Introduction
Hepatocellular carcinoma (HCC) is one of the common
cancers in Asia and Africa. The incidence of HCC is increasing in
Europe and the United States (1).
Although HCC can be cured at the early stage by surgical resection,
most patients can not be diagnosed at the early stage since tumors
are asymptomatic (2). Current
treatment options for HCC patients at the late stage include
chemotherapy, chemoembolization, ablation, and proton beam therapy.
These treatment options remain disappointed in clinic. HCC patients
will relapse and rapidly progress to the advanced stages with
vascular invasion and multiple metastases, which lead to a low
5-year survival rate of less than 7% (3). HCC patients who have surgically
resectable localized tumors show a better prognosis. However, even
these patients have a dismal 5-year survival rate of 15 to 39%
(4). Clearly, there is an urgent
need to search for new therapies for this lethal disease.
We have reported that chrysanthemum indicum
extract (CIE), a Chinese herbal extraction, exerts a significantly
inhibitory effect on HCC cells (MHCC97H) in previous studies
(5,6). One particular point to stress is that
CIE appeares to have no cytotoxic effect on normal liver cells,
highlighting an advantage of the herbal treatment. Herbal medicine
flavonoids have recently received increasing attention because of
the beneficial effects of anti-tumor and as chemopreventive agents
(2–5). Baicalein
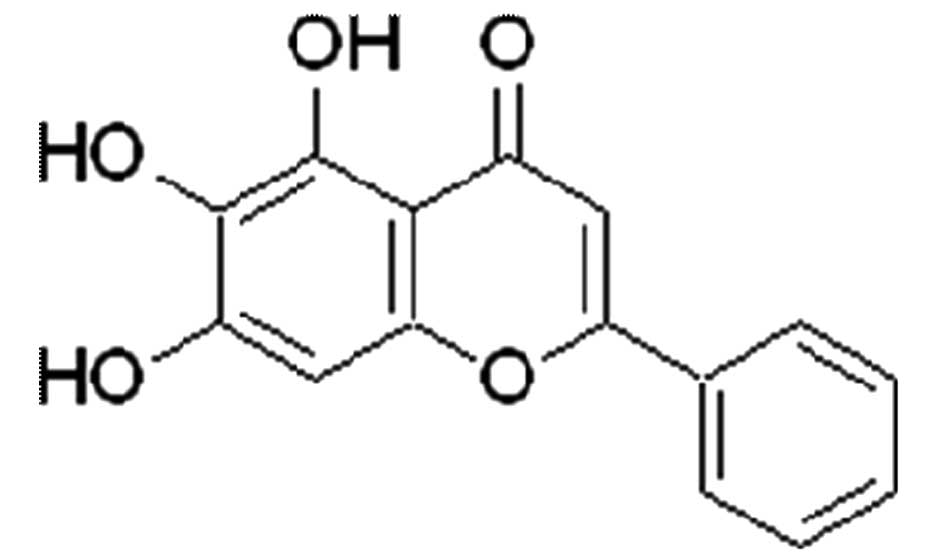
(5,6,7-trihydroxy-2-phenyl-4H-1-benzopyran-4-one) is a purified
flavonoid with defined chemical structure (Fig. 1) and is extracted from the roots of
Scutellaria baicalensis or Scutellaria radix.
Although the anti-tumor activity of Baicalein in HCC has
been reported in vitro (7,8),
little is known about the underlying mechanisms of action on HCC,
as well as the anti-tumor effect in vivo.
Previously genetic and expression profiling analyses
of human HCC have led to the identification of key oncogenes and
tumor-suppressor genes in liver carcinogenesis (9). They are mostly associated with the
mitogen-activated protein kinase (MAPK) pathway (9). Constitutively activated extracellular
signal-regulated kinases (ERK) have been shown to increase
proliferation of human HCC cells (10). So far there is no report that
investigates the effects of Baicalein on ERK in HCC. In this
study, we have investigated the effects of Baicalein on HCC
cells in vitro and in vivo, especially the effects of
Baicalein on ERK in HCC. We have demonstrated that
inhibition of MAPK/ ERK signaling and induction of apoptosis by
Baicalein treatment are critical mechanisms by which
Baicalein inhibits HCC growth.
Materials and methods
Reagents
Baicalein was purchased from Sigma-Aldrich
Co. (St. Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM)
was purchased from Invitrogen (Carlsbad, CA, USA). Fetal bovine
serum (FBS), penicillin, and streptomycin were ordered from
Gibco-BRL (Rockville, MD, USA). The apoptosis detection kit was
from Nanjing KeyGen Biotech. Co. Ltd. (Nanjing, China). MTT
(3-(4,5-dimethyl-2-thiazole)-2,5-diphenyltetrazolium bromide) was
purchased from Sigma Chemicals Co. (St. Louis, MO, USA). Caspase
inhibitor z-VAD-fmk and anti-cytochrome c were purchased
from Beyotime (Haimen, China), anti-MEK1 and anti-Phospho-MEK1
(Thr386) anti-Phospho (Thr386) MEK1 (p-MEK1) were from Millipore
Co. (Billerica, MA, USA). Anti-ERK1/2, anti-Phospho (Thr202/Tyr204)
ERK1/2 (p-ERK1/2), anti-Phospho-MEK1/2 (p-MEK1/2), anti-caspase-3,
anti-Bad and anti-Phospho-Bad(Ser112) antibodies were purchased
from Cell Signaling Technology Inc. (Danvers, MA, USA).
Animals
Male BALB/c nude mice (4-week-old) were purchased
from the Beijing Experimental Animal Center and maintained in the
Laboratory Animal Center of Xi’an Jiaotong University, in
accordance with the University Institutional Animal Care and Use
Committee. HepG2 cells (2×106) suspended in
200 μl of DMEM were injected subcutaneously into the right
inguinal area of the 6-week-old male nude mice. All animals
developed palpable tumors. Mice were divided into two groups (n=6
per group): group I, treatment with vehicle DMSO as the control
group; group II, treatment with 20 mg/kg/day Baicalein via
oral adminitration. Treatments were started one week after the
injection of HepG2 HCC cells. Resulting tumors were
measured using a vernier caliper every two days following the tumor
cell injections, and tumor volumes were calculated using the
formula: volume = (length x width2)/2 and expressed as
mean size ± standard error.
Cell culture
Human HCC cell lines (HepG2, BEL-7402,
SMMC-7721) and human normal liver cell line (HL-7702) were
purchased from Shanghai Institute of Cell Biology (Shanghai,
China). Cells were cultured in DMEM supplemented with 10% FBS, 100
U/ml penicillin, 100 μg/ml streptomycin, and 2 mmol/l
glutamine. All cells were incubated at 37°C with 5%
CO2.
Construction of expression plasmids and
transfection
The full-length pcDNA3.1 (Invitrogen, Paisley, UK)
MEK1 vector was made by cloning of the full-length PCR product of
MEK1 with KOD® DNA polymerase (Toyobo, Osaka, Japan).
All the plasmid sequences were confirmed by DNA sequencing. For
transient transfection experiments, cells were plated 24 h before
transfection in a 6-well plate at a density of 2×105.
Lipofectamine 2000 (Invitrogen) was used to perform transfection
with 4.0 μg pcDNA3.1(+)-MEK1 vector or 4.0 μg
pcDNA3.1(+) empty vector (as a negative control) according to the
manufacturer’s protocol.
Assessment of cell viability and
apoptosis
Cell viability was determined by a colorimetric
3-(4,5-dimethylthiazol-2-yl) 2,5-diphenyltetrazolium bromide (MTT)
assay as previous reported (11).
In brief, after treatment of cells with or without the indicated
agent and/or serum for 48 h, the cells were washed twice with PBS
and incubated with 0.5 mg/ml MTT (Sigma) for 4 h. The reagent was
absorbed by living cells and eventually formed an insoluble blue
formazan product. After the incubation period, cells were washed
with PBS, solubilized with dimethyl sulfoxide (DMSO), and
quantified using a microplate reader at the absorbance of 550 nm.
The inhibition rate was determined using SPSS software (version
17.0, SPSS Inc, Chicago, IL, USA).
Apoptotic and/or necrotic cells were evaluated by
Annexin V binding and propidium iodide (PI) uptake using an Annexin
V-FITC/PI kit as previously described (12). Briefly, tumor cells were plated at
a density of 1×105 cells per well into 6-well plates for
24 h. The cells were treated with various concentrations of
Baicalein (0, 40, 80 and 120 μM) and incubated at
37°C for 24 and 48 h. The cells were washed with cold PBS and
resuspended in Annexin V binding buffer. The cells were stained
with Annexin V-FITC for 15 min, washed, and then stained with PI.
The samples were analyzed by flow cytometer with CellQuest
software.
Detection of mitochondrial membrane
potential (MMPΔΨm)
Loss of MMPΔΨm was assessed by flow cytometry, using
a fluorescent indicator Rh123, as previously described (13,14).
Briefly, cells were treated with Baicalein at different
concentrations (0, 20, 40 and 60 μM) for 24 h. Then, Rh123
working solution was added to the culture at a final concentration
of 2 μg/ml and then incubated in the dark at 37°C for 30
min. Cells were then washed with PBS, and fluorescence of Rh123 was
detected immediately using a FACSCalibur, at an excitation
wavelength of 488 nm and an emission wavelength of 525 nm.
Caspase-3 and caspase-9 activity
assay
Cell lysates were prepared by incubating
2×106 cells/ml in extraction buffer (25 mM Tris-HCl, pH
7.5, 20 mM MgCl2, and 150 mM NaCl, 1% Triton X-100, 25
μg/ml leupeptin, and 25 μg/ml aprotinin) for 30 min
on ice. Lysates were centrifuged at 12,000 x g for 15 min. Cellular
extracts (30 μg) were then incubated in a 96-well microtitre
plate with 20 ng Ac-DEVD-pNA (caspase-3 activity) or Ac-LEHD-pNA
(caspase-9 activity) (Beyotime) for 2 h at 37°C. Caspase activity
was measured by cleavage of the Ac-DEVD-pNA or Ac-LEHD-pNA
substrate to pNA, the absorbance of which was measured at 405 nm.
Relative caspase activity was calculated as a ratio of emission of
treated cells to untreated cells.
Western blot analysis
Western blot analysis was executed as previously
described (15). Whole-cell
extracts were prepared from Baicalein-treated or
control-treated cells cultured in 6-well plates. After incubation,
cells were harvested and resuspended in lysis buffer, washed with
ice-cold PBS and lysed in extraction buffer (40 mmol/l Tris-HCl, pH
7.5, 150 mmol/l KCl, 1 mmol/l EDTA, 1% Triton X-100, 100 mmol/l
NaVO3, 1 mmol/l PMSF) supplemented with the protease
inhibitor cocktail. The protein (50 μg) was separated on 10%
SDS-PAGE and transferred onto PVDF membranes. The membranes were
blocked with 5% non-fat milk in Tris-buffered saline (TBS) at 37°C,
and then incubated with rabbit anti-MEK1 antibody (1:1,000), rabbit
anti-p-MEK1 antibody (1:1,000), mouse anti-ERK1/2 antibody
(1:1,000), rabbit anti-p-ERK1/2 antibody (1:1,000), rabbit anti-Bad
antibody (1:1,000), rabbit anti-p-Bad antibody (1:1,000) or mouse
anti-β-actin antibody (1:500) in TBS containing 5% non-fat milk for
12 h at 4°C. Horseradish peroxidase-linked anti-mouse IgG (1:5,000)
or horseradish peroxidase-linked anti-rabbit IgG (1:5,000) was used
as a secondary antibody (in TBS containing 5% non-fat milk for 3 h
at room temperature), and antigen-antibody complexes were detected
using an enhanced chemiluminescence kit (Amersham, ECL Plus,
Freiburg, Germany). Densitometry values for western blot analysis
and antibody array experiments were estimated by the ImageQuant TL
software (GE Healthcare, Buckinghamshire, UK) and expressed as
arbitrary units (a.u.). Multiple film exposures were used to verify
the linearity of the samples analyzed and to avoid saturation of
the film.
Immunohistochemical procedures
The expressions and intracellular localizations of
MAPK/ERK in HCC and mice xenograft were examined
immunohistochemically. Antigen retrieval was performed by microwave
oven for 15 min in TEG buffer (10 mM Tris, 0.5 mM ethylene glycol
tetraacetic acid, pH 9.0). Incubation with primary antibody for 60
min at room temperature was followed by detection of the primary
antibody using the AdvanceTM HRP system (Dako). The
chromogen 3,3′-diaminobenzidine was applied and all the staining
was performed using the Autostainer Plus Link Instrument (Dako).
After washing, the slides were counterstained with Meyer’s
hematoxylin for 30 sec. The following antibodies were used:
p-MEK1/2 (dilution factor 1:100), pERK1/2 (dilution factor 1:100),
PCNA (dilution factor 1:100). All antibodies mentioned above were
from Cell Signaling Technology.
Terminal dUTP nick end labeling (TUNEL)
analysis
Xenograft tumors were resected and fixed in formalin
for 24 h, and imbedded in paraffin and 5-micron of sections were
prepared. TUNEL assay was performed using an apoptag peroxidase
in situ apoptosis detection kit (Chemicon International,
Temecula, CA, USA). Briefly, the sections were digested using
proteinase K and the endogenous peroxidase activity was blocked
using 3% hydrogen peroxide in PBS. The sections were then placed in
equilibration buffer and incubated with working strength of TdT
enzyme in a humidifying chamber at 37°C for 1 h. The reaction was
terminated with a stop/wash buffer provided with the kit. The
apoptotic nuclei were stained by direct immunoperoxidase detection
of digioxigenin-labeled DNA in test sections.
Statistical analysis
Data are presented as the mean ± standard errors
from at least three independent experiments and analyzed using
Student’s t-test. p<0.05 was considered statistically
significant. All statistical tests and corresponding p-values were
two sided.
Results
Baicalein preferentially inhibits HCC
cells and spares normal liver cells
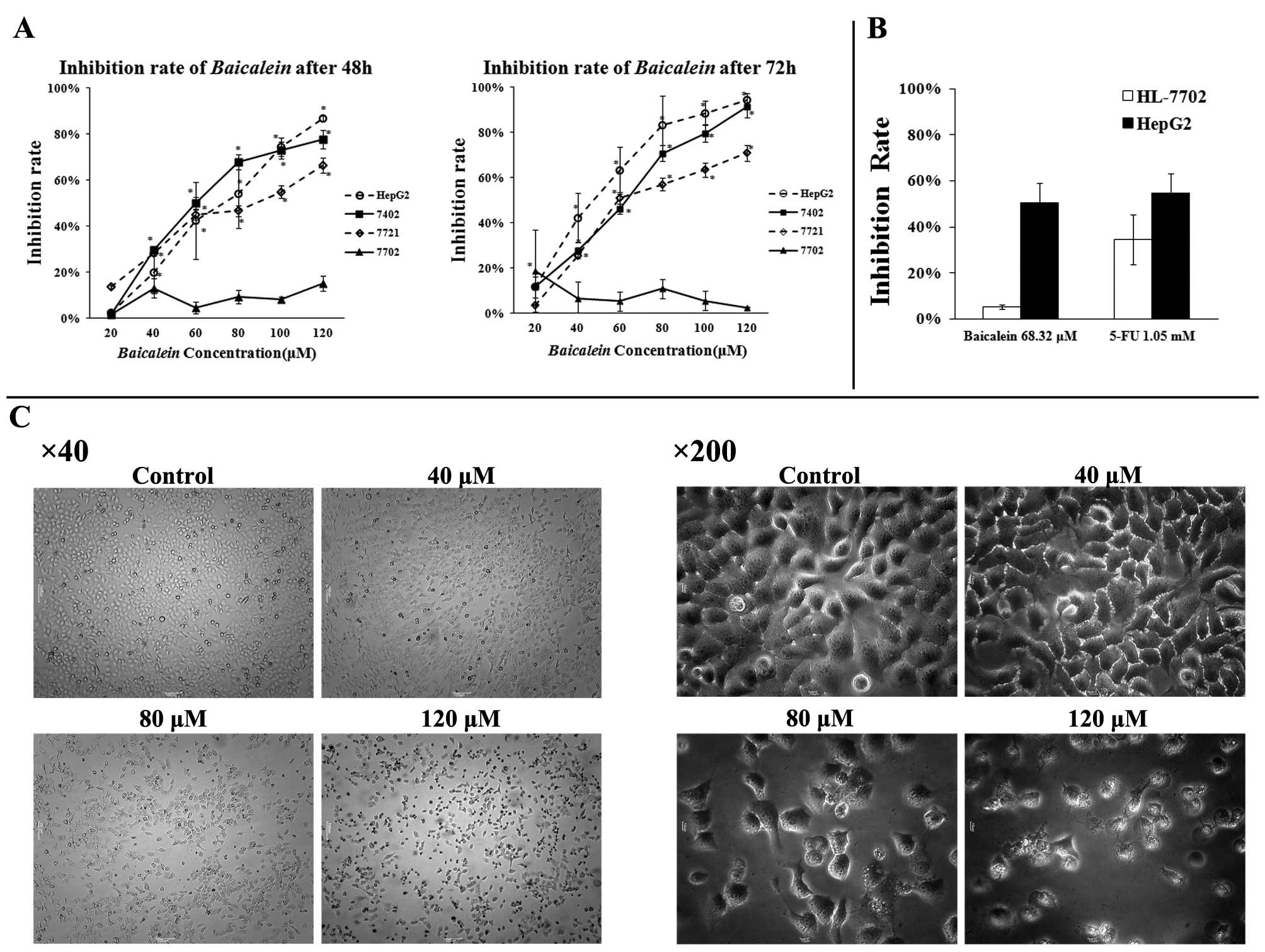
In order to investigate whether or not
Baicalein has any differential cytotoxicity to HCC and
normal liver cells as CIE does (5,6). We
examined the cytotoxic activity of Baicalein in three HCC
lines (HepG2, BEL-7402 and SMMC-7721) and one normal
liver cell line (HL-7702). The anti-tumor effects of
Baicalein were examined by an MTT assay after treatment with
20 to 120 μM of Baicalein for 48 or 72 h. As shown in
Fig. 2A, the viability of
HepG2, BEL-7402 and SMMC-7721 cells was significantly
reduced by Baicalein treatment in a time- and dose-dependent
manner, whereas the normal liver cell line (HL-7702) was hardly
affected. To further examine the cytotoxicity of Baicalein
in HepG2 HCC cells and normal liver HL-7702 cells, 5-FU
was used as a treatment benchmark in comparison with
Baicalein. While 5-FU had a 50% inhibiting concentration
(IC50) of 1.05 mM in HepG2 cells,
Baicalein had an IC50 value of 68.32 μM
(Fig. 2B). As shown in Fig. 2B, the inhibitory effect of
Baicalein on HL-7702 normal liver cells at its
IC50 concentration was significantly lower than that of
5-FU at its IC50 concentration. Baicalein had an
inhibition rate of 5.43%±1.00%, whereas 5-FU inhibited more than
35% on normal liver cells at its IC50 concentration in
normal liver cells (Fig. 2B).
Baicalein reduces mitochondrial
transmembrane potential and induces intrinsic apoptosis
To explore the mechanisms by which Baicalein
inhibits HCC growth, HepG2 HCC cells were first examined
by phase contrast microscopy for any apoptotic characteristics
after incubation with Baicalein at different concentrations
(0, 40, 80, 120 μM) for 24 h. As shown in Fig. 2C, the control-treated cells showed
a typical polygonal and intact appearance, whereas the
Baicalein-treated cells displayed cellular shrinkage (40, 80
μM), rounding (120 μM), poor adherence (120
μM) and round floating shapes (120 μM). To determine
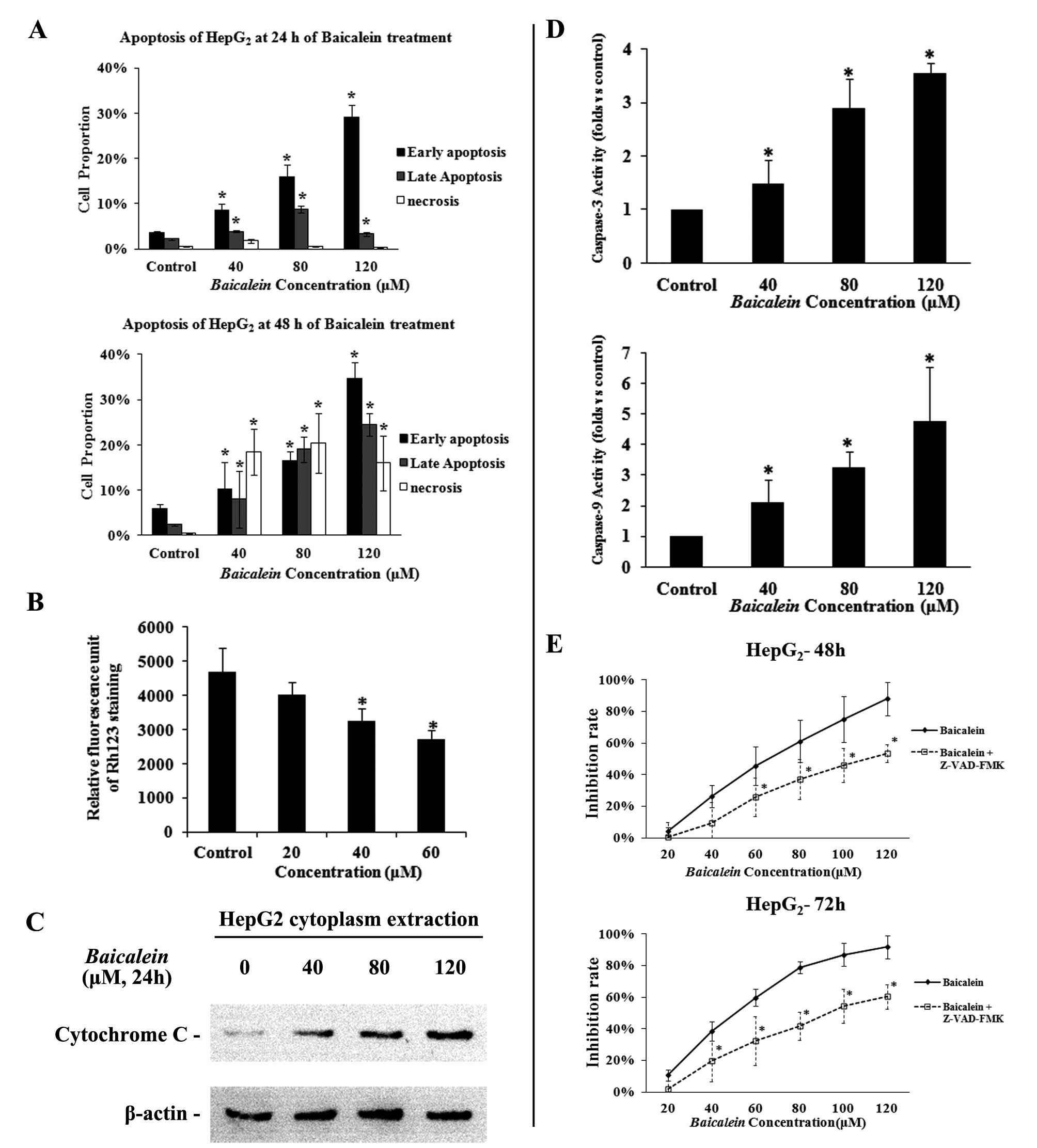
the effect of Baicalein on apoptosis in detail,
HepG2 HCC cells were treated with different
concentrations of Baicalein (40, 80 or 120 μM), for
24 h and then subjected to an Annexin V analysis on flow cytometry.
As shown in Fig. 3A,
Baicalein induced marked apoptosis in HepG2 cells
in a concentration-dependent manner. After short treatment for 24
h, the numbers of early apoptotic cells accompanied some late
apoptotic cells were significantly increased in the
Baicalein-treated HepG2 cells when compared with
control-treated HepG2 cells (Fig. 3A). After longer treatment for 48 h,
the numbers of late apoptotic and necrotic cells were also
dramatically increased along with early apoptotic cells in the
Baicalein-treated HepG2 cells (Fig. 3A).
The decrease of mitochondrial transmembrane
potential (MMPΔΨm) has been reported as an early event of apoptosis
(16) and can be detected by the
decline of rhodamine 123 fluorescence. To determine whether or not
Baicalein-induced apoptosis involves the MMPΔΨm, we used a
fluorescent indicator Rh123 to detect the MMPΔΨm in
HepG2 cells that were treated with 20–60 μM of
Baicalein for 24 h. As shown in Fig. 3B, after exposure to different doses
of Baicalein, the cells exhibited dose-dependent decline of
Rho123 staining. At the doses of 40 and 60 μM,
Baicalein-treated cells had significant lower values of
rhodamine 123 fluorescence (3,258.11±355.90, 2,705.45±276.17) than
the control (4,703.24±698.91, p<0.05), further indicating that
Baicalein can induce apoptosis in liver cancer cells
(Fig. 3B).
To further determine whether apoptosis induced by
Baicalein was a mitochondrial-dependent pathway, we tested
whether cytochrome c could be released from the mitochondria
into the cytoplasm. As shown in Fig.
3C, levels of cytochrome c release from the mitochondria
increased dose-dependently in the Baicalein-treated
HepG2 cells at concentrations ranging from 40 to 120
μM. To further investigate whether apoptosis induced by
Baicalein was a caspase-dependent pathway, we tested whether
mitochondrial-related caspases were activated by Baicalein
treatment. Our research showed that caspase-9 and caspase-3
activities were highly increased dose-dependently on exposure to
Baicalein in HepG2 cells (Fig. 3E). Furthermore, we treated
HepG2 cells with Baicalein in the presence of 10
M pan-caspase inhibitor (z-VAD-fmk) or DMSO (as a control). MTT
assay showed that z-VAD-fmk partially attenuated
Baicalein-induced inhibition on HepG2 cells
(Fig. 3D), suggesting that
apoptosis induction is an important cause for
Baicalein-induced growth inhibition in HCC.
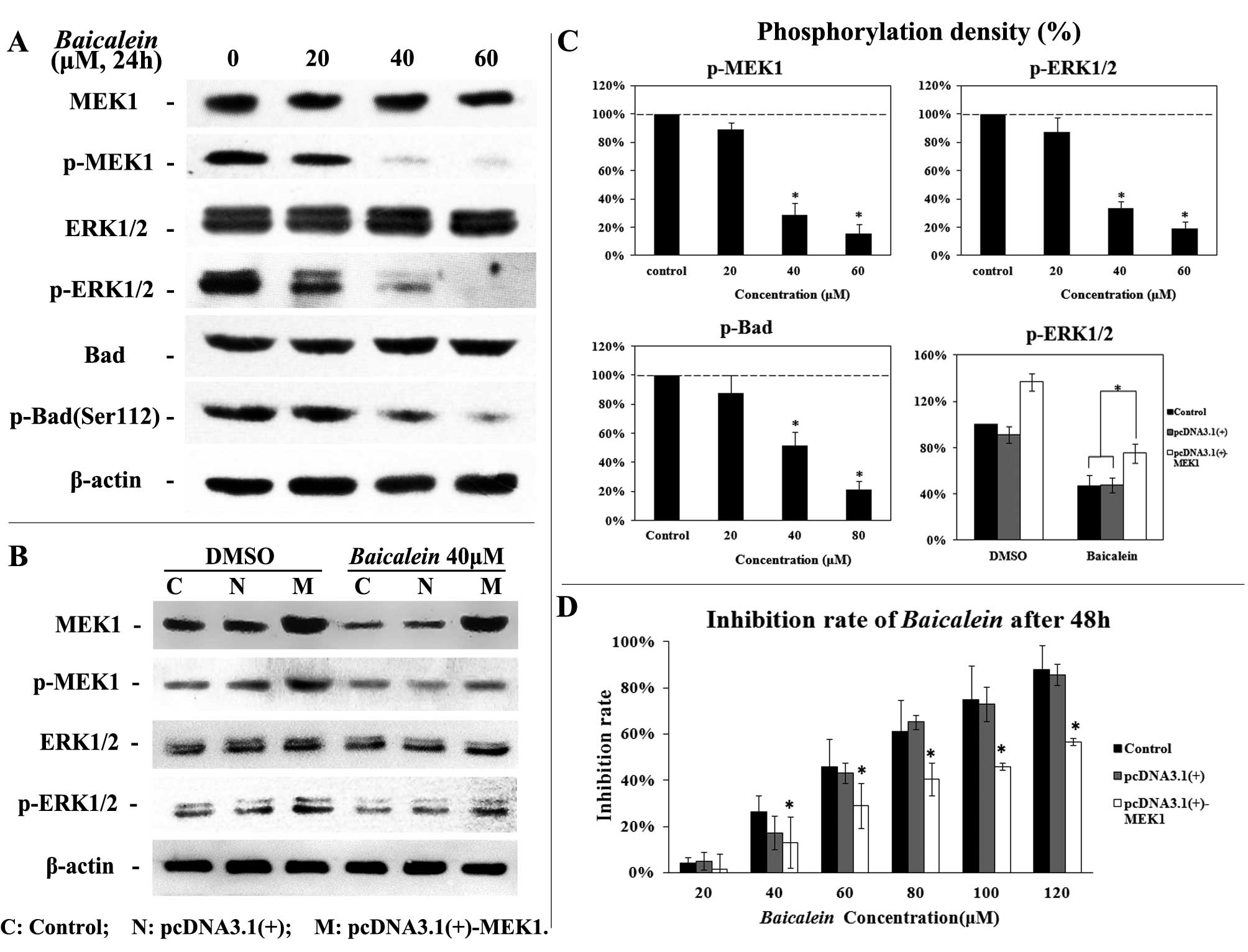
Baicalein inhibits MEK/ERK signaling in
vitro
Western blot analysis has been utilized to evaluate
the effect of Baicalein on phosphorylation levels of MEK and
ERK in HepG2 cells. As shown in Fig. 4A and 4C, Baicalein inhibited
MEK1 and ERK1/2 phosphorylation at a concentration-dependent manner
in HepG2 cells. The phosphorylation level of Bad (Ser
112), which is an anti-apoptosis protein activated by the MEK/ERK
pathway in tumor cells (17), was
also measured 24 h after Baicalein treatment.
Baicalein reduced levels of phosphorylated Bad of Ser 112 in
a dose-dependent manner (Fig. 4A and
4C).
Roles of MEK-ERK signaling in Baicalein
activity
To determine whether this Baicalein-induced
growth inhibition depends on the MEK-ERK pathway, HepG2
cells were transfected with a plasmid pcDNA3.1(+)-MEK1 expressing
human MEK1. Ectopic expression of MEK1 led to an enhanced activity
of MEK-ERK pathway indicated by increased phosphorylation of MEK1
and ERK1/2 (18) (Fig. 4B). Importantly, HepG2
cells with ectopic expression of MEK1 (higher MEK-ERK activity)
became relatively resistant to Baicalein-induced growth
inhibition (Fig. 4).
Overexpression of MEK1 partially attenuated
Baicalein-induced inhibition of ERK1/2 phosphorylation
(Fig. 4C). Overexpression of MEK1,
in part, blocked Baicalein-induced growth inhibition in
vitro (Fig. 4D). These data
suggest that inhibition of MEK-ERK is one of critical mechanism by
which Baicalein inhibits HCC cells.
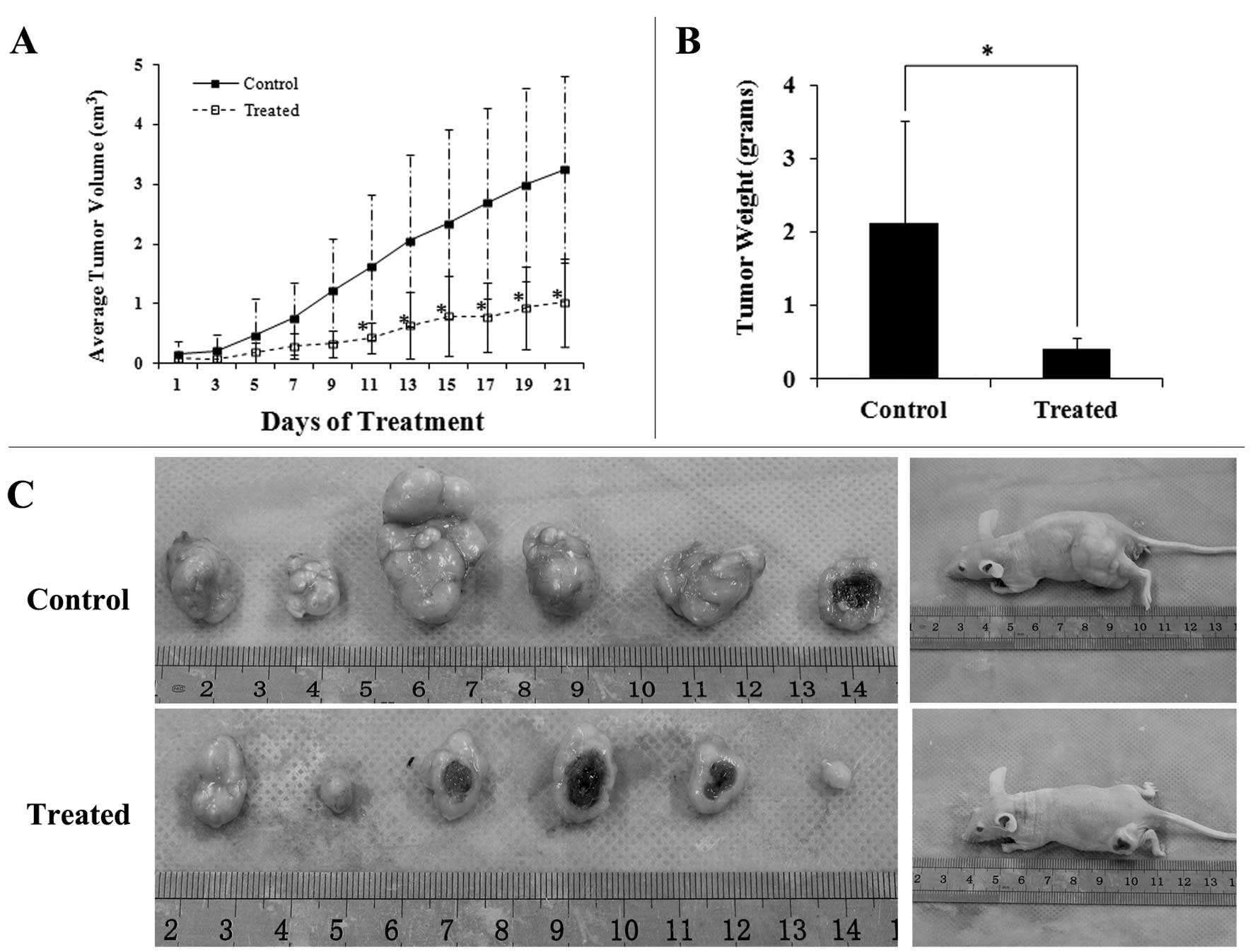
Baicalein suppresses HCC xenograft
growth, inhibits MEK-ERK phosphorylation, and induces apoptosis in
vivo
In the animal study, the control group received
diluent vehicle treatment only, whereas the treatment group
received Baicalein 20 mg/kg/day. This in vivo dosage
was selected by our pilot experiments that showed significant tumor
inhibition but without significant side effects. The mice were
treated with Baicalein daily for 21 days. As shown in
Fig. 5A, Baicalein-treated
mice exhibited a statistically significant tumor volume reduction
(p<0.01) compared with the control group. The average tumor
volume of control and treatment group were 3.25±0.56 cm3
and 1.02±0.40 cm3, respectively. After treatment for 21
days, the mice were sacrificed, xenograft tumors were resected and
the tumor weight of xenograft were measured. As shown in Fig. 5B and 5C, tumor sizes and weights in
Baicalein-treated mice were dramatically smaller than those
in control-treated mice. The control-treated mice had a median
tumor weight of 2.12 g, whereas the Baicalein-treated mice
had a median tumor weights of 0.41 g (Fig. 5B).
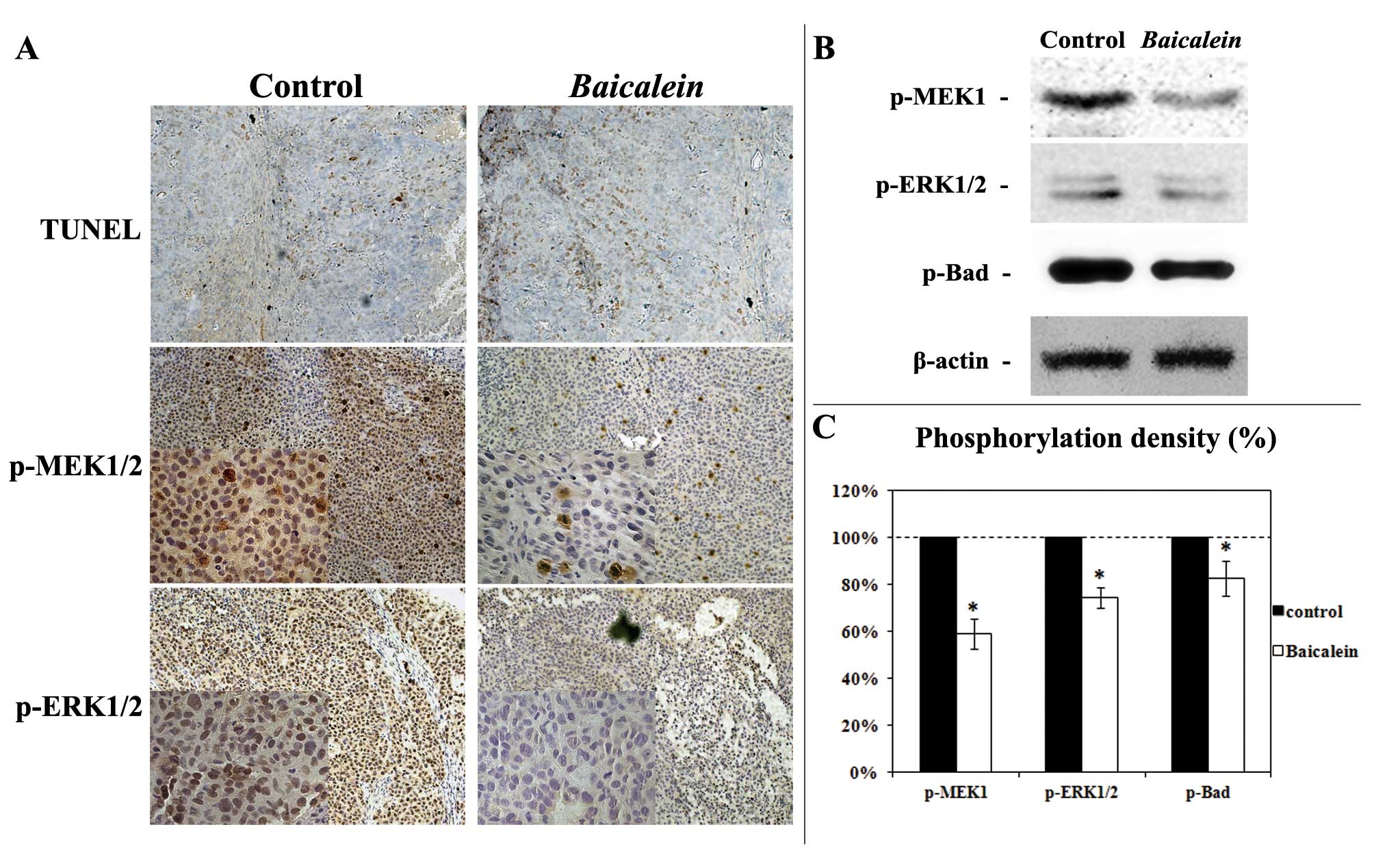
In order to confirm the in vitro observation
of Baicalein-induced apoptosis (Fig. 3), Baicalein-induced
apoptosis in xenograft tumors was evaluated with the terminal
deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.
As shown in Fig. 6A,
Baicalein-treated tumors had greater TUNEL-postive cells
than control-treated tumors. In agreement with the in vitro
observations, p-MEK1/2 and p-ERK1/2 expression were markedly
inhibited in Baicalein-treated tumors as illustrated by
immunohistochemical analysis (Fig.
6A) and western blot analysis (Fig. 6B and 6C). MEK-ERK signaling
associated Bad phosphorylation (Serine 112) was also decreased by
Baicalein treatment (Fig. 6B
and 6C). Above data confirm the in vitro results and
show that Baicalein treatment can significantly suppress HCC
tumor growth and MEK-ERK signaling, and can induce apoptosis in
vivo.
Discussion
Baicalein alone, or in combination with
other herbs, has recently been shown to have cytostatic effect on
several cancer cell lines in vitro (7,19)
and also in vivo (20,21).
Baicalein has shown the advantage of inhibiting the growth
of cancer cells while leaving normal cells relatively unaffected in
several studies (22,23). In this report, we confirmed that
Baicalein had anti-cancer effect against HCC cells in
vitro. We have further demonstrated that Baicalein had
much lower cytotoxicity to normal liver cells in comparison with
5-FU. 5-FU can be benefitially used for hepatic arterial infusion
chemotherapy (24) or
intra-peritoneal administration (25) as treatment for HCC. However,
toxicity issue limits its clinical application. Our data showed
that Baicalein had greater effect on HCC cells but less
toxicity on normal liver cells than 5-FU. Thus, Baicalein is
potentially more acceptable than 5-FU in clinic and deserves
further clinical trials. To our knowledge, this is the first study
to evaluate the potential of Baicalein in vivo treatment of
HCC xenografts. Significant reduction of tumor mass was observed
after a 3-week treatment. The in vivo effect of
Baicalein on HCC tumors strongly support Baicalein as
a potential new chemodrug for anti-HCC treatment.
Whether Baicalein inhibits HCC cells via
apoptosis induction is still controversial. A high proportion of
necrotic HCC cells after Baicalein treatment was observed by
Matsuzaki et al (8).
Baicalein was also reported to induce caspase-related
apoptosis in cancer cells (26).
In the present study, we confirmed a pro-apoptotic effect of
Baicalein on HCC cells by using several methods. Although it
was widely reported that mostly chemotherapy reagent induced
mitochondrial signaling apoptosis (27), the mechanisms and pathways involved
in Baicalein-induced apoptosis on HCC cells are still
unclear. Our data illustrated a decrease of MMP ΔΨm and released of
cytochrome c from mitochondria, and the following activation
of caspase-9 and caspase-3, suggesting a mitochondrial
signaling-related apoptosis was induced by Baicalein in HCC
cells. We have further demonstrated that induction of apoptosis is
important for Baicalein effect. z-VAD-fmk is a pan-caspases
inhibitor which could nullify caspases activity (28). Our results confirmed that z-VAD-fmk
did blocked Baicalein effects, suggesting that
caspase-dependent apoptotic pathways were involved in
Baicalein-induced inhibition on HCC cells.
The exact molecular mechanism by which
Baicalein inhibits cell growth is still not known. There are
few studies suggesting that MEK-ERK pathway could be the downstream
signaling in response to Baicalein (29–31).
Our study has demonstrated that inhibition of MEK-ERK pathway is
critical for Baicalein action in HCC. The experiments of MEK
overexpression showed that without inhibition of MEK-ERK pathway,
Baicalein-induced growth inhibition was significantly
attenuated. In fact, extracellular signal-regulated kinase (ERK)
kinase (MEK)/ERK cascade plays critical roles in the development of
HCC (32). ERK is a
serine/threonine kinase that can be activated by hepatocyte growth
factor (HGF) (33) and its
receptor the c-Met proto-oncogene (34). ERK is activated in HepG2
cells after treatment with HGF and constitutive expression of
Ha-Ras (35,36). ERK inhibitor is suggested as a
potential anti-HCC agent (37–39).
Sorafenib is the first targeted therapy drug that has demonstrated
an improved overall survival benefit in patients with advanced HCC
(40–42). Sorafenib can inhibit tumor cell
proliferation in vitro by targeting the Raf/MEK/ ERK
signaling pathway at the level of Raf kinase (43) and by targeting angiogenesis
(44). p-ERK could be a useful
biomarker predictive of sensitivity to sorafenib (45), suggesting the critical role of the
MAPK/ERK signaling in HCC. Again, our study supports the notion
that down-regulation of the MAPK/ERK activity is beneficial in HCC
treatment.
Bad is a pro-apoptotic protein and its function is
modulated by phosphorylation at two sites, Ser-112 and Ser-136
(46,47). the MAPK-activated pp90-ribosomal S6
kinase family can catalyze the phosphorylation of Bad Ser-112
(48). The Ras-MAPK pathway is
involved in the phosphorylation of Bad Ser-112 and its function
related to dissociation of Bad from Bcl-xL (49). Underphosphorylated Bad interacts
with anti-apoptotic Bcl-2 members and anchors on the mitochondria
to induce apoptosis whereas phosphorylated Bad is sequestered in
the cytoplasm by 14-3-3 proteins that attenuate Bad induced
apoptosis (50). Our results
indicate that Baicalein downregulates the phosphorylation
level of Bad, suggesting that Bad is one of the downstream targets
of Baicalein-induced inhibition of ERK.
Baicalein-induced apoptosis in hepatocellular cells could be
through Bad-related regulation, which needs to be further
determined.
Taken together, this study found that
Baicalein is an effective anti-HCC agent with low
cytotoxicity to normal liver cells. This study provides evidence to
show that inhibition of MAPK-ERK signaling and induction of
intrinsic apoptosis are the critical mechanisms by which
Baicalein inhibits HCC growth.
Acknowledgements
This study was supported by a grant
from Program for Changjiang Scholars and Innovative Research Team
in University (PCSIRT: 1171) and the Kwang-Hua Education Foundation
of Xi’an Jiaotong University.
References
|
1.
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: The impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Zhang Y, Wang S, Li D, et al: A systems
biology-based classifier for hepatocellular carcinoma diagnosis.
PLoS One. 6:e224262011. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Almogy G, Lieberman S, Gips M, et al:
Clinical outcomes of surgical resections for primary liver sarcoma
in adults: results from a single centre. Eur J Surg Oncol.
30:421–427. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Livraghi T, Makisalo H and Line PD:
Treatment options in hepatocellular carcinoma today. Scand J Surg.
100:22–29. 2011.PubMed/NCBI
|
|
5.
|
Li ZF, Wang ZD, Ji YY, et al: Induction of
apoptosis and cell cycle arrest in human HCC MHCC97H cells with
Chrysanthemum indicum extract. World J Gastroenterol.
15:4538–4546. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Wang ZD, Huang C, Li ZF, et al:
Chrysanthemum indicum ethanolic extract inhibits invasion of
hepatocellular carcinoma via regulation of MMP/TIMP balance as
therapeutic target. Oncol Rep. 23:413–421. 2010.
|
|
7.
|
Motoo Y and Sawabu N: Antitumor effects of
saikosaponins, baicalin and baicalein on human hepatoma cell lines.
Cancer Lett. 86:91–95. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Matsuzaki Y, Kurokawa N, Terai S,
Matsumura Y, Kobayashi N and Okita K: Cell death induced by
baicalein in human hepatocellular carcinoma cell lines. Jpn J
Cancer Res. 87:170–177. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: from genes to environment. Nat Rev Cancer.
6:674–687. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Gailhouste L, Ezan F, Bessard A, et al:
RNAi-mediated MEK1 knock-down prevents ERK1/2 activation and
abolishes human hepatocarcinoma growth in vitro and in vivo. Int J
Cancer. 126:1367–1377. 2010.PubMed/NCBI
|
|
11.
|
Scudiero DA, Shoemaker RH, Paull KD, et
al: Evaluation of a soluble tetrazolium formazan assay for
cell-growth and drug sensitivity in culture using human and other
tumor-cell lines. Cancer Res. 48:4827–4833. 1988.PubMed/NCBI
|
|
12.
|
Yang GY, Liao J, Kim K, Yurkow EJ and Yang
CS: Inhibition of growth and induction of apoptosis in human cancer
cell lines by tea polyphenols. Carcinogenesis. 19:611–616. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Tang W, Liu HW, Zhao WM, Wei DZ and Zhong
JJ: Ganoderic acid T from Ganoderma lucidum mycelia induces
mitochondria mediated apoptosis in lung cancer cells. Life Sci.
80:205–211. 2006.
|
|
14.
|
Li L, Lu QH, Shen YW and Hu X: Schisandrin
B enhances doxorubicin-induced apoptosis of cancer cells but not
normal cells. Biochem Pharmacol. 71:584–595. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Li ZF, Jiang A, Zhang S, et al: miR-615-3p
promotes the phagocytic capacity of splenic macrophages by
targeting ligand-dependent nuclear receptor corepressor in
cirrhosis-related portal hypertension. Exp Biol Med (Maywood).
236:672–680. 2011. View Article : Google Scholar
|
|
16.
|
Brunnemann C, Weiger TM, Langeluddecke C,
et al: Ethanol depolarizes the membrane potential and changes the
cell volume of pituitary tumor cells (Gh3). Alcohol Clin Exp Res.
34:125a. 2010.
|
|
17.
|
Fang XJ, Yu SX, Eder A, et al: Regulation
of BAD phosphorylation at serine 112 by the Ras-mitogen-activated
protein kinase pathway. Oncogene. 18:6635–6640. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Deak JC and Templeton DJ: Regulation of
the activity of MEK kinase 1 (MEKK1) by autophosphorylation within
the kinase activation domain. Biochem J. 322:185–192.
1997.PubMed/NCBI
|
|
19.
|
Ikemoto S, Sugimura K, Yoshida N, et al:
Antitumor effects of Scutellariae radix and its components
baicalein, baicalin, and wogonin on bladder cancer cell lines.
Urology. 55:951–955. 2000.
|
|
20.
|
Ye F, Wu J, Dunn T, Yi J, Tong XD and
Zhang D: Inhibition of cyclooxygenase-2 activity in head and neck
cancer cells by genistein. Cancer Lett. 211:39–46. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Yu J, Liu H, Lei J, Tan W, Hu X and Zou G:
Antitumor activity of chloroform fraction of Scutellaria
barbata and its active constituents. Phytother Res. 21:817–822.
2007. View Article : Google Scholar
|
|
22.
|
Du GJ, Han G, Zhang S, et al: Baicalin
suppresses lung carcinoma and lung metastasis by SOD mimic and
HIF-1 alpha inhibition. Eur J Pharmacol. 630:121–130. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Chen CH, Huang LLH, Huang CC, Lin CC, Lee
Y and Lu FJ: Baicalein, a novel apoptotic agent for hepatoma cell
lines: a potential medicine for hepatoma. Nutr Cancer. 38:287–295.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Han KH, Kim BK, Park JY, et al: Long-term
clinical outcomes of hepatic arterial infusion chemotherapy with
cisplatin with or without 5-fluorouracil in locally advanced
hepatocellular carcinoma. J Cancer Res Clin. 137:659–667. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Hoffman RM, Rashidi B, An ZL, et al:
Efficacy of intra-hepatectomy 5-FU on recurrence and metastasis of
human hepatocellular carcinoma in nude mice. Int J Cancer.
91:231–235. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Bose Dasgupta S, Das BB, Sengupta S, et
al: The caspase-independent algorithm of programmed cell death in
Leishmania induced by baicalein: the role of LdEndoG, LdFEN-1 and
LdTatD as a DNA ‘degradesome’. Cell Death Differ. 15:1629–1640.
2008.PubMed/NCBI
|
|
27.
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Casares N, Pequignot MO, Tesniere A, et
al: Caspase-dependent immunogenicity of doxorubicin-induced tumor
cell death. J Exp Med. 202:1691–1701. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Li HB, Jiang Y and Chen F: Separation
methods used for Scutellaria baicalensis active components.
J Chromatogr B Analyt Technol Biomed Life Sci. 812:277–290. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Ma Z, Otsuyama K, Liu SQ, et al:
Baicalein, a component of Scutellaria radix from
Huang-Lian-Jie-Du-Tang (HLJDT), leads to suppression of
proliferation and induction of apoptosis in human myeloma cells.
Blood. 105:3312–3318. 2005.PubMed/NCBI
|
|
31.
|
Ye F, Che YF, McMillen E, et al: The
effect of Scutellaria baicalensis on the signaling network
in hepatocellular carcinoma cells. Nutr Cancer. 61:530–537.
2009.
|
|
32.
|
Hui LJ, Min LH and He BK:
Mitogen-activated protein kinases in hepatocellular carcinoma
development. Semin Cancer Biol. 21:10–20. 2011. View Article : Google Scholar
|
|
33.
|
Lu SC, Yang HP, Magilnick N, Noureddin M
and Mato JM: Effect of hepatocyte growth factor on methionine
adenosyl-transferase genes and growth is cell density-dependent in
HepG2 cells. J Cell Physiol. 210:766–773. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Dong JH, Xie B, Xing RX, et al:
Down-regulation of c-Met expression inhibits human HCC cells growth
and invasion by RNA interference. J Surg Res. 162:231–238. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Tsukada Y, Miyazawa K and Kitamura N: High
intensity ERK signal mediates hepatocyte growth factor-induced
proliferation inhibition of the human hepatocellular carcinoma cell
line HepG2. J Biol Chem. 276:40968–40976. 2001.
View Article : Google Scholar
|
|
36.
|
Han JH, Tsukada Y, Hara E, Kitamura N and
Tanaka T: Hepatocyte growth factor induces redistribution of
p21(CIP1) and p27(KIP1) through ERK-dependent p16(INK4a)
up-regulation, leading to cell cycle arrest at G(1) in
HepG2 hepatoma cells. J Biol Chem. 280:31548–31556.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
O’Neil BH, Goff LW, Kauh JSW, et al: Phase
II study of the mitogen-activated protein kinase 1/2 inhibitor
selumetinib in patients with advanced hepatocellular carcinoma. J
Clin Oncol. 29:2350–2356. 2011.PubMed/NCBI
|
|
38.
|
Klein PJ, Schmidt CM, Wiesenauer CA, et
al: The effects of a novel MEK inhibitor PD184161 on MEK-ERK
signaling and growth in human liver cancer. Neoplasia. 8:1–8. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Kuo TC, Lu HP and Chao CCK: The tyrosine
kinase inhibitor sorafenib sensitizes hepatocellular carcinoma
cells to taxol by suppressing the HURP protein. Biochem Pharmacol.
82:184–194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Shen Y, Hsu C, Hsu C, et al: A phase II
study of sorafenib in combination with tegafur/uracil (UFT) for
Asian patients with advanced hepatocellular carcinoma (HCC). J Clin
Oncol. 27:45892009.
|
|
41.
|
Abou-Alfa GK, Schwartz L, Ricci S, et al:
Phase II study of sorafenib in patients with advanced
hepatocellular carcinoma. J Clin Oncol. 24:4293–4300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Wang SH, Huang X, Li Y, et al: RN181
suppresses hepatocellular carcinoma growth by inhibition of the
ERK/MAPK pathway. Hepatology. 53:1932–1942. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Takezawa K, Okamoto I, Yonesaka K, et al:
Sorafenib inhibits non-small cell lung cancer cell growth by
targeting B-RAF in KRAS wild-type cells and C-RAF in KRAS mutant
cells. Cancer Res. 69:6515–6521. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Wilhelm SM, Adnane L, Newell P, Villanueva
A, Llovet JM and Lynch M: Preclinical overview of sorafenib, a
multikinase inhibitor that targets both Raf and VEGF and PDGF
receptor tyrosine kinase signaling. Mol Cancer Ther. 7:3129–3140.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Zhang Z, Zhou XY, Shen HJ, Wang DX and
Wang YH: Phosphorylated ERK is a potential predictor of sensitivity
to sorafenib when treating hepatocellular carcinoma: evidence from
an in vitro study. BMC Med. 7:412009. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Zha JP, Harada H, Yang E, Jockel J and
Korsmeyer SJ: Serine phosphorylation of death agonist BAD in
response to survival factor results in binding to 14-3-3 not
BGL-X(L). Cell. 87:619–628. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Datta SR, Dudek H, Tao X, et al: Akt
phosphorylation of BAD couples survival signals to the
cell-intrinsic death machinery. Cell. 91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Bonni A, Brunet A, West AE, Datta SR,
Takasu MA and Greenberg ME: Cell survival promoted by the Ras-MAPK
signaling pathway by transcription-dependent and -independent
mechanisms. Science. 286:1358–1362. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Scheid MP, Schubert KM and Duronio V:
Regulation of bad phosphorylation and association with Bcl-x(L) by
the MAPK/Erk kinase. J Biol Chem. 274:31108–31113. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Xing HM, Zhang SS, Weinheimer C, Kovacs A
and Muslin AJ: 14-3-3 proteins block apoptosis and differentially
regulate MAPK cascades. EMBO J. 19:349–358. 2000. View Article : Google Scholar : PubMed/NCBI
|