Introduction
Influenza A virus (IAV) surface antigens are highly
prone to mutation, leading to the emergence of new subtypes
(1). These mutations make it
difficult for humans to effectively prevent influenza, thereby
triggering large-scale outbreaks of influenza-like diseases
(1). For instance,
population-based hospitalization surveillance data from the United
States demonstrates that H1N1pdm09 accounted for nearly one-quarter
of laboratory-confirmed influenza-associated hospitalizations
during the 2010-11 and 2018-19 influenza seasons (2). More critically, hospitalized
patients with H1N1pdm09 infection exhibited a markedly elevated
risk of developing severe outcomes, including ICU admission and
in-hospital mortality, compared with those infected with H3N2
(2). The 2009 A/H1N1pdm09
pandemic led to the disappearance of the old seasonal A/H1N1. Since
then we have had the COVID pandemic, so the way in which we
approach airborne viruses has changed considerably. Importantly,
not only A/H3N2 and B lineages but also A/H1N1pdm09 showed
near-complete disappearance from global circulation during the
COVID-19 pandemic, probably due to non-pharmaceutical interventions
and behavioral changes (3).
However, unlike B/Yamagata, A/H1N1pdm09 re-emerged after the
relaxation of non-pharmaceutical interventions, highlighting its
resilience and continued role in seasonal influenza dynamics. Upon
IAV infection, the virus initially causes respiratory tract
infections, leading to congestion and edema of the upper
respiratory mucosa and extensive epithelial cell shedding and
necrosis (4). As the virus
spreads, the lung tissue undergoes significant infiltration by
inflammatory cells, with congestion, dilation, thickening and
possible fusion of the alveolar walls (5). Additionally, bronchial epithelial
cells may also undergo shedding and necrosis, ultimately resulting
in lung tissue damage (5). In
this process, the host immune response is often excessively
activated, releasing large amounts of inflammatory cytokines such
as interleukin (IL-)6, IL-10, tumor necrosis factor-α (TNF-α) and
interferon-γ (IFN-γ) (6). The
overproduction of these cytokines is one of the key pathological
mechanisms underlying influenza virus-induced pneumonia. In
particular, excessive production of TNF-α and IFN-γ not only
activates a number of mononuclear macrophages but also exacerbates
lung tissue damage (7). Although
vaccination is the most effective method for preventing influenza,
the efficacy of vaccines may gradually diminish due to viral
mutations (7). Currently, drugs
such as oseltamivir phosphate and amantadine are used to inhibit
viral replication (8). However,
a critical therapeutic gap persists: Although antiviral agents
(such as the neuraminidase inhibitor oseltamivir and M2 inhibitors
such as amantadine) can restrict viral replication, they are
inadequate for modulating the dysregulated immune response of a
host once systemic inflammation and cytokine storms are initiated
(9,10). For example, studies have
confirmed that while Tamiflu suppresses viral replication, its
efficacy in regulating immune dysregulation (such as excessive
release of TNF-α and IFN-γ) in patients with severe H1N1 pneumonia
remains limited (11,12). Some patients continue to exhibit
persistent viral replication and elevated inflammatory cytokine
levels during treatment, demonstrating that single-agent antiviral
therapy cannot halt disease progression. Consequently, there is an
urgent need to shift therapeutic strategies from solely antiviral
interventions to method for the comprehensive regulation of both
viral replication and the host immune response, particularly by
targeting the coexisting viral burden and excessive inflammation in
patients with viral sepsis.
Innate immunity is the first line of defense against
pathogen invasion, and inflammasome activation is a crucial
component of the innate immune response (13). Absent in melanoma 2 (AIM2) is a
cytosolic DNA sensor and a member of the PYHIN family that is
capable of recognizing bacterial, viral, or host-derived cytosolic
double-stranded DNA (13). Upon
activation, AIM2 first recruits procaspase-1 through the adaptor
protein ASC, leading to self-cleavage and activation of
procaspase-1 (13). Activated
caspase-1 then cleaves pro-IL-1β and pro-IL-18, generating mature
IL-1β and IL-18, which are subsequently secreted extracellularly.
Simultaneously, caspase-1 cleaves gasdermin D (GSDMD), producing
the pore-forming N-terminal fragment GSDMD-NT, which forms pores in
the cell membrane, inducing pyroptosis (14). This process regulates the
inflammatory response and helps defend against pathogen infection
and stress-induced damage. However, excessive activation of the
AIM2 inflammasome may lead to an overly robust immune response,
causing tissue damage and chronic inflammatory diseases (15). Under chronic inflammatory
conditions, the sustained activation of AIM2 may contribute to the
maintenance of chronic inflammation, increase the recruitment of
immune cells, and promote the secretion of proinflammatory
cytokines, ultimately leading to tissue damage and organ
dysfunction (13). Therefore,
modulating AIM2 activity may be a promising therapeutic strategy
for treating various inflammatory diseases.
Triptolide (TP), the principal active component
isolated from Tripterygium wilfordii (Celastraceae), exerts
significant biological effects, including immunosuppressive,
anti-inflammatory, antitumor and antifertility effects (16). A number of pharmacological and
clinical studies have shown that TP plays a crucial role in
regulating the immune system, particularly in inhibiting excessive
immune responses and alleviating autoimmune diseases such as
rheumatoid arthritis and nephritis (16-19). TP exerts its effects by targeting
lymphocytes, inhibiting the overactivation of immune cells, and
reducing immune-mediated inflammation, thereby improving disease
symptoms and minimizing tissue damage (20). In the context of H1N1-induced
pneumonia, viral infection typically triggers an exaggerated immune
response, leading to the infiltration of inflammatory cells and the
excessive release of inflammatory cytokines, such as IL-6 and TNF-α
(21,22). This overactive immune response
not only fails to effectively clear the virus but also exacerbates
lung tissue damage, potentially leading to severe pneumonia and
organ failure (21,22). Given the notable
immunosuppressive and anti-inflammatory properties of TP, it was
hypothesized that TP could specifically attenuate the dysregulated
immune response and cytokine storm in H1N1 influenza-induced
pneumonia, thereby reducing inflammatory lung damage and improving
outcomes. Current therapies fail to concurrently address viral
replication and dysregulated inflammation; however, the present
study identified TP as a novel dual-function agent capable of both
suppressing influenza virus proliferation and mitigating excessive
host immune responses. Crucially, it revealed, for the first time
to the best of the authors' knowledge, that TP exerts these effects
by inhibiting the AIM2 inflammasome pathway, a key driver of
pyroptosis and inflammation amplification in severe viral
pneumonia.
Materials and methods
Cell culture
Immortalized human bronchial epithelial cells
(HBEpiCs) were purchased from Shanghai Zhongqiao Xinzhou
Biotechnology Co., Ltd. (cat. no. ZQ0001). The cell line was
derived from normal bronchial epithelium obtained from a
54-year-old male patient who underwent lobectomy for squamous cell
carcinoma. These cells were immortalized by infection with the
recombinant retrovirus LXSN16E6E7, which carries the E6 and E7
genes of human papillomavirus type 16, followed by selection in
medium containing 0.4 mg/ml G418. Consequently, these cells should
be referred to as immortalized human bronchial epithelial cells
rather than primary cells. These cells serve as a representative
in vitro model for airway epithelial cells, which constitute
the first line of defense against respiratory pathogens. The
bronchial epithelium plays a pivotal role in mucociliary clearance
and acts as a physical and immunological barrier, initiating innate
immune responses upon pathogen invasion. THP-1 cells, a human
monocytic cell line, obtained from Wuhan Procell Life Science &
Technology Co., Ltd., was derived from a patient with acute
monocytic leukemia. THP-1 cells are widely used as models for human
monocytes/macrophages in immunological studies because of their
ability to differentiate into macrophage-like cells upon
stimulation. They are instrumental in investigating
monocyte/macrophage functions, signaling pathways, and inflammatory
responses. HBEpiCs were cultured in bronchial epithelial cells
complete culture medium (cat. no. ZQ-1322; Shanghai Zhongqiao
Xinzhou Biotechnology Co., Ltd.), while THP-1 cells were cultured
in RPMI-1640 supplemented with 10% fetal bovine serum (HyClone;
Cytiva) and 1% penicillin-streptomycin (100X; Beijing Solarbio
Science & Technology Co., Ltd.). All cultures were maintained
at 37°C in a humidified atmosphere with 5% CO2.
The influenza A/PR/8/34 (H1N1) strain was obtained
from the Pathogen Detection and Biosafety Laboratory of the
Shanghai Public Health Clinical Center. The blank control group was
treated with complete culture medium. The H1N1 virus infection
group was infected with H1N1 virus at a multiplicity of infection
(MOI) of 1.0 for 24 h to establish an HBEpiC model of H1N1 virus
infection.
PMA induction
THP-1 cells (1×106 cells/ml) were seeded
in a six-well plate and cultured for 24 h. Then, phorbol
12-myristate 13-acetate (PMA; MedChemExpress) was added to the
culture medium at a final concentration of 100 nM. The cells were
then incubated at 37°C with 5% CO2 for an additional 24
h to induce the differentiation of THP-1 cells into adherent
macrophage-like cells.
TP intervention
After HBEpiCs were infected with H1N1 for 24 h, the
cells were treated with various concentrations of TP (5, 10, or 20
nM; Beijing Solarbio Science & Technology Co., Ltd.) for an
additional 24 h. Cell supernatants or cells were then collected for
subsequent experiments.
Enzyme-linked immunosorbent assay
(ELISA)
Cell supernatants were collected and analyzed using
Human TNF-α High Sensitivity ELISA Kit [cat. no. EK182HS; Hangzhou
Multi Sciences (Lianke) Biotech Co., Ltd.], Human IL-8 ELISA Kit
[cat. no. EK108; Hangzhou Multi Sciences (Lianke) Biotech Co.,
Ltd.], a human IL-1β ELISA kit [EH0185; Hangzhou Multi Sciences
(Lianke) Biotech Co., Ltd.] and IL-6 [cat. no. EK1217; Hangzhou
Multi Sciences (Lianke) Biotech Co., Ltd.] according to the
manufacturer's instructions.
For the mouse experiments, all mice were
anesthetized, and tracheal intubation was performed through the
midline of the neck. A total of 30 mice (n=6 in each group) were
used in the present study. The trachea was isolated, and the right
main bronchus was ligated. Two milliliters of physiological saline
were slowly injected into and then removed from the left lung using
a syringe, and this procedure was repeated three times to collect
bronchoalveolar lavage fluid (BALF). The BALF was centrifuged at
3,000 × g for 10 min at 4°C, and the supernatant was collected.
TNF-α, IFN-γ, and IL-6, IL-10 levels in the BALF were measured
using Mouse Tumor necrosis factor α, TNF-α ELISA KIT (cat. no.
CSB-E04741m; Cusabio Technology, LLC), Mouse Interferon γ,
IFN-γ/interferon, gamma/Ifng ELISA Kit (cat. no. CSB-E04578m;
Cusabio Technology, LLC) and Mouse IL-10 ELISA Kit (cat. no.
U96-1517E; YOBIBIO (Shanghai) Biotechnology Co., Ltd.).
TP attenuates H1N1-triggered pneumonia
pathogenesis in a co-culture model
In the present study, the primary aim was to
investigate the therapeutic effect of TP on H1N1-induced pneumonia,
with particular focus on the two key cell types that play critical
roles in the development and progression of viral pneumonia:
alveolar epithelial cells and immune cells. HBEpiC cells were used
as an in vitro model of human bronchial/alveolar epithelial
cells, which are the primary targets for H1N1 virus infection in
the respiratory tract. Infection of these cells initiates viral
replication and triggers local inflammatory responses, leading to
epithelial damage and impaired lung function. THP-1 cells are a
well-established human monocytic cell line commonly used to
simulate monocytes/macrophages in vitro. These cells are
representative of the innate immune cells recruited to the lungs
during influenza infection, where they participate in the antiviral
response and contribute to the release of pro-inflammatory
cytokines, which can amplify lung inflammation and tissue injury.
By using HBEpiC and THP-1 cells, the present study aimed to
recreate an in vitro co-culture context that mimicked the
interactions between infected epithelial cells and responding
immune cells, thus allowing it to evaluate whether TP can modulate
both virus-induced epithelial damage and the excessive
immune-inflammatory response that underlies the pathogenesis of
H1N1-induced pneumonia.
Adhesion assay between THP-1 cells and
HBEpiCs
An adhesion assay was performed as an integral
component of the investigation into the interactions between
HBEpiCs and monocytes (THP-1) during H1N1 influenza virus
infection. This assay aimed to simulate and quantify the
recruitment and adhesion of immune cells to the alveolar
epithelium, a critical step in the initiation and propagation of
pulmonary inflammation during viral infections. THP-1 cells were
induced with PMA for 24 h as previously described. Simultaneously,
HBEpiCs were transfected with a GFP-tagged fluorescent adenovirus
(30 MOI; Charles River Laboratories). After 24 h, the transfected
HBEpiCs were collected. These GFP-labeled HBEpiCs were then added
to the pretreated THP-1 cells and both cell types were cocultured
for an additional 24 h. In the TP intervention group, TP at
concentrations of 5, 10 and 20 nM (Beijing Solarbio Science &
Technology Co., Ltd.) was added for another 24 h of culture. The
adhesion between THP-1 cells and HBEpiCs was observed and evaluated
under a fluorescence microscope (20×; Olympus Corporation).
Cell migration assay
A cell migration assay was used to investigate the
chemotactic response of monocyte-derived THP-1 cells to soluble
factors secreted by H1N1-infected HBEpiCs. This approach aimed to
simulate the in vivo recruitment of immune cells to sites of
viral infection, a critical component of the inflammatory response
in H1N1-induced pneumonia. THP-1 cells were seeded into the upper
chamber of a Transwell insert and induced with phorbol myristate
acetate (PMA) for 24 h as aforementioned. HBEpiCs were infected
with H1N1 virus, and the culture supernatant was collected after 24
h. The supernatant was then placed in the lower chamber of the
Transwell insert. After 24 h of incubation, the migration of THP-1
cells to the lower chamber was observed. Migrated cells were
counted and quantified via microscopy. The number of cells that
migrated through the membrane was used as an indicator of cell
migration.
Reverse transcription-quantitative (RT-q)
PCR
Viral RNA quantification (absolute
quantification)
Lung tissue stored at −80°C was homogenized in
liquid nitrogen to obtain a fine powder. RNA was extracted using
Triquick Reagent (cat. no. R1100; Beijing Solarbio Science &
Technology Co., Ltd.) and stored at −80°C. DEPC-treated water
(DEPC-H2O) was used as a negative control. For absolute
quantification of the viral load, a standard curve was generated
using positive controls prepared by serially diluting a standard
sample with a known concentration to final concentrations of
1×107, 1×106, 1×105,
1×104 and 1×103 copies/ml. For each reaction
using the H1N1 Subtype Influenza A (Avian Influenza) Dual RT-PCR
Detection Kit (cat. no. LM82068SRP; Shanghai Lianmai Biological
Engineering Co., Ltd.) 18 μl of master mix, 1 μl of
internal control and 1 μl of RT-PCR enzyme mixture were
added. The mixture was vortexed and centrifuged briefly at 3,000 ×
g. A total of 20 μl of the resulting mixture was transferred
to a PCR tube, followed by the addition of 5 μl of sample
nucleic acid extract, DEPC-H2O (negative control), or
diluted positive control (standard curve points). The samples were
then centrifuged and immediately subjected to PCR amplification:
Reverse transcription: 50°C for 15 min; initial denaturation: 95°C
for 5 min; cycling program (number of cycles: 40): Denaturation:
95°C for 15 sec; annealing/extension: 58°C for 60 sec). The cycle
threshold (Cq) values obtained for the samples and the standard
curve points were recorded. The viral nucleic acid concentration
(copies/ml) in each sample was determined by interpolating its Cq
value onto the standard curve (log10 copies/ml vs. Cq
value) generated from the diluted positive controls (23).
Cellular gene expression analysis
(relative quantification)
In addition, total RNA was collected and extracted
from the cell samples. First-strand cDNA synthesis was performed
using the Invitrogen; Thermo Fisher Scientific, Inc. SuperScript IV
UniPrime Kit (Invitrogen; Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. Subsequently, quantitative
real-time PCR was performed using NovoStart® SYBR
High-Sensitivity qPCR SuperMix (Novoprotein Scientific Inc.).
Relative gene expression levels were determined via the comparative
Cq (ΔΔCq) method (23). The
expression of the target gene(s) was normalized to that of GAPDH.
The sequences of primers used in the present study: AIM2-F:
TAGCGCCTCACGTGTGTTAG, AIM2-R: TCGGGGTTTCACCAGCTTTT; Caspase-1-F:
GGGAGTGTGGGAAGGTTGAG, Caspase-1-R: GTGCTCTGCACTGACCAGAA; GSDMD-F:
CCTCTCCATGATGAGGTGCC, GSDMD-R: CCCAGCAGGTAGACAACAGG; GAPDH-F:
CCAGCAAGAGCACAAGAGGA, GAPDH-R: GGGGAGATTCAGTGTGGTGG.
Western blot analysis
The proteins were extracted using RIPA buffer (high;
Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China) supplemented with 1 mM PMSF (Beijing Solarbio Science &
Technology Co., Ltd.) and 1X protease inhibitor cocktail (cat. no.
A8260; Beijing Solarbio Science & Technology Co., Ltd.). Cell
or tissue lysates were collected and centrifuged at 15,000 × g for
10 min at 4°C. Protein concentrations were determined via a BCA
assay kit. Proteins (10 μg/lane) were separated by 12%
SDS-PAGE and transferred onto a PVDF membrane at 4°C. The membrane
was blocked with 5% bovine serum albumin at room temperature for 1
h, followed by overnight incubation with primary antibodies
targeting AIM2 (cat. no. TB7010S), caspase-1 (cat. no. TA5418S),
GSDMD (cat. no. TA4012S) and β-actin (cat. no. P30002; 1:1,000; all
from Abmart Pharmaceutical Technology Co., Ltd.). The membrane was
then incubated with ActivAb Goat Anti-Rabbit IgG/HRP secondary
antibody (1:5,000, cat. no. SE134, Beijing Solarbio Science &
Technology Co., Ltd.) at 37°C for 2 h. Protein bands were
visualized using an enhanced chemiluminescence reagent (Beijing
Solarbio Science & Technology Co., Ltd.), and the signal
intensities of the target bands were quantified using ImageJ
software (version number: 1.8.0, National Institutes of
Health).
Dual-immunofluorescence staining
To investigate the assembly of the AIM2 inflammasome
during viral infection, the present study performed
dual-immunofluorescence staining for ASC (an adaptor protein) and
AIM2 (a cytosolic DNA sensor). The colocalization of these two
proteins (visualized as overlapping fluorescent signals) indicates
the formation of the ASC-AIM2 inflammasome complex, a key event in
innate immune activation. THP-1 cells and HBEpiCs were seeded at a
density of 1×105 cells/ml onto glass slides and cultured
in a 6-well plate for 24 h. The cells were fixed at room
temperature using 4% paraformaldehyde (PFA; Beijing Solarbio
Science & Technology Co., Ltd.) in PBS for 10 min, followed by
three 5-min washes with PBS. Blocking was performed at room
temperature for 1 h. Blocking was performed using QuickBlock
Blocking Buffer for Immunol Staining (P0260; Beyotime Institute of
Biotechnology) at room temperature for 15 min. Primary antibodies
targeting ASC and AIM2 were added, and the samples were incubated
overnight at 4°C. Then, fluorescently labeled anti-rabbit or
anti-mouse secondary antibodies (Beijing Solarbio Science &
Technology Co., Ltd.) were applied and incubated with the samples
at room temperature for 1 h. Following secondary antibody
incubation and washing, the cells were stained with DAPI (1
μg/ml in PBS) for 5 min at room temperature to visualize
cell nuclei. The cells were washed three times with PBS for 5 min
each. Finally, anti-fade mounting medium (Beijing Solarbio Science
& Technology Co., Ltd.) was applied to the slides, and the
cells were observed under a fluorescence microscope at a
magnification of ×20.
LDH release cytotoxicity assay
Cells in both experimental and control groups were
seeded into 96-well plates at 100 μl per well and allowed to
grow until reaching approximately 70-80% confluency before
treatment. The following controls were included: A blank control
(medium only, no cells), a spontaneous release control (untreated
cells), and a maximum release control (cells fully lysed by
addition of lysis buffer as per manufacturer's instructions). After
treatment, 50 μl of the supernatant from each well was
transferred to a new plate, and an equal volume of LDH reaction mix
from the Lactate Dehydrogenase (LDH) Activity Assay Kit (cat. no.
E-BC-K046-M; Elabscience Bionovation Inc.) was added. The reaction
was incubated at room temperature in the dark for 20 min, then
stopped with stop solution. Absorbance was measured at 450 nm using
a microplate reader. Background absorbance (blank) was subtracted
from all sample values to yield corrected OD readings.
Ad-AIM2 transfection
To confirm AIM2 as a key functional target through
which TP exerts its immunosuppressive effects, a rescue experiment
was performed in which AIM2 was overexpressed in THP-1-derived
macrophages. Ad-AIM2 was constructed by Hanheng Biotechnology
(Shanghai) Co., Ltd. After 24 h of PMA induction in THP-1 cells,
the cells were transfected with a viral vector containing Ad-AIM2
at 30 MOI for 24 h. Following transfection, subsequent experiments
were conducted as aforementioned.
In vivo mouse model of H1N1 influenza
virus infection and TP treatment
To evaluate the therapeutic efficacy of TP against
influenza virus infection in vivo, a murine model of H1N1
influenza virus-induced pneumonia was established. Specific
pathogen-free (SPF)-grade male C57BL/6 mice (8 weeks old; weighing
20-22 g; n=30) were purchased from Cyagen Biosciences (Guangzhou)
Co., Ltd. The mice were housed in an SPF barrier facility at
Guangzhou University of Chinese Medicine under controlled
environmental conditions consisting of a temperature of 22±2°C, a
humidity of 55±10%, and a 12-h light/dark cycle (lights on at 7:00
am). The animals were provided standard laboratory rodent chow and
autoclaved water ad libitum. The mice were acclimatized to
the housing conditions for at least 7 days prior to any
experimental procedures. All experimental procedures were performed
in accordance with the guidelines of the Institutional Animal Care
and Use Committee and approved by the Committee on the Ethics of
Animal Experiments of Guangzhou University of Chinese Medicine
(approval no. 2024055R).
Virus and infection
The influenza A virus strain A/Puerto Rico/8/1934
(H1N1; PR8) was obtained from the American Type Culture Collection.
The virus was propagated in Madin-Darby canine kidney (MDCK) cells
and purified. The viral titer was determined by plaque assay. For
infection, the mice were lightly anesthetized via inhalation of
isoflurane (4% induction, 2% maintenance) and intranasally
inoculated with 100 μl of a viral suspension containing
1×105 plaque-forming units (PFUs) of PR8 virus (diluted
in sterile saline to a concentration of 1×106 PFU/ml).
For model establishment, control mice were inoculated intranasally
with an equivalent volume (100 μl) of sterile saline under
identical anesthesia conditions.
Model confirmation
At 6 days post-infection, all mice in the
virus-infected group (n=5) and saline control group (n=5) were
sacrificed by cervical dislocation under deep anesthesia (5%
isoflurane inhalation, with the absence of the pedal reflex
confirmed). Lung tissues were collected for histopathological
analysis by hematoxylin and eosin (H&E) staining. Successful
model establishment was defined as >90% of infected mice
exhibiting characteristic viral pneumonia features (inflammatory
cell infiltration, hemorrhage, atelectasis, interstitial edema, and
alveolar wall thickening), with control mice showing no significant
pathology.
Group assignment and treatment
Following confirmation of successful model
establishment, H1N1-infected mice were randomly divided into five
experimental groups (n=5 per group): i) H1N1 control (model
control): infected, treated with vehicle (sterile saline via oral
gavage); ii) Tamiflu: infected, treated with Tamiflu (oseltamivir
phosphate, 20 mg/kg via oral gavage; this dose was selected on the
basis of its well-established efficacy in murine models of H1N1
infection, as reported in previous studies (24,25), and aligns with doses commonly
used to demonstrate significant antiviral activity and survival
benefit in this context); iii) L-TP: infected, treated with
low-dose TP (5 μg·kg-1·day-1 via
intraperitoneal injection); iv) M-TP: infected, treated with
medium-dose TP (10 μg·kg-1·day-1 via
intraperitoneal injection); and v) H-TP: infected, treated with
high-dose TP (20 μg·kg-1·day-1 via
intraperitoneal injection) (26). vi) Additionally, the initial
saline control (uninfected control) group (n=5) was maintained as
an uninfected, untreated control. Treatments commenced at 48 h
post-infection and were administered twice daily (morning and
afternoon) by oral gavage for 5 consecutive days. The gavage volume
for all treatments was standardized at 0.2 ml per 10 g of body
weight. Animals in the uninfected control group and H1N1 control
group received equivalent volumes of sterile saline via oral
gavage.
Terminal procedures and sample
collection
At the end of the treatment period (day 7), all mice
were weighed and sacrificed. Prior to sacrifice, mice were deeply
anesthetized with isoflurane (4% induction, 2% maintenance).
Following sufficient anesthesia, sacrifice was performed by
cervical dislocation or another approved method in accordance with
ethical guidelines. The lungs were aseptically removed. The left
lung lobe was fixed in 4% PFA at room temperature for 15 min
subsequent histopathological analysis. The right lung lobes were
snap-frozen in liquid nitrogen and stored at −80°C for further
biochemical/molecular analyses. For immediate analysis, a portion
of frozen lung tissue from the right lung was homogenized in 1 ml
of ice-cold sterile saline using a tissue homogenizer. The
homogenate was centrifuged at 3,000 × g and 4°C for 10 min, and the
supernatant was collected and stored at −80°C until analysis.
Assessment parameters
The lung index was calculated as follows: (lung wet
weight/body weight) ×100%. The lung index inhibition rate for the
treatment groups was calculated using the following formula: lung
index inhibition rate (%)=[(mean lung index_H1N1 control-mean lung
index_treatment group)/(mean lung index_H1N1 control-mean lung
index_uninfected control)] ×100%.
H&E staining
A total of 30 mice from each group (n=5 in each
group) were sacrificed, and their lungs were dissected. The left
lung was fixed in 4% paraformaldehyde (Beijing Solarbio Science
& Technology Co., Ltd.), dehydrated in a graded ethanol series
and embedded in paraffin. Sections with a thickness of 4 μm
were prepared. H&E staining was performed at room temperature
(~25°C), as per the standard protocol provided with the kit
(Beijing Solarbio Science & Technology Co., Ltd.). The
pathological changes in the lung tissue were then observed under a
light microscope. Two well-established semiquantitative
histopathological scoring systems were used to assess lung injury
in our study. Specifically, the first was reported by Zeldin et
al (27): This method is
used to evaluate lung injury based on parameters such as
perivascular edema, inflammatory cell infiltration, and alveolar
damage. Each parameter is scored on a scale from 0 to 4, with
higher scores indicating more severe injury. The second was
reported by Matute-Bello et al (28) and is recommended by the American
Thoracic Society: This system assesses five key histological
features, including alveolar and interstitial neutrophil
infiltration, hyaline membrane formation, proteinaceous debris in
airspaces, and alveolar septal thickening. Each feature is scored
from 0 to 2 across multiple high-power fields, and the total score
provides a quantitative measure of lung injury severity.
Statistical analysis
All experiments were independently repeated three
times. Statistical analysis of the experimental data was performed
via GraphPad Prism 8.0 software. Data that followed a normal
distribution are presented as the mean ± standard deviation.
Intergroup comparisons were performed using one-way ANOVA, followed
by Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
H1N1 infection induces cytotoxicity and
modulates inflammatory responses in HBEpiCs and THP-1 cells
The present study infected HBEpiCs with H1N1 and
observed a concentration-dependent decrease in cell viability
following infection (Fig. 1A).
Moreover, the levels of the inflammatory cytokines IL-1β, IL-6,
TNF-α, and IL-8 in HBEpiCs increased following H1N1 infection
(Fig. 1B). Furthermore, the
supernatant from H1N1-infected HBEpiCs was collected and added to
THP-1 cell cultures. The results revealed a gradual reduction in
THP-1 cell viability in response to increasing H1N1 concentration
(Fig. 1C). Similarly, the levels
of IL-1β, IL-6, TNF-α, and IL-8 in THP-1 cells also increased
following H1N1 infection (Fig.
1D). Additionally, the adhesion between THP-1 cells and HBEpiCs
was assessed and it was observed that the number of THP-1 cells
adhering to HBEpiCs increased with increasing H1N1 concentration
(Fig. 1E). Moreover, the
migration capacity of THP-1 cells increased as the H1N1
concentration increased (Fig.
1F). These findings suggested a potential interaction between
these cells under inflammatory conditions induced by the virus.
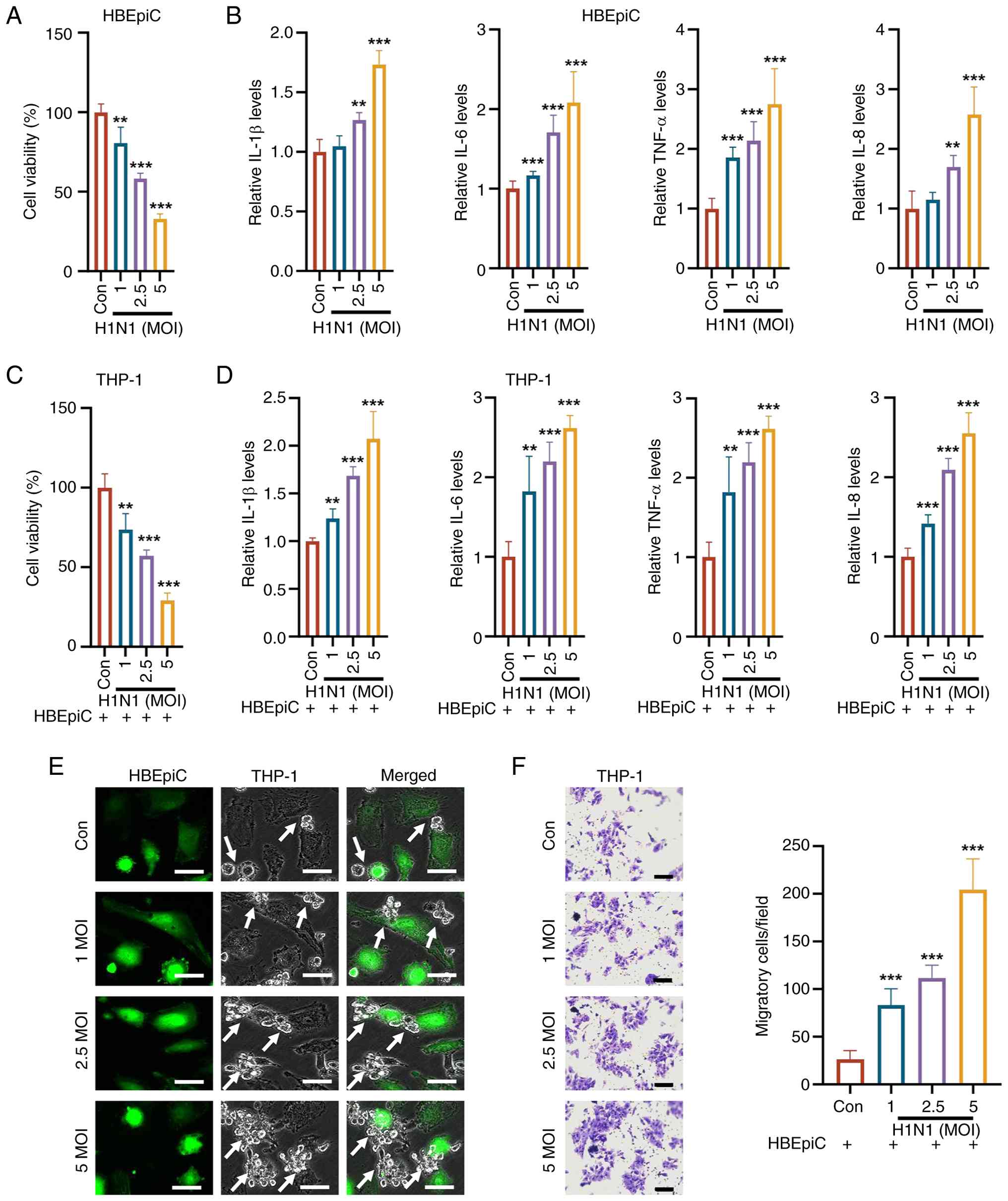 | Figure 1H1N1 infection affects cell
viability, inflammatory cytokine secretion and interactions between
HBEpiCs and THP-1 cells. (A) CCK-8 assay revealed that HBEpiC
viability decreased in a concentration-dependent manner following
H1N1 infection. (B) ELISA revealed that the levels of IL-1β, IL-6,
TNF-α, and IL-8 in HBEpiCs decreased with increasing H1N1
infection. (C) CCK-8 assay indicated that supernatants from
H1N1-infected HBEpiC cultures reduced the viability of THP-1 cells
in a dose-dependent manner. (D) ELISA results suggested that the
levels of inflammatory cytokines (IL-1β, IL-6, TNF-α and IL-8) in
THP-1 cells were decreased following exposure to supernatants from
H1N1-infected HBEpiC cultures. (E) Cell adhesion assay revealed
that the number of THP-1 cells adhering to HBEpiCs increased with
increasing H1N1 concentration (scale bar, 10 μm). Arrow
indicates THP-1 cells that remain attached to the surface of
HBEpiCs, highlighting the adhesion interaction between the two cell
types. (F) Transwell assay suggested that H1N1 infection enhanced
the migration capacity of THP-1 cells, with increased migration
observed at higher virus concentrations (scale bar, 50 μm).
The data are presented as the mean ± standard deviation;
**P<0.01, ***P<0.001 vs. control. H1N1,
influenza A; HBEpiCs, human bronchial epithelial cells; ELISA,
enzyme-linked immunosorbent assay; IL, interleukin; TNF-α, tumor
necrosis factor-α; Con, control; MOI, multiplicity of
infection. |
H1N1 infection activates AIM2 signaling
in THP-1 cells but not in HBEpiCs
AIM2 regulation plays a crucial role in the
pathogenesis of H1N1-induced lung diseases, serving as an important
component of the antiviral response while potentially exacerbating
pathological damage (29).
Therefore, the present study focused on investigating whether H1N1
infection alters the AIM2 signaling pathway in alveolar epithelial
cells and immune cells. The RT-qPCR results revealed that H1N1
infection did not affect AIM2 mRNA levels in HBEpiCs but markedly
increased AIM2 mRNA levels in THP-1 cells (Fig. 2A). Additionally, lactate
dehydrogenase (LDH) release was assessed and found that H1N1
infection did not affect the LDH release rate in HBEpiCs but
markedly increased the release of LDH-1 in THP-1 cells (Fig. 2B). Furthermore,
dual-immunofluorescence staining revealed the formation of a
complex between ASC and AIM2 in THP-1 cells (Fig. 2C). Compared with the control,
H1N1 infection did not markedly alter the protein levels of AIM2,
caspase-1, or GSDMD in HBEpiCs (Fig.
2D). However, in THP-1 cells, H1N1 infection led to increased
protein levels of AIM2, caspase-1, and GSDMD (Fig. 2E). Following infection of HBEpiCs
with H1N1 virus at an MOI of 5 for 24 h, no significant cell death
was observed among HBEpiCs (Fig.
2F). The supernatants from both the control and H1N1-infected
HBEpiC groups were subsequently collected and used to treat THP-1
cells for an additional 24 h. Compared with that in the control
(Con) group, the death rate in the treated THP-1 cell group was
markedly greater (Fig. 2G).
Overall, H1N1 infection activated the AIM2 signaling pathway and
increased cell death among THP-1 cells, whereas it did not activate
the AIM2 signaling pathway or induce cell death among HBEpiCs.
These results highlighted the differential responses of different
cell types to H1N1 infection. Immune cells exert their effects
through AIM2-mediated inflammatory responses, whereas epithelial
cells may use other mechanisms for antiviral defense. However, the
factors they release can still influence immune cell behavior,
further exacerbating the immune response and cell death.
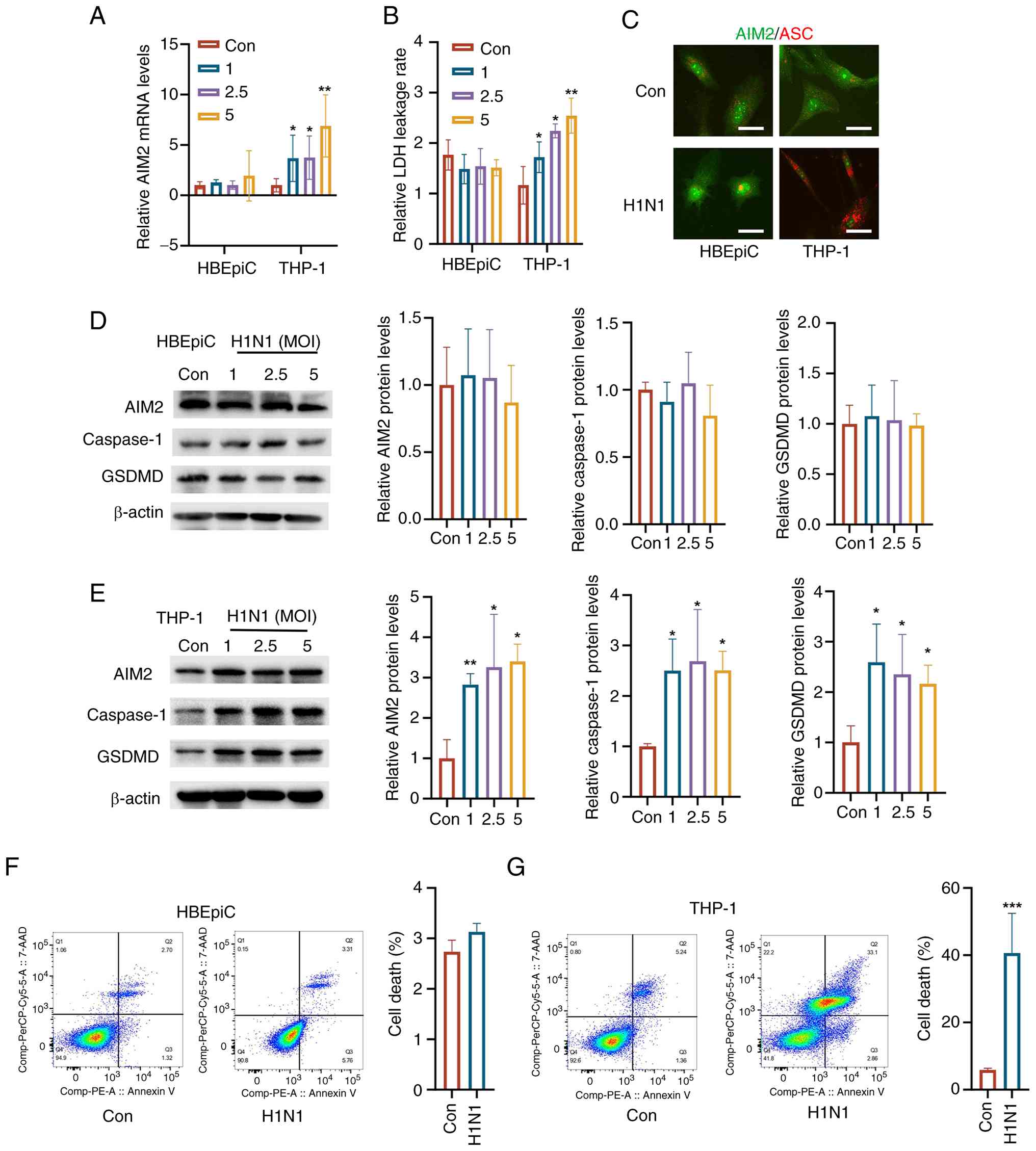 | Figure 2Differential activation of AIM2
signaling in THP-1 cells and HBEpiCs following H1N1 infection. (A)
RT-qPCR analysis revealed that H1N1 infection did not alter AIM2
mRNA levels in HBEpiCs but markedly increased AIM2 mRNA levels in
THP-1 cells. (B) LDH release assay indicated that H1N1 infection
did not affect LDH release in HBEpiCs but markedly increased LDH-1
release in THP-1 cells. (C) Dual-immunofluorescence staining
demonstrated the formation of ASC-AIM2 complexes in THP-1 cells
post-H1N1 infection (scale bar, 10 μm). (D) Western blot
analysis revealed no significant changes in AIM2, caspase-1, or
GSDMD protein levels in HBEpiCs following H1N1 infection but
increased expression of AIM2, caspase-1 and GSDMD in THP-1 cells
following H1N1 infection (E). (F) Flow cytometry analysis revealed
no significant cell death in HBEpiCs following infection, (G)
whereas THP-1 cells treated with the supernatant from H1N1-infected
HBEpiC cultures presented markedly increased cell death compared
with the control. The data are presented as the mean ± standard
deviation; *P<0.05, **P<0.01,
***P<0.001 vs. control. AIM2, absent in melanoma 2;
HBEpiCs, human bronchial epithelial cells; H1N1, influenza A;
RT-qPCR, reverse transcription-quantitative PCR; GSDMD, gasdermin
D; Con, control; MOI, multiplicity of infection. |
TP inhibits the inflammatory response and
immune cell activity in H1N1-Infected HBEpiCs and THP-1 cells
The concentration gradient of TP (5, 10, 20, 30 nM)
was designed based on established literature evidence and
fundamental biological differences between cell models. Zhou et
al (29) demonstrated that
10 nM TP markedly reduces viability in immortalized human bronchial
epithelial cells (BEAS-2B) within 24 h in an acute lung injury
model. Additionally, in vitro experiments with primary mouse
lung fibroblasts showed that TP concentrations ranging from 10-40
nM gradually inhibited cell viability, with concentrations >40
nM exhibiting marked cytotoxicity (30). Therefore, 20 and 40 nM were
selected or subsequent experiments to balance efficacy and
cytotoxicity. Consequently, 5, 10, 20 and 30 nM were selected for
the preliminary dose-response experiments to evaluate antiviral
efficacy while minimizing cytotoxic effects. Cell viability assays
indicated that 5, 10, and 20 nM TP did not markedly affect HBEpiC
viability compared with the control, whereas 30 nM TP led to a
noticeable decrease in cell viability (Fig. 3A). Based on these findings, 5,
10, and 20 nM TP were selected for subsequent experiments for
assessing antiviral efficacy. However, following TP treatment, the
levels of IL-1β, IL-6, TNF-α, and IL-8 in HBEpiCs were markedly
reduced (Fig. 3B). By contrast,
when TP was applied to THP-1 cells pretreated with the supernatant
from H1N1-infected HBEpiC cultures, the viability of the THP-1
cells decreased (Fig. 3C).
Additionally, the levels of IL-1β, IL-6, TNF-α, and IL-8 in THP-1
cells were markedly lower after TP treatment (Fig. 3D). Moreover, the adhesion of
THP-1 cells to HBEpiCs induced by H1N1 infection was reduced in a
dose-dependent manner with increasing TP concentrations (Fig. 3E). Similarly, when the
supernatant collected from H1N1-infected HBEpiC cultures were
treated with TP, the migration capacity of the THP-1 cells was also
markedly reduced (Fig. 3F).
These findings suggested that TP, as an immunomodulator, may
regulate the inflammatory response by inhibiting the overactivation
of immune cells.
 | Figure 3TP modulates the inflammatory
response and immune cell activity in H1N1-infected HBEpiCs and
THP-1 cells. (A) No significant changes were observed in HBEpiCs
treated with various concentrations of TP (5, 10 and 20 nM)
following H1N1 infection compared with the control. (B) After TP
treatment, the levels of the inflammatory cytokines IL-1β, IL-6,
TNF-α and IL-8 in HBEpiCs were markedly lower than those in the
untreated group. (C) The viability of THP-1 cells pretreated with
H1N1-infected HBEpiC culture supernatant decreased after TP
treatment. (D) The levels of IL-1β, IL-6, TNF-α, and IL-8 in THP-1
cells were markedly lower after TP treatment. (E) The adhesion of
THP-1 cells to HBEpiCs induced by H1N1 infection decreased in a
dose-dependent manner with increasing TP concentration (scale bar,
10 μm). Arrow indicates THP-1 cells that remain attached to
the surface of HBEpiCs, highlighting the adhesion interaction
between the two cell types. (F) The migration capacity of THP-1
cells was markedly reduced when the supernatant from H1N1-infected
HBEpiC cultures was treated with TP (scale bar, 50 μm). The
data are presented as the mean ± standard deviation;
*P<0.05, **P<0.01,
***P<0.001 vs. control. TP, triptolide; H1N1,
influenza A; HBEpiCs, human bronchial epithelial cells; IL,
interleukin; TNF-α, tumor necrosis factor-α; Con, control. |
TP suppresses AIM2 signaling, induces
pyroptosis in THP-1 cells, and alleviates inflammatory damage to
HBEpiCs
Following H1N1 infection of HBEpiCs, TP was
administered and the culture supernatant was collected to pretreat
THP-1 cells. In HBEpiCs, TP treatment did not affect the mRNA level
of AIM2, whereas in THP-1 cells, the mRNA expression of AIM2 was
markedly reduced by TP treatment (Fig. 4A). Additionally, TP treatment of
HBEpiCs did not alter the LDH leakage rate, but in THP-1 cells, TP
reduced the LDH leakage rate (Fig.
4B). Furthermore, dual-fluorescence staining of THP-1 cells
revealed a decrease in the formation of ASC and AIM2 complexes
(Fig. 4C). In HBEpiCs, TP
treatment did not markedly affect the protein expression of AIM2,
caspase-1, or GSDMD (Fig. 4D).
However, in THP-1 cells, TP treatment decreased the protein
expression of AIM2, caspase-1, and GSDMD (Fig. 4E). Additionally, at 20 nM, TP did
not affect the death rate of HBEpiCs but markedly mitigated the
increased death rate of THP-1 cells induced by H1N1 infection
(Fig. 4F and G). These results
indicated that TP exerted differential effects on different cell
types and that its anti-inflammatory action is mediated primarily
by the inhibition of AIM2-mediated cell death and inflammatory
responses.
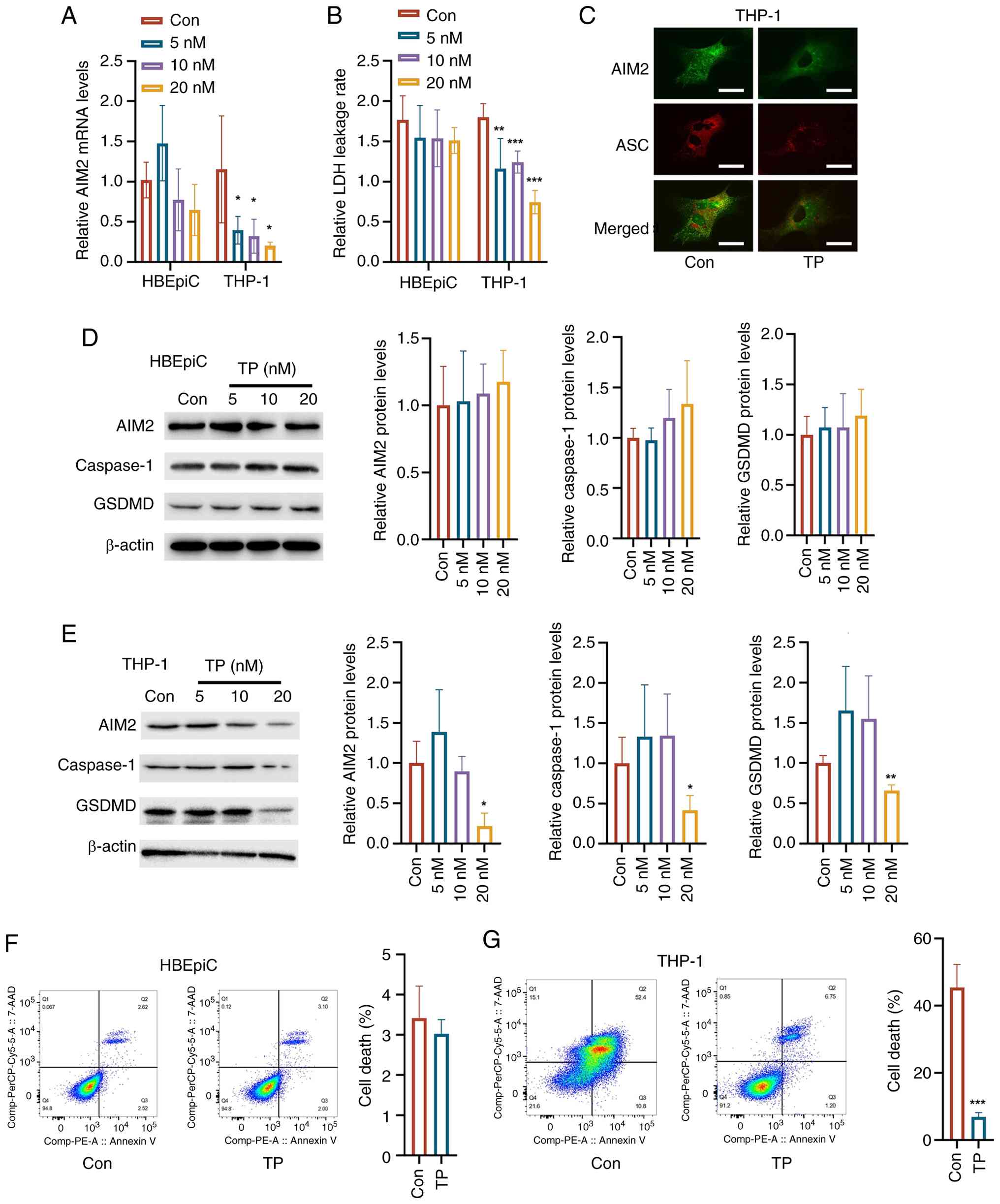 | Figure 4Effects of TP on AIM2 signaling, LDH
leakage, and pyroptosis in HBEpiCs and THP-1 cells. (A) RT-qPCR
analysis revealed that TP treatment did not alter AIM2 mRNA
expression in HBEpiCs but markedly reduced AIM2 mRNA levels in
THP-1 cells. (B) TP did not affect the LDH leakage rate in HBEpiCs
but markedly reduced LDH leakage in THP-1 cells. (C)
Dual-fluorescence staining revealed that TP treatment decreased the
formation of ASC-AIM2 complexes in THP-1 cells (scale bar, 10
μm). (D) Western blot analysis indicated that TP treatment
did not markedly affect the expression of these proteins in
HBEpiCs. (E) TP treatment reduced the protein expression of AIM2,
caspase-1, and GSDMD in THP-1 cells. (F) Flow cytometry revealed no
difference in cell death between HBEpiCs treated with 20 nM TP and
the control. (G) In THP-1 cells infected with H1N1 and treated with
20 nM TP, a significant reduction in cell death was identified
compared with that in the H1N1 infection group. The data are
presented as the mean ± standard deviation; *P<0.05,
**P<0.01, ***P<0.001 vs. control. TP,
triptolide; AIM2, absent in melanoma 2; LDH, lactate dehydrogenase;
HBEpiCs, human bronchial epithelial cells; RT-qPCR, reverse
transcription-quantitative PCR; H1N1, influenza A; GSDMD, gasdermin
D; Con, control. |
AIM2 overexpression reverses the
immunosuppressive effects of TP in THP-1 cells
To confirm that AIM2 is a key target of TP in THP-1
cells, AIM2 was overexpressed in THP-1 cells. The data showed that
transfection with Ad-AIM2 markedly upregulated the expression of
AIM2 in THP-1 cells compared with that of Ad-NC (Fig. 5A). HBEpiCs were incubated with
H1N1 and the supernatant was collected to treat AIM2-overexpressing
THP-1 cells with or without TP treatment. Compared with those in
THP-1 cells without AIM2 overexpression, AIM2 protein levels were
elevated in AIM2-overexpressing THP-1 cells (Fig. 5B). In THP-1 cells, after TP
treatment, the LDH leakage rate was lower than that in the control
group of cells treated with H1N1 alone. However, overexpression of
AIM2 increased the LDH leakage rate (Fig. 5C). Moreover, AIM2 overexpression
reversed the reduction in adhesion between THP-1 cells and HBEpiCs
induced by TP treatment (Fig.
5D). Furthermore, AIM2 overexpression enhanced the migration
ability of THP-1 cells compared with that of control cells treated
with H1N1 alone (Fig. 5E).
Notably, AIM2 overexpression even reversed the decrease in THP-1
cell migration induced by TP treatment (Fig. 5E). Additionally, AIM2
overexpression reversed the reduction in the levels of various
inflammatory cytokines caused by TP treatment in THP-1 cells
(Fig. 5F). These findings
suggested that TP exerts its immunosuppressive effects by
modulating AIM2 signaling, thereby preventing overactivation of
immune cells and reducing inflammation.
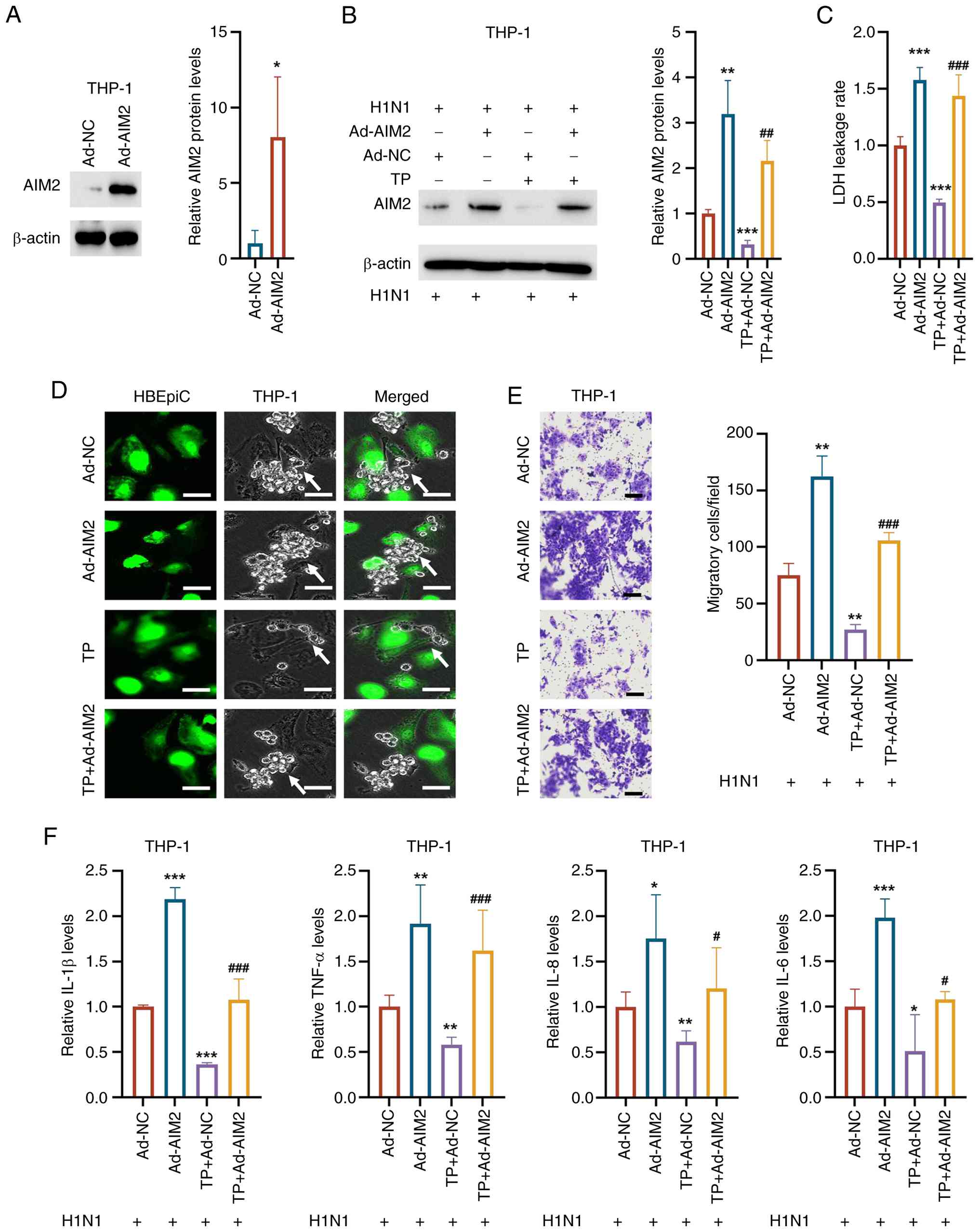 | Figure 5AIM2 overexpression reverses the
immunosuppressive effects of TP in THP-1 cells. (A) Transfection
with Ad-AIM2 upregulated the expression of AIM2 compared with that
of Ad-NC in THP-1 cells. (B) AIM2 protein levels were elevated in
THP-1 cells overexpressing AIM2 compared with control THP-1 cells.
(C) In THP-1 cells, AIM2 overexpression increased the LDH leakage
rate. (D) AIM2 overexpression reversed the TP-induced reduction in
adhesion between THP-1 cells and HBEpiCs (scale bar, 10 μm).
(E) AIM2 overexpression enhanced the migration ability of THP-1
cells compared with that of H1N1-treated control cells and reversed
the TP-induced decrease in THP-1 cell migration (scale bar, 50
μm). (F) AIM2 overexpression reversed the TP-induced
reduction in inflammatory cytokine levels. The data are presented
as the mean ± standard deviation; *P<0.05,
**P<0.01, ***P<0.001 vs. Con;
#P<0.05, ##P<0.01,
###P<0.001 vs. Ad-AIM2. AIM2, absent in melanoma 2;
TP, triptolide; LDH, lactate dehydrogenase; HBEpiCs, human
bronchial epithelial cells; H1N1, influenza A; GSDMD, gasdermin D;
IL, interleukin; TNF-α, tumor necrosis factor-α; Con, control. |
TP alleviates lung injury in a murine
H1N1 pneumonia model
To further investigate the effect of TP in
vivo, a murine pneumonia model induced by H1N1 infection was
established. Histological analysis with H&E staining revealed
that, compared with that of model mice, the lung tissue of normal
mice presented a clear structure with no thickening of the alveolar
septa, no infiltration of inflammatory cells in the stroma, and no
abnormal changes in bronchial structure. By contrast, mice in the
model group showed varying degrees of alveolar septal thickening
and even consolidation of lung tissue. Compared with those in the
model group, mice in the H-TP and Tamiflu treatment groups
exhibited mild thickening of the alveolar septa, with markedly
fewer histopathological changes. Additionally, compared with those
in the model group, mice in the M-TP and L-TP groups presented
varying degrees of improvement in the lung parenchyma, interstitial
tissue and bronchial lesions (Fig.
6A). Furthermore, compared with those in the control group
(0.1412±0.0035 g; 20.56±1.54 g), mice in the H1N1 treatment group
presented increased lung weight and decreased body weight
(0.1808±0.0022 g; 17.70±1.65 g) (Fig. 6B and C). The lung weight was
markedly lower in the Tamiflu, M-TP and H-TP groups (0.1724±0.0030
g; 0.1700±0.0027 g; 0.1676±0.0122 g) than in the H1N1 group
(0.1808±0.0022 g; Fig. 6B).
However, there were no significant differences in body weight among
the four treatment groups (18.84±1.81 g; 17.62±1.02 g; 18.52±1.50
g; 19.42±2.93 g) compared with the H1N1 group (17.70±1.65 g;
Fig. 6C). The present study also
analyzed the lung index, which was elevated following H1N1
infection (0.01029±0.00089) compared with that in the control group
(0.00690±0.00050). After treatment in the four groups, the lung
index did not fully recover (Tamiflu: 0.00922±0.00093; L-TP:
0.01044±0.00063; M-TP: 0.00922±0.00059; and H-TP: 0.00879±0.00140;
Fig. 6D). Notably, the lung
viral load was markedly lower in the Tamiflu (1.95±0.50), L-TP
(4.90±0.45), M-TP (4.61±0.47), and H-TP (3.76±0.47) treatment
groups compared with the H1N1 group (7.34±0.16), with the Tamiflu
group showing the most prominent reduction (Fig. 6E). Additionally, the levels of
inflammatory cytokines in the lung tissue were elevated following
H1N1 infection (IL-6: 331.4±114.1 pg/ml; IL-10: 200.8±2.4 pg/ml;
TNF-α: 82.0±15.2 pg/ml; IFN-γ: 169.4±3.2 pg/ml) compared with those
in the control group (IL-6: 5.2±14.3 pg/ml; IL-10: 2.2±0.8 pg/ml;
TNF-α: 3.6±1.8 pg/ml; IFN-γ: 8.2±1.3 pg/ml), but treatment with
Tamiflu (IL-6: 106.8±25.2 pg/ml; IL-10: 39.6±12.3 pg/ml; TNF-α:
46.8±1.9 pg/ml; IFN-γ: 77.8±27.7 pg/ml), L-TP (IL-6: 199.0±47.3
pg/ml; IL-10: 95.8±13.1 pg/ml; TNF-α: 40.8±12.6 pg/ml; IFN-γ:
82.8±29.8 pg/ml), M-TP (IL-6: 128.4±50.2 pg/ml; IL-10: 55.4±13.9
pg/ml; TNF-α: 32.4±15.2 pg/ml; IFN-γ: 73.8±23.4 pg/ml), and H-TP
(IL-6: 91.0±18.8 pg/ml; IL-10: 32.8±16.2 pg/ml; TNF-α: 32.6±8.8
pg/ml; IFN-γ: 45.6±18.6 pg/ml) markedly reduced these levels
(Fig. 6F-I). Overall, TP
demonstrated immune modulation at different doses, alleviating lung
damage induced by H1N1 and reducing immune injury by regulating
inflammatory responses and viral replication.
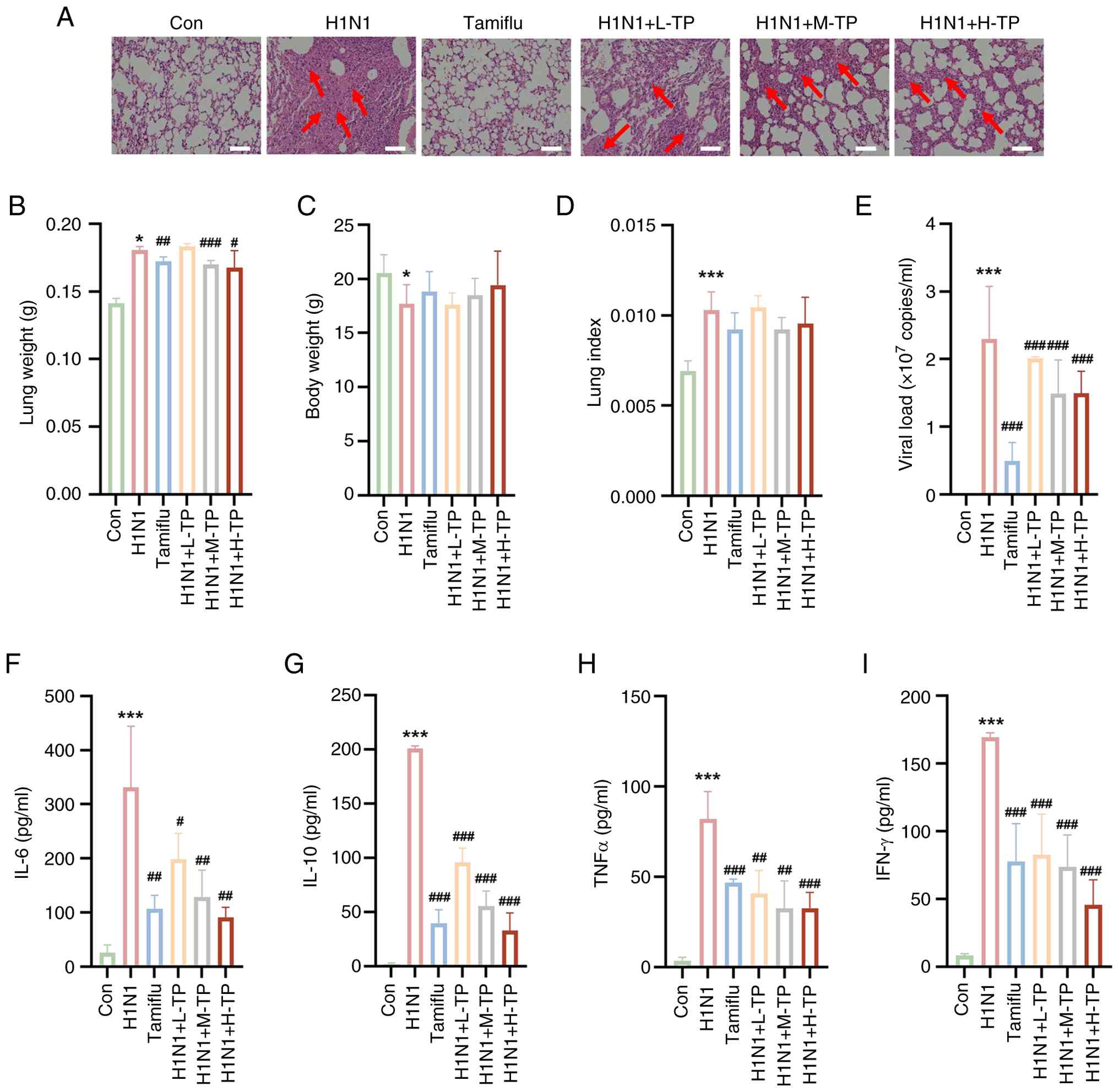 | Figure 6TP modulates the immune response and
alleviates lung injury in a murine H1N1 pneumonia model. (A)
Representative images of hematoxylin and eosin stained sections
(magnification, ×20; red arrows demarcate regions of pulmonary
consolidation; scale bar, 100 μm). (B) H1N1 infection led to
increased lung weight compared with that in the control group,
whereas lung weight was markedly reduced in the Tamiflu, L-TP,
M-TP, and H-TP groups. (C) No significant changes in body weight
were observed among the treatment groups. (D) The lung index was
decreased in the H1N1 group and did not fully recover in the four
treatment groups. (E) The Tamiflu, L-TP, M-TP, and H-TP treatment
groups presented markedly reduced viral loads compared with the
H1N1 group, with the Tamiflu group exhibiting the most significant
reduction. (F-I) Following H1N1 infection, inflammatory cytokine
levels were elevated, but treatment with Tamiflu, L-TP, M-TP, and
H-TP markedly reduced these levels in a dose-dependent manner. The
data are expressed as the mean ± standard deviation;
*P<0.05, **P<0.01,
***P<0.001, vs. Con; #P<0.05,
##P<0.01, ###P<0.001 vs. H1N1;
$$P<0.01 vs. H1N1+L-TP. TP, triptolide; H1N1,
influenza A; Con, control. |
Discussion
Limitations of current Tamiflu
therapy
Tamiflu is a commonly used antiviral medication for
treating influenza A (H1N1), which works by inhibiting viral
neuraminidase to slow viral replication, helping to alleviate
symptoms and shorten the course of the disease (31). However, Tamiflu has several
limitations. Its therapeutic efficacy is optimal within 48 h of
symptom onset (31). Missing
this window may result in reduced effectiveness. Some H1N1 strains
may develop resistance, especially in immunocompromised patients
(31). Additionally, Tamiflu
primarily helps with symptom relief (32). It has limited efficacy in
patients who have developed severe complications, such as acute
respiratory distress syndrome (ARDS) or severe pneumonia (32). Tamiflu may also cause side
effects, such as nausea and vomiting (32,33). Therefore, relying solely on
Tamiflu may not fully address the complex pathology of H1N1
pneumonia. TP, an immunomodulatory agent, exerts multiple effects,
including anti-inflammatory, immunosuppressive and antiviral
effects (16,34). Unlike Tamiflu, TP not only acts
as an antiviral drug but also offers unique advantages in immune
modulation and inflammation control. Therefore, the combined use of
Tamiflu and TP could provide a more comprehensive treatment
approach for H1N1-induced pneumonia, especially for critically ill
patients, and warrants further investigation.
Pathophysiological mechanisms of
H1N1-induced pneumonia
H1N1 infection activates the interaction between
alveolar epithelial cells and immune cells, triggering a series of
immune responses that ultimately lead to the development of
pneumonia (35). Initially, the
H1N1 virus infects alveolar epithelial cells, which recognize the
virus through pattern recognition receptors (such as Toll-like
receptors) and activate an immune response, releasing a number of
proinflammatory cytokines, including IL-6, IL-8 and TNF-α (35). These cytokines recruit immune
cells, such as neutrophils and macrophages, to the site of
infection (36). In addition,
infected alveolar epithelial cells secrete alert factors that
activate immune cells such as dendritic cells and macrophages,
which further amplify the inflammatory response (36). These immune cells also release
interferons to inhibit viral replication but may exacerbate local
inflammation (37). As immune
cells are overactivated, a cytokine storm occurs, and the release
of cytokines (such as IL-1β, IL-6 and TNF-α) increases vascular
permeability, leading to the infiltration of more immune cells into
the lung tissue, thereby triggering a more severe inflammatory
response and tissue damage (38). This excessive immune response and
local tissue damage not only destroy alveolar epithelial cells and
endothelial cells but also lead to fluid accumulation in the lungs,
impairing normal gas exchange, which ultimately leads to ARDS and
progresses to pneumonia (39).
Prolonged inflammation and immune cell infiltration may further
result in pulmonary fibrosis, severely impairing lung function.
This series of processes collectively contributes to the
development of severe pneumonia and respiratory failure caused by
H1N1 infection (39,40). Consistent with these findings,
after H1N1 infection, the levels of inflammatory factors in THP1
cells and HBEpiCs cultured in vitro increased in the present
study. Furthermore, the adhesion between these two cell types and
the migration capacity of the THP-1 cells increased. Increased cell
adhesion and migration may represent a response of the immune
system to combat viral invasion. The increased chemotaxis of immune
cells helps direct more immune cells to the site of infection,
increasing local immune responses to counter the spread of the
virus.
Cell type-specific activation of AIM2
inflammasome in H1N1 immunopathology
During the immune response to H1N1 infection, AIM2
recognizes viral RNA or DNA to activate immune cells, particularly
macrophages and dendritic cells, initiating the host antiviral
immune response (41). However,
when the activation of AIM2 is excessive, it may exacerbate the
inflammatory response and lead to severe lung damage (41,42). Overactivation of AIM2 not only
induces the release of proinflammatory cytokines, such as IL-1β and
IL-18, but may also trigger a cytokine storm, further intensifying
local inflammation (43). This
excessive immune response results in lung tissue damage, disruption
of the alveolar structure and impairment of gas exchange function
(43). AIM2 activates caspase-1
to trigger the formation of the inflammasome, which facilitates the
maturation and secretion of IL-1β and IL-18 (44). In this process, the activation of
caspase-1 is critical for the expansion of the inflammatory
response, as it not only participates in the activation of
proinflammatory cytokines but also cleaves GSDMD protein, leading
to the formation of GSDMD pores (45). GSDMD is a membrane-penetrating
protein that forms pores in the cell membrane, resulting in cell
rupture and the release of additional proinflammatory factors, thus
exacerbating the immune response and causing local tissue damage
(45). Overactivation of AIM2,
caspase-1, and GSDMD plays a crucial role in the development of
pneumonia induced by H1N1 infection. First, the interactions
between these molecules lead to the occurrence of a cytokine storm,
particularly in severe H1N1 infections (46). The cytokine storm increases
vascular permeability and causes extensive immune cell infiltration
into lung tissue, worsening lung damage and potentially leading to
life-threatening complications such as acute respiratory failure.
Second, sustained inflammation can result in chronic lung tissue
damage, including destruction of alveolar structures and disruption
of gas exchange, eventually leading to lung fibrosis and further
impairment of lung function. Therefore, the overactivation of AIM2,
caspase-1, and GSDMD in H1N1-induced pneumonia contributes to the
severity of pneumonia through excessive immune responses and cell
death, complicating treatment. In the present study, H1N1 infection
activated AIM2 signaling in THP-1 cells, whereas AIM2 signaling in
HBEpiCs remained unchanged. These findings suggested that the
activation of AIM2 in immune cells could be a critical mechanism
underlying H1N1-induced pneumonia, with excessive immune activation
potentially leading to aggravated inflammation and tissue
damage.
Dual antiviral and immunomodulatory roles
of TP
An increasing number of studies have demonstrated
that TP has significant antiviral effects. For example, TP inhibits
the replication of HSV-1 in a dose-dependent manner without showing
cytotoxicity, suggesting that its action may involve specific
cellular components rather than broad toxicity (47). Moreover, TP has been shown to
enhance the oncolytic activity of vesicular stomatitis virus by
suppressing antiviral immune responses, thereby delaying tumor
growth and prolonging survival in mouse models (47). The present study investigated
whether TP could ameliorate pneumonia induced by the H1N1 influenza
virus. In vitro and in vivo experiments both
confirmed that TP effectively reduced inflammatory responses in
alveolar epithelial cells and immune cells. Additionally, in
vivo studies revealed that TP partially inhibited the
transcription of the H1N1 virus. These findings validated the
potent antiviral and anti-inflammatory properties of TP. Notably,
TP demonstrated low cytotoxicity under the tested conditions
especially in alveolar epithelial cells, since treating HBEpiCs
with TP at concentrations of 5, 10 and 20 nM did not markedly
affect cell viability compared with the control. However, TP did
have some cytotoxic effects on immune cells, such as THP-1 cells,
which suggested that its immune-modulating effects might involve a
trade-off in immune cell viability. This finding warrants further
investigation into the optimal dosing of TP for therapeutic use,
ensuring that its beneficial antiviral and anti-inflammatory
effects are maximized while minimizing toxicity to immune
cells.
Next, the present study explored the regulatory
effects of TP on the AIM2 signaling pathway in HBEpiCs and THP-1
cells. The results showed that TP treatment did not markedly alter
the AIM2 signaling pathway in HBEpiCs. This may reflect the fact
that HBEpiCs maintain stable AIM2 signal activation, primarily
exerting their antiviral effects rather than modulating the
inflammatory response to mediate cell damage (48). By contrast, TP exerted a
significant inhibitory effect in THP-1 cells, as evidenced by
decreased mRNA and protein expression of AIM2 and reduced formation
of ASC-AIM2 complexes. Excessive activation of the AIM2 signaling
pathway often leads to an overreaction of immune cells, resulting
in the excessive release of inflammatory factors (such as IL-1β,
IL-6, and TNF-α) (49). Thus, TP
reversed the inflammatory response in THP-1 cells by suppressing
the AIM2 signaling pathway. Further experiments demonstrated that
TP not only reduced the levels of inflammatory factors in THP-1
cells but also attenuated the overactivation of immune cells and
suppressed the excessive inflammatory response induced by these
cells. Importantly, when AIM2 was overexpressed in THP-1 cells, the
regulatory effects of TP on inflammatory factors and other
indicators were markedly blocked, suggesting that the suppression
of AIM2 signaling is a key mechanism of action of TP. In summary,
TP alleviates the inflammatory burden in HBEpiCs by inhibiting AIM2
signaling in THP-1 cells.
Study limitations and future
directions
There are several limitations to the present study
that warrant consideration. First, the in vitro experiments
used HBEpiC and THP-1 cell lines to model airway epithelial and
immune responses, respectively. While these cell lines are valuable
for controlled studies, they may not fully replicate the complexity
of primary human cells or the in vivo environment. Second,
the in vivo assessments employed a murine model of
H1N1-induced pneumonia to evaluate the effects of TP. However,
differences between murine and human immune systems, including
variations in cytokine profiles and immune cell populations, may
limit the direct applicability of these findings to human
physiology. Building upon these findings, future research
directions should include the incorporation of more complex models,
such as human organoids or nonhuman primate models, to improve the
simulation of human responses and evaluate the efficacy and safety
of TP in settings that more closely resemble human physiology.
Moreover, the present study did not delineate the specific
molecular pathways through which TP exerts its antiviral effects on
H1N1. Although TP showed efficacy in inhibiting viral replication,
the exact targets and mechanisms, such as potential interference
with viral RNA synthesis or modulation of the host cell translation
machinery, were not directly investigated in the present study. To
build upon the findings of the present study, future studies should
aim to elucidate the molecular mechanisms by which TP inhibits H1N1
replication. This includes investigating whether TP directly
affects viral RNA polymerase activity, impedes the assembly of
viral ribonucleoprotein complexes, or modulates host factors
involved in viral replication and protein synthesis. Understanding
these mechanisms will provide deeper insights into the antiviral
properties of TP and support its potential development as a
therapeutic agent against influenza.
Based on its findings, the present study
demonstrated that TP selectively modulated the AIM2 signaling
pathway in immune cells, abolishing excessive activation of
macrophages and neutrophils while preserving alveolar epithelial
cell viability. This cell type-specific targeting markedly reduced
pathogenic immune cell-epithelial cell interactions, thereby
alleviating H1N1-induced lung immunopathology. Crucially, the dual
capacity of TP to suppress viral replication and mitigate immune
system dysregulation provides a novel therapeutic strategy for
severe viral pneumonia. As a selective immune modulator, it may
synergize with existing antivirals to bridge the critical gap
between viral clearance and inflammation control, thereby
ultimately minimizing organ damage in high-risk patients.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YC, HW, and XW performed the experiments and wrote
the paper. LC performed the experiments and interpreted the data.
XF, RQ, DJ, ML and WX performed part of the animal experiments. WZ
supervised the project, interpreted the data and wrote the paper.
All authors confirm the authenticity of all the raw data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All the experimental procedures for the mice were
performed in accordance with the guidelines of the Institutional
Animal Care and Use Committee. The protocol was approved by the
Committee on the Ethics of Animal Experiments of Guangzhou
University of Chinese Medicine (approval no. 2024055R).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural Science
Foundation of China (grant no. 82405094), the Shenzhen Natural
Science Foundation (grant no. JCYJ20240813152345060) and the
Science and Technology Planning Project of Shenzen Municipality
(grant no. JSGG20220226090203006).
References
|
1
|
Li X, Gu M, Zheng Q, Gao R and Liu X:
Packaging signal of influenza A virus. Virol J. 18:362021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dong Y, Wang L, Burgner DP, Miller JE,
Song Y, Ren X, Li Z, Xing Y, Ma J, Sawyer SM and Patton GC:
Infectious diseases in children and adolescents in China: Analysis
of national surveillance data from 2008 to 2017. BMJ.
369:m10432020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen Z, Tsui JL, Cai J, Su S, Viboud C, du
Plessis L, Lemey P, Kraemer MUG and Yu H: Disruption of seasonal
influenza circulation and evolution during the 2009 H1N1 and
COVID-19 pandemics in Southeastern Asia. Nat Commun. 16:4752025.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ghorbani A, Ngunjiri JM and Lee CW:
Influenza A virus subpopulations and their implication in
pathogenesis and vaccine development. Annu Rev Anim Biosci.
8:247–267. 2020. View Article : Google Scholar
|
|
5
|
Park J, Fong Legaspi SL, Schwartzman LM,
Gygli SM, Sheng ZM, Freeman AD, Matthews LM, Xiao Y, Ramuta MD,
Batchenkova NA, et al: An inactivated multivalent influenza A virus
vaccine is broadly protective in mice and ferrets. Sci Transl Med.
14:eabo21672022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pinto RM, Bakshi S, Lytras S, Zakaria MK,
Swingler S, Worrell JC, Herder V, Hargrave KE, Varjak M,
Cameron-Ruiz N, et al: BTN3A3 evasion promotes the zoonotic
potential of influenza A viruses. Nature. 619:338–347. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang L, Wang J, Ma X, Sun L, Hao C and
Wang W: Inhibition of influenza a virus infection by natural
stilbene piceatannol targeting virus hemagglutinin. Phytomedicine.
120:1550582023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu J, Li H, Jia J, Huang Z, Liu S, Zheng
Y, Mu S, Deng X, Zou X, Wang Y, et al: Pandemic influenza A (H1N1)
virus causes abortive infection of primary human T cells. Emerg
Microbes Infect. 11:1191–1204. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng Y, He D, Zuo W, Wang W, Wu K, Wu H,
Yuan Y, Huang Y, Li H, Lu Y, et al: Influenza A virus dissemination
and infection leads to tissue resident cell injury and dysfunction
in viral sepsis. EBioMedicine. 116:1057382025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meng X, Zhu Y, Yang W, Zhang J, Jin W,
Tian R, Yang Z and Wang R: HIF-1α promotes virus replication and
cytokine storm in H1N1 virus-induced severe pneumonia through
cellular metabolic reprogramming. Virol Sin. 39:81–96. 2024.
View Article : Google Scholar :
|
|
11
|
Rewar S, Mirdha D and Rewar P: Treatment
and prevention of pandemic H1N1 influenza. Ann Glob Health.
81:645–653. 2015. View Article : Google Scholar
|
|
12
|
Lampejo T: Influenza and antiviral
resistance: An overview. Eur J Clin Microbiol Infect Dis.
39:1201–1208. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu DW and Tate MD: Taking AIM at
influenza: The role of the AIM2 inflammasome. Viruses. 16:15352024.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang H, Luo J, Alcorn JF, Chen K, Fan S,
Pilewski J, Liu A, Chen W, Kolls JK and Wang J: AIM2 inflammasome
is critical for influenza-induced lung injury and mortality. J
Immunol. 198:4383–4393. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schattgen SA, Gao G, Kurt-Jones EA and
Fitzgerald KA: Cutting edge: DNA in the lung microenvironment
during influenza virus infection tempers inflammation by engaging
the DNA sensor AIM2. J Immunol. 196:29–33. 2016. View Article : Google Scholar
|
|
16
|
Gao J, Zhang Y, Liu X, Wu X, Huang L and
Gao W: Triptolide: Pharmacological spectrum, biosynthesis, chemical
synthesis and derivatives. Theranostics. 11:7199–7221. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fang WY, Tseng YT, Lee TY, Fu YC, Chang
WH, Lo WW, Lin CL and Lo YC: Triptolide prevents LPS-induced
skeletal muscle atrophy via inhibiting NF-κB/TNF-α and regulating
protein synthesis/degradation pathway. Br J Pharmacol.
178:2998–3016. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Han R, Rostami-Yazdi M, Gerdes S and
Mrowietz U: Triptolide in the treatment of psoriasis and other
immune-mediated inflammatory diseases. Br J Clin Pharmacol.
74:424–436. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang N, Min X, Ma N, Zhu Z, Cao B, Wang Y,
Yong Q, Huang J and Li K: The negative impact of triptolide on the
immune function of human natural killer cells. Pharmaceuticals
(Basel). 16:4582023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu W, Li Y, Zhao J and Wang Y, Li Y and
Wang Y: The mechanism of triptolide in the treatment of connective
tissue disease-related interstitial lung disease based on network
pharmacology and molecular docking. Ann Med. 54:541–552. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu C, Wu X, Bing X, Qi W, Zhu F, Guo N,
Li C, Gao X, Cao X, Zhao M and Xia M: H1N1 influenza virus
infection through NRF2-KEAP1-GCLC pathway induces ferroptosis in
nasal mucosal epithelial cells. Free Radic Biol Med. 204:226–242.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wei Z, Gao R, Sun Z, Yang W, He Q, Wang C,
Zhang J, Zhang X, Guo L and Wang S: Baicalin inhibits influenza A
(H1N1)-induced pyroptosis of lung alveolar epithelial cells via
caspase-3/GSDME pathway. J Med Virol. 95:e287902023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Du HX, Zhou HF, Yang JH, Lu YY, He Y and
Wan HT: Preliminary study of Yinhuapinggan granule against H1N1
influenza virus infection in mice through inhibition of apoptosis.
Pharm Biol. 58:979–991. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Paukner S, Kimber S, Cumper C, Rea-Davies
T, Sueiro Ballesteros L, Kirkham C, Hargreaves A, Gelone SP,
Richards C and Wicha WW: In vivo immune-modulatory activity of
lefamulin in an influenza virus A (H1N1) infection model in mice.
Int J Mol Sci. 25:54012024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wan YS, You Y, Ding QY, Xu YX, Chen H,
Wang RR, Huang YW, Chen Z, Hu WW and Jiang L: Triptolide protects
against white matter injury induced by chronic cerebral
hypoperfusion in mice. Acta Pharmacol Sin. 43:15–25. 2022.
View Article : Google Scholar
|
|
27
|
Zeldin DC, Wohlford-Lenane C, Chulada P,
Bradbury JA, Scarborough PE, Roggli V, Langenbach R and Schwartz
DA: Airway inflammation and responsiveness in prostaglandin H
synthase-deficient mice exposed to bacterial lipopolysaccharide. Am
J Respir Cell Mol Biol. 25:457–465. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matute-Bello G, Downey G, Moore BB,
Groshong SD, Matthay MA, Slutsky AS and Kuebler WM; Acute Lung
Injury in Animals Study Group: An official American thoracic
society workshop report: Features and measurements of experimental
acute lung injury in animals. Am J Respir Cell Mol Biol.
44:725–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao J, Peng S, Shan X, Deng G, Shen L, Sun
J, Jiang C, Yang X, Chang Z, Sun X, et al: Inhibition of AIM2
inflammasome-mediated pyroptosis by Andrographolide contributes to
amelioration of radiation-induced lung inflammation and fibrosis.
Cell Death Dis. 10:9572019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou J, Li S, Yang Y, Zhou C, Wang C and
Zeng Z: Triptolide alleviates acute lung injury by reducing
mitochondrial dysfunction mediated ferroptosis through the
STAT3/p53 pathway. Free Radic Biol Med. 230:79–94. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yoo ES: Study of specific oligosaccharide
structures related with swine flu (H1N1) and avian flu, and tamiflu
as their remedy. J Microbiol Biotechnol. 21:449–454. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zima V, Albiñana CB, Rojíková K, Pokorná
J, Pachl P, Řezáčová P, Hudlicky J, Navrátil V, Majer P, Konvalinka
J, et al: Investigation of flexibility of neuraminidase 150-loop
using tamiflu derivatives in influenza A viruses H1N1 and H5N1.
Bioorg Med Chem. 27:2935–2947. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gu C, Chen Y, Li H, Wang J and Liu S:
Considerations when treating influenza infections with oseltamivir.
Expert Opin Pharmacother. 25:1301–1316. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hou W, Liu B and Xu H: Triptolide:
Medicinal chemistry, chemical biology and clinical progress. Eur J
Med Chem. 176:378–392. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ling L, Ren A, Lu Y, Zhang Y, Zhu H, Tu P,
Li H and Chen D: The synergistic effect and mechanisms of
flavonoids and polysaccharides from Houttuynia cordata on
H1N1-induced pneumonia in mice. J Ethnopharmacol. 302:1157612023.
View Article : Google Scholar
|
|
36
|
Fontes CA, Dos Santos AAS and De Oliveira
SA: High-resolution computed tomography enhances the diagnosis and
follow-up of influenza A (H1N1) virus-associated pneumonia. J
Infect Dev Ctries. 14:317–320. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Himaal Dev GJ, Venkategowda PM, Sutar AR
and Shankar V: Intestinal mucormycosis in an adult with H1N1
pneumonia on extracorporeal membrane oxygenation. Ann Card Anaesth.
24:92–94. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ding W, Li R, Song T, Yang Z, Xu D, Huang
C, Shen S, Zhong N, Lai K and Deng Z: AMG487 alleviates influenza A
(H1N1) virus-induced pulmonary inflammation through decreasing
IFN-γ-producing lymphocytes and IFN-γ concentrations. Br J
Pharmacol. 181:2053–2069. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang S, Sun F, Zhu J, Qi J, Wang W, Liu
Z, Li W, Liu C, Liu X, Wang N, et al: Phillyrin ameliorates
influenza a virus-induced pulmonary inflammation by antagonizing
CXCR2 and inhibiting NLRP3 inflammasome activation. Virol J.
20:2622023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Arranz-Herrero J, Presa J, Rius-Rocabert
S, Utrero-Rico A, Arranz-Arija JÁ, Lalueza A, Escribese MM, Ochando
J, Soriano V and Nistal-Villan E: Determinants of poor clinical
outcome in patients with influenza pneumonia: A systematic review
and meta-analysis. Int J Infect Dis. 131:173–179. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Han Y, Ge C, Ye J, Li R and Zhang Y:
Demethyleneberberine alleviates Pseudomonas aeruginosa-induced
acute pneumonia by inhibiting the AIM2 inflammasome and oxidative
stress. Pulm Pharmacol Ther. 83:1022592023. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tseng YH, Chen IC, Li WC and Hsu JH:
Regulatory cues in pulmonary fibrosis-with emphasis on the AIM2
inflammasome. Int J Mol Sci. 24:108762023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cho SJ, Hong KS, Jeong JH, Lee M, Choi
AMK, Stout-Delgado HW and Moon JS: DROSHA-dependent AIM2
inflammasome activation contributes to lung inflammation during
idiopathic pulmonary fibrosis. Cells. 8:9382019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cho SJ, Moon JS, Nikahira K, Yun HS,
Harris R, Hong KS, Huang H, Choi AMK and Stout-Delgado H:
GLUT1-dependent glycolysis regulates exacerbation of fibrosis via
AIM2 inflammasome activation. Thorax. 75:227–236. 2020. View Article : Google Scholar :
|
|
45
|
Zhang T, Du H, Feng S, Wu R, Chen T, Jiang
J, Peng Y, Ye C and Fang R: NLRP3/ASC/caspase-1 axis and serine
protease activity are involved in neutrophil IL-1β processing
during Streptococcus pneumoniae infection. Biochem Biophys Res
Commun. 513:675–680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fang R, Tsuchiya K, Kawamura I, Shen Y,
Hara H, Sakai S, Yamamoto T, Fernandes-Alnemri T, Yang R,
Hernandez-Cuellar E, et al: Critical roles of ASC inflammasomes in
caspase-1 activation and host innate resistance to Streptococcus
pneumoniae infection. J Immunol. 187:4890–4899. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Aliabadi N, Jamalidoust M, Pouladfar G,
Ziyaeyan A and Ziyaeyan M: Antiviral activity of triptolide on
herpes simplex virus in vitro. Immun Inflamm Dis. 10:e6672022.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ben Yebdri F, Van Grevenynghe J, Tang VA,
Goulet ML, Wu JH, Stojdl DF, Hiscott J and Lin R:
Triptolide-mediated inhibition of interferon signaling enhances
vesicular stomatitis virus-based oncolysis. Mol Ther. 21:2043–2053.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tian J, Liu Y, Gao W, Shi X, Cheng F and
Xie B: NETs activate AIM2 to mediate synovial fibroblast pyroptosis
and promote acute gouty arthritis development. Immunol Lett.
275:1070072025. View Article : Google Scholar : PubMed/NCBI
|














