Patients with cancer face a substantially elevated
risk of venous thromboembolism (VTE), with incidence rates 4-7-fold
higher than those observed in the normal population (1,2).
Furthermore, the coexistence of VTE further worsens the clinical
outcomes of patients with cancer (3). While thrombotic complications have
historically been associated mainly with solid tumors,
hematological malignancies have typically been characterized by
bleeding tendencies and overt disseminated intravascular
coagulation (DIC). However, contemporary data indicate that the
incidence of VTE in patients with hematological malignancies is
comparable to that observed in patients with solid tumors at high
risk of thrombosis (4). The high
incidence of thrombosis is closely related to the hypercoagulable
state of the tumor itself, treatment-related factors (such as
chemotherapy and central venous catheterization), and the patient's
own factors (such as advanced age and long-term bed rest) (5). Additionally, a common treatment
course for patients with hematological malignancies consists of
aggressive chemotherapy followed by hematopoietic stem cell (HSC)
transplantation. This regimen frequently causes long-lasting and
severe pancytopenia (4). Given
these challenges, there is a paucity of accumulated clinical
expertise concerning the prevention and treatment of thrombotic
events in individuals with hematological malignancies.
Through the extrusion of neutrophil extracellular
traps (NETs), which are thread-like structures composed of
chromatin, neutrophils are able to eliminate invading microbes
(6). NET formation leads to a
unique cell death process called NETosis (7). Previous studies have shown that
intravascular NETs promote coagulation and thrombosis by activating
the clotting cascade and interacting with platelets, playing a key
role in immunothrombosis (8-11). Excessive immunothrombosis
triggers thromboinflammation, driven by endothelial dysfunction,
dysregulated coagulation, complement activation, platelet
activation and leukocyte recruitment (12). In sterile inflammation, such as
in hematological malignancies, NET-mediated thrombosis can cause
tissue ischemia. For example, the Jak2V617F mutation in
myeloproliferative neoplasms (MPNs) was shown to increase
thrombosis by enhancing NET formation (13). Disrupting NETs [for example with
deoxyribonuclease (DNase)] can reduce clotting and thrombotic
events (14). Understanding the
mechanisms of thromboinflammation is crucial, as current
anticoagulants only partially prevent thrombosis while increasing
bleeding risk, particularly in thrombocytopenic hematological
malignancies (15). The present
review explores the role of NETs in thrombosis and potential
therapeutic strategies.
Beyond their well-established role in entrapping and
neutralizing a wide spectrum of pathogens (including bacteria,
fungi, viruses and parasites), NETs also help confine microbial
dissemination to local sites (6). However, excessive or dysregulated
NET formation can drive immune-related diseases (16). Structurally, NETs consist of a
DNA scaffold associated with antimicrobial proteins such as
myeloperoxidase (MPO), neutrophil elastase (NE), defensins,
cathepsin G, lactoferrin, matrix metalloproteinase-9, pentraxins,
peptidoglycan-recognizing proteins and LL-37 (a 37-amino acid
peptide) (6,17).
NETs are activated through diverse pathways,
including protein kinase C (PKC) activation [such as phorbol
12-myristate 13-acetate (PMA)-induced reactive oxygen species (ROS)
production], microbial stimuli (bacterial/viral pathogens) and
immunological/tumor-related factors (including immune complexes,
chemokines, complement components and damage-associated molecular
patterns). Notably, stimulus-specific variations in NET protein
composition may lead to functional heterogeneity, warranting
further investigation (9).
Several reported mechanisms contribute to the
induction of NET formation in individuals with hematological
malignancies. In a previous study on CD5+ B-cell chronic
lymphoblastic leukemia, Sangaletti et al (18) observed that splenic neutrophils
from lpr/lpr/Sparc−/− mice enhanced the generation of
B-cell activating factors, resulting in a more notable tendency
toward NET formation. Previous research has shown that, in patients
with MPNs, platelets rapidly adhere to and stimulate neutrophils,
thereby driving NET production (19). Increased interleukin (IL)-8 in
patients with diffuse large B-cell lymphoma (DLBCL) was
demonstrated to interact with C-X-C motif chemokine receptor 2
(CXCR2) expressed on neutrophils, leading to NET induction through
the coordinated action of the Src, p38 and ERK pathways. In a DLBCL
mouse model, the use of corresponding inhibitors was shown to
eliminate NETosis triggered by C-X-C motif chemokine ligand
(CXCL)1/CXCL2 within neutrophils (20); NETosis triggered by tumor
necrosis factor (TNF) together with interferon-γ (IFN-γ) has been
widely reported in classical Hodgkin lymphoma (cHL) (18,21). These findings suggest that
hematological malignancies and infection-induced NETs share various
upstream triggers while also being driven by their respective
tumor-related stimuli.
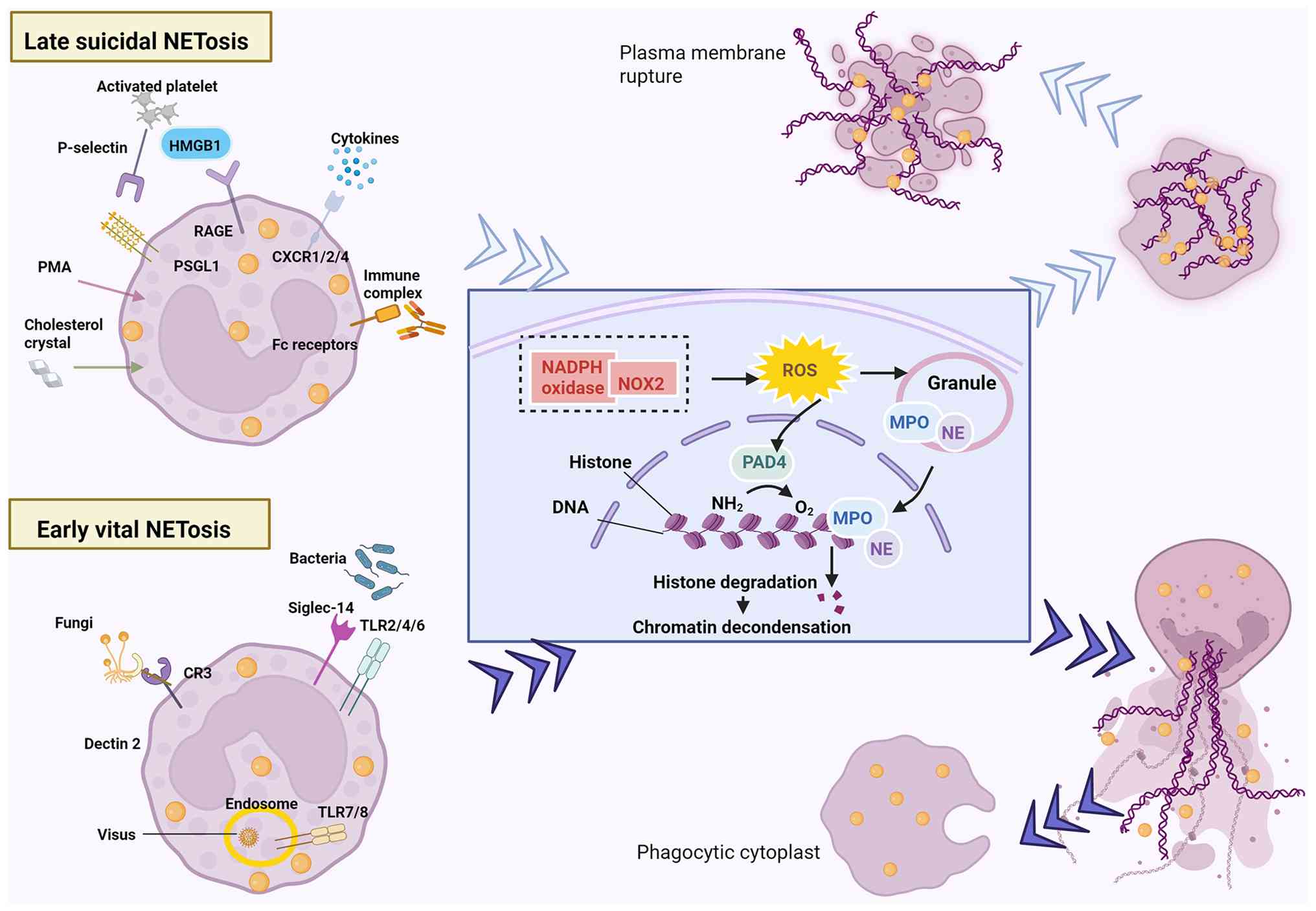
Upon activation, neutrophils adhere to vascular
endothelial cells (ECs) and transfer their granular components
(such as MPO and elastase) into the nucleus, where they work in
concert with peptidylarginine deiminase (PAD)4 to promote chromatin
decondensation (22,23). Stimulus-dependent NETosis occurs
via two pathways: Suicidal (membrane rupture) or vital (intact
membrane), both releasing chromatin-bound granular proteins
extracellularly (24-26). The vital NETosis or alternative
pathway is caused by pathogens, including bacteria, fungi, viruses
and protozoa (24,27). NETs can be rapidly secreted
within minutes of stimulation, while maintaining anucleate
phagocytes that remain functional and retain the ability to clear
microbes and respond to chemotactic signals (24). The classic suicidal NETosis
pathway is triggered by inflammatory mediators (such as IL-8 and
TNF-α), platelet activation, autoantibodies, or cholesterol
crystals (24,27). The generation of ROS by reduced
nicotinamide adenine dinucleotide phosphate (NADPH) oxidase serves
as an essential prerequisite for this distinct cell death mechanism
(28). Following neutrophil
activation (3-8 h, membrane rupture enables extracellular NET
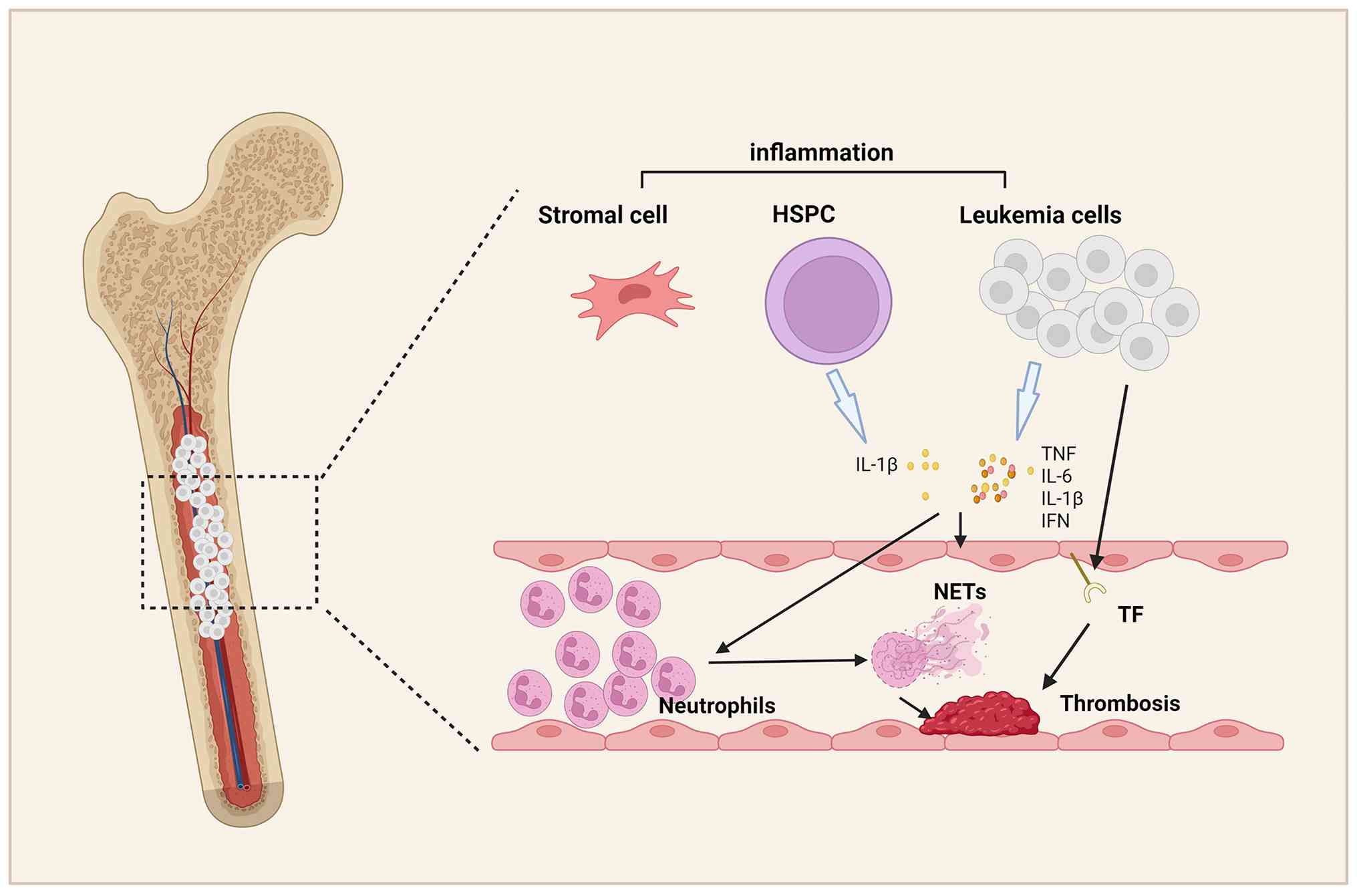
extrusion, culminating in lytic cell death (Fig. 1).
The concept of 'immunothrombosis', which was
introduced by Engelmann and Massberg (15), describes the physiological
antimicrobial function of NET-mediated thrombosis. However,
dysregulated NET formation drives pathological thrombosis (Fig. 2). Emerging research has
established NETs as pivotal mediators in venous thrombosis,
fundamentally advancing the current understanding of thrombotic
pathogenesis (29,30).
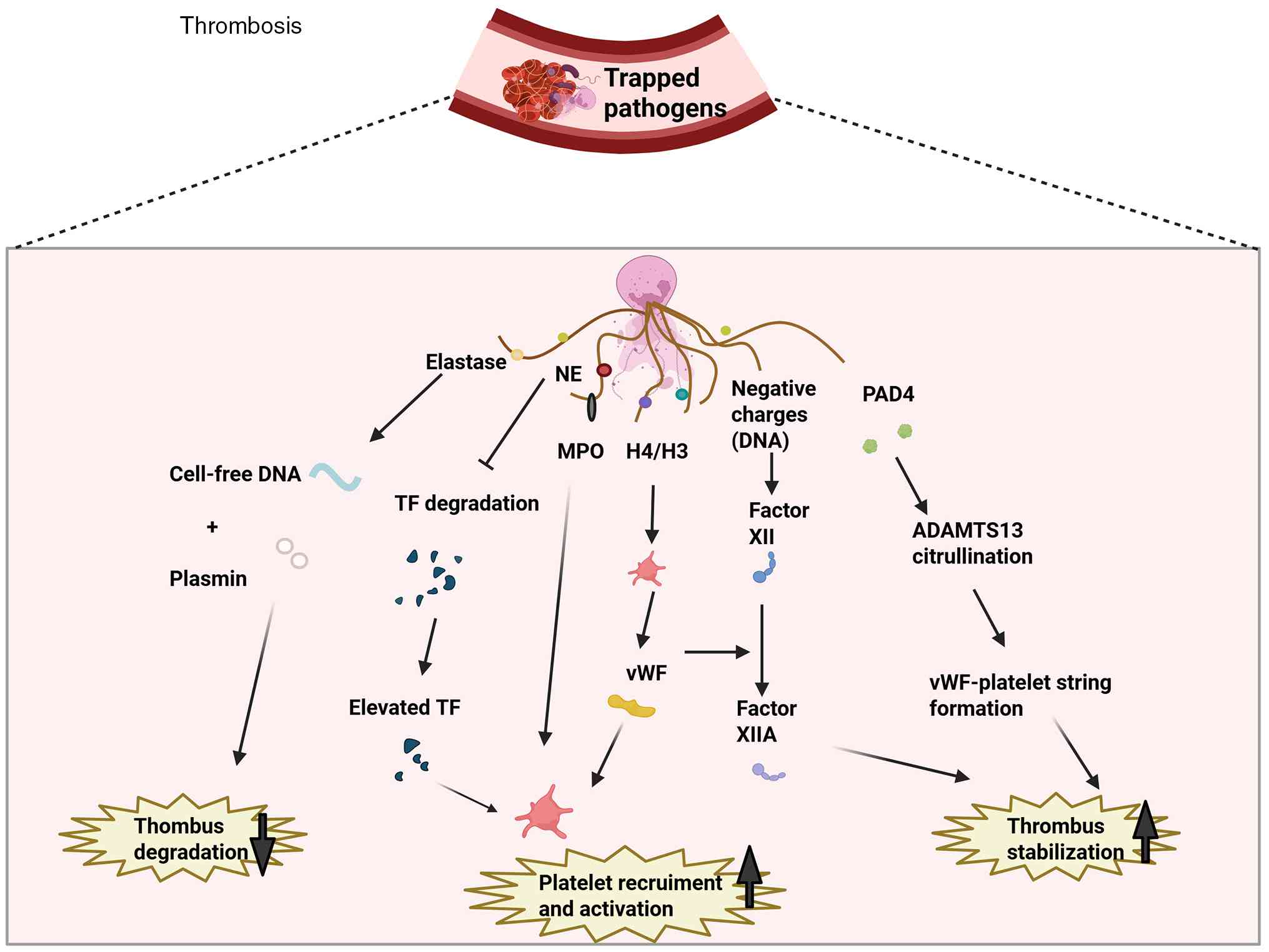
In addition to mediating antimicrobial activity,
NETs contribute to thrombosis by supplying a scaffold that
facilitates a potent cohesive reaction (8). NETs harbor a range of prothrombotic
molecules (such as anionic DNA scaffold, cathelicidin, MPO,
histones H3/H4 and NE). Each of these components contributes to
clot formation by engaging separate mechanisms, namely platelet
activation and aggregation, thrombin synthesis, and tissue factor
(TF) export (10,11,31-33). NETs possess the ability to
trigger the coagulation cascade. Circulating cell-free DNA (cfDNA)
triggers the intrinsic coagulation cascade by activating FXII, a
plasma serine protease (34).
This event sets off sequential activation of multiple coagulation
factors, leading ultimately to fibrin deposition and thrombus
formation. As factors that inhibit coagulation, TF pathway
inhibitors (TFPIs) can be cleaved by NEs bound to NETs. Therefore,
TF increases and supports exogenous coagulation pathways (35). NETs can also interact with
platelets. Histones are the most abundant proteins in NETs.
Previous studies have shown that histone H3 can activate platelets
(8,36). Activated platelets release
platelet factor 4, platelet activating factor and von Willebrand
factor (vWF), all of which act as soluble mediators that promote
NET generation (37). NETs bind
to platelet-secreted vWF, and this interaction serves to augment
platelet adhesion and aggregation, along with the production of
fibrin and subsequent thrombosis (32,38), thereby establishing a
self-amplifying cycle wherein platelets are activated by NETs. In
addition, NET-derived PAD4 is capable of citrullinating a
disintegrin and metalloproteinase with a thrombospondin type 1
motif, member 13 (ADAMTS13). This post-translational modification
diminishes the activity of the enzyme, which in turn facilitates
the assembly of ultra-large vWF-platelet strings and promotes
microvascular thrombus formation following vascular damage
(38).
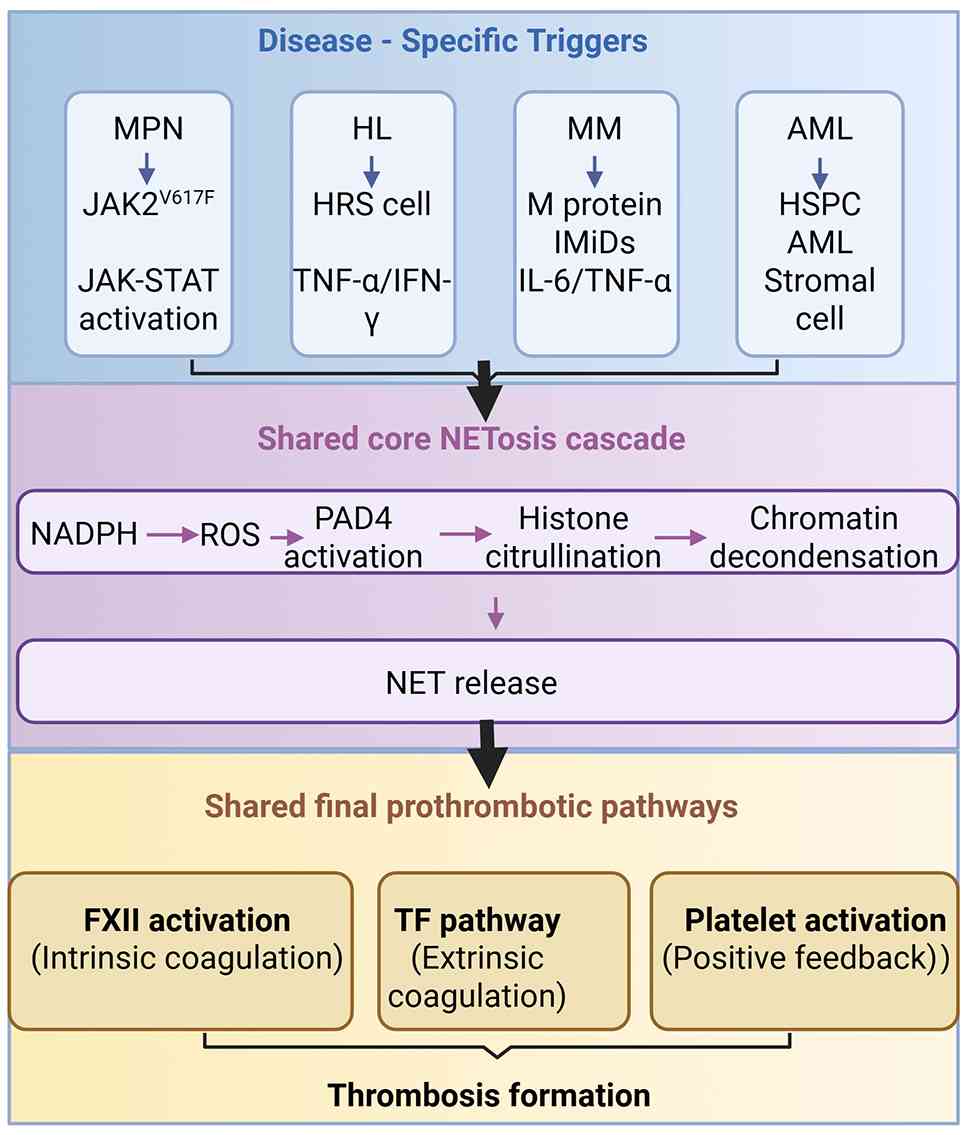
In summary, NETs promote thrombosis through multiple
mechanisms. To establish an integrated molecular framework linking
these mechanisms to hematological malignancies, a three-tiered
model is proposed, as illustrated in Fig. 3. In the upper tier,
disease-specific triggers [Jak2V617F signaling in MPN, inflammatory
cytokines in multiple myeloma (MM)/acute myeloid leukemia (AML),
and the inflammatory microenvironment in HL] activate NADPH
oxidase-dependent ROS production via distinct proximal mechanisms.
In the middle tier, this ROS production drives PAD4-mediated
histone citrullination and NET release. In the lower tier, NETs
promote thrombosis via the FXII-driven intrinsic pathway,
NE-mediated TFPI cleavage to facilitate the extrinsic TF pathway,
and histone-induced platelet/vWF activation that creates a positive
feedback loop. This framework distinguishes disease-specific
heterogeneity from a unifying prothrombotic mechanism downstream of
NETosis, highlighting that therapeutic targeting of the downstream
NETosis pathway could offer broad antithrombotic benefit across
multiple hematologic cancers.
VTE, a serious complication in hematological
malignancies, significantly increases morbidity and mortality with
varying risk across cancer types. Emerging evidence associated NETs
to thrombotic events in MPN, HL, chronic myeloid leukemia, acute
lymphoblastic leukemia (ALL) and AML. Deciphering the
NET-thrombosis interplay in these malignancies may reveal novel
therapeutic approaches to reduce thrombosis risk and improve
outcomes. Based on the unified molecular framework described in
Fig. 3, the following
subsections will elaborate, by disease type, the evidence for each
molecular pathway in different hematological malignancies.
MPN is a clonal disorder originating from HSCs,
marked by the overproliferation of one or more myeloid lines in the
bone marrow (BM) (39). In a
previous European collaborative study on low-dose aspirin, the
cumulative incidence of fatal and nonfatal thrombosis in patients
with polycythemia vera (PV) was 5.5% (40). The Gruppo Italiano Studio
Paliticemia conducted a study involving 1,213 patients and reported
that 19% of them experienced thrombotic events (41). Patients with essential
thrombocythemia (ET) experience thrombotic complications at a rate
of 2-4% per patient each year, a frequency that parallels the 2.33
events per person-year observed in primary myelofibrosis (PMF)
(42). Both a prior history of
thrombosis and the Jak2V617F mutation serve as important predictors
of thrombotic risk, with established value in clinical risk
assessment (36,43). However, there is still a lack of
biomarkers for predicting the risk of thrombosis in an individal,
especially in younger age groups. The high incidence, complex
management and high mortality of thrombus make it a worthy clinical
concern in patients with MPN (44). It is necessary to identify new
risk factors for thrombosis.
Multiple studies have examined the procoagulant role
of NETs among patients with MPN. Using a murine MPN model driven by
the Jak2V617F mutation, Wolach et al (13) observed increased NET formation
and a predisposition to spontaneous thrombosis in lung tissues.
Ruxolitinib, which targets JAK2 and is already in clinical use, was
found to abrogate NET production and attenuate thrombosis through
suppression of JAK-STAT signaling. Additionally, PAD4 was found to
be essential for Jak2V617F-induced NET production and thrombotic
events in vivo. In summary, that study suggested that
Jak2V617F may promote thrombosis in MPN by enhancing NET formation.
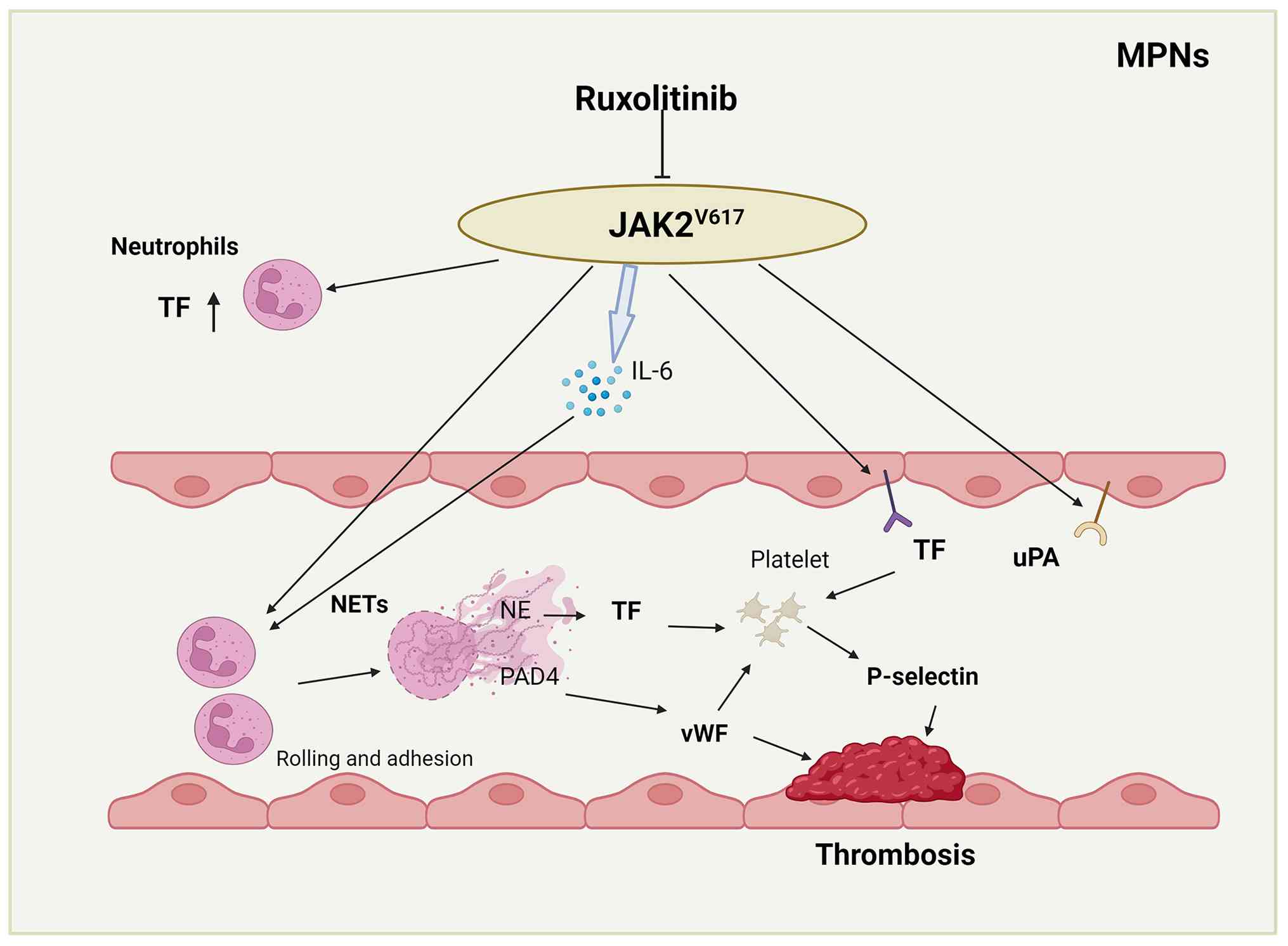
According to previous research, essential
thrombocythemia-associated thrombosis arises in part from
hyperactivation of platelets and white blood cells, a state that is
itself closely associated with whether the patient harbors a JAK2
mutation. Among individuals harboring the Jak2V617F mutation or
having experienced prior thrombotic events, the levels of TF,
P-selectin and vWF were all higher than in those without a history
of thrombosis (45,46). Suppressing the JAK-STAT pathway
reduces the release of pro-adhesive and procoagulant P-selectin and
vWF, as well as the pro-inflammatory cytokine IL-6 (47). Therefore, it can be hypothesized
that JAK2 mutations activate platelets through NETs, promote the
elevation of TF and vWF, and induce high expression of P-selectin
in platelets, thereby increasing thrombotic events in patients with
MPN. Ruxolitinib suppresses NET-induced thrombosis by restricting
the secretion of IL-6. This hypothesis is presented in Fig. 4. MPO-DNA is a specific marker of
NETs that can be measured in plasma. Guy et al (48) found that the MPO-DNA
concentration in patients with MPN exhibiting thrombosis was
significantly higher than that in the control group, supporting the
pathogenic role of NET formation in thrombosis. Notably, these
patients with MPN had a history of thrombosis, particularly portal
vein thrombosis. However, no differences were found when comparing
patients with MPN with thrombosis with those without thrombosis.
According to Marin Oyarzún et al (49), only nucleosome levels were
elevated. However, nucleosomes lack specificity as NET biomarkers,
as they can also be generated through other cell death processes
such as apoptosis or necrosis.
Meanwhile, the aforementioned studies have yielded
some conflicting findings. In a study by Guy et al (48), 52 patients with newly diagnosed
MPN and 54 healthy controls (free of prior thrombotic bleeding
episodes) were examined. Neutrophils isolated from the MPN group
exhibited a greater propensity for NET formation compared with
those obtained from healthy subjects. However, two separate studies
conducted by Wolach et al (13) and Marin Oyarzún et al
(49) failed to demonstrate that
the presence of Jak2V617F in unstimulated neutrophils leads to
increased NETosis. The reasons for these discrepancies may be
attributed to differences in patient inclusion criteria, treatment
status, sample size and MPN subtype heterogeneity across the
studies. First, regarding treatment-related confounding, in the two
studies by Wolach et al (13) and Marin Oyarzún et al
(49), the majority of enrolled
patients were undergoing cytoreductive regimens along with JAK
inhibitor therapy. These interventions exert myelosuppressive
effects and increase the risk of opportunistic infections, leading
to neutropenia and altered neutrophil functional status, thereby
affecting NET release. Second, regarding cohort characteristics,
according to Guy et al (48), half of the patients enrolled in
their study had experienced prior thrombotic events. Relative to
the data obtained from the other two aforementioned studies, this
observation highlights the association between NET release and
thrombosis. Third, regarding sample size limitations, the sample
sizes of the aforementioned studies were relatively limited (each
study included <70 cases), which may be insufficient to detect
true intergroup differences, and limits the statistical power of
multivariable and subgroup analyses. Fourth, regarding MPN subtype
heterogeneity, MPN encompasses three main subtypes, namely PV, ET
and PMF, each with distinct disease characteristics. Patients with
PV frequently present with erythrocytosis and elevated blood
viscosity; patients with ET are characterized by megakaryocyte
hyperplasia and thrombocytosis; and patients with PMF exhibit more
pronounced BM fibrosis and an inflammatory cytokine storm. These
subtype differences may lead to heterogeneity in baseline NETosis
levels and their contribution to thrombotic risk. However, due to
the limited sample sizes of the aforementioned studies, no
subtype-stratified analyses were performed. Differences in the
proportions of MPN subtypes included across studies may also
represent an important source of result variability.
Although the aforementioned studies show minor
discrepancies in their results, they all consistently demonstrate
that NETs promote thrombosis in MPN. Future larger-scale and more
precisely stratified studies are needed to further strengthen this
preliminary evidence (Table
I).
Thrombosis incidence in AML ranges from 2.09 to
8.60% across studies, with variations attributed to diagnostic
methods and patient populations (50-52). While disease aggressiveness
correlates with VTE risk and poorer outcomes (53), thrombocytopenia in patients with
AML has limited their inclusion in thrombosis trials, resulting in
scarce management guidelines. This evidence gap underscores the
need for an improved mechanistic understanding of thrombosis in
AML.
According to previous studies, NET-TF interplay
serves as a key driver of thrombus formation. Hell et al
(54) conducted a study on 28
patients with malignant diseases accompanied by overt DIC (ISTH
score >5), including 15 patients with AML. The authors defined
plasma DNA and nucleosomes as parameters related to NETs formation,
and measured TF activity in microvesicles (MV-TF) as well as other
conventional coagulation indicators. The results showed that
NETs-related parameters were closely associated with changes in
MV-TF and conventional coagulation parameters. Doubling in plasma
DNA levels was linked to a 7.6% decrease in fibrinogen, a 41%
increase in D-dimer, a 15.3% reduction in platelets and a 3.9%
shortening in prothrombin time. A 10% increase in nucleosomes was
associated with a 3.1% decrease in fibrinogen, a 112.7% increase in
D-dimer, a 5.0% reduction in platelets and a 1.0% shortening in
prothrombin time. Furthermore, upon administration of chemotherapy
to patients with AML, NETs parameters and MV-TF activity
significantly decreased (nucleosomes and MV-TF activity returned to
normal levels after 1 week and 1 month, respectively), and
coagulation indicators improved. According to these findings, NETs
and TF-carrying MVs are interconnected, and this association may
explain why thrombosis frequently complicates AML. Ostafin et
al (55) conducted a study
on 45 children with leukemia (including 33 patients with ALL, 8
patients with AML and 4 patients with T-cell ALL). During the
progression of acute leukemia in children, unlike in adults,
neutrophils demonstrate a compromised ability to extrude NETs.
Consequently, infectious complications become more probable,
whereas thrombotic risk is not similarly elevated. The currently
available research is limited, and therefore larger multicenter
studies with bigger sample sizes are needed for further validation
(Table I).
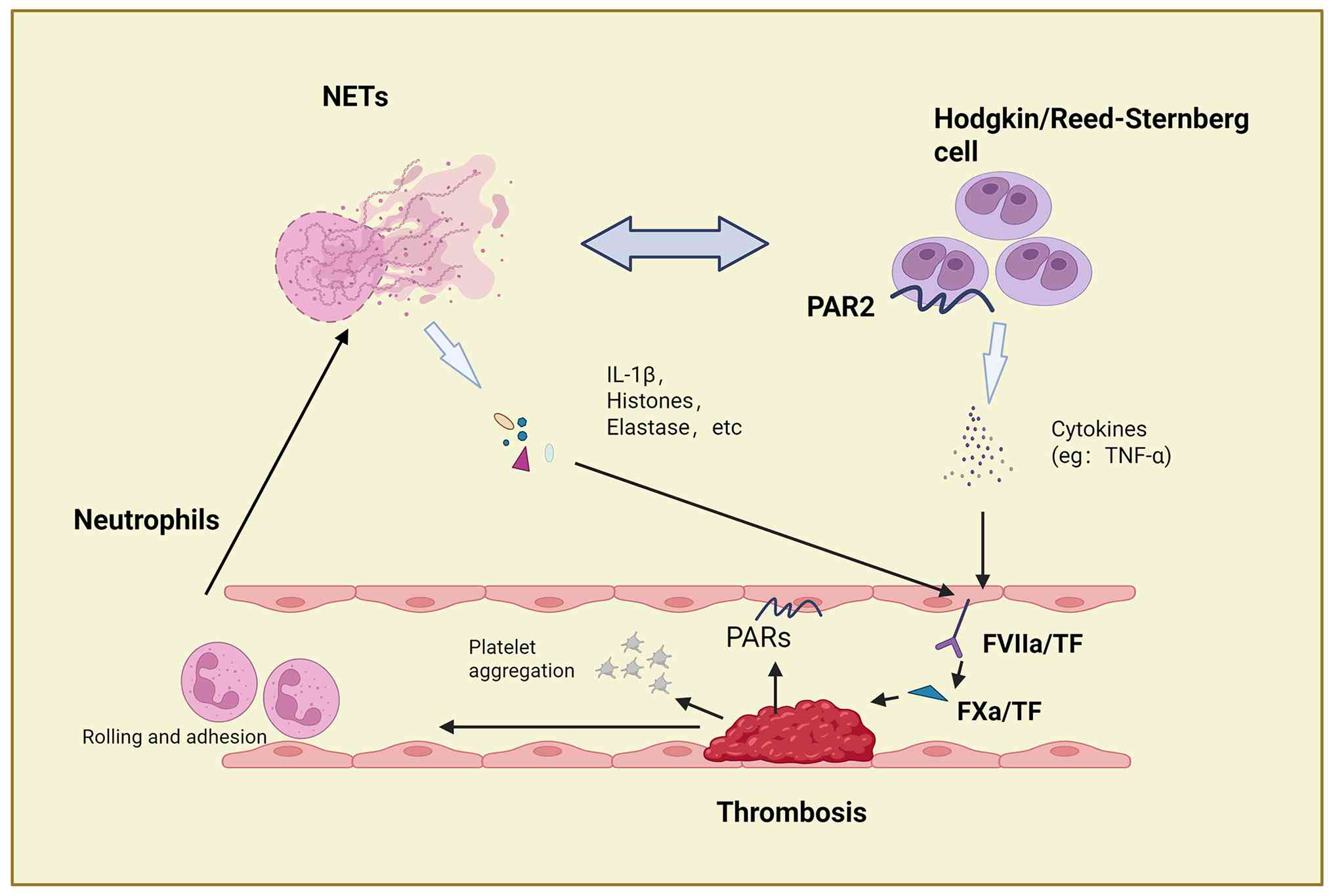
In the German Hodgkin Study Group trials (n=5,773),
patients with HL showed a 3.3% thrombosis incidence (193 events)
(62). Given the significant
mortality impact of thrombosis in this curable malignancy, improved
risk prediction is clinically crucial. Current tools such as the
Khorana score inadequately predict VTE risk in HL (63), highlighting the need for refined
risk assessment to guide timely thromboprophylaxis and improve
outcomes.
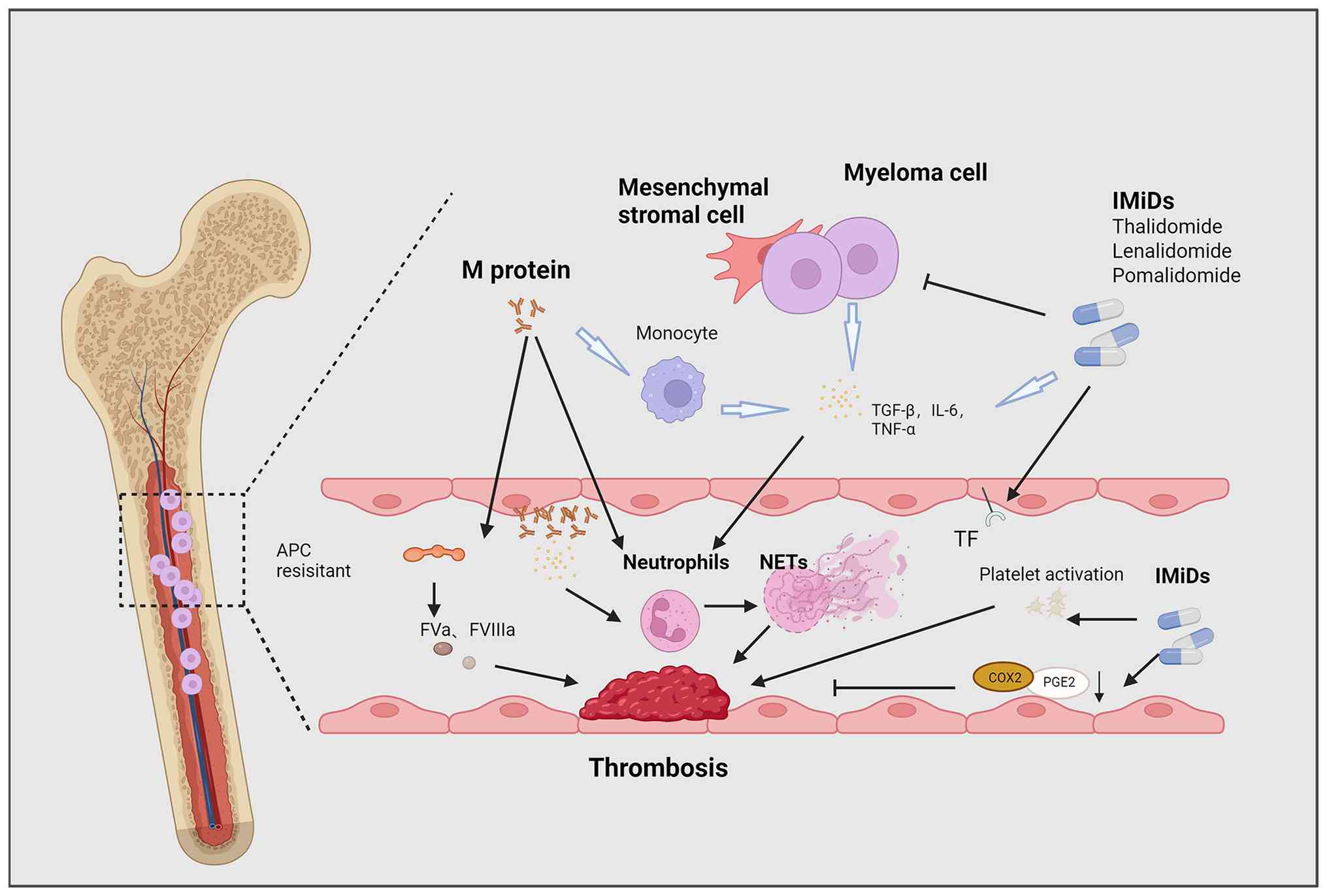
VTE occurs in >10% of patients with MM, driven by
disease-specific factors (such as M-protein hyperviscosity,
proinflammatory cytokines, newly diagnosed disease, renal
impairment, and chromosome 11 abnormalities) (4,68-71) and hemostatic alterations
(including elevated FVIII, vWF, fibrinogen, and thrombin
generation) (72-74). Despite existing International
Myeloma Working Group and European risk stratification guidelines
(2014-2015) (75,76), current models cannot reliably
distinguish between low-, intermediate- and high-risk patients, and
validated predictive biomarkers remain lacking, thus highlighting
the need for deeper mechanistic insights to improve VTE risk
assessment in MM.
The California Cancer Registry analyzed 2,482 cases
of ALL from 1993 to 1995, accounting for 4.5% of the cohort
(51). Ziegler et al
(50) studied 185 cases of ALL
and found a thrombosis incidence of 2.09%. Adolescents with ALL
face an elevated thrombotic risk compared with other age groups. A
meta-analysis comprising 17 prospective studies (1,752
participants) reported that 5.2% of pediatric patients with ALL
experienced thrombotic complications (83). VTE occurrence, meanwhile,
elevates the likelihood of mortality within the first year by 40%
(51). The incomplete
understanding of ALL-associated thrombosis mechanisms and the
absence of standardized thromboprophylaxis underscore the need for
personalized prevention strategies tailored to patient-specific
risk profiles.
Notably, four common issues emerge from the
literature on the aforementioned diseases (1), including i) sample size: The
majority of studies are single-center exploratory analyses
(n<50), lacking multicenter, prospective, large-cohort
validation (n>200), as well as prospective stratification by
treatment stage (newly diagnosed, post-chemotherapy, or
post-transplantation) and disease subtype; ii) NET marker
standardization: cfDNA and nucleosomes are easily measurable but
lack specificity; MPO-DNA and H3Cit exhibit high specificity, yet
their detection methods (ELISA, immunofluorescence, and flow
cytometry) have not been standardized. At the same time, as NET
research has advanced, the understanding of traditional indicators
has evolved. Free plasma DNA and nucleosomes have been discussed as
non-exclusive indicators of NET formation, since they can likewise
derive from other modes of cell death, namely cancer-associated or
treatment-induced apoptosis and necrosis (49). To the best of our knowledge, no
study has to date performed a parallel comparison of the predictive
efficacy of cfDNA, nucleosomes, MPO-DNA and H3Cit for thrombotic
events within the same cohort, nor has a unified positive threshold
been established. In future research, a combined detection strategy
is recommended, and the specificity of plasma markers should be
verified whenever possible by immunocytochemistry or flow cytometry
to guarantee the robustness of the findings; iii) therapeutic
confounding: The majority of studies have not adequately controlled
for or performed stratified analyses on the direct effects of
cytoreductive therapy, JAK inhibitors, IMiDs, asparaginase or
GM-CSF on NETosis. An ideal study design should involve dynamic
sample collection before treatment (baseline), during treatment
(acute phase) and after treatment (remission phase) to distinguish
the contributions of the disease itself vs. therapeutic
interventions to NET formation; and iv) lack of causal validation:
Existing studies are predominantly correlational in nature and lack
causal chain validation (such as using PAD4 inhibitors, DNase I or
PAD4-knockout mice in disease models to demonstrate that NETs lead
to thrombosis). Furthermore, in vitro experiments often
employ non-physiological strong stimuli such as PMA to induce
NETosis, which may differ mechanistically from NETosis induced by
tumor-related stimuli in vivo (characterized by
low-concentration, chronic stimulation).
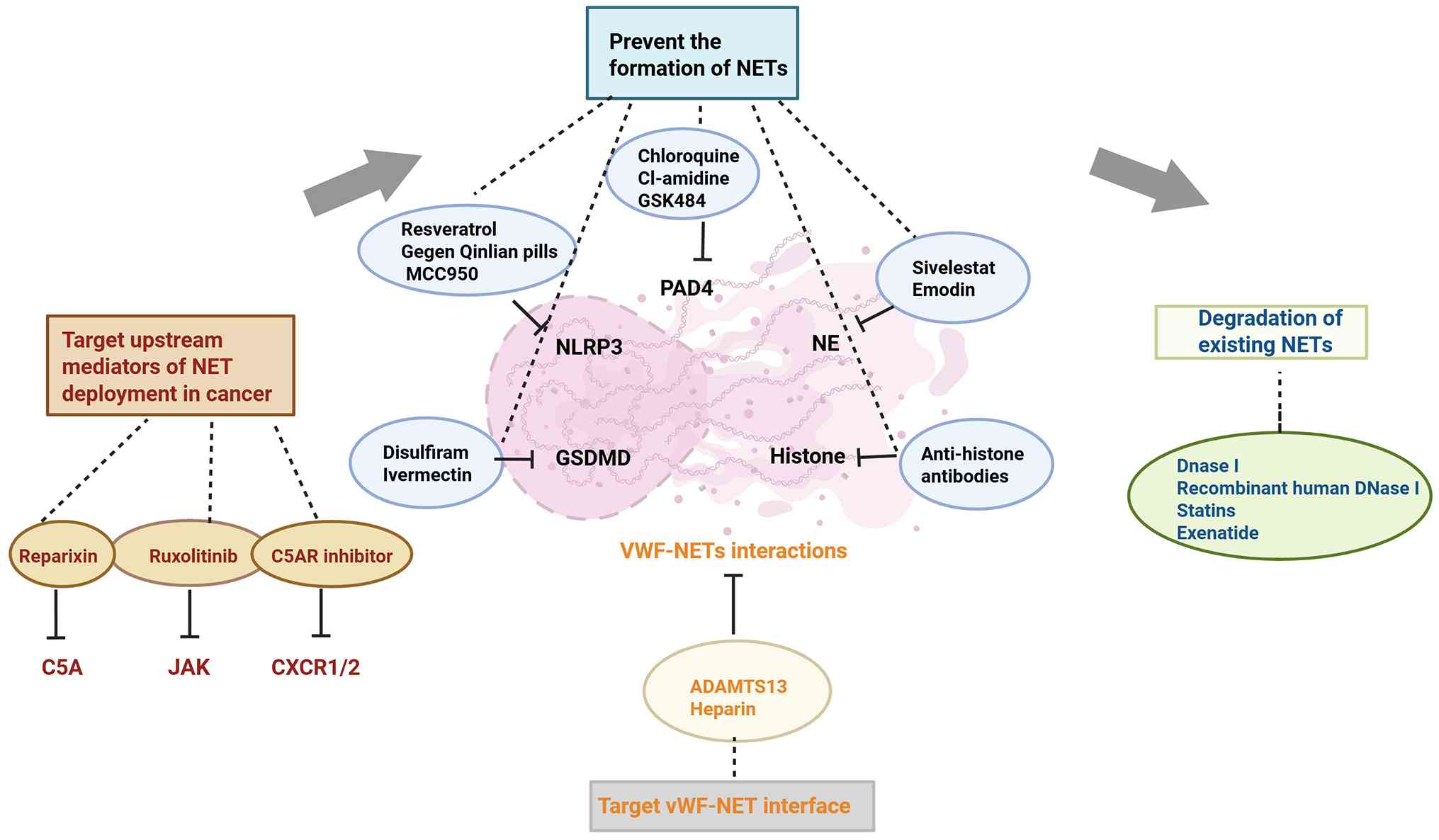
In hematological malignancies, the development of
thrombosis typically is associated with worse prognosis. As one of
the emerging mechanisms of thrombosis, NETs may be potential
intervention targets for the treatment of hematological
malignancies (Fig. 8).
Therapeutic targeting of NET formation pathways,
particularly through PAD inhibition, has shown promising
antithrombotic effects in preclinical studies. In vivo, PAD
gene deletion and inhibition of NET formation resulted in similar
phenotypes in various disease models (9). Previous studies have examined
Cl-amidine, a comparatively new PAD-targeting agent, in diverse
inflammatory disease settings, including atherosclerosis (86), lupus, diabetes mellitus and
endometritis (87-89). Knight et al (86) reported that, in atherosclerotic
mice, pharmacologically targeting NET formation with Cl-amidine was
shown to lessen both atherosclerotic plaque load and arterial
thrombotic events. Novotny et al (90) treated mice with arterial
thrombosis with Cl-amidine and showed inhibition of NET formation,
reduced thrombosis and reduced disease-associated tissue damage.
Additionally, two more specific PAD4 inhibitors, GSK199 and GSK484,
have been reported to suppress NET production in models of
arthritis, heparin-induced settings and a murine coronavirus system
(91-93). Previous studies on GSK484
intervention showed significant inhibition of thrombosis. In
addition, an important aminoquinoline compound, chloroquine, is
extensively employed in treating diverse illnesses, including
malaria, lupus erythematosus and rheumatoid arthritis, and
additionally functions as a PAD4 inhibitor (94). Dyer et al (95) found that hydroxychloroquine (HCQ)
could reduce deep vein thrombotic burden in traumatized mice.
Notably, Petri et al (96) found that it inhibited thrombosis
in patients with systemic lupus erythematosus (SLE). Thrombosis
occurred in 38 (5.1%) of 739 patients with SLE. Patients with mean
HCQ blood levels ≥1,068 ng/ml had 69% fewer thrombotic events
(P=0.024). However, published reports describing other PAD
inhibitors in human applications are lacking, despite that the fact
that these agents possess the capacity to modulate thrombosis.
The assembly process of the NLR family pyrin
domain-containing 3 (NLRP3) inflammasome also requires PAD4
(97). NLRP3, a multimeric
protein complex, recognizes endogenous or pathogen-associated
signals and stimulates the secretion of pro-inflammatory cytokines
(98). Activation of NETosis
requires the involvement of the NLRP3 inflammasome. Gegen Qinlian
pills (99) and resveratrol
(100) have been shown to
reduce thrombosis by inhibiting NLRP3 signaling or downregulating
NLRP3 expression, respectively. MCC950, a synthetic compound,
represents the most powerful selective NLRP3 inhibitor yet
described (101) and was
demonstrated to ameliorate thrombin-induced platelet aggregation
(102). Therefore, inhibiting
NETosis through NLRP3 targeting could open novel avenues for
antithrombotic therapy.
The release of NETs following NLRP3 inflammasome
activation is orchestrated by gasdermin D (GSDMD), a protein known
for its pore-forming activity in pyroptosis (103,104). Approved as a therapy for
alcohol use disorder, disulfiram, which inhibits GSDMD and exerts
its clinical effect through suppression of aldehyde dehydrogenase
(105), has been shown to
downregulate innate immunity and the complement/coagulation
pathways (106). Another
GSDMD-targeting agent, ivermectin, was found to diminish microclot
development via interference with SARS-CoV-2 binding to fibrinogen,
a finding supported by molecular docking and dynamics simulations
(107).
Therapeutic targeting of NET-associated cytotoxic
components shows clinical potential. Sivelestat, an inhibitor of
NE, has received regulatory approval in Japan and South Korea as a
therapy for acute respiratory distress syndrome (108). Zhou et al (109) demonstrated that NETs induce
endothelial cytotoxicity and procoagulant transformation, effects
attenuated by 25% with Sivelestat treatment. Zhang et al
(110) demonstrated that NETs
trigger endothelial procoagulant activation through vWF/plasminogen
activator inhibitor-1 (PAI-1) release, a process inhibited by
Sivelestat to potentially enhance fibrinolysis. Meanwhile, the NE
inhibitor α-1 antitrypsin (prolastin-C) is under clinical
evaluation for bronchiectasis (NCT05582798). Prolastin-C represents
the sole NET-formation inhibitor currently in clinical trials.
Meanwhile, histones drive endothelial dysfunction and prothrombotic
responses (111), with
anti-histone antibodies showing efficacy in reducing pulmonary
microthrombi in murine trauma models (112), supporting histone targeting as
a viable therapeutic strategy.
N-acetylcysteine (NAC) is a drug with antioxidant,
expectorant and anti-inflammatory effects. The dependence of
NETosis on ROS has led to the observation that NAC inhibits NET
formation in vitro (113). NAC has been explored as a
potential therapy for various thrombotic conditions, including
thrombotic thrombocytopenic purpura (NCT01808521). In that phase I
clinical trial, the treatment was administered as an initial
intravenous injection of 150 mg/kg over 60 min. If this dose was
tolerated, it was then followed by a 17-h intravenous infusion at
the same dose. According to Craver et al (19), NAC prolonged the survival of
Jak2V617F mice while leaving both blood cell parameters and spleen
enlargement unaffected. NAC was shown to decrease thrombus
formation when tested in an animal model of acute pulmonary
thrombosis. Analysis of in vitro platelet activation
revealed that NAC decreased the formation of platelet-leukocyte
aggregates in Jak2V617F mice (19). Additionally, NAC suppressed NET
generation in primary human neutrophils derived from patients with
MPN and from healthy individuals (19). These findings demonstrate that
NAC exerts antithrombotic effects in Jak2V617F mice. Furthermore,
they provide a preclinical rationale for continued evaluation of
NAC as a potential agent to lower thrombotic risk in MPN.
Drugs targeting molecules such as EPCR, BTK, SIRT
and NADPH have demonstrated potential in inhibiting NET formation
and ameliorating thrombosis (19,114-123) (Table II). However, clinical trials for
these drugs have not yet been initiated.
In addition, statins could reduce the levels of
macrophages, neutrophils and NETs in a murine model of venous
thrombosis, and decrease thrombus burden through profibrinolytic,
anticoagulant and antiplatelet effects (127). As a GLP-1 agonist, exenatide
exhibits multiple activities: It reduces circulating NET marker
concentrations (128), inhibits
platelet aggregation in vitro and suppresses thrombus
formation in vivo, suggesting its potential to reduce
NET-associated thrombosis (129).
Targeting the upstream mediators of cancer NET
formation also has the potential to ameliorate coagulation.
Previous research has shown that CXCR2 has a promoting effect on
tumorigenesis (130).
Furthermore, CXCR2 can bind to IL-8 to recruit and activate
neutrophils and promote NET formation (131). Alsabani et al (132) found that NET formation was
associated with fibrin deposition (r=0.702) and lung injury
(r=0.692) in a mouse model of sepsis. The application of the
CXCR1/2 inhibitor reparixin was demonstrated to inhibit NETs and
thrombosis, thereby reducing multiorgan injury and mortality.
Complement component 5a (C5A) promotes NET
formation, whereas complement component 5a receptor 1 (C5AR1)
blockers are authorized for use in anti-neutrophil cytoplasmic
antibody-related small-vessel vasculitis (133). Zhao et al (134) found that inhibiting the
C5A/C5AR axis resolves cholesterol crystals and attenuates renal
vascular thrombosis in mice. Although the role of this axis in
malignancy remains unclear, its inhibition may slow disease
progression in animal models by targeting NETs (Table III).
NETs function as a scaffold, which facilitates the
adherence of platelets and red blood cells, and also serves to
concentrate clotting-relevant proteins. Interactions with NETs can
be mediated in several ways. Under inflammatory conditions,
ultra-large vWF fibers are formed, which are anchored to the
endothelial lining and serve as intermediaries that facilitate
NET-driven remodeling of fibrin networks. Finally, the fibers exert
a procoagulant effect by stabilizing the thrombus (135). Grässle et al (37) reported that heparin interferes
with DNA-vWF binding, thereby blocking adhesion to white blood
cells. Fuchs et al (32)
found that heparin prevented the formation of blood clots in
baboons by affecting the binding of NETs to fibrin chains.
Similarly, vWF can be lysed by the metalloproteinase ADAMTS13,
which has been shown to improve ischemic brain injury in
vivo (136). Denorme et
al (137) observed that
ADAMTS13 broke down TPA-resistant clots in a
concentration-responsive manner in a murine stroke model, reducing
the area of cerebral infarction. In summary, interfering with the
vWF-NET interface may serve as a promising strategy for developing
antithrombotic treatments (Table
III).
In conclusion, directing therapeutic efforts toward
NETs may offer a viable approach to address thrombosis in the
context of hematological malignancies. However, these are
frequently complicated by cytopenias and coagulation disorders,
which may predispose patients to bleeding complications and
platelet-related safety risks following NET-targeted therapies.
Although no adverse effects on platelet counts or organ function
were observed in NAC-treated MPN mouse models, future
investigations should prioritize the assessment of these potential
risks. Furthermore, given that monotherapy may not adequately
mitigate thrombotic risk, the combination of NET-targeted agents
with conventional anticoagulants warrants exploration as a
promising approach. The two modalities are mechanistically
complementary, potentially exerting synergistic antithrombotic
effects by targeting distinct steps in thrombus formation. However,
concurrent administration of two antithrombotic agents may also
increase bleeding risk, particularly in patients with
thrombocytopenia. The efficacy and safety of such combination
therapy require systematic evaluation in future animal studies.
Thrombosis remains a life-threatening complication
in hematological malignancies, with emerging evidence implicating
NETs as key mediators through multiple prothrombotic mechanisms.
While the involvement of NETs in hypercoagulability across MPN,
AML, MM, HL and ALL is established, their disease-specific
mechanisms require further elucidation. Current NET-targeting
strategies, including formation inhibition, degradation enhancement
and mediator blockade, show preclinical promise but demand rigorous
clinical validation, particularly regarding bleeding risks in
patients with thrombocytopenia.
Critical challenges persist in NET quantification
and biomarker development. Future directions should prioritize i)
standardized NET detection assays, ii) mechanistic studies on
NET-driven thrombosis, iii) therapeutic optimization through
targeted NETosis inhibition or combination approaches and iv)
integration of NET biomarkers into risk stratification models.
Addressing these priorities through collaborative research will
accelerate the translation of NET-focused strategies into clinical
practice, offering novel avenues for thrombosis management in
hematological malignancies.
Not applicable.
KL and ZM designed, conducted the literature review
and drafted the manuscript. QC, YS and JM contributed to
supervision, funding acquisition, and manuscript writing, review
and editing. LY contributed to the conceptualization of the review,
as well as manuscript, writing, review and editing. CW contributed
to the study design, supervision, funding acquisition,
conceptualization, project administration, and manuscript writing,
review and editing. Data authentication is not applicable. All
authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
This work was financially supported by grants from the Huai'an
First People's Hospital Innovation Team Project (grant no.
YCT202306), the Affiliated Huai'an No.1 People's Hospital of
Nanjing Medical University (grant no. GQ202406), the Northern
Jiangsu Clinical Medicine Research Institute's 2024 Projects (grant
nos. HAKY202400324, HAKY202400218 and HAKY202400216).
|
1
|
Grilz E, Posch F, Nopp S, Königsbrügge O,
Lang IM, Klimek P, Thurner S, Pabinger I and Ay C: Relative risk of
arterial and venous thromboembolism in persons with cancer vs
persons without cancer-a nationwide analysis. Eur Heart J.
42:2299–2307. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ashrani AA, Gullerud RE, Petterson TM,
Marks RS, Bailey KR and Heit JA: Risk factors for incident venous
thromboembolism in active cancer patients: A population based
case-control study. Thromb Res. 139:29–37. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sørensen HT, Mellemkjaer L, Olsen JH and
Baron JA: Prognosis of cancers associated with venous
thromboembolism. N Engl J Med. 343:1846–1850. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Falanga A and Marchetti M: Venous
thromboembolism in the hematologic malignancies. J Clin Oncol.
27:4848–4857. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Elshoury A, Schaefer JK, Lim MY, Skalla DP
and Streiff MB: Update on guidelines for the prevention of
cancer-associated thrombosis. J Natl Compr Canc Netw. 20:2022.
|
|
6
|
Brinkmann V, Reichard U, Goosmann C,
Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y and Zychlinsky A:
Neutrophil extracellular traps kill bacteria. Science.
303:1532–1535. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fuchs TA, Abed U, Goosmann C, Hurwitz R,
Schulze I, Wahn V, Weinrauch Y, Brinkmann V and Zychlinsky A: Novel
cell death program leads to neutrophil extracellular traps. J Cell
Biol. 176:231–241. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brill A, Fuchs TA, Chauhan AK, Yang JJ, De
Meyer SF, Köllnberger M, Wakefield TW, Lämmle B, Massberg S and
Wagner DD: von Willebrand factor-mediated platelet adhesion is
critical for deep vein thrombosis in mouse models. Blood.
117:1400–1407. 2011. View Article : Google Scholar
|
|
9
|
Wang H, Kim SJ, Lei Y, Wang S, Wang H,
Huang H, Zhang H and Tsung A: Neutrophil extracellular traps in
homeostasis and disease. Signal Transduct Target Ther. 9:2352024.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
von Köckritz-Blickwede M, Goldmann O,
Thulin P, Heinemann K, Norrby-Teglund A, Rohde M and Medina E:
Phagocytosis-independent antimicrobial activity of mast cells by
means of extracellular trap formation. Blood. 111:3070–3080. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yousefi S, Gold JA, Andina N, Lee JJ,
Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich
GJ and Simon HU: Catapult-like release of mitochondrial DNA by
eosinophils contributes to antibacterial defense. Nat Med.
14:949–953. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stark K and Massberg S: Interplay between
inflammation and thrombosis in cardiovascular pathology. Nat Rev
Cardiol. 18:666–682. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wolach O, Sellar RS, Martinod K,
Cherpokova D, McConkey M, Chappell RJ, Silver AJ, Adams D,
Castellano CA, Schneider RK, et al: Increased neutrophil
extracellular trap formation promotes thrombosis in
myeloproliferative neoplasms. Sci Transl Med. 10:eaan82922018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McDonald B, Davis RP, Kim SJ, Tse M, Esmon
CT, Kolaczkowska E and Jenne CN: Platelets and neutrophil
extracellular traps collaborate to promote intravascular
coagulation during sepsis in mice. Blood. 129:1357–1367. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Engelmann B and Massberg S: Thrombosis as
an intravascular effector of innate immunity. Nat Rev Immunol.
13:34–45. 2013. View Article : Google Scholar
|
|
16
|
Papayannopoulos V: Neutrophil
extracellular traps in immunity and disease. Nat Rev Immunol.
18:134–17. 2018. View Article : Google Scholar
|
|
17
|
Hirose T, Hamaguchi S, Matsumoto N,
Irisawa T, Seki M, Tasaki O, Hosotsubo H, Yamamoto N, Yamamoto K,
Akeda Y, et al: Presence of neutrophil extracellular traps and
citrullinated histone H3 in the bloodstream of critically ill
patients. PLoS One. 9:e1117552014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sangaletti S, Tripodo C, Vitali C,
Portararo P, Guarnotta C, Casalini P, Cappetti B, Miotti S,
Pinciroli P, Fuligni F, et al: Defective stromal remodeling and
neutrophil extracellular traps in lymphoid tissues favor the
transition from autoimmunity to lymphoma. Cancer Discov. 4:110–129.
2014. View Article : Google Scholar
|
|
19
|
Craver BM, Ramanathan G, Hoang S, Chang X,
Mendez Luque LF, Brooks S, Lai HY and Fleischman AG:
N-acetylcysteine inhibits thrombosis in a murine model of
myeloproliferative neoplasm. Blood Adv. 4:312–321. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nie M, Yang L, Bi X, Wang Y, Sun P, Yang
H, Liu P, Li Z, Xia Y and Jiang W: Neutrophil extracellular traps
induced by IL8 promote diffuse large B-cell lymphoma progression
via the TLR9 signaling. Clin Cancer Res. 25:1867–1879. 2019.
View Article : Google Scholar
|
|
21
|
Francischetti IMB, Alejo JC, Sivanandham
R, Davies-Hill T, Fetsch P, Pandrea I, Jaffe ES and Pittaluga S:
Neutrophil and eosinophil extracellular traps in Hodgkin lymphoma.
Hemasphere. 5:e6332021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li P, Li M, Lindberg MR, Kennett MJ, Xiong
N and Wang Y: PAD4 is essential for antibacterial innate immunity
mediated by neutrophil extracellular traps. J Exp Med.
207:1853–1862. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, Li M, Stadler S, Correll S, Li P,
Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, et al: Histone
hypercitrullination mediates chromatin decondensation and
neutrophil extracellular trap formation. J Cell Biol. 184:205–213.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jorch SK and Kubes P: An emerging role for
neutrophil extracellular traps in noninfectious disease. Nat Med.
23:279–287. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Desai J, Mulay SR, Nakazawa D and Anders
HJ: Matters of life and death. How neutrophils die or survive along
NET release and is 'NETosis'=necroptosis? Cell Mol Life Sci.
73:2211–2219. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zawrotniak M and Rapala-Kozik M:
Neutrophil extracellular traps (NETs)-formation and implications.
Acta Biochim Pol. 60:277–284. 2013. View Article : Google Scholar
|
|
27
|
Ferrer-Marín F, Cuenca-Zamora EJ,
Guijarro-Carrillo PJ and Teruel-Montoya R: Emerging role of
neutrophils in the thrombosis of chronic myeloproliferative
neoplasms. Int J Mol Sci. 22:11432021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stoiber W, Obermayer A, Steinbacher P and
Krautgartner WD: The role of reactive oxygen species (ROS) in the
formation of extracellular traps (ETs) in humans. Biomolecules.
5:702–723. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thakur M, Junho CVC, Bernhard SM,
Schindewolf M, Noels H and Döring Y: NETs-induced thrombosis
impacts on cardiovascular and chronic kidney disease. Circ Res.
132:933–949. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laridan E, Martinod K and De Meyer SF:
Neutrophil extracellular traps in arterial and venous thrombosis.
Semin Thromb Hemost. 45:86–93. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Scharrig E, Carestia A, Ferrer MF, Cédola
M, Pretre G, Drut R, Picardeau M, Schattner M and Gómez RM:
Neutrophil extracellular traps are involved in the innate immune
response to infection with leptospira. PLoS Negl Trop Dis.
9:e00039272015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fuchs TA, Brill A, Duerschmied D,
Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW,
Hartwig JH and Wagner DD: Extracellular DNA traps promote
thrombosis. Proc Natl Acad Sci USA. 107:15880–15885. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brill A, Fuchs TA, Savchenko AS, Thomas
GM, Martinod K, De Meyer SF, Bhandari AA and Wagner DD: Neutrophil
extracellular traps promote deep vein thrombosis in mice. J Thromb
Haemost. 10:136–144. 2012. View Article : Google Scholar
|
|
34
|
von Brühl ML, Stark K, Steinhart A,
Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A,
Coletti R, Köllnberger M, et al: Monocytes, neutrophils, and
platelets cooperate to initiate and propagate venous thrombosis in
mice in vivo. J Exp Med. 209:819–835. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Massberg S, Grahl L, von Bruehl ML,
Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov
K, Khandagale AB, et al: Reciprocal coupling of coagulation and
innate immunity via neutrophil serine proteases. Nat Med.
16:887–896. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Grinfeld J, Nangalia J, Baxter EJ, Wedge
DC, Angelopoulos N, Cantrill R, Godfrey AL, Papaemmanuil E, Gundem
G, MacLean C, et al: Classification and personalized prognosis in
myeloproliferative neoplasms. N Engl J Med. 379:1416–1430. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Carestia A, Kaufman T, Rivadeneyra L,
Landoni VI, Pozner RG, Negrotto S, D'Atri LP, Gómez RM and
Schattner M: Mediators and molecular pathways involved in the
regulation of neutrophil extracellular trap formation mediated by
activated platelets. J Leukoc Biol. 99:153–162. 2016. View Article : Google Scholar
|
|
38
|
Grässle S, Huck V, Pappelbaum KI,
Gorzelanny C, Aponte-Santamaría C, Baldauf C, Gräter F,
Schneppenheim R, Obser T and Schneider SW: von Willebrand factor
directly interacts with DNA from neutrophil extracellular traps.
Arterioscler Thromb Vasc Biol. 34:1382–1389. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Falanga A and Marchetti M: Thrombosis in
myeloproliferative neoplasms. Semin Thromb Hemost. 40:348–358.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Marchioli R, Finazzi G, Landolfi R, Kutti
J, Gisslinger H, Patrono C, Marilus R, Villegas A, Tognoni G and
Barbui T: Vascular and neoplastic risk in a large cohort of
patients with polycythemia vera. J Clin Oncol. 23:2224–2232. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
No authors listed. Polycythemia vera: The
natural history of 1213 patients followed for 20 years. Gruppo
italiano studio policitemia. Ann Intern Med. 123:656–664. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Carobbio A, Thiele J, Passamonti F, Rumi
E, Ruggeri M, Rodeghiero F, Randi ML, Bertozzi I, Vannucchi AM,
Antonioli E, et al: Risk factors for arterial and venous thrombosis
in WHO-defined essential thrombocythemia: An international study of
891 patients. Blood. 117:5857–5859. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tefferi A, Lasho TL, Guglielmelli P, Finke
CM, Rotunno G, Elala Y, Pacilli A, Hanson CA, Pancrazzi A,
Ketterling RP, et al: Targeted deep sequencing in polycythemia vera
and essential thrombocythemia. Blood Adv. 1:21–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Barbui T, Finazzi G and Falanga A:
Myeloproliferative neoplasms and thrombosis. Blood. 122:2176–2184.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Arellano-Rodrigo E, Alvarez-Larrán A,
Reverter JC, Villamor N, Colomer D and Cervantes F: Increased
platelet and leukocyte activation as contributing mechanisms for
thrombosis in essential thrombocythemia and correlation with the
JAK2 mutational status. Haematologica. 91:169–175. 2006.PubMed/NCBI
|
|
46
|
Arellano-Rodrigo E, Alvarez-Larrán A,
Reverter JC, Colomer D, Villamor N, Bellosillo B and Cervantes F:
Platelet turnover, coagulation factors, and soluble markers of
platelet and endothelial activation in essential thrombocythemia:
Relationship with thrombosis occurrence and JAK2 V617F allele
burden. Am J Hematol. 84:102–108. 2009. View Article : Google Scholar
|
|
47
|
Beckman JD, DaSilva A, Aronovich E, Nguyen
A, Nguyen J, Hargis G, Reynolds D, Vercellotti GM, Betts B and Wood
DK: JAK-STAT inhibition reduces endothelial prothrombotic
activation and leukocyte-endothelial proadhesive interactions. J
Thromb Haemost. 21:1366–1380. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Guy A, Favre S, Labrouche-Colomer S,
Deloison L, Gourdou-Latyszenok V, Renault MA, Riviere E and James
C: High circulating levels of MPO-DNA are associated with
thrombosis in patients with MPN. Leukemia. 33:2544–2548. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Marin Oyarzún CP, Carestia A, Lev PR,
Glembotsky AC, Castro Ríos MA, Moiraghi B, Molinas FC, Marta RF,
Schattner M and Heller PG: Neutrophil extracellular trap formation
and circulating nucleosomes in patients with chronic
myeloproliferative neoplasms. Sci Rep. 6:387382016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ziegler S, Sperr WR, Knöbl P, Lehr S,
Weltermann A, Jäger U, Valent P and Lechner K: Symptomatic venous
thromboembolism in acute leukemia. Incidence, risk factors, and
impact on prognosis. Thromb Res. 115:59–64. 2005. View Article : Google Scholar
|
|
51
|
Ku GH, White RH, Chew HK, Harvey DJ, Zhou
H and Wun T: Venous thromboembolism in patients with acute
leukemia: Incidence, risk factors, and effect on survival. Blood.
113:3911–3917. 2009. View Article : Google Scholar
|
|
52
|
Vu K, Luong NV, Hubbard J, Zalpour A,
Faderl S, Thomas DA, Yang D, Kantarjian H and Kroll MH: A
retrospective study of venous thromboembolism in acute leukemia
patients treated at the University of Texas MD Anderson cancer
center. Cancer Med. 4:27–35. 2015. View Article : Google Scholar :
|
|
53
|
Poh C, Brunson A, Keegan T, Wun T and
Mahajan A: Incidence of upper extremity deep vein thrombosis in
acute leukemia and effect on mortality. TH Open. 4:e309–e317. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hell L, Thaler J, Martinod K, Ay C, Posch
F, Wagner DD and Pabinger I: OC-16-Neutrophil extracellular traps
and tissue factor-bearing microvesicles: A liaison dangereuse
causing overt DIC in cancer patients? Thromb Res. 140(Suppl 1):
S174–S175. 2016. View Article : Google Scholar
|
|
55
|
Ostafin M, Ciepiela O, Pruchniak M,
Wachowska M, Ulińska E, Mrówka P, Głodkowska-Mrówka E and Demkow U:
Dynamic changes in the ability to release neutrophil extracellular
traps in the course of childhood acute leukemias. Int J Mol Sci.
22:8212021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mei Y, Ren K, Liu Y, Ma A, Xia Z, Han X,
Li E, Tariq H, Bao H, Xie X, et al: Bone marrow-confined IL-6
signaling mediates the progression of myelodysplastic syndromes to
acute myeloid leukemia. J Clin Invest. 132:e1526732022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang TY, Dutta R, Benard B, Zhao F, Yin R
and Majeti R: IL-6 blockade reverses bone marrow failure induced by
human acute myeloid leukemia. Sci Transl Med. 12:eaax51042020.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tobler A, Moser B, Dewald B, Geiser T,
Studer H, Baggiolini M and Fey MF: Constitutive expression of
interleukin-8 and its receptor in human myeloid and lymphoid
leukemia. Blood. 82:2517–2525. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Corradi G, Bassani B, Simonetti G,
Sangaletti S, Vadakekolathu J, Fontana MC, Pazzaglia M, Gulino A,
Tripodo C, Cristiano G, et al: Release of IFNγ by acute myeloid
leukemia cells remodels bone marrow immune microenvironment by
inducing regulatory T cells. Clin Cancer Res. 28:3141–3155. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhong C, Wang R, Hua M, Zhang C, Han F, Xu
M, Yang X, Li G, Hu X, Sun T, et al: NLRP3 inflammasome promotes
the progression of acute myeloid leukemia via IL-1β pathway. Front
Immunol. 12:6619392021. View Article : Google Scholar
|
|
61
|
Basiorka AA, McGraw KL, Eksioglu EA, Chen
X, Johnson J, Zhang L, Zhang Q, Irvine BA, Cluzeau T, Sallman DA,
et al: The NLRP3 inflammasome functions as a driver of the
myelodysplastic syndrome phenotype. Blood. 128:2960–2975. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Borchmann S, Müller H, Hude I, Fuchs M,
Borchmann P and Engert A: Thrombosis as a treatment complication in
Hodgkin lymphoma patients: A comprehensive analysis of three
prospective randomized German Hodgkin Study Group (GHSG) trials.
Ann Oncol. 30:1329–1334. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ay C, Dunkler D, Marosi C, Chiriac AL,
Vormittag R, Simanek R, Quehenberger P, Zielinski C and Pabinger I:
Prediction of venous thromboembolism in cancer patients. Blood.
116:5377–5382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Thålin C, Hisada Y, Lundström S, Mackman N
and Wallén H: Neutrophil extracellular traps: Villains and targets
in arterial, venous, and cancer-associated thrombosis. Arterioscler
Thromb Vasc Biol. 39:1724–1738. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Aldinucci D, Celegato M and Casagrande N:
Microenvironmental interactions in classical Hodgkin lymphoma and
their role in promoting tumor growth, immune escape and drug
resistance. Cancer Lett. 380:243–252. 2016. View Article : Google Scholar
|
|
66
|
Ruf W, Rothmeier AS and Graf C: Targeting
clotting proteins in cancer therapy-progress and challenges. Thromb
Res. 140(Suppl 1): S1–S7. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Francischetti IMB, Seydel KB and Monteiro
RQ: Blood coagulation, inflammation, and malaria. Microcirculation.
15:81–107. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Leebeek FWG: Update of thrombosis in
multiple myeloma. Thromb Res. 140(Suppl 1): S76–S80. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Cavo M, Zamagni E, Cellini C, Tosi P,
Cangini D, Cini M, Valdrè L, Palareti G, Masini L, Tura S and
Baccarani M: Deep-vein thrombosis in patients with multiple myeloma
receiving first-line thalidomide-dexamethasone therapy. Blood.
100:2272–2273. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Esmon CT: Possible involvement of
cytokines in diffuse intravascular coagulation and thrombosis.
Baillieres Clin Haematol. 7:453–468. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zangari M, Barlogie B, Thertulien R,
Jacobson J, Eddleman P, Fink L, Fassas A, Van Rhee F, Talamo G, Lee
CK and Tricot G: Thalidomide and deep vein thrombosis in multiple
myeloma: Risk factors and effect on survival. Clin Lymphoma.
4:32–35. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Auwerda JJA, Sonneveld P, de Maat MPM and
Leebeek FWG: Prothrombotic coagulation abnormalities in patients
with paraprotein-producing B-cell disorders. Clin Lymphoma Myeloma.
7:462–466. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Minnema MC, Fijnheer R, De Groot PG and
Lokhorst HM: Extremely high levels of von Willebrand factor antigen
and of procoagulant factor VIII found in multiple myeloma patients
are associated with activity status but not with thalidomide
treatment. J Thromb Haemost. 1:445–449. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Petropoulou AD, Gerotziafas GT, Samama MM,
Hatmi M, Rendu F and Elalamy I: In vitro study of the
hypercoagulable state in multiple myeloma patients treated or not
with thalidomide. Thromb Res. 121:493–497. 2008. View Article : Google Scholar
|
|
75
|
Palumbo A, Rajkumar SV, San Miguel JF,
Larocca A, Niesvizky R, Morgan G, Landgren O, Hajek R, Einsele H,
Anderson KC, et al: International myeloma working group consensus
statement for the management, treatment, and supportive care of
patients with myeloma not eligible for standard autologous
stem-cell transplantation. J Clin Oncol. 32:587–600. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Terpos E, Kleber M, Engelhardt M, Zweegman
S, Gay F, Kastritis E, van de Donk NW, Bruno B, Sezer O, Broijl A,
et al: European myeloma network guidelines for the management of
multiple myeloma-related complications. Haematologica.
100:1254–1266. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Nielsen T, Kristensen SR, Gregersen H,
Teodorescu EM and Pedersen S: Prothrombotic abnormalities in
patients with multiple myeloma and monoclonal gammopathy of
undetermined significance. Thromb Res. 202:108–118. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zavidij O, Haradhvala NJ, Mouhieddine TH,
Sklavenitis-Pistofidis R, Cai S, Reidy M, Rahmat M, Flaifel A,
Ferland B, Su NK, et al: Single-cell RNA sequencing reveals
compromised immune microenvironment in precursor stages of multiple
myeloma. Nat Cancer. 1:493–506. 2020. View Article : Google Scholar
|
|
79
|
Jourdan M, Cren M, Robert N, Bolloré K,
Fest T, Duperray C, Guilloton F, Hose D, Tarte K and Klein B: IL-6
supports the generation of human long-lived plasma cells in
combination with either APRIL or stromal cell-soluble factors.
Leukemia. 28:1647–1656. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
de Jong MME, Chen L, Raaijmakers MHGP and
Cupedo T: Bone marrow inflammation in haematological malignancies.
Nat Rev Immunol. 24:543–558. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zangari M, Berno T, Zhan F, Tricot G and
Fink L: Mechanisms of thrombosis in paraproteinemias: The effects
of immunomodulatory drugs. Semin Thromb Hemost. 38:768–779. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Li W, Garcia D, Cornell RF, Gailani D,
Laubach J, Maglio ME, Richardson PG and Moslehi J: Cardiovascular
and thrombotic complications of novel multiple myeloma therapies: A
review. JAMA Oncol. 3:980–988. 2017. View Article : Google Scholar
|
|
83
|
Caruso V, Iacoviello L, Di Castelnuovo A,
Storti S, Mariani G, de Gaetano G and Donati MB: Thrombotic
complications in childhood acute lymphoblastic leukemia: A
meta-analysis of 17 prospective studies comprising 1752 pediatric
patients. Blood. 108:2216–2222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Kim TY, Gu JY, Jung HS, Koh Y, Kim I and
Kim HK: Elevated extracellular trap formation and contact system
activation in acute leukemia. J Thromb Thrombolysis. 46:379–385.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kumar R, Katare PB, Lentz SR, Modi AJ,
Sharathkumar AA and Dayal S: Thrombotic potential during pediatric
acute lymphoblastic leukemia induction: Role of cell-free DNA. Res
Pract Thromb Haemost. 5:e125572021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Knight JS, Luo W, O'Dell AA, Yalavarthi S,
Zhao W, Subramanian V, Guo C, Grenn RC, Thompson PR, Eitzman DT and
Kaplan MJ: Peptidylarginine deiminase inhibition reduces vascular
damage and modulates innate immune responses in murine models of
atherosclerosis. Circ Res. 114:947–956. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Shen Y, You Q, Wu Y and Wu J: Inhibition
of PAD4-mediated NET formation by cl-amidine prevents diabetes
development in nonobese diabetic mice. Eur J Pharmacol.
916:1746232022. View Article : Google Scholar
|
|
88
|
Knight JS, Subramanian V, O'Dell AA,
Yalavarthi S, Zhao W, Smith CK, Hodgin JB, Thompson PR and Kaplan
MJ: Peptidylarginine deiminase inhibition disrupts NET formation
and protects against kidney, skin and vascular disease in
lupus-prone MRL/lpr mice. Ann Rheum Dis. 74:2199–2206. 2015.
View Article : Google Scholar :
|
|
89
|
Shen W, Oladejo AO, Ma X, Jiang W, Zheng
J, Imam BH, Wang S, Wu X, Ding X, Ma B and Yan Z: Inhibition of
neutrophil extracellular traps formation by Cl-amidine alleviates
lipopolysaccharide-induced endometritis and uterine tissue damage.
Animals (Basel). 12:11512022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Novotny J, Chandraratne S, Weinberger T,
Philippi V, Stark K, Ehrlich A, Pircher J, Konrad I, Oberdieck P,
Titova A, et al: Histological comparison of arterial thrombi in
mice and men and the influence of Cl-amidine on thrombus formation.
PLoS One. 13:e01907282018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Jang EE, Wagner M and Sinclair J: National
survey on essential communication skills to address language
demands in Canadian nursing practice. J Adv Nurs. 82:347–365. 2026.
View Article : Google Scholar
|
|
92
|
Salzmann M, Gibler P, Haider P, Brekalo M,
Plasenzotti R, Filip T, Nistelberger R, Hartmann B, Wojta J,
Hengstenberg C, et al: Neutrophil extracellular traps induce
persistent lung tissue damage via thromboinflammation without
altering virus resolution in a mouse coronavirus model. J Thromb
Haemost. 22:188–198. 2024. View Article : Google Scholar
|
|
93
|
Perdomo J, Leung HHL, Ahmadi Z, Yan F,
Chong JJH, Passam FH and Chong BH: Neutrophil activation and
NETosis are the major drivers of thrombosis in heparin-induced
thrombocytopenia. Nat Commun. 10:13222019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ivey AD, Matthew Fagan B, Murthy P, Lotze
MT, Zeh HJ, Hazlehurst LA, Geldenhuys WJ and Boone BA: Chloroquine
reduces neutrophil extracellular trap (NET) formation through
inhibition of peptidyl arginine deiminase 4 (PAD4). Clin Exp
Immunol. 211:239–247. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Dyer MR, Alexander W, Hassoune A, Chen Q,
Brzoska T, Alvikas J, Liu Y, Haldeman S, Plautz W, Loughran P, et
al: Platelet-derived extracellular vesicles released after trauma
promote hemostasis and contribute to DVT in mice. J Thromb Haemost.
17:1733–1745. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Petri M, Konig MF, Li J and Goldman DW:
Association of higher hydroxychloroquine blood levels with reduced
thrombosis risk in systemic lupus erythematosus. Arthritis
Rheumatol. 73:9972021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Münzer P, Negro R, Fukui S, di Meglio L,
Aymonnier K, Chu L, Cherpokova D, Gutch S, Sorvillo N, Shi L, et
al: NLRP3 inflammasome assembly in neutrophils is supported by PAD4
and promotes NETosis under sterile conditions. Front Immunol.
12:6838032021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Broz P and Dixit VM: Inflammasomes:
Mechanism of assembly, regulation and signalling. Nat Rev Immunol.
16:407–420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wei X, Zhang B, Wei F, Ding M, Luo Z, Han
X and Tan X: Gegen Qinlian pills alleviate carrageenan-induced
thrombosis in mice model by regulating the HMGB1/NF-κB/NLRP3
signaling. Phytomedicine. 100:1540832022. View Article : Google Scholar
|
|
100
|
Fei J, Qin X, Ma H, Zhang X, Wang H, Han
J, Yu C and Jiang J: Resveratrol ameliorates deep vein
thrombosis-induced inflammatory response through inhibiting
HIF-1α/NLRP3 pathway. Inflammation. 45:2268–2279. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Harrison D, Boutard N, Brzozka K, Bugaj M,
Chmielewski S, Cierpich A, Doedens JR, Fabritius CRY, Gabel CA,
Galezowski M, et al: Discovery of a series of ester-substituted
NLRP3 inflammasome inhibitors. Bioorg Med Chem Lett. 30:1275602020.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Zhang W, Zhang Y, Han L, Bo T, Qi Z, Zhong
H, Xu H, Hu L, Chen S and Zhang S: Double-stranded DNA enhances
platelet activation, thrombosis, and myocardial injury via cyclic
GMP-AMP synthase. Cardiovasc Res. 121:353–366. 2025. View Article : Google Scholar
|
|
103
|
Yang S, Feng Y, Chen L, Wang Z, Chen J, Ni
Q, Guo X, Zhang L and Xue G: Disulfiram accelerates diabetic foot
ulcer healing by blocking NET formation via suppressing the
NLRP3/Caspase-1/GSDMD pathway. Transl Res. 254:115–127. 2023.
View Article : Google Scholar
|
|
104
|
Silva CMS, Wanderley CWS, Veras FP, Sonego
F, Nascimento DC, Gonçalves AV, Martins TV, Cólon DF, Borges VF,
Brauer VS, et al: Gasdermin D inhibition prevents multiple organ
dysfunction during sepsis by blocking NET formation. Blood.
138:2702–2713. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y,
Zhao J, Ruan J, Luo X, Lou X, Bai Y, et al: FDA-approved disulfiram
inhibits pyroptosis by blocking gasdermin D pore formation. Nat
Immunol. 21:736–745. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Adrover JM, Carrau L, Daßler-Plenker J,
Bram Y, Chandar V, Houghton S, Redmond D, Merrill JR, Shevik M,
tenOever BR, et al: Disulfiram inhibits neutrophil extracellular
trap formation and protects rodents from acute lung injury and
SARS-CoV-2 infection. JCI Insight. 7:e1573422022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Vottero P, Tavernini S, Santin AD, Scheim
DE, Tuszynski JA and Aminpour M: Computational prediction of the
interaction of ivermectin with fibrinogen. Int J Mol Sci.
24:114492023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Tagami T, Tosa R, Omura M, Fukushima H,
Kaneko T, Endo T, Rinka H, Murai A, Yamaguchi J, Yoshikawa K, et
al: Effect of a selective neutrophil elastase inhibitor on
mortality and ventilator-free days in patients with increased
extravascular lung water: A post hoc analysis of the PiCCO
pulmonary edema study. J Intensive Care. 2:672014. View Article : Google Scholar
|
|
109
|
Zhou P, Li T, Jin J, Liu Y, Li B, Sun Q,
Tian J, Zhao H, Liu Z, Ma S, et al: Interactions between neutrophil
extracellular traps and activated platelets enhance procoagulant
activity in acute stroke patients with ICA occlusion. EBioMedicine.
53:1026712020. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Zhang S, Cao Y, Du J, Liu H, Chen X, Li M,
Xiang M, Wang C, Wu X, Liu L, et al: Neutrophil extracellular traps
contribute to tissue plasminogen activator resistance in acute
ischemic stroke. FASEB J. 35:e218352021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Fuchs TA, Bhandari AA and Wagner DD:
Histones induce rapid and profound thrombocytopenia in mice. Blood.
118:3708–3714. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Abrams ST, Zhang N, Manson J, Liu T, Dart
C, Baluwa F, Wang SS, Brohi K, Kipar A, Yu W, et al: Circulating
histones are mediators of trauma-associated lung injury. Am J
Respir Crit Care Med. 187:160–169. 2013. View Article : Google Scholar :
|
|
113
|
Patel S, Kumar S, Jyoti A, Srinag BS,
Keshari RS, Saluja R, Verma A, Mitra K, Barthwal MK, Krishnamurthy
H, et al: Nitric oxide donors release extracellular traps from
human neutrophils by augmenting free radical generation. Nitric
Oxide. 22:226–234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Chen J, Zhou C, Fang W, Yin J, Shi J, Ge
J, Shen L, Liu SM and Liu SJ: Identification of endothelial protein
C receptor as a novel druggable agonistic target for
reendothelialization promotion and thrombosis prevention of eluting
stent. Bioact Mater. 41:485–498. 2024.PubMed/NCBI
|
|
115
|
Méndez D, Arauna D, Fuentes F,
Araya-Maturana R, Palomo I, Alarcón M, Sebastián D, Zorzano A and
Fuentes E: Mitoquinone MitoQ) inhibits platelet activation steps by
reducing ROS levels. Int J Mol Sci. 21:61922020. View Article : Google Scholar
|
|
116
|
Wang SB, Jang JY, Chae YH, Min JH, Baek
JY, Kim M, Park Y, Hwang GS, Ryu JS and Chang TS: Kaempferol
suppresses collagen-induced platelet activation by inhibiting NADPH
oxidase and protecting SHP-2 from oxidative inactivation. Free
Radic Biol Med. 83:41–53. 2015. View Article : Google Scholar
|
|
117
|
Osmanoglu Ö, Gupta SK, Almasi A, Yagci S,
Srivastava M, Araujo GHM, Nagy Z, Balkenhol J and Dandekar T:
Signaling network analysis reveals fostamatinib as a potential drug
to control platelet hyperactivation during SARS-CoV-2 infection.
Front Immunol. 14:12853452023. View Article : Google Scholar
|
|
118
|
Strich JR, Ramos-Benitez MJ, Randazzo D,
Stein SR, Babyak A, Davey RT, Suffredini AF, Childs RW and Chertow
DS: Fostamatinib inhibits neutrophils extracellular traps induced
by COVID-19 patient plasma: A potential therapeutic. J Infect Dis.
223:981–984. 2021. View Article : Google Scholar :
|
|
119
|
Huang ZS, Zeng CL, Zhu LJ, Jiang L, Li N
and Hu H: Salvianolic acid A inhibits platelet activation and
arterial thrombosis via inhibition of phosphoinositide 3-kinase. J
Thromb Haemost. 8:1383–1393. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Rysenga CE, May-Zhang L, Zahavi M, Knight
JS and Ali RA: Taxifolin inhibits NETosis through activation of
Nrf2 and provides protective effects in models of lupus and
antiphospholipid syndrome. Rheumatology (Oxford). 63:2006–1015.
2024. View Article : Google Scholar
|
|
121
|
Smith CW, Campos J, Brown HC, Jooss NJ,
Ivanova VS, Harbi M, Garcia-Quintanilla L, Jossi S, Perez-Toledo M,
Rookes K, et al: Selective Btk inhibition by PRN1008/PRN473 blocks
human CLEC-2, and PRN473 reduces venous thrombosis formation in
mice. Blood Adv. 8:5557–5570. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Xu S, Fan F, Liu H, Cheng S, Tu M and Du
M: Novel anticoagulant peptide from lactoferrin binding thrombin at
the active site and exosite-I. J Agric Food Chem. 68:3132–3139.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Ryan TAJ, Hooftman A, Rehill AM, Johansen
MD, Brien ECO, Toller-Kawahisa JE, Wilk MM, Day EA, Weiss HJ,
Sarvari P, et al: Dimethyl fumarate and 4-octyl itaconate are
anticoagulants that suppress tissue factor in macrophages via
inhibition of type I Interferon. Nat Commun. 14:35132023.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Weber AG, Chau AS, Egeblad M, Barnes BJ
and Janowitz T: Nebulized in-line endotracheal dornase alfa and
albuterol administered to mechanically ventilated COVID-19
patients: A case series. Mol Med. 26:912020. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Gollomp K, Kim M, Johnston I, Hayes V,
Welsh J, Arepally GM, Kahn M, Lambert MP, Cuker A, Cines DB, et al:
Neutrophil accumulation and NET release contribute to thrombosis in
HIT. JCI Insight. 3:e994452018. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Várady CBS, Oliveira AC, Monteiro RQ and
Gomes T: Recombinant human DNase I for the treatment of
cancer-associated thrombosis: A pre-clinical study. Thromb Res.
203:131–137. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Kessinger CW, Kim JW, Henke PK, Thompson
B, McCarthy JR, Hara T, Sillesen M, Margey RJ, Libby P, Weissleder
R, et al: Statins improve the resolution of established murine
venous thrombosis: Reductions in thrombus burden and vein wall
scarring. PLoS One. 10:e01166212015. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Chen D, Li Q, Liang H, Huang L, Zhou H,
Zheng X and Wang Z: Exenatide enhanced the antitumor efficacy on
PD-1 blockade by the attenuation of neutrophil extracellular traps.
Biochem Biophys Res Commun. 619:97–103. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Cameron-Vendrig A, Reheman A, Siraj MA, Xu
XR, Wang Y, Lei X, Afroze T, Shikatani E, El-Mounayri O, Noyan H,
et al: Glucagon-like peptide 1 receptor activation attenuates
platelet aggregation and thrombosis. Diabetes. 65:1714–1723. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Highfill SL, Cui Y, Giles AJ, Smith JP,
Zhang H, Morse E, Kaplan RN and Mackall CL: Disruption of
CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy.
Sci Transl Med. 6:237ra672014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Pedersen F, Waschki B, Marwitz S, Goldmann
T, Kirsten A, Malmgren A, Rabe KF, Uddin M and Watz H: Neutrophil
extracellular trap formation is regulated by CXCR2 in COPD
neutrophils. Eur Respir J. 51:17009702018. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Alsabani M, Abrams ST, Cheng Z, Morton B,
Lane S, Alosaimi S, Yu W, Wang G and Toh CH: Reduction of NETosis
by targeting CXCR1/2 reduces thrombosis, lung injury, and mortality
in experimental human and murine sepsis. Br J Anaesth. 128:283–293.
2022. View Article : Google Scholar
|
|
133
|
Kessenbrock K, Krumbholz M, Schönermarck
U, Back W, Gross WL, Werb Z, Gröne HJ, Brinkmann V and Jenne DE:
Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med.
15:623–625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Zhao D, Han C, Mammadova-Bach E,
Watanabe-Kusunoki K, Bandeira Honda TS, Li Y, Li C, Li Q, Long H,
Lyubenov L, et al: Inhibition of complement factor C5a or C5aR for
cholesterol crystal embolism-related vascular thrombosis with
microvascular injury and its consequences. Kidney Int. 106:819–825.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Goerge T, Niemeyer A, Rogge P, Ossig R,
Oberleithner H and Schneider SW: Secretion pores in human
endothelial cells during acute hypoxia. J Membr Biol. 187:203–211.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Zhao BQ, Chauhan AK, Canault M, Patten IS,
Yang JJ, Dockal M, Scheiflinger F and Wagner DD: von Willebrand
factor-cleaving protease ADAMTS13 reduces ischemic brain injury in
experimental stroke. Blood. 114:3329–3334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Denorme F, Langhauser F, Desender L,
Vandenbulcke A, Rottensteiner H, Plaimauer B, François O, Andersson
T, Deckmyn H, Scheiflinger F, et al: ADAMTS13-mediated thrombolysis
of t-PA-resistant occlusions in ischemic stroke in mice. Blood.
127:2337–2345. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Zhang YR, Liu YR, Tang ZS, Song ZX, Zhang
JW, Chang BJ, Zhao ML and Xu J: Rheum officinale Baill. Treats
zebrafish embryo thrombosis by regulating NOS3 expression in the
arginine biosynthesis pathway. Phytomedicine. 99:1539672022.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Amadio P, Colombo GI, Tarantino E,
Gianellini S, Ieraci A, Brioschi M, Banfi C, Werba JP, Parolari A,
Lee FS, et al: BDNFVal66met polymorphism: A potential bridge
between depression and thrombosis. Eur Heart J. 38:1426–1435.
2017.
|