Introduction
Glioblastoma (GBM) is the most common type of
malignant tumor and one of the most fatal and least successfully
treated solid tumors. Evidence from hematopoietic malignancies and
solid tumors (including brain, breast, colorectal, and head and
neck cancers) strongly support the hypothesis that a subpopulation
of cancer cells exists in each tumor which has greater potential of
cancer initiation and repopulation (1). Though the origin of glioma-initiating
cells (GICs) is not yet clearly defined, GICs exhibit similar
properties to normal neural stem cells, such as enhanced potential
for proliferation, angiogenesis, and invasion. Moreover, it appears
that GICs contribute to radiation and therapeutic resistance and
are likely responsible for GBM tumor recurrence (1).
The prognosis for GBM patients has not improved in
recent decades, thereby underscoring the difficulties and
challenges in effectively treating this lethal tumor. The
fundamental problem of GBMs is their highly infiltrative nature and
the aggressive invasion of GBM cells into the normal brain tissue
(1). To facilitate cell motility,
invading cells need to change cell-cell adhesion properties,
rearrange the extracellular matrix (ECM) environment, and
reorganize their cytoskeletons (2). Integrins, the transmembrane receptor
molecules, link the ECM to the intracellular actin cytoskeleton of
the cell. Integrins do not possess intrinsic catalytic activity.
Thus, the signals initiated by ECM-integrin interactions are
transduced into cells through activation of integrin-associated
proteins (3). These adaptor
molecules, such as focal adhesion kinase (FAK), α-actinin, talin,
tensin, paxillin, and vinculin, connect integrins to filamentous
actin in the cytoskeleton. This initiates an ‘outside-in’ signaling
cascade, which mediates dynamic cell behavior and results in
forward gliding of the cell body (4).
Protein kinase C (PKC), which belongs to a family of
serinethreonine kinases that catalyze numerous biochemical
reactions critical to the function of many cellular constituents,
are over-expressed or hyperactive in malignant brain tumors
(5). These kinases play a crucial
role in the regulation of various integrin-dependent cellular
functions. Studies conducted to demonstrate the relationship
between PKC activation and modulation of integrin-dependent
functions indicate that adhesion, spreading, and metastasis are
partly regulated by the action of PKCs (6). Phorbol esters are well-known
activators of PKC isoforms, and they induce immediate effects on
the cytoskeleton, such as cell spreading and ruffling, which
indicates that one or several forms of PKC isoforms promote changes
in the cytoskeleton that facilitate or drive cell spreading and
migration (7).
uPAR associates with several members of the integrin
family and is also involved in the initiation of several
intracellular signal transduction pathways that involve
cytoskeletal components, cytosolic kinases, and transmembrane
kinases. Reducing uPAR expression in human glioma cells leads to
changes in cell morphology, decreased cell diffusion, and
cytoskeletal disorganization (8).
Cathepsin B has also been proposed to mediate the dissemination of
cancer cells by degrading components of the ECM or by activating
other proteases that are capable of degrading the ECM (9). In addition, uPAR occupancy has been
implicated in the expression of cathepsin B. Cathepsin B has also
been shown to have an active role in the initiation of proteolytic
cascade involving uPA, plasminogen, and plasmin (9).
The present study attempts to elucidate the effects
of uPAR and cathepsin B on the cytoskeletal organization of glioma
cells, which leads to invasion into normal brain. Our results
demonstrate that shRNA-mediated down-regulation of uPAR and
cathepsin B induces cytoskeletal disorganization in both GICs and
non-GICs by disrupting the PKC-integrin complex and thereby
down-regulating the association of FAK with α-actinin and vinculin
in both in vitro and in vivo models.
Materials and methods
Ethics statement
The Institutional Animal Care and Use Committee of
the University of Illinois College of Medicine at Peoria (Peoria,
IL) approved all surgical interventions and postoperative animal
care. The consent was written and approved. The approved protocol
number is 851 and is dated November 20, 2009.
Cell lines
In the present study, we used U251 glioma cells
obtained from ATCC (American Type Culture Collection, Manassas, VA)
and 5310 glioma xenograft cells kindly provided by Dr David James
(University of California-San Francisco, San Francisco, CA). U251
and 5310 cells were cultured in DMEM medium and RPMI-1640 medium
respectively, supplemented with 10% FBS (Gibco-BRL, Grand Island,
NY) and 1% penicillin/streptomycin (Lonza, Walkersville, MD) at
37°C and 5% CO2.
Isolation of glioma-initiating cells
U251 and 5310 cells were cultured in their
respective media for 18–24 h. Thereafter, 25% of the culture medium
was replaced with an equal volume of knock-out DMEM (Gibco-BRL)
containing 10% knock-out serum replacement (Gibco-BRL), 1%
penicillin/streptomycin, recombinant human epidermal growth factor
(rhEGF, 20 ng/ml; Millipore, Billerica, MA), basic fibroblast
growth factor (bFGF, 20 ng/ml; Millipore), leukemia inhibitory
factor (LIF, 10 ng/ml; Millipore), B27 (1X, Gibco-BRL), N2 (1X,
Gibco-BRL), and L-glutamine (2 mM; Fisher Scientific, Manassas,
VA), and then incubated at 37°C and 5% CO2. This
procedure was repeated until adherent, sphere-like structures were
visible under a microscope. Then, the cells were dissociated,
washed and incubated with PE-conjugated CD133 antibody (Miltenyl
Biotech, Bergisch Gladbach, Germany) at a dilution of 1:10 in
phosphate-buffered saline-bovine serum albumin for 30 min at 4°C.
Cells incubated with isotype IgG antibody were used as a control.
Dead cells were analyzed and excluded using trypan blue at 1:1,000
(FL3 channel). Expression level analysis and sorting were done on
FACScan and FACSAria, respectively (BD Biosciences, San Jose, CA).
CD133+ (GICs) and CD133− (non-GICs) cells
were collected and cultured in their respective media. When GIC
spheres reached a size of more than 100 cells (>120 μm),
they were mechanically dissociated and passaged for further
experiments.
Transfection, radiation and inhibitor
treatments
All transfections were carried out in 100 mm culture
plates using FuGene HD reagent as per the manufacturer’s protocol
(Roche, Indianapolis, IN). U251 and 5310 non-GICs or GICs were
transfected with scrambled vector (pSV) or a bicistronic construct
of uPAR and cathepsin B (pCU) or siRNA against integrin β1 (SCBT,
Santa Cruz, CA). Either 48 h (non-GIC) or 24 h (GIC) after
transfection, the cells were treated with 10 Gy radiation using an
RS 2000 biological irradiator (Rad Source Technologies Inc., Boca
Raton, FL) X-ray unit operated at 150 kV/25 mA. Cells were then
incubated for another 24 or 48 h, respectively. For inhibitor
studies, cells seeded in 6-well plates were treated with rottlerin
(200 μM, Calbiochem, San Diego, CA) and U0126 (10 μM,
Promega, Madison, WI) for 24 h.
Immunofluorescent assays
GICs grown in 4-well chamber slides (Nalge Nunc
International, Naperville, IL) were fixed with 4% buffered
formalin, labeled with anti-CD133, anti-CD44, anti-Nestin,
anti-Sox-2, anti-Ki67, anti-Tuj1, or anti-GFAP (1:100 dilution)
primary antibodies, incubated at 4°C overnight, and stained with
Alexa Fluor-conjugated secondary antibodies. DAPI
(4,6-diamidino-2-phenylindole) was used for nuclear staining.
Samples were photographed using an Olympus IX71 fluorescence
microscope (Melville, NY).
Wound healing assays
For wound healing assay, the non-GICs and GICs
treated with SV, pUC, SV+10 Gy, or pUC+10 Gy were grown in Ibidi
silicon culture inserts (Ibidi, Verona, WI) as per the
manufacturer’s instructions. Briefly, the culture inserts were
placed in 2-well slides coated with fibronectin (2 μg/ml),
and 5000 cells were added into each well of the inserts. The cells
were allowed to grow for 24 h. Then, the inserts were removed, and
the cell patches were overlayed with culture medium. The cells were
allowed to migrate for about 16 h. After the incubation,
immunofluorescence was performed as described above using PKC ζ and
integrin β1 antibodies, and cells were analyzed with a confocal
microscope (Olympus BX61 Fluoview, Minneapolis, USA). Overlay of
images was carried out using SPOT advanced software (Windows
version 4.0.8).
Reverse transcription PCR
Total RNA was isolated using Trizol reagent
(Invitrogen, Carlsbad, CA), and cDNA was produced using
Transcriptor First Strand cDNA Synthesis Kit (Roche). Both tasks
were performed according to the manufacturer’s instructions.
The following primers were used for PCR analysis.
CD133 sense: 5′-ccaagttctacctcatgtttgg-3′, antisense:
5′-accaacagggaga ttgcaaagc-3′; CD44 sense:
5′-aatccctgctaccaatatggact-3′, anti-sense:
5′-agccttcagaatgatttgggtc-3′; Nestin sense: 5′-ggcgcacctca
agatgtcc-3′, antisense: 5′-cttggggtcctgaaagctg-3′; Sox-2 sense:
5′-tggacagttacgcgcacat-3′, antisense: 5′-cgagtaggacatgctgtaggt-3′;
Ki67 sense: 5′-ggatgttgccaaaatagttgctg-3′, antisense: 5′-tccgattc
cgattataccgtttc-3′; Tuj-1 sense: 5′-ctgctcgcagctggagtgag-3′,
anti-sense: 5′-cataaatactgcaggagggc-3′; GFAP sense: 5′-aggtccatgtgg
agcttgac-3′, antisense: 5′-caactggcgtcggtagtcg-3′; β-actin sense:
5′-gtcgtaccactggcattgt-3′, antisense:
5′-cagctgtggtggtgaagct-3′.
Western blotting and
immunoprecipitation
For immunoblot analysis, cells were washed with
ice-cold 1X PBS and resuspended in radioimmunoprecipitation assay
buffer with added protease inhibitors (10 μg/ml aprotinin,
10 μg/ml leupeptin, and 0.5 mM PMSF) and phosphatase
inhibitors (1 mM sodium fluoride and 1 mM sodium orthovanadate).
The cell lysates were analyzed by fractioning equal amounts of
protein using SDS-PAGE. The following antibodies were used: uPAR,
cathepsin B, CD133, CD44, Nestin, Sox-2, Ki67, Tuj-1, β-actin, PKC
θ, integrin α2, integrin α5, integrin β1, FAK, pFAK
(Tyr397), pFAK (Tyr925), vinculin, α-actinin,
Rac-1, Cdc42, ERK, pERK, and GAPDH (all from SCBT, Santa Cruz, CA).
We also used antibodies for PKC δ, pPKC θ/δ, PKC ζ, and
pCdc42/Rac-1 (all from Cell Signaling Technology, Danvers, MA). We
obtained GFAP from Dakocytomation (Carpinteria, CA) and Ras10 from
Millipore. Immunoblots were then incubated with species-specific
HRP-conjugated secondary antibodies, and the signals were detected
using the ECL western blot detection system (Pierce, Rockford,
IL).
Integrin β1 and FAK were immunoprecipitated from 300
μg of total protein using anti-integrin β1 antibody (2
μg) and anti-FAK antibody (2 μg) and protein A plus G
agarose beads (20 μg) as described earlier (10). Integrin β1 pulled down proteins
were immunoblotted for pPKC θ/δ and PKC ζ, and FAK pulled down
proteins were immunoblotted for α-actinin and vinculin.
Immunohistochemical analysis
Stereotactic implantation of U251 non-GICs and U251
GICs (1×105 cells) was carried out as previously
described (11). Seven days after
tumor implantation, mice were treated with pUC (450 μg per
animal) using Alzet mini-pumps at a flow rate of 0.25 μl/h.
Then, radiation was given in two doses (5 Gy on days 8 and 10).
When chronic symptoms were observed in the control group, mice were
euthanized by cardiac perfusion using 10% buffered formalin, and
paraffin sections were prepared. For immunohistochemical analysis,
tumor sections were deparaffinized, blocked with 10% normal goat
serum for 1 h, and then incubated with primary antibody (1:100
dilution in 10% goat serum) overnight at 4°C in a humidified
chamber. Next, they were washed with 1X PBS, incubated with the
Alexa Fluor-conjugated secondary antibody (fluorescent) for 1 h at
room temperature in the dark, and visualized under a confocal
microscope. DAPI was used for nuclear staining.
Results
Enrichment, isolation, and
characterization of glioma-initiating cells
We enriched the population of glioma-initiating
cells by progressively increasing the knock-out DMEM containing
growth factors until many adherent, neurosphere-like structures of
U251 and 5310 cells were observed in the culture plates. This
usually occurred one week after the initial medium replacement.
These spheres were collected, dissociated, conjugated with CD133,
and the positive population was sorted out. The results showed that
U251 and 5310 cells contained 14.65% and 19.69% of
CD133+ cells respectively (Fig. 1A). Further, these isolated cells
were grown in the glioma stem cell medium, and then they were
characterized for stem-cell markers by western blot analysis,
RT-PCR, and immunofluorescence assay (Fig. 1B, D and E). Densitometric analysis
of the western blots revealed that there was a considerable
increase in the protein expression levels of CD133 (6.4-fold in
U251 GICs, 4.4-fold in 5310 GICs), CD44 (2.2-fold in U251 GICs,
2.4-fold in 5310 GICs), Nestin (5.4-fold in U251 GICs, 2.3-fold in
5310 GICs) and Sox-2 (2.7-fold in U251 GICs, 5.5-fold in 5310 GICs)
when compared to that of their non-GIC counterparts (Fig. 1C). U251 GICs and 5310 GICs also
expressed the proliferation marker Ki67 and the lineage markers
GFAP and Tuj-1.
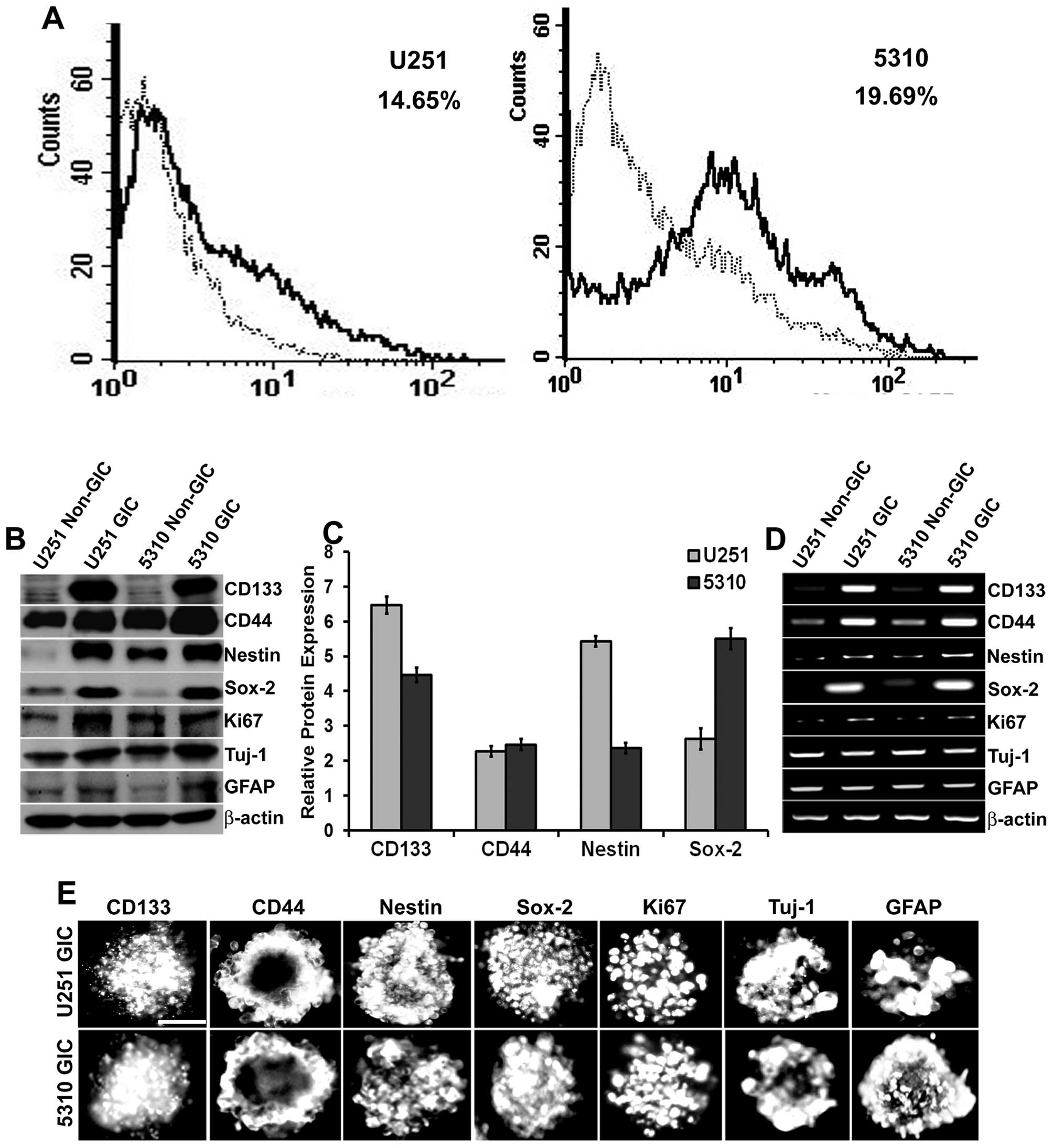 | Figure 1Isolation and characterization of GICs
in U251 glioblastoma cells and 5310 xenograft cells. A, U251 and
5310 cells were enriched with knockout DMEM supplemented with
growth factors as described in Materials and methods. After a
considerable amount of sphere formation was observed, cells were
collected, dissociated, and labeled with CD133 for isolation of
positive cells using IgG-stained cells as a negative control. The
CD133+ population is indicated as a black line over the
dotted line, representing the total population in the histogram. B,
Western blot analysis of non-GICs and GICs showing the expression
of stem cell markers CD133, CD44, Nestin, Sox-2, proliferation
marker Ki67, and the lineage markers GFAP and Tuj-1. C,
Densitometric analysis indicating the increase in the protein
expression levels of CD133, CD44, Nestin, and Sox-2 markers in GICs
as compared to that of their non-GIC counterparts. D, Total RNA was
extracted from the non-GICs and GICs and the mRNA expression levels
of the stem cell, proliferation, and lineage markers were
determined by RT-PCR analysis. E, GICs were further characterized
by immunofluorescence analysis. GICs were immunoprocessed with the
respective antibodies and then conjugated with species-specific
Alexa Fluor 594-conjugated secondary antibodies and visualized
under a microscope. Scale bar, 200 μm. |
Radiation enhanced the expression of uPAR
and cathepsin B in both non-GICs and GICs
Several investigators have posited that
cancer-initiating cells are radio-resistant and that they
contribute to the poor treatment outcomes. Thus, the effect of
radiation on the expression levels of uPAR and cathepsin B was
investigated by western blot analysis (Fig. 2A). For U251 non-GICs and 5310
non-GICs, the protein expression levels of uPAR and cathepsin B
were increased 24 h after initial radiation treatment, and these
increases were dose- and time-dependent. In the U251 GIC and 5310
GIC populations, increases in the expression of uPAR and cathepsin
B were significant only after 48 h following radiation treatment
(Fig. 2A). Based on these results,
further experiments were carried out at 24 h, 10 Gy radiation for
non-GICs and 48 h, 10 Gy radiation for GICs.
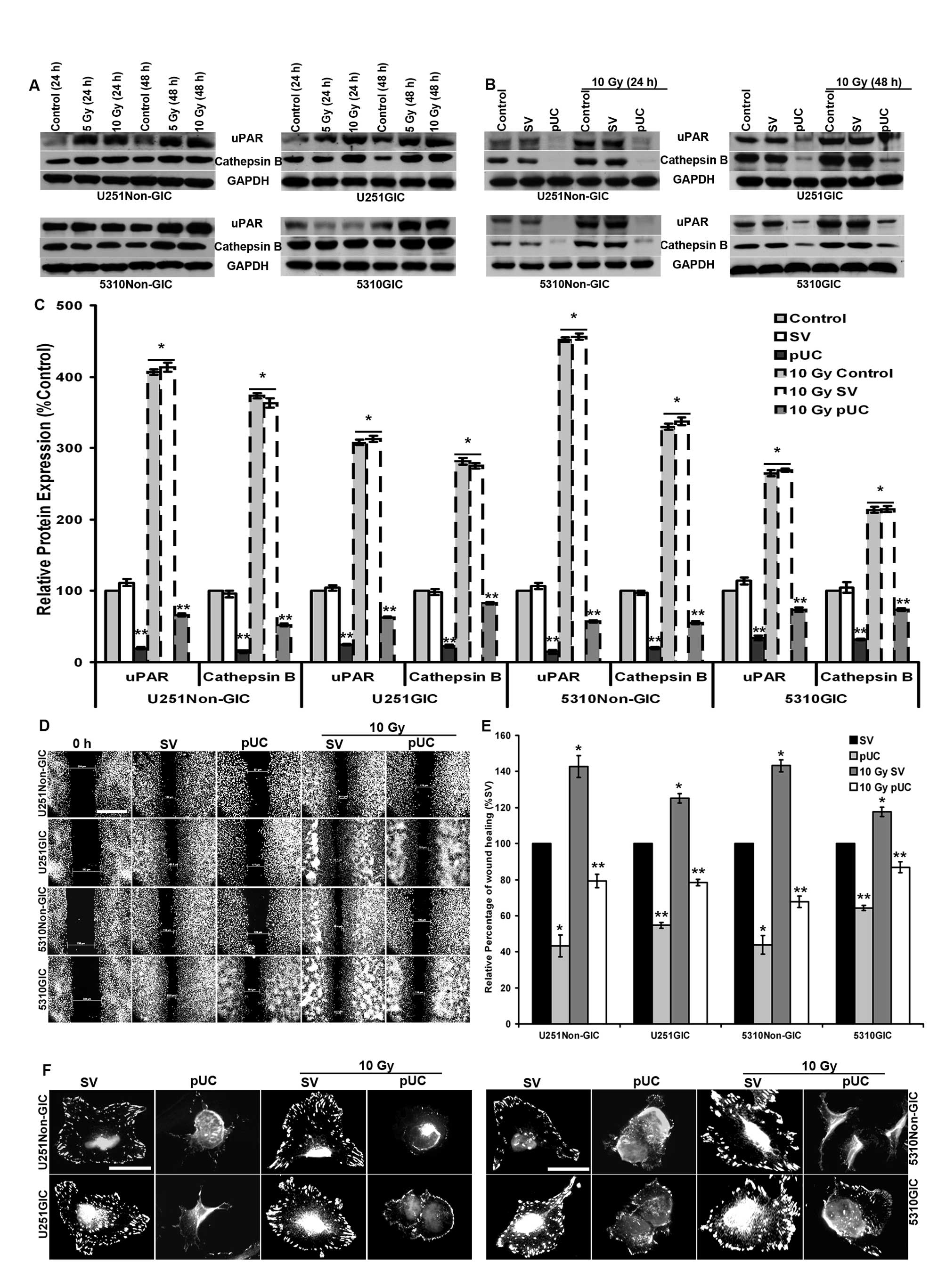 | Figure 2Effect of radiation and pUC on
migration and cytoskeleton of non-GICs and GICs. A, U251 and 5310
non-GICs and GICs were irradiated with 5 and 10 Gy at 24 and 48 h.
Cell lysates were extracted and analyzed by SDS-PAGE to determine
the expression levels of uPAR and cathepsin B with and without
radiation. GAPDH served as a loading control. All the results are
representative of three individual experiments. B, U251 and 5310
non-GICs and GICs were transfected with pSV and pUC with and
without irradiation for 24 and 48 h. Cell lysates were prepared and
analyzed for uPAR and cathepsin B expression levels using western
blotting. C, The protein expression levels of uPAR and cathepsin B
in U251 and 5310 cells treated with pUC and radiation alone and in
combination were analyzed by densitometric analysis and are
depicted in the graph as relative protein expression (control of
each set as 100%). D, Wound healing assay was performed with pSV
and pUC and/or radiation treatments in U251 and 5310 non-GICs and
GICs as described in Materials and methods. The cells were then
fixed, stained with DAPI, and imaged using a fluorescence
microscope (Olympus IX71, USA). Scale bar, 500 μm. E, The
migration and wound healing capacities of U251 and 5310 non-GICs
and GICs treated under the previously described conditions were
measured using a microscope, analyzed, and are graphically
represented as a relative percentage of wound healing (migration of
pSV-treated samples as 100%). F, U251 and 5310 non-GICs and GICs
treated with pSV and pUC with and without radiation were grown on
fibronectin-coated 4-well chamber slides. The cells were then fixed
with 4% buffered formalin, stained with vinculin antibody, and
incubated with DAPI for a brief period of time before mounting.
Scale bar, 200 μm. Values are mean ± SD of three different
experiments (*p<0.05, **p<0.01, in
comparison with the control). |
pUC treatment of non-GICs and GICs reduced the
protein expression levels of uPAR considerably (80.3%-U251
non-GICs, 75.2%-U251 GICs, 85.5%–5310 non-GICs, and 65.7%–5310
GICs) and cathepsin B (85.5%-U251 non-GICs, 77.6%-U251 GICs,
80.2%–5310 non-GICs, and 67.8%–5310 GICs) when compared to controls
(Fig. 2B and C). Treating non-GICs
with 24-h radiation and GICs with 48-h radiation augmented the
expression of uPAR (approximately 3-fold-U251 non-GICs, 2-fold-U251
GICs, 3.5-fold-5310 non-GICs, and 1.7-fold-5310 GICs) and cathepsin
B (∼2.7-fold-U251 non-GICs, 1.8-fold-U251 GICs, 2.3-fold-5310
non-GICs, and 1.1-fold-5310 GICs) (Fig. 2B and C). Further, the combination
treatment of pUC and 10 Gy radiation inhibited the expression of
uPAR (∼83.8%-U251 non-GICs, 79.6%-U251 GICs, 87.4%–5310 non-GICs,
and 72.5%–5310 GICs) and cathepsin B (∼85.9%-U251 non-GICs,
80.8%-U251 GICs, 83.4%–5310 non-GICs, and 72.6%–5310 GICs) when
compared to that of their respective irradiated controls (Fig. 2B and C).
Down-regulation of uPAR and cathepsin B
inhibits migration and induces cytoskeletal disorganization of
non-GICs and GICs
Wound healing assay was carried out using Ibidi
silicon culture inserts, and the variations in the migration of
non-GICs and GICs treated with either pUC and radiation alone and
in combination were measured. The irradiated cells migrated more
(42.7%-U251 non-GICs, 25%-U251 GICs, 43.2%–5310 non-GICs, and
17.6%–5310 GICs) when compared to the pSV-treated (control)
samples, normalized to 100% (Fig. 2D
and E). pUC treatment effectively reduced the migration
capacity of glioma cells (56.8%-U251 non-GICs, 45.4%-U251 GICs,
56.2%–5310 non-GICs, and 35.8%–5310 GICs) when compared to
pSV-treated cells, and it also efficiently inhibited the
radiation-induced increase in the migration of the cells
(44.5%-U251 non-GICs, 32.2%-U251 GICs, 52.7%–5310 non-GICs, and
24.8%–5310 GICs versus pSV-treated irradiated cells). Thus, pUC
treatment significantly retarded the wound healing capacity of
non-GICs and GICs in both the cell lines irrespective of radiation
exposure.
One of the key facilitators of cell motility is the
cytoskeletal organization within the cell. Since pUC treatment
reduced the migratory capacity of the glioma cells, the effect of
treatments on the cytoskeletons of non-GICs and GICs were
investigated. U251 and 5310 cells were stained with vinculin (an
essential cytoskeletal molecule) antibody and then imaged (Fig. 2F). The cells treated with uPAR and
cathepsin B shRNA had disorganized cytoskeletons, the integrity of
cell shape was lost, and the cells became rounded while pSV and pSV
+ 10 Gy treated cells showed a distinct and well-defined
morphology.
uPAR and cathepsin B knockdown inhibited
radiation-induced PKC-integrin signaling
To elucidate the roles of uPAR and cathepsin B in
the PKC-integrin-dependent signal transduction, the expression of
uPAR and cathepsin B were simultaneously knocked down using shRNA
alone or in combination with radiation treatment in U251 and 5310
non-GICs and GICs. Western blotting revealed that the expression
levels of PKC θ, PKC δ, pPKC θ/δ, PKC ζ, integrin β1, integrin α2,
and integrin α5 were reduced significantly with pUC treatment, but
their levels increased with radiation treatment in both non-GICs
and GICs to that of the controls (Fig.
3A). The combination treatment of pUC and 10 Gy efficiently
inhibited the radiation-induced increase in the protein levels of
these molecules.
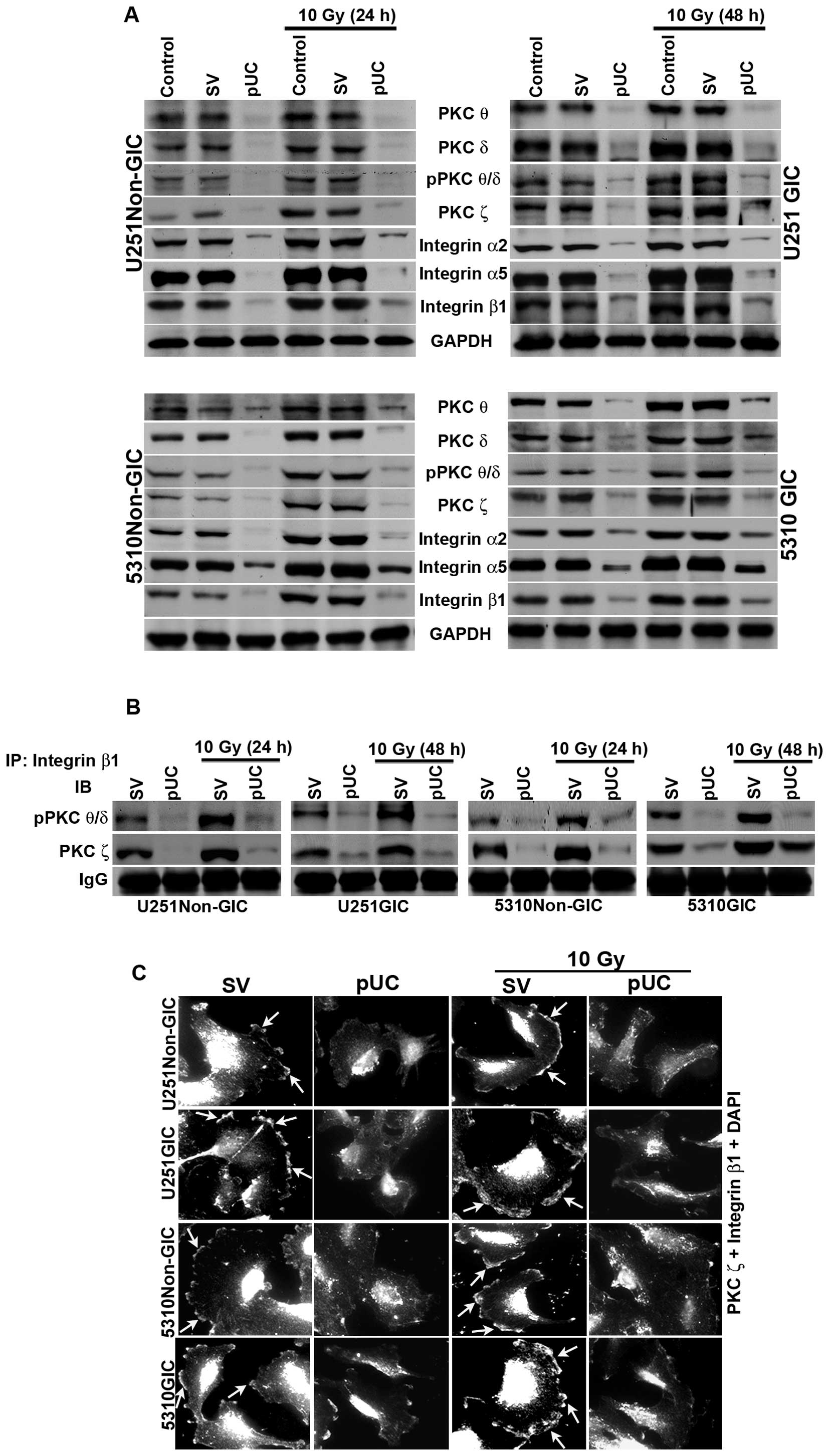 | Figure 3Effect of pUC and radiation alone and
in combination on PKC and integrin levels in U251 and 5310 non-GICs
and GICs. A, Western blot analysis of the cell lysates of U251 and
5310 non-GICs and GICs showing the protein expression levels of PKC
θ, PKC δ, pPKC θ/δ, PKC ζ, integrin β1, integrin α2, and integrin
α5 after treatment with pUC and radiation alone or in combination.
B, Total protein (300 μg) was collected from pSV, pUC, pSV +
10 Gy and pUC + 10 Gy samples of U251 and 5310 non-GICs and GICs
and immunoprecipitated with integrin β1 antibody (2 μg) and
protein A plus G agarose beads (20 μg) overnight at 4°C. The
precipitates were washed with lysis buffer and the integrin β1
pulled down protein was immunoblotted for pPKC θ/δ and PKC ζ. C,
Co-localization of PKC ζ and integrin β1 was carried out with pSV
and pUC with and without 10 Gy (24 h for non-GICs and 48 h for
GICs). The cells were allowed to migrate on 4-well chamber slides
for about 16 h after growing them in Ibidi culture inserts for 24 h
after treatments. The cells were fixed, stained with primary
antibody overnight at 4°C, counter-stained with species-specific
Alexa Fluor-conjugated secondary antibodies, nuclear stained with
DAPI, mounted, and imaged under a confocal microscope. Arrows
indicating the co-localization of PKC ζ and integrin β1. |
Co-immunoprecipitation studies of the total cell
lysates revealed that only the phosphorylated forms of PKC θ and
PKC δ interacted with integrin β1 along with PKC ζ. The bulk of
pPKC θ/δ and PKC ζ co-immunoprecipated with integrin β1 in the
pSV-treated samples of both non-GICs and GICs (Fig. 3B). Radiation treatment further
increased the amount of co-immuno precipitation of integrin β1/pPKC
θ/δ and integrin β1/PKC ζ in U251 and 5310 non-GICs and GICs.
Simultaneous inhibition of uPAR and cathepsin B by shRNA treatment
reduced the interaction between integrin β1/pPKC θ/δ and integrin
β1/PKC ζ and, more importantly, a significant reduction in the
radiation-induced increase in the interaction between these
molecules was observed. However, neither integrin α2 nor integrin
α5 interacted with any of the PKCs (data not shown). Further,
interaction between integrin β1 and PKC ζ was confirmed by
immunocytochemical analysis of U251 and 5310 non-GICs and GICs
(Fig. 3C). In pSV-treated cells
and pSV-treated irradiated cells, a profound co-localization of
integrin β1 and PKC ζ was observed at the leading edge of the
migrating cell and this co-localization was significantly inhibited
in the pUC-treated and pUC + 10 Gy-treated cells (Fig. 3C).
ECM-integrin interaction signal is
influenced by PKCs and vice versa
It is well established that integrins are the main
receptors of ECM. To investigate the influence of ECM-integrin
interaction on PKCs as well as the influence of PKCs on the
ECM-integrin interaction signal, the non-GICs and GICs were grown
on the culture plates coated with collagen (2 μg/ml) and
fibronectin (2 μg/ml). Then, the cells were treated with
either the PKC inhibitor rottlerin (200 μM) or integrin β1
siRNA and compared with cells grown on non-coated culture plates.
Western blotting indicated that the expression of pPKC θ/δ, PKC ζ
and integrin β1 increased along with integrin α2 on collagen-coated
plates (Fig. 4A) and integrin α5
on fibronectin-coated plates (Fig.
4B) in non-GICs and GICs when compared to their counterparts
grown on non-coated plates. Rottlerin treatment as well as integrin
β1 siRNA treatment inhibited the protein expression levels of pPKC
θ/δ, PKC ζ and integrin β1 in U251 and 5310 non-GICs and GICs on
both coated and non-coated plates. Fig. 4 shows that integrin β1 siRNA and
rottlerin did not have any effect on the expression levels of
integrin α2 and integrin α5.
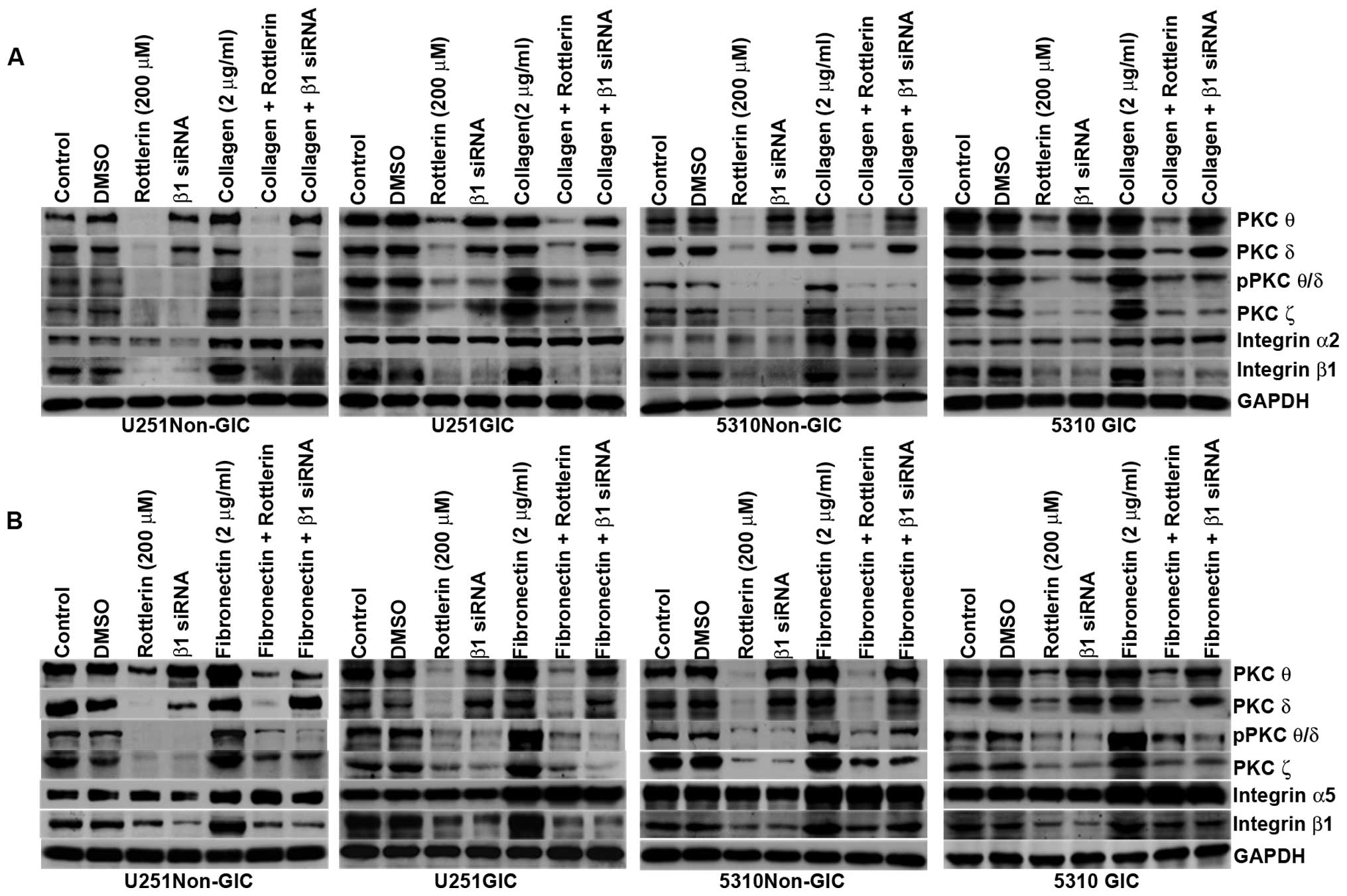 | Figure 4PKCs regulate the ECM-integrin
interaction signal and vice versa in U251 and 5310 non-GICs and
GICs. A, SDS-PAGE was conducted with the cell lysates of control,
DMSO-treated, rottlerin (200 μM) and integrin β1
siRNA-treated U251 and 5310 non-GICs and GICs grown in non-coated
and collagen-coated culture dishes. The cell lysates were
immunoblotted with PKC θ, PKC δ, pPKC θ/δ, PKC ζ, integrin β1 and
integrin α2. B, SDS-PAGE was conducted with the cell lysates of
control, DMSO-treated, rottlerin (200 μM) and integrin β1
siRNA-treated U251 and 5310 non-GICs and GICs grown in non-coated
and fibronectin-coated culture dishes. The cell lysates were
immunoblotted for PKC θ, PKC δ, pPKC θ/δ, PKC ζ, integrin β1 and
integrin α5. |
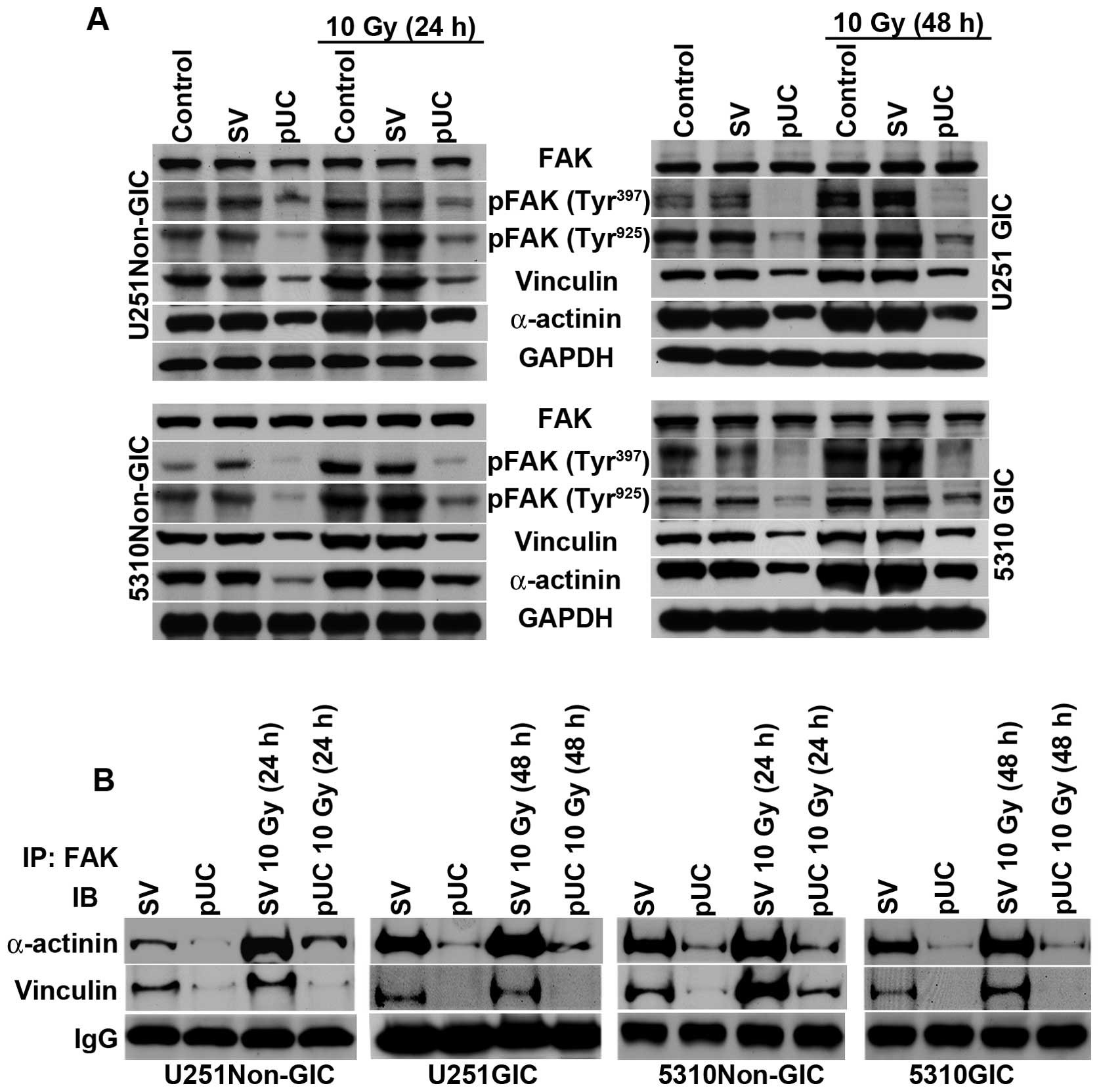
uPAR and cathepsin B depletion by shRNA
treatment disrupted FAK interaction with cytoskeletal
molecules
The cytoplasmic tails of β integrins reportedly
facilitate FAK activation and provide a mechanical linkage between
integrins and cytoskeletal molecules (3). In the present study, the effect of
uPAR and cathepsin B down-regulation on PKC integrated integrin
β1-mediated FAK activation of cyto skeletal molecules was studied.
Western blotting revealed that phosphorylation of FAK at tyrosine
residues 397 and 925 and the expression levels of vinculin and
α-actinin were significantly upregulated following radiation
treatment (Fig. 5A). Non-GICs and
GICs treated with pUC showed a substantial decrease in the
expression of these molecules in non-irradiated as well as
irradiated cells.
FAK co-immunoprecipitated with both vinculin and
α-actinin (Fig. 5B), which
indicates that the PKC/integrin signal was communicated to the
cytoskeletal molecules through the association with FAK. Radiation
treatment of non-GICs and GICs further augmented the interactions
between FAK/vinculin and FAK/α-actinin when compared to pSV-treated
cells. Depletion of uPAR and cathepsin B inhibited the interaction
of FAK with vinculin and α-actinin and also blocked the
radiation-induced interaction between FAK and the cytoskeletal
molecules (Fig. 5B).
FAK/vinculin and FAK/α-actinin
interaction in uPAR and cathepsin B-depleted cells
FAK is involved in the regulation of several
cellular events including cell survival, motility, and adhesion.
pUC treatment effectively reduced the expression of FAK signaling
molecules Rac-1, Cdc42, pCdc42/Rac-1, Ras, and pERK, and the
treatment also reduced radiation-induced expression of these
molecules (Fig. 6A).
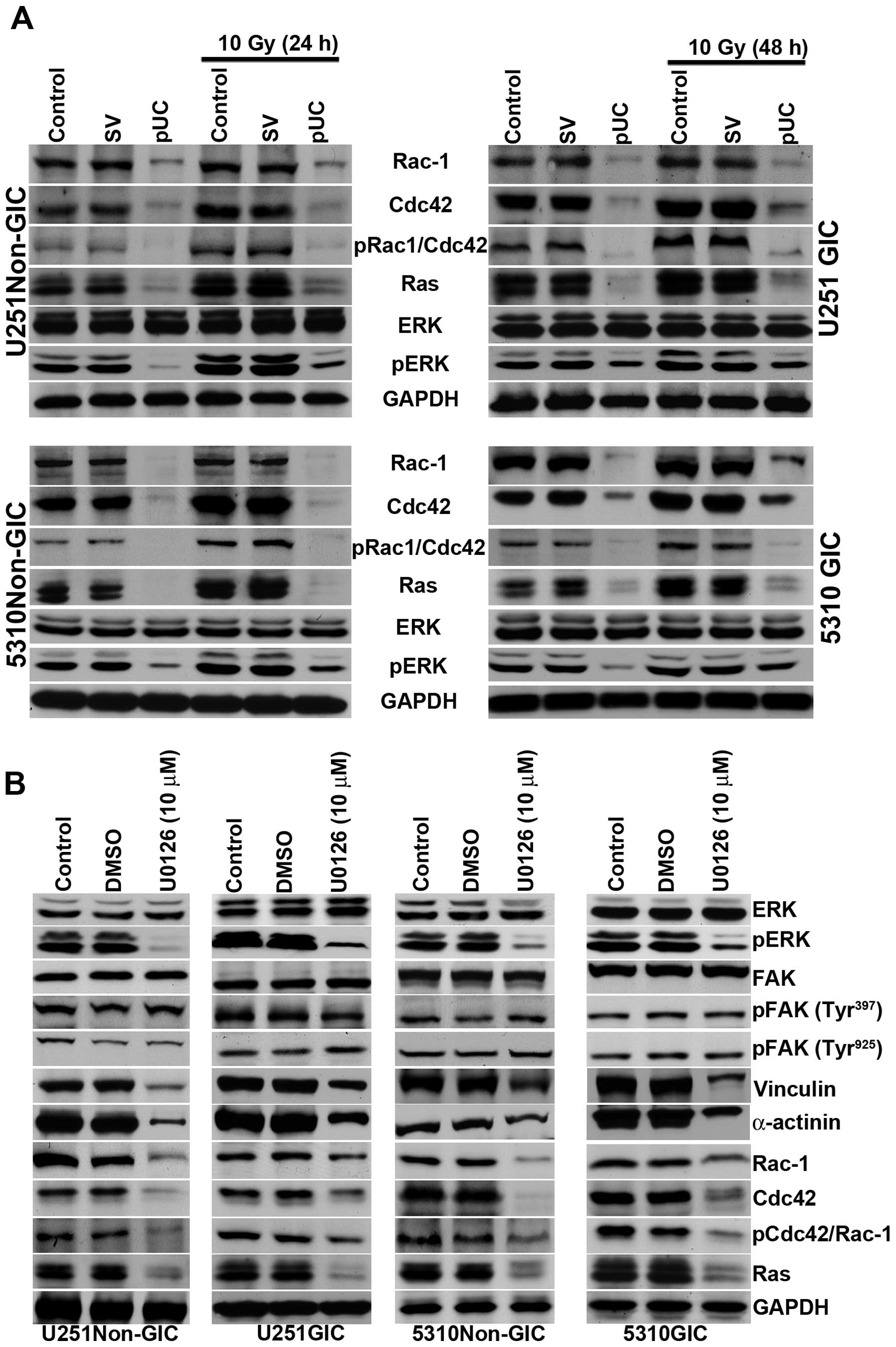 | Figure 6pUC inhibits the FAK migratory
signaling pathway and inhibition of ERK can further influence the
FAK signal. A, Cell lysates of control, pSV-transfected, and
pUC-transfected U251 and 5310 non-GICs and GICs with and without
irradiation were collected. Western blot analysis was performed for
FAK migratory signaling molecules Rac-1, Cdc42, pCdc42/Rac-1, Ras,
ERK, and pERK using their specific antibodies. GAPDH served as a
loading control. B, The total protein lysates from U251 and 5310
non-GICs and GICs were collected from U0126 (10 μM) and DMSO
treatments. SDS-PAGE was conducted as described in Materials and
methods. Western blotting was performed to determine the protein
expression levels of ERK, pERK, FAK, pFAK (Tyr397), pFAK
(Tyr925), vinculin, α-actinin, Rac-1, Cdc42,
pCdc42/Rac-1, and Ras using their specific antibodies. |
Phosphorylated ERK translocates into the nucleus of
the cell and regulates the activity of various downstream
substrates involved in a multitude of cellular responses including
cytoskeletal changes and gene transcription. To determine if there
is any effect of ERK on FAK signaling, cells were treated with an
ERK inhibitor, U0126 (10 μM); there was no detectable effect
of U0126 on FAK or its phosphorylation. However, the U0126
treatment did reduce the expression of vinculin, α-actinin, Ras,
Rac-1, and Cdc42 (Fig. 6B). This
suggests that the down-regulation of pERK by uPAR and cathepsin B
knockdown further induces a feedback effect, causing an extreme
reduction in the expression levels of the molecules responsible for
cytoskeletal rearrangement.
pUC treatment reduces the interaction
between integrin β1/PKCs and FAK/cytoskeletal molecules with and
without radiation in vivo
To determine the in vivo effect of shRNA
treatment alone or in combination with radiation on the interaction
of the signaling molecules, fluorescent immunohistochemical
analysis was carried out on the brain tissue sections of the mice
implanted with U251 non-GICs and U251 GICs. Results showed that
integrin β1 strongly co-localized with pPKC θ/δ and PKC ζ and
radiation further augmented this co-localization in U251 non-GICs
and U251 GICs (Fig. 7A and B). In
accordance with the in vitro studies, pUC treatment
decreased the interaction between PKCs and integrin β1. Moreover,
pUC treatment in combination with radiation blocked the
radiation-induced PKC/integrin signaling when compared to
appropriate irradiated controls. In vivo, brain sections
also revealed a prominent interaction between FAK and both
α-actinin (Fig. 7C) and vinculin
(Fig. 7D), which was further
increased by radiation. The pUC treatment effectively hindered the
radiation-induced association of FAK with those cytoskeletal
molecules.
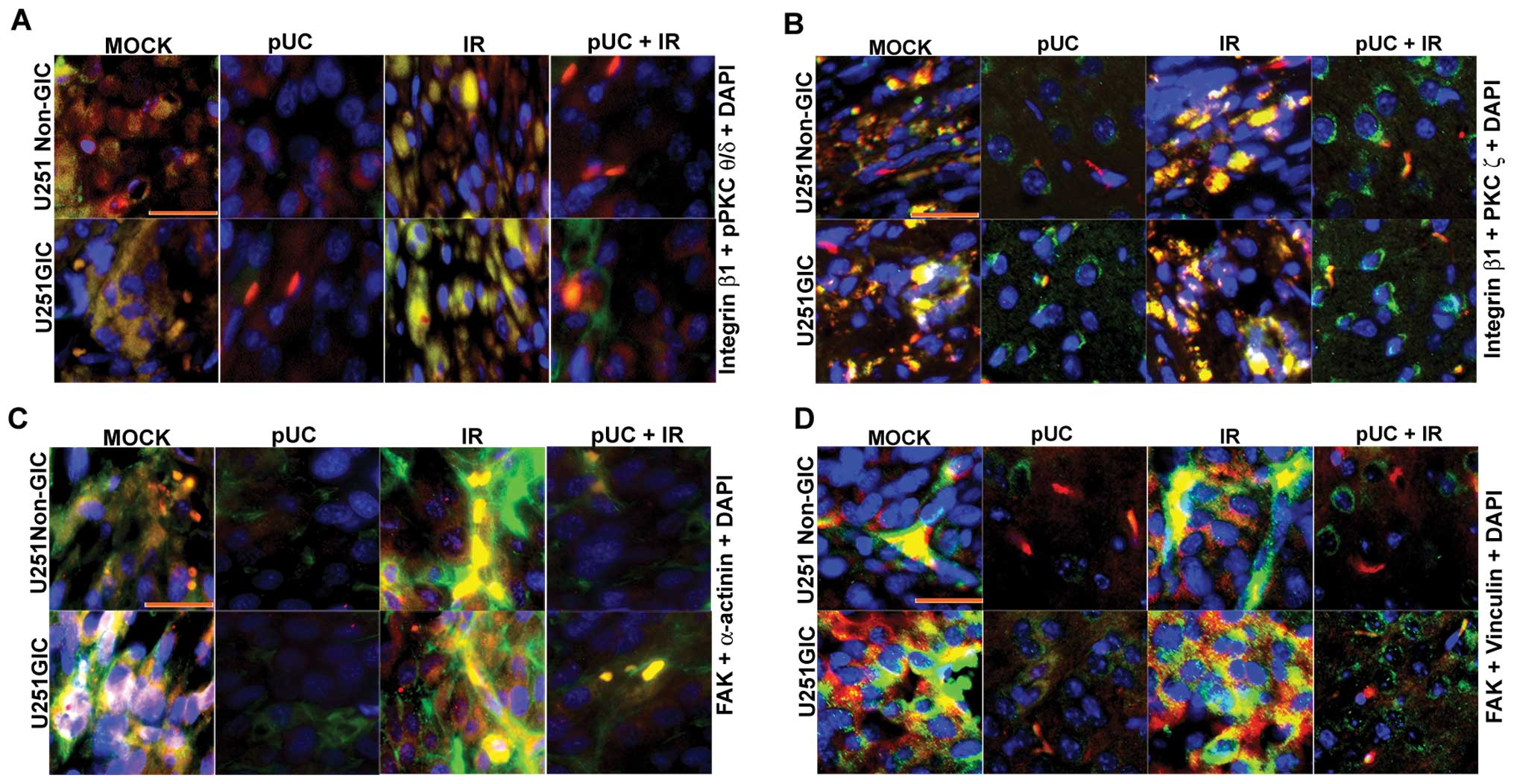 | Figure 7Effect of pUC and radiation treatment
on pre-established intracranial tumors. U251 non-GICs and GICs were
implanted intracranially into nude mice and treated with 450
μg of pUC seven days after implantation. Radiation treatment
was given in two doses (5 Gy at days 8 and 10). When chronic
symptoms were observed, the mice were sacrificed, and their brains
were collected and embedded in paraffin. Paraffin-embedded sections
were deparaffinized, antigen retrieved, and co-localization studies
were carried out. The brain sections were incubated overnight with
primary antibodies in 10% goat serum at 4°C in a humidified
chamber, counterstained with Alexa Fluor-conjugated secondary
antibodies, and incubated with DAPI for nuclear staining before
mounting. A, Co-localization of integrin β1 (green), pPKC θ/δ (red)
and DAPI (blue) in the paraffin sections of the nude mice
established with U251 non-GICs and GICs and treated with shRNA and
radiation alone or in combination. B, Interaction between integrin
β1 (green) and PKC ζ (red) in the various in vivo
combination treatments. DAPI (blue) was used for nuclear staining.
C, In vivo brain sections of immunocompromised mice
implanted with U251 non-GICs and GICs were labeled with FAK (green)
and α-actinin (red) and processed for immunofluorescence. The
sections were incubated with DAPI for a brief period of time for
nuclear staining. D, Mock, pUC-treated, irradiated, and pUC +
irradiated brain sections of the nude mice pre-established with
U251 non-GICs and GICs were immunoprocessed and labeled with FAK
(green), vinculin (red) and DAPI (blue) in order to observe the
co-localization of FAK and vinculin in in vivo samples. |
Discussion
There is increasing evidence that cancerous tumors
might contain their own stem cells. The presence of this small
subpopulation of slowly dividing cancer stem cells might explain
why so many cancers recur after treatment with radiation or
cytotoxic drugs, even when most of the malignant cells seem to be
killed by the therapy; these surviving cells may be not only
resistant to therapy but also essential for the malignancy
(12). Advocates for the cancer
stem cell model have suggested that therapy should be directed
against these stem cells and the remainder of the tumor cells
should eventually wither away. However, it has also been proposed
that the differentiated cells can dedifferentiate into stem
cell-like cancer cells. Thus, a combined therapy targeting both the
bulk of the tumor and the glioma stem cells will be necessary to
obtain a successful long-term cure of glioma (13). Therefore, for cancer therapy to be
curative, it is crucial to identify and study cancer stem
cells.
Establishing cell lines from tumors that retain
glioma-initiating stem cell properties would provide a valuable and
accurate model for the human disease. It would also give insight
into the origin of tumor heterogeneity and enable more refined
analysis of molecular mechanisms that regulate transformation,
self-renewal, commitment, and differentiation (14). In the present study, we isolated
GICs from the U251 cell line and the 5310 xenograft cell line with
a CD133 surface marker using fluorescence-activated cell sorting
(FACS) and then characterized the cells with other stem cell
markers like CD44, Nestin, and Sox-2. CD133 has been used to
isolate populations of cancer stem cells with enhanced stem cell
phenotypes from multiple types of brain cancer and these
CD133+ cells from glioblastoma are capable of
multi-lineage differentiation and had a high capacity for
neurosphere formation (15,16).
Normally, cellular migration is under strict control
but, in transformed tumor cells, control mechanisms are disturbed
so that cells are able to migrate and to invade the surrounding
tissue. Key determinants of the metastatic potential of tumor cells
are matrix invasion, changes in cell shape, cell movement, and
motility. uPAR and cathepsin B, which are both overex-pressed in
glioma, function either individually or in combination to degrade
the ECM, thereby facilitating metastasis (17). We elucidated the roles of uPAR and
cathepsin B in the regulation of interaction between PKCs and
integrins and their subsequent effect on the cytoskeletal molecules
through FAK signaling by simultaneous down-regulation of uPAR and
cathepsin B alone or in combination with radiation.
Ionizing radiation is one of the most common and
effective treatments for malignant brain tumors. However, the
tumors frequently recur or progress as focal complexes after
treatment with ionizing radiation, suggesting the existence of a
critical, radiation-resistant subpopulation with potent tumorigenic
activity (18). The fact that some
glioblastoma cells are resistant to radiation lends support to this
concept. We observed increases in the levels of uPAR and cathepsin
B with radiation treatment in a dose-dependent manner for non-GICs
and in a dose- and time-dependent manner in GICs, indicating that
GICs are more resistant than their matched non-GIC counterparts.
The changes in the cytoskeletal organization and distribution of
actin and actin-binding proteins are necessary for several cellular
processes, such as focal adhesion formation, cell motility, and
cell invasion (19). Therefore,
the cytoskeleton is the key cellular machinery responsible for
cellular movement. Treating the glioma cells with pUC alone or in
combination with radiation, severely disorganized the cytoskeleton
and also disrupted cell shape, thereby inhibiting the cells’
migratory capacity when compared to their respective non-irradiated
and irradiated controls.
Members of the PKC family of serine/threonine
kinases are key components of signal transduction pathways that
have been linked to carcinogenesis and cancer progression.
Recently, it has been reported that cell signaling involving PKC δ
is crucial for radiation-induced expansion of glioma-initiating
cell populations and acquisition of resistance to anti-cancer
treatments (18). Treating
non-GICs and GICs with radiation therapy further augmented the
expression levels of PKC θ, PKC δ, pPKC θ/δ, and PKC ζ, indicating
that radiation treatment is making these cells more resistant.
Treating glioma cells with pUC prior to radiation inhibited the
induction of the expression of these molecules.
Interaction between the ECM and cell surface
integrins leads to intracellular signaling events that affect cell
migration, proliferation and survival, which in the context of
neoplastic cells, can translate directly into malignant phenotype
(20). Carriero et al
reported that binding of uPA to uPAR regulates integrins in a
PKC-dependent manner in MDA-MB-231 and MCF-7 breast carcinoma cell
lines (21), and Rigot et
al reported that integrin engagement by PKCs was required for
the migration of HT29-D4 colon carcinoma cells (6). The studies here indicated that pPKC
θ/δ and PKC ζ co-immunoprecipitated with integrin β1 and the
interaction between these molecules was strengthened following
radiation exposure, whereas pUC treatment efficiently inhibited the
interaction in non-irradiated and irradiated samples of non-GICs
and GICs.
To further confirm the interaction between PKCs and
integrin β1 and to study the effect of ECM-integrin interaction,
collagen- and fibronectin-coated plates were used to culture the
cells. The GICs were grown as adherent cultures on these coated
plates. Then, the cells were treated with inhibitors, either
rottlerin (200 μM) or integrin β1 siRNA. The addition of ECM
proteins induced increase in the levels of integrin α2, integrin
α5, integrin β1, pPKC θ/δ and PKC ζ. We observed that the
inhibition of PKCs by rottlerin inhibited the expression of
integrin β1 and the inhibition of integrin β1 reduced the levels of
pPKC θ/δ and PKC ζ but neither the inhibitor nor the siRNA had any
effect on the expression levels of integrin α2 and integrin α5.
These results clearly demonstrate that PKCs and integrin β1
interact with each other and blocking of any of these molecules may
not render an effective downstream signal.
It is well known that integrin binding to ECM
transduces the signal through FAK, which is believed to be central
in the orchestration of this signal to the downstream effectors
that ultimately control the events important for cell motility,
including cytoskeletal reorganization and contraction, focal
adhesion disassembly, and maturation (22). It has also been shown that
PKC-dependent activation of FAK and Src regulates the actin
cytoskeleton of SH-SY5Y neuroblastoma cells (23). Thus, integrins as well as PKCs may
influence FAK signaling to the downstream molecules. Since pUC
treatment disrupted the complex formation of PKCs and integrins,
the effect of the shRNA treatment on FAK and the downstream
cytoskeletal molecules was chosen for further study. Western
blotting showed that there was a decrease in the protein content of
pFAK (Tyr397), pFAK (Tyr925), vinculin, and
α-actinin in the nonirradiated and irradiated cell lysates of pUC
treated non-GICs and GICs. Chen et al conducted studies that
suggested that cell shape depends on focal adhesion assembly,
knocking out focal adhesion proteins, such as FAK, vinculin, and
α-actinin, predictably resulted in the cells failing to spread
normally (24). The
immmunoprecipitation analysis further showed that the depletion of
uPAR and cathepsin B from the cells not only reduced pPKC
θ/δ/integrin β1 and PKC ζ/integrin β1 complexes but also reduced
the interaction of FAK with vinculin and α-actinin.
Integrin engagement and its clustering at focal
adhesions transduces signals for the activation of FAK, Src, ERK,
and/or Akt. Rho family GTPases appear poised to contribute to these
integrin-mediated signals, particularly signals that control
cytoskeletal organization (25).
Rho family members, such as Cdc42, Rac1, and RhoA, are part of the
Ras superfamily of proteins, cycling between active GTP-bound and
inactive GDP-bound states. Clark et al reported that
adhesive interactions stimulate the formation of punctate focal
complexes at the cell periphery during cell spreading, and cell
spreading is dependent on Cdc42 and Rac, which are responsible for
membrane ruffling mechanisms (25). In the present study, we observed
that the key regulatory molecules of cytoskeletal organization and
migration, such as Rac, Cdc42, Ras and ERK, were down-regulated
with pUC treatment. MAP kinases (ERK1 and ERK2) have been
recognized as a major system through which cells transduce a
variety of extracellular signals, and thus, they represent the
convergence point for many signaling pathways. It is now clear that
these kinases can promote cell migration on the ECM (6). Consequently, we thought that ERK in
turn might play a role in the FAK-mediated signaling; to confirm
this hypothesis, we used U0126 (10 μM), an ERK inhibitor.
Western blot analysis of cell lysates treated with U0126 showed a
decrease in the expression levels of vinculin, α-actinin, Rac,
Cdc42, and Ras, but there was no significant effect on FAK or FAK
phosphorylation, thus confirming that ERK can regulate FAK
signaling downstream from FAK. Further, this feedback effect of ERK
might cause an extreme reduction in the cytoskeletal organization
and migratory signals.
In vivo studies demonstrated that interaction
between pPKC θ/δ/integrin β1, PKC ζ/integrin β1, FAK/vinculin and
FAK/α-actinin increased with radiation treatment, which
demonstrates the radio-resistance of these glioma cells. Moreover,
the increase in the interaction of the aforementioned molecules was
larger in GICs when compared to their non-GIC counterparts; these
interactions and expression levels of these molecules were
inhibited with pUC and pUC + radiation treatments. Carriero et
al reported that HT1080 human fibrosarcoma and MCF-7 human
breast carcinoma cells respond to urokinase by exhibiting
remarkable cytoskeletal rearrangements that are mediated by αvβ5
and require protein kinase C activity (21). Parsons et al identified a
direct link between PKC α and integrin β1 that is critical for
directed tumor cell migration of human breast carcinoma cells
(26). Follicular thyroid
carcinoma cells treated with rottlerin exhibited decreased protein
levels of integrin β1, FAK, and focal adhesion complex components
(e.g., vinculin, α-actinin), and reduced activity of GTPases
supporting an integrin/focal adhesion/cytoskeleton signaling in the
migration arrest induced by rottlerin (27). In summary, simultaneous
down-regulation of uPAR and cathepsin B by shRNA with and without
radiation treatment disrupted the PKC and integrin complex and
subsequently blocked the interaction between FAK and the
cytoskeletal molecules vinculin and α-actinin, which regulate the
actin cytoskeleton of the cell.
Acknowledgements
The authors wish to thank Shellee
Abraham for manuscript preparation, and Diana Meister and Sushma
Jasti for manuscript review. Funding: This research was supported
by award number CA116708 (J.S.R.) from the National Cancer
Institute. Contents are solely the responsibility of the authors
and do not necessarily represent the official views of National
Institutes of Health.
References
|
1
|
Huang Z, Cheng L, Guryanova OA, Wu Q and
Bao S: Cancer stem cells in glioblastoma - molecular signaling and
therapeutic targeting. Protein Cell. 1:638–655. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang CY, Fong YC, Lee CY, et al: CCL5
increases lung cancer migration via PI3K, Akt and NF-kappaB
pathways. Biochem Pharmacol. 77:794–803. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mitra SK and Schlaepfer DD:
Integrin-regulated FAK-Src signaling in normal and cancer cells.
Curr Opin Cell Biol. 18:516–523. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wolf K and Friedl P: Molecular mechanisms
of cancer cell invasion and plasticity. Br J Dermatol. 154(Suppl
1): 11–15. 2006. View Article : Google Scholar
|
|
5
|
Da Rocha AB, Mans DR, Regner A and
Schwartsmann G: Targeting protein kinase C: new therapeutic
opportunities against high-grade malignant gliomas? Oncologist.
7:17–33. 2002.PubMed/NCBI
|
|
6
|
Rigot V, Lehmann M, Andre F, Daemi N,
Marvaldi J and Luis J: Integrin ligation and PKC activation are
required for migration of colon carcinoma cells. J Cell Sci.
111:3119–3127. 1998.PubMed/NCBI
|
|
7
|
Larsson C: Protein kinase C and the
regulation of the actin cytoskeleton. Cell Signal. 18:276–284.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chintala SK, Mohanam S, Go Y, et al:
Altered in vitro spreading and cytoskeletal organization in human
glioma cells by down-regulation of urokinase receptor. Mol
Carcinog. 20:355–365. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rao JS: Molecular mechanisms of glioma
invasiveness: the role of proteases. Nat Rev Cancer. 3:489–501.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gopinath S, Malla RR, Gondi CS, et al:
Co-depletion of cathepsin B and uPAR induces G0/G1 arrest in glioma
via FOXO3a mediated p27 upregulation. PLoS One. 5:e116682010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Malla RR, Gopinath S, Gondi CS, et al:
Cathepsin B and uPAR knockdown inhibits tumor-induced angiogenesis
by modulating VEGF expression in glioma. Cancer Gene Ther.
18:419–434. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kondo T, Setoguchi T and Taga T:
Persistence of a small subpopulation of cancer stem-like cells in
the C6 glioma cell line. Proc Natl Acad Sci USA. 101:781–786. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gilbert CA and Ross AH: Glioma stem cells:
cell culture, markers and targets for new combination therapies.
Cancer Stem Cells Theories and Practice. Shostak S: InTech; pp.
79–104. 2011
|
|
14
|
Pollard SM, Yoshikawa K, Clarke ID, et al:
Glioma stem cell lines expanded in adherent culture have
tumor-specific phenotypes and are suitable for chemical and genetic
screens. Cell Stem Cell. 4:568–580. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Singh SK, Clarke ID, Terasaki M, et al:
Identification of a cancer stem cell in human brain tumors. Cancer
Res. 63:5821–5828. 2003.PubMed/NCBI
|
|
16
|
Singh SK, Hawkins C, Clarke ID, et al:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gondi CS, Kandhukuri N, Kondraganti S, et
al: RNA interference-mediated simultaneous down-regulation of
urokinase-type plasminogen activator receptor and cathepsin B
induces caspase-8-mediated apoptosis in SNB19 human glioma cells.
Mol Cancer Ther. 5:3197–3208. 2006. View Article : Google Scholar
|
|
18
|
Kim MJ, Kim RK, Yoon CH, et al: Importance
of PKCdelta signaling in fractionated-radiation-induced expansion
of glioma-initiating cells and resistance to cancer treatment. J
Cell Sci. 124:3084–3094. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Donald CD, Cooper CR, Harris-Hooker S,
Emmett N, Scanlon M and Cooke DB III: Cytoskeletal organization and
cell motility correlates with metastatic potential and state of
differentiation in prostate cancer. Cell Mol Biol (Noisy-le-grand).
47:1033–1038. 2001.PubMed/NCBI
|
|
20
|
Uhm JH, Gladson CL and Rao JS: The role of
integrins in the malignant phenotype of gliomas. Front Biosci.
4:D188–D199. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carriero MV, Del Vecchio S, Capozzoli M,
et al: Urokinase receptor interacts with alpha(v)beta5 vitronectin
receptor, promoting urokinase-dependent cell migration in breast
cancer. Cancer Res. 59:5307–5314. 1999.PubMed/NCBI
|
|
22
|
Gunther W, Skaftnesmo KO, Arnold H and
Terzis AJ: Molecular approaches to brain tumour invasion. Acta
Neurochir (Wien). 145:1029–1036. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bruce-Staskal PJ and Bouton AH:
PKC-dependent activation of FAK and src induces tyrosine
phosphorylation of Cas and formation of Cas-Crk complexes. Exp Cell
Res. 264:296–306. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen CS, Alonso JL, Ostuni E, Whitesides
GM and Ingber DE: Cell shape provides global control of focal
adhesion assembly. Biochem Biophys Res Commun. 307:355–361. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clark EA, King WG, Brugge JS, Symons M and
Hynes RO: Integrin-mediated signals regulated by members of the rho
family of GTPases. J Cell Biol. 142:573–586. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Parsons M, Keppler MD, Kline A, et al:
Site-directed perturbation of protein kinase C-integrin interaction
blocks carcinoma cell chemotaxis. Mol Cell Biol. 22:5897–5911.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin CJ, Lin CY, Chen Y, Huang SH and Wang
SM: Rottlerin inhibits migration of follicular thyroid carcinoma
cells by PKCdelta-independent destabilization of the focal adhesion
complex. J Cell Biochem. 110:428–437. 2010.
|