Introduction
Breast cancer is the most frequently diagnosed
cancer in women and is one of the leading causes of cancer
mortality worldwide (1).
Approximately 4,000 new cases of female breast cancer are diagnosed
and approximately 1,000 women succumb to the disease each year in
Korea (2). Candidate molecular
pathways of cancer therapy emphasize signaling networks that
control cell proliferation or survival (3,4).
However, the involvement of signaling networks in breast cancer
therapy is not clearly understood. There is an urgent need to
develop new strategies for breast cancer therapy.
Recently, certain studies demonstrated that
autophagic cell death plays an important role in regulating cell
death in breast cancer cells with acquired-resistance to various
treatments (5–7). Autophagy is a catabolic pathway
whereby cytoplasmic proteins and organelles are sequestered in
vacuoles and delivered to lysosomes for degradation and recycling
(8). The induction of autophagy
has also been observed in malignant cells following treatment with
histone deacetylase (HDAC) inhibitors. HDACs comprise a superfamily
of proteins involved in a wide range of cellular functions,
including regulation of transcription, cell proliferation and cell
death (9,10). HDACs are divided into four classes
(I–IV) and regulate the expression and activity of numerous
proteins involved in cancer. Sirtuins (also known as SIRTs) are
NAD+-dependent class III HDACs (11). In mammals, seven SIRT homologues
have been identified that primarily possess HDACs (SIRT1, SIRT2,
SIRT3 and SIRT5) or monoribosyltransferase activity (SIRT4 and
SIRT6), which target histone and various non-histone proteins in
distinct subcellular locations (12,13).
As SIRT1 blocks senescence, cell differentiation and
stress-induced apoptosis, and promotes cell growth, angio genesis
and vasodilation (14), SIRT1
overexpression can enhance tumor growth and promote cell survival
in response to stress and drug resistance; moreover, SIRT1 is
upregulated in a spectrum of cancers (15,16).
SIRT1 can induce chromatin silencing through the deacetylation of
histones H1, H3 and H4 (17) and
can modulate cell survival by regulating the Ku autoantigen, Ku70
(18), NF-κB (19), FOXO proteins (20,21)
and p300 (22). The putative role
of SIRT1 in cancer biology was first postulated when p53 was
identified as a direct substrate (23). Acetylated Lys382 within p53 has
been identified as a direct target of SIRT1, whereby p53
deacetylation has been shown to induce apoptosis. Therefore, SIRT1
has been implicated in the initiation and progression of various
malignancies.
Conversely, the specific SIRT1 inhibitor, sirtinol,
does not inhibit class I and II HDACs (24,25).
The first known SIRT inhibitors can be classified into two groups:
substances that inhibit NAD+-dependent reactions in
general, such as nicotinamide (26,27)
and SIRT-specific inhibitors, such as spitomicin (24), sirtinol (28), cambinol (29), dihydrocoumarin (30) and certain indoles (31). Their common feature is that they
have antitumor properties; however, their molecular mechanisms of
action vary and are not yet fully understood.
In the present study, we focused on the anticancer
effect of sirtinol, a molecule which has a potent inhibitory effect
on SIRT1. We also focused on the effect of sirtinol on the tumor
suppressor, p53, and its acetylation status. The role of
acetylation as a protein post-translational modification,
independent of histone modification, may also play a critical role
in cell fate and thus, tumorigenesis. Additionally, the anticancer
effects of sirtinol on cell viability, cell cycle regulation and
modulation of apoptosis- and autophagy-related molecules were
investigated.
Materials and methods
Reagents
Sirtinol was purchased from Sigma-Aldrich (St.
Louis, MO, USA). Culture medium and its supplements including
antibiotics and fetal bovine serum (FBS) were purchased from Gibco
Invitrogen Corp. (Carlsbad, CA, USA). Primary antibodies against
Atg5, Atg7, β-actin, beclin-1, cleaved caspase 7, caspase 7,
cleaved caspase 9, Cdc2, cyclin A, cyclin B1, cyclin D1, cyclin E,
cytochrome c, HDACs, LC3B and p21, and horseradish
peroxidase-conjugated secondary antibody were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Bax, Bcl-2, Cdk4,
Cdk6, Cdk2, histone H1, poly(ADP-ribose) polymerase (PARP), p53,
Ac-p53, and p27 were purchased from Santa Cruz Biotechnology (Santa
Cruz, CA, USA); The Annexin V-FITC apoptosis detection kit I was
from BD Biosciences (San Diego, CA, USA). All other chemicals were
purchased from Sigma-Aldrich. Sirtinol was dissolved in dimethyl
sulfoxide (DMSO) and stored at −20°C until use. Sirtinol was
diluted to the appropriate concentrations in culture medium
containing 1% FBS. The final concentration of DMSO was <0.1%
(vol/vol), and was also present in the corresponding control.
Cell lines and culture medium
MCF-7 human breast cancer cells were purchased from
the American Type Culture Collection (Manassas, VA, USA). The cells
were grown in Dulbecco’s modified Eagle’s medium (Gibco, Rockville,
MD, USA) containing 10% heat-inactivated FBS. Cells were maintained
as monolayers in a humidified atmosphere containing 5%
CO2 at 37°C and the culture medium was replaced every
two days. After 48 h of incubation, the culture medium was replaced
with treatment medium containing the desired concentrations of
chemicals.
Cell viability assay
Cell viability was determined using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT,
5 mg/ml, Sigma). The cultures were initiated in 96-well plates at a
density of 2.5×103 cells per well. After 48 h of
incubation, the cells were treated with various concentrations of
sirtinol and cultured for 24 or 48 h. At the end of the treatment
period, 100 μl of 1X MTT reagent were added to each well and
incubated for 4 h at 37°C in the dark. After incubation, the
supernatant was aspirated and formazan crystals were dissolved in
100 μl of DMSO at 37°C for 15 min with gentle agitation. The
absorbance per well was measured at 540 nm using the VersaMax
Microplate reader (Molecular Devices Corp., CA, USA). Data from
three independent experiments were analyzed and then normalized to
the absorbance of wells containing medium only (0%) and untreated
cells (100%). IC50 values were calculated from sigmoidal
dose-response curves using SigmaPlot 10.0 software.
Protein extraction and western blot
analysis
MCF-7 cells were treated with sirtinol (2, 10, or 50
μM) for 48 h. Cells were harvested by trypsinization and
washed twice with cold phosphate-buffered saline (PBS). For total
protein isolation, cells were suspended in PRO-PREP™ protein
extract solution (Intron, Seongnam, Korea) and protein
concentrations were measured using the protein assay kit (Bio-Rad,
Hercules, CA, USA) according to the manufacturer’s instructions.
Equivalent amounts of proteins were resolved and electrophoresed
using SDS-PAGE on a 6–15% gel. Gels were transferred to
polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica,
MA, USA) and membranes were blocked with blocking buffer (TNA
buffer containing 5% skim milk) for 1 h. Subsequently, the
membranes were incubated with primary antibodies at 4°C overnight.
After washing for 1 h with TNA buffer (10 mM Tris-Cl, pH 7.6, 100
mM NaCl and 0.5% Tween-20), the membranes were incubated with
horseradish peroxidaseconjugated anti-mouse or anti-rabbit antibody
(1:10,000, Santa Cruz Biotechnology) for 30 min at room temperature
and then washed for 1 h with TNA buffer. The blots were developed
using an enhanced chemiluminescence (ECL)-plus kit (Amersham
Biosciences, Buckinghamshire, UK).
Flow cytometry analysis
The cells were treated with various concentrations
of sirtinol (2, 10, or 50 μM) for 48 h. The total number of
cells, including the ones in suspension and those adhered on the
walls, were harvested separately for sub-G1 or other cell cycle
stages, respectively, and washed in 1% bovine serum albumin (BSA)
before fixing in 95% ice-cold ethanol containing 0.5% Tween-20 for
1 h at −20°C. The cells (1×106) were washed in 1% BSA,
stained with cold propidium iodide (PI) staining solution (10
μg/ml PI and 100 μg/ml RNase in PBS) and incubated in
the dark for 30 min at room temperature. Data acquisition and
analysis were carried out using a flow cytometry system
(Becton-Dickinson, San Jose, CA, USA).
4′,6-Diamidino-2-phenylindole (DAPI)
staining
Morphological changes in the nuclear chromatin of
apoptotic cells were identified by staining with the DNA binding
dye DAPI. Cells were grown in 6-well plates at a density of
1×105 cells per well followed by the desired treatment.
After 48 h of incubation, the cells were washed with cold PBS,
fixed with methanol for 30 min, rewashed and stained with 200
μl of DAPI solution (1 μg/ml) at 37°C for 30 min.
After the staining solution was removed, the apoptotic cells were
visualized using a fluorescence confocal microscope at ×400
magnification.
Annexin V-FITC binding assay
The Annexin V-FITC binding assay was performed
according to manufacturer’s instructions using the Annexin V-FITC
detection kit I (BD Biosciences). The cells were treated with
sirtinol (2, 10, or 50 μM) for 48 h. The total number of
cells was counted by trypsinization and washed twice with cold PBS.
The cell pellet was resuspended with 100 μl of binding
buffer at a density of 1×103 cells per ml and incubated
with 5 μl of FITC-conjugated Annexin V and 5 μl of PI
for 15 min at room temperature in the dark. A total of 400
μl of 1X binding buffer was added to each sample tube and
the samples were immediately analyzed by FACSCalibur
(Becton-Dickinson).
Caspase activity analysis
The cultures were initiated in 6-well plates at a
density of 1×105 cells per well. Cells were allowed to
attach for 48 h and exposed to SAHA for 48 h. Caspase 8 and caspase
9 activities in the cell lysate were measured using colorimetric
assay kits (Biovision Ins., CA, USA) as described in the
manufacturer’s protocol. The kits used in this study utilized
synthetic tetrapeptides labeled with p-nitroanilide (pNA).
Briefly, fifty microliters (100 μg) of cell lysate were
incubated with 50 μl of 2× reaction buffer and 2 μl
of IETD-pNA for capase 8 or LEHD-pNA for caspase 9 at 37°C for 2 h.
A reading was then taken from a spectrophotometer at 405 nm with a
VERS Amax Microplate Reader (Molecular Devices Corp.). In case of
caspase 8 activity, a reading was taken from a fluorescence
microtiter plate reader. Caspase 7 activities in the cell lysate
were measured using caspase 7 immunoassay kits (Biovision Ins., CA,
USA) as described in the manufacturer’s protocol. Briefly, the
assay utilizes caspase 7 polyclonal antibody to capture activated
caspase 7 from cell lysates. Substrate DEVD-AFC is then added that
is cleaved proportionally to the amount of activated caspase 7 in
the cell lysate. The cleavage generates free AFC which is then
analyzed fluorometrically (Ex./Em. = 400/505 nm) using a
fluorescence plate reader. The assay ensures absolute specific
detection of caspase 7. Other known caspases and non-specific
proteases are not detected.
Acridine orange staining
MCF-7 cells were seeded in T-25 flasks and at 70%
confluence the cells were treated with sirtinol for 48 h. At the
appropriate time-points, cells were incubated with acridine orange
(1 μg/ml) in serum-free medium at 37°C for 15 min. The
acridine orange was removed and fluorescent micrographs were
obtained using an inverted fluorescence microscope (Olympus FV10i;
Olympus, Tokyo, Japan). The cytoplasm and nucleus of the stained
cells fluoresced bright green, whereas the acidic autophagic
vacuoles fluoresced bright red. Cells were treated with 200 nmol/l
bafilomycin A1 for 30 min before the addition of acridine orange to
inhibit the acidification of autophagic vacuoles. To quantify the
development of acidic vesicular organelles (AVOs), sirtinol-treated
or control cells were stained with acridine orange (1 μg/ml)
for 15 min and removed from the plate with trypsin-EDTA, and
collected in phenol red-free growth medium. Green (510–530 nm) and
red (650 nm) fluorescence emission from 1×104 cells
illuminated with blue (488 nm) excitation light were measured with
a FACSCalibur.
Monodansylcadaverine (MDC) incorporation
assay
Autophagic vacuoles were also detected with MDC by
incubating the cells with MDC (50 μmol/l) in PBS at 37°C for
10 min. After incubation, the cells were washed four times with
cold PBS and fixed with 3.75% paraformaldehyde in PBS. The cells
were immediately analyzed by fluorescence microscopy using an
inverted microscope (Olympus FV10i) equipped with a filter system
(excitation wavelength, 380 nm; emission filter, 525 nm).
Results
Sirtinol suppresses the growth of MCF-7
breast cancer cells
We first assessed the cytotoxicity of sirtinol in
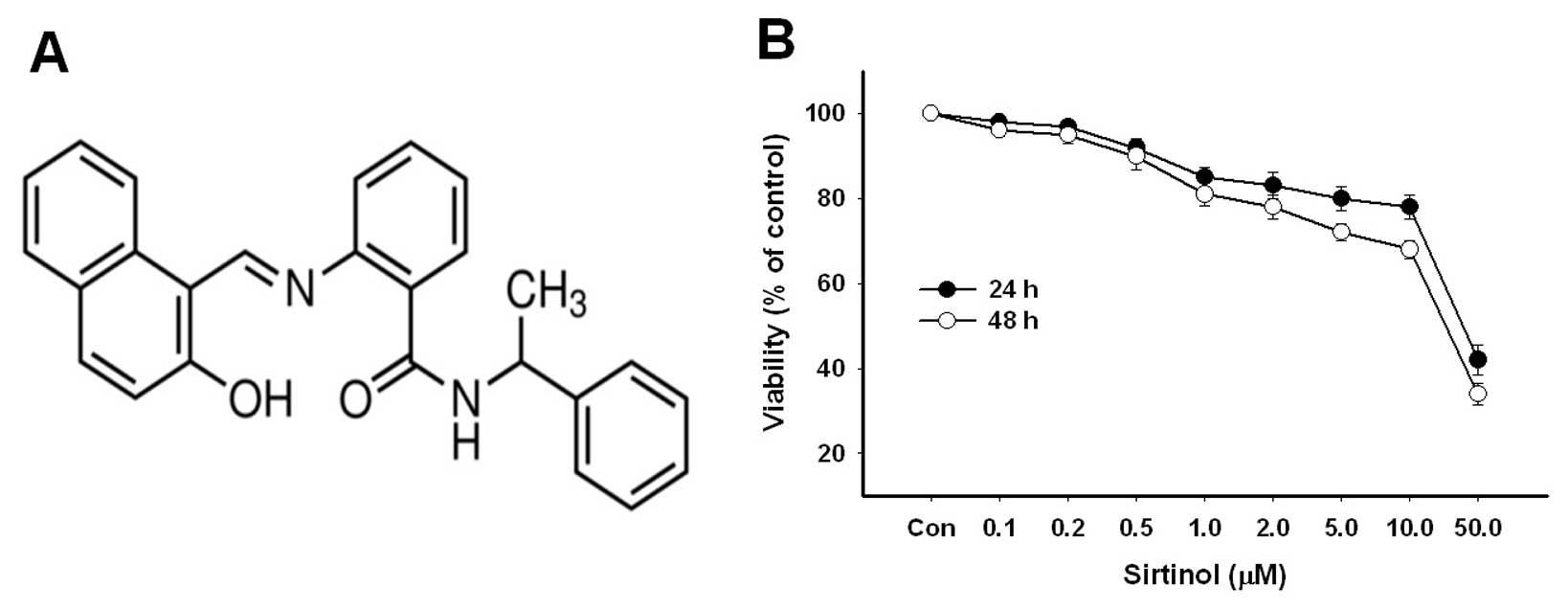
MCF-7 cells by using the MTT assay. The chemical structure of
sirtinol is shown in Fig. 1A.
Sirtinol significantly reduced the growth of MCF-7 cells in a
concentration- and time-dependent manner. The IC50
values of sirtinol were 48.6 μM and 43.5 μM after 24
and 48 h of treatment, respectively (Fig. 1B).
Sirtinol decreases SIRT protein
expression in MCF-7 cells
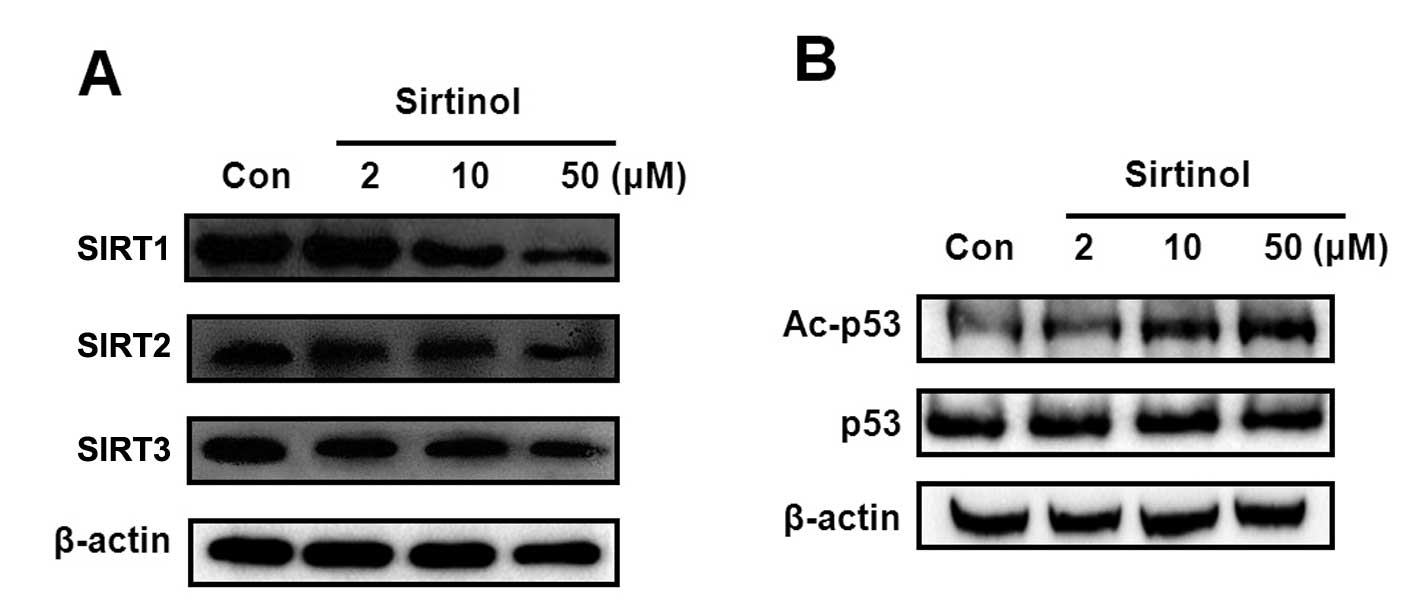
The expression levels of SIRT1, SIRT2 and SIRT3 were
measured by western blot analysis. The experiment revealed that
sirtinol significantly decreased SIRT1 expression (Fig. 2A). Mammalian SIRT1 is the direct
homologue of yeast SIRT2 and belongs to the
NAD+-dependent HDAC family and has been implicated as a
regulator of a variety of important biological processes, such as
aging, metabolism and stress resistance (32). To further investigate the distinct
effects of the SIRT inhibitor on cell fate, we measured the
acetylated status of the SIRT1/2 target, p53. Sirtinol
significantly increased the acetylated p53 level, whereas p53
acetylation was also accompanied by an induction of p53 stability
(Fig. 2B).
Sirtinol affects cell cycle regulation in
MCF-7 cells
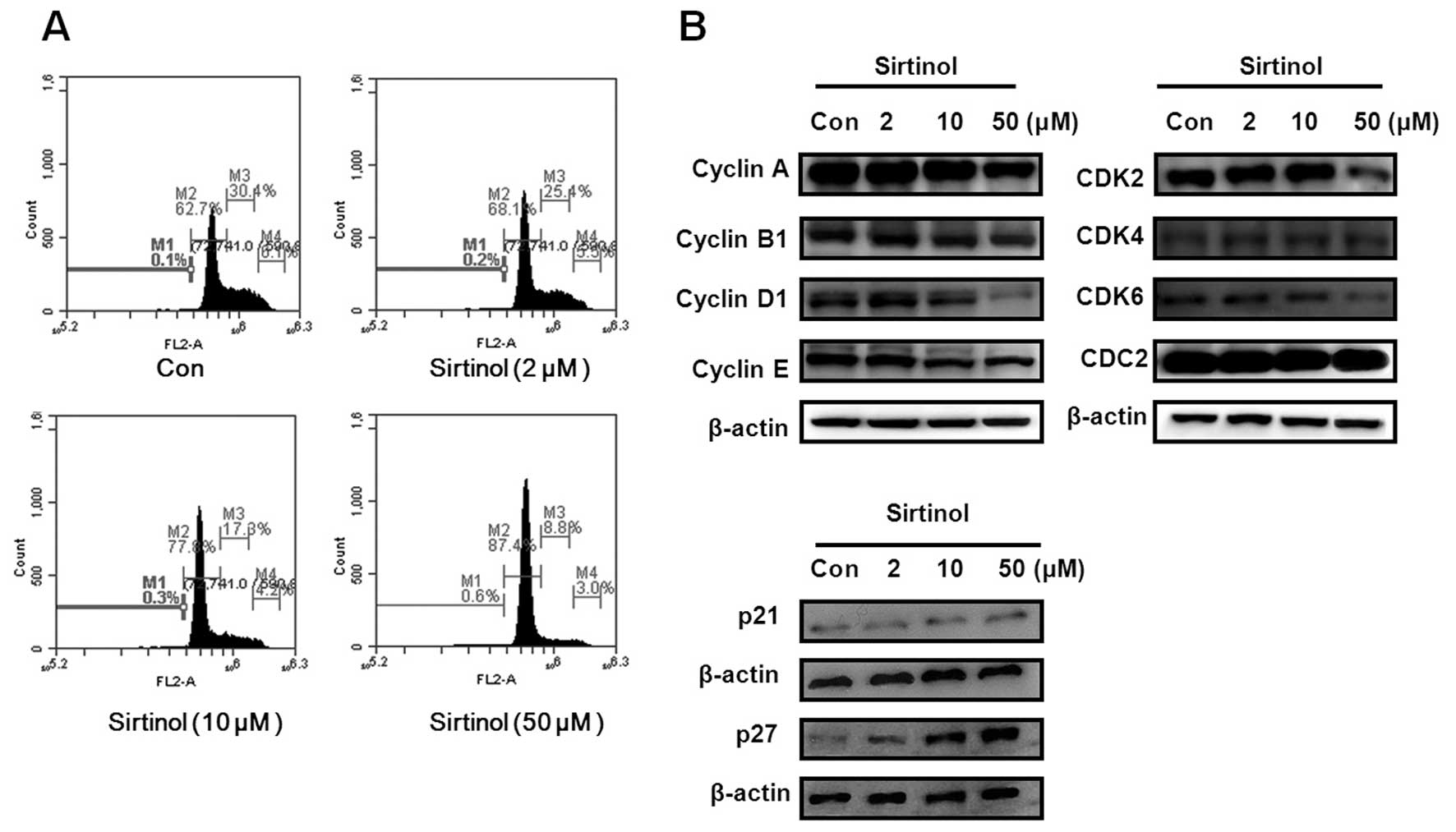
To confirm that sirtinol affects cell cycle
regulation in MCF-7 cells, we measured the cell cycle distribution
using flow cytometry analyses. Sirtinol significantly induced G1
phase arrest at 48 h (Fig. 3A).
The percentage of the sub-G1 population, which is indicative of
apoptosis, increased after treatment with sirtinol in MCF-7 cells.
We also examined the expression levels of cell cycle-regulated
proteins by western blot analysis. Sirtinol significantly decreased
the expression of cyclin B1, cyclin D1, CDK2 and CDK6, indicating
that these molecules are closely associated with the G1 cell cycle
check-point. Furthermore, sirtinol significantly increased p21 and
p27 expression in MCF-7 cells (Fig.
3B).
Sirtinol induces apoptosis in MCF-7
cells
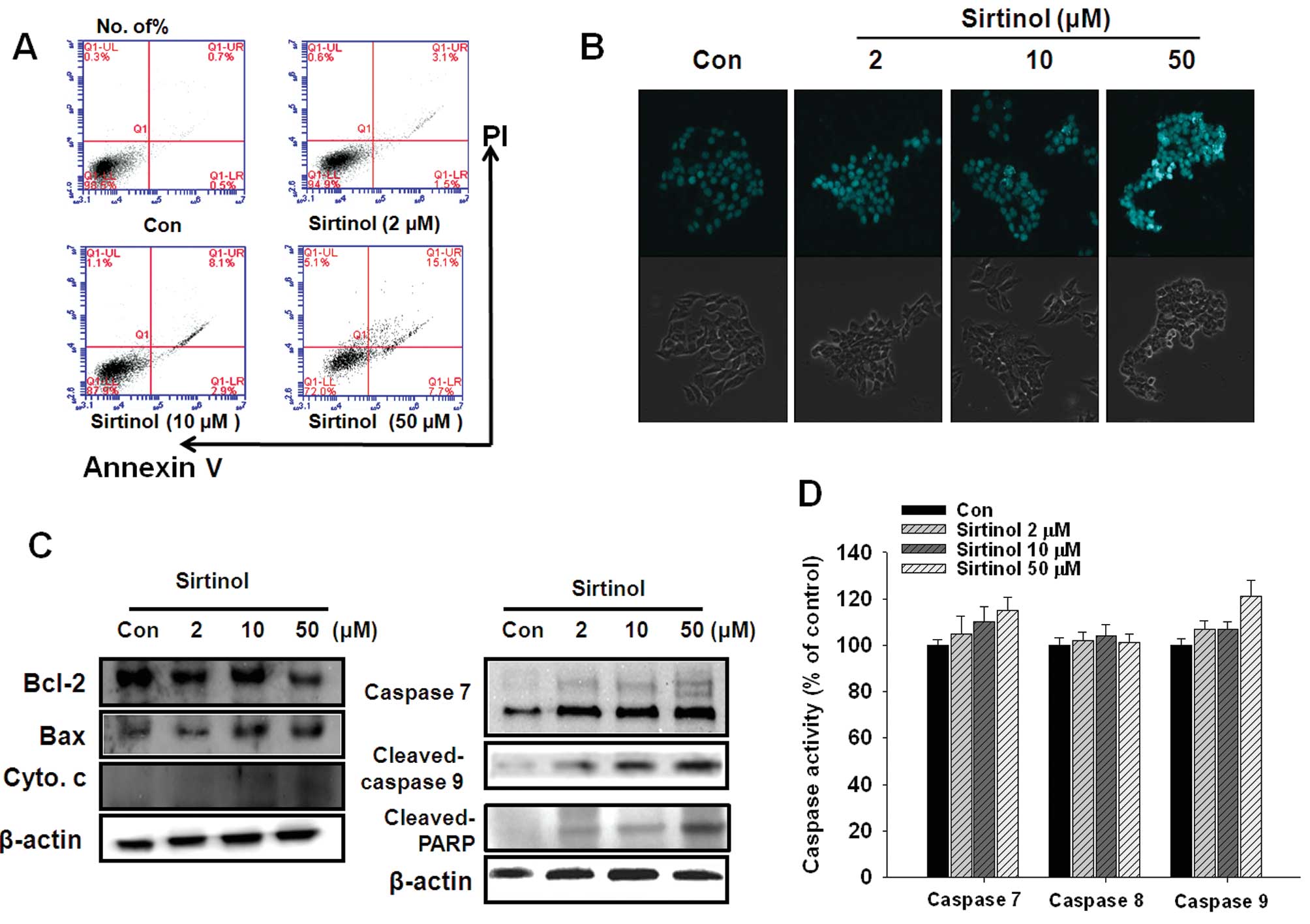
To elucidate the mechanism underlying the cytotoxic
effect of sirtinol on MCF-7 cells, apoptotic cell death was
measured by Annexin V-FITC assay, DAPI staining and western blot
analysis. Sirtinol (10 and 50 μM) significantly increased
the population of late-stage apoptotic cells (Fig. 4A). DAPI staining was used to
confirm the effect of sirtinol on apoptosis in MCF-7 cells.
Sirtinol increased the number of apoptotic nuclei (condensed or
fragmented chromatin) compared with the control culture, which
showed enhanced fluorescence staining with DAPI (Fig. 4B). To determine the apoptotic
pathway involved, we measured the expression of apoptosis-related
protein levels by western blot analysis. Sirtinol (50 μM)
significantly increased the cleavage of PARP, release of cytochrome
c and the expression of Bax, and decreased the expression of
Bcl-2 in MCF-7 cells (Fig. 4C).
The activities of caspase 7 and caspase 9 were slightly increased
in MCF-7 cells treated with sirtinol; however, caspase 8 activity
was not altered (Fig. 4D).
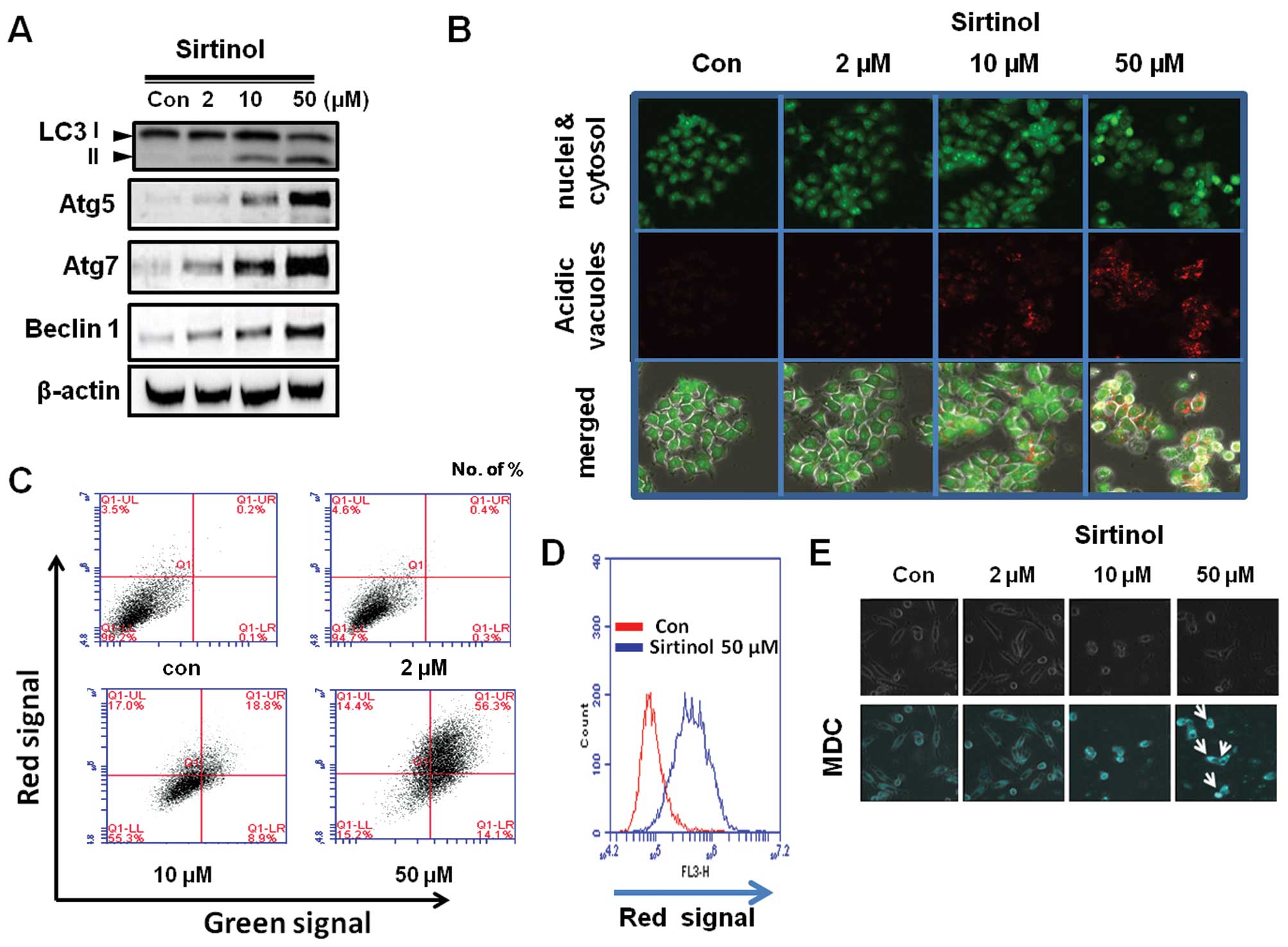
Sirtinol induces autophagy in MCF-7
cells
To evaluate autophagic cell death induced by
sirtinol, western blot analysis, acridine orange and MDC staining
were performed. The conversion of the soluble form of LC3-I to the
autophagic vesicle-associated form, LC3-II, is considered a
specific marker of autophagosome promotion. Sirtinol significantly
increased the level of LC3-II and decreased the unconjugated LC3-I
levels. Moreover, similar to the LC3-II expression pattern, the
level of beclin-1, known as Atg6, was increased by sirtinol
treatment (Fig. 5A).
The induction of autophagy was confirmed by acridine
orange and MDC staining. The vital dyes, acridine orange and MDC,
are commonly used to study autophagy. Acridine orange is a
lysotropic dye that accumulates in acidic organelles in a
pH-dependent manner. At a neutral pH, acridine orange is a
hydrophobic green fluorescent molecule. However, within acidic
vesicles, acridine orange becomes protonated and trapped within the
organelle and forms aggregates that emit bright red fluorescence
(33). MDC is another popular
autofluorescent marker that preferentially accumulates in
autophagic vacuoles. While acridine orange staining in lysosomes is
primarily due to ion trapping, MDC accumulation in auotphagic
vacuoles is due to a combination of ion trapping and specific
interactions with vacuole membrane lipids (34,35).
With acridine orange staining, control cells primarily exhibited
green fluorescence with minimal red fluorescence, indicating a lack
of AVOs; however, sirtinol-treated cells showed a fold-increase in
red fluorescent AVOs at 48 h (Fig.
5B). Flow cytometric analysis after acridine orange staining
also showed an increase in red fluorescence intensity with sirtinol
treatment, indicating an enhancement of AVOs (Fig. 5C). Histogram profiles show the mean
fluorescence intensity of the control and drug-treated cells
(Fig. 5D). Similar results were
observed with MDC staining (Fig.
5E). There was significant autophagic vesicle formation in
MCF-7 cells exposed to sirtinol. The morphological characteristics
demonstrated that sirtinol induced autophagy in MCF-7 cells.
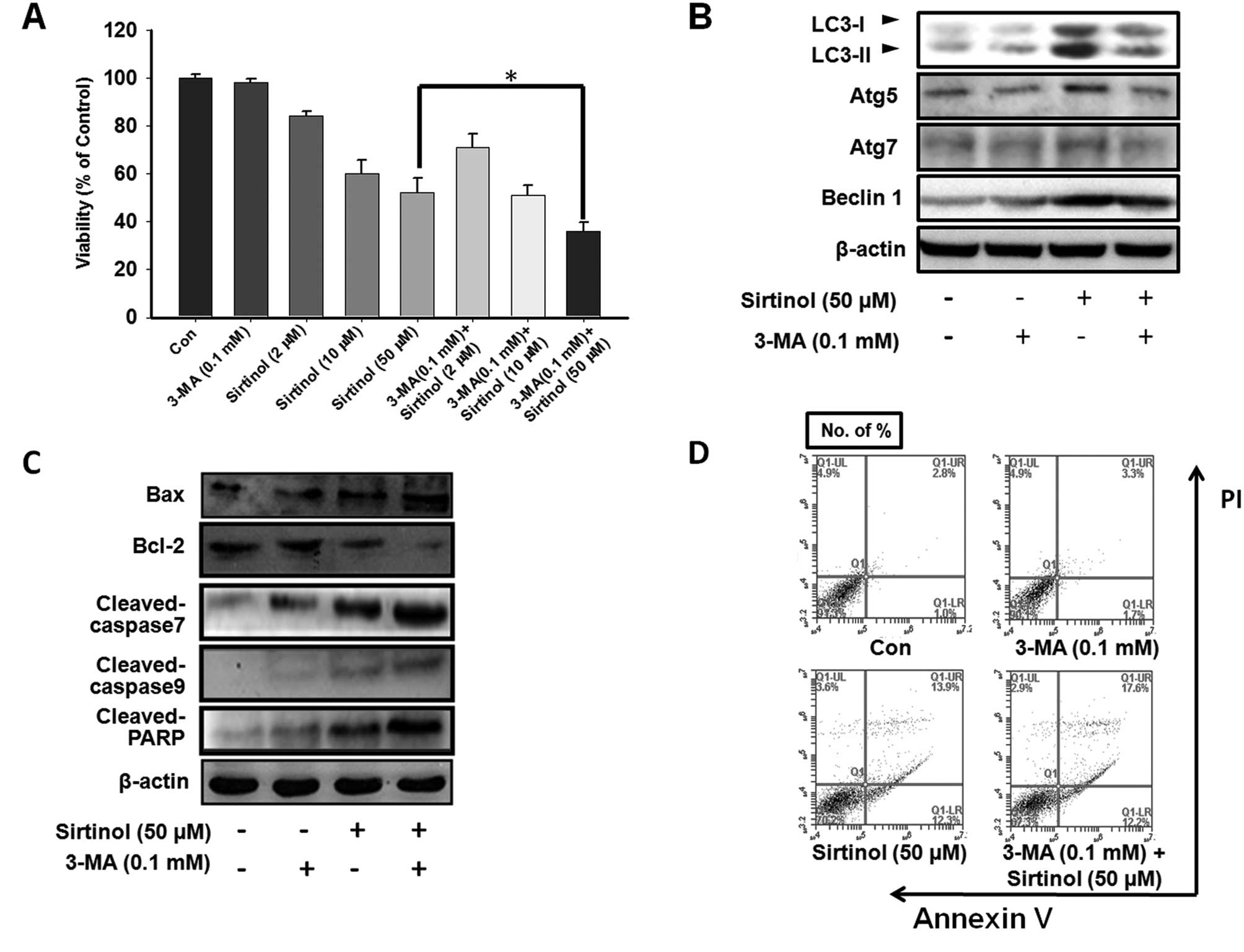
Inhibition of autophagy sensitizes the
sirtinol-induced apoptosis in MCF-7 cells
To explore whether the inhibition of autophagy
sensitizes MCF-7 cells to sirtinol-induced apoptotic cell death, we
used 3-methyladenine (3-MA), a well-known inhibitor of autophagy
(36). 3-MA blocks autopha gic
cell death by inhibiting phosphoinositide kinase-3 (PI3K), an
enzyme required for autophagy. 3-MA alone had no toxic effect on
MCF-7 cells. Following pre-treatment with 3-MA (0.1 mM), sirtinol
significantly reduced cell viability in a dose-dependent manner
(Fig. 6A). Western blot analysis
was performed to assess the expression levels of autophagy- or
apoptosis-related protein levels after 3-MA treatment. As shown in
Fig. 6B, LC3-II, beclin 1, Atg5
and Atg7 levels were decreased in MCF-7 cells pre-treated with 3-MA
compared with those in cells treated with sirtinol alone (Fig. 6B). In addition, 3-MA enhanced
sirtinol-induced apoptosis. As shown in Fig. 6C, sirtinol increased the Bax level
and decreased the Bcl-2 level in 3-MA pre-treated MCF-7 cells. The
expression levels of caspase 7 and caspase 9 were also altered by
sirtinol in the 3-MA-pre-treated MCF-7 cells (Fig. 6C). To confirm that apoptosis was
affected by the inhibition of autophagy, cells were subjected to
FITC-Annexin V/PI double-staining, followed by flow cytometric
analysis to quantify the apoptotic cell populations. Sirtinol
significantly increased the number of apoptotic cells among MCF-7
cells pre-treated with 3-MA (Fig.
6D).
Discussion
SIRTs could be one of the lost links between aging
and cancer (37). SIRT1 is the
most widely examined member of the SIRT family and is known to
modulate cell proliferation, differentiation, apoptosis, as well as
migration and invasion (13,14).
SIRT1 functions in controlling cellular senescence and specific
cancer cell types overexpress SIRT1 (15,16).
In this study, we investigated the hypothesis that SIRT is
overexpressed in breast cancer cells and that its inhibition would
have anticancer effects on human breast cancer. To determine the
mechanisms of action by which the SIRT inhibitor, sirtinol, exerts
its anticancer effects, we examined its action on apoptotic and
autophagic cell death pathways.
First, we measured cell viability and performed cell
cycle analysis. We found that sirtinol significantly increased
cytotoxicity in a concentration-dependent manner and significantly
induced G1 phase arrest in MCF-7 cells. SIRT1/2 expression was
significantly reduced by sirtinol treatment, although the effect on
SIRT1 expression was more pronounced. Previous studies have
demonstrated that class III HDAC inhibitors can also induce
senescent-like growth arrest in breast cancer cells (38). SIRT1 has a more prominent role in
controlling cell growth and survival as it exists in the same
intracellular compartments as most of the cell cycle and death
regulators (39). In this study,
we used MCF-7 breast cancer cells as they have substantial levels
of SIRT1, as well as functional p53, which is a target for
acetylation by SIRT1 and SIRT2. The tumor suppressor, p53, can
exert anti-proliferative effects, such as growth arrest, apoptosis
and cellular senescence, in response to various types of stressors.
As expected, sirtinol significantly increased the levels of
acetylated p53 in MCF-7 cells compared with the control culture.
Therefore, sirtinol reduced the SIRT1-mediated deacetylation of p53
and increased p53 transcription-dependent cell cycle arrest and
apoptosis in MCF-7 cells. These results are similar to those from a
previous report indicating that the inhibition of SIRT1 allows the
activation of p53 and BAX gene expression, which induces cell cycle
arrest and apoptosis (39).
To explore the mechanism responsible for the
anticancer effects of sirtinol, the apoptotic cell death were
assessed. Flow cytometric analysis revealed that sirtinol markedly
induced apoptosis and subsequently increased the sub-G1 phase cell
population. We investigated the exact downstream mechanism of
apoptotic cell deaths induced by sirtinol. Sirtinol increased
cytochrome c release into the cytoplasm, upregulated the
pro-apoptotic protein, Bax, downregulated the anti-apoptotic
protein, Bcl-2, and induced PARP cleavage in MCF-7 breast cancer
cells. These results were confirmed by Annexin V-FITC assay and
DAPI staining. However, while sirtinol potently induced apoptotic
cell death in MCF-7 cells, the results concerning autophagy were
more significant. Autophagy is becoming an important area of cancer
research. Autophagy plays a role in both the promotion and
prevention of cancer, and its role may be altered during tumor
progression. The inhibition of autophagy may allow the conti nuous
growth of pre-cancerous cells and autophagy can act as a suppressor
of cancer (26); then, as a tumor
grows, cancer cells may need autophagy to survive nutrient-limiting
and low-oxygen conditions, especially in the internal region of the
tumor that is poorly vascularized. In addition, autophagy may
protect certain cancer cells against ionizing radiation, possibly
by removing damaged macromolecules or organelles, such as
mitochondria (40). According to
our results, the autophagic process caused MCF-7 cell death
following sirtinol treatment. These results were confirmed by the
sirtinol induction of AVOs in the cytoplasm stained by acridine
orange and MDC. Likewise, increases in LC3-II levels and other
autophagy-related molecules were observed after sirtinol treatment
compared with the control cells and these results correlated
closely with their cytotoxic effects.
In truth, whether autophagy promotes cell death or
protects cancer cell survival is circumstantial. In this study,
sirtinol induced MCF-7 cell death. The inhibition of the early
stages of autophagy by the specific inhibitor, 3-MA, resulted in
accelerated apoptotic cell death, as revealed by Annexin V/PI
staining. Autophagy and apoptosis share many common inducers;
however, the current knowledge on the molecular intersections
between the autophagic and apoptotic pathways is incomplete and
fragmented. It may therefore be necessary to further elucidate the
relationship between autophagy and apoptosis following sirtinol
treatment in MCF-7 cells. In the present study, we found that
sirtinol simultaneously induced p53-mediated apoptosis and
caspase-independent autophagy in MCF-7 cells. The inhibition of
autophagy by 3-MA-sensitized cells to sirtinol-induced apoptotic
cell death, which suggests that the anticancer effects of sirtinol
are mainly the result of apoptosis.
Taken together, these results confirm that sirtinol,
which decreased the expression of SIRT1 in MCF-7 breast cancer
cells, induced cell death effectively, causing cell cycle arrest in
the G1 phase and apoptotic cell death, while at the same time
inducing autophagic cell death in MCF-7 cells. This evokes hope
that a strategy for cancer treatment may be developed based on
SIRTs inhibitors.
Acknowledgements
This study was supported by The Health
Fellowship Foundation grants funded by Yuhan Corporation.
References
|
1.
|
International Agency for Research on
Cancer (IARC): World Cancer Report. http://globocan.iarc.fr/factsheets/populations/factsheet.asp?uno=900,
2008.
|
|
2.
|
Yoo KY, Kang D, Park SK, et al:
Epidemiology of breast cancer in Korea: occurrence, high-risk
groups, and prevention. J Korean Med Sci. 17:1–6. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Esteve JM and Knecht E: Mechanisms of
autophagy and apoptosis: Recent developments in breast cancer
cells. World J Biol Chem. 26:232–238. 2011. View Article : Google Scholar
|
|
4.
|
Cook KL, Shajahan AN and Clarke R:
Autophagy and endocrine resistance in breast cancer. Expert Rev
Anticancer Ther. 11:1283–1294. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Aguilar H, Solé X, Bonifaci N, et al:
Biological reprogramming in acquired resistance to endocrine
therapy of breast cancer. Oncogene. 29:6071–6083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Yue W, Fan P, Wang J, Li Y and Santen RJ:
Mechanisms of acquired resistance to endocrine therapy in
hormone-dependent breast cancer cells. J Steroid Biochem Mol Biol.
106:102–110. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Notte A, Leclere L and Michiels C:
Autophagy as a mediator of chemotherapy-induced cell death in
cancer. Biochem Pharmacol. 82:427–434. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Glick D, Barth S and Macleod KF:
Autophagy: cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Blander G and Guarente L: The Sir2 family
of protein deacetylases. Annu Rev Biochem. 73:417–435. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Frye RA: Characterization of five human
cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins
(sirtuins) metabolize NAD and may have protein
ADP-ribosyltransferase activity. Biochem Biophys Res Commun.
260:273–279. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Liszt G, Ford E, Kurtev M and Guarente L:
Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J
Biol Chem. 280:21313–21320. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Liu T, Liu PY and Marshall GM: The
critical role of the class III histone deacetylase SIRT1 in cancer.
Cancer Res. 69:1702–1705. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Fraga MF and Esteller M: Epigenetics and
aging: the targets and the marks. Trends Gene. 23:413–418. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Lim CS: Human SIRT1: a protein biomarker
for tumorigenesis? Cell Bio Int. 31:636–637. 2007. View Article : Google Scholar
|
|
17.
|
Vaquero A, Scher MB, Lee DH, et al: SirT2
is a histone deacetylase with preference for histone H4 Lys 16
during mitosis. Genes Dev. 20:1256–1261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Jeong J, Juhn K, Lee H, et al: SIRT1
promotes DNA repair activity and deacetylation of Ku70. Exp Mol
Med. 39:8–13. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Yeung F, Hoberg JE, Ramsey CS, et al:
Modulation of NF-κB-dependent transcription and cell survival by
the SIRT1 deacetylase. EMBO J. 23:2369–2380. 2004.
|
|
20.
|
Brunet A, Sweeney LB, Sturgill JF, et al:
Stress-dependent regulation of FOXO transcription factors by the
SIRT1 deacetylase. Science. 303:2011–2015. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Motta MC, Divecha N, Lemieux M, et al:
Mammalian SIRT represses forkhead transcription factors. Cell.
116:551–563. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Bouras T, Fu M, Sauve AA, et al: SIRT1
deacetylation and repression of p300 involves lysine residues
1020/1024 within the cell cycle regulatory domain 1. J Bio Chem.
280:10264–10276. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Vaziri H, Dessain SK, Ng Eaton E, et al:
Hsir2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell.
107:149–159. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Bedalov A, Gatbonton T, Irvine WP,
Gottschling DE and Simon JA: Identification of a small molecule
inhibitor of Sir2p. Proc Natl Acad Sci USA. 98:15113–15118. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Grozinger CM, Chao ED, Blackwell HE,
Moazed D and Schreiber SL: Identification of a class of small
molecule inhibitors of the sirtuin family of NAD-dependent
deacetylases by phenotypic screening. J Biol Chem. 276:38837–38843.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Avalos JL, Bever KM and Wolberger C:
Mechanism of sirtuin inhibition by nicotinamide: altering the
NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol Cell.
17:855–868. 2005.
|
|
27.
|
Bitterman KJ, Anderson RM, Cohen HY,
Latorre-Esteves M and Sinclair DA: Inhibition of silencing and
accelerated aging by nicotinamide, a putative negative regulator of
yeast sir2 and human SIRT1. J Biol Chem. 277:45099–45107. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Ota H, Akishita M, Eto M, Iijima K, Kaneki
M and Ouchi Y: Sirt1 modulates premature senescence-like phenotype
in human endothelial cells. Mol Cell Cardiol. 43:571–579. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Heltweg B, Gatbonton T, Schuler AD, et al:
Antitumor activity of a small-molecule inhibitors of human silent
information regulator 2 enzymes. Cancer Res. 66:4368–4377. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Olaharski AJ, Rine J, Marshall BL, et al:
The flavoring agent dihydrocoumarin reverses epigenetic silencing
and inhibits sirtuin deacetylases. PLoS Genet. 1:e772005.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Napper AD, Hixon J, McDonagh T, et al:
Discovery of indoles as potent and selective inhibitors of the
deacetylase SIRT1. J Med Chem. 48:8045–8054. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Saunders LR and Verdin E: Sirtuins:
critical regulators at the crossroads between cancer and aging.
Oncogene. 26:5489–5504. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Millot C, Millot JM, Morjani H, Desplaces
A and Manfait M: Characterization of acidic vesicles in
multidrug-resistant and sensitive cancer cells by acridine orange
staining and confocal micro-spectrofluorometry. J Histochem
Cytochem. 45:1255–1264. 1997. View Article : Google Scholar
|
|
34.
|
De Duve C, de Barsy T, Poole B, Trouet A,
Tulkens P and van Hoof F: Commentary. Lysosomotropic agents.
Biochem Pharmacol. 23:2495–2531. 1974.
|
|
35.
|
Paglin S, Hollister T, Delohery T, et al:
A novel response of cancer cells to radiation involves autophagy
and formation of acidic vesicles. Cancer Res. 61:439–444.
2001.PubMed/NCBI
|
|
36.
|
Petiot A, Ogier-Denis E, Blommaart EF,
Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol
3-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem. 275:992–998. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Fraga MF, Agrelo R and Esteller M:
Cross-talk between aging and cancer: the epigenetic language. Ann
NY Acad Sci. 1100:60–74. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Mahlknecht U and Hoelzer D: Histone
acetylation modifiers in the pathogenesis of malignant disease. Mol
Med. 6:623–644. 2000.PubMed/NCBI
|
|
39.
|
Michishita E, Park JY, Burneskis JM,
Barrett JC and Horikawa I: Evolutionarily conserved and
nonconserved cellular localizations and functions of human SIRT
proteins. Mol Biol Cell. 16:4623–4635. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Arico S, Petiot A, Bauvy C, et al: The
tumor suppressor PTEN positively regulates macroautophagy by
inhibiting the phosphatidylinositol 3-kinase/protein kinase B
pathway. J Biol Chem. 276:35243–35246. 2001. View Article : Google Scholar : PubMed/NCBI
|