Introduction
Latest statistics show that obesity affects more
than 1/3 of the adult population in the United States, with higher
incidences in women (1,2). With postmenopausal breast cancer
incidence and mortality rates closely correlated with obesity
(3), it is imperative to explore
the underlying molecular mechanisms of this connection, as an
increasing number of female patients will be in the obese
postmenopausal group in the near future. Obesity is associated with
concentration changes in the endocrine secretion profile of white
adipose tissue. These changes, amongst others, contribute to the
link between obesity and hyperinsulinaemia (4). Hyperinsulinaemia is consistently
linked with increased breast cancer risk and mortality. An
epidemiological study demonstrated a positive correlation between
increased insulin resistance and increased risk of all cancers,
even in non-obese individuals (5).
A meta-analysis indicated an increased risk of development of and
mortality from breast cancer in diabetic women (6). Another meta-analysis, examining
C-peptide concentrations and breast cancer risk, showed a positive
correlation in case/control studies (7). A large cohort study additionally
observed a positive correlation between hyperinsulinaemia and
breast cancer in their non-fasting postmenopausal sub-group
(8). Additionally, fasting insulin
concentrations at a young age were directly related to increased
breast cancer incidences in later life (9). Similarly, in a case control study of
Chilean women, insulin resistance was identified as a risk factor
for developing breast cancer in postmenopausal women (10). Interestingly, in contrast to
overall obesity, assessed by BMI, hyperinsulinaemia has also been
linked to premenopausal breast cancer (11,12),
although this association was not consistently observed (13). Additionally, recent findings
suggest that the combined effects of obesity and insulin resistance
elevate certain biochemical markers generally associated with an
increased risk of developing premenopausal breast cancer (14).
Previous studies have indicated that the main effect
of obesity on breast cancer incidence rates is mediated by
increasing oestrogen levels in obese postmenopausal women (15,16).
While the connection between obesity and oestrogen receptor
(ER)-negative breast cancer incidence rates is lower than between
obesity and ER-positive breast cancer, this association still
indicates a connection between obesity and breast cancer,
independent of oestrogen. Given the known interaction between the
insulin receptor (IR) and oestrogen (17), this study used ER-negative cell
lines.
In in vitro studies, high insulin
concentrations increased cell growth in a number of cell lines
(18,19), including breast cancer cell lines
(20,21). The breast cancer cell line, MCF-7,
is a well-established model of an ER-positive breast cancer.
Stimulation with up to 100 nM insulin has been shown to increase
the ability of these cells to incorporate leucine and thymidine
(20), increase fatty acid
synthesis (22), increase cell
cycle progression (23), protect
against apoptosis (24),
downregulate protein degradation and promote an increase in cell
size (25). Treatment of
MDA-MB-231 cells and other ER-negative breast cancer cell lines
with up to 1 μM insulin did not increase cell proliferation
or affect apoptosis (21,26). Conversely, MDA-MB-231 breast cancer
cells have an elevated IR content as they carry an uncommon IR gene
amplification (27), indicating
that insulin may play a role in the metabolism of this cell
line.
In this study, the influence of high insulin
concentrations (100 nM) on several molecular aspects in breast
cancer cells and breast epithelial cells was assessed. MDA-MB-231
and SK-BR-3 breast cancer cells were used to identify the impact of
hyperinsulinaemia on oestrogen-independent breast cancer
progression. MCF-10A cells were used representatively of normal
breast epithelial cells to examine the impact of hyperinsulinaemia
on breast cancer aetiology. Changes in insulin receptor
phosphorylation, cell proliferation, activation of cell signalling
pathways, changes in cell cycle and early apoptosis were
determined.
Materials and methods
Materials
Human Caucasian breast adenocarcinoma cells,
MDA-MB-231 (Cat no. 92020424, passage no. 36; European Collection
of Cell Cultures, Salisbury, UK) and SK-BR-3 [American Type Culture
Collection (ATCC) no. HTB-30, passage no. 28; ATCC, Manassas, VA,
USA], were routinely cultured in RPMI-1640 medium (containing 25 mM
HEPES, 1X glutamax) [Gibco (Invitrogen), Paisley, UK; Cat no.
72400], as recommended by the supplier and described previously
(28). Human Caucasian breast
epithelial cells, MCF-10A (ATCC no. CRL-10317, passage no. 102;
ATCC), were cultured in DMEM/F-12 medium [Bio-Whittaker UK (Lonza
Biologics), Slough, UK; Cat no. BE12-7199] as recommended by the
supplier and described previously (28). The serum-free medium used was
RPMI-1640 (25 mM HEPES, 1X glutamax) (Gibco) supplemented with 100
U/ml penicillin and 100 μg/ml streptomycin (Gibco) for
MDA-MB 231 and SK-BR-3 cells and DMEM/F12 (Bio-Whittaker)
supplemented with 100 U/ml penicillin and 100 μg/ml
streptomycin for MCF-10A cells. Cell culture conditions were 37°C
in humidified air containing 5% CO2.
Cell proliferation assay
Cell proliferation was detected using a colorimetric
Cell Proliferation ELISA kit (Roche Diagnostics, Penzberg, Germany;
Cat no. 11 647 229 001), which assesses DNA replication by
measuring bromodeoxyuridine (BrdU) incorporation. Cells
(5×103/well of each cell line) were plated in 96-well
plates (Fisher Scientific; Cat no. 167008) with 100 μl/well
growth medium and incubated for 24 h at 37°C. Cells were washed
once in 100 μl/well sterile PBS and incubated in serum-free
medium for 24 h. Cells were then washed as described above and
treated for 24 or 48 h with 100 nM insulin in 100 μl/well
serum-free medium. During treatment, the medium was supplemented
with 10 μM BrdU for the final 24 h of treatment.
Incorporated BrdU was detected according to the manufacturer’s
instructions and colour development was quantified on a
μQuant Microplate Spectrophotometer (BioTek, Potton, UK) by
measuring absorption at 450 nm with a reference wavelength of 690
nm. Three experiments were performed for each cell line and each
time-point. Each experiment consisted of six replicates for each
treatment, i.e. six wells for the control and six wells for the
treatment groups.
Insulin receptor phosphorylation
assay
In 60-mm2 tissue culture dishes (Fisher
Scientific; Cat no. 150288), 1×106 cells of each of the
three cell lines were plated and incubated in 3 ml growth medium
for 24 h at 37°C. Cells were starved as described above and treated
with 100 nM insulin for 2 min at 37°C. IR phosphorylation was
determined using the DuoSet IC human phospho insulin receptor kit
(R&D Systems, Abingdon, UK; Cat no. DYC2718) following the
manufacturer’s instructions. In brief, cell lysate was prepared
after treatment and added to a 96-well ELISA plate coated with 100
μl/well of supplied capture antibody, targeting the IR
β-subunit, at 8 μg/ml. Detection was achieved by adding the
supplied secondary antibody (100 μl/well), tagged with
horseradish-peroxidase (HRP) and diluted 1:1,000, and by adding 100
μl of a stable peroxide solution and tetramethylbenzidine
solutions (R&D Systems; Cat no. DY999), mixed 1:1, to each
well. Absorption was quantified at 450 nm with a correction
wavelength of 540 nm on a μQuant microplate
spectrophotometer (BioTek). Three experiments were performed for
each cell line.
Phospho-kinase ELISA
Cell-based ELISA Phospho-AKT (S473) Immunoassay (Cat
no. KCB887) and Phospho-ERK1/ERK2 (T202/Y204) Immunoassay (Cat no.
KCB1018) were purchased from R&D Systems. Cells were grown and
starved as before and treated as indicated in Fig. 3. The phosphorylation of protein
kinase B (PKB/AKT) and extracellular-regulated kinase (ERK)1/2 was
then assessed following the manufacturer’s instructions. In brief,
after fixing, the cells were incubated with phospho-AKT- or
phospho-ERK1/2-specific mouse antibodies in conjunction with
anti-total AKT- or anti-total ERK1/2-specific rabbit antibodies,
respectively. Phosphorylated and total protein was detected with
species-specific secondary antibodies tagged with HRP and alkaline
phosphatase (AP), respectively. Two different fluorescent
substrates were used for quantification. Fluorescence was measured
on a Fluoroskan Ascent microplate reader (Lab Systems, Hull, UK)
with excitation at 544 nm and emission at 590 nm (phosphorylated
protein) and excitation at 355 nm and emission at 460 nm (total
protein). For each cell line three experiments were performed to
assess AKT phosphorylation and three to assess ERK1/2
phosphorylation after insulin treatment. Each experiment included
two replicates for each treatment time and the control.
Flow cytometry
Cell cycle
Changes in the cell distribution across cell cycle
stages were assessed by measurement of DNA content in the cells.
The DNA specific dye used was propidium iodide (PI) (Sigma, Cat no.
P4170). Cells were plated at 5×105 cells/well in
six-well plates with 3 ml growth medium and incubated for 24 h at
37°C. Cells were starved for 24 h and then treated with 100 nM
insulin for 24 h. Cells were harvested, stained and analysed as
previously described (28). Three
experiments were performed for each cell line, containing two
replicates for the control and treatment groups.
Apoptosis
The Annexin V-FITC/7-AAD apoptosis kit (Beckman
Coulter; Cat no. IM3614) was used to examine apoptosis following
insulin treatment. The cells were grown (1×106/dish) and
treated with 100 nM insulin in 60-mm2 dishes as
described above. After the cells had been treated and collected,
100 μl of 1X binding buffer (supplied), 10 μl Annexin
V-FITC (supplied) and 20 μl 7-AAD dye (supplied) were added
to each tube. The samples were incubated on ice in the dark for 15
min. The samples were then diluted 1:5 prior to analysis. Flow
cytometry of the Annexin V- and 7-AAD-stained cells was performed
using a Coulter Epics XLMCL flow cytometer (Beckman Coulter). Data
were analysed using EXPO-32 Software (Applied Cytometry Systems,
Sheffield, UK). Apoptotic cells were captured by linear FL-2 vs.
area plots, with a cell line-specific upper cut-off point. Necrotic
cells were similarly captured by linear FL-4 vs. area plots.
Numbers were assessed after 10,000 events. Three experiments were
performed for each cell lines with two replicates for the control
and treatment groups.
RT-PCR
Cells were grown (1×106/dish) and treated
with 100 nM insulin in 60-mm2 dishes as described above.
After the cells were treated, total-RNA was extracted using TRIzol
(Invitrogen; Cat no. 15596). The extracted RNA (1 ng) was
reverse-transcribed in a 20 μl volume containing 50 mM
Tris-HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 10 mM
dithiothreitol (all from Invitrogen; Cat no. 18080-044), 1 mM of
each dNTP (Roche; Cat no. 11969064001), 100 μg/ml BSA (New
England Biolabs; Cat no. B9001S), 25 μg/ml random primers
(Promega; Cat no. C1181), 40 units RNaseOUT (Invitrogen; Cat no.
10777-019), 80 units SuperScript® III Reverse
Transcriptase (Invitrogen; Cat no. 18080-044) for 10 min at 25°C,
followed by 52 min at 42°C and 15 min at 72°C. The resulting cDNA
(4 μl) was amplified with primers specific for cyclin D
(forward primer, GCTCGAGCCCGTGAAAAAGA; reverse primer,
CTCCGCCTCTGGCATTTTG) or cyclin E (forward primer,
TTACCCAAACTCAACGTGCAA; reverse primer, GCTCAAAGTGCTGATCCC) and
β-actin (forward primer, CATGTACGTTGCTATCCAGGC; reverse primer,
CTCCTTAATGTCACGCACGAT) as the loading control, in a 20 μl
volume containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.75 mM
MgCl2, 1 unit of TaqDNA polymerase (all from Sigma; Cat
no. D4545-250UN) and 1 μM of each primer. Following a hot
start (95°C) and 4 min at 94°C, 25 cycles for cyclin D and 33
cycles for cyclin E with 1 min at 94°C, 2 min at gene-specific
annealing temperature (60°C for cyclin D, 59°C for cyclin E and
68°C for β-actin) and 2 min at 72°C were performed. This was
followed by a 10-min final extension step at 72°C. PCR-products
were separated on 1% agarose gels, images captured by GelDoc CCD
imaging (Bio-Rad) and quantified by Quantity-One software
(Bio-Rad).
Results
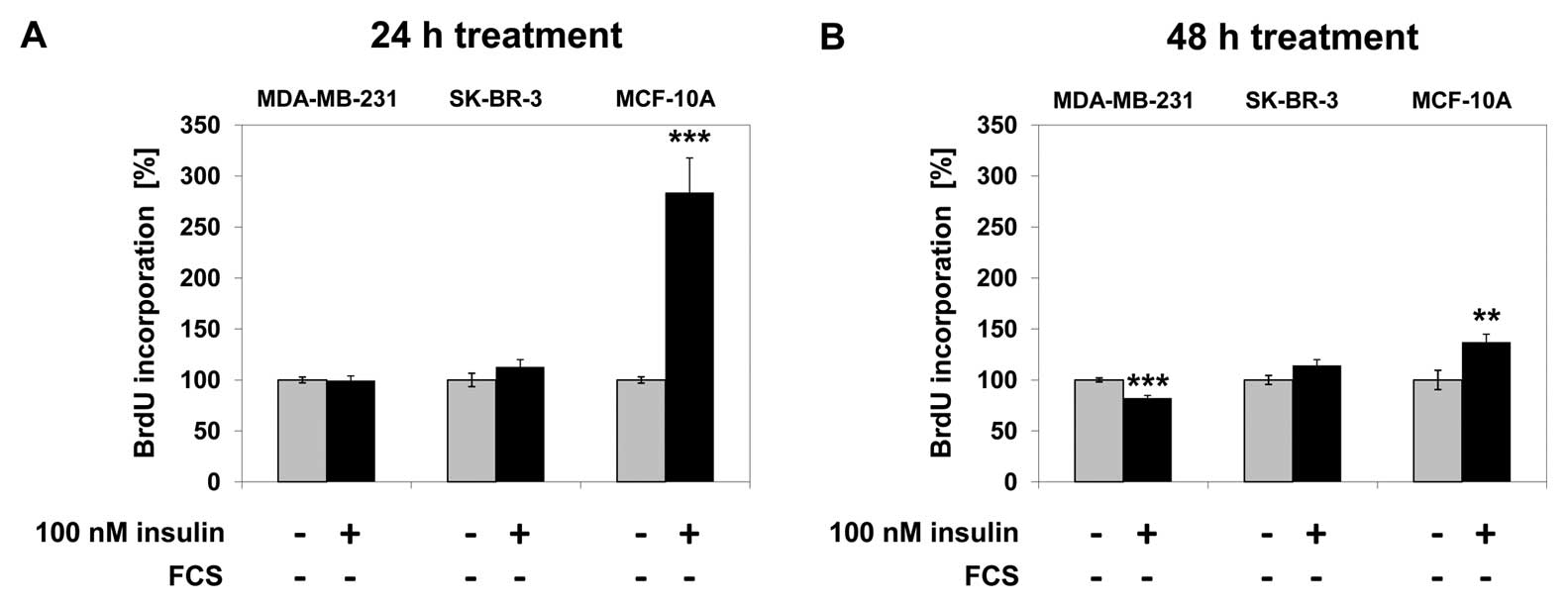
Effect of insulin on cell
proliferation
In MDA-MB-231 cells, 100 nM insulin treatment for 24
h did not increase cell proliferation; after 48 h of insulin
treatment cell proliferation decreased by 18% (p<0.001) compared
to the untreated control. Cell proliferation did not change in
SK-BR-3 cells after 24 or 48 h of treatment with 100 nM insulin. In
MCF-10A cells, proliferation increased by 184% (p<0.001) after
24 h of insulin treatment and by 34% (p<0.001) after 48 h of
treatment compared to the untreated control (Fig. 1).
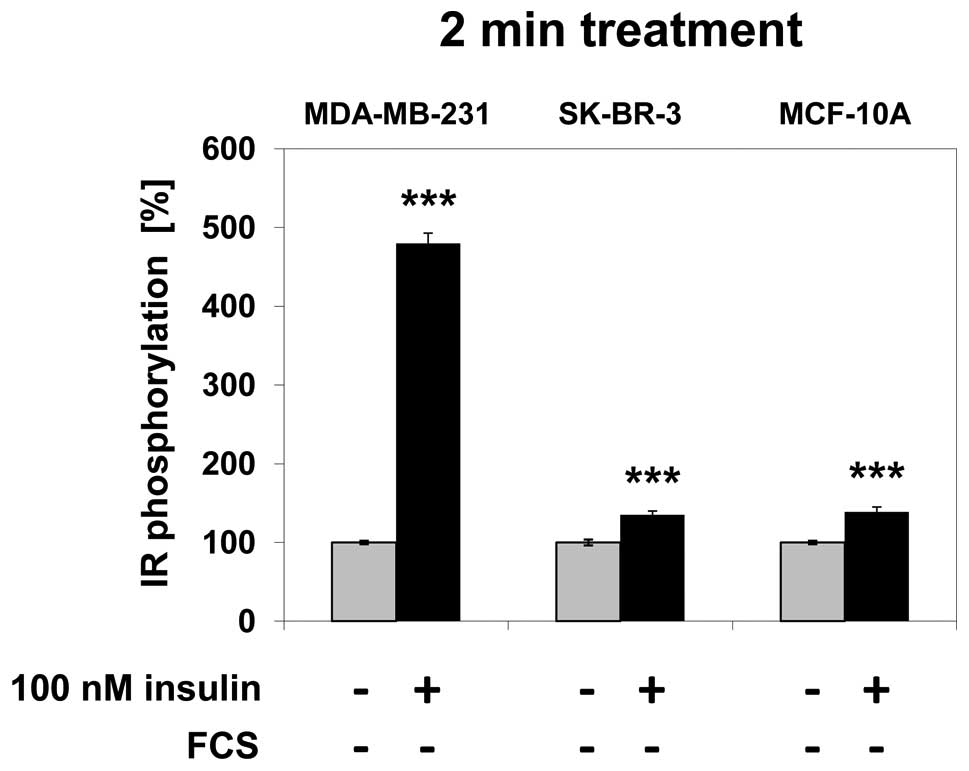
Effect of insulin treatment on IR
phosphorylation
In MDA-MB-231 cells, IR phosphorylation increased by
380% (p<0.001) following 2 min of treatment with 100 nM insulin
compared to the untreated control. In SK-BR-3 cells, the increase
in IR phosphorylation was 35% (p<0.001) and 38% (p<0.001) in
MCF-10A cells after 2 min of treatment compared to the untreated
control (Fig. 2).
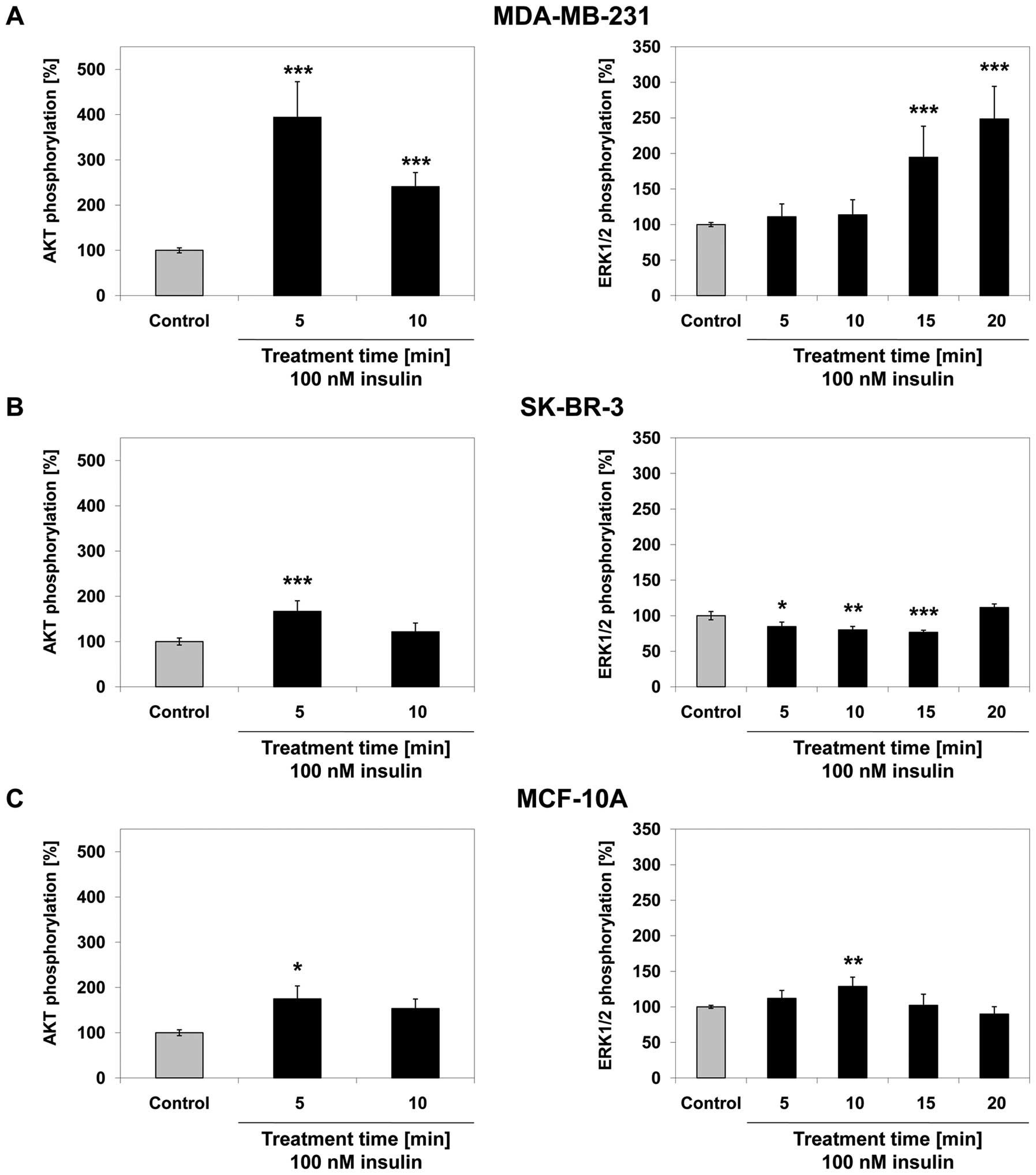
Effect of insulin on cell signalling
pathways
In MDA-MB-231 cells, AKT phosphorylation increased
by 294% after 5 min (p<0.001) and by 141% after 10 min
(p<0.001) of treatment with 100 nM insulin compared to the
untreated control. ERK1/2 phosphorylation did not change
significantly after 5 or 10 min of insulin treatment. The
phosphorylation of ERK1/2 increased by 95% after 15 min
(p<0.001) and by 148% after 20 min (p<0.001) of treatment
with 100 nM insulin (Fig. 3A)
compared to the untreated control. In SK-BR-3 cells, AKT
phosphorylation increased by 67% after 5 min (p<0.001) of
treatment with 100 nM insulin and no significant change in
AKT-phosphorylation was observed after 10 min of treatment. The
phosphorylation of ERK1/2 decreased by 15, 20 and 23% after
treatment with 100 nM insulin for 5 (p=0.016), 10 (p=0.002) and 15
min (p<0.001), respectively, but did not significantly change
after 20 min of insulin treatment compared to the untreated control
(Fig. 3B). In MCF-10A cells, AKT
phosphorylation increased by 75% after 5 min (p= 0.022) of
treatment with 100 nM insulin and no statistically significant
change was observed after 10 min of insulin treatment. The
phosphorylation of ERK1/2 increased by 29% after 10 min (p=0.002)
of treatment with 100 nM insulin compared to the untreated control.
No statistically significant change was observed at any other
time-point (Fig. 3C).
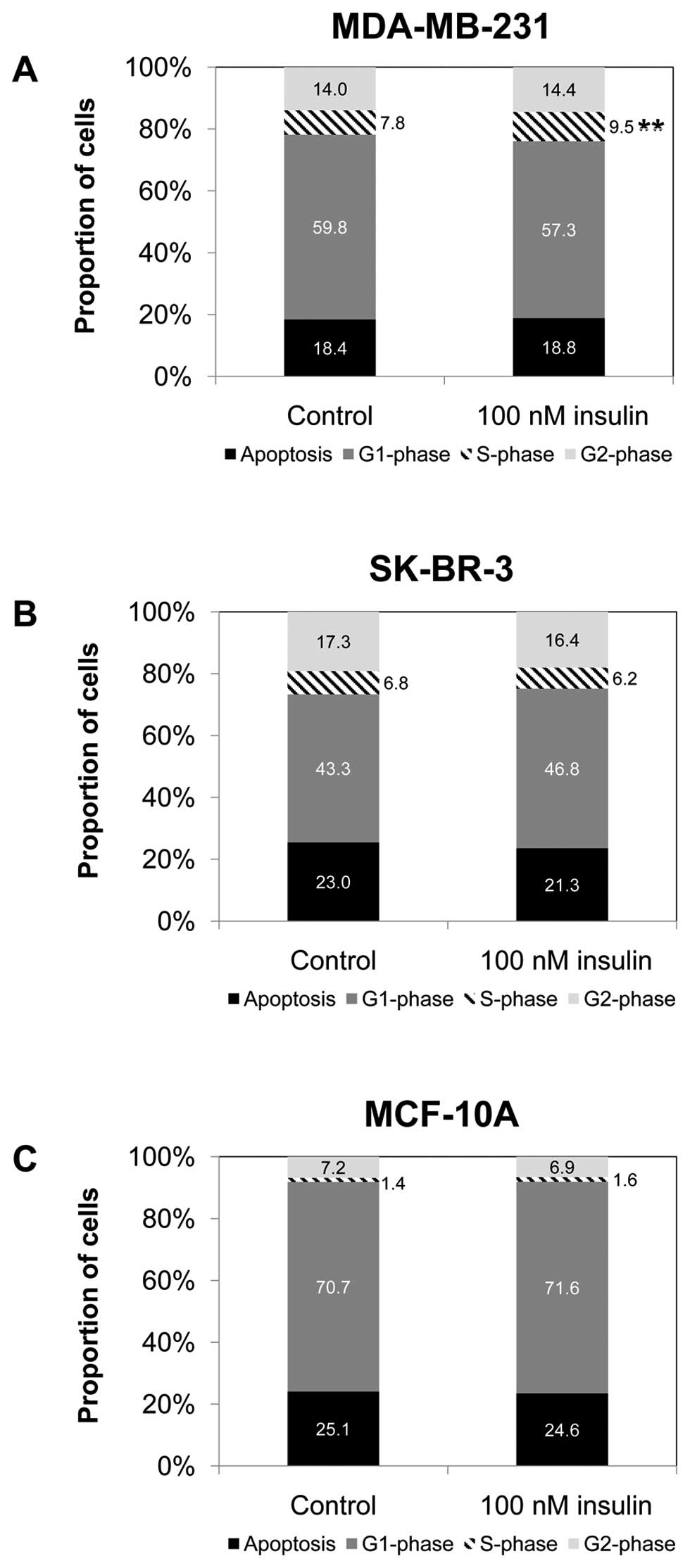
Effect of insulin on cell cycle
In MDA-MB-231 cells, the S-phase population
increased by 1.7 percentage points after 24 h (p=0.002) of
treatment with 100 nM insulin compared to the S-phase population of
the control cells (Fig. 4A). In
SK-BR-3 and MCF-10A cells, treatment with 100 nM insulin did not
significantly change the distribution of the cell population across
the cell cycle phase (Fig. 4B and
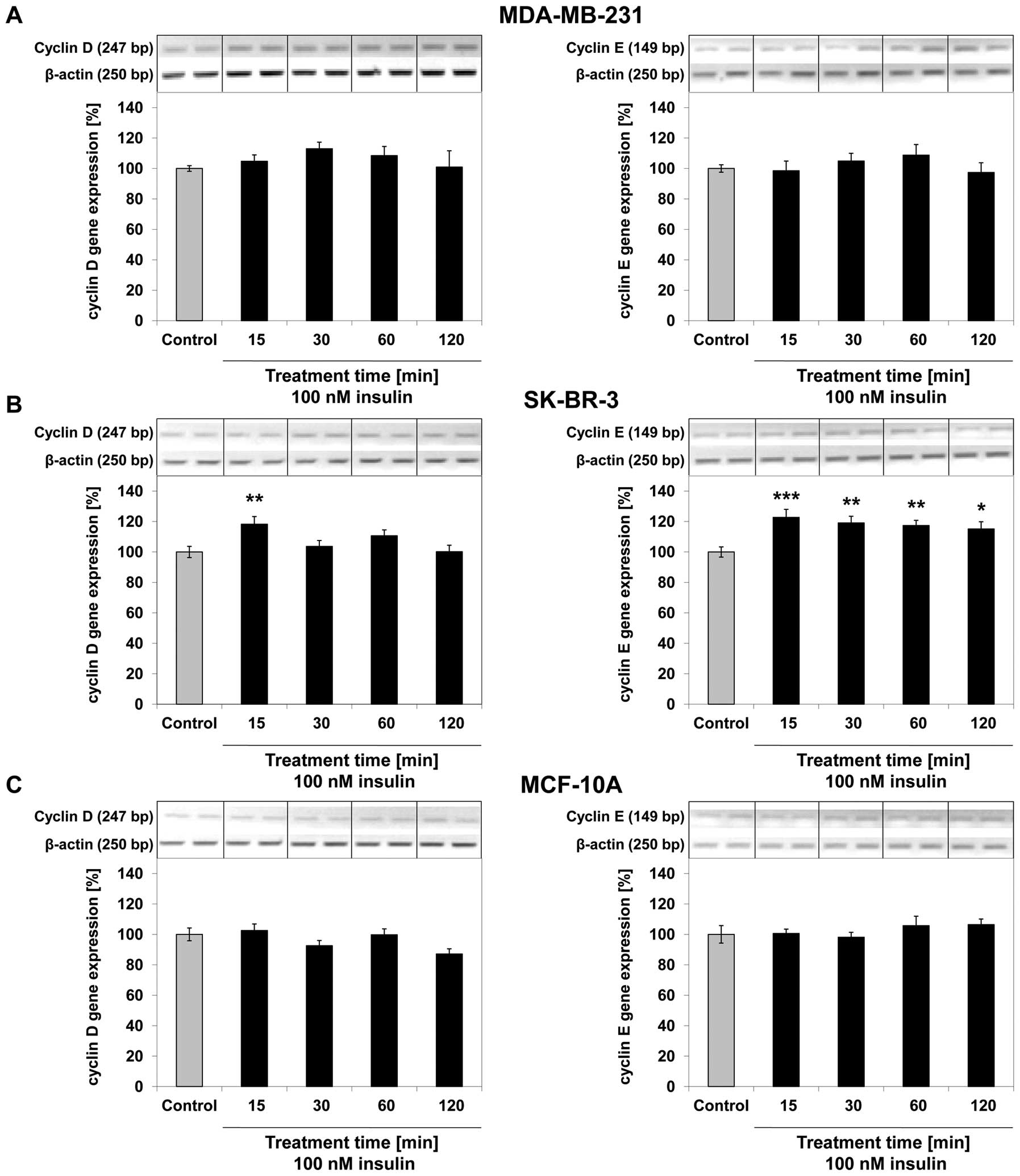
C). In MDA-MB-231 and MCF-10A cells, cyclin D and cyclin E gene
expression did not significantly change after up to 120 min of
treatment with 100 nM insulin compared to the untreated control
(Fig. 5A and C). In SK-BR-3 breast
cancer cells, cyclin D gene expression increased by 18% after 15
min (p=0.005) of treatment with 100 nM insulin compared to the
untreated control. Cyclin E gene expression increased by 22% after
15 min (p<0.001), by 19% after 30 min (p=0.002), by 17% after 60
min (p=0.003) and by 15% after 120 min (p=0.012) of treatment with
100 nM insulin compared with the untreated control (Fig. 5B).
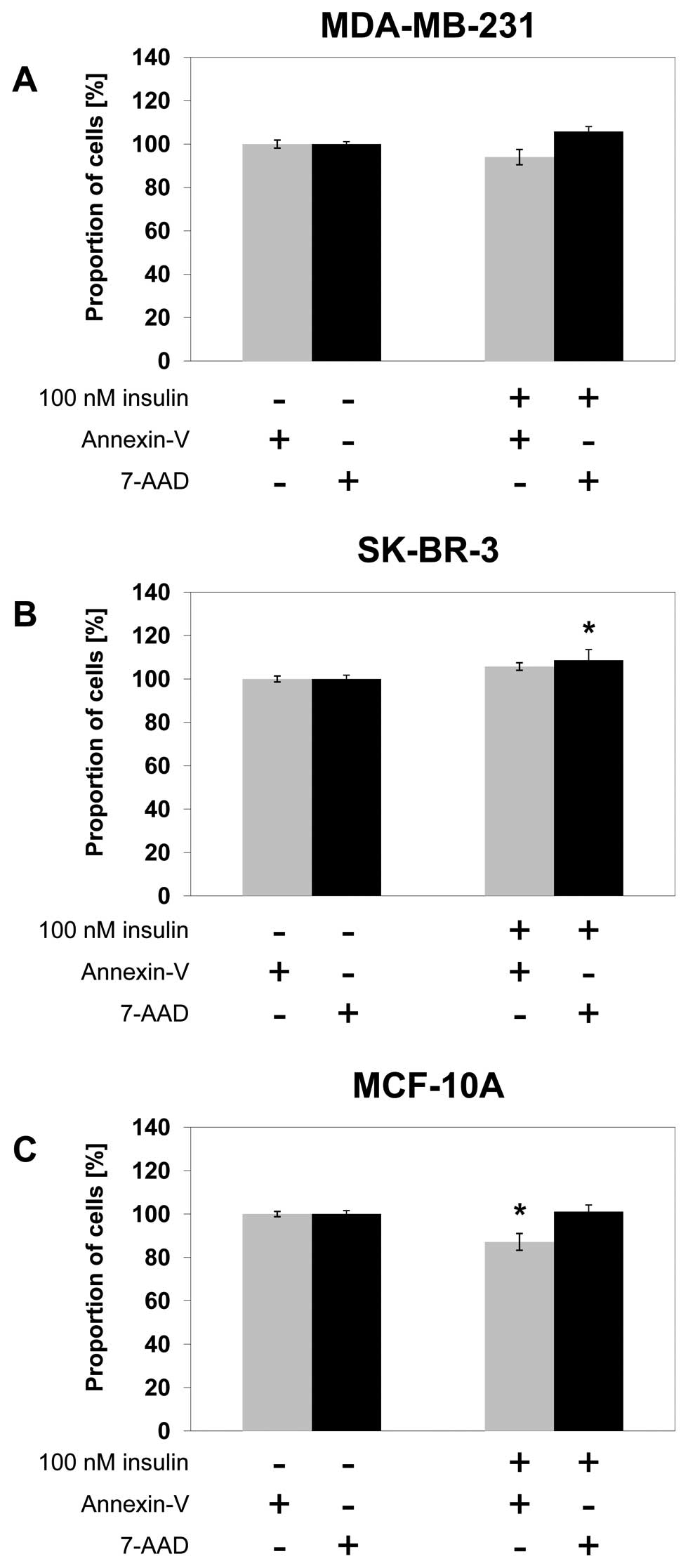
Effect of insulin on apoptosis
In MDA-MB-231 breast cancer cells, treatment with
100 nM insulin did not significantly change Annexin V detection or
7-AAD staining after 24 h of treatment (Fig. 6A). In SK-BR-3 breast cancer cells,
Annexin V detection did not change significantly after 24 h of
treatment with 100 nM insulin. The percentage of cells with high
levels of 7-AAD staining increased significantly by 9% after 24 h
(p=0.024) of 100 nM insulin treatment (Fig. 6B). In MCF-10A breast epithelial
cells, Annexin V detection decreased significantly by 13% after 24
h (p=0.037) of treatment with 100 nM insulin compared to the
untreated control. The amount of cells with high levels of 7-AAD
staining did not change significantly after 24 h of treatment with
100 nM insulin (Fig. 6C).
Discussion
The epidemiological link between obesity and breast
cancer lacks a fundamental molecular mechanism explaining this
connection. Insulin may be a molecular mediator of the
obesity-breast cancer connection (29). Previous in vitro studies
have indicated that insulin increases cell proliferation in
ER-positive, but not in ER-negative breast cancer cells (20,21,26,30).
On the other hand, epidemiologically insulin and C-peptide levels
have been linked to an increased risk of ER-negative breast cancer
(31). This suggests an additional
mode of action of insulin to increase breast cancer risk. Thus, in
the present study, we investigated the effect of insulin on
ER-negative cell lines to elucidate the impact of insulin
independently of ER. Previously, when MDA-MB-231 breast cancer
cells were treated with 100 nM insulin, no increase in cell
proliferation after 24 h was observed (21). Similarly, in the present study, we
observed no increase in cell proliferation in this cell line after
24 h of treatment and a statistically significant decrease after 48
h of treatment, which may not be of physiological importance. In a
previous study, it was concluded that the overexpression of
glycoprotein PC-1 led to an inhibition of IR-autophosphorylation
and consequently to a complete inhibition of insulin signalling in
MDA-MB-231 breast cancer cells (32). Conversely, in the present study, we
observed an increase in IR, AKT and ERK1/2 phosphorylation after
insulin treatment, suggesting that insulin signalling in these
cells is intact. Additionally a slight but statistically
significant increase in the S-phase population was observed after
insulin treatment, suggesting a potential effect of insulin on cell
cycle progression. This observation however, was not supported by
changes in cyclin D or cyclin E gene expression, indicating that
the precise impact of insulin on the cell cycle in MDA-MB-231
breast cancer cells remains unclear. There was also no indication
that insulin reduced apoptosis in these cells. A comparison to
SK-BR-3 breast cancer cells and MCF-10A breast epithelial cells
indicated that insulin signalling was amplified in these cells.
However, this signal did not translate into significantly altered
physiological changes. Thus, insulin may play a role in MDA-MB-231
cell metabolism, but not in increasing cell proliferation and in
enhancing cell cycle progression or in decreasing apoptosis.
A second ER-negative breast cancer cell line,
SK-BR-3, also showed no indications of increased cell proliferation
after treatment with 100 nM insulin for up to 48 h. Further
similarities to the MDA-MB-231 breast cancer cell line were the
increase in IR and AKT phosphorylation. This increase, while
statistically significant, did not reach the same level as in the
MDA-MB-231 breast cancer cell line. This may be explained by the
previously observed IR gene amplification in MDA-MB-231 breast
cancer cells, which translated into the increased phosphorylation
of IR and IR targets, such as AKT and ERK1/2, when compared to the
other cell lines (33).
Additionally, MDA-MB-231 breast cancer cells have mutated Raf and
Ras genes [members of the mitogen-activated protein kinase (MAPK)
pathway], which may explain the difference in phosphorylation
levels between the two breast cancer cell lines (34). Interestingly, ERK1/2
phosphorylation decreased with the insulin treatment of SK-BR-3
cells. This is in contrast to both MDA-MB-231 and MCF-10A cells,
which showed increased ERK1/2 phosphorylation after insulin
treatment, suggesting that this decrease is unique to the SK-BR-3
cells. An explanation may be that insulin interferes with an
underlying autocrine stimulus in these cells, which utilizes the
MAP-kinase pathway but becomes inhibited as insulin signalling
initiates changes in the same pathway. There was a significant
increase in cyclin D and cyclin E gene expression after insulin
treatment in SK-BR-3 breast cancer cells, indicating that insulin
may increase cell cycle progression. As in MDA-MB-231 breast cancer
cells, however, this observation was not supported by changes in
cell population distribution across the cell cycle stages, which
remained unaffected by insulin treatment of these cells. From these
results, increased cell cycle progression cannot be predicted with
a high degree of confidence in these cells. Furthermore, there was
a small yet significant increase in necrosis with insulin
treatment, which on its own would suggest cytotoxicity of insulin
in these cells. Physiologically, these changes of increased cyclin
expression and increased necrosis may be negligible, especially as
they are seemingly contradictory and not supported by the
additional analyses. Thus, similar to MDA-MB-231 breast cancer
cells, high levels of insulin did not induce a uniform
proliferative effect, even though insulin signalling seemed
intact.
The most interesting question arising from the
results presented in this study is what the physiological effect of
insulin on ER-negative breast cancer cells is, given that insulin
signalling is intact and for MDA-MB-231 breast cancer cells even
amplified. While our results did not validate a previous
explanation for insulin resistance in MDA-MB-231 breast cancer
cells, it is still likely that ER-negative breast cancer cells may
have acquired insulin resistance. If the condition of ER negativity
in cells was regarded as an enhancer of breast cancer progression
and not an ab initio determinant, it could be speculated
that acquired insulin resistance may be an additional feature of
breast cancer progression. This would explain the connection of
induced cell proliferation in ER-positive but not in ER-negative
cells.
MCF-10A breast epithelial cells were also analysed
following insulin treatment. Similar to the breast cancer cell
lines, normal insulin signalling, IR phosphorylation and AKT and
ERK1/2 phosphorylation, were intact. The increase in the
phosphorylation of these pathways was similar to that observed for
the SK-BR-3 but not the MDA-MB-231 breast cancer cell line, further
emphasizing the highly aberrant insulin signalling in MDA-MB-231
breast cancer cells. In contrast to the effects in both breast
cancer cell lines, however, insulin treatment induced cell
proliferation in MCF-10A cells and a significant decrease in
apoptosis. These results suggest a significant physiological impact
of insulin on cell growth and reduction of apoptosis in these
non-malignant epithelial cells. Clearly, there is sufficient
evidence to suggest a significant growth-promoting and
survival-inducing effect of insulin on MCF-10A breast epithelial
cells. This is a novel finding and may suggest that insulin could
play a role in the aetiology of postmenopausal breast cancer
attributable to obesity.
Epithelial cells rather than breast cancer cells may
be more susceptible to insulin-induced proliferative pressure.
Thus, insulin may have a greater impact on breast cancer aetiology
than on breast cancer progression (at least for progression of
ER-negative breast cancers). This may provide a novel molecular
explanation for the frequently proposed, but unsatisfactorily
explained role of insulin in the promotion of increased
postmenopausal breast cancer risk in obese women.
Abbreviations:
|
IR
|
insulin receptor
|
|
ER
|
oestrogen receptor
|
|
AKT
|
protein kinase B
|
|
ERK
|
extracellular-regulated kinase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
PI3-K
|
phosphoinositol-3 kinase
|
Acknowledgements
This study was supported by a Robert
Gordon University Research and Development Initiative postgraduate
research student fellowship, NHS Endowment Trust and Breast Cancer
Campaign.
References
|
1
|
Flegal KM, Carroll MD, Kuczmarski RJ and
Johnson CL: Overweight and obesity in the United States: prevalence
and trends, 1960–1994. Int J Obes Relat Metab Disord. 22:39–47.
1998.
|
|
2
|
Flegal KM, Carroll MD, Ogden CL and Curtin
LR: Prevalence and trends in obesity among US adults, 1999–2008.
JAMA. 303:235–241. 2010.
|
|
3
|
Calle EE, Rodriguez C, Walker-Thurmond K
and Thun MJ: Overweight, obesity, and mortality from cancer in a
prospectively studied cohort of U.S. adults. N Engl J Med.
348:1625–1638. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Antuna-Puente B, Feve B, Fellahi S and
Bastard JP: Adipokines: the missing link between insulin resistance
and obesity. Diabetes Metab. 34:2–11. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Facchini FS, Hua N, Abbasi F and Reaven
GM: Insulin resistance as a predictor of age-related diseases. J
Clin Endocrinol Metab. 86:3574–3578. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Larsson SC, Mantzoros CS and Wolk A:
Diabetes mellitus and risk of breast cancer: a meta-analysis. Int J
Cancer. 121:856–862. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pisani P: Hyper-insulinaemia and cancer,
meta-analyses of epidemiological studies. Arch Physiol Biochem.
114:63–70. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Verheus M, Peeters PH, Rinaldi S, Dossus
L, Biessy C, Olsen A, Tjonneland A, Overvad K, Jeppesen M,
Clavel-Chapelon F, Tehard B, Nagel G, Linseisen J, Boeing H,
Lahmann PH, Arvaniti A, Psaltopoulou T, Trichopoulou A, Palli D,
Tumino R, Panico S, Sacerdote C, Sieri S, van Gils CH,
Buenode-Mesquita BH, Gonzalez CA, Ardanaz E, Larranaga N, Garcia
CM, Navarro C, Quiros JR, Key T, Allen N, Bingham S, Khaw KT,
Slimani N, Riboli E and Kaaks R: Serum C-peptide levels and breast
cancer risk: results from the European Prospective Investigation
into Cancer and Nutrition (EPIC). Int J Cancer. 119:659–667. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gunter MJ, Hoover DR, Yu H,
Wassertheil-Smoller S, Rohan TE, Manson JE, Li J, Ho GY, Xue X,
Anderson GL, Kaplan RC, Harris TG, Howard BV, Wylie-Rosett J, Burk
RD and Strickler HD: Insulin, insulin-like growth factor-I, and
risk of breast cancer in postmenopausal women. J Natl Cancer Inst.
101:48–60. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Garmendia ML, Pereira A, Alvarado ME and
Atalah E: Relation between insulin resistance and breast cancer
among Chilean women. Ann Epidemiol. 17:403–409. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Del Giudice ME, Fantus IG, Ezzat S,
McKeown-Eyssen G, Page D and Goodwin PJ: Insulin and related
factors in premenopausal breast cancer risk. Breast Cancer Res
Treat. 47:111–120. 1998.PubMed/NCBI
|
|
12
|
Goodwin PJ, Ennis M, Pritchard KI, Trudeau
ME, Koo J, Madarnas Y, Hartwick W, Hoffman B and Hood N: Fasting
insulin and outcome in early-stage breast cancer: results of a
prospective cohort study. J Clin Oncol. 20:42–51. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eliassen AH, Tworoger SS, Mantzoros CS,
Pollak MN and Hankinson SE: Circulating insulin and c-peptide
levels and risk of breast cancer among predominately premenopausal
women. Cancer Epidemiol Biomarkers Prev. 16:161–164. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Alokail MS, Al-Daghri NM, Al-Attas OS and
Hussain T: Combined effects of obesity and type 2 diabetes
contribute to increased breast cancer risk in premenopausal women.
Cardiovasc Diabetol. 8:332009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Purohit A, Newman SP and Reed MJ: The role
of cytokines in regulating estrogen synthesis: implications for the
etiology of breast cancer. Breast Cancer Res. 4:65–69. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stephenson GD and Rose DP: Breast cancer
and obesity: an update. Nutr Cancer. 45:1–16. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lanzino M, Morelli C, Garofalo C, Panno
ML, Mauro L, Ando S and Sisci D: Interaction between estrogen
receptor alpha and insulin/IGF signaling in breast cancer. Curr
Cancer Drug Targets. 8:597–610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Straus DS: Growth-stimulatory actions of
insulin in vitro and in vivo. Endocr Rev. 5:356–369. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Whittaker J, Okamoto AK, Thys R, Bell GI,
Steiner DF and Hofmann CA: High-level expression of human insulin
receptor cDNA in mouse NIH 3T3 cells. Proc Natl Acad Sci USA.
84:5237–5241. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Osborne CK, Bolan G, Monaco ME and Lippman
ME: Hormone responsive human breast cancer in long-term tissue
culture: effect of insulin. Proc Natl Acad Sci USA. 73:4536–4540.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Costantino A, Milazzo G, Giorgino F, Russo
P, Goldfine ID, Vigneri R and Belfiore A: Insulin-resistant
MDA-MB231 human breast cancer cells contain a tyrosine kinase
inhibiting activity. Mol Endocrinol. 7:1667–1676. 1993.PubMed/NCBI
|
|
22
|
Monaco ME and Lippman ME: Insulin
stimulation of fatty acid synthesis in human breast cancer in long
term tissue culture. Endocrinology. 101:1238–1246. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gross GE, Boldt DH and Osborne CK:
Perturbation by insulin of human breast cancer cell cycle kinetics.
Cancer Res. 44:3570–3575. 1984.PubMed/NCBI
|
|
24
|
Geier A, Beery R, Haimshon M, Hemi R and
Lunenfeld B: Serum and insulin inhibit cell death induced by
cycloheximide in the human breast cancer cell line MCF-7. In Vitro
Cell Dev Biol. 28A:415–418. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Faridi J, Fawcett J, Wang L and Roth RA:
Akt promotes increased mammalian cell size by stimulating protein
synthesis and inhibiting protein degradation. Am J Physiol
Endocrinol Metab. 285:E964–E972. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Godden J, Leake R and Kerr DJ: The
response of breast cancer cells to steroid and peptide growth
factors. Anticancer Res. 12:1683–1688. 1992.PubMed/NCBI
|
|
27
|
Papa V, Milazzo G, Goldfine ID, Waldman FM
and Vigneri R: Sporadic amplification of the insulin receptor gene
in human breast cancer. J Endocrinol Invest. 20:531–536. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Weichhaus M, Broom I and Bermano G: The
molecular contribution of TNF-α in the link between obesity and
breast cancer. Oncol Rep. 25:477–483. 2011.
|
|
29
|
Lorincz AM and Sukumar S: Molecular links
between obesity and breast cancer. Endocr Relat Cancer. 13:279–292.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Osborne CK, Monaco ME, Lippman ME and Kahn
CR: Correlation among insulin binding, degradation, and biological
activity in human breast cancer cells in long-term tissue culture.
Cancer Res. 38:94–102. 1978.PubMed/NCBI
|
|
31
|
Hirose K, Toyama T, Iwata H, Takezaki T,
Hamajima N and Tajima K: Insulin, insulin-like growth factor-I and
breast cancer risk in Japanese women. Asian Pac J Cancer Prev.
4:239–246. 2003.PubMed/NCBI
|
|
32
|
Belfiore A, Costantino A, Frasca F,
Pandini G, Mineo R, Vigneri P, Maddux B, Goldfine ID and Vigneri R:
Overexpression of membrane glycoprotein PC-1 in MDA-MB231 breast
cancer cells is associated with inhibition of insulin receptor
tyrosine kinase activity. Mol Endocrinol. 10:1318–1326.
1996.PubMed/NCBI
|
|
33
|
Papa V, Pezzino V, Costantino A, Belfiore
A, Giuffrida D, Frittitta L, Vannelli GB, Brand R, Goldfine ID and
Vigneri R: Elevated insulin receptor content in human breast
cancer. J Clin Invest. 86:1503–1510. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hollestelle A, Elstrodt F, Nagel JH,
Kallemeijn WW and Schutte M: Phosphatidylinositol-3-OH kinase or
RAS pathway mutations in human breast cancer cell lines. Mol Cancer
Res. 5:195–201. 2007. View Article : Google Scholar : PubMed/NCBI
|