Introduction
Malignant pleural mesothelioma (MPM) is an
aggressive neoplasm that arises from mesothelial cells. It was
reported that asbestos, iron, and simian virus 40 were linked to
the etiology of MPM (1–4). MPM was once considered a rare
disease, but its incidence is increasing worldwide (5).
Current aggressive multimodality therapy for MPM
(consisting of surgical resection, cytotoxic chemotherapy, and
radiation) offers survival benefits for only a small subset of
patients in early stages of the disease (6). Recently, the multi-targeted
antifolate pemetrexed has been approved as the front-line agent in
combination with cisplatin for the treatment of MPM (7). However, most of the patients relapsed
within a year after starting treatments. Therefore, new and more
effective therapies are necessary to improve the prognosis of this
disease.
The mitogen-activated protein (MAP) kinase kinase
(MEK)-extracellular signal-regulated kinase (ERK) pathway and the
phosphatidylinositol 3-kinase (PI3K)-Akt pathway play critical
roles in the regulation of cell proliferation, growth,
differentiation and survival (8–10).
These pathways are activated in many types of solid tumor models,
including MPM (8,9,11–13).
It was also reported that inhibition of these pathways affected the
proliferation of MPM cell lines in vitro(14,15).
However, no report has demonstrated growth-inhibitory effects of
these agents on MPM cells in vivo.
In the present study, we examined whether the MEK or
the PI3K inhibitor affected the growth of MPM cells in vitro
and in vivo. Furthermore, we evaluated the possibility that
combined use of the MEK inhibitor and the PI3K inhibitor might
enhance MPM treatment.
Materials and methods
Cell cultures
The human mesothelioma cell line EHMES-10 was
established from the pleural effusion of a patient with MPM in our
institution (16,17). MSTO211H was purchased from the
American Type Culture Collection (Manassas, VA, USA). These cell
lines were cultured in RPMI-1640 medium (Nikken Bio Medical
Laboratories, Kyoto, Japan) supplemented with 10% fetal bovine
serum (Hyclone, Logan, UT, USA), penicillin (100 U/ml) and
streptomycin (50 mg/ml) in a 37°C humidified incubator with 5%
carbon dioxide.
Reagents and inhibitors
MEK inhibitor U0126 and PI3K inhibitor LY294002 were
purchased from LC Laboratories (Woburn, MA, USA). For in
vitro experiments, these agents were dissolved in
dimethylsulfoxide (DMSO) (Sigma-Aldrich Co., St. Louis, MO, USA)
and were added to cells in medium with a final DMSO concentration
of 1.0%. For in vivo studies, these agents were prepared as
a suspension in a vehicle consisting of 40% DMSO in
phosphate-buffered saline (PBS) (Wako Pure Chemical Industries,
Osaka, Japan). Rabbit polyclonal antibodies against ERK1/2,
phospho-ERK1/2, Akt, phospho-Akt, p27kip1, cyclin E, cyclin D1,
p70S6K, phospho-p70S6K, S6, phospho-S6, p90 ribosomal S6 kinase
(p90RSK), phospho-p90RSK, glycogen synthase kinase-3β (GSK3β),
phospho-GSK3β, Bad, phospho-Bad, poly(ADP-ribose) polymerase
(PARP), procaspase 3, hypoxia-inducible factor 1α (HIF1α), and
β-actin were purchased from Cell Signaling Technology (Danvers, MA,
USA). Rabbit polyclonal antibody against vascular endothelial
growth factor (VEGF) was purchased from Millipore Co. (Tokyo,
Japan). Mouse monoclonal antibody against CD31/platelet/endothelial
cell adhesion molecule-1 was purchased from BD Pharmingen (Tokyo,
Japan) for in vivo immunohistochemical study. Mouse
monoclonal antibody against CD31 (PECAM-1) was purchased from Cell
Signaling Technology for in vivo western blot analysis.
Horseradish peroxidase conjugated goat anti-rabbit IgG and horse
anti-mouse IgG were purchased from Cell Signaling Technology.
Cell proliferation assay
The cell proliferation assay reagent WST-1
(4-[3-(4-lodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene
disulfonate) (Roche Diagnostics GmbH, Mannheim, Germany) was used
to assess the effect of U0126 or LY294002 on cell growth. MPM cells
(1×104 cells/well) were plated in 96-well plates (Nunc,
Roskilde, Denmark) and were exposed to various concentrations of
test agents dissolved in DMSO. Controls received DMSO vehicle at a
concentration equal to that of drug treated cells. After drug
treatment for 72 h, 10 μl of WST-1 reagent were added to
each well. Absorbance was measured at 450 nm with a reference
wavelength at 690 nm by an E max precision microplate reader
(Molecular Devices, Tokyo, Japan).
Cell cycle analysis
MPM cells, treated with or without test agents for
24 h, were trypsinized and collected, and the cell nuclei were
stained using the CycleTest Plus DNA Reagent Kit (Becton-Dickinson,
San Jose, CA, USA). Cells were subjected to FACScan analysis, and
cell cycle profiles were determined using ModFitLT software
(Becton-Dickinson, San Diego, CA, USA). This analysis was carried
out independently three times.
DNA fragmentation assay
We examined DNA fragmentation to assess apoptosis in
EHMES-10 or MSTO211H cells. Cells were treated with either U0126 or
LY294002 or a combination of both for 24 h. DNA fragmentation was
evaluated using the Cell Death Detection ELISA kit (Roche Molecular
Biochemical, Indianapolis, IN, USA) as previously reported
(18).
Western blot analysis
Cultured cells were treated with lysis buffer [25 mM
Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X, 50 mM NaF and 1 mM
Na3VO4] containing proteinase inhibitor
cocktail (Roche Diagnostics GmbH). Tumor tissue samples were
homogenized in lysis buffer. Insoluble materials were removed by
centrifugation at 4°C for 15 min 15,000 × g. Protein concentration
was determined using a Bio Rad Protein Assay Kit (Bio Rad
Laboratories, Hercules, CA, USA).
Proteins were separated on 7.5 to 15% polyacrylamide
gels (Bio Rad Laboratories). After electrophoresis, the protein was
transferred to a nitrocellulose membrane and detected by
immunoblotting using SNAP i.d. Protein Detection System (Millipore
Co.) as previously described (19). This analysis was carried out
independently three times.
Experimental animals
Male severe combined immunodeficient (SCID) mice
(six to eight weeks old) were obtained from Clea Japan (Osaka,
Japan), fed autoclaved standard pellets and water, and maintained
under specific pathogen-free conditions throughout this study. All
of the protocols involving SCID mice were approved by the
guidelines established by the Ehime University Committee on Animal
Care and Use.
Orthotopic implantation model
Cultured EHMES-10 cells were harvested, washed twice
and re-suspended in PBS. The SCID mice were inoculated in the
thoracic cavity with the tumor cells (3×106
cells/mouse), as previously described (17,20).
Seven days after inoculation, mice were randomized into eight
groups (n=7 mice/group) to receive vehicle alone (DMSO + PBS),
U0126 alone (20, 30 and 40 mg/kg), LY294002 alone (12.5, 25 and 50
mg/kg) and a combination of U0126 (30 mg/kg) and LY294002 (25
mg/kg). These agents were administrated intraperitoneally twice a
week. Mice were sacrificed on day 30 after tumor cell inoculation.
The tumor tissue was excised and weighed, and the volume of pleural
effusion was measured. We also measured the body weights and serum
levels of total protein (TP), blood urea nitrogen (BUN), creatinine
(Cre), aspartate amino transferase (AST) and alanine
aminotransferase (ALT), and evaluated the degree of dermatopathy as
a measure of side effects.
Immunohistochemistry
Paraffin-embedded tissues were subjected to
immunohistochemistry with anti-phospho-ERK1/2 monoclonal antibody,
phospho-Akt monoclonal antibody, or anti-p27kip1 monoclonal
antibody. For in situ apoptosis detection, we used terminal
deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL
assay) with the In situ Apoptosis Detection Kit (Takara
Biomedicals, Ohtsu, Japan). Frozen tissue sections were used for
identification of endothelial cells using rat anti-mouse
CD31/platelet/endothelial cell adhesion molecule-1 monoclonal
antibody. Immunohistochemical procedures were performed using the
Envision™ Systems (Dako, Glostrup, Denmark) method, as previously
described (21). Phospho-ERK1/2-
or phospho-Akt- or p27kip1-positive cells were visualized with
Fuchsin+ substrate-chromogen (Dako). Antibodies against
TUNEL assay or CD31 localization were detected using a peroxidase
reaction with 3-diaminobenzidine (Dako).
Statistical analysis
In vitro study data are presented as means ±
SD, and were analyzed using ANOVA followed by Dunnett’s t-test.
In vivo data were expressed as median values and ranges. The
Mann-Whitney U test was used to compare groups. The Kaplan-Meier
method was used to evaluate the survival analysis and comparisons
were made using a log-rank test. Drug interactions were analyzed by
the Chou and Talalay method using the CalcuSyn software program
(version 2.0; Biosoft, Cambridge, UK). The combination index (CI)
was simulated from each level of fractional affect. According to
this method, a CI<0.3, 0.3–0.7, 0.7–0.9, 0.9–1.1, 1.1–1.45,
1.45–3.3 and >3.3 indicates highly synergistic, synergistic,
moderate to slight synergistic, nearly additive, slight to moderate
antagonistic, antagonistic and strong antagonistic, respectively.
Differences between groups are considered statistically significant
at P<0.05.
Results
Growth inhibition of MPM cells by U0126
and/or LY294002 treatment
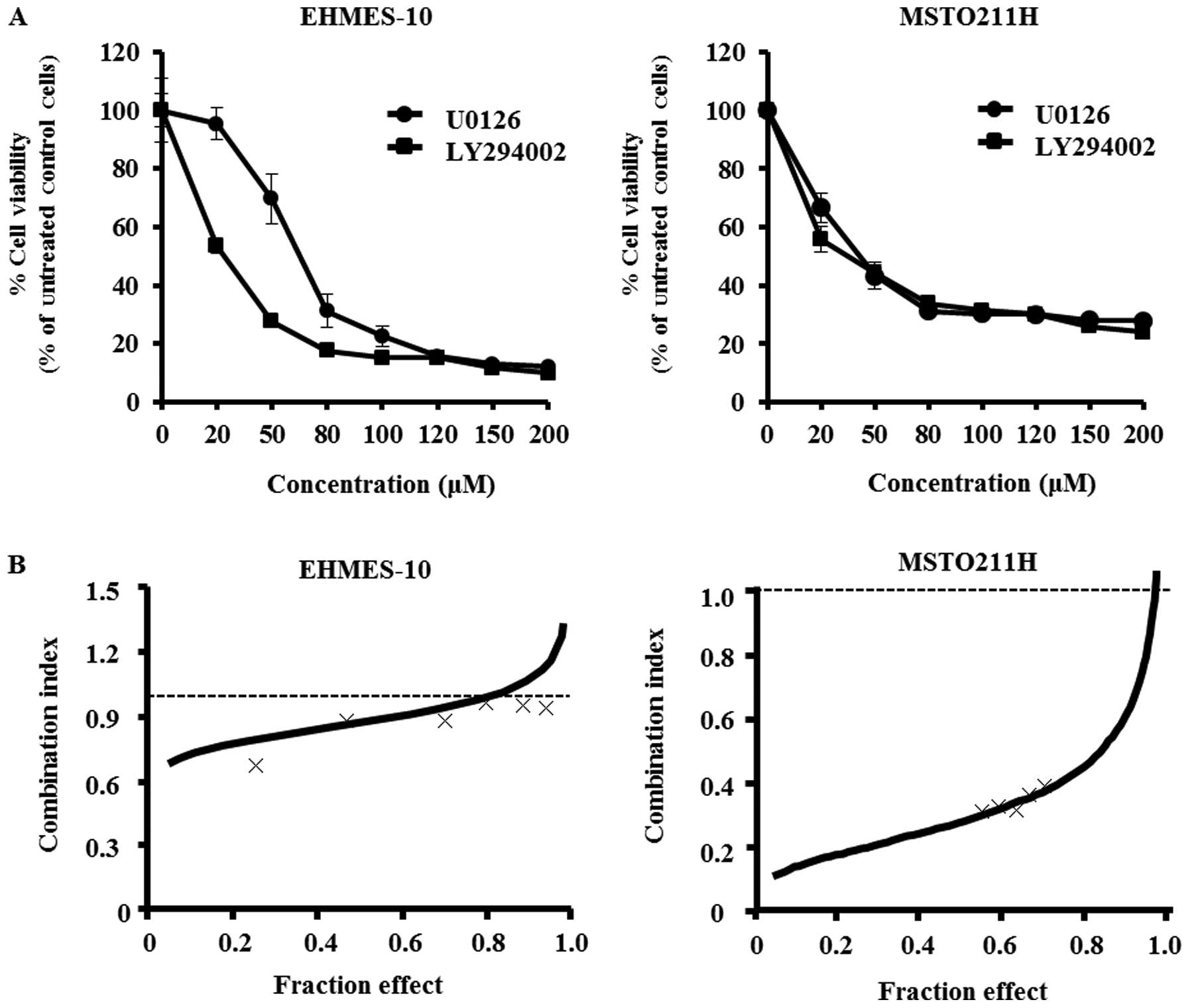
The effects of U0126 or LY294002 at concentrations
ranging from 20 to 200 μM on the proliferation of EHMES-10
or MSTO211H cells were determined with the WST-1 assay. Each agent
inhibited MPM cell growth in a dose-dependent manner (Fig. 1A). The IC50 values for
U0126 and LY294002 against EHMES-10 cells were 66.8 μM and
20.7 μM, respectively. Moreover, the IC50 values
for U0126 and LY294002 against MSTO211H cells were 39.0 μM
and 29.9 μM, respectively.
We evaluated the effect of combining treatments with
U0126 and LY294002. The ratio of IC50 values for U0126
and LY294002 against EHMES-10 cells was approximately 3:1 while the
ratio was 4:3 against MSTO211H cells. Therefore, the two MPM cell
lines were exposed to varying concentrations of U0126 and LY294002
at fixed ratios of 3:1 or 4:3, as appropriate. Cell viability was
then assessed by the WST-1 assay. The averaged CIs for EHMES-10
cells and MSTO211H cells were 1.017 and 0.54, which indicates a
nearly additive effect and a synergistic effect, respectively
(Fig. 1B).
Induced G1 cell cycle arrest of MPM cells
after treatment with U0126 and/or LY294002
To investigate the mechanisms of growth inhibition
of MPM cells by U0126 or LY294002 treatment, we performed cell
cycle analysis of EHMES-10 cells or MSTO211H cells treated with 80
μM U0126 and/or 80 μM LY294002. Treatment with U0126
or LY294002 for 24 h significantly increased the G1-phase
populations compared to control in both MPM cell lines (all,
P<0.05) (Fig. 2A). In addition,
U0126 alone and LY294002 alone significantly increased the
percentage of MPM cells in the sub-G1 phase, indicative of cell
apoptosis, compared to control (all, P<0.05). Combining
treatment with U0126 with that of LY294002 led to a significant
increase in the sub-G1 phase population in both cell lines compared
to control or individual drugs (all, P<0.01).
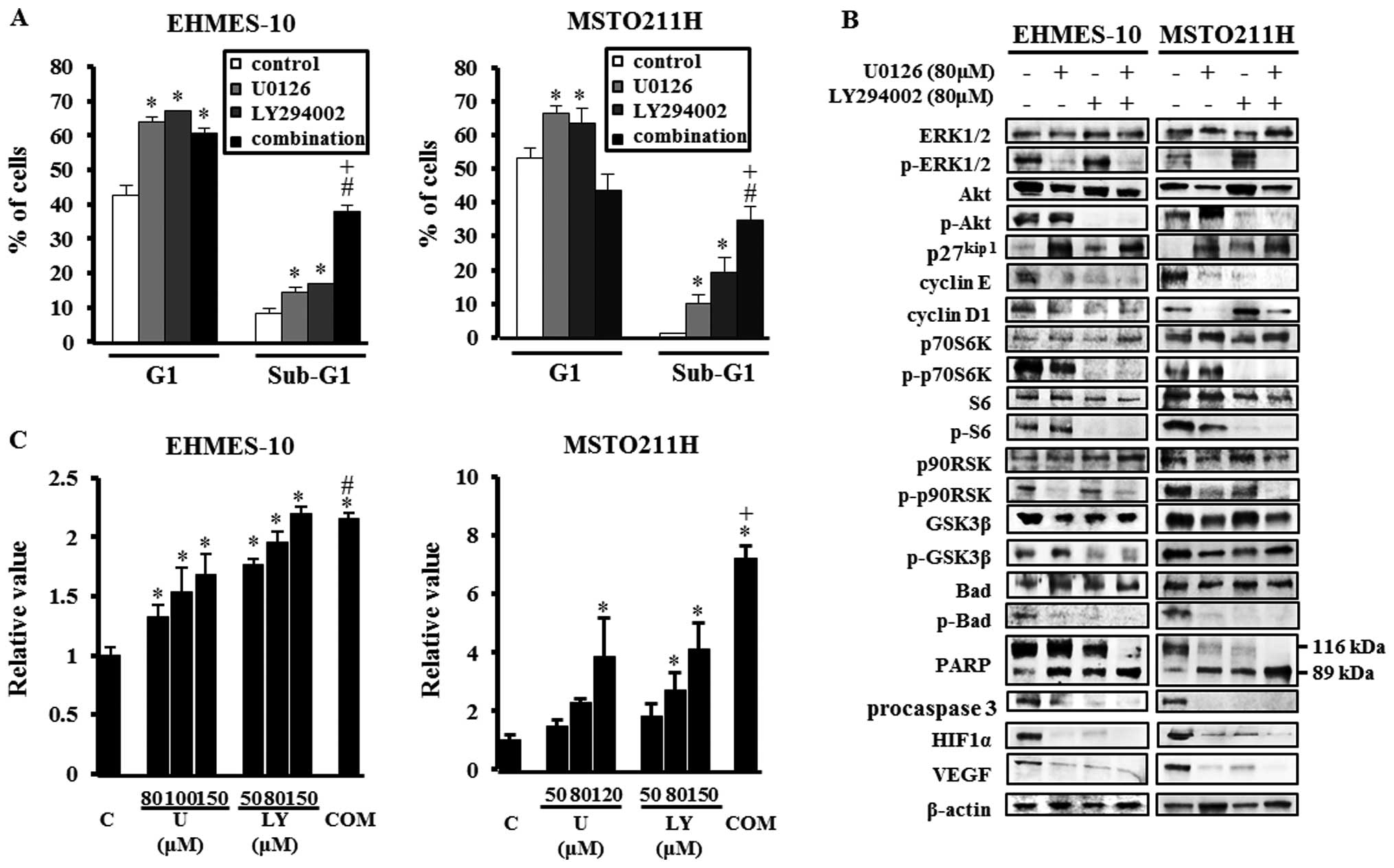 | Figure 2The mechanisms by which U0126 or
LY294002 inhibited growth of EHMES-10 cells and MSTO211H cells
in vitro. (A) The effects of U0126 and/or LY294002 on the
cell cycle profile. After treatment with 80 μM U0126 and/or
80 μM LY294002 for 24 h, MPM cells were collected, fixed,
strained with propidium iodide and analyzed by flow cytometry. Data
shown are representative of three independent experiments.
*P<0.05, compared to control; #P<0.01,
compared to control; +P<0.01, compared to individual
drug. (B) Effects of U0126 and/or LY294002 on the expression of
phospho-extracellular signal-regulated kinase (ERK)1/2 (p-ERK1/2),
phospho-Akt (p-Akt), p27kip1, cyclin E, cyclin D1, phospho-p70S6K
(p-p70S6K), phospho-S6 (p-S6), phospho-p90 ribosomal S6 kinase
(p90RSK) (p-p90RSK), phospho-glycogen synthase kinase-3β (GSK3β)
(p-GSK3β), phospho-Bad (p-Bad), poly (ADP-ribose) polymerase
(PARP), procaspase 3, hypoxia-inducible factor 1α (HIF1α) and
vascular endothelial growth factor (VEGF). Tumor cells were treated
with or without U0126 (80 μM), and/or LY294002 (80
μM) for 24 h. Then, cells were lysed, and the indicated
proteins were detected by immunoblotting. Data shown are
representative of three independent experiments. (C) Cytoplasmic
histone-associated DNA fragments determined by ELISA-based
quantification. MPM cells were treated with U0126 or LY294002 or a
combination of 80 μM U0126 and 80 μM LY294002 for 24
h. C, control; U, U0126; LY, LY294002; COM, combination of 80
μM U0126 and 80 μM LY294002; *P<0.01,
compared to control; #P<0.01, compared to treatment
with 80 μM U0126 alone; +P<0.01, compared to
the individual drug. |
We also analyzed the expression of cell cycle
regulatory proteins after treatment with U0126 and/or LY294002 in
both EHMES-10 cells and MSTO211H cells. Both agents increased
p27kip1 expression and decreased cyclin E expression in both cell
lines (Fig. 2B). A decrease of
cyclin D1 expression was observed in treatment with either U0126 or
LY294002 in EHMES-10 cells, and following treatment with U0126 in
MSTO211H cells.
Induction of apoptosis by U0126 and/or
LY294002 treatment
We assessed the ability of U0126 and LY294002 to
induce apoptosis in MPM cells. DNA fragmentation analysis showed
that treatment with U0126 or LY294002 alone induced apoptosis in
EHMES-10 cells and in MSTO211H cells in a dose-dependent manner.
Furthermore, the combined treatment with 80 μM U0126 and 80
μM LY294002 significantly increased the number of apoptotic
cells compared to control and to treatment with U0126 alone in
EHMES-10 cells (all, P<0.01) and compared to control and
treatment with the individual drug in MSTO211H cells (all,
P<0.01) (Fig. 2C). Western blot
analysis showed that treatments with U0126 and/or LY294002
increased the level of 89 kDa cleaved PARP and decreased the levels
of phospho-Bad and procaspase 3 in both cell lines (Fig. 2B).
Signaling alterations induced by
treatment with U0126 and/or LY294002
To investigate the effects of U0126 and LY294002 on
intercellular signaling, MPM cells were treated with U0126 or
LY294002 or a combination of both. As shown in Fig. 2B, U0126 blocked the phosphorylation
of ERK1/2 and p90RSK, and decreased HIF1α and VEGF expression in
EHMES-10 cells. On the other hand, treatment with LY294002
suppressed the phosphorylation of Akt, p70S6K, S6 and GSK3β, and
inhibited HIF1α and VEGF expression in EHMES-10 cells. Use of the
combination treatment inhibited the phosphorylation of all of the
above proteins and decreased the level of HIF1α and VEGF in
EHMES-10 cells. Signaling alterations in MSTO211H cells tended to
be similar to those in EHMES-10 cells, except for phospho-GSK3β
alteration.
Antitumor activity of U0126 and/or
LY294002 in EHMES-10 cell xenografts
To assess the in vivo therapeutic efficacy of
U0126 and/or LY294002, SCID mice bearing EHMES-10 xenografts were
treated with vehicle, U0126, LY294002, or a combination of U0126
and LY294002 as described in Materials and methods. Administration
of 30 or 40 mg/kg of U0126, or 50 mg/kg of LY294002, or use of
combined therapy with 30 mg/kg of U0126 and 25 mg/kg of LY294002
significantly prolonged the survival time of EHMES-10 cell-bearing
SCID mice compared to the control group (all, P<0.01) (Fig. 3A–C). The combination therapy more
effectively prolonged the survival time compared to treatment with
either individual drug, although statistical significance was not
obtained (Fig. 3C).
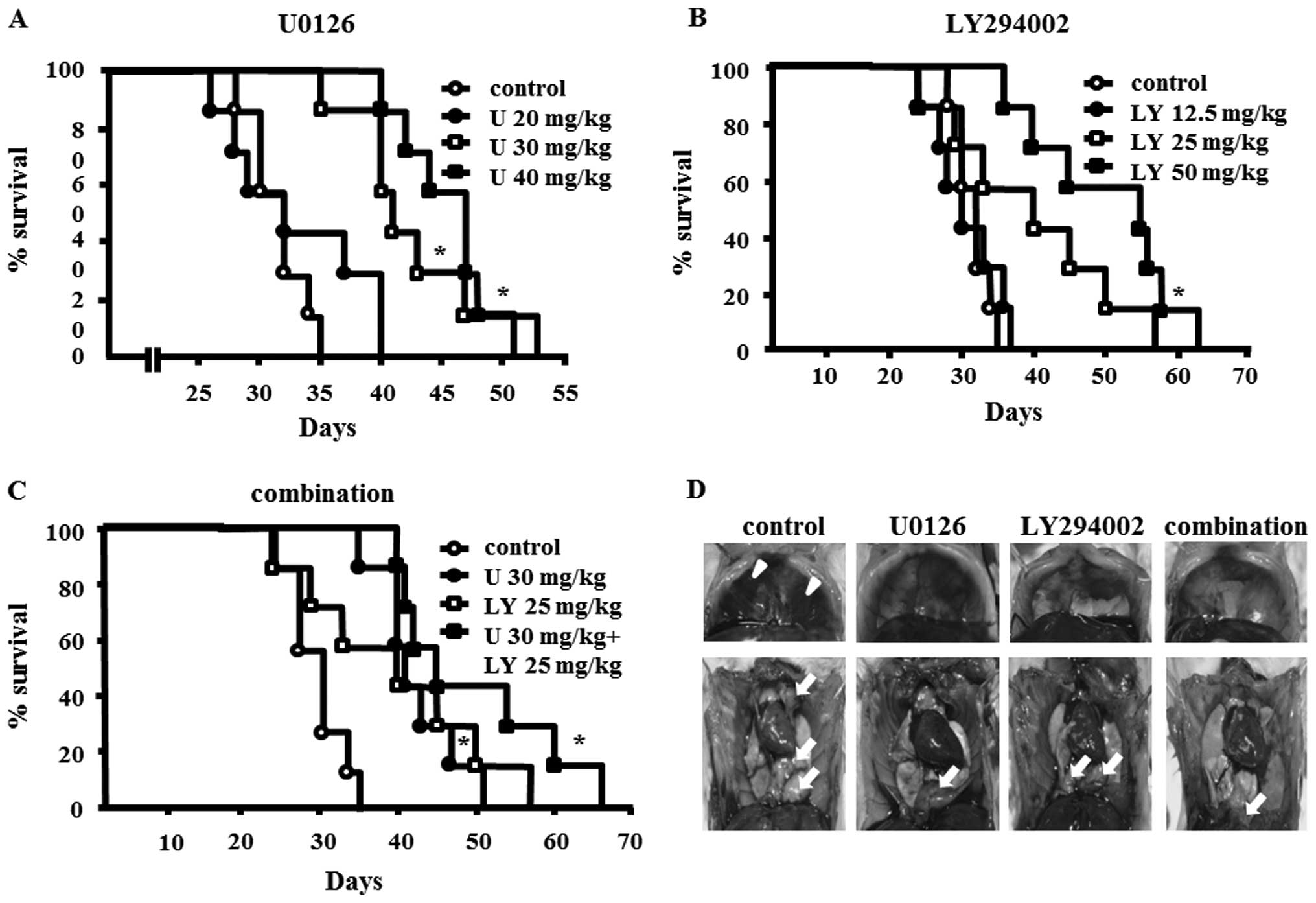 | Figure 3Effects of U0126 or LY294002 or the
combination of U1026 and LY294002 on severe combined
immunodeficiency (SCID) mice bearing EHMES-10 cells. (A–C) Survival
times of EHMES-10 cell-bearing SCID mice treated with U0126,
LY294002, or the combination of U0126 and LY294002. EHMES-10 cells
(3×106) were inoculated into the thoracic cavity of SCID
mice. Seven days after inoculation, SCID mice were randomized into
eight groups (n=7 mice/group) to receive vehicle (DMSO + PBS),
U0126 (20, 30 and 40 mg/kg), or LY294002 (12.5, 25 and 50 mg/kg),
or a combination of U0126 (30 mg/kg) and LY294002 (25 mg/kg). U,
U0126; LY, LY294002; *P<0.01, compared to control.
(D) Formation of thoracic tumors and pleural effusion by EHMES-10
cells with or without test agents. Mice were sacrificed on day 30
and thoracic tumors and pleural effusions were evaluated.
Arrowheads, pleural effusions; arrows, thoracic tumors. |
We also evaluated the effect of U0126 and/or
LY294002 on the production of thoracic tumors and pleural effusion
in EHMES-10 cell-bearing SCID mice (Fig. 3D, Table I). U0126 and/or LY294002
significantly inhibited tumor growth and pleural effusion
production compared to control (all, P<0.05). The combination
therapy more effectively inhibited tumor growth compared to
treatments with individual drugs, although statistical significance
was not obtained.
 | Table IEffects of U0126, LY294002 and
combination therapy on thoracic tumor and pleural effusion produced
by MPM cells in SCID mice. |
Table I
Effects of U0126, LY294002 and
combination therapy on thoracic tumor and pleural effusion produced
by MPM cells in SCID mice.
| Thoracic tumor
| Pleural effusion
|
|---|
| Incidence | Weight (mg) | Incidence | Volume
(μl) |
|---|
| Control | 5/5 | 487.7
(162.8–907.1) | 5/5 | 150
(50.0–280.0) |
| U0126 | | | | |
| 30 mg/kg | 4/5 | 31.3
(0–59.2)a | 1/5 | 0 (0–100)a |
| 40 mg/kg | 3/5 | 14.6
(0–191.3)a | 0/5 | 0 (−)a |
| LY294002 | | | | |
| 25 mg/kg | 3/5 | 69.1
(0–481.6)a | 1/5 | 0 (0–10)a |
| 50 mg/kg | 2/5 | 0 (0–41)a | 0/5 | 0 (−)a |
| Combination | 3/5 | 7.5 (0–8.6)a | 0/5 | 0 (−)a |
Immunohistochemical staining and western
blot analysis to clarify the antitumor mechanisms of U0126 and/or
LY294002 in vivo
Immunohistochemical analysis showed that treatment
with U0126 or with LY294002 reduced phospho-ERK1/2-positive tumor
cells or phospho-Akt-positive tumor cells, respectively (Fig. 4A). Furthermore, treatment with an
individual drug increased the number of p27kip1-positive tumor
cells and TUNEL assay-positive tumor cells, and decreased the
number of CD31-positive endothelial cells. The combination therapy
group showed decreased phospho-ERK1/2 and phospho-Akt activities
and CD31-positive endothelial cells, and increased p27kip1-positive
tumor cells and TUNEL assay-positive tumor cells. The effect of the
inhibitors delivered in combination was more pronounced than the
drugs applied individually in the analyses of p27kip1 and TUNEL
assay.
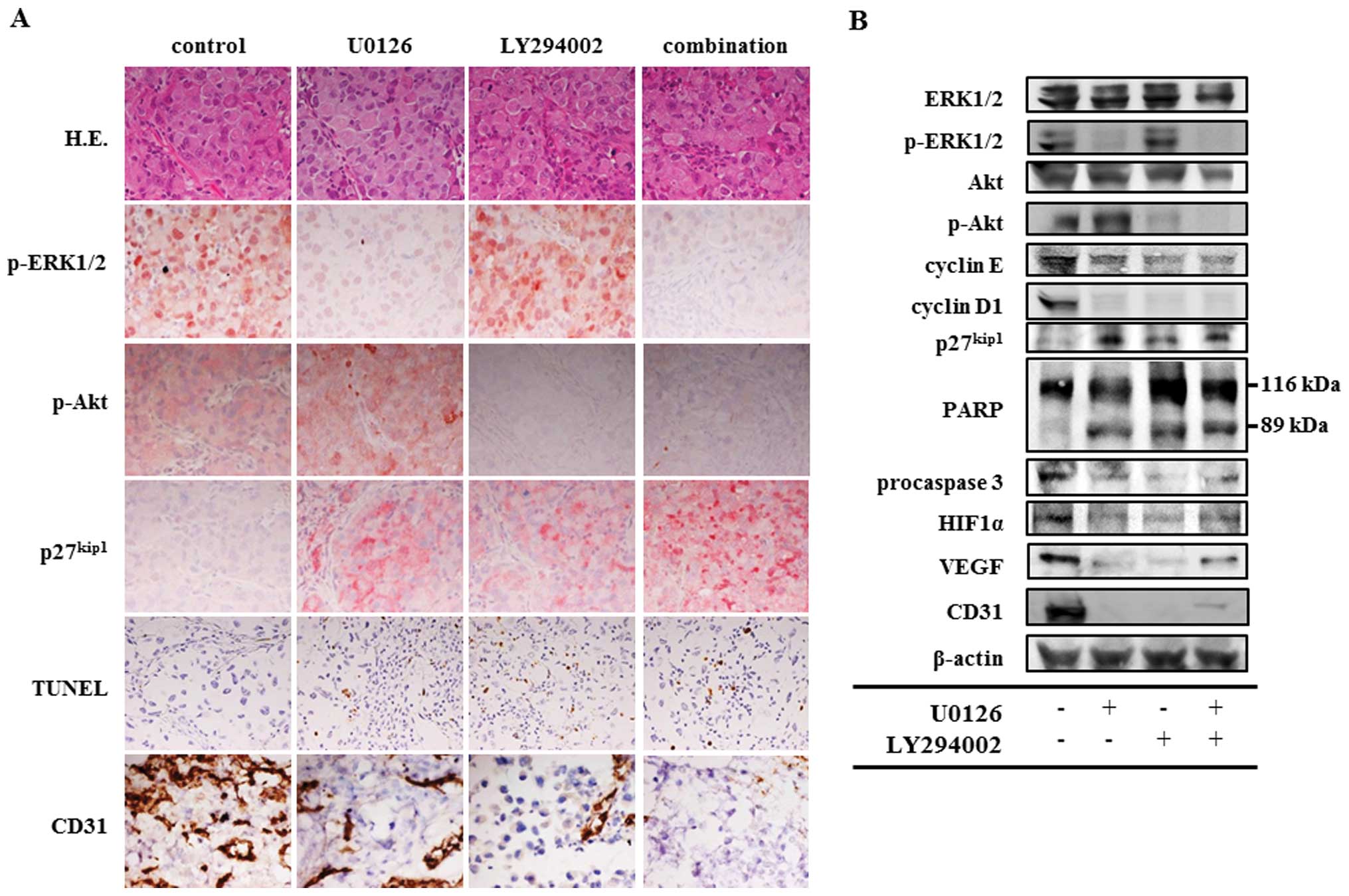 | Figure 4Histological and western blot
analysis of thoracic tumors produced by EHMES-10 cells. SCID mice
bearing EHMES-10 cells were treated with 30 mg/kg U0126, 25 mg/kg
LY294002 and a combination of 30 mg/kg U0126 and 25 mg/kg LY294002.
Mice were sacrificed on day 30 after tumor cell inoculation. (A)
Thoracic tumors were analyzed by H&E and immunohistochemistry
of phospho-extracellular signal-regulated kinase (ERK)1/2
(p-ERK1/2), phospho-Akt (p-Akt), p27kip1, TUNEL and CD31.
Magnification, ×400. (B) Western blots analysis showing modulation
of phospho-extracellular signal-regulated kinase (ERK)1/2
(p-ERK1/2), phospho-Akt (p-Akt), cyclin E, cyclin D1, p27kip1,
poly(ADP-ribose) polymerase (PARP), procaspase 3, hypoxia-inducible
factor 1α (HIF1α), vascular endothelial growth factor (VEGF) and
CD31 after treatment with U0126 and/or LY294002. |
Western blot analysis showed that treatment with
U0126, LY294002 and a combination of these agents inhibited ERK1/2
phosphorylation, Akt phosphorylation, and the phosphorylation of
ERK1/2 and Akt, respectively (Fig.
4B). In treatment with U0126, LY294002 and the combination, we
observed inhibited expression of cyclin E, cyclin D1, procaspase 3,
HIF1α, VEGF, and CD31, and increased expression of p27kip1 and 89
kDa cleaved PARP.
Side effects of treatment with U0126
and/or LY294002
To examine the side effects of treatment with U0126
and/or LY294002, the body weights and serum levels of TP, BUN, Cre,
AST and ALT were determined at the end of therapy on day 30
(Table II). Dermatopathy was also
evaluated during the treatment with these agents. Side effects were
not observed after the administration of these agents.
| Table IISide effects of treatment with U0126
or LY294002 or combination therapy after 30 days. |
Table II
Side effects of treatment with U0126
or LY294002 or combination therapy after 30 days.
| Weight (g) | TP (g/dl) | BUN (mg/dl) | Cre (mg/dl) | AST (IU/l) | ALT (IU/l) |
|---|
| Control | 24.8
(19.7–25.7) | 4.6 (4.5–4.8) | 17.1
(14.8–18.5) | 0.15
(0.10–0.21) | 55 (48–66) | 36.5 (23–42) |
| U30 mg/kg | 24.1
(23.5–27.2) | 4.3 (4.0–5.0) | 22.2
(15.4–24.0) | 0.10
(0.08–0.15) | 48 (44–64) | 30 (20–60) |
| U40 mg/kg | 24.9
(23.3–25.9) | 5.0 (4.4–6.6) | 19.1
(15.3–22.1) | 0.07
(0.05–0.12)a | 75 (53–111) | 45 (31–80) |
| LY25 mg/kg | 25.4
(24.4–26.3) | 4.3 (3.9–5.6) | 20.2
(17.9–23.0) | 0.11
(0.06–0.20) | 46 (37–117) | 19 (15–72) |
| LY50 mg/kg | 24.4
(22.7–26.3) | 4.4 (3.9–5.6) | 21.0
(16.4–23.2) | 0.09
(0.07–0.12)a | 51 (46–92) | 25 (16–42) |
| Combination | 23.5
(22.5–23.7) | 4.9 (4.6–5.0) | 24.9
(18.0–25.7) | 0.06
(0.04–0.07)a | 63.5 (62–102) | 34 (30–79) |
Discussion
The Ras pathway is one of the most frequently
deregulated pathways in cancer (22). Ras signals through multiple
effector pathways, including the RAF/MEK/ERK and PI3K/Akt signaling
cascades. A previous study reported that these pathways were
frequently activated in MPM (13).
Therefore, downregulation of these pathways might contribute to the
inhibition of tumor development and progression. In this study, we
showed that treatment with MEK and PI3K inhibitors, U0126 and
LY294002, inhibited MPM cell growth via cell cycle arrest,
apoptosis, and inhibition of tumor angiogenesis in vitro and
in vivo. In addition, each drug prolonged the survival time
of SCID mice bearing EHMES-10 cells. When drugs were applied in
combination, survival times were longer than those achieved with
the individual treatments.
Treatment with a MEK inhibitor or a PI3K inhibitor
has been shown to inhibit the growth of many types of cancer cells,
including MPM cell lines, via induction of cell cycle arrest and
apoptosis (14,15,23–25).
In the present study, MEK and PI3K inhibitors suppressed growth of
MPM cells in a dose-dependent manner. Flow cytometric analysis
showed that the treatment with MEK or PI3K inhibitors achieved G1
cell cycle arrest of MPM cells. DNA fragmentation analysis showed
apoptosis of MPM cells following treatment with these agents. It
was reported that treatment of MPM with a MEK inhibitor induced
p27kip1 upregulation (26). A PI3K
inhibitor induced p27kip1 upregulation and inhibition of
phosphorylation of p70S6K and S6 (14,26).
The present study showed similar results. In addition, treatment of
MPM cells with a MEK inhibitor downregulated cyclin E and cyclin
D1, and inhibited phosphorylation of p90RSK and Bad. Inhibition of
cyclin E and phospho-Bad expression was observed in treatment with
a PI3K inhibitor for MPM cells. Treatment of EHMES-10 cells with a
PI3K inhibitor reduced cyclin D1 and phospho-GSK3β expression.
Our study demonstrated the efficacy and the
mechanisms of action of a MEK inhibitor and a PI3K inhibitor for
MPM not only in vitro but also in in vivo
experiments. All previous reports of MEK inhibitors and PI3K
inhibitors for MPM cells have been limited to showing the
inhibitory effects and mechanisms in vitro(14,26).
Our study showed that treatment with a MEK inhibitor or a PI3K
inhibitor prolonged the survival time of EHMES-10 cells-bearing
SCID mice. Tumor weight and pleural effusion at day 30 were reduced
by these treatments. Furthermore, the immunohistochemical and
western blot analyses of thoracic tumors suggested that the MEK and
the PI3K inhibitors induced cell cycle arrest and cell apoptosis,
which was compatible with the results of the in vitro
study.
Treatments with the MEK inhibitor or the PI3K
inhibitor might be associated with inhibition of angiogenesis in
MPM cells. More than 60% of patients with MPM commonly present with
a pleural effusion associated with breathlessness, often
accompanied by chest wall pain, which compromises their quality of
life (27). Angiogenesis has
significant effects on the development of a pleural effusion and
ascites (28,29). It was also reported that treatment
with a MEK or a PI3K inhibitor suppressed proangiogenic cytokine
production in melanoma and MPM cells (15,23).
Our study showed that these agents significantly inhibited pleural
effusion production and CD31 protein expression and decreased
CD31-positive endothelial cells compared to controls. In addition,
western blot analysis showed that treatment of these agents
decreased the expression of HIF1α and VEGF, both of which play an
essential role in tumor angiogenesis and progression, in
vitro and in vivo.
Combination therapy with the MEK and PI3K inhibitors
might be more rational than an individual drug for MPM. It was
reported that antitumor activity of a MEK inhibitor or a PI3K
inhibitor induces activation of the other pathway (30). Moreover, several reports
demonstrated that inhibition of both cascades results in greater
antitumor activity (15,23). In the present study, the
combination therapy with MEK and PI3K inhibitors was also more
effective compared to that of individual drugs both in vitro
and in vivo.
Treatment with a MEK inhibitor and a PI3K inhibitor
might be well tolerated. For example, treatment with the MEK
inhibitor CI-1040 caused only mild or moderate toxicities such as
diarrhea, nausea, asthenia, rash, and anorexia in patients with
advanced non-small cell lung, breast, colon, and pancreatic cancers
(31). In addition, Hu et
al reported that daily intraperitoneal administration of
LY294002 at a dose of 100 mg/kg caused body weight loss and dry
skin in mice with ovarian cancer (32). However, in the following study,
side effects were not shown by reduction of LY294002 administration
to three days per week (33). In
the present study, side effects were not observed using a
combination of MEK and PI3K inhibitors.
In conclusion, our study demonstrates that in MPM
cells our selected MEK and PI3K inhibitors functioned via cell
cycle arrest, induction of apoptosis, and inhibition of tumor
angiogenesis, both in vivo and in vitro. In addition,
combining the MEK inhibitor with the PI3K inhibitor had additive or
synergistic effects in vitro. Combination therapy with MEK
and PI3K inhibitors may represent a promising novel therapeutic
strategy in the treatment of MPM.
Acknowledgements
The authors thank Mr. K. Kameda (Ehime
University, Ehime, Japan) for technical assistance with this
study.
References
|
1
|
Broaddus VC: Asbestos, the mesothelial
cell and malignancy: a matter of life or death. Am J Respir Cell
Mol Biol. 17:657–659. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Morinaga K, Kishimoto T, Sakatani M, Akira
M, Yokoyama K and Sera Y: Asbestos-related lung cancer and
mesothelioma in Japan. Ind Health. 39:65–74. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dufresne A, Bégin R, Churg A and Massé S:
Mineral fiber content of lungs in patients with mesothelioma
seeking compensation in Quebec. Am J Respir Crit Care Med.
153:711–718. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Light RW: Primary tumors of the pleura:
malignant mesotheliomas, solitary fibrous tumors, body cavity
lymphoma, and pyothorax-associated lymphoma. Pleural Diseases. 4th
edn. Lippincott Williams and Wilkins; Philadelphia, PA: pp.
135–150. 2001
|
|
5
|
Britton M: The epidemiology of
mesothelioma. Semin Oncol. 29:18–25. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zellos L and Sugarbaker DJ: Current
surgical management of malignant pleural mesothelioma. Curr Oncol
Rep. 4:354–360. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vogelzang NJ, Rusthoven JJ, Symanowski J,
Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S,
Manegold C, Niyikiza C and Paoletti P: Phase III study of
pemetrexed in combination with cisplatin versus cisplatin alone in
patients with malignant pleural mesothelioma. J Clin Oncol.
21:2636–2644. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Manning BD and Cantley LC: AKT/PKB
signaling: navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marte BM and Downward J: PKB/Akt:
connecting phosphoinositide 3-kinase to cell survival and beyond.
Trends Biochem Sci. 22:355–358. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sebolt-Leopold JS, Dudley DT, Herrera R,
Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges
A, Przybranowski S, Leopold WR and Saltiel AR: Blockade of the MAP
kinase pathway suppresses growth of colon tumors in vivo. Nat Med.
5:810–816. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hoshino R, Chatani Y, Yamori T, Tsuruo T,
Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J and Kohno
M: Constitutive activation of the 41-/43-kDa mitogen-activated
protein kinase signaling pathway in human tumors. Oncogene.
18:813–822. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Melo M, Gerbase MW, Curran J and Pache
JC: Phosphorylated extracellular signal-regulated kinases are
significantly increased in malignant mesothelioma. J Histochem
Cytochem. 54:855–861. 2006.PubMed/NCBI
|
|
14
|
Altomare DA, You H, Xiao GH, Ramos-Nino
ME, Skele KL, De Rienzo A, Jhanwar SC, Mossman BT, Kane AB and
Testa JR: Human and mouse mesotheliomas exhibit elevated AKT/PKB
activity, which can be targeted pharmacologically to inhibit tumor
cell growth. Oncogene. 24:6080–6089. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cole GW, Alleva AM, Zuo JT, Sehgal SS,
Yeow WS, Schrump DS and Nguyen DM: Suppression of pro-metastasis
phenotypes expression in malignant pleural mesothelioma by the PI3K
inhibitor LY294002 or the MEK inhibitor U0126. Anticancer Res.
26:809–822. 2006.PubMed/NCBI
|
|
16
|
Yokoyama A, Kohno N, Fujino S, Hamada H,
Inoue Y, Fujioka S and Hiwada K: Origin of heterogeneity of
interleukin-6 (IL-6) levels in malignant pleural effusions. Oncol
Rep. 1:507–511. 1994.PubMed/NCBI
|
|
17
|
Nakataki E, Yano S, Matsumori Y, Goto H,
Kakiuchi S, Muguruma H, Bando Y, Uehara H, Hamada H, Kito K,
Yokoyama A and Sone S: Novel orthotopic implantation of human
malignant pleural mesothelioma (EHMES-10 cells) highly expressing
vascular endothelial growth factor and its receptor. Cancer Sci.
97:183–191. 2006. View Article : Google Scholar
|
|
18
|
Rak-Mardyla A and Gregoraszczuk EL: ERK
1/2 and PI-3 kinase pathways as a potential mechanism of ghrelin
action on cell proliferation and apoptosis in the porcine ovarian
follicular cells. J Physiol Pharmacol. 61:451–458. 2010.PubMed/NCBI
|
|
19
|
Ciofani G, Ricotti L, Danti S, Moscato S,
Nesti C, D’Alessandro D, Dinucci D, Chiellini F, Pietrabissa A,
Petrini M and Menciassi A: Investigation of interactions between
poly-L-lysine-coated boron nitride nanotubes and C2C12 cells:
up-take, cytocompatibility, and differentiation. Int J
Nanomedicine. 5:285–298. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hamaguchi N, Hamada H, Miyoshi S, Irifune
K, Ito R, Miyazaki T and Higaki J: In vitro and in vivo therapeutic
efficacy of the PPAR-γ agonist troglitazone in combination with
cisplatin against malignant pleural mesothelioma cell growth.
Cancer Sci. 101:1955–1964. 2010.
|
|
21
|
Soga Y, Komori H, Miyazaki T, Arita N,
Terada M, Kamada K, Tanaka Y, Fujino T, Hiasa Y, Matsuura B, Onji M
and Nose M: Toll-like receptor 3 signaling induces chronic
pancreatitis through the Fas/Fas ligand-mediated cytotoxicity.
Tohoku J Exp Med. 217:175–184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wee S, Jagani Z, Xiang KX, Loo A, Dorsch
M, Yao YM, Sellers WR, Lengauer C and Stegmeier F: PI3K pathway
activation mediates resistance to MEK inhibitors in KRAS mutant
cancers. Cancer Res. 69:4286–4293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bedogni B, Welford SM, Kwan AC,
Ranger-Moore J, Saboda K and Powell MB: Inhibition of
phosphatidylinositol-3-kinase and mitogen-activated protein kinase
kinase 1/2 prevents melanoma development and promotes melanoma
regression in the transgenic TPRas mouse model. Mol Cancer Ther.
5:3071–3077. 2006. View Article : Google Scholar
|
|
24
|
Furuya F, Lu C, Willingham MC and Cheng
SY: Inhibition of phosphatidylinositol 3-kinase delays tumor
progression and blocks metastatic spread in a mouse model of
thyroid cancer. Carcinogenesis. 28:2451–2458. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McCubrey JA, Steelman LS, Abrams SL, Lee
JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA,
D’Assoro AB, Salisbury JL, Mazzarino MC, Stivala F and Libra M:
Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant
transformation and drug resistance. Adv Enzyme Regul. 46:249–279.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mukohara T, Civiello G, Johnson BE and
Janne PA: Therapeutic targeting of multiple signaling pathways in
malignant pleural mesothelioma. Oncology. 68:500–510. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Robinson BW and Lake RA: Advances in
malignant mesothelioma. N Engl J Med. 353:1591–603. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li Q, Yano S, Ogino H, Wang W, Uehara H,
Nishioka Y and Sone S: The therapeutic efficacy of anti vascular
endothelial growth factor antibody, bevacizumab, and pemetrexed
against orthotopically implanted human pleural mesothelioma cells
in severe combined immunodeficient mice. Clin Cancer Res.
13:5918–5925. 2007. View Article : Google Scholar
|
|
29
|
Yano S, Shinohara H, Herbst RS, Kuniyasu
H, Bucana CD, Ellis LM and Fidler IJ: Production of experimental
malignant pleural effusions is dependent on invasion of the pleura
and expression of vascular endothelial growth factor/vascular
permeability factor by human lung cancer cells. Am J Pathol.
157:1893–1903. 2000. View Article : Google Scholar
|
|
30
|
Rexer BN, Ghosh R and Arteaga CL:
Inhibition of PI3K and MEK: It is all about combinations and
biomarkers. Clin Cancer Res. 15:4518–4520. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rinehart J, Adjei AA, Lorusso PM,
Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury
P, Kaldjian EP, Gulyas S, Mitchell DY, Herrera R, Sebolt-Leopold JS
and Meyer MB: Multicenter phase II study of the oral MEK inhibitor,
CI-1040, in patients with advanced non-small-cell lung, breast,
colon, and pancreatic cancer. J Clin Oncol. 22:4456–4462. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hu L, Zaloudek C, Mills GB, Gray J and
Jaffe RB: In vivo and in vitro ovarian carcinoma growth inhibition
by a phosphatidylinositol 3-kinase inhibitor (LY294002). Clin
Cancer Res. 6:880–886. 2000.PubMed/NCBI
|
|
33
|
Hu L, Hofmann J, Lu Y, Mills GB and Jaffe
RB: Inhibition of phosphatidylinositol 3′-kinase increases efficacy
of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer
Res. 62:1087–1092. 2002.
|