Introduction
Hepatocellular carcinoma (HCC) is one of the most
aggressive human types of cancer in the world and the second
leading cause of cancer mortality; with few curative treatment
options by surgical resection and/or transplantation. Current
predictions suggest that new cases are increasing steeply, rising
to 748,300, with 695,900 deaths every year (1). This is due to the fact that only 15%
of patients qualify for tumor resection and patients who undergo
hepatectomy are reported to develop new tumors in their residual
liver due to the aggressive features of HCC, which include poor
liver function and metastasis (2).
However, no major advances have been made in the past decades.
Therefore, HCC remains a serious problem and more effective
prevention and treatment strategies are urgently needed.
5-fluorouracil (5-FU), a pyrimidine based analog,
has been widely used as an anticancer drug for the treatment of
gastrointestinal, breast, head and neck, and ovarian cancers. Due
to its clastogenic nature, 5-FU is converted to
5-fluoro-2-deoxyuridylate monophosphate (FdUMP), and it
competitively inhibits DNA synthesis by inhibiting the enzyme
thymidylate synthase (3,4). 5-FU induced DNA damage activates
various signaling pathways to enhance or inhibit apoptotic cell
death. 5-FU by itself or in conjunction with andrographolide
induces apoptosis and inhibition of cell proliferation through a
p53 mediated pathway in human colon cancer cells (5,6).
Apoptosis is a form of cell death that involves the
consecutive action of a number of intracellular signaling pathways.
Resistance to apoptotic induction is an important cause of failure
in chemotherapy; therefore, strengthening apoptosis induction might
improve the effect of anticancer therapy. It has been suggested
that there are several apoptotic pathways in cells responsive to
death stimuli. Previous studies (7,8) have
concentrated on the endoplasmic reticulum (ER) as a third
subcellular compartment implicated in apoptotic execution.
Accumulation of misfolded proteins and changes in Ca2+
homeostasis in ER results in ER stress and eventually leads to
apoptotic cell death (7). In
general, autophagy can activate both pro-survival mechanisms as
well as cell death programs, especially if autophagy activated
following ER stress is a pro-survival response to restore the ER
homeostasis by clearing the unfolded aggregates (8). A wide range of chemotherapeutic
agents have been identified and are used to activate ER stress
along with autophagy in carcinoma cells (8). Heat shock proteins function as
molecular chaperones in regulating cellular homeostasis and promote
cell survival response. Inhibition of autophagy and molecular
chaperones might be a suitable pharmacological target to promote
apoptosis in tumor cells. In this study, we have evaluated the
effect of 5-FU to elicit ER stress-induced apoptosis in Sk-Hep1
cells. The results suggest that 5-FU induces ER stress and
activates the intrinsic apoptotic cell death pathway by
downregulating GRP78 and autophagy in hepatocarcinoma Sk-Hep1
cells, explaining a multiple role of 5-FU as a therapeutic agent
targeting human HCC.
Materials and methods
Cell culture and reagents
Human Sk-Hep1 cells (American Type Culture
Collection, Manassas, VA, USA) were cultured in EMEM supplemented
with 10% FBS (v/v) (HyClone, Logan, UT, USA) and penicillin (100
U/ml)/streptomycin (100 μg/ml) (PAA Laboratories GmbH, Pasching,
Austria). Cultures were maintained in a humidified incubator at
37°C in 5% CO2. 5-FU, dimethyl sulfoxide and propidium
iodide were purchased from Sigma Aldrich (St. Louis, MO, USA).
EZ-Cytox Cell Viability Assay Solution (WST-1) was purchased from
Daeil Lab Service (Jong-No, Seoul, Korea). Lysis buffer was
obtained from Intron Biotechnology (Gyeonggi, Korea). Hoechst 33342
was purchased from Cell Signaling Technology (Danvers, MA, USA) and
Fluo3-AM was from Invitrogen (Grand Island, NY, USA). Enhanced
chemiluminescent (ECL) detection solutions were obtained from
Pierce (Rockford, IL, USA). Anti-HSP70, anti-HSP27,
anti-CHOP/GADD153, and anti-AIF were obtained from Santa Cruz
Biotechnology, Inc (Santa Cruz, CA, USA). All other antibodies were
obtained from Cell Signaling Technology.
Cell viability assay
Sk-Hep1 cells in exponential growth phase at a
density of 1×104 cells were resuspended in 100 μl of
EMEM medium and seeded on 96-well plates in triplicate. Following
overnight incubation, 5-FU at various concentrations was added.
Cells were incubated for 24 h, and 10 μl of WST-1 solution was
added and incubated for an additional 3 h. The absorbance of the
reaction was measured using an ELISA reader (Molecular Devices,
Sunnyvale CA, USA) at 460 nm. Cell viability was calculated
according to the following formula: Cell viability (%) = A460
(sample)/A460 (control) x 100.
In situ labeling of apoptotic cells
Cells were cultured in a cover glass bottom culture
dish for 24 h and treated with graded concentrations of 5-FU for
another 24 h. Following incubation, cells were washed twice with
PBS, stained with Hoechst 33342 (1 μg/ml) for 15 min at 37°C, and
then fixed for 15 min with 4% paraformaldehyde. The cells were
examined under an ECLIPSE 50i fluorescence microscope (Nikon,
Tokyo, Japan).
Flow cytometry
Briefly, cells were harvested by trypsinization and
fixed with 70% ethanol overnight at 4°C. Then, the cells were
resuspended in PBS buffer containing 0.2 mg/ml RNase A and
incubated for 1 h at 37°C. The cells were then stained with 40
μg/ml propidium iodide at room temperature for 30 min under dark
conditions. The distribution of subgenomic DNA was analyzed using a
flow cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA).
Western blot analysis
After treatment with 5-FU, cells were washed twice
with ice-cold PBS and the reaction was terminated by the addition
of 100 μl lysis buffer containing 50 mM Tris-Cl (pH 7.5), 150 mM
NaCl, 1 mM DTT, 0.5% NP-40, 1% Triton X-100, 1% deoxycholate, 0.1%
SDS, and protease inhibitors (PMSF, EDTA, aprotinin, leupeptin,
prostatin A). Western blotting was performed using 30 μg of protein
separated by electrophoresis in a 12% polyacrylamide gel,
transferred to a nitrocellulose membrane, and immuno-reacted with
the indicated antibodies.
Fluo3-AM calcium assay
Cells were cultured in a cover glass bottom culture
dish for 24 h, treated with graded concentrations of 5-FU for
another 24 h, and then stained with Fluo3-AM (1.5 μM) for 30 min at
37°C. Cells were washed twice with PBS and examined under an
Eclipse 50i fluorescence microscope (Nikon).
Immunofluorescence
Cells grown on a cover glass bottom culture dish
were treated with 5-FU, washed with PBS, fixed with 4%
paraformaldehyde for 15 min at room temperature, and then blocked
for 1 h in PBS containing 0.3% Triton X-100. Then, the cells were
stained with 1:800 diluted anti-p53 and anti-β-actin polyclonal
antibodies, and incubated for 16 h at 4°C. Cells were washed with
PBS and incubated for 1 h with Alexa Fluor-488-conjugated secondary
antibody and Alexa Fluor-555-conjugated secondary antibody (Cell
Signaling Technology). To visualize the nucleus, cells were
counterstained with 1 μg/ml DAPI (Roche, Pleasanton, CA, USA).
After mounting, the cells were analyzed with an Eclipse 50i
fluorescence microscope.
Results
5-FU treatment results in apoptotic cell
death of Sk-Hep1 cells
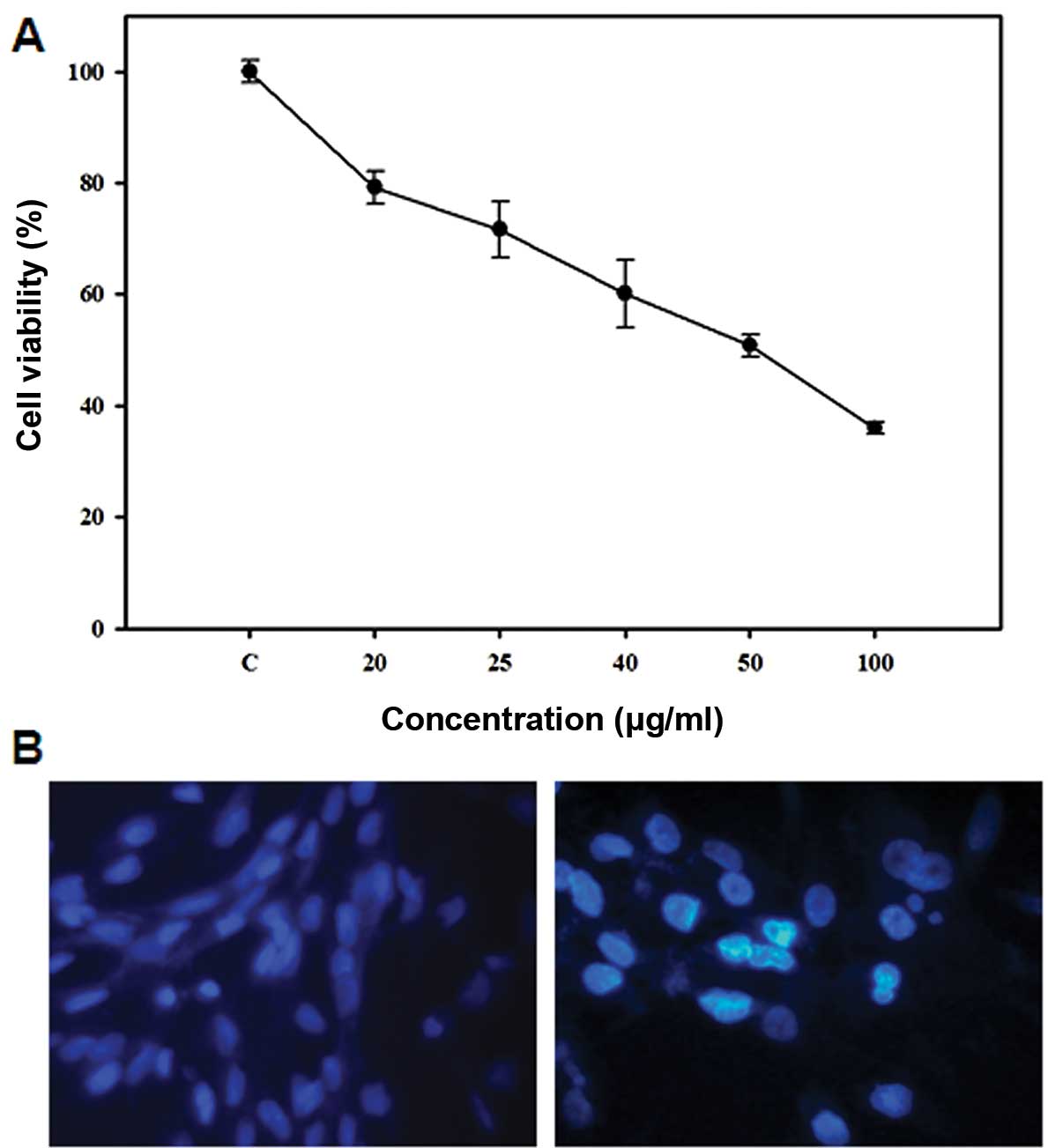
A dose response study was conducted to investigate
the effect of 5-FU on the growth of Sk-Hep1 HCC cells. As shown in
Fig. 1A, 5-FU inhibited Sk-Hep1
cell proliferation after 24 h exposure, with an IC50
value of 50 μg/ml. In order to determine the mode of cell death
induced by 5-FU, we examined the nuclear morphology of cells using
Hoechst 33342, a DNA-specific fluorescent dye. When cells were
treated with 50 μg/ml 5-FU for 24 h, the Sk-Hep1 cells exhibited
condensed and fragmented nuclei similar to apoptotic cell
morphology (Fig. 1B), indicating
that 5-FU induces apoptotic cell death in Sk-Hep1 HCC cells.
5-FU induces ER stress in Sk-Hep1
cells
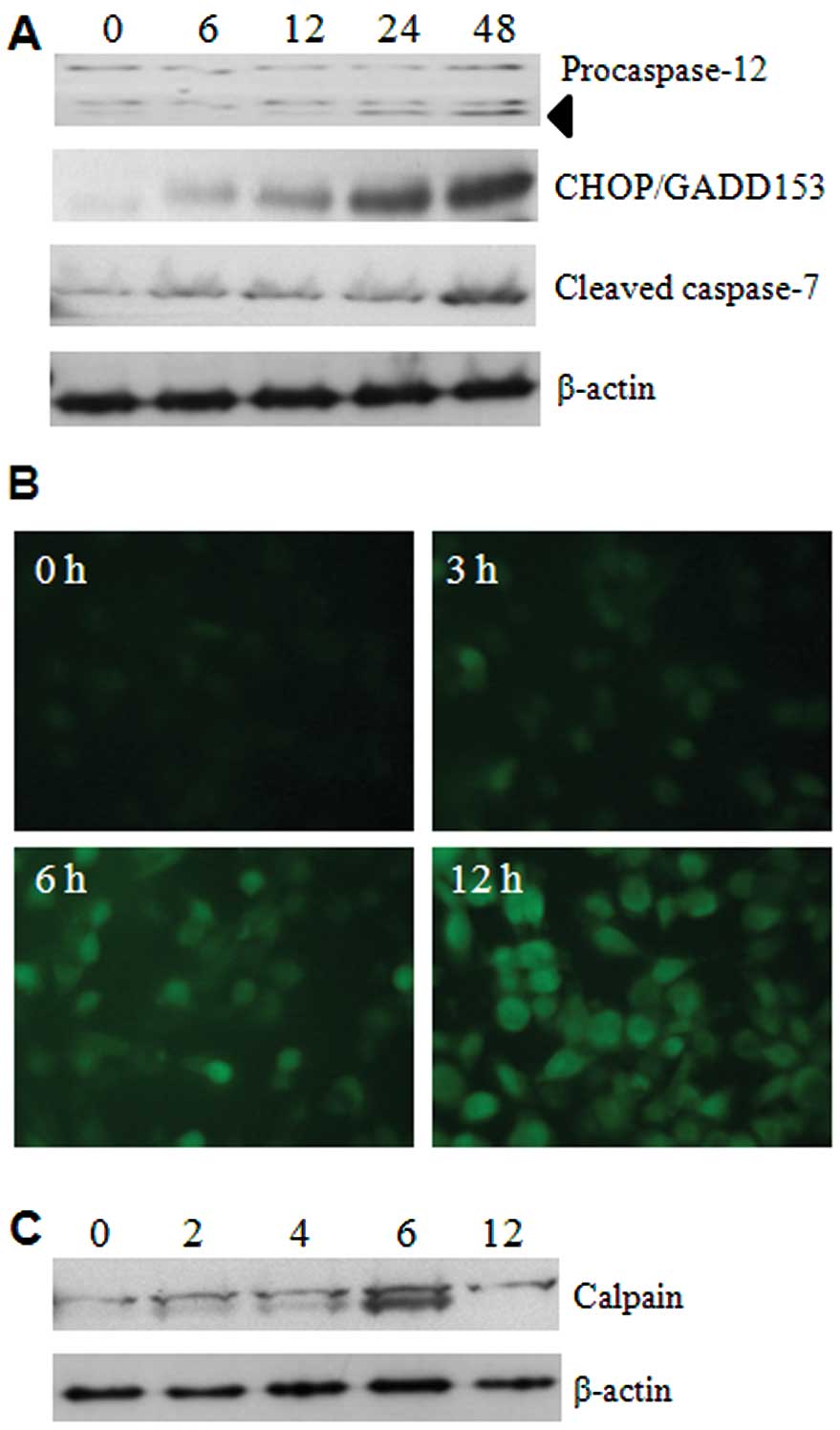
To determine whether 5-FU has possible effects on
ER, several ER-specific mediators were analysed. Initially, we
focused on the expression levels of caspase-12 and caspase-7, using
respective antibodies, and the results showed that activation
occurred from 24 h of 5-FU treatment (Fig. 2A). This suggests that cleavages of
these two caspases are palpable in ER stress. We next examined the
expression of CHOP/GADD153, a transcription factor induced by
cellular stresses such as UV light and ER stress (9). The expression of CHOP/GADD153 was
dramatically increased in a time-dependent manner by 5-FU treatment
(Fig. 2A). These data indicate
that 5-FU induces ER stress and it may initiate apoptotic cell
death in Sk-Hep1 cells.
As depletion of Ca2+ homeostasis in the
ER lumen can modify numerous cellular responses, and especially
accumulation of misfolded proteins. The mobilization of
intracellular Ca2+ was examined by Fluo3-AM (a green
fluorescent Ca2+ sensor). This analysis shows that 5-FU
increases the intracellular Ca2+ mobilization in a
time-dependent manner (Fig. 2B).
Moreover, the activation of calpain, an indicator of
Ca2+ homeostasis disruption, was also detected in its
active form of about 75 kDa products (Fig. 2C).
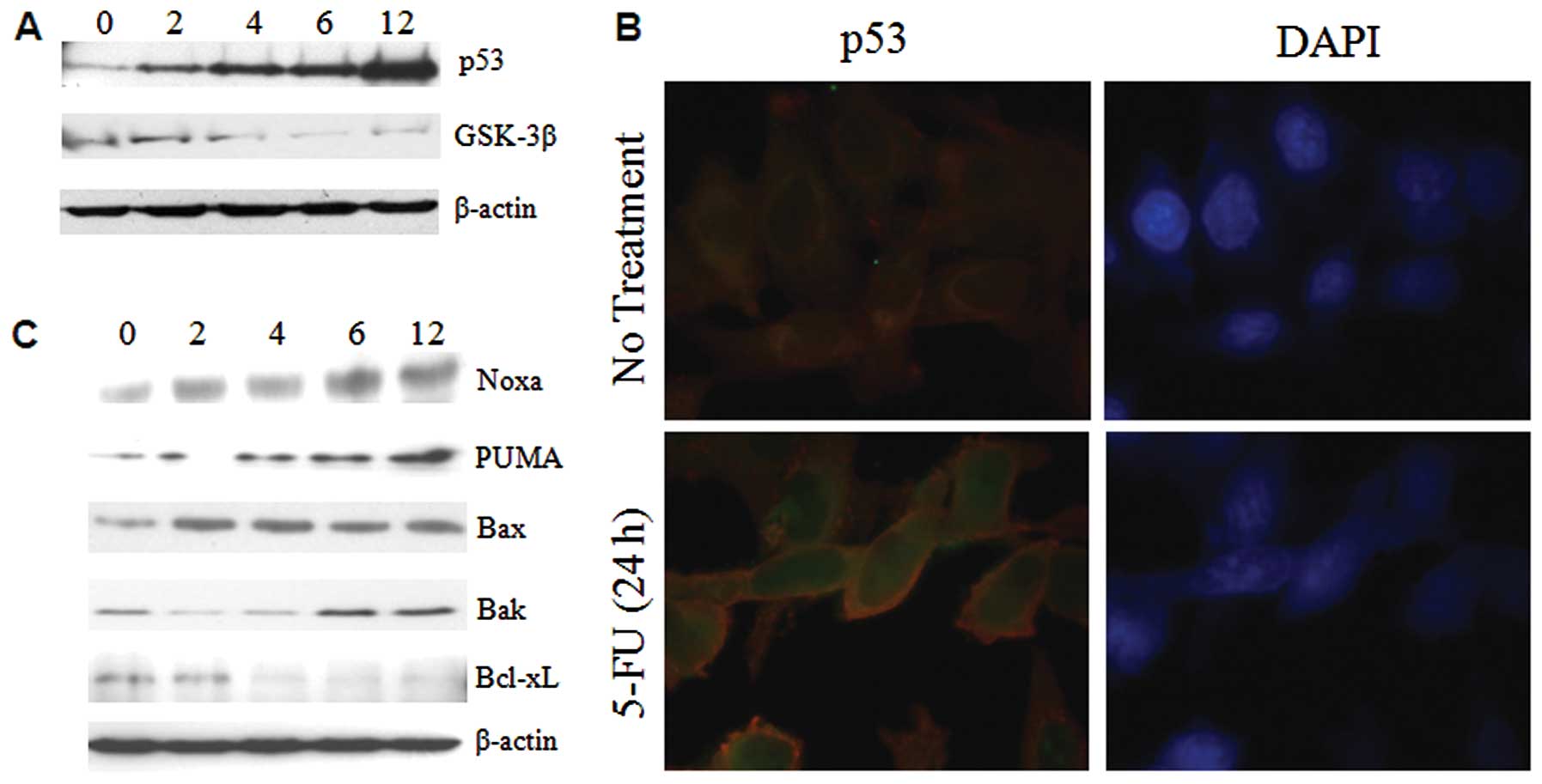
ER stress enhances the expression of p53
and bcl-2 family proteins
The tumor suppressor p53 plays a pivotal role in
cells by enhancing apoptotic genes and inducing inhibition of the
cell cycle in response to various forms of stress. It has been
shown that ER stress induces increased accumulation of p53 in MEF
(10). On the other hand, p53
activity was inhibited in ER stressed WI-38 and HT1080 cells
(11). To identify the expression
levels of p53 in ER under stressed conditions, Sk-Hep1 cells were
treated with 5-FU and ER stress was found to induce large changes
in the expression pattern of p53 within 6–12 h in Sk-Hep1 cells
(Fig. 3A). To further investigate
the nuclear retention of p53, the 5-FU exposed Sk-Hep1 cells were
subjected to immunofluorescence studies. The results showed that
p53 was intact in the nucleus (Fig.
3B), thus substantiating that 5-FU-induced ER stress promotes
nuclear accumulation and stabilization of this tumor suppressor
protein in hepatoma cells.
The Bcl-2 family proteins PUMA and Noxa are targets
of p53 and are involved in ER stress-induced apoptosis (12). Recent evidence reveals that Bax and
Bak can also be localized in ER and activated in response to ER
stress (13). Western blotting was
used to test whether the induction of a subset of Bcl-2 family
proapoptotic proteins in 5-FU induced ER stress, and we observed
that p53 targeted proteins (PUMA and Noxa) and Bak were increased
time-dependently in Sk-Hep1 cells, whereas Bax expression remains
unchanged (Fig. 3C). Taken
together, these findings demonstrate that in Sk-Hep1, induction of
ER stress by 5-FU causes overexpression, and nuclear stabilization
of p53 promotes the expression of proapoptotic Bcl-2 family
proteins.
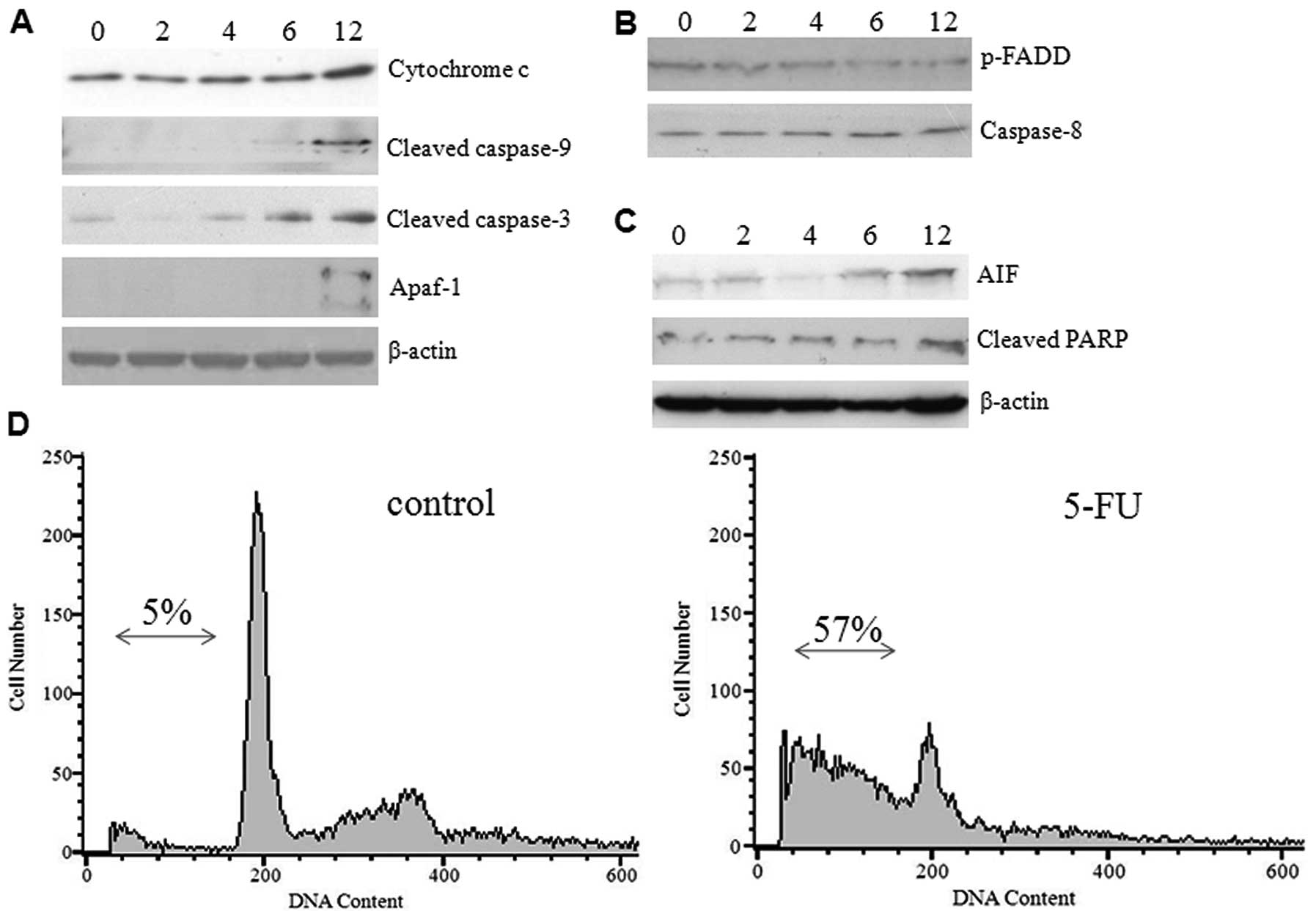
ER stress activates the intrinsic
mitochondrial pathway
The transition of ER stress to an apoptotic response
is not clearly understood, but it does appear to be dependent on
caspases and proteins of the Bcl-2 family. The release of
cytochrome c from mitochondria and activation of initiator
caspase-9 which occurred during ER stress-induced apoptosis suggest
the involvement of an intrinsic cell death pathway (14). To investigate whether 5-FU-induced
ER stress causes the release of cytochrome c and other
apoptogenic proteins from mitochondria, we examined the expression
levels of caspases and proapoptotic proteins by western blot
analysis using commercially available antibodies. Increased protein
expression was detected for cytochrome c, cleaved caspase-9,
and Apaf-1 (molecular core of the apoptosome) in a time-dependent
manner. The effector caspase-3 was found in active form in a
gradient manner (Fig. 4A). In
contrast, the expression of phospho-FADD and caspase-8 were
decreased gradually (Fig. 4B)
suggesting that 5-FU-induced ER stress did not activate the death
receptor pathway. The hepatoma cells exposed to 5-FU expressed
significant amounts of cleaved PARP and AIF (Fig. 4C), thus confirming programmed cell
death, which was previously observed (15). These results confirm that
5-FU-induced ER stress promotes the mitochondrial cell death
pathway in Sk-Hep1 cells by over-expressing the proapoptotic
proteins. To further substantiate our results, we examined the
effect of 5-FU-induced cell death by FACS analysis and demonstrated
a higher population of Sk-Hep1 cells in the sub-G1 phase (∼57%), as
opposed to the control cells (∼5%) (Fig. 4D). We also noted that treatment of
Sk-Hep1 cells with 5-FU was able to elicit a G1/S arrest (Fig. 4D).
ER stress downregulates the expression of
GRP78 and autophagy
Glucose regulated protein 78 (GRP78) is an ER
chaperone protein with Ca2+ binding and antiapoptotic
properties; it is required for protein folding, assembly of
membrane and secretary proteins, and to mitigate ER stress. It has
been shown that GRP78 protect tumor cells and prevent apoptosis by
interfering with caspase activation in many different cell types in
response to a variety of cellular stresses and a range of
chemotherapeutic agents (16,17).
Autophagy is a process of degradation of long-lived proteins and
sub-cellular organelles through lysosomal machinery, frequently
activated in response to an adverse environment or stress (8). Accumulation of terminally misfolded
protein aggregates in ER can be effectively removed by autophagy
(18). To better understand the
5-FU-induced ER stress and its consequent intrinsic apoptosis, we
administered 5-FU to the Sk-Hep1 cells and performed western blot
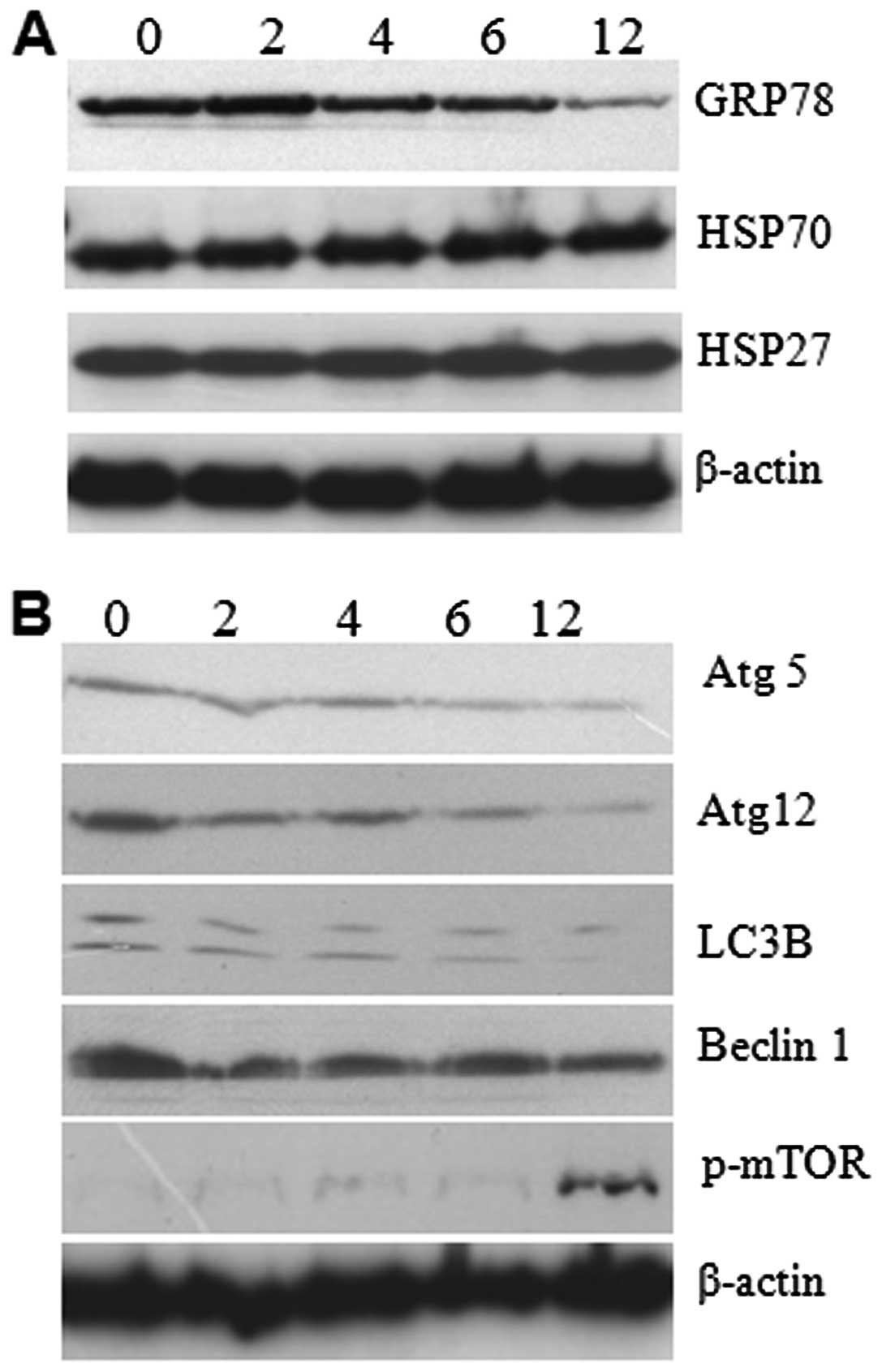
analysis for GRP78 and autophagy proteins. The results showed that
expressions of GRP78 decreased gradually, which explains how
prolonged ER stress can cause failure in the ER recovery process
and lead to cell death (Fig. 5A).
In addition to GRP78, we studied the expression level of heat shock
proteins by western analysis. The result reveals no changes in the
expression level suggesting that 5-FU-induced ER stress did not
affect the expression of heat shock proteins (Fig. 5A). The autophagy related LC3, Atg5,
Atg12, and Beclin1 expressions were gradually decreased in a
time-dependent manner (Fig. 5B).
In contrast, we found enhanced phospho-mTOR in 5-FU-treated cells
with optimum expression at 12 h of exposure (Fig. 5B). It seems that the enhanced
phospho-mTOR resulted in the downregulation of autophagy-related
LC3, Atg5, Atg12, and Beclin1 expression. It suggests that
downregulation of the expression of autophagy-related proteins
promote apoptosis in Sk-Hep1 cells due to accumulation of misfolded
protein aggregates.
Discussion
The key focus of molecular cancer investigation is
the development of new therapeutic strategies and the design of
drugs to target the genetic and biochemical genesis of malignant
transformation. To achieve this goal, many conventional and
experimental chemotherapeutic drugs have been used to stimulate ER
stress along with autophagy in cancer cells (8). Among the chemotherapeutic agents,
5-FU remains the most widely used drug and is used in many cancer
treatments. In this study, we have shown that 5-FU-induced ER
stress enhances the expression of p53 and Bcl-2 family proteins but
downregulates the expression of GRP78 and autophagy related
proteins. Based on the results, co-communications between
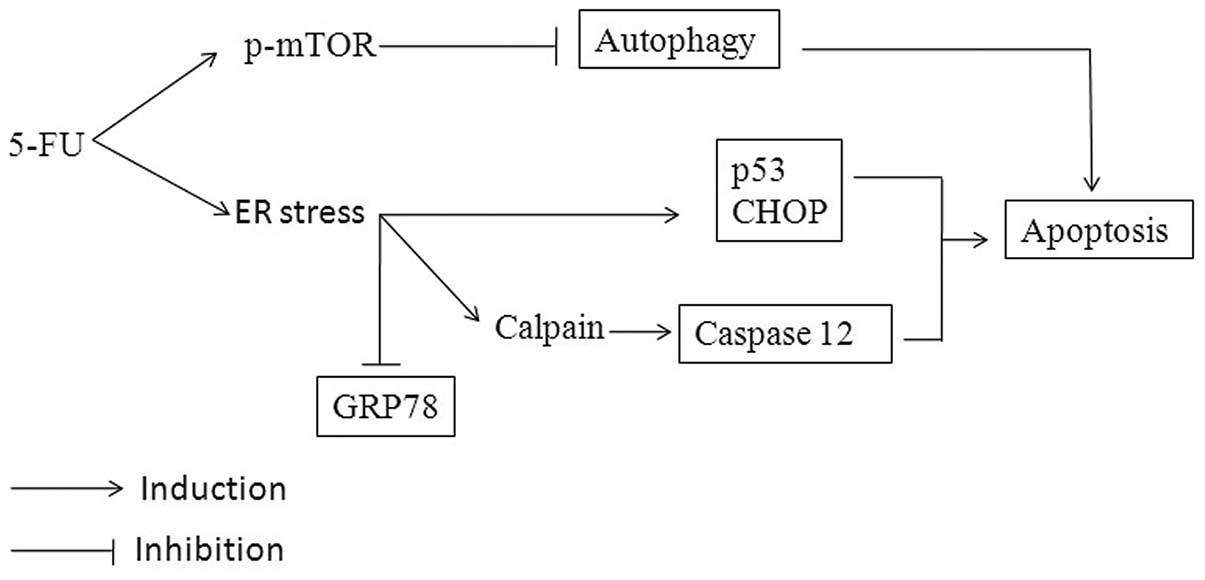
5-FU-induced ER stress and apoptosis are summarized in Fig. 6.
In this study, 5-FU profoundly induced the
disruption of Ca2+ homeostasis and consequent expression
of ER stress mediators. It has also been documented that
Ca2+ elevation promotes mobilization of calpain to the
ER surface, resulting in activation of caspase-12 (19). In addition, cleaved caspase-7 is
suggested to be an upstream initiator caspase responsible for
caspase-12 activation (14). As
previously demonstrated, 5-FU induced Ca2+ mobilization
and calpain expression indicates perturbation in Ca2+
homeostasis and ER stress activation. Furthermore, prolonged ER
stress causes activation of caspase-7 and −12, which indicates that
both effectors are possibly attributed to ER stress-induced
apoptosis. Consistent with prior reports (20,21),
the results showed that the expression level of CHOP/GADD153 was
low at basal condition, but was elevated in response to
5-FU-induced ER stress (Fig. 2A),
suggesting that prolonged stress initiates progression of
apoptosis.
5-FU-induced ER stress increases the upregulation of
Bcl-2 family proapoptotic (Bax, Bak, PUMA and Noxa) proteins, which
indicates a possible crosstalk between ER and mitochondria. In
addition, p53 expression was dramatically increased within 12 h
following 5-FU treatment, suggesting that it might be due to the
alteration in calcium homeostasis. Defects in nuclear retention of
p53 inactivate its function in certain solid tumors, including
hepatocellular carcinoma (22) and
this inactivation is associated with metastasis and poor prognosis.
It has been reported the inactivation of p53 by GSK-3β mediated
phosphorylation in ER stressed cells rescues the cells from
apoptosis. GSK-3β mediated p53 phosphorylation in the nucleus
facilitating nucleocytoplasmic export and degradation (11). Here, 5-FU-induced ER stress
decreased the activity and binding of GSK-3β to p53. Consistent
with western blotting and immunofluorescence data (Fig. 3A and B), 5-FU-induced ER stress
provokes nuclear retention of p53 by downregulating GSK-3β.
Activation of upstream ER stress modulators (CHOP and Bcl-2 family
proteins) and effectors (caspase-12), ultimately direct to the
activation of caspases, resulting in the sequential dismantling of
the cell. Western blot analysis demonstrated increased cytosolic
levels of cytochrome c, active initiator caspase-9, and
Apaf-1 in 5-FU treated cells, suggesting that release of
mitochondrial intermembrane space proteins into the cytosol forms
apoptosomes in association with caspase-9 and Apaf-1. Furthermore,
the expression of active downstream effector caspase-3 was also
found to increase gradually. In this study, we determined increased
levels of AIF and cleaved PARP, signifying that the active forms of
AIF and PARP are involved in large scale DNA fragmentation and
other cellular proteins (23). DNA
fragmentation and apoptotic cell death were further confirmed by
flow cytometry, and the sub-G1 content drastically increased up to
57% and cell cycle arrest was also observed. As confirmed by
various reports, our data also ensures that Bcl-2 family proteins
play a central role in regulating ER stress-induced apoptosis by
promoting the release of apoptogenic factors (10,12,13,24,25).
Prolonged ER stress causes initiation of cell death
and the biochemical mechanism for the switch from an unfolded
protein response (UPR) to apoptosis remains unclear. UPR provokes
the induction of chaperone proteins which increases the protein
folding capacity of the ER. The ER lumen houses a large array of
molecular chaperones, among which, GRP78 is the best characterized
and a member of the highly conserved HSP70 protein family. GRP78
can inhibit apoptosis by binding to caspase-7 and −12. It acts as a
Ca2+-binding protein to preserve ER Ca2+
homeostasis and also as a chaperone to limit the aggregation of
misfolded proteins (16,26). Several studies have reported that
GRP78 levels are increased in various types of cancer, including
HCC (27). Increased expression of
GRP78 enhances the protein folding capacity of ER and is associated
with a pro-survival response. In fact, the diminished expression of
GRP78 with siRNA activates the UPR and apoptosis in glioma and HeLa
cells (28,29). A lower level of GRP78 was
associated with an increase in CHOP expression and activation of
executioner caspases-3 and −7 in a heterozygous mouse mammary model
(30). The data presented here
show that HCC constitutively overexpressed GRP78 and it was
modulated by 5-FU-induced ER stress in Sk-Hep1 cells. This
downregulation promotes the activation of caspase-7, −12 and
CHOP/GADD153, leading to apoptotic cell death.
This study also demonstrated that 5-FU-induced ER
stress promotes apoptotic cell death by suppressing the protective
autophagy in human HCC Sk-Hep1 cells. In general, ER stress and
depletion of Ca2+ homeostasis induces the accumulation
of misfolded proteins in the lumen. The accumulated unfolded
proteins are refolded by eIF2α through phosphorylation, or cleared
by proteosomal degradation (31).
Initial evidence suggests that autophagy could act as a potential
degradation system for unfolded proteins accumulated in the ER
(18). Recent reports have shown
that autophagy is activated in response to ER stress for cell
survival by degradation of both soluble and insoluble aggregates.
Polyglutamine (poly-Q)-induced ER stress activates autophagosome
formation with LC3 conversion from LC3-I to LC3-II via
PERK-dependent eIF2α phosphorylation (31). Many ER stress inducing agents
activate autophagy to eliminate polyubiquitinated unfolded protein
aggregates in prostate and colon cancers, and autophagy induced by
same chemicals does not confer resistance in normal colon cells and
non-transformed murine embryonic fibroblasts (32). 5-FU-induced autophagy protects
cells from apoptosis and deactivation by 3-methyladenine or Atg7
siRNA induces apoptosis in colon cancer cells (33,34).
Our study demonstrates that the 5-FU-induced ER stress promotes
apoptotic cell death by suppressing the protective autophagy
response in human HCC Sk-Hep1 cells.
In summary, it is suggested that 5-FU induces
apoptotic signaling through a series of consecutive actions in
Sk-Hep1 cells. It causes ER stress, which is characterized by the
increase of CHOP/GADD153, p53 and caspase-12 activation, and
Ca2+ mobilization. These expressions modulate GRP78 and
autophagy activity, followed by interaction and activation of
mitochondrial mediated apoptosis. The present study proposes that
the induction of ER stress and downregulation of GRP78 and
autophagy may be the major contributors for 5-FU-induced apoptosis
in HCC Sk-Hep1 cells.
Abbreviations:
|
HCC
|
hepatocellular carcinoma;
|
|
EMEM
|
Eagle’s minimal essential medium;
|
|
FBS
|
fetal bovine serum;
|
|
WST-1
|
2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium,
monosodium salt;
|
|
PBS
|
phosphate-buffered saline
|
Acknowledgements
This work was supported by the Pukyong
National University Research Abroad Fund in 2010
(PS-2010-0012000201004500). We thank Dr J. Venkatesan, Department
of Chemistry, Pukyong National University, Korea, for his technical
help during FACS analysis, and Dr P. Gopal and Dr T. Jebasingh
(Madurai Kamaraj University, India) for their critical reading of
our manuscript.
References
|
1.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2.
|
Roayaie S, Blume I, Thung S, et al: A
system of classifying microvascular invasion to predict outcome
after resection in patients with hepatocellular carcinoma.
Gastroenterology. 137:850–855. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Sampath D, Rao V and Plunkett W:
Mechanisms of apoptosis induction by nucleoside analogs. Oncogene.
22:9063–9074. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Longley D, Harkin D and Johnston P:
5-Fluorouracil: mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Yang L, Wu D, Luo K, Wu S and Wu P:
Andrographolide enhances 5-fluorouracil-induced apoptosis via
caspase-8-dependent mitochondrial pathway involving p53
participation in hepatocellular carcinoma (SMMC-7721) cells. Cancer
Lett. 276:180–188. 2009. View Article : Google Scholar
|
|
6.
|
Osaki M, Tatebe S, Goto A, Hayashi H,
Oshimura M and Ito H: 5-Fluorouracil (5-FU) induced apoptosis in
gastric cancer cell lines: role of the p53 gene. Apoptosis.
2:221–226. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Rao R, Ellerby H and Bredesen D: Coupling
endoplasmic reticulum stress to the cell death program. Cell Death
Differ. 11:372–380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Tom V, Maria S, Guillermo V and Patrizia
A: Linking ER stress to autophagy: potential implications for
cancer therapy. Int J Cell Biol. 2010:9305092010.PubMed/NCBI
|
|
9.
|
Wang X, Lawson B, Brewer J, et al: Signals
from the stressed endoplasmic reticulum induce C/EBP-homologous
protein (CHOP/GADD153). Mol Cell Biol. 16:4273–4280.
1996.PubMed/NCBI
|
|
10.
|
Li J, Lee B and Lee A: Endoplasmic
reticulum-stress induced apoptosis: Multiple pathways and
activation of PUMA and NOXA by p53. J Biol Chem. 281:7260–7270.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Qu L, Huang S, Baltzis D, et al:
Endoplasmic reticulum stress induces p53 cytoplasmic localization
and prevents p53-dependent apoptosis by a pathway involving
glycogen synthase kinase-3. Genes Dev. 18:261–277. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Shibue T, Suzuki S, Okamoto H, et al:
Differential contribution of Puma and Noxa in dual regulation of
p53-mediated apoptotic pathways. EMBO J. 25:4952–4962. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Zong W, Li C, Hatzivassiliou G, et al: Bax
and Bak can localize to the endoplasmic reticulum to initiate
apoptosis. J Cell Biol. 162:59–69. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Rao R, Hermel E, Castro-Obregon S, et al:
Coupling endoplasmic reticulum stress to the cell death program.
Mechanism of caspase activation. J Biol Chem. 276:33869–33874.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Yu S, Andrabi S, Wang H, et al:
Apoptosis-inducing factor mediates poly (ADP-ribose)(PAR)
polymer-induced cell death. Proc Natl Acad Sci USA.
103:18314–18319. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Rao R, Peel A, Logvinova A, et al:
Coupling endoplasmic reticulum stress to the cell death program:
role of the ER chaperone GRP78. FEBS Lett. 514:122–128. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lee A: GRP78 induction in cancer:
therapeutic and prognostic implications. Cancer Res. 67:3496–3499.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Ogata M, Hino S, Saito A, et al: Autophagy
is activated for cell survival after endoplasmic reticulum stress.
Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Nakagawa T and Yuan J: Cross-talk between
two cysteine protease families: activation of caspase-12 by calpain
in apoptosis. J Cell Biol. 150:887–894. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Ron D and Habener J: CHOP, a novel
developmentally regulated nuclear protein that dimerizes with
transcription factors C/EBP and LAP and functions as a
dominant-negative inhibitor of gene transcription. Genes Dev.
6:439–453. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
McCullough K, Martindale J, Klotz L, Aw T
and Holbrook N: Gadd153 sensitizes cells to endoplasmic reticulum
stress by down-regulating Bcl2 and perturbing the cellular redox
state. Mol Cell Biol. 21:1249–1259. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Ueda H, Ullrich SJ, Gangemi JD, et al:
Functional inactivation but not structural mutation of p53 causes
liver cancer. Nat Genet. 9:41–47. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Susin S, Lorenzo H, Zamzami N, et al:
Molecular characterization of mitochondrial apoptosis-inducing
factor. Nature. 397:441–446. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Zhang X, Chen J, Graham S, et al:
Intranuclear localization of apoptosis inducing factor (AIF) and
large scale dna fragmentation after traumatic brain injury in rats
and in neuronal cultures exposed to peroxynitrite. J Neurochem.
82:181–191. 2002. View Article : Google Scholar
|
|
25.
|
Andrabi S, Kim N, Yu S, et al: Poly
(ADP-ribose)(PAR) polymer is a death signal. Proc Natl Acad Sci
USA. 103:18308–18313. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Reddy R, Mao C, Baumeister P, Austin R,
Kaufman R and Lee A: Endoplasmic reticulum chaperone protein GRP78
protects cells from apoptosis induced by topoisomerase inhibitors.
J Biol Chem. 278:20915–20924. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Healy SJM, Gorman AM, Mousavi-Shafaei P,
Gupta S and Samali A: Targeting the endoplasmic reticulum-stress
response as an anticancer strategy. Eur J Pharmacol. 625:234–246.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Pyrko P, Schönthal A, Hofman F, Chen T and
Lee A: The unfolded protein response regulator GRP78/BiP as a novel
target for increasing chemosensitivity in malignant gliomas. Cancer
Res. 67:9809–9816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Suzuki T, Lu J, Zahed M, Kita K and Suzuki
N: Reduction of GRP78 expression with siRNA activates unfolded
protein response leading to apoptosis in HeLa cells. Arch Biochem
Biophys. 468:1–14. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Dong D, Ni M, Li J, et al: Critical role
of the stress chaperone GRP78/BiP in tumor proliferation, survival,
and tumor angiogenesis in transgene-induced mammary tumor
development. Cancer Res. 68:498–505. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Kouroku Y, Fujita E, Tanida I, et al: ER
stress (PERK//eIF2[alpha] phosphorylation) mediates the
polyglutamine-induced LC3 conversion, an essential step for
autophagy formation. Cell Death Differ. 14:230–239. 2007.
|
|
32.
|
Ding W, Ni H, Gao W, et al: Differential
effects of endoplasmic reticulum stress-induced autophagy on cell
survival. J Biol Chem. 282:4702–4710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Li J, Hou N, Faried A, Tsutsumi S,
Takeuchi T and Kuwano H: Inhibition of autophagy by 3-MA enhances
the effect of 5-FU-induced apoptosis in colon cancer cells. Ann
Surg Oncol. 16:761–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Li J, Hou N, Faried A, Tsutsumi S and
Kuwano H: Inhibition of autophagy augments 5-fluorouracil
chemotherapy in human colon cancer in vitro and in vivo model. Eur
J Cancer. 46:1900–1909. 2010. View Article : Google Scholar : PubMed/NCBI
|