Genetically engineered stem cell (GESTEC)-based
therapy for treating breast cancer
Breast cancer is the most frequently diagnosed
cancer and the leading cause of cancer mortality in women,
accounting for 23% of all cancer cases and 14% of all cases of
cancer mortality (1). The breasts
are composed of fat, glandular, and connective (fibrous) tissues,
and contain several lobes which are divided into lobules that end
in milk glands. Tiny ducts run from the glands, converge, and end
in the nipple. Breast cancer changes the size or shape of the
breast and can be separated into two histopathological categories:
ductal and lobular carcinoma (2).
Additionally, these carcinomas are further divided into in
situ and invasive carcinomas according to whether the tumor is
confined to the glandular component of the organ or whether it has
invaded the stroma (3). Ductal
carcinoma represents 80% of breast cancer cases and presumably
originates from malignant epithelial cells within the ducts or
tubes that carry milk to the nipple from the breast (4). Lobular carcinoma is a less common
form of breast cancer that commences in the milk-producing lobules
of the breast (5). This type of
carcinoma is composed of acini filled with a small, round,
polygonal or cuboidal cells (6).
Breast cancer progression includes five stages
defined according to tumor size, spread to the lymph nodes, and
metastasis (spread to a more distant part of the body) (7). Stage 0 is a pre-cancerous state in
which the cancerous cells have not spread outside of the
milk-producing lobules or ducts. Lesions in this stage are also
referred to as ductal carcinoma in situ (DCIS) and lobular
carcinoma in situ (LCIS) (8). DCIS is generally categorized into the
five most common architectural subtypes, including papillary,
micropapillary, cribriform solid, and comedo (9). Stages I to III are characterized by
lesions within the breast or regional lymph nodes; these stages are
based on the size of the tumor and the spread to the lymph nodes
(10). Finally, stage IV is
metastatic cancer that has spread to other organs of the body
(i.e., lungs, bones, liver, or brain) (11). Although breast cancer is the most
frequently diagnosed cancer and the leading cause of cancer
mortality in women, if detected during the early stages it can be
treated successfully by surgery or chemotherapy (12).
Variable risk factors for breast cancer include
gender, age, female hormone exposure, ethnicity, obesity, family
history of breast cancer, genetic risk factors, and many abnormal
conditions of the breast (13,14).
Being female is the main risk factor for developing breast cancer
since women have significantly more breast cells than men.
Nevertheless, men can also develop breast cancer but they account
for <1% of all breast cancer cases (15). Clinically, breast cancer in men is
similar to that in women and is also affected by hormonal, genetic,
and environmental factors (16).
In women, cells in the breast are exposed to
growth-stimulating female hormones including estrogen (E2) and
progesterone (P4) (17). E2
stimulates breast cell division which can increase the risk of
permanent DNA damage (18). The
growth factor transforming growth factor-α (TGF-α) can also affect
cell division, and overexpression of this factor is associated with
increased cell division in breast cancer (19).
The risk of developing breast cancer increases with
age and doubles every 10 years until menopause (20). Age is the strongest risk factor for
breast cancer after gender (21).
There are also numerous genetic risk factors for breast cancer.
Numerous cases of cancer begin when one or more genes in a cell
mutate, thereby producing an abnormal protein or no protein at all
(22). Production of an abnormal
protein and lack of protein production may cause cells to divide
uncontrollably and become cancerous (23). The normal function of genetic risk
factor-associated genes is the suppression of tumorigenesis. Genes
associated with a high risk of developing breast cancer include
BRCA1, BRCA2, p53, PTEN, STK11,
CHEK2, and ATM(24,25).
Finally, various other factors such as medical history, life style,
dense breast tissue, alcohol intake, and smoking can promote the
development of breast cancer (26).
Mutation of breast cancer type 1 and 2
susceptibility proteins (BRCA1 and BRCA2) cause most hereditary
breast or ovarian cancer syndromes. BRCA gene-associated
mutations might also be caused by Li-Fraumeni-like syndrome (LFS)
(27,28). Mutation of these genes confers a
43–84% risk of breast cancer by the age of 50–70 in women (29,30).
It is now clear that the normal protein products of BRCA1 and BRCA2
are tumor suppressors (31).
BRCA1 is located on chromosome 17. The BRCA1 protein acts as
a hub protein that promotes genomic stability and DNA repair by its
involvement in homologous recombination and nucleotide excision
repair, DNA damage response and cell cycle check point control,
chromatin remodeling, transcriptional regulation, and protein
ubiquitylation (32). BRCA2
is located on chromosome 13. The BRCA2 protein plays an important
role in maintaining genomic stability via homologous recombination,
both during meiosis and repair of double-strand breaks (33). Both BRCA1 and BRCA2 mutations have
been found more often in patients with high grade breast cancer
compared to age-matched control patients (34). However, tumors with BRCA1 mutations
have high mitotic counts and ones with BRCA2 mutations mostly
contain less tubular structures. Furthermore, BRCA2 mutations are
associated with a more extensive intraductal component than BRCA1
mutations, and increase the risks for certain childhood tumors
(35).
Activated CHEK2 phosphorylates critical cell cycle
proteins that results in the stabilization of p53 and the
inhibition of Cdc25C phosphatase, leading to G1 cell-cycle arrest
along with the prevention of entry into mitosis and the activation
of DNA repair (64). This kinase
also phosphorylates BRCA1, resulting in get back DNA damage
(65). Mutations in the
CHEK2 gene, including truncation variant 1100delC, have been
reported to increase breast cancer risk by up to two-fold and may
vary according to the Li-Fraumeni syndrome as well as breast cancer
(66). Susceptibility to cancer
due to this gene variation was first described in 1999, and the
products of the CHEK2 and ATM genes are now known to
be involved in p53 inactivation (67).
Breast cancer is almost always treated with surgery,
chemotherapy, radiotherapy, and hormone therapy. Surgical
procedures, called mastectomy or lumpectomy, have a role in
treating most patients with breast cancer (68). During these procedures, the
cancerous lesions are removed from the breast along with some of
the surrounding tissue. For this reason, the number of patients who
receive breast implants after undergoing a mastectomy has increased
(69). After performing surgery to
treat breast cancer, radiation is used as an adjuvant treatment
depending on the disease stage (70).
Hormonal therapy, including administration of
tamoxifen, raloxifene (a selective estrogen receptor modulator,
SERM), and aromatase inhibitors (AIs), increases the survival rate
of hormone-sensitive breast cancer patients (71). Treatment of breast cancer patients
with AIs is more effective than tamoxifen although patients
receiving AIs have a higher prevalence of osteoporosis, bone
fractures, and musculoskeletal symptoms, particularly joint pain
and stiffness (72).
Chemotherapy is given to slow or stop the growth of
cancer cells. For this, 5-fluorouracil (5-FU), cyclophosphamide,
methotrexate, anthracyclines, trastuzumab, and taxanes are
primarily used (73). If the
breast cancer is positive for human epidermal growth factor
receptor 2 (HER-2), it is treated with trastuzumab (herceptin)
which targets the HER-2 oncogene (74). 5-FU has also been the preferred
chemotherapeutic agent for treating a majority of solid tumors,
including gastric and colon cancers (75). However, serious side-effects such
as alopecia, anesthesia, diarrhea, and arthralgia, as well as high
dose requirements have limited the use of these chemotherapeutic
agents (76).
Gene-direct enzyme/prodrug therapy (GEPT), or
suicide gene therapy, aims to improve the therapeutic efficacy of
conventional cancer radio- and chemotherapy without side-effects
(77,78). This system is a novel approach with
the potential to selectively eradicate tumor cells (79). For this, an exogenous suicide
enzyme gene is delivered to tumor cells (80). GEPT systems most often involve the
use of a viral vector (adeno-, lenti-, or retroviral vectors) to
deliver a gene not normally found in mammalian cells that produces
enzymes which, when expressed, can convert a relatively non-toxic
prodrug into a toxic agent (81,82).
GEPT systems involve two separate events: direct
cell death and cell death via the bystander effect (83). The viral vectors transfected into
the target tumor cells induced cell death (84). Direct cell death is caused by
expression of the viral DNA in the targeted tumor cells (85). Next, cell death via the bystander
effect is induced by the gene transfer of a viral or bacterial
enzyme into targeted tumor cells. The enzymes convert an inactive
prodrug into a short-lived toxic metabolite, leading to the death
of cells surrounding the targeted tumor cells (86). Prodrugs can be defined as
pharmacologically inactive derivatives which require chemical
transformation for the release or conversion into the active drug
(87). A suicide enzyme converts
the administered non-toxic prodrug into an active drug which
subsequently kills tumor cells but not normal tissues (88). Several types of suicidal genes have
been studied and used for therapeutic purposes (82).
An advantage of these GEPT systems derives from the
local bystander effect through which more wide-spread cell death is
achieved without the need to express the gene in all cells
(89). This is due to the ability
of the toxic metabolite to diffuse freely across cells membranes or
via gap junctions (90).
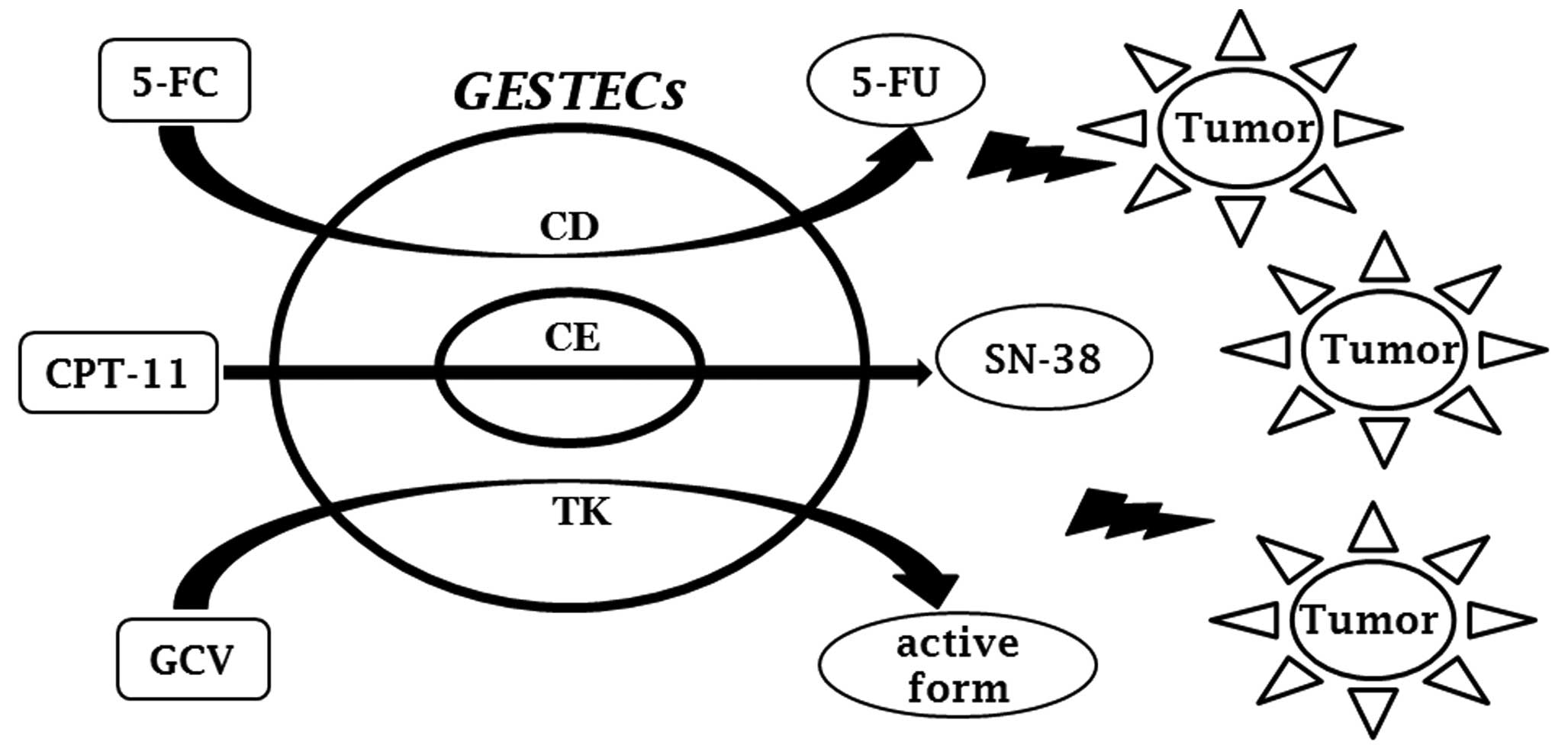
Currently, a large number of enzyme/prodrug systems have been
developed for GEPT. These include the cytosine
deaminase/5-fluorocytosine (CD/5-FC), carboxyl esterase/irinotecan
(CE/CPT-11), and thymidine kinase/ganciclovir (TK/GCV) systems
(91).
The CD/5-FC system is very effective for treating
human cancers as non-toxic 5-FC systemically administered can be
converted into the cytotoxic agent 5-FU by the CD gene
product located in the vicinity of the cancer (99). Deamination of the 5-FC prodrug by
CD results in the formation of two toxic metabolites:
5-fluorodeoxyuridine monophosphate (FdUMP) and 5-fluorouridine
triphosphate (FURTP). FdUMP is a potent inhibitor of thymidylate
synthetase (TS) which is an enzyme essential for DNA synthesis.
This compound impairs DNA synthesis and promotes apoptosis in
bacteria and tumor cells (100,101).
CE enzyme is a serine esterase found in a variety of
tissues from numerous mammalian species (102). This enzyme plays a critical role
in increasing the solubility and bio-availability of therapeutic
agents (103,104). It is cleaved into the bulky
piperidino sidechain of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]
carbonyl-oxycamptothecin (irinotecan or CPT-11). The anti-cancer
agent CPT-11 is a prodrug that is activated by CE to generate the
active form 7-ethyl-10-hydroxycamptothecin (SN-38) (105,106). SN-38 is a strong mammalian
topoisomerase I inhibitor that is 1,000-fold more potent than
CPT-11. This agent induces the accumulation of double-strand DNA
breaks in actively dividing cancer cells (107).
The most common GEPT uses the herpes simplex type-1
thymidine kinase enzyme (HSV-TK) in conjunction with a variety of
guanosine-based prodrugs, compounds originally developed as
antiviral agents (108). The
HSV-TK enzyme converts to the prodrug into its monophosphate form,
GCV, which is then further converted into the toxic triphosphates
form, an intermediary metabolite, by cellular enzymes (109,110). These actions cause cell death by
inhibiting the incorporation of dGTP into DNA without preventing
progression through the S-phase; chain elongation is also inhibited
(111).
Toxicity of anticancer agents to normal cells is a
major limitation of breast cancer therapy (112). Therefore, stem cells have
recently received a great deal of attention for their clinical and
therapeutic potential to treat breast cancer. Stem cells are
capable of continuous self-renewal and differentiation (113,114). A variety of stem cells, such as
neural stem cells (NSCs), neural progenitor cells, and mesenchymal
stem cells (MSCs) from bone marrow or adipose tissue, have been
found to exert tumor-tropism effects (115). This ability makes these cells
attractive for use as targeted delivery vectors for antitumor
therapies (87,88,99,116–117). The tumor-tropism effects of stem
cells are mediated by multiple cell-surface and secreted proteins,
and candidate cytokines/receptors including stromal cell-derived
factor-1 (SDF-1)/CXCR4, stem cell factor (SCF)/c-Kit, hepatocyte
growth factor (HGF)/Met, vascular endothelial growth factor
(VEGF)/VEGF receptor (VEGFR), monocyte chemoattractant protein-1
(MCP-1)/CCP, and high-mobility group box1 (HMGB1)/RAGE (87,88,99,117,118). In addition, NSCs appear to
migrate to cancer cells more efficiently compared to MSCs. Although
both NSCs and MSCs have a tumor tropic effect, NSCs (50–100% of
total cell number) were proven to display greater tropism towards
tumor cells than MSCs (40–75% of total cell number) (119).
The field of NSC research in recent years has seen
major advances and efforts have been made to develop their use in
potential stem cell-based transplantation therapies (120). NSCs can be used to generate all
major mature neural cell types such as neurons, oligodendrocytes,
glial cells and cells of neuronal lineages (121). The fetal brain, characterized by
active neurogenesis, is thought to be a promising source of
therapeutic NSCs (122). Previous
studies have shown that NSCs derived from human fetal telencephalon
can be used for GESTEC-based therapy for treating several different
cancers as well as brain diseases (87,88,99).
As this is based on a GEPT system, it involves the expression of
several suicide enzymes (Fig. 1).
In previous studies, GESTECs were immortalized by using retrovirus
v-myc and suicide genes such as CD, CE, and
TK. Therapeutic efficacy has been assessed by monitoring
tumor-tropism in a brain cancer animal model (123).
Breast cancer is the leading cause of cancer related
mortality among women worldwide. Several gene mutations lead to the
development of breast cancer including ductal and lobular breast
carcinoma. Chemo-, hormone-, and radiotherapies are used to treat
breast cancer but these therapies are associated with many
side-effects. For this reason, GEPT systems have been examined as a
novel anticancer therapeutic approach with the potential to
selectively eradicate tumor cells. Prodrugs used for GEPT are
primarily antimetabolites that require cell cycling (S phase) to
induce cytotoxicity and are not active against normal cells. These
systems may involve the use of NSCs which express suicide genes and
have the ability to selectively migrate to tumors. In summary,
GESTECs using GEPT systems may be an effective new modality for
treating breast cancer as well as brain tumors without inducing
injurious effects commonly associated with more conventional
anticancer therapies.
This study was supported by two
National Research Foundation of Korea (NRF) grants funded by the
Ministry of Education, Science and Technology (MEST) of the Korean
Government (no. 2010-0003093 and 2011-0015385).
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Vincent-Salomon A and Thiery JP: Host
microenvironment in breast cancer development:
epithelial-mesenchymal transition in breast cancer development.
Breast Cancer Res. 5:101–106. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Middleton LP, Tressera F, Sobel ME, Bryant
BR, Alburquerque A, Grases P and Merino MJ: Infiltrating
micropapillary carcinoma of the breast. Mod Pathol. 12:499–504.
1999.PubMed/NCBI
|
|
4
|
Goldstein NS, Vicini FA, Kestin LL and
Thomas M: Differences in the pathologic features of ductal
carcinoma in situ of the breast based on patient age. Cancer.
88:2553–2560. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li CI, Anderson BO, Porter P, Holt SK,
Daling JR and Moe RE: Changing incidence rate of invasive lobular
breast carcinoma among older women. Cancer. 88:2561–2569. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lakhani SR, Audretsch W, Cleton-Jensen AM,
Cutuli B, Ellis I, Eusebi V, Greco M, Houslton RS, Kuhl CK, Kurtz
J, et al: The management of lobular carcinoma in situ (LCIS). Is
LCIS the same as ductal carcinoma in situ (DCIS)? Eur J Cancer.
42:2205–2211. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cianfrocca M and Goldstein LJ: Prognostic
and predictive factors in early-stage breast cancer. Oncologist.
9:606–616. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lehman CD, Gatsonis C, Kuhl CK, Hendrick
RE, Pisano ED, Hanna L, Peacock S, Smazal SF, Maki DD, Julian TB,
et al: MRI evaluation of the contralateral breast in women with
recently diagnosed breast cancer. N Engl J Med. 356:1295–1303.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Skinner KA and Silverstein MJ: The
management of ductal carcinoma in situ of the breast. Endocr Relat
Cancer. 8:33–45. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Singletary SE and Connolly JL: Breast
cancer staging: working with the sixth edition of the AJCC Cancer
Staging Manual. CA Cancer J Clin. 56:37–50. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Askoxylakis V, Thieke C, Pleger ST, Most
P, Tanner J, Lindel K, Katus HA, Debus J and Bischof M: Long-term
survival of cancer patients compared to heart failure and stroke: a
systematic review. BMC Cancer. 10:1052010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lv H, Pan G, Zheng G, Wu X, Ren H, Liu Y
and Wen J: Expression and functions of the repressor element 1
(RE-1)-silencing transcription factor (REST) in breast cancer. J
Cell Biochem. 110:968–974. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vogel KJ, Atchley DP, Erlichman J, Broglio
KR, Ready KJ, Valero V, Amos CI, Hortobagyi GN, Lu KH and Arun B:
BRCA1 and BRCA2 genetic testing in Hispanic patients: mutation
prevalence and evaluation of the BRCAPRO risk assessment model. J
Clin Oncol. 25:4635–4641. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Olsson H: Tumour biology of a breast
cancer at least partly reflects the biology of the
tissue/epithelial cell of origin at the time of initiation - a
hypothesis. J Steroid Biochem Mol Biol. 74:345–350. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giordano SH, Cohen DS, Buzdar AU, Perkins
G and Hortobagyi GN: Breast carcinoma in men: a population-based
study. Cancer. 101:51–57. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Iredale R, Brain K, Williams B, France E
and Gray J: The experiences of men with breast cancer in the United
Kingdom. Eur J Cancer. 42:334–341. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yager JD and Davidson NE: Estrogen
carcinogenesis in breast cancer. N Engl J Med. 354:270–282. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nass SJ and Davidson NE: The biology of
breast cancer. Hematol Oncol Clin North Am. 13:311–332. 1999.
View Article : Google Scholar
|
|
19
|
Antonova L, Aronson K and Mueller CR:
Stress and breast cancer: from epidemiology to molecular biology.
Breast Cancer Res. 13:2082010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McTiernan A: Behavioral risk factors in
breast cancer: can risk be modified? Oncologist. 8:326–334. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Friedenreich CM, Courneya KS and Bryant
HE: Influence of physical activity in different age and life
periods on the risk of breast cancer. Epidemiology. 12:604–612.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
MacConaill LE, Campbell CD, Kehoe SM, Bass
AJ, Hatton C, Niu L, Davis M, Yao K, Hanna M, Mondal C, et al:
Profiling critical cancer gene mutations in clinical tumor samples.
PLoS One. 4:e78872009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martin AM and Weber BL: Genetic and
hormonal risk factors in breast cancer. J Natl Cancer Inst.
92:1126–1135. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Plak K, Czarnecka AM, Krawczyk T, Golik P
and Bartnik E: Breast cancer as a mitochondrial disorder (Review).
Oncol Rep. 21:845–851. 2009.PubMed/NCBI
|
|
25
|
Lynch HT, Silva E, Snyder C and Lynch JF:
Hereditary breast cancer: part I. Diagnosing hereditary breast
cancer syndromes. Breast J. 14:3–13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Singletary SE: Rating the risk factors for
breast cancer. Ann Surg. 237:474–482. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kadouri L, Hubert A, Rotenberg Y,
Hamburger T, Sagi M, Nechushtan C, Abeliovich D and Peretz T:
Cancer risks in carriers of the BRCA1/2 Ashkenazi founder
mutations. J Med Genet. 44:467–471. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Campeau PM, Foulkes WD and Tischkowitz MD:
Hereditary breast cancer: new genetic developments, new therapeutic
avenues. Hum Genet. 124:31–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
King MC, Marks JH and Mandell JB: Breast
and ovarian cancer risks due to inherited mutations in BRCA1 and
BRCA2. Science. 302:643–646. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thorslund T and West SC: BRCA2: a
universal recombinase regulator. Oncogene. 26:7720–7730. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Welcsh PL and King MC: BRCA1 and BRCA2 and
the genetics of breast and ovarian cancer. Hum Mol Genet.
10:705–713. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Narod SA and Foulkes WD: BRCA1 and BRCA2:
1994 and beyond. Nat Rev Cancer. 4:665–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Thorslund T, Esashi F and West SC:
Interactions between human BRCA2 protein and the meiosis-specific
recombinase DMC1. EMBO J. 26:2915–2922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Verhoog LC, Berns EM, Brekelmans CT,
Seynaeve C, Meijers-Heijboer EJ and Klijn JG: Prognostic
significance of germline BRCA2 mutations in hereditary breast
cancer patients. J Clin Oncol. 18:119S–124S. 2000.PubMed/NCBI
|
|
35
|
Loman N, Johannsson O, Bendahl P, Dahl N,
Einbeigi Z, Gerdes A, Borg A and Olsson H: Prognosis and clinical
presentation of BRCA2-associated breast cancer. Eur J Cancer.
36:1365–1373. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lim LY, Vidnovic N, Ellisen LW and Leong
CO: Mutant p53 mediates survival of breast cancer cells. Br J
Cancer. 101:1606–1612. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bergamaschi D, Gasco M, Hiller L, Sullivan
A, Syed N, Trigiante G, Yulug I, Merlano M, Numico G, Comino A, et
al: p53 polymorphism influences response in cancer chemotherapy via
modulation of p73-dependent apoptosis. Cancer Cell. 3:387–402.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Geisler S, Lonning PE, Aas T, Johnsen H,
Fluge O, Haugen DF, Lillehaug JR, Akslen LA and Borresen-Dale AL:
Influence of TP53 gene alterations and c-erbB-2 expression on the
response to treatment with doxorubicin in locally advanced breast
cancer. Cancer Res. 61:2505–2512. 2001.PubMed/NCBI
|
|
39
|
Beroud C and Soussi T: p53 gene mutation:
software and database. Nucleic Acids Res. 26:200–204. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vousden KH and Lane DP: p53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007. View Article : Google Scholar
|
|
41
|
Olivier M, Petitjean A, Marcel V, Petre A,
Mounawar M, Plymoth A, de Fromentel CC and Hainaut P: Recent
advances in p53 research: an interdisciplinary perspective. Cancer
Gene Ther. 16:1–12. 2009. View Article : Google Scholar
|
|
42
|
Hohenstein P and Giles RH: BRCA1: a
scaffold for p53 response? Trends Genet. 19:489–494. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Coutant C, Rouzier R, Qi Y, Lehmann-Che J,
Bianchini G, Iwamoto T, Hortobagyi GN, Symmans WF, Uzan S, Andre F,
de The H and Pusztai L: Distinct p53 gene signatures are needed to
predict prognosis and response to chemotherapy in ER-positive and
ER-negative breast cancers. Clin Cancer Res. 17:2591–2601. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yamashita H, Nishio M, Toyama T, Sugiura
H, Zhang Z, Kobayashi S and Iwase H: Coexistence of HER2
over-expression and p53 protein accumulation is a strong prognostic
molecular marker in breast cancer. Breast Cancer Res. 6:R24–30.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
45
|
Feki A and Irminger-Finger I: Mutational
spectrum of p53 mutations in primary breast and ovarian tumors.
Crit Rev Oncol Hematol. 52:103–116. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang J, Ren Y, Wang L, Li B, Chen Y, Zhao
W, Xu W, Li T and Dai F: PTEN mutation spectrum in breast cancers
and breast hyperplasia. J Cancer Res Clin Oncol. 136:1303–1311.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wallace JA, Li F, Leone G and Ostrowski
MC: Pten in the breast tumor microenvironment: modeling
tumor-stroma coevolution. Cancer Res. 71:1203–1207. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tokunaga E, Oki E, Kimura Y, Yamanaka T,
Egashira A, Nishida K, Koga T, Morita M, Kakeji Y and Maehara Y:
Coexistence of the loss of heterozygosity at the PTEN locus and
HER2 overexpression enhances the Akt activity thus leading to a
negative progesterone receptor expression in breast carcinoma.
Breast Cancer Res Treat. 101:249–257. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Marty B, Maire V, Gravier E, Rigaill G,
Vincent-Salomon A, Kappler M, Lebigot I, Djelti F, Tourdes A,
Gestraud P, et al: Frequent PTEN genomic alterations and activated
phosphatidylinositol 3-kinase pathway in basal-like breast cancer
cells. Breast Cancer Res. 10:R1012008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shaw RJ and Cantley LC: Ras, PI(3)K and
mTOR signalling controls tumour cell growth. Nature. 441:424–430.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
DeGraffenried LA, Fulcher L, Friedrichs
WE, Grunwald V, Ray RB and Hidalgo M: Reduced PTEN expression in
breast cancer cells confers susceptibility to inhibitors of the PI3
kinase/Akt pathway. Ann Oncol. 15:1510–1516. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tate G, Suzuki T and Mitsuya T: Mutation
of the PTEN gene in a human hepatic angiosarcoma. Cancer Genet
Cytogenet. 178:160–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Petrocelli T and Slingerland JM: PTEN
deficiency: a role in mammary carcinogenesis. Breast Cancer Res.
3:356–360. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lee JS, Kim HS, Kim YB, Lee MC, Park CS
and Min KW: Reduced PTEN expression is associated with poor outcome
and angiogenesis in invasive ductal carcinoma of the breast. Appl
Immunohistochem Mol Morphol. 12:205–210. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Steelman LS, Navolanic PM, Sokolosky ML,
Taylor JR, Lehmann BD, Chappell WH, Abrams SL, Wong EW, Stadelman
KM, Terrian DM, et al: Suppression of PTEN function increases
breast cancer chemotherapeutic drug resistance while conferring
sensitivity to mTOR inhibitors. Oncogene. 27:4086–4095. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Tanaka M, Koul D, Davies MA, Liebert M,
Steck PA and Grossman HB: MMAC1/PTEN inhibits cell growth and
induces chemosensitivity to doxorubicin in human bladder cancer
cells. Oncogene. 19:5406–5412. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bates RC, Edwards NS, Burns GF and Fisher
DE: A CD44 survival pathway triggers chemoresistance via lyn kinase
and phosphoinositide 3-kinase/Akt in colon carcinoma cells. Cancer
Res. 61:5275–5283. 2001.PubMed/NCBI
|
|
58
|
Nevanlinna H and Bartek J: The CHEK2 gene
and inherited breast cancer susceptibility. Oncogene. 25:5912–5919.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shaag A, Walsh T, Renbaum P, Kirchhoff T,
Nafa K, Shiovitz S, Mandell JB, Welcsh P, Lee MK, Ellis N, et al:
Functional and genomic approaches reveal an ancient CHEK2 allele
associated with breast cancer in the Ashkenazi Jewish population.
Hum Mol Genet. 14:555–563. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Abraham RT: PI 3-kinase related kinases:
‘big’ players in stress-induced signaling pathways. DNA Repair.
3:883–887. 2004.
|
|
61
|
Angele S, Romestaing P, Moullan N,
Vuillaume M, Chapot B, Friesen M, Jongmans W, Cox DG, Pisani P,
Gerard JP and Hall J: ATM haplotypes and cellular response to DNA
damage: association with breast cancer risk and clinical
radiosensitivity. Cancer Res. 63:8717–8725. 2003.PubMed/NCBI
|
|
62
|
McKinnon PJ: ATM and ataxia
telangiectasia. EMBO Rep. 5:772–776. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Milne RL: Variants in the ATM gene and
breast cancer susceptibility. Genome Med. 1:122009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lee JH and Paull TT: Direct activation of
the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science.
304:93–96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Consortium TCBCC-C: CHEK2*1100delC and
susceptibility to breast cancer: a collaborative analysis involving
10,860 breast cancer cases and 9,065 controls from 10 studies. Am J
Hum Genet. 74:1175–1182. 2004.
|
|
66
|
Kilpivaara O, Vahteristo P, Falck J,
Syrjakoski K, Eerola H, Easton D, Bartkova J, Lukas J, Heikkila P,
Aittomaki K, et al: CHEK2 variant I157T may be associated with
increased breast cancer risk. Int J Cancer. 111:543–547. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kleibl Z, Havranek O, Novotny J, Kleiblova
P, Soucek P and Pohlreich P: Analysis of CHEK2 FHA domain in Czech
patients with sporadic breast cancer revealed distinct rare genetic
alterations. Breast Cancer Res Treat. 112:159–164. 2008. View Article : Google Scholar
|
|
68
|
Le GM, O’Malley CD, Glaser SL, Lynch CF,
Stanford JL, Keegan TH and West DW: Breast implants following
mastectomy in women with early-stage breast cancer: prevalence and
impact on survival. Breast Cancer Res. 7:R184–193. 2005.PubMed/NCBI
|
|
69
|
Brown SL: Epidemiology of silicone-gel
breast implants. Epidemiology. 13:S34–39. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Bese NS, Kiel K, El-Gueddari Bel K,
Campbell OB, Awuah B and Vikram B: Radiotherapy for breast cancer
in countries with limited resources: program implementation and
evidence-based recommendations. Breast J. 12:S96–102.
2006.PubMed/NCBI
|
|
71
|
Goss PE, Ingle JN, Martino S, Robert NJ,
Muss HB, Piccart MJ, Castiglione M, Tu D, Shepherd LE, Pritchard
KI, et al: Randomized trial of letrozole following tamoxifen as
extended adjuvant therapy in receptor-positive breast cancer:
updated findings from NCIC CTG MA.17. J Natl Cancer Inst.
97:1262–1271. 2005. View Article : Google Scholar
|
|
72
|
Crew KD, Greenlee H, Capodice J, Raptis G,
Brafman L, Fuentes D, Sierra A and Hershman DL: Prevalence of joint
symptoms in postmenopausal women taking aromatase inhibitors for
early-stage breast cancer. J Clin Oncol. 25:3877–3883. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zoli W, Ulivi P, Tesei A, Fabbri F,
Rosetti M, Maltoni R, Giunchi DC, Ricotti L, Brigliadori G, Vannini
I and Amadori D: Addition of 5-fluorouracil to
doxorubicin-paclitaxel sequence increases caspase-dependent
apoptosis in breast cancer cell lines. Breast Cancer Res.
7:R681–689. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
McArthur HL, Mahoney KM, Morris PG, Patil
S, Jacks LM, Howard J, Norton L and Hudis CA: Adjuvant trastuzumab
with chemotherapy is effective in women with small, node-negative,
HER2-positive breast cancer. Cancer. 117:5461–5468. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Piccart M: The role of taxanes in the
adjuvant treatment of early stage breast cancer. Breast Cancer Res
Treat. 79(Suppl 1): S25–34. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Weiss RB, Woolf SH, Demakos E, Holland JF,
Berry DA, Falkson G, Cirrincione CT, Robbins A, Bothun S, Henderson
IC and Norton L: Natural history of more than 20 years of
node-positive primary breast carcinoma treated with
cyclophosphamide, methotrexate, and fluorouracil-based adjuvant
chemotherapy: a study by the Cancer and Leukemia Group B. J Clin
Oncol. 21:1825–1835. 2003.
|
|
77
|
Hedley D, Ogilvie L and Springer C:
Carboxypeptidase-G2-based gene-directed enzyme-prodrug therapy: a
new weapon in the GDEPT armoury. Nat Rev Cancer. 7:870–879. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Nawa A, Tanino T, Luo C, Iwaki M, Kajiyama
H, Shibata K, Yamamoto E, Ino K, Nishiyama Y and Kikkawa F: Gene
directed enzyme prodrug therapy for ovarian cancer: could GDEPT
become a promising treatment against ovarian cancer? Anticancer
Agents Med Chem. 8:232–239. 2008. View Article : Google Scholar
|
|
79
|
Greco O and Dachs GU: Gene directed
enzyme/prodrug therapy of cancer: historical appraisal and future
prospectives. J Cell Physiol. 187:22–36. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
McKeown SR, Ward C and Robson T:
Gene-directed enzyme prodrug therapy: a current assessment. Curr
Opin Mol Ther. 6:421–435. 2004.PubMed/NCBI
|
|
81
|
Voeks D, Martiniello-Wilks R, Madden V,
Smith K, Bennetts E, Both GW and Russell PJ: Gene therapy for
prostate cancer delivered by ovine adenovirus and mediated by
purine nucleoside phosphorylase and fludarabine in mouse models.
Gene Ther. 9:759–768. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Anderson WF: Gene therapy scores against
cancer. Nat Med. 6:862–863. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
83
|
Chung-Faye GA, Kerr DJ, Young LS and
Searle PF: Gene therapy strategies for colon cancer. Mol Med Today.
6:82–87. 2000. View Article : Google Scholar
|
|
84
|
Schepelmann S, Hallenbeck P, Ogilvie LM,
Hedley D, Friedlos F, Martin J, Scanlon I, Hay C, Hawkins LK,
Marais R and Springer CJ: Systemic gene-directed enzyme prodrug
therapy of hepatocellular carcinoma using a targeted adenovirus
armed with carboxypeptidase G2. Cancer Res. 65:5003–5008. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chung-Faye GA, Chen MJ, Green NK, Burton
A, Anderson D, Mautner V, Searle PF and Kerr DJ: In vivo gene
therapy for colon cancer using adenovirus-mediated, transfer of the
fusion gene cytosine deaminase and uracil
phosphoribosyltransferase. Gene Ther. 8:1547–1554. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hernandez-Alcoceba R, Sangro B and Prieto
J: Gene therapy of liver cancer. World J Gastroenterol.
12:6085–6097. 2006.
|
|
87
|
Yi BR, O SN, Kang NH, Hwang KA, Kim SU,
Jeung EB, Kim YB, Heo GJ and Choi KC: Genetically engineered stem
cells expressing cytosine deaminase and interferon-β migrate to
human lung cancer cells and have potentially therapeutic anti-tumor
effects. Int J Oncol. 39:833–839. 2011.
|
|
88
|
Yi BR, Kang NH, Hwang KA, Kim SU, Jeung EB
and Choi KC: Antitumor therapeutic effects of cytosine deaminase
and interferon-β against endometrial cancer cells using genetically
engineered stem cells in vitro. Anticancer Res. 31:2853–2861.
2011.
|
|
89
|
Dachs GU, Hunt MA, Syddall S, Singleton DC
and Patterson AV: Bystander or no bystander for gene directed
enzyme prodrug therapy. Molecules. 14:4517–4545. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Pfeifer A and Verma IM: Gene therapy:
promises and problems. Annu Rev Genomics Hum Genet. 2:177–211.
2001. View Article : Google Scholar
|
|
91
|
Shah K: Mesenchymal stem cells engineered
for cancer therapy. Adv Drug Deliv Rev. 64:739–748. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ramnaraine M, Pan W, Goblirsch M, Lynch C,
Lewis V, Orchard P, Mantyh P and Clohisy DR: Direct and bystander
killing of sarcomas by novel cytosine deaminase fusion gene. Cancer
Res. 63:6847–6854. 2003.PubMed/NCBI
|
|
93
|
Nyati MK, Symon Z, Kievit E, Dornfeld KJ,
Rynkiewicz SD, Ross BD, Rehemtulla A and Lawrence TS: The potential
of 5-fluorocytosine/cytosine deaminase enzyme prodrug gene therapy
in an intrahepatic colon cancer model. Gene Ther. 9:844–849.
2002.PubMed/NCBI
|
|
94
|
Ireton GC, Black ME and Stoddard BL:
Crystallization and preliminary X-ray analysis of bacterial
cytosine deaminase. Acta Crystallogr D Biol Crystallogr.
57:1643–1645. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ireton GC, Black ME and Stoddard BL: The
1.14 A crystal structure of yeast cytosine deaminase: evolution of
nucleotide salvage enzymes and implications for genetic
chemotherapy. Structure. 11:961–972. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Denny WA: Prodrugs for Gene-Directed
Enzyme-Prodrug Therapy (Suicide Gene Therapy). J Biomed Biotechnol.
2003:48–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ireton GC, McDermott G, Black ME and
Stoddard BL: The structure of Escherichia coli cytosine deaminase.
J Mol Biol. 315:687–697. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Fuchita M, Ardiani A, Zhao L, Serve K,
Stoddard BL and Black ME: Bacterial cytosine deaminase mutants
created by molecular engineering show improved
5-fluorocytosine-mediated cell killing in vitro and in vivo. Cancer
Res. 69:4791–4799. 2009. View Article : Google Scholar
|
|
99
|
Kim KY, Kim SU, Leung PC, Jeung EB and
Choi KC: Influence of the prodrugs 5-fluorocytosine and CPT-11 on
ovarian cancer cells using genetically engineered stem cells:
tumor-tropic potential and inhibition of ovarian cancer cell
growth. Cancer Sci. 101:955–962. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Chen JK, Hu LJ, Wang D, Lamborn KR and
Deen DF: Cytosine deaminase/5-fluorocytosine exposure induces
bystander and radiosensitization effects in hypoxic glioblastoma
cells in vitro. Int J Radiat Oncol Biol Phys. 67:1538–1547. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Miller CR, Williams CR, Buchsbaum DJ and
Gillespie GY: Intratumoral 5-fluorouracil produced by cytosine
deaminase/5-fluorocytosine gene therapy is effective for
experimental human glioblastomas. Cancer Res. 62:773–780.
2002.PubMed/NCBI
|
|
102
|
Humerickhouse R, Lohrbach K, Li L, Bosron
WF and Dolan ME: Characterization of CPT-11 hydrolysis by human
liver carboxylesterase isoforms hCE-1 and hCE-2. Cancer Res.
60:1189–1192. 2000.PubMed/NCBI
|
|
103
|
Bencharit S, Morton CL, Howard-Williams
EL, Danks MK, Potter PM and Redinbo MR: Structural insights into
CPT-11 activation by mammalian carboxylesterases. Nat Struct Biol.
9:337–342. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bodor N and Buchwald P: Soft drug design:
general principles and recent applications. Med Res Rev. 20:58–101.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wadkins RM, Morton CL, Weeks JK, Oliver L,
Wierdl M, Danks MK and Potter PM: Structural constraints affect the
metabolism of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]
carbonyloxycamptothecin (CPT-11) by carboxylesterases. Mol
Pharmacol. 60:355–362. 2001.PubMed/NCBI
|
|
106
|
Wierdl M, Morton CL, Weeks JK, Danks MK,
Harris LC and Potter PM: Sensitization of human tumor cells to
CPT-11 via adenoviral-mediated delivery of a rabbit liver
carboxylesterase. Cancer Res. 61:5078–5082. 2001.PubMed/NCBI
|
|
107
|
Kojima A, Hackett NR, Ohwada A and Crystal
RG: In vivo human carboxylesterase cDNA gene transfer to activate
the prodrug CPT-11 for local treatment of solid tumors. J Clin
Invest. 101:1789–1796. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
van Dillen IJ, Mulder NH, Vaalburg W, de
Vries EF and Hospers GA: Influence of the bystander effect on
HSV-tk/GCV gene therapy. A review. Curr Gene Ther. 2:307–322.
2002.PubMed/NCBI
|
|
109
|
Gerolami R, Cardoso J, Lewin M, Bralet MP,
Sa Cunha A, Clement O, Brechot C and Tran PL: Evaluation of HSV-tk
gene therapy in a rat model of chemically induced hepatocellular
carcinoma by intratumoral and intrahepatic artery routes. Cancer
Res. 60:993–1001. 2000.PubMed/NCBI
|
|
110
|
Fillat C, Carrio M, Cascante A and Sangro
B: Suicide gene therapy mediated by the Herpes Simplex virus
thymidine kinase gene/Ganciclovir system: fifteen years of
application. Curr Gene Ther. 3:13–26. 2003.PubMed/NCBI
|
|
111
|
Huang Q, Pu P, Xia Z and You Y: Exogenous
wt-p53 enhances the antitumor effect of HSV-TK/GCV on C6 glioma
cells. J Neurooncol. 82:239–248. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Cowen RL, Williams JC, Emery S, Blakey D,
Darling JL, Lowenstein PR and Castro MG: Adenovirus vector-mediated
delivery of the prodrug-converting enzyme carboxypeptidase G2 in a
secreted or GPI-anchored form: High-level expression of this active
conditional cytotoxic enzyme at the plasma membrane. Cancer Gene
Ther. 9:897–907. 2002. View Article : Google Scholar
|
|
113
|
Muller FJ, Snyder EY and Loring JF: Gene
therapy: can neural stem cells deliver? Nat Rev Neurosci. 7:75–84.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Kim SU, Jeung EB, Kim YB, Cho MH and Choi
KC: Potential tumor-tropic effect of genetically engineered stem
cells expressing suicide enzymes to selectively target invasive
cancer in animal models. Anticancer Res. 31:1249–1258. 2011.
|
|
115
|
Tang Y, Shah K, Messerli SM, Snyder E,
Breakefield X and Weissleder R: In vivo tracking of neural
progenitor cell migration to glioblastomas. Hum Gene Ther.
14:1247–1254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Power AT and Bell JC: Cell-based delivery
of oncolytic viruses: a new strategic alliance for a biological
strike against cancer. Mol Ther. 15:660–665. 2007.PubMed/NCBI
|
|
117
|
Kim KY, Yi BR, Lee HR, Kang NH, Jeung EB,
Kim SU and Choi KC: Stem cells with fused gene expression of
cytosine deaminase and interferon-β migrate to human gastric cancer
cells and result in synergistic growth inhibition for potential
therapeutic use. Int J Oncol. 40:1097–1104. 2012.PubMed/NCBI
|
|
118
|
Aboody KS, Najbauer J and Danks MK: Stem
and progenitor cell-mediated tumor selective gene therapy. Gene
Ther. 15:739–752. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Gutova M, Najbauer J, Frank RT, Kendall
SE, Gevorgyan A, Metz MZ, Guevorkian M, Edmiston M, Zhao D, Glackin
CA, et al: Urokinase plasminogen activator and urokinase
plasminogen activator receptor mediate human stem cell tropism to
malignant solid tumors. Stem Cells. 26:1406–1413. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Bithell A and Williams BP: Neural stem
cells and cell replacement therapy: making the right cells. Clin
Sci. 108:13–22. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Kim SK, Kim SU, Park IH, Bang JH, Aboody
KS, Wang KC, Cho BK, Kim M, Menon LG, Black PM and Carroll RS:
Human neural stem cells target experimental intracranial
medulloblastoma and deliver a therapeutic gene leading to tumor
regression. Clin Cancer Res. 12:5550–5556. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Sohur US, Emsley JG, Mitchell BD and
Macklis JD: Adult neurogenesis and cellular brain repair with
neural progenitors, precursors and stem cells. Philos Trans R Soc
Lond B Biol Sci. 361:1477–1497. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Kim SU, Park IH, Kim TH, Kim KS, Choi HB,
Hong SH, Bang JH, Lee MA, Joo IS, Lee CS and Kim YS: Brain
transplantation of human neural stem cells transduced with tyrosine
hydroxylase and GTP cyclohydrolase 1 provides functional
improvement in animal models of Parkinson disease. Neuropathology.
26:129–140. 2006. View Article : Google Scholar
|
|
124
|
Joo KM, Park IH, Shin JY, Jin J, Kang BG,
Kim MH, Lee SJ, Jo MY, Kim SU and Nam DH: Human neural stem cells
can target and deliver therapeutic genes to breast cancer brain
metastases. Mol Ther. 17:570–575. 2009. View Article : Google Scholar : PubMed/NCBI
|