Introduction
Neuropathic pain, a debilitating chronic pain
condition triggered by damage or dysfunction of the somatosensory
nervous system, affects millions of individuals worldwide (1,2).
Characterized by spontaneous pain, hyperalgesia and allodynia, this
disorder often resists conventional analgesics, with current
therapeutic options offering limited efficacy and frequent adverse
effects (3-5). As such, unraveling the intricate
pathophysiological mechanisms underlying neuropathic pain is
critical for identifying novel therapeutic targets.
Mitochondrial biogenesis is pivotal for regulating
mitochondrial DNA (mtDNA) replication, oxidative phosphorylation
and ATP production, processes essential to sustaining mitochondrial
function and homeostasis (6,7).
It is orchestrated by nuclear and mitochondrial genes, with
peroxisome proliferative activated receptor γ coactivator 1α
(PGC-1α) serving as the master transcriptional co-activator
(8). PGC-1α upregulates nuclear
respiratory factor 1 (NRF1) and mitochondrial transcription factor
A (TFAM), thereby driving expression of mtDNA and assembly of
respiratory-chain complexes (9).
Impaired mitochondrial biogenesis contributes to diverse diseases,
including obesity (10),
doxorubicin-induced cardiotoxicity (11), cerebral ischemia-reperfusion
injury (12) and
neurodegenerative disorders (13,14). The authors' previous studies also
indicated that peripheral nerve injury-induced neuropathic pain
suppresses mitochondrial biogenesis, while enhancing this process
alleviates pain hypersensitivity (15,16). However, the specific mechanisms
linking mitochondrial biogenesis to neuropathic pain remain
incompletely understood.
The mitochondrial unfolded protein response
(UPRmt) represents a mitochondria-to-nucleus signaling
pathway and acts as a cellular survival mechanism that gets
triggered when the balance of mitochondrial protein homeostasis is
perturbed (17-19). Its core function lies in
preserving mitochondrial functionality by upregulating the
transcription of chaperone proteins and proteases, which
collectively regulate processes including protein folding, complex
assembly and targeted degradation (18). Key molecular players involved in
this regulatory network include the 10 kDa heat shock protein
(Hsp10), 60 kDa heat shock protein (Hsp60), the proteolytic subunit
of the ATP-dependent Clp protease (ClpP) and the mitochondrial Lon
protease homolog (LonP1) (17).
It can be activated by diverse factors through multiple regulatory
pathways, such as activating transcription factor 5 (ATF5), a
homolog of nematode ATFS-1 (18). Impaired UPRmt elevates
reactive oxygen species (ROS) levels, triggering oxidative stress
and contributing to diseases such as osteoarthritis (OA) (20), Alzheimer's disease (21), cerebral ischemia (22) and traumatic brain injury
(23). A recent study showed
that enhancing UPRmt with nicotinamide riboside (NR)
improves mitochondrial function, reduces chondrocyte death, and
alleviates OA pain; these effects notably were diminished in
chondrocyte-specific ATF5-knockout mice (24). Additionally, it was found that
the ATF5-mediated UPRmt alleviated intervertebral disc
degeneration via promoting mitophagy (25). However, the mechanisms linking
UPRmt to neuropathic pain remain poorly understood.
C1q-tumor necrosis factor-related protein-3 (CTRP3),
a member of the adipokine CTRP family, has drawn attention for its
pleiotropic cytoprotective effects (26,27). Beyond metabolic regulation, it
exerts anti-inflammatory, antioxidant and tissue-protective
properties in diverse pathological contexts, including
cardiovascular disease (28),
metabolic disorders (29) and
intestinal inflammation disease (30). Recent research shows CTRP3
mitigates mitochondrial dysfunction and oxidative stress in
pathological cardiac hypertrophy by activating the UPRmt
(31). Another study indicated
that CTRP3 alleviates neurological deficits in cerebral ischemic
stroke by promoting mitochondrial biogenesis (32). However, its specific role in pain
hypersensitivity during neuropathic pain remains largely
uninvestigated.
SIRT1, a NAD+-dependent deacetylase
belonging to the sirtuin family, regulates mitochondrial
homeostasis by interacting with key transcriptional regulators
(15,33). Recent studies show that aberrant
SIRT1 expression reduces PGC-1α levels and impairs mitochondrial
biogenesis (15,34). It may also activate the
ATF5-mediated UPRmt, protecting mitochondria from
proteotoxic damage and mitigating oxidative stress (31,35). However, whether SIRT1 mediates
CTRP3's regulatory effects on pain hypersensitivity, mitochondrial
biogenesis, and UPRmt in neuropathic pain remains
unclear.
Against this backdrop, the present study sought to
investigate the role of CTRP3 in neuropathic pain and explore its
potential mechanisms of action. Using a rat model of spared nerve
injury (SNI), it was examined whether CTRP3 regulates pain
hypersensitivity by modulating mitochondrial function, and whether
this effect is mediated through the SIRT1-PGC-1α/ATF5 signaling
axis. By clarifying these relationships, it was aimed to shed light
on novel therapeutic strategies for neuropathic pain targeting
CTRP3 and its downstream pathways.
Materials and methods
Animals
Male Sprague-Dawley (SD) rats aged 6-7 weeks
(weighing 250-280 g; total number: n=315 rats) were purchased from
the experimental animal center of Tongji Hospital, Tongji Medical
College, Huazhong University of Science and Technology. All rats
were housed in the specific pathogen-free animal facility, under
controlled environmental conditions: Temperature maintained at
23±1°C, relative humidity at 50±10%, and a 12/12-h light/dark
cycle. Rats were group-housed (2 per cage) in polypropylene cages
with autoclaved wood chip bedding and provided with standard rodent
chow and sterile water ad libitum. All experimental
procedures were approved by the Experimental Animal Care and Use
Committee of Tongji Hospital, Tongji Medical College, Huazhong
University of Science and Technology (approval no. TJH-202106615;
Wuhan, China). The study was conducted in accordance with the
ARRIVE guidelines to ensure transparency and reproducibility. All
experiments were carried out under blinded conditions. Rats were
randomly assigned to distinct groups, with syringes holding
different drugs randomly coded by an independent researcher;
injections were then administered strictly according to these
codes. Behavioral assessments were conducted without the assessors
knowing which treatment each group received.
Neuropathic pain model
Neuropathic pain was induced in rats using the SNI
model, as previously described (15). Rats were anesthetized with
isoflurane (induction, 5%; maintenance, 2-3%; cat. no. R510-22-10,
RWD) and placed on a 37°C heating pad. The left hindlimb was shaved
and disinfected, followed by a 1.5-2-cm incision along the thigh to
expose the sciatic nerve and its three branches (common peroneal,
tibial and sural). The common peroneal and tibial nerves were
ligated with 6-0 silk sutures (5 mm apart), transected, and 2 mm of
distal stumps were removed; the sural nerve was spared. Muscles and
skin were sutured with 4-0 absorbable and 5-0 nylon sutures,
respectively. After surgery, rats were allowed to recover in
individual cages with free access to food and water. Sham-operated
rats underwent the same exposure but without nerve
ligation/transections.
Pain behavioral testing
Mechanical pain sensitivity was evaluated using the
von Frey test (36). All tests
were performed by a blinded experimenter in a quiet room. Rats were
acclimatized to Plexiglas chambers on a wire mesh floor for 30 min
before testing. Von Frey filaments (1.0-15.0 g) were applied
vertically to the ipsilateral hindpaw plantar surface for 5 sec. A
positive response (paw withdrawal, flinching, or licking) prompted
using a weaker filament; no response prompted a stronger one. Using
the up-down method, paw withdrawal threshold (PWT, 50% withdrawal
threshold) was calculated after 6 measurements. Each paw was tested
3 times (5-min intervals), and the mean was recorded. Cold
hyperalgesia was assessed using the acetone drop method, as
previously described (37).
Prior to testing, rats were individually housed in transparent
plastic observation chambers with a wire-mesh floor and allowed a
30-min habituation period to minimize environmental stress.
Following acclimatization, a 0.1-ml syringe was used to form an
acetone bubble, which was gently applied to the plantar surface of
the left hind paw. This procedure was repeated three times per paw,
with a 5-min interval between consecutive applications to avoid
sensory adaptation. During each trial, the cumulative duration of
paw-related nocifensive behaviors, including licking, biting, and
lifting the paw off the mesh floor, was recorded. The paw
withdrawal cold duration (PWCD) for each rat was determined as the
mean value derived from six independent trials.
Intrathecal catheterization
Intrathecal catheterization was performed as
previously described (38). Rats
were anesthetized with isoflurane (induction, 5%; maintenance,
2-3%) and placed in a stereotaxic frame in a prone position. The
atlanto-occipital membrane was exposed via a midline incision at
the base of the skull. A PE-10 polyethylene catheter filled with
sterile saline was inserted through the membrane into the
subarachnoid space at the lumbar enlargement (L5-L6 level). The
catheter was secured to the surrounding tissues with 4-0 silk
sutures, and the free end was exteriorized at the back of the neck,
sealed with a stainless-steel obturator, and protected by a plastic
cap. Successful catheter placement was verified by injecting 10
μl of sterile saline containing 2% lidocaine. A transient
(30-60 sec) bilateral hindlimb paralysis indicated correct
positioning. Rats were allowed a 7-day recovery period before
experimental use.
Drug administration
Recombinant CTRP3 (rCTRP3; cat. no. 9398-TN; Novus
Biologicals, LLC) and EX-527 (a selective SIRT1 inhibitor; cat. no.
HY-15452; MedChemExpress) were administered via intrathecal
injection. rCTRP3 was dissolved in sterile physiological saline to
prepare working solutions at concentrations corresponding to three
doses: 10 μg/rat, 30 μg/rat and 90 μg/rat.
Intrathecal injections were performed daily for 7 consecutive days
starting from 7 day after SNI surgery. The injection volume was
adjusted to 5 μl per rat, followed by 5 μl PBS to
ensure complete delivery. To investigate the role of SIRT1 in
CTRP3-mediated effects, the selective SIRT1 inhibitor EX-527 was
used. EX-527 was dissolved in 10% dimethyl sulfoxide and
administered at a fixed dose of 1.5 μg per rat. The dose of
EX-527 was chosen according to a previous study (15). Intrathecal injection of EX-527
was performed 30 min prior to rCTRP3 administration in the
respective experimental groups. At the end point of experiment,
rats were euthanized by intraperitoneal injection of an overdose of
pentobarbital sodium at a dose of 150 mg/kg body weight (39). Death was confirmed by
respiratory/cardiac arrest, dilated pupils, and absent corneal
reflex.
Intrathecal injection of small
interfering RNA (siRNA)
siRNAs targeting PGC-1α (PGC-1α siRNA) and ATF5
(ATF5 siRNA), along with non-targeting scrambled siRNA (negative
control), were used to silence the expression of these genes in the
spinal cord. All siRNAs were purchased from TsingKe Biological
Technology. The sequences of siRNAs are listed in Table SI. To verify the knockdown
efficiency and specificity of PGC-1α siRNA and ATF5 siRNA,
independent in vitro validation was performed in PC12 cells.
Cells were transfected with PGC-1α siRNA or ATF5 siRNA using
Lipofectamine 3000 transfection reagent (cat. no. L3000008;
Invitrogen; Thermo Fisher Scientific, Inc.), as previously
described (40). The knockdown
efficiency was verified by reverse transcription-quantitative PCR
(RT-qPCR). For in vivo, the siRNA was dissolved in
RNase-free water at 2.5 μg/μl and mixed with the
transfection reagent branched polyethyleneimine (PEI; cat. no.
408727; MilliporeSigma). Intrathecal injection of siRNA in a total
volume of 5 μl was initiated 5-11 days post SNI, and the
knockdown efficiency of PGC-1α and ATF5 was verified by RT-qPCR
(38).
RT-qPCR
Total RNA was extracted from the L4-L6 segments of
the spinal cord using RNA Extraction kit (cat. no. R701-01; Vazyme
Biotech Co., Ltd.) following the manufacturer's protocol (41). The complementary DNA (cDNA) was
synthesized using a reverse transcription reagent kit (cat. no.
R212-01; Vazyme Biotech Co., Ltd.), following the manufacturer's
protocols. Amplifications were run on a Real-Time PCR System
(Applied Biosystems QuantStudio 6; Thermo Fisher Scientific, Inc.).
The reaction mixture (20 μl total volume) contained 10
μl of SYBR Green Supermix (cat. no. 172515; Bio-Rad
Laboratories, Inc.), 0.4 μl of forward primer, 0.4 μl
of reverse primer, 2 μl of cDNA template, and 7.2 μl
of nuclease-free water. The thermal cycling conditions were as
follows: Initial denaturation at 95°C for 30 sec, followed by 40
cycles of denaturation at 95°C for 5 sec and annealing/extension at
60°C for 30 sec. Primers for target genes and the reference gene
(β-actin) were synthesized by TsingKe Biological Technology. The
primer sequences are listed in Table SII. Relative mRNA expression
levels were calculated using the 2−ΔΔCq method, with
β-actin serving as the internal reference to normalize target gene
expression (42).
Immunofluorescence (IF) staining
IF staining was conducted according to a previous
study (43). Spinal cord
segments (L4-L6) were harvested and immediately fixed in 4%
paraformaldehyde (PFA) in 0.01 M phosphate-buffered saline (PBS) at
4°C for 24 h. After fixation, tissues were transferred to 30%
sucrose solution (in 0.01 M PBS) for cryoprotection at 4°C, with
solution changes every 24 h until tissues fully sank. Transverse
frozen sections (20-μm thickness) were cut using a cryostat
microtome (Leica Microsystems GmbH). To permeabilize cell membranes
and block non-specific binding, sections were incubated in blocking
buffer (5% bovine serum albumin (Wuhan Boster Biological
Technology, Ltd.) + 0.3% Triton X-100 in 0.01 M PBS) at 25°C for 1
h. Primary antibodies were diluted in blocking buffer and applied
to sections, which were then incubated overnight at 4°C in a
humidified chamber. The following day, sections were washed with
0.01 M PBS and incubated with a mixture of second antibodies at
25°C for 1 h in the dark. The antibodies used in IF staining are
listed in Table SIII. Images
were acquired using a fluorescence microscope (Olympus
Corporation). The results were expressed as the number of
double-positive cells per square millimeter and quantified using
ImageJ 1.8 software (National Institutes of Health), as previously
described (38).
Western blotting (WB)
WB analysis was performed as previously described
(15). Spinal cord tissues
(L4-L6 segments) were homogenized in RIPA (Wuhan Boster Biological
Technology, Ltd.; main components: 1% TritonX-100, 1% Sodium
deoxycholate, 0.1% SDS) lysis buffer (containing 1% protease and
phosphatase inhibitor cocktail) on ice, at a ratio of 1 g tissue to
10 ml buffer. After centrifugation at 12,000 × g for 15 min at 4°C,
the supernatant was collected, and protein concentration was
determined using a BCA protein assay kit (Thermo Fisher Scientific,
Inc.). Equal amounts of protein (50 μg per sample) were
separated by 10% SDS-PAGE and transferred to polyvinylidene
difluoride membranes. Membranes were blocked with 5% non-fat milk
in Tris-buffered saline with 0.1% Tween-20 (TBST) for 1 h at 25°C,
then incubated overnight at 4°C with primary antibodies. After
washing with 10% TBST, the membranes were incubated with second
antibodies for 1 h at 25°C. The antibodies used in WB are listed in
Table SIV. Protein bands were
visualized using an enhanced chemiluminescence (ECL) detection kit
(Millipore) and imaged with a ChemiDoc XRS+ system (Bio-Rad
Laboratories, Inc.). Band intensities were quantified using
ImageLab software 5.2 (Bio-Rad Laboratories, Inc.), with target
protein expression normalized to β-actin.
mtDNA copy number quantification
The mtDNA copy number quantification was performed
as previously described (15).
Total DNA was extracted from L4-L6 spinal cord segments using a
Tissue Genomic DNA Extraction Kit [Elk (Wuhan) Biotechnology Co.,
Ltd.] following the manufacturer's protocol, and mtDNA copy number
was determined by RT-qPCR using primers specific for mitochondrial
genes and a nuclear reference gene. Relative mtDNA copy number was
assessed by the mitochondrial encoded NADH dehydrogenase 1 (ND1)
and was normalized to nuclear-encoded β-actin. The primer sequences
are listed in Table SII.
Dihydroethidium (DHE) staining
Superoxide anions were detected using DHE staining,
following the previously described method (36,44). Frozen sections (20-μm
thickness) of the L4-L6 spinal cord segments were prepared. These
sections were incubated with 1.5 μmol/ml DHE (cat. no.
HY-D0079; MedChemExpress) at 25°C for 50 min in a dark environment.
Subsequently, the sections were stained with
4',6-diamidino-2-phenylindole (DAPI) at a concentration of 0.001
mg/ml at 25°C for 10 min in the dark. Images were acquired using a
fluorescence microscope (Olympus Corporation). For DHE staining
analysis, the number of DHE-positive cells per square millimeter
was counted, and the data were processed using ImageJ 1.8 software
(National Institutes of Health).
Biochemical assays for oxidative stress
and antioxidant markers
Oxidative stress parameters and antioxidant indices
were measured using commercial kits, following procedures similar
to those previously described (36). Tissue samples were homogenized
and centrifuged at 16,000 × g for 30 min at 4°C, with the
supernatants collected for subsequent biochemical assays. The
resulting supernatant was collected for subsequent analyses, with
protein concentrations determined via BCA assay to normalize the
results. Levels of oxidative damage markers, including
malondialdehyde (MDA) and protein carbonyl (PCO), were quantified
using the corresponding assay kits respectively, in strict
accordance with the manufacturers' protocols. For antioxidant
indices, glutathione (GSH) levels, superoxide dismutase (SOD)
activity and glutathione peroxidase (GSH-PX) activity were assessed
using the corresponding assay kits respectively, following the
manufacturer's instructions. The commercial assay kits used in
biochemical assays are listed in Table SV.
Mitochondrial membrane potential (MMP)
measurement
Mitochondria were isolated from rat spinal cord
tissues using a mitochondrial isolation kit (cat. no. ab110168;
Abcam) according to the manufacturer's instructions. Briefly,
tissue samples were homogenized in ice-cold isolation buffer, and
the homogenate was centrifuged at low speed to remove debris. The
supernatant was then centrifuged at high speed to obtain the
mitochondrial pellet, which was washed and resuspended in assay
buffer. For MMP detection, the isolated mitochondria were incubated
with 5 μM JC-1 dye (cat. no. C2003S; Beyotime Institute of
Biotechnology) at 37°C for 30 min in the dark. Fluorescence
intensities were measured by flow cytometry (BD FACSCanto II; BD
Biosciences), and MMP was calculated as the ratio of red
fluorescence (aggregated JC-1) to green fluorescence (monomeric
JC-1) using FlowJo software (version 10.8.1; Tree Star, Inc.).
Measurement of ATP levels
ATP levels were measured using an ATP Assay Kit
(cat. no. S0026; Beyotime Institute of Biotechnology), as
previously described (45).
Briefly, spinal cord tissues were homogenized in lysis buffer, and
the homogenate was centrifuged at 12,000 × g for 5 min at 4°C. The
supernatant was collected and mixed with ATP detection working
solution, and the luminescence intensity was determined using a
microplate reader. The ATP concentration was calculated according
to a standard curve. Protein concentration was measured using a BCA
Protein Assay Kit, and ATP levels were normalized to total protein
content and expressed as nmol/mg protein.
Statistical analysis
All data are presented as the mean ± standard error
of the mean (SEM). Statistical analyses were performed using
GraphPad Prism 9.0 software (Dotmatics). Normality distribution was
evaluated using the Shapiro-Wilk test. For comparisons between two
groups, unpaired Student's t-test was used. For multiple group
comparisons, one-way analysis of variance (ANOVA) was applied,
followed by Bonferroni's post hoc test for multiple comparison
correction. Behavioral data were analyzed using two-way
repeated-measures ANOVA, with group (row factor) and time (column
factor) as the main factors, and interaction between group and time
was also tested. P<0.05 was considered to indicate a
statistically significant difference. Effect sizes were reported as
eta squared (η2, representing partial eta squared) for
all main effects, interactions, and two-sample t-tests. Relevant
statistical results have been incorporated into the following
section.
Results
Expression of CTRP3 in the spinal cord
following SNI
A neuropathic pain model was established using the
SNI procedure. As depicted in Fig.
1A [Interaction: F (3, 72)=17.45, P<0.0001,
η2=0.4211; Time factor: F (3, 72)=20.39, P<0.0001,
η2=0.4593; Group factor: F (1, 72)=207.9, P<0.0001,
η2=0.7428] and Fig.
1B [Interaction: F (3, 72)=24.09, P<0.0001,
η2=0.5010; Time factor: F (3, 72)=25.93, P<0.0001,
η2=0.5193; Group factor: F (1, 72)=194.6, P<0.0001,
η2=0.7299], behavioral pain testing results indicated
that, relative to sham-operated controls, rats receiving SNI
displayed a statistically significant decrease in PWT and a
concurrent increase in PWCD over the time window of post-operative
day 3 to day 14, confirming the successful induction of neuropathic
pain hypersensitivity. To assess changes in CTRP3 expression,
RT-qPCR and WB analyses were performed on spinal cord tissues. The
results revealed that SNI led to a downregulation of both CTRP3
mRNA [Fig. 1C: F (3, 16)=47.17,
P<0.0001, η2=0.8984] and protein [Fig. 1D: F (3, 16)=51.68, P<0.0001,
η2=0.9065] levels in the spinal cord compared with the
sham-operated group. Furthermore, IF staining was employed to
determine the cellular localization of CTRP3 in the spinal dorsal
horn. As shown in Fig. 1E-I
[Fig. 1G: t (8)=14.17, P<0.0001,
η2=0.9617; Fig. 1H: t
(8)=2.194, P=0.0595,
η2=0.3757; Fig. 1I: t
(8)=1.965, P=0.0850,
η2=0.3255], CTRP3 was predominantly localized in neurons
(identified by NeuN) within the spinal dorsal horn. Notably, the
number of CTRP3-positive neurons was significantly reduced post-SNI
relative to the sham group. These findings suggest that neuronal
CTRP3 in the spinal cord may play a critical role in the
development of SNI-induced neuropathic pain in male rats.
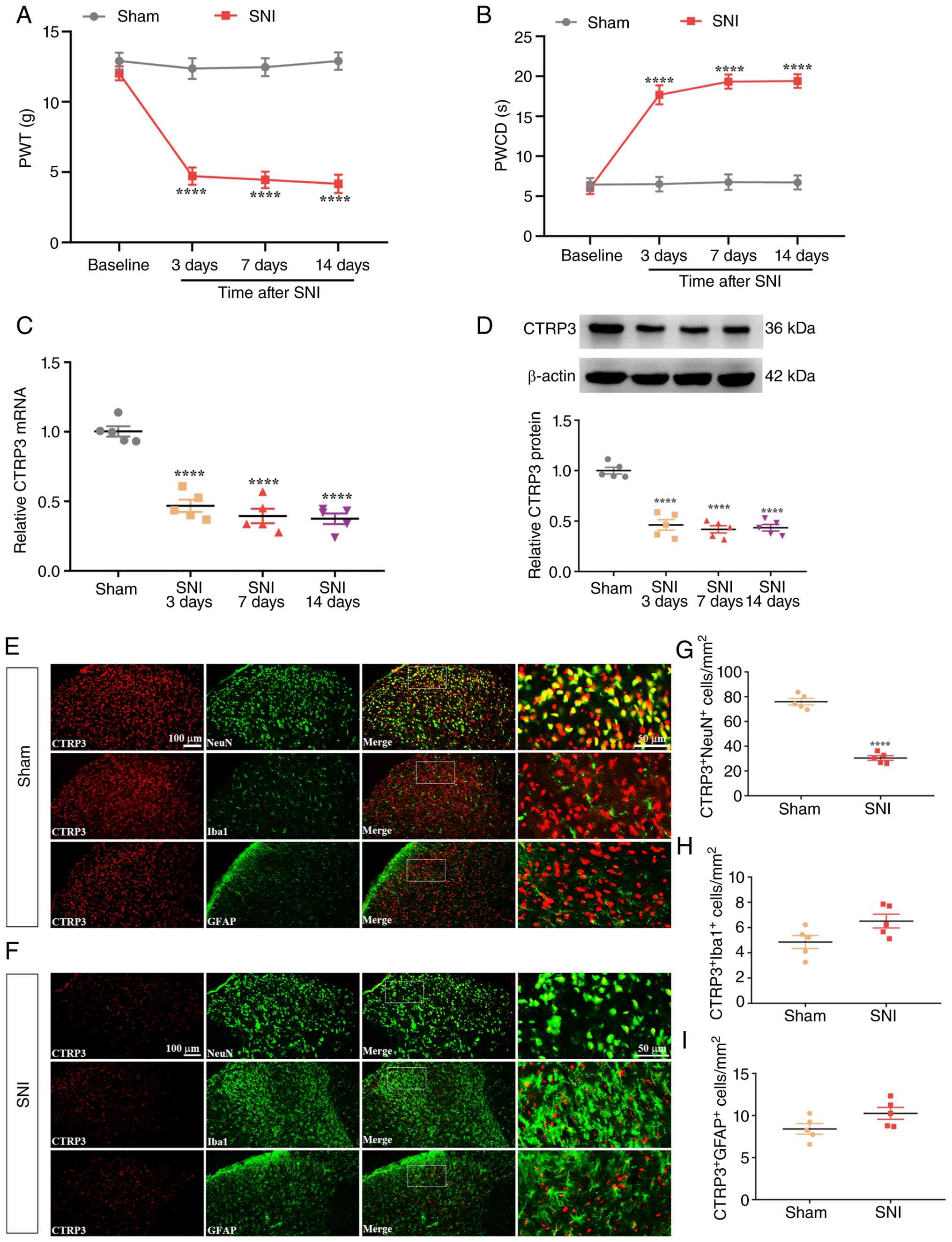 | Figure 1Spinal CTRP3 expression is
downregulated following SNI. (A and B) Compared with sham-operated
control rats, rats subjected to SNI exhibited a significant
reduction in PWT and a prominent elevation in PWCD from post-SNI
day 3 to day 14. ****P<0.0001 vs. sham group, n=10
rats/group. (C and D) RT-qPCR and WB analyses showed a marked
decrease in CTRP3 mRNA and protein levels in the spinal cord of SNI
rats relative to sham controls. (E-I) IF staining results
demonstrated that SNI led to reduced CTRP3 expression in neurons
within the spinal dorsal horn of rats. ****P<0.0001
vs. sham group, n=5 rats/group. SNI, spared nerve injury; PWT, paw
withdrawal threshold; PWCD, paw withdrawal cold duration; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction; WB,
western blotting; IF, immunofluorescence; CTRP3, C1q-tumor necrosis
factor-related protein 3. |
Treatment with rCTRP3 alleviates pain
hypersensitivity in SNI rats mediated by SIRT1
To further investigate the role of CTRP3 in
neuropathic pain, intrathecal injections of rCTRP3 at doses of 10,
30, or 90 μg were administered from day 7 to day 11
post-nerve injury. As shown in Fig.
2A [Interaction: F (20, 270)=3.781, P<0.0001,
η2=0.2187; Time factor: F (5, 270)=45.32, P<0.0001,
η2=0.4562; Group factor: F (4, 270)=115.9, P<0.0001,
η2=0.6319] and Fig.
2B [Interaction: F (20, 270)=4.618, P<0.0001,
η2=0.2548; Time factor: F (5, 270)=41.57, P<0.0001;
η2=0.4350; Group factor: F (4, 270)=124.8, P<0.0001,
η2=0.6489], repeated intrathecal administration of
rCTRP3 at 30 and 90 μg significantly restored the reduced
PWT and abrogated the elevated PWCD relative to the SNI + vehicle
group. These results demonstrated that repeated rCTRP3
administration could mitigate established mechanical allodynia and
cold hyperalgesia in SNI rats. Additionally, IF staining and WB
analyses were used to assess spinal SIRT1 expression. IF staining
results revealed that the number of SIRT1-positive neurons was
decreased after SNI compared with the sham group, while treatment
with rCTRP3 (30 and 90 μg) reversed this reduction in SIRT1
expression in spinal dorsal horn neurons of SNI rats [Fig. 2C and D: F (4, 20)=13.83,
P<0.0001, η2=0.7344]. Consistently, WB results showed
that rCTRP3 administration (30 and 90 μg) blocked the nerve
injury-induced downregulation of SIRT1 in the spinal cord, relative
to the SNI + vehicle group [Fig.
2E: F (4, 20)=10.86, P<0.0001, η2=0.6847]. These
findings confirm that CTRP3 alleviates SNI-induced mechanical
allodynia and cold hyperalgesia and suggest that neuronal SIRT1 in
the spinal cord may mediate this analgesic effect in SNI rats.
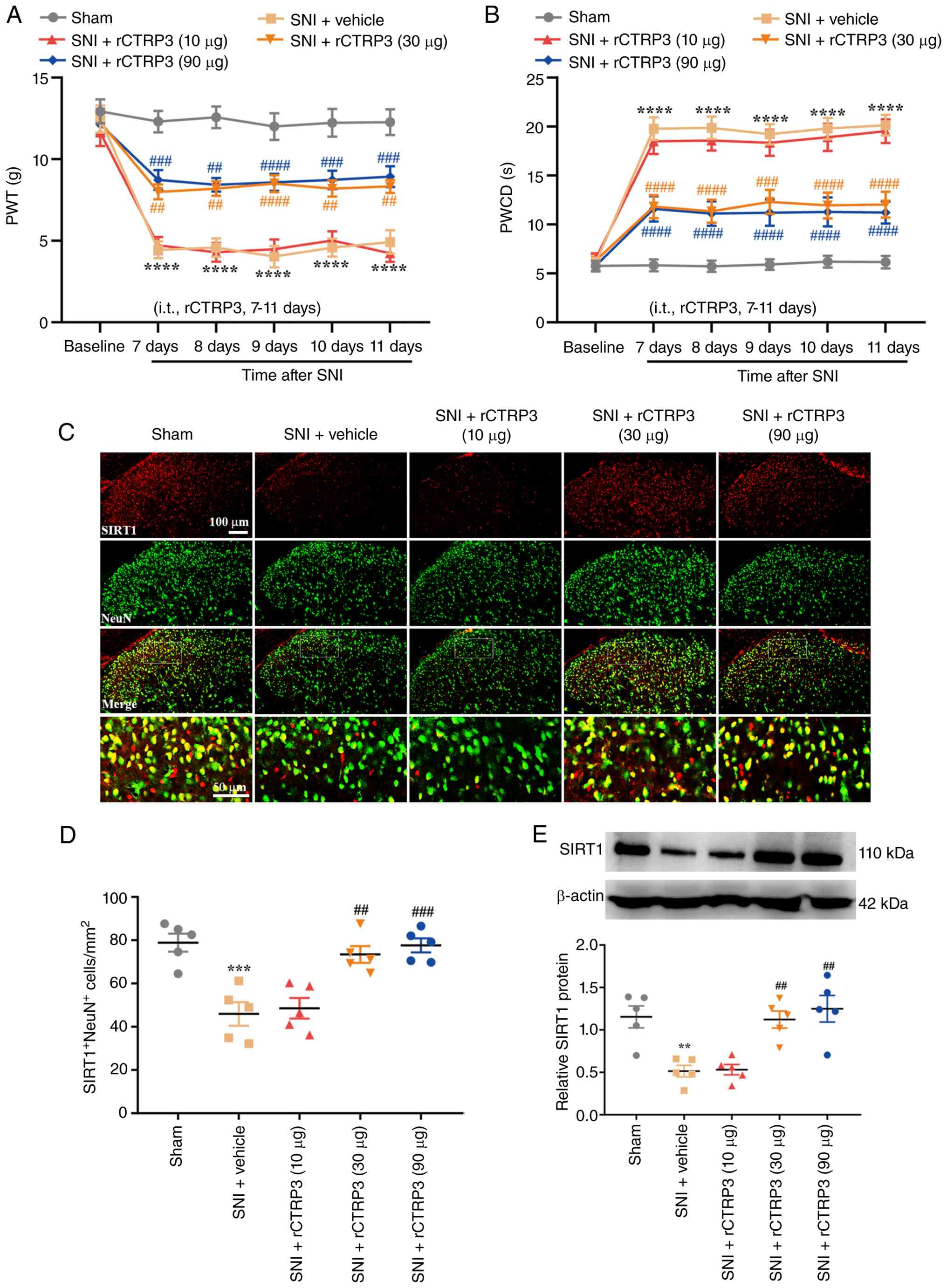 | Figure 2Intrathecal administration of rCTRP3
alleviates pain hypersensitivity and activates spinal SIRT1 in SNI
rats. (A and B) Behavioral assessments showed that repeated
intrathecal injection of rCTRP3 (30 or 90 μg) significantly
reversed the SNI-induced reductions in PWT and increases in PWCD.
****P<0.0001 vs. sham group; ##P<0.01,
###P<0.001 and ####P<0.0001 vs. SNI +
vehicle group; n=10 rats/group. (C and D) IF staining results
revealed that the number of SIRT1-positive neurons was lower in SNI
rats than in the sham group, whereas treatment with rCTRP3 (30 and
90 μg) reversed this decrease in SIRT1 expression in spinal
dorsal horn neurons of SNI rats. (E) WB analysis demonstrated that
SIRT1 levels in the spinal cord were reduced in SNI rats, and this
downregulation was markedly attenuated by rCTRP3 treatment at doses
of 30 or 90 μg. **P<0.01 and
***P<0.001 vs. sham group; ##P<0.01 and
###P<0.001, vs. SNI + vehicle group; n=5 rats/group.
rCTRP3, recombinant complement C1q tumor necrosis factor-related
protein 3; SNI, spared nerve injury; PWT, paw withdrawal threshold;
PWCD, paw withdrawal cold duration; IF, immunofluorescence; SIRT1,
sirtuin 1; WB, western blotting. |
Intrathecal injection with rCTRP3
promotes mitochondrial biogenesis in SNI rats
Mitochondrial biogenesis is a key process for
sustaining mitochondrial homeostasis. It was hypothesized that the
analgesic effect of rCTRP3 may involve the regulation of
mitochondrial biogenesis. To test this, targets associated with
mitochondrial biogenesis were examined, including PGC-1α (a
critical inducer of mitochondrial biogenesis), NRF1, TFAM and mtDNA
copy number. As demonstrated in Fig.
3A and B [F (4, 20)=12.33, P<0.0001,
η2=0.7115], IF staining revealed that the number of
PGC-1α-positive neurons in the spinal dorsal horn was reduced after
SNI compared with the sham group, whereas treatment with rCTRP3 (30
and 90 μg) reversed this decrease in PGC-1α expression in
SNI rats. Consistently, WB analysis demonstrated that rCTRP3
administration (30 and 90 μg) blocked the nerve
injury-induced downregulation of PGC-1α [Fig. 3C: F (4, 20)=16.94, P<0.0001,
η2=0.7721], NRF1 [Fig.
3D: F (4, 20)=14.79, P<0.0001, η2=0.7473] and
TFAM [Fig. 3E: F (4, 20)=12.68,
P<0.0001, η2=0.7172] in the spinal cord, relative to
the SNI + vehicle group. Additionally, mtDNA copy number was
measured to assess mitochondrial biogenesis activity. As revealed
in Fig. 3F [F (4, 20)=18.68, P<0.0001,
η2=0.7888], rCTRP3 treatment (30 and 90 μg)
reversed the reduction in spinal mtDNA copy number observed in SNI
rats. These findings confirm that CTRP3 enhances PGC-1α-dependent
mitochondrial biogenesis in the spinal cord of SNI-induced
rats.
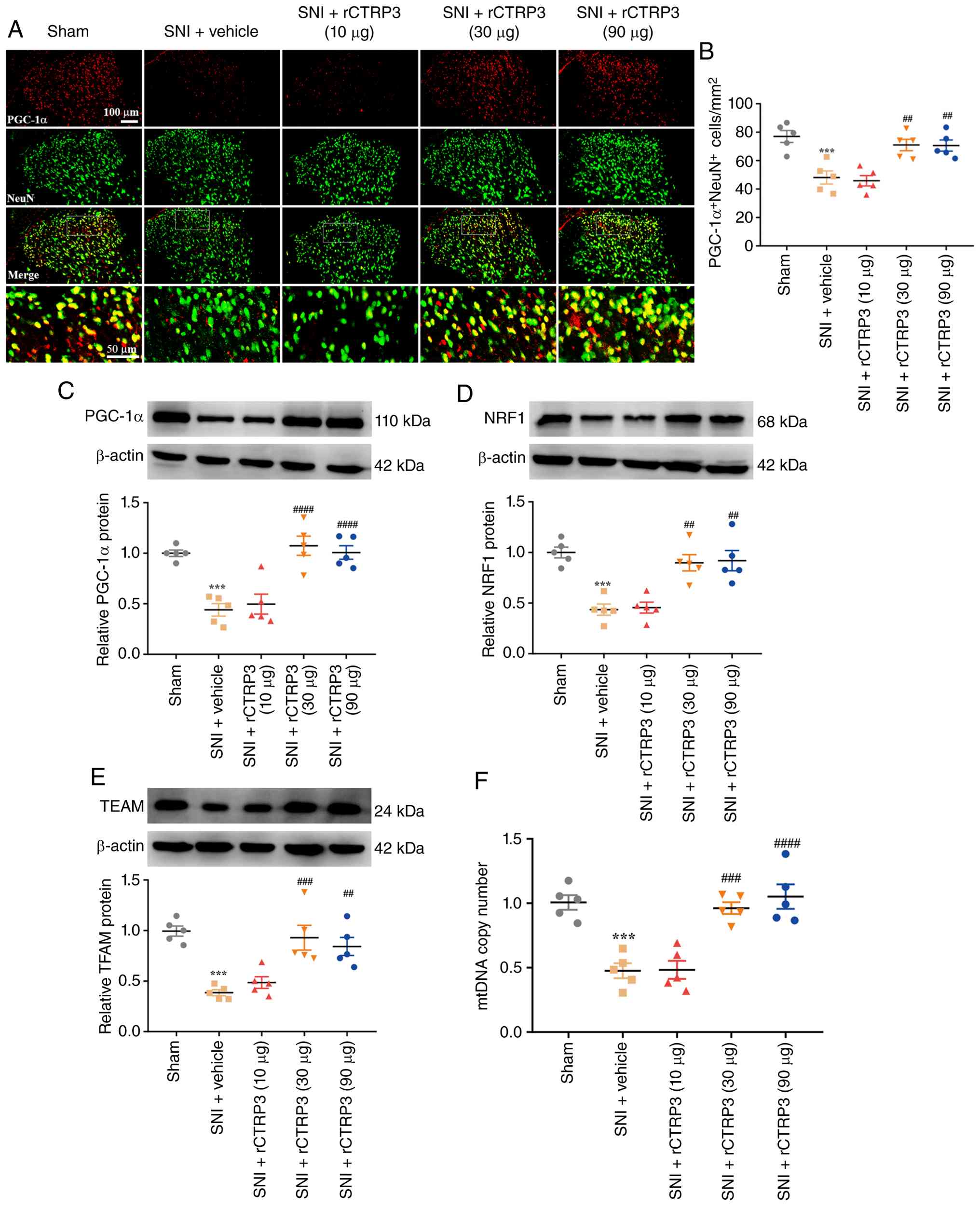 | Figure 3rCTRP3 promotes PGC-1α-mediated
mitochondrial biogenesis in the spinal cord of SNI rats. (A and B)
IF staining showed a significant decrease in neuronal PGC-1α levels
in SNI rats; this reduction was notably reversed by intrathecal
administration of rCTRP3 (30 and 90 μg). (C-E) WB analysis
demonstrated that rCTRP3 treatment (30 and 90 μg) blocked
the nerve injury-induced downregulation of PGC-1α, NRF1 and TFAM in
the spinal cord, compared with the SNI + vehicle group. (F) rCTRP3
administration (30 and 90 μg) reversed the reduction in
spinal mtDNA copy number observed in SNI rats.
***P<0.001 vs. sham group; ##P<0.01,
###P<0.001 and ####P<0.0001 vs. SNI +
vehicle group; n=5 rats/group. rCTRP3, recombinant complement C1q
tumor necrosis factor-related protein 3; PGC-1α, peroxisome
proliferator-activated receptor gamma coactivator 1-alpha; SNI,
spared nerve injury; IF, immunofluorescence; WB, western blotting;
NRF1, nuclear respiratory factor 1; TFAM, mitochondrial
transcription factor A; mtDNA, mitochondrial DNA. |
Intrathecal delivery of rCTRP3
facilitates UPRmt in SNI rats
The UPRmt is a vital mechanism for
maintaining mitochondrial protein homeostasis and redox balance. It
was hypothesized that CTRP3 may exert its analgesic effects partly
through the regulation of UPRmt. To explore this,
UPRmt-related targets were analyzed, including ATF5 (a
key inducer of UPRmt), chaperone proteins (Hsp60) and
proteases (ClpP, LonP1). As shown in Fig. 4A and B [F (4, 20)=10.88, P<0.0001,
η2=0.6851], IF staining indicated that the number of
ATF5-positive neurons in the spinal dorsal horn was decreased after
SNI compared with the sham group, while rCTRP3 treatment (30 and 90
μg) reversed this reduction in ATF5 expression in SNI rats.
Consistently, RT-qPCR results demonstrated that rCTRP3
administration (30 and 90 μg) blocked the nerve
injury-induced downregulation of ATF5 [Fig. 4C: F (4, 20)=19.39, P<0.0001,
η2=0.7950], ClpP [Fig.
4D: F (4, 20)=15.12, P<0.0001, η2=0.7515], Hsp60
[Fig. 4E: F (4, 20)=13.06,
P<0.0001, η2=0.7231] and LonP1 [Fig. 4F: F (4, 20)=11.67, P<0.0001,
η2=0.7001] in the spinal cord, relative to the SNI +
vehicle group. These results confirm that CTRP3 promotes
ATF5-mediated UPRmt in the spinal cord of SNI-induced
rats.
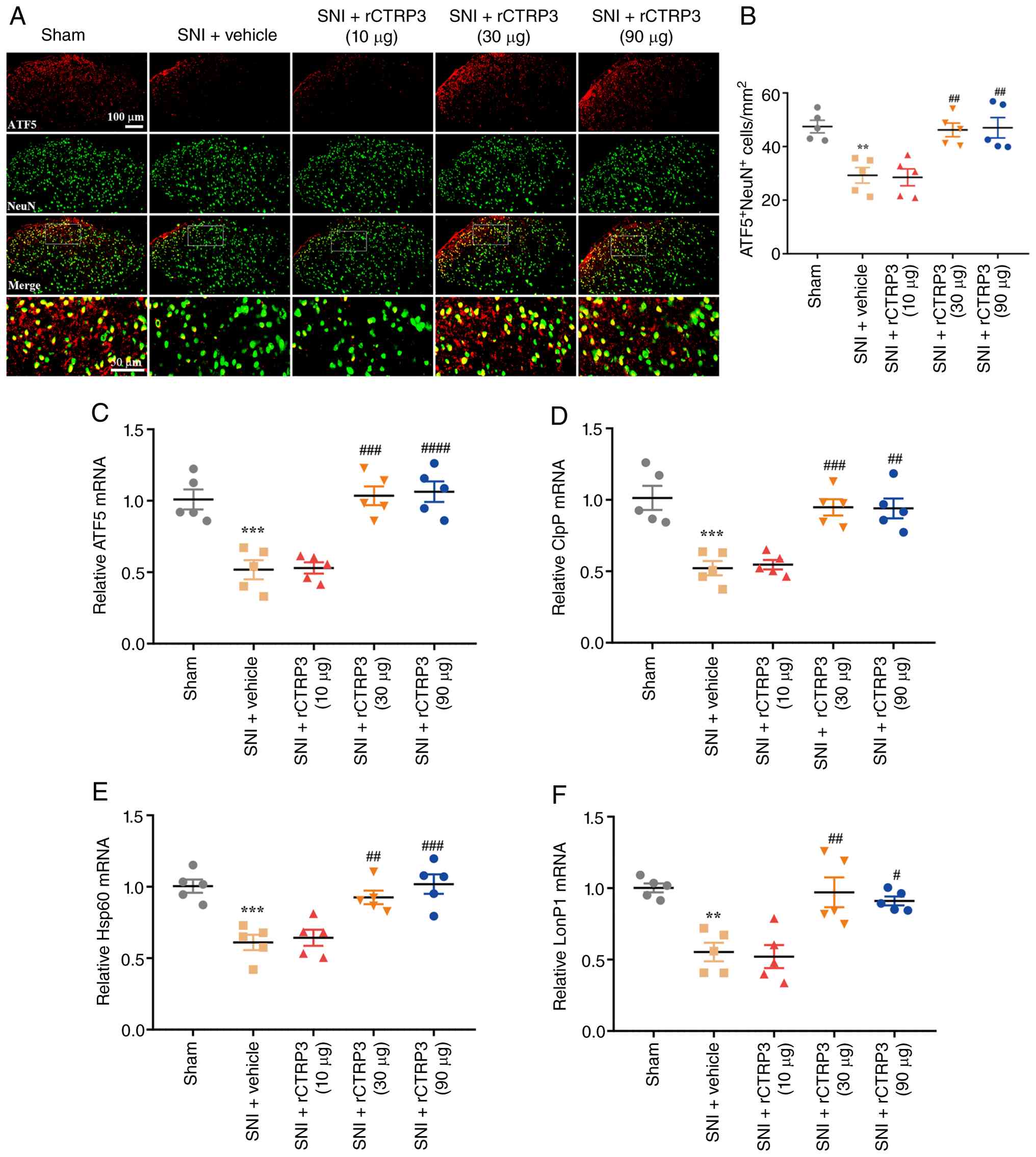 | Figure 4rCTRP3 promotes ATF5-induced
UPRmt in the spinal cord of SNI rats. (A and B) IF
staining showed a notable decrease in spinal neuronal ATF5 levels
in SNI rats; this reduction was significantly reversed by
intrathecal administration of rCTRP3 (30 and 90 μg). (C-F)
RT-qPCR analysis demonstrated that rCTRP3 treatment (30 and 90
μg) blocked the nerve injury-induced downregulation of ATF5,
ClpP, Hsp60 and LonP1 in the spinal cord, compared with the SNI +
vehicle group. **P<0.01 and ***P<0.001
vs. sham group; #P<0.05, ##P<0.01,
###P<0.001 and ####P<0.0001 vs. SNI +
vehicle group; n=5 rats/group. rCTRP3, recombinant complement C1q
tumor necrosis factor-related protein 3; ATF5, activating
transcription factor 5; UPRmt, mitochondrial unfolded
protein response; SNI, spared nerve injury; IF, immunofluorescence;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; ClpP, caseinolytic mitochondrial matrix peptidase
proteolytic subunit; Hsp60, heat shock protein 60; LonP1, Lon
protease 1. |
Administration of rCTRP3 mitigates spinal
mitochondrial dysfunction and oxidative stress in SNI rats
To further verify the protective effects of rCTRP3
on mitochondrial function, MMP and intracellular ATP production
were directly measured. As illustrated in Fig. S1A-C, SNI induced a marked
reduction in MMP [F (4, 20)=31.33, P<0.0001,
η2=0.8624] and ATP levels [F (4, 20)=14.26, P<0.0001,
η2=0.7404] in the spinal cord, both of which were
significantly restored by intrathecal administration of rCTRP3 at
30 and 90 μg. These data provide direct functional evidence
that rCTRP3 preserves mitochondrial bioenergetic capacity in the
setting of neuropathic pain. Redox balance in the spinal cord was
evaluated using DHE staining and biochemical assays. As revealed in
Fig. 5A and B [F (4, 20)=69.93, P<0.0001,
η2=0.9333], DHE staining results revealed that
administration of rCTRP3 (30 and 90 μg) reduced the number
of DHE-positive cells in the spinal cord of SNI rats, which was
elevated in the SNI + vehicle group. Consistent with this,
biochemical assay data demonstrated that rCTRP3 treatment (30 and
90 μg) reversed the SNI-induced increases in oxidative
damage markers, including MDA [Fig.
5C: F (4, 20)=16.82, P<0.0001, η2=0.7709] and PCO
[Fig. 5D: F (4, 20)=16.88,
P<0.0001, η2=0.7715] in the spinal cord.
Additionally, rCTRP3 intervention (30 and 90 μg)
significantly increased the level of reduced GSH [Fig. 5E: F (4, 20)=11.99, P<0.0001,
η2=0.7056] and enhanced the activity of antioxidant
enzymes, including GSH-PX [Fig.
5F: F (4, 20)=13.09, P<0.0001, η2=0.7236] and SOD
[Fig. 5G: F (4, 20)=10.52,
P<0.0001, η2=0.6779] in the spinal cord of SNI rats.
These findings confirm that CTRP3 can alleviate SNI-induced
oxidative stress in the spinal cord of rats.
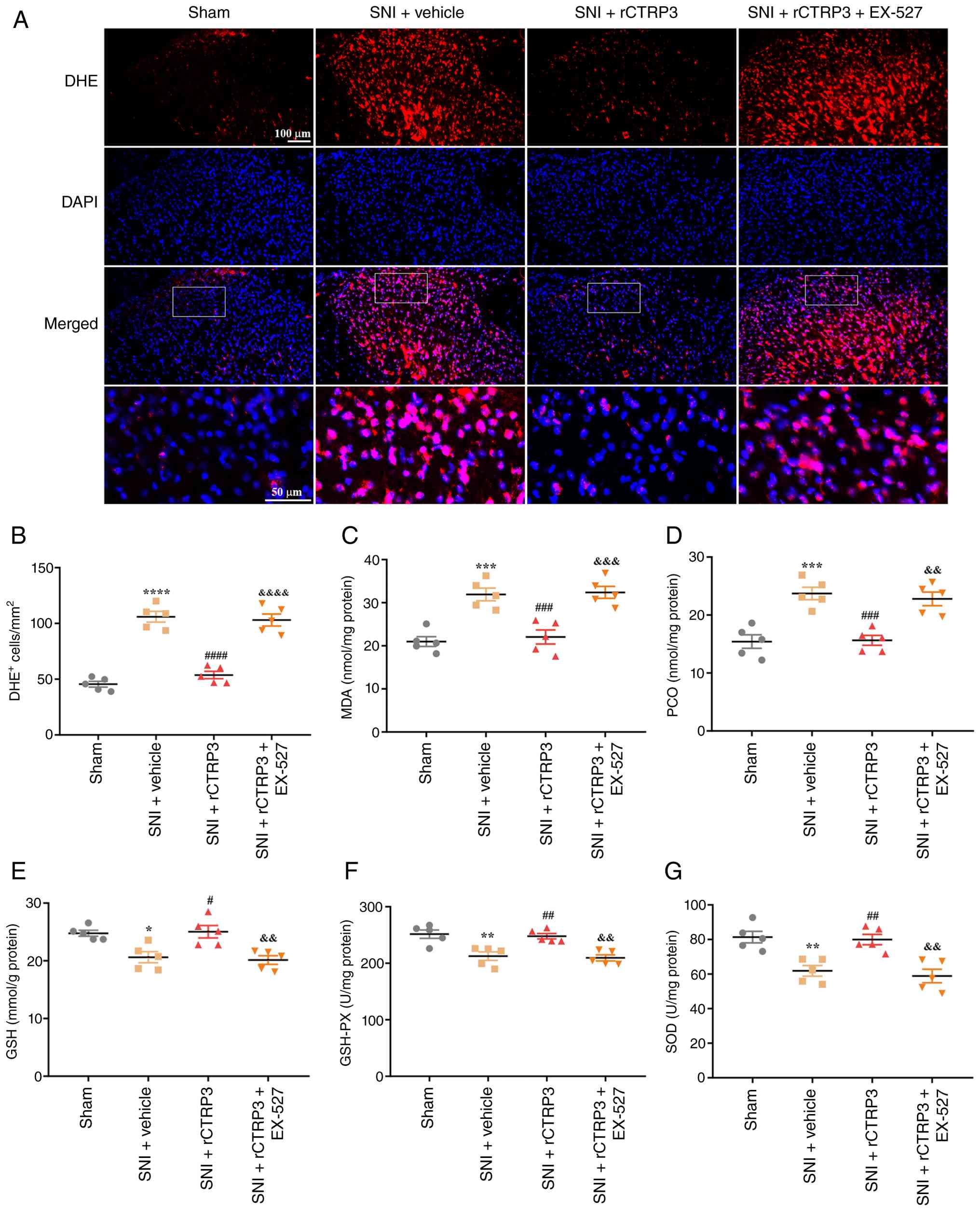
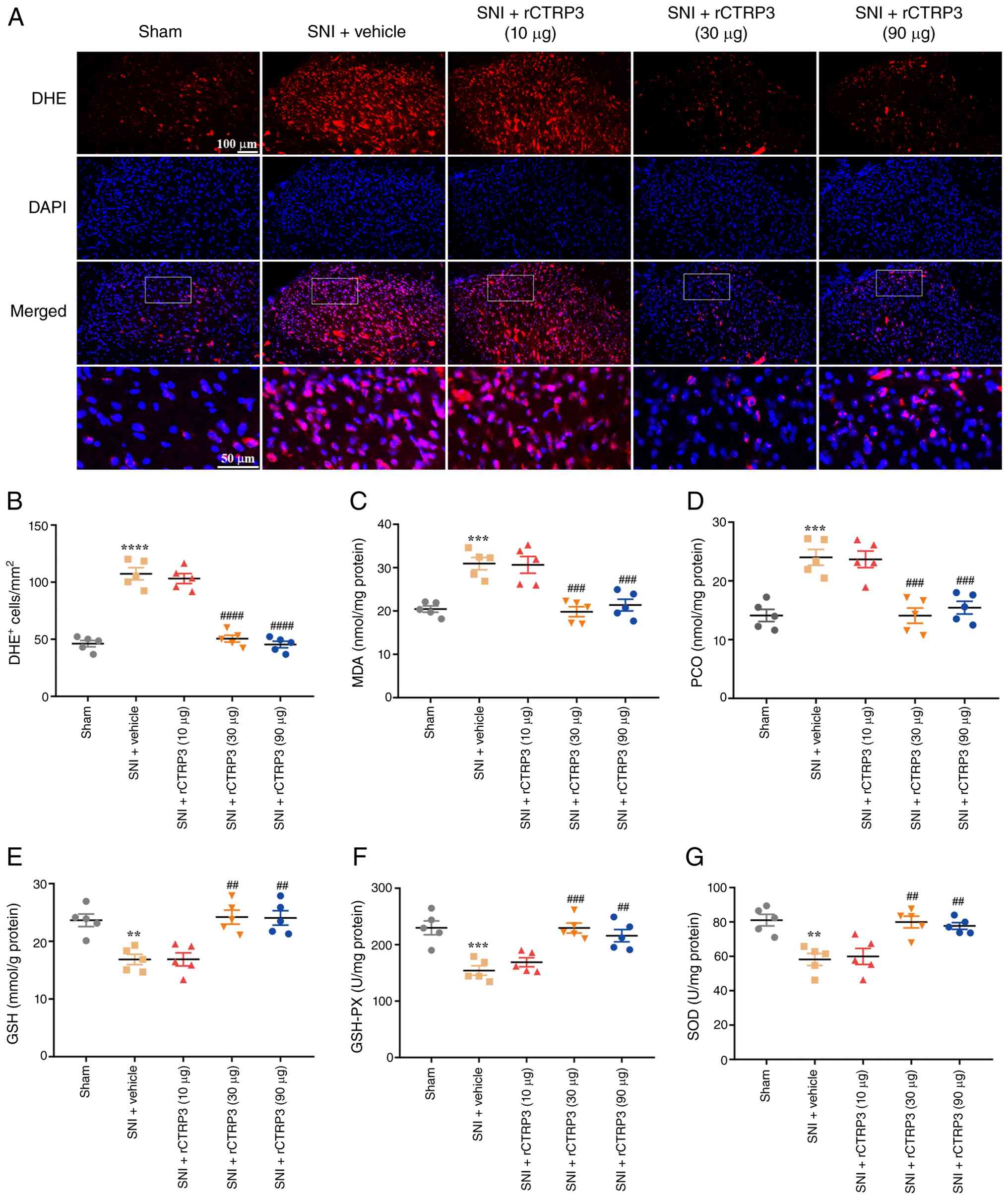 | Figure 5rCTRP3 administration attenuates
SNI-induced oxidative stress in the rat spinal cord. (A and B) DHE
staining showed that rCTRP3 treatment (30 and 90 μg)
significantly reduced the increased number of DHE-positive cells in
the spinal dorsal horn of SNI rats. (C and D) Biochemical assays
revealed that SNI elevated the spinal content of MDA and PCO;
intrathecal injection of rCTRP3 (30 and 90 μg) reversed this
elevation. (E and F) rCTRP3 (30 and 90 μg) restored the
reduced concentrations of reduced GSH and activity of GSH-PX in the
spinal cord of SNI rats. (G) Administration of rCTRP3 (30 and 90
μg) reversed the decreased activity of SOD in the spinal
cord of SNI rats. **P<0.01, ***P<0.001
and ****P<0.0001 vs. sham group;
##P<0.01, ###P<0.001 and
####P<0.0001 vs. SNI + vehicle group; n=5 rats/group.
rCTRP3, recombinant complement C1q tumor necrosis factor-related
protein 3; SNI, spared nerve injury; DHE, dihydroethidium; MDA,
malondialdehyde; PCO, protein carbonyl; GSH, glutathione; GSH-PX,
glutathione peroxidase; SOD, superoxide dismutase. |
SIRT1 antagonist abolishes
rCTRP3-mediated amelioration of pain hypersensitivity and
mitochondrial biogenesis in SNI rats
To investigate whether SIRT1 mediates the analgesic
effect of rCTRP3 in SNI rats, the SIRT1 antagonist EX-527 was
employed. Results demonstrated that rCTRP3 treatment restored the
reduced PWT [Fig. 6A:
Interaction: F (15, 216)=6.652, P<0.0001, η2=0.3160;
Time factor: F (5, 216)=44.13, P<0.0001, η2=0.5053;
Group factor: F (3, 216)=148.5, P<0.0001, η2=0.6734]
and attenuated the elevated PWCD [Fig. 6B: Interaction: F (15, 216)=7.944,
P<0.0001, η2=0.3556; Time factor: F (5, 216)=51.85,
P<0.0001, η2=0.5455; Group factor: F (3, 216)=175.3,
P<0.0001, η2=0.7088] in SNI rats; however,
co-administration of EX-527 abolished this analgesic effect of
rCTRP3. These data indicated that CTRP3 alleviates mechanical
allodynia and cold hyperalgesia by activating spinal SIRT1 in SNI
rats. Furthermore, to explore the mechanism by which CTRP3
regulates mitochondrial biogenesis, EX-527 was used in subsequent
experiments. IF staining revealed that EX-527 blocked the
rCTRP3-induced increase in PGC-1α expression in spinal dorsal horn
neurons of SNI rats [Fig. 6C and
D: F (3, 16)=16.84, P<0.0001, η2=0.7594].
Consistently, WB analysis identified that EX-527 abolished the
rCTRP3-mediated upregulation of PGC-1α [Fig. 6E: F (3, 16)=27.96, P<0.0001,
η2=0.8398], NRF1 [Fig.
6F: F (3, 16)=17.25, P<0.0001, η2=0.7639] and
TFAM [Fig. 6G: F (3, 16)=9.562,
P=0.0007, η2=0.6419] in the spinal cord, compared with
the SNI + rCTRP3 group. Additionally, rCTRP3 treatment reversed the
reduction in mtDNA copy number in SNI rats, but this effect was
blocked by EX-527 [Fig. 6H: F
(3, 16)=44.02, P<0.0001, η2=0.8919]. These results
confirm that CTRP3 alleviates mechanical allodynia and cold
hyperalgesia and promotes PGC-1α-mediated mitochondrial biogenesis
in the spinal cord of SNI rats through SIRT1 activation.
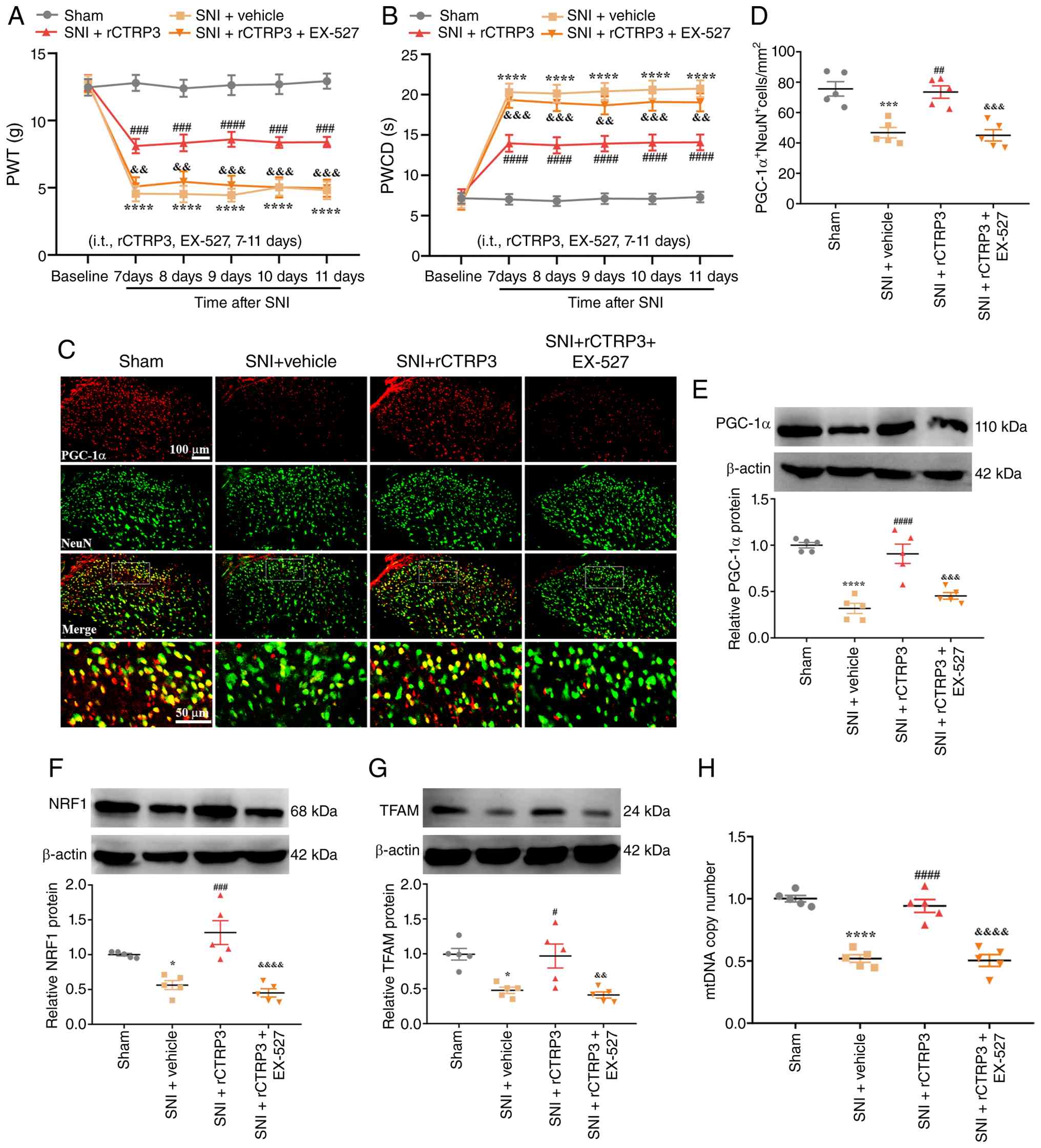 | Figure 6SIRT1 antagonist abrogates
rCTRP3-mediated amelioration of pain hypersensitivity and
PGC-1α-dependent mitochondrial biogenesis in SNI rats. (A and B)
Intrathecal injection of EX-527 (a SIRT1 antagonist) reversed the
rCTRP3-induced increases in PWT and reductions in PWCD in SNI rats.
****P<0.0001 vs. sham group; ###P<0.001
and ####P<0.0001 vs. SNI + vehicle group;
&&P<0.01 and
&&&P<0.001 vs. SNI + rCTRP3 group; n=10
rats/group. (C and D) IF staining revealed that EX-527 blocked the
rCTRP3-induced upregulation of PGC-1α expression in spinal dorsal
horn neurons of SNI rats. (E-G) WB analysis demonstrated that
EX-527 abolished the rCTRP3-mediated elevation of PGC-1α, NRF1 and
TFAM levels in the spinal cord of SNI rats. (H) Administration of
rCTRP3 reversed the reduction in mtDNA copy number in SNI rats, but
this protective effect was inhibited by EX-527.
*P<0.05, ***P<0.001 and
****P<0.0001 vs. sham group; #P<0.05,
##P<0.01, ###P<0.001 and
####P<0.0001 vs. SNI + vehicle group;
&&P<0.01,
&&&P<0.001 and
&&&&P<0.0001 vs. SNI + rCTRP3 group;
n=5 rats/group. SIRT1, sirtuin 1; rCTRP3, recombinant complement
C1q tumor necrosis factor-related protein 3; SNI, spared nerve
injury; EX-527, a selective SIRT1 antagonist; PWT, paw withdrawal
threshold; PWCD, paw withdrawal cold duration; IF,
immunofluorescence; WB, Western blotting; PGC-1α, peroxisome
proliferator-activated receptor gamma coactivator 1-alpha; NRF1,
nuclear respiratory factor 1; TFAM, mitochondrial transcription
factor A; mtDNA, mitochondrial DNA. |
SIRT1 antagonist blocks rCTRP3-induced
enhancement of UPRmt in SNI rats
To investigate the mechanism by which CTRP3
regulates the UPRmt, the SIRT1 antagonist EX-527 was
utilized in the present study. IF staining results revealed that
EX-527 inhibited the rCTRP3-induced increase in ATF5 expression in
spinal dorsal horn neurons of SNI rats [Fig. 7A and B: F (3, 16)=15.36,
P<0.0001, η2=0.7423]. Consistently, RT-qPCR analysis
demonstrated that EX-527 abolished the rCTRP3-mediated upregulation
of ATF5 [Fig. 7C: F (3,
16)=24.98, P<0.0001, η2=0.8240], ClpP [Fig. 7D: F (3, 16)=14.60, P<0.0001;
η2=0.7324], Hsp60 [Fig.
7E: F (3, 16)=17.52, P=0.0007, η2=0.7666] and LonP1
[Fig. 7F: F (3, 16)=15.18,
P<0.0001, η2=0.7400] in the spinal cord, relative to
the SNI + rCTRP3 group. These findings demonstrate that CTRP3
promotes ATF5-mediated UPRmt in the spinal cord of SNI
rats through SIRT1 activation.
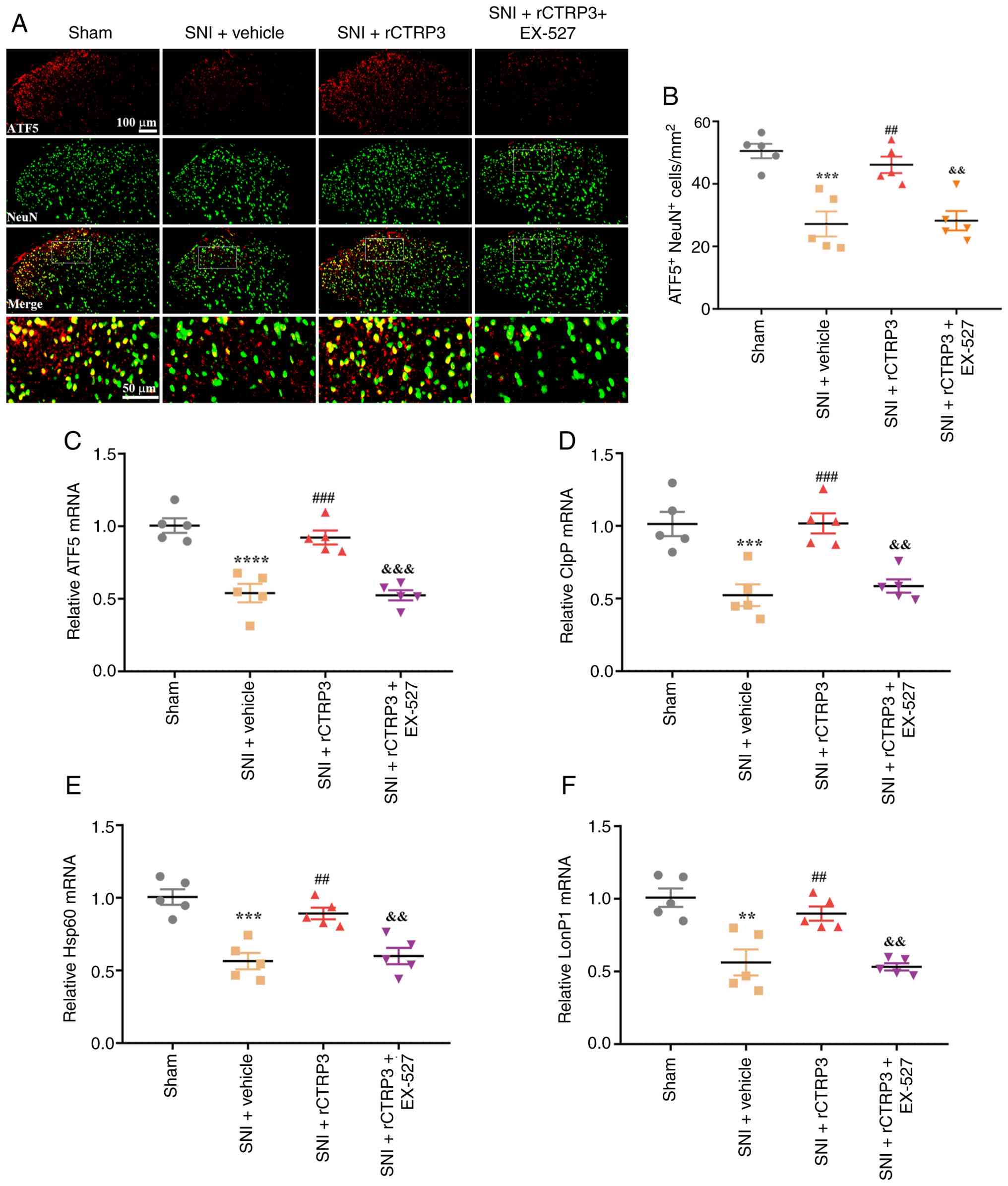 | Figure 7SIRT1 antagonist blocks
rCTRP3-induced enhancement of ATF5-mediated UPRmt in SNI
rats. (A and B) IF staining results showed that EX-527 inhibited
the rCTRP3-induced upregulation of ATF5 expression in spinal dorsal
horn neurons of SNI rats. (C-F) RT-qPCR analysis demonstrated that
EX-527 abolished the rCTRP3-mediated elevation of ATF5, ClpP, Hsp60
and LonP1 expression in the spinal cord, compared with the SNI +
rCTRP3 group. **P<0.01, ***P<0.001 and
****P<0.0001 vs. sham group; ##P<0.01
and ###P<0.001 vs. SNI + vehicle group;
&&P<0.01 and
&&&P<0.001 vs. SNI + rCTRP3 group; n=5
rats/group. SIRT1, sirtuin 1; rCTRP3, recombinant complement C1q
tumor necrosis factor-related protein 3; SNI, spared nerve injury;
EX-527, selective SIRT1 inhibitor; IF, immunofluorescence; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction; ATF5,
activating transcription factor 5; UPRmt, mitochondrial
unfolded protein response; ClpP, caseinolytic mitochondrial matrix
peptidase proteolytic subunit; Hsp60, heat shock protein 60; LonP1,
Lon protease 1. |
SIRT1 inhibitor abolishes rCTRP3-mediated
alleviation of mitochondrial dysfunction and oxidative stress in
SNI rats
Moreover, the beneficial effects of rCTRP3 on
restoring MMP [F (3, 16)=20.21, P<0.0001,
η2=0.7912] and ATP production [F (3, 16)=12.61, P=0.0002,
η2=0.7028] were completely abolished by pretreatment
with the SIRT1 inhibitor EX-527 (Fig. S2A-C), indicating that SIRT1
activity is essential for rCTRP3 to maintain mitochondrial membrane
integrity and energy production. As demonstrated in Fig. 8A and B [F (3, 16)=58.50, P<0.0001,
η2=0.9164], DHE staining revealed that compared with the
SNI + rCTRP3 group, treatment with EX-527 reversed the
rCTRP3-induced reduction in the number of DHE-positive cells in the
spinal cord of SNI rats. Furthermore, biochemical assay results
demonstrated that EX-527 abolished the rCTRP3-mediated
downregulation of oxidative damage markers, including MDA [Fig. 8C: F (3, 16)=18.93, P<0.0001,
η2=0.7802] and PCO [Fig.
8D: F (3, 16)=17.45, P<0.0001, η2=0.7659] in the
spinal cord of SNI rats. Notably, while rCTRP3 increased the level
of GSH [Fig. 8E: F (3,
16)=9.383, P=0.0008, η2=0.6376] and enhanced the
activity of antioxidant enzymes such as GSH-PX [Fig. 8F: F (3, 16)=12.27, P=0.0002,
η2=0.6970] and SOD [Fig.
8G: F (3, 16)=12.34, P=0.0002, η2=0.6983] in the
spinal cord of SNI rats, these beneficial effects were reversed by
the SIRT1 antagonist. These findings confirm that CTRP3 mitigates
spinal oxidative stress in SNI rats through SIRT1 activation.
 | Figure 8SIRT1 inhibitor abrogates
rCTRP3-induced attenuation of spinal oxidative stress in SNI rats.
(A and B) DHE staining revealed that, compared with the SNI +
rCTRP3 group, EX-527 treatment reversed the rCTRP3-induced decrease
in the number of DHE-positive cells in the spinal cord of SNI rats.
(C and D) Biochemical assay results demonstrated that EX-527
eliminated the rCTRP3-mediated reduction in spinal levels of MDA
and PCO in SNI rats. (E-G) rCTRP3 treatment increased the spinal
level of reduced GSH and enhanced the activity of GSH-PX and SOD in
SNI rats; however, these protective effects were counteracted by
the SIRT1 antagonist (EX-527). *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001 vs. sham group; #P<0.05,
##P<0.01, ###P<0.001 and
####P<0.0001 vs. SNI + vehicle group;
&&P<0.01,
&&&P<0.001 and
&&&&P<0.0001 vs. SNI + rCTRP3 group;
n=5 rats/group. SIRT1, sirtuin 1; rCTRP3, recombinant complement
C1q tumor necrosis factor-related protein 3; SNI, spared nerve
injury; EX-527, a selective SIRT1 inhibitor; DHE, dihydroethidium;
MDA, malondialdehyde; PCO, protein carbonyl; GSH, glutathione;
GSH-PX, glutathione peroxidase; SOD, superoxide dismutase. |
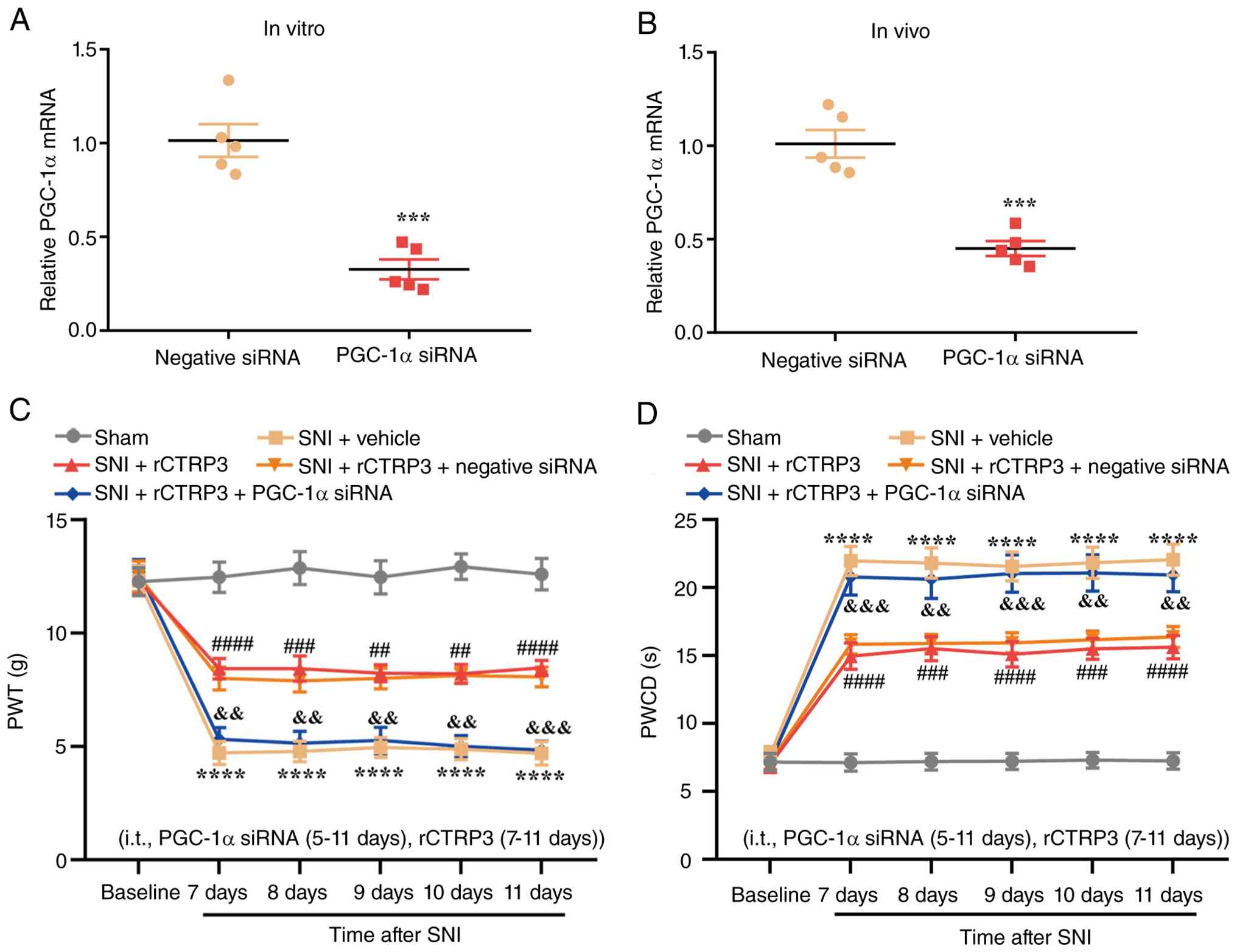
PGC-1α siRNA abolishes rCTRP3-mediated
amelioration of pain hypersensitivity, mitochondrial biogenesis and
ATF5-dependent UPRmt in SNI rats
To establish whether PGC-1α serves as a critical
downstream mediator of rCTRP3-induced analgesia in SNI rats, PGC-1α
expression in the spinal cord was specifically silenced using
siRNA. RT-qPCR analysis confirmed that PGC-1α siRNA transfection
induced significant downregulation of PGC-1α mRNA in PC12 cells
in vitro without additional stimulation [Fig. 9A: t (8)=6.717, P=0.0002,
η2=0.8494]. Consistently, intrathecal injection of
PGC-1α siRNA also significantly inhibited the endogenous
transcription of spinal PGC-1α in SNI rats compared with the
negative control siRNA group [Fig.
9B: t (8)=6.654, P=0.0002,
η2=0.8470]. Subsequent behavioral testing demonstrated
that intrathecal administration of rCTRP3 significantly elevated
the reduced PWT and reduced the prolonged PWCD in SNI-operated
rats; however, these analgesic effects were completely abrogated by
co-delivery of PGC-1α siRNA [Fig. 9B
and C; Fig. 9B: Interaction:
F (20, 270)=6.262, P<0.0001, η2=0.3169; Time factor:
F (5, 270)=62.06, P<0.0001, η2=0.5347; Group factor:
F (4, 270)=143.9, P<0.0001, η2=0.6807; Fig. 9C: Interaction: F (20, 270)=6.418,
P<0.0001, η2=0.3222; Time factor: F (5, 270)=80.32,
P<0.0001, η2=0.5980; Group factor: F (4, 270)=168.2,
P<0.0001, η2=0.7137]. These results indicate that
PGC-1α activation is essential for rCTRP3 to alleviate mechanical
allodynia and cold hyperalgesia in the context of neuropathic
pain.
To further clarify the regulatory role of PGC-1α in
mitochondrial biogenesis triggered by rCTRP3, IF staining and WB
analysis were performed. IF staining revealed that PGC-1α siRNA
significantly blocked the rCTRP3-mediated upregulation of PGC-1α in
neurons of the spinal dorsal horn [Fig. 10A and B: F (4, 20)=14.12,
P<0.0001, η2=0.7385]. In line with this observation,
WB analysis demonstrated that PGC-1α silencing abolished the
rCTRP3-induced increases in PGC-1α [F (4, 20)=7.979, P=0.0005,
η2=0.6148], NRF1 [F (4, 20)=38.50, P<0.0001,
η2=0.8850] and TFAM [F (4, 20)=19.60, P<0.0001,
η2=0.7967] protein levels in the spinal cord, as
compared with the SNI + rCTRP3 group (Fig. 10C-E). Moreover, rCTRP3 treatment
effectively reversed the SNI-induced reduction in mtDNA copy
number, and this beneficial effect was also negated by PGC-1α siRNA
intervention [Fig. 10F: F (4,
20)=16.59, P<0.0001, η2=0.7684].
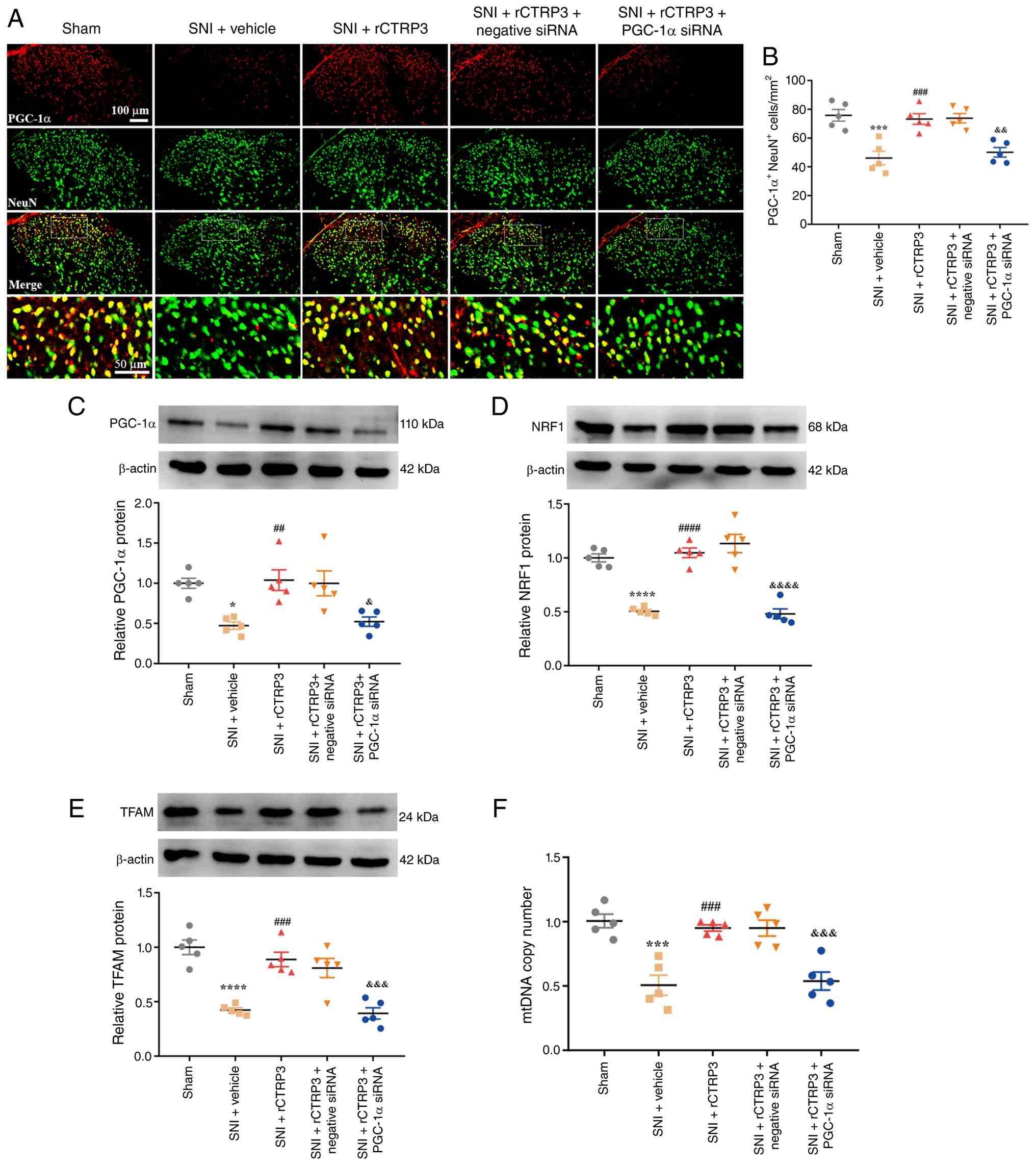 | Figure 10PGC-1α siRNA abrogates
rCTRP3-mediated enhancement of mitochondrial biogenesis in SNI
rats. (A and B) IF staining revealed that PGC-1α siRNA blocked the
rCTRP3-induced upregulation of PGC-1α expression in spinal dorsal
horn neurons of SNI rats. (C-E) WB analysis demonstrated that
PGC-1α siRNA eliminated the rCTRP3-mediated elevation of PGC-1α,
NRF1, and TFAM levels in the spinal cord, compared with the SNI +
rCTRP3 group. (F) rCTRP3 treatment reversed the reduction in mtDNA
copy number in SNI rats, and this regulatory effect was inhibited
by PGC-1α siRNA. *P<0.05, ***P<0.001
and ****P<0.0001 vs. sham group;
##P<0.01, ###P<0.001 and
####P<0.0001 vs. SNI + vehicle group;
&P<0.05, &&P<0.01,
&&&P<0.001 and
&&&&P<0.0001 vs. SNI + rCTRP3 group;
n=5 rats/group. PGC-1α, peroxisome proliferator-activated receptor
gamma coactivator 1-alpha; siRNA, small interfering RNA; rCTRP3,
recombinant complement C1q tumor necrosis factor-related protein 3;
SNI, spared nerve injury; IF, immunofluorescence; WB, Western
blotting; NRF1, nuclear respiratory factor 1; TFAM, mitochondrial
transcription factor A; mtDNA, mitochondrial DNA. |
Notably, it was further explored whether PGC-1α
could modulate ATF5-mediated UPRmt during rCTRP3
treatment. IF staining showed that PGC-1α siRNA significantly
inhibited the rCTRP3-evoked upregulation of ATF5 in spinal dorsal
horn neurons [Fig. S3A and B: F
(4, 20)=15.48, P<0.0001, η2=0.7558]. Consistently,
RT-qPCR analysis revealed that silencing PGC-1α completely reversed
the rCTRP3-mediated enhancement of ATF5 [F (4, 20)=13.22, P<0.0001,
η2=0.7256], ClpP [F (4, 20)=14.29, P<0.0001,
η2=0.7408], Hsp60 [F (4, 20)=13.33, P<0.0001,
η2=0.7273] and LonP1 [F (4, 20)=14.17, P<0.0001,
η2=0.7391] mRNA expression in the spinal cord (Fig. S3C-F). Collectively, these
findings demonstrate that PGC-1α functions as a key molecular hub
that enables rCTRP3 to simultaneously promote mitochondrial
biogenesis and ATF5-dependent UPRmt, ultimately
mitigating neuropathic pain hypersensitivity.
PGC-1α siRNA blocks rCTRP3-induced
attenuation of spinal mitochondrial dysfunction and oxidative
stress in SNI rats
Similarly, silencing PGC-1α via siRNA also blocked
the rCTRP3-induced restoration of MMP [F (4, 20)=24.04, P<0.0001,
η2=0.8278] and ATP content [F (4, 20)=15.84, P<0.0001,
η2=0.7601] in the spinal cord of SNI rats (Fig. S4A-C), supporting that
PGC-1α-dependent mitochondrial biogenesis is required for rCTRP3 to
sustain normal mitochondrial function. As depicted in Fig. 11A and B [F (4, 20)=52.53, P<0.0001,
η2=0.9131], DHE staining analysis showed that compared
with the SNI + rCTRP3 group, administration of PGC-1α siRNA
counteracted the rCTRP3-induced decrease in the number of
DHE-positive cells in the spinal cord of SNI rats. In addition,
biochemical assay data revealed that PGC-1α siRNA eliminated the
rCTRP3-driven downregulation of oxidative damage indicators,
specifically MDA [Fig. 11C: F
(4, 20)=18.60, P<0.0001, η2=0.7881] and PCO [Fig. 11D: F (4, 20)=18.20, P<0.0001,
η2=0.7845] in the spinal cord of SNI rats. Of note,
although rCTRP3 elevated the level of GSH [Fig. 11E: F (4, 20)=14.86, P<0.0001,
η2=0.7483] and improved the activity of antioxidant
enzymes including GSH-PX [Fig.
11F: F (4, 20)=11.54, P<0.0001, η2=0.6977] and
SOD [Fig. 11G: F (4, 20)=22.86,
P<0.0001, η2=0.8205] in the spinal cord of SNI rats,
these favorable changes were reversed by PGC-1α siRNA intervention.
These results indicate that CTRP3 alleviates spinal oxidative
stress in SNI rats through the activation of PGC-1α.
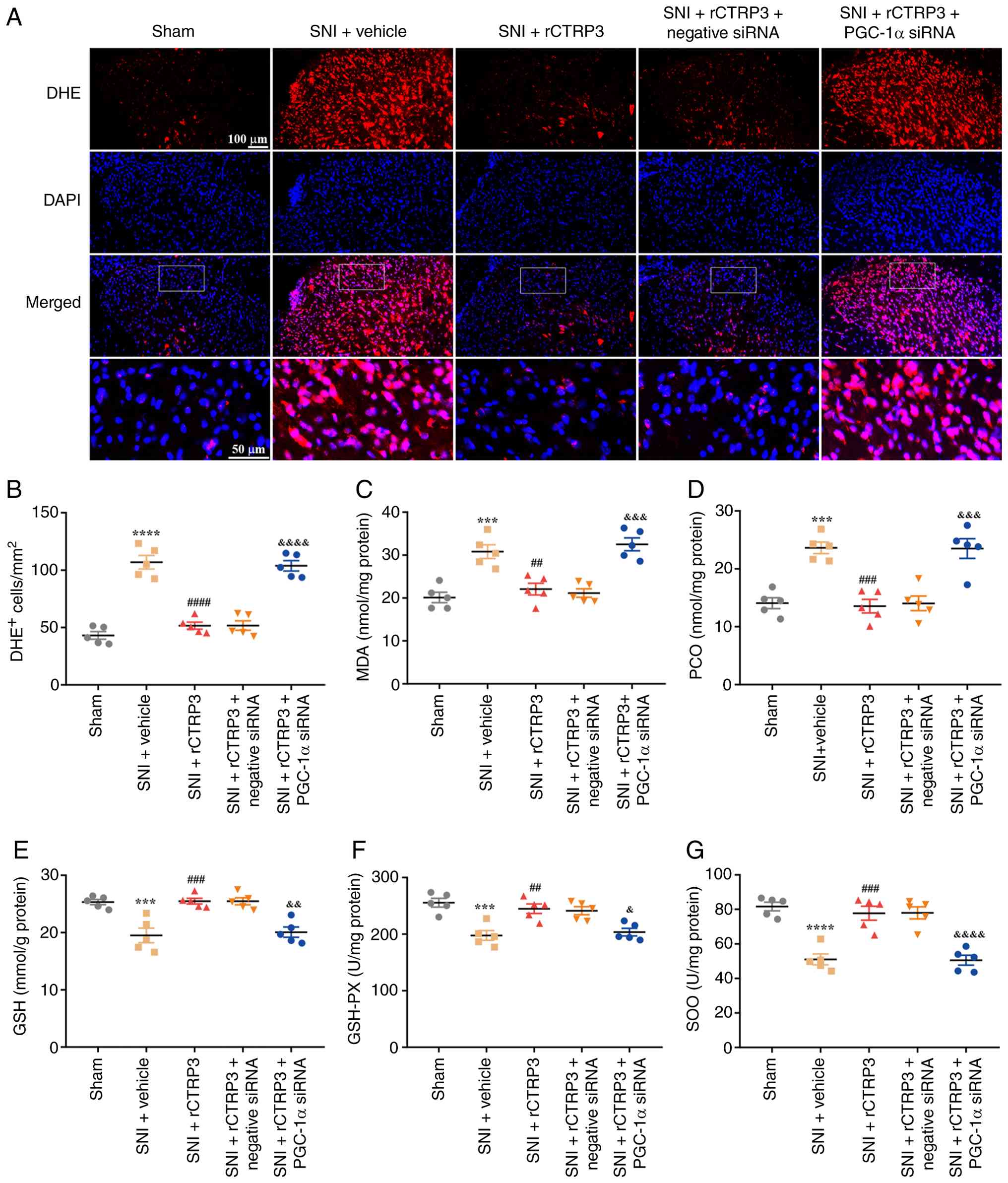 | Figure 11PGC-1α siRNA blocks rCTRP3-induced
reduction of spinal oxidative stress in SNI rats. (A and B) DHE
staining analysis showed that compared with the SNI + rCTRP3 group,
administration of PGC-1α siRNA reversed the rCTRP3-induced decrease
in the number of DHE-positive cells in the spinal cord of SNI rats.
(C and D) Biochemical assay results revealed that PGC-1α siRNA
abolished the rCTRP3-mediated downregulation of MDA and PCO, in the
spinal cord of SNI rats. (E-G) Administration of rCTRP3 increased
the level of reduced GSH and enhanced the activity of GSH-PX and
SOD in the spinal cord of SNI rats, but these beneficial changes
were counteracted by PGC-1α siRNA intervention.
***P<0.001 and ****P<0.0001 vs. sham
group; ##P<0.01, ###P<0.001 and
####P<0.0001 vs. SNI + vehicle group;
&P<0.05, &&P<0.01,
&&&P<0.001 and
&&&&P<0.0001 vs. SNI + rCTRP3 group;
n=5 rats/group. PGC-1α, peroxisome proliferator-activated receptor
gamma coactivator 1-alpha; siRNA, small interfering RNA; rCTRP3,
recombinant complement C1q tumor necrosis factor-related protein 3;
SNI, spared nerve injury; DHE, dihydroethidium; MDA,
malondialdehyde; PCO, protein carbonyl; GSH, glutathione; GSH-PX,
glutathione peroxidase; SOD, superoxide dismutase. |
ATF5 siRNA reverses rCTRP3-mediated
attenuation of pain hypersensitivity, UPRmt activation
and PGC-1α-dependent mitochondrial biogenesis in SNI rats
To define the functional contribution of ATF5 to
rCTRP3-evoked analgesic effects, siRNA was used to specifically
knock down ATF5 expression in the spinal cord of SNI rats. RT-qPCR
analysis confirmed that ATF5 siRNA transfection induced significant
downregulation of ATF5 mRNA in PC12 cells in vitro without
additional stimulation [Fig.
12A, t (8)=9.359,
P<0.0001, η2=0.9163]. Consistently, intrathecal
injection of ATF5 siRNA also significantly inhibited the endogenous
transcription of spinal ATF5 in SNI rats compared with the negative
control siRNA group [Fig. 12B,
t (8)=6.954, P=0.0001,
η2=0.8581]. Behavioral assessments revealed that rCTRP3
treatment significantly restored PWT and shortened PWCD in SNI
rats, whereas combined administration of ATF5 siRNA fully abolished
these pain-relieving effects [Fig.
12B and C; Fig. 12B:
Interaction: F (20, 270)=4.874, P<0.0001, η2=0.2652;
Time factor: F (5, 270)=49.97, P<0.0001, η2 =0.4807;
Group factor: F (4, 270)=121.0, P<0.0001, η2=0.6419;
Fig. 12C: Interaction: F (20,
270)=7.286, P<0.0001, η2=0.3505; Time factor: F (5,
270)=91.13, P<0.0001, η2=0.6279; Group factor: F (4,
270)=196.1, P<0.0001, η2=0.7439]. These data confirm
that ATF5 activation is required for rCTRP3 to exert its
anti-nociceptive actions in neuropathic pain.
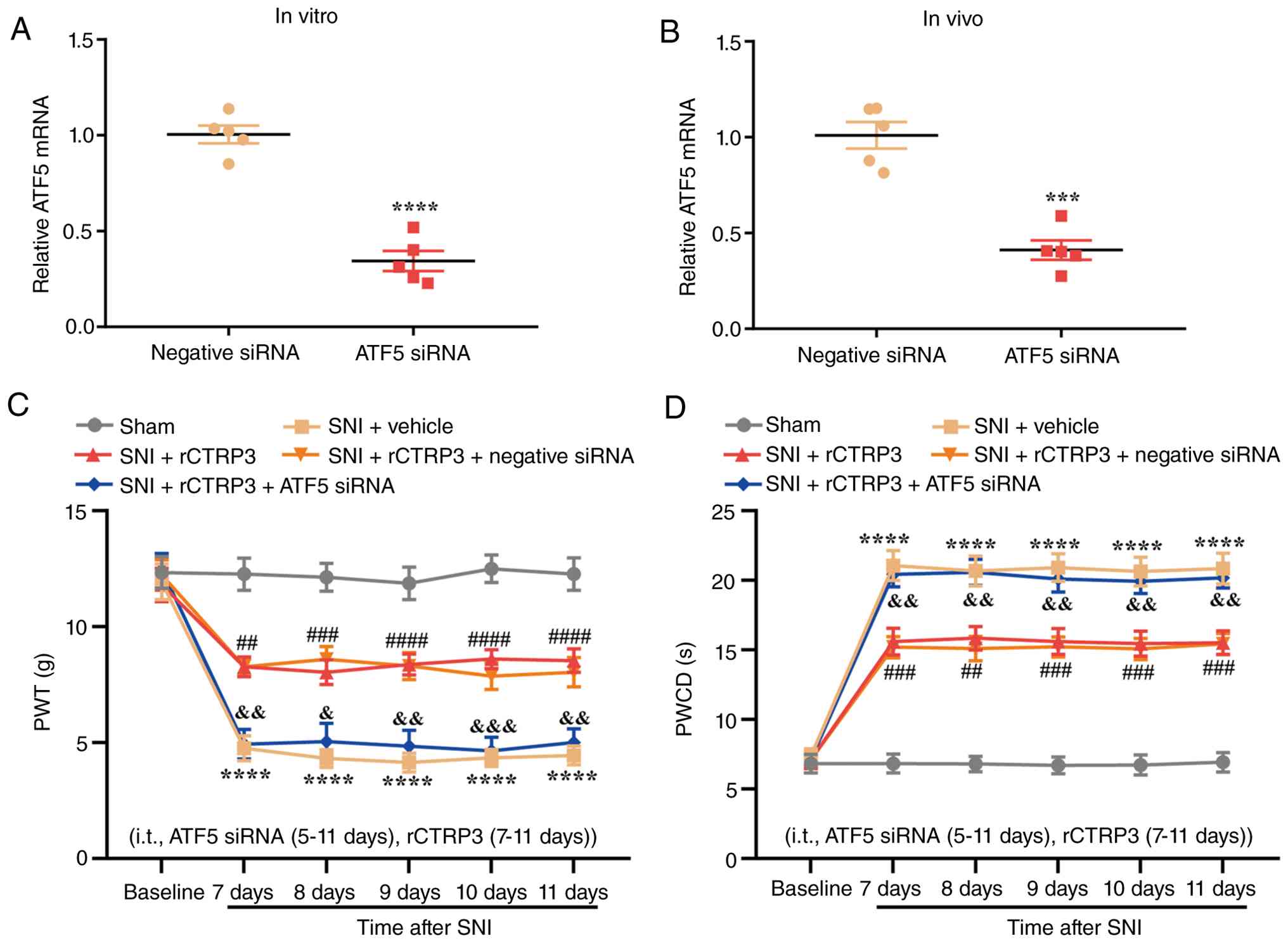 | Figure 12ATF5 siRNA abrogates rCTRP3-mediated
alleviation of pain hypersensitivity in SNI rats. (A) RT-qPCR
analysis confirmed that ATF5 siRNA transfection induced significant
downregulation of ATF5 mRNA in PC12 cells.
****P<0.0001 vs. negative siRNA group; n=5. (B)
RT-qPCR results confirmed that ATF5 siRNA effectively inhibited
ATF5 expression in the spinal cord. ***P<0.001 vs.
negative siRNA group; n=5 rats/group. (C and D) Behavioral
assessments demonstrated that rCTRP3 treatment elevated PWT and
downregulated PWCD in SNI rats; however, co-administration of ATF5
siRNA reversed this analgesic effect of rCTRP3.
****P<0.0001 vs. sham group; ##P<0.01,
###P<0.001 and ####P<0.0001 vs. SNI +
vehicle group; &P<0.05,
&&P<0.01 and
&&&P<0.001 vs. SNI + rCTRP3 group; n=10
rats/group. ATF5, activating transcription factor 5; siRNA, small
interfering RNA; rCTRP3, recombinant complement C1q tumor necrosis
factor-related protein; SNI, spared nerve injury; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; PWT, paw
withdrawal threshold; PWCD, paw withdrawal cold duration. |
The impact of ATF5 silencing on rCTRP3-mediated
regulation of UPRmt was next examined. IF staining
showed that ATF5 siRNA significantly suppressed the rCTRP3-induced
elevation of ATF5 in neurons of the spinal dorsal horn [Fig. 13A and B: F (4, 20)=11.67,
P<0.0001, η2=0.7001]. Correspondingly, RT-qPCR
analysis demonstrated that ATF5 knockdown abolished the
rCTRP3-mediated upregulation of ATF5 [F (4, 20)=14.71, P<0.0001,
η2=0.7464], ClpP [F (4, 20)=14.10, P<0.0001,
η2=0.7383], Hsp60 [F (4, 20)=8.704, P=0.0003,
η2=0.6351] and LonP1 [F (4, 20)=18.67, P<0.0001,
η2=0.7888] transcripts in the spinal cord, relative to
the SNI + rCTRP3 group (Fig.
13C-F). These results confirm that ATF5 is indispensable for
rCTRP3 to trigger the UPRmt in the spinal cord after
nerve injury.
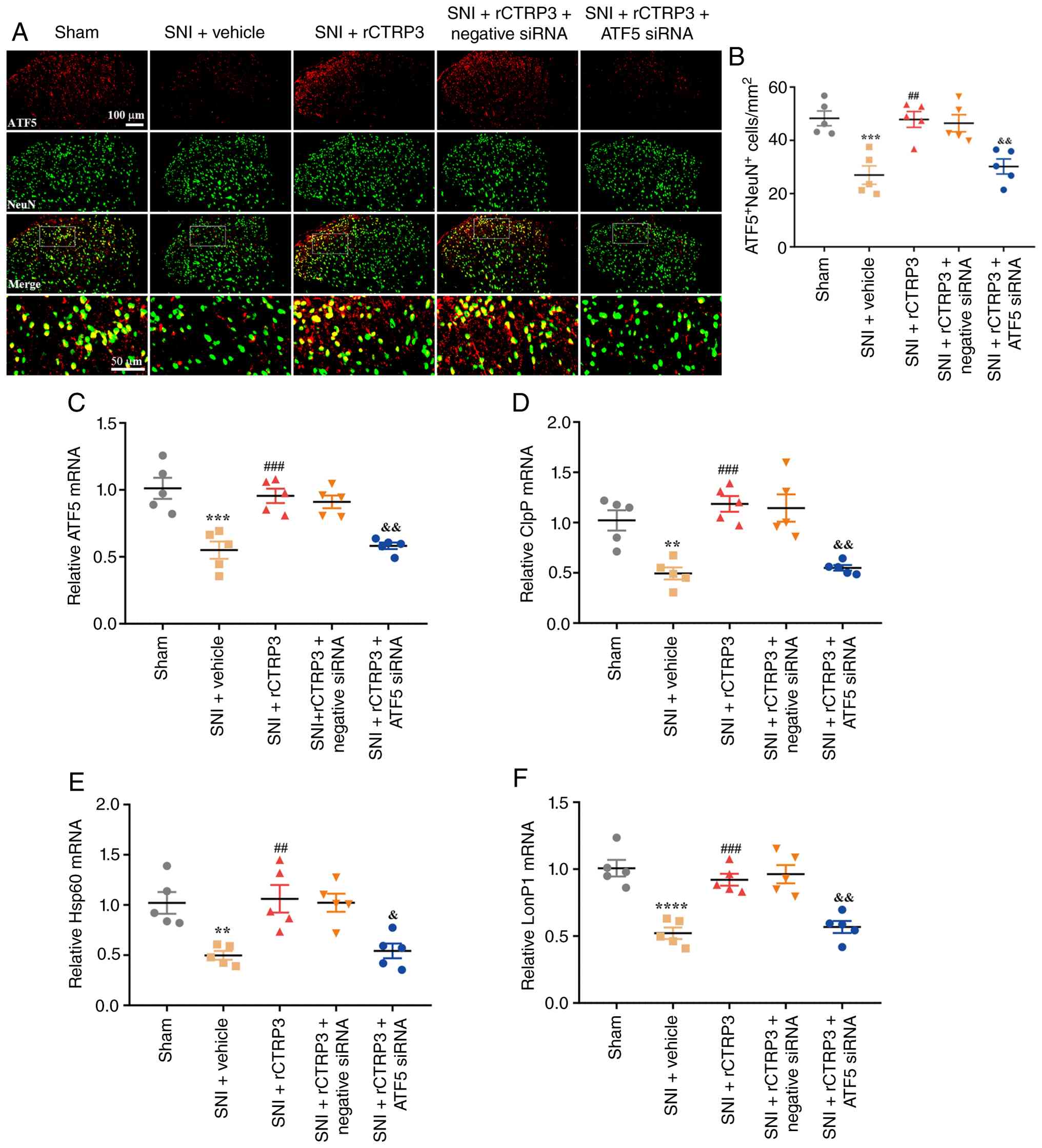 | Figure 13ATF5 siRNA abrogates rCTRP3-mediated
enhancement of UPRmt in SNI rats. (A and B) IF staining
revealed that ATF5 siRNA inhibited the rCTRP3-induced upregulation
of ATF5 expression in spinal dorsal horn neurons of SNI rats. (C-F)
RT-qPCR analysis demonstrated that ATF5 siRNA eliminated the
rCTRP3-mediated elevation in the expression levels of ATF5, ClpP,
Hsp60 and LonP1 in the spinal cord, relative to the SNI + rCTRP3
group. **P<0.01, ***P<0.001 and
****P<0.0001 vs. sham group; ##P<0.01
and ###P<0.001 vs. SNI + vehicle group;
&P<0.05 and &&P<0.01 vs.
SNI + rCTRP3 group; n=5 rats/group. ATF5, activating transcription
factor 5; siRNA, small interfering RNA; rCTRP3, recombinant
complement C1q tumor necrosis factor-related protein;
UPRmt, mitochondrial unfolded protein response; SNI,
spared nerve injury; IF, immunofluorescence; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; ClpP,
caseinolytic mitochondrial matrix peptidase proteolytic subunit;
Hsp60, heat shock protein 60; LonP1, Lon protease 1. |
To test whether ATF5 also modulates mitochondrial
biogenesis downstream of rCTRP3, key molecules involved in this
process were assessed. IF staining revealed that ATF5 siRNA
significantly inhibited the rCTRP3-mediated increase in PGC-1α
expression in spinal dorsal horn neurons [Fig. S5A and B: F (4, 20)=14.20,
P<0.0001, η2=0.7395]. In agreement, WB analysis
showed that ATF5 silencing significantly reversed the
rCTRP3-induced upregulation of PGC-1α [F (4, 20)=15.32, P<0.0001,
η2=0.7539], NRF1 [F (4, 20)=11.57, P<0.0001,
η2=0.6982] and TFAM [F (4, 20)=15.69, P<0.0001,
η2=0.7583] proteins in the spinal cord (Fig. S5C-E). Taken together, these
results demonstrate that ATF5 acts as a critical downstream
effector that supports rCTRP3-induced UPRmt and
cooperatively enhances PGC-1α-dependent mitochondrial biogenesis,
thereby contributing to the alleviation of neuropathic pain.
ATF5 siRNA abrogates rCTRP3-mediated
restoration of mitochondrial function and reduction of oxidative
stress in SNI rats
In parallel, ATF5 siRNA eliminated the
rCTRP3-induced restoration of MMP [F (4, 20)=12.34, P<0.0001,
η2=0.7116] and ATP production [F (4, 20)=16.38, P<0.0001,
η2=0.7661] (Fig.
S6A-C), demonstrating that ATF5-mediated UPRmt also
contributes to the improved mitochondrial bioenergetics induced by
rCTRP3. DHE staining analysis indicated that compared with the SNI
+ rCTRP3 group, administration of ATF5 siRNA reversed the
rCTRP3-induced decrease in the number of DHE-positive cells in the
spinal cord of SNI rats [Fig. 14A
and B: F (4, 20)=65.07, P<0.0001, η2=0.9286].
Moreover, biochemical assay results revealed that ATF5 siRNA
eliminated the rCTRP3-mediated downregulation of oxidative damage
markers, including MDA [Fig.
14C: F (4, 20)=17.76, P<0.0001, η2=0.7803] and
PCO [Fig. 14D: F (4, 20)=9.924,
P=0.0001, η2=0.6650] in the spinal cord of SNI rats.
Notably, while rCTRP3 increased the level of GSH [Fig. 14E: F (4, 20)=10.71, P<0.0001,
η2=0.6818] and enhanced the activity of antioxidant
enzymes such as GSH-PX [Fig.
14F: F (4, 20)=13.68, P<0.0001, η2=0.7323] and
SOD [Fig. 14G: F (4, 20)=18.68,
P<0.0001, η2=0.7888] in the spinal cord of SNI rats,
these beneficial changes were counteracted by ATF5 siRNA
intervention. These results indicate that CTRP3 mitigates spinal
oxidative stress in SNI rats through ATF5 activation.
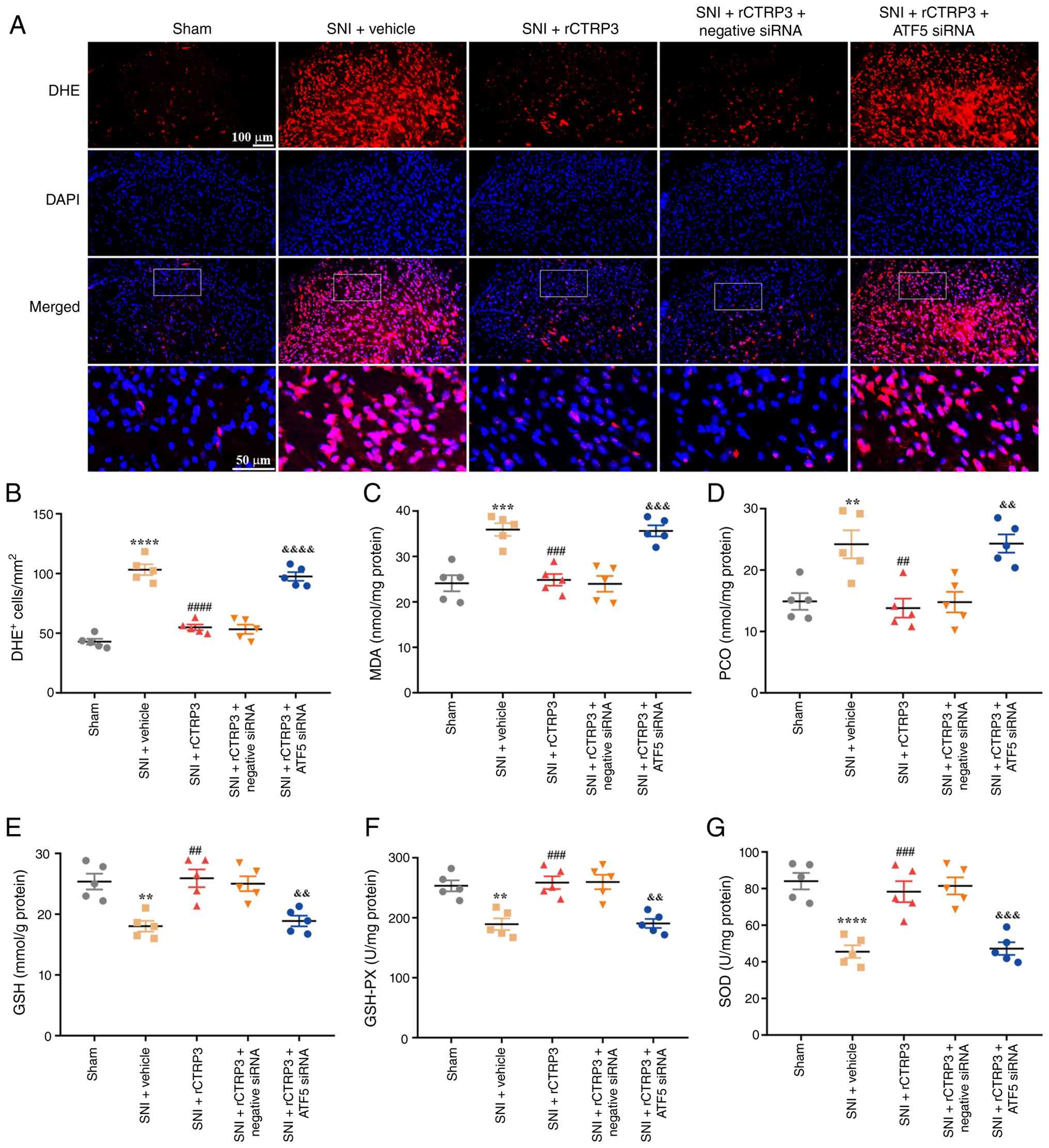 | Figure 14ATF5 siRNA abrogates rCTRP3-induced
reduction of spinal oxidative stress in SNI rats. (A and B) DHE
staining analysis indicated that compared with the SNI + rCTRP3
group, administration of ATF5 siRNA counteracted the rCTRP3-induced
decrease in the number of DHE-positive cells in the spinal cord of
SNI rats. (C and D) Biochemical assay data revealed that ATF5 siRNA
abolished the rCTRP3-mediated downregulation of MDA and PCO in the
spinal cord of SNI rats. (E-G) rCTRP3 treatment increased the level
of reduced GSH, and enhanced the activity of GSH-PX and SOD in the
spinal cord of SNI rats, but these favorable changes were reversed
by ATF5 siRNA intervention. **P<0.01,
***P<0.001 and ****P<0.0001 vs. sham
group; ##P<0.01, ###P<0.001 and
####P<0.0001 vs. SNI + vehicle group;
&&P<0.01,
&&&P<0.001 and
&&&&P<0.0001 vs. SNI + rCTRP3 group;
n=5 rats/group. ATF5, activating transcription factor 5; siRNA,
small interfering RNA; rCTRP3, recombinant complement C1q tumor
necrosis factor-related protein; SNI, spared nerve injury; DHE,
dihydroethidium; MDA, malondialdehyde; PCO, protein carbonyl; GSH,
glutathione; GSH-PX, glutathione peroxidase; SOD, superoxide
dismutase. |
Discussion
In the present study, the effects of CTRP3 on
neuropathic pain, mitochondrial biogenesis, and UPRmt,
as well as the underlying mechanisms, were investigated using a
male rat model of SNI. The results showed the following: i) SNI
significantly downregulated CTRP3 expression in spinal neurons; ii)
intrathecal delivery of rCTRP3 relieved mechanical allodynia and
cold hyperalgesia in rats with SNI; iii) rCTRP3 intervention
promoted PGC-1α-dependent mitochondrial biogenesis, ATF5-initiated
UPRmt, and reduced oxidative stress in the spinal cord;
iv) pharmacological suppression of SIRT1 using EX-527, and
siRNA-induced knockdown of PGC-1α or ATF-5, both reversed rCTRP3's
effects on pain hypersensitivity, mitochondrial biogenesis,
UPRmt and oxidative stress in the spinal cord. The
present research reveals that CTRP3 alleviates mechanical allodynia
and cold hyperalgesia in male SNI rats by activating spinal SIRT1,
which in turn enhances PGC-1α-mediated mitochondrial biogenesis and
ATF5-induced UPRmt (Fig.
15).
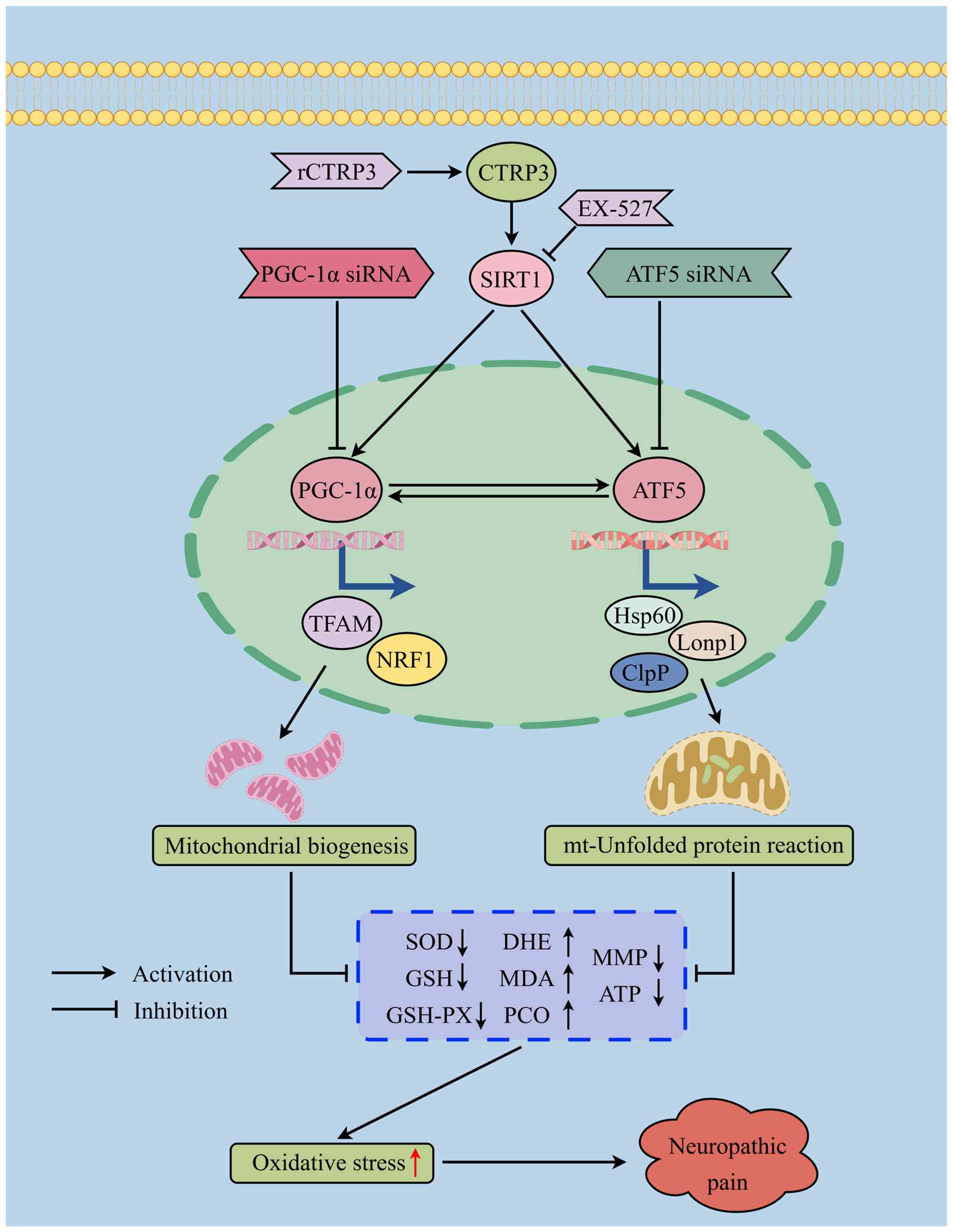 | Figure 15Schematic illustration of the role of
CTRP3 in neuropathic pain. CTRP3 alleviates pain hypersensitivity
and spinal oxidative stress in SNI rats by activating spinal SIRT1,
thereby enhancing PGC-1α/ATF5-induced mitochondrial biogenesis and
UPRmt. CTRP3, complement C1q tumor necrosis
factor-related protein 3; SNI, spared nerve injury; SIRT1, sirtuin
1; PGC-1α, peroxisome proliferator-activated receptor gamma
coactivator 1-alpha; ATF5, activating transcription factor 5;
UPRmt, mitochondrial unfolded protein response; MDA,
malondialdehyde; PCO, protein carbonyl; MMP, mitochondrial membrane
potential; GSH, glutathione; GSH-PX, glutathione peroxidase; SOD,
superoxide dismutase; DHE, dihydroethidium; ClpP, caseinolytic
mitochondrial matrix peptidase proteolytic subunit; Hsp60, heat
shock protein 60; LonP1, Lon protease 1; siRNA, small interfering
RNA. |
Existing studies have documented that CTRP3
participates in multiple biological processes, including
inflammatory responses (30),
glycolipid metabolism (29),
oxidative stress regulation (26) and apoptotic pathways (46). For instance, in inflammatory
bowel disease, CTRP3 has been shown to reduce the phosphorylation
of the NF-κB subunit p65 as well as the levels of proinflammatory
cytokines such as tumor necrosis factor-α and interleukin-6, with
this effect mediated through SIRT1 activation (30). In the field of cardiovascular
research, CTRP3 exerts protective effects against apoptotic and
oxidative stress-induced damage and alleviates myocardial
ischemia/reperfusion injury via the LAMP1/JIP2/JNK signaling
pathway (46). Additionally,
CTRP3 can directly suppress inflammatory reactions in psoriatic
keratinocytes by inhibiting the phosphorylation of signal
transducer and activator of transcription 3 (STAT3) through
LAMP1-dependent mechanisms (27). However, the specific role of
CTRP3 in neuropathic pain remains incompletely understood. The
present study found that SNI led to a significant downregulation of
CTRP3 expression in spinal neurons. Although the precise mechanism
by which peripheral nerve injury reduces spinal CTRP3 expression
remains to be fully clarified, multiple transcriptional regulators
may modulate CTRP3 expression under pathological conditions.
Peripheral nerve injury triggers a cascade of pro-inflammatory,
oxidative, ER stress, and hypoxic signals in the spinal cord, which
may activate a series of negative transcriptional regulators (FOXO4
and KLF10) (47,48), and inhibitory microRNAs
(miR-495-3p, miR-409-3p and miR-186-5p) (49-51) to reduce CTRP3 expression in the
spinal cord. Furthermore, repeated intrathecal administration of
rCTRP3 at doses of 30 and 90 μg significantly mitigated the
reduction in PWT and attenuated the elevation in PWCD observed in
male SNI rats. These results indicate that CTRP3 effectively
alleviates mechanical allodynia and cold hyperalgesia in
SNI-induced rats.
Mitochondrial biogenesis refers to the cellular
process of generating new mitochondria to fulfill energy
requirements and preserve organelle quality (6). This process is coordinated by both
nuclear and mitochondrial genes, with PGC-1α acting as a key
transcriptional co-activator that upregulates NRF-1/2 and TFAM,
thereby driving the expression of mtDNA and the assembly of
respiratory chain complexes (7).
Impaired mitochondrial biogenesis has been implicated in the
pathogenesis of various diseases (9,15,36,52,53). For example, it has been reported
that cardiomyocyte G protein-coupled receptor containing
leucine-rich repeats 6 (LGR6) alleviates ferroptosis in diabetic
cardiomyopathy by promoting mitochondrial biogenesis (52). Additionally, a study found that
PGC-1α-mediated mitochondrial biogenesis enhances the recovery and
survival of neuronal cells from degenerative damage (53). Our previous research also
demonstrated that SNI-induced neuropathic pain suppresses
mitochondrial biogenesis, while enhancing this process alleviates
pain hypersensitivity (15,16,36). However, the specific mechanisms
connecting mitochondrial biogenesis to neuropathic pain remain
incompletely clarified. Another study indicated that CTRP3
mitigates neurological deficits in cerebral ischemic stroke by
promoting PGC-1α-mediated mitochondrial biogenesis (32). Consistent with these findings,
the present study revealed that PGC-1α siRNA blocked the
rCTRP3-induced increase in PGC-1α expression in spinal dorsal horn
neurons of SNI rats. WB analysis showed that PGC-1α siRNA abolished
the rCTRP3-mediated upregulation of PGC-1α, NRF1 and TFAM in the
spinal cord. Moreover, while rCTRP3 treatment reversed the
reduction in mtDNA copy number in SNI rats, this effect was
inhibited by PGC-1α siRNA. Furthermore, behavioral assessments
demonstrated that rCTRP3 treatment ameliorated pain
hypersensitivity in SNI rats, whereas co-administration of PGC-1α
siRNA reversed this analgesic effect of rCTRP3. These results
indicate that CTRP3 alleviates mechanical allodynia and cold
hyperalgesia and promotes mitochondrial biogenesis in the spinal
cord of SNI rats through PGC-1α activation.
Upon exposure to pathological stimuli, a hallmark
of mitochondrial impairment is the perturbation of mitochondrial
protein homeostasis, accompanied by the accumulation of unfolded or
misfolded proteins within mitochondria. This accumulation not only
elevates ROS levels but also triggers oxidative stress-induced
injury, which in turn further compromises protein integrity and
folding processes. Notably, activation of the UPRmt acts
as a protective mechanism for cell survival, facilitating proper
protein folding or promoting the degradation of misfolded proteins.
In mammalian cells, UPRmt can be activated by multiple
factors through diverse regulatory mechanisms, including via ATF5-a
homolog of the ATFS-1 gene. ATF5 activation promotes the
transcription of proteases (for example, ClpP and Lonp1) and heat
shock proteins (for example, HSP60 and HSP10), which collectively
aid in the correct folding or degradation of unfolded proteins,
thereby restoring mitochondrial protein homeostasis. Impaired
UPRmt has been linked to the pathogenesis of various
diseases. For example, a previous study demonstrated that enhancing
UPRmt with NR improved mitochondrial function, reduced
chondrocyte death, and alleviated OA pain-effects that were
markedly diminished in chondrocyte-specific Atf5 knockout mice
(24). Additionally,
ATF5-mediated UPRmt has been shown to alleviate
intervertebral disc degeneration by promoting mitophagy. However,
strategies for safely and effectively regulating UPRmt
in pain hypersensitivity remain unclear, largely due to poorly
understood underlying mechanisms. Another study indicated that
CTRP3 mitigates mitochondrial dysfunction and oxidative stress
injury in pathological cardiac hypertrophy by activating
ATF5-induced UPRmt (31). Consistent with this, the findings
of the present study showed that ATF5 silencing via siRNA blocked
the rCTRP3-induced upregulation of ATF5 in spinal dorsal horn
neurons of SNI rats. Furthermore, RT-qPCR analysis revealed that
ATF5 siRNA abolished the rCTRP3-mediated increases in spinal
expression of proteases (ClpP and Lonp1) and heat shock proteins
(Hsp60). Furthermore, the current data also demonstrated that
rCTRP3 treatment dampened pain hypersensitivity in SNI rats,
whereas co-administration of ATF5 siRNA reversed this analgesic
effect. These data suggest that CTRP3 alleviates pain
hypersensitivity and enhances UPRmt in the spinal cord
of SNI rats through ATF5 activation.
Next, the signaling pathways underlying CTRP3's
analgesic effects and its regulatory role in mitochondrial function
were investigated. SIRT1, a NAD+-dependent protein
deacetylase, has been widely studied in the field of neuropathic
pain (54). It counteracts
oxidative stress damage and mitochondrial dysfunction through
multiple molecular or signaling pathways, while also exerting
protective effects in neuropathic pain (55,56). Recent studies have shown that
aberrant SIRT1 expression reduces PGC-1α-mediated mitochondrial
biogenesis (34), and suppresses
ATF5-induced UPRmt (35). Furthermore, it has been reported
that CTRP3 activates PGC-1α-induced mitochondrial biogenesis
(32) and ATF5-mediated
UPRmt (31), thereby
protecting mitochondria from proteotoxic damage and alleviating
oxidative stress. A recent study demonstrated that CTRP3 mitigates
mitochondrial dysfunction and enhances ATF5-induced
UPRmt in pathological cardiac hypertrophy by activating
SIRT1 (31). Another study
indicated that CTRP3 alleviated neurological deficits and promoted
PGC-1α-mediated mitochondrial biogenesis in cerebral ischemic
stroke by upregulating SIRT1 (32). Consistent with these findings,
the present study showed that rCTRP3 treatment alleviated pain
hypersensitivity in SNI rats, whereas co-administration of the
SIRT1 antagonist EX-527 reversed this analgesic effect. IF staining
revealed that EX-527 blocked the rCTRP3-induced upregulation of
PGC-1α in spinal dorsal horn neurons of SNI rats. Moreover, while
rCTRP3 treatment reversed the reduction in mtDNA copy number in SNI
rats, this effect was abolished by EX-527. The present study also
found that EX-527 inhibited the rCTRP3-induced increase in ATF5
expression in spinal dorsal horn neurons of SNI rats. Furthermore,
RT-qPCR analysis demonstrated that EX-527 eliminated the
rCTRP3-mediated upregulation of ClpP, Hsp60 and LonP1 in the spinal
cord of rats with SNI. These results confirm that CTRP3 alleviates
mechanical allodynia and cold hyperalgesia and promotes
PGC-1α-mediated mitochondrial biogenesis and ATF5-induced
UPRmt in the spinal cord of SNI rats through SIRT1
activation.
Furthermore, previous studies have demonstrated
that PGC-1α promotes ATF5-dependent UPRmt (57), whereas ATF5 enhances the
expression of PPARγ (58), a key
positive transcriptional regulator of PGC-1α. In line with these
observations, the present study revealed that PGC-1α silencing not
only blocked rCTRP3-induced mitochondrial biogenesis but also
significantly suppressed ATF5 and its downstream
UPRmt-related markers. Similarly, ATF5 knockdown not
only abolished rCTRP3-mediated UPRmt activation but also
significantly reduced PGC-1α and its downstream targets involved in
mitochondrial biogenesis. Collectively, these findings confirm that
PGC-1α and ATF5 mutually regulate each other downstream of SIRT1
during the analgesic action of CTRP3. This reciprocal interaction
accounts for the similar reversal effects on neuropathic pain and
oxidative stress observed following inhibition of SIRT1, PGC-1α, or
ATF5. Our proposed signaling axis (CTRP3→SIRT1→PGC-1α/ATF5) is
further supported, wherein PGC-1α and ATF5 function as
interdependent downstream effectors rather than discrete parallel
pathways.
Notably, it was previously reported by the authors
that CTRP9 alleviates SNI-induced neuropathic pain in mice by
modulating AdipoR1/AMPK/NF-κB signaling to suppress
neuroinflammation and promote microglial M2 polarization (43). The present study extends these
findings by demonstrating that CTRP3 exerts potent analgesic
effects in rats via a distinct mechanism centered on
SIRT1/PGC-1α/ATF5-mediated mitochondrial homeostasis,
UPRmt and oxidative stress inhibition. Several critical
inter-species and molecular differences deserve emphasis. First,
CTRP3 was downregulated in the spinal cord after SNI in rats,
whereas CTRP9 was upregulated in mice, suggesting divergent
expression patterns of CTRP family members in response to
peripheral nerve injury across species. Second, the previous mouse
study focused on microglial polarization and neuroinflammation,
while the current rat study highlights mitochondrial dysfunction,
mitochondrial biogenesis, UPRmt and oxidative stress as
core pathological events. Third, the downstream signaling differs
substantially: CTRP9 acted through AdipoR1/AMPK/NF-κB in mice,
whereas CTRP3 signals via SIRT1/PGC-1α/ATF5 in rats. These
cross-species comparisons indicate that CTRP3 and CTRP9 function as
complementary analgesic regulators with overlapping protective
effects but distinct molecular cascades and expression patterns.
Such species-specific regulatory features are critical for
preclinical translation and suggest that CTRP3-targeted therapy may
be more suitable for mitochondrial and oxidative stress-related
pathological pain conditions in higher mammals.
Although the present study provides important
insights into the role of CTRP3 in neuropathic pain, mitochondrial
biogenesis and UPRmt, several aspects warrant further
exploration in future research. First, the study utilized rCTRP3,
but its specificity and potential off-target effects in the context
of neuropathic pain and mitochondrial function were not fully
validated. Additional studies employing genetic knockdown or
knockout approaches could help more definitively confirm the role
of CTRP3. Second, the long-term effects of rCTRP3 treatment,
including potential tolerance development or secondary changes over
time, were not investigated. A longer follow-up period would
provide a more comprehensive understanding of CTRP3's therapeutic
potential. Additionally, only male rats were used in the present
study, further studies are needed to explore potential sex
differences in CTRP3 expression and function, as sex is a known
modifier of neuropathic pain susceptibility and treatment
response.
In summary, the present study demonstrates that
CTRP3 mitigates pain hypersensitivity and spinal oxidative stress
in male SNI rats by activating spinal SIRT1, thereby enhancing
PGC-1α/ATF5-mediated mitochondrial biogenesis and UPRmt.
CTRP3 may thus represent a novel therapeutic target for the
management of neuropathic pain.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
TL designed the study. LZ provided experimental
support. WM analyzed interpreted the data. TL and WM wrote the
manuscript. TL and WM confirm the authenticity of all the raw data.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The present study was approved (approval no.
TJH-202106615) by the Experimental Animal Care and Use Committee of
Tongji Hospital, Tongji Medical College, Huazhong University of
Science and Technology (Wuhan, China). All procedures were
conducted in compliance with the Animal Research Reporting of in
Vivo Experiments (ARRIVE) guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
The authors would like to thank the Laboratory
Animal Center of Tongji Hospital, Tongji Medical College, Huazhong
University of Science and Technology for their assistance in
raising rats.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 82501492, 82471289 and
82271291).
References
|
1
|
Borbjerg MK, Wegeberg AM, Nikontovic A,
Mørch CD, Arendt-Nielsen L, Ejskjaer N, Brock C, Vestergaard P and
Røikjer J: Understanding the impact of diabetic peripheral
neuropathy and neuropathic pain on quality of life and mental
health in 6,960 people with diabetes. Diabetes Care. 48:588–595.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Soliman N, Moisset X, Ferraro MC, de
Andrade DC, Baron R, Belton J, Bennett DLH, Calvo M, Dougherty P,
Gilron I, et al: Pharmacotherapy and non-invasive neuromodulation
for neuropathic pain: A systematic review and meta-analysis. Lancet
Neurol. 24:413–428. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Su Y, Verkhratsky A and Yi C: Targeting
connexins: Possible game changer in managing neuropathic pain?
Trends Mol Med. 30:642–659. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ward J, Grinstead A, Kemp A, Kersten P,
Schmid AB and Ridehalgh C: A meta-analysis exploring the efficacy
of neuropathic pain medication for low back pain or spine-related
leg pain: Is efficacy dependent on the presence of neuropathic
pain? Drugs. 84:1603–1636. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Malcangio M and Sideris-Lampretsas G: How
microglia contribute to the induction and maintenance of
neuropathic pain. Nat Rev Neurosci. 26:263–275. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu L, Li Y, Chen G and Chen Q: Crosstalk
between mitochondrial biogenesis and mitophagy to maintain
mitochondrial homeostasis. J Biomed Sci. 30:862023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang L, Xin C, Wang S, Zhuo S, Zhu J, Li
Z, Liu Y, Yang L and Chen Y: Lactate transported by MCT1 plays an
active role in promoting mitochondrial biogenesis and enhancing TCA
flux in skeletal muscle. Sci Adv. 10:eadn45082024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jamwal S, Blackburn JK and Elsworth JD:
PPARγ/PGC1α signaling as a potential therapeutic target for
mitochondrial biogenesis in neurodegenerative disorders. Pharmacol
Ther. 219:1077052021. View Article : Google Scholar
|
|
9
|
Yang D, Sun Y, Wen P, Chen Y, Cao J, Sun X
and Dong Y: Chronic stress-induced serotonin impairs intestinal
epithelial cell mitochondrial biogenesis via the AMPK-PGC-1α axis.
Int J Biol Sci. 20:4476–4495. 2024. View Article : Google Scholar
|
|
10
|
Dumesic PA, Wilensky SE, Bose S, Van
Vranken JG, Gygi SP and Spiegelman BM: RBM43 controls PGC1α
translation and a PGC1α-STING signaling axis. Cell Metab.
37:742–757.e8. 2025. View Article : Google Scholar
|
|
11
|
Lin H, Shao X, Gu H, Yu X, He L, Zhou J,
Zhong Z, Guo S, Li D, Chen F, et al: Akkermansia muciniphila
ameliorates doxorubicin-induced cardiotoxicity by regulating
PPARα-dependent mitochondrial biogenesis. NPJ Biofilms Microbiomes.
11:862025. View Article : Google Scholar
|
|
12
|
Lan X, Wang Q, Liu Y, You Q, Wei W, Zhu C,
Hai D, Cai Z, Yu J, Zhang J and Liu N: Isoliquiritigenin alleviates
cerebral ischemia-reperfusion injury by reducing oxidative stress
and ameliorating mitochondrial dysfunction via activating the Nrf2
pathway. Redox Biol. 77:1034062024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang W, Zhao F, Ma X, Perry G and Zhu X:
Mitochondria dysfunction in the pathogenesis of Alzheimer's
disease: Recent advances. Mol Neurodegener. 15:302020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng Q, Liu H, Zhang H, Han Y, Yuan J,
Wang T, Gao Y and Li Z: Ameliorating mitochondrial dysfunction of
neurons by biomimetic targeting nanoparticles mediated
mitochondrial biogenesis to boost the therapy of Parkinson's
disease. Adv Sci (Weinh). 10:e23007582023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang L, Tan X, Song F, Li D, Wu J, Gao S,
Sun J, Liu D, Zhou Y and Mei W: Activation of G-protein-coupled
receptor 39 reduces neuropathic pain in a rat model. Neural Regen
Res. 19:687–696. 2024. View Article : Google Scholar
|
|
16
|
Zhang LQ, Zhou YQ, Li JY, Sun J, Zhang S,
Wu JY, Gao SJ, Tian XB and Mei W: 5-HT1F receptor
agonist ameliorates mechanical allodynia in neuropathic pain via
induction of mitochondrial biogenesis and suppression of
neuroinflammation. Front Pharmacol. 13:8345702022. View Article : Google Scholar
|
|
17
|
Zhou Z, Fan Y, Zong R and Tan K: The
mitochondrial unfolded protein response: A multitasking giant in
the fight against human diseases. Ageing Res Rev. 81:1017022022.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shpilka T and Haynes CM: The mitochondrial
UPR: Mechanisms, physiological functions and implications in
ageing. Nat Rev Mol Cell Biol. 19:109–120. 2018. View Article : Google Scholar
|
|
19
|
Sutandy FXR, Gößner I, Tascher G and Münch
C: A cytosolic surveillance mechanism activates the mitochondrial
UPR. Nature. 618:849–854. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zu X, Chen S, Li Z, Hao L, Fu W, Zhang H,
Yin Z, Wang Y and Wang J: SPI1 activates mitochondrial unfolded
response signaling to inhibit chondrocyte senescence and relieves
osteoarthritis. Bone Res. 13:472025. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xiong X, Hou J, Zheng Y, Jiang T, Zhao X,
Cai J, Huang J, He H, Xu J, Qian S, et al: NAD+-boosting
agent nicotinamide mononucleotide potently improves mitochondria
stress response in Alzheimer's disease via ATF4-dependent
mitochondrial UPR. Cell Death Dis. 15:7442024. View Article : Google Scholar
|
|
22
|
An H, Zhou B, Hayakawa K, Durán Laforet V,
Park JH, Nakamura Y, Mandeville ET, Liu N, Guo S, Yu Z, et al:
ATF5-mediated mitochondrial unfolded protein response
(UPRmt) protects neurons against oxygen-glucose
deprivation and cerebral ischemia. Stroke. 55:1904–1913. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang Y, Lu D, Wang M, Liu G, Feng Y, Ren
Y, Sun X, Chen Z and Wang Z: Endoplasmic reticulum stress and the
unfolded protein response: Emerging regulators in progression of
traumatic brain injury. Cell Death Dis. 15:1562024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou Z, Lu J, Yang M, Cai J, Fu Q, Ma J
and Zhu L: The mitochondrial unfolded protein response
(UPRmt) protects against osteoarthritis. Exp Mol Med.
54:1979–1990. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu WN, Zheng HL, Yang RZ, Sun YF, Peng BR,
Liu C, Song J, Jiang SD and Zhu LX: The mitochondrial UPR induced
by ATF5 attenuates intervertebral disc degeneration via cooperating
with mitophagy. Cell Biol Toxicol. 40:162024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Q, Piao J, Li Y, Tu H, Lv D, Hu L,
Zhang R and Zhong Z: Bone marrow-mesenchymal stem cells alleviate
microglial Pyroptosis after intracerebral hemorrhage in rat by
secreting C1q/tumor necrosis factor-related protein 3. Exp Neurol.
364:1143872023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xue K, Shao S, Fang H, Ma L, Li C, Lu Z
and Wang G: Adipocyte-derived CTRP3 exhibits anti-inflammatory
effects via LAMP1-STAT3 axis in psoriasis. J Invest Dermatol.
142:1349–1359.e8. 2022. View Article : Google Scholar
|
|
28
|
Liu N, Gong Z, Li Y, Xu Y, Guo Y, Chen W,
Sun X, Yin X and Liu W: CTRP3 inhibits myocardial fibrosis through
the P2X7R-NLRP3 inflammasome pathway in SHR rats. J Hypertens.
42:315–328. 2024. View Article : Google Scholar
|
|
29
|
Yan Z, Cao X, Wang C, Liu S, Li Y, Lu G,
Yan W, Guo R, Zhao D, Cao J and Xu Y: C1q/tumor necrosis
factor-related protein-3 improves microvascular endothelial
function in diabetes through the AMPK/eNOS/NO• signaling pathway.
Biochem Pharmacol. 195:1147452022. View Article : Google Scholar
|
|
30
|
Yu H, Zhang Z, Li G, Feng Y, Xian L,
Bakhsh F, Xu D, Xu C, Vong T, Wu B, et al: Adipokine C1q/tumor
necrosis factor-related protein 3 (CTRP3) attenuates intestinal
inflammation via sirtuin 1/NF-κB signaling. Cell Mol Gastroenterol
Hepatol. 15:1000–1015. 2023. View Article : Google Scholar
|
|
31
|
Shi L, Tan Y, Zheng W, Cao G, Zhou H, Li
P, Cui J, Song Y, Feng L, Li H, et al: CTRP3 alleviates
mitochondrial dysfunction and oxidative stress injury in
pathological cardiac hypertrophy by activating UPRmt via the
SIRT1/ATF5 axis. Cell Death Discov. 10:532024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao J, Qian T and Wang W: CTRP3 activates
the AMPK/SIRT1-PGC-1α pathway to protect mitochondrial biogenesis
and functions in cerebral ischemic stroke. Neurochem Res.
45:3045–3058. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luo Y, Ali T, Hu Y, Gong Q, Zheng C, Li L,
Li S and Hao L: Erythropoietin (EPO) alleviates chronic
stress-induced depression by modulating SIRT1-mediated
mitochondrial function. J Neuroimmune Pharmacol. 20:732025.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu Y, Yang H, Luo N, Fu Y, Qiu F, Pan Z,
Li X, Jian W, Yang X, Xue Q, et al: An Fgr kinase inhibitor
attenuates sepsis-associated encephalopathy by ameliorating
mitochondrial dysfunction, oxidative stress, and neuroinflammation
via the SIRT1/PGC-1α signaling pathway. J Transl Med. 21:4862023.
View Article : Google Scholar
|
|
35
|
Li H, Chen D, Zhang X, Chen M, Zhi Y, Cui
W, Li S, Xu F, Tan Y, Zhou H, et al: Screening of an FDA-approved
compound library identifies apigenin for the treatment of
myocardial injury. Int J Biol Sci. 19:5233–5244. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
He Q, Zhou Y, Wu L, Huang L, Yuan Y,
Flores JJ, Luo X, Tao Y, Chen X, Kanamaru H, et al: Inhibition of
acid-sensing receptor GPR4 attenuates neuronal ferroptosis via
RhoA/YAP signaling in a rat model of subarachnoid hemorrhage. Free
Radic Biol Med. 225:333–345. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu Z, Xie W, Feng Y, Wang Y, Li X, Liu J,
Xiong Y, He Y, Chen L, Liu G and Wu Q: Positive interaction between
GPER and β-alanine in the dorsal root ganglion uncovers potential
mechanisms: Mediating continuous neuronal sensitization and
neuroinflammation responses in neuropathic pain. J
Neuroinflammation. 19:1642022. View Article : Google Scholar
|
|
38
|
Zhang LQ, Gao SJ, Sun J, Li DY, Wu JY,
Song FH, Liu DQ, Zhou YQ and Mei W: DKK3 ameliorates neuropathic
pain via inhibiting ASK-1/JNK/p-38-mediated microglia polarization
and neuroinflammation. J Neuroinflammation. 19:1292022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao Y, Zhang Y, Lei T, Zhang S, Nan K,
Zhang X and Fan LH: ZEB1 maintains mitochondrial fission and
macrophage efferocytosis by restraining MFN2, thereby limiting
inflammation and improving tendon-bone healing. Int J Mol Med.
57:1342026. View Article : Google Scholar
|
|
40
|
Li L, Bai L, Yang K, Zhang J, Gao Y, Jiang
M, Yang Y, Zhang X, Wang L, Wang X, et al: KDM6B epigenetically
regulated-interleukin-6 expression in the dorsal root ganglia and
spinal dorsal horn contributes to the development and maintenance
of neuropathic pain following peripheral nerve injury in male rats.
Brain Behav Immun. 98:265–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fan CY, McAllister BB, Stokes-Heck S,
Harding EK, Pereira de Vasconcelos A, Mah LK, Lima LV, van den
Hoogen NJ, Rosen SF, Ham B, et al: Divergent sex-specific
pannexin-1 mechanisms in microglia and T cells underlie neuropathic
pain. Neuron. 113:896–911.e9. 2025. View Article : Google Scholar
|
|
42
|
Zhang L, Dai X, Li D, Wu J, Gao S, Song F,
Liu L, Zhou Y, Liu D and Mei W: MFG-E8 ameliorates nerve
injury-induced neuropathic pain by regulating microglial
polarization and neuroinflammation via integrin β3/SOCS3/STAT3
pathway in mice. J Neuroimmune Pharmacol. 19:492024. View Article : Google Scholar
|
|
43
|
Liu T, Zhang L and Mei W: CTRP9 attenuates
peripheral nerve injury-induced mechanical allodynia and thermal
hyperalgesia through regulating spinal microglial polarization and
neuroinflammation mediated by AdipoR1 in male mice. Cell Biol
Toxicol. 40:912024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yingze Y, Zhihong J, Tong J, Yina L, Zhi
Z, Xu Z, Xiaoxing X and Lijuan G: NOX2-mediated reactive oxygen
species are double-edged swords in focal cerebral ischemia in mice.
J Neuroinflammation. 19:1842022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang H, Zhang N, Yang X, Zhang J, Ge X,
Wang L, Wang S and Wen Y: DL-3-n-butylphthalide protects
mitochondria against ischemia/hypoxia damage via suppressing
GCN5L1-mediated Drp1 acetylation in neurons and mouse brains. CNS
Neurosci Ther. 31:e706822025. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Song Y, Zhang Y, Wan Z, Pan J, Gao F, Li
F, Zhou J and Chen J: CTRP3 alleviates myocardial
ischemia/reperfusion injury in mice through activating
LAMP1/JIP2/JNK pathway. Int Immunopharmacol. 107:1086812022.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zeng X, Peng Y, Wang Y and Kang K:
C1q/tumor necrosis factor-related protein-3 (CTRP3) activated by
forkhead box O4 (FOXO4) down-regulation protects retinal pericytes
against high glucose-induced oxidative damage through nuclear
factor erythroid 2-related factor 2 (Nrf2)/Nuclear factor-kappaB
(NF-κB) signaling. Bioengineered. 13:6080–6091. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zeng Y, Xu Y, Pan Y and Guo H: KLF10
knockdown negatively regulates CTRP3 to improve OGD/R-induced brain
microvascular endothelial cell injury and barrier dysfunction
through Nrf2/HO-1 signaling pathway. Tissue Cell. 82:1021062023.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen Y, Liu W, Chen M, Sun Q, Chen H and
Li Y: Up-regulating lncRNA OIP5-AS1 protects neuron injury against
cerebral hypoxia-ischemia induced inflammation and oxidative stress
in microglia/macrophage through activating CTRP3 via sponging
miR-186-5p. Int Immunopharmacol. 92:1073392021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wu T, Tang C, Fan J and Tao J:
Administration of rTMS alleviates stroke-induced cognitive deficits
by modulating miR-409-3p/CTRP3/AMPK/Sirt1 axis. J Mol Neurosci.
72:507–515. 2022. View Article : Google Scholar
|
|
51
|
Zhao G, Zhang L, Qian D, Sun Y and Liu W:
miR-495-3p inhibits the cell proliferation, invasion and migration
of osteosarcoma by targeting C1q/TNF-related protein 3. Onco
Targets Ther. 12:6133–6143. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao M, Shen Z, Zheng Z, Xu Y, Zhang J,
Liu J, Peng S, Wan J, Qin JJ and Wang M: Cardiomyocyte LGR6
alleviates ferroptosis in diabetic cardiomyopathy via regulating
mitochondrial biogenesis. Metabolism. 159:1559792024. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
You W, Knoops K, Berendschot TTJM,
Benedikter BJ, Webers CAB, Reutelingsperger CPM and Gorgels TGMF:
PGC-1a mediated mitochondrial biogenesis promotes recovery and
survival of neuronal cells from cellular degeneration. Cell Death
Discov. 10:1802024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Song FH, Liu DQ, Zhou YQ and Mei W: SIRT1:
A promising therapeutic target for chronic pain. CNS Neurosci Ther.
28:818–828. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xu JW, Xu X, Ling Y, Wang YC, Huang YJ,
Yang JZ, Wang JY and Shen X: Vincamine as an agonist of
G-protein-coupled receptor 40 effectively ameliorates diabetic
peripheral neuropathy in mice. Acta Pharmacol Sin. 44:2388–2403.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Pal A, Sharan L, Das A, Paul S, Babu SS,
Das S, Banerjee S and Kumar A: Vitexin mitigates AIM2
inflammasome-mediated mitochondrial dysfunction and
neuroinflammation in chronic constriction injury induced neuropathy
model. Int Immunopharmacol. 158:1148772025. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang B, Tan Y, Zhang Z, Feng P, Ding W,
Wang Q, Liang H, Duan W, Wang X, Yu S, et al: Novel PGC-1α/ATF5
axis partly activates UPRmt and mediates
cardioprotective role of tetrahydrocurcumin in pathological cardiac
hypertrophy. Oxid Med Cell Longev. 2020:91870652020.
|
|
58
|
Zhao Y, Zhang YD, Zhang YY, Qian SW, Zhang
ZC, Li SF, Guo L, Liu Y, Wen B, Lei QY, et al: p300-dependent
acetylation of activating transcription factor 5 enhances C/EBPβ
transactivation of C/EBPα during 3T3-L1 differentiation. Mol Cell
Biol. 34:315–324. 2014. View Article : Google Scholar :
|