Introduction
The growth and function of a normal prostate is
mainly controlled via endocrine and paracrine factors. In this
context, androgens play a major role by regulating the expression
of specific genes to maintain the prostate homeostasis (1). When a prostate cancer (PCa) develops,
the majority of the tumors remain initially androgen-dependent and
thus, the first-line treatment of PCa is based on androgen
ablation. However, in many cases the tumor cells progress to a
hormone refractory state, generating androgen-independent tumors
with increased proliferation and invasion capacity. In this
situation, the therapeutic options are limited and the prognosis is
poor (2). The proliferation of the
androgen-independent PCa cells is mediated principally by autocrine
factors, such as the epidermal growth factor and their receptors,
which may interact with the androgen receptor in absence of
androgen ligand binding, constituting an essential signaling
pathway for tumor growth, invasion and metastasis (3,4).
The human epidermal growth factor receptor (HER)
family includes four type 1 transmembrane receptors: EGFR, HER2
(ErbB2), HER3 (ErbB3) and HER4 (ErbB4). They consist of a large
extracellular ligand-binding region, a transmembrane segment and an
intracellular tyrosine kinase domain. Stimulation through ligand
binding induces the homodimerization or heterodimerization of a
receptor with another family member at the plasma membrane,
resulting in the phosphorylation of specific tyrosine residues in
their cytoplasmic tail that leads to the activation of several
signaling cascades, predominantly the ERK1/2 and the
phosphatidylinositol 3-kinase (PI3K)/Akt pathways (5). These pathways control multiple
biological processes like cellular proliferation, differentiation,
survival, migration, and angiogenesis (6). Of the four receptors, HER2 and HER3
are dependent proteins: the HER2 extracellular domain lacks
ligand-binding capacity, while HER3 has a very weak kinase activity
(7). However, HER2-HER3
heterodimers are highly functional and constitute the most active
signaling dimer in this family. These receptors are activated by a
number of ligands that are EGF-related peptide growth factors. They
are classified in three groups depending on their receptor binding
specificity: epidermal growth factor (EGF), amphiregulin (AR),
epigen, and transforming growth factor-α (TGF-α) bind exclusively
to EGFR; betacellulin (BTC), epiregulin (EPR) and heparin-binding
EGF (HB-EGF) bind to EGFR and HER4; and the neuregulin (NRG1-4)
family members, NRG1 and NRG2, bind to HER3 and HER4, whereas NRG3
and NRG4 bind exclusively to HER4 (8). Importantly, HER ligands diversify the
pattern of receptor activation and can direct different biological
responses, even when they bind to the same receptor (9). However, the HER ligands share a
significant degree of functional redundancy, as HB-EGF is the only
ligand whose absence results in postnatal lethality (10).
The HER family, especially EGFR and HER2, are
frequently deregulated in cancer cells as a result of
overexpression, mutations, or increased ligand production (11,12).
In general terms, their enhanced expression in the tumor correlates
with a worse clinical outcome (13,14).
In the recent years, intense efforts have been focused on the
development of therapeutic strategies to block EGFR and HER2
signaling. Two families of HER-directed agents have demonstrated
clinical activity and are currently in use for the treatment of
cancer: monoclonal antibodies (MAbs) directed against the
extracellular domain of the receptors and small molecule tyrosine
kinase inhibitors (TKIs) that bind to the ATP-binding site of the
tyrosine kinase domain of the receptors (15–17).
However, the marked differences in the specificity and clinical
efficiency of the HER inhibitors in different carcinomas as well as
the development of resistances have limited the efficacy of these
drugs (18,19). In PCa, EGFR, HER2 and HER3
expression levels are increased as the disease progresses from
localized to metastasic and to the androgen-independent state and
these receptors have long been implicated in the survival of
androgen-independent prostate cancer cells (20–23).
Thus, the HER family members have been considered as potential
therapeutic targets in prostate cancer. However, despite the
blockade of EGFR or HER2 having demonstrated an ability to inhibit
the proliferation of human prostate tumor cells (24–26),
the clinical results have revealed only slight benefit of the
HER-targeted therapy in patients with androgen-independent PCa both
when EGFR inhibitors were administered as monotherapy or in
association with antiandrogens or chemotherapeutics (27–33).
The molecular mechanism underlying the low efficiency of EGFR
inhibitors in human PCa remains to be elucidated.
As tumor cells co-express several HER receptors and
ligands (34), the blockade of a
single HER receptor function can be compensated by the cell both
increasing the expression of alternative HER receptors or
upregulating the production of HER-ligands, establishing autocrine
growth factor loops that maintain downstream signaling and cellular
proliferation. In a previous study, we described the capacity of a
subset of breast cancer cell lines to compensate for the loss of
EGFR receptor function after gefitinib treatment by upregulating
the expression of genes coding for EGF-related ligands, especially
EPR, NRG and EGF (35). The
changes in the expression pattern remarkably correlated with the
intrinsic degree of sensitivity of the breast cancer cells to the
antiproliferative effects of gefitinib. Similar findings were
described for a cetuximab-resistant non-small cell lung cancer,
which overexpressed HER family ligands, especially EGF, AR, HB-EGF
and BTC (36), and for human
breast cancer cells selected for resistance to trastuzumab, which
presented an amplified ligand-induced activation of HER receptors
(37). The overexpression of
neuregulin has also been involved in the resistance of breast
cancer cells to EGFR-TKIs (38,39).
In addition, emerging studies are demonstrating that the
reactivation of HER3 is a prominent mechanism of resistance to the
current TKIs targeting EGFR and HER2 (40–42).
In this work, we were interested in examining the
capacity of androgen-independent PCa cells to modulate the
expression of the HER family members as an intrinsic mechanism of
resistance to EGFR-targeted therapies. To this end, we have
analyzed the changes in the expression pattern of different HER
receptors and ligands in two androgen-independent PCa cell lines in
response to the treatment with the EGFR-directed antibody cetuximab
and two EGFR TKIs, gefitinib and erlotinib. We have also
established an erlonitib-resistant cell line in order to further
determine the relevance of this molecular mechanism in the
resistance of prostate cancer cells to EGFR TKIs.
Materials and methods
Reagents
Cetuximab (Erbitux®, Merck-Serono,
Darmstadt, Germany) and trastuzumab (Herceptin®, Roche,
Basel, Switzerland) were kindly provided by the pharmacy of the
Catalan Institute of Oncology (Hospital Dr Josep Trueta, Girona,
Spain). Erlotinib (Tarceva®) was provided by Roche
(London, UK) and gefitinib (Iressa®) was obtained from
Astrazeneca (London, UK). For immunofluorescence assays, the
monoclonal antibodies against human EGFR, HER2, HER3, and HER4 were
obtained from Calbiochem (San Diego, CA, USA). The Alexa-Fluor
488-conjugated goat anti-mouse IgG antibody (Invitrogen, Carlsbad,
CA, USA) was used as a secondary reagent. The rabbit primary
antibodies for EGFR, HER2, HER3, HER4, and β-actin immunoblotting
were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The mouse
monoclonal antibody against phospho HER2 (Tyr1248) was from Thermo
Fisher (Fremont, CA, USA). The rabbit monoclonal antibodies against
phospho-AktSer473 and phospho HER3 and the rabbit
polyclonal antibodies against Akt, ERK1/2, and phospho HER1, as
well as the mouse monoclonal antibody against Phospho ERK 1/2 were
all from Cell Signaling Technology (Danvers, MA, USA). The mouse
monoclonal antibody against NRG-1β was from R&D Systems
(Minneapolis, MN, USA). The peroxidase-conjugated secondary
antibodies were from Calbiochem. For neutralizing assays, a
monoclonal antibody against HER3 (clone H3.105.5) was obtained from
Calbiochem and the monoclonal antibody against NRG-1 (clone C-18)
was from R&D Systems.
Cell lines
The PC3 and DU145 cell lines were obtained from the
American Tissue Culture Collection (Rockville, MD, USA). The cells
were maintained in Dulbecco’s modified Eagle’s medium supplemented
with 10% fetal bovine serum and 1% penicillin-streptomycin
(Gibco-BRL, Grand Island, NY, USA) at 37°C in a humidified
atmosphere containing 5% CO2. The cells were passaged
twice a week. The erlotinib DU145-resistant cell line (DUErR) was
established by continuously exposing the cells to increasing
concentrations of erlotinib. Initially the cells were treated with
their corresponding inhibitory concentration 50 (IC50)
of erlotinib (2.5 μM) for one month. Subsequently, the
erlotinib concentration in the culture medium was increased every
month to 5, 10 and 15 μM, and then the cells were maintained
in continuous culture with the maximum achieved dose of erlotinib
for three additional months. Control parental cells were cultured
in parallel and exposed to the phosphate-buffered saline (PBS)
(Gibco-BRL) vehicle.
Growth assays
Exponentially growing cells were seeded at a density
of 1.5×105 cells per dish in 100-mm diameter dishes. The
cells were allowed to attach and grow for 72 h in culture medium
and then gefitinib (15 μM), erlotinib (15 μM),
cetuximab (500 μg/ml) or a PBS vehicle alone (control cells)
were added. Just before the treatments and at 24, 48 and 72 h, the
cells were trypsinized and counted manually in a haemocytometer
using the trypan blue dye exclusion test. Three independent counts
were made from each treatment. All experiments were conducted in
triplicate.
Cell proliferation assays
To assess the cytotoxicity of EGFR-inhibitors in
DU145, PC3 and DUErR cells, aliquots of 4,000 cells were seeded in
96-well plates. Three days later, the cells were treated with
concentrations ranging from 0 to 15 μM of erlotinib, 0 to 15
μM of gefitinib, and 0 to 500 μg/ml of cetuximab for
72 h. Then, the treatments were removed, the cells were washed with
PBS and incubated for 3 h with 100 μl of fresh culture
medium together with 10 μl of MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
(Sigma-Aldrich, St. Louis, MO, USA). The medium was discarded and
dimethyl sulfoxide (DMSO) (Sigma-Aldrich) was added to each well to
dissolve the purple formazan crystals. Plates were agitated at room
temperature for 10 min and the absorbance of each well was
determined with an absorbance microplate reader (ELx800, BioTek,
Winooski, VT, USA) at a wavelength of 570 nm. Three replicates were
used for each experiment. The cell viability was determined as a
percentage of the untreated control cells, by dividing the mean
absorbance of each treatment by the mean absorbance of the
untreated cells. The concentration that reduces cell viability by
50% (IC50) was established for gefitinib and erlotinib,
while the concentration that decreases cell viability by 30%
(IC30) was determined for cetuximab. Cell proliferation
assays were also performed in order to evaluate the influence of
different HER-blocking antibodies on the growth of parental DU145
cells and DUErR cell, after treating the cells for 72 h with either
15 μM of erlotinib, 10 μg/ml of trastuzumab, a
humanized monoclonal antibody against the extracellular domain of
HER2, 10 μg/ml of the anti-HER3 blocking antibody H3.105.5,
10 μg/ml of an anti-neuregulin-1 antibody or by different
combinations of these compounds.
Quantitative real-time PCR analysis
DU145 and PC3 cells were treated for 24 h with a
concentration analogous to the IC30 for cetuximab (350
μg/ml and 500 μg/ml, respectively) or to the
IC50 for erlotinib and gefitinib (2,5 μM and 15
μM, respectively) or with vehicle alone as a control. DUErR
cells exposed to either erlotinib (15 μM) were also
analysed. The cells were washed with PBS and immediately
trypsinized. The total-RNA from each sample was isolated using the
RNeasy mini kit (Qiagen, Venlo, The Netherlands). The RNA
concentration was measured using an ND-1000 spectrophotometer
(NanoDrop Technologies, Wilmington, DE, USA) at 260 nm, and 1
μg of each RNA was reverse-transcribed into complementary
DNA (cDNA) using the High Capacity cDNA Archive Kit (Applied
Biosystems, Foster City, CA, USA). The expression of the HER family
members EGFR, HER2, HER3, HER4, EGF, TGF-α, AR, BTC, EPR, HB-EGF,
and NRG-1 was quantified by real-time PCR using a pre-designed,
gene-specific TaqMan® probe and primer sets
(TaqMan® Gene Expression assays, Applied Biosystems).
Quantitative PCR was performed in a standard 96-well plate format
using the TaqMan One-Step Universal Master Mix (Applied Biosystems)
and the 7300 Real-Time PCR system (Applied Biosystems). Cycling
conditions were 95°C for 10 min, followed by 40 cycles at 95°C for
15 sec and 60°C for 1 min. All samples were tested in triplicate.
The relative quantification of the mRNA level (μg/ml) of HER
receptors and ligands was carried out by preparing a standard curve
using known dilutions of its corresponding standard RNA. Then, the
mRNA level was normalized to the mRNA level of the housekeeping
gene TATA box binding (TBP) protein.
Immunofluorescence analysis
The quantification of HER receptor expression on the
cell membrane was performed using flow cytometry. The cells were
analyzed by double immunofluorescence using monoclonal antibodies
against human EGFR, HER2, HER3 and HER4 (Calbiochem). The cells
were incubated for 30 min at 4°C with the primary antibody. After
washing with PBS, the cells were incubated for 30 min at 4°C in the
presence of the Alexa-Fluor 488-conjugated goat anti-mouse IgG
antibody. Fluorescence was analyzed using a FACSCalibur flow
cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA,
USA) equipped with CellQuest™ software (Becton Dickinson).
Fluorescence intensity was represented on a four orders of
magnitude log scale (1–10,000). In each experiment 10,000 cells
were analyzed.
Western blot analysis
For western blot analyses, parental DU145 cells
(exposed to either medium alone or supplemented with erlotinib (2.5
μM)) and DUErR cells (continuously exposed to erlotinib (15
μM)) were collected and lysed with ice-cold lysis buffer
containing 1 mM EDTA, 150 mM NaCl, 100 μg/ml PMSF, 50 mM
Tris-HCl (pH 7.5) and protease and phosphatase inhibitor cocktails
(Sigma-Aldrich). Protein concentrations were determined by
Lowry-based Bio-Rad assay (Bio-Rad Laboratories, Hercules, CA,
USA). Equal amounts of protein extracts were electrophoresed on an
8% SDS-PAGE gel, transferred to nitrocellulose membranes and
blocked for 1 h at room temperature in a blocking buffer containing
2.5% powdered skim milk in PBS-T (10 mM Tris-HCl, pH 8.0, 150 mM
NaCl and 0.05% Tween-20) to prevent non-specific antibody binding.
Blots were incubated overnight at 4°C with the corresponding
primary antibody diluted in blocking buffer. After washes in PBS-T,
blots were incubated for 1 h with the corresponding secondary
antibody, and revealed with a commercial kit (West Pico
Chemiluminescent Substrate, Pierce Biotechnology, Rockford, IL,
USA). Blots were reprobed with an antibody for β-actin to control
the protein loading and transfer.
Statistical analysis
The statistical analysis was performed with the SPSS
statistical software for Windows (version 15.0; SPSS Inc., Chicago,
IL, USA). Quantitative variables were expressed as mean and
standard error (SE). The normality of the data was tested using the
Kolmogorov-Smirnov test. The differences between data with normal
distribution and homogeneous variances were analyzed using the
parametric Student’s t-test, otherwise the non-parametric
Mann-Whitney U test was applied. A value of P<0.05 was
considered significant.
Results
Basal expression of HER family receptors
and ligands in prostate cancer cells
The basal expression of the four HER receptors and
seven different ligands (EGF, TGF-α, AR, BTC, EREG, NRG-1 and
HB-EGF) was determined according to their mRNA levels in two
androgen-independent human carcinoma cell lines, DU145 and PC3
cells, which are derived from metastases of prostate cancer, to
brain and bone, respectively. Real-time PCR analyses revealed mRNA
expression for the four HER receptors in both cell lines, although
minimal constitutive levels of the HER4 mRNA were detected
(Fig. 1). In the DU145 cells, HER2
was the receptor with the most prominent mRNA expression, followed
by EGFR and HER3. EGFR, HER2 and HER3 showed similar mRNA levels in
the PC3 cells. HER2 mRNA expression was markedly higher in the
DU145 cells than in the PC3 cells, while EGFR expression was only
moderately increased. The receptors expression on the cell surface
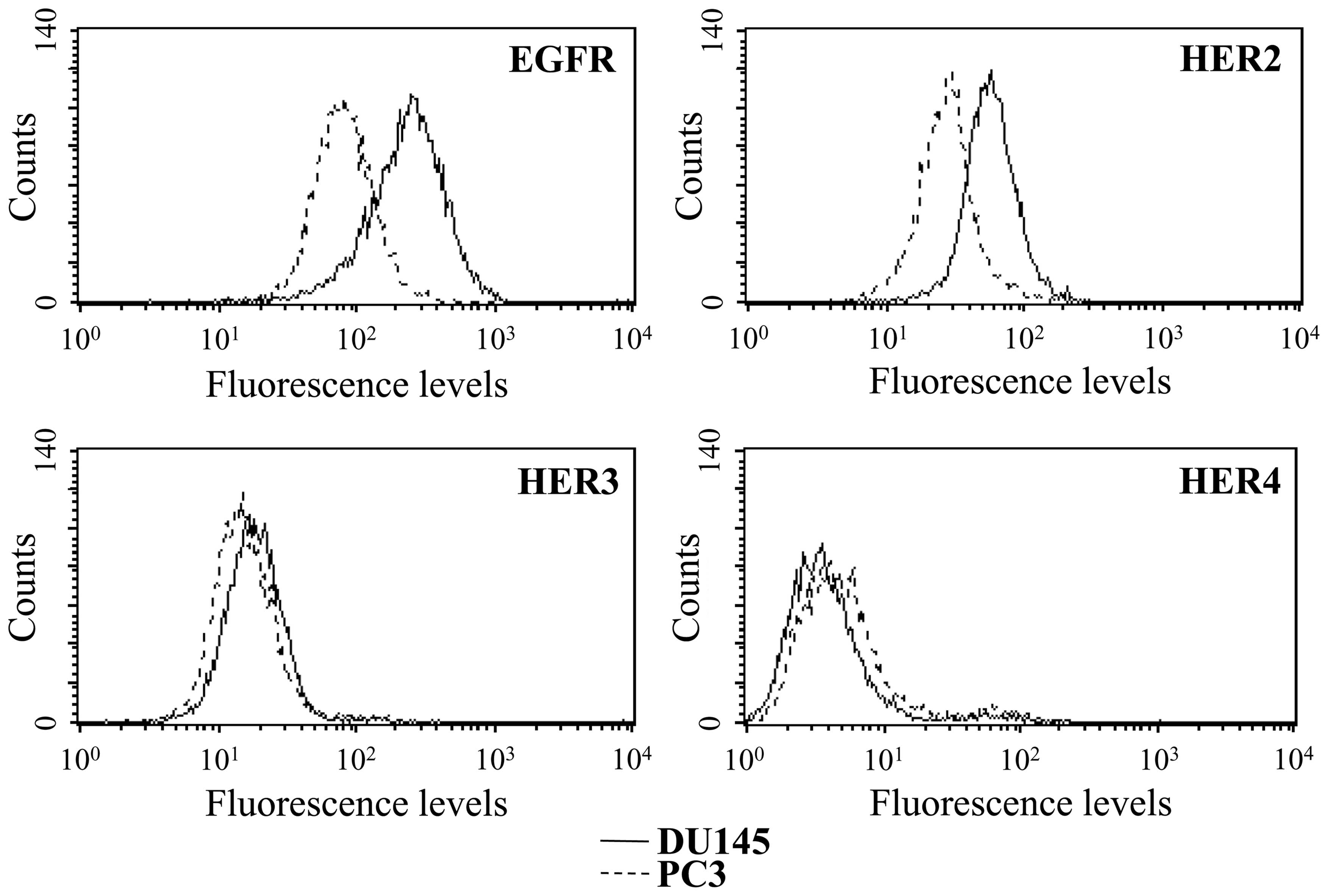
was additionally analyzed by flow cytometry (Fig. 2) confirming higher protein levels
of both EGFR and HER2 in the DU145 cells than in the PC3 cells. The
membrane expression of HER3 was parallel in both cell lines. In
accordance with the low mRNA levels, a very weak HER4 expression on
the cell surface was observed in both cell lines.
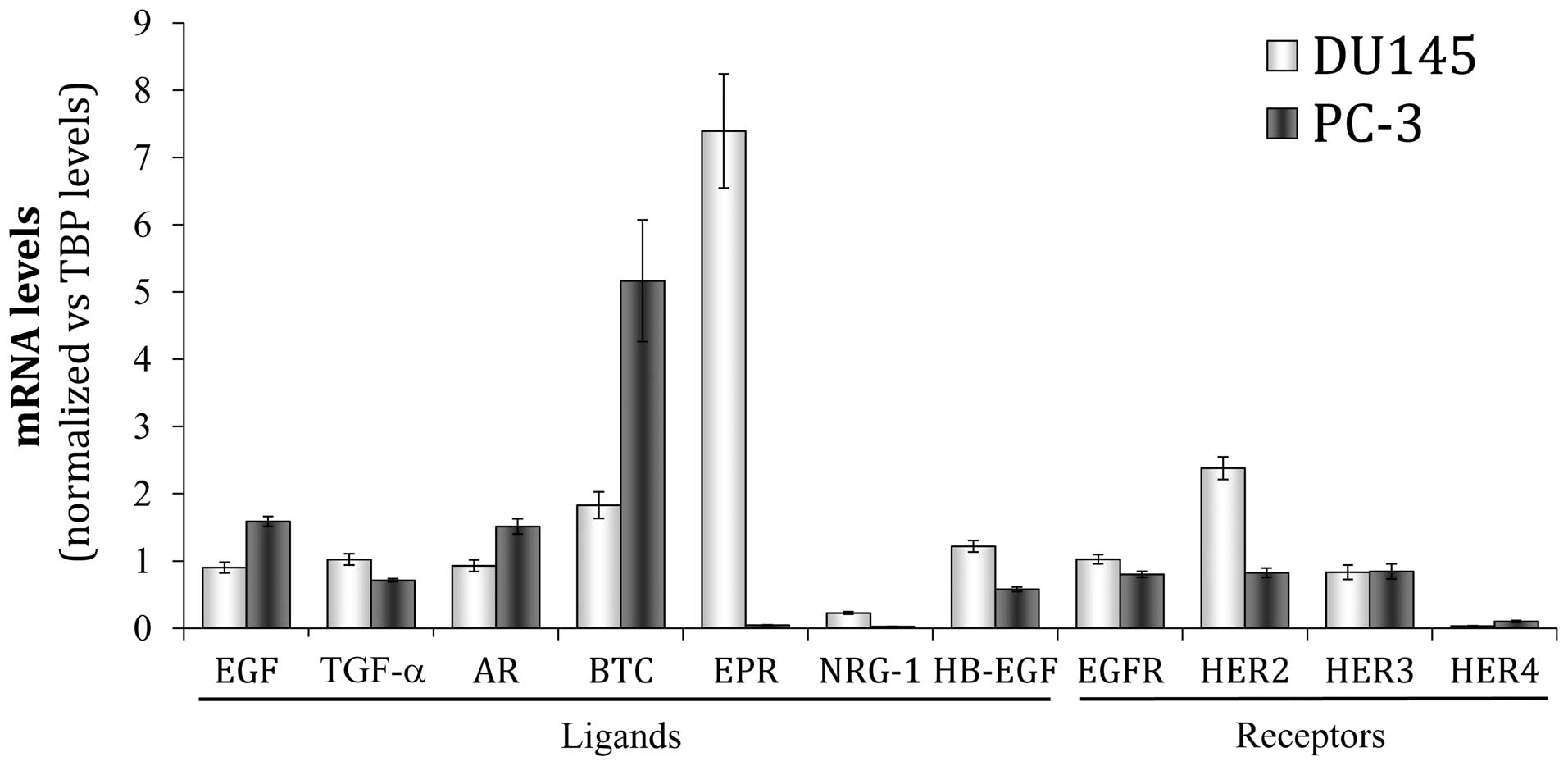 | Figure 1Gene expression pattern of HER family
members in DU145 and PC3 cells. The basal mRNA level of each HER
receptor (EGFR, HER2, HER3, HER4) and ligand (EGF, epidermal growth
factor; TGF-α, transforming growth factor-α; AR, amphiregulin; BTC,
betacellulin; EPR, epiregulin; NRG-1, neuregulin-1; HB-EGF,
heparin-binding EGF) was assessed by real-time PCR. Each value was
normalized versus the corresponding mRNA level of the TATA box
binding protein (TBP) constitutive gene. The bars represent the
mean value ± SE of three independent quantifications. |
Regarding the ligands, a basal gene expression of
all HER ligands analyzed was detected in both cell lines (Fig. 1). Interestingly, each cell line
showed a particular expression pattern with a primarily expressed
HER ligand: EPR was over-expressed in DU145 cells, while BTC was
the principal ligand in PC3 cells. This is a typical feature of
nearly all the epithelial cancer cells, which characteristically
deregulate the expression of one or more members of the HER family
(43). Notably, neuregulin-1 was
the ligand with the lowest mRNA levels in both cell lines,
particularly in PC3 cells.
Sensitivity of the prostate cancer cells
to EGFR inhibitors
The sensitivity of the prostate cancer cells to the
EGFR inhibitors was established according to the concentration of
each drug required to decrease the cell viability by 50%
(IC50) for the TKIs erlotinib and gefitinib or by 30%
(IC30) in the case of the monoclonal antibody cetuximab,
as the maximal concentration of cetuximab tested (500 μg/ml)
did not reduce the cell viability by 50% in any cell line. As
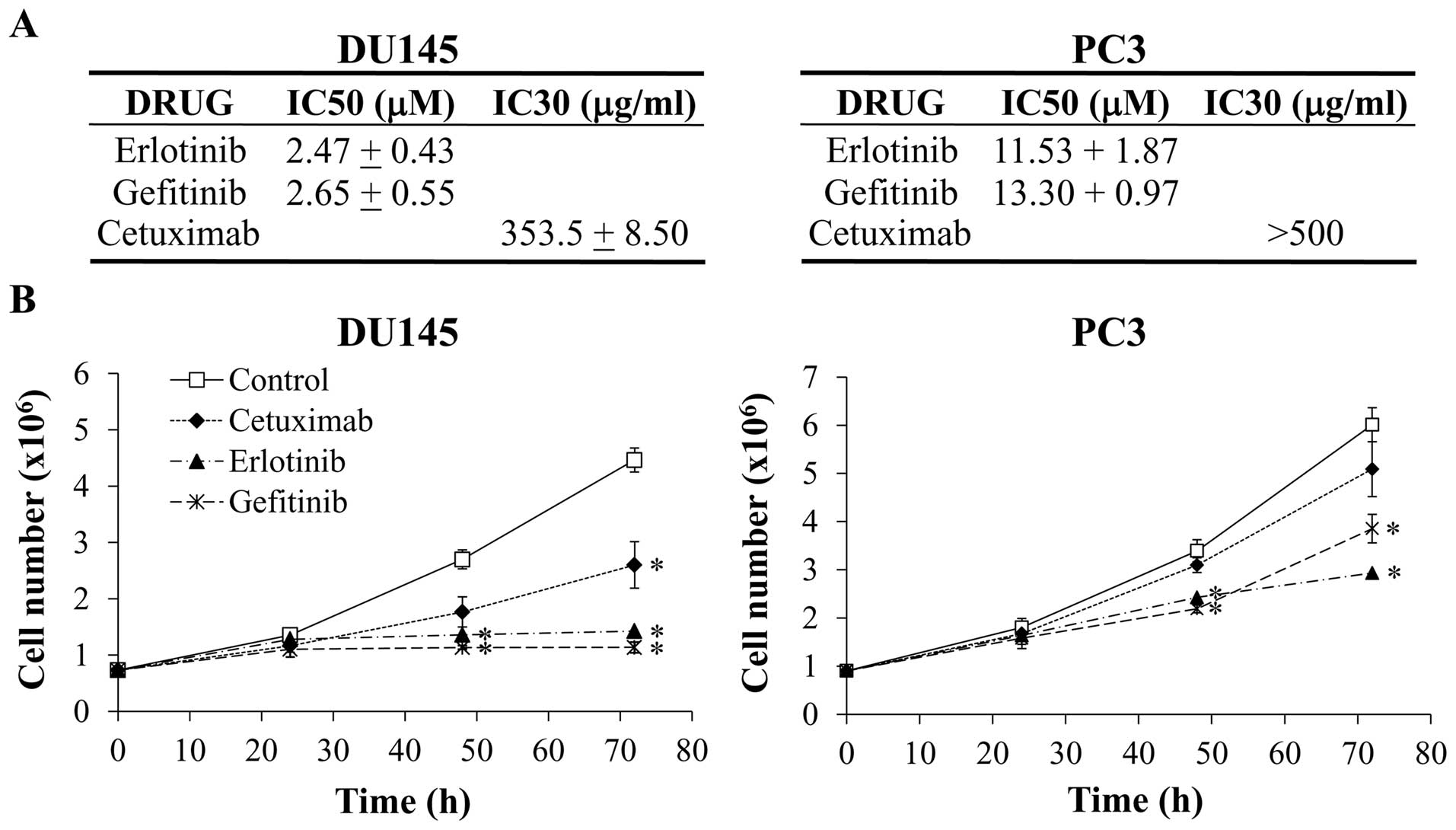
detailed in Fig. 3A, the
IC50 values for both TKIs were around 2.5 μM in
DU145 cells while values were 4 to 5-folds higher in PC3 cells. The
IC30 of cetuximab was 353 μg/ml in DU145 and
higher than 500 μg/ml in the PC3. According to these
results, the DU145 cells were more sensitive to the three anti-EGFR
agents than PC3 cells. These observations were confirmed in the
cell growth assays, where both cell lines were treated for up to 72
h with an elevated concentration of each compound: 15 μM for
erlotinib and gefitinib and 500 μg/ml for cetuximab.
Analysis of the growth curves (Fig.
3B) showed that the proliferation rate of DU145 cells was
significantly reduced by the three EGFR inhibitors. At 72 h, the
cell number was about four times lower in the erlotinib- and
gefitinib-treated DU145 cells than in untreated cells, while the
cetuximab effect on cell growth was more moderated. A more
attenuated effect of the treatments in the PC3 cells growth was
observed.
Effect of the EGFR inhibitors on the
expression pattern of HER receptors and ligands
The effect of EGFR inhibitors on the endogenous
expression of HER receptors and ligands was determined at the mRNA
levels 24 h after treating the cells with a concentration analogous
to the IC30 for cetuximab, to the IC50 for
erlotinib and gefitinib, or with vehicle alone as a control.
Fig. 4 represents the variations
in the gene expression compared with the corresponding untreated
cells. Significant changes were observed in the DU145 cells, which
overexpressed a subset of HER family members, while only minor
differences were observed in the PC3 cells. The treatment with
cetuximab and the TKIs caused similar effects on each gene
expression profile; although in most cases gefitinib induced the
most marked changes. Remarkably, the inhibition of EGFR in the
DU145 cell line induced the mRNA overexpression of three HER
ligands that display different binding affinities: EGF, BTC and
NRG-1. While EGF exclusively binds EGFR, BTC is a potent survival
factor that activates both EGFR and HER4 (44) and NRG-1 is a HER3 and HER4 ligand
that acts as a strong mitogenic factor (45). No changes were detected in the mRNA
levels of epiregulin, which is the most predominantly expressed HER
ligand in DU145 cells. Along with these changes, the mRNA level of
HER2 and HER3 were moderately increased in the DU145 cells, while
EGFR levels were reduced. Particularly, HER4 expression was
upregulated four-fold after EGFR blockade. HER4 levels were also
significantly increased but to a lesser extent, after the treatment
of the PC3 cells with the TKIs.
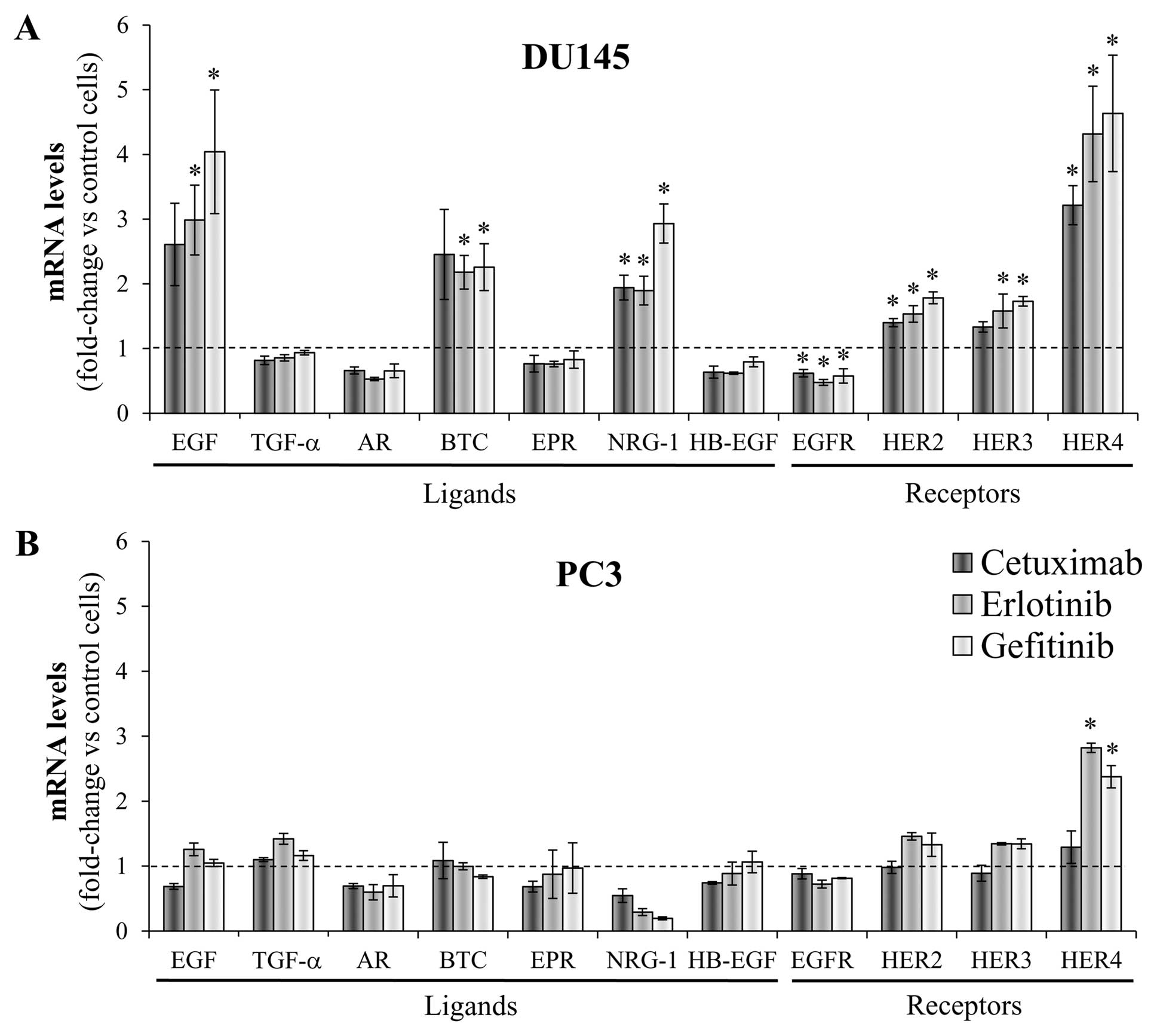 | Figure 4Changes in the gene expression of HER
receptors and ligands after EGFR inhibition. (A) DU145 and (B) PC3
cells were treated with either the corresponding IC30 of
cetuximab (350 μg/ml and 500 μg/ml, respectively),
the corresponding IC50 of erlotinib (2.5 μM and
15 μM, respectively) and gefitinib (2.5 μM and 15
μM, respectively) or vehicle as a control. After 24 h, the
mRNA level of each HER receptor (EGFR, HER2, HER3, HER4) and ligand
(EGF, epidermal growth factor; TGF-α, transforming growth factor-α;
AR, amphiregulin; BTC, betacellulin; EPR, epiregulin; NRG-1,
neuregulin-1; HB-EGF, heparin-binding EGF) was assessed by
real-time PCR in each cell line and values were normalized against
the corresponding mRNA expression of the TBP constitutive gene.
Then, the change in the expression of each particular gene induced
by the treatments was determined by comparing the normalized values
in treated cells against the corresponding normalized value in the
control cells. The bars indicate the mean fold change ± SE of three
independent quantifications (*P<0.05 vs. control
cells). Bars over the dotted line indicate an increase in the gene
expression compared to the control cells, while bars under the
dotted line represent impaired gene expression after the
treatment. |
Expression of the HER receptors and
ligands in erlotinib-resistant DUErR cells
In order to analyze if the overexpression of
alternative HER family members might be involved in the acquired
resistance to EGFR inhibitors in PCa, an erlotinib-resistant DU145
cell line (DUErR) was established by exposing the cells to
increasing concentrations (up to 15 μM) of erlotinib during
three months followed by three additional months of cell culture in
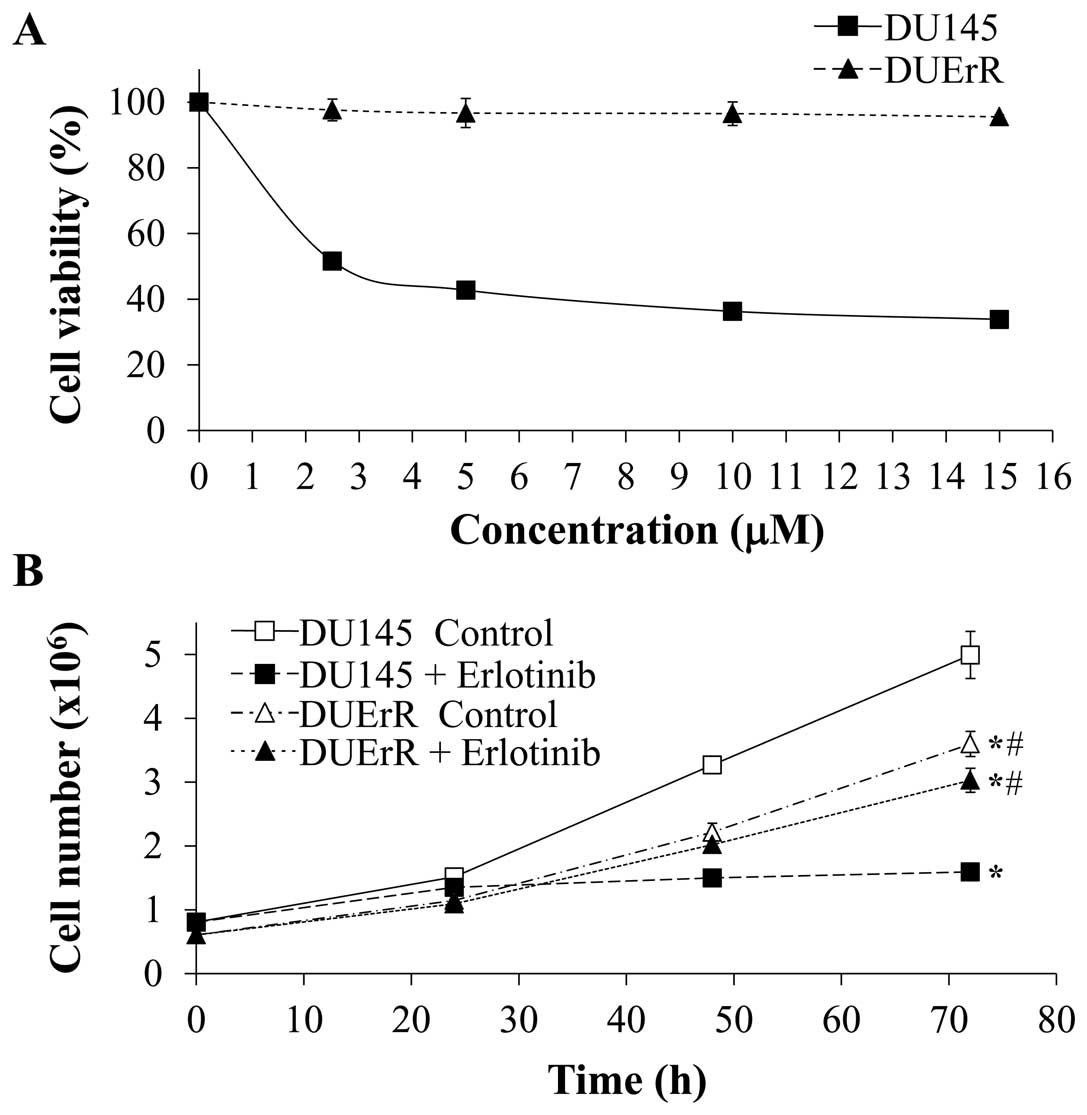
the maximal concentration of erlotinib. As represented in Fig. 5, the viability of DUErR cells was
not affected by increasing concentrations of erlotinib (up to 15
μM) in the cell culture (Fig.
5A). DUErR proliferation rate was similar when the cells were
cultured in the presence of erlotinib (15 μM) or vehicle
alone (PBS) (Fig. 5B). In
contrast, the viability and proliferation of the parental DU145
cells was markedly inhibited by erlotinib. Notably, the DUErR cell
line presented a slower proliferation rate compared to the parental
DU145 cells (Fig. 5B). DUErR cells
were also cross-resistant to gefitinib (data not shown).
Next, the mRNA expression profile of HER receptors
and ligands was determined by real-time PCR in the DUErR cells and
the differences from the parental control DU145 cells were
assessed. As shown in Fig. 6, in
erlotinib-exposed DUErR cells the NRG-1 mRNA expression was
significantly amplified (more than 2-fold) and TGF-α expression was
almost doubled compared to parental DU145 control cells. HB-EGF
expression was significantly impaired in these cells. Regarding HER
receptors, a 2-fold increase in HER2 and HER3 mRNA levels was
detected in the erlotinib-exposed DUErR cells while slight
differences in HER4 mRNA levels were observed compared with control
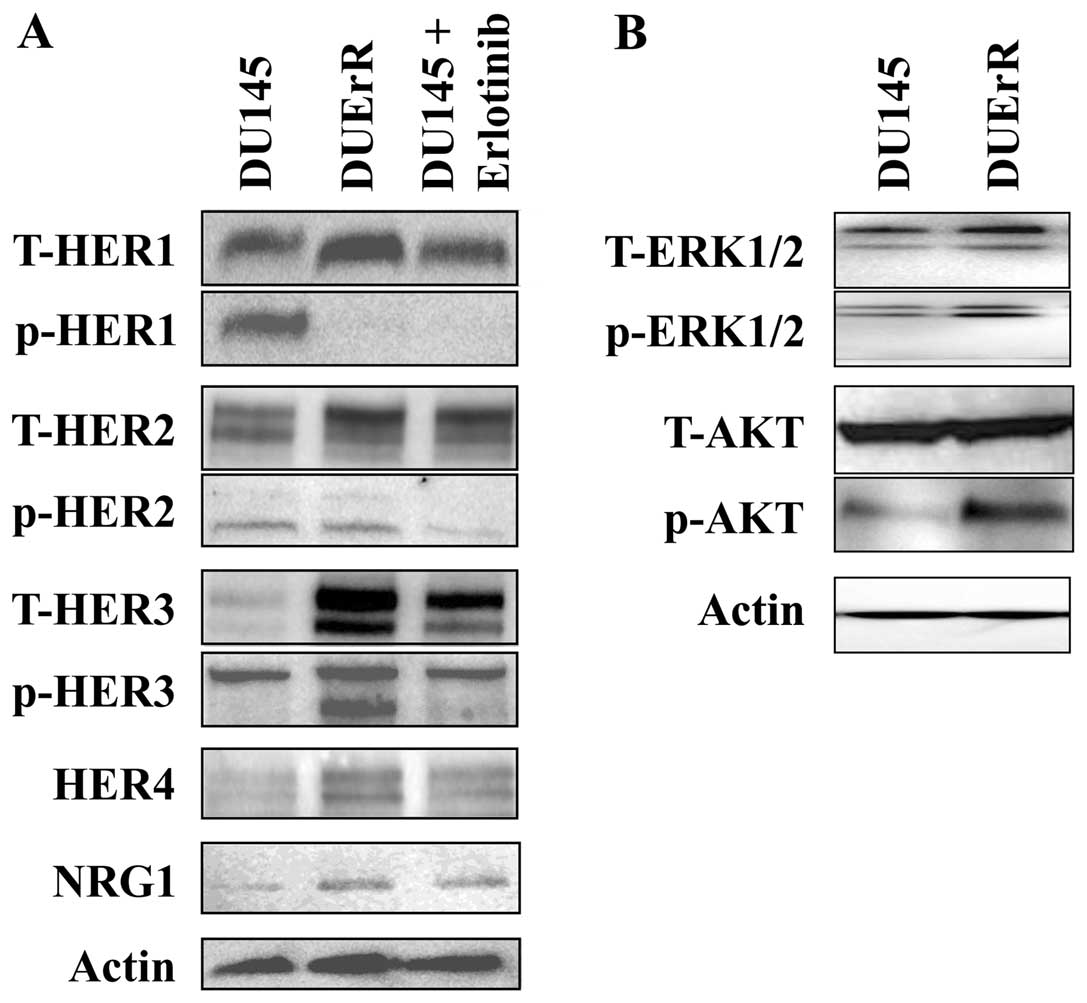
DU145 cells. Western blot analyses confirmed an upregulation of
HER2 and HER3 protein expression in erlotinib-treated DUErR cells
and a weak expression of HER4 (Fig.
7A).
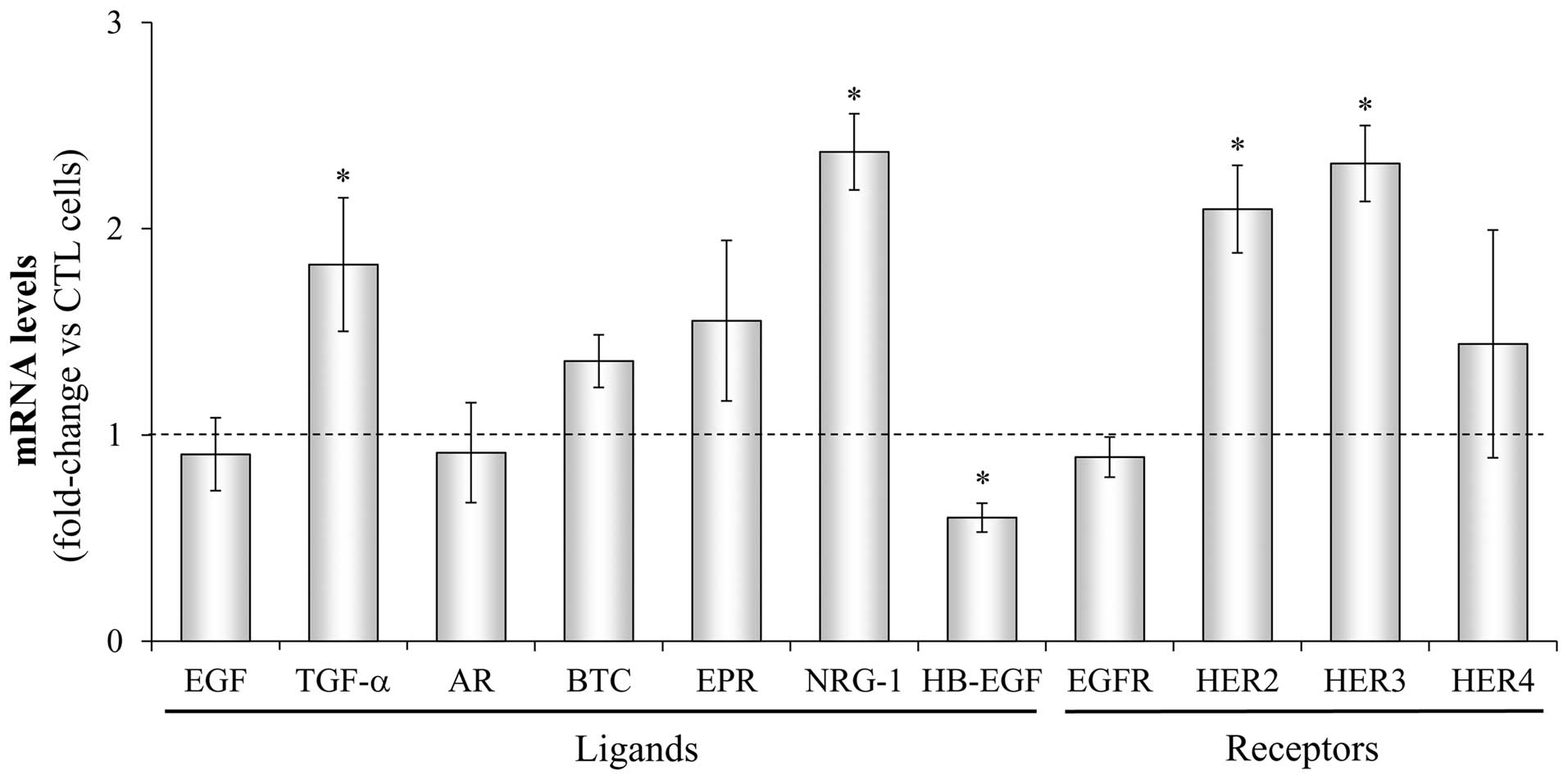 | Figure 6Changes in the gene expression of HER
receptors and ligands in DUErR cells. The gene expression of each
HER receptor (EGFR, HER2, HER3, HER4) and ligand (EGF, epidermal
growth factor; TGF-α, transforming growth factor-α; AR,
amphiregulin; BTC, betacellulin; EPR, epiregulin; NRG, neuregulin;
HB-EGF, heparin-binding EGF) was determined by real-time PCR in
parental DU145 cells (control) and in erlotinib-resistant DUErR
cells (cultured in medium containing 15 μM erlotinib). Each
value was normalized against the corresponding mRNA level of the
TBP constitutive gene. Then, the change in the expression of each
particular gene was assessed in DUErR cells compared to the control
cells. The bars represent the mean fold-change ± SE of three
independent quantifications (*P<0.05 vs. control
cells). Bars over the dotted line indicate an increase in the gene
expression compared to the control cells, while bars under the line
represent impaired gene expression after the treatment. |
HER receptors activation and downstream
signaling activity in DUErR cells
The activation of EGFR, HER2, and HER3 receptors and
the downstream signaling proteins, Akt and ERK1/2, was evaluated in
control DU145 cells, erlotinib-treated DU145 cells and
erlotinib-exposed DUErR cells. As presented in Fig. 7A, erlotinib treatment completely
abolished the phosphorylation of EGFR both in DU145 cells and in
DUErR cells. In DUErR cells, together with the overexpression of
HER2 and HER3, we detected a marked increased in HER3
phosphorylation and a more attenuated activation of HER2. HER3
overactivation was associated with enhanced levels of activated Akt
in DUErR cells, while slight effects were observed in ERK1/2
phosphorylation (Fig. 7B). In
contrast, after the acute treatment of DU145 cells for 72 h with
erlotinib, the phosphorylation of HER3 was not increased compared
to control cells (Fig. 7A). Thus,
the signaling through the HER3/PI3K/Akt pathway was selectively
activated after the prolonged exposure of DU145 cells to erlotinib,
providing a resistance mechanism to the EGFR TKI.
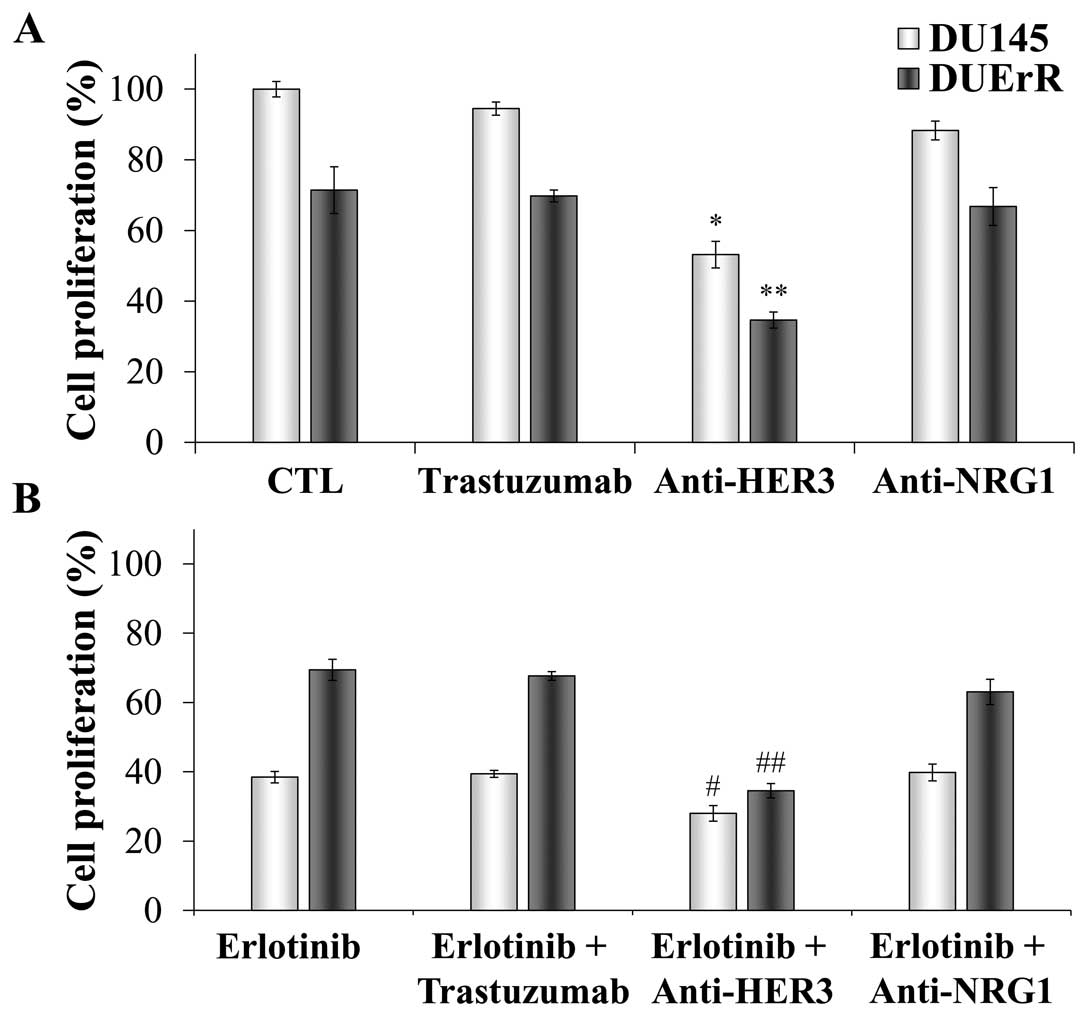
Effect of HER2 and HER3 inhibition on the
proliferation of parental and erlotinib-resistant DU145 cells
In order to further determine the relative
contribution of the HER receptors in the growth function of
parental and erlotinib-resistant DU145 cells, both cell lines were
exposed to HER2 and HER3 inactivating antibodies (trastuzumab and
H3.105.5, respectively) at a dose of 10 μg/ml for 72 h
(46), in presence or absence of
erlotinib. As shown in Fig. 8A,
the proliferation rate of both cell lines was unaffected by HER2
inhibition. Trastuzumab also failed to decrease the cell
proliferation when EGFR signaling was inhibited by erlotinib
(Fig. 8B). In contrast, the cell
growth was significantly reduced to half when HER3 was blocked both
in parental and in erlotinib resistant cell lines (Fig. 8A), revealing a key role for HER3 in
mediating the proliferation of these androgen-independent prostate
cells. In addition, the combination of erlotinib and the HER3
blocking antibody lead to a 25% decrease in the proliferation of
parental DU145 cells compared with DU145 cells exposed to erlotinib
alone. Notably, this combination resulted in a 50% reduction in the
growth of DUErR cells, restoring the cytotoxic activity of
erlotinib in these cells. These results confirm a key role for HER3
in sustaining the proliferation of prostate cells when the
signaling through EGFR is inhibited. The contribution of
neuregulin-1, which is a HER3 ligand, in activating the cell
proliferation was also analyzed as our previous results
demonstrated that both neuregulin-1 gene and protein expression was
increased after the acute and prolonged exposure of DU145 cells to
erlotinib. However, after incubating the cells with an antibody
against neuregulin-1, we observed a very moderate reduction on cell
proliferation. These results suggest a relative role for
neuregulin-1 in promoting the cell survival, probably because other
ligands, like neuregulin-2, may be involved in activating the
signaling through HER3 in these cells. In addition, a recent work
describe that HER3 may be activated in a neuregulin-1 independent
manner in DU145 cells (22).
Discussion
EGFR expression is involved in PCa progression and
correlates with androgen independence and metastasis of prostate
cancer cells (3,4,47).
However, the results of clinical trials involving EGFR inhibitors
have revealed a reduced efficacy in PCa patients (27–29,35).
The cooperative nature of the HER family might play an important
role in the low effectiveness of drugs targeting EGFR as the
co-expression of different HER members in the cancer cell can
activate parallel signaling pathways to circumvent the inhibition
of this specific receptor (6,48).
Actually, analysis of clinical data has revealed that patients
overexpressing two or more HER family members develop more
aggressive malignancies than patients that overexpress a single
receptor (49–52).
In the present study, we were interested in
determining if the endogenous expression of alternative HER family
members might contribute to the inefficacy of EGFR inhibitors in
PCa cells. To this end, the expression pattern of EGFR, HER2, HER3,
and HER4 as well as seven ligands of the HER family was assessed in
the DU145 and PC3 androgen-independent human prostate carcinoma
cell lines at basal level and after the treatment with the
EGFR-directed antibody cetuximab and the EGFR TKIs gefitinib and
erlotinib. Both cell lines are EGFR-positive, however PC3 cells
lack the phosphatase and tensin homolog tumor (PTEN) gene
expression, which is a tumor suppressor protein that down-regulates
the PI3K/Akt pathway. The loss of PTEN results in a continuous
signaling though the PI3K/Akt pathway independently of the upstream
receptor activation (53–55). The absence of the PTEN gene is a
common feature of PCa, as about 50% of prostate tumors lack PTEN
expression (56). Our results
showed that the sensitivity to EGFR inhibitors, determined as
IC50 or IC30 values as well as the inhibition
of the cell growth, was markedly higher in the PTEN-positive DU145
cells than in the PC3 cells. These results are in agreement with a
previous study, where the sensitivity against erlotinib was
strongly dependent on PTEN presence in PCa cells (26). In that study, the efficacy of
erlotinib was inversely related to the EGFR/HER2 ratio; however, we
did not find this association. Our data indicate a direct
association between EGFR expression levels and the effectiveness of
the treatments.
Our findings have demonstrated the constitutive
co-expression of different HER receptors and ligands at the mRNA
level in both PCa cell lines. The expression profile of the HER
ligands was cell type-specific. Epiregulin was the predominant
ligand in DU145 cells while betacellulin was the main ligand in PC3
cells. Both are EGFR ligands, which is the most largely expressed
receptor at protein level in both cell lines. Thus, tumor cell
proliferation could be sustained autonomously by the autocrine
production of growth factors. In fact, both cell lines were able to
proliferate independently of the exogenous supply of growth factors
in the cell culture (data not shown). Highly increased expression
of epiregulin and amphiregulin, together with TGF-α and HB-EGF, has
been previously described in androgen-independent prostate cancer
compared to normal prostate epithelial cells or androgen-sensitive
cancer cells (57,58). Epiregulin and amphiregulin
expression also increased after androgen deprivation in an
androgen-sensitive prostate cancer xenograft, indicating that these
growth factors might contribute to the tumor progression to an
androgen-independent stage (59).
In addition, the expression of epiregulin in breast cancer cells
together with further three genes was identified as a key
determinant for pulmonary metastasis (60). Epiregulin and amphiregulin gene
expression has also been associated with the development of liver
metastasis in colorectal cancer patients (61). Our group and others have recently
reported that an elevated gene expression of epiregulin and
amphiregulin in cancer cells might be a predictive marker of the
therapeutic response to cetuximab (62,63).
In keeping with this observation, the epiregulin-overexpressing
DU145 cells were markedly more sensitive to cetuximab than the PC3
cell line.
In response to inhibition of EGFR signaling by
cetuximab, gefitinib and erlotinib treatments, the gene expression
of EGF, betacellulin and neuregulin ligands along with the HER2,
HER3 and HER4 receptors was significantly increased in the DU145
cells. Interestingly, the alternative binding affinities of these
ligands might activate all possible HER heterodimers to compensate
the loss of EGFR function and preserve the EGFR downstream cell
signaling. In contrast, slight differences in the mRNA levels of
HER family members were observed in the PC3 cell line, which only
upregulated HER4 expression, indicating that the autocrine growth
factor loops are attenuated when the signaling activity through the
EGFR pathways is constitutively activated independently of ligand
binding. While the upregulation of HER2 and HER3 was moderate in
DU145 cells, the mRNA levels of HER4 were increased by four times
after EGFR inhibition. A few studies have analyzed the role of HER4
receptor in PCa, and the conclusions are controversial. Some
investigators have correlated the expression of HER4 with enhanced
prostate cell proliferation and migratory capacity (64,65),
but others have reported that HER4 has a tumor-suppressive effect
(66,67). HER4 activity has also been related
to acquired resistance to anti-EGFR therapies, as the signaling
through the PI3K/Akt survival pathway can be alternatively
activated by HER4 (39). Although
we have observed a marked increase in HER4 mRNA levels shortly
after EGFR inhibition, the low protein expression of HER4 in DU145
cells suggest a reduced contribution of this receptor to overcome
EGFR blockade.
There is growing evidence that tumor cells might
circumvent the effects of EGFR inhibitors through the promotion of
HER3/HER2 heterodimerization and the activation of the PI3K/Akt
pathway (38,45,61,68–70).
Our results confirm the relevance of HER3 in mediating resistance
to EGFR TKIs in androgen-independent prostate cancer cells. The
erlotinib-resistant (DUErR) cells, as well as the parental DU145
cells, showed a strong dependence on HER3 for proliferation, as
HER3 neutralization lead to a significant reduction in the growth
of both cell lines. The prolonged exposure of DU145 cells to
erlotinib induced a reprogramming of HER family members expression
and HER2 and HER3 levels were significantly upregulated, along with
neuregulin-1, which is a HER3 natural ligand, and TGF-α, which
exclusively binds EGFR. Ligand-activated HER3 might be then
transactivated by heterodimerization with HER2, or even with
relatively low levels of active EGFR, leading to an enhanced
activity through the PI3K/Akt survival pathway and, thus providing
a plausible escape mechanism to the EGFR blockade. Importantly,
HER3 inhibition rendered the DUErR cells sensitive to erlotinib,
indicating a key role for this receptor in driving the resistance
to EGFR-targeted therapies in PCa cells.
HER ligands have also been considered as targets for
cancer therapy, as the loss of HER receptors function can be
balanced by the endogenous expression of their cognate ligands by
cancer cells. However the redundancy of ligands for each receptor
may limit the effectiveness of this strategy, leading to a general
consensus that inhibiting the receptor function is more effective
than inhibiting multiple ligands. Our results are in keeping with
this hypothesis, as the proliferation of the cells was prominently
reduced after blocking the HER3 receptor, while targeting a
particular ligand, neuregulin-1, revealed a minor effectiveness in
reducing the cell growth.
In conclusion, our results show the ability of the
androgen-independent PCa cells to rapidly modulate the expression
of HER receptors and ligands to sustain the cell proliferation
after EGFR blockage and a central role for HER3 in mediating
resistance to EGFR inhibition. This molecular mechanism might be
especially relevant in the physiological conditions, as the
proliferation of malignant prostate cells is markedly dependent on
the autocrine release of growth factors as well as the expression
of their corresponding receptors in the tumor microenvironment.
Therefore, the clinical efficacy of the current therapeutic
approaches targeting EGFR in prostate cancer might be improved by
the dual inhibition of EGFR and HER3 receptors.
Acknowledgements
We acknowledge the Instituto de Salud
Carlos III (grants RD06/0020/0041, RD06/0020/0028), Universitat de
Girona (grant PUG2007A/10) and Generalitat de Catalunya (grant
2009SGR208) for providing funding for this project. D.C.S. and C.P.
acknowledge their fellowships from Ministerio de Educación y
Ciencia (grant AP2007-01953) and Universitat de Girona (grant
BR08/19), respectively.
References
|
1.
|
Zhu ML and Kyprianou N: Androgen receptor
and growth factor signaling cross-talk in prostate cancer cells.
Endocr Relat Cancer. 15:841–849. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Pomerantz M and Kantoff P: Advances in the
treatment of prostate cancer. Annu Rev Med. 58:205–220. 2007.
View Article : Google Scholar
|
|
3.
|
Traish AM and Morgentaler A: Epidermal
growth factor receptor expression escapes androgen regulation in
prostate cancer: a potential molecular switch for tumour growth. Br
J Cancer. 101:1949–1956. 2009. View Article : Google Scholar
|
|
4.
|
Shah RB, Ghosh D and Elder JT: Epidermal
growth factor receptor (ErbB1) expression in prostate cancer
progression: correlation with androgen independence. Prostate.
66:1437–1444. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Normanno N, De Luca A, Bianco C, et al:
Epidermal growth factor receptor (EGFR) signaling in cancer. Gene.
366:2–16. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Shi F, Telesco SE, Liu Y, Radhakrishnan R
and Lemmon MA: ErbB3/HER3 intracellular domain is competent to bind
ATP and catalyze autophosphorylation. Proc Natl Acad Sci USA.
107:7692–7697. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Pinkas-Kramarski R, Shelly M, Glathe S,
Ratzkin BJ and Yarden Y: Neu differentiation factor/neuregulin
isoforms activate distinct receptor combinations. J Biol Chem.
271:19029–19032. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Wilson KJ, Gilmore JL, Foley J, Lemmon MA
and Riese DJ II: Functional selectivity of EGF family peptide
growth factors: implications for cancer. Pharmacol Ther. 122:1–8.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Iwamoto R, Yamazaki S, Asakura M, et al:
Heparin-binding EGF-like growth factor and ErbB signaling is
essential for heart function. Proc Natl Acad Sci USA.
100:3221–3226. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Miyamoto S, Fukami T, Yagi H, Kuroki M and
Yotsumoto F: Potential for molecularly targeted therapy against
epidermal growth factor receptor ligands. Anticancer Res.
29:823–830. 2009.PubMed/NCBI
|
|
12.
|
Hynes NE and Lane HA: ERBB receptors and
cancer: the complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Salomon DS, Brandt R, Ciardiello F and
Normanno N: Epidermal growth factor-related peptides and their
receptors in human malignancies. Crit Rev Oncol Hematol.
19:183–232. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Normanno N, Bianco C, De Luca A, Maiello
MR and Salomon DS: Target-based agents against ErbB receptors and
their ligands: a novel approach to cancer treatment. Endocr Relat
Cancer. 10:1–21. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Scaltriti M and Baselga J: The epidermal
growth factor receptor pathway: a model for targeted therapy. Clin
Cancer Res. 12:5268–5272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Baselga J: Targeting tyrosine kinases in
cancer: the second wave. Science. 312:1175–1178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Bianco R, Troiani T, Tortora G and
Ciardiello F: Intrinsic and acquired resistance to EGFR inhibitors
in human cancer therapy. Endocr Relat Cancer. 12(Suppl 1):
S159–S171. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Marshall J: Clinical implications of the
mechanism of epidermal growth factor receptor inhibitors. Cancer.
107:1207–1218. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Benavente S, Huang S, Armstrong EA, et al:
Establishment and characterization of a model of acquired
resistance to epidermal growth factor receptor targeting agents in
human cancer cells. Clin Cancer Res. 15:1585–1592. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Di Lorenzo G, Tortora G, D’Armiento FP, et
al: Expression of epidermal growth factor receptor correlates with
disease relapse and progression to androgen-independence in human
prostate cancer. Clin Cancer Res. 8:3438–3444. 2002.PubMed/NCBI
|
|
21.
|
Craft N, Shostak Y, Carey M and Sawyers
CL: A mechanism for hormone-independent prostate cancer through
modulation of androgen receptor signaling by the HER-2/neu tyrosine
kinase. Nat Med. 5:280–285. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Soler M, Mancini F, Meca-Cortes O, et al:
HER3 is required for the maintenance of neuregulin-dependent and
-independent attributes of malignant progression in prostate cancer
cells. Int J Cancer. 125:2565–2575. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ricciardelli C, Jackson MW, Choong CS, et
al: Elevated levels of HER-2/neu and androgen receptor in
clinically localized prostate cancer identifies metastatic
potential. Prostate. 68:830–838. 2008. View Article : Google Scholar
|
|
24.
|
Vicentini C, Festuccia C, Gravina GL,
Angelucci A, Marronaro A and Bologna M: Prostate cancer cell
proliferation is strongly reduced by the epidermal growth factor
receptor tyrosine kinase inhibitor ZD1839 in vitro on human cell
lines and primary cultures. J Cancer Res Clin Oncol. 129:165–174.
2003.
|
|
25.
|
Dhupkar P, Dowling M, Cengel K and Chen B:
Effects of anti-EGFR antibody cetuximab on androgen-independent
prostate cancer cells. Anticancer Res. 30:1905–1910.
2010.PubMed/NCBI
|
|
26.
|
Festuccia C, Gravina GL, Biordi L, et al:
Effects of EGFR tyrosine kinase inhibitor erlotinib in prostate
cancer cells in vitro. Prostate. 69:1529–1537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Canil CM, Moore MJ, Winquist E, et al:
Randomized phase II study of two doses of gefitinib in
hormone-refractory prostate cancer: a trial of the National Cancer
Institute of Canada-Clinical Trials Group. J Clin Oncol.
23:455–460. 2005. View Article : Google Scholar
|
|
28.
|
Gravis G, Bladou F, Salem N, et al:
Results from a monocentric phase II trial of erlotinib in patients
with metastatic prostate cancer. Ann Oncol. 19:1624–1628. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Wilding G, Soulie P, Trump D, Das-Gupta A
and Small E: Results from a pilot phase I trial of gefitinib
combined with docetaxel and estramustine in patients with
hormone-refractory prostate cancer. Cancer. 106:1917–1924. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Ziada A, Barqawi A, Glode LM, et al: The
use of trastuzumab in the treatment of hormone refractory prostate
cancer; phase II trial. Prostate. 60:332–337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Whang YE, Armstrong AJ, Rathmell WK, et
al: A phase II study of lapatinib, a dual EGFR and HER-2 tyrosine
kinase inhibitor, in patients with castration-resistant prostate
cancer. Urol Oncol. Mar 9–2011.(Epub ahead of print).
|
|
32.
|
Sridhar SS, Hotte SJ, Chin JL, et al: A
multicenter phase II clinical trial of lapatinib (GW572016) in
hormonally untreated advanced prostate cancer. Am J Clin Oncol.
33:609–613. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Nabhan C, Lestingi TM, Galvez A, et al:
Erlotinib has moderate single-agent activity in chemotherapy-naive
castration-resistant prostate cancer: final results of a phase II
trial. Urology. 74:665–671. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Yotsumoto F, Yagi H, Suzuki SO, et al:
Validation of HB-EGF and amphiregulin as targets for human cancer
therapy. Biochem Biophys Res Commun. 365:555–561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Ferrer-Soler L, Vazquez-Martin A, Brunet
J, Menendez JA, De Llorens R and Colomer R: An update of the
mechanisms of resistance to EGFR-tyrosine kinase inhibitors in
breast cancer: gefitinib (Iressa) -induced changes in the
expression and nucleo-cytoplasmic trafficking of HER-ligands
(Review). Int J Mol Med. 20:3–10. 2007.
|
|
36.
|
Li C, Iida M, Dunn EF, Ghia AJ and Wheeler
DL: Nuclear EGFR contributes to acquired resistance to cetuximab.
Oncogene. 28:3801–3813. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Ritter CA, Perez-Torres M, Rinehart C, et
al: Human breast cancer cells selected for resistance to
trastuzumab in vivo over-express epidermal growth factor receptor
and ErbB ligands and remain dependent on the ErbB receptor network.
Clin Cancer Res. 13:4909–4919. 2007. View Article : Google Scholar
|
|
38.
|
Hutcheson IR, Knowlden JM, Hiscox SE, et
al: Heregulin beta1 drives gefitinib-resistant growth and invasion
in tamoxifen-resistant MCF-7 breast cancer cells. Breast Cancer
Res. 9:R502007. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Kong A, Calleja V, Leboucher P, Harris A,
Parker PJ and Larijani B: HER2 oncogenic function escapes EGFR
tyrosine kinase inhibitors via activation of alternative HER
receptors in breast cancer cells. PLoS One. 3:e28812008. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Amin DN, Campbell MR and Moasser MM: The
role of HER3, the unpretentious member of the HER family, in cancer
biology and cancer therapeutics. Semin Cell Dev Biol. 21:944–950.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Baselga J and Swain SM: Novel anticancer
targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer.
9:463–475. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Sergina NV, Rausch M, Wang D, et al:
Escape from HER-family tyrosine kinase inhibitor therapy by the
kinase-inactive HER3. Nature. 445:437–441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Mendelsohn J: Antibody-mediated EGF
receptor blockade as an anticancer therapy: from the laboratory to
the clinic. Cancer Immunol Immunother. 52:342–346. 2003.PubMed/NCBI
|
|
44.
|
Pinkas-Kramarski R, Lenferink AE, Bacus
SS, et al: The oncogenic ErbB-2/ErbB-3 heterodimer is a surrogate
receptor of the epidermal growth factor and betacellulin. Oncogene.
16:1249–1258. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Montero JC, Rodriguez-Barrueco R, Ocana A,
Diaz-Rodriguez E, Esparis-Ogando A and Pandiella A: Neuregulins and
cancer. Clin Cancer Res. 14:3237–3241. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Dua R, Zhang J, Nhonthachit P, Penuel E,
Petropoulos C and Parry G: EGFR over-expression and activation in
high HER2, ER negative breast cancer cell line induces trastuzumab
resistance. Breast Cancer Res Treat. 122:685–697. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Festuccia C, Angelucci A, Gravina GL, et
al: Epidermal growth factor modulates prostate cancer cell
invasiveness regulating urokinase-type plasminogen activator
activity. EGF-receptor inhibition may prevent tumor cell
dissemination. Thromb Haemost. 93:964–975. 2005.
|
|
48.
|
Viloria-Petit AM and Kerbel RS: Acquired
resistance to EGFR inhibitors: mechanisms and prevention
strategies. Int J Radiat Oncol Biol Phys. 58:914–926. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Koutsopoulos AV, Mavroudis D, Dambaki KI,
et al: Simultaneous expression of c-erbB-1, c-erbB-2, c-erbB-3 and
c-erbB-4 receptors in non-small-cell lung carcinomas: correlation
with clinical outcome. Lung Cancer. 57:193–200. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Onn A, Correa AM, Gilcrease M, et al:
Synchronous overexpression of epidermal growth factor receptor and
HER2-neu protein is a predictor of poor outcome in patients with
stage I non-small cell lung cancer. Clin Cancer Res. 10:136–143.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Chow NH, Chan SH, Tzai TS, Ho CL and Liu
HS: Expression profiles of ErbB family receptors and prognosis in
primary transitional cell carcinoma of the urinary bladder. Clin
Cancer Res. 7:1957–1962. 2001.PubMed/NCBI
|
|
52.
|
Wiseman SM, Makretsov N, Nielsen TO, et
al: Coexpression of the type 1 growth factor receptor family
members HER-1, HER-2, and HER-3 has a synergistic negative
prognostic effect on breast carcinoma survival. Cancer.
103:1770–1777. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Brader S and Eccles SA: Phosphoinositide
3-kinase signalling pathways in tumor progression, invasion and
angiogenesis. Tumori. 90:2–8. 2004.PubMed/NCBI
|
|
54.
|
Festuccia C, Muzi P, Millimaggi D, et al:
Molecular aspects of gefitinib antiproliferative and pro-apoptotic
effects in PTEN-positive and PTEN-negative prostate cancer cell
lines. Endocr Relat Cancer. 12:983–998. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Wang G, Reed E and Li QQ: Apoptosis in
prostate cancer: progressive and therapeutic implications (Review).
Int J Mol Med. 14:23–34. 2004.PubMed/NCBI
|
|
56.
|
Vlietstra RJ, van Alewijk DC, Hermans KG,
van Steenbrugge GJ and Trapman J: Frequent inactivation of PTEN in
prostate cancer cell lines and xenografts. Cancer Res.
58:2720–2723. 1998.PubMed/NCBI
|
|
57.
|
Zhu Z, Kleeff J, Friess H, et al:
Epiregulin is Up-regulated in pancreatic cancer and stimulates
pancreatic cancer cell growth. Biochem Biophys Res Commun.
273:1019–1024. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Torring N, Jorgensen PE, Sorensen BS and
Nexo E: Increased expression of heparin binding EGF (HB-EGF),
amphiregulin, TGF alpha and epiregulin in androgen-independent
prostate cancer cell lines. Anticancer Res. 20:91–95.
2000.PubMed/NCBI
|
|
59.
|
Torring N, Hansen FD, Sorensen BS, Orntoft
TF and Nexo E: Increase in amphiregulin and epiregulin in prostate
cancer xenograft after androgen deprivation-impact of specific HER1
inhibition. Prostate. 64:1–8. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Gupta GP, Nguyen DX, Chiang AC, et al:
Mediators of vascular remodelling co-opted for sequential steps in
lung metastasis. Nature. 446:765–770. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Watanabe T, Kobunai T, Yamamoto Y, et al:
Prediction of liver metastasis after colorectal cancer using
reverse transcription-polymerase chain reaction analysis of 10
genes. Eur J Cancer. 46:2119–2126. 2010. View Article : Google Scholar
|
|
62.
|
Oliveras-Ferraros C, Vall-Llovera AM,
Salip DC, et al: Evolution of the predictive markers amphiregulin
and epiregulin mRNAs during long-term cetuximab treatment of KRAS
wild-type tumor cells. Invest New Drugs. 30:846–852. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Baker JB, Dutta D, Watson D, et al: Tumour
gene expression predicts response to cetuximab in patients with
KRAS wild-type metastatic colorectal cancer. Br J Cancer.
104:488–495. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Ben-Yosef R, Starr A, Karaush V, et al:
ErbB-4 may control behavior of prostate cancer cells and serve as a
target for molecular therapy. Prostate. 67:871–880. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Vexler A, Lidawi G, Loew V, et al:
Anti-ERBb4 targeted therapy combined with radiation therapy in
prostate cancer. Results of in vitro and in vivo studies. Cancer
Biol Ther. 7:1090–1094. 2008.PubMed/NCBI
|
|
66.
|
Edwards J, Traynor P, Munro AF, Pirret CF,
Dunne B and Bartlett JM: The role of HER1-HER4 and EGFRvIII in
hormone-refractory prostate cancer. Clin Cancer Res. 12:123–130.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
67.
|
Center MM, Jemal A, Smith RA and Ward E:
Worldwide variations in colorectal cancer. CA Cancer J Clin.
59:366–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68.
|
De Alava E, Ocana A, Abad M, et al:
Neuregulin expression modulates clinical response to trastuzumab in
patients with metastatic breast cancer. J Clin Oncol. 25:2656–2663.
2007.PubMed/NCBI
|
|
69.
|
Motoyama AB, Hynes NE and Lane HA: The
efficacy of ErbB receptor-targeted anticancer therapeutics is
influenced by the availability of epidermal growth factor-related
peptides. Cancer Res. 62:3151–3158. 2002.PubMed/NCBI
|
|
70.
|
Normanno N, De Luca A, Maiello MR, et al:
The MEK/MAPK pathway is involved in the resistance of breast cancer
cells to the EGFR tyrosine kinase inhibitor gefitinib. J Cell
Physiol. 207:420–427. 2006. View Article : Google Scholar : PubMed/NCBI
|