Introduction
Recent studies have revealed that cancer stem cells
(CSCs) are a source of therapy resistance, disease recurrence, and
metastasis to other organs (1–3). At
least two types of CSCs, dormant (dCSC) and activated (aCSC), are
involved in tumor homeostasis, which are in contrast to two types
of stem cells, dormant and activated types in normal skin,
intestine and the hematopoietic system (4). Our previous study indicated that
CD13+CD90− dCSCs of hepatocellular carcinoma
survive in hypoxic areas in marginal regions in liver after therapy
(5). In
CD13+/CD90− dCSCs, the occurrence of
double-strand breaks (DSBs) in genomic DNA, a deleterious cellular
event, and damage-induced repairs that are necessary for cellular
survival (6), reduce after therapy
presumably due to CD13/aminopeptidase N functioning as a scavenger
of reactive oxygen species (ROS) (5) and partially due to error-prone repair
such as non-homologous end-joining (NHEJ) (6,7). In
a sharp contrast, CD13−/CD90+/−
dCSCs are sensitive to therapeutic insults from chemotherapeutic
agents, which is associated with ROS-induced cell death after
chemotherapy (8); however, damage
is typically repaired though high-fidelity, error-free homologous
recombination (HR) (6,7). Thus, dCSCs may be a cause of
accumulation of deleterious mutations and should be targeted in
therapy in terms of complete eradication of malignant cells,
although hibernation therapy (the induction of dormancy) may be a
viable option dependent on the patient’s condition (7). Chemotherapy results in a shift from
aCSCs to dCSCs and accumulation of dCSCs after treatment, and
dormancy may function as a type of refuge for the survival of
malignant cells. CD13 cells play a role in the inhibition of
increase in ROS and the resultant suppression of cell death during
the process of epithelial mesenchymal transition (EMT) of
metastatic CSCs (9). The exposure
to a CSC-specific inhibitor, ubenimex, resulted in considerable
eradication of malignant cells in vivo, indicating an
apparent benefit in the combination of conventional chemotherapy
and a CSC-specific inhibitor.
Here, we performed transcriptome analysis for coding
mRNAs and non-coding microRNAs (miRs) in
CD13+/CD90− dCSCs. This study allowed us to
identify several pathways, which play a role in fundamental
mechanisms in above-mentioned potentially malignant phenotype, and
provided further clues for identification of molecular targets in
therapeutic approaches for dCSCs.
Materials and methods
Cell cultures
Cell lines were maintained in Dulbecco’s modified
Eagle’s medium (DMEM; Nacalai Tesque, Kyoto, Japan) supplemented
with 10% fetal bovine serum (FBS) at 37°C in a 5% humidified
CO2 atmosphere.
Flow cytometric analysis and cell
sorting
The antibodies used were purchased from BD
Biosciences (Tokyo, Japan). In brief, cells were harvested with
trypsin and EDTA. Doublet cells were eliminated using FSC-A/FSC-H
and SSC-A/SSC-H. Dead and dying cells were eliminated with 7-AAD
(BD Pharmingen, San Jose, CA, USA). Isotype controls (BD
Biosciences) were used. FcR blocking was performed using an FcR
blocking reagent (Miltenyi Biotec, Bergisch Gladbach, Germany).
RNA
Total-RNA was extracted using TRIzol reagent
(Invitrogen/Life Technologies Japan, Tokyo, Japan). Reverse
transcription was performed with SuperScript III reverse
transcription kit (Invitrogen). qPCR was performed using the
LightCycler TaqMan Master Kit (Roche Diagnostics, Tokyo, Japan) for
cDNA amplification of target-specific genes. Purified cDNA from
mouse ES cells was used as a positive control for target genes. The
expression of mRNA copies was normalized to GAPDH (for mRNA) or
RNU48 (for miR) expression, as indicated. The RNA samples were
analyzed using SurePrint G3 Human GE 8×60K Microarray and the Human
miRNA Microarray 8×15K Rel.12.0 (Takara, Kyoto, Japan).
Statistical analysis
For continuous variables, results are expressed as
means ± SE. The relationship between the gene expression level and
cell count was analyzed by chi-square and Wilcoxon rank tests. All
data were analyzed using JMP software (SAS Institute, Cary, NC,
USA). P-values of <0.05 were considered statistically
significant.
Results and Discussion
A study of
CD13+/CD90− as dCSCs
CD13+/aminopeptidase N is expressed in
liver CSCs (5), where it is
involved in the reduction of ROS through the glutathione reductase
pathway. Considering that another independent study has shown
CD90+ as a candidate stem marker, critical to
tumorigenicity in mice in vivo and clinical outcomes of
patients (10), we indicated that
CD13+/CD90− cells exist in dormant phase of
cell cycle, whereas CD13+/CD90+ cells are
predominantly in the S phase and CD13−/CD90+
cells are in the G2/M phase (5).
In previous studies, we have elucidated that following exposure to
genotoxic insults, such as chemotherapy or radiation therapy,
CD13− cells shift to the CD13+ fraction in
dormant phase of cell cycle. In dCSCs, double stranded breaks
(DSBs) are repaired predominantly through the error-prone NHEJ
mechanism (6,7). In sharp contrast, the high-fidelity
HR-type repair proteins are increased in non-dormant CSCs compared
with NHEJ proteins, of which cells are usually sensitized through
chemoradiation therapy (6,7). Thus, after chemoradiation therapy,
NHEJ supposedly contributes to the generation of misrepair after
DSBs, which may cause chromosomal deletions, insertions, or
translocations, and subsequent genomic instability (11). Such genomic alterations lead to the
inactivation of tumor suppressor genes and activation of oncogenes,
which become more apparent during tumor development of primary
lesions, recurrence and metastasis (12). Nevertheless, there remains an
important issue to be addressed, i.e., the identification of
molecular mechanisms fundamental for initiation and development of
tumor tissues composed of stem cell hierarchy, and moreover, the
type of mechanism involved in CSC-based heterogeneous tumors. We
began with transcriptome assessment, i.e., the expression of mRNAs
and miRs and their association with
CD13+/CD90− cells in supporting or
maintaining CSC survival in the absence of genotoxic stimuli, which
may be beneficial in the study of the basal situation and may help
understand the differences between therapy-resistant clones and
de novo tumor-initiating cells.
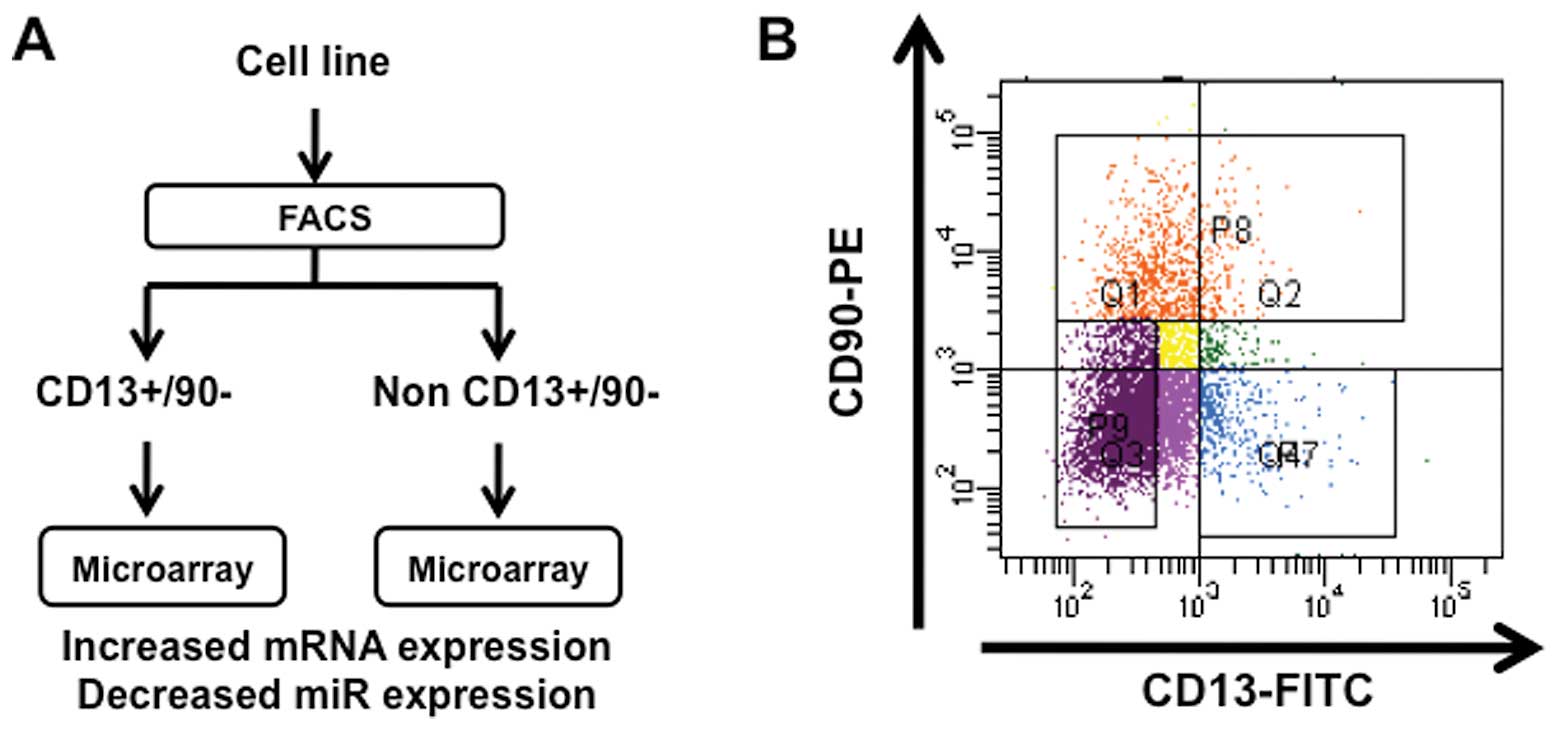
As shown in Fig. 1,
we separated liver cancer cells by FACS sorting into
CD13+CD90− dCSCs from other non-CSCs.
Considering that miRs play a role in the regulation of mRNA in its
stability and translation as an inhibitory regulation system, we
focused on increased expression of mRNA clones and decreased
expression of miR clones. The data of high-density array screening
indicated 17 clones of increased expression in
CD13+CD90− populations compared with unsorted
cells with more than 2-fold significant increase. The data were
almost consistent in CD13+/CD90− populations
compared with non-CD13+/CD90− cells (Table I). Next, we analyzed miR expression
and successfully isolated nine miRs in
CD13+/CD90− population compared with unsorted
cells with more than 4-fold significant decrease. The data were
almost consistent in CD13+/CD90− populations
compared with non-CD13+/CD90− cells (Table II).
 | Table ImRNAs expressed highly in
CD13+CD90− PLC cells. |
Table I
mRNAs expressed highly in
CD13+CD90− PLC cells.
| mRNA |
CD13+CD90−/unsorted |
CD13+CD90−/non-CD13+CD90− |
|---|
| Cadherin 6, type 2,
K-cadherin (CDH6) | 3.33 | 3.21 |
| Interleukin 8
(IL8) | 3.27 | 1.50 |
| Aldehyde
dehydrogenase 1 family, member A3 (ALDH1A3) | 3.26 | 2.61 |
| Endothelin 1
(EDN1) | 3.20 | 1.16 |
| Cytochrome P450,
family 1, subfamily B, polypeptide 1 (CYP1B1) | 3.16 | 2.06 |
| Tumor necrosis
factor, α-induced protein 6 (TNFAIP6) | 3.13 | 1.83 |
| Chemokine (C-X-C
motif) ligand 1 (CXCL1) | 3.08 | 1.25 |
| Vascular cell
adhesion molecule 1 (VCAM1) | 2.94 | 2.49 |
| L1 cell adhesion
molecule (L1CAM) | 2.72 | 2.40 |
| Wingless-type MMTV
integration site family member 2 (WNT2) | 2.66 | 1.10 |
| Carcinoembryonic
antigen-related cell adhesion molecule 3 (CEACAM3) | 2.65 | 1.99 |
| Chemokine (C-X-C
motif) ligand 3 (CXCL3) | 2.29 | 1.79 |
| Chemokine (C-C
motif) ligand 20 (CCL20) | 2.24 | 2.44 |
| Chemokine (C-X-C
motif) ligand 12 (CXCL12) | 2.11 | 1.53 |
| Tumor necrosis
factor (TNF superfamily, member 2) (TNF) | 2.07 | 1.59 |
| Chemokine (C-X-C
motif) ligand 5 (CXCL5) | 2.04 | 1.52 |
| Wingless-type MMTV
integration site family, member 7B (WNT7B) | 2.02 | 1.16 |
| Table IImiRs expressed highly in
CD13+CD90− PLC cells. |
Table II
miRs expressed highly in
CD13+CD90− PLC cells.
| miR |
CD13+CD90−/unsorted | Non
CD13+CD90−/unsorted | Function |
|---|
| hsa-miR-374a | −8.24 | 0.19 | Downregulated upon
cisplatin exposure |
| hsa-miR-489 | −7.17 | 0.41 | miR-489 inhibited
cell growth in all head and neck cancer cell lines |
| hsa-miR-223 | −6.69 | 0.16 | Reduced miR-223
expression in primary MEF leads to increased Fbw7 expression and
decreased cyclin-E activity |
| hsa-miR-101 | −6.68 | −0.21 | miR-101 could
sensitize hepatoma cell lines to both serum starvation- and
chemotherapeutic drug-induced apoptosis. Genomic loss of miR 101
leads to overexpression of histone methyltransferase EZH2 in
cancer |
| hsa-miR-9 | −6.29 | 0.70 | Directly repress
Lin28 |
| hsa-miR-378 | −6.21 | −0.06 | Novel target of the
c-Myc oncoprotein that is able to cooperate with activated Ras or
HER2 to promote cellular transformation |
| hsa-miR-182 | −6.14 | −0.68 | Antagonizing
miR-182 enhances BRCA1 protein levels and protects them from
IR-induced cell death |
| hsa-miR-421 | −5.69 | 0.20 | Overexpression of
miR-421 in pancreatic cancer cells promoted cell proliferation and
colony formation |
|
hsa-miR-125a-3p | −4.99 | 0.01 | Hypoxia regulated
microRNA |
Identification of regulatory
networks
By assessment of pairs of miRs and its putative
target mRNAs using prediction software (http://www.targetscan.org/; http://www.microrna.org/microrna/home.do), we
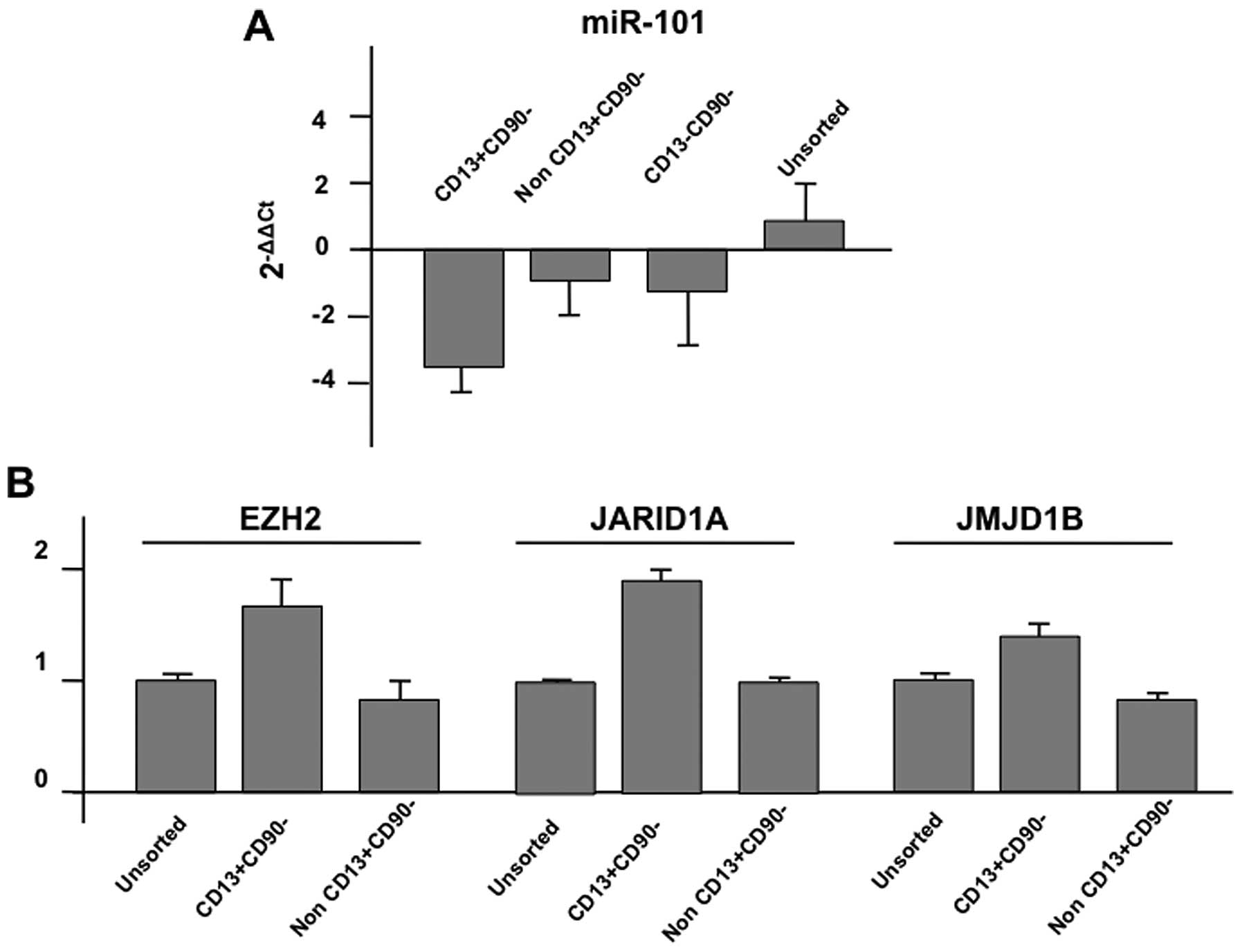
confirmed the data of the array by quantitative PCR. As shown in
the representative data, the expression of miR-101 was
downregulated in CD13+/CD90− cells compared
with non-CD13+/CD90− cells or
CD13−/CD90− cells; in sharp contrast, the
expression of putative targets, EZH2 (enhancer of zeste homolog 2;
http://www.genecards.org/cgi-bin/carddisp.pl?gene=EZH2&search=EZH2),
JARID1A; (http://www.genecards.org/cgi-bin/carddisp.pl?gene=KDM5A&search=JARID1A),
and JMJD1B (http://www.genecards.org/cgi-bin/carddisp.pl?gene=KDM3B&search=JMJD1B)
were increased (Fig. 2; summarized
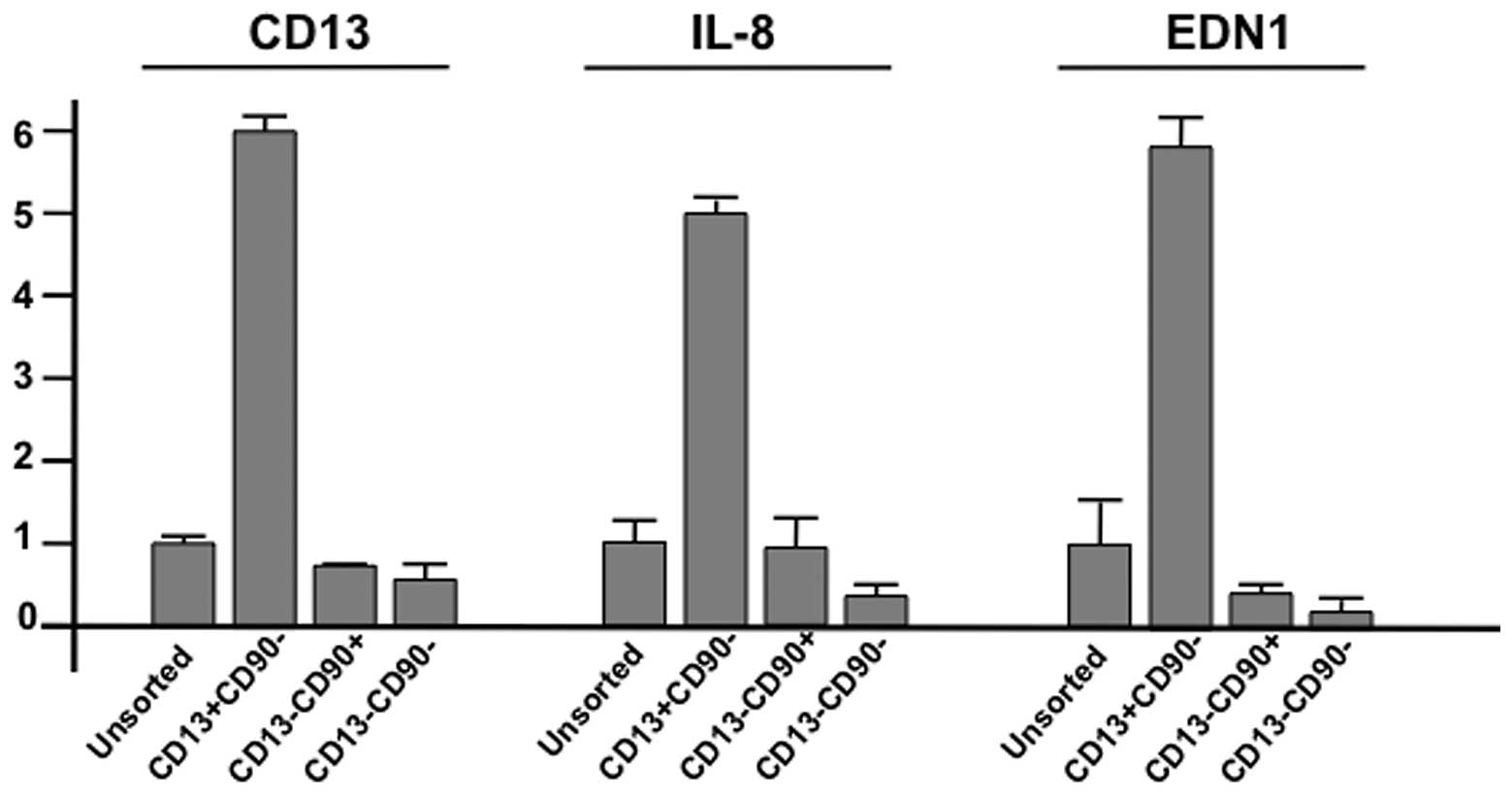
in Fig. 3). CD13 mRNA expression
was increased in CD13+/CD90− cells, but not
in CD13−/CD90+,
CD13−/CD90−, or unsorted cells. The
expression of interleukin-8 (IL-8; http://www.genecards.org/cgi-bin/carddisp.pl?gene=IL8&search=Interleukin+8)
and endothelin 1 (EDN1; http://www.genecards.org/cgi-bin/carddisp.pl?gene=EDN1&search=Endothelin+1)
was increased in CD13+/CD90− cells, but not
in others (Fig. 3; summarized in
Fig. 3). Through this study, we
were able to find at least three core regulatory networks for the
maintenance of dormant CD13+/CD90− cells, but
not in others, i.e., miR-182 pathways, miR-101 pathways,
cytokines/chemokines pathways (IL-8, CXCL5 [http://www.genecards.org/cgi-bin/carddisp.pl?gene=CXCL5&search=CXCL5],
CCL-20 [http://www.genecards.org/cgi-bin/carddisp.pl?gene=CCL20&search=CCL-20]),
growth factors and their receptor pathways [EDN1, Wnt2 (http://www.genecards.org/cgi-bin/carddisp.pl?gene=WNT2&search=Wnt2),
Wnt7B (http://www.gene-cards.org/cgi-bin/carddisp.pl?gene=WNT7B&search=Wnt7B),
and TNF (http://www.genecards.org/cgi-bin/carddisp.pl?gene=TNF&search=TNF)].
As noted in this study, we did not detect any apparent involvement
in DNA damage response machineries, except for an association
underlined by TP53INP1 (http://www.genecards.org/cgi-bin/carddisp.pl?gene=TP53INP1&search=TP53INP1)
and BRCA1 (http://www.genecards.org/cgi-bin/carddisp.pl?gene=BRCA1&search=BRCA1),
suggesting that (1) the damage
response in dCSCs is characteristic after exposure to genotoxic
stimuli, whereas in the absence of damage insults, they are not
apparent and (2) the DNA damage
response was regulated mainly by the modification of proteins such
as phosphorylation or ubiquitination, and the expression array
technology was less sensitive to pathway detection and other
networks may have been missed.
Significance of regulatory networks
(Fig. 4)
The EZH2 gene encodes a member of the Polycomb group
(PcG) family, which forms multimeric protein complexes involved in
maintaining a transcriptionally repressive state of genes over
successive cellular generations (13). Reportedly, the genomic loss of
miR-101, a putative tumor suppressor, leads to overexpression of
histone methyltransferase EZH2 in cancer (14,15),
hypoxia, and androgen-dependent conditions (16) as well as in gastric cancer
(17), pancreatic cancer (18), lung cancer (19) glioblastoma (20) and invasive squamous cell carcinoma
(21). Thus, PcG proteins are
critical epigenetic mediators of stem cell pluripotency and CSC
functions, which may be implicated in human cancer pathogenesis,
probably indicating candidacy for novel pharmacological targets of
cancer therapy. Recently, it was reported that the administration
of diflourinated-curcumin (CDF), a novel analogue of the turmeric
spice component curcumin, has antioxidant properties and inhibits
tumor growth through reduced expression of EZH2, Notch-1, CD44,
EpCAM, and Nanog and increased expression of let-7, miR-26a, and
miR-101 (18). These findings
indicated that CDF inhibited CSC growth by targeting an EZH2-miRNA
regulatory circuit for epigenetically controlled gene expression.
In the present study, we identified various miR-101 targets,
including JARID1A, JMJD1B, TP53INP1, and EZH2, suggesting that
these target molecules act together to maintain CSC dormancy, and
proposed the possible significance of the miR-101 pathway. We also
identified the miR-182 pathway, in which miR-182-mediated
downregulation of BRCA1 impacts DNA repair and sensitivity to
poly-ADP ribose polymerase (PARP) inhibitors (22). The overexpression of miR-182 was
reported in high-grade ovarian papillary serous carcinoma (23). Reportedly, aberrant miR-182
expression promotes melanoma metastasis by repressing FOXO3
(24). Taken together with our
study, the miR-182 pathway may be involved in the maintenance of
CSC function in a similar manner. As summarized in Fig. 4, we identified other cytokines,
chemokines, growth factors, receptors and pathways. These findings
may facilitate further studies on the regulatory mechanisms of the
dormant phase of gastrointestinal CSCs.
References
|
1.
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Visvader JE and Lindeman GJ: Cancer stem
cells in solid tumours: accumulating evidence and unresolved
questions. Nat Rev Cancer. 10:755–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Dewi DL, Ishii H, Kano Y, et al: Cancer
stem cell theory in gastrointestinal malignancies: recent progress
and upcoming challenges. J Gastroenterol. 46:1145–1157. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Li L and Clevers H: Coexistence of
quiescent and active adult stem cells in mammals. Science.
327:542–545. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Haraguchi N, Ishii H, Mimori K, et al:
CD13 is a therapeutic target in human liver cancer stem cells. J
Clin Invest. 120:3326–3339. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K
and Linn S: Molecular mechanisms of mammalian DNA repair and the
DNA damage checkpoints. Annu Rev Biochem. 73:39–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Nishikawa S, Ishii H, Haraguchi N, et al:
Genotoxic therapy stimulates error-prone DNA repair in dormant
hepatocellular cancer stem cells. Exp Ther Med. (In press).
|
|
8.
|
Haraguchi N, Ishii H, Nagano H, Doki Y and
Mori M: The future prospects and subject of the liver cancer stem
cells study for the clinical application. Gastroenterology. Feb
23–2011.(Epub ahead of print).
|
|
9.
|
Kim HM, Haraguchi N, Ishii H, et al:
Increased CD13 expression reduces reactive oxygen species,
promoting survival of liver cancer stem cells via an
epithelial-mesenchymal transition-like phenomenon. Ann Surg Oncol.
Aug 31–2011.(Epub ahead of print).
|
|
10.
|
Yang ZF, Ho DW, Ng MN, et al: Significance
of CD90+ cancer stem cells in human liver cancer. Cancer
Cell. 13:153–166. 2008.
|
|
11.
|
Weinstock DM, Richardson CA, Elliott B and
Jasin M: Modeling oncogenic translocations: distinct roles for
double-strand break repair pathways in translocation formation in
mammalian cells. DNA Repair (Amst). 5:1065–1074. 2006. View Article : Google Scholar
|
|
12.
|
Ishii H, Iwatsuki M, Ieta K, Ohta D,
Haraguchi N, Mimori K and Mori M: Cancer stem cells and
chemoradiation resistance. Cancer Sci. 99:1871–1877. 2008.
View Article : Google Scholar
|
|
13.
|
Chen H, Rossier C and Antonarakis SE:
Cloning of a human homolog of the Drosophila enhancer of zeste gene
(EZH2) that maps to chromosome 21q22.2. Genomics. 38:30–37. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Varambally S, Cao Q, Mani RS, et al:
Genomic loss of microRNA-101 leads to overexpression of histone
methyltransferase EZH2 in cancer. Science. 322:1695–1699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Friedman JM, Liang G, Liu CC, et al: The
putative tumor suppressor microRNA-101 modulates the cancer
epigenome by repressing the polycomb group protein EZH2. Cancer
Res. 69:2623–2629. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Cao P, Deng Z, Wan M, Huang W, et al:
MicroRNA-101 negatively regulates Ezh2 and its expression is
modulated by androgen receptor and HIF-1alpha/HIF-1beta. Mol
Cancer. 9:1082010. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Wang HJ, Ruan HJ, He XJ, et al:
MicroRNA-101 is down-regulated in gastric cancer and involved in
cell migration and invasion. Eur J Cancer. 46:2295–2303. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Bao B, Ali S, Banerjee S, Wang Z, et al:
Curcumin analogue CDF inhibits pancreatic tumor growth by switching
on suppressor microRNAs and attenuating EZH2 expression. Cancer
Res. 72:335–345. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Zhang JG, Guo JF, Liu DL, Liu Q and Wang
JJ: MicroRNA-101 exerts tumor-suppressive functions in non-small
cell lung cancer through directly targeting enhancer of zeste
homolog 2. J Thorac Oncol. 6:671–678. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Smits M, Nilsson J, Mir SE, et al: miR-101
is down-regulated in glioblastoma resulting in EZH2-induced
proliferation, migration, and angiogenesis. Oncotarget. 1:710–720.
2010.PubMed/NCBI
|
|
21.
|
Banerjee R, Mani RS, Russo N, et al: The
tumor suppressor gene rap1GAP is silenced by miR-101-mediated EZH2
overexpression in invasive squamous cell carcinoma. Oncogene.
30:4339–4349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Moskwa P, Buffa FM, Pan Y, et al:
miR-182-mediated downregulation of BRCA1 impacts DNA repair and
sensitivity to PARP inhibitors. Mol Cell. 41:210–220. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Liu Z, Liu J, Segura MF, et al: MiR182
overexpression in tumorigenesis of high-grade ovarian papillary
serous carcinoma. J Pathol. Feb 9–2012.(Epub ahead of print).
|
|
24.
|
Segura MF, Hanniford D, Menendez S, et al:
Aberrant miR-182 expression promotes melanoma metastasis by
repressing FOXO3 and microphthalmia-associated transcription
factor. Proc Natl Acad Sci USA. 106:1814–1819. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Dewi DL, Ishii H, Haraguchi N, et al:
Reprogramming of gastrointestinal cancer cells. Cancer Sci.
103:393–399. 2012. View Article : Google Scholar
|