability and efficacy of ETS-1 inhibitors, so as to clarify
whether they can be developed into clinically valuable anti-cancer
agents.
Conclusions and perspectives
In recent years, with the in-depth progression of
research into the pathogenesis of cardiovascular diseases, the
transcription factors ETS-1/ETS-2 have gradually emerged as a
research hotspot in this field. A growing body of evidence has
confirmed that ETS-1/ETS-2 are closely associated with
cardiovascular diseases and can regulate the occurrence and
development of such diseases through multiple mechanisms, thereby
providing an important theoretical basis and potential targets for
the early warning, targeted therapy and prognostic evaluation of
cardiovascular diseases (Table
I). However, as ubiquitously acting transcription factors,
ETS-1/ETS-2 are associated with intricate signaling pathways and
mechanisms of action. At present, research on their roles in the
cardiovascular system is still in its infancy and such studies are
confined to the scope of preclinical basic research. Large-sample,
long-term follow-up longitudinal cohort studies have not been
conducted, resulting in a lack of sufficient evidence-based medical
support for their application value in clinical settings.
Meanwhile, their downstream targets and extensive regulatory
networks have not been fully elucidated and further investigations
are therefore warranted. As previously proposed, no experimental
data have been reported regarding whether ETS-2 can prevent
ischemia-reperfusion (I/R) injury through specific mechanisms or by
regulating its downstream signaling pathways. Notably, the
translational application of ETS-1/ETS-2 for cardiovascular disease
therapy is confronted with multiple challenges: First, it is
difficult to achieve target-specific regulation. Since ETS-1/ETS-2
are widely expressed in various tissues and organs, they
participate in multiple physiological and pathological processes
such as cell proliferation, differentiation and apoptosis. Second,
regulatory networks are complex. ETS-1/ETS-2 exert their regulatory
functions through cross-talk with multiple signaling pathways and
the synergistic and antagonistic relationships among these pathways
have not been fully elucidated, making it difficult to accurately
identify key intervention nodes. Third, translational bridging
studies are lacking for clinical translation. Current research
mostly remains at the cellular and animal experimental levels,
which are quite different from human clinical pathological
conditions and there is a lack of intermediate translational
research data linking basic research to clinical application.
Fourth, the absence of longitudinal studies results in unclear
prognostic evaluation value.
 | Table ILink between cardiovascular diseases
and ETS-1/ETS-2. |
Table I
Link between cardiovascular diseases
and ETS-1/ETS-2.
| Authors, year | Cardiovascular
disease | Models | Target | Effects | (Refs.) |
|---|
| Jiang et al,
2022 | AS | Diabetic rat
model | ETS-1 | ETS-1 promotes
hyperglycemia-mediated endothelial inflammation and facilitates the
development of AS by upregulating PTP1B expression. | (80) |
| Zhan et al,
2005 | Vascular
inflammation and remodeling | Use of the male
ETS-1−/−mouse model | ETS-1 | Arterial wall
thickening and perivascular fibrosis were markedly reduced in
ETS-1−/− mice. Meanwhile, reduced expression of p21CIP,
PAI-1 and MCP-1 were all found in ETS-1−/− mice, leading
to a significant reduction in the recruitment of T cells and
macrophages to the vessel wall. | (82) |
| Feng et al,
2010 | Vascular
inflammation and remodeling | A mouse model of
balloon injury to the right common carotid artery | ETS-1 | Blockade of ETS-1
before carotid balloon injury not only reduced leukocyte
infiltration but also prevented the development of neointima after
balloon injury. | (85) |
| Cheng et al,
2011 | AS | ApoE−/−
mouse model | ETS-2 | ETS-2 levels
correlate with intraplaque microvessel formation and
proinflammatory cytokine levels in human vulnerable atherosclerotic
plaques and overexpression of ETS-2 promotes lesion growth with
neovascularization, hemorrhage and plaque instability. | (96) |
| Li et al,
2017 | AS | ApoE−/−
mouse model | ETS-1 | E2 increased the
expression level of miR-126 through ETS-1 in HUVEC and subsequently
miR-126-3p bound to its targets Spred1 and VCAM-1 mRNA, leading to
their degradation and silencing of protein expression and finally
promoting endothelial proliferation, migration and tube formation
and inhibiting monocyte adhesion as a means of improving
endothelial function and delaying the onset of AS. | (104) |
| Nie et al,
2016 | AS | ApoE−/−
mouse model | ETS-2 | Xiaoxianggou
reduced atherosclerotic plaque area and serum antioxidant LDL
autoantibodies in endogenous high AngII ApoE−/− mice by
increasing miR-203 expression and decreasing ETS-2 expression,
which were beneficial in preventing or treating the occurrence and
progression of AS. | (105) |
| Jiang et al,
2015 | AS | / | ETS-1 | TrkB can protect
endothelial integrity during atherogenesis by inducing and
activating ETS-1 expression. | (106) |
| Zhu et al,
2011 | AS | In vivo MIRI
mouse model | ETS-1 | HUVEC highly
expressing miR-155 co-targeted AT1R and ETS-1, miR-221/222 targeted
ETS-1 and indirectly regulated the expression of several
inflammatory molecules in ECs, thereby attenuating the adhesion of
Jurkat T cells to activated HUVEC and decreasing HUVEC migration as
a means to reduce endothelial cell inflammation and ameliorate
atherosclerosis. | (107) |
| Lee et al,
2021 | MIRI | In vivo MIRI
mouse model | ETS-1 | ADSC-CM reduces
cardiac apoptosis and fibrosis by decreasing the expression of PUMA
and ETS-1 mediated by miR-221/222/p38/NF-kB pathway, thereby
attenuating myocardial ischemia-reperfusion injury. | (112) |
| Lai et al,
2020 | MIRI | The MIRI rat
model | ETS-1 | Treatment with
ADSC-Exo prevented MIRI by markedly inhibiting the levels of PUMA
and ETS-1 through the miR-221/miR-222/PUMA/ETS-1 pathway, reducing
apoptosis and hypertrophy. | (113) |
| Guo et al,
2022 | MIRI | The MIRI rat
model | | KDM3A
overexpression leads to higher ETS-1 transcription, resulting in
reduced apoptosis, ROS, and inflammation, which plays a protective
role during myocardial injury. | (115) |
| Wang et al,
2014 | Cardiomyocyte
apoptosis | Diabetic mouse
model | ETS-1 | Inhibition of HMGB1
may prevent hyperglycemia-induced cardiomyocyte apoptosis by
downregulating ERK-dependent activation of ETS-1. | (121) |
| Zhan et al,
2005 | Myocardial
hypertrophy | ETS-1−/−
mouse model | ETS-1 | Vascular
proliferation, perivascular fibrosis and cardiac hypertrophy in
ETS-1−/− mice were markedly reduced in response to
systemic administration of Ang II. | (82) |
| Luo et al,
2021 | Myocardial
hypertrophy | Severe transverse
aortic constriction mouse model | ETS-2 | ETS-2 deficiency
attenuated cardiac hypertrophy in response to pressure overload,
while ETS-2 was found to play a key role in cardiac hypertrophy
driven by the calmodulin phosphatase/NFAT pathway. | (124) |
| Hao et al,
2015 | Myocardial
fibrosis | Myocardial fibrosis
rat model | ETS-1 | Knockdown of ETS-1
by siRNA markedly inhibited the induction of cell proliferation and
CTGF and PAI-1 expression by Ang II. | (125) |
| Xu et al,
2019 | Myocardial fibrosis
and myocardial hypertrophy |
Endothelium-specific ETS-1 knockout mice
bind Ang II-induced cardiac fibrosis model | ETS-1 | Deletion of
endothelial ETS-1attenuates Ang II-induced myocardial fibrosis by
inhibiting EMT and inhibition of ETS-1 may be a potential
therapeutic strategy to delay myocardial fibrosis. | (126) |
| Wang et al,
2022 | Myocardial
fibrosis | Autoimmune
myocarditis mouse model | ETS-1 | The use of
spironolactone markedly inhibited ETS-1 activation and
TGF-β1/smad2/3 phosphorylation and suppressed ETS-1-mediated
myocarditis-induced myocardial fibrosis. In addition, inhibition of
ETS-1 also reduced myocardial fibrosis by decreasing the expression
and activity of MMP-2 and MMP-9 in EAM mice. | (127) |
| Sun et al,
2023 | Myocardial
fibrosis | Myocardial fibrosis
mouse model | ETS-1 | Lnc TUG1 weakens
the inhibitory effect on ETS-1 through sponge adsorption of
miR-495-3p, thereby promoting the process of myocardial
fibrosis. | (128) |
| Li et al,
2025 | Myocardial
fibrosis | Myocardial
infarction rat model | ETS-1 | miR-1180-3p can be
delivered to CFs via Exo to attenuate TGF-β1-induced fibrosis by
inhibiting ETS-1 signaling. | (129) |
| Mattina et
al, 2009 | Jacobsen
syndrome | ETS-1-deficient
mouse model | ETS-1 | ETS-1 is a gene in
the critical region of Jacobsen syndrome and its deletion results
in ventricular septal defects and abnormal ventricular morphology
in mice. | (133) |
| Ye et al,
2010 | Jacobsen
syndrome | ETS-1-deficient
mouse model | ETS-1 | Deletion of ETS-1
leads to a decrease in migrating cells expressing Sox10 in E10.5
C57/B6 mouse embryos, which results in impaired cardiac neural
crest cell function and causes congenital heart defects. | (135) |
| Tan et al,
2023 | HF | HF model | ETS-2 | ETS-2 can promote
cardiomyocyte apoptosis and autophagy in heart failure by
regulating the lncRNA TUG1/miR-129-5p/ATG7 axis. | (140) |
| Deng et al,
2022 | HF | DOX-induced heart
failure in a rat model | ETS-1 | DNMT1 may inhibit
miR-152-3p expression by promoting methylation of miR-152-3p and
enhancing ETS-1 expression, which induces RHOH transcriptional
activation and inhibits mitochondrial autophagy, ultimately
contributing to the development of heart failure. | (143) |
Nevertheless, it is considered that with the
innovation in research techniques and the deepening of
interdisciplinary collaboration, systematically conducting
longitudinal cohort studies to clarify their clinical correlations,
leveraging precision regulation technologies to overcome the
challenge of target specificity and gradually improving the
mechanisms underlying their regulatory networks will effectively
address the current predicaments in research and application. This
will enable ETS-1/ETS-2 (the accession numbers of all identified
proteins are listed in Table
II) to emerge as a promising and reliable therapeutic target
for cardiovascular diseases, thereby providing more effective novel
strategies for clinical diagnosis and treatment.
| Table IIProteins and NCBI gene ID. |
Table II
Proteins and NCBI gene ID.
| Protein | NCBI gene ID |
|---|
| ETS-1 | 2113 |
| ETS-2 | 2114 |
| VE- cadherin | 100488458 |
| PUMA | 27113 |
| DLK1 | 8788 |
| VCAM-1 | 25361 |
| DKK1 | 22943 |
| PTP1B | 5770 |
| TrkB | 25054 |
| KDM3A | 55818 |
| HMGB1 | 3146 |
| Cadherin | 100144934 |
| NFAT | 6551030 |
| CHI3L1 | 1116 |
Availability of data and materials
Not applicable.
Author's contributions
SY and JZ were involved in the conceptualization of
the article and the writing of the manuscript. YY and YL provided
writing guidance and reviewed the manuscript. JY and HW made
substantial contributions to the selection of the topic, writing
guidance and financial support for the article. Data authentication
is not applicable. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, artificial
intelligence tools DeepL (version 2025; https://www.deepl.com/translator) and Doubao (version
2026; https://www.doubao.com) were used to
improve the readability and language of the manuscript or to
generate images, and subsequently, the authors revised and edited
the content produced by the artificial intelligence tools as
necessary, taking full responsibility for the ultimate content of
the present manuscript.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants to HW from the
National Natural Science Foundation of China (grant no. 82100270);
by grant no. 2021CFB202 from the Natural Science Foundation of
Hubei Province; by grant no. 2023PTCM06 from the Open Fund Project
of the Third level Laboratory of Traditional Chinese Medicine
Pharmacology Research, State Administration of Traditional Chinese
Medicine, Three Gorges University; by grant no. Q2021004 from the
Starting Foundation for Doctoral Research Project of Yichang
Central People's Hospital.
References
|
1
|
Virani SS, Alonso A, Benjamin EJ,
Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR,
Cheng S, Delling FN, et al: Heart disease and stroke
statistics-2020 update: A report from the American heart
association. Circulation. 141:e139–e596. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
In China TWCOTROCHAD; Hu SS: Report on
cardiovascular health and diseases in China 2021: An updated
summary. J Geriatr Cardiol. 20:399–430. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Global Burden of Cardiovascular Diseases
and Risks 2023 Collaborators: Global, regional, and national burden
of cardiovascular diseases and risk factors in 204 countries and
territories, 1990-2023. J Am Coll Cardiol. 86:2167–2243. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vaduganathan M, Mensah GA, Turco JV,
Fuster V and Roth GA: The global burden of cardiovascular diseases
and risk: A compass for future health. J Am Coll Cardiol.
80:2361–2371. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chong B, Jayabaskaran J, Jauhari SM, Chan
SP, Goh R, Kueh MTW, Li H, Chin YH, Kong G, Anand VV, et al: Global
burden of cardiovascular diseases: projections from 2025 to 2050.
Eur J Prev Cardiol. 32:1001–1015. 2025. View Article : Google Scholar
|
|
6
|
Zhou H, He L, Xu G and Chen L: Mitophagy
in cardiovascular disease. Clin Chim Acta. 507:210–218. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bravo-San Pedro JM, Kroemer G and Galluzzi
L: Autophagy and mitophagy in cardiovascular disease. Circ Res.
120:1812–1824. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bei Y, Das S, Rodosthenous RS, Holvoet P,
Vanhaverbeke M, Monteiro MC, Monteiro VVS, Radosinska J, Bartekova
M, Jansen F, et al: Extracellular vesicles in cardiovascular
theranostics. Theranostics. 7:4168–4182. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ding HS, Yang J, Chen P, Yang J, Bo SQ,
Ding JW and Yu QQ: The HMGB1-TLR4 axis contributes to myocardial
ischemia/reperfusion injury via regulation of cardiomyocyte
apoptosis. Gene. 527:389–393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sharrocks AD: The ETS-domain transcription
factor family. Nat Rev Mol Cell Biol. 2:827–837. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Adler D and Wernert N: ETS transcription
factors and prostate cancer: The role of the family prototype ETS-1
(review). Int J Oncol. 40:1748–1754. 2012.PubMed/NCBI
|
|
12
|
Leprince D, Gegonne A, Coll J, de Taisne
C, Schneeberger A, Lagrou C and Stehelin D: A putative second
cell-derived oncogene of the avian leukaemia retrovirus E26.
Nature. 306:395–397. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nagel S, Meyer C and Pommerenke C:
Establishment of the lymphoid ETS-code reveals deregulated ETS
genes in Hodgkin lymphoma. PLoS One. 18:e02880312023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thornton CEM, Hao J, Tamarapu PP and Landa
I: Multiple Ets factors participate in the transcriptional control
of tert mutant promoter in thyroid cancers. Cancers (Basel).
14:3572022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li C, Wang Z, Chen Y, Zhou M, Zhang H,
Chen R, Shi F, Wang C and Rui Z: Transcriptional silencing of ETS-1
abrogates epithelial-mesenchymal transition resulting in reduced
motility of pancreatic cancer cells. Oncol Rep. 33:559–565. 2015.
View Article : Google Scholar :
|
|
16
|
Li D, Chen Y, Mei H, Jiao W, Song H, Ye L,
Fang E, Wang X, Yang F, Huang K, et al: Ets-1 promoter-associated
noncoding RNA regulates the NONO/ERG/Ets-1 axis to drive gastric
cancer progression. Oncogene. 37:4871–4886. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shao Z, Li Y, Dai W, Jia H, Zhang Y, Jiang
Q, Chai Y, Li X, Sun H, Yang R, et al: ETS-1 induces
Sorafenib-resistance in hepatocellular carcinoma cells via
regulating transcription factor activity of PXR. Pharmacol Res.
135:188–200. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gupta N, Song H, Wu W, Ponce RK, Lin YK,
Kim JW, Small EJ, Feng FY, Huang FW and Okimoto RA: The CIC-ERF
co-deletion underlies fusion-independent activation of ETS family
member, ETV1, to drive prostate cancer progression. Elife.
11:e770722022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Heinemann V, Stintzing S, Modest DP,
Giessen-Jung C, Michl M and Mansmann UR: Early tumour shrinkage
(ETS) and depth of response (DpR) in the treatment of patients with
metastatic colorectal cancer (mCRC). Eur J Cancer. 51:1927–1936.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou X, Zhou R, Zhou H, Li Q, Hong J, Meng
R, Zhu F, Zhang S, Dai X, Peng G, et al: ETS-1 induces
endothelial-like differentiation and promotes metastasis in
non-small cell lung cancer. Cell Physiol Biochem. 45:1827–1839.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nazir SU, Kumar R, Dil-Afroze, Rasool I,
Bondhopadhyay B, Singh A, Tripathi R, Singh N, Khan A, Tanwar P, et
al: Differential expression of Ets-1 in breast cancer among North
Indian population. J Cell Biochem. 120:14552–14561. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Delattre O, Zucman J, Plougastel B,
Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau
G, et al: Gene fusion with an ETS DNA-binding domain caused by
chromosome translocation in human tumours. Nature. 359:162–165.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, Huang Z, Sun M, Huang W and Xia L:
ETS transcription factors: Multifaceted players from cancer
progression to tumor immunity. Biochim Biophys Acta Rev Cancer.
1878:1888722023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oikawa T and Yamada T: Molecular biology
of the Ets family of transcription factors. Gene. 303:11–34. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Seth A and Watson DK: ETS transcription
factors and their emerging roles in human cancer. Eur J Cancer.
41:2462–2478. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cooper CD, Newman JA and Gileadi O: Recent
advances in the structural molecular biology of Ets transcription
factors: Interactions, interfaces and inhibition. Biochem Soc
Trans. 42:130–138. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zeng B, Knapp EM, Skaritanov E, Oramas R
and Sun J: ETS transcription factors regulate precise matrix
metalloproteinase expression and follicle rupture in Drosophila.
Development. 151:dev2022762024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu C, Boisclair Lachance JF, Ludwig MZ and
Rebay I: A context-dependent bifurcation in the Pointed
transcriptional effector network contributes specificity and
robustness to retinal cell fate acquisition. PLoS Genet.
16:e10092162020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Findlay VJ, Larue AC, Turner DP, Watson PM
and Watson DK: Understanding the role of ETS-mediated gene
regulation in complex biological processes. Adv Cancer Res.
119:1–61. 2013.PubMed/NCBI
|
|
30
|
Liu F and Patient R: Genome-wide analysis
of the zebrafish ETS family identifies three genes required for
hemangioblast differentiation or angiogenesis. Circ Res.
103:1147–1154. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Garrett-Sinha LA, Dahl R, Rao S, Barton KP
and Simon MC: PU.1 exhibits partial functional redundancy with
Spi-B, but not with Ets-1 or Elf-1. Blood. 97:2908–2912. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fry EA and Inoue K: Aberrant expression of
ETS1 and ETS2 proteins in cancer. Cancer Rep Rev. 2: View Article : Google Scholar : 2018.PubMed/NCBI
|
|
33
|
Hollenhorst PC, Mcintosh LP and Graves BJ:
Genomic and biochemical insights into the specificity of ETS
transcription factors. Annu Rev Biochem. 80:437–471. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang BS, Hauser CA, Henkel G, Colman MS,
Van Beveren C, Stacey KJ, Hume DA, Maki RA and Ostrowski MC:
Ras-mediated phosphorylation of a conserved threonine residue
enhances the transactivation activities of c-Ets1 and c-Ets2. Mol
Cell Biol. 16:538–547. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kabbout M, Dakhlallah D, Sharma S, Bronisz
A, Srinivasan R, Piper M, Marsh CB and Ostrowski MC: MicroRNA 17-92
cluster mediates ETS1 and ETS2-dependent RAS-oncogenic
transformation. PLoS One. 9:e1006932014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sreeramaneni R, Chaudhry A, Mcmahon M,
Sherr CJ and Inoue K: Ras-Raf-Arf signaling critically depends on
the Dmp1 transcription factor. Mol Cell Biol. 25:220–232. 2005.
View Article : Google Scholar :
|
|
37
|
Watson DK, Mcwilliams-Smith MJ, Nunn MF,
Duesberg PH, O'Brien SJ and Papas TS: The ets sequence from the
transforming gene of avian erythroblastosis virus, E26, has unique
domains on human chromosomes 11 and 21: Both loci are
transcriptionally active. Proc Natl Acad Sci USA. 82:7294–7298.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Langer SJ, Bortner DM, Roussel MF, Sherr
CJ and Ostrowski MC: Mitogenic signaling by colony-stimulating
factor 1 and ras is suppressed by the ets-2 DNA-binding domain and
restored by myc overexpression. Mol Cell Biol. 12:5355–5362.
1992.PubMed/NCBI
|
|
39
|
Nunn MF, Seeburg PH, Moscovici C and
Duesberg PH: Tripartite structure of the avian erythroblastosis
virus E26 transforming gene. Nature. 306:391–395. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Iotsova V, Crépieux P, Montpellier C,
Laudet V and Stehelin D: TATA-less promoters of some Ets-family
genes are efficiently repressed by wild-type p53. Oncogene.
13:2331–2337. 1996.PubMed/NCBI
|
|
41
|
Laitem C, Leprivier G, Choul-Li S, Begue
A, Monte D, Larsimont D, Dumont P, Duterque-Coquillaud M and
Aumercier M: Ets-1 p27: A novel Ets-1 isoform with
dominant-negative effects on the transcriptional properties and the
subcellular localization of Ets-1 p51. Oncogene. 28:2087–2099.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bories JC, Willerford DM, Grévin D,
Davidson L, Camus A, Martin P, Stéhelin D and Alt FW: Increased
T-cell apoptosis and terminal B-cell differentiation induced by
inactivation of the Ets-1 proto-oncogene. Nature. 377:635–638.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Muthusamy N, Barton K and Leiden JM:
Defective activation and survival of T cells lacking the Ets-1
transcription factor. Nature. 377:639–642. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Higuchi T, Bartel FO, Masuya M, Deguchi T,
Henderson KW, Li R, Muise-Helmericks RC, Kern MJ, Watson DK and
Spyropoulos DD: Thymomegaly, microsplenia, and defective
homeostatic proliferation of peripheral lymphocytes in p51-Ets1
isoform-specific null mice. Mol Cell Biol. 27:3353–3366. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang C, Kam RK, Shi W, Xia Y, Chen X, Cao
Y, Sun J, Du Y, Lu G, Chen Z, et al: The Proto-oncogene
transcription factor Ets1 regulates neural crest development
through histone deacetylase 1 to mediate output of bone
morphogenetic protein signaling. J Biol Chem. 290:21925–21938.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shiu YT and Jaimes EA: Transcription
factor ETS-1 and reactive oxygen species: Role in vascular and
renal injury. Antioxidants (Basel) 2018. 7:842018.
|
|
47
|
Ghysdael J, Gegonne A, Pognonec P, Dernis
D, Leprince D and Stehelin D: Identification and preferential
expression in thymic and bursal lymphocytes of a c-ets
oncogene-encoded Mr 54,000 cytoplasmic protein. Proc Natl Acad Sci
USA. 83:1714–1718. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Garrett-Sinha LA: Review of Ets1
structure, function, and roles in immunity. Cell Mol Life Sci.
70:3375–3390. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lincoln DW II and Bove K: The
transcription factor Ets-1 in breast cancer. Front Biosci.
10:506–511. 2005. View
Article : Google Scholar
|
|
50
|
Zhang X, Wu D, Aldarouish M, Yin X, Li C
and Wang C: ETS-1: A potential target of glycolysis for metabolic
therapy by regulating glucose metabolism in pancreatic cancer. Int
J Oncol. 50:232–240. 2017. View Article : Google Scholar
|
|
51
|
Pritchard M, Reeves RH, Dierssen M,
Patterson D and Gardiner KJ: Down syndrome and the genes of human
chromosome 21: current knowledge and future potentials. Report on
the Expert workshop on the biology of chromosome 21 genes: Towards
gene-phenotype correlations in down syndrome. Washington D.C.,
September 28-October 1, 2007. Cytogenet Genome Res. 121:67–77.
2008. View Article : Google Scholar
|
|
52
|
Mavrothalassitis GJ, Watson DK and Papas
TS: The human ETS-2 gene promoter: Molecular dissection and
nuclease hypersensitivity. Oncogene. 5:1337–1342. 1990.PubMed/NCBI
|
|
53
|
Pitarresi JR, Liu X, Sharma SM, Cuitiño
MC, Kladney RD, Mace TA, Donohue S, Nayak SG, Qu C, Lee J, et al:
Stromal ETS2 regulates chemokine production and immune cell
recruitment during acinar-to-ductal metaplasia. Neoplasia.
18:541–552. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Baran CP, Fischer SN, Nuovo GJ, Kabbout
MN, Hitchcock CL, Bringardner BD, McMaken S, Newland CA,
Cantemir-Stone CZ, Phillips GS, et al: Transcription factor ets-2
plays an important role in the pathogenesis of pulmonary fibrosis.
Am J Respir Cell Mol Biol. 45:999–1006. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liao YL, Hu LY, Tsai KW, Wu CW, Chan WC,
Li SC, Lai CH, Ho MR, Fang WL, Huang KH and Lin WC: Transcriptional
regulation of miR-196b by ETS2 in gastric cancer cells.
Carcinogenesis. 33:760–769. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kabbout M, Garcia MM, Fujimoto J, Liu DD,
Woods D, Chow CW, Mendoza G, Momin AA, James BP, Solis L, et al:
ETS2 mediated tumor suppressive function and MET oncogene
inhibition in human non-small cell lung cancer. Clin Cancer Res.
19:3383–3395. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang H, Huang R, Guo W, Qin X, Yang Z,
Yuan Z, Wei Y, Mo C, Zeng Z, Luo J, et al: RNA-binding protein
CELF1 enhances cell migration, invasion, and chemoresistance by
targeting ETS2 in colorectal cancer. Clin Sci (Lond).
134:1973–1990. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Satpathy C, Kumar Mishra T, Singh S and
Jha AK: Reverse cardio-oncology: A budding concept. Indian Heart J.
75:398–402. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yuan Y, Mei Z, Qu Z, Li G, Yu S, Liu Y,
Liu K, Shen Z, Pu J, Wang Y, et al: Exosomes secreted from
cardiomyocytes suppress the sensitivity of tumor ferroptosis in
ischemic heart failure. Signal Transduct Target Ther. 8:1212023.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lau ES, Paniagua SM, Liu E, Jovani M, Li
SX, Takvorian K, Suthahar N, Cheng S, Splansky GL, Januzzi JL Jr,
et al: Cardiovascular risk factors are associated with future
cancer. JACC CardioOncol. 3:48–58. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Karlstaedt A, Moslehi J and de Boer RA:
Cardio-onco-metabolism: Metabolic remodelling in cardiovascular
disease and cancer. Nat Rev Cardiol. 19:414–425. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sturgeon KM, Deng L, Bluethmann SM, Zhou
S, Trifiletti DM, Jiang C, Kelly SP and Zaorsky NG: A
population-based study of cardiovascular disease mortality risk in
US cancer patients. Eur Heart J. 40:3889–3897. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu G and Zhang BF: Age-specific
cardiovascular disease-related mortality among patients with major
gastrointestinal cancers: A SEER population-based study. Cancer
Med. 12:17253–17265. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Xia Y, Lin M, Huang J and Fan L:
Cardiovascular disease related death among patients with esophagus
cancer: A population-based competing risk analysis. Front Oncol.
12:9767112022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cao L, Wang X, Yan Y, Ning Z, Ma L and Li
Y: Analysis of competing risks of cardiovascular death in patients
with hepatocellular carcinoma: A population-based study. Medicine
(Baltimore). 102:e367052023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Guan T, Monteiro O, Chen D, Luo Z, Chi K,
Li Z, Liang Y, Lu Z, Jiang Y, Yang J, et al: Long-term and
short-term cardiovascular disease mortality among patients of 21
non-metastatic cancers. J Adv Res. 69:215–224. 2025. View Article : Google Scholar :
|
|
67
|
Wang C, Wang Z, Yang J, Zhang S, Zhang P
and Yang Y: Lung cancer and risk of cardiovascular mortality. Front
Cardiovasc Med. 11:14919122024. View Article : Google Scholar
|
|
68
|
Sabaté-Tormos M, Bardají A, Peiró OM,
Carrasquer A, Cediel G and Ferreiro JL: Cancer and myocardial
injury in patients with suspected acute coronary syndrome.
Cardiooncology. 11:212025.PubMed/NCBI
|
|
69
|
Lie-Venema H, Gittenberger-De Groot AC,
Van Empel LJ, Boot MJ, Kerkdijk H, de Kant E and DeRuiter MC: Ets-1
and Ets-2 transcription factors are essential for normal coronary
and myocardial development in chicken embryos. Circ Res.
92:749–756. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhou P, Zhang Y, Sethi I, Ye L, Trembley
MA, Cao Y, Akerberg BN, Xiao F, Zhang X, Li K, et al: GATA4
regulates developing endocardium through interaction with ETS1.
Circ Res. 131:e152–e168. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wang L, Lin L, Qi H, Chen J and Grossfeld
P: Endothelial Loss of ETS1 impairs coronary vascular development
and leads to ventricular non-compaction. Circ Res. 131:371–387.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Adachi Y, Ueda K, Nomura S, Ito K, Katoh
M, Katagiri M, Yamada S, Hashimoto M, Zhai B, Numata G, et al:
Beiging of perivascular adipose tissue regulates its inflammation
and vascular remodeling. Nat Commun. 13:51172022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhang G, Qin Q, Zhang C, Sun X, Kazama K,
Yi B, Cheng F, Guo ZF and Sun J: NDRG1 signaling is essential for
endothelial inflammation and vascular remodeling. Circ Res.
132:306–319. 2023. View Article : Google Scholar
|
|
74
|
Bai C, Su M, Zhang Y, Lin Y, Sun Y, Song
L, Xiao N, Xu H, Wen H, Zhang M, et al: Oviductal glycoprotein 1
promotes hypertension by inducing vascular remodeling through an
interaction with MYH9. Circulation. 146:1367–1382. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Liu H, Sun M, Wu N, Liu B, Liu Q and Fan
X: TGF-β/Smads signaling pathway, Hippo-YAP/TAZ signaling pathway,
and VEGF: Their mechanisms and roles in vascular remodeling related
diseases. Immun Inflamm Dis. 11:e10602023. View Article : Google Scholar
|
|
76
|
Gomez-Salinero JM, Itkin T, Houghton S,
Badwe C, Lin Y, Kalna V, Dufton N, Peghaire CR, Yokoyama M, Wingo
M, et al: Cooperative ETS transcription factors enforce adult
endothelial cell fate and cardiovascular homeostasis. Nat
Cardiovasc Res. 1:882–899. 2022. View Article : Google Scholar
|
|
77
|
Guo Z, Li L, Luo J, Xue W, Bian S, Zhu Y,
Huo D, Yang W, Ma J, Hao Y, et al: The Ets2 super-enhancer
modulates endothelial-mesenchymal transition during cardiac ageing.
Cardiovasc Res. 121:2892–2908. 2026. View Article : Google Scholar
|
|
78
|
Tharakan B, Hunter FA, Muthusamy S,
Randolph S, Byrd C, Rao VN, Reddy ESP and Childs EW: ETS-Related
gene activation preserves adherens junctions and permeability in
microvascular endothelial cells. Shock. 57:309–315. 2022.
View Article : Google Scholar
|
|
79
|
Oettgen P: Regulation of vascular
inflammation and remodeling by ETS factors. Circ Res. 99:1159–1166.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Jiang L, Liang J, Wang T, Meng F and Duan
W: ETS proto-oncogene 1 modulates PTP1B expression to participate
in high glucose-mediated endothelial inflammation. Acta Biochim
Biophys Sin (Shanghai). 54:565–573. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Harel S, Sanchez V, Moamer A,
Sanchez-Galan JE, Abid Hussein MN, Mayaki D, Blanchette M and
Hussain SNA: ETS1, ELK1, and ETV4 transcription factors regulate
angiopoietin-1 signaling and the angiogenic response in endothelial
cells. Front Physiol. 12:6836512021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhan Y, Brown C, Maynard E, Anshelevich A,
Ni W, Ho IC and Oettgen P: Ets-1 is a critical regulator of Ang
II-mediated vascular inflammation and remodeling. J Clin Invest.
115:2508–2516. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Baldus S, Heeschen C, Meinertz T, Zeiher
AM, Eiserich JP, Münzel T, Simoons ML and Hamm CW; CAPTURE
Investigators: Myeloperoxidase serum levels predict risk in
patients with acute coronary syndromes. Circulation. 108:1440–1445.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Miller AP, Xing D, Feng W, Fintel M, Chen
YF and Oparil S: Aged rats lose vasoprotective and
anti-inflammatory actions of estrogen in injured arteries.
Menopause. 14:251–260. 2007. View Article : Google Scholar
|
|
85
|
Feng W, Xing D, Hua P, Zhang Y, Chen YF,
Oparil S and Jaimes EA: The transcription factor ETS-1 mediates
proinflammatory responses and neointima formation in carotid artery
endoluminal vascular injury. Hypertension. 55:1381–1388. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Dejana E, Taddei A and Randi AM: Foxs and
Ets in the transcriptional regulation of endothelial cell
differentiation and angiogenesis. Biochim Biophys Acta.
1775:298–312. 2007.PubMed/NCBI
|
|
87
|
Di M, Zhang Y, Zeng R, Liu X, Chen W and
Zhang M, Zhang C, Li M and Zhang M: The pro-angiogenesis effect of
miR33a-5p/Ets-1/DKK1 signaling in ox-LDL induced HUVECs. Int J Biol
Sci. 17:4122–4139. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yuan D, Chu J, Lin H, Zhu G, Qian J, Yu Y,
Yao T, Ping F, Chen F and Liu X: Mechanism of homocysteine-mediated
endothelial injury and its consequences for atherosclerosis. Front
Cardiovasc Med. 9:11094452023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Xu K, Saaoud F, Yu S, Drummer C IV, Shao
Y, Sun Y, Lu Y, Sun J, Yu J, Jiang X, et al: Monocyte adhesion
assays for detecting endothelial cell activation in vascular
inflammation and atherosclerosis. Methods Mol Biol. 2419:169–182.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Desideri G and Ferri C: Endothelial
activation. Sliding door to atherosclerosis. Curr Pharm Des.
11:2163–2175. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Xiang P, Chen Q, Chen L, Lei J, Yuan Z, Hu
H, Lu Y, Wang X, Wang T, Yu R, et al: Metabolite Neu5Ac triggers
SLC3A2 degradation promoting vascular endothelial ferroptosis and
aggravates atherosclerosis progression in ApoE−/−mice.
Theranostics. 13:4993–5016. 2023. View Article : Google Scholar
|
|
92
|
Wang C, Li Z, Liu Y and Yuan L: Exosomes
in atherosclerosis: Performers, bystanders, biomarkers, and
therapeutic targets. Theranostics. 11:3996–4010. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Li H, Sun K, Zhao R, Hu J, Hao Z, Wang F,
Lu Y, Liu F and Zhang Y: Inflammatory biomarkers of coronary heart
disease. Front Biosci (Schol Ed). 10:185–196. 2018. View Article : Google Scholar
|
|
94
|
Zhang L, Wu X and Hong L: Endothelial
reprogramming in atherosclerosis. Bioengineering (Basel) 2024.
11:3252024.
|
|
95
|
Santiago FS and Khachigian LM: Ets-1
stimulates platelet-derived growth factor A-chain gene
transcription and vascular smooth muscle cell growth via
cooperative interactions with Sp1. Circ Res. 95:479–487. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Cheng C, Tempel D, Den Dekker WK, Haasdijk
R, Chrifi I, Bos FL, Wagtmans K, van de Kamp EH, Blonden L, Biessen
EA, et al: Ets2 determines the inflammatory state of endothelial
cells in advanced atherosclerotic lesions. Circ Res. 109:382–395.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Stankey CT, Bourges C, Haag LM,
Turner-Stokes T, Piedade AP, Palmer-Jones C, Papa I, Silva Dos
Santos M, Zhang Q, Cameron AJ, et al: A disease-associated gene
desert directs macrophage inflammation through ETS2. Nature.
630:447–456. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Higashida T, Kanno H, Nakano M, Funakoshi
K and Yamamoto I: Expression of hypoxia-inducible angiogenic
proteins (hypoxia-inducible factor-1alpha, vascular endothelial
growth factor, and E26 transformation-specific-1) and plaque
hemorrhage in human carotid atherosclerosis. J Neurosurg.
109:83–91. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Katsume A, Okigaki M, Matsui A, Che J,
Adachi Y, Kishita E, Yamaguchi S, Ikeda K, Ueyama T, Matoba S, et
al: Early inflammatory reactions in atherosclerosis are induced by
proline-rich tyrosine kinase/reactive oxygen species-mediated
release of tumor necrosis factor-alpha and subsequent activation of
the p21Cip1/Ets-1/p300 system. Arterioscler Thromb Vasc Biol.
31:1084–1092. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Hopkins PN: Molecular biology of
atherosclerosis. Physiol Rev. 93:1317–1542. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Simoncini T, Scorticati C, Mannella P,
Fadiel A, Giretti MS, Fu XD, Baldacci C, Garibaldi S, Caruso A,
Fornari L, et al: Estrogen receptor alpha interacts with Galpha13
to drive actin remodeling and endothelial cell migration via the
RhoA/Rho kinase/moesin pathway. Mol Endocrinol. 20:1756–1771. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Jiang P, Xu J, Zheng S, Huang J, Xiang Q,
Fu X and Wang T: 17beta-estradiol down-regulates
lipopolysaccharide-induced MCP-1 production and cell migration in
vascular smooth muscle cells. J Mol Endocrinol. 45:87–97. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Arnal JF, Laurell H, Fontaine C, Billon A,
Calippe B, Lenfant F and Gourdy P: Estrogen receptor actions on
vascular biology and inflammation: Implications in vascular
pathophysiology. Climacteric. 12(Suppl 1): S12–S17. 2009.
View Article : Google Scholar
|
|
104
|
Li P, Wei J, Li X, Cheng Y, Chen W, Cui Y,
Simoncini T, Gu Z, Yang J and Fu X: 17β-Estradiol enhances vascular
endothelial Ets-1/miR-126-3p expression: The possible mechanism for
attenuation of atherosclerosis. J Clin Endocrinol Metab.
102:594–603. 2017. View Article : Google Scholar
|
|
105
|
Nie W, Zhang X, Yan H, Li S, Zhu W, Fan F
and Zhu J: Xiaoxianggou attenuates atherosclerotic plaque formation
in endogenous high Ang II ApoE(−/−) mice via the
inhibition of miR-203 on the expression of Ets-2 in endothelial
cells. Biomed Pharmacother. 82:173–179. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Jiang H, Huang S, Li X, Li X, Zhang Y and
Chen ZY: Tyrosine kinase receptor B protects against coronary
artery disease and promotes adult vasculature integrity by
regulating Ets1-mediated VE-cadherin expression. Arterioscler
Thromb Vasc Biol. 35:580–588. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhu N, Zhang D, Chen S, Liu X, Lin L,
Huang X, Guo Z, Liu J, Wang Y, Yuan W and Qin Y: Endothelial
enriched microRNAs regulate angiotensin II-induced endothelial
inflammation and migration. Atherosclerosis. 215:286–293. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
GBD 2019 Risk Factors Collaborators:
Global burden of 87 risk factors in 204 countries and territories,
1990-2019: A systematic analysis for the Global Burden of Disease
Study 2019. Lancet. 396:1223–1249. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Hausenloy DJ, Botker HE, Engstrom T,
Erlinge D, Heusch G, Ibanez B, Kloner RA, Ovize M, Yellon DM and
Garcia-Dorado D: Targeting reperfusion injury in patients with
ST-segment elevation myocardial infarction: trials and
tribulations. Eur Heart J. 38:935–941. 2017.
|
|
110
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Cai W, Liu L, Shi X, Liu Y, Wang J, Fang
X, Chen Z, Ai D, Zhu Y and Zhang X: Alox15/15-HpETE aggravates
myocardial ischemia-reperfusion injury by promoting cardiomyocyte
ferroptosis. Circulation. 147:1444–1460. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Lee TL, Lai TC, Lin SR, Lin SW, Chen YC,
Pu CM, Lee IT, Tsai JS, Lee CW and Chen YL: Conditioned medium from
adipose-derived stem cells attenuates ischemia/reperfusion-induced
cardiac injury through the microRNA-221/222/PUMA/ETS-1 pathway.
Theranostics. 11:3131–3149. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Lai TC, Lee TL, Chang YC, Chen YC, Lin SR,
Lin SW, Pu CM, Tsai JS and Chen YL: MicroRNA-221/222 Mediates
ADSC-Exosome-Induced cardioprotection against ischemia/reperfusion
by targeting PUMA and ETS-1. Front Cell Dev Biol. 8:5691502020.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Pei YH, Chen J, Wu X, He Y, Qin W, He SY,
Chang N, Jiang H, Zhou J, Yu P, et al: LncRNA PEAMIR inhibits
apoptosis and inflammatory response in PM2.5 exposure aggravated
myocardial ischemia/reperfusion injury as a competing endogenous
RNA of miR-29b-3p. Nanotoxicology. 14:638–653. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Guo X, Zhang BF, Zhang J, Liu G, Hu Q and
Chen J: The histone demthylase KDM3A protects the myocardium from
ischemia/reperfusion injury via promotion of ETS1 expression.
Commun Biol. 5:2702022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Ali H, Braga L and Giacca M: Cardiac
regeneration and remodelling of the cardiomyocyte cytoarchitecture.
FEBS J. 287:417–438. 2020. View Article : Google Scholar
|
|
117
|
Mckinsey TA, Foo R, Anene-Nzelu CG,
Travers JG, Vagnozzi RJ, Weber N and Thum T: Emerging epigenetic
therapies of cardiac fibrosis and remodelling in heart failure:
From basic mechanisms to early clinical development. Cardiovasc
Res. 118:3482–3498. 2023. View Article : Google Scholar :
|
|
118
|
Liu D, Richardson G, Benli FM, Park C, de
Souza JV, Bronowska AK and Spyridopoulos I: Inflammageing in the
cardiovascular system: mechanisms, emerging targets, and novel
therapeutic strategies. Clin Sci (Lond). 134:2243–2262. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Takefuji M, Wirth A, Lukasova M, Takefuji
S, Boettger T, Braun T, Althoff T, Offermanns S and Wettschureck N:
G(13)-mediated signaling pathway is required for pressure
overload-induced cardiac remodeling and heart failure. Circulation.
126:1972–1982. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Mccarroll CS, He W, Foote K, Bradley A,
Mcglynn K, Vidler F, Nixon C, Nather K, Fattah C, Riddell A, et al:
Runx1 deficiency protects against adverse cardiac remodeling after
myocardial infarction. Circulation. 137:57–70. 2018. View Article : Google Scholar :
|
|
121
|
Wang WK, Lu QH, Zhang JN, Wang B, Liu XJ,
An FS, Qin WD, Chen XY, Dong WQ, Zhang C, et al: HMGB1 mediates
hyperglycaemia-induced cardiomyocyte apoptosis via ERK/Ets-1
signalling pathway. J Cell Mol Med. 18:2311–2320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Heineke J and Molkentin JD: Regulation of
cardiac hypertrophy by intracellular signalling pathways. Nat Rev
Mol Cell Biol. 7:589–600. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Molkentin JD, Lu JR, Antos CL, Markham B,
Richardson J, Robbins J, Grant SR and Olson EN: A
calcineurin-dependent transcriptional pathway for cardiac
hypertrophy. Cell. 93:215–228. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Luo Y, Jiang N, May HI, Luo X, Ferdous A,
Schiattarella GG, Chen G, Li Q, Li C, Rothermel BA, et al:
Cooperative Binding of ETS2 and NFAT Links Erk1/2 and calcineurin
signaling in the pathogenesis of cardiac hypertrophy. Circulation.
144:34–51. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Hao G, Han Z, Meng Z, Wei J, Gao D, Zhang
H and Wang N: Ets-1 upregulation mediates angiotensin II-related
cardiac fibrosis. Int J Clin Exp Pathol. 8:10216–10227.
2015.PubMed/NCBI
|
|
126
|
Xu L, Fu M, Chen D, Han W, Ostrowski MC,
Grossfeld P, Gao P and Ye M: Endothelial-specific deletion of Ets-1
attenuates Angiotensin II-induced cardiac fibrosis via suppression
of endothelial-to-mesenchymal transition. BMB Rep. 52:595–600.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Wang WK, Wang B, Cao XH and Liu YS:
Spironolactone alleviates myocardial fibrosis via inhibition of
Ets-1 in mice with experimental autoimmune myocarditis. Exp Ther
Med. 23:3692022. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Sun Y, Shan X, Guo J, Liu X and Ma D:
CHI3L1 promotes myocardial fibrosis via regulating lncRNA
TUG1/miR-495-3p/ETS1 axis. Apoptosis. 28:1436–1451. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Li C, Zheng C, Pu Y, Zhou H, Li Y, Wang W,
Chen X, Zhang C and Chen Y: Vericiguat enhances the therapeutic
efficacy of mesenchymal stem cells-derived exosomes in acute
myocardial infarction through microRNA-1180-3p/ETS1 pathway. Cell
Signal. 125:1115122025. View Article : Google Scholar
|
|
130
|
Yamashita D, Muramatsu H, Narita A,
Wakamatsu M, Tsumura Y, Sajiki D, Maemura R, Yamamori A, Imaya M,
Narita K, et al: Hematological abnormalities in Jacobsen syndrome:
Cytopenia of varying severities and morphological abnormalities in
peripheral blood and bone marrow. Haematologica. 108:3438–3443.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Jacobsen P, Hauge M, Henningsen K, Hobolth
N, Mikkelsen M and Philip J: An (11;21) translocation in four
generations with chromosome 11 abnormalities in the offspring. A
clinical, cytogenetical, and gene marker study. Hum Hered.
23:568–585. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Chen S, Wang R, Zhang X, Li L, Jiang Y,
Liu R and Zhang H: Ultrasonographic findings and prenatal diagnosis
of Jacobsen syndrome: A case report and review of the literature.
Medicine (Baltimore). 99:e186952020. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Mattina T, Perrotta CS and Grossfeld P:
Jacobsen syndrome. Orphanet J Rare Dis. 4:92009. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Pierpont ME, Brueckner M, Chung WK, Garg
V, Lacro RV, McGuire AL, Mital S, Priest JR, Pu WT, Roberts A, et
al: Genetic basis for congenital heart disease: Revisited: A
scientific statement from the American heart association.
Circulation. 138:e653–e711. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Ye M, Coldren C, Liang X, Mattina T,
Goldmuntz E, Benson DW, Ivy D, Perryman MB, Garrett-Sinha LA and
Grossfeld P: Deletion of ETS-1, a gene in the Jacobsen syndrome
critical region, causes ventricular septal defects and abnormal
ventricular morphology in mice. Hum Mol Genet. 19:648–656. 2010.
View Article : Google Scholar
|
|
136
|
Ye M, Yin Y, Fukatsu K and Grossfeld P:
Evidence that deletion of ETS-1, a gene in the Jacobsen syndrome
(11q-) cardiac critical region, causes congenital heart defects
through impaired cardiac neural crest cell function. Etiology and
Morphogenesis of Congenital Heart Disease: From Gene Function and
Cellular Interaction to Morphology. Nakanishi T, Markwald RR,
Baldwin HS, et al: Springer Copyright; Tokyo: pp. 361–369. 2016,
View Article : Google Scholar
|
|
137
|
Mazurek JA and Jessup M: Understanding
heart failure. Heart Fail Clin. 13:1–19. 2017. View Article : Google Scholar
|
|
138
|
Inglis SC, Clark RA, Dierckx R,
Prieto-Merino D and Cleland JG: Structured telephone support or
non-invasive telemonitoring for patients with heart failure.
Cochrane Database Syst Rev. 2015:Cd0072282015.PubMed/NCBI
|
|
139
|
Li J, Su H, Zhu Y, Cao Y and Ma X: ETS2
and microRNA-155 regulate the pathogenesis of heart failure through
targeting and regulating GPR18 expression. Exp Ther Med.
19:3469–3478. 2020.PubMed/NCBI
|
|
140
|
Tan L, Xiong D, Zhang H, Xiao S, Yi R and
Wu J: ETS2 promotes cardiomyocyte apoptosis and autophagy in heart
failure by regulating lncRNA TUG1/miR-129-5p/ATG7 axis. FASEB J.
37:e229372023. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Chaturvedi P, Kalani A, Givvimani S, Kamat
PK, Familtseva A and Tyagi SC: Differential regulation of DNA
methylation versus histone acetylation in cardiomyocytes during
HHcy in vitro and in vivo: An epigenetic mechanism. Physiol
Genomics. 46:245–255. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Moyzis AG, Sadoshima J and Gustafsson ÅB:
Mending a broken heart: The role of mitophagy in cardioprotection.
Am J Physiol Heart Circ Physiol. 308:H183–H192. 2015. View Article : Google Scholar
|
|
143
|
Deng Z, Yao J, Xiao N, Han Y, Wu X, Ci C,
Chen K and Geng X: DNA methyltransferase 1 (DNMT1) suppresses
mitophagy and aggravates heart failure via the
microRNA-152-3p/ETS1/RhoH axis. Lab Invest. 102:782–793. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Sizemore GM, Pitarresi JR, Balakrishnan S
and Ostrowski MC: The ETS family of oncogenic transcription factors
in solid tumours. Nat Rev Cancer. 17:337–351. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Wang S, Wan L, Zhang X, Fang H, Zhang M,
Li F and Yan D: ETS-1 in tumor immunology: Implications for novel
anti-cancer strategies. Front Immunol. 16:15263682025. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Furlan A, Vercamer C, Heliot L, Wernert N,
Desbiens X and Pourtier A: Ets-1 drives breast cancer cell
angiogenic potential and interactions between breast cancer and
endothelial cells. Int J Oncol. 54:29–40. 2019.
|
|
147
|
Vishnoi K, Ke R, Viswakarma N, Srivastava
P, Kumar S, Das S, Singh SK, Principe DR, Rana A and Rana B: Ets1
mediates sorafenib resistance by regulating mitochondrial ROS
pathway in hepatocellular carcinoma. Cell Death Dis. 13:5812022.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Li Q, Yang L, Han K, Zhu L, Zhang Y, Ma S,
Zhang K, Yang B and Guan F: Ets2 knockdown inhibits tumorigenesis
in esophageal squamous cell carcinoma in vivo and in vitro.
Oncotarget. 7:61458–61468. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Zabuawala T, Taffany DA, Sharma SM,
Merchant A, Adair B, Srinivasan R, Rosol TJ, Fernandez S, Huang K,
Leone G and Ostrowski MC: An ets2-driven transcriptional program in
tumor-associated macrophages promotes tumor metastasis. Cancer Res.
70:1323–1333. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Jie Y, Liu G, E M, Li Y, Xu G, Guo J, Li
Y, Rong G, Li Y and Gu A: Novel small molecule inhibitors of the
transcription factor ETS-1 and their antitumor activity against
hepatocellular carcinoma. Eur J Pharmacol. 906:1742142021.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Hörberg J, Carlesso A and Reymer A:
Mechanistic insights into ASO-RNA complexation: Advancing antisense
oligonucleotide design strategies. Mol Ther Nucleic Acids.
35:1023512024. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Szlachta K, Kuscu C, Tufan T, Adair SJ,
Shang S, Michaels AD, Mullen MG, Fischer NL, Yang J, Liu L, et al:
CRISPR knockout screening identifies combinatorial drug targets in
pancreatic cancer and models cellular drug response. Nat Commun.
9:42752018. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Wang H, Chu F, Zhijie L, Bi Q, Lixin L,
Zhuang Y, Xiaofeng Z, Niu X, Zhang D, Xi H and Li BA: MTBP enhances
the activation of transcription factor ETS-1 and promotes the
proliferation of hepatocellular carcinoma cells. Front Oncol.
12:9850822022. View Article : Google Scholar : PubMed/NCBI
|